 Kei Murata
Kei Murata Nobutsugu Numasawa
Nobutsugu Numasawa Nobuharu Iwasawa
Nobuharu Iwasawa- Department of Chemistry, Tokyo Institute of Technology, Tokyo, Japan
The improved catalytic conditions and detailed reaction mechanism of the visible-light driven hydrocarboxylation of alkenes with CO2 by the Rh(I) and photoredox dual catalysts were investigated. The use of the benzimidazoline derivative, BI(OH)H, as a sacrificial electron donor was found to increase the yield of the hydrocarboxylated product by accelerating the reduction process. In addition, the incorporation of the cyclometalated Ir(III) complex as a second photosensitizer with [Ru(bpy)3]2+ photosensitizer also resulted in the promotion of the reduction process, supporting that the catalytic cycle includes two photochemical elementary processes: photoinduced electron and energy transfers.
Introduction
Catalytic hydrocarboxylation of unsaturated hydrocarbons with CO2 is one of the promising methods for the CO2 fixation (Luan and Ye, 2018; Yan et al., 2018; for recent reviews, see: Tortajada et al., 2018). The most common strategy to accomplish the hydrocarboxylation is to utilize a metal hydride complex as an active species. However, in these reactions, more than a stoichiometric amount of highly active, metallic reductants such as ZnEt2, AlEt3, or hydrosilanes are usually required to promote the reduction process in the catalytic cycle (Takaya and Iwasawa, 2008; Williams et al., 2008; Fujihara et al., 2011; Li et al., 2011; Hayashi et al., 2015; Wang et al., 2015; Zhu et al., 2015; Kawashima et al., 2016). In order to realize a more efficient and environmentally-friendly system, the reaction which necessitates just a catalytic amount of metallic reagents is highly desirable. Meanwhile, the photochemical reduction process has been widely employed in the field of artificial photosynthesis, such as photocatalytic hydrogen generations (for review, see Esswein and Nocera, 2007) and CO2 reductions (for reviews, see Morris et al., 2009; Doherty et al., 2010; Takeda and Ishitani, 2010) in homogeneous systems. In these reactions, transition-metal catalysts are combined with redox photosensitizers and sacrificial electron donors to drive the multielectron transfer processes under visible-light irradiation. When the electron transfer is accompanied by the proton transfer, metal hydrides can act as an active species in the catalytic cycle (for reviews, see Stoll et al., 2015; Adams et al., 2018). Although transition-metal/photoredox dual catalysis has been actively studied in the field of organic synthesis (for recent reviews, see Fabry and Rueping, 2016; Skubi et al., 2016; Twilton et al., 2017), few examples have been reported for catalytic organic transformations driven by photochemically-generated metal hydrides (Ghosh et al., 2015).
We recently developed the visible-light driven hydrocarboxylation of alkenes with CO2 for the first time by means of the photochemical generation of Rh(I) hydride species (Murata et al., 2017). 4-Cyanostyrene was transformed to the branched hydrocarboxylated product by using a Rh(I) hydride or chloride complex as a carboxylation catalyst, [Ru(bpy)3]2+ as a photoredox catalyst, iPr2NEt as a sacrificial electron donor, with visible-light irradiation under CO2 atmosphere at room temperature (Figure 1). The photoredox catalysis made it possible to take electrons from tertiary amines and drive the reduction process without using a metallic reductant. Since then, several photoredox-catalyzed hydrocarboxylation reactions of unsaturated hydrocarbons with CO2 have been reported by other groups (Seo et al., 2017b; Hou et al., 2018b; Meng et al., 2018). Concomitantly, difunctionalizations of alkenes such as thiocarboxylation (Ye et al., 2017), carbocarboxylation and silylcarboxylation (Yatham et al., 2017; Hou et al., 2018a) have also been developed by incorporating an appropriate radical precursor with CO2. Furthermore, in addition to unsaturated hydrocarbons, various substrates such as aryl and alkyl halides (Meng et al., 2017; Shimomaki et al., 2017), amines (Seo et al., 2017a), imines and enamides (Fan et al., 2018; Ju et al., 2018) have been carboxylated with CO2 by photoredox catalysis so far (for review, see Yeung, 2019). These examples demonstrated wide applicability of the photoinduced electron transfer to carboxylation reactions.
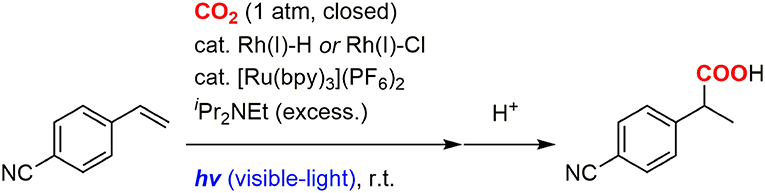
Figure 1. The photocatalytic hydrocarboxylation of 4-cyanostyrene.
On the basis of our previous experiments, the reaction mechanism of the hydrocarboxylation by Rh(I) and photoredox dual catalysts was proposed as shown in Figure 2. Initially, the hydrometallation of a styrene derivative by Rh(I) hydride species A gave the Rh(I) benzyl species B (i), and the visible-light promoted nucleophilic addition to CO2 afforded the Rh(I) carboxylate species C (ii). Then, the reductive quenching cycle of [Ru(bpy)3]2+ with iPr2NEt mediated 2-electron, 2-proton transfers afforded the Rh(III) dihydride carboxylate species D (iii), followed by the base-promoted liberation of the carboxylated product to regenerate the active species A (iv). Although this reaction demonstrated fundamental aspects of the application of photochemical reduction processes to catalytic carboxylation reactions, there still has been room for improvement from the viewpoint of the applicability in organic synthesis: (i) The efficiency of the reaction was not very high. Good yield was obtained with 4-cyanostyrene and moderate yields were obtained with several other substrates. (ii) A large excess amount of a tertiary amine and long reaction time (>24 h) were necessary for completion of the reaction even for the reactive substrates, (iii) A significant amount of the hydrogenated product was produced as a byproduct. In order to resolve these problems, further screenings of the catalytic conditions were desired.
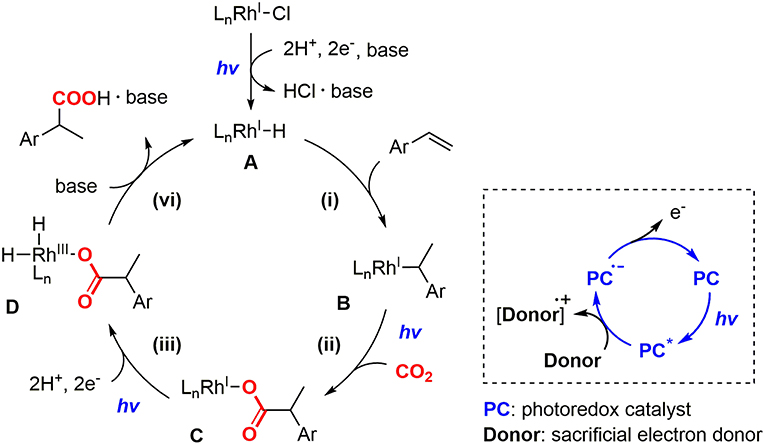
Figure 2. The proposed reaction mechanism of the photocatalytic hydrocarboxylation with the schematic representation of the reductive quenching cycle of a photoredox catalyst.
Herein, we explored the improved conditions of the visible-light driven hydrocarboxylation, and the catalytic efficiency was analyzed based on the detailed mechanistic study with a series of stoichiometric reactions of the rhodium intermediates. Through the investigation, the hydrocarboxylation was successfully improved by the alteration of the sacrificial electron donor or the incorporation of the second photosensitizer. The mechanistic study suggested that the promotion of the photochemical reduction process was crucial for the enhancement of the catalytic reaction.
Results and Discussion
Screening of Reaction Conditions
On the basis of our previous experiments in terms of the screening of catalytic conditions and the observation of the reaction intermediates under the catalytic conditions, the followings were demonstrated: (i) As a carboxylation catalyst, Rh(I) hydride or chloro complexes with triarylphosphines were applicable. In particular, the μ-chloro bridged Rh(I) dimer [Rh(P(4-CF3C6H4)3)2Cl]2 (4) was the most effective catalyst. (ii) When 4 was employed under the catalytic conditions, the resting state was the corresponding Rh(I) carboxylate complex, indicating that the rate-determining step was its transformation to the Rh(I) hydride species. This result suggested that the promotion of the reduction process was crucial for the improvement of the catalytic reaction. According to these considerations, the reaction conditions were screened in terms of the photoredox catalyst and sacrificial electron donor, which would have taken an important part in the reductive quenching cycle.
Photoredox Catalyst
Photoredox catalysts were initially screened by performing the reaction of 4-cyanostyrene (1a) in a mixture of 2.0 mol% of a photosensitizer, 3.5 mol% of 4 and 4.0 equiv. of iPr2NEt under a CO2 atmosphere at room temperature (Table 1). In the case of [Ru(bpy)3](PF6)2 ( = +0.77 V, = −1.33 V vs. SCE) (Kalyanasundaram, 1982) as a photoredox catalyst, the hydrocarboxylated (2a) and hydrogenated (3a) products were obtained in 54 and 25% yields, respectively, after visible-light irradiation for 24 h (Table 1, entry 1). A small amount of polymerized product of 1a was also produced as byproduct. Though no other photosensitizers overcame this activity, the yield of 2a was found to be strongly dependent on the photoredox catalyst. For instance, when [Ru(bpz)3](PF6)2 ( = +1.45 V, = −0.80 V vs. SCE) (Crutchley and Lever, 1980) or fac-Ir(ppy)3 ( = +0.31 V, = −2.19 V vs. SCE) (Flamigni et al., 2007) was employed, the yields substantially decreased compared to [Ru(bpy)3](PF6)2 (Table 1, entries 3, 6). On the other hand, when using [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) ( = +1.21 V, = −1.37 V vs. SCE) or [Ir(ppy)2(dtbbpy)](PF6) ( = +0.66 V, = −1.51 V vs. SCE) (Lowry et al., 2005), moderate yields were obtained (Table 1, entry 4, 5). These results indicated that both sufficient oxidizing ability of the excited state and reducing ability of the one-electron reduced species are at least necessary for the photosensitizer. However, the detailed dependency was not simple, as other factors such as absorption properties, excited-state energies and photochemical stability of the photosensitizer could also affect the catalytic performances. Meanwhile, the screenings of additives with [Ru(bpy)3]2+ photosensitizer demonstrated that the addition of Cs2CO3 as an inorganic base significantly improved the yield of the hydrocarboxylated product by suppressing the formation of the hydrogenated byproduct: the yield of 2a increased to 67% while the yield of 3a decreased to 1% (Table 1, entry 11).
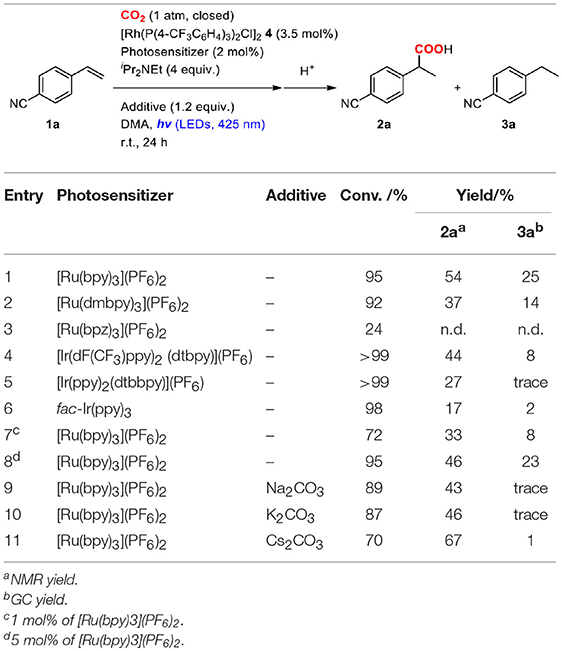
Table 1. Optimization of the reaction conditions of the hydrocarboxylation of 4-cyanostyrene.
Sacrificial Electron Donor and Additives
Sacrificial electron donors were then screened in the presence of an excess amount of Cs2CO3. In order to highlight the reactivity, a less reactive alkene, 3,5-bis(trifluoromethyl)styrene (1b), was used as a substrate. The reactions of 1b were performed in a mixture of 2.0 mol% of [Ru(bpy)3](PF6)2, 3.5 mol% of 4, 4.0 equiv. of sacrificial electron donor and 1.2 equiv. of Cs2CO3 under a CO2 atmosphere at room temperature (Table 2). When iPr2NEt was employed as a sacrificial electron donor, 32% yield of the hydrocarboxylated product (2b) and a trace amount of the hydrogenated product (3b) were obtained after visible-light irradiation for 12 h (Table 2, entry 1). Although the use of TEOA (triethanolamine) slightly increased the yield of 2b, the formation of 3b became pronounced probably due to the increase of proton concentration (Table 2, entry 3). On the other hand, the use of BI(OH)H (1,3-dimethyl-2-(o-hydroxyphenyl)-2,3-dihydro-1H-benzo[d]imidazole) successfully accelerated the hydrocarboxylation and increased the yield of 2b considerably with maintaining the low yield of 3b (Table 2, entry 5). Furthermore, the incorporation of BI(OH)H made it possible to reduce the amounts of the photoredox catalyst and the sacrificial electron donor: the use of only 1.0 mol% of [Ru(bpy)3](PF6)2 and 2.0 equiv. of BI(OH)H gave 70% yield of 2b (Table 2, entry 7). When the reaction was performed under an Ar atmosphere in the presence of Cs2CO3, no hydrocarboxylated product 2b was obtained. This result confirmed that the carbonate did not work as a source of CO2 in the present reaction (Table 2, entry 9). BI(OH)H has been known to work as a 2-electron, 2-proton donor with high reducing ability in redox photosensitizing reactions (Hasegawa et al., 2005, 2006; Tamaki et al., 2015). Since a tertiary amine contributed to the reductive quenching cycle of [Ru(bpy)3]2+, the increase in the yield of 2b was attributed to the promotion of the reduction process of the Rh(I) carboxylate species, which was the rate-determining step in the hydrocarboxylation. These results indicated that the redox property of a sacrificial electron donor is one of the crucial factors for the efficient promotion of the reaction.
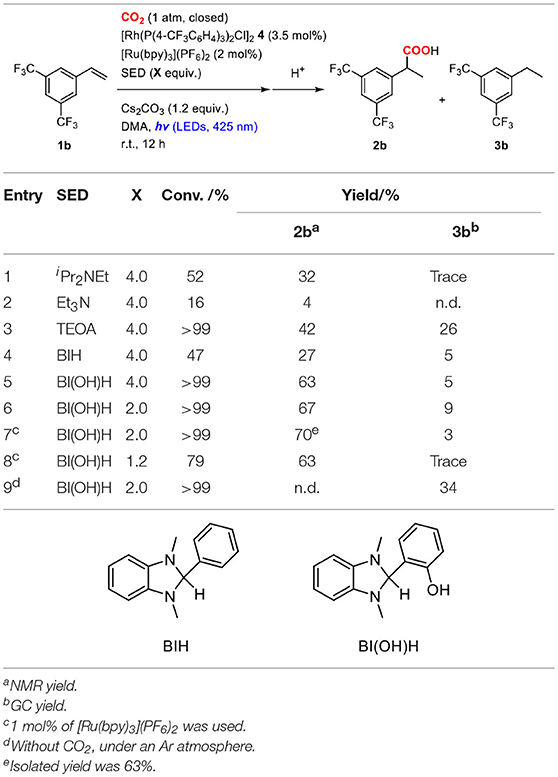
Table 2. Optimization of the reaction conditions of the hydrocarboxylation of 3,5-bis(trifluoromethyl)styrene.
Generality of the Hydrocarboxylation Under the Improved Conditions
Based on the improved conditions using 1b as discussed above, the generality of the hydrocarboxylation was examined using various alkene substrates. 1.0 mol% of [Ru(bpy)3](PF6)2, 3.5 mol% of 4, 2.0 equiv. of BI(OH)H and 1.2 equiv. of Cs2CO3 were employed for the hydrocarboxylation (Table 3). In the cases of using styrenes with an electron-withdrawing group such as 1a and 4-methoxycarbonyl styrene (1d), the reaction was almost completed after irradiation for 12 h, and the yields of the corresponding hydrocarboxylated products were significantly improved compared with those obtained in the previous conditions where 2.0 mol% of [Ru(bpy)3](PF6)2 and 4.0 equiv. of iPr2NEt were employed. Moreover, 4-trifluoromethyl styrene (1c) and non-substituted styrene (1e), which exhibited quite low reactivities in the previous conditions, did react to afford significant amounts of the corresponding hydrocarboxylated products though the yields were still not sufficiently high. The yields of the hydrocarboxylated products were also improved in the case of alkyl acrylates (1f and 1g). Consequently, the introduction of BI(OH)H electron donor with Cs2CO3 base successfully resulted in the increase in the yields of the present hydrocarboxylation reaction.
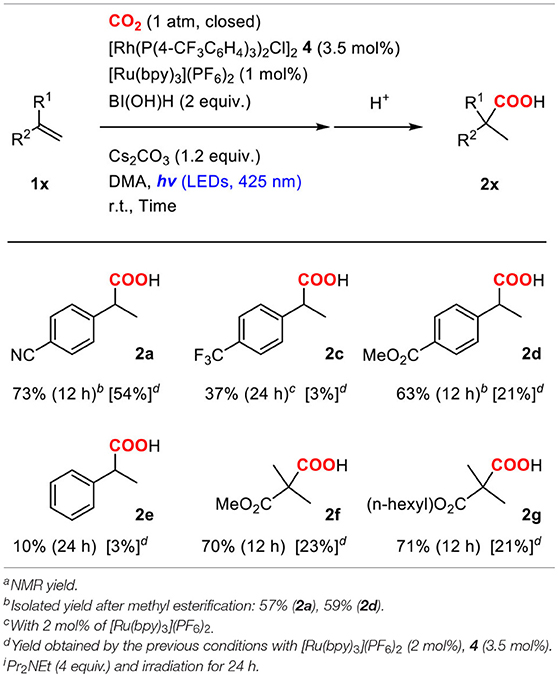
Table 3. Generality of the hydrocarboxylation of alkenesa.
Mechanistic Study
In order to reveal the reaction mechanism of the photocatalytic hydrocarboxylation, the stoichiometric reactions of the possible rhodium intermediates, which corresponded to each elementary step in the proposed catalytic cycle, were examined.
Rh Hydride Formation
Initially, the Rh(I) hydride formation step was investigated using Rh(PPh3)2(OAc) (5) as a model complex of the Rh(I) carboxylate intermediate. The DMA solution of 5 was irradiated by visible-light in the presence of a catalytic amount of [Ru(bpy)3](PF6)2, an excess amount of iPr2NEt and 1.5 equivalent of PPh3. After visible-light irradiation for 6 h, the Rh(I) monohydride complex, Rh(PPh)3H (6) was successfully obtained in 57% yield with the recovery of ca. 30% of 5 (based on 1H NMR using an internal standard) (Figure 3A; Supplementary Figure 1). Control experiments demonstrated that [Ru(bpy)3](PF6)2, iPr2NEt, and visible-light were all essential for the transformation. Since hydrogen evolution was not evident during the reaction, the contribution of gaseous hydrogen was excluded. Thus, this transformation was considered to proceed via (i) stepwise 2-electron, 2-proton transfers from the tertiary amine by the photoredox catalysis to give the Rh(III) dihydride carboxylate (7), and (ii) the base-assisted elimination of the carboxylic acid to give 6. In terms of step (i), the similar mechanisms have been proposed in photocatalytic hydrogen generation systems by a Rh(I) catalyst (Stoll et al., 2015). The initial single electron transfer to the protonated form of 5 would give the Rh(II) carboxylate monohydride, and the following electron and proton transfers or disproportionation of the two Rh(II) hydride species would give 7 (Figure 4). The presence of the Rh(III) dihydride intermediate was also supported by the fact that Rh(PCy3)2(OAc) (5′) was transformed to Rh(PCy3)2(OAc)(H)2 (7′) almost quantitatively under the similar conditions although the reaction was relatively slow (Figure 3B; Supplementary Figure 2). In this case, PCy3 ligands with strong σ-donation were considered to stabilize the Rh(III) dihydride intermediate to inhibit the following elimination reaction.
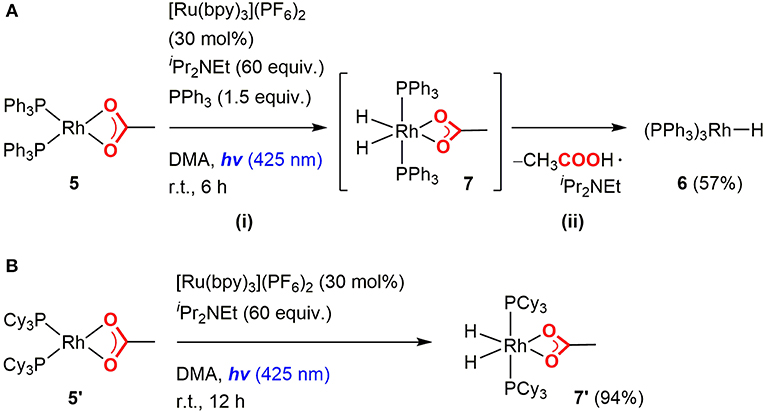
Figure 3. Rh hydride formation from the Rh(I) acetate complexes with (A) triphenylphosphines and (B) tricyclohexylphosphines.
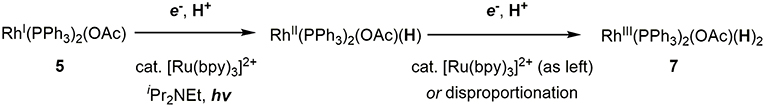
Figure 4. Proposed mechanism for the formation of the Rh(III) dihydride complex.
In order to confirm the carboxylic acid elimination step (ii), the reactivity of 7 was investigated in the presence of base. 7 was alternatively synthesized by the hydrogenation of 5 with H2, and was treated with an excess amount of iPr2NEt in the presence of PPh3 in C6D6. The reaction readily gave a mixture of 7 and 6 with liberation of [iPr2NHEt]+[CH3COO]−. Furthermore, addition of a small excess amount of [iPr2NHEt]+[CH3COO]− to the C6D6 solution of 6 resulted in the quantitative formation of 7. These results demonstrated that 7 was in equilibrium with 6 in the presence of iPr2NEt and PPh3 (Figure 5). When the treatment of 7 with iPr2NEt was similarly conducted in DMA, 6 was detected as a sole rhodium species in the reaction mixture, indicating that the equilibrium was almost completely shifted to the product side owing to the solvent effect of DMA.
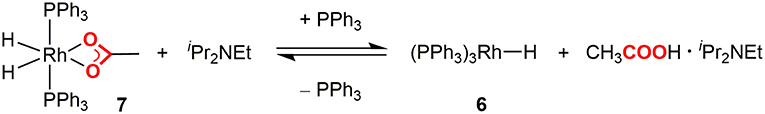
Figure 5. Equilibrium between the Rh(III) dihydride and Rh(I) monohydride complexes.
The possible mechanisms for generation of the hydrogenated product were (i) 2-electron, 2-proton transfers to the Rh(I) benzyl intermediate by the photoredox catalysis to give Rh(III) benzyl dihydride intermediate, which would undergo reductive elimination of the hydrogenated product, and (ii) the alkene insertion to the Rh(III) dihydride intermediate and the successive reductive elimination. Both pathways could be inhibited by lowering proton concentrations, as proton transfers would become inefficient in the former, and the competing carboxylic acid elimination from the dihydride complex would be promoted in the latter. Therefore, the inhibition of the hydrogenated product formation by the addition of Cs2CO3 was attributed to the decrease of the proton concentration in the catalytic system.
The photochemical formation of Rh(I) monohydride species was also feasible by using Rh(I) chloride complex as a Rh(I) source. It was demonstrated by the fact that Wilkinson's type complex Rh(PPh3)3Cl was converted to Rh(PPh)3H (6) by visible-light irradiation in the presence of a catalytic amount of [Ru(bpy)3](PF6)2 and an excess amount of iPr2NEt. Therefore, the Rh(I) chloride complex was confirmed to work as a precursor of the Rh(I) hydride active species.
Hydrometalation and Carboxylation
Since the Rh(I) monohydride species was successfully generated from the Rh(I) carboxylate species by photoredox catalysis, the hydrometallation and subsequent carboxylation processes were then investigated to complete the catalytic cycle. Treatment of Rh(PPh)3H (6) with an excess amount of 1a at room temperature readily formed the Rh(I) benzyl species, Rh(PPh3)2(η3-CHCH3(4-CNC6H4)) (8), almost quantitatively with the liberation of a PPh3 ligand (Figure 6i). The benzyl ligand in 8 was found to possess η3-coordination to the Rh(I) center based on NMR spectroscopic data (Werner et al., 1994). However, the attempt for isolation of 8 was not successful due to the presence of an equilibrium with 6. Therefore, in situ generated 8 was directly used for the carboxylation step.
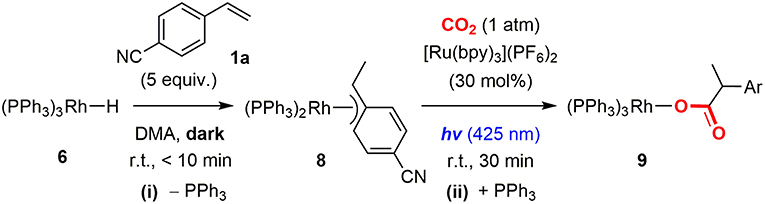
Figure 6. Hydrometallation of Rh(I) hydride complex and successive carboxylation of the Rh(I) π-benzyl complex with CO2.
To investigate the carboxylation process with CO2, a DMA solution of a 1:1 mixture of in-situ generated 8 and PPh3 was exposed to the atmospheric pressure of CO2 under various conditions. The carboxylation did not proceed under dark even by heating, which was against our expectations based on the general reactivity of organorhodium(I) complexes with CO2 (Ukai et al., 2006; Mizuno et al., 2011; Suga et al., 2014; Kawashima et al., 2016). Quite interestingly, when the mixture was irradiated by visible-light for 30 min in the presence of 30 mol% of [Ru(bpy)3](PF6)2, 8 was successfully converted to the Rh(I) carboxylate complex, Rh(PPh3)3(η1-O2CCHCH3(4-CNC6H4)) (9), almost quantitatively (Figure 6ii). 31P{1H} NMR spectroscopy confirmed the clean formation of 9: a pair of the doublet of doublet signals attributed to 8 completely disappeared with the PPh3 signal, and the doublet of doublet and doublet of triplet signals attributed to 9 appeared in 2: 1 ratio by visible-light irradiation (Figure 7). The control experiments demonstrated that CO2, [Ru(bpy)3](PF6)2 and visible-light were all essential for the carboxylation, suggesting that the nucleophilic addition of 8 to CO2 was facilitated by the photosensitization of [Ru(bpy)3]2+. The luminescence quenching experiment demonstrated that the excited state of [Ru(bpy)3]2+ was effectively quenched by 8 (Figure 8). The quenching constant was determined to be Kq = 2.07 × 103, which was much larger than that by iPr2NEt (Kq = 1.56 × 102, Supplementary Figure 3A). This result indicates that either photoinduced electron transfer or triplet-triplet energy transfer to 8 contributed to the quenching (Campagna et al., 2007; Arias-Rotondo and McCusker, 2016; Strieth-Kalthoff et al., 2018). However, the photoinduced electron transfer mechanism was unlikely in this case since (i) the carboxylation of 8 proceeded with a catalytic amount of [Ru(bpy)3](PF6)2 even in the absence of the sacrificial electron donor, and (ii) the cyclic voltammogram of 8 showed no significant redox peak within the window where the oxidative quenching of [Ru(bpy)3]2+ was possible. Therefore, the photoinduced triplet-triplet energy transfer from the excited [Ru(bpy)3]2+ to 8 was considered to be the most likely process in this carboxylation process. Although not very common, several examples on the photocatalytic organic transformations mediated by the triplet-triplet energy transfer were previously reported (Ikezawa et al., 1986; Osawa et al., 2001; Islangulov and Castellano, 2006; Lu and Yoon, 2012; Farney and Yoon, 2014).
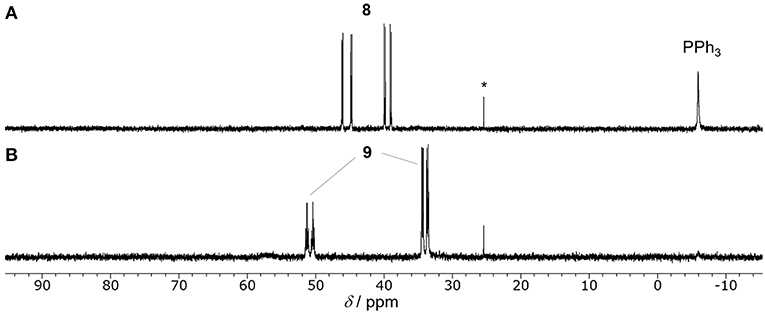
Figure 7. 31P{1H} NMR spectra (202 MHz, DMA, −15°C) of the reactions of 8 under CO2 atmosphere in the presence of [Ru(bpy)3](PF6)2 (30 mol%) (A) after kept in dark, (B) after visible-light irradiation (λirr. = 425 nm) for 30 min. *(O=)PPh3.
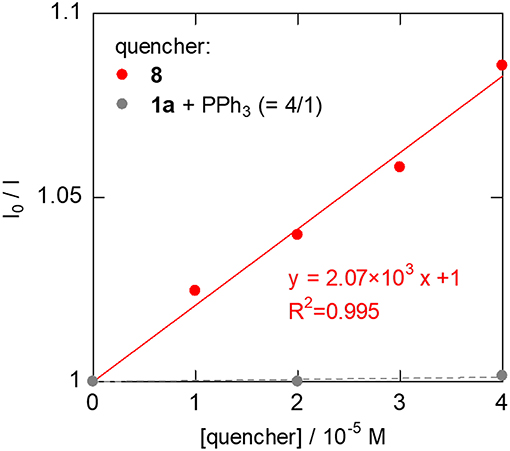
Figure 8. Luminescent quenching experiments of [Ru(bpy)3]2+ (1.0 × 10−5 M, deaerated DMF, r.t., λex. = 450 nm). 8 was generated in situ by the addition of 5 equiv. of 1a to the DMF solution of 6.
In order to investigate the detailed effect of the photoinduced energy transfer, the electronic structure analyses were performed in terms of the ground (S0) and the lowest excited triplet (T1) states of 8 based on DFT/TD-DFT methods. The calculated energy level of the T1 state of 8 (1.07 eV, based on the comparison between the S0 and T1 optimized geometries) was much lower than that of [Ru(bpy)3]2+ (2.17 eV), indicating that the triplet-triplet energy transfer from the excited [Ru(bpy)3]2+ to 8 was feasible. In terms of the optimized structures, a notable difference was found on the coordination manner of the benzyl ligand between the S0 and T1 geometries. In the S0 optimized structure, the η3-coodination of the benzyl ligand was represented by the similar three Rh-C distances, which coincided with the results of the 1H NMR observation (Figure 9A; Table 4). On the other hand, in the T1 optimized structure, while the Rh-C1 (benzyl carbon) distance remained unchanged, the Rh-C2/C3 distances significantly elongated compared with those of the S0 structure. These results indicated that the benzyl ligand changed its coordination-mode from η3-type (α-benzyl) to η1-type (σ-benzyl) in the T1 state. According to the analysis on the electronic transition characters, the T1 state was mainly contributed by the transitions of HOMO → LUMO (88%) and HOMO → LUMO+16 (4%) (Supplementary Table 2). The molecular orbital distribution indicated that LUMO and LUMO+16 mainly localized on the Rh (dπ) and benzyl ligand (π*) while the HOMO localized on the Rh (dσ) center (Figure 9B). As these LUMOs partially possessed the antibonding character on the Rh-C2/C3 bonds, the photoexcitation induced the dissociation of these Rh-C bonds, which resulted in the isomerization to the σ-benzyl species.
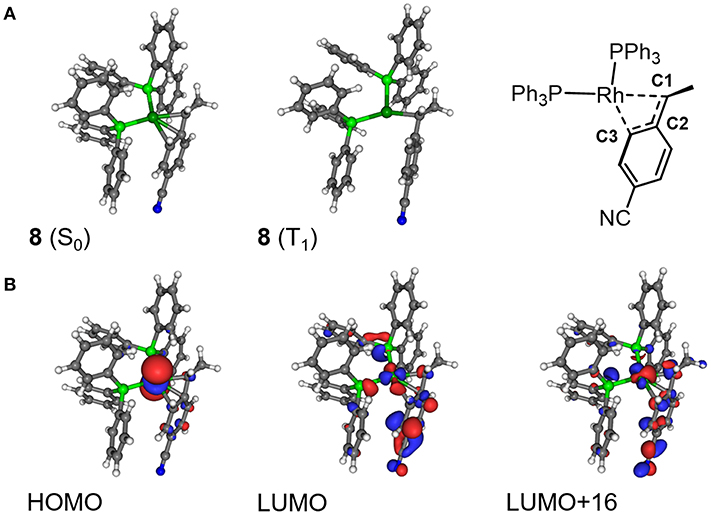
Figure 9. (A) Optimized structures of 8 in the ground (S0) state and lowest excited triplet (T1) state, and (B) the selected molecular orbitals of 8 (S0).
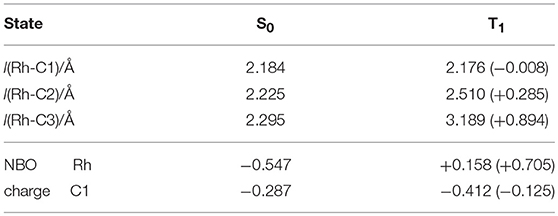
Table 4. Selected bond lengths and NBO natural charges in the optimized geometries of 8(S0) and 8(T1).
Concerning the acceleration of the carboxylation step, one possibility is the generation of the coordination site by taking σ-benzyl structure in the T1 state, which would promote the following carboxylation by facilitating coordination of CO2 to Rh center. Indeed, a similar thermal process has been proposed as a plausible mechanism for the carboxylation of organorhodium(I) complexes (Darensbourg et al., 1987). Another possibility is the direct nucleophilic addition of the benzyl carbon to CO2 in the T1 state. The NBO analysis demonstrated that the natural charge on the C1 atom significantly shifted to the negative side while that on the Rh atom shifted to the positive side in the T1 state. The increase of the electron density on the C1 atom in the T1 state would result in the acceleration of the nucleophilic addition to CO2. Therefore, the structural and/or electronic factors associated with the transition to the T1 state are thought to contribute to the carboxylation of 8.
The effect of the photoactivation of the Rh(I) π-benzyl complex was also supported by the reactivity of the Rh(I) σ-alkyl complex with CO2. When a mixture of 6 and an excess amount of methyl acrylate (1f) was subjected to a CO2 atmosphere for 3 h even under dark, the quantitative formation of Rh(PPh3)3(η1-O2CCHCH3(CO2CH3)) (10) was indicated by 31P{1H} NMR spectroscopy. The carboxylation of 1f was confirmed by the fact that the corresponding hydrocarboxylated product (2f) was obtained from the reaction mixture. This result indicated that the photosensitization by [Ru(bpy)3]2+ was not essential in this case. Thus, the major role of the excitation was thought to be the transformation from π-benzyl to σ-benzyl complexes to generate a coordination site and to make them more nucleophilic.
Addition of the Second Photosensitizer
The above mechanistic study revealed that a photosensitizer played two key roles in the hydrocarboxylation cycle: one is a “photoredox catalyst” to reduce the Rh(I) carboxylate species, and the other is a “triplet photosensitizer” to promote carboxylation of the Rh(I) benzyl species. With a single photosensitizer, the excited state of the photosensitizer was quenched by either a tertiary amine for the electron transfer or a Rh(I) benzyl species for the energy transfer, and these two processes competed during the reaction. Since the former was related to the rate-determining step when using iPr2NEt as a sacrificial electron donor, the incorporation of the second photosensitizer possessing suitable redox properties for the reductive quenching cycle was expected to facilitate the catalytic reaction.
On the basis of the idea, 2.0 mol% of a cyclometalated Ir(III) complex was added as a second photosensitizer to a mixture of 1b, 3.5 mol% of 4, 2.0 mol% of [Ru(bpy)3](PF6)2 and 4.0 equiv. of iPr2NEt, and the solution was irradiated under CO2 atmosphere at room temperature (Table 5). To excite both photosensitizers, a wide range of UV-visible-light (380–750 nm) was applied to the reactions. As expected, the addition of the second photosensitizer was found to be effective. For instance, when [Ir(ppy)2(dtbbpy)](PF6) was added, the reaction was completed after irradiation for only 6 h, and the yield of 2b was increased more than five-fold compared to that of the reaction without the second photosensitizer (Table 5, entry 2). According to the redox properties of [Ir(ppy)2(dtbbpy)](PF6), the acceleration of the reaction was thought to be attributed mainly to the high reducing ability of the one-electron reduced species to promote the reduction process. The yield of 2b further increased when incorporating [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) as a second photosensitizer (Table 5, entry 3), and its concentration could be reduced to 1.0 mol% without lowering the yield (Table 5, entry 4). This result was assumed to be due to the high oxidizing ability of the excited state in addition to the sufficient reducing ability of the one-electron reduced species. The excited state of [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) was found to be able to work as an energy transfer agent of 8 based on the luminescence quenching experiment (Kq = 2.76 × 104, Supplementary Figure 3B). However, it is considered to contribute to the reaction mainly as an electron transfer agent under the catalytic conditions owing to the efficient quenching by the sacrificial electron donor. On the other hand, the addition of fac-Ir(ppy)3 resulted in only a small acceleration, which was probably attributable to the inferior oxidizing ability in the excited state (Table 5, entry 6). These results demonstrate that photosensitizers possessing both high oxidizing ability of the excited state and high reducing ability of the one-electron reduced species are advantageous as a second photosensitizer. The positive result on the addition of the two appropriate photosensitizers reflected the fact that the catalytic cycle was composed of the multiple photochemical processes, and the acceleration of the reduction process led to the enhancement of the catalytic activity when using iPr2NEt as a sacrificial electron donor.
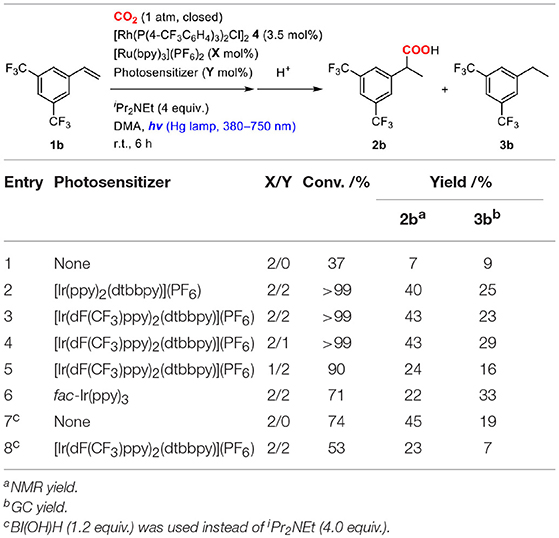
Table 5. Screening of the reaction conditions of the hydrocarboxylation of 3,5-bis(trifluoromethyl)styrene with a second photosensitizer.
Rate-Determining Step in the Hydrocarboxylation With BI(OH)H
The previous experiments demonstrated that the rate-determining step of the catalytic cycle was the reduction process of the Rh(I) carboxylate species 9 when employing iPr2NEt as a sacrificial electron donor. In order to investigate the contribution of BI(OH)H to the catalytic cycle, the similar examination was carried out using BI(OH)H as a sacrificial electron donor instead of iPr2NEt. Interestingly, the resting-state was found to be Rh(I) π-benzyl intermediate 8 when a mixture of 1a, catalytic amounts of 6 and [Ru(bpy)3](PF6)2, and 1.2 equiv. of BI(OH)H was irradiated by visible-light under CO2 atmosphere (Supplementary Figure 4). In that case, 9 was not detectable even after prolonged irradiation, indicating that the rate-determining step obviously altered from the reduction process to the carboxylation process by changing the sacrificial electron donor. This result was also supported by the fact that the acceleration of the reaction by the addition of a second photosensitizer was not observed in the case of the reaction using BI(OH)H as a sacrificial electron donor (Table 5, entry 8). These observations suggested that the use of BI(OH)H strongly accelerated the reductive quenching cycle of [Ru(bpy)3]2+ to promote the reduction process.
Conclusion
In this study, the improved catalytic conditions of the visible-light driven hydrocarboxylation by Rh(I) and [Ru(bpy)3]2+ catalysts were explored, and the detailed reaction mechanism was investigated. On the basis of the stoichiometric reactions of the possible rhodium intermediates, the proposed catalytic cycle was confirmed to be composed of (i) the hydrometallation of alkenes by Rh(I) monohydride species, (ii) the photochemical carboxylation of the Rh(I) benzyl species with CO2, (iii) the photoinduced 2-electron, 2-proton transfers to the Rh(I) carboxylate species, and (iv) the base-assisted carboxylic acid elimination. One strategy for the enhancement of the catalytic reaction was to employ BI(OH)H possessing superior reducing ability as a sacrificial electron donor instead of iPr2NEt. It successfully improved the efficiency of the reaction, which had been major challenges in the previous catalytic conditions. The alteration of the resting-state by changing the sacrificial electron donor indicated that the addition of BI(OH)H significantly promoted the reduction process of the Rh(I) carboxylate species through the enhancement of the reductive quenching efficiency of [Ru(bpy)3]2+. Another strategy for the enhancement of the efficiency was to add the second photosensitizer in charge of the reductive quenching cycle. The acceleration of the catalytic reaction by the addition of the appropriate cyclometalated Ir(III) complex together with [Ru(bpy)3]2+ supported this hypothesis. These two effective strategies suggested that the promotion of the reduction processes was a key to enhance the catalytic activity in the present system. In addition to expand the versatility of the present hydrocarboxylation, this study would provide fundamental insights into the catalytic organic transformations by transition-metal/photoredox dual catalysis.
Experimental
General
All operations were carried out under an argon atmosphere unless otherwise noted. 1H, 13C, and 31P NMR spectra were recorded on Bruker DRX-500, JEOL ECZ-500, ECX-400, and ECS-400 spectrometers. 31P and 19F NMR chemical shifts were calibrated using external 85% H3PO4 (δ: 0.0 ppm) and neat C6F6 (δ: −164.9 ppm), respectively. IR spectra were recorded on an SC-100-VIR with an ATR PRO450-S accessory (JASCO Co., Ltd.). Emission spectra were recorded on an FP-6500 spectrofluorometer (JASCO Co., Ltd.). FAB-MS and FD-MS spectra were recorded on a JEOL JMS-700 spectrometer and a JMS-T100 spectrometer, respectively. Gas chromatography (GC-FID / TCD) was recorded on a Shimadzu GC-2010 spectrometer. Analytical thin-layer chromatography (TLC) was performed with a glass plate coated with silica gel (Wakogel B-5F). Visible-light irradiation was performed with a Relyon Twin LED Light (3W × 2, λirr. = 425 ± 15 nm), and UV-visible-light irradiation was performed with an USHIO Optical Modulex OPM2-502XQ (500 W Xe lamp) with a super cold filter ZSC0750 (ASAHI Spectra Inc.).
THF, toluene, pentane and diethyl ether were purified by a solvent purification system by Glass Contour. Dehydrated dimethylacetamide (DMA) and dimethylformamide (DMF) were purchased from Kanto Chemical Co., Inc., degassed by argon bubbling and stored in a glovebox. Tertiary amines were distilled, degassed three times by freeze-pump-thaw method and stored under N2. Solvents for NMR measurements were dried over molecular sieves, degassed three times by freeze-pump-thaw method and stored under N2. All other solvents were distilled, degassed by argon bubbling and stored in a glovebox. CO2 and H2 gases were purchased from Taiyo Nippon Sanso Corporation. [Rh(coe)2Cl]2 (Van der Ent et al., 1990), P(4-CF3C6H4)3 (Suomalainen et al., 2001), Rh(PPh3)2(OAc) (5) (Grushin et al., 1995), [Rh(cod)(OAc)]2 (Chatt and Venanzi, 1957), Rh(PPh3)3H (6) (Annibale and Song, 2014), [Ru(bpy)3](PF6)2 (Damrauer et al., 1997), [Ru(dmbpy)3](PF6)2 (Damrauer et al., 1997), [Ru(bpz)3](PF6)2 (Schultz et al., 2015), Ir(ppy)2(dtbbpy)(PF6) (Tellis et al., 2014), [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (Slinker et al., 2004), fac-Ir(ppy)3 (Tamayo et al., 2003), and diisopropylethylammonium acetate (Anouti et al., 2008) were prepared according to the published methods. 4-Cyanostyrene (1a) was prepared by Wittig reaction of 4-cyanobenzaldehyde (Falk et al., 2013). Other chemicals were purchased and used as received.
Photocatalytic Reactions
For screening conditions with alkenes (1a–1g), a DMA solution (0.6 mL) of an alkene (0.060 mmol), [Rh(P(4-CF3C6H4)3)2Cl]2 (4, 4.5 mg, 0.0021 mmol), photoredox catalyst(s), sacrificial electron donor and inorganic base (defined amounts) was prepared in a glass tube (φ 2.0 cm, 18 cm) under an argon atmosphere. Then the headspace gas was replaced by an atmospheric pressure of CO2, and the reaction vessel was put in a water bath placed at a distance of 10 mm from light sources. The mixture was irradiated with visible-light from blue LED lamp (λirr. = 425 nm, two sockets) or UV-visible-light from Xe lamp (λirr. = 380–800 nm) for defined time in the closed system. The product mixture was analyzed by 1H NMR and GC to determine the NMR yield of the hydrocarboxylated product (2a–2g) and the GC yield of the hydrogenated product (3a, 3b), respectively (internal standard: 1,1,2,2-tetrachloroethane).
For isolation of the methyl esters of the hydrocarboxylated products (2a, 2b, 2d), a DMA solution (1.2 mL) of a styrene (0.12 mmol), 4 (9.0 mg, 0.0042 mmol), [Ru(bpy)3](PF6)2 (1.0 mg, 0.0012 mmol), BI(OH)H (58 mg, 0.24 mmol), and Cs2CO3 (47 mg, 0.14 mmol) was prepared in a glass tube (φ 2.0 cm, 18 cm), and irradiated with visible-light from blue LED lamp (λirr. = 425 nm, three sockets) for defined time after replacement of the headspace gas by an atmospheric pressure of CO2. After irradiation, the reaction mixture was diluted with diethyl ether and extracted with H2O three times. The combined aqueous layer was acidified by 1N HCl aq., and then extracted with diethyl ether three times. The combined organic layer was dried over MgSO4, filtered and evaporated under reduced pressure to give the hydrocarboxylated product. Then, the product was dissolved in Et2O-MeOH, and TMSCHN2 (excess) was added at 0°C. The mixture was stirred at 0°C for 30 min and the solvent was removed under reduced pressure. The crude product was purified by preparative TLC (AcOEt/n-hexane = 1/5) to give the corresponding methyl-esterified product.
Preparations of Rhodium Complexes and Their Stoichiometric Reactions
Preparation of [Rh(P(4-CF3C6H4)3)2Cl]2 (4)
A solution of P(4-CF3C6H4)3 (200 mg, 0.429 mmol) in toluene (2 mL) was added dropwise to a solution of [Rh(coe)2Cl]2 (77 mg, 0.107 mmol) in toluene (2 mL) and the mixture was stirred at room temperature overnight. After removal of solvent under reduced pressure, the crude product was dissolved in THF and then pentane was added to induce precipitation. The precipitates were collected to give the target product (218 mg, 0.102 mmol, 95% yield). 1H NMR (500 MHz, THF-d8, r.t., δ/ppm): δ 7.73–7.67 (m, 24 H, PAr3), 7.39 (d, J = 8 Hz, 24 H, PAr3). 13C{1H} NMR (125 MHz, r.t., THF-d8, δ/ppm): δ 139.3 (vt, N = 22 Hz, PAr3), 135.7 (s, PAr3), 132.1 (q, JC−F = 33 Hz, PAr3), 125.0 (s, PAr3), 124.6 (q, JC−F = 272 Hz, -CF3). 31P{1H} NMR (202 MHz, THF-d8, r.t., δ/ppm): δ 53.4 (d, J = 194 Hz). 19F NMR (471 MHz, THF-d8, r.t., δ/ppm): δ −60.6 (s). ESI-MS: m/z = 1035 [M/2 − Cl]+. Anal. Found (calcd for C84H48Cl2F36P4Rh2): C, 46.94 (47.11); H, 2.28 (2.26).
Preparation of Rh(PCy3)2(OAc) (5′)
[Rh(cod)(OAc)]2 (60 mg, 0.111 mmol) and PCy3 (125 mg, 0.444 mmol) were suspended in DMA (4 mL), and the mixture was irradiated with UV-visible-light (500 W Xe lamp, λirr. = 380–800 nm) at room temperature with vigorous stirring for 30 h. The precipitate was filtered, washed with DMA and cold pentane (−35°C), and then recrystallized from a minimum volume of pentane at −35°C to yield the target compound (98 mg, 0.135 mmol, 61%). IR (KBr): ν(OCOas) = 1,528, ν(OCOsym) = 1,445 cm−1. 1H NMR (500 MHz, C6D6, r.t., δ/ppm): δ 2.35 – 1.24 (m, 69 H, PCy3 and O2CCH3). 13C{1H} NMR (125 MHz, r.t., C6D6, δ/ppm): δ 188.2 (s, O2CCH3), 35.9 (vt, N = 10 Hz, PCy3), 31.1 (s, PCy3), 28.4 (vt, N = 5 Hz, PCy3), 27.3 (s, PCy3), 24.9 (s, O2CCH3). 31P{1H} NMR (202 MHz, C6D6, r.t., δ/ppm): δ 59.1 (d, JP−Rh = 198 Hz). HR-MS (FAB): m/z = 722.3817 [M]+ (calcd for [C38H69O2P2Rh]+: 722.3828).
Preparation of an Authentic Sample of Rh(PCy3)2(OAc)(H)2 (7′)
[Rh(cod)(OAc)]2 (60 mg, 0.111 mmol) and PCy3 (125 mg, 0.444 mmol) were dissolved in THF (3 mL), and the mixture was stirred under H2 (1 atm) at room temperature overnight. After removal of solvent, the crude product was dissolved in toluene and filtered through Celite®. The resultant solid after evaporation was washed with cold diethyl ether (−35°C) to give the target compound (113 mg, 0.156 mmol, 70%). IR (KBr): ν(RhH) = 2,143, ν(OCOas) = 1,551, ν(OCOsym) = 1,436 cm−1. 1H NMR (500 MHz, C6D6, r.t., δ/ppm): δ 2.20 – 1.22 (m, 69 H, PCy3 and O2CCH3), −23.7 (dt, JH−Rh = 24, JH−P = 15 Hz, 2 H, Rh-H) 13C{1H} NMR (125 MHz, r.t., C6D6, δ/ppm): δ 180.2 (s, O2CCH3), 35.8 (vt, JC−P = 10 Hz, PCy3), 30.5 (s, PCy3), 28.3 (vt, N = 5 Hz, PCy3), 27.1 (s, PCy3), 24.8 (s, O2CCH3). 31P{1H} NMR (202 MHz, C6D6, r.t., δ/ppm): δ 51.1 (d, JP−Rh = 115 Hz). HR-MS (FD): m/z = 724.3975 (calcd for [C38H71O2P2Rh]+: 724.3984).
Preparation of Rh(PPh3)2(η3-CHCH3(4-CNC6H4)) (8)
To a solution of Rh(PPh3)3H (6) (5.4 mg, 0.0060 mmol) in THF-d8 (0.6 mL) in a J. Young NMR tube was added 4-cyanostyrene (1a) (1.6 μL, 0.012 mmol) at room temperature. Rh(PPh3)2(η3-CHCH3(4-CNC6H4)) (8) formed almost quantitatively. 1H NMR (500 MHz, THF-d8, −10°C, δ/ppm): δ 7.47 – 7.00 (m, 30 H, PPh3), 6.81 (brd, J = 8 Hz, 1 H, Ar), 6.57 (brd, J = 7 Hz, 1 H, Ar), 6.10 (brd, J = 8 Hz, 1 H, Ar), 4.89 (brd, J = 7 Hz, 1 H, Ar), 2.47 – 2.40 (m, 1 H, -CHCH3), 0.92 – 0.87 (m, 3 H, -CHCH3). 31P{1H} NMR (202 MHz, THF-d8, −10°C, δ/ppm): δ 46.2 (dd, JP−Rh = 263 Hz, JP−P = 31 Hz), 39.6 (dd, JP−Rh = 178 Hz, JP−P = 31 Hz). HR-MS (FAB): m/z = 757.1539 (calcd for [C45H38NP2Rh]+: 757.1535).
Preparation of Rh(PPh3)3(η1-O2CCHCH3(4-CNC6H4)) (9)
To a solution of Rh(PPh3)3H (6) (60 mg, 0.090 mmol) in toluene (3 mL) was added dropwise a solution 2-(4-cyanophenyl)propionic acid (16 mg, 0.090 mmol) in toluene (2 mL), and the mixture was stirred at room temperature for 4 h. After removal of solvent under reduced pressure, the crude product was dissolved in toluene and then pentane was added to induce precipitation. The precipitates were collected to give the target product as a 9: 1 mixture with Rh(PPh3)2(η2-O2CCHCH3(4-CNC6H4)) which was formed by dissociation of PPh3 from 9 (59 mg). 9 IR (KBr): ν(CN) = 2,224, ν(OCOas) = 1,604, ν(OCOsym) = 1,342 cm−1. 1H NMR (500 MHz, THF-d8, −60°C, δ/ppm): δ 7.56 – 6.79 (m, 47 H, PPh3, O2CCHCH3Ar), 6.30 (brd, J = 8 Hz, 2 H, O2CCHCH3Ar), 1.65 (brq, J = 7 Hz, 1 H, O2CCHCH3Ar), 0.25 (brd, J = 7 Hz, 3 H, O2CCHCH3Ar). 13C{1H} NMR (125 MHz, r.t., THF-d8, −30°C, δ/ppm): δ 176.6 (s, O2CCHCH3Ar), 150.8 (s, O2CCHCH3Ar), 135.7 (vt, N = 6 Hz, PPh3), 131.1 (s, O2CCHCH3Ar), 129.6 (s, O2CCHCH3Ar), 129.2 (s, PPh3), 127.8 (s, PPh3), 127.5 (s, PPh3), 127.4 (s, PPh3), 119.8 (s, -CN), 108.9 (s, O2CCHCH3Ar), 47.8 (s, O2CCHCH3Ar), 18.1 (s, O2CCHCH3Ar). 31P{1H} NMR (202 MHz, THF-d8, −30°C, δ/ppm): δ 51.5 (dt, JP−Rh = 174 Hz, JP−P = 41 Hz), 34.9 (dd, JP−Rh = 153 Hz, JP−P = 41 Hz). HR-MS (FAB): m/z = 801.1453 [M−(PPh3)]+ (calcd for [C46H38NO2P2Rh]+: 801.1433). Rh(PPh3)2(η2-O2CCHCH3(4-CNC6H4)) 31P{1H} NMR (202 MHz, THF-d8, −30°C, δ / ppm): δ 57.6 (brd, JP−Rh = 210 Hz).
Redox-Photosensitized Reaction of Rh(PPh3)2(OAc) (5)
Rh(PPh3)2(OAc) (5) (2.7 mg, 0.0040 mmol), [Ru(bpy)3](PF6)2 (1.0 mg, 0.0012 mmol), PPh3 (1.5 mg, 0.0060 mmol), iPr2NEt (41 μL, 0.24 mmol) and DMA (0.6 mL) were added in a glass tube with a magnetic stirrer. The mixture was irradiated with visible-light (λirr. = 425 nm) at room temperature for 6 h. The solvent was removed under reduced pressure, and the resulting solid was analyzed by 1H and 31P NMR spectroscopies (THF-d8, internal standard: mesitylene). The Rh(I) monohydride species corresponding to Rh(PPh3)3H (6) formed in 57% yield (based on the Rh-hydride signal) and ca. 30% of the starting material 5 remained in the product mixture. 6 was highly fluxional in the reaction solution at room temperature in the presence of triphenylphosphine. 1H NMR (500 MHz, THF-d8, δ/ppm, r.t.): −8.45 (brd, JH−Rh = 13 Hz, Rh-H). 31P NMR (202 MHz, THF-d8, δ/ppm, r.t.): 41 (br). The intensity of the Rh-hydride signal was significantly increased at room temperature when Rh(PPh3)3H (6) synthesized alternatively was added to the reaction mixture, also supporting the formation of 6. In addition, when the solution was cooled to −90°C, the signals attributed to Rh(PPh3)4H were observed instead, indicating 6 was converted to Rh(PPh3)4H at low temperature. 1H NMR (500 MHz, THF-d8, δ/ppm, −90°C): −13.5 (dq JH−Rh = 118, JH−P = 15 Hz, Rh-H). 31P NMR (202 MHz, THF-d8, δ/ppm, −90°C): 33.4 (dm, JP−Rh = 112), 30.2 (dd, JP−Rh = 162, JP−P = 32 Hz). The spectrosopic data was analogous to the values reported previously (Dewhirst et al., 1968; Strauss and Shriver, 1978).
Redox-Photosensitized Reaction of Rh(PCy3)2(OAc) (5′)
Rh(PCy3)2(OAc) (5′) (2.9 mg, 0.0040 mmol), [Ru(bpy)3](PF6)2 (1.0 mg, 0.0012 mmol), iPr2NEt (41 μL, 0.24 mmol) and DMA (0.6 mL) were added in a glass tube with a magnetic stirrer. The mixture was irradiated with visible-light (λirr. = 425 nm) at room temperature for 12 h. The solvent was removed under reduced pressure, and the resulting solid was analyzed by 1H and 31P NMR spectroscopies (C6D6, internal standard: mesitylene). Rh(PCy3)(OAc)(H)2 (7′) was formed in 94% yield (based on the rhodium hydride signal). The spectroscopic feature well agreed with the complex 7′ synthesized alternatively (vide supra). No other Rh hydride signal was observed even by addition of 1 equiv. of PCy3 to the reaction mixture.
Reaction of Rh(PPh3)2(OAc)(H)2 (7) and iPr2NEt
A solution of Rh(PPh3)2(OAc) (5) (2.7 mg, 0.0040 mmol) in C6D6 (0.6 mL) was put in a J. Young NMR tube under hydrogen atmosphere at room temperature. After 2 h, Rh(PPh3)2(OAc)(H)2 (7) formed in situ. 1H NMR (500 MHz, C6D6, δ/ppm, r.t.): 7.86 – 7.00 (m, PPh3), −20.8 (dt, JH−Rh = 22 Hz, JH−P = 17 Hz, Rh-H). 31P{1H} NMR (202 MHz, C6D6, δ/ppm, r.t.): 41.4 (d, JP−Rh = 121 Hz). Then, the reaction solution was degassed by freeze-pump-thaw method 3 times to remove the hydrogen gas. Addition of 1 equiv. of PPh3 (1.5 mg, 0.0060 mmol) and 80 equiv. of iPr2NEt (41 μL, 0.24 mmol) gave Rh(PPh3)3H (6) in 58% yield (based on the rhodium hydride signal). Ca. 20% of 7 remained in the reaction mixture. The spectroscopic data of 6 was identical with that of the sample prepared by the photosensitizing reaction of Rh(PPh3)2(OAc) (5). The same reaction was also performed in DMA. In this case, 7 converted fully after addition of PPh3 and iPr2NEt, and 6 was obtained as a major product.
Reaction of Rh(PPh3)3H (6) and [iPr2NHEt]+[CH3COO]−
To a solution of Rh(PPh3)3H (6) (3.6 mg, 0.0040 mmol) in C6D6 (0.6 mL) was added diisopropylethylammonium acetate (1.1 mg, 0.0060 mmol) and the mixture was stirred at room temperature for 10 min. Rh(PPh3)2(OAc)(H)2 (7) formed quantitatively. The spectroscopic data of the product in C6D6 was identical with those of the sample prepared by the hydrogenation of Rh(PPh3)2(OAc) (5).
Redox-Photosensitized Reaction of Rh(PPh3)3Cl
Rh(PPh3)3Cl (3.7 mg, 0.0040 mmol), [Ru(bpy)3](PF6)2 (1.0 mg, 0.0012 mmol), iPr2NEt (41 μL, 0.24 mmol) and DMF-d7 (0.6 mL) were added in a J. Young NMR tube. The mixture was irradiated with visible-light (λirr. = 425 nm) at room temperature for 6 h. Rh(PPh3)3H (6) formed in 52% yield (based on the Rh-hydride signal) and ca. 40% of the starting material remained in the reaction mixture. 1H NMR (500 MHz, δ/ppm, −50°C): −8.48 (ddt, JH−Rh = 101 Hz, JH−P = 18 Hz, JH−P = 15 Hz, Rh-H). 31P NMR (202 MHz, δ/ppm, −50°C): 43.7 (dd, JP−Rh = 170 Hz, JP−P = 25 Hz), 38.9 (dm, JP−Rh = 149 Hz). The spectrosopic data was analogous to the values reported previously (Dewhirst et al., 1968; Strauss and Shriver, 1978).
Reaction of Rh(PPh3)3H (6) With 4-Cyanostyrene (1a) and CO2
To a solution of Rh(PPh3)3H (6) (3.6 mg, 0.0040 mmol) in DMA (0.6 mL) was added 4-cyanostyrene (1a) (2.6 μL, 0.020 mmol) in a J. Young NMR tube at room temperature. Rh(PPh3)2(η3-CHCH3(4-CNC6H4)) (8) was generated in situ almost quantitatively. Then, [Ru(bpy)3](PF6)2 (1.0 mg, 0.0012 mmol) was added, and the mixture was irradiated with visible-light (λirr. = 425 nm) under a CO2 atmosphere (1 atm) at room temperature for 30 min. Rh(PPh3)3(η1-O2CCHCH3(4-CNC6H4)) (9) was formed almost quantitatively, as the spectroscopic data well agreed with the complex synthesized alternatively.
Reaction of Rh(PPh3)3H (6) With Methyl Acrylate (1f) and CO2
To a solution of Rh(PPh3)3H (6) (3.6 mg, 0.0040 mmol) and [Ru(bpy)3](PF6)2 (1.0 mg, 0.0012 mmol) in DMA (0.6 mL) was added methyl acrylate (1f) (1.8 μL, 0.020 mmol) in a J. Young NMR tube at room temperature. The mixture was exposed to a CO2 atmosphere (1 atm) under dark at room temperature for 3 h. The observed 31P{1H} NMR spectral feature was analogous to that of 9, indicating the formation of Rh(PPh3)3(η1-O2CCHCH3(CO2CH3)) (10). 31P{1H} NMR (162 MHz, DMA, −15°C, δ/ppm): δ 50.6 (dt, JP−Rh = 174 Hz, JP−P = 44 Hz), 33.5 (dd, JP−Rh = 153 Hz, JP−P = 44 Hz). The reaction solution was treated with NaHCO3 aq., and then acidified with 1N HCl aq. The organic layer was extracted with diethyl ether three times, dried over MgSO4, filtered and evaporated under reduced pressure to give methyl methylmalonate in 54% yield (based on 6).
Observation of the Reaction Intermediates Under Catalytic Conditions
Rh(PPh3)3H (6) (3.6 mg, 0.0040 mmol), [Ru(bpy)3](PF6)2 (1.0 mg, 0.0012 mmol), BI(OH)H (17.3 mg, 0.072 mmol), 4-cyanostyrene (1a) (7.7 μL, 0.060 mmol) and DMA (0.6 mL) were added in a J. Young NMR tube. The mixture was irradiated with visible-light (λirr. = 425 nm) under CO2 atmosphere (1 atm) at room temperature for 30 min. The 31P{1H} NMR spectra were observed at −15°C before and after irradiation, and only Rh(PPh3)2(η3-CHCH3(4-CNC6H4)) (8) was observed as a resting-state in both spectra.
Theoretical Study
Theoretical calculations were performed at the DFT level with the Gaussian 09 package. The geometry optimizations were performed using the mPW1PW91 functional (Adamo and Barone, 1998). The LanL2DZ basis set was used for all atoms and extended by a polarization function (except for H) (Dunning and Hay, 1976; Wadt and Hay, 1985a,b). To address solvation effects, the conductor-like polarizable continuum model (CPCM, N,N-Dimethylacetamide) (Tomasi et al., 2005) was used for the ground and excited states. For validation, vibrational frequencies were calculated for the ground and excited states. The orbital plots as well as the graphical representations were performed using Molekel (Varetto, 2009). Natural bond orbital (NBO) analysis was used to predict and interpret the computational results (Glendening et al., 2001). Total ZPE energies and cartesian coordinates of computed structures are given in Supplementary Table 3.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
NN and KM performed the experiments. KS instructed the experiments of NN. KM performed the theoretical calculations. NI and JT supervised the project. KM and NI wrote the paper.
Funding
The authors are grateful for the financial supports by an ACT-C program JPMJCR12Y3 from JST and JSPS KAKENHI Grant no. 15H05800, 17H06143, and 24245019.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MA declared a shared affiliation, with no collaboration, with the authors NN, KS, JT, NI, to the handling editor at the time of review.
Acknowledgments
The authors also thank Prof. Osamu Ishitani for valuable discussions on the use of photosensitizers and sacrificial electron donors.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00371/full#supplementary-material
Abbreviations
bpy, 2,2′-bipyridyl; dtbbpy, 4,4′-di-tert-butyl-2,2′-bipyridyl; bpz, 2,2′-bipyrazine; ppy, 2-phenylpyridiine; dF(CF3)ppy, 3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl]phenyl; SCE, saturated calomel electrode; BIH, 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole; BI(OH)H, 1,3-dimethyl-2-(o-hydroxyphenyl)-2,3-dihydro-1H-benzo[d]imidazole; SED, sacrificial electron donor; PC, photoredox catalyst; SCE, saturated calomel electrode; vt, virtual coupling. For the abbreviations of the rhodium complexes, see Supplementary Table 1.
References
Adamo, C., and Barone, V. (1998). Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: the mPW and mPW1PW models. J. Chem. Phys. 108, 664–675. doi: 10.1063/1.475428
Adams, R. E., Grusenmeyer, T. A., Griffith, A. L., and Schmehl, R. H. (2018). Transition metal hydride complexes as mechanistic models for proton reduction catalysis. Coord. Chem. Rev. 362, 44–53. doi: 10.1016/j.ccr.2018.02.014
Annibale, V. T., and Song, D. (2014). Reaction of dinuclear rhodium 4,5-diazafluorenyl-9-carboxylate complexes with H2 and CO2. Organometallics 33, 2776–2783. doi: 10.1021/om500278a
Anouti, M., Caillon-Caravanier, M., Le Floch, C., and Lemordant, D. (2008). Alkylammonium-based protic ionic liquids part I: preparation and physicochemical characterization. J. Phys. Chem. B 112, 9406–9411. doi: 10.1021/jp803483f
Arias-Rotondo, D. M., and McCusker, J. K. (2016). The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 45, 5803–5820. doi: 10.1039/c6cs00526h
Campagna, S., Puntoriero, F., Nastasi, F., Bergamini, G., and Balzani, V. (2007). Photochemistry and photophysics of coordination compounds: ruthenium. Topp. Curr. Chem. 280, 117–214. doi: 10.1007/128_2007_133
Chatt, J., and Venanzi, L. M. (1957). 955. Olefin co-ordination compounds. Part VI. Diene complexes of rhodium(I). J. Chem. Soc. 1957, 4735–4741. doi: 10.1039/JR9570004735
Crutchley, R. J., and Lever, A. B. P. (1980). Ruthenium(II) tris(bipyrazyl) dication - a new photocatalyst. J. Am. Chem. Soc. 102, 7128–7129. doi: 10.1021/ja00543a053
Damrauer, N. H., Boussie, T. R., Devenney, M., and McCusker, J. K. (1997). Effects of intraligand electron delocalization, steric tuning, and excited-state vibronic coupling on the photophysics of aryl-substituted bipyridyl complexes of Ru(II). J. Am. Chem. Soc. 119, 8253–8268. doi: 10.1021/ja971321m
Darensbourg, D. J., Grotsch, G., Wiegreffe, P., and Rheingold, A. L. (1987). Insertion reactions of carbon dioxide with square-planar rhodium alkyl and aryl complexes. Inorg. Chem. 26, 3827–3830. doi: 10.1021/ic00269a043
Dewhirst, K. C., Keim, W., and Reilly, C. A. (1968). Preparation and nuclear magnetic resonance spectra of hydridophosphine complexes of ruthenium and rhodium. Inorg. Chem. 7, 546–551. doi: 10.1021/ic50061a033
Doherty, M. D., Grills, D. C., Muckerman, J. T., Polyansky, D. E., and Fujita, E. (2010). Toward more efficient photochemical CO2 reduction: Use of scCO2 or photogenerated hydrides. Coord. Chem. Rev. 254, 2472–2482. doi: 10.1016/j.ccr.2009.12.013
Dunning, T. H., and Hay, P. J. (1976). “Gaussian basis sets for molecular calculations in methods of electronic structure theory,” in Modern Theoretical Chemistry, Vol. 3, ed H. F. Schaefer III (Plenum Press), 1–28.
Esswein, A. J., and Nocera, D. G. (2007). Hydrogen production by molecular photocatalysis. Chem. Rev. 107, 4022–4047. doi: 10.1021/cr050193e
Fabry, D. C., and Rueping, M. (2016). Merging visible light photoredox catalysis with metal catalyzed C–H activations: On the role of oxygen and superoxide ions as oxidants. Acc. Chem. Res. 49, 1969–1979. doi: 10.1021/acs.accounts.6b00275
Falk, A., Göderz, A.-L., and Schmalz, H.-G. (2013). Enantioselective nickel-catalyzed hydrocyanation of vinylarenes using chiral phosphine-phosphite ligands and TMS-CN as a source of HCN. Angew. Chem. Int. Ed. 52, 1576–1580. doi: 10.1002/anie.201208082
Fan, X., Gong, X., Ma, M., Wang, R., and Walsh, P. J. (2018). Visible light-promoted CO2 fixation with imines to synthesize diaryl α-amino acids. Nat. Chem. 9:4936. doi: 10.1038/s41467-018-07351-2
Farney, E. P., and Yoon, T. P. (2014). Visible-light sensitization of vinyl azides by transition-metal photocatalysis. Angew. Chem. Int. Ed. 53, 793–797. doi: 10.1002/anie.201308820
Flamigni, L., Barbieri, A., Sabatini, C., Ventura, B., and Barigelletti, F. (2007). Photochemistry and photophysics of coordination compounds: iridium. Top. Curr. Chem. 281, 143–203. doi: 10.1007/128_2007_131
Fujihara, T., Xu, T., Semba, K., Terao, J., and Tsuji, Y. (2011). Copper-catalyzed hydrocarboxylation of alkynes using carbon dioxide and hydrosilanes. Angew. Chem. Int. Ed. 50, 523–527. doi: 10.1002/anie.201006292
Ghosh, T., Slanina, T., and Knig, B. (2015). Visible light photocatalytic reduction of aldehydes by Rh(III)–H: a detailed mechanistic study. Chem. Sci. 6, 2027–2034. doi: 10.1039/C4SC03709J
Glendening, E. D., Badenhoop, J. K., Reed, A. E., Carpenter, J. E., Bohmann, J. A., Morales, C. M., et al. (2001). NBO 5.0, Theoretical Chemistry Institute. Madison, WI: University of Wisconsin.
Grushin, V. V., Kuznetsov, V. F., Bensimon, C., and Alper, H. (1995). A simple and convenient preparation of [(Ph3P)4Rh2(μ-OH)2] and its reactions with C-H, O-H, and M-H acids. Organometallics 14, 3927–3932. doi: 10.1021/om00008a044
Hasegawa, E., Seida, T., Chiba, N., Takahashi, T., and Ikeda, H. (2005). Contrastive photoreduction pathways of benzophenones governed by regiospecific deprotonation of imidazoline radical cations and additive effects. J. Org. Chem. 70, 9632–9635. doi: 10.1021/jo0514220
Hasegawa, E., Takizawa, S., Seida, T., Yamaguchi, A., Yamaguchi, N., Chiba, N., et al. (2006). Photoinduced electron-transfer systems consisting of electron-donating pyrenes or anthracenes and benzimidazolines for reductive transformation of carbonyl compounds. Tetrahedron 62, 6581–6588. doi: 10.1016/j.tet.2006.03.061
Hayashi, C., Hayashi, T., and Yamada, T. (2015). Cobalt-catalyzed reductive carboxylation of α,β-unsaturated compounds with carbon dioxide. Bull. Chem. Soc. Jpn. 88, 862–870. doi: 10.1246/bcsj.20150043
Hou, J., Ee, A., Cao, H., Ong, H.-W., Xu, J.-H., and Wu, J. (2018a). Visible-light-mediated metal-free difunctionalization of alkenes with CO2 and silanes or C(sp3)-H alkanes. Angew. Chem., Int. Ed. 57, 17220–17224. doi: 10.1002/anie.201811266
Hou, J., Ee, A., Feng, W., Xu, J.-H., Zhao, Y., and Wu, J. (2018b). Visible-light-driven alkyne hydro-/carbocarboxylation using CO2 via iridium/cobalt dual catalysis for divergent heterocycle synthesis. J. Am. Chem. Soc. 140, 5257–5263. doi: 10.1021/jacs.8b01561
Ikezawa, H., Kutal, C., Yasufuku, K., and Yamazaki, H. (1986). Direct and sensitized valence photoisomerization of a substituted norbornadiene. Examination of the disparity between singlet- and triplet-state reactivities. J. Am. Chem. Soc. 108, 1589–1594. doi: 10.1021/ja00267a032
Islangulov, R. R., and Castellano, F. N. (2006). Photochemical upconversion: anthracene dimerization sensitized to visible light by a RuII Chromophore. Angew. Chem. Int. Ed. 45, 5957–5959. doi: 10.1002/anie.200601615
Ju, T., Fu, Q., Ye, J.-H., Zhang, Z., Liao, L.-L., Yan, S.-S., et al. (2018). Selective and catalytic hydrocarboxylation of enamides and imines with CO2 to generate α,α-disubstituted α-amino acids. Angew. Chem. Int. Ed. 57, 13897–13901. doi: 10.1002/anie.201806874
Kalyanasundaram, K. (1982). Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coord. Chem. Rev. 46, 159–244. doi: 10.1016/0010-8545(82)85003-0
Kawashima, S., Aikawa, K., and Mikami, K. (2016). Rhodium-catalyzed hydrocarboxylation of olefins with carbon dioxide. Eur. J. Org. Chem. 2016, 3166–3170. doi: 10.1002/ejoc.201600338
Li, S., Yuan, W., and Ma, S. (2011). Highly regio- and stereoselective three-component nickel-catalyzed syn-hydrocarboxylation of alkynes with diethyl zinc and carbon dioxide. Angew. Chem. Int. Ed. 50, 2578–2582. doi: 10.1002/anie.201007128
Lowry, M. S., Goldsmith, J. I., Slinker, J. D., Rohl, R., Pascal, R. A., Malliaras, G. G., et al. (2005). Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium(III) complex. Chem. Mater. 17, 5712–5719. doi: 10.1021/cm051312+
Lu, Z., and Yoon, T. P. (2012). Visible light photocatalysis of [2+2] styrene cycloadditions by energy transfer. Angew. Chem. Int. Ed. 51, 10329–10332. doi: 10.1002/anie.201204835
Luan, Y.-X., and Ye, M. (2018). Transition metal-mediated or catalyzed hydrocarboxylation of olefins with CO2. Tetrahedron Lett. 59, 853–861. doi: 10.1016/j.tetlet.2018.01.035
Meng, Q.-Y., Wang, S., Huff, G. S., and König, B. (2018). Ligand-controlled regioselective hydrocarboxylation of styrenes with CO2 by combining visible light and nickel catalysis. J. Am. Chem. Soc. 140, 3198–3201. doi: 10.1021/jacs.7b13448
Meng, Q.-Y., Wang, S., and König, B. (2017). Carboxylation of aromatic and aliphatic bromides and triflates with CO2 by dual visible-light–nickel catalysis. Angew. Chem. Int. Ed. 56, 13426–13430. doi: 10.1002/anie.201706724
Mizuno, H., Takaya, J., and Iwasawa, N. (2011). Rhodium(I)-catalyzed direct carboxylation of arenes with CO2 via chelation-assisted C-H bond activation. J. Am. Chem. Soc. 133, 1251–1253. doi: 10.1021/ja109097z
Morris, A. J., Meyer, G. J., and Fujita, E. (2009). Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc. Chem. Res. 42, 1983–1994. doi: 10.1021/ar9001679
Murata, K., Numasawa, N., Shimomaki, K., Takaya, J., and Iwasawa, N. (2017). Construction of a visible light-driven hydrocarboxylation cycle of alkenes by the combined use of Rh(I) and photoredox catalysts. Chem. Commun. 53, 3098–3101. doi: 10.1039/C7CC00678K
Osawa, M., Hoshino, M., and Wakatsuki, Y. (2001). A Light-harvesting tert-phosphane ligand bearing a ruthenium(II) polypyridyl complex as substituent. Angew. Chem. Int. Ed. 40, 3472–3474. doi: 10.1002/1521-3773(20010917)40:18<3472::AID-ANIE3472>3.0.CO;2-W
Schultz, D. M., Sawicki, J. M., and Yoon, T. P. (2015). An improved procedure for the preparation of Ru(bpz)3(PF6)2 via a high-yielding synthesis of 2,2'-bipyrazine. Beilstein J. Org. Chem. 11, 61–65. doi: 10.3762/bjoc.11.9
Seo, H., Katcher, M. H., and Jamison, T. F. (2017a). Photoredox activation of carbon dioxide for amino acid synthesis in continuous flow. Nat. Chem. 9, 453–456. doi: 10.1038/nchem.2690
Seo, H., Liu, A., and Jamison, T. F. (2017b). Direct β-selective hydrocarboxylation of styrenes with CO2 enabled by continuous flow photoredox catalysis. J. Am. Chem. Soc. 139, 13969–13972. doi: 10.1021/jacs.7b05942
Shimomaki, K., Murata, K., Martin, R., and Iwasawa, N. (2017). Visible-light-driven carboxylation of aryl halides by the combined use of palladium and photoredox catalysts. J. Am. Chem. Soc. 139, 9467–9470. doi: 10.1021/jacs.7b04838
Skubi, K. L., Blum, T. R., and Yoon, T. P. (2016). Dual catalysis strategies in photochemical synthesis. Chem. Rev. 116, 10035–10074. doi: 10.1021/acs.chemrev.6b00018
Slinker, J. D., Gorodetsky, A. A., Lowry, M. S., Wang, J., Parker, S., Rohl, R., et al. (2004). Efficient yellow electroluminescence from a single layer of a cyclometalated iridium complex. J. Am. Chem. Soc. 126, 2763–2767. doi: 10.1021/ja0345221
Stoll, T., Castillo, C. E., Kayanuma, M., Sandroni, M., Daniel, C., Odobel, F., et al. (2015). Photo-induced redox catalysis for proton reduction to hydrogen with homogeneous molecular systems using rhodium-based catalysts. Coord. Chem. Rev. 304, 20–37. doi: 10.1016/j.ccr.2015.02.002
Strauss, S. H., and Shriver, D. F. (1978). Lewis acid-influenced ethylene hydrogenation by rhodium(I) complexes. Inorg. Chem. 17, 3069–3074. doi: 10.1021/ic50189a020
Strieth-Kalthoff, F., James, M. J., Teders, M., Pitzer, L., and Glorius, F. (2018). Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev. 47, 7190–7202. doi: 10.1039/c8cs00054a
Suga, T., Mizuno, H., Takaya, J., and Iwasawa, N. (2014). Direct carboxylation of simple arenes with CO2 through a rhodium-catalyzed C–H bond activation. Chem. Commun. 50, 14360–14363. doi: 10.1039/C4CC06188H
Suomalainen, P., Reinius, H. K., Riihimäki, H., Laitinen, R. H., Jääskeläinen, S., Haukka, M., et al. (2001). Hydroformylation of 1-hexene and propene with in situ formed rhodium phosphine catalysts. J. Mol. Catal. A. Chem. 169, 67–78. doi: 10.1016/S1381-1169(01)00038-3
Takaya, J., and Iwasawa, N. (2008). Hydrocarboxylation of allenes with CO2 catalyzed by silyl pincer-type palladium complex. J. Am. Chem. Soc. 130, 15254–15255. doi: 10.1021/ja806677w
Takeda, H., and Ishitani, O. (2010). Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies. Coord. Chem. Rev. 254, 346–354. doi: 10.1016/j.ccr.2009.09.030
Tamaki, Y., Koike, K., and Ishitani, O. (2015). Highly efficient, selective, and durable photocatalytic system for CO2 reduction to formic acid. Chem. Sci. 6, 7213–7221. doi: 10.1039/C5SC02018B
Tamayo, A. B., Alleyne, B. D., Djurovich, P. I., Lamansky, S., Tsyba, I., Ho, N. N., et al. (2003). Synthesis and characterization of facial and meridional tris-cyclometalated iridium(III) complexes. J. Am. Chem. Soc. 125, 7377–7387. doi: 10.1021/ja034537z
Tellis, J. C., Primer, D. N., and Molander, G. A. (2014). Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 345, 433–436. doi: 10.1126/science.1253647
Tomasi, J., Mennucci, B., and Cammi, R. (2005). Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094. doi: 10.1021/cr9904009
Tortajada, A., Juliá-Hernández, F., Börjesson, M., Moragas, T., and Martin, R. (2018). Transition-metal-catalyzed carboxylation reactions with carbon dioxide. Angew. Chem. Int. Ed. 57, 15948–15982. doi: 10.1002/anie.201803186
Twilton, J., Le, C., Zhang, P., Shaw, M. H., Evans, R. W., and MacMillan, D. W. C. (2017). The merger of transition metal and photocatalysis. Nat. Rev. Chem. 1:0052. doi: 10.1038/s41570-017-0052
Ukai, K., Aoki, M., Takaya, J., and Iwasawa, N. (2006). Rhodium(I)-catalyzed carboxylation of aryl- and alkenylboronic esters with CO2. J. Am. Chem. Soc. 128, 8706–8707. doi: 10.1021/ja061232m
Van der Ent, A., Onderdelinden, A. L., and Schunn, R. A. (1990). Chlorobis(cyclooctene)rhodium(I) and –iridium(I) complexes. Inorg. Synth. 28, 90–92. doi: 10.1002/9780470132593.ch23
Wadt, W. R., and Hay, P. J. (1985a). Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 82, 284–298. doi: 10.1063/1.448800
Wadt, W. R., and Hay, P. J. (1985b). Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 299–310. doi: 10.1063/1.448975
Wang, X., Nakajima, M., and Martin, R. (2015). Ni-catalyzed regioselective hydrocarboxylation of alkynes with CO2 by using simple alcohols as proton sources. J. Am. Chem. Soc. 137, 8924–8927. doi: 10.1021/jacs.5b05513
Werner, H., Schäfer, M., Nürnberg, O., and Wolf, J. (1994). η3-Allyl and η3-benzyl rhodium complexes: synthesis, structure dynamic, and reactions with carboxylic acids. Chem. Ber. 127, 27–38. doi: 10.1002/cber.19941270106
Williams, C. M., Johnson, J. B., and Rovis, T. (2008). Nickel-catalyzed reductive carboxylation of styrenes using CO2. J. Am. Chem. Soc. 130, 14936–14937. doi: 10.1021/ja8062925
Yan, S.-S., Fu, Q., Liao, L.-L., Sun, G.-Q., Ye, J.-H., Gong, L., et al. (2018). Transition metal-catalyzed carboxylation of unsaturated substrates with CO2. Coord. Chem. Rev. 374, 439–463. doi: 10.1016/j.ccr.2018.07.011
Yatham, V. R., Shen, Y., and Martin, R. (2017). Catalytic intermolecular dicarbofunctionalization of styrenes with CO2 and radical precursors. Angew. Chem. Int. Ed. 56, 10915–10919. doi: 10.1002/anie.201706263
Ye, J.-H., Miao, M., Huang, H., Yan, S.-S., Yin, Z.-B., Zhou, W.-J., et al. (2017). Visible-light-driven iron-promoted thiocarboxylation of styrenes and acrylates with CO2. Angew. Chem. Int. Ed. 56, 15416–15420. doi: 10.1002/anie.201707862
Yeung, C. S. (2019). Photoredox catalysis as a strategy for CO2 incorporation: direct access to carboxylic acids from a renewable feedstock. Angew. Chem. Int. Ed. 58, 5492–5502. doi: 10.1002/anie.201806285
Keywords: carboxylation, CO2 fixation, photoredox catalyst, rhodium catalyst, visible light
Citation: Murata K, Numasawa N, Shimomaki K, Takaya J and Iwasawa N (2019) Improved Conditions for the Visible-Light Driven Hydrocarboxylation by Rh(I) and Photoredox Dual Catalysts Based on the Mechanistic Analyses. Front. Chem. 7:371. doi: 10.3389/fchem.2019.00371
Received: 07 March 2019; Accepted: 06 May 2019;
Published: 22 May 2019.
Edited by:
Soumyajit Roy, Indian Institute of Science Education and Research Kolkata, IndiaReviewed by:
Munetaka Akita, Tokyo Institute of Technology, JapanYang Yang, University of Central Florida, United States
Copyright © 2019 Murata, Numasawa, Shimomaki, Takaya and Iwasawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nobuharu Iwasawa, bml3YXNhd2FAY2hlbS50aXRlY2guYWMuanA=
†Present Address: Kei Murata, Institute of Industrial Science, The University of Tokyo, Tokyo, Japan