 Bing Qiu
Bing Qiu Wan Wang1,2
Wan Wang1,2 Xinzheng Yang
Xinzheng Yang- 1Beijing National Laboratory for Molecular Sciences, State Key Laboratory for Structural Chemistry of Unstable and Stable Species, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry Chinese Academy of Sciences, Beijing, China
- 2University of Chinese Academy of Sciences, Beijing, China
Inspired by the catalytic mechanism and active site structure of lactate racemase, three scorpion-like SCS nickel pincer complexes were proposed as potential catalysts for transfer hydrogenation of ketones and imines with ammonia-borane (AB) as the hydrogen source. Density functional theory calculations reveal a stepwise hydride and proton transfer mechanism for the dehydrocoupling of AB and hydrogenation of N-methylacetonimine, and a concerted proton-coupled hydride transfer process for hydrogenation of acetone, acetophenone, and 3-methyl-2-butanone. Among all proposed Ni complexes, the one with symmetric NH2 group on both arms of the SCS pincer ligand has the lowest free energy barrier of 15.0 kcal/mol for dehydrogenation of AB, as well as total free energy barriers of 17.8, 18.2, 18.0, and 18.6 kcal/mol for hydrogenation of acetone, N-methylacetonimine, acetophenone, and 3-methyl-2-butanone, respectively.
Introduction
Compared with direct hydrogenation, transfer hydrogenation (TH) avoids the use of hazardous molecular hydrogen and high pressure equipment by adopting low-cost and safe hydrogen provider compounds (Gladiali and Alberico, 2006; Ikariya and Blacker, 2007). Transition-metal-catalyzed TH has attracted increasing attentions in pharmaceutical, agrochemical, fragrance and other fine chemical industries as a powerful and practical way for the production of valuable chiral alcohols and amines (Blaser et al., 2003, 2007; Klingler, 2007; Saudan, 2007; Hansen et al., 2009).
The first TH reaction is Meerwein-Pondorf-Verley (MPV) reduction of ketone reported in mid-1920s (Meerwein and Schmidt, 1925; Verley, 1925; Ponndorf, 1926). In MPV reduction, direct TH happens through a cyclic six-membered transition state with alcohol and the carbonyl coordinated to the aluminum center. However, the low enantio-selectivity and undesired side reactions are well-known drawbacks in MPV reduction. In early 1980s, Matteoli et al. (1981) reported the first catalytic asymmetric transfer hydrogenation (ATH) reaction, in which a ruthenium complex H4Ru4(CO)8[(–)-DIOP]2 was used as the catalyst with secondary alcohols or indoline as the hydrogen source, for hydrogenation of prochiral ketones. To date, although significant progress has been made in transition metal-catalyzed TH and ATH reactions (Gopalaiah, 2013; Zuo et al., 2013, 2016; Pellissier and Clavier, 2014; Li et al., 2015; Morris, 2015; Wang and Astruc, 2015; Zuo and Morris, 2015), most reported catalysts are based on expensive and toxic noble metals, such as Rh, Ir, Ru, etc. (Gopalaiah, 2013; Pellissier and Clavier, 2014; Li et al., 2015; Morris, 2015; Wang and Astruc, 2015). The replacement of high-cost and toxic precious metals with abundant and environmentally benign base metals in catalysts for efficient TH and ATH reactions has attracted increasing attention in recent years, and several iron catalysts have been reported (Zhou et al., 2011; Gopalaiah, 2013; Zuo et al., 2013, 2016; Pellissier and Clavier, 2014; Li et al., 2015; Lu et al., 2015; Morris, 2015; Zuo and Morris, 2015; Smith et al., 2017). For example, Gao (Li et al., 2015) and Morris (Zuo et al., 2013; Morris, 2015; Zuo and Morris, 2015) groups reported tetradentate PNNP iron catalysts for ATH of acetophenone with high enantioselectivities. Morris and co-workers (Smith et al., 2017) reported unsymmetrical iron P-NH-P' complexes for asymmetric hydrogenation of aryl ketones with ee values >90%. Morris (Zuo et al., 2013; Morris, 2015) and Beller (Zhou et al., 2011; Lu et al., 2015) groups developed iron catalyzed asymmetric hydrogenation of imines and found high enantioselectivities for the reductions of N-phosphinyl ketimines.
In contrast to the encouraging progress archived in iron catalysts, ATH of ketones catalyzed by cobalt complexes have rather low enantioselectivities (Morris, 2015). Only a few Ni catalysts have been reported so far (Hamada et al., 2008; Hibino et al., 2009; Dong et al., 2012; Yang et al., 2014; Xu et al., 2015). In 2008, Hamada and co-workers (Hamada et al., 2008) applied nickel-bisphosphine complexes to catalyze asymmetric hydrogenation of α-amino-β-keto ester hydrochlorides and achieved high diastereo- and enantioselectivities (88–93% ee) for the production of anti-β-hydroxy-α-amino esters. They also used the same catalyst for asymmetric hydrogenation of substituted aromatic α-aminoketone hydrochlorides to produce β-aminoalcohols and found excellent diastereo- and enantioselectivities (Hibino et al., 2009). In 2012, Dong et al. (2012) reported Ni(II) complexes chelated by PNO ligands for catalytic ATH of aromatic ketones with 2-propanol as the hydrogen source and obtained optical alcohols up to 84% ee under mild conditions. In 2014, Yang et al. (2014) reported a highly active Ni(OAc)2/Binapine catalyst for the synthesis of α- and β-amino acid trough ATH of olefins with formic acid. Later on, they reported a highly active NiCl2(dme)/Binapine catalysts for ATH of hydrazones and other ketimines (Xu et al., 2015).
In a typical TH process, low-cost compounds such as 2-propanol (Haack et al., 1997) and formic acid (Fujii et al., 1996), are used as hydrogen donors. However, reversibility is a major drawback while 2-propanol is used as the hydrogen source. Excessive 2-propanol are usually required for the formation of alcohol. Formic acid releases stable CO2 after its dehydrogenation and does not suffer from equilibrium problems but can only be used for a limited range of stable complexes. Compared to alcohols, amine-boranes are easy to handle, irreversible, and potentially recyclable hydrogen donors (Nixon et al., 2011). Therefore, amine-boranes, especially ammonia-borane (AB), are promising to serve as solid hydrogen surrogates in TH reactions (Nixon et al., 2011). In recent years, Pagano et al. (2015) reported cobalt catalyzed dehydrocoupling of AB and TH of alkenes and alkynes. Fu et al. (2016), Shao et al. (2016), and Ai et al. (2019) reported the first cobalt-catalyzed selective TH of alkynes, and the only example of cobalt-catalyzed TH of nitriles with dehydrocoupling of AB under mild conditions. Yang et al. (2010, 2011a,b) recently reported direct TH of polarized C=N, C=C, and C=O bonds in imines, olefins, ketones, and aldehydes from AB. Li et al. (2016, 2017), Meng et al. (2018) reported frustrated Lewis pair catalyzed ATH of imines with AB and ATH of 2,3-disubstituted quinoxalines with AB. Ding et al. (2017) reported TH of N-heterocycles from AB prompted by B(C6F5)3. Korytiakova et al. (2017) reported Cu(I) catalyzed transfer semihydrogenation of alkyne and conjugate transfer hydrogenation of enoates with the dehydrocoupling of AB. Barrios-Francisco and García (2010) reported semihydrogenation of alkynes from AB catalyzed by Ni(0) complexes. Chong et al. (2014) reported 1,3,2-diazaphospholenes-catalyzed metal-free TH of N=N double bond using AB. Although some iron and nickel catalysts for acceptorless dehydrogenation of AB have also been developed (Keaton et al., 2007; Yang and Hall, 2008; Zimmerman et al., 2009a,b; Vogt et al., 2011; Bhunya et al., 2016; Lunsford et al., 2016; Rossin and Peruzzini, 2016; Chakraborty et al., 2017; Coles et al., 2017), effective base metal catalyst for TH of ketones using AB under mild conditions was rarely reported.
Inspired by the catalytic mechanism and the active site structure of lactate racemase (Desguin et al., 2015), as well as previously reported SCS nickel pincer complexes (Meguro et al., 2008; Xu et al., 2017), we recently proposed and computationally predicted several promising scorpion-like SCS nickel pincer complexes for catalytic lactate racemization and ATH of 1-acetonaphthone based on density functional theory (DFT) calculations (Qiu and Yang, 2017; Qiu et al., 2019). In this paper, we further examined three SCS nickel pincer complexes as potential catalysts for fast dehydrocoupling of AB and TH of ketones and imines.
Results and Discussion
Figure 1 shows our previously proposed scorpion-like SCS nickel pincer complexes 1A, 1B, and 1C with imidazole tails. 1A and 1B have symmetric arms but different sizes of amino groups in the SCS ligand, which are similar to the Ni complexes with pincer SCS ligands synthesized by Hu and co-workers (Xu et al., 2017). 1C is a mimic of the active site of lactate racemase with a carbonyl arm in the SCS pincer ligand.
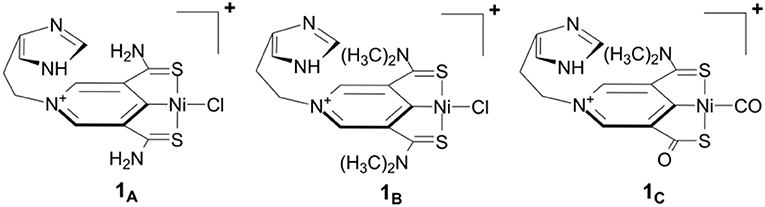
Figure 1. Scorpion-like (SCS)Ni pincer complexes proposed by Qiu and Yang (2017).
Scheme 1 is the proposed reaction cycle for the dehydrocoupling of AB and hydrogenation of acetone to 2-propanol catalyzed by 1A. Figure 2 reports the corresponding free energy profile. The optimized structures of key intermediates and transition states in the reaction are displayed in Figure 3. The reaction cycle, free energy profile, and key structures of the TH of N-methylacetonimine catalyzed by 1Aare shown in Scheme 2, Figures 4, 5, respectively.
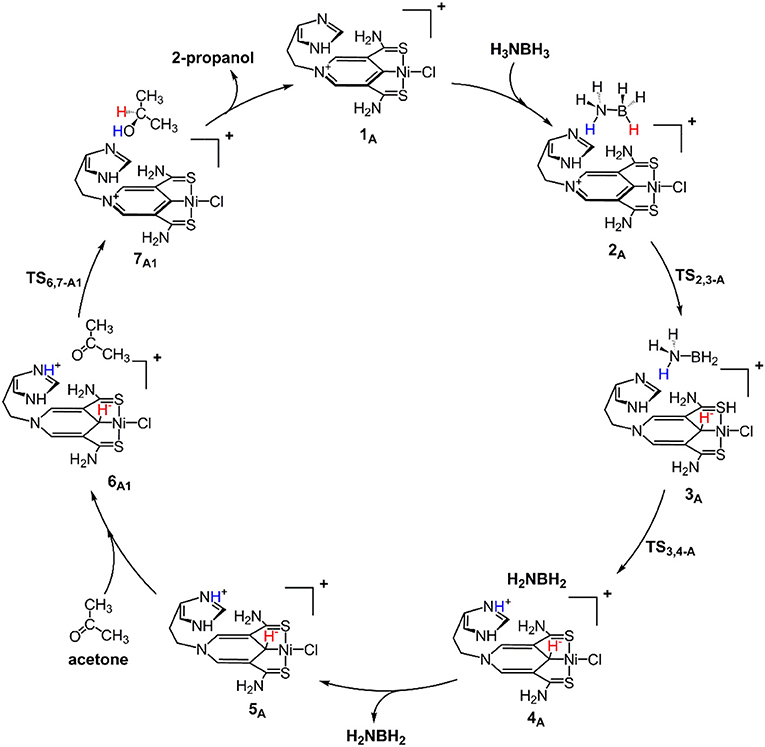
Scheme 1. Proposed mechanism for the dehydrocoupling of AB and TH of acetone catalyzed by 1A.
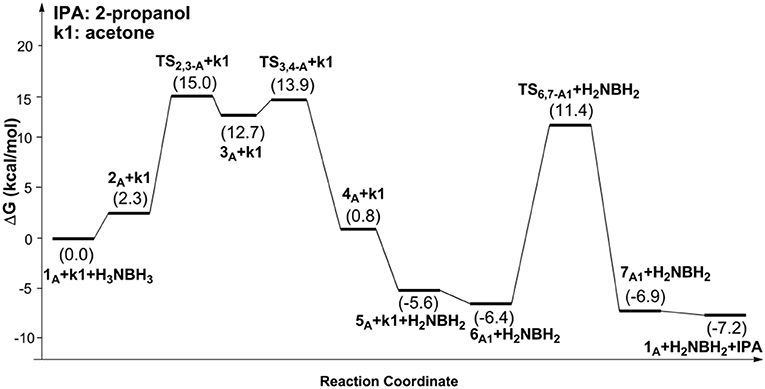
Figure 2. Free energy profile for the dehydrocoupling of AB and TH of acetone catalyzed by 1A.
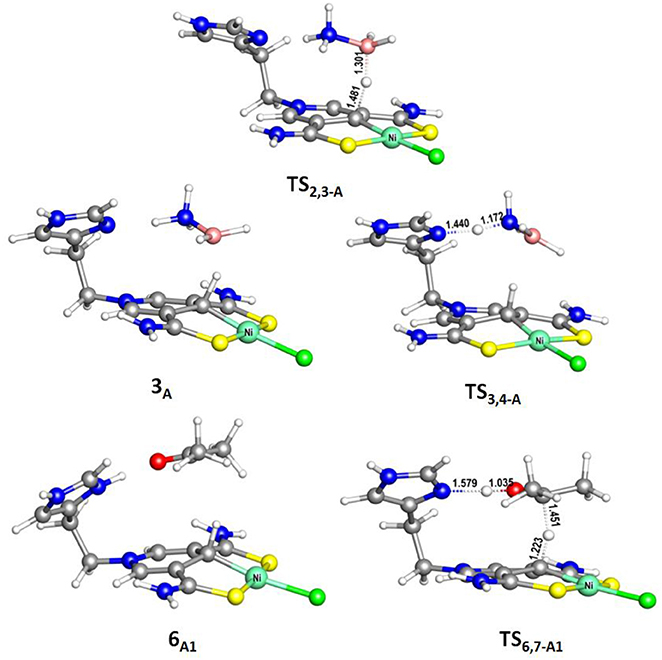
Figure 3. Optimized structures of 6A1, TS2,3−A (495i cm−1), TS3,4−A (388i cm−1), and TS6,7−A1 (510i cm−1). Bond lengths are in angstrom.
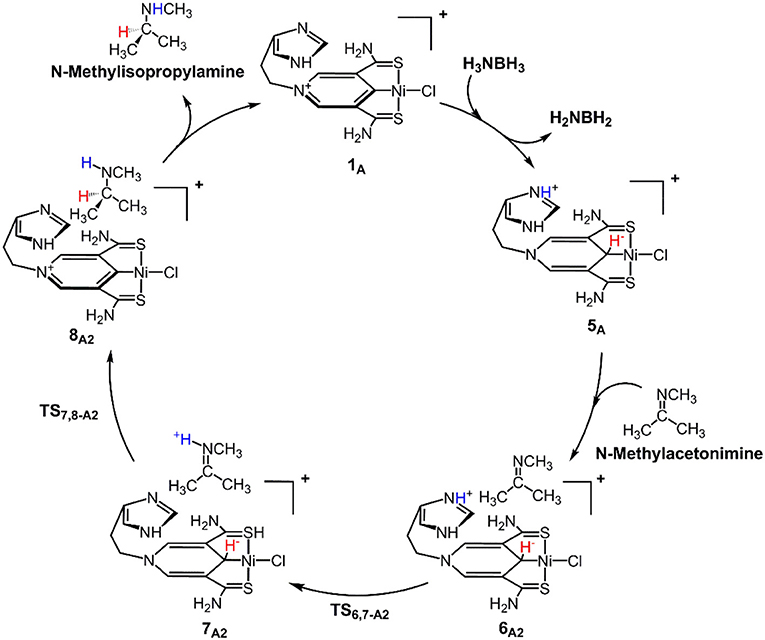
Scheme 2. 1A catalyzed TH of N-methylisopropylamine with stepwise proton and hydride transfers.
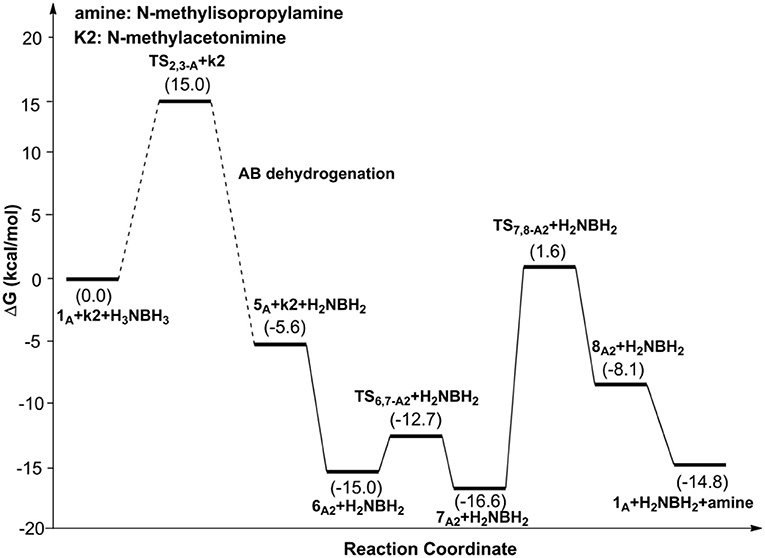
Figure 4. Free energy profile for the TH of N-methylacetonimine catalyzed by 1A.
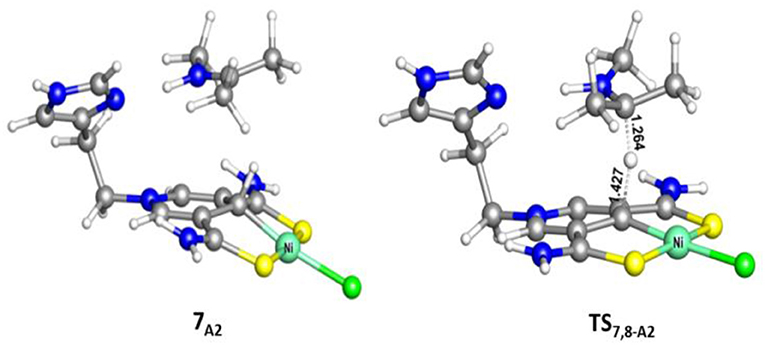
Figure 5. Optimized structures of 7A2 and TS7,8−A2 (700i cm−1). Bond lengths are in angstrom.
The transfer hydrogenation reaction starts with dehydrocoupling of AB. When an AB molecule approaches 1A, a hydride on borane could be transferred to the sp2 carbon coordinated to nickel through transition state TS2,3−A (ΔG = 15.0 kcal/mol) and forms an unstable intermediate 3A. An ammonia proton then transfers to the imidazole nitrogen through transition state TS3,4−A (ΔG = 13.9 kcal/mol). The exchange of acetone and H2NBH2 in 4A forms a 7.2 kcal/mol stable intermediate 6A1. Then the proton and hydride obtained from AB transfer from the pincer ligand in 6A1 to acetone in one-step through TS6,7−A1 (Figure 3) with a free energy barrier of 17.8 kcal/mol for the formation of 2-propanol.
Different from the hydrogenation of acetone, the proton and hydride are transferred from the pincer ligand in 6A2 to N-methylacetonimine in a stepwise way with two transition states, TS6,7−A2 and TS7,8−A2. The total free energy barrier of the hydrogenation of N-methylacetonimine is 18.2 kcal/mol (7A2 → TS7,8−A2).
Table 1 lists calculated free energy barriers of the hydrogenations of acetone and N-methylacetonimine (ΔGacetone and ΔGN−methylacetonimine) catalyzed by 1A, 1B, and 1C. The difference of those relative free energies are less than 4 kcal/mol, which indicates a rather weak steric effect with different functional groups in the pincer ligand.

Table 1. Free energy barriers of the TH of acetone and N-methylacetonimine with AB catalyzed by 1A, 1B, and 1C.
Figures 6, 7 report free energy profiles for 1A catalyzed TH of acetophenone to 1-phenyl-ethanol and TH of 3-methyl-2-butanone to 3-methyl-2-butanol, respectively. Some key structures in those catalytic reactions are displayed in Figure 8.
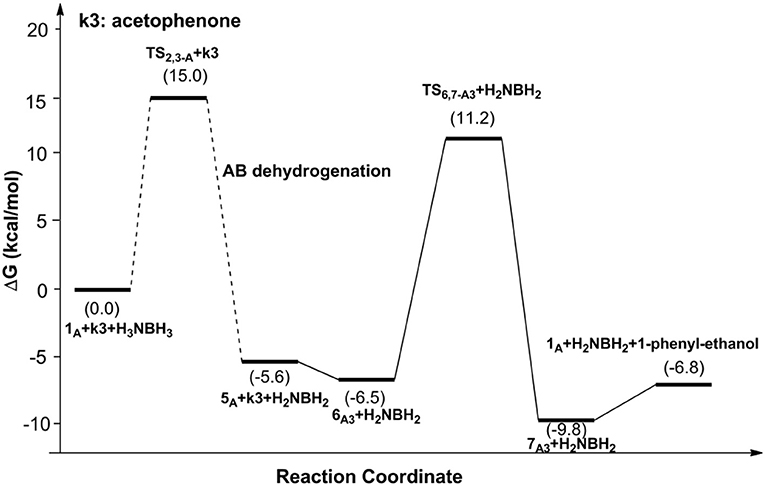
Figure 6. Free energy profile for the TH of acetophenone catalyzed by 1A.
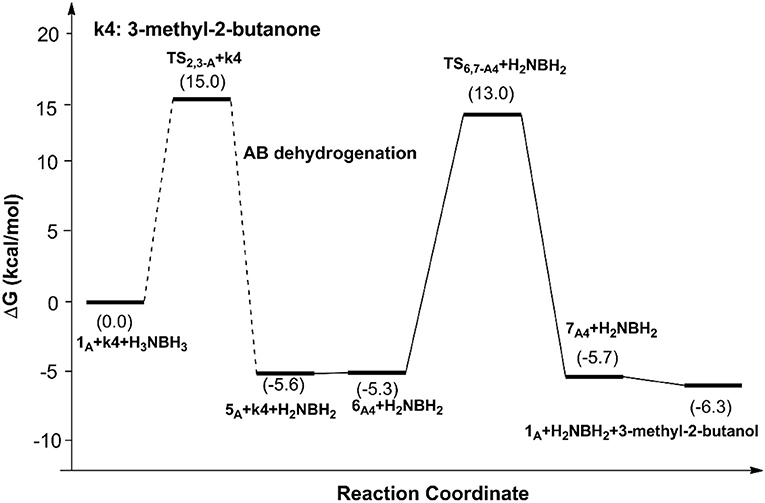
Figure 7. Free energy profile for the TH of 3-methyl-2-butanone catalyzed by 1A.
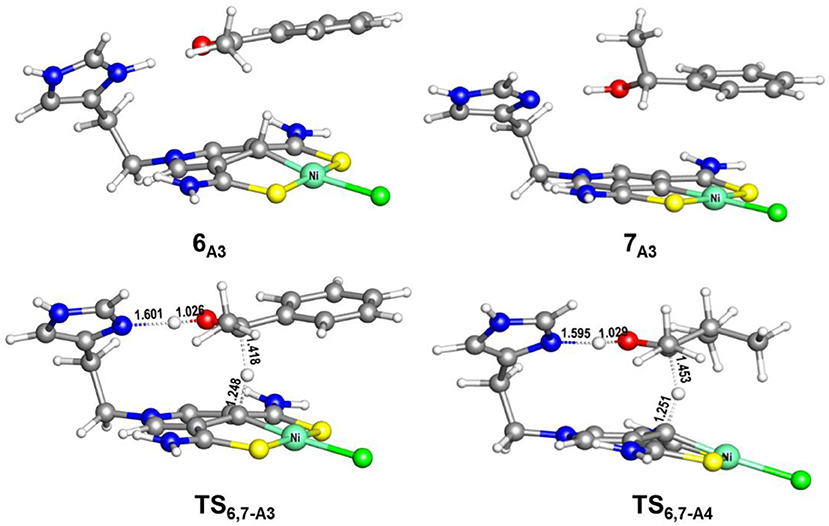
Figure 8. Optimized structures of 6A3, 7A3, TS6,7−A3 (485i cm−1), and TS6,7−A4 (663i cm−1). Bond lengths are in angstrom.
The AB dehydrocoupling process has the same free energy barrier of 15.0 kcal/mol in Figures 6, 7. The hydrogenation of acetophenone goes through a concerted one-step proton and hydride transfer transition state TS6,7−A3 (Figure 8). Although TS6,7−A3 is 17.7 kcal/mol higher than 7A3, the total free energy barrier of this catalytic TH reaction is 18.0 kcal/mol (7A3 → TS2,3−A) after considering the barrier of AB dehydrogenation.
The THs of acetophenone and 3-methyl-2-butanone catalyzed by 1A, 1B, and 1C have similar mechanisms but slightly different energy barriers, which are listed in Table 2. We can see the free energy barriers of 1B and 1C are higher because of the stronger steric effects of dimethyl groups in them.
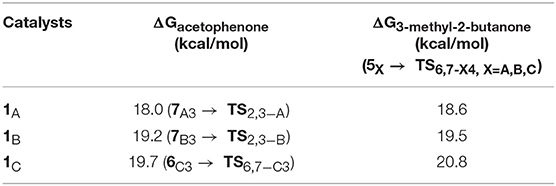
Table 2. Free energy barriers of the TH of acetophenone and 3-methyl-2-butanone with AB catalyzed by 1A, 1B, and 1C.
It is worth to note that the role of the imidazole groups in those scorpion-like SCS nickel pincer complexes are proton reservoirs facilitating proton transfer in the dehydrocoupling of AB and hydrogenation of C=O and C=N bonds. The ethylene group connecting the pyridinium ring and the imidazole group ensures the adjustability of the imidazole group's position for the hydrogenation of different ketones and imines. The substituents on the arms of the SCS ligand can slightly influence the reaction barriers through their steric effects. We believe the stepwise hydride and proton transfers in hydrogenation of 3-methyl-2-butanone are caused by the weak polarity of the C=N bond in it.
Conclusions
In summary, we computationally examined three scorpion-like SCS nickel pincer complexes, 1A, 1B, and 1C, with different steric effects as potential catalysts for catalytic TH of ketones and imines. Our calculations reveal stepwise hydride and proton transfer processes for the dehydrocoupling of AB and hydrogenation of imine, and a proton coupled hydride transfer process for hydrogenation of ketones. Among three examined Ni complexes, 1A with symmetric NH2 groups in the pincer ligand has the lowest free energy barriers of 17.8, 18.2, 18.0, and 18.6 kcal/mol for transfer hydrogenations of acetone, N-methylacetonimine, acetophenone, and 3-methyl-2-butanone, respectively. 1B and 1C have slightly higher barriers for the same reactions because of their stronger steric effects. Such low barriers and exothermicities shown in calculated free energy profiles indicate that those (SCS)Ni pincer complexes are promising catalyst candidates for efficient TH of ketones and amines under mild conditions. The steric effects of the substituents on the arms of the pincer ligand have very weak influence on the energy barriers of the catalytic reactions. Our computational predictions not only provide prototypical base metal catalysts for dehydrocoupling of AB and transfer hydrogenation of ketones and imines, but also shed a light for further development of cost-effective catalysts for the hydrogenation of polarized double bonds. Further design of base metal complexes with SCS pincer ligands for more hydrogenation and dehydrogenation reactions is underway.
Computational Details
The Gaussian 09 suite of ab initio programs (Frisch et al., 2010) was employed to perform all DFT calculations at the ultrafine (99,590) numerical integration level for the ωB97X-D (Chai and Head-Gordon, 2008) functional with Stuttgart relativistic effective core potential (ECP10MDF) basis set (Martin and Sundermann, 2001) and all-electron 6-31+G(d,p) basis set (Hehre et al., 1972; Hariharan and Pople, 1973; Francl et al., 1982) for Ni and all other atoms, respectively. Without other specific instruction, all structures in this paper were fully optimized in THF by using the integral equation formalism polarizable continuum model (IEFPCM) (Tomasi et al., 2005) with SMD (Marenich et al., 2009) atomic radii solvent corrections. The ground states of all structures were confirmed as singlet through comparison with optimized high-spin analogs. Thermal corrections were considered under 298.15 K and 1 atm pressure through frequency calculations using the same method on optimized structures. The optimized structures were confirmed to have no imaginary vibrational mode for intermediates and only one imaginary vibrational mode for each transition state, which was further confirmed by intrinsic reaction coordinate (IRC) calculations to ensure proper stationary points were connected. The 3D molecular structures displayed in the text were drawn by using the JIMP2 program (Manson et al., 2006). We also evaluated the performance of various density functionals and the influence of solvent effect corrections for this Ni catalytic system. The calculation results and discussions are provided in the Supplementary Materials (Tables S1 and S2).
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
XY proposed the computational catalyst design project and the catalytic transfer hydrogenation reactions. BQ designed and computed all of the catalysts with the help of XY. BQ and WW wrote the paper with the help of XY.
Funding
This work was supported by the National Natural Science Foundation of China (21873107, 21673250). We are also grateful to Institute of Chemistry Chinese Academy of Sciences for funding.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00627/full#supplementary-material
References
Ai, W., Zhong, R., Liu, X., and Liu, Q. (2019). Hydride transfer reactions catalyzed by cobalt complexes. Chem. Rev. 119, 2876–2953. doi: 10.1021/acs.chemrev.8b00404
Barrios-Francisco, R., and García, J. J. (2010). Semihydrogenation of alkynes in the presence of Ni (0) catalyst using ammonia-borane and sodium borohydride as hydrogen sources. Appl. Catal. A 385, 108–113. doi: 10.1016/j.apcata.2010.06.052
Bhunya, S., Malakar, T., Ganguly, G., and Paul, A. (2016). Combining protons and hydrides by homogeneous catalysis for controlling the release of hydrogen from ammonia-borane: present status and challenges. ACS Catal. 6, 7907–7934. doi: 10.1021/acscatal.6b01704
Blaser, H.-U., Malan, C., Pugin, B., Spindler, F., Steiner, H., and Studer, M. (2003). Selective hydrogenation for fine chemicals: recent trends and new developments. Adv. Synth. Catal. 345, 103–151. doi: 10.1002/adsc.200390000
Blaser, H.-U., Pugin, B., Spindler, F., and Thommen, M. (2007). From a chiral switch to a ligand portfolio for asymmetric catalysis. Acc. Chem. Res. 40, 1240–1250. doi: 10.1021/ar7001057
Chai, J.-D., and Head-Gordon, M. (2008). Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. doi: 10.1039/B810189B
Chakraborty, U., Demeshko, S., Meyer, F., Rebreyend, C., de Bruin, B., Atanasov, M., et al. (2017). Electronic structure and magnetic anisotropy of an unsaturated cyclopentadienyl iron(I) complex with 15 valence electrons. Angew. Chem. Int. Ed. 56, 7995–7999. doi: 10.1002/anie.201702454
Chong, C. C., Hirao, H., and Kinjo, R. (2014). A concerted transfer hydrogenolysis: 1,3,2-diazaphospholene-catalyzed hydrogenation of N=N bond with ammonia-borane. Angew. Chem. Int. Ed. 53, 3342–3346. doi: 10.1002/anie.201400099
Coles, N. T., Mahon, M. F., and Webster, R. L. (2017). Phosphine- and amine-borane dehydrocoupling using a three-coordinate iron(II) β-diketiminate precatalyst. Organometallics 36, 2262–2268. doi: 10.1021/acs.organomet.7b00326
Desguin, B., Zhang, T., Soumillion, P., Hols, P., Hu, J., and Hausinger, R. P. (2015). A tethered niacin-derived pincer complex with a nickel-carbon bond in lactate racemase. Science 349, 66–69. doi: 10.1126/science.aab2272
Ding, F., Zhang, Y., Zhao, R., Jiang, Y., Bao, R. L. Y., Lin, K., et al. (2017). B(C6F5)3-promoted hydrogenations of N-heterocycles with ammonia borane. Chem. Commun. 53, 9262–9264. doi: 10.1039/C7CC04709F
Dong, Z. R., Li, Y. Y., Yu, S. L., Sun, G. S., and Gao, J. X. (2012). Asymmetric transfer hydrogenation of ketones catalyzed by nickel complex with new PNO-type ligands. Chin. Chem. Lett. 23, 533–536. doi: 10.1016/j.cclet.2012.02.005
Francl, M. M., Pietro, W. J., Hehre, W. J., Binkley, J. S., Gordon, M. S., DeFrees, D. J., et al. (1982). Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 77, 3654–3665. doi: 10.1063/1.444267
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2010). Gaussian 09, revision C.01, Gaussian, Inc., Wallingford CT.
Fu, S., Chen, N.-Y., Liu, X., Shao, Z., Luo, S.-P., and Liu, Q. (2016). Ligand-controlled cobalt-catalyzed transfer hydrogenation of alkynes: stereodivergent synthesis of Z- and E-alkenes. J. Am. Chem. Soc. 138, 8588–8594. doi: 10.1021/jacs.6b04271
Fujii, A., Hashiguchi, S., Uematsu, N., Ikariya, T., and Noyori, R. (1996). Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid-triethylamine mixture. J. Am. Chem. Soc. 118, 2521–2522. doi: 10.1021/ja954126l
Gladiali, S., and Alberico, E. (2006). Asymmetric transfer hydrogenation: chiral ligands and applications. Chem. Soc. Rev. 35, 226–236. doi: 10.1039/B513396C
Gopalaiah, K. (2013). Chiral iron catalysts for asymmetric synthesis. Chem. Rev. 113, 3248–3296. doi: 10.1021/cr300236r
Haack, K. J., Hashiguchi, S., Fujii, A., Ikariya, T., and Noyori, R. (1997). The catalyst precursor, catalyst, and intermediate in the RuII-promoted asymmetric hydrogen transfer between alcohols and ketones. Angew. Chem. Int. Ed. 36, 285–288. doi: 10.1002/anie.199702851
Hamada, Y., Koseki, Y., Fujii, T., Maeda, T., Hibino, T., and Makino, K. (2008). Catalytic asymmetric hydrogenation of α-amino-β-keto ester hydrochlorides using homogeneous chiral nickel-bisphosphine complexes through DKR. Chem. Commum. 46, 6206–6208. doi: 10.1039/B816524F
Hansen, K. B., Hsiao, Y., Xu, F., Rivera, N., Clausen, A., Kubryk, M., et al. (2009). Highly efficient asymmetric synthesis of sitagliptin. J. Am. Chem. Soc. 131, 8798–8804. doi: 10.1021/ja902462q
Hariharan, P. C., and Pople, J. A. (1973). The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta. 28, 213–222. doi: 10.1007/BF00533485
Hehre, W. J., Ditchfield, R., and Pople, J. A. (1972). Self-consistent molecular orbital methods. XII. Further extensions of gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56, 2257–2261. doi: 10.1063/1.1677527
Hibino, T., Makino, K., Sugiyama, T., and Hamada, Y. (2009). Homogeneous chiral nickel-catalyzed asymmetric hydrogenation of substituted aromatic α-aminoketone hydrochlorides through dynamic kinetic resolution. Chem. Cat. Chem. 1, 237–240. doi: 10.1002/cctc.200900084
Ikariya, T., and Blacker, A. J. (2007). Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res. 40, 1300–1308. doi: 10.1021/ar700134q
Keaton, R. J., Blacquiere, J. M., and Baker, R. T. (2007). Base metal catalyzed dehydrogenation of ammonia-borane for chemical hydrogen storage. J. Am. Chem. Soc. 129, 1844–1845. doi: 10.1021/ja066860i
Klingler, F. D. (2007). Asymmetric hydrogenation of prochiral amino ketones to amino alcohols for pharmaceutical use. Acc. Chem. Res. 40, 1367–1376. doi: 10.1021/ar700100e
Korytiakova, E., Thiel, N. O., Pape, F., and Teichert, J. F. (2017). Copper (I)-catalysed transfer hydrogenations with ammonia borane. Chem. Commun. 53, 732–735. doi: 10.1039/C6CC09067B
Li, S., Li, G., Meng, W., and Du, H. (2016). A frustrated lewis pair catalyzed asymmetric transfer hydrogenation of imines using ammonia borane. J. Am. Chem. Soc. 138, 12956–12962. doi: 10.1021/jacs.6b07245
Li, S., Meng, W., and Du, H. (2017). Asymmetric transfer hydrogenations of 2, 3-disubstituted quinoxalines with ammonia borane. Org. Lett. 19, 2604–2606. doi: 10.1021/acs.orglett.7b00935
Li, Y. Y., Yu, S. L., Shen, W. Y., and Gao, J. X. (2015). Iron-, cobalt-, and nickel-catalyzed asymmetric transfer hydrogenation and asymmetric hydrogenation of ketones. Acc. Chem. Res. 48, 2587–2598. doi: 10.1021/acs.accounts.5b00043
Lu, L. Q., Li, Y., Junge, K., and Beller, M. (2015). Relay iron/chiral brønsted acid catalysis: enantioselective hydrogenation of benzoxazinones. J. Am. Chem. Soc. 137, 2763–2768. doi: 10.1021/jacs.5b00085
Lunsford, A. M., Blank, J. H., Moncho, S., Haas, S. C., Muhamad, S., Brothers, E. N., et al. (2016). Catalysis and mechanism of H2 release from amine-boranes by diiron complexes. Inorg. Chem. 55, 964–973. doi: 10.1021/acs.inorgchem.5b02601
Manson, J., Webster, C. E., and Hall, M. B. (2006). JIMP2, version 0.091, A Free Program for Visualizing and Manipulating Molecules. Texas A&M University, College Station, TX.
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396. doi: 10.1021/jp810292n
Martin, J. M. L., and Sundermann, A. (2001). Correlation consistent valence basis sets for use with the Stuttgart-Dresden-Bonn relativistic effective core potentials: the atoms Ga-Kr and In-Xe. J. Chem. Phys. 114, 3408–3420. doi: 10.1063/1.1337864
Matteoli, U., Frediani, P., Bianchi, M., Botteghi, C., and Gladiali, S. (1981). Asymmetric homogeneous catalysis by ruthenium complexes. J. Mol. Catal. 198, 73–80. doi: 10.1016/0304-5102(81)85035-3
Meerwein, H., and Schmidt, R. (1925). Ein neues verfahren zur reduktion von aldehyden und ketonen. Liebigs Ann. Chem. 444, 221–238. doi: 10.1002/jlac.19254440112
Meguro, H., Koizumi, T., Yamamoto, T., and Kanbara, T. (2008). Synthesis, structure, and quaternization and complexation reactions of κ3SCS pincer palladium complexes having 3,5-pyridinediyl unit. J. Organomet. Chem. 693, 1109–1116. doi: 10.1016/j.jorganchem.2007.12.033
Meng, W., Feng, X., and Du, H. (2018). Frustrated lewis pairs catalyzed asymmetric metal-free hydrogenations and hydrosilylations. Acc. Chem. Res. 51, 191–201. doi: 10.1021/acs.accounts.7b00530
Morris, R. H. (2015). Exploiting metal-ligand bifunctional reactions in the design of iron asymmetric hydrogenation catalysts. Acc. Chem. Res. 48, 1494–1502. doi: 10.1021/acs.accounts.5b00045
Nixon, T. D., Whittlesey, M. K., and Williams, J. M. J. (2011). Ruthenium-catalysed transfer hydrogenation reactions with dimethylamine borane. Tetrahedron Lett. 52, 6652–6654. doi: 10.1016/j.tetlet.2011.10.039
Pagano, J. K., Stelmach, J. P. W., and Waterman, R. (2015). Cobalt-catalyzed ammonia borane dehydrocoupling and transfer hydrogenation under aerobic conditions. Dalton Trans. 44, 12074–12077. doi: 10.1039/C5DT00108K
Pellissier, H., and Clavier, H. (2014). Enantioselective cobalt-catalyzed transformations. Chem. Rev. 114, 2775–2823. doi: 10.1021/cr4004055
Ponndorf, W. (1926). Der reversible Austausch der Oxydationsstufen zwischen Aldehyden oder Ketonen einerseits und primären oder sekundären Alkoholen anderseits. Angew. Chem. 39, 138–143. doi: 10.1002/ange.19260390504
Qiu, B., Wang, W., and Yang, X. (2019). Computational design of SCS nickel pincer complexes for asymmetric transfer hydrogenation of 1-acetonaphthone. Catalysts 9:101. doi: 10.3390/catal9010101
Qiu, B., and Yang, X. (2017). A bio-inspired design and computational prediction of scorpion-like SCS nickel pincer complexes for lactate racemization. Chem. Commun. 53, 11410–11413. doi: 10.1039/C7CC06416K
Rossin, A., and Peruzzini, M. (2016). Ammonia-borane and amine-borane dehydrogenation mediated by complex metal hydrides. Chem. Rev. 116, 8848–8872. doi: 10.1021/acs.chemrev.6b00043
Saudan, L. A. (2007). Hydrogenation processes in the synthesis of perfumery ingredients. Acc. Chem. Res. 40, 1309–1319. doi: 10.1021/ar700140m
Shao, Z., Fu, S., Wei, M., Zhou, S., and Liu, Q. (2016). Mild and selective cobalt-catalyzed chemodivergent transfer hydrogenation of nitriles. Angew. Chem. Int. Ed. 55, 14653–14657. doi: 10.1002/anie.201608345
Smith, S. A. M., Lagaditis, P. O., Lupke, A., Lough, A. J., and Morris, R. H. (2017). Unsymmetrical iron P-NH-P′ catalysts for the asymmetric pressure hydrogenation of aryl ketones. Chem. Eur. J. 23, 7212–7216. doi: 10.1002/chem.201701254
Tomasi, J., Mennucci, B., and Cammi, R. (2005). Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094. doi: 10.1021/cr9904009
Verley, A. (1925). Exchange of functional groups between two molecules. Exchange of alcohol and aldehyde groups. Bull. Soc. Chim. Fr. 37, 537–542.
Vogt, M., Bruin, B., Berke, H, Trincado, M., and Grutzmacher, H. (2011). Amino olefin nickel(I) and nickel(0) complexes as dehydrogenation catalysts for amine boranes. Chem. Sci. 2, 723–727. doi: 10.1039/C0SC00483A
Wang, D., and Astruc, D. (2015). The golden age of transfer hydrogenation. Chem. Rev. 115, 6621–6686. doi: 10.1021/acs.chemrev.5b00203
Xu, H., Yang, P., Chuanprasit, P., Hirao, H., and Zhou, J. (2015). Nickel-catalyzed asymmetric transfer hydrogenation of hydrazones and other ketimines. Angew. Chem. Int. Ed. 54, 5112–5116. doi: 10.1002/anie.201501018
Xu, T., Wodrich, M. D., Scopelliti, R., Corminboeuf, C., and Hu, X. (2017). Nickel pincer model of the active site of lactate racemase involves ligand participation in hydride transfer. Proc. Natl. Acad. Sci. U.S.A. 114, 1242–1245. doi: 10.1073/pnas.1616038114
Yang, P., Xu, H., and Zhou, J. (2014). Nickel-catalyzed asymmetric transfer hydrogenation of olefins for the synthesis of α- and β-amino acids. Angew. Chem. Int. Ed. 53, 12210–12213. doi: 10.1002/anie.201407744
Yang, X., Fox, T., and Berke, H. (2011a). Facile metal free regioselective transfer hydrogenation of polarized olefins with ammonia borane. Chem. Commun. 47, 2053–2055. doi: 10.1039/C0CC03163A
Yang, X., Fox, T., and Berke, H. (2011b). Ammonia borane as a metal free reductant for ketones and aldehydes: a mechanistic study. Tetrahedron 67, 7121–7127. doi: 10.1016/j.tet.2011.06.104
Yang, X., and Hall, M. B. (2008). The catalytic dehydrogenation of ammonia-borane involving an unexpected hydrogen transfer to ligated carbene and subsequent carbon-hydrogen activation. J. Am. Chem. Soc. 130, 1798–1799. doi: 10.1021/ja0751328
Yang, X., Zhao, L., Fox, T., Wang, Z. X., and Berke, H. (2010). Transfer hydrogenation of imines with ammonia-borane: a concerted double-hydrogen-transfer reaction. Angew. Chem. Int. Ed. 49, 2058–2062. doi: 10.1002/anie.200906302
Zhou, S., Fleischer, S., Junge, K., and Beller, M. (2011). Cooperative transition-metal and chiral brønsted acid catalysis: enantioselective hydrogenation of imines to form amines. Angew. Chem. Int. Ed. 50, 5120–5124. doi: 10.1002/anie.201100878
Zimmerman, P. M., Paul, A., and Musgrave, C. B. (2009a). Catalytic dehydrogenation of ammonia borane at Ni monocarbene and dicarbene catalysts. Inorg. Chem. 48, 5418–5433. doi: 10.1021/ic900417z
Zimmerman, P. M., Zhang, Z., and Musgrave, C. B. (2009b). The role of free N-heterocyclic carbene (NHC) in the catalytic dehydrogenation of ammonia-borane in the nickel NHC system. Angew. Chem., Int. Ed. 48, 2201–2205. doi: 10.1002/anie.200803211
Zuo, W. W., Lough, A. J., Li, Y. F., and Morris, R. H. (2013). Amine(imine)diphosphine iron catalysts for asymmetric transfer hydrogenation of ketones and imines. Science 342, 1080–1083. doi: 10.1126/science.1244466
Zuo, W. W., and Morris, R. H. (2015). Synthesis and use of an asymmetric transfer hydrogenation catalyst based on iron(II) for the synthesis of enantioenriched alcohols and amines. Nat. Protoc. 10, 241–257. doi: 10.1038/nprot.2015.012
Zuo, W. W., Prokopchuk, D. E., Lough, A. J., and Morris, R. H. (2016). Details of the mechanism of the asymmetric transfer hydrogenation of acetophenone using the amine(imine)diphosphine iron precatalyst: the base effect and the enantiodetermining step. ACS Catal. 6, 301–314. doi: 10.1021/acscatal.5b01979
Keywords: lactate racemase, SCS nickel pincer, ammonia-borane, density functional theory, transfer hydrogenation, ketones, amines
Citation: Qiu B, Wang W and Yang X (2019) Computational Prediction of Ammonia-Borane Dehydrocoupling and Transfer Hydrogenation of Ketones and Imines Catalyzed by SCS Nickel Pincer Complexes. Front. Chem. 7:627. doi: 10.3389/fchem.2019.00627
Received: 06 July 2019; Accepted: 30 August 2019;
Published: 13 September 2019.
Edited by:
Nino Russo, Department of Chemistry and Chemical Technologies, University of Calabria, ItalyReviewed by:
Emilia Sicilia, Department of Chemistry and Chemical Technologies, University of Calabria, ItalyShi-Lu Chen, Beijing Institute of Technology, China
Copyright © 2019 Qiu, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinzheng Yang, eHlhbmcmI3gwMDA0MDtpY2Nhcy5hYy5jbg==