 Andrea Lombardi
Andrea Lombardi Fernando Pirani
Fernando Pirani Massimiliano Bartolomei3
Massimiliano Bartolomei3 Cecilia Coletti
Cecilia Coletti Antonio Laganà
Antonio Laganà- 1Dipartimento di Chimica, Biologia e Biotecnologie, Università di Perugia, Perugia, Italy
- 2Consortium for Computational Molecular and Materials Sciences (CMS)2, Perugia, Italy
- 3Instituto de Física Fundamental, Consejo Superior de Investigaciones Científicas, Madrid, Spain
- 4Dipartimento di Farmacia, Università “G. d'Annunzio” Chieti-Pescara, Chieti, Italy
- 5CNR ISTM-UOS Perugia, Perugia, Italy
- 6Master-UP srl, Perugia, Italy
A full dimensional Potential Energy Surface (PES) of the CO + N2 system has been generated by extending an approach already reported in the literature and applied to N2-N2 (Cappelletti et al., 2008), CO2-CO2 (Bartolomei et al., 2012), and CO2-N2 (Lombardi et al., 2016b) systems. The generation procedure leverages at the same time experimental measurements and high-level ab initio electronic structure calculations. The procedure adopts an analytic formulation of the PES accounting for the dependence of the electrostatic and non-electrostatic components of the intermolecular interaction on the deformation of the monomers. In particular, the CO and N2 molecular multipole moments and electronic polarizabilities, the basic physical properties controlling the behavior at intermediate and long-range distances of the interaction components, were made to depend on relevant internal coordinates. The formulated PES exhibits substantial advantages when used for structural and dynamical calculations. This makes it also well suited for reuse in Open Molecular Science Cloud services.
1. Introduction
The presence of significant traces of CO in gaseous systems in which molecular nitrogen N2 is an abundant component is a frequent situation in astrochemistry, plasma chemistry and combustion. Besides obvious implications for combustion and CO2 plasmas, CO plays an important role in planetary atmospheric chemistry (He et al., 2017), and it has been detected in Titan, Triton, and Pluto as a stable minor constituent (Lellouch et al., 2010), contributing to the energy balance of the planet and the chemistry of small organic molecule formation. For example, recent studies consider the contribution of CO to the formation and role of atmospheric hazes present in a number of solar system and exoplanetary atmospheres (He et al., 2017) and, particularly, hydrocarbon hazes, which have substantial radiative heating and cooling effects in such atmospheres, as observed for Saturn's moon Titan (Tomasko et al., 2008; He et al., 2017; Hörst et al., 2018) and for Jupiter's stratosphere (Zhang et al., 2015) (not to mention the role of CO in the Earth's atmosphere chemistry). Organic hazes are particularly interesting due to astrobiological implications, such as the potential for Titan's atmosphere to contain the building blocks of life (Hörst, 2012; Fabiano et al., 2017). It was recently shown in a series of atmosphere simulation experiments using gas mixtures of CO, CH4, and N2 (Hörst et al., 2018) that the inclusion of CO has a dramatic effect on the gas phase chemistry as well as on the density and composition of the solid material that is formed. In this respect, CO can actually be seen as the smallest molecule that could serve as a source of oxygen and carbon in space and planetary atmospheres.
The knowledge of the intermolecular interactions involving carbon monoxide and nitrogen (as well as methane) is generally required in the above mentioned fields to correctly assess their role in the chemical kinetics of gaseous environments. A fundamental issue is the accurate representation of the set of energy transfer processes involving CO in the various kinetic models used to study the gaseous mixtures.
Collisions between molecules promote transfer of energy amongst translational, rotational and vibrational degrees of freedom, determining the molecular state population. Dynamics simulations based upon quantum, classical and semiclassical scattering calculations permit one to obtain cross sections and thermal or state-to-state rate coefficients (Lombardi et al., 2013, 2015, 2016a; Celiberto et al., 2016). The resulting data, refined and complemented by experimental data (whenever available) can be used to set up relevant parts of kinetic models (Kustova and Kremer, 2014), routinely used in many research fields to simulate complex environments such as combustion mixtures, plasmas, atmospheres and molecular clouds in in the interstellar medium (Bacmann et al., 2002). The energy transfer processes in each collision event, being generally strongly state-specific, are sensitive to the initial quantum states of the involved molecules. Therefore, energy exchange kinetics is better accounted for by appropriate sets of state-to-state rate coefficients, rather than averaged. Indeed, state-to-state coefficients are entirely necessary when non-equilibrium conditions prevail. In those cases, the collection of reliable energy transfer cross sections and rate coefficients is the true accuracy-determining step of the modeling (Kustova and Nagnibeda, 2012; Kustova and Kremer, 2014).
In turn, the accuracy of whatever rate coefficient obtained by calculations depends on the realism of the underlying dynamics simulations, which essentially means a very accurate description of the intermolecular interactions, occurring at long- and mid-range distances, with the energy exchanges being to a large extent due to such forces. Finally, the need for accuracy has to be contrasted with the computational demand of campaigns of massive state-to-state coefficient calculations. In this respect, flexible and easy to reuse potential energy surface formulations play an important role in the cooperative assemblage of simulations of chemical processes based on atomistic approaches. However, most of the structural and dynamics data available in popular data banks, of interest in several fields, including astrochemistry, combustion and plasmas, lack solid validation and are sometimes even totally arbitrary. A typical weakness of the mentioned data consists of the fact that even those originating from high-level ab initio calculations might be lacking sufficient accuracy in the long-range region. For this purpose, we currently check the suitability of the proposed PESs for carrying out calculations of the detailed probabilities, cross sections and rate coefficients for inelastic processes of the title systems.
Our work, however, due to the general high demand of computational resources that kinetics and dynamics simulations generate, has also the broader prospective aim of building MOlecular Simulator Enabled Cloud Services (MOSEX) (Vitillaro and Laganà, 2018) meant to provide the Molecular Science Community with a Cloud service (Laganà et al., 2018b) by offering the provision of accurate estimates of molecular properties generated in (often collaborative) experiments and validated by high-level theory (also often collaborative) simulations.
Accordingly, the purpose of the present work is threefold. First is to illustrate the development of an accurate full dimensional PES describing the intermolecular interactions of CO-N2 systems. This is done by extending a well established semiempirical approach, based on a bond-bond description of the interactions (see e.g., Cappelletti et al., 2008; Faginas Lago et al., 2013 and references therein).
Second is to discuss a dynamical preliminary validation of the proposed PES through application to the case of the basic Vibration-Vibration (VV) energy exchange processes of CO in nitrogen-containing mixtures: CO(1)+N2(0) → CO(0)+N2(1) and N2(1)+CO2(0) → N2(0)+CO2(1).
Third is to report on the progress made in providing calculated scattering properties as a service to the community using the so-called Grid Empowered Molecular Simulator (GEMS) (Laganà et al., 2010; Manuali et al., 2010; Rampino et al., 2012a) through the activities of the Virtual Organization (VO) COMPCHEM (Laganà et al., 2010) first and of the Chemistry Molecular and Materials Science and Technologies (CMMST) Virtual Research Community (VRC) (Laganà, 2012) later.
The paper is therefore organized as follows. Section 2 is devoted to the description, formulation and optimization of the proposed CO+N2 PES. Section 3 is devoted to illustrating the quantum-classical calculations and related results. Section 4 is devoted to illustrating the implemented Open Molecular Science Cloud prototype.
2. The Potential Energy Surface
In this section we discuss the formulation of the PES of the CO-N2 system used for the calculations and briefly illustrate as well its optimization and extension to flexible monomers. The approach we use is based on the so called bond-bond method (Cappelletti et al., 2008), a valuable feature of which is the choice of expressing the potential function parameters in terms of bond properties and parameters characterizing the internal molecular structure, such as charge distributions and polarizabilities. Since the energy functions depend on parameters having a well-defined physical meaning, these are portable in different molecular environments as building blocks of force fields of, in principle, whatever complexity, as testified by the application of the method to a variety of systems (see e.g., Albertí et al., 2011; Albertí and Faginas Lago, 2012; Bartolomei et al., 2012; Lombardi et al., 2012, 2016b; Falcinelli et al., 2013; Pacifici et al., 2013; Faginas-Lago et al., 2014a,b, 2015; Yeamin et al., 2014). It is worth mentioning that this aspect of the method is a pivotal element for our strategy of developing in order to provide rich and diverse data sets of molecular properties as a cloud service.
2.1. The Representation of the PES
The intermolecular potential energy, Vinter, of the two interacting molecules, CO and N2, is formulated as a combination of two effective interaction components:
where VvdW and Velect represent the van der Waals (size repulsion plus dispersion-attraction) and the electrostatic interaction components, respectively. Velect originates from the anisotropic molecular charge distributions of the two bodies, which asymptotically tend to the sum of the (permanent) quadrupole-(permanent) quadrupole and dipole-quadrupole interactions. Both VvdW and Velect depend on the distance R between the centers of mass of the two molecules (say a for CO and b for N2), and on the Jacobi angular coordinates Θa, Θb and Φ describing the a − b mutual orientation as well (see Figure 1). In the present work we will consider a series of limiting configurations of the interacting molecules, specifically (Θa,Θb,Φ)=(90°, 90°, 0°), (90°, 90°, 90°), (90°, 0°, 0°), (0°, 90°, 0°), (180°, 90°, 0°), (0°, 0°, 0°) and (180°, 0°, 0°), which will be referred to as H, X, Ta, Tb1, Tb2, I1 and I2.
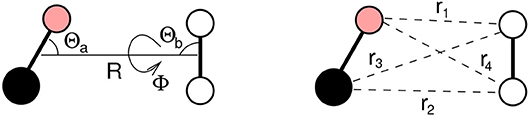
Figure 1. CO-N2 dimer with the R, Θa, Θb, Φ coordinates, defining the distance between the molecules and the mutual orientation, and the ri coordinates defining the distance for all atom-atom interacting pairs.
The van der Waals term, VvdW of Equation (1), is expressed as a sum of the noncovalent contributions as follows,
where ri is the distance between different interaction centers of the involved molecules, which here are chosen to coincide with the constituting atoms (see Figure 1). Accordingly, the sum of Equation (2) runs over all four atom pairs of the CO-N2 complex (see Figure 1). It must be emphasized here that in the present work the bond-bond formulation is replaced by the pseudoatom-pseudoatom one in order to properly account for the variation of the anisotropy with the atom-atom distance and the polarizability consistent with the diatom (Pirani et al., 2019).
The explicit form of is obtained using the Improved Lennard-Jones (ILJ) potential function (Pirani et al., 2008):
where xi is the reduced distance defined as
and ε and Rm are, respectively, the well depth and position of the corresponding pair interaction. Note that the ILJ function (Pirani et al., 2008) is more realistic than the original Lennard-Jones (12,6) one, with a much more accurate size repulsion (first term within the square brackets) and the long range dispersion attraction tail (second term within the square brackets) (Pirani et al., 2004; Lombardi and Palazzetti, 2008).
The exponent n in Equation (3) is expressed as a function of xi using the following empirical equation (Pirani et al., 2004):
in which β is a parameter depending on the nature and the hardness of the interacting particles. For the present system, β has been set equal to 8 (a value typical of van der Waals interactions in neutral-neutral systems) for all atom-atom pairs.
The relevant ε and Rm parameters are directly related to the molecular polarizability of the involved partners, and a zeroth-order estimate of them can be obtained from correlation formulas (Pirani et al., 2001, 2019; Cappelletti et al., 2008) which exploit “effective” atomic polarizability.
In this way a tentative full dimensional PES is generated and related ε and Rm parameters can be fine-tuned by fitting experimental data and by comparing model predictions with accurate ab initio electronic structure calculations (see below).
The Velect term of Equation (1) has been formulated as a sum of Coulomb potentials as follows:
with qja and qkb being point charges (located on the monomers a and b, respectively, and having values consistent with the corresponding calculated molecular dipole and quadrupoles) and rjk being the distance between them.
Such a formulation of Velect must be used for cases in which the molecular dimensions are not negligible with respect to the intermolecular distance R (Maitland et al., 1987). For both monomers a linear distribution of charges as that used in a previous work, see Lombardi et al. (2016b), has been adopted, which consists of three charges placed on the atoms and on the molecule center of mass.
For charge values qi (as well as for the corresponding position ri), we took for N2 the values reported in Lombardi et al. (2016b) while for CO we obtained them by exploiting corresponding dipole and quadrupole moment calculations (see below) together with simple geometrical considerations (see Appendix A). A comparison of the van der Waals and electrostatic interaction contributions to the intermolecular potential energy, for the various configurations of the two interacting molecules is reported in Figure S1 of the Supplementary Material.
2.2. The Optimization of the PES
The above-mentioned adopted values of ε and Rm (hereinafter called “predicted”) were fine-tuned by carrying out a comparison with ab-initio estimates of the intermolecular part of the interaction and an analysis of the second virial coefficient data (see next section). Predicted and optimized values for the case of rigid monomers are given in Table 1 together with the other parameters used to formulate the intermolecular potential.
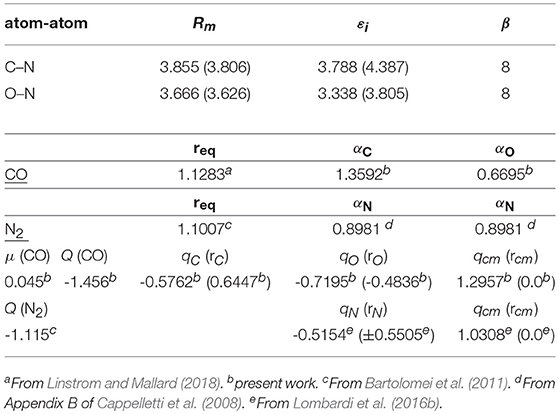
Table 1. Optimized Rm (Å) and ε (meV) parameters and (within parentheses) predicted values estimated for rigid momomers at the equilibrium bond length req (Å) and by considering atomic “effective” polarizabilities αx values (Å3).
In Figure 2 the main features of the intermolecular potential Vinter calculated for seven selected geometries of the interacting system at the CCSD(T) level of theory (see next section) are reported. Although such results will be discussed in more detail later, it is worth pointing out here that, as shown by the figure, the stability ranking of the investigated geometries on the empirical PES model globally agrees well with that of the ab initio values.
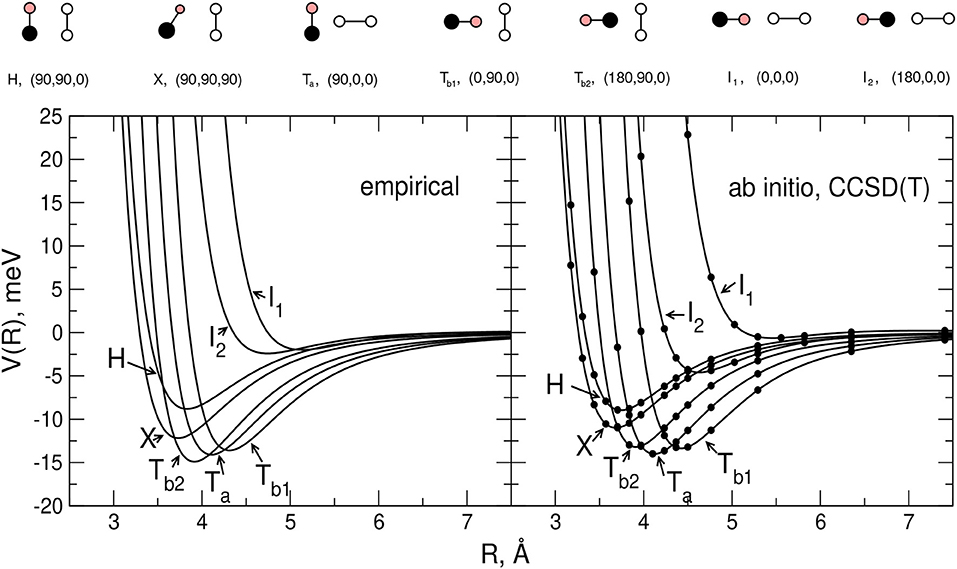
Figure 2. Comparison between the empirical and ab-initio PES for the selected H,X,Ta,Tb1,Tb2,I1, and I2 configurations of the CO-N2 system (rigid monomers). For each reported configuration the C, O, and N atoms are depicted as pink, black and white circles, respectively, and (Θa,Θb,Φ) represent the Jacobi angular coordinates in degrees. Left hand side panel: present model empirical PES. Right hand side panel: present ab initio calculations (see section 2.2).
In order to carry out the fine tuning of the parameters of the semiempirical functional representation of the PES, we followed the procedure already employed in Bartolomei et al. (2012); Lombardi et al. (2016b), and we first investigated the dimer formation for two rigid monomers (with bond lengths and angles fixed at their equilibrium values). In this perspective, we performed CCSD(T) intermolecular potential calculations for the H, X, Ta, Tb1, Tb2, I1, and I2 configurations of the dimer (see section 2.1), as a function of the distance R of the two monomers. Results are reported in Figure 2 where they are compared with the corresponding empirical PES energy profiles. The supermolecular CCSD(T) energies were calculated using the MOLPRO package (Werner et al., 2006) and corrected using the counterpoise method (Boys and Bernardi, 1970; van Lenthe et al., 1987) in order to remove the basis set superposition error. For all supermolecular calculations, the Dunning's aug-cc-pVTZ basis set (Kendall et al., 1992) was used together with the bond function set [3s3p2d1f] developed by Tao Tao and Pan (1992) and placed on the midpoint of the intermolecular distance R that was varied in the range 2.5–8 Å. In order to assess the convergence of the supermolecular energies with the basis set, additional calculations using the aug-cc-pVQZ plus bond functions (see above) basis set were also performed for the usual dimer configurations around their minima. The corresponding results, compared in Table 2 with those obtained using the less extended basis set, show deviations of about 0.1–0.2 meV. The C-O and N2 bond lengths of the linear monomers were set equal to 1.1283 Å (Linstrom and Mallard, 2018) and 1.1007 Å (Bartolomei et al., 2011), respectively.
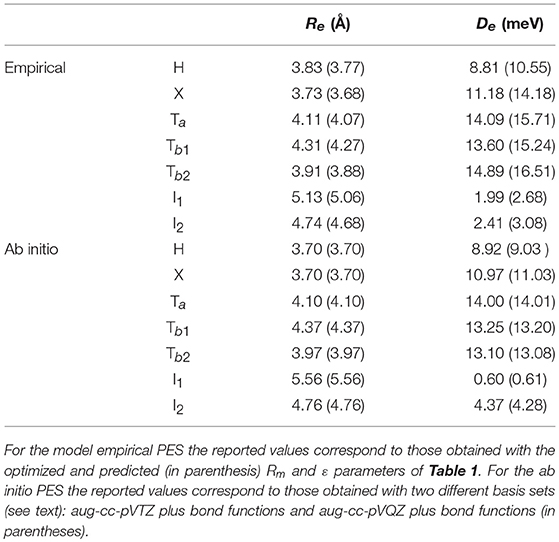
Table 2. Equilibrium distance (Re) and binding energy (De) for the present rigid monomers empirical and ab initio PESs considering selected geometries of the CO-N2 dimer (see Figure 2).
In order to carry out a comparison with the flexible monomer empirical potential (to be described below), further CCSD(T) calculations were carried out by considering a maximum variation of 10% in the bond length of one of the two monomers. Such small deformations did not alter the single determinant character (Lee and Taylor, 1989) of the used wavefunctions and allowed the use of the CCSD(T) method also for the deformed arrangements. The ab initio interaction profiles related to flexible monomers, reported in the following, have been compared with the results of the model potential in Figures 6, 7, to prove the validity of the model predictions.
In order to introduce into the empirical model potential the dependence of the electrostatic component Velect (see Equation 1) on monomer deformations, the variation of the CO and N2 molecular multipole moments with the internal coordinates (and consequently, also that of the related charge distributions) was introduced. As for N2, the molecular multipoles and the related point charge dependence as those adopted in Lombardi et al. (2016b) were used. In the case of CO, as previously done for CO2 (Bartolomei et al., 2012) and N2 (Lombardi et al., 2016b), multireference ACPF (Averaged Coupled Pair Functional) calculations were performed as a function of the stretching for the molecular dipole and quadrupole moments by following the guidelines reported in Bartolomei et al. (2011). In particular, the molecular orbitals in the ACPF calculations were all taken to be these natural ones from the complete active space self-consistent field (CASSCF) reference wave functions. Therefore, the considered active space (CAS) was assumed to distribute 10 electrons in the orbitals indicated as (2, 3, 4)σg(2, 3, 4)σu(1, 2)πu(1, 2)πg. The 1σg1σu core molecular orbitals were fully optimized, while being constrained to be doubly occupied and excluded from the used CAS. The Dunning's aug-cc-pV5Z basis set (Kendall et al., 1992) was employed and the calculations were performed using the MOLPRO package (Werner et al., 2006).
The parameters of the rigid monomers defining the empirical PES were optimized in order to best fit at the same time the measured second virial coefficients and the ab initio interaction energies. It is worth pointing out here that, given the physical ground of the procedure, both the number of parameters allowed to vary in the fit and their interval of variation are rather small. As an example, the long-range dispersion attraction coefficient values, defined as were allowed to vary within a 10% interval of their initial value (see also Appendix A of Cappelletti et al., 2008). Moreover, it is also worth pointing out here that, due to some inter-dependencies, the final best fit values of the adjustable parameters usually differ only by a few percent from the initial ones (see Table 1 for ε and Rm). The main features (equilibrium distance Re and binding energy De for selected dimer geometries) of the optimized and predicted empirical PES's are reported in Table 2, and it can be seen that the small variations of the optimized ϵ and Rm (see Table 1) lead in general to a less attractive potential. Table 2 shows also that while optimized and predicted parameter values are both able to satisfactorily reproduce the relative stability of the limiting configurations (see Table 2), the former are closer to ab initio results, especially for the most attractive dimer geometries. However, it can also be noticed that the model PES is not capable of predicting the Ta configuration as the most stable of those here considered; nevertheless, it has to be stressed that the ab initio energy difference between the Ta, Tb1, and Tb2 configurations is quite low and less than 1 meV (see Table 2) and that the very few parameters of the model PES do not allow the description of this very fine behavior.
As anticipated above, to validate and optimize the rigid rotor PES, second virial coefficient (B(T)) values, including first quantum correction Bq1(T) to the classical estimate Bcl(T) (Pack, 1978) (B(T) = Bcl(T)+Bq1(T)), were also computed. The calculations evidence that quantum corrections are smaller than 1% even for temperature values as low as 200 K. A comparison of calculated B(T) values with experimental measurements (Jaeschke et al., 1988; McElroy and Buchanan, 1995) over the temperature range 273 < T < 350 K is given in Figure 3.
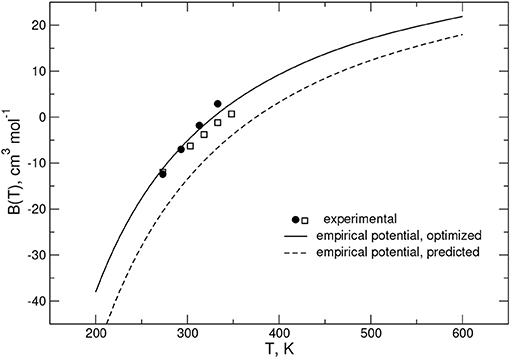
Figure 3. Second virial coefficients for the CO-N2 system: solid circles and open squares refer to experimental data from Jaeschke et al. (1988) and McElroy and Buchanan (1995), respectively; calculations obtained from the empirical PES are shown as solid and dashed black lines and correspond to the optimized and predicted potentials, respectively (see Table 2).
The figure shows that the predicted PES results underestimate the experimental data in the considered range of temperature, while those referring to the optimized PES lie between the two sets of the measured values. This behavior suggests that, in agreement with the above reported analysis for the ab initio energy values, the predicted PES provides an interaction that is too attractive, which indeed can be properly corrected by tuning the involved ε and Rm parameters.
A further check of the reliability of the empirical PES is reported in Figure 4, where the related spherical averaged potential is compared with an accurate ab initio estimate at the MP4 level and obtained from Karimi-Jafari et al. (2011). Again, it has to be pointed out that although the predicted PES shows a 15% deeper well, it becomes practically negligible after optimization.
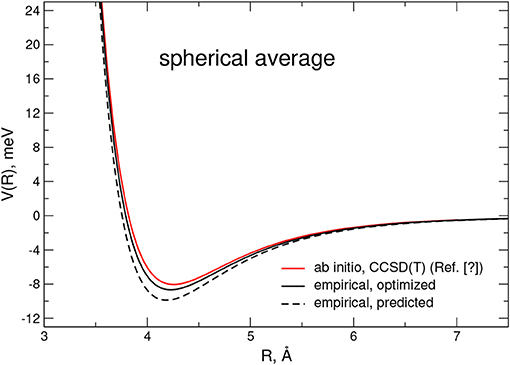
Figure 4. Spherical averaged interaction potential for the CO-N2 dimer as obtained from ab initio computations (Surin et al., 2018) at the CCSD(T) level of theory and from the present empirical PES. Estimations from the the optimized and predicted potentials are depicted as black solid and dashed lines, respectively.
2.3. Extension of the PES to Consider Flexible Monomers
The parametrization of the empirical PES has been generalized by properly introducing the dependence of VvdW and Velec (see Equation 1) on the molecular elongation. Specifically, the variation of the electric multipole affecting Velec has been obtained as indicated above, while the change of the molecular polarizability has been taken into account for the modulation of the VvdW potential parameters.
For the N2 monomer deformation, the empirical bond length dependence of the molecular polarizability α as reported in Appendix B of Cappelletti et al. (2008) is used, while for the corresponding point charges distribution dependence the analytic formulas given in Appendix A of Lombardi et al. (2016b) are employed.
In the case of the CO monomer we used the empirical bond length dependence of the polarizablity α as reported in Appendix A, which is shown here in the lower panel of Figure 5 together with previous ab initio estimates. In addition, for the CO permanent dipole quadrupole dependence on the bond length, an appropriate representation can be obtained from the analysis of present ab initio results reported in the upper panels of Figure 5. To this end, ab initio data were fitted using suitable analytic functions providing the radial dependence of point charges on the CO bond length r, as shown in detail in Appendix A.
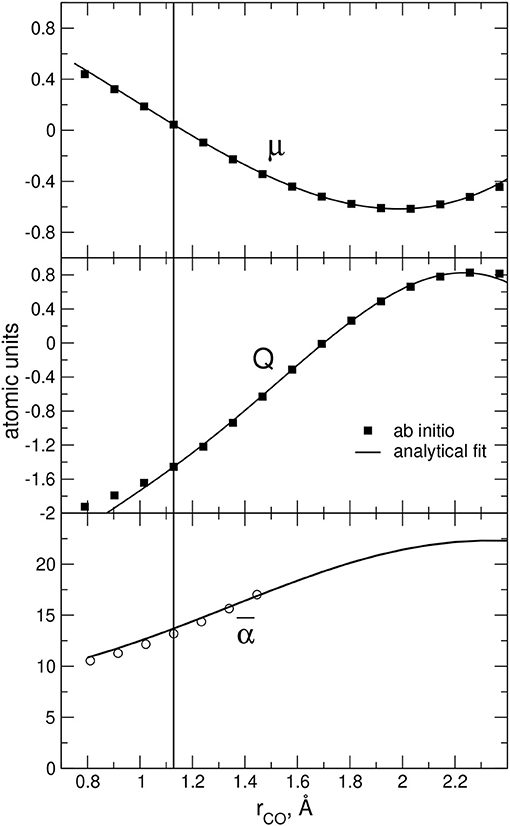
Figure 5. CO electric dipole (μ) and quadrupole (Q) moments as well as molecular polarizability () plotted as a function of the interatomic distance r. Full squares refer to present ab initio values obtained as detailed in the text, while solid lines correspond to analytic fits (see text and Appendix A). Open circles in the lowest panel refer to previous ab initio estimations at the CCSD(T) level from Maroulis (1996). The vertical line indicates the CO equilibrium distance.
The effect of the stretching of a single monomer on the complex interaction predicted by the adopted potential formulation is illustrated in Figures 6, 7. In all cases the plots of the CCSD(T) values, obtained for rigid and flexible molecules, are also reported for comparison: it can be appreciated that the elongation of one monomer provokes the same trends in both PESs for the stability of the considered interaction profiles, confirming therefore the reliability of the adopted empirical model.
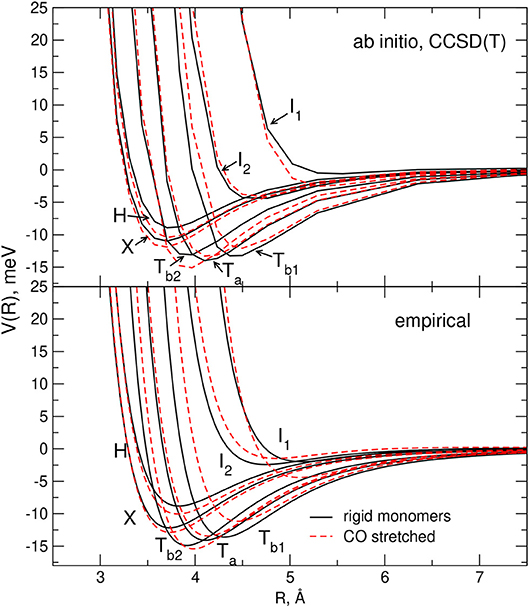
Figure 6. Comparison of the interaction energies for selected dimer geometries of a pair of rigid monomers (black lines) and of a rigid N2 plus a stretched CO (the bond length has been elongated of 10% with respect to equilibrium) monomer (red dashed lines). Empirical PES results are plotted in the lower panel while those corresponding to the ab initio calculations are plotted in the upper panel.
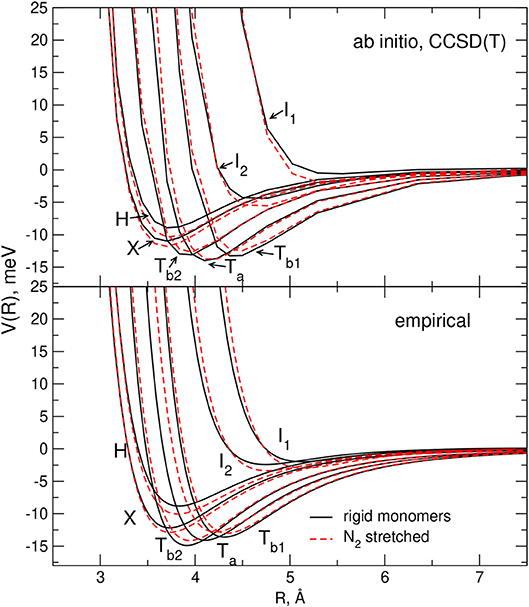
Figure 7. Comparison of the interaction energies for selected dimer geometries of a pair of rigid monomers (black lines) and of a rigid CO plus a stretched N2 (the bond length has been elongated of 10% with respect to equilibrium) monomer (red dashed lines). Empirical PES results are plotted in the lower panel while those corresponding to the ab initio calculations are plotted in the upper panel.
3. Quantum Classical Calculations for Vibrational Energy Transfer Processes
As a premise to this section, we remark that, when collisions involve more than three atoms, it is impractical to ground realistic simulations and the related systematic computations on full dimensional quantum methods, solving the associated Schrödinger equations, since their efficiency is strongly dependent by the number of degrees of freedom. Although work is constantly being done to achieve the feasibility of exact quantum calculations (e.g., through new coordinates and basis sets Aquilanti et al., 2000, 2002, 2004a,b, 2006; Sevyuk et al., 2005; Castro Palacio et al., 2007; Barreto et al., 2011, 2012; Palazzetti et al., 2011), these remain mainly limited to three-atom systems. For energy transfer, reactive and photodissociating systems involving triatomic and larger molecules, or for the simulation of more complex environments such as gaseous mixtures and flows, classical trajectories are widely employed to interpret experimental results, saving computing time (see e.g., Lombardi et al., 2010; Aquilanti et al., 2011; Palazzetti et al., 2013; Nakamura et al., 2015).
Semiclassical methods are also available (Laganà et al., 2003; Faginas-Lago and Laganá, 2005; Lago et al., 2005; Faginas-Lago et al., 2006, 2010; Faginas et al., 2008; Rampino et al., 2012b), but these won't be considered here.
On the other hand, an efficient and accurate method for calculating cross sections and rate coefficients for the exchange of vibrational quanta of energy upon inelastic collisions for the process:
is the Quantum Classical (QC) one introduced and developed by G.D. Billing (see Billing, 1984a, 1987) by combining quantum mechanics treatments (for selected bound degrees of freedom) with classical mechanics ones (for the remainder).
The QC method is still one of the most efficient tools to calculate large numbers of accurate rate coefficients for processes involving vibrational energy transfer. The detailed description of the method goes beyond the aim of the present work. The formulation used here is essentially the one described in detail in Coletti and Billing (1999), Coletti and Billing (2000), Coletti and Billing (2002), Billing et al. (2003), and Fioccola et al. (2017), to which the reader is referred for the relevant mathematical derivation. In this paper we give only the formulation of the basic properties.
3.1. The Main Features of the Adopted Quantum Classical Method
According to the QC approach, the quantum mechanical time-dependent Schrödinger equation for the nuclear motion is solved for the degrees of freedom of the system playing the most relevant role in vibration-to-vibration (VV) quantum energy exchange, vibrations and roto-vibrational couplings, by a coupled equations method to obtain the quantum transition amplitudes , where v and v′ are the initial vibrational quantum numbers of the diatoms. The vibrational wavefunction is initialized as the product of the Morse functions for the two infinitely separated diatoms, whose parameters are given in Table 3, and is expanded as:
where Ev and are the vibrational energies of the oscillators in their initial states. The remaining degrees of freedom are treated classically by integrating the corresponding set of Hamilton equations of motion in an effective potential defined as the Ehrenfest average of the interaction potential.
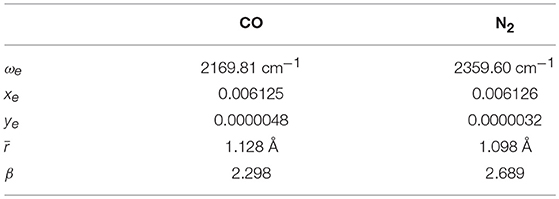
Table 3. Morse and vibrational parameters for CO and N2.
The quantum transition amplitudes can then be used to calculate either specific state-to-state vibrational/rotational transitions cross sections, , or Monte Carlo averaged cross sections over the Boltzmann distribution of the initial rotational angular momenta j and j′ for CO and N2, respectively:
where T0 is an arbitrary reference temperature, which cancels out in the formulation of rate constants (Equation 9), ICO and IN2 are the moments of inertia of the diatoms, U is the classical energy, obtained by subtracting from total energy the vibrational energy of the two diatoms, , jmax and are the upper limit for the randomly chosen rotational quantum numbers for the diatoms, lmax the upper limit for the angular momentum and μ is the reduced mass of the system.
From such averaged cross sections, rate coefficients for vibrational relaxation (see e.g., Coletti and Billing, 2002; Billing et al., 2003) can be derived as follows:
where ϵmin = 0 for exothermic and ϵmin = ΔE for endothermic processes and is the symmetrized classical energy (Billing, 1984b):
which has been introduced to restore, in an approximate fashion, the quantum mechanical detailed balance principle (Billing, 1984a,b, 1987).
In the present treatment the anharmonic vibrational energy is formulated as:
where ωei is the wavenumber for the i-th oscillator and xei and yei are the anharmonicity constants (see Table 3 for the values employed in the calculation for N2 and CO).
3.2. Results and Discussion
QC rate coefficients have been computed for the exothermic exchange of a single vibrational quantum of energy between the first excited vibrational level of N2 (N2(1)) and the ground vibrational level vi = 0 of CO (CO(0))
and for the endothermic exchange of a single vibrational quantum of energy between the first excited vibrational level vi = 1 of CO (CO(1)) and the ground vibrational level of N2 (N2(0))
and in the temperature range 80–3,000 K, by running trajectories at 15 initial values of total classical energy comprised between 50 cm−1 and 15,000 cm−1, with a more frequent sampling driven toward lower energies. For each energy value, 2,000 trajectories were considered, which should ensure an accuracy for rate constants of ~ 20% at lower temperatures and ~ 15% at higher ones.
For each trajectory, the impact parameter was randomly chosen between 0 and 10 Å, and the initial separation between the colliding partners was set equal to 15 Å. In the expansion (7) a total of 36 vibrational states was included, i.e., those consisting of a band of energy ΔE up to 14,000 cm−1.
Figures 8, 9 show the dependence of the computed QC rate coefficients on the temperature for the exothermic CO(0)+N2(1) → CO(1)+N2(0) and the endothermic CO(1)+N2(0) → CO(0)+N2(1) processes, respectively.
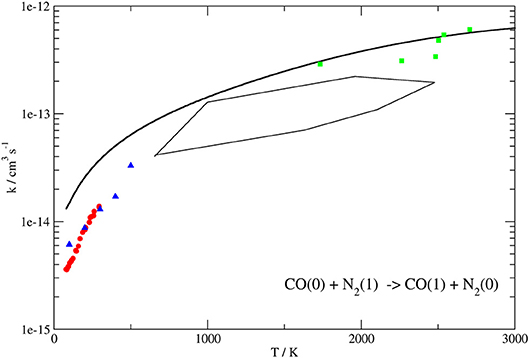
Figure 8. Rate coefficients (logarithmic scale) plotted as a function of temperature for the CO(0)+N2(1) → CO(1)+N2(0) transition. Present work results (solid line) compared to the experimental ones up to 300 K of Allen and Simpson (1980) (red circles), those obtained by laser-induced fluorescence (blue triangles) (Mastrocinque et al., 1976) and the high temperature ones of Sato et al. (1969) (green squares). The close area represents rate coefficients obtained in von Rosenberg et al. (1972) by shock wave in the range 1,000–2,000 K.
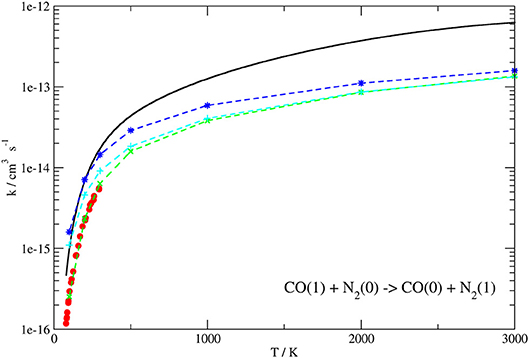
Figure 9. Rate coefficients (logarithmic scale) plotted as a function of temperature for the CO(1)+N2(0) → CO(0)+N2(1) transition. Present work results (solid line) compared to the experimental ones up to 300 K of Allen and Simpson (1980) (red circles) and those calculated in Kurnosov et al. (2003) using different potentials (dashed lines with points).
For comparison, the same Figures show the experimental data (Sato et al., 1969; von Rosenberg et al., 1972; Mastrocinque et al., 1976; Allen and Simpson, 1980), when available, together with previous theoretical results (Kurnosov et al., 2003).
Rate coefficients computed at some selected temperature values are also reported in Tables 4, 5.
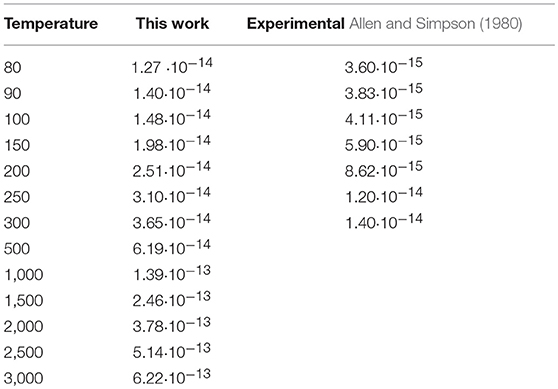
Table 4. Experimental and calculated rate constants, in cm3 s−1, for the exothermic CO(0)+N2(1) → CO(1)+N2(0)+187.45 cm−1 process.

Table 5. Experimental and calculated rate constants, in cm3 s−1, for the endothermic CO(1)+N2(0) → CO(0)+N2(1)-187.45 cm−1 process.
The behavior of the QC rate coefficient plots computed on our PES shown in Figures 8, 9 for both processes agrees well with the experimental ones over the whole temperature range. More in detail, deviations by a maximum factor of 3 are found at the lowest values of T, where the experimental data are more bound to be affected by lower accuracy, although the corresponding slope is well reproduced. Indeed the calculated and experimental value of β in the Arrhenius expression for the T range 80–300 K differs for little less than 10%.
In the case of the exothermic VV exchange, where experimental data are available in a wider range of temperatures up to 3,000 K, the behavior at low temperatures is the same as for the endothermic process, but the deviation between calculated and experimental rate coefficients is found to decrease to ~ 20% at temperatures larger than 1,000 K (Figure 8). Thus, the agreement between theoretical and experimental data is rather good in the whole temperature range. Agreement at higher temperatures (superior than in previous theoretical determination) is an indication that the basic contributions to the interaction are well described. The slightly larger discrepancy at low temperature can be attributed either to the lower accuracy of low-temperature experiments or to the neglecting of a proper quantum treatment for rotations which might play a role in the vibrational energy exchange process at low collision energies.
4. The Prototyping of the Open Molecular Science Cloud Service
In this section the present implementation of the above-mentioned MOSEX [MOlecular Simulator Enabled Cloud Services (MOSEX) (Vitillaro and Laganà, 2018)], which aimed to support research in computational simulations of molecular scattering processes, is illustrated with particular focus on related progress in the following:
a) networking and networked software applications,
b) assembling cloud computing infrastructures,
c) developing a sustainable operational model.
4.1. Networking and Networked Software Applications
Networking and assembling networked software applications for the Molecular science community begun within COST (www.cost.eu/) Action D23 (METACHEM) and D37 (Grid Computing in Chemistry: GRIDCHEM). In METACHEM the activities of various Molecular Science research laboratories were networked on a shared computing platform made of a geographically distributed cluster of heterogeneous computers operating as a single virtual parallel machine (Foster and Kesselman, 1999). In the following Action GRIDCHEM Grid solutions and paradigms for molecular science research developed by D23 were consolidated on the grid. GRIDCHEM leveraged the creation and the use of distributed computing infrastructures (the “Grid”) to drive collaborative computer modeling and simulation in chemistry toward “new frontiers in complexity and a new regime of time-to-solution” (GRIDCHEM, 2006–2010). At more infrastructural level, the European projects EGEE (Enabling Grids for E-sciencE, https://cordis.europa.eu/project/rcn/87264_en.html), first, and the EGI (European Grid Infrastructure, https://en.wikipedia.org/wiki/European-Grid-Infrastructure), next, provided various disciplines, including Molecular Science, with a world class level platform for computational collaborations. In particular, during EGEE-III, the first pilot computational application called GEMS (Laganà et al., 2010)—designed to enable the distributed calculation of cross sections and rate coefficients starting from the ab initio computation of the electronic structure of the molecular system followed by the fitting of the computed ab initio points using a combination of analytic formulae into a proper functional representation of the interaction in short, intermediate and long-range regions—was implemented. This allows at present routine computation of reactive and nonreactive properties of elementary systems based on the desired number of molecular geometries (Storchi et al., 2006) and collisional paths (Gervasi and Laganà, 2004) by distributing them on the grid. The scheme of the most recent evolution of GEMS toward the atomistic simulation of the kinetics of more complex systems, together with the related “data analysis and validation” and “open archive and reuse” in a cloud service perspective, is sketched in Figure 10.
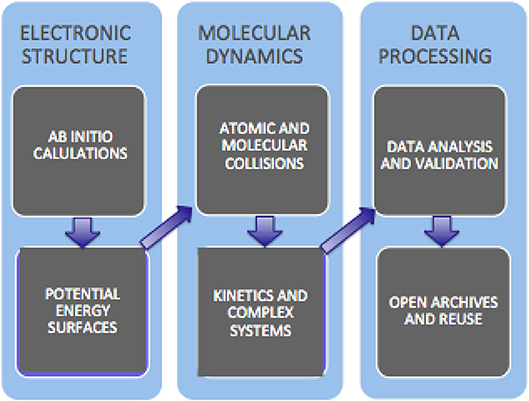
Figure 10. Block diagram of the present evolution of GEMS.
The molecular science community has been accordingly organized into the COMPCHEM (Laganà et al., 2010) VO first and in the CMMST (Chemistry, Molecular and Materials Science a Technologies) VRC (Virtual Research Community) (https://wiki.egi.eu/wiki/Towards_a_CMMST_VRC) later. On a national basis, then, the different disciplinary communities were gathered in Joint Research Units (JRU), for example in Italy with IGI (http://www.italiangrid.it), in order to give a higher local momentum to networked applications. In this spirit, the following activities were pursued in networked Molecular Sciences:
• orchestrate the initiatives of the e-infrastructure experts and of the disciplinary researchers so as to enable an effective intra- and trans-community networked implementation and coordination of a collaborative/competitive (synergistic) research environment by allowing a selection of compute resources based on quality parameters;
• compose higher level of complexity chained applications through the coordinated usage of distributed hardware and software, the creation of specialized web portals and workflows facilitating the production of data and innovative know how, the direct re-use of the produced data and knowledge in education, training and further research, the rewarding of the work done on behalf of the community by proactive members;
• produce and provide computational services useful to enhancing multi-scale treatments necessary to reproduce the observables of realistic systems in the areas of energy, environment, materials, pharmacology, biology, biotechnologies, medicine, etc., by means of state-of-the-art first principle electronic structure and nuclei dynamics computations, high level of accuracy multiscale design of complex molecular systems, knowledge management for training and education in sciences and technologies;
• turn (in collaboration with partner SMEs), the versatility of the adopted e-infrastructure tools, the richness of the developed CMMST knowledge and the credit mechanism supporting the synergistic operating into a business model, enabling an efficient transfer of the activities to the market, thereby ensuring business sustainability by leveraging synergistic models.
4.2. Assembling Cloud Computing Infrastructures
The cloud-oriented computing infrastructure developed for our calculations is an embryonic platform of the Beowulf type running under the OpenStack platform sketched in the lower side of Figure 3 of Vitillaro and Laganà (2018) named HERLA (https://en.wikipedia.org/wiki/Beowulf_cluster). HERLA has been established at the Dipartimento di Chimica, Biologia e Biotecnologia (DCBB) of the University of Perugia. The platform consists of a couple of HPC clusters, (CG/training) and (FE/research) running Scientific Linux 6.x with two distinct access nodes. The clusters are connected using NIS in a single-image system and are used first for students' training (CG) and second for scientists' research (FE). The management of Herla is presently carried out by the CMS2 Consortium (http://www.cms-2.org/index.php) of the University of Perugia, CNR-ISTM - Perugia unit and the two companies Master-UP s.r.l. and Molecular Horizon s.r.l. Recently, cloud images of HERLA (VHERLA) have been created and deployed in a storage system (CEPH located at DCBB). This effort was meant to support as well the activities of the School on Open Science Cloud (SOSC17) held in Perugia on June 2017 in collaboration with the Department of Physics and Geology (DFG) and INFN Perugia (running under the INFN OpenStack platform sketched in the lhs upper side of Figure 3 of Vitillaro and Laganà, 2018).
Later, as sketched in the rhs side of Figure 4 of Vitillaro and Laganà (2018), the images of VHERLA were allocated to the OpenStack GARR Cloud platform in Palermo within the project “cnr-istm” and were used to install the version VHERLA(GARR-CLOUD) hscw (http://hscw.herla.unipg.it/ganglia/?p=2&c=FrontEnd) bearing the following features: Access node hscw (2 core, 4Gb RAM, 200Gb storage) and Cluster (7 nodes, 96 cores, 360Gb RAM, 700Gb storage). The Access node hscw, can be reached at the IP address [90.147.189.20] via SSH and the following 7 nodes, 96core, 380Gb, 512Gb scratch are defined at Torque(PBS)/MAUI as [Intel Xeon E3-12xx v2(Ivy Bridge)/2.6Ghz]. This allowed the generation of a virtual cluster for Molecular Sciences that has also been used for the training of the Students of the XIII EM TCCM (European Master in Theoretical Chemistry and Computational Modeling) Intensive Course (http://www-old.chm.unipg.it/chimgen/mb/theo2/TCCM2018/EM- TCCM2018/EM-TCCM/Welcome.html) with details at the following reference web page: http://hscw.herla.unipg.it URL.
4.3. Developing a Sustainable Operational Model
As to the operational model we have already developed a sustainable open collaborative user/producer (Prosumer), whose prototype has been first implemented for running the ECTN (European Chemistry Thematic Network (http://ectn.eu/)) EChemTest e-tests based on the use of the educational LibreEOL (https://echemtest.libreeol.org) and GLOREP (https://glorep.unipg.it/) services Laganà et al., 2018a. An important feature of the Prosumer model adopted for EChemTest was the containment of costs by leveraging the fact that the ECTN member HEIs running e-test Self Evaluation Sessions (SES)s for the assessment of the Chemistry competences of their own students already act at the same time as consumers of EChemTest services and as producers of Question and Answers (Q&A)s, assessors of students' competences, designers and developers of e-learning materials (a typical cluster of the horizontal type) for the harmonization at the European level of the assessment of Molecular Science competences. This behavior is quite usual in education and research activities in which knowledge is a common good to be at the same time produced and consumed.
The only additional action needed to the end of making the prosumer model sustainable was to assign to a company the role of market spinner. For EChemTest this role was taken by Master-Up s.r.l. thanks to its nature as a former spinoff of the University of Perugia (started in the year 2004 out of the aggregation of some members of the Chemistry Department and the Mathematics and Informatics Department who were experts in molecular dynamics simulations and computer science) devoted to design, production and marketing services for technological innovation as its main goal. As a matter of fact, the mission of Master-Up has been since the very beginning the design and development of cloud services for molecular sciences and technologies. In the particular case of EChemTest, the Prosumer model has led to the production in the year 2018 of 2622 SESs, with an increase of 5 percent over the previous year. Further efforts have also been spent in the period up to January 2019 for the implementation of the already mentioned MOlecular Simulator Enabled Cloud Services (MOSEX), a European Open Science Cloud Pilot designed as a follow-up of the EGI COMPCHEM VO activities for the following applications:
1. Molecular electronic structure and dynamical properties programs of GEMS (including the QC ones reported reported in this paper)
2. Drug design programs (QSPR and 3D QSPR models)
3. Distributed repository of molecular science data (including the IOCHEM (https://www.iochem-bd.org/) software for the organization, publication and storage of molecular information on materials)
4. Dissemination and evaluation of molecular knowledge through GLOREP (Distributed repository of shared Learning Objects and educational tools) for the assemblage of Molecular Science multimedia Learning Objects.
5. Conclusions
In this work we have presented an accurate full dimensional potential energy surface for the CO-N2 system, characterized by an extension of the bond-bond formulation of the intermolecular interactions. The PES, intended for use in dynamics simulations aimed at calculating sets of accurate state-to-state rate coefficients, has been validated by performing Quantum-Classical dynamics simulations of the CO-N2 vibrational energy exchange, with outcoming rate coefficients in substantial agreement with experimental data on a wide range of temperatures. The generation of large pools of kinetics data and accurate molecular properties to feed databases employed in astrochemical models, plasma chemistry and combustion studies, is a current challenge for theoretical and computational chemistry, with a strong multidisciplinary character. Here we suggest to set up cloud-oriented computing infrastructures based on collaborative computer modeling and simulations to gather communities and boost efforts.
Author Contributions
AaL and AoL conceived and supervised the study and wrote the manuscript. FP and MB formulated the PES and performed ab initio calculations. CC performed dynamics simulations based on the Quantum Classical method, obtaining the theoretical rate coefficients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank MIUR and Perugia University for financial support through the AMIS project (Dipartimenti di Eccellenza 2018–2022). Financial support from VII FP (Egi-Inspire, Phys4Entry) and COST (Action CM901) is acknowledged. AaL and FP thank the Italian MIUR for funding through the program PRIN 2015 (contract 2015F59J3R_002). AaL also thanks the Dipartimento di Chimica, Biologia e Biotecnologie for funding under the program Fondo Ricerca di Base 2017. The work was also financially supported by Fondazione Cassa Risparmio Perugia (Codice Progetto: 2015.0331.021 Ricerca Scientifica e Tecnologica). MB was supported by the Ministerio de Ciencia, Innovación y Universidades (Spain, grant FIS2017-84391-C2-2-P). Calculations have been made possible by the support of the Virtual Organization COMPCHEM and allocated computing time from the OU Supercomputing Center for Education & Research (OSCER) at the University of Oklahoma (OU).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00309/full#supplementary-material
References
Albertí, M., and Faginas Lago, N. (2012). Ion size influence on the ar solvation shells of M+-C6F6 clusters (m = na, k, rb, cs). J. Phys. Chem. A 116, 3094–3102. doi: 10.1021/jp300156k
Albertí, M., Faginas-Lago, N., Laganà, A., and Pirani, F. (2011). A portable intermolecular potential for molecular dynamics studies of NMA–NMA and NMA H2O aggregates. Phys. Chem. Chem. Phys. 13:8422. doi: 10.1039/c0cp01763a
Allen, D., and Simpson, C. J. S. M. (1980). Vibrational energy exchange between co and the isotopes of N2 between 300 k and 80 k. Chem. Phys. 45, 203–211. doi: 10.1016/0301-0104(80)85068-3
Aquilanti, V., Beddoni, A., Cavalli, S., Lombardi, A., and Littlejohn, R. (2000). Collective hyperspherical coordinates for polyatomic molecules and clusters. Mol. Phys. 98, 1763–1770. doi: 10.1080/00268970009483380
Aquilanti, V., Beddoni, A., Lombardi, A., and Littlejohn, R. (2002). Hyperspherical harmonics for polyatomic systems: basis set for kinematic rotations. Int. J. Quantum. Chem. 89, 277–291. doi: 10.1002/qua.10278
Aquilanti, V., Carmona Novillo, E., Garcia, E., Lombardi, A., Sevryuk, M. B., and Yurtsever, E. (2006). Invariant energy partitions in chemical reactions and cluster dynamics simulations. Comput. Mat. Sci. 35, 187–191. doi: 10.1016/j.commatsci.2004.11.008
Aquilanti, V., Grossi, G., Lombardi, A., Maciel, G. S., and Palazzetti, F. (2011). Aligned molecular collisions and a stereodynamical mechanism for selective chirality. Rendiconti Lincei 22:125. doi: 10.1007/s12210-011-0123-7
Aquilanti, V., Lombardi, A., and Littlejohn, R. (2004a). Hyperspherical harmonics for polyatomic systems: basis set for collective motions. Theor. Chem. Accounts 111, 400–406. doi: 10.1007/s00214-003-0526-3
Aquilanti, V., Lombardi, A., Sevryuk, M. B., and Yurtsever, E. (2004b). Phase-space invariants as indicators of the critical behavior of nanoaggregates. Phys. Rev. Lett. 93:113402. doi: 10.1103/PhysRevLett.93.113402
Bacmann, A., Lefloch, B., Ceccarelli, C., Castets, A., Steinacker, J., and Loinard, L. (2002). The degree of co depletion in pre-stellar cores. A&A 389, L6–L10. doi: 10.1051/0004-6361:20020652
Barreto, P., Albernaz, A. F., Capobianco, A., Palazzetti, F., Lombardi, A., and Grossi, G., et al. (2012). Potential energy surfaces for interactions of H2O with H2, N2 and O2: a hyperspherical harmonics representation, and a minimal model for the H2O-rare-gas-atom systems. Comp. Theor. Chem. 990, 53–61. doi: 10.1016/j.comptc.2011.12.024
Barreto, P. R. P., Albernaz, A. F., Palazzetti, F., Lombardi, A., Grossi, G., and Aquilanti, V. (2011). Hyperspherical representation of potential energy surfaces: intermolecular interactions in tetra-atomic and penta-atomic systems. Phys. Script. 84:028111. doi: 10.1088/0031-8949/84/02/028111
Bartolomei, M., Carmona-Novillo, E., Hernández, M. I., Campos-Martínez, J., and Hernández-Lamoneda, R. (2011). Long-range interaction for dimers of atmospheric interest: dispersion, induction and electrostatic contributions for O2–O2, N2–N2 and O2–N2. J. Comp. Chem. 32:279. doi: 10.1002/jcc.21619
Bartolomei, M., Pirani, F., Laganà, A., and Lombardi, A. (2012). A full dimensional grid empowered simulation of the CO2 + CO2 processes. J. Comp. Chem. 33:1806. doi: 10.1002/jcc.23010
Billing, G., Coletti, C., Kurnosov, A. K., and Napartovich, A. (2003). Sensitivity of molecular vibrational dynamics to energy exchange rate constants. J. Phys. B 36, 1175–1192. doi: 10.1088/0953-4075/36/6/308
Billing, G. D. (1984a). Rate constants and cross sections for vibrational transitions in atom-diatom and diatom-diatom collisions. Comp. Phys. Comm. 32, 45–62. doi: 10.1016/0010-4655(84)90007-9
Billing, G. D. (1984b). The semiclassical treatment of molecular roto/vibrational energy transfer. Comput. Phys. Rept. 1, 239–296. doi: 10.1016/0167-7977(84)90006-6
Billing, G. D. (1987). Rate constants for vibrational transitions in diatom-diatom collisions. Comp. Phys. Comm. 44, 121–136. doi: 10.1016/0010-4655(87)90022-1
Boys, S. F., and Bernardi, F. (1970). The calculation of small molecular interactions by the differences of separate total energies. some procedures with reduced errors. Mol. Phys. 19:553. doi: 10.1080/00268977000101561
Cappelletti, D., Pirani, F., Bussery-Honvault, B., Gomez, L., and Bartolomei, M. (2008). A bond–bond description of the intermolecular interaction energy: the case of weakly bound N2–H2 and N2–N2 complexes. Phys. Chem. Chem. Phys. 10, 4281–4293. doi: 10.1039/b803961e
Castro Palacio, J. C., Velasquez Abad, L., Lombardi, A., Aquilanti, V., and Soneira, J. R. (2007). Normal and hyperspherical mode analysis of no-doped kr crystals upon rydberg excitation of the impurity. J. Chem. Phys. 126:174701. doi: 10.1063/1.2730786
Celiberto, R., Armenise, I., Cacciatore, M., Capitelli, M., Esposito, F., and Gamallo, P., et al. (2016). Atomic data for spacecraft re-entry plasmas. Plasma Sources Sci. Technol. 25:033004. doi: 10.1088/0963-0252/25/3/033004
Coletti, C., and Billing, G. D. (1999). Isotopic effects on vibrational energy transfer in co. J. Chem. Phys. 111, 3891–3897. doi: 10.1063/1.479692
Coletti, C., and Billing, G. D. (2000). Rate constants for energy transfer in carbon monoxyde. J. Chem. Phys. 113, 4869–4875. doi: 10.1063/1.1289251
Coletti, C., and Billing, G. D. (2002). Vibrational energy transfer in molecular oxygen collisions. Chem. Phys. Lett. 356, 14–22. doi: 10.1016/S0009-2614(02)00279-8
Fabiano, F., Lòpez Puertas, M., Adriani, A., Moriconi, M. L., D'Aversa, E., and Funke, B., et al. (2017). Co concentration in the upper stratosphere and mesosphere of titan from vims dayside limb observations at 4.7 μm. Icarus 293, 119–131. doi: 10.1016/j.icarus.2017.04.014
Faginas Lago, N., Albertí, M., laganà, A., and Lombardi, A. (2013). “Water (H2O)m or benzene (C6H6)n aggregates to solvate the K+,” in Computational Science and Its Applications - ICCSA 2013, volume 7971 of Lecture Notes in Computer Science (Berlin; Heidelberg: Springer), 1–15.
Faginas, N., Huarte-Larranaga, F., and Laganà, A. (2008). Full dimensional quantum versus semiclassical reactivity for the bent transition state reaction n + N2. Chem. Phys. Lett. 464, 249–255. doi: 10.1016/j.cplett.2008.09.008
Faginas-Lago, N., Albertí, M., Costantini, A., Laganá, A., Lombardi, A., and Pacifici, L. (2014a). An innovative synergistic grid approach to the computational study of protein aggregation mechanisms. J. Mol. Model. 20:2226. doi: 10.1007/s00894-014-2226-4
Faginas-Lago, N., Albertí, M., Laganà, A., Lombardi, A., Pacifici, L., and Costantini, A. (2014b). “The molecular stirrer catalytic effect in methane ice formation,” in Computational Science and Its Applications-ICCSA 2014, volume 8579 of Lecture Notes in Computer Science, eds B. Murgante and S. E. A. Misra (Cham: Springer), 585–600.
Faginas-Lago, N., Costantini, A., and Huarte-Larrañaga, F. (2010). Direct calculation of the rate coefficients on the grid: exact quantum versus semiclassical results for n+ n2. Int. J. Quant. Chem. 110, 422–431. doi: 10.1002/qua.22273
Faginas-Lago, N., and Laganá, A. (2005). A comparison of semiclassical ivr and exact quantum collinear atom diatom transition probabilities for mixed reactive and non reactive regimes. AIP Conf. Proc. 762:920. doi: 10.1063/1.1941652
Faginas-Lago, N., Laganà, A., Gargano, R., and Barreto, P. (2006). On the semiclassical initial value calculation of thermal rate coefficients for the n + n reaction. J. Chem. Phys. 125:114311. doi: 10.1063/1.2345363
Faginas-Lago, N., Lombardi, A., Albertí, M., and Grossi, G. (2015). Accurate analytic intermolecular potential for the simulation of Na+ and K+ ion hydration in liquid water. J. Mol. Liquids 204, 192–197. doi: 10.1016/j.molliq.2015.01.029
Falcinelli, S., Rosi, M., Candori, P., Vecchiocattivi, F., Bartocci, A., and Lombardi, A., et al. (2013). “Modeling the intermolecular interactions and characterization of the dynamics of collisional autoionization processes,” in International Conference on Computational Science and Its Applications (Berlin; Heidelberg: Springer), 69–83.
Fioccola, S., Pirani, F., Bartolomei, M., and Coletti, C. (2017). “Influence of the intermolecular potential energy on N2-N2 inelastic collisions: a quantum-classical study,” in Computational Science and Its Applications-ICCSA 2017, volume 10408 of Lecture Notes in Computer Science, ed O. E. A. Gervasi (Cham: Springer), 281–296.
Foster, I., and Kesselman, C. (1999). The Grid: Blueprint for a New Computing Infrastructure. San Francisco, CA: Morgan Kaufmann Publ.
Gervasi, O., and Laganà, A. (2004). Simbex: a portal for the a priori simulation of crossed beam experiments. Fut. Generat. Comput. Syst. 20, 703–716. doi: 10.1016/j.future.2003.11.028
GRIDCHEM (2006–2010). E-science and Learning Approaches in Molecular Sciences (2006–2010). COST Action D37 Grid Computing in Chemistry: GRIDCHEM.
He, C., Horst, S. M., Riemer, S., Sebree, J. A., Pauley, N., and Vuitton, V. (2017). Carbon monoxide affecting planetary atmospheric chemistry. Astrophys. J. Lett. 841:L31. doi: 10.3847/2041-8213/aa74cc
Hörst, S. M., Yoon, Y. H., Ugelow, M. S., Parker, A. H., Li, R., and de Gouw, J. A., et al. (2018). Laboratory investigations of titan haze formation: in situ measurement of gas and particle composition. Icarus 301, 136–151. doi: 10.1016/j.icarus.2017.09.039
Hörst, S. M. E. A. (2012). Formation of amino acids and nucleotide bases in a titan atmosphere simulation experiment. Astrobiology 12, 809–817. doi: 10.1089/ast.2011.0623
Jaeschke, M., Audibert, S., van Caneghem, P., Humphreys, A. E., Janssen-van Rosmalen, R., and Pellei, Q., et al. (1988). High accuracy compressibility factor calculation of natural gases and similar mixtures by use of a truncated virial equation. GERG Tech. Monogr. TM2 162.
Karimi-Jafari, M. H., Maghari, A., and Farjamnia, A. (2011). Intermolecular potential energy surface of the N2-CO dimer: Ab initio investigation and analytical representation. J. Phys. Chem. A 115, 1143–1151. doi: 10.1021/jp111279t
Kendall, R. A., Dunning, J. T. H., and Harrison, R. J. (1992). Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 96:4989. doi: 10.1063/1.462569
Kurnosov, A., Cacciatore, M., and Billing, G. (2003). State-to-state rate constant calculations for v-v energy transfer in co–N2 collisions. J. Phys. Chem. 107, 2403–2410. doi: 10.1021/jp0218239
Kustova, E., and Nagnibeda, E. (2012). Kinetic model for multi-temperature flows of reacting carbon dioxide mixtures. Chem. Phys. 398, 111–117. doi: 10.1016/j.chemphys.2011.05.019
Kustova, E. V., and Kremer, G. M. (2014). Chemical reaction rates and non-equilibrium pressure of reacting gas mixtures in the state-to-state approach. Chem. Phys. 445, 82–94. doi: 10.1016/j.chemphys.2014.10.019
Laganà, A., Costantini, A., Gervasi, O., Lago, N. F., Manuali, C., and Rampino, S. (2010). Compchem: progress towards gems a grid empowered molecular simulator and beyond. J. Grid. Comput. 8, 571–586. doi: 10.1007/s10723-010-9164-x
Laganà, A., Crocchianti, S., Faginas Lago, N., Pacifici, L., and Ferraro, G. (2003). A nonorthogonal coordinate approach to atom-diatom parallel reactive scattering calculations. Collect. Czech. Chem. Commun. 68, 307–330. doi: 10.1135/cccc20030307
Laganà, A., Gervasi, O., Tasso, S., Perri, D., and Franciosa, F. (2018a). “The ECTN Virtual Education Community prosumer model for promoting and assessing chemical knowledge,” in Computational Science and Its Applications-ICCSA 2018, Vol. 10964, Springer, Cham of Lecture Notes in Computer Science, ed O. E. A. Gervasi (Cham: Springer), 533–548.
Laganà, A., Pirani, F., Faginas-Lago, N., Vitillaro, G., and Garcia, E. (2018b). “Process driven potentials for Open Molecular Science Cloud computational services: the nitrogen case study,” in Rendiconti Accademia Nazionale delle Scienze detta dei XL, Memorie di Scienze Fisiche e Naturali, 136o, Vol. XLII, Parte II, eds F. P. Alazzetti, and A. Lombardi (Rome: Accademia dei XL), 47–59.
Lago, N. F., Laganà, A., Garcia, E., and Gimenez, X. (2005). “Thermal rate coefficients for the N + N2 reaction: quasiclassical, semiclassical and quantum calculations,” in Computational Science and Its Applications - ICCSA 2005. Lecture Notes in Computer Science, Vol. 3480, eds O. Gervasi, M. L. Gavrilova, V. Kumar, A. Laganà, H. P. Lee, Y. Mun, D. Taniar, and C. J. Kenneth Tan (Berlin; Heidelberg: Springer).
Lee, T. J., and Taylor, P. R. (1989). A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 36(Suppl. 23):199. doi: 10.1002/qua.560360824
Lellouch, E., de Bergh, C., Sicardy, B., Ferron, S., and Küfl, H.-U. (2010). Detection of CO in triton's atmosphere and the nature of surface-atmosphere interactions. A&A 512:L8. doi: 10.1051/0004-6361/201014339
Linstrom, P. J., and Mallard, W. G. (2018). Nist Chemistry Webbook, Nist Standard Reference Database.
Lombardi, A., Faginas-Lago, N., Gaia, G., and Palazzetti, F. (2016a). “Collisional energy exchange in CO2–N2 gaseous mixtures,” in Computational Science and Its Applications - ICCSA 2016, volume 9786 of Lecture Notes in Computer Science, ed O. E. A. Gervasi (Cham: Springer), 246–257.
Lombardi, A., Faginas-Lago, N., Pacifici, L., and Grossi, G. (2015). Energy transfer upon collision of selectively excited CO2 molecules: state-to-state cross sections and probabilities for modeling of atmospheres and gaseous flows. J. Chem. Phys. 143:034307. doi: 10.1063/1.4926880
Lombardi, A., Laganá, A., Pirani, F., Palazzetti, F., and Faginas-Lago, N. (2013). “Carbon oxides in gas flows and earth and planetary atmospheres: state-to-state simulations of energy transfer and dissociation reactions,” in Computational Science and Its Applications - ICCSA 2013, volume 7972 of Lecture Notes in Computer Science, eds B. Murgante, S. Misra, M. Carlini, C. Torre, H.-Q. Nguyen, D. Taniar, B. Apduhan, and O. Gervasi (Berlin; Heidelberg: Springer), 17–31.
Lombardi, A., Lago, N. F., Laganà, A., Pirani, F., and Falcinelli, S. (2012). “A bond-bond portable approach to intermolecular interactions: simulations for n-methylacetamide and carbon dioxide dimers,” in International Conference on Computational Science and Its Applications (Berlin; Heidelberg: Springer), 387–400.
Lombardi, A., Maciel, G. S., Palazzetti, F., Grossi, G., and Aquilanti, V. (2010). Alignment and chirality in gaseous flows. J. Vacuum Soc. Japan 53, 645–653. doi: 10.3131/jvsj2.53.645
Lombardi, A., and Palazzetti, F. (2008). A comparison of interatomic potentials for rare gas nanoaggregates. J. Mol. Struct. 852, 22–29. doi: 10.1016/j.theochem.2007.12.011
Lombardi, A., Pirani, F., Laganá, A., and Bartolomei, M. (2016b). Energy transfer dynamics and kinetics of elementary processes (promoted) by gas-phase CO2-N2 collisions: selectivity control by the anisotropy of the interaction. J. Comp. Chem. 37, 1463–1475. doi: 10.1002/jcc.24359
Maitland, G. C., Rigby, M., Smith, E. B., and Wakeham, W. A. (1987). Intermolecular Forces. Oxford: Clarendon Press.
Manuali, C., Laganà, A., and Rampino, S. (2010). Grif: a grid framework for a web service approach to reactive scattering. Comput. Phys. Commun. 181, 1179–1185. doi: 10.1016/j.cpc.2010.03.001
Maroulis, G. (1996). Electric polarizability and hyperpolarizability of carbon monoxide. J. Phys. Chem. 100, 13466–13473. doi: 10.1021/jp960412n
Mastrocinque, G., Chakroun, A., Doyenette, L., Gueguen, H., Margottin-Maclou, M., and Henry, L. (1976). Vibrational energy transfer from the v=1 level of carbon monoxide to the v=1 level of the two isotopic species of nitrogen 14N2 and 145N2. Chem. Phys. Lett. 39, 347–349. doi: 10.1016/0009-2614(76)80092-9
McElroy, J. P., and Buchanan, S. (1995). Excess and unlike interaction second virial coefficients and excess enthalpy of mixing of (carbon monoxide + pentane). J. Chem. Eng. Data 40:452. doi: 10.1021/je00018a021
Nakamura, M., Tsai, P.-Y., Kasai, T., Lin, K.-C., Palazzetti, F., and Lombardi, A., et al. (2015). Dynamical, spectroscopic and computational imaging of bond breaking in photodissociation: roaming and role of conical intersections. Faraday Discuss. 177, 77–98. doi: 10.1039/C4FD00174E
Pacifici, L., Verdicchio, M., Lago, N. F., Lombardi, A., and Costantini, A. (2013). A high-level ab initio study of the n2+ n2 reaction channel. J. Comput. Chem. 34, 2668–2676. doi: 10.1002/jcc.23415
Pack, R. T. (1978). Anisotropic potentials and the damping of rainbow and diffraction oscillations in differential cross sections. Chem. Phys. Lett. 55:197. doi: 10.1016/0009-2614(78)87001-8
Palazzetti, F., Munusamy, E., Lombardi, A., Grossi, G., and Aquilanti, V. (2011). Spherical and hyperspherical representation of potential energy surfaces for intermolecular interactions. Int. J. Quantum. Chem. 111, 318–332. doi: 10.1002/qua.22688
Palazzetti, F., Tsai, P.-Y., Lombardi, A., Nakamura, M., Che, D.-C., and Kasai, T., et al. (2013). Aligned molecules: chirality discrimination in photodissociation and in molecular dynamics. Rendiconti Lincei 24, 299–308. doi: 10.1007/s12210-013-0248-y
Pirani, F., Albertí, M., Castro, A., Moix, M., and Cappelletti, D. (2004). Atom-bond pairwise additive representation for intermolecular potential energy surfaces. Chem. Phys. Lett. 394, 37–44. doi: 10.1016/j.cplett.2004.06.100
Pirani, F., Cappelletti, D., Falcinelli, S., Cesario, D., Nunzi, F., and Belpassi, L., et al. (2019). Selective emergence of halogen bond in ground and excited states of noble-gas-chlorine systems. Angew. Chem. Int. Ed. 58, 4195–4199. doi: 10.1002/anie.201812889
Pirani, F., Cappelletti, D., and Liuti, G. (2001). Range, strength and anisotropy of intermolecular forces in atom-molecule systems: an atom-bond pairwise additive approach. Chem. Phys. Lett. 350:286. doi: 10.1016/S0009-2614(01)01297-0
Pirani, P., Brizi, S., Roncaratti, L., Casavecchia, P., Cappelletti, D., and Vecchiocattivi, F. (2008). Beyond the lennard-jones model: a simple and accurate potential function probed by high resolution scattering data useful for molecular dynamics simulations. Phys. Chem. Chem. Phys. 10:5489. doi: 10.1039/b808524b
Rampino, S., Faginas-Lago, N., Laganà, A., and Huarte-Larrañaga, F. (2012a). An extension of the grid empowered molecular simulator gems to quantum reactive scattering. J. Comp. Chem. 33, 707–714. doi: 10.1002/jcc.22878
Rampino, S., Faginas-Lago, N., Laganà, A., and Huarte-Larrañaga, F. (2012b). An extension of the grid empowered molecular simulator to quantum reactive scattering. J. Comput. Chem. 33, 708–714.
Sato, Y., Tsuchiya, S., and Kuratani, K. (1969). Shockwave study of vibrational energy exchange between diatomic molecules. J. Chem. Phys. 50:1911. doi: 10.1063/1.1671306
Sevyuk, M. B., Lombardi, A., and Aquilanti, V. (2005). Hyperangular momenta and energy partitions in multidimensional many-particle classical mechanics: the invariance approach to cluster dynamics. Phys. Rev. A 72:033201. doi: 10.1103/PhysRevA.72.033201
Storchi, L., Tarantelli, F., and Laganà, A. (2006). “Computing molecular energy surfaces on the grid,” in Computational Science and Its Applications-ICCSA 2006, volume 3980 of Lecture Notes in Computer Science, ed O. E. A. Gervasi (Berlin; Heidelberg: Springer), 675–683.
Surin, L. A., Tarabukin, I. V., Schlemmer, S., Kalugina, Y. N., and van der Avoird, A. (2018). Ab initio potential and rotational spectra of the CO-N2 complex. J. Chem. Phys. 148:044313. doi: 10.1063/1.5013115
Tao, F. M., and Pan, Y. K. a. (1992). Møller-plesset perturbation investigation of the he2 potential and the role of midbond basis functions. J. Chem. Phys. 97:4989. doi: 10.1063/1.463852
Tomasko, M. G., Bèzard, B., Doose, L., Engel, S., Karkoschka, E., and Vinatier, S. (2008). Heat balance in titan's atmosphere. Planet. Space Sci. 56, 648–659. doi: 10.1016/j.pss.2007.10.012
van Lenthe, J. H., van Duijneveldt-van de Rijdt, J. G. C. M., and van Duijneveldt, F. B. (1987). Weakly bonded systems. Adv. Chem. Phys 69:521. doi: 10.1002/9780470142943.ch9
Vitillaro, G., and Laganà, A. (2018). Vherla: A Virtual Molecular Science Data Center Allocated on the Garr Cloud. Available online at: http://services.chm.unipg.it/ojs/index.php/virtlcomm/article/view/203
von Rosenberg, C. M., Bray, K. N. C., and Pratt, N. H. (1972). Shock tube vibrational relaxation measurements: N2 relaxation by H2O and the co–N2 vv rate. J. Chem. Phys. Lett. 56:3230. doi: 10.1063/1.1677684
Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R, and Schütz, M. (2006). A Package of Ab initio Programs. Molpro, Version 2006.1
Yeamin, M. B., Faginas-Lago, N., Albertí, M., Cuesta, I. G., Sanchez-Marin, J., and Sanchez de Meras, A. M. J. (2014). Multi-scale theoretical investigation of molecular hydrogen adsorption over graphene: coronene as a case study. RSC Adv. 4, 54447–54453. doi: 10.1039/C4RA08487J
Zhang, X., West, R. A., Irwin, P. G. J., Nixon, C. A., and Yung, Y. L. (2015). Aerosol influence on energy balance of the middle atmosphere of Jupiter. Nat. Commun. 6:10231. doi: 10.1038/ncomms10231
Appendix A
Dependence of the CO Polarizability and Permanent Multipoles on the Monomer Bond Length
The radial dependence of the CO average polarizability (in Å3) is defined by the following empirical equations given in terms of n, an effective bond order, r, the bond length, and req its equilibrium value (see Table 1):
where the effective bond order reads
The dependence of on r is reported in the bottom panel of Figure 5 where a comparison with previous ab initio estimates is also provided.
The atomic polarizabilities related to the C and O atoms are obtained as
while that for the N atom (αN) is assumed to be half the N2 molecular polarizability which is obtained from the Appendix B of Cappelletti et al. (2008).
The atomic polarizabilities of the involved molecular partners are then used to obtain the Rm and ε interaction parameters for each atoma-atom pairb as follows Pirani et al. (2001):
with = 1.382 αC, = 1.382 αO and = 1.65 αN, and
being
an efficient dispersion coefficient depending on the numerical coefficients NC, NO and NN with the meaning of the total atomic effective electron numbers which assume the 4, 6, and 4.4 values, respectively.
The radial dependence of the CO molecular dipole (μ(r)) is formulated as
where μ(req) represents the dipole moment at req.
That of the CO molecular quadrupole (Q(r)) is expressed as
being Q(req) the quadrupole moment at req.
Such polynomial representations provide results in atomic units which reproduce the ab initio data illustrated in the top and intermediate panels of Figure 5.
From the above relationships we obtain the molecular charge distribution as
and
where qO, qC, and qcm are point charges on the oxygen and carbon atoms and CO center of mass (CM), respectively, and rO, rC are atom positions with respect to CM.
Keywords: intermolecular interactions, molecular energy transfer, rate constants, molecular beams, astrochemistry, carbon monoxide, planetary atmospheres
Citation: Lombardi A, Pirani F, Bartolomei M, Coletti C and Laganà A (2019) Full Dimensional Potential Energy Function and Calculation of State-Specific Properties of the CO+N2 Inelastic Processes Within an Open Molecular Science Cloud Perspective. Front. Chem. 7:309. doi: 10.3389/fchem.2019.00309
Received: 08 February 2019; Accepted: 18 April 2019;
Published: 22 May 2019.
Edited by:
Xi Zhang, Shenzhen University, ChinaReviewed by:
Thuat Thanh Trinh, Norwegian University of Science and Technology, NorwaySugata Chowdhury, National Institute of Standards and Technology (NIST), United States
Copyright © 2019 Lombardi, Pirani, Bartolomei, Coletti and Laganà. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Lombardi, YW5kcmVhLmxvbWJhcmRpQHVuaXBnLml0