
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Chem., 24 May 2018
Sec. Medicinal and Pharmaceutical Chemistry
Volume 6 - 2018 | https://doi.org/10.3389/fchem.2018.00178
 Judite R. M. Coimbra1,2
Judite R. M. Coimbra1,2 Daniela F. F. Marques1,2
Daniela F. F. Marques1,2 Salete J. Baptista1,2,3
Salete J. Baptista1,2,3 Cláudia M. F. Pereira2,4
Cláudia M. F. Pereira2,4 Paula I. Moreira2,5
Paula I. Moreira2,5 Teresa C. P. Dinis2,6
Teresa C. P. Dinis2,6 Armanda E. Santos2,6*
Armanda E. Santos2,6* Jorge A. R. Salvador1,2*
Jorge A. R. Salvador1,2*Alzheimer's disease (AD) is a severe neurodegenerative disorder and the most common type of dementia in the elderly. The clinical symptoms of AD include a progressive loss of memory and impairment of cognitive functions interfering with daily life activities. The main neuropathological features consist in extracellular amyloid-β (Aβ) plaque deposition and intracellular Neurofibrillary tangles (NFTs) of hyperphosphorylated Tau. Understanding the pathophysiological mechanisms that underlie neurodegeneration in AD is essential for rational design of neuroprotective agents able to prevent disease progression. According to the “Amyloid Cascade Hypothesis” the critical molecular event in the pathogenesis of AD is the accumulation of Aβ neurotoxic oligomers. Since the proteolytic processing of Amyloid Precursor Protein (APP) by β-secretase (beta-site APP cleaving enzyme 1, BACE1) is the rate-limiting step in the production of Aβ, this enzyme is considered a major therapeutic target and BACE1 inhibitors have the potential to be disease-modifying drugs for AD treatment. Therefore, intensive efforts to discover and develop inhibitors that can reach the brain and effectively inhibit BACE1 have been pursued by several groups worldwide. The aim of this review is to highlight the progress in the discovery of potent and selective small molecule BACE1 inhibitors over the past decade.
Dementia has a tremendous emotional impact on the individual and his family, as well as an immense percussion at the economic level. Dementia was recognized by WHO in 2017 as a public health priority, thereafter a global intervention aiming to achieve physical, mental and social wellbeing for people with dementia, their caregivers and families, both for present and future generations, was planned (WHO, 2017).
Alzheimer's disease (AD), which accounts for 60–70% of all dementias, is clinically characterized by a gradual loss of memory and cognitive abilities, as well as by altered behavior, ultimately causing disability and dependency (Masters et al., 2015). Essential areas to cognitive processes in the brain are particularly damaged during the disease development, leading to early synapse loss, neuronal dysfunction, and death culminating in brain atrophy, which are responsible for the manifestation of the disease (Querfurth and LaFerla, 2010). An accumulation of misfolded proteins—amyloid-β (Aβ) and Tau protein—marks the AD brain. The “Amyloid Cascade Hypothesis” proposes that the cerebral appearance of aggregates of oligomeric Aβ plays a central role for AD neuropathogenicity (Karran et al., 2011; Selkoe and Hardy, 2016). These neurotoxic agents may interact with many neuronal receptors triggering a neurodegenerative cascade of events that are responsible for mitochondria dysfunction (Hauptmann et al., 2006; Carvalho et al., 2015), endoplasmic reticulum stress (Correia et al., 2015; Plácido et al., 2015), oxidative stress (Carvalho et al., 2016), DNA damage, neuroinflammation, among other deleterious mechanisms. The Aβ oligomers can also disrupt normal kinase and phosphatase activity, resulting in the hyperphosphorylation of Tau protein and subsequent Neurofibrillary tangles (NFTs) formation (Resende et al., 2008).
The current pharmacological therapeutics for AD only attenuate the symptoms, and do not affect the mechanisms underlying disease progression. Disease-modifying approaches have recently become the focus of AD research, aiming to prevent the neurodegenerative process at early phases before clinical manifestations (Bachurin et al., 2017; Cummings et al., 2017). The main directions include the search for drugs that are aimed at blocking the pathogenic steps responsible for the appearance of the pathological structures of AD—Aβ plaques and NTFs. Thus, several research groups are seeking to develop small molecules that can act as anti-amyloidogenic agents via inhibiting Aβ production and aggregation and promoting its removal (Kulshreshtha and Piplani, 2016).
The BACE1 is the transmembrane aspartyl protease that cleaves APP at the β-site. The sequential proteolytic cleavage of APP by BACE1 and γ-secretase leads to the production and release of Aβ peptide in the brain. Therefore, amyloidogenic secretases are key therapeutic targets being currently explored for AD-modifying intervention. Several studies report that BACE1 inhibitors hold great potential as a potential strategy in decreasing Aβ brain concentrations, thus preventing the progression of AD (Citron, 2002; De Strooper et al., 2010). Since BACE1 identification in 1999 (Vassar, 1999), inhibitors covering many different structural classes have been extensively reported in the literature (Ghosh et al., 2012; Ghosh and Osswald, 2014; Menting and Claassen, 2014; Yan and Vassar, 2014).
Here we will provide a summary overview of the recent advances in structural evolution of BACE1 inhibitor classes in order to improve their potency and selectivity.
BACE1 inhibitors can be identified into two main categories, peptidomimetics and nonpeptidics, and further subclassification can be made based on the core functional groups that interact with the catalytic dyad (Ghosh and Osswald, 2014). The development of OM99-2, an inhibitor based on the APP cleavage site sequence, demonstrated for the first time the druggability of BACE1. Therefore, the elucidation of the enzyme-ligand complex crystal structure gave much insight into the binding mode of inhibitors in the BACE1 binding pocket (Ghosh et al., 2001).
The development of effective BACE1 inhibitors faces many challenges. First, due to the location of BACE1 in the brain and in the lumen of endosomes, inhibitors need to cross the blood-brain barrier (BBB) and neuronal membranes to access the target (Ben Halima et al., 2016). Although peptidomimetic inhibitors are highly potent in vitro, their inherent poor drug properties (reduced brain permeability, short half-life and reduced oral availability) account for their low in vivo efficacy. In fact, one of the major issues in the design of recent generations of inhibitors is the transition from peptidomimetics to brain penetrant small molecules (Oehlrich et al., 2014).
Furthermore, concerning the BACE1 inhibitors' discovery process, there are some important structural features of the enzyme that should be wisely taken into consideration, namely the catalytic aspartic dyad, structural flexibility, and the large binding pocket (Dislich and Lichtenthaler, 2012; Yuan et al., 2013). Due to the large catalytic pocket of BACE1 the inhibitors are required to be large enough to interact within the active site, yet these inhibitors should be small enough to exhibit suitable drug-like properties (Vassar, 2016). Moreover, to prevent cross-inhibition toxic effects, it is especially important to ensure that BACE1 inhibitors are selective over other aspartic proteases because these enzymes are closely related to each other since the two catalytic aspartic acid residues are conserved across the class (Ghosh and Osswald, 2014). Additional challenges to overcome include the susceptibility to P-glycoprotein (P-gp) efflux and the inhibition of hERG (human Ether-A-Go-Go ion channel) related to cardiotoxicity, as well as the inhibition of cytochrome enzymes to avoid drug interactions by BACE1 inhibitors (Brodney et al., 2015). Beyond APP processing, BACE1 has several cellular substrates and its physiological role is required for optimal cognitive function. The BACE1-null mice present neuronal phenotypes including synaptic dysfunction, retinopathy, epileptic seizures, hypomyelination, etc., that appear to be related to the abolished cleavage of BACE1 substrates. Therefore, BACE1 inhibitors might exhibit mechanism-based toxicity (Barão et al., 2016). Although more studies are needed to further understand BACE1 biological functions and the consequences of its chronic inhibition, a careful titration of drug dosage should be considered when using BACE1 inhibitors.
Hence, the focus has been searching for inhibitors that can effectively and selectively inhibit BACE1 in vivo. Some of them have shown benefits in preclinical studies and six drugs, JNJ-54861911, CNP520, LY3202626, Elenbecestat, Lanabecestat, and Verubecestat, are currently being tested in human clinical trials on different types of populations, including patients with mild-to-moderate AD and in patients at risk for AD (Yan, 2016; Mullard, 2017). Verubecestat was the first inhibitor to progress to clinical phase 3 and the best positioned drug to confirm the safety and efficacy of BACE1 inhibition. However, Merck has halted its pivotal trial in patients with mild-to-moderate AD and the trial in patients with prodromal AD due to ineffectiveness in cognitive decline reduction and unlikely positive benefit/risk outcome, respectively (Staff, 2017; Merck, 2018). Consequently, these unfortunate drawback raises questions about the clinical use of BACE1 inhibitors as potential anti-AD drugs.
One the other hand, recent evidence obtained in an animal model where deletion of BACE1 was performed at the adult stage to mimic BACE1 inhibition in AD patients, demonstrates that sequential and gradual BACE1 inhibition can completely reverse amyloid pathology. However, caution is needed in translating these findings to the clinical practice since BACE1 is required for cognitive functions and BACE1 inhibitors are expected to be used for chronic treatment (Hu et al., 2018). Another recent study, using a longitudinal approach with two-photon microscopy to monitor the impact of pharmacological BACE1 inhibition on early Aβ plaque deposition in an AD mouse model, demonstrated that it slows down progression of initial Aβ plaque formation but was less effective toward existing plaques (Peters et al., 2018). Therefore, the timing of intervention with BACE1 inhibitors in clinical trials may be adjusted according to the emerging data to achieve optimal therapeutic efficacy for AD treatment.
In addition, a diversity of complex medicinal chemistry approaches, such as high throughput and fragment-based screening, X-ray crystallography, nuclear magnetic resonance (NMR) and computational methods, have been directed in recent years toward the development of a wide variety of scaffolds of small BACE1 inhibitors. Also, the knowledge of BACE1 structure has helped to conduct a structure-based (SB) design of novel BACE1 inhibitors, and promising findings have been obtained, some of them described herein.
Small molecule derivatives with aminohydantoin scaffold have been reported as BACE1 inhibitors, including the disubstituted pyridinyl aminohydantoins with low nanomolar potency and high selectivity for BACE1 (Malamas et al., 2010a,b,c, 2011; Zhou et al., 2010) and the small and rigid spirocyclic aminohydantoin analogs, which were designed through a structure-activity relationship (SAR) approach to improve their in vivo activity (Hunt et al., 2013). Furthermore, derivatives of 7-tetrahydropyran-2-yl chromans and 8-tetrahydropyran-2-yl chromans were also prepared. Using structure-based design these chemotypes were substituted with three different aspartate-binding groups: aminohydantoin, aminooxazolines, and aminothiazolines, in an attempt to modulate potency, selectivity, efflux, and permeability. In the first chemotype, the aminohydantoin analogs were the most potent and selective against Cathepsin-D (Thomas et al., 2014a,b).
The potent and brain penetrant class of iminohydantoin BACE1 inhibitor was revealed via a fragment-based NMR screening followed by X-ray crystallography (Zhu et al., 2010). Later, a new set of iminohydantoin analogs identified through a SB approach, showed selectivity and high-affinity for BACE1, and a low level of central Aβ after oral administration in rats (Cumming et al., 2012). More recently, iminohydantoin spiropiperidine methyl-substituted derivatives were identified by molecular modeling methods used to explore available binding pockets adjacent to the ligand binding. As a result, the addition of a placed methyl group maintained the required pKa of the piperidine nitrogen and occupied a small hydrophobic pocket, stabilizing the ligand into the active site. Compound 1 (listed in Table 1) showed an improvement in BACE1 inhibitory potency and good selectivity against Cathepsin-D (Egbertson et al., 2015).
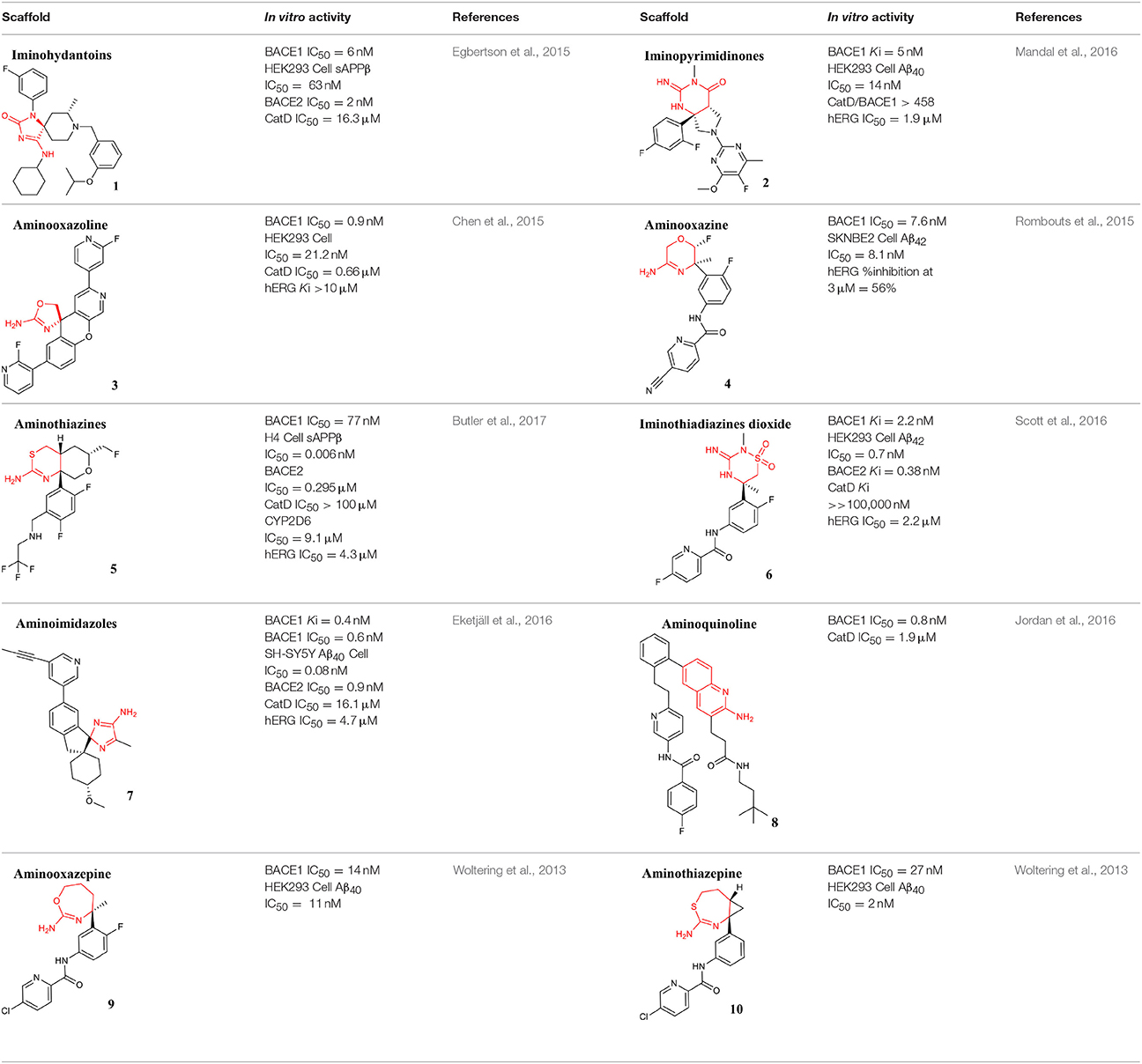
Table 1. Representative structures of potent and selective BACE1 inhibitors scaffolds.
A new class of orally bioavailable and BBB permeable BACE1 inhibitors with iminopyrimidinone scaffold was obtained by SB optimization of iminohydantoin chemotype. The best compound of these series potently reduced Aβ levels when administered orally to rats (Stamford et al., 2012). Mandal and collaborators have also identified potent BACE1 inhibitors with iminopyrimidinone core (Mandal et al., 2012). Thus, seeking to identify novel, highly soluble, permeable, and selective inhibitors minimizing off-target liabilities, a set of fused pyrrolidine iminopyrimidinone derivatives were recently prepared using a SB approach. As a result of these successful efforts, the difluorophenyl compound 2 (listed in Table 1), which represents a novel molecular shape, was identified. This compound has been shown to strongly lower central Aβ levels following oral administration of a single dose in preclinical assays and to display good selectivity over Cathepsin-D, without significant inhibitory effect toward a range of human cytochrome enzymes, thus having minimal risk of interaction with other drugs (Mandal et al., 2016).
Over the last years, a variety of aminooxazoline derivatives, including a class of aminooxazoline xanthenes have been reported as BACE1 inhibitors (Huang et al., 2012; Epstein et al., 2014). Moreover, a class of aminooxazoline 4-aza substituted on the xanthene core shows good BACE1 potency while reducing hERG binding affinity (Dineen et al., 2014). A further optimization based on SB design with balance of physicochemical parameters conducted to 3-aza-4-fluoroxanthene analogs. It was found that the incorporation of a nitrogen atom into the 3-position of the xanthene scaffold improved BACE1 potency, reduced hERG binding affinity and improved cell-permeability (Cheng et al., 2015). Further efforts though SAR generated several potent and orally active BACE1 inhibitors. The compound 3 (listed in Table 1) was identified as the most attractive candidate within this series, exhibiting low in vivo clearance, good oral bioavailability, and reduced potential for cardiovascular liabilities in preclinical species (Chen et al., 2015).
Efforts to obtain fluorine-containing aminooxazines derivatives resulted in a class characterized by high in vivo activity and good drug properties. Through an extensive fluorine scan study it was revealed the impact of fluorine to lower the pKa and to change the pharmacological profile of this class of BACE1 inhibitors (Hilpert et al., 2013). Recently, the effort to reduce the amidine pKa while optimizing interactions with the BACE1 active site, allowed to modulate cell-permeation ability and P-gp efflux. This enabled the discovery of orally bioavailable and centrally active 1,4-oxazines derivatives that robustly lowered brain Aβ levels. Compound 4 (listed in Table 1) was identified as the most promising within this series (Rombouts et al., 2015).
Two potent aminothiazine BACE1 inhibitors developed by Eli Lilly, LY2811376, and LY2886721, advanced into the clinics with a good pharmacokinetic profile. Unfortunately, these inhibitors have been discontinued due to preclinical retinal toxicity and to clinical abnormal liver biochemistry, respectively (May et al., 2011, 2015).
Recently, a lot of strategies have been implemented to disclose novel and potent aminothiazine BACE1 inhibitors, namely the truncation of the S3 substituent of the biaryl aminothiazine, which enables oral bioavailability and brain penetration (Wu et al., 2016b), and the preparation of fused bicyclic series of furo[2,3-d][1,3]thiazinamine derivatives (Wu et al., 2016a). Additional series of truncated and fused thioamidines were reported as efficiently selective in inhibiting BACE1 and with suitable pharmacokinetic profile (Butler et al., 2015). Further development guided by the crystal structures of CYP2D6 and BACE1 complexed with inhibitor and its corresponding metabolic product, led to improved potency, central efficacy, low clearance, and reduced hERG activity and risk of drug-drug interactions (Brodney et al., 2015). To this end, new derivatives with an aminomethyl linker that engages Gly230, a key residue in the BACE1 binding pocket and display high potency and improved brain penetration were disclosed. Therefore, a strategy that was implemented to prevent the drug-drug interactions predicted with basis on CYP2D6 binding affinities, led to the design of compound 5 (listed in Table 1), which exhibits robust in vivo efficacy (Butler et al., 2017).
Iminothiadiazine dioxides core derivatives have revealed high inhibitory activity against BACE1. Efforts have culminated in the discovery of the clinical Verubecestat (compound 6 listed in Table 1) that significantly lowered central Aβ levels in preclinical and in humans after oral administration. Despite not being selective over BACE2, it has highly selectivity compared to Cathepsin-D (Scott et al., 2016).
Structures with high potency and selectivity for BACE1 with aminoimidazole scaffold were firstly reported in 2009 (Malamas et al., 2009). The effort to improve cell-permeability and to reduce efflux properties in a new series of bicyclic aminoimidazoles BACE1 inhibitors was described to achieve in vivo brain efficacy (Swahn et al., 2012). The design of new amino-2H-imidazoles derivatives was performed to obtain new inhibitors with high activity, good drug properties, and low effect on hERG in combination with great in vivo exposure (Gravenfors et al., 2012). The clinical BACE1 inhibitor Lanabecestat developed by AstraZeneca (compound 7 listed in Table 1) belongs to the aminoimidazole class and is characterized as potent, highly permeable, orally active, BBB-penetrant with a very slow off-rate from BACE1, resulting in prolongation of the observed effect (Eketjäll et al., 2016).
A library of potent BACE1 inhibitors with 2-aminoquinoline core was designed through a complex strategy, including fragment-based screening and further SAR development supported by X-ray crystallography and molecular modeling studies (Cheng et al., 2011). In an effort to improve potency and selectivity of this class of compounds a fragment-linking approach using 19F NMR spectroscopy was conducted. This technique revealed to be useful in the identification of a second-site fragment that binds to a specific pocket of BACE1, which ultimately increased the potency while maintaining reasonable ligand efficiency with improved selectivity over Cathepsin-D. Compound 8 (listed in Table 1) exhibited a high-fold gain in its affinity and in selectivity for BACE1 (Jordan et al., 2016).
A series of seven-membered aminooxazepine and aminothiazepine derivatives was described as BACE1 inhibitors (Blass, 2012) and the effect of physicochemical properties, basicity and P-gp efflux on their activity was evaluated. It was demonstrated that compounds less basic, P-gp free and more lipophilic can achieve a high exposure profile. The 2-aminooxazepine derivatives were identified as promising brain penetrants, and compound 9 (listed in Table 1) showed high BACE1 inhibitory potency. Moreover, a series of cyclopropyl-fused oxazepine and thiazepine analogs was also prepared (Blass, 2013). The fusion of the cyclopropyl was shown to lower pKa of the functional group and the orientation of the ligand into the binding pocket. The cyclopropyl-fused 1,3-thiazepine compound 10 (listed in Table 1) is highly active in in vitro enzymatic and cellular studies and more potent than the oxygen analog cyclopropyl-fused-1,4-homo-oxazepine (Woltering et al., 2013).
Recently, the identification of new structural classes of compounds with BACE1 inhibitory activity at micromolar level via in silico methods was reported (listed in Table 2). Jain and colleagues identified derivatives with sulfonyl-amino-acetamide and alkylidene hydrazinecarboximidamide cores acting on BACE1. Compounds 11 and 12 were demonstrated as the most active of each chemotype, respectively (Jain et al., 2016a,b). De Tran and collaborators reported (3S,4S)-4-aminopyrrolidine-3-ol derivatives as potent BACE1 inhibitors and selective over BACE2 and Cathepsin-D. Compound 13 was identified as the most promising derivative (De Tran et al., 2016). In 2017, several new chemical structures acting as BACE1 inhibitors were described, among them—quinazolinone-based hydrazone derivative 14 (Haghighijoo et al., 2017), 2-substituted-thio-N-(4-substituted-thiazol/1H-imidazol-2-yl)acetamide derivative 15 (Yan et al., 2017), imidazopyridine derivative 16 (Azimi et al., 2017) and 2-oxopiperazine derivative 17 (Ghosh et al., 2017), were those that showed the best BACE1 inhibitory activity. These findings can be promising for further advance in the field of BACE1 inhibitors.
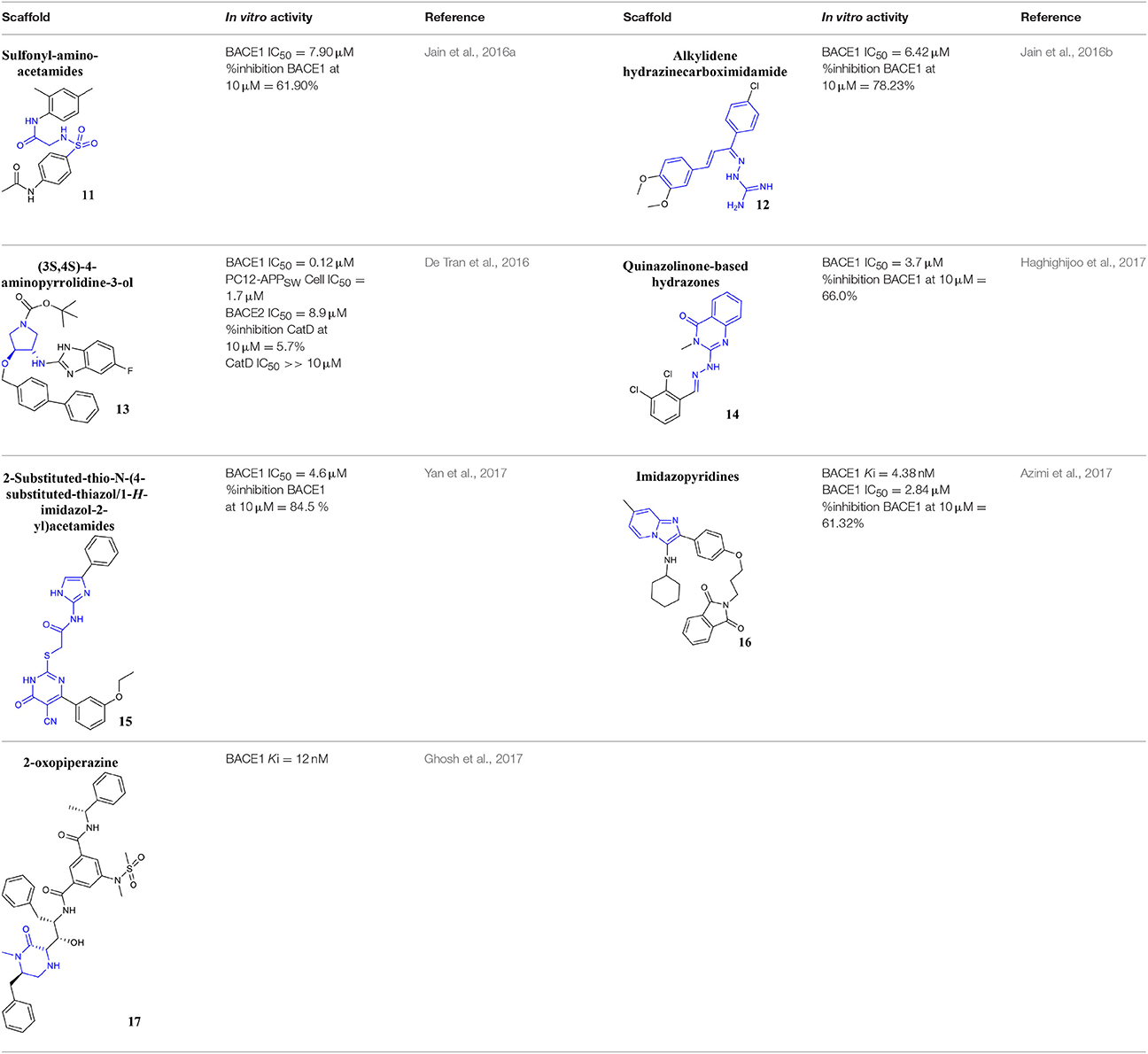
Table 2. Recent reported structures with BACE1 inhibitory activity.
The therapeutic potential of BACE1 inhibition has been investigated in the last decade. However, even though inhibitors efficiently lower brain Aβ levels, clinical trials still do not demonstrate improvements in cognitive function when administered to mild-to-moderate AD patients, calling into question the real worth of these potential anti-AD drugs and the clinical trials design. Emerging evidences suggest that the optimal timing for treatment with BACE1 inhibitors should be as early as possible. The optimal time point for a treatment with BACE1 inhibitors is a crucial issue that possibly explains some of the previous failures. The advances in technological diagnostic tools allowing better identification of patients at risk of developing AD, will enable clinical trials at preclinical stage when BACE1 inhibitors are expected to have the largest impact on amyloid plaques (Voytyuk et al., 2018).
Moreover, taking into account the complexity of AD, recent studies report multitarget approaches centered on BACE1, whose ligands are developed as small molecules that can modulate both BACE1 and other AD-related targets within synergistically pathways (Prati et al., 2018). Noteworthy, as BACE1 biological functions may be affected upon inhibition, further studies are required to develop efficient strategies to minimize the side-effects associated with long-term use of BACE1 inhibitors (Yan, 2017).
Indeed, although design and development of BACE1 inhibitors have proved extremely challenging, there is a substantial body of evidence that BACE1 inhibitors can act as disease-modifying agents and their use should be pursued to achieve benefits for AD patients.
JC, DM, SB, CP, PM, and TD drafted the work and wrote the manuscript. AS and JS revised the manuscript. All authors approved the manuscript in its final form for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, MB, and handling Editor declared their shared affiliation.
This work was supported by PT2020 (Programa Operacional do Centro 2020) through the COMPETE 2020 Programme (Operational Programme for Competitiveness and Internationalization, project CENTRO-01-0247-FEDER-003269, drugs2CAD and financially support by FEDER (European Regional Development Fund). This work was also financed by the European Regional Development Fund (ERDF), through the Centro 2020 Regional Operational Programme: project CENTRO-01-0145-FEDER-000012-HealthyAging2020, the COMPETE 2020—Operational Programme for Competitiveness and Internationalisation, and the Portuguese national funds via FCT—Fundação para a Ciência e a Tecnologia, I.P.: project POCI-01-0145-FEDER-007440.
Azimi, S., Zonouzi, A., Firuzi, O., Iraji, A., Saeedi, M., Mahdavi, M., et al. (2017). Discovery of imidazopyridines containing isoindoline-1,3-dione framework as a new class of BACE1 inhibitors: design, synthesis and SAR analysis. Eur. J. Med. Chem. 138, 729–737. doi: 10.1016/j.ejmech.2017.06.040
Bachurin, S. O., Bovina, E. V., and Ustyugov, A. A. (2017). Drugs in clinical trials for Alzheimer's Disease: the major trends. Med. Res. Rev. 37, 1186–1225. doi: 10.1002/med.21434
Barão, S., Moechars, D., Lichtenthaler, S. F., and De Strooper, B. (2016). BACE1 physiological functions may limit its use as therapeutic target for Alzheimer's Disease. Trends. Neurosci. 39, 158–169. doi: 10.1016/j.tins.2016.01.003
Ben Halima, S., Mishra, S., Raja, K. M. P., Willem, M., Baici, A., Simons, K., et al. (2016). Specific inhibition of β-secretase processing of the alzheimer disease amyloid precursor protein. Cell Rep. 14, 2127–2141. doi: 10.1016/j.celrep.2016.01.076
Blass, B. (2012). 5-amino-oxazepine and 5-amino-thiazepine compounds as β-secretase antagonists and methods of use: patent highlight. ACS Med. Chem. Lett. 3, 873–874. doi: 10.1021/ml300323y
Blass, B. (2013). Cyclopropyl-Fused 1,3-Thiazepines as BACE1 and BACE2 inhibitors. ACS Med. Chem. Lett. 4, 379–380. doi: 10.1021/ml400060e
Brodney, M. A., Beck, E. M., Butler, C. R., Barreiro, G., Johnson, E. F., Riddell, D., et al. (2015). Utilizing structures of CYP2D6 and BACE1 complexes to reduce risk of drug-drug interactions with a novel series of centrally efficacious BACE1 inhibitors. J. Med. Chem. 58, 3223–3252. doi: 10.1021/acs.jmedchem.5b00191
Butler, C. R., Brodney, M. A., Beck, E. M., Barreiro, G., Nolan, C. E., Pan, F., et al. (2015). Discovery of a series of efficient, centrally efficacious BACE1 inhibitors through structure-based drug design. J. Med. Chem. 58, 2678–2702. doi: 10.1021/jm501833t
Butler, C. R., Ogilvie, K., Martinez-Alsina, L., Barreiro, G., Beck, E. M., Nolan, C. E., et al. (2017). Aminomethyl-derived β secretase (BACE1) inhibitors: engaging Gly230 without an anilide functionality. J. Med. Chem. 60, 386–402. doi: 10.1021/acs.jmedchem.6b01451
Carvalho, C., Correia, S. C., Cardoso, S., Plácido, A. I., Candeias, E., Duarte, A. I., et al. (2015). The role of mitochondrial disturbances in Alzheimer, Parkinson and Huntington diseases. Expert Rev. Neurother. 15, 867–884. doi: 10.1586/14737175.2015.1058160
Carvalho, C., Correia, S. C., Perry, G., Castellani, R. J., and Moreira, P. I. (2016). Cerebrovascular and mitochondrial abnormalities in Alzheimer's disease: a brief overview. J. Neural. Transm. 123, 107–111. doi: 10.1007/s00702-015-1367-7
Chen, J. J., Liu, Q., Yuan, C., Gore, V., Lopez, P., Ma, V., et al. (2015). Development of 2-aminooxazoline 3-azaxanthenes as orally efficacious beta-secretase inhibitors for the potential treatment of Alzheimer's disease. Bioorganic Med. Chem. Lett. 25, 767–774. doi: 10.1016/j.bmcl.2014.12.092
Cheng, Y., Brown, J., Judd, T. C., Lopez, P., Qian, W., Powers, T. S., et al. (2015). An orally available BACE1 inhibitor that affords robust CNS Aβ reduction without cardiovascular liabilities. ACS Med. Chem. Lett. 6, 210–215. doi: 10.1021/ml500458t
Cheng, Y., Judd, T. C., Bartberger, M. D., Brown, J., Chen, K., Fremeau, R. T. Jr., et al. (2011). From fragment screening to in vivo efficacy: optimization of a series of 2-aminoquinolines as potent inhibitors of β-site amyloid precursor protein cleaving enzyme 1 (BACE1). J. Med. Chem. 54, 5836–5857. doi: 10.1021/jm200544q
Citron, M. (2002). Beta-secretase as a target for the treatment of Alzheimer's disease. J. Neurosci. Res. 70, 373–379. doi: 10.1002/jnr.10393
Correia, S. C., Resende, R., Moreira, P. I., and Pereira, C. M. (2015). Alzheimer's disease-related misfolded proteins and dysfunctional organelles on autophagy menu. DNA Cell Biol. 34, 261–273. doi: 10.1089/dna.2014.2757
Cumming, J. N., Smith, E. M., Wang, L., Misiaszek, J., Durkin, J., Pan, J., et al. (2012). Structure based design of iminohydantoin BACE1 inhibitors: identification of an orally available, centrally active BACE1 inhibitor. Bioorganic Med. Chem. Lett. 22, 2444–2449. doi: 10.1016/j.bmcl.2012.02.013
Cummings, J., Lee, G., Mortsdorf, T., Ritter, A., and Zhong, K. (2017). Alzheimer's disease drug development pipeline: 2017. TRCI 3, 367–384. doi: 10.1016/j.trci.2017.05.002
De Strooper, B., Vassar, R., and Golde, T. (2010). The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 6, 99–107. doi: 10.1038/nrneurol.2009.218
De Tran, Q., Bepary, S., Lee, G. H., Cho, H., Park, W. K., and Lim, H. J. (2016). Synthesis of (3S,4S)-4-aminopyrrolidine-3-ol derivatives and biological evaluation for their BACE1 inhibitory activities. Bioorganic Med. Chem. Lett. 26, 51–54. doi: 10.1016/j.bmcl.2015.11.033
Dineen, T. A., Chen, K., Cheng, A. C., Derakhchan, K., Epstein, O., Esmay, J., et al. (2014). Inhibitors of beta-site amyloid precursor protein cleaving enzyme (BACE1): identification of (S)-7-(2-fluoropyridin-3-yl)-3-((3-methyloxetan-3-yl)ethynyl)-5'H-spiro[chromeno[2,3-b]pyridine-5,4'-oxazol]-2'-amine (AMG-8718). J. Med. Chem. 57, 9811–9831. doi: 10.1021/jm5012676
Dislich, B., and Lichtenthaler, S. F. (2012). The membrane-bound aspartyl protease BACE1: molecular and functional properties in Alzheimer's Disease and beyond. Front. Physiol. 3:16. doi: 10.3389/fphys.2012.00008
Egbertson, M., McGaughey, G. B., Pitzenberger, S. M., Stauffer, S. R., Coburn, C. A., Stachel, S. J., et al. (2015). Methyl-substitution of an iminohydantoin spiropiperidine β-secretase (BACE-1) inhibitor has a profound effect on its potency. Bioorganic Med. Chem. Lett. 25, 4812–4819. doi: 10.1016/j.bmcl.2015.06.082
Eketjäll, S., Janson, J., Kaspersson, K., Bogstedt, A., Jeppsson, F., Fälting, J., et al. (2016). AZD3293: a novel, orally active BACE1 inhibitor with high potency and permeability and markedly slow off-rate kinetics. J. Alzheimers Dis. 50, 1109–1123. doi: 10.3233/JAD-150834
Epstein, O., Bryan, M. C., Cheng, A. C., Derakhchan, K., Dineen, T. A., Hickman, D., et al. (2014). Lead optimization and modulation of hERG activity in a series of aminooxazoline xanthene β-site amyloid precursor protein cleaving enzyme (BACE1) inhibitors. J. Med. Chem. 57, 9796–9810. doi: 10.1021/jm501266w
Ghosh, A. K., and Osswald, H. L. (2014). BACE1 (β-secretase) inhibitors for the treatment of Alzheimer's disease. Chem. Soc. Rev. 43, 6765–6813. doi: 10.1039/C3CS60460H
Ghosh, A. K., Bilcer, G., Harwood, C., Kawahama, R., Shin, D., Hussain, K. A., et al. (2001). Structure-based design: potent inhibitors of human brain memapsin 2 (β-secretase). J. Med. Chem. 44, 2865–2868. doi: 10.1021/jm0101803
Ghosh, A. K., Brindisi, M., and Tang, J. (2012). Developing β-secretase inhibitors for treatment of Alzheimer's disease. J. Neurochem. 120(Suppl 1.), 71–83. doi: 10.1111/j.1471-4159.2011.07476.x
Ghosh, A. K., Brindisi, M., Yen, Y. C., Cárdenas, E. L., Ella-Menye, J. R., Kumaragurubaran, N., et al. (2017). Design, synthesis, and X-ray structural studies of BACE-1 inhibitors containing substituted 2-oxopiperazines as P1'-P2' ligands. Bioorganic Med. Chem. Lett. 27, 2432–2438. doi: 10.1016/j.bmcl.2017.04.011
Gravenfors, Y., Viklund, J., Blid, J., Ginman, T., Karlstrom, S., Kihlstrom, J., et al. (2012). New aminoimidazoles as beta-secretase (BACE-1) inhibitors showing amyloid-β (Abeta) lowering in brain. J. Med. Chem. 55, 9297–9311. doi: 10.1021/jm300991n
Haghighijoo, Z., Firuzi, O., Hemmateenejad, B., Emami, S., Edraki, N., and Miri, R. (2017). Synthesis and biological evaluation of quinazolinone-based hydrazones with potential use in Alzheimer's disease. Bioorg. Chem. 74, 126–133. doi: 10.1016/j.bioorg.2017.07.014
Hauptmann, S., Keil, U., Scherping, I., Bonert, A., Eckert, A., and Müller, W. E. (2006). Mitochondrial dysfunction in sporadic and genetic Alzheimer's disease. Exp. Gerontol. 41, 668–673. doi: 10.1016/j.exger.2006.03.012
Hilpert, H., Guba, W., Woltering, T. J., Wostl, W., Pinard, E., Mauser, H., et al. (2013). Beta-Secretase (BACE1) inhibitors with high in vivo efficacy suitable for clinical evaluation in Alzheimer's disease. J. Med. Chem. 56, 3980–3995. doi: 10.1021/jm400225m
Hu, X., Das, B., Hou, H., He, W., and Yan, R. (2018). BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J. Exp. Med. 215, 927–940. doi: 10.1084/jem.20171831
Huang, H., La, D. S., Cheng, A. C., Whittington, D. A., Patel, V. F., Chen, K., et al. (2012). Structure- and property-based design of aminooxazoline xanthenes as selective, orally efficacious, and CNS penetrable BACE inhibitors for the treatment of Alzheimer's disease. J. Med. Chem. 55, 9156–9169. doi: 10.1021/jm300598e
Hunt, K. W., Cook, A. W., Watts, R. J., Clark, C. T., Vigers, G., Smith, D., et al. (2013). Spirocyclic β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors: from hit to lowering of cerebrospinal fluid (CSF) amyloid β in a higher species. J. Med. Chem. 56, 3379–3403. doi: 10.1021/jm4002154
Jain, P., Wadhwa, P. K., Gunapati, S., and Jadhav, H. R. (2016a). Design, synthesis and in vitro evaluation studies of sulfonyl-amino-acetamides as small molecule BACE-1 inhibitors. Bioorganic Med. Chem. 24, 2567–2575. doi: 10.1016/j.bmc.2016.04.023
Jain, P., Wadhwa, P. K., Rohilla, S., and Jadhav, H. R. (2016b). Rational design, synthesis and in vitro evaluation of allylidene hydrazinecarboximidamide derivatives as BACE-1 inhibitors. Bioorganic Med. Chem. Lett. 26, 33–37. doi: 10.1016/j.bmcl.2015.11.044
Jordan, J. B., Whittington, D. A., Bartberger, M. D., Sickmier, E. A., Chen, K., Cheng, Y., et al. (2016). Fragment-linking approach using (19)F NMR spectroscopy to obtain highly potent and selective inhibitors of β-secretase. J. Med. Chem. 59, 3732–3749. doi: 10.1021/acs.jmedchem.5b01917
Karran, E., Mercken, M., and De Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712. doi: 10.1038/nrd3505
Kulshreshtha, A., and Piplani, P. (2016). Current pharmacotherapy and putative disease-modifying therapy for Alzheimer's disease. Neurol. Sci. 37, 1403–1435. doi: 10.1007/s10072-016-2625-7
Malamas, M. S., Barnes, K., Johnson, M., Hui, Y., Zhou, P., Turner, J., et al. (2010a). Di-substituted pyridinyl aminohydantoins as potent and highly selective human beta-secretase (BACE1) inhibitors. Bioorg. Med. Chem. 18, 630–639. doi: 10.1016/j.bmc.2009.12.007
Malamas, M. S., Erdei, J., Gunawan, I., Barnes, K., Hui, Y., Johnson, M., et al. (2011). New pyrazolyl and thienyl aminohydantoins as potent BACE1 inhibitors: exploring the S2' region. Bioorg Med. Chem. Lett. 21, 5164–5170. doi: 10.1016/j.bmcl.2011.07.057
Malamas, M. S., Erdei, J., Gunawan, I., Barnes, K., Johnson, M., Hui, Y., et al. (2009). Aminoimidazoles as potent and selective human beta-secretase (BACE1) inhibitors. J. Med. Chem. 52, 6314–6323. doi: 10.1021/jm9006752
Malamas, M. S., Erdei, J., Gunawan, I., Turner, J., Hu, Y., Wagner, E., et al. (2010b). Design and synthesis of 5,5′-disubstituted aminohydantoins as potent and selective human beta-secretase (BACE1) inhibitors. J. Med. Chem. 53, 1146–1158. doi: 10.1021/jm901414e
Malamas, M. S., Robichaud, A., Erdei, J., Quagliato, D., Solvibile, W., Zhou, P., et al. (2010c). Design and synthesis of aminohydantoins as potent and selective human beta-secretase (BACE1) inhibitors with enhanced brain permeability. Bioorganic Med. Chem. Lett. 20, 6597–6605. doi: 10.1016/j.bmcl.2010.09.029
Mandal, M., Wu, Y., Misiaszek, J., Li, G., Buevich, A., Caldwell, J. P., et al. (2016). Structure-based design of an iminoheterocyclic β-site amyloid precursor protein cleaving Enzyme (BACE) inhibitor that lowers central Aβ in nonhuman primates. J. Med. Chem. 59, 3231–3248. doi: 10.1021/acs.jmedchem.5b01995
Mandal, M., Zhu, Z., Cumming, J. N., Liu, X., Strickland, C., Mazzola, R. D., et al. (2012). Design and validation of bicyclic iminopyrimidinones as beta amyloid cleaving enzyme-1 (BACE1) inhibitors: conformational constraint to favor a bioactive conformation. J. Med. Chem. 55, 9331–9345. doi: 10.1021/jm301039c
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer's disease. Nat. Rev. Dis. Primers 1, 1–18. doi: 10.1038/nrdp.2015.56
May, P. C., Dean, R. A., Lowe, S. L., Martenyi, F., Sheehan, S. M., Boggs, L. N., et al. (2011). Robust central reduction of amyloid-β in humans with an orally available, non-peptidic beta-secretase inhibitor. J. Neurosci. 31, 16507–16516. doi: 10.1523/JNEUROSCI.3647-11.2011
May, P. C., Willis, B. A., Lowe, S. L., Dean, R. A., Monk, S. A., Cocke, P. J., et al. (2015). The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 35, 1199–1210. doi: 10.1523/JNEUROSCI.4129-14.2015
Menting, K. W., and Claassen, J. A. (2014). Beta-secretase inhibitor; a promising novel therapeutic drug in Alzheimer's disease. Front. Aging Neurosci. 6:165. doi: 10.3389/fnagi.2014.00165
Merck, S. (2018). Merck Announces Discontinuation of APECS Study Evaluating Verubecestat (MK-8931) for the Treatment of People with Prodromal Alzheimer's Disease. Available online at: http://investors.merck.com/news/press-release-details/2018/Merck-Announces-Discontinuation-of-APECS-Study-Evaluating-Verubecestat-MK-8931-for-the-Treatment-of-People-with-Prodromal-Alzheimers-Disease/default.aspx (Accessed February 25, 2018).
Mullard, A. (2017). BACE inhibitor bust in Alzheimer trial. Nat. Rev. Drug Discov. 16:5. doi: 10.1038/nrd.2017.43
Oehlrich, D., Prokopcova, H., and Gijsen, H. J. (2014). The evolution of amidine-based brain penetrant BACE1 inhibitors. Bioorganic Med. Chem. Lett. 24, 2033–2045. doi: 10.1016/j.bmcl.2014.03.025
Peters, F., Salihoglu, H., Rodrigues, E., Herzog, E., Blume, T., Filser, S., et al. (2018). BACE1 inhibition more effectively suppresses initiation than progression of β-amyloid pathology. Acta Neuropathol. 135, 695–710. doi: 10.1007/s00401-017-1804-9
Plácido, A. I., Oliveira, C. R., Moreira, P. I., and Pereira, C. M. (2015). Enhanced amyloidogenic processing of amyloid precursor protein and cell death under prolonged endoplasmic reticulum stress in brain endothelial cells. Mol. Neurobiol. 51, 571–590. doi: 10.1007/s12035-014-8819-1
Prati, F., Bottegoni, G., Bolognesi, M. L., and Cavalli, A. (2018). BACE-1 inhibitors: from recent single-target molecules to multitarget compounds for Alzheimer's Disease. J. Med. Chem. 61, 619–637. doi: 10.1021/acs.jmedchem.7b00393
Querfurth, H., and LaFerla, F. (2010). Alzheimer's disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Resende, R., Ferreiro, E., Pereira, C., and Oliveira, C. R. (2008). ER stress is involved in Aβ-induced GSK-3β activation and tau phosphorylation. J. Neurosci. Res. 86, 2091–2099. doi: 10.1002/jnr.21648
Rombouts, F. J., Tresadern, G., Delgado, O., Martinez-Lamenca, C., Van Gool, M., Garcia-Molina, A., et al. (2015). 1,4-Oxazine β-Secretase 1 (BACE1) inhibitors: from hit generation to orally bioavailable brain penetrant leads. J. Med. Chem. 58, 8216–8235. doi: 10.1021/acs.jmedchem.5b01101
Scott, J. D., Li, S. W., Brunskill, A. P., Chen, X., Cox, K., Cumming, J. N., et al. (2016). Discovery of the 3-imino-1,2,4-thiadiazinane 1,1-dioxide derivative verubecestat (MK-8931)-A β-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer's Disease. J. Med. Chem. 59, 10435–10450. doi: 10.1021/acs.jmedchem.6b00307
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Staff, R. (2017). Alzheimer's Drug Fails in Another Crushing Disappointment [Online]. Available online at: https://www.scientificamerican.com/article/alzheimer-rsquo-s-drug-fails-in-another-crushing-disappointment/ (Accessed February 25, 2018).
Stamford, A. W., Scott, J. D., Li, S. W., Babu, S., Tadesse, D., Hunter, R., et al. (2012). Discovery of an orally available, brain penetrant BACE1 inhibitor that affords robust CNS Aβ reduction. ACS Med. Chem. Lett. 3, 897–902. doi: 10.1021/ml3001165
Swahn, B. M., Holenz, J., Kihlström, J., Kolmodin, K., Lindström, J., Plobeck, N., et al. (2012). Aminoimidazoles as BACE-1 inhibitors: the challenge to achieve in vivo brain efficacy. Bioorganic Med. Chem. Lett. 22, 1854–1859. doi: 10.1016/j.bmcl.2012.01.079
Thomas, A. A., Hunt, K. W., Newhouse, B., Watts, R. J., Liu, X., Vigers, G., et al. (2014a). 8-Tetrahydropyran-2-yl chromans: highly selective β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors. J. Med. Chem. 57, 10112–10129. doi: 10.1021/jm5015132
Thomas, A. A., Hunt, K. W., Volgraf, M., Watts, R. J., Liu, X., Vigers, G., et al. (2014b). Discovery of 7-tetrahydropyran-2-yl chromans: β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors that reduce amyloid β-protein (Aβ) in the central nervous system. J. Med. Chem. 57, 878–902. doi: 10.1021/jm401635n
Vassar, R. (1999). Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Vassar, R. (2016). BACE1 inhibition as a therapeutic strategy for Alzheimer's disease. J. Sport Health Sci. 5, 388–390. doi: 10.1016/j.jshs.2016.10.004
Voytyuk, I., De Strooper, B., and Chavez-Gutierrez, L. (2018). Modulation of gamma- and β-secretases as early prevention against Alzheimer's Disease. Biol. Psychiatry 83, 320–327. doi: 10.1016/j.biopsych.2017.08.001
WHO, S. (2017). World Health Organization. Global Action Plan on the Public Health Response to Dementia 2017–2025.
Woltering, T. J., Wostl, W., Hilpert, H., Rogers-Evans, M., Pinard, E., Mayweg, A., et al. (2013). BACE1 inhibitors: a head group scan on a series of amides. Bioorganic Med. Chem. Lett. 23, 4239–4243. doi: 10.1016/j.bmcl.2013.05.003
Wu, Y. J., Guernon, J., Rajamani, R., Toyn, J. H., Ahlijanian, M. K., Albright, C. F., et al. (2016a). Discovery of furo[2,3-d][1,3]thiazinamines as beta amyloid cleaving enzyme-1 (BACE1) inhibitors. Bioorganic Med. Chem. Lett. 26, 5729–5731. doi: 10.1016/j.bmcl.2016.10.055
Wu, Y. J., Guernon, J., Shi, J., Marcin, L., Higgins, M., Rajamani, R., et al. (2016b). Discovery of S3-truncated, C-6 heteroaryl substituted aminothiazine β-site APP cleaving enzyme-1 (BACE1) inhibitors. J. Med. Chem. 59, 8593–8600. doi: 10.1021/acs.jmedchem.6b01012
Yan, G., Hao, L., Niu, Y., Huang, W., Wang, W., Xu, F., et al. (2017). 2-Substituted-thio-N-(4-substituted-thiazol/1H-imidazol-2-yl)acetamides as BACE1 inhibitors: synthesis, biological evaluation and docking studies. Eur. J. Med. Chem. 137, 462–475. doi: 10.1016/j.ejmech.2017.06.020
Yan, R. (2016). Stepping closer to treating Alzheimer's disease patients with BACE1 inhibitor drugs. Transl. Neurodegener. 5, 1–11. doi: 10.1186/s40035-016-0061-5
Yan, R. (2017). Physiological functions of the β-site amyloid precursor protein cleaving enzyme 1 and 2. Front. Mol. Neurosci. 10:97. doi: 10.3389/fnmol.2017.00097
Yan, R., and Vassar, R. (2014). Targeting the β secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 13, 319–329. doi: 10.1016/S1474-4422(13)70276-X
Yuan, J., Venkatraman, S., Zheng, Y., McKeever, B. M., Dillard, L. W., and Singh, S. B. (2013). Structure-based design of β-site APP cleaving enzyme 1 (BACE1) inhibitors for the treatment of Alzheimer's disease. J. Med. Chem. 56, 4156–4180. doi: 10.1021/jm301659n
Zhou, P., Li, Y., Fan, Y., Wang, Z., Chopra, R., Olland, A., et al. (2010). Pyridinyl aminohydantoins as small molecule BACE1 inhibitors. Bioorganic Med. Chem. Lett. 20, 2326–2329. doi: 10.1016/j.bmcl.2010.01.136
Keywords: Alzheimer's Disease (AD), amyloid-β (Aβ), BACE1, inhibitors, small molecules, drug discovery and development
Citation: Coimbra JRM, Marques DFF, Baptista SJ, Pereira CMF, Moreira PI, Dinis TCP, Santos AE and Salvador JAR (2018) Highlights in BACE1 Inhibitors for Alzheimer's Disease Treatment. Front. Chem. 6:178. doi: 10.3389/fchem.2018.00178
Received: 14 March 2018; Accepted: 04 May 2018;
Published: 24 May 2018.
Edited by:
Simone Brogi, University of Siena, ItalyReviewed by:
Mohammad Hassan Baig, Yeungnam University, South KoreaCopyright © 2018 Coimbra, Marques, Baptista, Pereira, Moreira, Dinis, Santos and Salvador. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Armanda E. Santos, YWVzYW50b3NAY2kudWMucHQ=
Jorge A. R. Salvador, c2FsdmFkb3JAY2kudWMucHQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.