 Xue-Ru Zou1
Xue-Ru Zou1 Lan-Chang Liang
Lan-Chang Liang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. , 13 December 2018
Sec. Catalysis and Photocatalysis
Volume 6 - 2018 | https://doi.org/10.3389/fchem.2018.00607
This article is part of the Research Topic Advanced Catalysts in Ring-Opening Polymerization of Cyclic Esters View all 7 articles
Aluminum complexes containing [RP(O)(2-O-3,5-tBu2C6H2)2]2− [R = tBu (3a), Ph (3b)] have been synthesized, structurally characterized, and their reactivity studied in comparison with those of their [RP(2-O-3,5-tBu2C6H2)2]2− [R = tBu (2a), Ph (2b)] analogs. Treating AlMe3 with one equiv of H2[3a-b] in THF at 0°C affords quantitatively [3a-b]AlMe, subsequent reactions of which with benzyl alcohol in THF at 25°C generate {[3a-b]Al(μ2-OCH2Ph)}2. The methyl [3a-b]AlMe and the benzyloxide {[3a-b]Al(μ2-OCH2Ph)}2 are all active for catalytic ring-opening polymerization (ROP) of ε-caprolactone and rac-lactide (rac-LA). Controlled experiments reveal that {[3a]Al(μ2-OCH2Ph)}2 is competent in living polymerization. Kinetic studies indicate that [3a]AlMe, in the presence of benzyl alcohol, catalyzes ROP of rac-LA at a rate faster than [3b]AlMe and [2a]AlMe(THF) by a factor of 1.8 and 23.6, respectively, highlighting the profound reactivity enhancement in ROP catalysis by varying the P-substituents of these biphenolate complexes of aluminum.
The search for efficient catalyst precursors or initiators for catalytic ring-opening polymerization (ROP) of cyclic esters continues to constitute an active area of exploratory chemistry (Kamber et al., 2007; Thomas, 2010; Hillmyer and Tolman, 2014; Sarazin and Carpentier, 2015). In this regard, metal complexes containing chelating biphenolate ligands have attracted significant attention. These complexes are intriguing as their catalytic activities are finely tunable by judiciously varying the peripheral substituents on the two phenolate rings and/or the bridge in between. While most studies concentrate on metal complexes of tetradentate biphenolate ligands such as ONNO (Ovitt and Coates, 2000; Zhong et al., 2002; Hormnirun et al., 2006; Zelikoff et al., 2009; Chen et al., 2012; Gao et al., 2015; Jones et al., 2015; Kirk et al., 2016; MacDonald et al., 2016; McKeown et al., 2016; Robert et al., 2017; Pang et al., 2018), OSSO (Buffet and Okuda, 2011; Buffet et al., 2011), ONSO (Stopper et al., 2012), and ONOX (X = OR, NR2) (Alcazar-Roman et al., 2003; Gendler et al., 2006; Tang and Gibson, 2007; Phomphrai et al., 2010; Wichmann et al., 2012) as exemplified in Figure 1, parallel research centered upon tridentate counterparts is relatively rare (Chmura et al., 2006; Chang and Liang, 2007; Hsu and Liang, 2010; Liang et al., 2011, 2013a,b,c,d,e; Huang et al., 2013; Klitzke et al., 2014a,b; Chang et al., 2016).
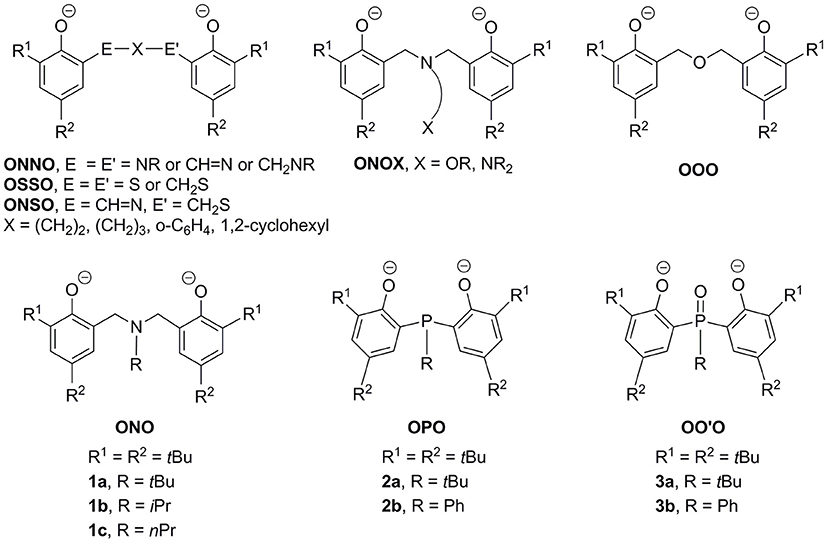
Figure 1. Representative examples of chelating biphenolate ligands.
It has been documented that complexes of tridentate OOO (Huang et al., 2013), ONO (1a-c) (Liang et al., 2013a,b,c,d,e), and OPO (2a-b) (Chang and Liang, 2007; Hsu and Liang, 2010; Liang et al., 2011; Chang et al., 2016) are active catalyst precursors for ROP of ε-caprolactone (ε-CL) or lactides (LAs). Studies on complexes of 1 and 2 have revealed that substituents at the pnictogen donor have decisive impacts on ROP catalysis if living polymerization is the goal. Of note are aluminum complexes of 2a that polymerize ε-CL and rac-LA in a living manner to produce well-defined poly(ε-caprolactone) (PCL) and poly(rac-lactide) (PLA), respectively (Chang et al., 2016). In contrast, analogous complexes of 1a give instead low molecular weight oligo(ε-caprolactone) or PCL with a somewhat broad molecular weight distribution (Liang et al., 2013b). Constitutionally, complexes of 1a and 2a are much alike as both are pnictogen biphenolate derivatives carrying a pnictogen-bound tert-butyl group. Having an extra benzylic methylene moiety in each arm, however, the former ligand, upon complexation, forms 6-membered chelating rings that are therefore less rigid than the 5-membered chelating rings derived from the latter. In an effort to better understand the effects of chelating ring size (Lee et al., 2017) and ligand rigidity (Liang et al., 2003a,b, 2005a,b, 2006; Huang and Liang, 2004; MacLachlan and Fryzuk, 2005; Liang, 2006; MacLachlan et al., 2007; Hung et al., 2014) on ROP catalysis, we turn our attention to the oxidative forms of 2 that would persist with the same rigidity but enlarge the chelating rings to be 6-membered. Such biphenolate phosphinoxide derivatives OO'O are distinguished from the ether-bridged OOO types that are intrinsically more flexible in ligand backbone and lack of the opportunities of changing substituents at the bridge donor. Note that complexes of OO'O types of ligands are relatively undeveloped (Tanke et al., 1991; Siefert et al., 2000; Paine et al., 2004; He et al., 2008; Zhang et al., 2013; Taniyama et al., 2014). In this contribution, we aim to demonstrate the syntheses of the first examples of OO'O complexes of aluminum and their enhanced catalytic activities in comparison with those of 2 with respect to ROP of ε-CL and rac-LA.
The protio ligand precursor H2[3b] is known (Siefert et al., 2000). Its tert-butyl analog H2[3a] can be readily prepared as an off-white solid in high yield from oxidation of H2[2a] with hydrogen peroxide in THF under ambient conditions. Its solution NMR data are consistent with a structure having time-averaged Cs symmetry. The diagnostic signals of this compound involve the downfield shift of its phosphorus atom at 65 ppm in comparison with that of H2[2a] at −60 ppm (Hsu and Liang, 2010) and the singlet resonance of its hydroxy protons at 12.19 ppm in comparison with the doublet resonance of those in H2[2a] at 7.61 ppm with JHP = 12 Hz (Hsu and Liang, 2010). The lack of OH⋯ P internuclear coupling and the downfield shift of the hydroxy protons in H2[3a] are apparently a consequence of the 6-membered OH⋯ O = P hydrogen bonding. Such intramolecular hydrogen bonding is also confirmed by the solid state structure of H2[3a] established by an X-ray diffraction study (Figure S1, Table S1).
Protonolysis of AlMe3 with one equiv of H2[3a-b] in THF at 0°C yields nearly quantitative [3a-b]AlMe (Figure 2). Interestingly, these methyl complexes are not THF-bound as evidenced by their 1H NMR spectra. This result is reminiscent of 4-coordinate [1a–c]AlMe (Liang et al., 2013b) but in contrast to 5-coordinate [2a-b]AlMe(THF) (Chang et al., 2016), ascribable to the hardness similarity of O (from phosphinoxide in 3) to N (from 1) rather than P (from 2) in consideration of the distinct preferences of these hard and soft donors to bind to a hard aluminum (Fryzuk et al., 1996, 1998; Liang et al., 2004, 2010; Lee and Liang, 2005, 2009; Su and Liang, 2018). As a result, the solution structures of [3a-b]AlMe and [1a–c]AlMe should be much alike. Subsequent reactions of either isolated or in situ prepared [3a-b]AlMe with one equiv of benzyl alcohol in THF at 25°C afford {[3a-b]Al(μ2-OCH2Ph)}2 as colorless crystals.
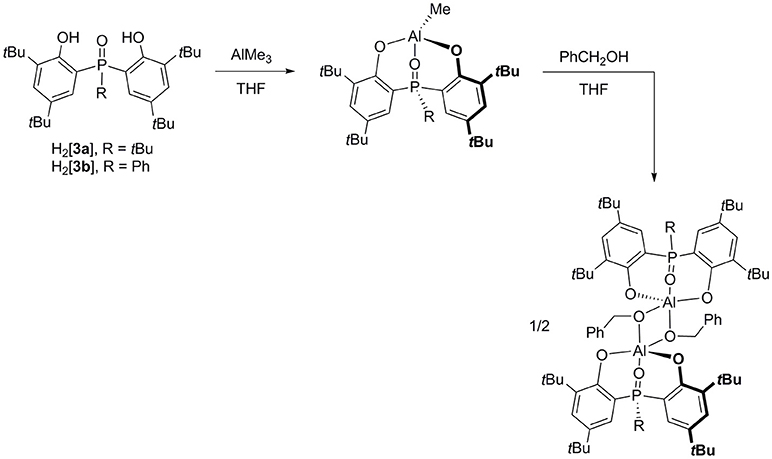
Figure 2. Synthesis of biphenolate phosphinoxide complexes of aluminum.
The solution NMR data of [3a-b]AlMe and {[3a-b]Al(μ2-OCH2Ph)}2 are indicative of a mirror plane symmetry that makes the two phenolate rings in 3 chemically equivalent as evidenced by the observation of two distinct singlet resonances for arylated tert-butyl groups in the 1H NMR spectra. The methylene groups in the benzyloxide ligands of {[3a-b]Al(μ2-OCH2Ph)}2 exhibit a singlet resonance in the 1H NMR spectra at ca. 5.7 ppm, a chemical shift that is similar to that found for {[2a-b]Al(μ2-OCH2Ph)}2 (Chang et al., 2016). A 1H NMR NOE difference experiment of {[3a]Al(μ2-OCH2Ph)}2 was conducted with selective irradiation on the methylene groups, resulting in NOE enhancements of 5.14% for the arylated tert-butyl groups ortho to the phenolate oxygen atoms and 3.21% for the P-bound tert-butyl group. Note that these tert-butyl groups are spatially far away from each other due to the inherent geometry of the facially tridentate 3a. The concurrent NOE enhancements on these tert-butyl groups thus strongly implicate a dimeric structure of {[3a]Al(μ2-OCH2Ph)}2 in solution. Consistent with the oxidized characteristics of the phosphorus atom in phosphinoxide derivatives, the 31P chemical shifts of [3a-b]AlMe and {[3a-b]Al(μ2-OCH2Ph)}2 are significantly downfield shifted from those of their corresponding analogs of 2 (Chang et al., 2016).
An attempt to characterize [3a]AlMe by X-ray crystallography led instead to the structure of [3a]Al2Me4 that is an O-bound AlMe3 adduct of [3a]AlMe (Figure 3). We attribute this unexpected result to unintentional imbalance of reaction stoichiometry that gives a trace amount of highly crystalline [3a]AlMe•AlMe3. Following this lead, we attempted the reactions of [3a]AlMe with one equiv of AlMe3 or H2[3a] with two equiv of AlMe3. Unfortunately, these reactions result ultimately in a mixture of equal molar [3a]AlMe and AlMe3 as evidenced by 1H and 31P{1H} NMR spectra of reaction aliquots. Subsequent attempts to crystallographically characterize [3a]AlMe have thus far been unsuccessful. Nevertheless, the structure of [3a]AlMe•AlMe3 confirms the 4-coordinate nature for the aluminum center of the [3a]AlMe moiety that has a distorted tetrahedral coordination core similar to [1a–c]AlMe (Liang et al., 2013b). The bond distances and angles of [3a]AlMe•AlMe3 are unexceptional.
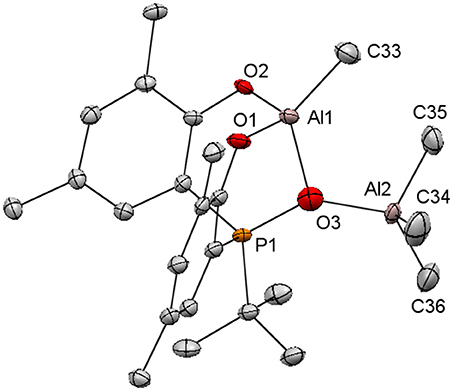
Figure 3. Molecular structure of [3a]AlMe•AlMe3 with thermal ellipsoids drawn at the 35% probability level. All hydrogen atoms and the methyl groups in arylated tert-butyls are omitted for clarity. Selected bond distances (Å) and angles (deg): Al(1)-O(2) 1.749(3), Al(1)-O(1) 1.764(3), Al(1)-C(33) 1.933(5), Al(1)-O(3) 1.934(4), Al(2)-O(3) 1.753(4), Al(2)-C(34) 1.844(6), Al(2)-C(36) 1.872(6), Al(2)-C(35) 1.872(5), O(3)-P(1) 1.623(4), O(2)-Al(1)-O(1) 110.75(17), O(2)-Al(1)-C(33) 111.5(2), O(1)-Al(1)-C(33) 108.6(2), O(2)-Al(1)-O(3) 98.48(17), O(1)-Al(1)-O(3) 99.39(16), C(33)-Al(1)-O(3) 127.0(2), O(3)-Al(2)-C(34) 107.6(3), O(3)-Al(2)-C(36) 121.0(2), C(34)-Al(2)-C(36) 107.5(3), O(3)-Al(2)-C(35) 107.2(2), C(34)-Al(2)-C(35) 111.6(3), C(36)-Al(2)-C(35) 101.8(3), P(1)-O(3)-Al(2) 141.8(2), P(1)-O(3)-Al(1) 103.2(2), Al(2)-O(3)-Al(1) 112.5(2).
Colorless crystals of {[3a]Al(μ2-OCH2Ph)}2 suitable for X-ray diffraction analysis were grown by layering pentane on top of a concentrated THF solution at −35°C. Figure 4 depicts its solid state structure. Consistent with the NOE study, this complex is a dimer, composed of two [3a]Al(OCH2Ph) units bridged with the benzyloxide ligands. With the coordination of the facially tridentate 3a, the aluminum atoms in {[3a]Al(μ2-OCH2Ph)}2 are therefore 5-coordinate. Its coordination geometry is best described as distorted trigonal bipyramidal, having the phosphinoxide donor and one of the bridging benzyloxide ligands disposed at the axial positions [O(3)-Al(1)-O(4A) = 165.10(8)°]. The axial Al-OCH2Ph bond distances of 1.8726 (17) Å are longer than those disposed equatorially [1.8372 (17) Å]. This indicates that the equatorial benzyloxide ligands are more anionic in nature whereas those at the axial positions are more dative. Consistent with this result, [2a]AlMe(THF), though constitutionally different, holds a methyl ligand equatorially, and a THF axially (Chang et al., 2016).
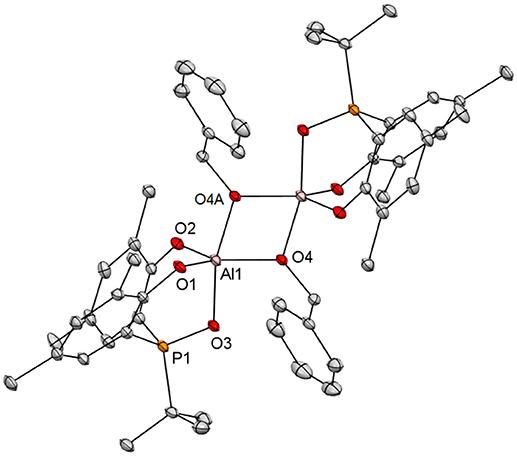
Figure 4. Molecular structure of {[3a]Al(μ2-OCH2Ph)}2 with thermal ellipsoids drawn at the 35% probability level. All hydrogen atoms and the methyl groups in arylated tert-butyls are omitted for clarity. Selected bond distances (Å) and angles (deg): Al(1)-O(2) 1.7766(18), Al(1)-O(1) 1.7890(18), Al(1)-O(4) 1.8372(17), Al(1)-O(3) 1.8414(17), Al(1)-O(4A) 1.8726(17), Al(1)-Al(1A) 2.9360(14), O(3)-P(1) 1.5274(16), O(2)-Al(1)-O(1) 109.46(9), O(2)-Al(1)-O(4) 117.94(9), O(1)-Al(1)-O(4) 132.10(9), O(2)-Al(1)-O(3) 94.74(8), O(1)-Al(1)-O(3) 92.52(8), O(4)-Al(1)-O(3) 90.14(8), O(2)-Al(1)-O(4A) 95.13(8), O(1)-Al(1)-O(4A) 94.67(8), O(4)-Al(1)-O(4A) 75.37(8), O(3)-Al(1)-O(4A) 165.10(8), Al(1)-O(4)-Al(1A) 104.63(8), P(1)-O(3)-Al(1) 114.84(10).
The P-bound tert-butyl groups in [2a]AlMe(THF) and {[2a]Al(μ2-OCH2Ph)}2 are known to sterically repulse their equatorial methyl and benzyloxide ligands away from the ideal positions with the P-Al-C and P-Al-O angles of 105.51(12)° and 107.25(9)°, respectively (Chang et al., 2016). Such steric repulsion is apparently eased by moving the tert-butyl-bound phosphorus atom to the β position in {[3a]Al(μ2-OCH2Ph)}2 as evidenced by the O(3)-Al(1)-O(4) angle of 90.14(8)°. With the incorporation of the rigid o-phenylene backbone and the 6-membered chelating rings in {[3a]Al(μ2-OCH2Ph)}2, the O(1)-Al(1)-O(3) [92.52(8)°] and O(2)-Al(1)-O(3) [94.74(8)°] angles are notably wider than the corresponding angles (80.35(9)° and 84.17(9)°) in the 5-membered chelating rings of {[2a]Al(μ2-OCH2Ph)}2 (Chang et al., 2016). As such, the tert-butyl groups ortho to the phenolate oxygen atoms in the former are spatially closer to the axial benzyloxide ligands than those in the latter. This should in principle encourage dissociation of the datively bonded benzyloxide ligands and formation of transient monomeric [3a]Al(OCH2Ph) for subsequent substrate coordination. The similarity of Al(1)-O(4A) distance [1.8726(17) Å] to that in {[2a]Al(μ2-OCH2Ph)}2 [1.878(2) Å] (Chang et al., 2016) implicates surprisingly little difference in trans influence invoked by P = O and P for axial benzyloxide ligand dissociation from these aluminum complexes.
Similar to aluminum derivatives of 1 (Liang et al., 2013b) and 2 (Chang et al., 2016), complexes [3a-b]AlMe and {[3a-b]Al(μ2-OCH2Ph)}2 are all active for catalytic ROP of ε-CL and rac-LA. To establish parallel comparison on reactivity of these pnictogen derivatives, the catalysis of [3]AlMe and {[3]Al(μ2-OCH2Ph)}2 was examined under conditions identical to those employed for [1]AlMe (Liang et al., 2013b), [2]AlMe(THF) (Chang et al., 2016), and {[2]Al(μ2-OCH2Ph)}2 (Chang et al., 2016). To simplify tabulated discussion and to make consistency with other mononuclear species, the benzyloxide complexes are presented as a monomer. Table 1 summarizes their catalytic activities with ε-CL. In the presence of one equiv of benzyl alcohol, [3a]AlMe reacts slightly faster than [3b]AlMe with 100 equiv of ε-CL under the conditions employed (entry 1 vs. 3) though both reactions complete in 2 h (entries 2 and 4). The observed number averaged molecular weights (Mn's), however, are generally smaller than those expected. Studies on Mn's of these PCLs by 1H NMR spectroscopy also give similar results (entry 2, 5.1 kg/mol; entry 4, 9.5 kg/mol). Both [3a]AlMe and [3b]AlMe are more reactive than [2a]AlMe(THF) and [2b]AlMe(THF) (entries 1 and 3 vs. 5 and 6) due apparently to the discrepancy of 3 and 2 that invokes THF coordination and retards ROP. Though [1a]AlMe, [2a]AlMe(THF), and [3a]AlMe are all tert-butylated at their pnictogen atom, the polydispersity indexes (PDIs) of PCLs produced from [2a]AlMe(THF) and [3a]AlMe are smaller than those from [1a]AlMe (entries 1–2 and 5 vs. 7–8). All in all, [3a]AlMe is therefore a superior catalyst precursor to [2a]AlMe(THF) that in turn outperforms [1a]AlMe in this catalysis. These results underscore the decisive role that the biphenolate bridges play in ROP catalysis.
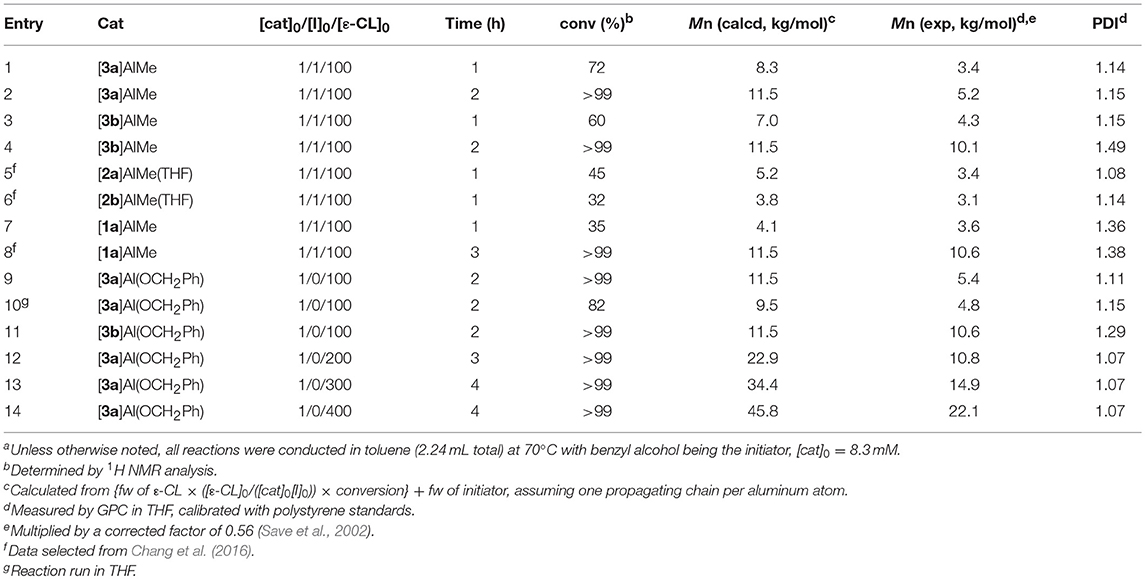
Table 1. ROP of ε-CL by catalytic [3a-b]AlMe and [3a-b]Al(OCH2Ph)a.
Similar to that generated in situ, [3a]Al(OCH2Ph) finishes polymerization of 100 equiv of ε-CL in toluene at 70°C in 2 h, producing quantitatively PCL having comparable Mn and PDI (entry 9 vs. 2). End group analysis by 1H NMR spectroscopy reveals a benzyl ester functionality, implicating that this ROP proceeds with a coordination-insertion mechanism that involves ε-CL coordination to the transient monomeric [3a]Al(OCH2Ph) (vide supra) followed by insertion of this ε-CL into the Al-OCH2Ph bond, allowing ε-CL to ring-open by cleaving its acyl-oxygen bond for chain propagation. Consistent with this rationale, the same reaction conducted in THF proceeds relatively slower (entry 10). Nevertheless, the PCL thus produced has a satisfactorily small PDI, indicating that the interfering THF coordination is reversible and does not much induce undesirable side reactions.
In contrast, PCL produced from catalytic [3b]Al(OCH2Ph) has a relatively larger PDI (entry 11), reminiscent of that acquired from [3b]AlMe as compared with [3a]AlMe (entries 4 vs. 2). Complexes [3a]AlMe and [3a]Al(OCH2Ph) thus outperform [3b]AlMe and [3b]Al(OCH2Ph) in this catalysis. Interestingly, [3a]Al(OCH2Ph) polymerizes ε-CL in a living fashion. The PCLs thus produced (entries 9 and 12–14) have Mn's linearly proportional to the consumed monomer-to-catalyst ratios (Figure 5) while maintaining consistently small PDIs.
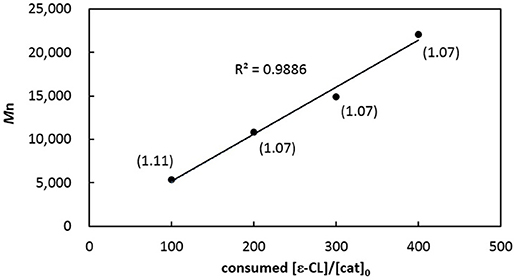
Figure 5. Linear plot of corrected Mn of PCLs vs. monomers consumed to [3a]Al(OCH2Ph) ratios (entries 9 and 12–14 in Table 1). Numbers shown in parentheses indicate their corresponding PDIs.
Table 2 summarizes ROP results with respect to rac-LA. In the presence of one equiv of benzyl alcohol, [3a]AlMe and [3b]AlMe complete polymerization of 100 equiv of rac-LA in toluene at 70°C in 6 h, producing PLAs quantitatively (entries 1–2). The former complex is again a superior catalyst precursor to the latter in view of the smaller PDI derived. These reactions are faster than those by catalytic [2a]AlMe(THF) and [2b]AlMe(THF) (entries 3–4) (Chang et al., 2016). Interestingly, [3a]Al(OCH2Ph) is also competent in living ROP of rac-LA (entries 5–8), affording PLAs having Mn's directly proportional to the consumed monomer-to-catalyst ratios (Figure 6) while keeping their PDIs consistently small. In contrast, PLA produced by catalytic [3b]Al(OCH2Ph) has a relatively larger PDI (entry 9). A reaction run in THF is again slow but does not change PDI much (entry 10). Catalysis run at room temperature results in slow reaction and low conversion (entry 11). In the presence of one equiv of poly(ethylene glycol) methyl ether (Mn 2000, denoted MePEG2000 in Table 2), [3a]AlMe polymerizes rac-LA to give PEG-b-PLA copolymers with satisfactorily small PDIs (entries 12–13).
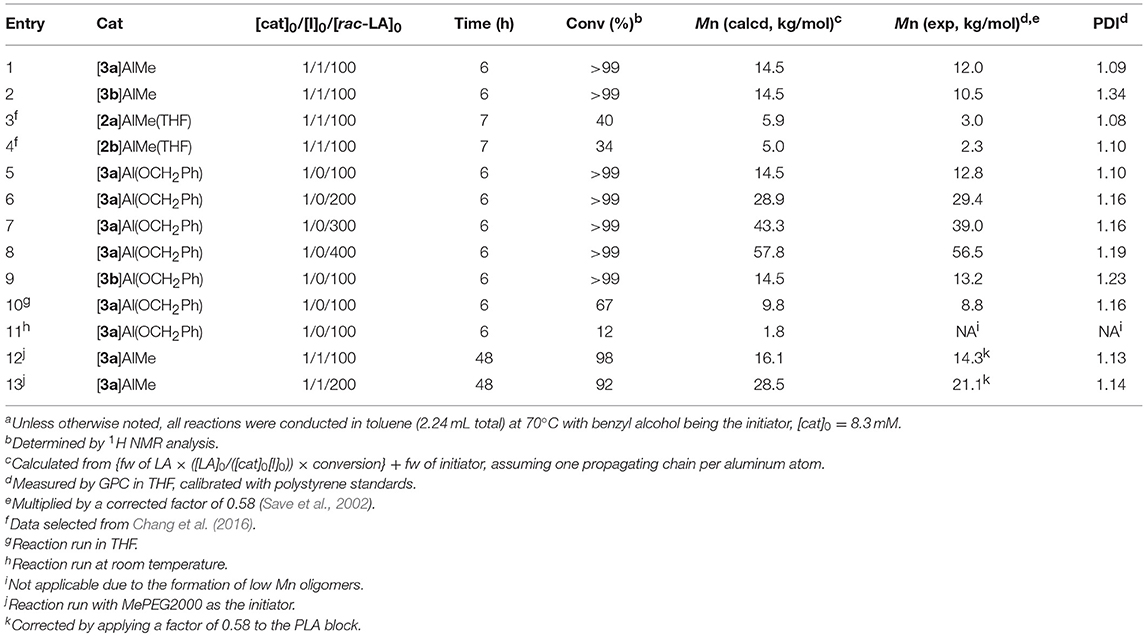
Table 2. ROP of rac-LA by catalytic [3a-b]AlMe and [3a-b]Al(OCH2Ph)a.
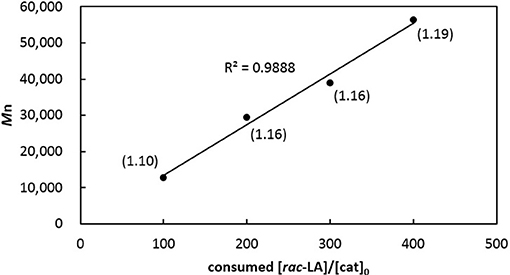
Figure 6. Linear plot of corrected Mn of PLAs vs. monomers consumed to [3a]Al(OCH2Ph) ratios (entries 5–8 in Table 2). Numbers shown in parentheses indicate their corresponding PDIs.
Kinetics of rac-LA polymerization by catalytic [3a]Al(OCH2Ph) was studied. Monitoring the reaction progress by 1H NMR spectroscopy reveals linear semilogarithmic plots for rac-LA consumptions vs. time (Figure 7), indicating a pseudo-first order dependence of the polymerization rates on the concentrations of rac-LA, i.e., –d[rac-LA]/dt = kobs[rac-LA]1, where kobs = kp[catalyst]x and kp = propagation rate constant. A plot of the observed rate constants vs. concentrations of [3a]Al(OCH2Ph) shows a linear dependence of the former on the latter (Figure 8), thus giving x = 1. The overall rate law of this catalysis is therefore expressed as –d[rac-LA]/dt = kp[catalyst][rac-LA], where kp = 1.47 (9) × 10−2 L mol−1 s−1 at 70°C.
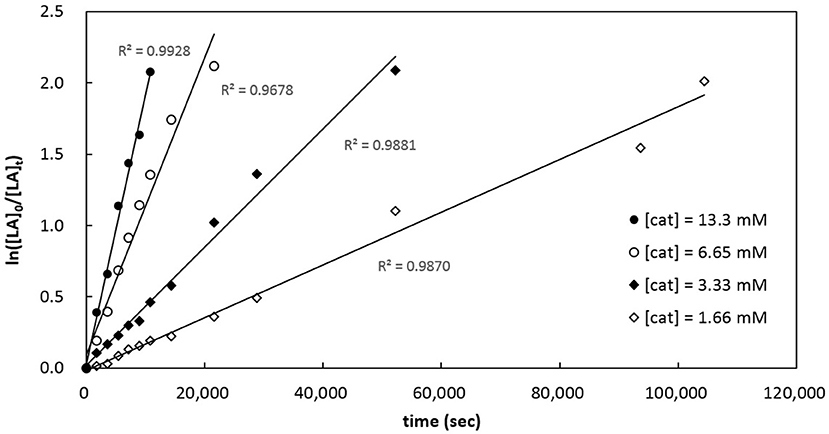
Figure 7. Semilogarithmic plots of rac-LA conversion with time employing catalytic [3a]Al(OCH2Ph) in toluene-d8 at 70°C. [rac-LA]0 = 208 mM; i, [cat]0 = 13.3 mM, kobs = 1.89 (7) × 10−4 s−1; ii, [cat]0 = 6.65 mM, kobs = 1.04 (7) × 10−4 s−1; iii, [cat]0 = 3.33 mM, kobs = 4.15 (15) × 10−5 s−1; iv, [cat]0 = 1.66 mM, kobs = 1.85 (6) × 10−5 s−1.
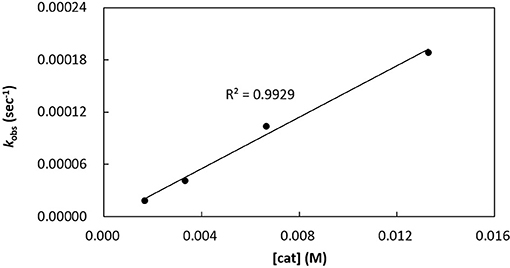
Figure 8. Plot of kobs vs. concentrations of [3a]Al(OCH2Ph) for ROP of rac-LA in toluene-d8 at 70°C; [rac-LA]0 = 208 mM.
To kinetically quantify the P-substituent effect, we turn our attention to the relative ROP rates of rac-LA by catalytic [3a]AlMe, [3b]AlMe, [2a]AlMe(THF), and [2b]AlMe(THF) in the presence of one equiv of benzyl alcohol. Figure 9 depicts their semilogarithmic plots of rac-LA conversions with time. As a result, the reactivity of these catalyst precursors follows the order of [3a]AlMe > [3b]AlMe > [2a]AlMe(THF) > [2b]AlMe(THF). In this catalysis, [3a]AlMe is more reactive than [3b]AlMe by 1.8 times and [2a]AlMe(THF) is more reactive than [2b]AlMe(THF) by 2.0 times. More importantly, [3a]AlMe is more reactive than [2a]AlMe(THF) by 23.6 times and [3b]AlMe is more reactive than [2b]AlMe(THF) by 26.1 times. Collectively, tert-butyl is a superior P-substituent to phenyl and P = O is a superior biphenolate bridge to P in view of offering higher reactivity in this ROP catalysis.
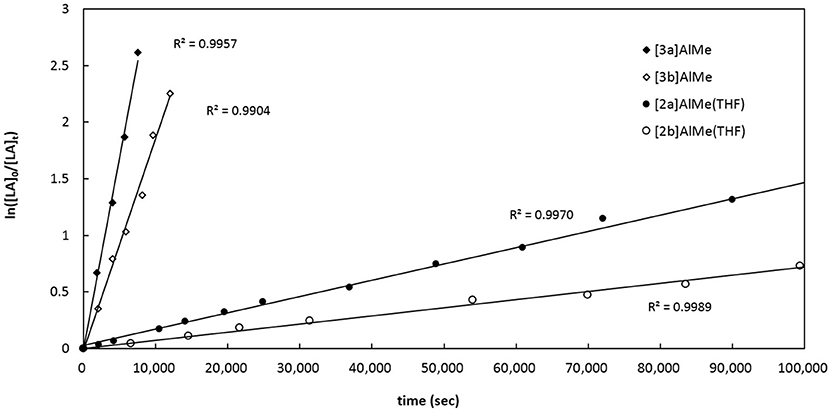
Figure 9. Semilogarithmic plots of rac-LA conversion with time employing (i) [3a]AlMe, kobs = 3.40 (13) × 10−4 s−1 (ii) [3b]AlMe, kobs = 1.87 (8) × 10−4 s−1 (iii) [2a]AlMe(THF), kobs = 1.44 (2) × 10−5 s−1 (iv) [2b]AlMe(THF), kobs = 7.19 (7) × 10−6 s−1. Conditions: [cat]0 = [PhCH2OH]0 = 6.7 mM, [rac-LA] = 670 mM, toluene-d8, 70°C.
We have prepared the first examples of biphenolate phosphinoxide complexes of aluminum and characterized their solution and solid state structures by NMR spectroscopy and X-ray crystallography, respectively. The coordination chemistry of these complexes is compared with those of their amine 1 (Liang et al., 2013b) and phosphine 2 (Chang et al., 2016) counterparts, so are their catalytic activities with respect to ROP of ε-CL and rac-LA. In addition to the inherent discrepancies of neutral donors in 1, 2, and 3, the 6-membered chelating rings rendered by the rigid 3 are advantageous to enhance substantially the reactivity of aluminum complexes in comparison with those derived from 1 and 2 as demonstrated by their relative ROP rates. Of particular note is also the competence of 3a complexes in living ROP of ε-CL and rac-LA.
Unless otherwise specified, all experiments were performed under nitrogen using standard Schlenk or glovebox techniques. Compounds H2[tBuP(2-O-3,5-tBu2C6H2)2] (H2[2a]) (Hsu and Liang, 2010) and H2[PhP(O)(2-O-3,5-tBu2C6H2)2] (H2[3b]) (Siefert et al., 2000) were prepared according to literature procedures. ε-CL was dried over CaH2 (1 wt%) at 80°C for 0.5 h and distilled under reduced pressure. rac-LA was purified by recrystallization (four times) from mixtures of toluene and ethyl acetate. All other chemicals were obtained from commercial vendors and used as received. All solvents were reagent grade or better and purified by standard methods. All NMR spectra were recorded at room temperature in specified solvents unless otherwise noted. Chemical shifts (δ) are listed as parts per million downfield from tetramethylsilane and coupling constants (J) are listed in hertz. Routine coupling constants are not listed. 1H NMR spectra are referenced using the residual solvent peak at δ 7.16 for C6D6 or δ 2.09 for toluene-d8 (the most upfield signal). 13C NMR spectra are referenced using the internal solvent peak at δ 128.39 for C6D6. The assignment of the carbon atoms for all new compounds is based on the DEPT 13C NMR spectroscopy. 31P NMR spectra are referenced externally using 85% H3PO4 at δ 0. The NOE data were obtained with a 1H NMR NOEDIF experiment. GPC analyses were carried out at 45°C with HPLC grade THF supplied at a constant flow rate of 1.0 mL/min. Molecular weights (Mn and Mw) were determined by interpolation from calibration plots established with polystyrene standards. Mass spectra were recorded on a Finnigan MAT 95XL Mass Spectrometer. Elemental analysis was performed on a Heraeus CHN-O Rapid analyzer.
Crystallographic data for H2[3a], [3a]AlMe•AlMe3, and {[3a]Al(μ2-OCH2Ph)}2 are available in Supplementary Material. Data were collected on a diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.7107 Å). Structures were solved by direct methods and refined by full matrix least squares procedures against F2 using SHELXL-97 (Sheldrick, 1998). All full-weight non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions. CCDC 1540207, 1540209, 1540210 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
To a THF solution (10 mL) of H2[2a] (500 mg, 1.0 mmol) was added H2O2 (0.23 mL, 30% in aqueous solution, 2.0 mmol, 2 equiv) under ambient conditions. The solution was stirred at room temperature for 3 h and evaporated to dryness under reduced pressure. The solid thus obtained was washed with acetonitrile (4 mL) to afford the product as an off-white solid; yield 450 mg (87%). Colorless crystals suitable for X-ray diffraction analysis were grown from a concentrated THF solution at −35°C. 1H NMR (C6D6, 300 MHz) δ 12.19 (s, 2, ArOH), 7.62 (s, 2, ArH), 7.55 (d, 2, JHP = 12, ArH), 1.52 (s, 18, ArCMe3), 1.27 (s, 18, ArCMe3), 1.20 (d, 9, 3JHP = 15, PCMe3). 31P{1H} NMR (C6D6, 121.5 MHz) δ 65.16. 13C{1H} NMR (C6D6, 75 MHz) δ 162.1 (s, C), 140.2 (d, JCP = 11.3, C), 139.1 (d, JCP = 6.8, C), 129.2 (s, ArH), 125.2 (d, JCP = 9.8, ArH), 109.1 (d, JCP = 89.3, C), 36.9 (d, JCP = 67.5, PCMe3), 35.8 (s, ArCMe3), 34.5 (s, ArCMe3), 31.8 (s, ArCMe3), 30.0 (s, ArCMe3), 24.5 (s, PCMe3). Anal. Calcd for C32H51O3P: C, 74.66; H, 9.99. Found: C, 74.65; H, 9.94. MS (EI): calcd for C32H51O3P m/z 514.4, found m/z 514.5.
A THF solution (6 mL) of AlMe3 (0.20 mL, 2 M in toluene, 0.4 mmol) was chilled to 0°C. To this was added a pre-chilled THF solution (6 mL) of H2[3a] (206.2 mg, 0.4 mmol) at 0°C. The reaction solution was stirred at room temperature for 1 h and evaporated to dryness under reduced pressure. The solid thus obtained was washed with pentane (2 mL) to afford the product as an off-white solid; yield 215.2 mg (97%). 1H NMR (C6D6, 300 MHz) δ 7.58 (d, 2, JHH = 3.0, Ar), 7.41 (dd, 2, JHP = 15.0 and JHH = 3.0, Ar), 1.54 (s, 18, ArCMe3), 1.36 (d, 9, 3JHP = 15, PCMe3), 1.19 (s, 18, ArCMe3), 0.01 (s, 3, AlMe). 31P{1H} NMR (C6D6, 121.5 MHz) δ 61.37. 13C{1H} NMR (C6D6, 75 MHz) δ 163.9 (s, C), 141.6 (d, JCP = 6.8, C), 139.1 (d, JCP = 12.8, C), 129.4 (s, CH), 122.5 (d, JCP = 12.8, CH), 114.1 (d, JCP = 90.0, C), 35.9 (s, ArCMe3), 35.2 (d, JCP = 68.3, PCMe3), 34.3 (s, ArCMe3), 31.6 (s, ArCMe3), 29.7 (s, ArCMe3), 26.1 (s, PCMe3), −16.4 (s, AlMe). Anal. Calcd for C33H52AlO3P: C, 71.44; H, 9.45. Found: C, 71.37; H, 9.38. MS (EI): calcd for C33H52AlO3P m/z 554.4, found m/z 554.5.
The procedures were all identical to those of [3a]AlMe except using H2[3b] in the place of H2[3a], affording the product as an off-white solid; yield 95%. 1H NMR (C6D6, 300 MHz) δ 7.70 (m, 2, Ar), 7.64 (d, 2, JHH = 1.2, Ar), 7.14 (d, 2, JHH = 1.2, Ar), 7.02 (m, 1, Ar), 6.92 (m, 2, Ar), 1.62 (s, 18, ArCMe3), 1.13 (s, 18, ArCMe3), 0.05 (s, 3, AlMe). 31P{1H} NMR (C6D6, 121.5 MHz) δ 55.06. 13C{1H} NMR (C6D6, 75 MHz) δ 164.4 (s, C), 141.4 (s, C), 139.4 (d, JCP = 7.5, C), 134.1 (d, JCP = 6.8, CH), 129.8 (s, CH), 129.0 (d, JCP = 6.8, CH), 128.4 (s, CH), 125.1 (d, JCP = 6.8, CH), 112.8 (d, JCP = 62.3, C), 35.9 (s, CMe3), 34.2 (s, CMe3), 31.5 (s, CMe3), 29.7 (s, CMe3), −16.5 (s, AlMe). Anal. Calcd for C35H48AlO3P: C, 73.13; H, 8.42. Found: C, 72.75; H, 8.26.
A THF solution of [3a]AlMe was prepared in situ as describe above from the reaction of H2[3a] (206.2 mg, 0.4 mmol) and AlMe3 (0.20 mL, 2 M in toluene, 0.4 mmol). To this was added PhCH2OH (43.2 mg, 0.4 mmol). The reaction solution was stirred at room temperature overnight and evaporated to dryness under reduced pressure. The solid thus obtained was washed with pentane (4 mL × 2) to afford the product as an off-white solid; yield 168.4 mg (65%). 1H NMR (C6D6, 300 MHz) δ 7.76 (d, 4, JHH = 7.5, Ar), 7.57 (d, 4, JHH = 2.1, Ar), 7.38 (dd, 4, JHP = 13.5 and JHH = 2.1, Ar), 7.21 (t, 4, JHH = 7.5, Ar), 7.01 (t, 2, JHH = 7.2, Ar), 5.72 (s, 4, OCH2Ph), 1.54 (s, 36, ArCMe3), 1.18 (s, 36, ArCMe3), 1.12 (d, 18, JHP = 14.7, PCMe3). 31P{1H} NMR (C6D6, 121.5 MHz) δ 56.74. 13C{1H} NMR (C6D6, 75 MHz) δ 164.9 (s, C), 143.0 (s, ipso-OCH2Ph), 141.4 (d, JCP = 6.8, C), 137.9 (d, JCP = 12.9, C), 128.8 (s, CH), 128.3 (s, CH), 125.8 (s, para-OCH2Ph), 125.6 (s, CH), 122.4 (d, JCP = 12.6, CH), 114.01 (d, JCP = 91.5, C), 67.5 (s, OCH2Ph), 35.9 (s, ArCMe3), 35.2 (d, JCP = 71.2, PCMe3), 34.2 (s, ArCMe3), 31.6 (s, ArCMe3), 30.1 (s, ArCMe3), 25.8 (s, PCMe3). Anal. Calcd for (C39H56AlO4P)2: C, 72.40; H, 8.73. Found: C, 72.06; H, 8.47.
The procedures were all identical to those of {[3a]Al(μ2-OCH2Ph)}2 except using H2[3b] in the place of H2[3a], affording the product as an off-white solid; yield 68%. 1H NMR (toluene-d8, 300 MHz) δ 7.60 (m, 8, Ar), 7.25 (m, 4, Ar), 6.90–7.09 (m, 16, Ar), 5.65 (s, 4, OCH2Ph), 1.55 (s, 36, ArCMe3), 1.13 (s, 36, ArCMe3). 31P{1H} NMR (toluene-d8, 121.5 MHz) δ 51.75. 13C{1H} NMR (C6D6, 75 MHz) δ 165.5 (s, C), 143.4 (s, ipso-OCH2Ph), 141.5 (d, JCP = 7.7, C), 138.2 (d, JCP = 13.4, C), 133.9 (d, JCP = 10.4, CH), 132.9 (s, CH), 129.2 (s, CH), 128.4 (s, CH), 127.6 (s, CH), 126.0 (s, CH), 125.2 (d, JCP = 14.3, CH), 125.0 (d, JCP = 12.2, CH), 112.9 (d, JCP = 104.1, C), 67.7 (s, OCH2Ph), 35.9 (s, ArCMe3), 34.1 (s, ArCMe3), 31.5 (s, ArCMe3), 30.2 (s, ArCMe3). Anal. Calcd for (C41H52AlO4P)2: C, 73.85; H, 7.86. Found: C, 73.51; H, 7.65.
A toluene solution (1 mL) containing an alcohol initiator (PhCH2OH or MePEG2000) where appropriate and monomer (ε-CL or rac-LA having prescribed [monomer]0/[catalyst]0 ratios) was heated in an oil bath at 70°C. To this was added a toluene solution (1.24 mL) of catalyst [3a-b]AlMe (0.0187 mmol) or {[3a-b]Al(μ2-OCH2Ph)}2 (0.00935 mmol). The reaction solution was stirred at 70°C for a period of prescribed time and quenched with a methanol solution of HCl. The solid thus precipitated was washed with hexane, isolated, and dried under reduced pressure until constant weights.
The procedures were similar to those described above except that the reactions were conducted in toluene-d8. The monomer conversion was monitored over time by 1H NMR spectrometry.
All authors made substantial contributions to this work. X-RZ and Y-NC conducted experiments, analyzed results, and tabulated data. K-WH participated in the development and discussion of this work. L-CL conceived the project, directed the investigations, and composed the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Ministry of Science and Technology of Taiwan (MOST 106-2113-M-110-003 and MOST 107-2113-M-110-018) and King Abdullah University of Science and Technology (KAUST) for financial support. Y-NC thanks MOST for a postdoctoral fellowship (MOST 104-2811-M-110-001).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00607/full#supplementary-material
X-ray crystallographic data for H2[3a], [3a]AlMe•AlMe3, and {[3a]Al(μ2-OCH2Ph)}2.
Alcazar-Roman, L. M., O'Keefe, B. J., Hillmyer, M. A., and Tolman, W. B. (2003). Electronic influence of ligand substituents on the rate of polymerization of epsilon-caprolactone by single-site aluminium alkoxide catalysts. Dalton Trans. 33, 3082–3087. doi: 10.1039/B303760F
Buffet, J. C., Martin, A. N., Kol, M., and Okuda, J. (2011). Controlled stereoselective polymerization of lactide monomers by group 4 metal initiators that contain an (OSSO)-type tetradentate bis(phenolate) ligand. Polym. Chem. 2, 2378–2384. doi: 10.1039/c1py00266j
Buffet, J. C., and Okuda, J. (2011). Group 4 metal initiators for the controlled stereoselective polymerization of lactide monomers. Chem. Commun. 47, 4796–4798. doi: 10.1039/c1cc10149h
Chang, Y.-N., Lee, P.-Y., Zou, X.-R., Huang, H.-F., Chen, Y.-W., and Liang, L.-C. (2016). Aluminum complexes containing biphenolate phosphine ligands: synthesis and living ring-opening polymerization catalysis. Dalton Trans. 45, 15951–15962. doi: 10.1039/C6DT02143C
Chang, Y.-N., and Liang, L.-C. (2007). Preparation and structural characterization of group 1 metal complexes containing a chelating biphenolato phosphine ligand. Inorg. Chim. Acta 360, 136–142. doi: 10.1016/j.ica.2006.07.050
Chen, H. L., Dutta, S., Huang, P. Y., and Lin, C. C. (2012). Preparation and characterization of aluminum alkoxides coordinated on salen-type ligands: highly stereoselective ring-opening polymerization of rac-lactide. Organometallics 31, 2016–2025. doi: 10.1021/om201281w
Chmura, A. J., Davidson, M. G., Jones, M. D., Lunn, M. D., and Mahon, M. F. (2006). Group 4 complexes of amine bis(phenolate)s and their application for the ring opening polymerisation of cyclic esters. Dalton Trans. 2006, 887–889. doi: 10.1039/B513345A
Fryzuk, M. D., Giesbrecht, G. R., Olovsson, G., and Rettig, S. J. (1996). Synthesis and characterization of four- and five-coordinate organoaluminum complexes incorporating the amido diphosphine ligand system N(SiMe2CH2PiPr2)2. Organometallics 15, 4832–4841. doi: 10.1021/om9604583
Fryzuk, M. D., Giesbrecht, G. R., and Rettig, S. J. (1998). Pyramidal inversion at phosphorus facilitated by the presence of proximate Lewis acids. Coordination chemistry of group 13 elements with the macrocyclic bis(amidophosphine) ligand P2N2 (P2N2 = PhP(CH2SiMe2NSiMe2CH2)2PPh). Inorg. Chem. 37, 6928–6934. doi: 10.1021/ic9805978
Gao, B., Li, D. N., Li, Y. H., Duan, Q., Duan, R. L., and Pang, X. (2015). Ring-opening polymerization of lactide using chiral salen aluminum complexes as initiators: high productivity and stereoselectivity. N. J. Chem. 39, 4670–4675. doi: 10.1039/C5NJ00469A
Gendler, S., Segal, S., Goldberg, I., Goldschmidt, Z., and Kol, M. (2006). Titanium and zirconium complexes of dianionic and trianionic amine - Phenolate-type ligands in catalysis of lactide polymerization. Inorg. Chem. 45, 4783–4790. doi: 10.1021/ic052120j
He, L.-P., Liu, J.-Y., Pan, L., and Li, Y.-S. (2008). Ethylene polymerization of the new titanium complexes bearing a phosphine oxide-bridged bisphenolato ligand. J. Polym. Sci. A 46, 7062–7073. doi: 10.1002/pola.23012
Hillmyer, M. A., and Tolman, W. B. (2014). Aliphatic polyester block polymers: renewable, degradable, and sustainable. Acc. Chem. Res. 47, 2390–2396. doi: 10.1021/ar500121d
Hormnirun, P., Marshall, E. L., Gibson, V. C., Pugh, R. I., and White, A. J. (2006). Study of ligand substituent effects on the rate and stereoselectivity of lactide polymerization using aluminum salen-type initiators. Proc. Natl. Acad. Sci. U.S.A. 103, 15343–15348. doi: 10.1073/pnas.0602765103
Hsu, Y.-L., and Liang, L.-C. (2010). Alkali metal complexes of a tert-butylphosphine-bridged biphenolate ligand. Organometallics 29, 6201–6208. doi: 10.1021/om100495e
Huang, M.-H., and Liang, L.-C. (2004). Amido pincer complexes of palladium: synthesis, structure, and catalytic Heck reaction. Organometallics 23, 2813–2816. doi: 10.1021/om049888g
Huang, Y., Wang, W., Lin, C. C., Blake, M. P., Clark, L., Schwarz, A. D., et al. (2013). Potassium, zinc, and magnesium complexes of a bulky OOO-tridentate bis(phenolate) ligand: synthesis, structures, and studies of cyclic ester polymerisation. Dalton Trans. 42, 9313–9324. doi: 10.1039/c3dt50135c
Hung, Y.-T., Chen, M.-T., Huang, M.-H., Kao, T.-Y., Liu, Y.-S., and Liang, L.-C. (2014). Catalytic Sonogashira couplings mediated by an amido pincer complex of palladium. Inorg. Chem. Front. 1, 405–413. doi: 10.1039/c3qi00086a
Jones, M. D., Brady, L., McKeown, P., Buchard, A., Schäfer, P. M., Thomas, L. H., et al. (2015). Metal influence on the iso- and hetero-selectivity of complexes of bipyrrolidine derived salan ligands for the polymerisation of rac-lactide. Chem. Sci. 6, 5034–5039. doi: 10.1039/C5SC01819F
Kamber, N. E., Jeong, W., Waymouth, R. M., Pratt, R. C., Lohmeijer, B. G., and Hedrick, J. L. (2007). Organocatalytic ring-opening polymerization. Chem. Rev. 107, 5813–5840. doi: 10.1021/cr068415b
Kirk, S. M., Kociok-Kohn, G., and Jones, M. D. (2016). Zirconium vs aluminum salalen initiators for the production of biopolymers. Organometallics 35, 3837–3843. doi: 10.1021/acs.organomet.6b00718
Klitzke, J. S., Roisnel, T., Kirillov, E., Casagrande, O., and Carpentier, J.-F. (2014a). Discrete O-Lactate and β-alkoxybutyrate aluminum pyridine–bis(naphtholate) complexes: models for mechanistic investigations in the ring-opening polymerization of lactides and β-lactones. Organometallics 33, 5693–5707. doi: 10.1021/om401214q
Klitzke, J. S., Roisnel, T., Kirillov, E., Casagrande, O., and Carpentier, J.-F. (2014b). Yttrium– and aluminum–bis(phenolate)pyridine complexes: catalysts and model compounds of the intermediates for the stereoselective ring-opening polymerization of racemic lactide and β-butyrolactone. Organometallics 33, 309–321. doi: 10.1021/om401047r
Lee, C.-L., Lin, Y.-F., Jiang, M.-T., Lu, W.-Y., Vandavasi, J. K., Wang, L.-F., et al. (2017). Improvement in aluminum complexes bearing schiff bases in ring-opening polymerization of ε-caprolactone: a five-membered-ring system. Organometallics 36, 1936–1945. doi: 10.1021/acs.organomet.7b00068
Lee, P.-Y., and Liang, L.-C. (2009). Synthesis and structural characterization of five-coordinate aluminum complexes containing diarylamido diphosphine ligands. Inorg. Chem. 48, 5480–5487. doi: 10.1021/ic802030d
Lee, W.-Y., and Liang, L.-C. (2005). Organoaluminium complexes incorporating an amido phosphine chelate with a pendant amine arm. Dalton Trans. 1952–1956. doi: 10.1039/b502873f
Liang, L.-C. (2006). Metal complexes of chelating diarylamido phosphine ligands. Coord. Chem. Rev. 250, 1152–1177. doi: 10.1016/j.ccr.2006.01.001
Liang, L.-C., Chen, F.-Y., Huang, M.-H., Cheng, L.-C., Li, C.-W., and Lee, H. M. (2010). Aluminium complexes of bidentate N,O- and N,N-ligands derived from oxidative functionalization of amido phosphines: synthesis, structure and reactivity. Dalton Trans. 39, 9941–9951. doi: 10.1039/c0dt00418a
Liang, L.-C., Chien, C.-C., Chen, M.-T., and Lin, S.-T. (2013a). Zirconium and hafnium complexes containing N-alkyl-substituted amine biphenolate ligands: unexpected ligand degradation and divergent complex constitutions governed by N-alkyls. Inorg. Chem. 52, 7709–7716. doi: 10.1021/ic400891b
Liang, L.-C., Chien, P.-S., and Huang, M.-H. (2005a). Catalytic suzuki coupling reactions by amido phosphine complexes of palladium. Organometallics 24, 353–357. doi: 10.1021/om0492395
Liang, L.-C., Chien, P.-S., and Huang, Y.-L. (2006). Intermolecular arene C-H activation by nickel(II). J. Am. Chem. Soc. 128, 15562–15563. doi: 10.1021/ja065505p
Liang, L.-C., Hsu, Y.-L., and Lin, S.-T. (2011). Group 4 complexes of a tert-butylphosphine-bridged biphenolate ligand. Inorg. Chem. 50, 3363–3372. doi: 10.1021/ic102194z
Liang, L.-C., Huang, M.-H., and Hung, C.-H. (2004). Aluminum complexes incorporating bidentate amido phosphine ligands. Inorg. Chem. 43, 2166–2174. doi: 10.1021/ic035373q
Liang, L.-C., Lee, W.-Y., and Hung, C.-H. (2003a). Amido phosphine complexes of zinc. Inorg. Chem. 42, 5471–5473. doi: 10.1021/ic0346228
Liang, L.-C., Lin, J.-M., and Hung, C.-H. (2003b). Nickel(II) complexes of bis(2-diphenylphosphinophenyl)amide. Organometallics 22, 3007–3009. doi: 10.1021/om030237e
Liang, L.-C., Lin, J.-M., and Lee, W.-Y. (2005b). Benzene C-H activation by platinum(II) complexes of bis(2-diphenylphosphinophenyl)amide. Chem. Commun. 2462–2464. doi: 10.1039/B501520K
Liang, L.-C., Lin, S.-T., and Chien, C.-C. (2013b). Aluminum complexes of tridentate amine biphenolate ligands containing distinct N-alkyls: synthesis and catalytic ring-opening polymerization. J. Chin. Chem. Soc. 60, 710–718. doi: 10.1002/jccs.201200559
Liang, L.-C., Lin, S.-T., and Chien, C.-C. (2013c). Lithium complexes of tridentate amine biphenolate ligands containing distinct N-alkyl substituents. Polyhedron 52, 1090–1095. doi: 10.1016/j.poly.2012.06.069
Liang, L.-C., Lin, S.-T., and Chien, C.-C. (2013d). Titanium complexes of tridentate aminebiphenolate ligands containing distinct N-alkyls: profound N-substituent effect on ring-opening polymerization catalysis. Inorg. Chem. 52, 1780–1786. doi: 10.1021/ic301551v
Liang, L.-C., Lin, S.-T., Chien, C.-C., and Chen, M.-T. (2013e). Zirconium and hafnium complexes containing N-alkyl substituted amine biphenolate ligands: coordination chemistry and living ring-opening polymerization catalysis. Dalton Trans. 42, 9286–9293. doi: 10.1039/C3DT50152C
MacDonald, J. P., Sidera, M., Fletcher, S. P., and Shaver, M. P. (2016). Living and immortal polymerization of seven and six membered lactones to high molecular weights with aluminum salen and salan catalysts. Eur. Polym. J. 74, 287–295. doi: 10.1016/j.eurpolymj.2015.11.032
MacLachlan, E. A., and Fryzuk, M. D. (2005). A new arene-bridged diamidophosphine ligand and its coordination chemistry with zirconium(IV). Organometallics 24, 1112–1118. doi: 10.1021/om049165x
MacLachlan, E. A., Hess, F. M., Patrick, B. O., and Fryzuk, M. D. (2007). New side-on bound dinitrogen complexes of zirconium supported by an arene-bridged diamidophosphine ligand and their reactivity with dihydrogen. J. Am. Chem. Soc. 129, 10895–10905. doi: 10.1021/ja073753v
McKeown, P., Davidson, M. G., Kociok-Köhn, G., and Jones, M. D. (2016). Aluminium salalens vs. salans: “Initiator Design” for the isoselective polymerisation of rac-lactide. Chem. Commun. 52, 10431–10434. doi: 10.1039/C6CC05795K
Ovitt, T. M., and Coates, G. W. (2000). Stereoselective ring-opening polymerization of rac-lactide with a single-site, racemic aluminum alkoxide catalyst: synthesis of stereoblock poly(lactic acid). J. Polym. Sci. A 38, 4686–4692. doi: 10.1002/1099-0518(200012)38:1+<4686::AID-POLA80>3.0.CO;2-0
Paine, T. K., Weyhermüller, T., Slep, L. D., Neese, F., Bill, E., Bothe, E., et al. (2004). Nonoxovanadium(IV) and oxovanadium(V) complexes with mixed O, X, O-donor ligands (X = S, Se, P., or PO). Inorg. Chem. 43, 7324–7338. doi: 10.1021/ic040052f
Pang, X., Duan, R. L., Li, X., Hu, C. Y., Wang, X. H., and Chen, X. S. (2018). Breaking the paradox between catalytic activity and stereoselectivity: rac-lactide polymerization by trinuclear salen-Al complexes. Macromolecules 51, 906–913. doi: 10.1021/acs.macromol.7b02662
Phomphrai, K., Chumsaeng, P., Sangtrirutnugul, P., Kongsaeree, P., and Pohmakotr, M. (2010). Reverse orders of reactivities in the polymerization of cyclic esters using N2O2 aluminium alkoxide complexes. Dalton Trans. 39, 1865–1871. doi: 10.1039/B919340E
Robert, C., Schmid, T. E., Richard, V., Haquette, P., Raman, S. K., Rager, M. N., et al. (2017). Mechanistic aspects of the polymerization of lactide using a highly efficient aluminum(III) catalytic system. J. Am. Chem. Soc. 139, 6217–6225. doi: 10.1021/jacs.7b01749
Sarazin, Y., and Carpentier, J. F. (2015). Discrete cationic complexes for ring-opening polymerization catalysis of cyclic esters and epoxides. Chem. Rev. 115, 3564–3614. doi: 10.1021/acs.chemrev.5b00033
Save, M., Schappacher, M., and Soum, A. (2002). Controlled ring-opening polymerization of lactones and lactides initiated by lanthanum isopropoxide, 1. general aspects and kinetics. Macromol. Chem. Phys. 203, 889–899. doi: 10.1002/1521-3935(20020401)203:5/6<889::AID-MACP889>3.0.CO;2-O
Siefert, R., Weyhermuller, T., and Chaudhuri, P. (2000). Isolation, structural and spectroscopic investigations of complexes with tridentate [OPO] and {OOO} donor ligands. J. Chem. Soc. Dalton Trans. 4656–4663. doi: 10.1039/B005693F
Stopper, A., Okuda, J., and Kol, M. (2012). Ring-opening polymerization of lactide with Zr complexes of {ONSO} ligands: from heterotactically inclined to isotactically inclined poly(lactic acid). Macromolecules 45, 698–704. doi: 10.1021/ma2023364
Su, W.-J., and Liang, L.-C. (2018). Elusive scorpionates: C3-symmetric, formally dianionic, facially tridentate ligands. Inorg. Chem. 57, 553–556. doi: 10.1021/acs.inorgchem.7b02884
Tang, Z. H., and Gibson, V. C. (2007). rac-Lactide polymerization using aluminum complexes bearing tetradentate phenoxy-amine ligands. Eur. Polym. J. 43, 150–155. doi: 10.1016/j.eurpolymj.2006.09.023
Taniyama, N., Ohki, Y., and Tatsumi, K. (2014). Synthesis of V/Fe/S clusters using vanadium(III) thiolate complexes bearing a phenoxide-based tridentate ligand. Inorg. Chem. 53, 5438–5446. doi: 10.1021/ic4030603
Tanke, R. S., Holt, E. M., and Crabtree, R. H. (1991). Ruthenium in an O-donor environment - properties and reactions of ETA-3-(RPO(C6H4O)2)2-, ETA-3-(CPCO(PO(OET)2)3)- and ETA-3-HC(POPH2)3 complexes of ruthenium. Inorg. Chem. 30, 1714–1719. doi: 10.1021/ic00008a009
Thomas, C. M. (2010). Stereocontrolled ring-opening polymerization of cyclic esters: synthesis of new polyester microstructures. Chem. Soc. Rev. 39, 165–173. doi: 10.1039/B810065A
Wichmann, O., Sillanpaa, R., and Lehtonen, A. (2012). Structural properties and applications of multidentate O,N,O,X ' aminobisphenolate metal complexes. Coord. Chem. Rev. 256, 371–392. doi: 10.1016/j.ccr.2011.09.007
Zelikoff, A. L., Kopilov, J., Goldberg, I., Coates, G. W., and Kol, M. (2009). New facets of an old ligand: titanium and zirconium complexes of phenylenediamine bis(phenolate) in lactide polymerisation catalysis. Chem. Commun. 6804–6806. doi: 10.1039/B915211C
Zhang, S.-W., Zhang, G.-B., Lu, L.-P., and Li, Y.-S. (2013). Novel vanadium(III) complexes with tridentate phenoxy-phosphine [O,P(= O),O] ligands: synthesis, characterization, and catalytic behavior of ethylene polymerization and copolymerization with 10-undecen-1-ol. J. Polym. Sci. A 51, 844–854. doi: 10.1002/pola.26441
Zhong, Z. Y., Dijkstra, P. J., and Feijen, J. (2002). (salen)Al -mediated, controlled and stereoselective ring-opening polymerization of lactide in solution and without solvent: synthesis of highly isotactic polylactide stereocopolymers from racemic D,L-lactide. Angew. Chem. Int. Ed. 41, 4510–4513. doi: 10.1002/1521-3773(20021202)41:23<4510::AID-ANIE4510>3.0.CO;2-L
Keywords: aluminum, biphenolate, ring-opening polymerization, lactone, lactide, catalyst
Citation: Zou X-R, Chang Y-N, Huang K-W and Liang L-C (2018) Enhanced Reactivity of Aluminum Complexes Containing P-Bridged Biphenolate Ligands in Ring-Opening Polymerization Catalysis. Front. Chem. 6:607. doi: 10.3389/fchem.2018.00607
Received: 27 September 2018; Accepted: 26 November 2018;
Published: 13 December 2018.
Edited by:
Hsuan-Ying Chen, Department of Medicinal and Applied Chemistry, Kaohsiung Medical University, TaiwanReviewed by:
Jui-Hsien Huang, National Changhua University of Education, TaiwanCopyright © 2018 Zou, Chang, Huang and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kuo-Wei Huang, aGt3QGthdXN0LmVkdS5zYQ==
Lan-Chang Liang, bGNsaWFuZ0BtYWlsLm5zeXN1LmVkdS50dw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.