 Amandine L. Flourat
Amandine L. Flourat Aurélien A. M. Peru1
Aurélien A. M. Peru1 Arnaud Haudrechy
Arnaud Haudrechy Jean-Hugues Renault
Jean-Hugues Renault Florent Allais
Florent Allais
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 09 December 2019
Sec. Green and Sustainable Chemistry
Volume 7 - 2019 | https://doi.org/10.3389/fchem.2019.00842
This article is part of the Research Topic From Biomass to Advanced Bio-Based Chemicals & Materials: A Multidisciplinary Perspective View all 17 articles
To investigate lignin degradation, scientists commonly use model compounds. Unfortunately, these models are most of the time simple β-O-4 dimers and do not sufficiently mimic the wide complexity of lignin structure (i.e., aliphatic side chains and robust C-C bonds). Herein, we present a methodology to access advanced lignin models through the first synthesis of two trimers of monolignol G—possessing side-chains and both robust β-5 bond and labile β-O-4 bond—via a chemo-enzymatic pathway. Key steps were (1) the C-C coupling via laccase-mediated oxidation, (2) the C-O coupling via a simple SN2 between a phenolate and a bromoketoester, and (3) a modified Upjohn dihydroxylation or a palladium-catalyzed hydrogenation. (β-5)-(β-O-4) dihydroxytrimer and dihydrotrimer of coniferyl alcohol (G) were obtained in good global yield, 9 and 20%, respectively, over nine steps starting from ferulic acid.
Lignins are the second most abundant biopolymer on Earth and the first potential source of aromatic compounds. However, due to their tridimensional architecture and the large diversity of linkages between their three principal constitutive units, they are under exploited. In order to provide a better understanding of this extremely complex biopolymer and design efficient degradation processes, models of lignin have been used for a long time. To the best of our knowledge, the first reported synthesis of lignin model compounds emerged in 1952 (Sen, 1952). Recent studies still involved lignin model compounds (Gao et al., 2018; Lee and Yang, 2018; Rinesch and Bolm, 2018; Shimizu et al., 2018). They mainly used (β-O-4) dimers without side chains as models to study oxidative cleavage of the ether bond, known as the non-condensed linkage in lignin. Not only such approach does not address the steric effect and electronical interaction due to the side chains, but it also bypasses the robust C-C bond in the lignins, the ones that are responsible for lignin recalcitrance to degradation. Even if (β-O-4) linkage is abundant, it seems to us important also to look at the impact of oxidative treatments on condensed bond, such as (β-5) C-C bond, in order to efficiently depolymerize lignins. Recently, Forsythe et al. have described an interesting route to obtain controlled oligomers containing (5-5), (β-5), and (β-O-4) linkages but always without functionalization of the side chain (Forsythe et al., 2013). In previous works, we have synthesized model compounds with elaborated side chains to (1) conduct electrochemical study (Cottyn et al., 2015), (2) develop analytical method and immunohistochemistry approaches (Mouterde et al., 2013), or (3) unveil the chemical reactivity of lignosulfonates (Broussard et al., 2016). Such models can be used for numerous applications such as determining energetic level for electronic excitation (e.g., photochemistry), determining preferential pathways of (de)lignification, or estimating the required energy to break specific C-C bonding and release monomers from lignin. Herein, we describe the first total chemo-enzymatic synthesis of both the (β-5)-(β-O-4) dihydroxytrimer of monolignol G and the naturally occurring dihydrotrimer (Ono et al., 1998; Ye et al., 2016; Jiang et al., 2017). Monolignol G is the most widespread in nature, as it is the main constituent of softwoods lignin (>90%) and is present in large amount in hardwood (30–40%) and grasses (35–50%). (Bouxin, 2011) These two trimers will allow comparison of reactivity with (β-5) and (β-O-4) dimers and determining the impact of side chain complexity in depolymerization for instance. The chosen synthetic methodology is a convergent pathway starting from ferulic acid and acetovanillone, involving (1) the enzymatic dimerization of coniferyl alcohol (G) to obtain the (β-5) C-C bond, (2) the formation of the (β-O-4) C-O linkage using nucleophilic substitution (SN2) between a phenolate and a bromoketoester, and (3) the dihydroxylation of the double bond to finalize the formation of (β-5)/(β-O-4) dihydroxytrimer of monolignol G, or the hydrogenation of this double bond to achieve the (β-5)/(β-O-4) dihydrotrimer of monolignol G (Scheme 1).
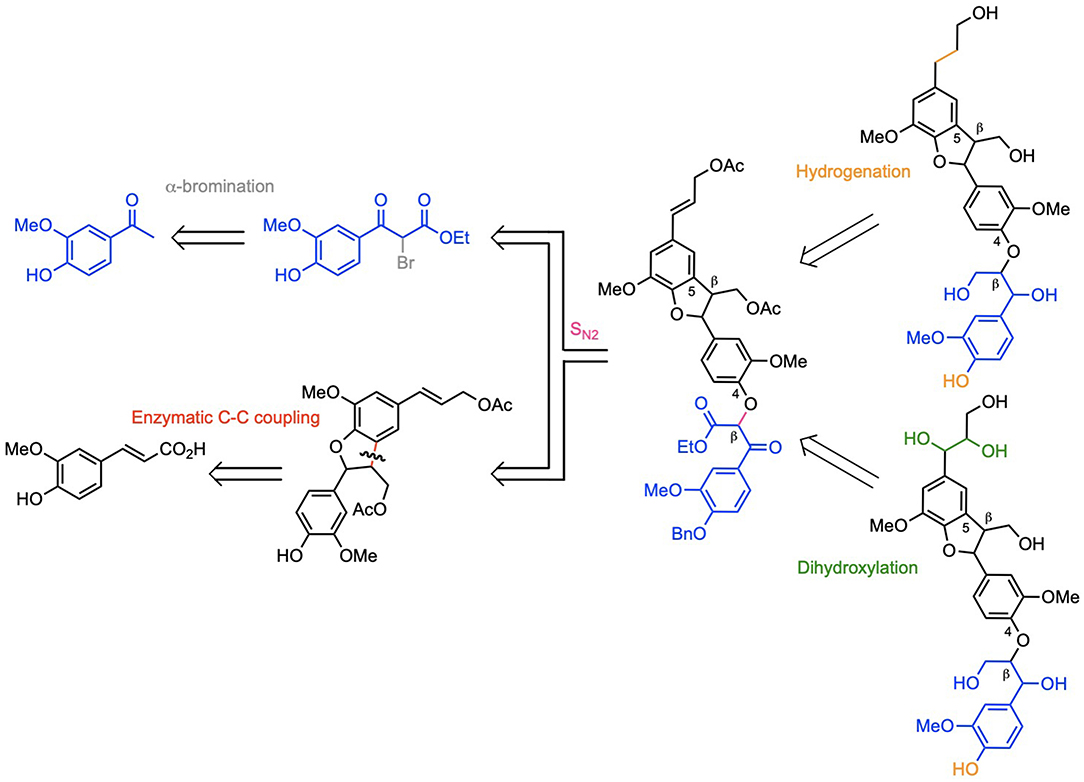
Scheme 1. Retrosynthetic routes towards dihydroxy-(β-5)-(β-O-4) trimer and naturally occurring dihydro-(β-5)-(β-O-4) trimer of coniferyl alcohol G.
The synthesis of the two targets started with the preparation of di-acetylated (β-5)-dimer 1 from ferulic acid (2), in five steps. Using the strategy previously reported for sinapyl alcohol (Jaufurally et al., 2016), 2 was transformed into ethyl ferulate (3) through a Fischer esterification. Subsequent DibalH-mediated reduction of 3 provided coniferyl alcohol (aka monolignol G) in 77% overall yield from 2. Enzymatic dimerization of G in the presence of laccase isolated from Trametes versicolor (EC.420-150-4) was performed in an emulsion of ethyl acetate and aqueous acetate buffer pH = 5 at room temperature. It is noteworthy to mention that a slow addition of laccase solution via a syringe pump allowed controlling the ratio between G and radical species in the reaction medium, thus limiting the formation of oligomers. Only trans diastereoisomer of β-5 dimer of G (4) was obtained as determined by NMR and according to Lou et al. (2018) (43% yield) (data shown in Supplementary Material). The two aliphatic alcohols and the phenol moiety were subsequently acetylated (Ac2O, pyridine), prior to specific deacetylation of the latter in the presence of piperazine in THF to provide compound 1 (92% yield, 30% overall yield in five steps starting from ferulic acid) (Scheme 2).
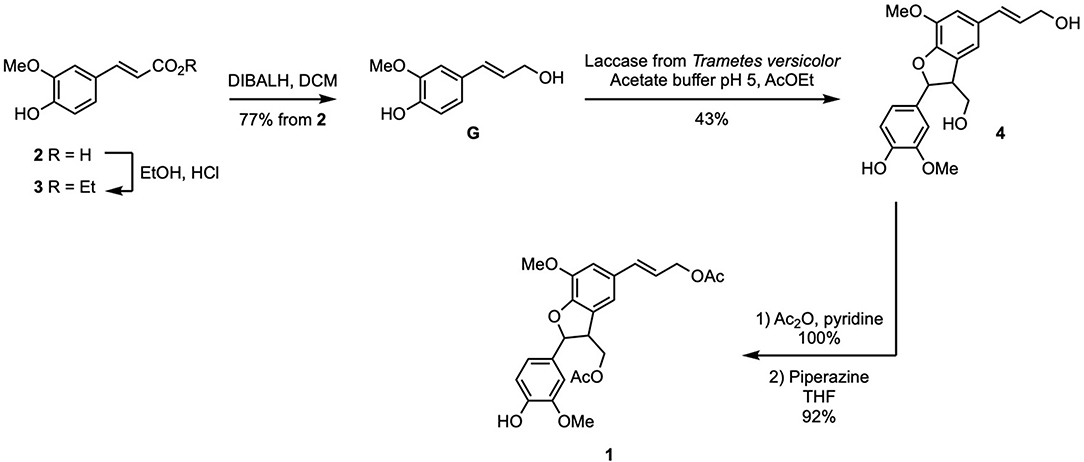
Scheme 2. Synthesis of 1 from ferulic acid (2).
In another hand, the synthesis of bromoketoester of guaiacol (6) was performed from acetovanillone (7) in three steps (Scheme 3). Benzylation of 7 (BnBr, KI, K2CO3, DMF, quant.) gave O-benzylacetovanillone that was then reacted with diethylcarbonate and sodium hydride to provide the corresponding β-ketoester (8) in 78% yield. The latter then underwent bromination (NBS, Amberlyst A15) giving 6 (79% yield, 62% overall yield from 7).
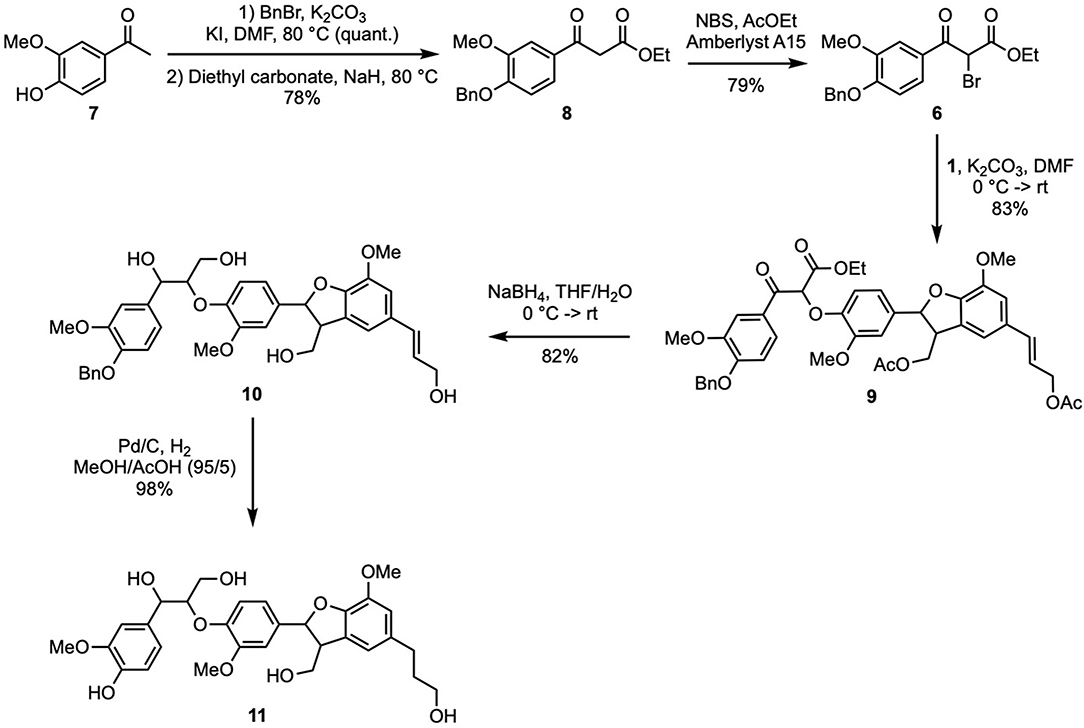
Scheme 3. Synthesis of naturally occurring dihydrotrimer (β-5)-(β-O-4) (11).
Formation of the β-O-4 bond was achieved through the coupling between 6 and 1 via a SN2 process as previously reported by Forsythe et al. (2013) and Kishimoto et al. (2006). It is worth mentioning that, to achieve (β-O-4) bond, such SN2 was preferred to the aldolization strategy we previously applied for the preparation of (5-5′)-(8′-O-4″) dihydrotrimer of ferulic acid (Mouterde et al., 2013), due to easier operation conditions and higher reported yields [49% in acetone (Lancefield and Westwood, 2015) and 97% in DMF (Kishimoto et al., 2006)]. Performing this reaction with potassium carbonate in DMF provided the per-protected (β-5)-(β-O-4) trimer of G (9) in 83% yield (Scheme 3). To access the desired targets, it was necessary to reduce the ketone and ester moieties. To do so, the procedure reported by Patil and Yan (2016) was applied (NaBH4, THF/H2O) and provided the intermediate 10 in 82% yield. Even if the molecular ion of these adducts cannot be identified in HRMS, probably because of a high reactivity leading to decomposition of the molecule, the disappearance of all peaks between 2.5 and 1.5 ppm and those at 4.24 and 1.21 ppm in the 1H NMR spectrum proved the cleavage of the acetates and esters, respectively (data shown in Supplementary Material). Moreover, 13C NMR spectrum further confirmed this result and proved also the reduction of the ketone as the peak at 190.0 ppm disappeared.
To access the dihydrotrimer 11, intermediate 10 was submitted to a palladium-catalyzed hydrogenation step in order to simultaneously cleave the benzyl protecting group and reduce the double bond. It was observed that performing the reaction in ethanol, resulted only in the reduction of the double bond, leaving the benzyl protective group untouched. The cleavage of recalcitrant benzyl moieties being facilitated in highly polar solvent and in the presence of an acid, the successful concomitant debenzylation and reduction of 10 was achieved by using a 95/5 mixture of methanol and acetic acid, providing dihydrotrimer (β-5)-(β-O-4) (11) in 98% yield (Scheme 3, 67% overall yield from 1, and 20% from ferulic acid in nine steps). It is worth mentioning that despite many attempt, no HRMS data could be obtained for 11. Nevertheless, 1H & 13C NMR analyses proved the reduction of the double bond and the cleavage of the benzyl protecting group.
Another pathway to intermediate 10 was designed to reduce the number of steps. Ethyl ferulate (3) was enzymatically dimerized using the same procedure applied for 4 leading to a phenolic diester (12) in similar yield. This procedure circumvented the reduction of 3 into G, the acetylation and the specific deacetylation. As the phenol moiety was free, the phenolate was directly generated by action of potassium carbonate and reacted with bromoketoester 6 to provide the desired ketotriester (13) in excellent yield (84%). The aluminum-mediated reduction (Dibal-H) of 13 proved troublesome due to extreme difficulty to recover the desired product from the quenched reaction mixture. Unfortunately, although there have been reports on the successful NaBH4-mediated reduction of aromatic/conjugated esters (Bianco et al., 1988; Boechat et al., 2004; da Costa et al., 2006) the NaBH4-mediated reduction of 13 proved unsuccessful even with longer reaction time (4 days) or addition of more NaBH4 (up to 20 equivalents) (Scheme 4).
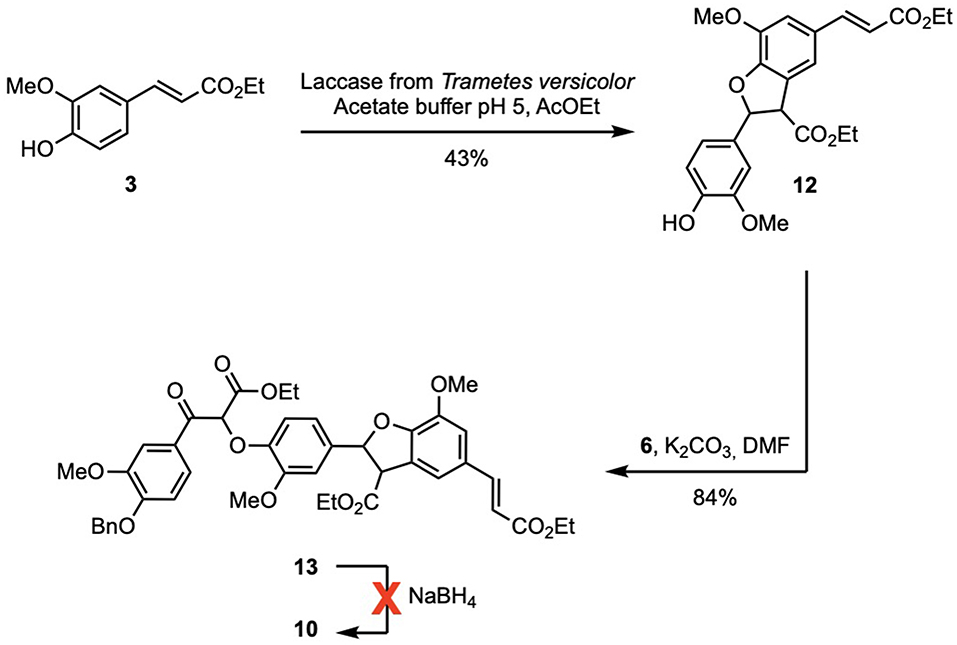
Scheme 4. Unsuccessful alternative pathway to 10.
Finally, in order to access naturally occurring (β-5)-(β-O-4) dihydroxytrimer 14, the double bond in 9 had to be converted into the corresponding 1,2-diol through a dihydroxylation step (Scheme 5). The use of a modified osmium tetroxide-based Upjohn approach (K2OsO4, NMO, EtOH/H2O, citric acid) (Moreaux et al., 2019) allowed the formation of compound 15 (58% yield). The latter was then deprotected in the presence of sodium borohydride to provide 16 in 65% yield. Compound 16 exhibiting a high polarity, its purification through flash chromatography had to be performed in reverse phase. Finally, dihydroxytrimer (β-5)-(β-O-4) 14 was obtained after the removal of the benzyl group using the same hydrogenation procedure as previously described. Efficient removal of acetic acid and ethanol was achieved through azeotropic distillation, providing pure 14 in excellent yield (99% overall yield starting from ferulic acid in nine steps).
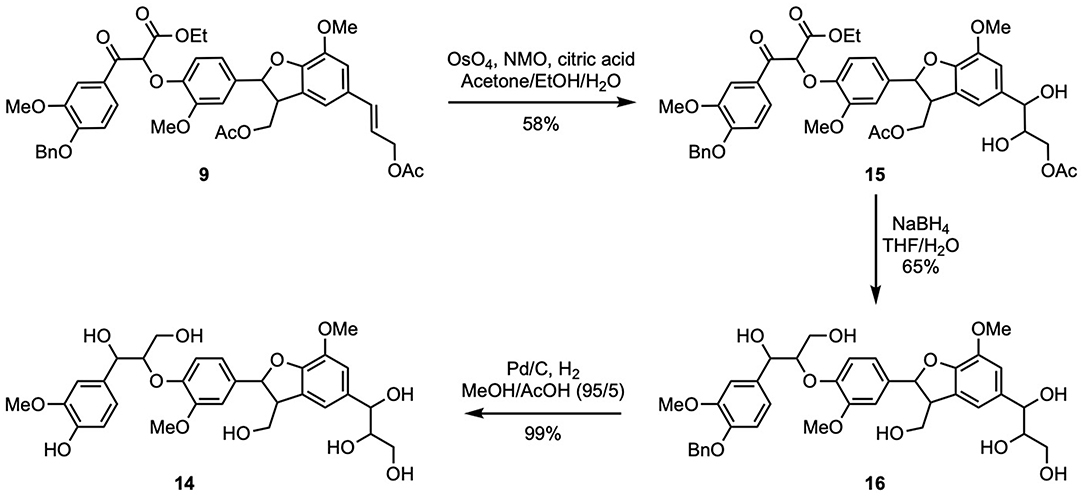
Scheme 5. Synthesis of naturally occurring (β-5)-(β-O-4) dihydroxytrimer (14).
Ferulic acid, 1 M diisobutyl aluminum hydride in dichloromethane, benzyl bromide, laccase from Trametes versicolor (776 U/g), piperazine, and sodium hydride were purchased from Sigma-Aldrich and used as received. Acetovanillone, sodium borohydride, pyridine, and diethylcarbonate were purchased from TCI and used as received. Palladium on carbon and anhydrous magnesium sulfate were purchased from Acros Organics and used as received. Deuterated solvents were purchased from Euriso-top. Other reagents, salts, and solvents were purchased from VWR.
DMF was dried using mBraun SPS 800. Evaporations were conducted under reduced pressure (Vario Vacuubrand pump) on Buchi R300. Flash chromatographies were performed on a Puriflash 4100 (Interchim) equipped with and pre-packed INTERCHIM PF-30SI-HP (30 μm silica gel) columns. IR analyses were performed on Cary 630 FTIR (Agilent). NMR analyses were recorded on a Bruker Fourier 300. 1H NMR spectra of samples were measured on a 300 MHz apparatus, chemicals shifts were reported in parts per million relative to solvent residual peak (CDCl3 δ = 7.26 ppm; DMSO-d6 δ = 2.50 ppm). 13C NMR spectra of samples were recorded at 75 MHz and calibrated on solvent peak (CDCl3 δ = 77.16 ppm; DMSO-d6 δ = 39.52 ppm).
Coniferyl alcohol G (3.0 g, 16.6 mmol) was dissolved in ethyl acetate (170 mL) at room temperature, then citrate/phosphate buffer 4.5 (170 mL) was added and the mixture was vigorously stirred (1,000 rpm). Laccase from Trametes versicolor (21 mg, 16.3 U/mmol) was dissolved in pH 4.5 buffer (50 mL) and added at 10 mL.h−1 to the mixture. At the end of the addition, layers were separated and aqueous layer was extracted twice with AcOEt (75 mL). Organic layers were then combined, washed with brine, dried over anhydrous MgSO4, filtered and concentrated to dryness. Product was purified by flash chromatography over silica gel in 10/90 cyclohexane/AcOEt, providing 4 as an orange foam (1.28 g, 43%).
IR (neat): ν = 3,332 (O-H), 2,926 + 2,869 (C=C), 1,610 + 1,516 + 1,496 + 1,461 (C=C arom) cm−1.
1H NMR (DMSO-d6, 300 MHz) δ = 9.02 (s, 1H, H23), 6.93 (m, 3H, H10 + H20 + H21), 6.75 (s, 2H, H6 + H17), 6.46 (d, 1H, J = 15.9 Hz, H4), 6.21 (dt, 1H, J = 15.9 Hz, J = 5.4 Hz, H3), 5.45 (d, 1H, J = 6.9 Hz, H15), 5.02 (t, 1H, J = 5.4 Hz, H14), 4.78, (t, 1H, J = 5.7 Hz, H1), 4.08 (td, 2H, J = 5.4 Hz, J = 1.2 Hz, H2), 3.79 (s, 3H, H22), 3.74 (s, 3H, H11), 3.58–3.74 (m, 2H, H13), 3.43 (m, 1H, H12) ppm.
13C NMR (DMSO-d6, 75 MHz) δ = 147.6 + 147.1 (Cq, C8 + C19), 146.4 + 143.7 (Cq, C7 + C18), 132.3 (Cq, C16), 130.5 (Cq, C5), 129.5 (Cq, C9), 129.0 (Cs, C4), 128.0 (Cs, C3), 118.5 + 115.3 (Cs, C6 + C17), 114.9 (Cs, C20), 110.3 (Cs, C10 + C21), 87.2 (Cs, C15), 62.9 (Cd, C13), 61.7 (Cd, C2), 55.6 (Ct, C11 + C22), 53.0 (Cs, C12) ppm.
HRMS (EI): Calcd for C20H22O6 [M]: 358.1416 and for C20H23 [M+H]+: 359.1489, found 359.1566.
4 (2.6 g, 7.25 mmol) was dissolved in pyridine (18 mL) and acetic anhydride (11 mL, 109 mmol, 5 equiv/function) was added. The reaction was stirred at room temperature overnight. Reaction medium was poured into acidified iced water. The precipitate was filtered off and rinsed with water until neutral pH. After drying, 5 was recovered in quantitative yield and used in the next step without further purification.
IR (neat): ν = 2,940 (C-C), 1,733 (C = O), 1,603 + 1,506 + 1,462 + 1,421 + 1,366 + 1,332 (C=C arom), 1,214 + 1,187 + 1,146 + 1,127 (C-O) cm−1.
1H NMR (CDCl3, 300 MHz) δ = 6.98 (m, 3H, H10 + H20 + H21) 6.88 (m, 2H, H6 + H17), 6.59 (m, 1H, H4), 6.15 (dt, 1H, J = 15.6 Hz, J = 6.6 Hz, H3), 6.54 (d, 1H, J = 6.6 Hz, H15), 4.70 (dd, 2H, J = 6.6 Hz, 1.2 Hz, H2), 4.26–4.47 (m, 2H, H13), 3.91 (s, 3H, H22), 3.78 (m, 4H, H15 + H11), 2.30 (s, 3H, H26), 2.09 (s, 3H, H24), 2.05 (s, 3H, H25) ppm.
13C NMR (CDCl3-d6) δ = 171.0 + 170.8 (Cq, C14 + C1), 169.0 (Cq, C23), 151.3 (Cq, C18), 148.2 (Cq, C8), 144.4 (Cq, C7), 139.7 + 139.4 (Cq, C16 + C19), 134.3 (Cs, C4), 130.7 (Cq, C5), 127.3 (Cq, C9), 122.9 (Cs, C20), 121.3 (Cs, C3), 118.2 (Cs, C21), 115.4 (Cs, C10), 110.6 (Cs, C17), 109.9 (Cs, C6), 88.1 (Cs, C15), 65.4 (Cd, C13), 65.2 (Cd, C2), 56.0 + 55.9 (Ct, C11 + C22), 50.5 (Cs, C12), 20.6–21.1 (Ct, C24 + C25 + C26) ppm.
HRMS (EI): Calcd for C26H28O9 [M]: 484.1733 and for C26H28O9Na+ [M+Na]+: 507.1631 found 507.1637.
5 (7.25 mmol) was dissolved in THF (70 mL) and piperazine (1.87 g, 21.75 mmol, 3 equiv) was added. Reaction mixture was stirred 4 h at room temperature. After completion, THF was removed under reduced pressure. The crude mixture was dissolved in AcOEt (50 mL), washed with HCl 1 M (3*15 mL) and brine (20 mL), dried over anhydrous MgSO4, filtered and concentrated. After purification by flash chromatography in 40/60 cyclohexane/AcOEt, 1 was recovered as colorless oil (2.95 g, 92%).
IR (neat): ν = 3,443 (OH), 2,939 (C-C), 1,732 (C = O), 1,601 + 1,515 + 1,493 + 1,460 + 1,423 + 1,364 + 1,331 (C=C arom), 1,216 + 1,144 (C-O) cm−1.
1H NMR (CDCl3, 300 MHz) δ = 6.88 (m, 5H, H6 + H10 + H17 + H20 + H21), 6.60 (d, 1H, J = 15.9 Hz, H4), 6.16 (dt, 1H, J = 15.9 Hz, J = 6.6 Hz, H3), 5.65 (s, 1H, H23), 5.47 (d, 1H, J = 7.5 Hz, H15), 4.71 (dd, J = 1.2 Hz, J = 6.6 Hz, 2H, H2) 4.37 (m, 2H, H13), 3.91 (s, 3H, H22), 3.86 (s, 3H, H11), 3.77 (m, 1H, H12), 2.10 (s, 3H, H24), 2.03 (s, 3H, H25) ppm.
13C NMR (CDCl3-d6, 75 MHz) δ = 171.1 (Cq, C14), 171.0 (Cq, C1), 148.4 (Cq, C8), 146.8 (Cq, C18), 146.0 (Cq, C19), 144.5 (Cq, C7), 134.5 (Cq, C16), 132.3 (Cs, C4), 130.6 (Cq, C5), 127.8 (Cq, C9), 121.2 (Cs, C3), 119.7 (Cs, C21), 115.4 (Cs, C20), 114.4 (Cs, C10), 110.6 (Cs, C17), 108.7 (Cs, C6), 89.0 (Cs, C15), 65.4 (Cd, C2 + C13), 56.1 (Ct, C11 + C22), 50.4 (Cs, C12), 21.2 (Ct, C24), 21.0 (Ct, C25) ppm.
HRMS (EI): Calcd for C24H26O8 [M]: 442.1628 and for C24H27 [M+H]+: 443.1700 found 443.1867.
Under N2, 1 (1.4 g, 3.16 mmol) was dissolved in dry DMF (10 mL) and K2CO3 (0.66 g, 4.75 mmol, 1.5 equiv) was added (reaction turned yellow). After cooling to 0°C, a solution of 6 (1.9 g, 4.75 mmol, 1.5 equiv) in DMF (9 mL) was added dropwise. Reaction mixture was stirred at room temperature for 3 h, then quenched with water (50 mL) and extracted with AcOEt (3*50 mL). Organic layers were combined, washed with brine, dried over anhydrous MgSO4, filtered and concentrated. After purification by flash chromatography in 5/5 cyclohexane/AcOEt, 9 was recovered as an oil (2.04 g, 83%).
IR (neat): ν = 2,953 (C-C), 1,734 (C = Oester), 1,676 (C = Oketone), 1,593 + 1,508 + 1,459 + 1,420 + 1,379 + 1,332 (C=Carom), 1,209 + 1,143 (C-O) cm−1.
1H NMR (CDCl3, 300 MHz) δ = 7.77 (dd, J = 8.4 Hz, J = 2.1 Hz, 1H, H31), 7.68 (d, J = 2.1 Hz, 1H, H27), 7.27–7.45 (m, 5H, H35 + H36 + H37), 6.74–6.94 (m, 6H, H6 + H10 + H17 + H20 + H21 + H30), 6.59 (d, J = 15.6 Hz, 1H, H4), 6.15 (dt, J = 15.6 Hz, J = 6.6 Hz, 1H, H3), 5.73 (s, 1H, H23), 5.47 (d, J = 6.9 Hz, 1H, H15), 5.23 (s, 2H, H33), 4.71 (d, J = 6.6 Hz, 2H, H2), 4.43 (m, 1H, H13a), 4.24 (m, 3H, H13b + H38), 3.92 (s, 3H, H32), 3.90 (s, 3H, H11), 3.75 (m, 4H, H22 + H12), 2.09 (s, 3H, H41), 2.01 (s, 3H, H40), 1.21 (t, J = 7.2 Hz, 3H, H39) ppm.
13C NMR (CDCl3, 75 MHz) δ = 190.0 (Cq, C25), 170.9–171.3 (Cq, C1 + C14), 167.0 (Cq, C24), 153.3 (Cs, C29), 150.7 (Cs, C18), 150.6 (Cs, C8), 149.5 (Cs, C28), 148.2 (Cs, C34), 146.3 (Cs, C19), 144.5 (Cs, C7), 136.2 (Cs, C5 + C16), 134.4 (Cs, C4), 130.7 (Cs, C9), 128.8 (Cs, C36), 128.3 (Cs, C26), 127.5 (Cs, C37), 127.3 (Cs, C35), 124.8 (Cs, C31), 121.3 (Cs, C3), 118.7 (Cs, C20), 118.3 (Cs, C21), 115.4 (Cs, C10), 112.1 (Cs, C30), 112.0 (Cs, C27), 110.5 (Cs, C17) 110.4 (Cs, C6), 88.4 (Cs, C15), 82.7 (Cs, C23), 70.9 (Cd, C33), 65.3 (Cd, C2 + C13), 62.3 (Cd, C38), 56.0–56.1 (Ct, C11 + C22 + C32), 50.4 (Cs, C12), 21.2 (Ct, C41), 20.9 (Ct, C40), 14.3 (Ct, C39) ppm.
HRMS (EI): Calcd for C43H44O13 [M]: 768.2782 and for C43H44O13Na+ [M+Na]+: 791.2680 found 791.2668.
9 (700 mg, 0.91 mmol) was dissolved in THF (7.6 mL) and water was added (2.4 mL). At 0°C, NaBH4 (344 mg, 9.1 mmol, 10 equiv) was added by portions. At the end of the addition, reaction mixture was stirred for two more hours at room temperature, then quenched by a saturated aqueous solution of NH4Cl (10 mL) and extracted with AcOEt (2*30 mL). Organic layers were combined, washed with brine, dried over anhydrous MgSO4, filtered and concentrated. After purification by flash chromatography in 100% AcOEt, 10 was recovered as an oil (432 mg, 82%).
IR (neat): ν = 3,346 (O-H), 2,934 (C-C), 1,595 + 1,508 + 1,459 + 1,420 + 1,379 + 1,326 (C=Carom), 1,257 + 1,217 + 1,137 (C-O) cm−1.
1H NMR (DMSO-d6, 300 MHz) δ = 7.30–7.44 (m, 5H, H35 + H36 + H37), 6.75–7.15 (m, 8H, H6 + H10 + H17 + H20 + H21 + H27 + H30 + H31), 6.60 (d, J = 15.6 Hz, 1H, H4), 6.21 (dt, J = 15.6 Hz, J = 6.6 Hz, 1H, H3), 5.49 (m, 1H, H15), 5.45 (m, 1H, H25), 5.05 (m, 3H, H33 + H39), 4.74 (m, 1H, H23), 4.64 (m, 2H, H2), 4.33 (m, 1H, H24a), 3.80 (s, 3H, H11), 3.73 (m, 4H, H32 + H1), 3.67 (s, 3H, H22), 3.60 (m, 3H, H13 + H24b), 3.42 (m, 1H, H12) ppm.
13C NMR (DMSO, 75 MHz) δ = 150.0 (Cq, C8), 148.9 (Cq, C19), 148.3 (Cq, C28), 148.1 (Cq, C18), 147.2 (Cq, C29), 144.2 (Cq, C7), 137.8 (Cq, C34), 135.7 (Cq, C16), 134.4 (Cq, C5), 134.3 (Cs, C4), 130.1 (Cq, C9), 129.9 (Cq, C26), 128.8 (Cs, C36 + C37), 128.2 (Cs, C35), 121.4 (Cs, C3), 119.6 (Cs, C31), 118.5 (Cs, C21), 116.0 (Cs, C10), 115.7 (Cs, C20), 113.2 (Cs, C30), 111.8 (Cs, C27), 111.0 (Cs, C17) 110.8 (Cs, C6), 87.5 (Cs, C15), 84.0 (Cs, C25), 71.9 (Cs, C23), 70.3 (Cd, C33), 65.1 (Cd, C2), 60.4 (Ct, C24), 60.2 (Ct, C13), 56.1 (Ct, C11 + C22), 55.8 (Ct, C32), 53.5 (Cs, C12) ppm.
10 (250 mg, 0.38 mmol) was dissolved in methanol/acetic acid (95/5) mixture (5 mL). Reaction medium was purged with nitrogen during 15 min, then palladium on charcoal (25 mg, 10% w/w) was added. The reaction was put under dihydrogen flux overnight at room temperature. Upon completion, reaction medium was purged with nitrogen during 20 min, filtered through PTFE syringe filter, rinsed twice with 5 mL of ethanol and concentrated. 11 was recovered as a foam (217 mg, 98%) without the need of further purification.
IR (neat): ν = 3,388 (O-H), 2,934 (C-C), 1,602 + 1,509 + 1,460 + 1,421 + 1,366 (C=Carom), 1,258 + 1,210 + 1,138 (C-O) cm−1.
1H NMR (DMSO-d6, 300 MHz) δ = 8.75 (s, 1H, H33), 7.02–6.64 (m, 8H, H6 + H10 + H17 + H20 + H21 + H27 + H30 + H31), 5.43 (m, 1H, H15), 5.28 (m, 1H, H25), 5.00 (s, 1H, H35), 4.70 (m, 1H, H23), 4.59 (s, 1H, H14), 4.44 (s, 1H, H1), 4.29 (m, 1H, H), 3.77–3.69 (m, 12H, H11 +H22 +H32 +H), 3.59 (m, 4H, H), 3.40 (m, 8H, H2 + H4), 1.69 (m, 2H, H3) ppm.
13C NMR (DMSO, 75 MHz) δ = 150.4, (Cq, C8), 148.2 (Cq, C19), 147.4 (Cq, C28), 146.1 (Cq, C18), 145.9 (Cq, C29), 143.9 (Cq, C7), 140.0 (Cq, C16), 134.6 (Cq, C5) 133.7 (Cq, C9), 128.2 (Cq, C26), 119.9 (Cs C21 + C31), 116.9 (Cs, C10 + C20), 112.9 (Cs, C30 + C27), 111.0 (Cs, C17 + C6), 95.5 (Cs, C15), 84.0 (Cs, C25), 72.0 (Cs, C23), 63.8 (Cd, C2), 63.5 (Ct, C24), 60.4 (Ct, C13), 56.1 (Cs, C11 + C22 + C32), 55.9 (Cs, C12) 31.8 (Cd, C3), 30.6 (Cd, C4) ppm.
9 (840 mg, 1.1 mmol) and citric acid (275 mg, 1.3 mmol) were dissolved in a mixture of acetone (3 mL), ethanol (2 mL) and water (1.5 mL). Osmium tetroxide solution 4% in water (170 μL, 27 μmol) was added and then N-methyl morpholine solution 0.5 M in water (270 μL, 1.3 mmol) was added too. The reaction media was stirred overnight, quenched with Na2S2O3 (10 mL) and extracted thrice with ethyl acetate (10 mL). Organic layers were combined washed with water and brine, dried over anhydrous MgSO4, filtered and concentrated. Flash purification was conducted on silica gel with cyclohexane/AcOEt (1/1) and then AcOEt (100%) to afford 512 mg of pure 15 as an oil (58%).
IR (neat): ν = 3,456 (O-H), 2,937 (C-C), 1,734 (C = O ester), 1,675 (C = O ketone), 1,592 + 1,508 + 1,454 + 1,419 + 1,368 (C=C arom), 1,209 + 1,135 (C-O) cm−1.
1H NMR (CDCl3, 300 MHz) δ = 7.77 (dd, J = 8.4 Hz, J = 2.1 Hz, 1H, H31), 7.68 (d, J = 2.1 Hz, 1H, H27), 7.31–7.44 (m, 5H, H35 + H36 + H37), 6.82–6.93 (m, 6H, H6 + H10 + H17 + H20 + H21 + H30), 5.73 (s, 1H, H23), 5.47 (d, J = 6.9 Hz, 1H, H15), 5.23 (s, 2H, H33), 4.57 (m, 1H, H), 4.39 (m, 1H, H), 4.21–4.35 (m, 3H, H13b + H38), 4.12 (m, 1H, H), 3.87–4.01 (m, 9H, H), 3.78 (m, 4H, H22 + H12), 2.10 (s, 3H, H41), 2.00 (s, 3H, H40), 1.21 (t, J = 7.2 Hz, 3H, H39) ppm.
13C NMR (CDCl3, 75 MHz) δ = 190.0 (Cq, C25), 170.9 and 171.4 (Cq, C1+ C14), 167.0 (Cq, C24), 153.3 (Cs, C29), 150.6 (Cs, C18), 149.5 (Cs, C8), 149.5 (Cs, C28), 148.2 (Cs, C), 146.3 (Cs, C19), 144.5 (Cs, C7), 136.2 (Cs, C34 + C5 + C16), 134.4 (Cs, C4), 130.7 (Cs, C9), 128.8 (Cs, C36), 128.3 (Cs, C26), 127.5 (Cs, C37), 127.4 (Cs, C35), 124.8 (Cs, C31), 118.8 (Cs, C20), 118.3 (Cs, C21), 115.0 (Cs, C10), 112.1 (Cs, C30), 112.0 (Cs, C27), 110.9 (Cs, C17) 110.4 (Cs, C6), 88.4 (Cs, C15), 82.7 (Cs, C23), 77.4, 74.6, 74.3 (Cd, C4), 70.9 (Cd, C33), 65.5+ 65.3 (Cd, C2+ C13), 62.3 (Cd, C38), 56.0–56.2 (Ct, C11 + C22 + C32), 50.6 (Cs, C12), 21.0 (Ct, C41), 20.9 (Ct, C40), 14.2 (Ct, C39) ppm.
HRMS (EI): Calcd for C43H46O15 [M]: 802.2837 and for C43H46O15Na+ [M+Na]+: 825.2726 found 825.2734.
15 (794 mg, 0.99 mmol) was dissolved in a THF/water mixture (8.2 mL/2.6 mL). At 0°C, sodium borohydride (375 mg, 9.9 mmol, 10 equiv) was added by portion. The reaction medium was stirred at room temperature during 18 h. THF was removed by distillation under reduce pressure and 2.5 mL of methanol was added. The crude mixture was purified by flash chromatography over C18-grafted silica gel with water/MeOH (1/1) and then water/ MeOH (1/4). 16 was recovered as a white foam (448 mg, 65%).
IR (neat): ν = 3,344 (O-H), 2,932 + 2,873 (C-C), 1,602 + 1,507 + 1,451 + 1,419 + 1,321 (C=C arom), 1,258 + 1,213 + 1,135 (C-O) cm−1.
1H NMR (DMSO, 300 MHz) δ = 7.31–7.41 (m, 5H, H35 + H36 + H37), 6.81–7.06 (m, 8H, H6 + H10 + H17 + H20 + H21 + H27 + H30 + H31), 5.38–5.47 (m, 2H, H23 +H41), 5.04 (s, 3H, H33 + H1), 4.99 (t, J = 4.5 Hz, 1H, H39), 4.76 (m, 1H, H3), 4.66 (m, 1H, H14), 4.53 (m, 1H, H38), 4.45 (m, 2H, H25 + H40), 4.33 (m, 1H, H1), 3.68–3.77 (m, 10H, H11 + H22 + H32 + H13a), 3.60 (m, 3H, H2 + H13b), 3.43–3.48 (m, 2H, H12 + H24a), 3.31 (m, 1H, H4), 3.19 (m, 1H, H24b) ppm.
13C NMR (DMSO, 75 MHz) δ = 149.6 (Cq, C7), 148.5 (Cq, C18), 147.8 (Cq, C8), 146.8 (Cq, C28), 146.3 (Cq, C29), 143.0–143.1 (Cq, C19), 137.4 (Cq, C34), 136.8 (Cq, C16), 135.3 (Cq, C26), 134.4 (Cs, C5), 128.4 (Cs, C36 +Cq, C9), 127.8 (Cs, C37 + C35), 119.2 (Cs, C31), 118.0 (Cs, C21), 115.1–115.4 (Cs, C10), 112.9 (Cs, C30), 11.4 (Cs, C17), 111.0–111.4 (C20), 110.4 (C6), 86.8 (Cs, C23), 83.5 (Cs, C15), 75.9 (Cs, C4), 72.8–73.0 (Cs, C25), 71.5 (Cs, C3), 69.9 (Cd, C33), 63.2 (Cd, C13), 62.7 (cd, C24), 60.0 (Cd, C2), 55.7 and 55.4 (Ct, C22 + C32 + C11), 53.5 (Cs, C12) ppm.
HRMS (EI): Calcd for C37H42O12 [M]: 678.7310 and for C37H42O12Na+ [M+Na]+: 701.2574 found 701.2565.
16 (325 mg, 0.48 mmol) was dissolved in a MeOH/AcOH (95/5) mixture (4.8 mL). The reaction medium was purged for 15 min under nitrogen flow before palladium on charcoal (32 mg, 10% w/w) was added. The mixture was then stirred under hydrogen flow overnight. Upon completion, reaction medium was purged with nitrogen during 20 min, filtered through PTFE syringe filter, rinsed twice with 5 mL of ethanol and concentrated. Crude product was submitted to azeotropic evaporation with toluene thrice, then thrice with water to eliminate acetic acid residues. Light yellow oil was recovered (99%).
IR (neat): ν = 3,324 (O-H), 2,933 (C-C), 1,601 + 1,507 + 1,451 + 1,420 (C=C), 1,261 + 1,214 + 1,134 (C-O) cm−1.
1H NMR (DMSO-d6, 300 MHz) δ = 6.52–7.01 (m, 13H, H6 + H10 + H17 + H20 + H21 + H27 + H30 +H31), 5.45 (m, 1H, H23), 4.70 (m, 3H, H3 + H37 + H36), 4.39 (m, 3H, H25), 4.24 (m, 2H, H15), 3.63–3.77 (m, 14H, H11 + H22 + H32 + H13a), 3.08–3.55 (m, 14H, H2 + H3 + H4 + H12 + H13b +H23 + H24) ppm.
13C NMR (DMSO, 75 MHz) δ = 149.6–145.5 (Cq, C7 + C18 + C8 + C28 + C29), 142.9–143.1 (Cq, C19), 136.8–136.9 (Cq, C16), 132.9–134.4 (Cq, C5 + C26), 128.2–128.4 (Cq, C9), 118.0–120.7 (Cs, C21 + C31), 114.6–115.5 (Cs, C10 + C30), 113.2–113.3 (Cs, C27), 110.4–111.4 (Cs, C17 + C20), 107.8–108.0 (Cs, C6), 86.8 (Cs, C23), 83.7 (Cs, C15), 75.9–76.1 (Cd, C4), 72.8–73.1 (Cs, C25), 71.5 (Cs, C3), 63.2–64.3 (Cd, C13), 62.6 (Cd, C24), 59.9–60.0 (Cd, C2) 55.4–55.7 (Ct, C11 + C22 + C32), 53.5 (Cs, C12) ppm.
HRMS (EI): Calcd for C30H36O12 [M]: 588.2207 and for C30H36O12Na+ [M+Na]+: 611.2104 found 611.2110.
The total convergent synthesis of the β-5/β-O-4 dihydroxytrimer of monolignol G (11) and that of the naturally occurring dihydrotrimer (10) have been successfully achieved in nine steps and in 9 and 20% yield, respectively, starting from ferulic acid using laccase-mediated dimerization, nucleophilic substitution (SN2), and dihydroxylation as key steps.
All datasets generated for this study are included in the article/Supplementary Material.
AF and FA wrote the publication. AP performed main bibliographical research. AF and AP designed and performed experimental work. AH, J-HR, and FA supervised this work and corrected the proof of article.
This work was fund through the financial support brought to URD ABI by the Grand Reims, the Conseil Général de la Marne and the Région Grand Est.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Grand Reims, Département de la Marne, and Grand Est for financial support. Authors also want to acknowledge analytical platform PLANET (Reims) for performing HRMS analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00842/full#supplementary-material
Bianco, A., Passacantilli, P., and Righi, G. (1988). Synthetic communications: an international journal for rapid communication of synthetic organic chemistry. Synth. Commun. 15, 1765–1771. doi: 10.1080/00397918808060931
Boechat, N., Santos Da Costa, J. C., De Souza Mendonça, J., De Oliveira, P. S. M., and De Souza, M. V. N. (2004). A simple reduction of methyl aromatic esters to alcohols using sodium borohydride-methanol system. Tetrahedron Lett. 45, 6021–6022. doi: 10.1016/j.tetlet.2004.06.034
Bouxin, F. (2011). Solvolyse Des Lignines: Production de Synthons Aromatiques de Faibles Masses. Available online at: https://www.theses.fr/2011REIMS004
Broussard, O., Petit, M., Elie, N., Baumberger, S., Arnaud, A., Ducrot, P.-H., et al. (2016). Monitoring of free phenol content in lignosulfonates by ClO2 titration and UV difference spectroscopy. Holzforschung 70, 719–724. doi: 10.1515/hf-2015-0111
Cottyn, B., Rivard, M., Majira, A., Beauhaire, J., Allais, F., Martens, T., et al. (2015). Comparative electrochemical study on monolignols and dimers relevant for the comprehension of the lignification process. Phytochem. Lett. 13, 280–285. doi: 10.1016/j.phytol.2015.07.002
da Costa, J. C. S., Pais, K. C., Fernandes, E. L., De Oliveira, P. S. M., Mendonça, J. S., De Souza, M. V. N., et al. (2006). Simple reduction of ethyl, isopropyl and benzyl aromatic esters to alcohols using sodium borohydride-methanol system. Arkivoc 1, 128–133. doi: 10.3998/ark.5550190.0007.115
Forsythe, W. G., Garrett, M. D., Hardacre, C., Nieuwenhuyzen, M., and Sheldrake, G. N. (2013). An efficient and flexible synthesis of model lignin oligomers. Green Chem. 15, 3031–3038. doi: 10.1039/c3gc41110a
Gao, R., Li, Y., Kim, H., Mobley, J. K., and Ralph, J. (2018). Selective oxidation of lignin model compounds. ChemSusChem 11, 2045–2050. doi: 10.1002/cssc.201800598
Jaufurally, A. S., Teixeira, A. R. S., Hollande, L., Allais, F., and Ducrot, P. H. (2016). Optimization of the laccase-catalyzed synthesis of (±)-syringaresinol and study of its thermal and antiradical activities. ChemistrySelect 1, 5165–5171. doi: 10.1002/slct.201600543
Jiang, H., Yang, L., Ma, G. X., Xing, X. D., Yan, M. L., Zhang, Y. Y., et al. (2017). New phenylpropanoid derivatives from the fruits of xanthium sibiricum and their anti-inflammatory activity. Fitoterapia 117, 11–15. doi: 10.1016/j.fitote.2016.12.007
Kishimoto, T., Uraki, Y., and Ubukata, M. (2006). Chemical synthesis of B-O-4 type artificial lignin. Org. Biomol. Chem. 4, 1343–1347. doi: 10.1039/b518005h
Lancefield, C. S., and Westwood, N. J. (2015). The synthesis and analysis of advanced lignin model polymers. Green Chem. 17, 4980–4990. doi: 10.1039/C5GC01334H
Lee, T. W., and Yang, J. W. (2018). Transition-metal-free, conversion of lignin model compounds to high-value aromatics: scope and chemoselectivity. Green Chem. 20, 3761–3771. doi: 10.1039/C8GC01886C
Lou, L. L., Yao, G. D., Wang, J., Zhao, W. Y., Wang, X. B., Huang, X. X., et al. (2018). Enantiomeric neolignans from picrasma quassioides exhibit distinctive cytotoxicity on hepatic carcinoma cells through ros generation and apoptosis induction. Bioorgan. Med. Chem. Lett. 28, 1263–1268. doi: 10.1016/j.bmcl.2018.03.043
Moreaux, M., Bonneau, G., Peru, A. A. M., Brunissen, F., Janvier, M., Haudrechy, A., et al. (2019). High-yielding diastereoselective syn-dihydroxylation of protected HBO: an access to D-(+)-Ribono-1,4-Lactone and 5-O-protected analogues. Eur. J. Organ. Chem. 7, 1600–1604. doi: 10.1002/ejoc.201801780
Mouterde, L. M. M., Flourat, A. L, Cannet, M. M. M., Ducrot, P.-H., and Allais, F. (2013). Chemoenzymatic total synthesis of a naturally occurring (5-5′)/(8′-O-4″) dehydrotrimer of ferulic acid. Eur. J. Organ. Chem. 1, 173–79. doi: 10.1002/ejoc.201201290
Ono, M., Masuoka, C., Ito, Y, and Nohara, T. (1998). Antioxidative constituents from viticis trifoliae fructus (fruit of vitex Rotundifolia L.). Food Sci. Technol. Int. Tokyo 4, 9–13. doi: 10.3136/fsti9596t9798.4.9
Patil, N. D., and Yan, N. (2016). Study of the nitroxyl radical catalyst in aerobic oxidative cleavage and functionalization of lignin model compounds. Catal. Commun. 84, 155–158. doi: 10.1016/j.catcom.2016.06.015
Rinesch, T., and Bolm, C. (2018). Cobalt-catalyzed oxidation of the β-O-4 bond in lignin and lignin model compounds. research-article. ACS Omega 3, 8386–8392. doi: 10.1021/acsomega.8b00994
Sen, B. (1952). Synthesis of lignin model compounds. J. Am. Chem. Soc. 74, 3445–3447. doi: 10.1021/ja01133a531
Shimizu, S., Posoknistakul, P., Akiyama, T., Yokoyama, T., and Matsumoto, Y. (2018). Effects of aromatic ring type on reactions subsequent to the β-O-4 bond cleavage of non-phenolic lignin model compounds under alkaline pulping conditions. J. Wood Sci. 64, 664–674. doi: 10.1007/s10086-018-1739-3
Keywords: lignin models, coniferyl alcohol, monolignol, trimers, total synthesis, C-C coupling, biocatalysis, laccase
Citation: Flourat AL, Peru AAM, Haudrechy A, Renault J-H and Allais F (2019) First Total Synthesis of (β-5)-(β-O-4) Dihydroxytrimer and Dihydrotrimer of Coniferyl Alcohol (G): Advanced Lignin Model Compounds. Front. Chem. 7:842. doi: 10.3389/fchem.2019.00842
Received: 16 August 2019; Accepted: 19 November 2019;
Published: 09 December 2019.
Edited by:
Marc Pera-Titus, UMI3464 Eco-Efficient Products and Processes Laboratory (E2P2L), Fudan University, ChinaReviewed by:
Simonetta Antonaroli, University of Rome Tor Vergata, ItalyCopyright © 2019 Flourat, Peru, Haudrechy, Renault and Allais. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amandine L. Flourat, YW1hbmRpbmUuZmxvdXJhdEBhZ3JvcGFyaXN0ZWNoLmZy; Florent Allais, ZmxvcmVudC5hbGxhaXNAYWdyb3BhcmlzdGVjaC5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.