 Luiza M. F. Gomes
Luiza M. F. Gomes Janaina C. Bataglioli
Janaina C. Bataglioli Jason R. Smith
Jason R. Smith Tim Storr
Tim Storr
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 03 December 2019
Sec. Inorganic Chemistry
Volume 7 - 2019 | https://doi.org/10.3389/fchem.2019.00838
This article is part of the Research TopicNew Strategies in Design and Synthesis of Inorganic PharmaceuticalsView all 7 articles
Alzheimer's disease (AD) is the most common form of dementia, leading to loss of cognition, and eventually death. The disease is characterized by the formation of extracellular aggregates of the amyloid-beta (Aβ) peptide and neurofibrillary tangles of tau protein inside cells, and oxidative stress. In this study, we investigate a series of Ru(III) complexes (Ru-N) derived from NAMI-A in which the imidazole ligand has been substituted for pyridine derivatives, as potential therapeutics for AD. The ability of the Ru-N series to bind to Aβ was evaluated by NMR and ESI-MS, and their influence on the Aβ peptide aggregation process was investigated via electrophoresis gel/western blot, TEM, turbidity, and Bradford assays. The complexes were shown to bind covalently to the Aβ peptide, likely via a His residue. Upon binding, the complexes promote the formation of soluble high molecular weight aggregates, in comparison to peptide precipitation for peptide alone. In addition, TEM analysis supports both amorphous and fibrillar aggregate morphology for Ru-N treatments, while only large amorphous aggregates are observed for peptide alone. Overall, our results show that the Ru-N complexes modulate Aβ peptide aggregation, however, the change in the size of the pyridine ligand does not substantially alter the Aβ aggregation process.
Dementias are disorders in which severe cognitive impairment occurs (Gaggelli et al., 2006; Crouch and Barnham, 2012; DeToma et al., 2012) affecting over 50 million people worldwide (Budimir, 2011; Crouch and Barnham, 2012; WHO, 2012). An increase in life expectancy is expected to lead to a sharp increase in the number of dementia cases over the next 20 years (Alzheimer's Association, 2019). Alzheimer's disease (AD), the most common type of dementia, represents 60–70% of dementia cases (Martin Prince et al., 2015), resulting in a significant burden to healthcare systems around the globe. AD is a neurodegenerative disease where protein misfolding and aggregation combined with oxidative stress causes neuronal cell death, leading to loss of cognition and eventually death (Crouch and Barnham, 2012; Rodriguez-Rodriguez et al., 2012; Lee et al., 2014). Currently, treatment strategies for most neurodegenerative diseases are very limited, and approved treatments for AD only ameliorate the symptoms at early to moderate stages of the disease, making this an important research area (Roberson and Mucke, 2006; Adlard et al., 2009; Citron, 2010; Finder, 2010; Selkoe, 2011; Hickey and Donnelly, 2012; Soto and Pritzkow, 2018; Savelieff et al., 2019).
The major neuropathological hallmarks of AD are the aggregation of two proteins, amyloid-β (Aβ) and tau, with the first forming aggregates (oligomers and plaques) in the extracellular environment of the brain, and the latter forming neurofibrillary tangles (NFTs) in neurons due to hyperphosphorylation and oxidative modifications of tau (Um et al., 2013). It is still unclear if these hallmarks are a cause or an effect of AD, however post-mortem examination of the brain in AD patients has shown that Aβ-plaques and NFTs are present (Querfurth and LaFerla, 2010). Interestingly, smaller, soluble Aβ oligomers have been more strongly linked to memory loss and progression of the disease in comparison to plaques. These species have been implicated in the initiation of the processes of oxidative stress, decreased cerebral blood flow, neuronal hyperactivity, synapse deterioration, and nerve cell death (McLean et al., 1999; Lesne et al., 2006; Watt et al., 2013; Heffern et al., 2014; Nortley et al., 2019; Zott et al., 2019).
As cofactors in metalloenzymes, metal ions such as Zn, Cu and Fe are central to many processes in healthy organisms. However, their dyshomeostasis has been observed in neurodegenerative diseases, such as AD (Curtain et al., 2001; Sung et al., 2006; Brown, 2009; Kepp, 2012; Savelieff et al., 2013; Hane and Leonenko, 2014; Ward et al., 2015). A high concentration of these metal ions are present in Aβ plaques (Savelieff et al., 2013), where they are found coordinated typically to His6, 13, or14 residues, although Asp1, Tyr10, and Glu11 have been shown to be involved in Aβ peptide metal binding (Miller et al., 2010, 2012; Parthasarathy et al., 2011; Hane and Leonenko, 2014; Heffern et al., 2014; Wineman-Fisher et al., 2016). This binding can modify the aggregation pattern of Aβ, disrupt normal metalloenzyme activity, and produce toxic reactive oxygen species (ROS) (Bousejra-ElGarah et al., 2011; Lakatos et al., 2012; Pithadia et al., 2012; Hane and Leonenko, 2014; Heffern et al., 2014; Leong et al., 2014; Ward et al., 2015).
A number of Pt (Barnham et al., 2008; Sasaki et al., 2012; Collin et al., 2013; Streltsov et al., 2013), Ru (Valensin et al., 2010; Messori et al., 2013; Jones et al., 2015), Co (Suh et al., 2007; Heffern et al., 2014; Derrick et al., 2017), and V (He et al., 2015) metal complexes have shown promise in interacting with the Aβ peptide and modifying its aggregation. For example, a series of Pt(II) phenanthroline complexes (Chart 1) were shown to bind to the peptide, modulating the aggregation and the neurotoxicity of Aβ (Barnham et al., 2008). The phenanthroline ligands were determined to facilitate π-π stacking interactions with Phe4, Tyr10, and Phe19 residues present in the hydrophobic region of the peptide, thus positioning the Pt(II) center in proximity to His residues (His6,13,and14) for covalent bond formation (Yao et al., 2004). In comparison, cisplatin (Chart 1) without large hydrophobic ligands, was shown to interact with Met35 (Barnham et al., 2008). For the Pt(II) phenanthroline complexes the modulation of aggregation was associated with almost complete rescuing of cell viability in primary cortical neurons, while cisplatin was inactive, demonstrating that the presence of phenanthroline ligands was essential for limiting Aβ toxicity. Barnham et al. have suggested that by coordinating to His residue(s) the Pt phenanthroline complexes inhibit the binding of ROS-generating metal ions to Aβ, such as Cu(II). This was demonstrated by a decrease in the production of H2O2 by Aβ-Cu in the presence of these complexes.
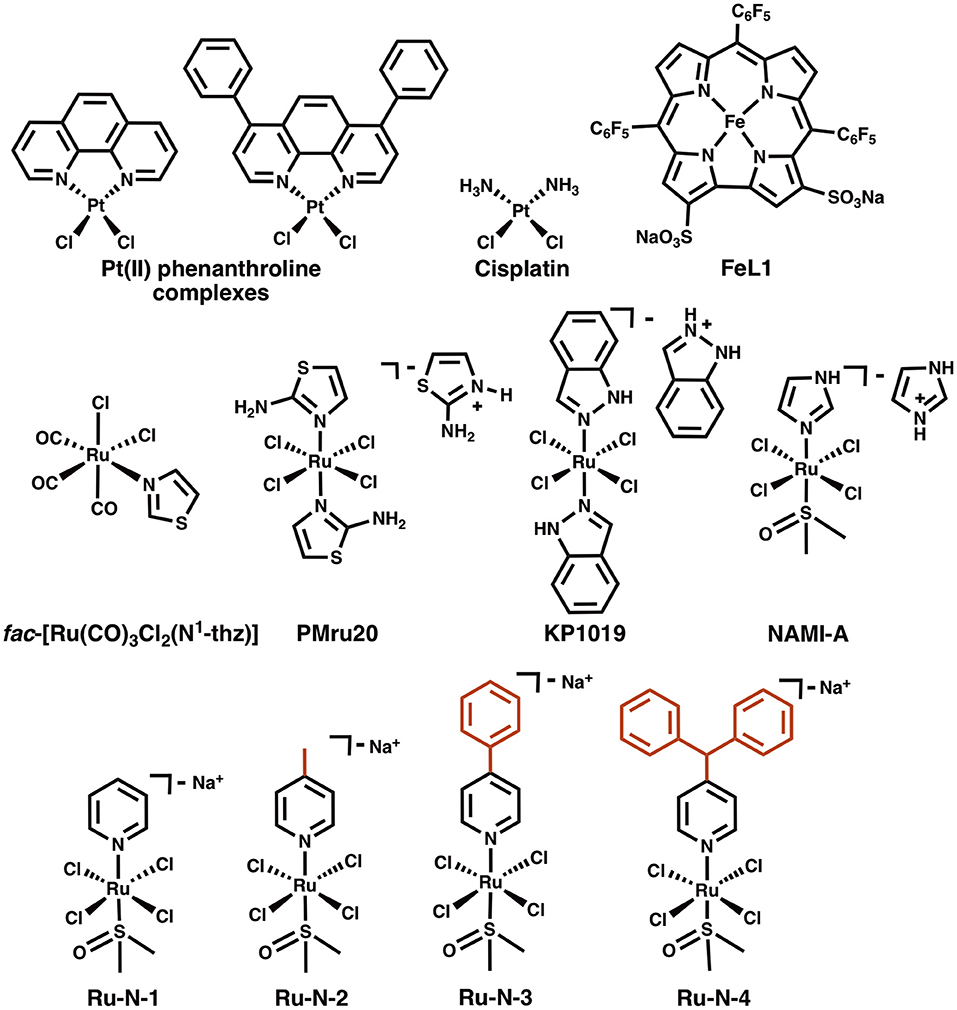
Chart 1. Structures of Pt(II) phenanthroline complexes, cisplatin, a Fe(III) corrole complex (FeL1), fac-[Ru(CO)3Cl2(N1-thz)], and Ru(III) complexes PMru20, KP1019, NAMI-A, and Ru(III) complexes derived from NAMI-A (Ru-N series).
Ru(III) complexes have been investigated in anticancer research based on their cytotoxicity, relatively slow ligand exchange rate (similar to Pt(II)) (Reedijk, 2008), accessible redox chemistry in physiological conditions, and the ability to tune targeting and pharmacokinetic properties via ligand design (Webb et al., 2013). Keppler et al. were the first to report the use of Ru(III) complexes with axial azole ligands as cancer therapeutics in the 1990's (Lipponer et al., 1996; Peti et al., 1999). One of the most promising agents studied by this group was KP1019 (Chart 1) (Henke et al., 2009), which was tested in a phase I clinical trial (Hartinger et al., 2008). Recently, an analog of this compound, NKP-1339, with a Na+ counterion to improve water solubility, has been the focus of further development, and has also completed a phase I clinical trial (Thompson et al., 2012; Trondl et al., 2014). A second type of structurally similar Ru(III) complexes were also developed during the same time period by Alessio and co-workers (Mestroni et al., 1994; Alessio et al., 2004). These compounds have an exchangeable DMSO ligand in place of one of the axial azoles of the Keppler-type complexes. Of these, the imidazole complex NAMI-A (Chart 1) has been the most studied. This compound demonstrates less cytotoxicity than KP1019, but displays a significant antimetastatic effect, thus NAMI-A -type complexes have also been a focus for development as anticancer agents (Bergamo and Sava, 2007; Webb et al., 2012; Alessio, 2017). NAMI-A was the first Ru(III) anticancer drug to be studied in humans (Alessio, 2017), and successfully completed a phase I clinical trial, although a phase II trial demonstrated that it is only moderately tolerated according to common toxicity criteria (CTC) (Leijen et al., 2015).
The concept of Ru complexes as AD treatment agents was introduced by Valensin et al. with the report of the interaction of fac-[Ru(CO)3Cl2(N1-thz)] (Chart 1) with Aβ (Valensin et al., 2010), showing that the complex loses N1-thz and both Cl− ligands and the unit binds to a His of the peptide. The anticancer agents PMru20 and KP1019 were also studied as a potential AD therapeutics (Messori et al., 2013). PMru20 protected rat cortical neurons from toxicity associated with both Aβ1−42 and the truncated Aβ25−35 (without His), likely by limiting peptide aggregation. KP1019 was shown to bind covalently to Aβ by modulating the peptide aggregation pattern of monomeric or pre-formed aggregates and forming soluble high-MW aggregates (Jones et al., 2015). KP1019 also limited Aβ toxicity in SH-SY5Y neuroblastoma cells.
A series of Ru(III) pyridine NAMI-A analogs (Ru-N, Chart 1) was reported by Walsby et al. to bind to human serum albumin (HSA), to which the use of suitable axial ligands enables tuning of the non-covalent interaction between the complexes and HSA (Webb et al., 2012). The Ru-N derivatives exhibited enhanced hydrophobic interactions with HSA when larger, more hydrophobic, axial pyridine-based ligands were incorporated into the NAMI-A type structure. As expected for these types of compounds, their axial DMSO ligand underwent rapid aqueous exchange at physiological pH, with loss of Cl− ligands also observed. These ligand exchange processes also promoted the formation of covalent interactions with HSA, likely to His residues. Based on these observations and the previous studies described above, we hypothesized that alteration of the axial ligand in the Ru-N series would influence the interaction of these complexes with the Aβ peptide, with more effective peptide binding for the larger, more hydrophobic derivatives. The interaction of these Ru(III) complexes with the Aβ peptide and the associated effect on peptide aggregation are described herein.
All common chemicals were purchased from Aldrich and used without further purification. All Ru complexes, Ru-N-1, Ru-N-2, Ru-N-3, and Ru-N-4 were synthesized as reported (Webb et al., 2012). The Aβ1−16, and Aβ1−42 peptides were purchased from 21st Century Biochemicals (Marlborough, MA, USA), and Cellmano Biotech Limited (Hefei, China), and monomerized before use according to a reported procedure (Sabate et al., 2003; Pachahara et al., 2012). Aβ1−16 was dissolved in double distilled H2O (ddH2O), while Aβ1−42 was dissolved in DMSO and ddH2O in a 1:1 mixture, unless stated otherwise. The stock peptide solution concentration was determined by absorbance with the use of a Thermo Nicolet UV nanodrop and an extinction coefficient of 1,410 and 1,450 M−1cm−1 at 280 nm for Aβ1−16, and Aβ1−42 respectively (Guilloreau et al., 2007; Coalier et al., 2013). Turbidity assays were measured using a Synergy 4 Multi-Detection microplate reader from BioTek. 1H NMR spectra were recorded on a Bruker AV-600 instrument. TEM images were obtained using an OSIRIS FEI scanning TEM (STEM) operating at 200 kV.
Deuterated phosphate buffered saline (PBS) (0.01 M Na2HPO3, 0.001 M KH2PO4, 0.14 M NaCl, 0.003 M KCl, pH 7.4) was prepared by removal of water by vacuum drying of PBS and dissolving the powder in D2O. Aβ1−16 was dissolved in deuterated PBS (0.01 M, pH 7.4), and Ru-N-1 and Ru-N-4 complexes were dissolved in DMSO-d6 and added to Aβ1−16 at 0.25 and 1 eq. at 10% of DMSO and the 1H NMR spectra were collected after approximately 15 min of incubation.
ESI-TOF-MS experiments were performed on an Agilent 6130 mass spectrometer connected to an Agilent 1260 HPLC system. Samples were analyzed by direct infusion (1-4 μL) of analyte into a mobile phase of 1:1 water:acetonitrile containing 5 mM ammonium acetate (pH unmodified), flowing at 0.3 mL/min and maintained at 30°C. All components of the mobile phase were mass spectrometry grade. Nitrogen drying gas was heated to 250°C, and run at 5 L/min with a nebulizing pressure of 15 psig. Voltages were: capillary 3 kV, fragmentor 175 V, skimmer 30 V, octopole 250 V. Samples were prepared as ~4 mg/mL of total protein (Aβ1−16) in ammonium carbonate (0.02 M, pH 9) buffer with 0 or 1 equivalents of the Ru-N complexes.
Aβ solutions with a concentration of 25 μM were prepared in PBS (0.01 M, pH 7.4) then incubated at 37°C with continuous agitation at 200 rpm to form aggregates in the presence of Ru-N complexes or pyridine ligands at 1 eq. Samples were collected at 3, 6, 11, and 24 hour time points. Concentration-dependent modulation of Aβ aggregation was also evaluated after 24 hour incubation for Ru-N-1 and Ru-N-4 (0.25, 0.5, 1, and 2 equivalents). Electrophoresis separation of peptide aggregates was completed using 8–16% Mini-PROTEAN® TGX Precast Gels from Bio-Rad, at 100 V for 100 min. The gels were then transferred to a nitrocellulose membrane for 1 hour at 100 V at 4°C, followed by blocking of the membrane in a 3% BSA solution in Tris-buffered saline (TBS) (0.02 M Tris, 0.15 M NaCl, 0.003 M KCl) for 1 h. The membrane was incubated in a solution (1:2,000 dilution) of 6E10 anti-Aβ primary antibody (Biolegends) overnight. After washing 5 × 5 min with TBS, the membrane was incubated in a solution containing the secondary antibody (Horseradish peroxidase, Caymen Chemicals) for 3 h. A Thermo Scientific SuperSignal® West Pico Chemiluminescent Substrate kit was used to visualize the Aβ species using a BioRad ChemiDocTM MP imaging system.
Samples were prepared from the Western blot assay after the 24 h incubation time at 37°C. TEM grids were prepared following previously reported methods (Jones et al., 2012). In order to increase hydrophilicity of the Formvar/Carbon 300-mesh grids (Electron Microscopy Sciences), the grids were glow discharged in a vacuum for 10 s. Drops of samples (10 μL) were placed onto a sheet of parafilm and the TEM grid was laid on the drop for 5 min. The grid was then placed on a drop of syringe-filtered 5% uranyl acetate and then immediately removed. This process was then repeated for a second drop of 5% uranyl acetate. Finally, the grid was placed on a third drop of 5% uranyl acetate and incubated for 1 min. Excess uranyl acetate was removed using a tissue between drops. The grid was allowed to air-dry for at least 15 min. Bright-field images were obtained on a FEI Tecnai Osiris STEM at 200 kV.
The turbidity assay was conducted in quadruplicate in flat-bottomed 96-well assay plates (Microtest, BD Falcon). Aβ1−42 peptide and Ru-N complexes had final concentrations of 10 μM. Ru-N complexes were dissolved in DMSO and further diluted to obtain the correct concentration. The absorbance at 500 nm was measured every 10 min for 3 h at 37°C under constant agitation using a Synergy 4 Fluorometer plate reader from BioTek. For the 20 h experiment, the samples were incubated at 37°C with constant agitation with a lid on to prevent evaporation and then the turbidity was measured.
The Bradford assay (Thermo Scientific) measures the absorbance at 595 nm of Coomassie brilliant blue G-250 as it binds to protein in duplicate in a flat-bottomed 96-well assay plate (Microtest, BD Falcon). Sixty microliter solutions of Aβ1−42 in the presence of 1 eq. of Ru-N complexes were incubated at 37°C for 24 h under constant agitation. A 30 μL sample was removed at the beginning of the experiment as the 0 hour time point, and kept frozen at −80°C until time for absorbance reading. Samples were centrifuged prior to reading of the assay to remove insoluble fibrils (Mok and Howlett, 2006). Measurements of absorbance used a Synergy 4 Fluorometer plate reader from BioTek. Samples were measured in duplicate, and statistics completed using the PRYSM program and ANOVA.
To evaluate the nature of the interactions between the Ru-N complexes and the Aβ peptide, 1H NMR of Aβ1−16 in the presence of paramagnetic (Ru(III), d5, S = ½) Ru-N-1 or Ru-N-4 were obtained at 0.25 and 1 equivalents (Figure 1). These complexes were selected as they exhibit the largest difference in pyridine ligand size. In addition, the non-aggregating Aβ1−16 peptide fragment was used, which includes the metal binding amino acid residues. Upon addition of either Ru(III) complex, all signals for the residues in Aβ1−16 exhibit a shift, suggesting that an interaction between peptide and complex is occurring. Interestingly there is a significant decrease in the intensity as well as a broadening of the signals of Aβ1−16 in the presence of 1 eq of the paramagnetic complexes. We do not observe a precipitate in the NMR tube in our experiments. The largest shift (ca. 0.1 ppm) observed is for the His resonance at 7.85 ppm, which suggests binding of a peptide His residue. This mode of coordination has also been reported for interaction of these complexes with HSA (Webb et al., 2012).
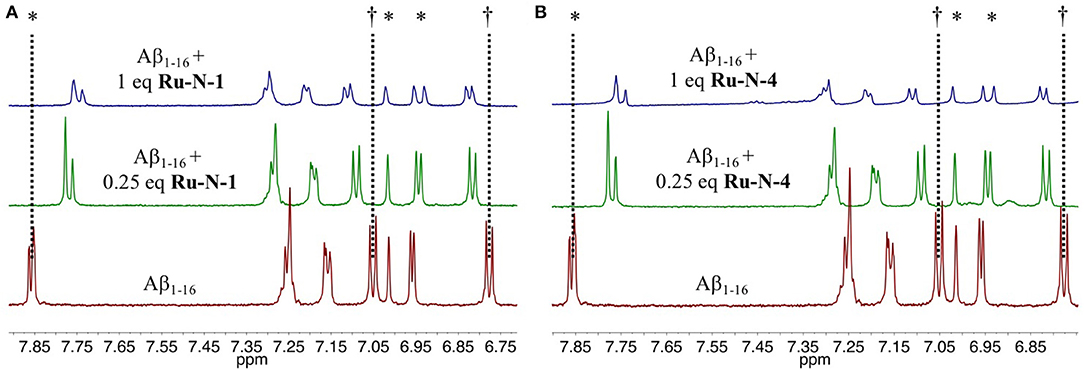
Figure 1. Changes in the 1H NMR spectra of Aβ1−16 in the presence of Ru-N derivatives. Shown are spectra obtained from 205 μM Aβ1−16, in pH 7.4 PBS/D2O buffer at 25°C (red) with addition of 0.25 eq. (green) and 1 eq (blue) of (A) Ru-N-1 or (B) Ru-N-4. *His6, His13 and His14. †Tyr10.
To investigate further the interaction between the complexes and the peptide, ESI mass spectrometry was performed on solutions of Aβ1−16 incubated with either Ru-N-1 or Ru-N-4. The mass spectra (Figure S1, Figures 2A,C, respectively) indicate the formation of the adducts [Ru-N-1(Aβ1−16)(CO3)]2− (m/z = 1167.5) and [Ru-N-4(Aβ1−16)(CO3)]2− (m/z = 1250.9), where carbonate () in the adducts is likely derived from the running buffer ([NH4]2+ [CO3]2−) used in the MS experiment. The characteristic Ru isotopic pattern was observed for both peaks (Figures 2B,D), and the masses of the adducts are consistent with loss of the DMSO ligand from each Ru complex, and subsequent coordination to the Aβ1−16 peptide.
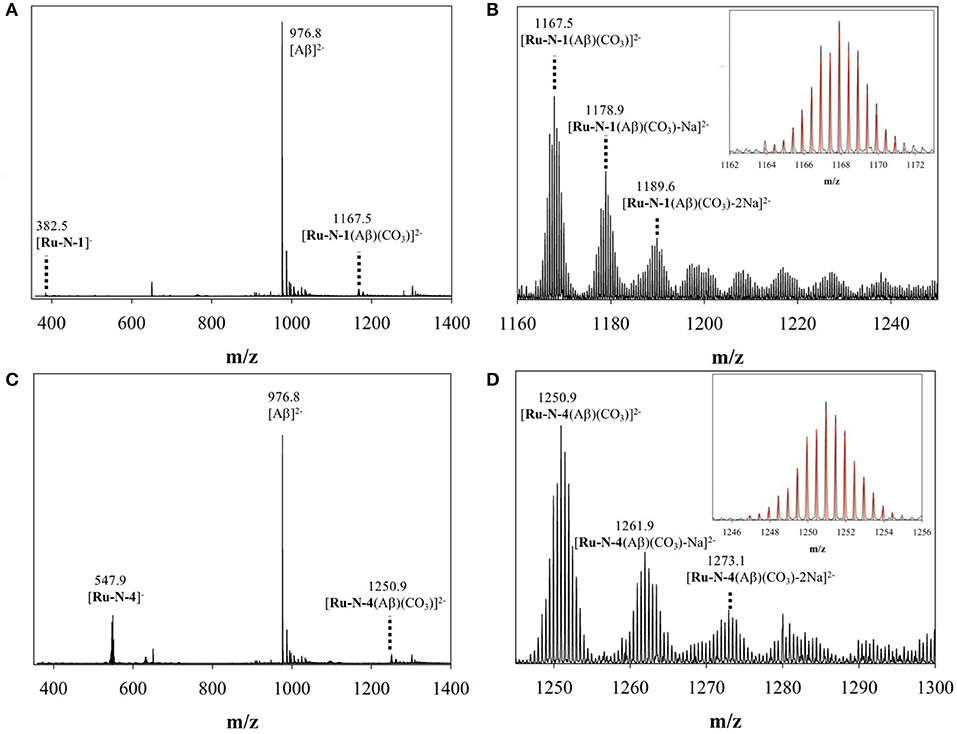
Figure 2. ESI-MS of (A) binding of Aβ1−16 to Ru-N-1; (B) zoomed region of [Ru-N-1(Aβ)(CO3)]2− with associated Na adducts; and (C) binding of Aβ1−16 to Ru-N-4; (D) zoomed region of [Ru-N-4(Aβ)(CO3)]2− with associated Na adducts. Across all experiments, species were observed as [Ru-N(0−1)(Aβ)(CO3)Na(0−6)]2−.
The time-dependent influence of the Ru-N derivatives on Aβ1−42 aggregation was analyzed via gel electrophoresis and western blotting, in combination with Transmission Electron Microscopy (TEM). The Aβ1−42 peptide was chosen for these experiments due to its high propensity for aggregation and significant neurotoxicity (Gong et al., 2003; Haass and Selkoe, 2007; Walsh and Selkoe, 2007; Jakob-Roetne and Jacobsen, 2009; Kepp, 2012; Nortley et al., 2019; Zott et al., 2019). At each time point (3, 6, 11, and 24 h) a 30 μL aliquot was removed from the stock incubation solution for each treatment and kept at −80°C until further analysis. An increase in high MW aggregates over time was observed for Aβ1−42 alone (Lane 1, Figure 3), with a significant decrease in soluble Aβ species at 24 h, as expected based on prior results (Jones et al., 2015; Gomes et al., 2019). The Ru-N derivatives do not significantly affect aggregation at the 3 h timepoint. However, at longer timepoints the complexes generate increased soluble higher molecular weight species in comparison to peptide alone. This effect is most pronounced for the complexes Ru-N-3 (lane 4) and Ru-N-4 (lane 5), which have the largest pyridine-derived ligands. The Ru(III) complexes containing smaller pyridine-derived ligands, such as Ru-N-1 (lane 2) and Ru-N-2 (lane 3), show a similar modulation of Aβ aggregation to NAMI-A (lane 6), which could reflect the similar properties of the pyridine, 6-methyl-pyridine, and imidazole ligands in this assay. Interestingly, the Na[Ru(DMSO)2Cl4] complex without an apical aza ligand also induces the formation of soluble high molecular weight Aβ species after 24 h incubation (Figure S2), however the molecular weight range is larger (~25–250 kDa), even in comparison to Ru-N-1. Overall, these results indicate that in comparison to the formation of insoluble peptide aggregates for peptide alone at 24 h, 1 eq. of the Ru-N complexes promotes the formation of soluble high molecular weight aggregates.
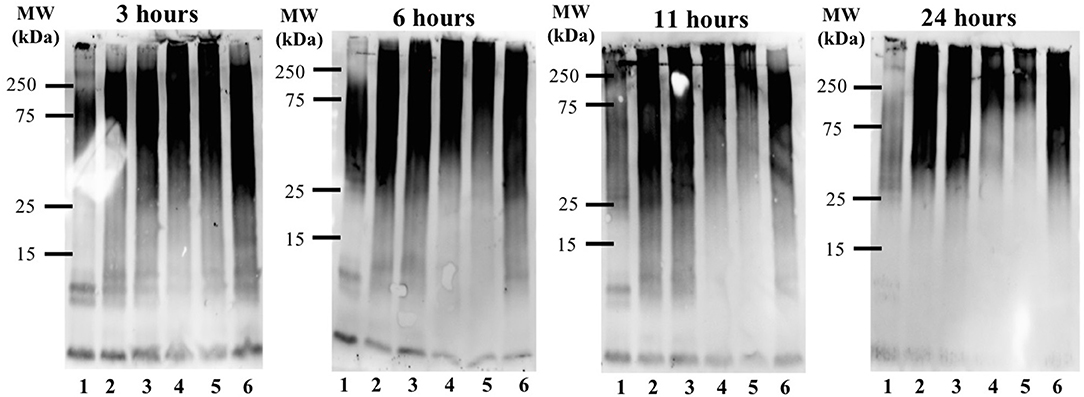
Figure 3. Influence of Ru-N derivatives on the aggregation profile of Aβ1−42. Gel electrophoresis/Western blot of 25 μM Aβ1−42 and 1 eq. of Ru-N derivatives in PBS buffer (0.01 M, pH 7.4) at incubation timepoints 3, 6, 11, and 24 h, with constant agitation at 37°C, using anti-Aβ antibody 6E10. Lane 1: Aβ1−42; lane 2: Aβ1−42 + Ru-N-1; lane 3: Aβ1−42 + Ru-N-2; lane 4: Aβ1−42 + Ru-N-3; lane 5: Aβ1−42 + Ru-N-4; lane 6: Aβ1−42 + NAMI-A.
In order to determine if the pyridine ligands alone can influence Aβ aggregation, Aβ1−42 aggregation was evaluated at 3, 6, 11, and 24 h by gel electrophoresis and western blotting in the presence of 1 eq. of the free pyridine ligands. As expected, a decrease in monomeric species and an increase in high MW species is observed for peptide alone over the incubation period (Figure S3). Interestingly, the presence of 1 eq. of the pyridine ligands does not significantly change the Aβ1−42 aggregation pattern (Figure S3), indicating that the Ru(III) complex, and not the pyridine ligand, is essential for influencing Aβ peptide aggregation.
TEM images (Figure 4 and Figure S5) of Aβ1−42 alone and in the presence of either Ru-N-1 or Ru-N-4 after incubation for 24 h show different morphologies for the three samples analyzed. Aβ1−42 incubated alone led to the formation of large amorphous aggregates, with no fibrils observed on the TEM grid. The presence of the Ru-N complexes led to an increase in fibril formation, with both Ru-N-1 and Ru-N-4 showing a mixture of fibrils and amorphous aggregates. The size of the amorphous aggregates are however much larger for peptide alone in comparison to treatment with the Ru-N derivatives (Figure S5). These results are consistent with the electrophoresis gels (Figure 3) that show that most of the peptide has formed large insoluble aggregates for peptide alone, while the Ru-N series show stabilization of smaller soluble aggregates. The concentration-dependent effect of the Ru complexes (Ru-N-1 and Ru-N-4) on Aβ1−42 aggregation was also investigated at the 24-h timepoint. In this case, the complexes Ru-N-1 and Ru-N-4 were added to Aβ1−42 at 0.25, 0.5, 1, and 2 eq. and the aggregation pattern investigated by gel electrophoresis and western blotting (Figure 5). Lane 1 shows high MW aggregates for peptide alone after 24 h of incubation. The presence of Ru-N-1 has a concentration-dependent effect on aggregation, with aggregates of 25 kDa and higher for 0.25, 0.5, and 1 eq., whereas 2 eq. leads to formation of aggregates of ca. 150 kDa and higher. Interestingly, Ru-N-4 shows a more pronounced concentration-dependent change in Aβ aggregation, with incubation of 1 eq. of Ru-N-4 resulting in aggregates of ca. 150 kDa or higher and 2 eq. affording aggregates higher than ca. 250 kDa in MW. These results indicate a greater shift to high MW aggregates for Ru-N-4 (incorporating the bulky pyridine-derived ligand) than for Ru-N-1.

Figure 4. Influence of Ru-N-1 and Ru-N-4 on the aggregation profile of Aβ1−42. TEM of Aβ1−42 alone (A), Aβ1−42 with 1 eq. of Ru-N-1 (B), and Aβ1−42 with 1 eq. of Ru-N-4 (C) incubated for 24 h with agitation at 37°C. Box shows where higher magnification image was obtained.
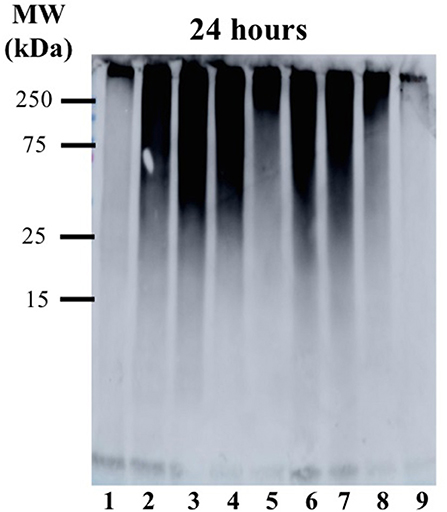
Figure 5. Gel electrophoresis/Western blot of 25 μM Aβ1−42 and different concentrations of Ru-N-1 and Ru-N-4 in PBS buffer (0.01 M, pH 7.4) at 24 h incubation with agitation at 37°C, using anti-Aβ antibody 6E10. Lane 1: Aβ1−42; lane 2: Aβ1−42 + 0.25 eq. Ru-N-1; lane 3: Aβ1−42 + 0.5 eq. Ru-N-1; lane 4: Aβ1−42 + 1 eq. Ru-N-1; lane 5: Aβ1−42 + 2 eq. Ru-N-1; lane 6: Aβ1−42 + 0.25 eq. Ru-N-4; lane 7: Aβ1−42 + 0.5 eq. Ru-N-4; lane 8: Aβ1−42 + 1 eq. Ru-N-4; lane 9: Aβ1−42 + 2 eq. Ru-N-4.
The Aβ aggregation process in solution can be studied by a number of different methods, including turbidity (Storr et al., 2007; Gomes et al., 2014; Barykin et al., 2018), dynamic light scattering (Davis et al., 2009; Nichols et al., 2015), and Thioflavin T (ThT) fluorescence (Barnham et al., 2008; Jones et al., 2015). We have previously shown that the Ru(III) complex KP1019 interferes with ThT fluorescence analysis (either by quenching or inhibition of ThT binding) (Jones et al., 2015), however, turbidity has been shown to be a reliable alternative for the investigation of peptide aggregation in the presence of compounds that disrupt ThT fluorescence (Cook and Martí, 2012). We thus employed turbidity measurements here to investigate the effect of Ru-N-1 and Ru-N-4 on Aβ1−42 aggregation in solution. The formation of peptide aggregates in solution over time leads to an increase in turbidity, and the degree of light scattering can be measured by UV-vis measurements (Gomes et al., 2014). The results of the time-dependent aggregation of Aβ1−42 in the presence of Ru-N by electrophoresis and western blot (Figure 3) show that the complexes appear to induce the formation of soluble higher MW aggregates after 24 h of incubation. At this time point the aggregation profiles for Ru-N-1 and Ru-N-4 differ indicating an effect of the axial ligands. In order to further evaluate the influence of the Ru-N series on peptide aggregation in solution, the turbidity of an Aβ1−42 solution was measured in quintuplicate in a 96-well plate over the course of 3 h in the presence and absence of Ru-N-1 and Ru-N-4 (Figure 6A). Aggregation was monitored at 500 nm as there is no absorption by either the Ru complexes or the peptide at this wavelength. As expected, an increase in turbidity was observed for the peptide alone over the 3-h incubation period. In the presence of the Ru(III) complexes an increase in turbidity was also observed, and at the 2 h timepoint the presence of Ru-N-1 and Ru-N-4 results in a significant increase in turbidity in comparison to peptide alone, but with no statistical difference between the two complexes. Due to water evaporation from the 96-well plate at longer measurement times, a lid was placed on the plate at 3 h, and a further single reading taken at the 20 h timepoint (Figure 6B). At the longer timepoint an approximate doubling of the turbidity is observed for solutions containing the Aβ1−42 peptide and either Ru-N-1 and Ru-N-4 complexes in comparison to peptide alone. Again, no statistical difference between the two complexes was observed (Chart 1). Overall, the higher turbidity reading for the Ru-N complexes in comparison to peptide alone is likely due to the formation of a large number of soluble aggregates for the former, while fewer insoluble peptide aggregates form for the latter. This conclusion is in accord with the gel studies, and TEM data. We next investigated the total amount of Aβ1−42 peptide after incubation with and without the Ru-N complexes via the Bradford assay. The Bradford assay measures the shift in the absorbance peak for the reagent Coomassie brilliant blue G-250 from 495 nm to 595 nm upon binding to the C-terminus of proteins (Bradford, 1976). Before measurement the samples were centrifuged to remove insoluble fibrils using an established protocol (Wang et al., 2002; Mok and Howlett, 2006). We analyzed the change in the concentration of Aβ1−42 between 0 and 24 h of incubation in the presence of the four Ru-N complexes. The peptide alone does not show a significant decrease in the amount of peptide at 24 h of incubation (Figure 7), suggesting that the aggregates at this stage are non-fibrillar. This result is in accord with TEM images showing only amorphous aggregates for peptide alone (Figure 4). In contrast, after 24 h of incubation in the presence of the Ru-N complexes, a decrease in the amount of peptide is observed, suggesting that fibrillar species had formed and were removed via centrifugation. This result is also in accord with fibril formation observed for Ru-N treatments by TEM. Overall, the complexes reduce the amount of measurable Aβ in the sample by 50% after 24 h, with no significant difference observed across the Ru-N series.
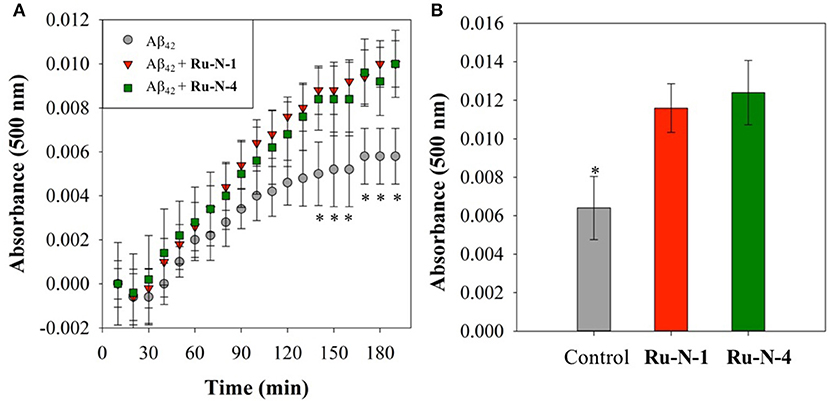
Figure 6. Turbidity assay data from 10 μM Aβ1−42 (gray) and with 1 eq. of Ru-N-1 (red) or Ru-N-4 (green) in PBS buffer (0.01 M, pH 7.4) with agitation at 37°C. (A) Change in absorbance during initial 3 h of incubation. (B) Change in absorbance at 20 h of incubation. *Statistically significant difference between Aβ1−42 only and in the presence of the complexes, p < 0.05 for (A) and p = 0.01 for (B). Calculated using JMP13 (Wilcoxon method).
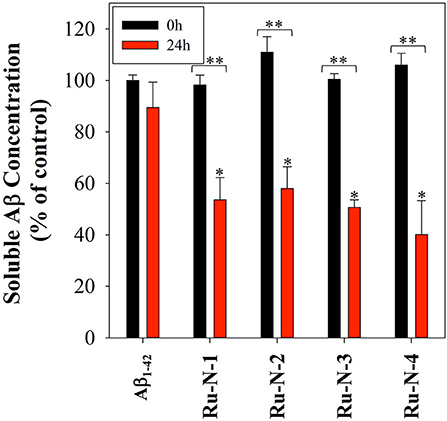
Figure 7. Bradford assay of 60 μM Aβ1−42 in the presence of 1 eq. of all four Ru-N complexes in PBS buffer (0.01 M, pH 7.4) at 0 hour (black) and after 24 h of incubation with agitation at 37°C (red). Samples were centrifuged at 14,000 g for 5 min prior to absorbance measurement. Statistically significant difference between *Aβ1−42 only and in the presence of the complexes (Ru-N-2, p = 0.0025, Ru-N-1 and Ru-N-3, p = 0.0005, and Ru-N-4, p < 0.0001) **0 and 24 h time point (Ru-N-1, p = 0.0003, and Ru-N-2, Ru-N-3, and Ru-N-4, p < 0.0001). Calculated using 2 way ANOVA.
The modulation of Aβ peptide aggregation in the presence of the Ru-N series of complexes, as well as the ability of these complexes to bind to the peptide, have been described in this study. The Ru-N series contains four NAMI-A derivatives with pyridyl ligands of different sizes (Chart 1), and it was hypothesized that an enhanced interaction with the Aβ peptide would occur for the larger more hydrophobic derivatives. Both non-covalent and covalent interactions of these complexes with HSA have been characterized by Walsby et al., using electron paramagnetic resonance (EPR) (Webb et al., 2012). HSA has 16 His residues, of which 5 His residues are available at the surface of the protein (Hnizda et al., 2008), providing binding sites for metal ions. The major species formed upon incubation of HSA with the Ru-N series are His adducts at both the axial and equatorial positions following the loss of DMSO or Cl ligands (Webb et al., 2012). Interestingly, the interaction of NAMI-A with a number of proteins including lysozyme (Messori and Merlino, 2014), carbonic anhydrase (Casini et al., 2010), and human H-chain ferritin (Ciambellotti et al., 2018) has been studied by X-ray crystallography (Alessio and Messori, 2019). In these studies all of the original ligands of NAMI-A are released, and the resulting Ru(III) center is bound to the protein via His, Asp, and Glu side-chains. However, it has been postulated that the process of crystal soaking, in which NAMI-A crystallizes with the protein, can lead to different binding/speciation in comparison to solution studies (Alessio, 2017). Ru-N-1 (also called AziRu) has also been reported to exchange all ligands when binding to lysozyme (Vergara et al., 2013a), and RNase A (Vergara et al., 2013b). The binding site for lysozyme involves His15, Arg14, and Asp87, while Ru-N-1 binds to RNase A via a single His. Interestingly, even though RNase A contains four solvent-exposed His, only one Ru-His adduct is formed per RNase molecule, to which water molecules complete the distorted octahedral coordination sphere.
The results of the ESI-MS studies herein are consistent with the prior work with HSA (Webb et al., 2012), showing adduct formation for both Ru-N-1 and Ru-N-4 with Aβ1−16 via loss of an exchangeable DMSO ligand. In addition, incubation of Ru-N-1 or Ru-N-4 with Aβ1−16 led to a shift and broadening of all of the 1H NMR signals of the Aβ peptide, suggesting an interaction between the Ru(III) complexes and Aβ1−16 (Figure 1). Similar line broadening of Aβ 1H NMR signals has been observed in the presence of Cu(II) (Eury et al., 2011) and an Fe(III) corrole complex (FeL1, Chart 1) (Gomes et al., 2019), along with the disappearance or shifting of the His resonances. This has been interpreted as binding of either Cu(II) or FeL1 to His residues present in the hydrophilic portion of the peptide. In another report (Valensin et al., 2010), broadening of the 1H NMR spectrum of Aβ1−28, and the almost complete disappearance of the aromatic signals for His and Tyr, was observed upon incubation of fac-[Ru(CO)3Cl2(N1-thz)] (Chart 1) with the peptide. These results supported Aβ1−28 His binding to the Ru(II) complex with ESI-MS verification of adduct formation (Valensin et al., 2010). Although all the peptide NMR signals shift upon interaction with the Ru-N complexes in this work, the peptide His resonance at 7.85 ppm undergoes the largest change (ca. 0.1 ppm), which is consistent with what has been observed for metal ions or complexes with Aβ (Eury et al., 2011; Gomes et al., 2019). Interestingly, weak signals attributed to the free pyridine ligand at 7.35 ppm and 7.45 ppm are observed upon addition of 1 eq. Ru-N-4 to Aβ1−16 (Figure 1), and these signals increase in intensity at 24 h for 0.25 eq of Ru-N-4 (Figure S4). Pyridine ligand loss is not observed for the Ru-N-1 complex (Figure 1 and Figure S3), suggesting that pyridine ligand exchange is enhanced for the more bulky hydrophobic Ru-N-4 complex. The presence of the free pyridine ligand of Ru-N-4 upon incubation with Aβ1−16 suggests further ligand exchange processes occur for this derivative in addition to DMSO exchange, similar to the reported X-ray studies (Casini et al., 2010; Messori and Merlino, 2014; Ciambellotti et al., 2018), and this difference between Ru-N-1 and Ru-N-4 may play a role in the peptide aggregation process (vide infra).
Several metal complexes have been reported to modulate the aggregation pattern of Aβ upon binding covalently to the peptide (Collin et al., 2013; Kenche et al., 2013; Heffern et al., 2014; Jones et al., 2015; Gomes et al., 2019). For example, the binding of the Fe(III) corrole complex FeL1 (Chart 1) to Aβ lead to the stabilization of low MW oligomeric species (Gomes et al., 2019), however, binding of KP1019 (Chart 1) led to decreased oligomer formation and an increase in high MW soluble aggregates (Jones et al., 2015). The Ru(III) complexes investigated in this study have a similar effect to that observed for KP1019, leading to the formation of soluble high MW aggregates in a concentration-dependent manner. Our results also show that the binding of the Ru(III) center is essential for the change in aggregation, since the ligands alone do not exhibit an effect on the aggregation process. The electrophoresis gel/western blot data suggests a greater influence on aggregation by the Ru-N complexes with larger, more hydrophobic ligands (Ru-N-3 and Ru-N-4). In addition, the complex without the apical Py ligand, leads to a range of soluble species after 24 h aggregation, with the gel results similar to NAMI-A. The fibrillar structures shown by TEM in the presence of Ru-N-1 and Ru-N-4 when compared to the amorphous aggregates for peptide alone, suggest that binding of the complexes to Aβ promotes fibrillization of the peptide.
Additionally, incubation of the Ru-N series with the Aβ peptide for 24 h, followed by centrifugation, leads to a 50% decrease in peptide concentration in comparison to peptide alone as determined by a Bradford assay. We employed a centrifugation protocol to remove insoluble fibrils (Wang et al., 2002; Mok and Howlett, 2006), and thus the reduction in peptide measured for the Ru-N treatments is likely due to the removal of fibrillar structures, as observed by TEM. Alternatively, the Bradford assay depends on Coomassie blue binding to basic amino acids (such as His), thus it is possible that Ru-N binding to the peptide leads to the observed reduction in signal. However, we would expect to see a reduction in signal in the initial measurements due to interaction of the Ru-N complexes with the peptide if this was the case.
Overall, the Ru-N series promote the formation of soluble high molecular weight aggregates at 24 h, while peptide alone leads to almost complete precipitation of the peptide. Only minor differences are observed across the Ru-N series, with the larger more hydrophobic derivatives (Ru-N-3 and Ru-N-4) narrowing the size distribution of the soluble aggregates to higher molecular weights (Figure 3). TEM analysis (Figure 4) of the insoluble aggregates shows that while incubation of peptide alone produces very large amorphous aggregates, Ru-N treatment results in both fibrils and amorphous aggregates, with the amorphous aggregates smaller in size in comparison to peptide alone. It is possible that by stabilizing soluble high molecular weight species, the Ru-N complexes slow down the rate of peptide precipitation, thereby promoting the formation of the more ordered fibrillar structures observed by TEM. Our results suggest that increasing the pyridine ligand size/hydrophobicity even further may afford fibrillar structures exclusively, which could ultimately have a protective effect in AD by promoting the formation of a stable insoluble peptide aggregate with limited potential to furnish toxic oligomeric species (Treusch et al., 2009; Iadanza et al., 2018; Mroczko et al., 2018).
This study highlights the ability of a series of Ru(III) complexes derived from NAMI-A to interact with the Aβ peptide and modify aggregation, a known hallmark of AD. It has been shown that the DMSO ligand of the Ru-N complexes can readily be exchanged in buffer (likely for H2O), which provides a binding site for His residues when incubated with proteins, such as HSA (Webb et al., 2013). Our NMR and ESI-MS results are in accord with the previous findings of binding of metal ions or complexes to Aβ and support a covalent interaction of the Ru-N complexes with His residues of the Aβ peptide. The effect of changing the size of the pyridine-derived ligands in the Ru-N series on Aβ aggregation was also investigated, and an increase in the size and hydrophobicity of the pyridine-derived ligand leads to larger-sized aggregates. The influence of Ru-N-3 and Ru-N-4 on peptide aggregation is demonstrated to be greater than that of the smaller complexes Ru-N-1 and Ru-N-2, with a more prominent induction of soluble high MW aggregates, as demonstrated by electrophoresis gel and western blotting. A concentration-dependent modulation of aggregation was demonstrated for Ru-N-1 and Ru-N-4, where addition of 2 equivalents of the first complex has a comparable effect on peptide aggregation as 1 equivalent of the latter. Interestingly, the aggregation of Aβ1−42 alone after 24 h shows only large amorphous aggregates by TEM, while the presence of 1 equivalent of either Ru-N-1 or Ru-N-4 shows formation of smaller amorphous aggregates as well as fibrils. However, investigation of the aggregation process in solution, by turbidity analysis, does not distinguish between the Ru-N complexes in terms of peptide aggregation. The Ru-N-1 and Ru-N-4 complexes exhibit increased turbidity in comparison to peptide alone at 3 and 24 h, consistent with formation of a greater number of aggregates in comparison to peptide alone. Interestingly, all four Ru-N complexes exhibit a ca. 50% decrease in peptide concentration in comparison to peptide alone via a Bradford assay. This result is likely due to the removal of insoluble fibrils in the Ru-N samples (observed by TEM) via centrifugation. In this work we have shown that the Ru-N series undergoes ligand exchange and covalent binding to the Aβ peptide, which leads to modulation of the peptide aggregation pathway, promoting the formation of high molecular weight aggregates in solution, with both amorphous and fibrillar aggregate morphology. Further investigation of the pharmacokinetic properties of the Ru-N complexes, and influence of these complexes on the toxicity of Aβ in cell assays, will provide insight into their therapeutic potential.
All datasets generated for this study are included in the article/Supplementary Material.
LG, JB, AJ, and JS: investigation. LG: writing—original draft preparation and visualization. TS and CW: writing—review and editing, supervision, and funding acquisition.
This research was funded by Discovery Grants from the Natural Sciences and Engineering Research Council (NSERC) (TS and CW), a Michael Smith Career Investigator Award (TS), and a New Investigator Grant from the Alzheimer's Association (NIRG-15-362537), Brain Canada (to TS). LG was funded by Science without borders (CAPES—Proc. n° 0711/13-6, Brazil) and AJ received a NSERC USRA scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00838/full#supplementary-material
Adlard, P. A., James, S. A., Bush, A. I., and Masters, C. L. (2009). beta-amyloid as a molecular therapeutic target in Alzheimer's disease. Drugs of Today 45, 293–304. doi: 10.1358/dot.2009.45.4.1353853
Alessio, E. (2017). Thirty years of the drug candidate NAMI-A and the myths in the field of ruthenium anticancer compounds: a personal perspective. Eur. J. Inorg. Chem. 2017, 1549–1560. doi: 10.1002/ejic.201600986
Alessio, E., and Messori, L. (2019). NAMI-A and KP1019/1339, two iconic ruthenium anticancer drug candidates face-to-face: a case story in medicinal inorganic chemistry. Molecules 24:E1995. doi: 10.3390/molecules24101995
Alessio, E., Mestroni, G., Bergamo, A., and Sava, G. (2004). Ruthenium antimetastatic agents. Curr. Top. Med. Chem. 4, 1525–1535. doi: 10.2174/1568026043387421
Alzheimer's Association. (2019). 2019 Alzheimer's disease facts and figures. Alzheimers. Dement. 15, 321–387. doi: 10.1016/j.jalz.2019.01.010
Barnham, K. J., Kenche, V. B., Ciccotosto, G. D., Smith, D. P., Tew, D. J., Liu, X., et al. (2008). Platinum-based inhibitors of amyloid-β as therapeutic agents for Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 105, 6813–6818. doi: 10.1073/pnas.0800712105
Barykin, E. P., Petrushanko, I. Y., Kozin, S. A., Telegin, G. B., Chernov, A. S., Lopina, O. D., et al. (2018). Phosphorylation of the amyloid-beta peptide inhibits zinc-dependent aggregation, prevents Na,K-ATPase inhibition, and reduces cerebral plaque deposition. Front. Mol. Neurosci. 11:302. doi: 10.3389/fnmol.2018.00302
Bergamo, A., and Sava, G. (2007). Ruthenium complexes can target determinants of tumour malignancy. Dalton Trans. 2007, 1267–1272. doi: 10.1039/b617769g
Bousejra-ElGarah, F., Bijani, C., Coppel, Y., Faller, P., and Hureau, C. (2011). Iron(II) Binding to Amyloid-β, the Alzheimer's Peptide. Inorg. Chem. 50, 9024–9030. doi: 10.1021/ic201233b
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Brown, D. R. (2009). Metal binding to alpha-synuclein peptides and its contribution to toxicity. Biochem. Biophys. Res. Commun. 380, 377–381. doi: 10.1016/j.bbrc.2009.01.103
Budimir, A. (2011). Metal ions, Alzheimer's disease and chelation therapy. Acta Pharmaceutica 61, 1–14. doi: 10.2478/v10007-011-0006-6
Casini, A., Temperini, C., Gabbiani, C., Supuran, C. T., and Messori, L. (2010). The X-ray structure of the adduct between NAMI-A and carbonic anhydrase provides insights into the reactivity of this metallodrug with proteins. ChemMedChem 5, 1989–1994. doi: 10.1002/cmdc.201000331
Ciambellotti, S., Pratesi, A., Severi, M., Ferraro, G., Alessio, E., Merlino, A., et al. (2018). The NAMI A – human ferritin system: a biophysical characterization. Dalton Trans. 47, 11429–11437. doi: 10.1039/C8DT00860D
Citron, M. (2010). Alzheimer's disease: strategies for disease modification. Nat. Rev. Drug Disc. 9, 387–398. doi: 10.1038/nrd2896
Coalier, K. A., Paranjape, G. S., Karki, S., and Nichols, M. R. (2013). Stability of early-stage amyloid-β(1-42) aggregation species. Biochim. Biophys. Acta 1834, 65–70. doi: 10.1016/j.bbapap.2012.08.017
Collin, F., Sasaki, I., Eury, H., Faller, P., and Hureau, C. (2013). Pt(II) compounds interplay with Cu(II) and Zn(II) coordination to the amyloid-β peptide has metal specific consequences on deleterious processes associated to Alzheimer's disease. Chem. Commun. 49, 2130–2132. doi: 10.1039/c3cc38537j
Cook, N. P., and Martí, A. A. (2012). Facile methodology for monitoring amyloid-β fibrillization. ACS Chem. Neurosci. 3, 896–899. doi: 10.1021/cn300135n
Crouch, P. J., and Barnham, K. J. (2012). Therapeutic redistribution of metal ions to treat Alzheimer's disease. Acc. Chem. Res. 45, 1604–1611. doi: 10.1021/ar300074t
Curtain, C. C., Ali, F., Volitakis, I., Cherny, R. A., Norton, R. S., Beyreuther, K., et al. (2001). Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 276, 20466–20473. doi: 10.1074/jbc.M100175200
Davis, T. J., Soto-Ortega, D. D., Kotarek, J. A., Gonzalez-Velasquez, F. J., Sivakumar, K., Wu, L., et al. (2009). Comparative study of inhibition at multiple stages of amyloid-β self-assembly provides mechanistic insight. Mol. Pharmacol. 76, 405–413. doi: 10.1124/mol.109.055301
Derrick, J. S., Lee, J., Lee, S. J. C., Kim, Y., Nam, E., Tak, H., et al. (2017). Mechanistic insights into tunable metal-mediated hydrolysis of amyloid-β peptides. J. Am. Chem. Soc. 139, 2234–2244. doi: 10.1021/jacs.6b09681
DeToma, A. S., Salamekh, S., Ramamoorthy, A., and Lim, M. H. (2012). Misfolded proteins in Alzheimer's disease and type II diabetes. Chem. Soc. Rev. 41, 608–621. doi: 10.1039/C1CS15112F
Eury, H., Bijani, C., Faller, P., and Hureau, C. (2011). Copper(II) coordination to amyloid beta: murine versus human peptide. Angew. Chem. Int. Ed. English 50, 901–905. doi: 10.1002/anie.201005838
Finder, V. H. (2010). Alzheimer's disease: a general introduction and pathomechanism. J. Alzheimers Dis. 22, S5–S19. doi: 10.3233/JAD-2010-100975
Gaggelli, E., Kozlowski, H., Valensin, D., and Valensin, G. (2006). Copper homeostasis and neurodegenerative disorders (Alzheimer's, prion, and Parkinson's diseases and amyotrophic lateral sclerosis). Chem. Rev. 106, 1995–2044. doi: 10.1021/cr040410w
Gomes, L. M. F., Mahammed, A., Prosser, K. E., Smith, J. R., Silverman, M. A., Walsby, C. J., et al. (2019). A catalytic antioxidant for limiting amyloid-beta peptide aggregation and reactive oxygen species generation. Chem. Sci. 10, 1634–1643. doi: 10.1039/C8SC04660C
Gomes, L. M. F., Vieira, R. P., Jones, M. R., Wang, M. C. P., Dyrager, C., Souza-Fagundes, E. M., et al. (2014). 8-Hydroxyquinoline Schiff-base compounds as antioxidants and modulators of copper-mediated Aβ peptide aggregation. J. Inorg. Biochem. 139, 106–116. doi: 10.1016/j.jinorgbio.2014.04.011
Gong, Y. S., Chang, L., Viola, K. L., Lacor, P. N., Lambert, M. P., Finch, C. E., et al. (2003). Alzheimer's disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422. doi: 10.1073/pnas.1834302100
Guilloreau, L., Combalbert, S., Sournia-Saquet, A., Mazarguil, H., and Faller, P. (2007). Redox chemistry of copper–amyloid-β: the generation of hydroxyl radical in the presence of ascorbate is linked to redox-potentials and aggregation state. Chembiochem 8, 1317–1325. doi: 10.1002/cbic.200700111
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hane, F., and Leonenko, Z. (2014). Effect of metals on kinetic pathways of amyloid-beta aggregation. Biomolecules 4, 101–116. doi: 10.3390/biom4010101
Hartinger, C. G., Jakupec, M. A., Zorbas-Seifried, S., Groessl, M., Egger, A., Berger, W., et al. (2008). KP1019, a new redox-active anticancer agent – preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 5, 2140–2155. doi: 10.1002/cbdv.200890195
He, L., Wang, X., Zhu, D., Zhao, C., and Du, W. (2015). Methionine oxidation of amyloid peptides by peroxovanadium complexes: inhibition of fibril formation through a distinct mechanism. Metallomics 7, 1562–1572. doi: 10.1039/C5MT00133A
Heffern, M. C., Velasco, P. T., Matosziuk, L. M., Coomes, J. L., Karras, C., Ratner, M. A., et al. (2014). Modulation of amyloid-beta aggregation by histidine-coordinating Cobalt(III) Schiff base complexes. Chembiochem 15, 1584–1589. doi: 10.1002/cbic.201402201
Henke, M. M., Richly, H., Drescher, A., Grubert, M., Alex, D., Thyssen, D., et al. (2009). Pharmacokinetic study of sodium trans[tetrachlorobis(1H-indazole)-ruthenate (III)]/-indazole hydrochloride (1:1.1) (FFC14A) in patients with solid tumors. Int. J. Clin. Pharmacol. Ther. 47, 58–60. doi: 10.5414/CPP47058
Hickey, J. L., and Donnelly, P. S. (2012). Diagnostic imaging of Alzheimer's disease with copper and technetium complexes. Coord. Chem. Rev. 256, 2367–2380. doi: 10.1016/j.ccr.2012.03.035
Hnizda, A., Santrucek, J., Sanda, M., Strohalm, M., and Kodicek, M. (2008). Reactivity of histidine and lysine side-chains with diethylpyrocarbonate – a method to identify surface exposed residues in proteins. J. Biochem. Biophys. Methods 70, 1091–1097. doi: 10.1016/j.jbbm.2007.07.004
Iadanza, M. G., Jackson, M. P., Hewitt, E. W., Ranson, N. A., and Radford, S. E. (2018). A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 19, 755–773. doi: 10.1038/s41580-018-0060-8
Jakob-Roetne, R., and Jacobsen, H. (2009). Alzheimer's disease: from pathology to therapeutic approaches. Angew. Chem. Int. Ed. English 48, 3030–3059. doi: 10.1002/anie.200802808
Jones, M. R., Mu, C., Wang, M. C. P., Webb, M. I., Walsby, C. J., and Storr, T. (2015). Modulation of the Aβ peptide aggregation pathway by KP1019 limits Aβ-associated neurotoxicity. Metallomics 7, 129–135. doi: 10.1039/C4MT00252K
Jones, M. R., Service, E. L., Thompson, J. R., Wang, M. C. P., Kimsey, I. J., DeToma, A. S., et al. (2012). Dual-function triazole-pyridine derivatives as inhibitors of metal-induced amyloid-beta aggregation. Metallomics 4, 910–920. doi: 10.1039/c2mt20113e
Kenche, V. B., Hung, L. W., Perez, K., Volitakes, I., Ciccotosto, G., Kwok, J., et al. (2013). Development of a platinum complex as an anti-amyloid agent for the therapy of Alzheimer's disease. Angew. Chem. Int. Ed. 52, 3374–3378. doi: 10.1002/anie.201209885
Kepp, K. P. (2012). Bioinorganic chemistry of Alzheimer's disease. Chem. Rev. 112, 5193–5239. doi: 10.1021/cr300009x
Lakatos, A., Gyurcsik, B., Nagy, N. V., Csendes, Z., Weber, E., Fulop, L., et al. (2012). Histidine-rich branched peptides as Cu(II) and Zn(II) chelators with potential therapeutic application in Alzheimer's disease. Dalton Trans. 41, 1713–1726. doi: 10.1039/C1DT10989H
Lee, S., Zheng, X., Krishnamoorthy, J., Savelieff, M. G., Park, H. M., Brender, J. R., et al. (2014). Rational design of a structural framework with potential use to develop chemical reagents that target and modulate multiple facets of Alzheimer's disease. J. Am. Chem. Soc. 136, 299–310. doi: 10.1021/ja409801p
Leijen, S., Burgers, S. A., Baas, P., Pluim, D., Tibben, M., van Werkhoven, E., et al. (2015). Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest. New Drugs 33, 201–214. doi: 10.1007/s10637-014-0179-1
Leong, S. L., Young, T. R., Barnham, K. J., Wedd, A. G., Hinds, M. G., Xiao, Z., et al. (2014). Quantification of copper binding to amyloid precursor protein domain 2 and its Caenorhabditis elegans ortholog. Implications for biological function. Metallomics 6, 105–116. doi: 10.1039/C3MT00258F
Lesne, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357. doi: 10.1038/nature04533
Lipponer, K. G., Vogel, E., and Keppler, B. K. (1996). Synthesis, characterization and solution chemistry of trans-indazoliumtetrachlorobis(Indazole)Ruthenate(III), a new anticancer ruthenium complex. IR, UV, NMR, HPLC investigations and antitumor activity. Crystal structures of trans-1-methyl-indazoliumtetrachlorobis-(1-Methylindazole)Ruthenate(III) and its hydrolysis product trans-monoaquatrichlorobis-(1-Methylindazole)-Ruthenate(III). Metal-Based Drugs 3, 243–260. doi: 10.1155/MBD.1996.243
Martin Prince, A. W., Guerchet, M., and Gemma-Claire Ali, Yu-Tzu Wu, Prina, M. (2015). World Alzheimer Report 2015: The Global Impact on Dementia. An Analysis of Prevalence, Incidence, Cost and Trends.
McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller, S. J., Smith, M. J., Beyreuther, K., et al. (1999). Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866. doi: 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M
Messori, L., Camarri, M., Ferraro, T., Gabbiani, C., and Franceschini, D. (2013). Promising in vitro anti-Alzheimer properties for a ruthenium(III) complex. ACS Med. Chem. Lett. 4, 329–332. doi: 10.1021/ml3003567
Messori, L., and Merlino, A. (2014). Ruthenium metalation of proteins: the X-ray structure of the complex formed between NAMI-A and hen egg white lysozyme. Dalton Trans. 43, 6128–6131. doi: 10.1039/c3dt53582g
Mestroni, G., Alessio, E., Sava, G., Pacor, S., Coluccia, M., and Boccarelli, A. (1994). Water-soluble ruthenium(III)-dimethyl sulfoxide complexes: chemical behaviour and pharmaceutical properties. Met. Based Drugs 1, 41–63. doi: 10.1155/MBD.1994.41
Miller, Y., Ma, B., and Nussinov, R. (2010). Zinc ions promote Alzheimer Aβ aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. U.S.A. 107, 9490–9495. doi: 10.1073/pnas.0913114107
Miller, Y., Ma, B., and Nussinov, R. (2012). Metal binding sites in amyloid oligomers: complexes and mechanisms. Coord. Chem. Rev. 256, 2245–2252. doi: 10.1016/j.ccr.2011.12.022
Mok, Y. F., and Howlett, G. J. (2006). Sedimentation velocity analysis of amyloid oligomers and fibrils. Methods Enzymol. 413, 199–217. doi: 10.1016/S0076-6879(06)13011-6
Mroczko, B., Groblewska, M., Litman-Zawadzka, A., Kornhuber, J., and Lewczuk, P. (2018). Amyloid β oligomers (AβOs) in Alzheimer's disease. J. Neural Transm. 125, 177–191. doi: 10.1007/s00702-017-1820-x
Nichols, M. R., Colvin, B. A., Hood, E. A., Paranjape, G. S., Osborn, D. C., and Terrill-Usery, S. E. (2015). Biophysical comparison of soluble amyloid-β(1–42) protofibrils, oligomers, and protofilaments. Biochemistry 54, 2193–2204. doi: 10.1021/bi500957g
Nortley, R., Korte, N., Izquierdo, P., Hirunpattarasilp, C., Mishra, A., Jaunmuktane, Z., et al. (2019). Amyloid β oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science 365:eaav9518. doi: 10.1126/science.aav9518
Pachahara, S. K., Chaudhary, N., Subbalakshmi, C., and Nagaraj, R. (2012). Hexafluoroisopropanol induces self-assembly of beta-amyloid peptides into highly ordered nanostructures. J. Peptide Sci. 18, 233–241. doi: 10.1002/psc.2391
Parthasarathy, S., Long, F., Miller, Y., Xiao, Y., McElheny, D., Thurber, K., et al. (2011). Molecular-level examination of Cu2+ binding structure for amyloid fibrils of 40-residue Alzheimer's β by solid-state NMR spectroscopy. J. Am. Chem. Soc. 133, 3390–3400. doi: 10.1021/ja1072178
Peti, W., Pieper, T., Sommer, M., Keppler, B. K., and Giester, G. (1999). Synthesis of tumor-inhibiting complex salts containing the anion trans-Tetrachlorobis(indazole)ruthenate(III) and crystal structure of the tetraphenylphosphonium salt. Eur. J. Inorganic Chem. 1999, 1551–1555. doi: 10.1002/(SICI)1099-0682(199909)1999:9<1551::AID-EJIC1551>3.0.CO;2-7
Pithadia, A. S., Kochi, A., Soper, M. T., Beck, M. W., Liu, Y. Z., Lee, S., et al. (2012). Reactivity of diphenylpropynone derivatives toward metal-associated amyloid-beta species. Inorg. Chem. 51, 12959–12967. doi: 10.1021/ic302084g
Querfurth, H. W., and LaFerla, F. M. (2010). Mechanisms of disease: Alzheimer's disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Reedijk, J. (2008). Metal-ligand exchange kinetics in platinum and ruthenium complexes. Significance for effectiveness as anticancer drugs. Platinum Metals Rev. 52:2. doi: 10.1595/147106708X255987
Roberson, E. D., and Mucke, L. (2006). 100 years and counting: Prospects for defeating Alzheimer's disease. Science 314, 781–784. doi: 10.1126/science.1132813
Rodriguez-Rodriguez, C., Telpoukhovskaia, M., and Orvig, C. (2012). The art of building multifunctional metal-binding agents from basic molecular scaffolds for the potential application in neurodegenerative diseases. Coord. Chem. Rev. 256, 2308–2332. doi: 10.1016/j.ccr.2012.03.008
Sabate, R., Gallardo, M., and Estelrich, J. (2003). An autocatalytic reaction as a model for the kinetics of the aggregation of beta-amyloid. Biopolymers 71, 190–195. doi: 10.1002/bip.10441
Sasaki, I., Bijani, C., Ladeira, S., Bourdon, V., Faller, P., and Hureau, C. (2012). Interference of a new cyclometallated Pt compound with Cu binding to amyloid-β peptide. Dalton Trans. 41, 6404–6407. doi: 10.1039/c2dt12177h
Savelieff, M. G., Lee, S., Liu, Y., and Lim, M. H. (2013). Untangling amyloid-beta, tau, and metals in Alzheimer's disease. ACS Chem. Biol. 8, 856–865. doi: 10.1021/cb400080f
Savelieff, M. G., Nam, G., Kang, J., Lee, H. J., Lee, M., and Lim, M. H. (2019). Development of multifunctional molecules as potential therapeutic candidates for Alzheimer's disease, parkinson's disease, and amyotrophic lateral sclerosis in the Last Decade. Chem. Rev. 119, 1221–1322. doi: 10.1021/acs.chemrev.8b00138
Selkoe, D. J. (2011). Resolving controversies on the path to Alzheimer's therapeutics. Nat. Med. 17, 1693–1693. doi: 10.1038/nm1211-1693d
Soto, C., and Pritzkow, S. (2018). Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 21, 1332–1340. doi: 10.1038/s41593-018-0235-9
Storr, T., Merkel, M., Song-Zhao, G. X., Scott, L. E., Green, D. E., Bowen, M. L., et al. (2007). Synthesis, characterization, and metal coordinating ability of multifunctional carbohydrate-containing compounds for Alzheimer's therapy. J. Am. Chem. Soc. 129, 7453–7463. doi: 10.1021/ja068965r
Streltsov, V. A., Chandana Epa, V., James, S. A., Churches, Q. I., Caine, J. M., Kenche, V. B., et al. (2013). Structural insights into the interaction of platinum-based inhibitors with the Alzheimer's disease amyloid-β peptide. Chem. Commun. 49, 11364–11366. doi: 10.1039/c3cc47326k
Suh, J., Yoo, S. H., Kim, M. G., Jeong, K., Ahn, J. Y., Kim, M.-S., et al. (2007). Cleavage Agents for Soluble Oligomers of Amyloid β Peptides. Angew. Chem. Int. Ed. 46, 7064–7067. doi: 10.1002/anie.200702399
Sung, Y. H., Rospigliosi, C., and Eliezer, D. (2006). NMR mapping of copper binding sites in alpha-synuclein. Biochim. Biophys. Acta 1764, 5–12. doi: 10.1016/j.bbapap.2005.11.003
Thompson, D. S., Weiss, G. J., Jones, S. F., Burris, H. A., Ramanathan, R. K., Infante, J. R., et al. (2012). NKP-1339: Maximum tolerated dose defined for first-in-human GRP78 targeted agent. J. Clin. Oncol. 30, 3033–3033. doi: 10.1186/2050-6511-13-S1-A82
Treusch, S., Cyr, D. M., and Lindquist, S. (2009). Amyloid deposits: Protection against toxic protein species? Cell Cycle 8, 1668–1674. doi: 10.4161/cc.8.11.8503
Trondl, R., Heffeter, P., Kowol, C. R., Jakupec, M. A., Berger, W., and Keppler, B. K. (2014). NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 5, 2925–2932. doi: 10.1039/C3SC53243G
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer a beta oligomer bound to cellular prion protein (vol 79, pg 887, 2013). Neuron 80, 531–531. doi: 10.1016/j.neuron.2013.10.001
Valensin, D., Anzini, P., Gaggelli, E., Gaggelli, N., Tamasi, G., Cini, R., et al. (2010). fac-{Ru(CO)3}2+ selectively targets the histidine residues of the β-amyloid peptide 1-28. Implications for New Alzheimer's Disease Treatments Based on Ruthenium Complexes. Inorg. Chem. 49, 4720–4722. doi: 10.1021/ic902593e
Vergara, A., D'Errico, G., Montesarchio, D., Mangiapia, G., Paduano, L., and Merlino, A. (2013a). Interaction of anticancer ruthenium compounds with proteins: high-resolution X-ray structures and raman microscopy studies of the adduct between hen egg white lysozyme and AziRu. Inorg. Chem. 52, 4157–4159. doi: 10.1021/ic4004142
Vergara, A., Russo Krauss, I., Montesarchio, D., Paduano, L., and Merlino, A. (2013b). Investigating the ruthenium metalation of proteins: X-ray structure and raman microspectroscopy of the complex between RNase A and AziRu. Inorg. Chem. 52, 10714–10716. doi: 10.1021/ic401494v
Walsh, D. M., and Selkoe, D. J. (2007). A beta Oligomers - a decade of discovery. J. Neurochem. 101, 1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x
Wang, H.-W., Pasternak, J. F., Kuo, H., Ristic, H., Lambert, M. P., Chromy, B., et al. (2002). Soluble oligomers of β amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 924, 133–140. doi: 10.1016/S0006-8993(01)03058-X
Ward, R. J., Dexter, D. T., and Crichton, R. R. (2015). Neurodegenerative diseases and therapeutic strategies using iron chelators. J. Trace Elements Med. Biol. 31, 267–273. doi: 10.1016/j.jtemb.2014.12.012
Watt, A. D., Villemagne, V. L., and Barnham, K. J. (2013). Metals, membranes, and amyloid-beta oligomers: key pieces in the Alzheimer's disease puzzle? J. Alzheimers Dis. 33 (Suppl. 1), S283–293. doi: 10.3233/JAD-2012-129017
Webb, M. I., Chard, R. A., Al-Jobory, Y. M., Jones, M. R., Wong, E. W., and Walsby, C. J. (2012). Pyridine analogues of the antimetastatic Ru(III) complex NAMI-A targeting non-covalent interactions with albumin. Inorg. Chem. 51, 954–966. doi: 10.1021/ic202029e
Webb, M. I., Wu, B., Jang, T., Chard, R. A., Wong, E. W., Wong, M. Q., et al. (2013). Increasing the bioavailability of Ru(III) anticancer complexes through hydrophobic albumin interactions. Chemistry 19, 17031–17042. doi: 10.1002/chem.201302671
WHO (2012). Dementia Cases Set to Triple by 2050 but Still Largely Ignored. Available online at: http://www.who.int/mediacentre/news/releases/2012/dementia_20120411/en/ (accessed December 12, 2013).
Wineman-Fisher, V., Bloch, D. N., and Miller, Y. (2016). Challenges in studying the structures of metal-amyloid oligomers related to type 2 diabetes, Parkinson's disease, and Alzheimer's disease. Coord. Chem. Rev. 327–428, 20–26. doi: 10.1016/j.ccr.2016.04.010
Yao, S. G., Cherny, R. A., Bush, A. I., Masters, C. L., and Barnham, K. J. (2004). Characterizing bathocuproine self-association and subsequent binding to Alzheimer's disease amyloid beta-peptide by NMR. J. Peptide Sci. 10, 210–217. doi: 10.1002/psc.539
Keywords: dementia, Alzheimer's disease, amyloid-beta peptide, Ru(III) complexes, peptide aggregation
Citation: Gomes LMF, Bataglioli JC, Jussila AJ, Smith JR, Walsby CJ and Storr T (2019) Modification of Aβ Peptide Aggregation via Covalent Binding of a Series of Ru(III) Complexes. Front. Chem. 7:838. doi: 10.3389/fchem.2019.00838
Received: 03 October 2019; Accepted: 18 November 2019;
Published: 03 December 2019.
Edited by:
Muhammad Hanif, The University of Auckland, New ZealandReviewed by:
Xiaohui Wang, Nanjing Tech University, ChinaCopyright © 2019 Gomes, Bataglioli, Jussila, Smith, Walsby and Storr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles J. Walsby, Y3dhbHNieUBzZnUuY2E=; Tim Storr, dGltX3N0b3JyQHNmdS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.