 Lei Guo
Lei Guo Zaki S. Safi2
Zaki S. Safi2- 1School of Material and Chemical Engieering, Tongren University, Tongren, China
- 2Chemistry Department, Faculty of Science, Al Azhar University-Gaza, Gaza City, Palestine
- 3Department of Chemistry, Faculty of Science, Cumhuriyet University, Sivas, Turkey
It is known that iron is one of the most widely used metals in industrial production. In this work, the inhibition performances of three thiophene derivatives on the corrosion of iron were investigated in the light of several theoretical approaches. In the section including DFT calculations, several global reactivity descriptors such as EHOMO, ELUMO, ionization energy (I), electron affinity (A), HOMO-LUMO energy gap (ΔE), chemical hardness (η), softness (σ), as well as local reactivity descriptors like Fukui indices, local softness, and local electrophilicity were considered and discussed. The adsorption behaviors of considered thiophene derivatives on Fe(110) surface were investigated using molecular dynamics simulation approach. To determine the most active corrosion inhibitor among studied thiophene derivatives, we used the principle component analysis (PCA) and agglomerative hierarchical cluster analysis (AHCA). Accordingly, all data obtained using various theoretical calculation techniques are consistent with experiments.
Introduction
One of the serious problems in industrial sector is the corrosion of metals or alloys, which causes great casualties and enormous property loss (Frankel et al., 2015; Li et al., 2015). The most environmentally-benign and cost-effective approach to prevent metals against corrosion in acid solutions is using of inhibitors (Raja et al., 2016). Organic molecules containing O, N, and/or S atoms are the most widely used and are considered to be effective corrosion inhibitors (Xhanari et al., 2017). It is generally assumed that they can be adsorbed at metal surface through some active groups like heteroatoms, triple bonds or aromatic rings (Kovačević and Kokalj, 2013; Ko and Sharma, 2017). Previous studies showed that most organic inhibitors decrease corrosion rate by adsorption on the substrate surface and the inhibition performance follows the sequence O < N < S < P (Loto et al., 2012). Bockris and Swinkels suggested that S and/or N atoms could easily adsorb on metal surface by replacing water molecules (Bockris and Swinkels, 1964). The interactions between the organic corrosion inhibitors and the metal substrates are generally divided into two types: physical adsorption and chemical adsorption. On the basis of the theory hard and soft acids and bases (HSAB) introduced by Pearson (Pearson and Songstad, 1967), molecules including sulfur atom in their molecular structures can be regarded as soft bases, which give easily electron to metal surfaces because they cannot resist to electron cloud polarization or deformation. Thiophene is a sulfur containing heterocyclic compound with the molecular formula C4H4S. The electron pairs on sulfur atom are delocalized in π-conjugated systems. Thiophene and its derivatives can be obtained from petroleum or coal and in general are well-known because of their therapeutic applications in medicinal chemistry. Many theoretical and experimental studies including the analyzing of inhibition efficiencies of thiophene and its derivatives are available in the literature (Benabdellah et al., 2011; Gece, 2013; Yadav et al., 2014). Especially, Fouda et al. (2014) synthesized three thiophene derivatives and investigated their anticorrosive effects against the corrosion of carbon steel using experimental methods such as weight loss, Tafel test, electrochemical frequency modulation, and electrochemistry impedance test. The molecular structures of mentioned molecules are given in Figure 1.
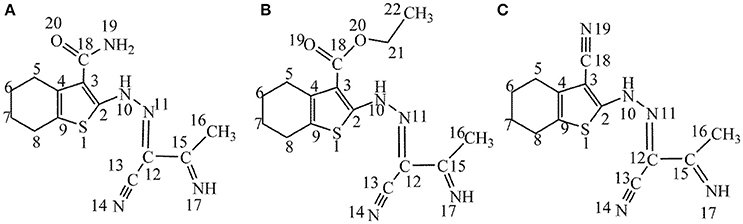
Figure 1. (A–C) Chemical molecular structures of studied thiophene derivatives (the number indicates atom-numbering).
It is well-known that reactivity in chemistry is a back-bone because it is well associated with reaction mechanisms. Therefore it allows us to understand chemical reactions, behavior of substance and improving synthesis procedures to obtain new materials such as corrosion inhibitors or drugs. Quantum chemistry calculations have proven to be very effective in evaluating the corrosion inhibition efficiency (Khalil, 2003; Obot et al., 2015; Taylor, 2015; Lgaz et al., 2018). They have been widely employed to interpret the experimental phenomena. Especially in recent years, conceptual density functional theory (CDFT) has been developed and applied to analyze the molecular activity of inhibitors (Geerlings et al., 2003; Liu, 2009). Furthermore, molecular dynamics simulation has become another effective way to explore the structure and bonding characteristics at inhibitor/metal interface (Khaled, 2008; Oguzie et al., 2013; Wang et al., 2016).
In the present work, three earlier mentioned thiophene derivatives (see Figure 1), namely, 2-[(2Z)-2-(1-cyano-2-iminopropylidene)hydrazinyl]-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxami de (A), ethyl-2-[(2Z)-2-(1-cyano-2-iminopropylidene)hydrazinyl]-4,5,6,7-tetrahydro-1-benzothiophe ne-3-carboxylate (B), as well as 2-[(2Z)-2-(1-cyano-2-iminopropylidene)hydrazinyl]-4,5,6,7-tetrahy dro-1-benzothiophene-3-carbonitrile(C), have been chosen to theoretically analyze their anticorrosive efficiencies. The objective of the work is to assess their anti-corrosive performances of studied molecules applying quantum chemical calculations, molecular dynamics simulation, principle component analysis (PCA) as well as agglomerative hierarchical cluster analysis (AHCA).
Computational Methods and Definitions
Gaussian Calculations
Gaussian 09 package program was used to the study of the isolated compounds using density functional theory (DFT), with the B3LYP functional (Wiberg, 2004) under the SDD, 6-31G and 6-31++G basis sets. For comparison, the calculations based on Hartree–Fock (HF) theory were also performed. Gauss View 5.0.8 program was employed to prepare the correlative calculation parameters for the compounds under probe.
As we all know, electrochemical corrosion generally happens in liquid environment. One of the most popular approaches to research solvent effect is to consider hydrogen bonded clusters of solvent molecules surrounding the solute molecules. Thus self-consistent reaction field (SCRF) theory, with Tomas's polarized continuum model (PCM) (Andzelm et al., 1995) was used to describe the solvent effect of water. This method describes the solvent as a structureless continuum with uniform dielectric permittivity, in which a molecular-shaped empty cavity is dug to host the solute (Scalmani et al., 2006; Aouniti et al., 2013). The reliability of PCM method to explore the solvent effect in the field of corrosion inhibitors has been validated by many researchers (Wazzan et al., 2016; Guo et al., 2017; Yang et al., 2017).
Global Reactivity Descriptors
Chemical reactivity can be simply defined as the tendency of a chemical matter to undergo chemical reaction with another chemical matter. It is well-known that the understanding of the nature of chemical interactions and the prediction of chemical reactivity of atoms, ions or molecules are some of the challenging issues in chemistry. In the CDFT, quantum chemical descriptors like electronegativity (χ), chemical hardness (η), and chemical potential (μ) are usually considered. μ and η are defined as the first derivative of the electronic energy and chemical potential with respect to the electron number (N) at constant external potential, v(r), respectively (Liu, 2009; Frau and Glossman-Mitnik, 2017).
Within the framework of finite differences approximation, the following expressions based on first vertical ionization energy and electron affinity values of chemical species are given (Madkour and Elshamy, 2016).
Softness that is known as a measure of the polarizability is mathematically defined as the multiplicative inverse of chemical hardness:
Ionization energies and electron affinities of molecules can be predicted via Koopman's Theorem (Bellafont et al., 2015), i.e., ionization energy and electron affinity values of a chemical species correspond to negative values of its HOMO and LUMO orbital energies, respectively. So we can write the following equations for the calculation of quantum chemical parameters like hardness, electronegativity and chemical potential.
Besides, global electrophilicity index (ω) introduced by Parr (Parr et al., 1999), nucleophilicity (ε), which is physically the inverse of the electrophilicity, the fraction of electrons (ΔN) transferred from (to) the inhibitor molecule to (from) the metal surface, the energy of back donation ΔEb-d, electronic charge accepting capability and the initial molecule-metal interaction energy Δψ, were calculated in terms of global hardness (η) and electronegativity (χ) as given in Equations (8–12).
Herein, χFe, χinh, ηFe, and ηinh represent the absolute electronegativity and hardness of iron and inhibitor molecule, correspondingly. In order to obtain the ΔN values, we adopted a theoretical value of χFe = 7.0 eV and ηFe = 0 by assuming that for a metallic bulk I = A since they are softer than the neutral metallic atoms (Zarrouk et al., 2014).
Fukui Functions
Evaluation of the Fukui functions has been employed to explore the local reactivity of the molecules. Yang and Mortier (1986) defined the Fukui function as the first derivative of the electronic density ρ(r) of a system with respect to the number of electrons (N) at a fixed external potential ν(r), as given Equation (13).
Roy et al. (1999) defined the electrophilic and nucleophilic Fukui functions for a site k in a molecule by using left and right derivatives with respect to the number of electrons as expressed via Equations (14–16).
where ρk(N), ρk(N–1), and ρk(N+1) are the gross electronic populations of the site k in neutral, cationic and anionic system, respectively.
As it is known, the concept of generalized philicity have been introduced by Chattaraj et al., they defined a local quantity called philicity associated with a site k in a molecule with the assistance of corresponding condensed-to-atom variants of Fukui function, as in Equation (17) (Parthasarathi et al., 2004).
where α = +, − and 0 corresponds to local philic quantities describing nucleophilic, electrophilic and radical attacks, respectively. In the light of Equation (17), the highest corresponds to the most electrophilic site in a molecule. In addition, Lee et al. (1988) proposed different local softness, which can be used to describe the reactivity of atoms in molecules, which can be defined as in Equation (18).
where α = +, − and 0 represents local softness quantities describing nucleophilic, electrophilic and radical attacks respectively.
Recently, Morell et al. (2006) put forward a dual descriptor, Δf (k), which is defined as the difference between the nucleophilic and electrophilic Fukui functions:
Likewise, the associated dual local softness have also been defined as expressed in Equation (20).
It is also defined as the condensed version of Δfk multiplied by the global softness σ. The multiphilic descriptor, Δωk, is defined as the difference between the nucleophilic and electrophilic condensed philicity functions. This parameter may be used as an index of selectivity toward nucleophilic attack, which can as well characterize an electrophilic attack and is given by Equation (21) (Padmanabhan et al., 2006).
If Δωk (or Δf (k)) > 0, the site k is favored for a nucleophilic attack, whereas if Δωk (or Δf (k)) < 0, the site k may be favored for an electrophilic attack.
Molecular Dynamic Simulation
Adsorption characteristics of studied thiophene derivatives on iron metal surface are investigated by molecular dynamic simulation employing the Forcite module in Materials Studio 8.0 software. As model iron surface, Fe(110) surface was considered because it possesses a density packed surface and is the most stable among three common iron substrates (Guo et al., 2014). The Fe(110) system was simulated through a repeated supercell containing a 5-layer slab of Fe with 80 atoms per layer, in a 8 × 10 two-dimensional periodicity. A vacuum region 40 Å thick was included between repeated surface slabs. To simulate the metal-inhibitor systems, COMPASS force field (Sun et al., 1998) was used. The simulations of three thiophene derivatives labeled as A, B, and C on iron surface were carried out to determine the optimal adsorption sites for these molecules. All simulations made in the study were performed in an NVT canonical ensemble at 298 K with a time step of 1.0 fs and a total simulation time of 1,000 ps. The operational temperature was monitored via the Andersen thermostat. In the calculations, vacuum media was preferred and five layers of iron atoms were used.
To calculate the adsorption energies (Eads) on Fe(110) surface of modeled of A, B, and C molecules, we used the Equation (22). Herein, it is important to note that binding energy (Ebinding) is defined by the negative value of adsorption energy as given in Equation (23).
where Ecomplex is the total energy of an inhibitor molecule and the metal surface system. EFe is described as the energy of iron surface without adsorption of any inhibitor molecule and Einh represents the energy of free inhibitor molecules.
Results and Discussion
Global Reactivity
As mentioned in the computation section, the investigated inhibitors were optimized by performing two different methods and using three different basis sets. The optimized electronic structures correspond to energy minima with no imaginary frequencies. According to frontier molecular orbital theory and in consistent with Fukui's theory, a high EHOMO value means the ability of a molecule to donate electrons to an assigned acceptor (metal surface in our case) with empty molecular orbital that facilitated the adsorption process and therefore indicated good inhibition performance (Obot et al., 2015). In contrast, ELUMO is related to electron affinity, which corresponds to a tendency for electron acceptance. Accordingly, the gap between energy levels of the molecules (ΔE = ELUMO-EHOMO) is a significant descriptor that need be calculated. It demonstrates inveterate electron donating ability and measures the interaction between inhibitor molecules and substrate surface. The optimized molecular structures, HOMO, LUMO, as well as the molecular electrostatic potential of the investigated molecules are showed in Figure 2.
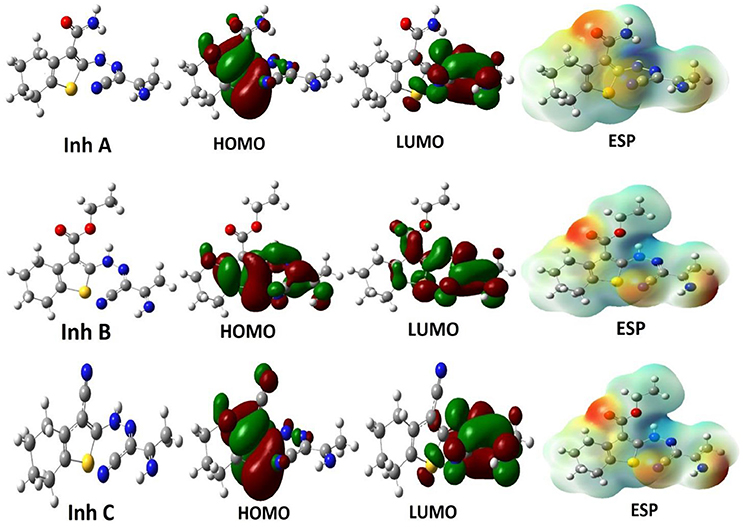
Figure 2. The optimized structures, HOMOs, LUMOs and electrostatic potential structures of studied inhibitor molecules at the DFT/B3LYP/6-31++G level of theory.
Many investigations (Khalil, 2003; Gece, 2013; Frau and Glossman-Mitnik, 2017) reported that the inhibition efficiency of inhibitors has been found to correlate with some other quantum chemical parameters like η, σ, χ, and dipole moment (DM). Besides, ω, ε, ΔN, ΔEb-d, Δψ, which have been calculated in terms of η and χ, are also highly useful descriptors in corrosion inhibition studies of organic molecules. Our computed quantum chemical parameters for three inhibitor molecules in gas phase and water phase are given in Tables 1, 2.
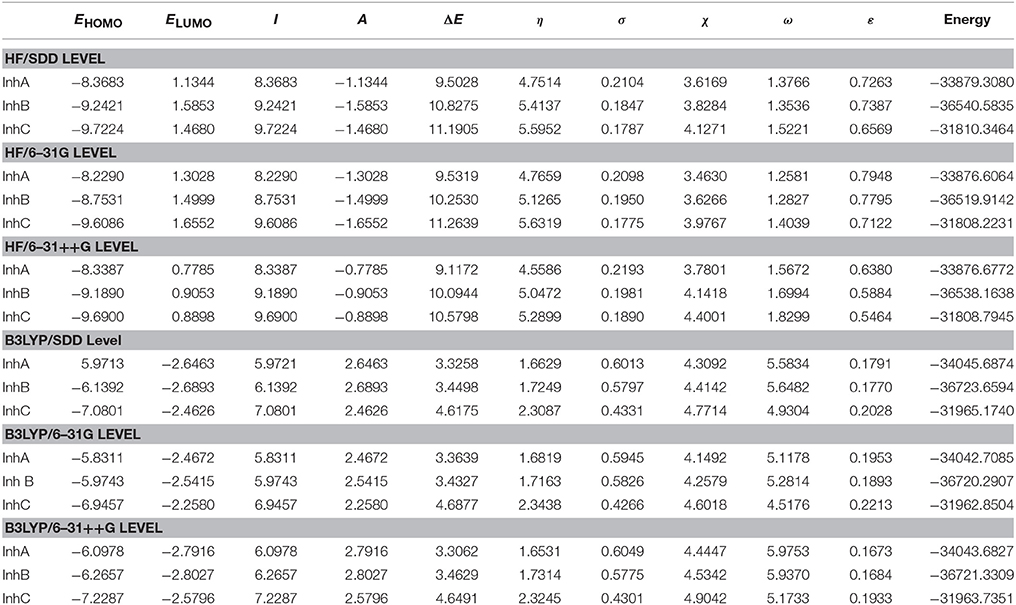
Table 1. Calculated quantum chemical parameters for three thiophene derivatives in gas phase (all in eV).
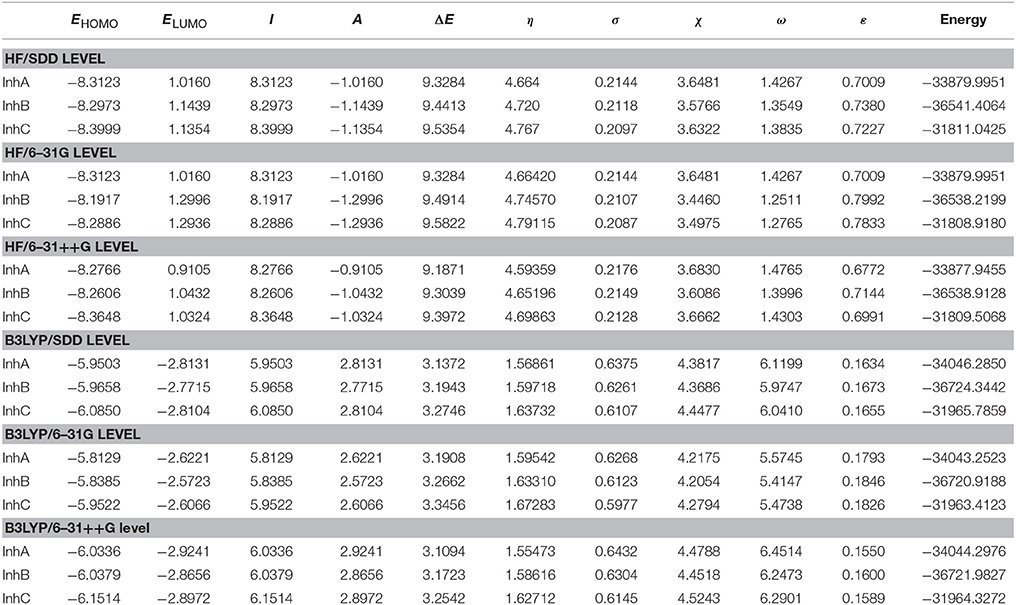
Table 2. Calculated quantum chemical parameters for three thiophene derivatives in aqueous phase (all in eV).
As given in Table 1, it is notable that inhibitor A has the highest EHOMO among all the studied inhibitors. This reflects that the electron-donating ability of Inhibitor A is strong. As is known, corrosion inhibitors with low ΔE values provide better inhibition performances. This is because that the excitation energy to remove an electron from the last occupied orbital will be low. It was also reported that a molecule with a low energy gap could be more polarizable, which is usually associated with a high chemical reactivity and low kinetic stability, termed as soft molecule (Madkour and Elroby, 2015). Jafari et al. (2013) also pointed that adsorption of inhibitor molecule onto a metallic surface occurs at the site of the molecule which has the greatest softness and lowest hardness. Our results in Tables 1, 2 show that all the elected levels inhibitor A has the lowest ΔE energy in both gas and aqueous phases, and hence the molecule could have a better inhibitive performance on the iron surface as corrosion inhibitor. Based on the above discussion, we can write the corrosion inhibition efficiency (in both gas and aqueous phases) order as: A > B > C. These results are in good agreement with the available experimental results (Fouda et al., 2014). However, from the results obtained for ELUMO in gas and aqueous phases the trend is irregular, which does not correlate well in the experimentally determination inhibition efficiency. So we claimed that LUMO energies of molecules may fail in terms of the explanation of their inhibition efficiencies.
Generally, chemical hardness (η) is the resistance against electron cloud polarization or deformation of chemical species. Thus, the η value of a molecule and its inhibition efficiency are inversely proportional to each other because a hard molecule is renitent to give electrons (Kaya et al., 2016). Lukovits et al. (2001) reported that η, σ, and ΔE are quantum chemical descriptors closely correlated with each other. As it is mentioned in computation part and according to Koopmans's theorem, both softness and hardness are obtained on the basis of HOMO and LUMO orbital energies. Hard molecules with high ΔE cannot act as good corrosion inhibitor. Nevertheless, soft molecules which have low ΔE could be excellent corrosion inhibitors since they can easily donate electrons to metals. Based on the results reported in Tables 1, 2, it is clear that the sequence of inhibitive efficiency for studied molecules based on their hardness, and softness values can be written as: A > B > C.
In the light of the simple charge transfer theory for donation and back-donation of charges proposed by Gomez et al. (2006), the electronic back-donation process can probably affect the interaction between the inhibitors and substrate surface. As given in Equation (11), when the electron transfer and back-donation processes occur simultaneously, the energy change is directly proportional to the hardness of the inhibitor molecule. The ΔEb−d indicates that since η > 0, then ΔEb−d < 0, and the charge transfer from a molecule, followed by a back-donation to the molecule, is energetically favored (Bedair, 2016). Based on this principle, it is available to compare the stabilization among the inhibitor molecules, because there will be an interaction with the same metal, it is obvious that ΔEb−d will decrease with the hardness increases. According to our results given in Table 3, as expected and in agreement with the experimental results (Fouda et al., 2014), the calculated ΔEb−d exhibit the tendency: A > B > C.
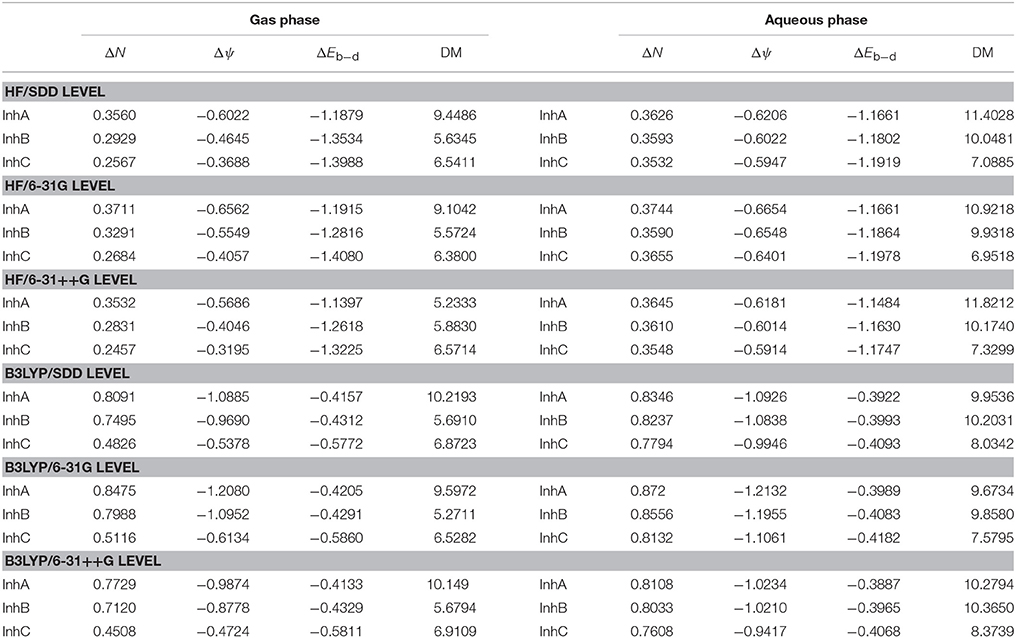
Table 3. Calculated ΔEb−d (back-donation), ΔN (the fraction of electrons transferred), Δψ (the initial molecule-metal interaction energy), and dipole moment (DM) values for studied thiophene derivatives in gas phase and aqueous phase.
Electrophilicity index (ω) represents the propensity of a molecule to receive electrons. Conversely, nucleophilicity (ε) indicates the tendency to donate or share electrons with others, it is defined as the inverse of electrophilicity (1/ω). It is generally assumed that a molecule that has a large electrophilicity value is ineffective against corrosion while a molecule that has a large nucleophilicity value is expected to an excellent candidate as corrosion inhibitor. Based on Tables 1, 2, it is evident that the inhibiors have low electrophilicity index values and are good nucleophiles. But there exists discrepancy in trend between the HF and B3LYP functionals for electrophilicity, which is due to the quadratic dependence on the electronegativity. In view of this, their molecular reactivity cannot be predicted accurately, it is necessary to take into account another additional criterion to determine their inhibitive capacity.
In this work, the number of electrons transferred (ΔN) of the between metal substrate and inhibitor molecules was calculated using Equation (10). The results are also gathered in Table 3. Based on the Sanderson's electronegativity equalization principle (Sanderson, 1983), the charge transfer process between metal and inhibitor will continue until their electronegativity values are equal with each other. In fact, ΔN can be regarded as a derived descriptor from the electronegativity/hardness equalization principle. It was pointed out that the positive value of electrons transferred (ΔN) indicates that the molecules act as electron donors. Based on the tabulated results (Table 3), we can see that molecule A has the largest number of the fraction of transferred electrons (ΔN) in both gas and aqueous phases, turn is, A > B > C, regardless of phase and the elected levels. These results agree well with the experimental results. Another important parameter is the initial molecule-metal interaction energy (Δψ), which has been introduced in Kokalj's work (Kovacevic and Kokalj, 2011). In our work, we have calculated (Δψ) for all the researched inhibitors and the results are given in Table 3. The results indicate that the trend of Δψ is also A > B > C.
Recently, some authors used the dipole moment as an indicator of corrosion inhibition efficiencies of molecules (Gece, 2008; Zarrouk et al., 2014). Several authors showed that corrosion inhibition efficiency increases with the increase of dipole moment (Stoyanova et al., 2002; Sahin et al., 2008). Considering the idea that increasing value of dipole moment facilitates the electron transport process. Yet others proposed the opposite correlation, that is, a low value of dipole moment favors the accumulation of inhibitor molecules on the metal surface and ultimately increasing the inhibition performances (Khalil, 2003; Lebrini et al., 2005). As shown in Table 3, the calculated the dipole moments for studied compounds are irregular. There is no any remarkable relationship between dipole moment and inhibition efficiency. Thus it cannot be used a priori to judge the inhibition effectiveness.
Overall, based on the global descriptors considered for each molecule shown in Tables 1–3, The order of chemical activities from high to low is: –C(=O)–NH2 > –C(=O)–OC2H5 > –C≡N substituents. Correspondingly, the inhibitive effectiveness order for the thiophene molecules is: InhA > InhB > InhC. This indicates that our calculated theoretical results are in agreement with experimental orders.
Local Reactivity
In order to have an understanding on the local reactivity of the thiophene derivatives, the Fukui indices for every atom in the inhibitors have been calculated at the B3LYP/6-31++G level. It is well known that an analysis of the Fukui indices and the local descriptors provides a more comprehensive information of the reactivity of the molecules under probe. To complete the picture, the local softness, local electrophilicity, and the dual descriptors have been also calculated for each atoms in the studied molecules.
Figure 3 represents graphically the local dual descriptors, Δfk, Δσk, and Δωk for the three compounds. It should be noted that the numbering of atoms, which is given in Figure 1 are employed in this analysis. Generally, the condensed Fukui functions can make us to distinguish each part of the inhibitor molecule in the light of its distinct chemical behavior with different substituent functional groups. Therefore, the site for nucleophilic attack will be the place where the value of f− is a maximum. Conversely, the site for electrophilic attack is controlled by the value of f+.
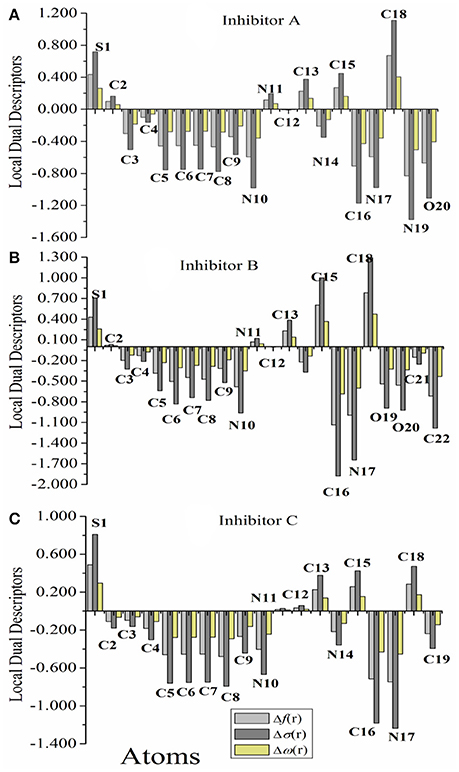
Figure 3. (A–C) Graphical representation of the local dual descriptors, Δf, Δσ, and Δω, based on Fukui Functions of the studied inhibitors A, B, and C (the atom-numbering is in correspond with Figure 1).
The results reported in Tables S1–S3 reveal that, for nucleophilic attack, the highest values of InhA are N19, N17, O20, and C16 atoms. For InhB, the most nucleophilic sites are N17, C16, and C22 atoms. For InhC, the most reactive sites are C16 and N17. This indicates the propensity to donate electrons to vacant molecular orbital on the iron surface to form coordinate bond. This agrees with the results of the computed HOMO density. For electrophilic attack, the highest values of the three studied inhibitors are S1, C18, C15, and C13, indicating that the sites most capable for an electrophilic attack that is through which the molecule accepts electrons to form feedback bonds with Fe(110) surface. This also conforms to the computed LUMO orbital density. These results are also supported by the values of the local dual indices (Δf, Δσ, and Δω), which indicate that these inhibitors have many active sites and most of these centers have values of the three descriptors of lower than 0, except some atoms, which found to be have values > 0 (see Figure 3), indicating an electrophilic centers. A close inspection would reveal that all the molecules had the back-donation process at their carbon atoms in agreement with the frontier orbital results obtained. According to these results, one can conclude that Inh A molecule will have many active centers to interact with iron substrate. These are most likely those areas that containing N and O atoms, which are the most possible sites for bonding to iron surface through donating electrons to the Fe 3d orbitals (Khaled, 2010). Also, it can be suggested that the binding between the surface of the metal with the InhA is stronger than that in the case of B and C, respectively. Finally, the above local descriptors reveal that the theoretical order for the variation of inhibition efficiencies of the investigative inhibitors agrees with the available experimental data and it is as follows: A > B > C.
PCA and AHCA Analysis
In this work, all calculated variables have been auto scaled to compare them at the same level. Thereafter, principal component analysis (PCA) was adopted to reduce the number of variables and select the most relevant ones, which are responsible for the reactivity of researched thiophene derivatives. After performing many tests, a good separation was obtained between more active and less active thiophene compounds using 11 variables: EHOMO, I, ΔE, χ, μ, σ, ω, ΔN, Δψ, and ΔEb−d. As indicated from PCA results, the first two principal components (F1 and F2) describe all of the overall variance as follows: F1 = 84.58% and F2 = 15.42%. The score plot of the variances is a reliable representation of the spatial distribution of the points for the data set studied after explaining almost all of the variances by the first two principal components.
In Figure 4A, the most informative score plot for the inhibitors is presented (F1 vs. F2). It is evident from the figure that PCA is responsible for the separation between more active InhA and InhB and less active Inh C where F1 > 0 for the more active compounds, and F1 < 0 for the less active one. These results are in a well agreement with the experimental results, the calculated global and local descriptors. Figure 4B shows AHCA analysis for the inhibitors under probe. The horizontal lines represent the inhibitors and the vertical lines the similarity values between pairs of inhibitors, an inhibitor and a group of inhibitors and among groups of inhibitors. It is noticed that AHCA results are very similar to those obtained with the PCA analysis, i.e., the studied inhibitors were grouped into two categories: More active inhibitors A and B and less active one C.
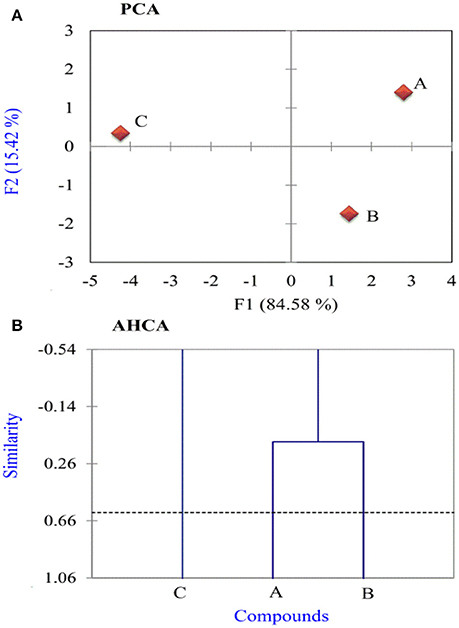
Figure 4. (A) Score and (B) dendrogram plots obtained for three studied inhibitors.
Molecular Dynamic Simulations
Molecular dynamic simulation approach is very important in terms of the explanation of the nature of the interactions between corrosion inhibitors and metallic surface. The optimized equilibrium adsorption configurations for A, B, and C molecules on Fe(110) surface are given in Figure 5. Adsorption energy is known as the energy released when inhibitor molecule was adsorbed on metal surface. As given in previous section, the binding energy is the negative value of the adsorption energy. Higher negative value of adsorption energy and higher positive values of binding energy represent the more stable and more strong interaction between metal surface and inhibitor molecule. In Table 4, calculated adsorption and binding energies as well as experimentally determined corrosion inhibition efficiencies for studied thiophene derivatives are presented. It is apparent that the binding energies of three derivatives on the Fe(110) substrate decrease in the order A > B > C, which is in consonance with the experimental inhibition efficiency orders (Fouda et al., 2014).
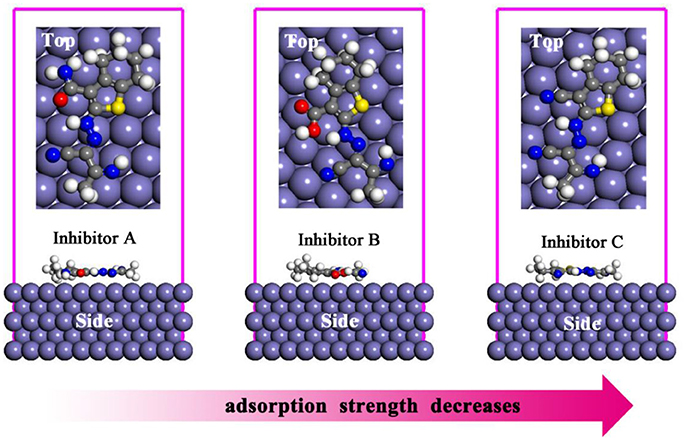
Figure 5. Side and top views of the most stable adsorption models of three inhibitors on Fe(110) surfaces.

Table 4. Experimental inhibition efficiency, IE(%), as well as the outputs and descriptors obtained from MD simulation for the adsorption of A, B, and C on Fe(110) surface.
Comparison Between Experimental and Theoretical Results
In this subsection, a comparison between experimental and theoretical results is presented. Our study shows that there is an excellent correlation between our theoretical results (global quantum descriptors and MDs results) with the experimental inhibition efficiency (IE%). Figures 6A,B shows graphical representation of the relationship between the linear correlation R obtained for the relationship between theoretical reactivity parameters calculated (in both gas and aqueous phases) using B3LYP and HF methods with 6-31++G basis sets for the studied inhibitors and their experimental IE%. The results of linear relationship coefficients are summarized in Table S4. However, the results obtained via B3LYP method are well correlated with the experimental results than those obtained by HF method.
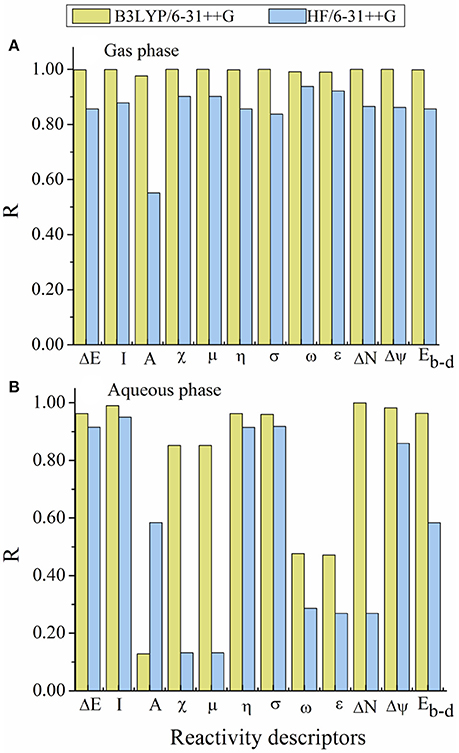
Figure 6. Graphical representation of the linear relationship coefficient (R) obtained between the calculated reactivity descriptors and the experimental inhibition efficiency (IE%). (A) in gas phase and (B) in aqueous phase.
As can be seen in Figure 6, there are a very excellent linear correlation between the experimental inhibition efficiency and the theoretical descriptors in gas phase for both methods in gas phase. It is apparent from the column graphs plotted that B3LYP method provided more accurate results compared to HF method. In addition, it can be said that calculation levels including 6-31++G basis set is more successful compared to other calculation levels in terms of the obtaining good agreement with experimental results. Finally, it can also be seen from Figure 7 that there exists closely correlation between experimental anticorrosion efficiencies and calculated binding energies with a high correlation coefficient of 1.00.
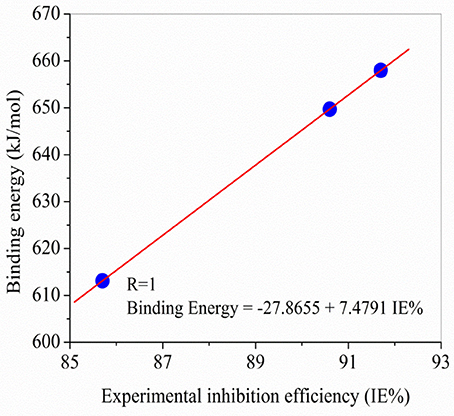
Figure 7. Comparisons made between experimental anticorrosion efficiencies and theoretical binding energy obtained in the study.
Conclusions
In this work, Hartree Fock as well as DFT calculations, molecular dynamic simulation, PCA, and AHCA were used to analyze the anticorrosive performances of some thiophene derivatives against the iron metal. Global and local reactivity descriptors were calculated in both gas and aqueous phase for three studied inhibitors. Within the framework of the theoretical results obtained in this study, the following conclusions can be written.
(1) DFT, molecular dynamic simulation, PCA, and AHCA results showed that corrosion inhibition efficiency ranking of studied molecules is given as: InhA > InhB > InhC.
(2) It is apparent from binding energies and adsorption energies calculated for studied thiophene derivatives, these molecules are very effective against the corrosion of iron.
(3) According the PCA and AHCA results, least active inhibitor among the studied molecules is inhibitor C.
(4) Both theoretical data and experimental results are compatible with inductive effect of functional groups appearing in the molecular structures of studied thiophene derivatives.
(5) Theoretical results obtained in this work have far-reaching significance to the rational design of novel thiophene derivatives as corrosion inhibitor.
Author Contributions
LG, ZS, and SK performed all the calculations. WS, BT, NA, and CK were involved in conception and design of the experiments. All authors were involved the drafting, revision and approval of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (21706195), the Cumhuriyet University Scientific Research Project Commission as a research project with F495 code, the Guizhou Province Science Fund for Excellent Young Scholars (QKHTC2017-5604), the Science and Technology Program of Guizhou Province (QKHJC2016-1149), the Provincial Key Disciplines of Chemical Engineering and Technology in Guizhou Province (ZDXK2017-8), and the Guizhou Provincial Department of Education Foundation (QJHKYZ2016-105).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00155/full#supplementary-material
References
Andzelm, J., Kölmel, C., and Klamt, A. (1995). Incorporation of solvent effects into density functional calculations of molecular energies and geometries. J. Chem. Phys. 103, 9312–9320. doi: 10.1063/1.469990
Aouniti, A., Khaled, K. F., and Hammouti, B. (2013). Correlation between inhibition efficiency and chemical structure of some amino acids on the corrosion of armco iron in molar HCl. Int. J. Electrochem. Sci. 8, 5925–5943.
Bedair, M. A. (2016). The effect of structure parameters on the corrosion inhibition effect of some heterocyclic nitrogen organic compounds. J. Mol. Liq. 219, 128–141. doi: 10.1016/j.molliq.2016.03.012
Bellafont, N. P., Illas, F., and Bagus, P. S. (2015). Validation of Koopmans' theorem for density functional theory binding energies. Phys. Chem. Chem. Phys. 17, 4015–4019. doi: 10.1039/C4CP05434B
Benabdellah, M., Yahyi, A., Dafali, A., Aouniti, A., Hammouti, B., and Ettouhami, A. (2011). Corrosion inhibition of steel in molar HCl by triphenyltin2-thiophene carboxylate. Arab. J. Chem. 4, 243–247. doi: 10.1016/j.arabjc.2010.06.055
Bockris, J. M., and Swinkels, D. (1964). Adsorption of n-decylamine on solid metal electrodes. J. Electrochem. Soc. 111, 736–743. doi: 10.1149/1.2426222
Fouda, A. S., Attia, A. A., and Negm, A. A. (2014). Some thiophene derivatives as corrosion inhibitors for carbon steel in hydrochloric acid. J. Metallurgy 2014, 1–15. doi: 10.1155/2014/472040
Frankel, G., Mauzeroll, J., Thornton, G., Bluhm, H., Morrison, J., Maurice, V., et al. (2015). Corrosion scales and passive films: general discussion. Faraday Discuss. 180, 205–232. doi: 10.1039/C5FD90045J
Frau, J., and Glossman-Mitnik, D. (2017). Conceptual DFT descriptors of amino acids with potential corrosion inhibition properties calculated with the latest minnesota density functionals. Front. Chem. 5:16. doi: 10.3389/fchem.2017.00016
Gece, G. (2008). The use of quantum chemical methods in corrosion inhibitor studies. Corros. Sci. 50, 2981–2992. doi: 10.1016/j.corsci.2008.08.043
Gece, G. (2013). Theoretical evaluation of the inhibition properties of two thiophene derivatives on corrosion of carbon steel in acidic media. Mater. Corros. 64, 940–944. doi: 10.1002/maco.201106482
Geerlings, P., De Proft, F., and Langenaeker, W. (2003). Conceptual density functional theory. Chem. Rev. 103, 1793–1873. doi: 10.1021/cr990029p
Gómez, B., Likhanova, N. V., Domínguez-Aguilar, M. A., Martínez-Palou, R., Vela, A., and Gazquez, J. L. (2006). Quantum chemical study of the inhibitive properties of 2-pyridyl-azoles. J. Phys. Chem. B 110, 8928–8934. doi: 10.1021/jp057143y
Guo, L., Ren, X., Zhou, Y., Xu, S., Gong, Y., and Zhang, S. (2017). Theoretical evaluation of the corrosion inhibition performance of 1,3-thiazole and its amino derivatives. Arab. J. Chem. 10, 121–130. doi: 10.1016/j.arabjc.2015.01.005
Guo, L., Zhu, S., Zhang, S., He, Q., and Li, W. (2014). Theoretical studies of three triazole derivatives as corrosion inhibitors for mild steel in acidic medium. Corros. Sci. 87, 366–375. doi: 10.1016/j.corsci.2014.06.040
Jafari, H., Danaee, I., Eskandari, H., and RashvandAvei, M. (2013). Electrochemical and theoretical studies of adsorption and corrosion inhibition of N,N'-bis(2-hydroxyethoxyacetophenone)-2,2-dimethyl-1,2-propanediimine on low carbon steel (API 5L Grade B) in acidic solution. Ind. Eng. Chem. Res. 52, 6617–6632. doi: 10.1021/ie400066x
Kaya, S., Guo, L., Kaya, C., Tüzün, B., Obot, I. B., Touir, R., et al. (2016). Quantum chemical and molecular dynamic simulation studies for the prediction of inhibition efficiencies of some piperidine derivatives on the corrosion of iron. J. Taiwan Inst. Chem. Eng. 65, 522–529. doi: 10.1016/j.jtice.2016.05.034
Khaled, K. F. (2008). Molecular simulation, quantum chemical calculations and electrochemical studies for inhibition of mild steel by triazoles. Electrochim. Acta 53, 3484–3492. doi: 10.1016/j.electacta.2007.12.030
Khaled, K. F. (2010). Studies of iron corrosion inhibition using chemical, electrochemical and computer simulation techniques. Electrochim. Acta 55, 6523–6532. doi: 10.1016/j.electacta.2010.06.027
Khalil, N. (2003). Quantum chemical approach of corrosion inhibition. Electrochim. Acta 48, 2635–2640. doi: 10.1016/S0013-4686(03)00307-4
Ko, X., and Sharma, S. (2017). Adsorption and self-assembly of surfactants on metal-water interfaces. J. Phys. Chem. B 121, 10364–10370. doi: 10.1021/acs.jpcb.7b09297
Kovacevic, N., and Kokalj, A. (2011). Analysis of molecular electronic structure of imidazole- and benzimidazole-based inhibitors: a simple recipe for qualitative estimation of chemical hardness. Corros. Sci. 53, 909–921. doi: 10.1016/j.corsci.2010.11.016
Kovačević, N., and Kokalj, A. (2013). The relation between adsorption bonding and corrosion inhibition of azole molecules on copper. Corros. Sci. 73, 7–17. doi: 10.1016/j.corsci.2013.03.016
Lebrini, M., Bentiss, F., Vezin, H., and Lagrenee, M. (2005). Inhibiting effects of some oxadiazole derivatives on the corrosion of mild steel in perchloric acid solution. Appl. Surf. Sci. 252, 950–958. doi: 10.1016/j.apsusc.2005.01.160
Lee, C. T., Yang, W. T., and Parr, R. G. (1988). Local softness and chemical reactivity in the molecules CO, SCN− and H2CO. J. Mol. Struct. Theochem 40, 305–313. doi: 10.1016/0166-1280(88)80397-X
Lgaz, H., Salghi, R., and Ali, I. H. (2018). Corrosion inhibition behavior of 9-hydroxyrisperidone as a green corrosion inhibitor for mild steel in hydrochloric acid: electrochemical, DFT and MD simulations studies. Int. J. Electrochem. Sci. 13, 250–264. doi: 10.20964/2018.01.26
Li, X., Zhang, D., Liu, Z., Li, Z., Du, C., and Dong, C. (2015). Share corrosion data. Nature 527, 441–442.
Liu, S. B. (2009). Conceptual density functional theory and some recent developments. Acta Phys. Chim. Sin. 25, 590–600. doi: 10.3866/PKU.WHXB20090332
Loto, C., Loto, R., and Popoola, A. (2012). Corrosion inhibition of thiourea and thiadiazole derivatives: a review. J. Mater. Environ. Sci. 3, 885–894.
Lukovits, I., Kalman, E., and Zucchi, F. (2001). Corrosion inhibitors - Correlation between electronic structure and efficiency. Corrosion 57, 3–8. doi: 10.5006/1.3290328
Madkour, L. H., and Elroby, S. K. (2015). Inhibitive properties, thermodynamic, kinetics and quantum chemical calculations of polydentate Schiff base compounds as corrosion inhibitors for iron in acidic and alkaline media. Int. J. Ind. Chem. 6, 165–184. doi: 10.1007/s40090-015-0039-7
Madkour, L. H., and Elshamy, I. H. (2016). Experimental and computational studies on the inhibition performances of benzimidazole and its derivatives for the corrosion of copper in nitric acid. Int. J. Ind. Chem. 7, 195–221. doi: 10.1007/s40090-015-0070-8
Morell, C., Grand, A., and Toro-Labbe, A. (2006). Theoretical support for using the Delta f(r) descriptor. Chem. Phys. Lett. 425, 342–346. doi: 10.1016/j.cplett.2006.05.003
Obot, I. B., Macdonald, D. D., and Gasem, Z. M. (2015). Density functional theory (DFT) as a powerful tool for designing new organic corrosion inhibitors. Part 1: an overview. Corros. Sci. 99, 1–30. doi: 10.1016/j.corsci.2015.01.037
Oguzie, E. E., Oguzie, K. L., Akalezi, C. O., Udeze, I. O., Ogbulie, J. N., and Njoku, V. O. (2013). Natural products for materials protection: corrosion and microbial growth inhibition using capsicum frutescens biomass extracts. ACS Sustain. Chem. Eng. 1, 214–225. doi: 10.1021/sc300145k
Padmanabhan, J., Parthasarathi, R., Subramanian, V., and Chattaraj, P. K. (2006). Chemical reactivity indices for the complete series of chlorinated benzenes: solvent effect. J. Phys. Chem. A 110, 2739–2745. doi: 10.1021/jp056630a
Parr, R. G., Von Szentpaly, L., and Liu, S. B. (1999). Electrophilicity index. J. Am. Chem. Soc. 121, 1922–1924. doi: 10.1021/ja983494x
Parthasarathi, R., Padmanabhan, J., Elango, M., Subramanian, V., and Chattaraj, P. K. (2004). Intermolecular reactivity through the generalized philicity concept. Chem. Phys. Lett. 394, 225–230. doi: 10.1016/j.cplett.2004.07.002
Pearson, R. G., and Songstad, J. (1967). Application of the principle of hard and soft acids and bases to organic chemistry. J. Am. Chem. Soc. 89, 1827–1836. doi: 10.1021/ja00984a014
Raja, P. B., Ismail, M., Ghoreishiamiri, S., Mirza, J., Ismail, M. C., Kakooei, S., et al. (2016). Reviews on corrosion inhibitors: a short view. Chem. Eng. Commun. 203, 1145–1156. doi: 10.1080/00986445.2016.1172485
Roy, R. K., Pal, S., and Hirao, K. (1999). On non-negativity of Fukui function indices. J. Chem. Phys. 110, 8236–8245. doi: 10.1063/1.478792
Sahin, M., Gece, G., Karci, F., and Bilgic, S. (2008). Experimental and theoretical study of the effect of some heterocyclic compounds on the corrosion of low carbon steel in 3.5% NaCl medium. J. Appl. Electrochem. 38, 809–815. doi: 10.1007/s10800-008-9517-3
Sanderson, R. T. (1983). Electronegativity and bond energy. J. Am. Chem. Soc. 105, 2259–2261. doi: 10.1021/ja00346a026
Scalmani, G., Frisch, M. J., Mennucci, B., Tomasi, J., Cammi, R., and Barone, V. (2006). Geometries and properties of excited states in the gas phase and in solution: theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 124:94107. doi: 10.1063/1.2173258
Stoyanova, A., Petkova, G., and Peyerimhoff, S. D. (2002). Correlation between the molecular structure and the corrosion inhibiting effect of some pyrophthalone compounds. Chem. Phys. 279, 1–6. doi: 10.1016/S0301-0104(02)00408-1
Sun, H., Ren, P., and Fried, J. R. (1998). The COMPASS force field: parameterization and validation for phosphazenes. Comput. Theor. Polym. Sci. 8, 229–246. doi: 10.1016/S1089-3156(98)00042-7
Taylor, C. D. (2015). Design and prediction of corrosion inhibitors from quantum chemistry I: can effective partition coefficients for organic corrosion inhibitors be determined from first-principles. J. Electrochem. Soc. 162, C340–C346. doi: 10.1149/2.0681507jes
Wang, Z., Lv, Q., Chen, S., Li, C., Sun, S., and Hu, S. (2016). Effect of interfacial bonding on interphase properties in SiO2/epoxy nanocomposite: a molecular dynamics simulation study. ACS Appl. Mater. Interfaces 8, 7499–7508. doi: 10.1021/acsami.5b11810
Wazzan, N. A., Obot, I. B., and Kaya, S. (2016). Theoretical modeling and molecular level insights into the corrosion inhibition activity of 2-amino-1,3,4-thiadiazole and its 5-alkyl derivatives. J. Mol. Liq. 221, 579–602. doi: 10.1016/j.molliq.2016.06.011
Wiberg, K. B. (2004). Basis set effects on calculated geometries: 6-311++G** vs. aug-cc-pVDZ. J. Comput. Chem. 25, 1342–1346. doi: 10.1002/jcc.20058
Xhanari, K., Finšgar, M., Knez Hrnčič, M., Maver, U., Knez, Ž., and Seiti, B. (2017). Green corrosion inhibitors for aluminium and its alloys: a review. RSC Adv. 7, 27299–27330. doi: 10.1039/C7RA03944A
Yadav, M., Behera, D., Sinha, R. R., and Yadav, P. N. (2014). Experimental and quantum studies on adsorption and corrosion inhibition effect on mild steel in hydrochloric acid by thiophene derivatives. Acta Metall. Sin. Engl. 27, 37–46. doi: 10.1007/s40195-013-0012-4
Yang, S. J., Qin, X. Y., He, R., Shen, W., Li, M., and Zhao, L. B. (2017). A density functional theory study on the thermodynamic and dynamic properties of anthraquinone analogue cathode materials for rechargeable lithium ion batteries. Phys. Chem. Chem. Phys. 19, 12480–12489. doi: 10.1039/C7CP01203A
Yang, W., and Mortier, W. J. (1986). The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 108, 5708–5711. doi: 10.1021/ja00279a008
Keywords: corrosion inhibition, molecular dynamics simulation, DFT, adsorption, principle component analysis
Citation: Guo L, Safi ZS, Kaya S, Shi W, Tüzün B, Altunay N and Kaya C (2018) Anticorrosive Effects of Some Thiophene Derivatives Against the Corrosion of Iron: A Computational Study. Front. Chem. 6:155. doi: 10.3389/fchem.2018.00155
Received: 20 January 2018; Accepted: 19 April 2018;
Published: 07 May 2018.
Edited by:
Ime Bassey Obot, King Fahd University of Petroleum and Minerals, Saudi ArabiaReviewed by:
Wenzhen Lai, Renmin University of China, ChinaDaniel Glossman-Mitnik, Centro de Investigación en Materiales Avanzados (CIMAV), Mexico
Copyright © 2018 Guo, Safi, Kaya, Shi, Tüzün, Altunay and Kaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Guo, Y3FnbGVpQDE2My5jb20=
Wei Shi, dHJ1d2Vpc2hpQDE2My5jb20=