Abstract
Development of a simple and easy handing process for preparation of multifunctional heterogenous catalysts and exploration of their applications in sequential organic transformation are of great significance in heterogeneous asymmetric catalysis. Herein, through the utilization of a anion–bonding strategy, we anchor conveniently both organic bases and chiral ruthenium complex into the nanopores of Me-FDU−12, fabricating a Lewis base/Ru bifunctional heterogeneous catalyst. As we envisaged, cyclic amine as a Lewis base promotes an intermolecular aza–Michael addition between enones and arylamines, affording γ-secondary amino ketones featuring with aryl motif, whereas ruthenium/diamine species as catalytic promoter boosts an asymmetric transfer hydrogenation of γ-secondary amino ketones to γ-secondary amino alcohols. As expected, both enhance synergistically the aza–Michael addition/asymmetric transfer hydrogenation one–pot enantioselective organic transformation, producing chiral γ-secondary amino alcohols with up to 98% enantioselectivity. Unique features, such as operationally simple one–step synthesis of heterogeneous catalyst, homo–like catalytic environment as well as green sustainable process make this heterogeneous catalyst an attracting in a practical preparation of optically pure pharmaceutical intermediates of antidepressants.
Introduction
With the extensive explorations of mesoporous silicas as supports in catalysis, various strategies have been employed to construct supported molecular catalysts(Song, 2005; Heitbaum et al., 2006; Yang et al., 2007; Margelefsky et al., 2008; Thomas and Raja, 2008; Bartók, 2010; Yu and He, 2012). Of all the well–established strategies, immobilization though a hydrogen–bonding have attracting a great deal of interest since it allow the facile anchoring of small molecular catalysts on a support with a suitable counterion of organometallic complexes (Fraile et al., 2009). This simple and practical approach endows the immobilized catalyst with an original catalytic environment as its homogeneous counterpart which is helpful for the catalytic reaction. These features are beneficial to construct highly efficient heterogeneous catalysts like their corresponding homogeneous counterparts, bridging the gap between homogeneous and heterogeneous catalysis. Recently, some remarkable works developed by several groups have utilized F3 anion to bond cationic complexes, fabricating various supported molecular catalysts with high catalytic efficiency (Bianchini et al., 2001; Raja et al., 2003; Rouzaud et al., 2003; O'Leary et al., 2004; Wang et al., 2006; McDonagh et al., 2007; Xu et al., 2012). These explorations overcome nicely the shortage of low catalytic efficiency of heterogeneous catalyst because of highly catalytic nature of cationic active species themselves, realizing superior catalytic performance relatively to their corresponding neutral counterparts. Similar to the F3 anion hydrogen–bonding method, recent reports also find that the anions are used for fabrication of highly recyclable catalysts (Jones et al., 2003; Wiench et al., 2009; Shu et al., 2015; Liao et al., 2017; Xia et al., 2017). An outstanding example was reported by Toste and co–workers (Shu et al., 2015). In this work, the Au– complexes (Ph3PAuBF4) were tethered within the nanochannels of SBA−15 material via a hydrogen–bonding method between the anion and the inner silanols of SBA−15. This as–made heterogeneous cationic gold (I) catalyst not only performs a superior regioselectivity to its corresponding homogeneous one in the cyclization of alkynoic acids. More importantly, through the suitable coordination with chiral phosphine ligands, the obtained chiral cationic gold (I) catalyst also shows the dramatic enhancement of enantioselectivities in the enantioselective lactonization reactions relative to its homogeneous one. In particular, high recyclability (11 runs) opens great potential in a practical application. Quite recently, we also take advantage of this anion–bonding method to bond Au/carbine complexes, together with a covalent–bonding of neutral Ru/diamine complexes, to fabricate a recyclable heterogeneous catalysts for the enantioselective synthesis of halohydrins through a hydration/asymmetric transfer hydrogenation cascade process (Xia et al., 2017). Therefore, further development of this easy handing hydrogen–bonding method and direct immobilization of dual active centers on mesoporous silica for multi–step sequential organic transformation is still of great significance in heterogeneous asymmetric catalysis.
Large–pore mesoporous silicas, like FDU−12 (Fan et al., 2005; Ma et al., 2010; Li et al., 2015), possess large size of regular mesopores in their silicate networks, which have great potential in assembly of various heterogeneous catalysts (Li et al., 2012; Yan et al., 2013; Chen et al., 2014). At first, large space of nanopores can accommodate large steric organometallic complexes or multiply active species, which are beneficial to fabricate bi– or multi–functionalized heterogeneous catalysts. Also, large space of mesopores can avoid the accumulation of multiple active species, which can reduce efficiently the cross–interactions of multiple active species and overcome the shortage of homogeneous catalysis. Furthermore, large size of regular mesopores benefits mass transport, which can lead to an improved catalytic efficiency relative to those small–sizes of mesoporous counterparts. More importantly, through a control of molecular catalysts in an adjacent position on a support, a potential cooperative effect may make some unfeasible or lowly efficient tandem reactions in homogeneous condition into possibility. Therefore, the utilization of advantages of large–pore mesoporous FDU−12 in fabrication of multifunctional catalyst and the exploration of its application in a tandem reaction to promote reactivity, enantioselectivity and stability are highly desirable.
In this contribution, we employ a large–pore size of FDU−12 as a support and assemble a Lewis base and a chiral ruthenium/diamine dual species within its regular nanopores via a anion–bonding approach, fabricating a bifunctional heterogeneous catalyst. This catalyst performs an efficient synthesis of valuable pharmaceutical intermediates of optically pure γ-secondary amino alcohols that had been explored extensively by various asymmetric catalysis in homogeneous conditions recently (Gao and Sharpless, 1988; Robertson et al., 1988; Kakei et al., 2004; Liu et al., 2005; Fujima et al., 2006; Geng et al., 2011; Träff et al., 2011; Wang et al., 2013a, 2015; Zhou et al., 2014; Hu et al., 2015; Wu et al., 2017). Benefits of this heterogeneous catalysis not only overcome the drawbacks of expensive transition–metal recycle and transition–metal contamination in homogeneous catalysis system, but also realize multi-step reactions to yield a series of synthetically useful amino alcohols under mild reaction conditions in environmental friendly fashion.
Experimental
Catalyst 1 preparation
In a typical route, (Step I: Synthesis of mesoporous FDU−12) directing template EO106PO70EO106 (Pluronic F127, 0.5 g), 1,3,5–trimethylbenzene (TMB, 0.6 g) and KCl (1.25 g) were added to a hydrochloric acid solution (50 mL, 1.0 M) at 15°C. After stirring for 1.0 h, tetraethyl orthosilicate (TEOS, 2.08 g) was slowly added to previous solution. The resulting mixture was vigorously stirred for another 24 h at 15°C. Subsequently, the obtained suspension was kept in a Teflon–lined autoclave at 170°C for 24 h under static conditions. After cooling to room temperature, the as-made FDU-12 was seperated through filtration and dried. (Step II: Preparation of mesoporous Me–FDU−12 by protection of the outer facial silanols) The collected FDU-12 (1.0 g) were dispersed in 25 mL anhydrous toluene, and HMDS [(CH3)3Si)2N] (10 mL, 0.050 mol) was then added. The suspension was kept and stirred for 12 h at room temperature. After filtration and rinse with excess water and ethanol, the collected solids was redistributed to 120 mL of ammonium nitrate solution (80 mg (1.0 mmol) in 120 mL (95%) of ethanol), and the cloudy solution was stirred at 60°C for 10 h. Once finsihed, the solids were filtered and washed with excess water and ethanol, and dried under vacuo, affording trimethysilylated Me–FDU−12 as a white powder (1.21 g). (Step III: Immobilization of (DABCO)BF4(1–(chloromethyl)−1,4–diazabicyclo[2.2.2]octanium tetrafluoroborate) and (MesityleneRuTsDPEN)(BF4) (TsDPEN = N–((S,S)−2–amino−1,2–diphenylethyl)−4–methylbenzenesulfonamide). The Me–FDU−12 (0.50 g) was firstly dispersed into dry CH2Cl2 (10.0 mL) within a round–bottom flask at 25°C, then the freshly prepared [MesityleneRuTsDPEN] BF4 (43.50 mg, 0.064 mmol) was rapidly added. The resulting red suspension was stirred for 12 h at 25°C. After that, the newly prepared (DABCO)BF4 (74.42 mg, 0.30 mmol) was added to previous solution, and the reaction solution was stirred at 25°C for another 12 h. Once finished, the crude solid catalyst was obtained via filteration. Finally, the unreacted starting materials was removed by Soxhlet extraction using dry CH2Cl2, furnishing desired (DABCO)BF4@(MesityleneRuTsDPEN)BF4@Me–FDU−12 (1) (0.57 g) as a faint–yellow powder. The Ru loadings was determined by inductively coupled plasma optical emission spectrometer (ICP–OES) analysis, showing that 10.40 mg (0.1020 mmol of Ru) per gram of catalyst. Elemental analysis for the fresh catalyst 1 (%):C 15.03, H 3.43, N 0.57, S 0.33.; and for the recycled catalyst after fifth run: C 14.45, H 3.35, N 0.48, S 0.29. 13C CP/MAS NMR (161.9 MHz): 149.2–121.7 (C of Ph and Ar groups), 108.4, 101.5 (CH3 of Arene groups), 73.6–67.3 (CH of –NCH–Ph), 66.2–59.3 (CH2 of –N+CH2- in DABCO moiety, and CH2 of –OCH2CH2O– groups in F127), 53.2, 46.3 (CH2 of N(CH2)3- in DABCO moiety), 31.1 (CH2 of –CH2Ph), 23.3 (CH3 of mesitylene), 19.4 (CH3 of –OCH(CH3)CH2O– in F127), 2.8 (CH3 of –Si(CH3)3) ppm; 29Si MAS NMR (79.4 MHz): Q3 (δ = −104.8 ppm), Q4 (δ = −114.3 ppm).
General procedure for 1-catlayzed aza–Michael addition/ATH one–pot enantioselective cascade reactions
In a typical procedure, catalyst 1 (19.61 mg, 2.0 μmol of Ru, according to ICP–OES analysis), HCO2Na (68.0 mg, 1.0 mmol), enones (0.10 mmol), amines (0.11 mmol) and 2.0 mL of co-solvents (H2O/iPrOH, v/v = 1/1) were successively added to a round bottom flask. The resulting mixture was heated to 40°C and stirred for 8–24 h until the fully consumption of enones. Upon completion of reaction (monitoring by TLC), the catalyst 1 was recycled via centrifugation (10,000 rpm). The transparent solution was extracted using ethyl acetate (3 × 3.0 mL). The combined organic phase were washed with brine (2 × 5 mL) and dried with Na2SO4. After removal of EtOAc under reduced evaporation, the crude residue was purified by flash column chromatography to release the desired products. The enantiomeric excess was determined by HPLC equipped with a Daicel chiralcel column (Ø 0.46 cm, L 25 cm) and an UV–Vis detector.
Results and discussion
Synthesis and structural characterization
The anion–bonding of DABCO molecules as well as chiral ruthenium/diamine complexes immobilized in the nanopores of trimethysilylated FDU−12 (Me–FDU−12), in an abbreviated form as (DABCO)BF4@(MesityleneRuTsDPEN)BF4@Me–FDU−12 (1), ((DABCO)BF4 = 1–(chloromethyl)−1,4–diazabicyclo[2.2.2] octanium tetrafluoroborate) (Li and Liu, 2004; Li et al., 2005; Chauhan et al., 2014; Wen et al., 2014; Ying et al., 2014), (MesityleneRuTsDPEN)BF4 (Hashiguchi et al., 1995; Ohkuma et al., 2007): where TsDPEN = N–((S,S)−2–amino−1,2–diphenylethyl)−4–methylbenzenesulfonamide), was performed as shown in Figure 1. The mesoporous Me–FDU−12 was firstly obtained according to the reported approach with slightly modification (Fan et al., 2005; Ma et al., 2010), where the outer facial silanols of FDU−12 were protected through the treatment with ((CH3)3Si)2N compounds. Continuous hydrogen–bonding of (DABCO)BF4 and (MesityleneRuTsDPEN)BF4 to the inner silanols of nanopores in Me–FUD−12 then led to the crude catalyst. Finally, the well-defined catalyst 1 was obtained through a strict Soxhlet extraction from its corresponding crude form (see Figures S1, S2 of ESI).
Figure 1
Figure 2, the solid–state 13C cross–polarization (CP)/magic angle spinning (MAS) NMR spectroscopy of as-synthesized catalyst 1, provided the direct evidence to the successful anchor of both anion functionalized chiral ruthenium/diamine complexes and DABCO–molecules species within the internal silanols of mesopores in Me–FDU−12 since both characteristic carbon signals could be observed clearly. In the part of DABCO–molecules, the characteristic peaks at 53.2 and 46.3 ppm were corresponded to the cyclic carbon atoms in DABCO moiety, which were similar to those attained with its homogeneous counterpart (Chauhan et al., 2014), suggesting the DABCO–functionality have been successfully incoporated in catalyst 1. In the part of chiral ruthenium/diamine–complexes, besides the general carbon peaks around 70 ppm for the carbon atoms of –NCH groups and around 130 ppm for the carbon atoms of aromatic groups from TsDPEN, the characteristic peaks found at 108.4 and 101.5 ppm could be attributed to the carbon of mesitylene, while that at 23.3 ppm belonged to the carbon atoms of the CH3 groups of mesitylene. These carbons signals are the same as those of its homogeneous (MesityleneRuTsDPEN)BF4 (Ohkuma et al., 2007), demonstrating its well–defined single–site ruthenium center in catalyst 1. Furthermore, to confirm DABCO molecules and chiral ruthenium/diamine–complexes dual active centers immobilized via the anions, the solid–state 19F MAS NMR spectrum of catalyst 1 was used to compare with those of two corresponding single immobilized DABCO molecules and chiral ruthenium/diamine–complexes on Me-FDU-12. It was found that the wide F signals between −147 and −151 ppm are assigned to F signals in anions interacting with dual centers via the hydrogen bonding since they are similar to those of the their corresponding single immobilied DABCO molecules or chiral ruthenium/diamine–complexes solid-state 19F NMR spectra (see Figure S3 of ESI for their solid–state and liquid–state 19F NMR spectra). The other signals (at −181.3, −155.1, −101.6, and −74.2 ppm) denoted by asterisks were responded to the general “bulk” species that often appeared in 19F MAS NMR spectroscopy.
Figure 2
Figures 3, 4 revealed the mesostructure and morphology of catalyst 1, which were characterized by scanning electron microscopy (SEM), nitrogen adsorption–desorption technique, transmission electron microscopy (TEM) with chemical mapping technique. The nitrogen adsorption–desorption isotherms of FDU−12, Me–FDU−12 and catalyst 1 presented the typical IV–type isotherms with H2 hysteresis loop (Figure 3), which are similar to those reported in the literatures, confirming its mesoporous structure(Fan et al., 2005; Ma et al., 2010). The SEM image of catalyst 1 disclosed the hexagonal arrays of uniform cages (Figure 4a) whereas the TEM images of catalyst 1 (Figures 4b,c) demonstrated the FDU−12–type face–centered–cubic mesostructures. Both confirmed that catalyst 1 retained the general morphological structure of FDU−12 after the immobilization of dual active species. Of significantly clear TEM image coupled with a chemical mapping (see Figure S4 of ESI) well demonstrated the uniformly distribution of ruthenium active centers within the inorganic silicate network of catalyst 1.
Figure 3
Figure 4
All these characterizations and analyses elucidated that the obtained heterogeneous catalyst 1 had an operationally simple prepared procedure, well-defined dual active species as well as uniformly distributed ruthenium active centers, which would have a positive impact on its catalytic activity discussed below.
Catalytic performance of catalyst 1
Cationic chiral N–sulfonylated diamine functionalized organometallic complexes, a type of catalytically efficient active species for asymmetric transfer hydrogenation (ATH) reaction, have obvious superiority in catalytic process with respect to their corresponding neutral ones since cationic complexes as real active species based on mechanism study could significantly favor asymmetric transfer hydrogenation process of ketones especially in an aqueous medium (Noyori and Hashiguchi, 1997; Liu et al., 2004a,b; Gladiali and Alberico, 2006; Wu et al., 2006, 2008; Ikariya and Blacker, 2007; Ohkuma et al., 2007; Wang et al., 2008, 2011, 2013b; Ding et al., 2013; Yang et al., 2014, 2016; Zhang et al., 2015). Therefore, direct immobilization of cationic ruthenium active species within the FDU−12 is beneficial to enhance ATH transformation. In particular, utilization of Me–FDU−12 as a support could ensure the dual active centers into the inner nanopores due the control of the protection step before the removal of surfactants during the synthetic procedure in the experiment part, where the potentially adjacent position of DABCO–molecule and chiral ruthenium/diamine–complex could promote cooperatively its catalytic performance.
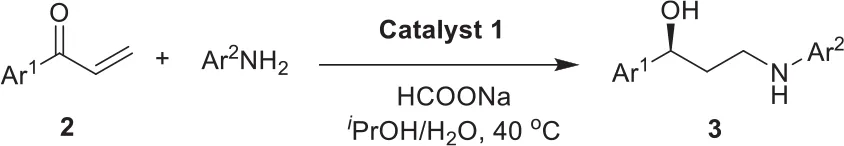
With the well-established bifunctional catalyst 1 in hand, we transferred our attention to its catalytic and enantioselective performance in intermolecular aza–Michael addition/ATH one–pot process through the use of tandem reaction of acrylophenone and phenylamine as an example at the beginning. Based on the preliminary optimization of reaction conditions (see Table S1 of ESI), the tandem reaction was carried out with the 2.0 mol% of ruthenium–loading in 1 as a catalyst, the HCOONa as a hydrogen resource in the mixed solvents (H2O/iPrOH, v/v = 1/1) at 40°C reaction temperature. It was found that the organic transformation of 1–phenylprop−2–enone and aniline could provide the target product 3a in 92% yield with 96% ee in 8 h. Such a yield was markedly higher than that of 79% yield obtained by the use of mixed homogeneous (DABCO)BF4 and homogeneous (MesityleneRuTsDPEN)BF4 as dual catalysts, even in a prolonged reaction time (Table 1, entry 1 and 2). This comparative analysis well demonstrated the existence of cross–interaction between (DABCO)BF4 and chiral (MesityleneRuTsDPEN)BF4 in the homogeneous catalytic conditions. Further products analysis found that about 19% of intermediate 1–phenyl−3–(phenylamino)propanone could not be converted, suggesting the DABCO part interfered in the reduction of (MesityleneRuTsDPEN)BF4. In other words, high catalytic activity of catalyst 1 demonstrated that the dual active species in catalyst 1 could overcome efficiently this kind of cross–interaction because the co-existent dual active had a potential site–isolated effect in the nanopores of catalyst 1. Meanwhile, the obtained 96% ee in this tandem reaction was comparable to that of 97% ee obtained in single–step ATH transformation of 1–phenyl−3–(phenylamino)propanone, disclosing that the chiral catalytic environment of single–site (MesityleneRuTsDPEN)BF4 species in catalyst 1 was not affected during the immobilization procedure. This judgment could be further confirmed by a comparison of the XPS spectra. It was found that catalyst 1 and its homogeneous counterpart ((MesityleneRuTsDPEN)BF4) had the similar Ru 3d5/2 electron binding energies (281.88 vs. 281.81 eV), as displayed in Figure 5. This finding confirmed the presence of the similar catalytic environment as both had the similar electronic environment of active ruthenium centers.
Table 1
 | ||||
|---|---|---|---|---|
| Entry | Ar1, Ar2(3) | Time (h) | %Yieldb | %eec |
| 1 | Ph, Ph (3a) | 8 | 92 | 96 |
| 2 | Ph, Ph (3a) | 24 | 79 | 95d |
| 3 | Ph, 4–ClPh (3b) | 10 | 89 | 95 |
| 4 | Ph, 3–ClPh (3c) | 10 | 81 | 94 |
| 5 | Ph, 2–ClPh (3d) | 10 | 92 | 97 |
| 6 | Ph, 4–BrPh (3e) | 10 | 91 | 96 |
| 7 | Ph, 4–NO2Ph (3f) | 10 | 83 | 98 |
| 8 | Ph, 3–NO2Ph (3g) | 10 | 85 | 96 |
| 9 | Ph, 3,4–Me2Ph (3h) | 12 | 76 | 96 |
| 10 | Ph, 3,5–Me2Ph (3i) | 12 | 73 | 95 |
| 11 | Ph, 3–Cl−4–MePh (3j) | 12 | 72 | 96 |
| 12 | Ph, 3–MeOPh (3k) | 12 | 81 | 96 |
| 13 | 4–FPh, Ph (3l) | 10 | 92 | 92 |
| 14 | 4–ClPh, Ph (3m) | 10 | 89 | 93 |
| 15 | 4–BrPh, Ph (3n) | 10 | 91 | 94 |
| 16 | 4–IPh, Ph (3o) | 10 | 88 | 94 |
| 17 | 4–MePh, Ph (3p) | 12 | 82 | 94 |
| 18 | 4–MeOPh, Ph (3q) | 12 | 80 | 95 |
The aza–Michael addition/ATH one–pot enantioselective tandem reactions to prepare chiral γ-secondary amino alcoholsa.
Reaction conditions: catalyst 1 (19.61 mg, 2.0 μmol of Ru based on ICP–OES analysis), enones (0.10 mmol), amines (0.11 mmol), HCOONa (1.0 mmol), 2.0 mL of iPrOH/H2O (v/v = 1: 1), and reaction time (8–24 h).
Isolated yields.
The ee was determined chiral HPLC analysis (see SI in Figures S5, S7).
Data were obtained using the mixed DABCO–siloxane and its homogeneous MesityleneRuTsDPEN as dual catalysts.
Figure 5
Promoted by the previous catalytic results, the as-synthesized heterogeneous catalyst 1 was thereafter examined using the aza–Michael addition/ATH enantioselective cascade reactions by systematically changing the substitution pattern of enones under the same reaction conditions, and the results were summarized in Table 1. It was found that all tandem reactions with the tested substrates could produce steadily the corresponding chiral products in satisfactory yields and excellent enantioselectivities. Gratifyingly, the electronic properties of substituents attached to the aromatic rings of Ar2 group affected slightly reactivity, where the reactions with electron–withdrawing substituents had inferior yields relative to those results obtained with electron–donating substituents (Entries 3–8, 9–12). However, their enantioselectivities were not affected significantly for all chiral products bearing electron–withdrawing and/or –donating substituents of Ar2 group had high ee values (Entries 3–12). Similarly, in the case of the substituents on the aromatic rings of Ar1 group, this phenomenon was also observed (Entries 13–18).
In order to confirm the tandem behavior and explore the catalytic nature of catalyst 1, the time course for one–pot organic transformation of acrylophenone and phenylamine to chiral γ-secondary amino alcohols 3a was investigated. It is worth noting that both the aza–Michael addition of aniline to acrylophenone (2a) and the ATH transformation of reaction intermediate A to chiral γ-secondary amino alcohols 3a occur simultaneously during the first 1 h, where the content of 1–phenylprop−2–enone (2a) rapidly decreases down to 9% and that of (S)−1–phenyl−3–(phenylamino)propanol (3a) rises to 12%, as outlined in Figure 6. Meanwhile, during the first hour, the maximum 79% yield of the intermediate 1–phenyl−3–(phenylamino)propanone (A) is observed. Subsequently, one–pot organic transformation of acrylophenone and phenylamine proceeds smoothly, resulting in (S)−1–phenyl−3–(phenylamino)propanol (3a) in 92% yield within 8 h. Such a time course reveals the catalytic nature of this aza–Michael addition/ATH enantioselective undergoes a tandem process.
Figure 6
As a heterogeneous catalyst, it was highly expected that it could be recovered by simple centrifugation and the recycled catalyst could retained highly reactivity even after several recycle. Indeed, the newly developed catalyst 1 could be easily separated and recovered using centrifugation techniques from reaction solution, providing the desired adduct 3a in 90% yield with 96% ee in fifth recycled experiment via the one-pot aza–Michael addition/ATH enantioselective tandem reaction of acrylophenone and aniline (Figure 7, also see Table S2 and Figure S7 of ESI). In order to explain the decreased reactivity after fifth recycle (at sixth run), an analysis of the Ru–leaching was carried out by the use of ICP–OES techniques. The result showed that the amount of Ru at fifth run was 9.59 mg (0.0941 mmol) per gram of 1, meaning that the leaching amount of Ru should be 7.8% with respect to the original value (10.40 mg, 0.1020 mmol of Ru per gram of catalyst). For comparison, ICP–OES analysis after fifth recycle (at sixth run) indicated that the amount of Ru was 9.21 mg (0.0904 mmol) per gram of 1 and 11.6% of Ru was lost, revealing the obviously decreased reactivity at sixth run was attributed to the large number of Ru–leaching in catalyst 1. In addition, the loss of (DABCO)BF4 molecules after fifth recycle was about 20% via a comparison of their element analyses.
Figure 7
Conclusions
In conclusions, by using a anion–bonding strategy, we develop a facile approach to one–steps assemble both DABCO–molecules and chiral ruthenium/diamine–complexes species into the inner surface of Me–FDU−12, constructing a bifunctional heterogeneous catalyst. As we envisaged, the DABCO–molecules in catalyst 1 as a Lewis base enables aza–Michael addition whereas chiral ruthenium/diamine–complex as chiral promoter catalyzes asymmetric transfer hydrogenation. Synergistic contribution of both functionality boost greatly one–pot enantioselective synthesis of a series of chiral γ-secondary amino alcohols from enones and amines in a tandem manner. This method described here offers an operationally simple synthetic procedure to fabricate bifunctional heterogeneous catalyst used for synthesis of valuable chiral pure γ-secondary amino alcohols.
Statements
Author contributions
LL is mainly responsible for the preparation of heterogeneous catalyst, corresponding characterization and part of catalytic reactions. DY and ZZ carried out the catalytic reactions. YS finished the time course experiment, and LZ is responsible for the chiral HPLC analysis. RL and GL are responsible for the characterization analysis and writing.
Acknowledgments
We are grateful to China National Natural Science Foundation (21672149), Ministry of Education of China (PCSIRT–IRT−16R49), and Shanghai Municipal Education Commission (14YZ074) for financial support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00272/full#supplementary-material
References
1
BartókM. (2010). Unexpected inversions in asymmetric reactions: reactions with chiral metal complexes, chiral organocatalysts, and heterogeneous chiral catalysts. Chem. Rev.110, 1663–1705. 10.1021/cr9002352
2
BianchiniC.BarbaroP.SantoV. D.GobettoR.MeliA.OberhauserW.et al. (2001). Immobilization of optically active rhodium-diphosphine complexes on porous silica via hydrogen bonding. Adv. Synth. Catal.343, 41–45. 10.1002/1615-4169(20010129)343:1<41::AID-ADSC41>3.0.CO;2-P
3
ChauhanM. S.YadavG. D.HussainF.SinghS. (2014). N-Fluorobenzenaminium tetrafluoroborate generated in situ by aniline and selectfluor as a reusable catalyst for the ring opening of epoxides with amines under microwave irradiation. Catal. Sci. Technol.4, 3945–3952. 10.1039/C4CY00609G
4
ChenC.LiS.XuS.LouH.XiaoL.FanJ. (2014). An ordered, extra-large mesoporous ceramic acid with strong Bronsted acid sites and excellent thermal/hydrothermal stability. Chem. Commun.50, 3457–3459. 10.1039/c4cc00222a
5
DingZ. Y.WangT.HeY. M.ChenF.ZhouH. F.FanQ. H.et al. (2013). Highly enantioselective synthesis of chiral tetrahydroquinolines and tetrahydroisoquinolines by ruthenium-catalyzed asymmetric hydrogenation in ionic liquid. Adv. Synth. Catal.355, 3727–3735. 10.1002/adsc.201300698
6
FanJ.YuC.LeiJ.ZhangQ.LiT.TuB.et al. (2005). Low-temperature strategy to synthesize highly ordered mesoporous silicas with very large pores. J. Am. Chem. Soc.127, 10794–10795. 10.1021/ja052619c
7
FraileJ. M.GarcíaJ. I.MayoralJ. A. (2009). Noncovalent immobilization of enantioselective catalysts. Chem. Rev.109, 360–417. 10.1021/cr800363y
8
FujimaY.IkunakaM.InoueT.MatsumotoJ. (2006). Synthesis of (S)-3-(N-Methylamino)-1-(2-thienyl)propan-1-ol: revisiting Eli Lilly's resolution-racemization-recycle synthesis of duloxetine for its robust processes. Org. Process Res. Dev.10, 905–913. 10.1021/op060118l
9
GaoY.SharplessK. B. (1988). Asymmetric synthesis of both enantiomers of tomoxetine and fluoxetine. selective reduction of 2,3-epoxycinnamyl alcohol with Red-Al. J. Org. Chem53, 4081–4084. 10.1021/jo00252a036
10
GengH.ZhangX.ChangM.ZhouL.WuW.ZhangX. (2011). Ruthenium-catalyzed asymmetric hydrogenation of β-keto-enamines: an efficient approach to chiral γ-amino alcohols. Adv. Synth. Catal.353, 3039–3043. 10.1002/adsc.201100305
11
GladialiS.AlbericoE. (2006). Asymmetric transfer hydrogenation: chiral ligands and applications. Chem. Soc. Rev.35, 226–236. 10.1039/B513396C
12
HashiguchiS.FujiiA.TakeharaJ.IkariyaT.NoyoriR. (1995). Asymmetric transfer hydrogenation of aromatic ketones catalyzed by chiral ruthenium(II) complexes. J. Am. Chem. Soc.117, 7562–7563. 10.1021/ja00133a037
13
HeitbaumM.GloriusF.EscherI. (2006). Asymmetric heterogeneous catalysis. Angew. Chem., Int. Ed.45, 4732–4762. 10.1002/anie.200504212
14
HuQ.ZhangZ.LiuY.ImamotoT.ZhangW. (2015). ZnCl2-Promoted asymmetric hydrogenation of β-secondary-amino ketones catalyzed by a P-chiral Rh–bisphosphine complex. Angew. Chem., Int. Ed.54, 2260–2264. 10.1002/anie.201411384
15
IkariyaT.BlackerA. J. (2007). Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res.40, 1300–1308. 10.1021/ar700134q
16
JonesM. D.RajaR.ThomasJ. M.JohnsonB. F. G.LewisD. W.RouzaudJ.et al. (2003). Enhancing the enantioselectivity of novel homogeneous organometallic hydrogenation catalysts. Angew. Chem.115, 4462–4467. 10.1002/ange.200250861
17
KakeiH.NemotoT.OhshimaT.ShibasakiM. (2004). Efficient synthesis of chiral α- and β-hydroxy amides: application to the synthesis of (R)-Fluoxetine. Angew. Chem., Int. Ed.43, 317–320. 10.1002/anie.200352431
18
LiJ.-H.LiuW.-J. (2004). Dabco as an inexpensive and highly efficient ligand for palladium-catalyzed Suzuki–Miyaura cross-coupling reaction. Org. Lett.6, 2809–2811. 10.1021/ol048907f
19
LiJ.-H.LiuW.-J.XieY.-X. (2005). Recyclable and reusable Pd(OAc)2/DABCO/PEG-400 system for Suzuki–Miyaura cross-coupling reaction. J. Org. Chem.70, 5409–5412. 10.1021/jo050353m
20
LiX.LiuX.MaY.LiM.ZhaoJ.XinH.et al. (2012). Engineering the formation of secondary building blocks within hollow interiors. Adv. Mater. Weinheim.24, 1424–1428. 10.1002/adma.201104167
21
LiY.YiJ.KrukM. (2015). Tuning of the temperature window for unit-cell and pore-size enlargement in face-centered-cubic large-mesopore silicas templated by swollen block copolymer micelles. Chem. Eur. J.21, 12747–12754. 10.1002/chem.201500189
22
LiaoH.ChouY.WangY.ZhangH.ChengT.LiuG. (2017). Multistep organic transformations over base-rhodium/diamine-bifunctionalized mesostructured silica nanoparticles. ChemCatChem9, 3197–3202. 10.1002/cctc.201700436
23
LiuD.GaoW.WangC.ZhangX. (2005). Practical synthesis of enantiopure γ-amino alcohols by rhodium-catalyzed asymmetric hydrogenation of β-secondary-amino ketones. Angew. Chem. Int. Ed.44, 1687–1689. 10.1002/anie.200462178
24
LiuP. N.DengJ. G.TuY. Q.WangS. H. (2004a). Highly efficient and recyclable heterogeneous asymmetric transfer hydrogenation of ketones in water. Chem. Commun.2070–2071. 10.1039/b408533g
25
LiuP. N.GuP. M.WangF.TuY. Q. (2004b). Efficient heterogeneous asymmetric transfer hydrogenation of ketones using highly recyclable and accessible silica-immobilized Ru-TsDPEN Catalysts. Org. Lett.6, 169–172. 10.1021/ol036065z
26
MaG.YanX.LiY.XiaoL.HuangZ.LuY.et al. (2010). Ordered nanoporous silica with periodic 30–60 nm pores as an effective support for gold nanoparticle catalysts with enhanced lifetime. J. Am. Chem. Soc.132, 9596–9597. 10.1021/ja1027524
27
MargelefskyE. L.ZeidanR. K.DavisM. E. (2008). Cooperative catalysis by silica-supported organic functional groups. Chem. Soc. Rev.37, 1118–1126. 10.1039/b710334b
28
McDonaghC.O'ConghaileP.Klein GebbinkR. J. M.O'LearyP. (2007). Electrostatic immobilisation of copper(I) and copper(II) bis(oxazolinyl)pyridine catalysts on silica: application to the synthesis of propargylamines via direct addition of terminal alkynes to imines. Tetrahedron Lett.48, 4387–4390. 10.1016/j.tetlet.2007.04.095
29
NoyoriR.HashiguchiS. (1997). Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res.30, 97–102. 10.1021/ar9502341
30
OhkumaT.TsutsumiK.UtsumiN.AraiN.NoyoriR.MurataK. (2007). Asymmetric Hydrogenation of α-chloro aromatic ketones catalyzed by η6-arene/TsDPEN–ruthenium(II) complexes. Org. Lett.9, 255–257. 10.1021/ol062661s
31
O'LearyP.KrosveldN. P.De JongK. P.van KotenG.Klein GebbinkR. J. M. (2004). Facile and rapid immobilization of copper(II) bis(oxazoline) catalysts on silica: application to Diels–Alder reactions, recycling, and unexpected effects on enantioselectivity. Tetrahedron Lett.45, 3177–3180. 10.1016/j.tetlet.2004.03.001
32
RajaR.ThomasJ. M.JonesM. D.JohnsonB. F. G.VaughanD. E. W. (2003). Constraining Asymmetric organometallic catalysts within mesoporous supports boosts their enantioselectivity. J. Am. Chem. Soc.125, 14982–14983. 10.1021/ja030381r
33
RobertsonD. W.KrushinskiJ. H.FullerR. W.LeanderJ. D. (1988). The absolute configurations and pharmacological activities of the optical isomers of fluoxetine, a selective serotonin-uptake inhibitor. J. Med. Chem.31, 1412–1417. 10.1021/jm00402a027
34
RouzaudJ.JonesM. D.RajaR.JohnsonB. F. G.ThomasJ. M.DuerM. J. (2003). Potent new heterogeneous asymmetric catalysts. Helv. Chim. Acta86, 1753–1759. 10.1002/hlca.200390145
35
ShuX.-Z.NguyenS. C.HeY.ObaF.ZhangQ.CanlasC.et al. (2015). Silica-supported cationic gold(I) complexes as heterogeneous catalysts for regio- and enantioselective lactonization reactions. J. Am. Chem. Soc.137, 7083–7086. 10.1021/jacs.5b04294
36
SongC. E. (2005). Immobilisation of chiral catalysts: easy recycling of catalyst and improvement of catalytic efficiencies. Annu. Rep. Prog. Chem. Sect. C.101, 143–173. 10.1039/b408828j
37
ThomasJ. M.RajaR. (2008). Exploiting nanospace for asymmetric catalysis: confinement of immobilized, single-site chiral catalysts enhances enantioselectivity. Acc. Chem. Res.41, 708–720. 10.1021/ar700217y
38
TräffA.LihammarR.BäckvallJ.-E. (2011). A chemoenzymatic dynamic kinetic resolution approach to enantiomerically pure (R)- and (S)-Duloxetine. J. Org. Chem.76, 3917–3921. 10.1021/jo2003665
39
WangC.WuX.XiaoJ. (2008). Broader, greener, and more efficient: recent advances in asymmetric transfer hydrogenation. Chem. Asian J.3, 1750–1770. 10.1002/asia.200800196
40
WangH.LiuX.XiaH.LiuP.GaoJ.YingP.et al. (2006). Asymmetric Diels–Alder reactions with hydrogen bonding heterogeneous catalysts and mechanistic studies on the reversal of enantioselectivity. Tetrahedron62, 1025–1032. 10.1016/j.tet.2005.09.148
41
WangJ.LiuD.LiuY.ZhangW. (2013a). Asymmetric hydrogenation of β-amino ketones with the bimetallic complex RuPHOX-Ru as the chiral catalyst. Org. Biomol. Chem.11, 3855–3861. 10.1039/C3OB40135A
42
WangJ.WangY.LiuD.ZhangW. (2015). Asymmetric hydrogenation of β-secondary amino ketones catalyzed by a ruthenocenyl phosphino-oxazoline-ruthenium complex (RuPHOX-Ru): the synthesis of γ-secondary amino alcohols. Adv. Synth. Catal.357, 3262-3272. 10.1002/adsc.201500653
43
WangL.ZhouQ.QuC.WangQ.CunL.ZhuJ.et al. (2013b). Efficient asymmetric transfer hydrogenation of N-sulfonylimines on water. Tetrahedron69, 6500–6506. 10.1016/j.tet.2013.05.064
44
WangT.ZhuoL.-G.LiZ.ChenF.DingZ.HeY.et al. (2011). Highly enantioselective hydrogenation of quinolines using phosphine-free chiral cationic ruthenium catalysts: scope, mechanism, and origin of enantioselectivity. J. Am. Chem. Soc.133, 9878–9891. 10.1021/ja2023042
45
WenL.-R.LanM.-C.YuanW.-K.LiM. (2014). DABCO-mediated isocyanide-based multicomponent reaction: synthesis of highly substituted cyclopentenes. Org. Biomol. Chem.12, 4628–4632. 10.1039/C4OB00012A
46
WienchJ. W.MichonC.EllernA.HazendonkP.IugaA.AngeliciR. J.et al. (2009). Solid-state NMR investigations of the immobilization of a salt of a palladium(II) complex on silica. J. Am. Chem. Soc.131, 11801–11810. 10.1021/ja902982u
47
WuJ.WangF.MaY.CuiX.CunL.ZhuJ.et al. (2006). Asymmetric transfer hydrogenation of imines and iminiums catalyzed by a water-soluble catalyst in water. Chem. Commun.1766–1768. 10.1039/b600496b
48
WuL.JinR.LiL.HuX.ChengT.LiuG. (2017). A Michael addition–asymmetric transfer hydrogenation one-pot enantioselective tandem process for syntheses of chiral γ-secondary amino alcohols. Org. Lett.19, 3047–3050. 10.1021/acs.orglett.7b00823
49
WuX.LiuJ.TommasoD. D.IggoJ. A.CatlowC. R. A.BacsaJ.et al. (2008). A multilateral mechanistic study into asymmetric transfer hydrogenation in water. Chem. Eur. J.14, 7699–7715. 10.1002/chem.200800559
50
XiaX.MengJ.WuH.ChengT.LiuG. (2017). Integration of multiple active sites on large-pore mesoporous silica for enantioselective tandem reactions. Chem. Commun.53, 1638–1641. 10.1039/C6CC09008G
51
XuY.ChengT.LongJ.LiuK.QianQ.GaoF.et al. (2012). An ion-pair immobilization strategy in rhodium-catalyzed asymmetric transfer hydrogenation of aromatic ketones. Adv. Synth. Catal.354, 3250–3258. 10.1002/adsc.201200320
52
YanX.WangX.TangY.MaG.ZouS.LiR.et al. (2013). Unusual loading-dependent sintering-resistant properties of gold nanoparticles supported within extra-large mesopores. Chem. Mater.25, 1556–1563. 10.1021/cm303816g
53
YangH.ZhangL.ZhongL.YangQ.LiC. (2007). Enhanced cooperative activation effect in the hydrolytic kinetic resolution of epoxides on [Co(salen)] catalysts confined in nanocages. Angew. Chem., Int. Ed.46, 6861–6865. 10.1002/anie.200701747
54
YangZ.ChenF.HeY.-M.YangN.FanQ.-H. (2014). Efficient asymmetric hydrogenation of quinolines in neat water catalyzed by chiral cationic Ru-diamine complexes. Catal. Sci. Technol.4, 2887–2890. 10.1039/C4CY00418C
55
YangZ.ChenF.HeY.YangN.FanQ. H. (2016). Highly enantioselective synthesis of indolines: asymmetric hydrogenation at ambient temperature and pressure with cationic ruthenium diamine catalysts. Angew. Chem., Int. Ed.55, 13863–13866. 10.1002/anie.201607890
56
YingA.LiuS.NiY.QiuF.XuS.TangW. (2014). Ionic tagged DABCO grafted on magnetic nanoparticles: a water-compatible catalyst for the aqueous aza-Michael addition of amines to α,β-unsaturated amides. Catal. Sci. Technol.4, 2115–2125. 10.1039/C4CY00232F
57
YuC.HeJ. (2012). Synergic catalytic effects in confined spaces. Chem. Commun.48, 4933–4940. 10.1039/c2cc31585h
58
ZhangJ.ChenF.HeY. M.FanQ. H. (2015). Asymmetric ruthenium-catalyzed hydrogenation of 2,6-disubstituted 1,5-naphthyridines: access to chiral 1,5-diaza-cis-decalins. Angew. Chem., Int. Ed.54, 4622–4625. 10.1002/anie.201411105
59
ZhouJ.-N.FangQ.HuY.-H.YangL.-Y.WuF.-F.XieL.-J.et al. (2014). Copper(II)-catalyzed enantioselective hydrosilylation of halo-substituted alkyl aryl and heteroaryl ketones: asymmetric of (R)-fluoxetine and (S)-duloxetine. Org. Biomol. Chem.12, 1009–1017. 10.1039/c3ob42214c
Summary
Keywords
asymmetric catalysis, heterogeneous catalyst, tandem reaction, silica, anion bonding
Citation
Li L, Yang D, Zhao Z, Song Y, Zhao L, Liu R and Liu G (2018) Boron Tetrafluoride Anion Bonding Dual Active Species Within a Large–Pore Mesoporous Silica for Two–Step Successive Organic Transformaion to Prepare Optically Pure Amino Alcohols. Front. Chem. 6:272. doi: 10.3389/fchem.2018.00272
Received
12 April 2018
Accepted
18 June 2018
Published
06 July 2018
Volume
6 - 2018
Edited by
Vasile I. Parvulescu, University of Bucharest, Romania
Reviewed by
Maryse Gouygou, UPR8241 Laboratoire de Chimie de Coordination (LCC), France; Matteo Guidotti, Consiglio Nazionale Delle Ricerche (CNR), Italy; Wenbo Wang, Lanzhou Institute of Chemical Physics (CAS), China
Updates
Copyright
© 2018 Li, Yang, Zhao, Song, Zhao, Liu and Liu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui Liu rliu@shnu.edu.cnGuohua Liu ghliu@shnu.edu.cn
This article was submitted to Green and Sustainable Chemistry, a section of the journal Frontiers in Chemistry
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.