 Xiaodong Ma
Xiaodong Ma Li Shen3†
Li Shen3† Xiaoqing Lv
Xiaoqing Lv
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 24 April 2019
Sec. Medicinal and Pharmaceutical Chemistry
Volume 7 - 2019 | https://doi.org/10.3389/fchem.2019.00236
This article is part of the Research Topic Medicinal and Pharmaceutical Chemistry Editor’s Pick 2021 View all 21 articles
A novel structural series of quinoline derivatives were designed, synthesized and biologically evaluated as PI3K/mTOR dual inhibitors upon incorporation of C-4 acrylamide fragment. Consequently, all of them exerted remarkable inhibition against PI3Kα with IC50 values ranging from 0.50 to 2.03 nM. Besides, they exhibited sub-micromolar to low micromolar anti-proliferative activity against both prostate cancer PC3 and colorectal cancer HCT116 cell lines. In subsequent profiling, 8i, a representative compound throughout this series, also significantly inhibited other class I PI3Ks and mTOR. In PC3 cells, it remarkably down-regulated the crucial biomarkers of PI3K/Akt/mTOR signaling, including phos-Akt (Ser473), phos-Akt (Thr308), phos-S6 ribosomal protein (Ser235/236), and phos-4E-BP1 (Thr37/46), at a concentration as low as 5 nM. Moreover, 8i displayed favorable metabolic stability with long elimination half-life in both human liver and rat liver microsomes. A further in vivo pharmacokinetic (PK) study demonstrated 8i possessed acceptable oral exposure, peak plasma concentration, and elimination half-life. Taken together, 8i, as a potent PI3K/mTOR dual inhibitor, merited further investigation and structural optimization.
Aberrant activation of the phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR, PAM) signaling is regarded as a crucial hallmark in a broad spectrum of human cancers (Folkes et al., 2008; Furet et al., 2013). Among the kinases relevant to the cascade, PI3Kα, a member of class I PI3K subfamily, has spurred significant pharmaceutical investment in exploring its inactivating agents, since the identification of oncogenic PIK3CA, the gene encoding its catalytic subunit (Ma and Hu, 2013; Ndubaku et al., 2013; Heffron et al., 2016; Yadav et al., 2016). Owing to the high sequence homology shared by the four class I PI3K members within the ATP-binding pocket, numerous clinically developed PI3Kα modulators also concurrently inhibit PI3Kβ, PI3Kγ, and PI3Kδ, thereby termed as pan-class I PI3K inhibitors. As a signaling effector located downstream of PI3K, mTOR is also intimately associated with cancer initiation and development (Yang et al., 2013; Pike et al., 2015). There is abundant evidence demonstrating that mTOR is relevant to multiple negative feedback loops, especially mTOR-S6K1-PI3K signaling (Liu et al., 2011; Ma et al., 2015). Consequently, the sole inhibition of mTOR may activate PI3K through S6K1 and IRS-1, thus restarting cancer development and mediating drug resistance (Liu et al., 2010). Meanwhile, mTOR inhibition has been testified to be beneficial for the sensitivity to PI3K inhibitors (Elkabets et al., 2013). In view of the evidence implicating both PI3K and mTOR in cancer, as well as the potential to obtain synergism and address drug resistance (Stauffer et al., 2008; Ma and Hu, 2013; Beaufils et al., 2017; Ma et al., 2018), simultaneous inhibition of PI3K and mTOR is expected to provide therapeutic advantages over mono-inhibition of PI3K or mTOR. So far, several PI3K/mTOR dual inhibitors have been advanced into clinical trials, as exemplified by GSK2126458 dactolisisb, bimiralisib, gedatolisib, apitolisib, voxtalisib, PF04691502, BGT226, GDC-0084, LY3023414, and VS-5584 (Garces and Stocks, 2019).
Quinoline represents a well-established template for constructing PI3K/mTOR dual inhibitors, and our efforts in search for PI3K signaling modulators belonging to this class have culminated in the discovery of numerous quinoline derivatives with attractive biological profiles (Zhang et al., 2017a,b). Although C-3 and C-4 positions of the quinoline template are oriented toward the entrance to PI3Kα active site, some surrounding residues can be exploited for producing additional interaction and enhancing the binding affinity. Previously, we have discovered some promising quinolines via exploring H-bond contact with residue Arg770 or Ser854 at this region upon structural elaboration at the C-3 position (Figure 1). To further broaden the chemical diversity of the quinoline-based PI3K/mTOR dual inhibitors, our recent medicinal chemistry efforts prioritize introduction of various acrylamide functionalities as the C-4 replacements for probing residue Gln859 at the entrance to the PI3Kα active site. The rationale for introducing the C-4 acrylamide functionality was based on the molecular docking analysis, which indicated its potential to confer H-bond interaction with residue Gln859. Moreover, a wide variety of terminal moieties of the C-4 acrylamide fragment were investigated for adjusting physicochemical properties. Hence, we herein communicate our work that has led to the discovery of a novel series of 4-acrylamido-quinoline derivatives as potent PI3K/mTOR dual inhibitors.
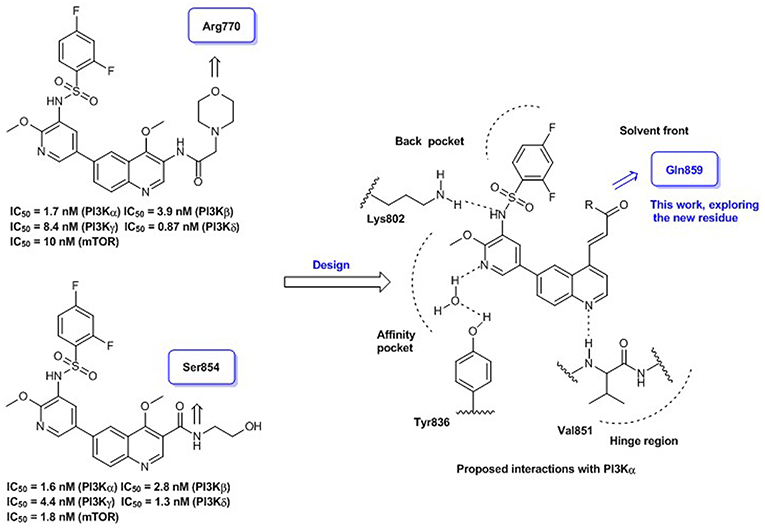
Figure 1. Quinoline-based PI3K/mTOR dual inhibitors obtained via probing residues at the entrance to PI3Kα active site: our previous and current work.
In this research, chemical reagents were commercially available, and, if necessary, pretreatment was carried out. With tetramethylsilane as the internal standard, 1H NMR and 13C NMR spectra were recorded on the 500 and 400 MHz instrument (Bruker Bioscience, Billerica, MA, USA), respectively. Chemical shifts (δ) were given in ppm and coupling constants (J) provided in hertz (Hz). ESI-MS data were measured on an Esquire-LC-00075 spectrometer, while HRMS data were collected by Waters Q-TOF Micromass. Column chromatography for the purification of intermediates or target compounds was performed using silica gel (200–300 mesh).
4-Bromoaniline (33.0 g, 193.02 mmol) was added to a three-neck round bottom flask with acetic acid (200 mL). After FeCl3 (32.0 g, 198.96 mmol) was added, the mixture was stirred at room temperature for 10 min. Subsequently, methyl vinyl ketone (17.0 mL, 209.71 mmol) was added dropwise over 30 min and the reaction maintained at 70°C for 3 h. Then, ZnCl2 (26.0 g, 194.22 mmol) was added and the mixture refluxed for 2 h. After cooling to room temperature, the mixture was evaporated under reduced pressure, basified with 1N NaOH solution, and extracted with EA. The combined organic extracts were dried over magnesium sulfate and concentrated to give the crude product, which was further purified by column chromatography (EA/PE = 1:5) to afford the title intermediate (6.78 g, 30.68 mmol; yield 16%) as a brown solid. 1H NMR (500 MHz, DMSO-d6): δ 8.79 (d, J = 4.5 Hz, 1H, Ar-H), 8.29 (d, J = 2.0 Hz, 1H, Ar-H), 7.96 (d, J = 9.0 Hz, 1H, Ar-H), 7.88 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.43 (d, J = 4.5 Hz, 1H, Ar-H), 2.67 (s, 3H, CH3). ESI-MS: m/z = 222 [M+H]+.
SeO2 (2.5 g, 22.34 mmol) was added to a solution of 6-bromo-4-methylquinoline (1.0 g, 4.52 mmol) in the mixture of dioxane/H2O (8/1, V/V) at room temperature. After being stirred at 100°C for 2 h, the reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was dissolved in EA and washed successively with saturated aqueous NaHCO3 and water. The organic phase was then dried with magnesium sulfate and concentrated in vacuo to afford a brown solid, which was purified by column chromatography (EA/PE = 1:5) to give 6-bromoquinoline-4-carbaldehyde (0.78 g, 3.32 mmol; yield 73%) as a light yellow solid. 1H NMR (500 MHz, DMSO-d6): δ 10.49 (s, 1H, CHO), 9.28 (d, J = 4.5 Hz, 1H, Ar-H), 9.18 (d, J = 2.0 Hz, 1H, Ar-H), 8.12 (d, J = 9.0 Hz, 1H, Ar-H), 8.11 (d, J = 4.5 Hz, 1H, Ar-H), 8.03 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H). ESI-MS: m/z = 236 [M+H]+.
Triethyl phosphonoacetate (350 mg, 1.56 mmol) was added to a suspension of NaH (100 mg, 60%, 2.50 mmol) in THF at 0°C, and the resultant mixture was stirred for 30 min. 6-bromoquinoline-4-carbaldehyde (300 mg, 1.28 mmol) was then added and the mixture stirred for 1 h at room temperature. The reaction mixture was cooled to 0°C, and ice water was added. After extracting with EA, the organic phase was washed with saturated aqueous NaHCO3, dried with magnesium sulfate and concentrated in vacuo to afford the crude product, which was further purified by column chromatography (EA/PE = 1:5) to give the title intermediate (342 mg, 1.12 mmol; yield 88%) as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 8.98 (d, J = 4.5 Hz, 1H, Ar-H), 8.48 (d, J = 2.0 Hz, 1H, Ar-H), 8.36 (d, J = 16.0 Hz, 1H, alkene hydrogen), 8.03 (d, J = 9.0 Hz, 1H, Ar-H), 7.97–7.95 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.93 (d, J = 4.5 Hz, 1H, Ar-H), 6.90 (d, J = 16.0 Hz, 1H, alkene hydrogen), 4.28 (q, J = 7.0 Hz, 2H, OCH2), 1.32 (t, J = 7.0 Hz, 3H, CH3). ESI-MS: m/z = 306 [M+H]+.
Ethyl (E)-3-(6-bromoquinolin-4-yl)acrylate (306 mg, 1.00 mmol) and 2.5 N NaOH (20 mL) were charged in a round bottom flask. The mixture was stirred under reflux for 2 h. After cooling to room temperature, the pH of the mixture was adjusted to 5 with 2 N HCl, and the resultant solid was filtered and washed with water. The filter cake was then dried under reduced pressure to afford the title intermediate (241 mg, 0.87 mmol; yield 87%) as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 12.89 (s, 1H, COOH), 8.97 (d, J = 4.5 Hz, 1H, Ar-H), 8.45 (d, J = 2.0 Hz, 1H, Ar-H), 8.29 (d, J = 16.0 Hz, 1H, alkene hydrogen), 8.02 (d, J = 9.0 Hz, 1H, Ar-H), 7.95 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.90 (d, J = 4.5 Hz, 1H, Ar-H), 6.80 (d, J = 16.0 Hz, 1H, alkene hydrogen). ESI-MS: m/z = 278 [M+H]+.
A solution of (E)-3-(6-bromoquinolin-4-yl)acrylic acid (1.0 equiv), EDCI (1.5 equiv) and HOBt (1.0 equiv) in dry DCM was stirred at room temperature for 2 h. TEA (3.0 equiv) and corresponding amine (2.0 equiv) were then added, and the resultant mixture was stirred for 1 h. After this, it was successively washed with 1N NaOH and water. The organic phase was dried with magnesium sulfate and concentrated in vacuo to afford the crude product, which was further purified by column chromatography to give the desired compounds.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and tert-butylamine (53 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (98 mg, 0.30 mmol; yield 83%). 1H NMR (500 MHz, DMSO-d6): δ 8.96 (d, J = 4.5 Hz, 1H, Ar-H), 8.40 (brs, 1H, NH), 8.10–7.98 (m, 3H, Ar-H × 2 + alkene hydrogen), 7.95 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.68 (d, J = 4.5 Hz, 1H, Ar-H), 6.91 (d, J = 15.5 Hz, 1H, alkene hydrogen), 1.37 (s, 9H, CH3 × 3). ESI-MS: m/z = 333 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and 2-methoxyethan-1-amine (54 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (76 mg, 0.23 mmol; yield 64%). 1H NMR (500 MHz, DMSO-d6): δ 8.96 (d, J = 4.5 Hz, 1H, Ar-H), 8.51 (brs, 1H, NH), 8.43 (brs, 1H, Ar-H), 8.09 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.02 (d, J = 9.0 Hz, 1H, Ar-H), 7.94 (brd, J = 9.0 Hz, 1H, Ar-H), 7.72 (d, J = 4.5 Hz, 1H, Ar-H), 6.91 (d, J = 15.5 Hz, 1H, alkene hydrogen), 3.48–3.40 (m, 4H, CH2 × 2), 3.30 (s, 3H, OCH3). ESI-MS: m/z = 335 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and 2-(methylamino)ethan-1-ol (54 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (88 mg, 0.26 mmol; yield 72%). 1H NMR (500 MHz, DMSO-d6): δ 8.97 (d, J = 4.5 Hz, 1H, Ar-H), 8.40 (d, J = 2.0 Hz, 0.36H, Ar-H), 8.39 (d, J = 2.0 Hz, 0.65H, Ar-H), 8.14 (d, J = 15.5 Hz, 0.66H, alkene hydrogen), 8.10 (d, J = 15.5 Hz, 0.38H, alkene hydrogen), 8.02 (d, J = 9.0 Hz, 1H, Ar-H), 7.97–7.87 (m, 2H, Ar-H), 7.46 (d, J = 15.5 Hz, 0.63H, alkene hydrogen), 7.43 (d, J = 15.5 Hz, 0.38H, alkene hydrogen), 4.88 (t, J = 5.5 Hz, 0.68H, OH), 4.76 (t, J = 5.5 Hz, 0.37H, OH), 3.63 – 3.56 (m, 3.28H, CH2), 3.51 (t, J = 5.5 Hz, 0.70H, CH2), 3.24 (s, 1H, CH3), 3.01 (s, 2H, CH3). ESI-MS: m/z = 335 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and cyclopropanamine (41 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (93 mg, 0.29 mmol; yield 81%). 1H NMR (500 MHz, CDCl3): δ 8.95 (d, J = 4.5 Hz, 1H, Ar-H), 8.50 (brs, 1H, NH), 8.42 (d, J = 2.0 Hz, 1H, Ar-H), 8.07 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.01 (d, J = 9.0 Hz, 1H, Ar-H), 7.97–7.91 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.71 (d, J = 4.5 Hz, 1H, Ar-H), 6.77 (d, J = 15.5 Hz, 1H, alkene hydrogen), 2.82 (m, 1H, CH), 0.73 (m, 2H, CH2), 0.53 (m, 2H, CH2). ESI-MS: m/z = 317 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and cyclobutanamine (51 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (102 mg, 0.31 mmol; yield 86%). 1H NMR (500 MHz, DMSO-d6): δ 8.96 (d, J = 4.5 Hz, 1H, Ar-H), 8.64 (d, J = 7.0 Hz, 1H, NH), 8.41 (brs, 1H, Ar-H), 8.07 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.02 (d, J = 9.0 Hz, 1H, Ar-H), 7.94 (brd, J = 9.0 Hz, 1H, Ar-H), 7.71 (d, J = 4.5 Hz, 1H, Ar-H), 6.80 (d, J = 15.5 Hz, 1H, alkene hydrogen), 4.35 (m, 1H, CH), 2.31–2.18 (m, 2H, CH2), 2.03–1.92 (m, 2H, CH2), 1.77–1.63 (m, 2H, CH2). ESI-MS: m/z = 331 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and pyrrolidine (51 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (95 mg, 0.29 mmol; yield 81%). 1H NMR (500 MHz, DMSO-d6): δ 8.98 (d, J = 4.5 Hz, 1H, Ar-H), 8.40 (d, J = 2.0 Hz, 1H, Ar-H), 8.15 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.02 (d, J = 9.0 Hz, 1H, Ar-H), 7.94 (m, 2H, Ar-H), 7.26 (d, J = 15.5 Hz, 1H, alkene hydrogen), 3.70 (d, J = 6.5 Hz, 2H, CH2), 3.46 (d, J = 6.5 Hz, 2H, CH2), 1.95 (m, 2H, CH2), 1.85 (m, 2H, CH2). ESI-MS: m/z = 331 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and piperidine (61 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (113 mg, 0.33 mmol; yield 92%). 1H NMR (500 MHz, DMSO-d6): δ 8.96 (d, J = 4.5 Hz, 1H, Ar-H), 8.40 (d, J = 2.0 Hz, 1H, Ar-H), 8.13 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.01 (d, J = 9.0 Hz, 1H, Ar-H), 7.97 (d, J = 4.5 Hz, 1H, Ar-H), 7.93 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.53 (d, J = 15.5 Hz, 1H, alkene hydrogen), 3.72–3.65 (m, 2H, CH2), 3.62–3.56 (m, 2H, CH2), 1.64 (m, 2H, CH2), 1.54 (m, 4H, CH2 × 2). ESI-MS: m/z = 345 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and morpholine (63 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (79 mg, 0.23 mmol; yield 64%). 1H NMR (500 MHz, DMSO-d6): δ 8.96 (d, J = 4.5 Hz, 1H, Ar-H), 8.41 (d, J = 2.0 Hz, 1H, Ar-H), 8.17 (d, J = 15.0 Hz, 1H, alkene hydrogen), 8.00 (d, J = 9.0 Hz, 1H, Ar-H), 7.96 (d, J = 4.5 Hz, 1H, Ar-H), 7.95–7.90 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.51 (d, J = 15.0 Hz, 1H, alkene hydrogen), 3.74 (m, 2H, CH2), 3.62 (m, 6H, CH2 × 3). ESI-MS: m/z = 347 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and N-methylpiperazine (72 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (81 mg, 0.23 mmol; yield 64%). 1H NMR (500 MHz, DMSO-d6): δ 8.96 (d, J = 4.5 Hz, 1H, Ar-H), 8.39 (d, J = 2.0 Hz, 1H, Ar-H), 8.15 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.00 (d, J = 9.0 Hz, 1H, Ar-H), 7.96 (d, J = 4.5 Hz, 1H, Ar-H), 7.92 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.53 (d, J = 15.5 Hz, 1H, alkene hydrogen), 3.77–3.69 (m, 2H, CH2), 3.65–3.58 (m, 2H, CH2), 2.40–2.30 (m, 4H, CH2 × 2), 2.21 (s, 3H, CH3). ESI-MS: m/z = 360 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and 1-isopropylpiperazine (92 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (61 mg, 0.16 mmol; yield 44%). 1H NMR (500 MHz, DMSO-d6): δ 8.97 (d, J = 4.5 Hz, 1H, Ar-H), 8.41 (brs, 1H, Ar-H), 8.15 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.01 (d, J = 9.0 Hz, 1H, Ar-H), 7.97 (d, J = 4.5 Hz, 1H, Ar-H), 7.91 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.53 (d, J = 15.5 Hz, 1H, alkene hydrogen), 3.70 (m, 2H, CH2), 3.60 (m, 2H, CH2), 2.70 (m, 1H, CH), 2.47 (m, 4H, CH2 × 2), 0.99 (s, 3H, CH3), 0.98 (s, 3H, CH3). ESI-MS: m/z = 388 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and 4-hydroxypiperidine (73 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (88 mg, 0.24 mmol; yield 67%). 1H NMR (500 MHz, DMSO-d6): δ 8.94 (d, J = 4.5 Hz, 1H, Ar-H), 8.38 (d, J = 2.0 Hz, 1H, Ar-H), 8.11 (d, J = 15.5 Hz, 1H, alkene hydrogen), 7.99 (d, J = 9.0 Hz, 1H, Ar-H), 7.95 (d, J = 4.5 Hz, 1H, Ar-H), 7.91 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.52 (d, J = 15.5 Hz, 1H, alkene hydrogen), 4.79 (d, J = 4.0 Hz, 1H, OH), 4.08–3.94 (m, 2H, CH2), 3.75 (m, 1H, CH2), 3.44–3.36 (m, 1H, CH2), 3.26–3.13 (m, 1H, CH2), 1.78 (m, 2H, CH2), 1.47–1.28 (m, 2H, CH2). ESI-MS: m/z = 361 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and 3-hydroxypiperidine (73 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (96 mg, 0.27 mmol; yield 75%). 1H NMR (500 MHz, DMSO-d6): δ 8.97 (d, J = 4.0 Hz, 1H, Ar-H), 8.41 (brs, 1H, Ar-H), 8.11 (dd, J = 15.5, 6.0 Hz, 1H, alkene hydrogen), 7.98 (m, 3H, Ar-H), 7.51 (t, J = 15.5 Hz, 1H, alkene hydrogen), 5.01 (d, J = 3.5 Hz, 0.48H, OH), 4.88 (d, J = 3.5 Hz, 0.57H, OH), 4.27 (m, 0.49H, CH), 3.99 (m, 0.49H, CH), 3.80 (m, 0.56H, CH2), 3.63 (m, 1H, CH2), 3.54–3.40 (m, 1.41H, CH2), 3.27–3.12 (m, 0.63H, CH2), 2.86–2.70 (m, 0.50H, CH2), 1.75 (m, 2H, CH2), 1.41 (m, 2H, CH2). ESI-MS: m/z = 361 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and piperidin-4-ylmethanol (83 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (72 mg, 0.19 mmol; yield 53%). 1H NMR (500 MHz, DMSO-d6): δ 8.96 (brs, 1H, Ar-H), 8.41 (brs, 1H, Ar-H), 8.13 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.06–7.88 (m, 3H, Ar-H), 7.54 (d, J = 15.5 Hz, 1H, alkene hydrogen), 4.52 (m, 2H, CH2), 4.28 (d, J = 12.5 Hz, 1H, OH), 3.28 (m, 2H, CH2), 3.10 (m, 1H, CH2), 2.70 (m, 1H, CH2), 1.80–1.61 (m, 3H, CH + CH2), 1.16–1.01 (m, 2H, CH2). ESI-MS: m/z = 375 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and 2-morpholinoethan-1-amine (94 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (85 mg, 0.22 mmol; yield 61%). 1H NMR (500 MHz, DMSO-d6): δ 8.96 (d, J = 4.5 Hz, 1H, Ar-H), 8.43 (d, J = 2.0 Hz, 1H, Ar-H), 8.37 (d, J = 5.0 Hz, 1H, NH), 8.08 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.02 (d, J = 9.0 Hz, 1H, Ar-H), 7.96 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.74 (d, J = 4.5 Hz, 1H, Ar-H), 6.90 (d, J = 15.5 Hz, 1H, alkene hydrogen), 3.63–3.55 (m, 4H, CH2 × 2), 3.37 (s, 2H, CH2), 2.48–2.34 (m, 6H, CH2 × 3). ESI-MS: m/z = 390 [M+H]+.
The title intermediate was prepared from 5 (100 mg, 0.36 mmol) and aniline (67 mg, 0.72 mmol) according to the general synthetic procedure A, as a white solid (68 mg, 0.19 mmol; yield 53%). 1H NMR (500 MHz, DMSO-d6): δ 10.45 (s, 1H, NH), 9.00 (d, J = 4.5 Hz, 1H, Ar-H), 8.49 (d, J = 2.0 Hz, 1H, Ar-H), 8.26 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.04 (d, J = 9.0 Hz, 1H, Ar-H), 7.99–7.93 (dd, J = 9.0, 2.0 Hz, 1H, Ar-H), 7.79 (d, J = 4.5 Hz, 1H, Ar-H), 7.75 (d, J = 8.0 Hz, 2H, Ar-H), 7.38 (t, J = 8.0 Hz, 2H, Ar-H), 7.13 (m, 1H, Ar-H), 7.07 (d, J = 15.5 Hz, 1H, alkene hydrogen). ESI-MS: m/z = 353 [M+H]+.
To a three-neck round bottom flask was added the aryl bromide (1.0 equiv), 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide 7 (1.0 equiv), Pd(dppf)2Cl2 (0.1 equiv), K2CO3 (3.0 equiv) and dioxane/H2O (3/1, V/V). The flask was fitted with a N2 inlet adaptor and purged with N2 for 15 min. The reaction mixture was then sealed under N2 atmosphere and stirred at 100°C for 10 h. The mixture was concentrated under reduced pressure, and the residue was dissolved in DCM, washed with water twice and dried over magnesium sulfate. The crude product was purified by column chromatography to yield the desired target compound. The copies of NMR and HRMS spectra are provided in the Supplementary Material.
The title compound was prepared from 6a (53 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (27 mg, 0.049 mmol; yield 31%). 1H NMR (500 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.94 (d, J = 4.5 Hz, 1H, Ar-H), 8.50 (brs, 1H, NH), 8.31 (d, J = 2.0 Hz, 1H, Ar-H), 8.24 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.15 (d, J = 9.0 Hz, 1H, Ar-H), 8.05 (m, 3H, Ar-H), 7.83 (m, 1H, Ar-H), 7.67 (d, J = 4.5 Hz, 1H, Ar-H), 7.62 (m, 1H, Ar-H), 7.26 (m, 1H, Ar-H), 6.96 (d, J = 15.5 Hz, 1H, alkene hydrogen), 3.70 (s, 3H, OCH3), 1.37 (s, 9H, CH3 × 3). 13C NMR (100 MHz, DMSO-d6): δ 165.41, 165.07 (dd, JC−F = 254.4, 12.3 Hz), 159.34 (dd, JC−F = 257.9, 12.9 Hz), 157.59, 150.51, 147.49, 142.78, 140.69, 134.87, 133.95, 132.81, 131.88 (d, JC−F = 11.0 Hz), 130.49, 129.09, 128.94, 128.55, 125.83, 125.12 (dd, JC−F = 16.1, 3.5 Hz), 120.92, 119.91, 118.64, 111.87 (dd, JC−F = 22.0, 3.4 Hz), 105.80 (t, JC−F = 26.1 Hz), 53.42, 22.62, 5.86. HRMS (ESI) m/z calcd for C28H27F2N4O4S [M + H]+ 553.1721, found 553.1725.
The title compound was prepared from 6b (53 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (18 mg, 0.032 mmol; yield 20%). 1H NMR (500 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.94 (d, J = 4.5 Hz, 1H, Ar-H), 8.53 (brs, 1H, Ar-H), 8.50 (t, J = 5.5 Hz, 1H, NH), 8.35 (d, J = 1.5 Hz, 1H, Ar-H), 8.30 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.16 (d, J = 9.0 Hz, 1H, Ar-H), 8.10–8.04 (m, 2H, Ar-H), 7.82 (m, 1H, Ar-H), 7.72 (d, J = 4.5 Hz, 1H, Ar-H), 7.60 (m, 1H, Ar-H), 7.25 (td, J = 8.5, 2.5 Hz, 1H, Ar-H), 6.96 (d, J = 15.5 Hz, 1H, alkene hydrogen), 3.70 (s, 3H, OCH3), 3.45 (m, 4H, CH2 × 2), 3.30 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6): δ 165.08 (dd, JC−F = 254.0, 11.7 Hz), 164.40, 159.34 (dd, JC−F = 257.5, 13.6 Hz), 157.57, 150.57, 147.50, 142.87, 140.65, 134.86, 133.95, 133.04, 131.88 (d, JC−F = 11.4 Hz), 130.50, 129.10, 129.06, 128.53, 125.82, 125.06 (dd, JC−F = 14.8, 4.0 Hz), 120.91, 119.82, 118.66, 111.87 (dd, JC−F = 25.6, 3.9 Hz), 105.80 (t, JC−F = 26.4 Hz), 70.57, 57.89, 53.42, 38.72. HRMS (ESI) m/z calcd for C27H25F2N4O5S [M + H]+ 555.1513, found 555.1510.
The title compound was prepared from 6c (53 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (20 mg, 0.036 mmol; yield 23%). 1H NMR (500 MHz, DMSO-d6): δ 8.90 (d, J = 4.5 Hz, 1H, Ar-H), 8.30 (d, J = 15.5 Hz, 0.44H, alkene hydrogen), 8.26 (d, J = 15.5 Hz, 0.56H, alkene hydrogen), 8.13 (brs, 1H, Ar-H), 8.10 (d, J = 8.5 Hz, 1H, Ar-H), 8.01–7.84 (m, 4H, Ar-H), 7.70 (brs, 1H, Ar-H), 7.52 (d, J = 15.5 Hz, 0.45H, alkene hydrogen), 7.49 (d, J = 15.5 Hz, 0.56H, alkene hydrogen), 7.29 (brs, 1H, Ar-H), 7.21 (m, 1H, Ar-H), 4.92 (brs, 0.62H, OH), 4.78 (t, J = 5.5 Hz, 0.44H, OH), 3.79 (s, 3H, OCH3), 3.63 (m, 3.22H, CH2), 3.53 (t, J = 6.0 Hz, 0.80H, CH2), 3.28 (s, 1H, CH3), 3.04 (s, 2H, CH3). HRMS (ESI) m/z calcd for C27H25F2N4O5S [M + H]+ 555.1513, found 555.1516.
The title compound was prepared from 6d (50 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (18 mg, 0.034 mmol; yield 21%). 1H NMR (500 MHz, CDCl3): δ 8.84 (brs, 1H, NH), 8.27 (d, J = 15.0 Hz, 1H, alkene hydrogen), 8.18–8.08 (m, 3H, Ar-H), 7.99 (d, J = 2.0 Hz, 1H, Ar-H), 7.91 (m, 1H, Ar-H), 7.82 (d, J = 8.5 Hz, 1H, Ar-H), 7.45 (brs, 1H, Ar-H), 7.28 (brs, 1H, Ar-H), 7.07 (m, 1H, Ar-H), 6.90 (m, 1H, Ar-H), 6.54 (d, J = 15.0 Hz, 1H, alkene hydrogen), 5.99 (brs, 1H, NH), 3.91 (s, 3H, OCH3), 2.87 (m, 1H, CH), 0.82 (m, 2H, CH2), 0.58 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6): δ 165.10 (dd, JC−F = 253.7, 12.0 Hz), 163.56, 159.35 (dd, JC−F = 257.6, 13.3 Hz), 157.51, 150.49, 147.53, 142.81, 140.84, 134.82, 133.81, 132.21, 131.91 (d, JC−F = 10.5 Hz), 130.49, 130.44, 129.11, 128.52, 125.86, 125.05 (dd, JC−F = 14.2, 3.7 Hz), 120.92, 119.82, 118.50, 111.89 (dd, JC−F = 22.1, 3.2 Hz), 105.84 (t, JC−F = 25.6 Hz), 53.42, 50.44, 28.45. HRMS (ESI) m/z calcd for C27H23F2N4O4S [M + H]+ 537.1408, found 537.1402.
The title compound was prepared from 6e (53 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (25 mg, 0.045 mmol; yield 28%). 1H NMR (500 MHz, DMSO-d6): δ 10.35 (s, 1H, NH), 8.94 (d, J = 4.5 Hz, 1H, Ar-H), 8.64 (d, J = 7.5 Hz, 1H, NH), 8.54 (d, J = 2.0 Hz, 1H, Ar-H), 8.34 (d, J = 1.5 Hz, 1H, Ar-H), 8.28 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.16 (d, J = 9.0 Hz, 1H, Ar-H), 8.08 (m, 2H, Ar-H), 7.82 (m, 1H, Ar-H), 7.71 (d, J = 4.5 Hz, 1H, Ar-H), 7.60 (m, 1H, Ar-H), 7.25 (m, 1H, Ar-H), 6.85 (d, J = 15.5 Hz, 1H, alkene hydrogen), 4.44–4.30 (m, 1H, CH), 3.70 (s, 3H, OCH3), 2.26 (m, 2H, CH2), 1.98 (m, 2H, CH2), 1.71 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6): δ 165.08 (dd, JC−F = 253.9, 11.6 Hz), 163.14, 159.34 (dd, JC−F = 257.5, 13.5 Hz), 157.57, 150.51, 147.51, 142.85, 140.73, 134.85, 133.97, 133.10, 131.88 (d, JC−F = 10.1 Hz), 130.50, 129.13, 129.11, 128.53, 125.82, 125.09 (dd, JC−F = 14.8, 3.3 Hz), 120.94, 119.82, 118.63, 111.86 (dd, JC−F = 22.5, 3.7 Hz), 105.81 (t, JC−F = 25.8 Hz), 53.42, 44.18, 30.27, 14.80. HRMS (ESI) m/z calcd for C28H25F2N4O4S [M + H]+ 551.1564, found 551.1571.
The title compound was prepared from 6f (53 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (21 mg, 0.038 mmol; yield 24%). 1H NMR (500 MHz, DMSO-d6): δ 10.37 (s, 1H, NH), 8.94 (d, J = 4.0 Hz, 1H, Ar-H), 8.52 (s, 1H, Ar-H), 8.34 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.33 (brs, 1H, Ar-H), 8.16 (d, J = 8.5 Hz, 1H, Ar-H), 8.08 (m, 2H, Ar-H), 7.91 (d, J = 4.0 Hz, 1H, Ar-H), 7.83 (m, 1H, Ar-H), 7.60 (m, 1H, Ar-H), 7.28 (d, J = 15.5 Hz, 1H, alkene hydrogen), 7.27 (m, 1H, Ar-H), 3.79–3.63 (m, 5H, OCH3 + CH2), 3.48 (t, J = 6.5 Hz, 2H, CH2), 1.94 (m, 2H, CH2), 1.89–1.81 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6): δ 165.09 (dd, JC−F = 254.5, 10.5 Hz), 162.87, 159.34 (dd, JC−F = 257.7, 13.2 Hz), 157.52, 150.39, 147.50, 142.76, 140.65, 134.81, 134.15, 133.78, 131.90 (d, JC−F = 11.0 Hz), 130.49, 129.07, 128.39, 127.15, 125.86, 125.07 (dd, JC−F = 14.5, 2.4 Hz), 120.80, 119.84, 119.03, 111.88 (dd, JC−F = 22.5, 2.6 Hz), 105.80 (t, JC−F = 26.6 Hz), 53.40, 46.18, 45.77, 25.60, 23.84. HRMS (ESI) m/z calcd for C28H25F2N4O4S [M + H]+ 551.1564, found 551.1568.
The title compound was prepared from 6g (55 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (12 mg, 0.021 mmol; yield 13%). 1H NMR (500 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.94 (d, J = 4.5 Hz, 1H, Ar-H), 8.53 (d, J = 2.5 Hz, 1H, Ar-H), 8.35 (s, 1H, Ar-H), 8.33 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.15 (d, J = 8.5 Hz, 1H, Ar-H), 8.11–8.05 (m, 2H, Ar-H), 7.96 (d, J = 4.5 Hz, 1H, Ar-H), 7.82 (m, 1H, Ar-H), 7.63–7.58 (m, 1H, Ar-H), 7.55 (d, J = 15.5 Hz, 1H, alkene hydrogen), 7.26 (m, 1H, Ar-H), 3.69 (s, 5H, OCH3 + CH2), 3.63–3.56 (m, 2H, CH2), 1.64 (m, 2H, CH2), 1.55 (m, 4H, CH2 × 2). 13C NMR (100 MHz, DMSO-d6): δ 165.09 (dd, JC−F = 252.6, 12.1 Hz), 163.75, 159.35 (dd, JC−F = 257.7, 13.0 Hz), 157.48, 150.33, 147.53, 142.70, 140.62, 134.78, 134.58, 133.63, 131.89 (d, JC−F = 10.8 Hz), 130.48, 129.09, 128.32, 125.96, 125.85, 125.11 (dd, JC−F = 14.5, 4.1 Hz), 120.74, 119.89, 118.87, 111.85 (dd, JC−F = 22.2, 3.0 Hz), 105.77 (t, JC−F = 26.0 Hz), 53.38, 46.34, 42.63, 26.44, 25.35, 24.06. HRMS (ESI) m/z calcd for C29H27F2N4O4S [M + H]+ 565.1721, found 565.1725.
The title compound was prepared from 6h (55 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (19 mg, 0.034 mmol; yield 21%). 1H NMR (500 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.95 (d, J = 4.5 Hz, 1H, Ar-H), 8.53 (d, J = 2.0 Hz, 1H, Ar-H), 8.39 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.35 (d, J = 1.5 Hz, 1H, Ar-H), 8.15 (d, J = 8.5 Hz, 1H, Ar-H), 8.10–8.05 (m, 2H, Ar-H), 7.95 (d, J = 4.5 Hz, 1H, Ar-H), 7.82 (m, 1H, Ar-H), 7.62–7.57 (m, 1H, Ar-H), 7.54 (d, J = 15.5 Hz, 1H, alkene hydrogen), 7.25 (td, J = 8.5, 2.5 Hz, 1H, Ar-H), 3.77 (m, 2H, CH2), 3.69 (s, 3H, OCH3), 3.65 (s, 6H, CH2 × 3). 13C NMR (100 MHz, DMSO-d6): δ 165.07 (dd, JC−F = 253.9, 12.1 Hz), 164.03, 159.34 (dd, JC−F = 256.5, 14.3 Hz), 157.57, 150.36, 147.53, 142.84, 140.49, 135.29, 134.81, 133.98, 131.88 (d, JC−F = 10.7 Hz), 130.49, 129.08, 128.41, 125.83, 125.24, 125.10 (dd, JC−F = 14.6, 3.7 Hz), 120.80, 119.83, 118.97, 111.86 (dd, JC−F = 22.1, 3.4 Hz), 105.79 (t, JC−F = 26.2 Hz), 66.36, 66.11, 53.40, 45.82, 42.15. HRMS (ESI) m/z calcd for C28H25F2N4O5S [M + H]+ 567.1513, found 567.1514.
The title compound was prepared from 6i (57 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (22 mg, 0.038 mmol; yield 24%). 1H NMR (500 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.94 (d, J = 4.5 Hz, 1H, Ar-H), 8.49 (d, J = 2.5 Hz, 1H, Ar-H), 8.36 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.34 (d, J = 2.0 Hz, 1H, Ar-H), 8.15 (d, J = 8.5 Hz, 1H, Ar-H), 8.08 (dd, J = 8.5, 2.0 Hz, 1H, Ar-H), 8.05 (d, J = 2.5 Hz, 1H), 7.96 (d, J = 4.5 Hz, 1H, Ar-H), 7.83 (m, 1H, Ar-H), 7.58 (m, 1H, Ar-H), 7.58 (d, J = 15.5 Hz, 1H, alkene hydrogen), 7.25 (td, J = 8.5, 2.5 Hz, 1H, Ar-H), 3.76 (m, 2H, CH2), 3.70 (s, 3H, OCH3), 3.65 (m, 2H, CH2), 2.45–2.35 (m, 4H, CH2 × 2), 2.25 (s, 3H, CH3). HRMS (ESI) m/z calcd for C29H28F2N5O4S [M + H]+ 580.1830, found 580.1819.
The title compound was prepared from 6j (62 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (25 mg, 0.041 mmol; yield 26%). 1H NMR (500 MHz, DMSO-d6): δ 10.34 (s, 1H, NH), 8.94 (d, J = 4.5 Hz, 1H, Ar-H), 8.48 (s, 1H), 8.35 (d, J = 15.5 Hz, 2H, Ar-H + alkene hydrogen), 8.15 (d, J = 9.0 Hz, 1H, Ar-H), 8.11–8.03 (m, 2H, Ar-H), 7.95 (d, J = 4.5 Hz, 1H, Ar-H), 7.82 (m, 1H, Ar-H), 7.56 (m, 2H, Ar-H + alkene hydrogen), 7.24 (m, 1H, Ar-H), 3.74 (m, 2H, CH2), 3.69 (s, 3H, OCH3), 3.64 (m, 2H, CH2), 2.74 (m, 1H, CH), 2.52 (m, 4H, CH2 × 2), 1.01 (s, 3H, CH3), 0.99 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6): δ 167.39 (dd, JC−F = 242.7, 13.7 Hz), 163.75, 159.31 (dd, JC−F = 255.8, 14.5 Hz), 157.52, 150.33, 147.50, 142.10, 140.52, 135.09, 134.94, 133.20, 131.86 (d, JC−F = 11.0 Hz), 130.47, 129.01, 128.40, 125.82, 125.52, 120.70, 118.96, 111.98–111.58 (dd, JC−F = 22.2, 4.4 Hz), 105.72 (t, JC−F = 25.3 Hz), 53.89, 53.32, 48.66, 47.76, 45.47, 41.90, 17.90. HRMS (ESI) m/z calcd for C31H32F2N5O4S [M + H]+ 608.2143, found 608.2135.
The title compound was prepared from 6k (58 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (25 mg, 0.043 mmol; yield 27%). 1H NMR (500 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.94 (d, J = 4.5 Hz, 1H, Ar-H), 8.54 (d, J = 2.0 Hz, 1H, Ar-H), 8.34 (d, J = 15.0 Hz, 1H, alkene hydrogen), 8.36 (d, J = 1.5 Hz, 1H, Ar-H), 8.15 (d, J = 8.5 Hz, 1H, Ar-H), 8.10–8.06 (m, 2H, Ar-H), 7.97 (d, J = 4.5 Hz, 1H, Ar-H), 7.85–7.79 (m, 1H, Ar-H), 7.64–7.59 (m, 1H, Ar-H), 7.57 (d, J = 15.0 Hz, 1H, alkene hydrogen), 7.25 (td, J = 8.5, 2.0 Hz, 1H, Ar-H), 4.80 (brs, 1H, OH), 4.04 (m, 2H, CH2), 3.77 (m, 1H, CH), 3.69 (s, 3H, OCH3), 3.45–3.39 (m, 1H, CH2), 3.24 (m, 1H, CH2), 1.79 (m, 2H, CH2), 1.44–1.35 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6): δ 165.14 (dd, JC−F = 252.7, 11.8 Hz), 163.78, 159.35 (dd, JC−F = 257.5, 13.5 Hz), 157.56, 150.37, 147.53, 142.88, 140.59, 134.77, 133.98, 131.89 (d, JC−F = 10.8 Hz), 130.48, 129.11, 128.39, 125.87, 125.85, 125.06 (dd, JC−F = 14.5, 3.8 Hz), 120.80, 119.76, 118.91, 111.87 (dd, JC−F = 22.1, 3.4 Hz), 105.80 (t, JC−F = 26.1 Hz), 65.45, 53.40, 42.83, 41.28, 34.94, 33.90. HRMS (ESI) m/z calcd for C29H27F2N4O5S [M + H]+ 581.1670, found 581.1666.
The title compound was prepared from 6l (58 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (21 mg, 0.036 mmol; yield 23%). 1H NMR (500 MHz, DMSO-d6): δ 10.35 (s, 1H, NH), 8.95 (d, J = 4.5 Hz, 1H, Ar-H), 8.53 (brs, 1H, Ar-H), 8.33 (d, J = 14.0 Hz, 2H, Ar-H + alkene hydrogen), 8.15 (d, J = 8.5 Hz, 1H, Ar-H), 8.08 (brd, J = 8.0 Hz, 2H, Ar-H), 8.03–7.92 (m, 1H, Ar-H), 7.83 (m, 1H, Ar-H), 7.63–7.49 (m, 2H, Ar-H + alkene hydrogen), 7.25 (m, 1H, Ar-H), 4.99 (d, J = 4.5 Hz, 0.52H, OH), 4.88 (d, J = 4.0 Hz, 0.51H, OH), 4.30 (m, 0.53H, CH), 4.02 (m, 0.53H, CH), 3.83 (m, 0.57H, CH2), 3.69 (s, 2H, OCH3), 3.62 (m, 1H, CH2), 3.57 (s, 1H, OCH3), 3.45 (m, 1.56H, CH2), 3.22 (m, 0.63H, CH2), 2.84–2.72 (m, 0.52H, CH2), 1.84 (m, 2H, CH2), 1.41 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6): δ 165.55 (dd, JC−F = 254.3, 11.7 Hz), 164.80, 164.46, 159.84 (dd, JC−F = 257.6, 13.5 Hz), 158.04, 150.87, 148.02, 143.21, 141.19, 141.07, 135.30, 134.89, 134.31, 132.40 (d, JC−F = 10.5 Hz), 130.98, 130.12, 130.07, 128.90, 126.65, 126.46, 126.34, 125.63 (dd, JC−F = 14.3, 2.7 Hz), 121.27, 120.46, 119.43, 119.28, 112.35 (dd, JC−F = 22.0, 3.2 Hz), 106.29 (t, JC−F = 26.1 Hz), 66.82, 65.65, 53.89, 52.90, 49.53, 45.98, 42.65, 33.54, 32.74, 25.40, 25.07, 24.36, 22.27. HRMS (ESI) m/z calcd for C29H27F2N4O5S [M + H]+ 581.1670, found 581.1668.
The title compound was prepared from 6m (60 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (28 mg, 0.047 mmol; yield 29%). 1H NMR (500 MHz, DMSO-d6): δ 10.35 (s, 1H, NH), 8.93 (d, J = 4.5 Hz, 1H, Ar-H), 8.54 (d, J = 2.0 Hz, 1H, Ar-H), 8.39–8.31 (m, 2H, Ar-H + alkene hydrogen), 8.15 (d, J = 9.0 Hz, 1H, Ar-H), 8.11–8.05 (m, 2H, Ar-H), 7.96 (d, J = 4.5 Hz, 1H, Ar-H), 7.81 (m, 1H, Ar-H), 7.57 (m, 2H, Ar-H + alkene hydrogen), 7.25 (m, 1H, Ar-H), 4.53 (m, 2H, CH2), 4.31 (d, J = 12.5 Hz, 1H, OH), 3.69 (s, 3H, OCH3), 3.28 (m, 2H, CH2), 3.13 (m, 1H, CH2), 2.71 (m, 1H, CH2), 1.76 (m, 3H, CH + CH2), 1.13 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6): δ 165.07 (dd, JC−F = 254.4, 12.2 Hz), 163.74, 159.34 (dd, JC−F = 257.6, 13.1 Hz), 157.55, 150.38, 147.52, 142.82, 140.61, 134.77, 134.72, 133.93, 131.89 (d, JC−F = 11.9 Hz), 130.48, 129.09, 128.39, 125.96, 125.84, 125.07 (dd, JC−F = 14.3, 4.5 Hz), 120.80, 119.82, 118.92, 111.86 (dd, JC−F = 22.0, 3.4 Hz), 105.80 (t, JC−F = 25.5 Hz), 65.46, 53.40, 45.35, 41.77, 38.41, 29.55, 28.42. HRMS (ESI) m/z calcd for C30H29F2N4O5S [M + H]+ 595.1827, found 595.1820.
The title compound was prepared from 6n (62 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (15 mg, 0.025 mmol; yield 16%). 1H NMR (500 MHz, DMSO-d6): δ 10.32 (s, 1H, NH), 8.94 (d, J = 4.5 Hz, 1H, Ar-H), 8.51 (brs, 1H, NH), 8.35 (m, 2H, Ar-H), 8.28 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.15 (d, J = 9.0 Hz, 1H, Ar-H), 8.07 (m, 2H, Ar-H), 7.82 (m, 1H, Ar-H), 7.73 (d, J = 4.5 Hz, 1H, Ar-H), 7.59 (m, 1H, Ar-H), 7.25 (m, 1H, Ar-H), 6.95 (d, J = 15.5 Hz, 1H, alkene hydrogen), 3.70 (s, 3H, OCH3), 3.59 (m, 4H, CH2 × 2), 3.39 (m, 2H, CH2), 2.44 (m, 6H, CH2 × 3). HRMS (ESI) m/z calcd for C30H30F2N5O5S [M + H]+ 610.1936, found 610.1913.
The title compound was prepared from 6o (56 mg, 0.16 mmol) and 7 (68 mg, 0.16 mmol) according to the general synthetic procedure B, as an off-white solid (15 mg, 0.026 mmol; yield 16%). 1H NMR (500 MHz, CDCl3): δ 10.44 (s, 1H, NH), 10.35 (s, 1H, NH), 8.97 (d, J = 4.5 Hz, 1H, Ar-H), 8.51 (brs, 1H, Ar-H), 8.46 (d, J = 15.5 Hz, 1H, alkene hydrogen), 8.40 (brs, 1H, Ar-H), 8.17 (d, J = 9.0 Hz, 1H, Ar-H), 8.09 (d, J = 9.0 Hz, 2H, Ar-H), 7.78 (m, 4H, Ar-H), 7.57 (m, 1H, Ar-H), 7.37 (t, J = 8.0 Hz, 2H, Ar-H), 7.24 (m, 1H, Ar-H), 7.15–7.07 (m, 2H, Ar-H + alkene hydrogen), 3.68 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6): δ 165.03 (dd, JC−F = 254.0, 11.8 Hz), 162.82, 159.35 (dd, JC−F = 257.3, 13.7 Hz), 157.60, 150.50, 147.56, 142.72, 140.47, 138.98, 135.00, 134.50, 133.87, 131.86 (d, JC−F = 10.6 Hz), 130.51, 129.23, 129.08, 128.83, 128.58, 125.82, 125.23 (dd, JC−F = 13.5, 4.9 Hz), 123.69, 120.97, 120.09, 119.42, 118.75, 111.81 (dd, JC−F = 22.1, 3.1 Hz), 105.76 (t, JC−F = 26.0 Hz), 53.39. HRMS (ESI) m/z calcd for C30H23F2N4O4S [M + H]+ 573.1408, found 573.1402.
The title compound was prepared from 6-bromoquinoline (25 mg, 0.12 mmol) and 7 (50 mg, 0.12 mmol) according to the general synthetic procedure B as an off-white solid (23 mg, 0.054 mmol; yield 45%). 1H NMR (500 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 8.93 (dd, J = 4.0, 1.5 Hz, 1H, Ar-H), 8.51 (d, J = 2.0 Hz, 1H, Ar-H), 8.45 (dd, J = 8.5, 1.0 Hz, 1H, Ar-H), 8.27 (d, J = 2.0 Hz, 1H, Ar-H), 8.12 (d, J = 9.0 Hz, 1H, Ar-H), 8.06 (m, 2H, Ar-H), 7.79 (m, 1H, Ar-H), 7.62–7.56 (m, 2H, Ar-H), 7.23 (m, 1H, Ar-H), 3.68 (s, 3H, OCH3). 13C NMR (125 MHz, DMSO-d6): δ 165.56 (dd, JC−F = 252.5, 11.8 Hz), 159.88 (dd, JC−F = 256.1, 13.4 Hz), 158.13, 151.23, 147.58, 143.16, 136.73, 134.78, 134.57, 132.36 (d, JC−F = 16.0 Hz), 130.24, 129.37, 128.65, 128.56, 126.65, 125.56 (dd, JC−F = 13.8, 3.6 Hz), 122.46, 120.22, 112.31 (dd, JC−F = 22.1, 3.1 Hz), 106.27 (t, JC−F = 25.8 Hz), 53.90. ESI-MS: m/z = 428 [M+H] +.
Kinase-Glo Luminescent Kinase assay (Promega) was performed to determine the PI3Kα inhibitory activity, while ADP-Glo Luminescent assay (Promega) was performed to identify the inhibitory activity against PI3Kβ, γ, or δ. The kinase buffer was composed of HEPES (50 mM, pH 7.5), MgCl2 (3 mM), EGTA (1 mM), NaCl (100 mM), CHAPS (0.03%) and DTT (2 mM). The serially diluted compound solutions (2.5 mL) were successively added to a 384-well plate. Afterwards, the solution of kinase (2.5 mL) in the kinase buffer and the solution of substrate (5 mL) in the kinase buffer containing PIP2 and ATP were added to each well of the assay plate for starting the reaction. Kinase buffer was added instead to the control well without enzyme. The final concentrations of PI3Kα, PI3Kβ, PI3Kγ, PI3Kδ, PIP2, and ATP in corresponding kinase reaction mixture were 1.65 nM, 4.8 nM, 7.6 nM, 5.7 nM, 50 mM and 25 mM, respectively. After being incubated at room temperature for 1 h, Kinase-Glo reagent (10 mL) was added to each well of the PI3Kα assay plate to quench the reaction. Subsequently, the mixture needs to be mixed briefly with centrifuge and shaken slowly for 15 min before being read on a plate reader for luminescence. As for PI3Kβ, γ, or δ, reaction mixture (5 mL) was transferred to a new 384-plate and ADP-Glo reagent (5 mL) was added for stopping the reaction. The mixture was treated briefly with centrifuge, shaken slowly, and equilibrated for 40 min. Kinase Detection reagent (10 mL) was added to each well. Before reading on a plate reader for luminescence, the resultant mixture was incubated at room temperature for 1 h. After calculating the percent inhibition from the RLU values, the curves were fitted in Graphpad Prism 5 to give IC50 values.
Lance Ultra assay was performed to determine the mTOR (1362-end, Millipore) inhibitory activity. The kinase buffer was composed of HEPES (50 mM, pH 7.5), MgCl2 (10 mM), EGTA (1 mM), MnCl2 (3 mM), Tween-20 (0.01%) and DTT (2 mM). The solution of substrate in the kinase buffer contained ULight-4E-BP1 peptide (Thr37/46, PE) and ATP. The kinase reaction mixture was prepared in a similar way to that for class I PI3K inhibition assays. The final concentrations of mTOR, ULight-4E-BP1 peptide and ATP in the kinase reaction mixture were 2.5 nM, 50 mM and 10.8 mM, respectively. After being incubated at room temperature for 1 h, kinase detection buffer (10 mL) containing EDTA and Eu-anti-phospho-4E-BP1 antibody (Thr37/46, PE) was added to each well of the assay plate. Before reading on a plate reader, the mixture needs to be mixed briefly with centrifuge and then allowed to equilibrate for 1 h. After calculating the percent inhibition from Lance signal, the curves were fitted in Graphpad Prism 5 to give IC50 values.
The anti-proliferative activity of compounds was evaluated by sulforhodamin B (SRB) assay. After being cultured for 10 h, cells in 96-well plates were subjected to 72 h-exposure to serially diluted compound solutions. Cells were washed with PBS and fixed with trichloroacetic acid (10%, W/V) at 4°C for 1 h. Then, cells were successively washed with PBS, stained for 0.5 h with SRB (0.4%, dissolved in 1% acetic acid), and washed by 1% acetic acid for 5 times. Protein-bound dye was extracted with unbuffered Tris base (10 mmol). After calculating the percent inhibition from absorbance, measured at 515 nm with a multiscan spectrum (Thermo Electron Co., Vantaa, Finland), the curves were fitted in Graphpad Prism 5 to give GI50 values.
The Western blot analysis for evaluating the capability to down-regulate phos-Akt (Ser473), phos-Akt (Thr308), phos-S6 ribosomal protein (Ser235/236), and phos-4E-BP1 (Thr37/46) in PC3 cells was performed according to the protocol disclosed in our previous study (Zhang et al., 2017a) with a minor modification. The cells were seeded into six-well plate at 1 × 106 cells per well, and then incubated at 37°C (5% CO2) overnight prior to drug treatment. Cells were treated with 8i and GSK2126458 at various concentrations and incubated at 37°C for 3 h.
The HLM and SD rat RLM utilized for this experiment were purchased from Corning and Xenotech, respectively. An appropriate amount of NADPH powder was weighed and dissolved in MgCl2 solution (10 mM). The microsome working solutions were prepared with potassium phosphate buffer (100 mM). Cold acetonitrile containing tolbutamide (100 ng/mL) and labetalol (100 ng/mL) as internal standard was used as the stop solution. Compound or control working solution (10 μL/well) was added to all plates (T0, T5, T10, T20, T30, T60, NCF60) except matrix blank. After microsome solution was dispensed to every plate (80 μL/well), the mixture was incubated at 37°C for about 10 min. Subsequently, potassium phosphate buffer (100 mM) was added to the plate NCF60 (10 μL/well), and the mixture was incubated at 37°C. As for other plates, the pre-warmed NADPH solution (10 μL/well) was added to start the reaction. At each time point, the stop solution (precooled in 4°C) was added to terminate the reaction (300 μL/well). After shaking and centrifuging, 100 μL supernatant was mixed with an appropriate amount of water for LC/MS/MS analysis.
SD rats were utilized for the PK study of 8i following oral gavage at the dosage of 5 mg/kg, and the oral dose was formulated in a homogenous opaque suspension of 0.5% methylcellulose. This experiment was performed with the protocol disclosed in our previous work (Zhang et al., 2017b). The study was carried out in accordance with institutional guidelines of the Animal Research Committee at Jiaxing University (log number JXU2015120812). The protocol was approved by the institution.
The co-crystal structure of PI3Kα in complex with BYL-719 (PDB code 4JPS) was selected as the template for the molecular docking analysis, which was conducted using Discovery Studio (Version 2.1). For preparing the ligands, 3D structures were generated, and their energy minimization performed. For preparing the receptor, the hydrogen atoms were added, and the CHARMm-force field was employed. The site sphere was chosen based on the binding location of BYL-719 in PI3Kα. BYL-719 was removed and 8i, as the ligand, was docked into the defined site. After analyzing the types of interactions between 8i and PI3Kα, the final binding conformation was determined according to the calculated energy.
The synthetic route for 4-acrylamido-quinolines 8a–o and C-4 unsubstituted quinoline derivative 10 is outlined in Scheme 1. The reaction of 4-bromoaniline 1 with methyl vinyl ketone in the presence of FeCl3 and ZnCl2 provided 4-methyl-6-bromoquinoline 2 (Phanumartwiwath et al., 2013). Following oxidation of 2 with SeO2 (Perez-Melero et al., 2004), the newly formed 6-bromoquinoline-4-carbaldehyde 3 was treated with triethyl phosphonoacetate, thereby leading to the generation of ethyl (E)-3-(6-bromoquinolin-4-yl)acrylate 4 (Lalonde et al., 2013). Afterwards, hydrolysis of 4 afforded (E)-3-(6-bromoquinolin-4-yl)acrylic acid 5, which was treated with corresponding amine in the presence of EDCI and HOBt to give the quinoline derivatives 6a-o. Subsequent Suzuki coupling of 6a–o with the borate 7, which was prepared with a procedure disclosed in our previous work (Lv et al., 2015), yielded corresponding target compounds 8a–o. Similarly, compound 10 was prepared by Suzuki coupling of 9 with 7.

Scheme 1. The synthetic route for target compounds 8a-o and 10. Reagents and conditions: (a) methyl vinyl ketone, FeCl3, CH3COOH, 70°C, 3 h; ZnCl2, reflux, 2 h; (b) SeO2, dioxane/H2O, 100°C, 2 h; (c) NaH, triethyl phosphonoacetate, THF, 0°C to rt, 1.5 h; (d) 2.5 N NaOH, rt, 2 h; 2 N HCl; (e) EDCI, HOBt, DCM, rt, 2 h; corresponding amine, TEA, 1 h, rt; (f) Pd(dppf)2Cl2, K2CO3, dioxane/H2O, 100°C, 10 h.
To validate the design rationale for the target compounds, they were evaluated for the PI3Kα inhibitory activity, as well as anti-proliferative efficacy against prostate cancer PC3 and colorectal cancer HCT116 cell lines (Table 1). According to the experimental results of the PI3Kα enzymatic assay, all the quinolines bearing acrylamides as the C-4 replacements exhibited potent PI3Kα inhibitory activity with IC50 values below 3 nM. Among them, compound 8i exerted the most potent PI3Kα inhibitory activity (IC50 = 0.5 nM), which was comparable to that of GSK2126458, the clinically investigated PI3K/mTOR dual inhibitor. Importantly, compounds 8a-o with various C-4 acrylamide substructures displayed over 7-fold improvement in the enzymatic activity compared to the C-4 unsubstituted counterpart 10, suggesting that the acrylamide replacements played an important role in boosting PI3Kα inhibitory activity. The structure-activity relationship was consistent with our docking analysis, which demonstrated that acrylamide substructure introduced at the C-4 position of the quinoline template had the potential to probe Gln859 via H-bond interaction, as exemplified by the modeling of 8i, a representative throughout this series, into the currently available ATP-binding pocket of PI3Kα (PDB code 4JPS) (Figure 2).
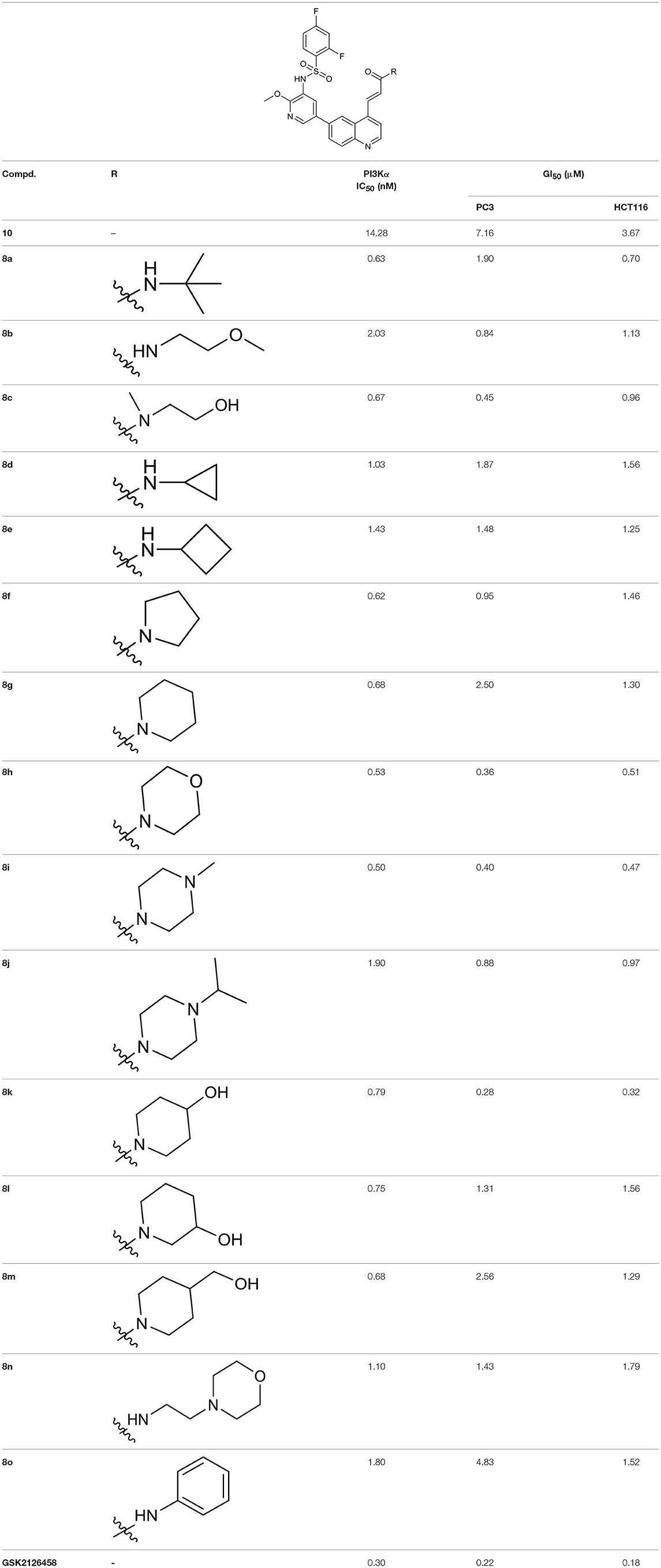
Table 1. PI3Kα inhibitory activity and anti-proliferative activity of compounds 8a-o and 10.
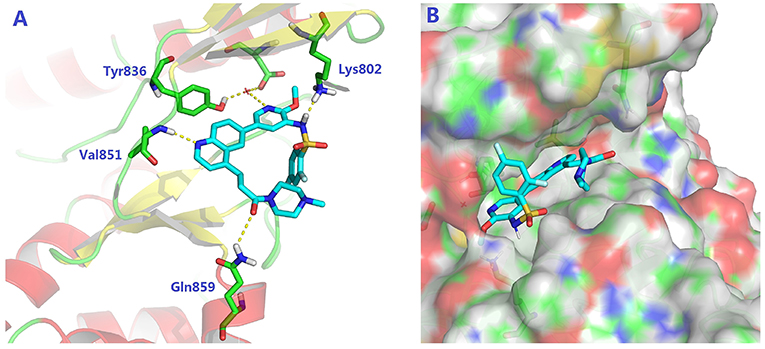
Figure 2. The molecular docking of compound 8i into PI3Kα, indicating the interaction of C-4 acrylamide substructure with Gln859. (A) the binding mode of 8i with the catalytic site; (B) the binding mode with surface area highlighted.
As illustrated by the results of the anti-proliferative assay (Table 1), all the 4-acrylamido-quinoline derivatives displayed sub-micromolar to low micromolar anti-proliferative activity against both PC3 and HCT116 cell lines. Four compounds in particular (8c, 8h, 8i, and 8k) exerted remarkable anti-proliferative activity against PC3 cell line with GI50 values below 0.5 μM, which was comparable to that of the positive reference GSK2126458. Besides, they also exhibited sub-micromolar GI50 values against HCT116 cell line.
Considering their favorable enzymatic and cellular activities, 8c and 8i, as the representatives throughout this series, were then evaluated for the inhibitory activity against other class I PI3Ks and mTOR with GSK2126458 as the positive control. As shown in Table 2, both compounds displayed strong inhibitory activity with IC50 values against all the tested enzymes ranging from 0.50 to 1.5 nM. This result suggested that compounds 8c and 8i were potent PI3K/mTOR dual inhibitors.
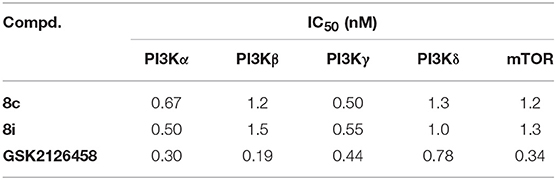
Table 2. Enzymatic activity of compounds 8c and 8i against PI3Ks and mTOR.
Subsequently, compound 8i was investigated for its capability to down-regulate the levels of phos-Akt (Ser473), phos-Akt (Thr308), phos-S6 ribosomal protein (Ser235/236), and phos-4E-BP1 (Thr37/46), four important biomarkers of PAM signaling, in PC3 cells. The suppressive effect of 8i was evaluated at the concentrations of 5, 25, 125, and 625 nM. As illustrated in Figure 3, at the concentration as low as 5 nM, 8i remarkably down-regulated all the four monitored biomarkers. In particular, the effect on the level of phos-Akt (Ser473) was stronger than that of GSK2126458. These results further demonstrated 8i can strongly down-regulate PAM signaling. Moreover, the concurrent down-regulation of the four biomarkers indicated the potential to fulfill synergism, as well as the advantage over mono-inhibition of mTOR in conquering the negative feedback loop.
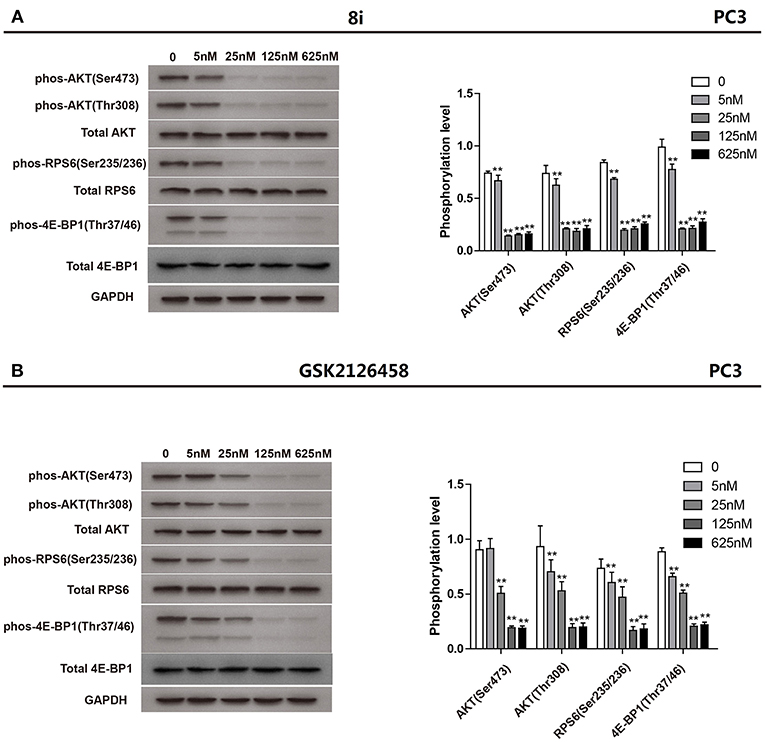
Figure 3. The suppressive effect of 8i (A) and GSK2126458 (B) on the phosphorylation of Akt, S6 Ribosomal protein, and 4E-BP1 in PC3 cells following 3 h-treatment: The levels of phos-Akt (Ser473), phos-Akt (Thr308), phos-S6 ribosomal protein (Ser235/236), and phos-4E-BP1 (Thr37/46) in different groups were determined via Western blot assay. The bar chart represented the quantification of the bands in the Western blot with the results shown as mean ± SD (n = 3 biological replicates). **p < 0.01 vs. control (cells incubated without 8i or GSK2126458).
The evaluation of the in vitro metabolic stability of 8i was then performed via incubation with human liver microsome (HLM) and rat liver microsome (RLM). Testosterone, diclofenac, and propafenone were utilized as the references. According to the experimental data shown in Table 3, 8i exhibited favorable stability in both HLM (T1/2 = 74.9 min) and RLM (T1/2 > 145 min), as illustrated by the long elimination half-lives.
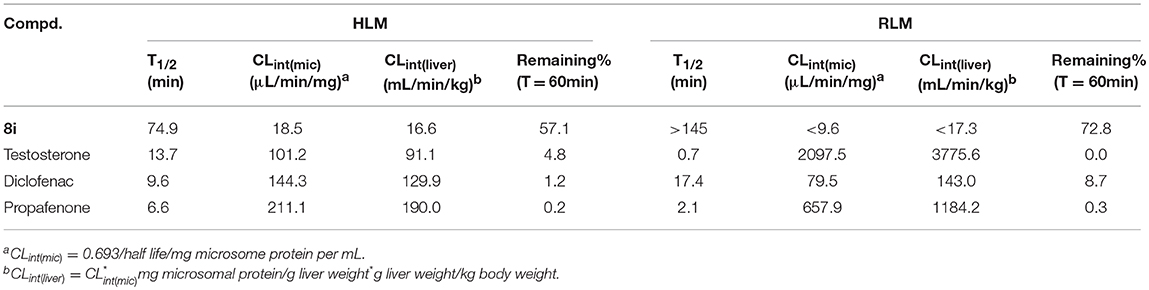
Table 3. The in vitro metabolic stability of compound 8i in HLM and RLM.
By virtue of its favorable in vitro potency and metabolic stability, 8i was further evaluated for its PK profiles in Sprague-Dawley (SD) rats following oral administration at 5 mg/kg (Figure 4). As a result, 8i showed acceptable plasma exposure (AUC0−t = 379 h•ng/mL; AUC0−∞ = 412 h•ng/mL), peak plasma concentration (Cmax = 249 ng/mL), and elimination half-life (t1/2 = 1.73 h).
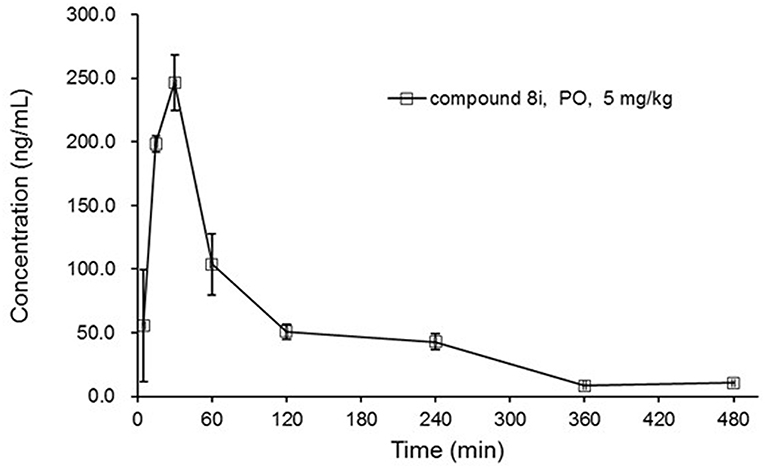
Figure 4. The concentration-time curve of 8i in SD rats: three animals for PO dose of 5 mg/kg.
In summary, a novel series of quinoline derivatives were designed and synthesized as PI3K/mTOR dual inhibitors, via introducing acrylamide fragment to the C-4 position for probing residue Gln859. According to the PI3Kα enzymatic assay, all of them were remarkably potent with IC50s at sub-nanomolar or low nanomolar level. Consistent with this, they exhibited attractive anti-proliferative efficacy against both PC3 and HCT116 cell lines. To our expectation, 8i, a representative throughout this series (IC50: PI3Kα, 0.50 nM; GI50s: PC3, 0.40 μM; HCT116, 0.47 μM), also significantly inhibited other class I PI3Ks and mTOR with all the IC50s at sub-nanomolar or low nanomolar level. In the PC3 cell line, 8i dramatically down-regulated the levels of phos-Akt (Ser473), phos-Akt (Thr308), phos-S6 ribosomal protein (Ser235/236), and phos-4E-BP1 (Thr37/46) at a concentration as low as 5 nM, indicating its considerable capability to ablate PAM signaling. Besides, the concurrent down-regulation of the four biomarkers was beneficial for fulfilling synergism and conquering the negative feedback loop released by mono-inhibition of mTOR. Moreover, 8i was metabolically stable in both HLM and RLM with long elimination half-life. A further in vivo PK study of 8i in SD rats following oral administration at 5 mg/kg illustrated 8i possessed acceptable oral exposure, peak plasma concentration, and elimination half-life. According to the above results, it can be concluded that introduction of 4-acrylamide substructure at the C-4 position of the quinoline core was a feasible way to attain potent PI3K/mTOR dual inhibitors with structural novelty. Besides, 8i, with acceptable in vitro and in vivo biological profiles, merited further investigation and structural optimization.
The study was carried out in accordance with institutional guidelines of the Animal Research Committee at Jiaxing University (log number JXU2015120812). The protocol was approved by the institution.
XL designed and supervised this research and performed the molecular simulation. The chemical synthesis and purification were predominantly conducted by XM and LS. XL and XM were involved in the data analysis and manuscript writing. JZ and GL critically evaluated the scientific work. SZ and BD participated in the biological evaluation of compounds. All authors approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer QH declared a shared affiliation, though no other collaboration, with several of the authors LS, JZ to the handling Editor.
The authors thank the financial support by the Public Welfare Technology Application Projects of Zhejiang Province (NO.2016C33089), National Natural Science Foundation of China (No.81503338), Natural Science Foundation of Zhejiang Province (No.LY18H280011), and Natural Science Foundation of Anhui Province (1808085QH261).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00236/full#supplementary-material
Beaufils, F., Cmiljanovic, N., Cmiljanovic, V., Bohnacker, T., Melone, A., Marone, R., et al. (2017). 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (PQR309), a Potent, Brain-Penetrant, Orally Bioavailable, Pan-Class I PI3K/mTOR inhibitor as clinical candidate in oncology. J. Med. Chem. 60, 7524–7538. doi: 10.1021/acs.jmedchem.7b00930
Elkabets, M., Vora, S., Juric, D., Morse, N., Mino-Kenudson, M., Muranen, et al. (2013). mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Sci. Transl. Med. 5:196ra99. doi: 10.1126/scitranslmed.3005747
Folkes, A. J., Ahmadi, K., Alderton, W. K., Alix, S., Baker, S. J., Box, G., et al. (2008). The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 51, 5522–5532. doi: 10.1021/jm800295d
Furet, P., Guagnano, V., Fairhurst, R. A., Imbach-Weese, P., Bruce, I., Knapp, M., et al. (2013). Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 23, 3741–3748. doi: 10.1016/j.bmcl.2013.05.007
Garces, A. E., and Stocks, M. E. (2019). Class 1 PI3K clinical candidates and recent inhibitor design strategies: a medicinal chemistry perspective. J. Med. Chem. doi: 10.1021/acs.jmedchem.8b01492.
Heffron, T. P., Heald, R. A., Ndubaku, C., Wei, B., Augistin, M., Do, S., et al. (2016). The Rational Design of Selective Benzoxazepin Inhibitors of the alpha-Isoform of Phosphoinositide 3-Kinase Culminating in the Identification of (S)-2-((2-(1-Isopropyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)oxy)propanamide (GDC-0326). J. Med. Chem. 59, 985–1002. doi: 10.1021/acs.jmedchem.5b01483
Lalonde, M. P., McGowan, M. A., Rajapaksa, N. S., and Jacobsen, E. N. (2013). Enantioselective formal aza-Diels-Alder reactions of enones with cyclic imines catalyzed by primary aminothioureas. J. Am. Chem. Soc. 135, 1891–1894. doi: 10.1021/ja310718f
Liu, Q., Chang, J. W., Wang, J., Kang, S. A., Thoreen, C. C., Markhard, A., et al. (2010). Discovery of 1-(4-(4-propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J. Med. Chem. 53, 7146–7155. doi: 10.1021/jm101144f
Liu, Q., Wang, J., Kang, S. A., Thoreen, C. C., Hur, W., Ahmed, T., et al. (2011). Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J. Med. Chem. 54, 1473–1480. doi: 10.1021/jm101520v
Lv, X., Ying, H., Ma, X., Qiu, N., Wu, P. B, et al. (2015). Design, synthesis and biological evaluation of novel 4-alkynyl-quinoline derivatives as PI3K/mTOR dual inhibitors. Eur. J. Med. Chem. 99, 36–50. doi: 10.1016/j.ejmech.2015.05.025
Ma, X., and Hu, Y. (2013). Targeting PI3K/Akt/mTOR cascade: the medicinal potential, updated research highlights and challenges ahead. Curr. Med. Chem. 20, 2991–3010. doi: 10.2174/09298673113209990124
Ma, X., Lv, X., Qiu, N., Yang, B., He, Q., and Hu, Y. (2015). Discovery of novel quinoline-based mTOR inhibitors via introducing intra-molecular hydrogen bonding scaffold (iMHBS): The design, synthesis and biological evaluation. Bioorg. Med. Chem. 23, 7585–7596. doi: 10.1016/j.bmc.2015.11.003
Ma, X., Lv, X., and Zhang, J. (2018). Exploiting polypharmacology for improving therapeutic outcome of kinase inhibitors (KIs): an update of recent medicinal chemistry efforts. Eur. J. Med. Chem. 143, 449–463. doi: 10.1016/j.ejmech.2017.11.049
Ndubaku, C. O., Heffron, T. P., Staben, S. T., Baumgardner, M., Blaquiere, N., Bradley, E., et al. (2013). Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a beta-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J. Med. Chem. 56, 4597–4610. doi: 10.1021/jm4003632
Perez-Melero, C., Maya, A. B., Rey, B., del Pelaez, R., Caballero, E., and Medarde, M. (2004). A new family of quinoline and quinoxaline analogues of combretastatins. Bioorg. Med. Chem. Lett. 14, 3771–3774. doi: 10.1016/j.bmcl.2004.04.098
Phanumartwiwath, A., Hornsby, T. W., Jamalis, J., Bailey, C. D., and Willis, C. L. (2013). Silyl migrations in D-xylose derivatives: total synthesis of a marine quinoline alkaloid. Org. Lett. 15, 5734–5737. doi: 10.1021/ol402760p
Pike, K. G., Morris, J., Ruston, L., Pass, S. L., Greenwood, R., Williams, E. J., et al. (2015). Discovery of AZD3147: a potent, selective dual inhibitor of mTORC1 and mTORC2. J. Med. Chem. 58, 2326–2349. doi: 10.1021/jm501778s
Stauffer, F., Maira, S. M., Furet, P., and Garcia-Echeverria, C. (2008). Imidazo[4,5-c]quinolines as inhibitors of the PI3K/PKB-pathway. Bioorg. Med. Chem. Lett. 18, 1027–1030. doi: 10.1016/j.bmcl.2007.12.018
Yadav, R. R., Guru, S. K., Joshi, P., Mahajan, G., Mintoo, M. J., Kumar, V., et al. (2016). 6-aryl substituted 4-(4-cyanomethyl) phenylamino quinazolines as a new class of isoform-selective PI3K-alpha inhibitors. Eur. J. Med. Chem. 122, 731–743. doi: 10.1016/j.ejmech.2016.07.006
Yang, H., Rudge, D. G., Koos, J. D., Vaidialingam, B., Yang, H. J., and Pavletich, N. P. (2013). mTOR kinase structure, mechanism and regulation. Nature 497, 217–223. doi: 10.1038/nature12122
Zhang, J., Lv, X., Ma, X., and Hu, Y. (2017a). Discovery of a series of N-(5-(quinolin-6-yl)pyridin-3-yl)benzenesulfonamides as PI3K/mTOR dual inhibitors. Eur. J. Med. Chem. 127, 509–520. doi: 10.1016/j.ejmech.2017.01.016
Keywords: 4-acrylamido-quinolines, PI3K/mTOR dual inhibitors, anti-proliferative activity, PI3K/Akt/mTOR signaling, metabolic stability, PK study
Citation: Ma X, Shen L, Zhang J, Liu G, Zhan S, Ding B and Lv X (2019) Novel 4-Acrylamido-Quinoline Derivatives as Potent PI3K/mTOR Dual Inhibitors: The Design, Synthesis, and in vitro and in vivo Biological Evaluation. Front. Chem. 7:236. doi: 10.3389/fchem.2019.00236
Received: 04 November 2018; Accepted: 25 March 2019;
Published: 24 April 2019.
Edited by:
Laurent G. Désaubry, Laboratoire de Cardio-Oncologie et Chimie Médicinale, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Francesco Ortuso, Università Degli Studi Magna Græcia di Catanzaro, ItalyCopyright © 2019 Ma, Shen, Zhang, Liu, Zhan, Ding and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqing Lv, bHhxZDFAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.