Abstract
During drug development, safety is always the most important issue, including a variety of toxicities and adverse drug effects, which should be evaluated in preclinical and clinical trial phases. This review article at first simply introduced the computational methods used in prediction of chemical toxicity for drug design, including machine learning methods and structural alerts. Machine learning methods have been widely applied in qualitative classification and quantitative regression studies, while structural alerts can be regarded as a complementary tool for lead optimization. The emphasis of this article was put on the recent progress of predictive models built for various toxicities. Available databases and web servers were also provided. Though the methods and models are very helpful for drug design, there are still some challenges and limitations to be improved for drug safety assessment in the future.
Introduction
Drug discovery and development is a long journey full of high risk. It is estimated that the attrition rate of drug candidates is up to 96% (Paul et al., 2010) and the average cost to develop a new drug reaches to 2.6 billion U.S. dollars in recent years (PhRMA, 2015). One of the major causes for the high attrition rate is drug safety, which accounts for 30% of drug failures (Giri and Bader, 2015). Even if a drug is approved in market, it could be withdrawn due to safety problems. Therefore, drug safety should be evaluated extensively as early as possible.
Usually, in vitro and in vivo tests are performed to investigate drug safety, including a variety of toxicities and adverse drug effects. In recent years, there are also some efforts to develop in vitro models such as “organ on a chip” to reduce cost (Huh et al., 2010, 2011). However, those approaches are still costly and time-consuming. In comparison of experimental approaches, computational methods have shown great advantages since they are green, fast, cheap, accurate, and most importantly they could be done before a compound is synthesized (Segall and Barber, 2014).
Till now, many computational models have been developed for drug safety assessment, which could be generally divided into three categories: qualitative classification, quantitative regression and read-across. As the first step of drug safety assessment, we only need to know a compound is toxic or non-toxic, highly toxic or slightly toxic, rather than its exact toxicity value, so classification models can be used. For a small number of chemical analogs, quantitative structure-toxicity relationship (QSTR) models can be derived for prediction of exact toxicity values. For those unique compounds, read-across is also a feasible approach to deduce certain toxicity endpoint from their similar structures with experimental toxicity values. These models have high accuracies especially in a local chemical space, and sometimes they can replace in vitro or in vivo assays for certain endpoints. Furthermore, structural alerts (SAs) can be derived from the models as keys for a compound to cause adverse effects on organs (Pizzo et al., 2015), which can be used in structural modification to reduce the risk by chemists.
In recent years, we have worked on drug safety assessment and developed a lot of predictive models for chemical toxicity with machine learning methods and structural alerts. A web server named admetSAR was also developed for publicly free access (Cheng et al., 2012b). In a previous paper published in 2013, we reviewed the advances and challenges of in silico prediction of chemical toxicity together with pharmacokinetic properties (Cheng et al., 2013a). Here, we would like to review the progress of in silico chemical toxicity prediction in recent 5 years, including methodologies of machine learning and structural alerts, and major toxicity endpoints in drug discovery and development (Figure 1). Available data sources and web servers were also mentioned. At last, challenges and future directions in this field were provided.
Figure 1
Model building with machine learning methods
The general procedure to build a predictive model contains roughly four steps: data collection, data description, model building, and model evaluation. Each step has its own requirements to guarantee the reliability and accuracy of the models.
Data collection
The quality of experimental data is the most important in model building. Currently, there are numerous well-defined data available online, which greatly facilitates the construction of computational models by machine learning methods. In Table 1, we listed some widely used databases, including those linking chemical structures with safety outcomes, protein targets and/or biological pathways.
Table 1
Data sources for prediction of chemical toxicity.
CTA, compound-toxicity association; MI, molecular interaction; SE, side effect; CPI, compound-protein interaction.
TOXNET is a comprehensive source that integrates several toxicity databases such as ToxLine and ChemIDplus (Fowler and Schnall, 2014). ACToR is a large database that aggregates data from thousands of public sources (Judson et al., 2008). DSSTox, a subset of ACToR, provides a high quality resource for toxicity prediction, including ToxCast and Tox21 data (Williams-DeVane et al., 2009). OECD established eChemPortal to provide chemical information including physicochemical properties, and toxicity. Many databases are contained in eChemPortal, such as ACToR and HSDB (Fonger et al., 2014). Some other toxicity databases include SuperToxic (Schmidt et al., 2009), T3DB (Wishart et al., 2015), and ToxBank (http://www.toxbank.net). We previously developed a web server admetSAR, which also contains toxicity data (Cheng et al., 2012b).
In addition to the phenotype data that are directly relevant to toxicity, databases on bioactivity, pathway and side effects are also important to toxicity prediction. Several bioactivity databases are free available, such as PubChem (Wang et al., 2009), ChEMBL (Gaulton et al., 2017), and BindingDB (Gilson et al., 2016). We developed a web server named MetaADEDB that integrates CTD (Davis et al., 2017), SIDER (Kuhn et al., 2010), and OFFSIDES (Tatonetti et al., 2012) with regard to the ADE of drugs (Cheng et al., 2013b,c).
Data description
There are two ways to represent chemical structures as numeric features which can be processed by machine learning methods. One way is to use molecular descriptors, which can be calculated from chemical structures, physicochemical or topological properties. Currently thousands of continuous and discrete molecular descriptors can be obtained via chemoinformatics toolkits such as PaDEL-Descriptor (Yap, 2011), OpenBabel (O'Boyle et al., 2011), CDKit (Steinbeck et al., 2003), RDKit (Landrum, 2017), or web servers like E-Dragon (Tetko et al., 2005), ChemBCPP (Dong et al., 2017a), and ChemDes (Dong et al., 2015). Using numeric features may result in overfitting when the size of training set is small (Xue et al., 2004). Hence, feature selection should be done before model building, to reduce the risk of overfitting and enhance the performance of model (Sun et al., 2017).
The other way is to use molecular fingerprints, which represent a molecule as a binary string, such as MACCS, PubChemFP, and KRFP (Klekota and Roth, 2008). In a molecular fingerprint, lists of substructures or other kinds of patterns are predefined. If a specified pattern presents in a molecule, the corresponding bit in the binary string is set to “1,” otherwise it will be set to “0.” Comparing to molecular descriptors, these binary features are more interpretable because each bit corresponds to a specific substructure. In addition to the common fingerprints, custom patterns can also be used to enhance the predictability of the models (Yang et al., 2017b).
Single-label model building
Machine learning methods are usually used to build the predictive models. There are many free and open access tools and development kits to fulfill this task. For example, Scikit-learn (Pedregosa et al., 2011) is a popular python toolkit for machine learning and TensorFlow (https://www.tensorflow.org) is a widely used python library for deep learning. WEKA (Frank et al., 2004), Orange (Demsar et al., 2013) and RapidMiner (https://rapidminer.com/) are machine learning toolboxes with GUI (Graph user interface).
Support vector machine (SVM), Random forest (RF), boost tree (BT), and k-nearest neighbor (kNN) are classic machine learning methods that are widely used in classification and regression models. SVM, also known as support vector classifier (SVC) or support vector regression (SVR) in particular tasks, is well-known for its high predictive performance and less risk of overfitting (Cortes and Vapnik, 1995). The basic idea of SVM is to construct a hyperplane in a high dimensional space with the largest distance to the nearest training data points (support vectors). RF and BT are derived from decision tree (Breiman, 2001; Elith et al., 2008). RF can be viewed as bagging many decision trees that use a random subset of features and combine them via a voting system. Different from RF, in which each tree is equal, BT dynamically adjusts the weight of each tree according to the mean error of prediction. kNN is one of the simplest algorithms (Cover and Hart, 1967). The creed of kNN is that compounds with similar structures have similar biological properties. In kNN, a sample is classified by the votes of the categories of its neighbors.
Sometimes, to enhance performance of prediction models, combination of these algorithms is applied. We developed a combined method using an artificial neural network (ANN) model to generate the final combination decision probability, which showed that the combined methods would be superior to “single” methods (Cheng et al., 2011b; Du et al., 2017; Sun et al., 2017).
Recently, deep learning (DL) has been applied in solving such challenging problems as computer vision and speech recognition (Deng et al., 2013; LeCun et al., 2015). Multilayer neural network (MNN) is one of the DL techniques. Different from common ANN that only has three layers including input layer, hidden layer and output layer (Shen et al., 2004), MNN contains more than one hidden layers and thus is more competent in large toxicological data with complex mechanisms. When the training set is large, it can perform better than ANN and above-mentioned classic machine learning methods (Mayr et al., 2016). However, more complex network indicates more weights to fit and more likely to be overfitting. Graph-convolutional networks (Duvenaud et al., 2015) and long short-term memory architectures (Altae-Tran et al., 2017) are recently developed to extract features from molecules based on atom features and show better performance in handling thousands of compounds or even more (Goh et al., 2017). DeepChem (https://deepchem.io) is an open source python library devoted to providing a high quality toolchain to facilitate the use of DL in drug discovery and other fields.
Multi-label model building
Unlike aforementioned single-label classification or regression models, multi-label classification (MLC) is a data mining approach in which each data instance can be assigned to multiple categories at once (Tsoumakas et al., 2010; Zhang and Zhou, 2014; Gibaja and Ventura, 2015). The demand for multi-label techniques is constantly growing in biology and genomics (Diplaris et al., 2005; Avila et al., 2009). The current algorithms used for this task are pretty new and many of them are still in an early stage of development.
There are three major approaches for multi-label learning: data transformation, method adaptation and ensembles of classifiers. The first one, including Binary Relevance (BR) (Godbole and Sarawagi, 2004), classifier chains (CC) (Read et al., 2011), and Label Powerset (LP) (Boutell et al., 2004), is to transform original multi-label dataset (MLD) to a set of binary datasets (BIDs) or one multi-class dataset (MCD) first, and then process them with traditional classification algorithms (Barot and Panchal, 2014). With the development of these frameworks for MLC, classification algorithms available for binary and multiclass data can be utilized as the underlying base classifier including SVM, ANN, decision tree, kNN, and so on. The second alternative aims for adapting existent algorithms to deal with multi-label data, such as multi-label C4.5 (Al-Otaibi et al., 2014), multi-label back-propagation (Zhang and Zhou, 2006), Rank-SVM (Wang et al., 2014), and multi-label kNN (Zhang and Zhou, 2007). Finally, the classification ensemble is also a widespread technique in multi-label field. For example, Ensemble of Classifier Chain (ECC) (Read et al., 2011), which consists of a set of CC with diverse label orders and then votes for the final prediction, is proposed to allow for the effect of chain order. Some other MLC methods based on the ensemble of multi-class classifiers were also proposed, such as EPS (Read et al., 2008), RAkEL (Tsoumakas and Vlahavas, 2007), and HOMER (Tsoumakas et al., 2008).
Model evaluation
For regression models, three evaluation metrics, namely Pearson product moment correlation coefficient (R2), mean absolute error (MAE) and root mean squared error (RMSE) are frequently used to estimate the performance of models. These parameters are defined as following:
where xi is the experimental value, yi is the predicted value, are their corresponding means and N is the number of samples.
For traditional single-label binary or multiple classification models, most of the performance metrics are calculated based on the count of true positive (TP), true negative (TN), false positive (FP), and false negative (FN). Accuracy, sensitivity and specificity metrics can be calculated as the following equations to represent the overall predictive ability, the predictive accuracy for positive samples and the predictive ability for negative ones:
In addition to these computed from binary partition of labels, metrics these calculated from a confidence degree of being positive are also used like area under the receiver operating characteristic curve (AUC).
Comparing to the single-label classification patterns, multi-label classifiers can have multiple outputs for an instance, of which the predictions can be fully or partially correct. The multi-label performance metrics introduced there can be classified into two groups, i.e., example-based and label-based metrics (Tsoumakas et al., 2007; Zhang and Zhou, 2014). Here, five example-based metrics (subset accuracy, Jaccard similarity coefficient, hamming-loss, micro-precision, micro-recall) are described with mathematical formulations below.
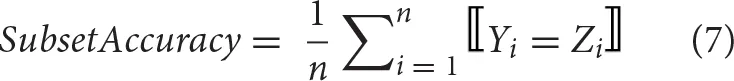
where Yi represents the real label-set of the ith instance, and Zi the predicted one. n is the number of instances and k is the number of labels.
Furthermore, another example-based metric named ranking loss can be used. The ranking loss metric portrays how many times an irrelevant label is ranked above a relevant one according to their probabilities belonging to each label. As for label-based metrics, micro-AUC is the most commonly used one. It is also a ranking based metric similar to ranking loss. However, different from the ranking loss that compares the ranks for each example, micros-AUC counts the number of all the relevant-irrelevant pairs meeting the condition that the relevant label is ranked above irrelevant one (in which the labels are not necessarily for the same example).
Methods for detecting structural alerts
Structural alerts (SAs) are key substructures responsible for certain toxicity. They are directly connected to toxicity and hence could be used for structural optimization by medicinal chemists to reduce the risk. In 1985, Ashby found strong associations between occurrence of some substructures or patterns and chemical mutagenicity to Salmonella, which was the first appearance of the concept of SA (Ashby and Tennant, 1988).
Till now, many methods and software have been developed for detecting SAs, such as SARpy (Ferrari et al., 2013), MoSS, Gaston, and MolFea. ToxAlerts is a web server that collects SAs defined by experts or identified by computational tools. It can predict toxicity according to the appearance of SAs (Sushko et al., 2012). Automatic detection of SAs by computational tools now becomes a hotspot as the development of cheminformatics and the explosion of available data (Lepailleur et al., 2013; Floris et al., 2017).
In a previous paper, we evaluated several methods for identification of SAs (Yang et al., 2017a). At present, the methods can be divided into three categories: fragment-based, graph-based, and fingerprint-based. Fragment-based methods, such as SARpy (Ferrari et al., 2013), cut the bonds of the molecules in dataset first to get all possible fragments. Then each fragment is evaluated according to their occurrence in toxic and non-toxic compounds. These methods have been used in detecting SAs for carcinogenicity (Golbamaki and Benfenati, 2016; Golbamaki et al., 2016). Graph-based approaches use subgraph searching algorithms, treating molecules as graphs that consist of a set of vertices and edges, to find the frequent patterns. MoSS uses depth-first search association rules to mine substructures (Borgelt and Berthold, 2002). Gaston is a stand-alone tool that uses a graph-based approach to obtain substructures from dataset (Kazius et al., 2006). Another graph-based method proposed by Ahlberg (Ahlberg et al., 2014) uses Atom Signature, a linear expression of a compound, to mined sub-signature as SAs. Fingerprint-based approaches do not obtain fragments from the dataset. Instead, the fragments are defined by different molecular fingerprints such as MACCS and SubFP (Shen et al., 2010). The selection of fingerprints may affect the final results of the identified SAs. Fingerprints such as Morgan, used by Bioalerts (Cortes-Ciriano, 2016) might lead to redundant SAs which are very similar and related to the same mechanism.
Information gain (IG) can also be used to evaluate the significance of a substructure. Compounds containing the substructure are categorized as toxic and others are categorized as non-toxic. IG is defined as the difference between the information entropy of original dataset and the weighted average information entropies of two datasets separated by a substructure (Sokolova and Szpakowicz, 2010). We previously used IG to detect privileged substructures whose occurrences have strong relevance to some endpoints (Shen et al., 2010).
Progress in toxicity prediction
Carcinogenicity and mutagenicity
Chemical carcinogenesis is of increasing importance in drug discovery for its serious effect on human health. Most of the predictive models use Carcinogenic Potency Database (CPDB) as the data source, which contains more than 1,500 chemicals with their labels (carcinogen or non-carcinogen) according to their TD50 values (Gold et al., 2005). Recently several publications shared their protocols to construct models to predict chemical carcinogenesis, including Naïve Bayes, kNN, probabilistic neural network, and SVM (Singh et al., 2013; Tanabe et al., 2013; Li et al., 2015; Zhang H. et al., 2016). Zhang et al. developed a web server, CarcinoPred-EL, for chemists to predict carcinogenicity online, in which Ensemble XGBoost was used to build the model (Zhang et al., 2017).
Due to its complicated mechanism and less available data, the predictive models based on phenotypic assays are not precise and reliable enough. It is an alternative to construct models based on in vitro assays. The mechanisms of carcinogenesis of chemicals can be categorized into: (1) genotoxicity, which are primarily caused by the mutagenicity of chemicals damaging DNA (Fan et al., in press); (2) non-genotoxic carcinogens acting through different specific mechanisms, which are more complicated (Golbamaki and Benfenati, 2016). Ames test devised by Bruce Ames is a well-known in vitro assay to detect mutagenic effects of chemicals. Currently more than 8,000 compounds with Ames mutagenicity are available. Both predictive models and structural alerts were promoted with these toxicity data in recent years (Kazius et al., 2005; Hansen et al., 2009; Xu et al., 2012; Yang et al., 2017a).
Acute oral toxicity
According to the exposure routes of chemicals, acute toxicity can be divided into oral, dermal and inhalation, among which acute oral toxicity is the most widely studied in computational prediction. It is often the first performed endpoint in drug discovery because any compounds causing acute toxicity will not be further considered for its strong hazardous to human health. Zhu et al. collected 7,385 compounds with LD50 values and built several models for prediction of chemical acute oral toxicity (Zhu et al., 2009). Based on the data set, several machine learning methods were developed and applied to construct classifiers and regression models to predict LD50 or their toxic categories (Li et al., 2014; Lei et al., 2016; Xu et al., 2017). Noticeably, the models built by Xu et al. have high performance in two test sets, more than 95% of accuracy for classification and 0.861 of R2 for regression, and the model is free available in web server (http://www.pkumdl.cn/DLAOT/DLAOThome.php).
Cardiotoxicity
Blockade of the hERG (human ether-a-go-go related gene) potassium channel is the main adverse effect with regard to cardiotoxicity (Gintant et al., 2016). Several in silico models were developed according to the in vitro hERG blockage test in early screening assays. Our group recently developed an in silico model that used chemical category approaches to predict hERG blockage (Zhang et al., 2016b), in which 1,570 unique compounds were collected from ChEMBL database and early studies (Doddareddy et al., 2010; Wang et al., 2012). In addition to machine learning methods, combination with multiple pharmacophores can improve the predictive capabilities and the model would be more interpretable (Wang et al., 2016).
However, as the simplified in vitro approaches for detection of cardiac safety are less specific, the in silico models will also output the false-positive predictions that may result in unwarranted attribution of novel drug candidates (Gintant et al., 2016). Other categories such as contractile and structural cardiotoxicity should be considered and more in vitro or in vivo data should be used to construct sophisticated models.
Hepatotoxicity
Chemical hepatotoxicity in drug discovery, also termed “drug induced liver injury (DILI),” is the leading cause for drug failure or withdrawn from the market (Schuster et al., 2005). Due to its complicated mechanism and inconsistency in diverse patients, experimental detection of hepatotoxicity in preclinical and clinical trials is difficult.
Computational approaches to predict DILI of compounds are widely applied for their low cost and high efficiency. Hewitt reviewed the in silico models on DILI prediction from 2000 to 2015, including statistics-based methods and expert systems (Hewitt and Przybylak, 2016). Chemical or hybrid descriptors as features, and different machine learning methods such as linear discriminant analysis and ANN were used in these models to predict general or specific endpoints related to hepatotoxicity (Hewitt and Przybylak, 2016). Zhu constructed a human hepatotoxicity database for QSTR models using post-market safety data originated from FDA adverse event reporting system (Zhu and Kruhlak, 2014). Our group previously used molecular fingerprints and machine learning methods to build classification models with a data set containing 1,317 diverse compounds (Zhang et al., 2016a). Xu et al. used a deep learning method called undirected graph recursive neural networks (UGRNN) that encodes molecules into an undirected graph to build QSTR models (Xu et al., 2015). The performance was excellent compared to other models, up to 0.955 of AUC. More recently, Mulliner et al. classified the complex pathology of hepatotoxicity into 21 endpoints at three levels, with a large data set comprising 3,712 compounds. Then the specific models were combined into an optimized global human hepatotoxicity that has high sensitivity of 68% and excellent specificity of 95% (Mulliner et al., 2016).
Respiratory toxicity
Respiratory toxicity is another toxicity category with complicated mechanisms. The most concerned endpoint is drug-induced interstitial lung disease (DILD), which can be classified into two categories in terms of their mechanisms: (1) cytotoxic lung injury and (2) immune-mediated (Matsuno, 2012). Another type of respiratory toxicity is respiratory sensitization, of which the mechanism is more complicated. There are still no good models for identification of respiratory sensitization (Mekenyan et al., 2014; Dik et al., 2015). The current QSTR studies tend to use phenotype data such as LD50, LC50 or symptoms such as asthma as endpoints to represent the respiratory toxicity of a chemical, and the built models performed well enough (Jarvis et al., 2015; Lei et al., 2017).
Irritation and corrosion
Risk assessment of eye and skin irritation/corrosion (EI/EC, SI/SC) is of importance in pharmaceutical and cosmetics industries. Though these endpoints might not be directly considered in drug discovery stage, in silico models for these endpoints are yet required since a lot of substances may cause irritation and corrosion and should be assessed, including the ocular and dermal pharmaceuticals and final products used in manufacturing, agriculture, and warfare (Wilhelmus, 2001; Kolle et al., 2017).
Verheyen et al. evaluated the existing QSTR models in Derek Nexus, Toxtree and Case Ultra for the prediction of skin and eye irritation/corrosion, and found that the performance of those models is unsatisfactory because of narrow applicability domain and low accuracy (Verheyen et al., 2017). However, using machine learning methods to predict eye injury was reported having high performance. For instance, Verma et al. build combined QSTR models by ANN and got 88% of sensitivity and 82% of specificity for EI (Verma and Matthews, 2015a), 96% of sensitivity and 91% of specificity for EC (Verma and Matthews, 2015b). Our group recently developed in silico models for EI/EC using machine learning methods and molecular fingerprints (Wang et al., 2017). In the paper, more positive data were manually collected from X-Mol (http://www.x-mol.com) and ChemIDplus and the performance is excellent, 94.6% of overall accuracy for EI and 95.9% for EC.
Endocrine disruption
Chemicals interacting with nuclear receptors such as estrogen and androgen receptors (ER and AR) as off-targets or exposed in environment may cause endocrine disruption. These chemicals, called endocrine disrupting chemicals (EDCs), may interfere with the normal functions of these endogenous steroid hormones and lead to adverse health consequences such as tissue or organ proliferation, reproductive disorders, metabolic disorders, or even cancers (Colborn, 1995; Chawla et al., 2001; Grün and Blumberg, 2007).
For the specific mechanisms such as binding to ER, using in silico models to predict the bioactivity of chemicals and evaluate their risk of being EDCs is preferred for its high accuracy and less cost. We previously built in silico models for AR and ER binding using molecular fingerprints and machine learning methods and the best performance in the test set was 0.84 and 0.79, respectively (Chen et al., 2014). The Tox21 project also includes nuclear receptors assays which involve more diverse compounds (Hsieh et al., 2015). DeepTox, the winner of the “Tox21 Data Challenge,” used deep neural network and obtained an excellent performance against other machine learning methods such as SVM (Mayr et al., 2016).
Previous studies on EDCs mainly focused on nuclear receptors. However, chemicals that do not directly interact with these receptors may also interfere through the pathway. For instance, aromatase (CYP19A1) is an important enzyme affecting the biosynthesis of estrogen and plays a key role in maintaining the balance between estrogen and androgen in many of the EDC-sensitive organs (Sonnet et al., 1998). Therefore, we recently built in silico models for prediction of aromatase inhibitors as potential EDCs using machine learning methods with molecular fingerprints (Du et al., 2017). The data used for training and test were collected from Tox21 and the best model had 0.84 of accuracy for the test set and 0.91 for the external validation set.
Eco-toxicity
Pharmaceuticals and their metabolites exposed to the environment may affect the ecosystem since they are designed to be bioactive to creature (Halling-Sørensen et al., 1998). For instance, chemicals with binding affinities to hormone receptors may be EDCs of fishes or concentrate in fish body and finally reach to high-level animal bodies (He et al., 2017). To evaluate the environmental persistence of a chemical, biodegradation half-life is widely used as a common criterion (Raymond et al., 2001). We previously categorized chemicals as ready biodegradability and not ready biodegradability according to their biological oxygen demand (BOD) with a threshold of 60% and built several classification models. The best model used kNN with molecular descriptors and had a AUC of 0.873 in test set (Cheng et al., 2012a).
Fishes are usually used as model species to evaluate aquatic toxicity and avian species are widely used as model species to evaluate the terrestrial toxicity. Our group previously collected LC50 data of three fish species from ECOTOX database and built several local and global models (Sun et al., 2015). Recently, we reported a model focusing on the aquatic toxicity of pesticides and found that the molecule fingerprints performed different between local and global models (Li et al., 2017). For the avian species, several in silico models were developed including classification (Zhang et al., 2015) and regression (Mazzatorta et al., 2006; Toropov and Benfenati, 2006). In addition to the endpoints mentioned above, another commonly used model species for eco-toxicology is Tetrahymena pyriformis (Sauvant et al., 1999). Cheng et al. collected 1,571 unique chemicals with toxicity to Tetrahymena pyriformis and built several models of which the best performance was 92.6% for validation set (Cheng et al., 2011a).
Software and web servers
Currently many software and web servers can predict chemical toxicity before synthesis. Drug design software suites such as Discovery Studio and Pipeline Pilot integrate toxicity prediction models to help filter compounds with risk of toxicity. But the endpoints are not as diverse as that in some toxicity-oriented commercial software including ADMET Predictor, Leadscope and Lhasa Derek, which take efforts primarily on predicting and alerting molecules with potential toxicity.
Free software or web servers are more preferred by academia, which can promote the development of high quality models and algorithms, and their applications in various fields including drug discovery. OCED Toolbox is an official suite for toxicity prediction and modeling using QSTR. Web servers are easier and lighter to use and will be preferred by outsiders of computational toxicology, such as medicinal chemists. Lazar is such a tool that can predict several toxicity endpoints with a user interface of drawing chemical structures (Maunz et al., 2013). ToxTree is an open source application that estimates toxic hazard by applying a decision tree approach (Patlewicz et al., 2008). Compared to QSTR-like models, ToxTree is more interpretable and the fragments (SAs) can guide the chemists in modification of the molecules. The performance of ToxTree, OECD Toolbox, and other commercial tools were compared in literature (Devillers and Mombelli, 2010; Mombelli and Devillers, 2010; Bhatia et al., 2015; Bhhatarai et al., 2016). Our group developed admetSAR that can also predict toxicity of compounds in SMILES format (Cheng et al., 2012b).
Web servers such as ChemSAR (Dong et al., 2017b) and ChemBench (Capuzzi et al., 2017) enable users to build custom models for particular use with machine learning methods and molecular descriptors. For chemists who have in-house data for some particular endpoints, it will be convenient to use these web servers to build predictive models to prioritize or substitute in vitro or in vivo tests.
Perspectives
Though in silico prediction of chemical toxicity has made a good progress in recent years, there are still some challenges and limitations to be improved. At first, data quality is still a big issue. Currently many toxicity data are obtained from high-throughput in vitro assays or in vivo tests on animals. For example, Tox21 and ToxCast provide the activity data of thousands of chemicals against hundreds of assays (Huang et al., 2016). While false positive and false negative data are inevitable in those assays, in vivo data from animals are also questionable to be used directly on humans. Therefore, more data from drug clinical trials and clinic applications are highly demanded.
Secondly, more computational methods should be developed to enhance the accuracy of the predictive models. For instance, read-across has gained wide attention recently because it can fill the gap of missing data (Shah et al., 2016). Meanwhile, some endpoints have complex mechanisms such as hepatotoxicity and respiratory toxicity, computational systems toxicology has emerged to use comprehensive data sources from gene to organ to understand the mechanisms of toxicity (Jack et al., 2013; Sauer et al., 2015). With the help of machine learning methods and cheminformatics techniques, more accurate models could be developed for toxicity prediction.
Thirdly, medicinal chemists are more interested in the relationship between substructures and chemical toxicity, which can guide the optimization of lead compounds. Using computational tools to identify SAs is a promising way. Current approaches of SA identification can only generate numerous but redundant substructures in terms of their frequency of occurrence, disregarding the chemical or biological mechanisms (Yang et al., 2017a). It is not difficult to obtain “potential” SAs for almost every endpoint with support of assay results, yet innovative protocol or framework is still required to further refine these substructures and explore the chemical mechanisms of toxicity.
Statements
Author contributions
YT, GL, and WL contributed conception and design of the study; HY wrote the first draft of the manuscript; HY and LS wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (Grant 2016YFA0502304), the National Natural Science Foundation of China (Grants 81373329 and 81673356) and the 863 Project (Grant 2012AA020308).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AhlbergE.CarlssonL.BoyerS. (2014). Computational derivation of structural alerts from large toxicology data sets. J. Chem. Inf. Model.54, 2945–2952. 10.1021/ci500314a
2
Al-OtaibiR.KullM.FlachP. (2014). LaCova: a tree-based multi-label classifier using label covariance as splitting criterion, in 2014 13th International Conference on Machine Learning and Applications, (Detroit, MI: Icmla), 74–79.
3
Altae-TranH.RamsundarB.PappuA. S.PandeV. (2017). Low data drug discovery with one-shot learning. ACS Cent. Sci.3, 283–293. 10.1021/acscentsci.6b00367
4
AshbyJ.TennantR. W. (1988). Chemical structure, Salmonella mutagenicity and extent of carcinogenicity as indicators of genotoxic carcinogenesis among 222 chemicals tested in rodents by the U.S. NCI/NTP. Mutat. Res.204, 17–115. 10.1016/0165-1218(88)90114-0
5
AvilaJ. L.GibajaE. L.VenturaS. (2009). Multi-label Classification with gene expression programming. Hybrid Artif. Intell. Syst.5572, 629–637. 10.1007/978-3-642-02319-4_76
6
BarotP.PanchalM. (2014). Review on various problem transformation methods for classifying multi-label data. Int. J. Data Min. Emerg. Technol.4, 45–52. 10.5958/2249-3220.2014.00001.9
7
BhatiaS.SchultzT.RobertsD.ShenJ.KromidasL.Marie ApiA. (2015). Comparison of Cramer classification between Toxtree, the OECD QSAR Toolbox and expert judgment. Regul. Toxicol. Pharmacol.71, 52–62. 10.1016/j.yrtph.2014.11.005
8
BhhataraiB.WilsonD. M.ParksA. K.CarneyE. W.SpencerP. J. (2016). Evaluation of TOPKAT, toxtree, and derek nexus in silico models for ocular irritation and development of a knowledge-based framework to improve the prediction of severe irritation. Chem. Res. Toxicol.29, 810–822. 10.1021/acs.chemrestox.5b00531
9
BorgeltC.BertholdM. R. (2002). Mining molecular fragments: finding relevant substructures of molecules, in Data Mining, (2002). ICDM 2003. Proceedings 2002 IEEE International Conference (Maebashi: IEEE), 51–58.
10
BoutellM. R.LuoJ. B.ShenX. P.BrownC. M. (2004). Learning multi-label scene classification. Pattern Recognit.37, 1757–1771. 10.1016/j.patcog.2004.03.009
11
BreimanL. (2001). Random forests. Mach. Learn.45, 5–32. 10.1023/A:1010933404324
12
CapuzziS. J.KimI. S.LamW. I.ThorntonT. E.MuratovE. N.PozefskyD.et al. (2017). Chembench: A publicly accessible, integrated cheminformatics portal. J. Chem. Inf. Model.57, 105–108. 10.1021/acs.jcim.6b00462
13
ChawlaA.RepaJ. J.EvansR. M.MangelsdorfD. J. (2001). Nuclear receptors and lipid physiology: opening the X-files. Science294, 1866–1870. 10.1126/science.294.5548.1866
14
ChenY.ChengF.SunL.LiW.LiuG.TangY. (2014). Computational models to predict endocrine-disrupting chemical binding with androgen or oestrogen receptors. Ecotoxicol. Environ. Saf.110, 280–287. 10.1016/j.ecoenv.2014.08.026
15
ChengF.IkenagaY.ZhouY.YuY.LiW.ShenJ.et al. (2012a). In silico assessment of chemical biodegradability. J. Chem. Inf. Model.52, 655–669. 10.1021/ci200622d
16
ChengF.LiW.LiuG.TangY. (2013a). In silico ADMET prediction: recent advances, current challenges and future trends. Curr. Top. Med. Chem.13, 1273–1289. 10.2174/15680266113139990033
17
ChengF.LiW.WangX.ZhouY.WuZ.ShenJ.et al. (2013b). Adverse drug events: database construction and in silico prediction. J. Chem. Inf. Model.53, 744–752. 10.1021/ci4000079
18
ChengF.LiW.WuZ.WangX.ZhangC.LiJ.et al. (2013c). Prediction of polypharmacological profiles of drugs by the integration of chemical, side effect, and therapeutic space. J. Chem. Inf. Model.53, 753–762. 10.1021/ci400010x
19
ChengF.LiW.ZhouY.ShenJ.WuZ.LiuG.et al. (2012b). admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model.52, 3099–3105. 10.1021/ci300367a
20
ChengF.ShenJ.YuY.LiW.LiuG.LeeP. W.et al. (2011a). In silico prediction of Tetrahymena pyriformis toxicity for diverse industrial chemicals with substructure pattern recognition and machine learning methods. Chemosphere82, 1636–1643. 10.1016/j.chemosphere.2010.11.043
21
ChengF.YuY.ZhouY.ShenZ.XiaoW.LiuG.et al. (2011b). Insights into molecular basis of cytochrome p450 inhibitory promiscuity of compounds. J. Chem. Inf. Model.51, 2482–2495. 10.1021/ci200317s
22
ColbornT. (1995). Environmental estrogens: health implications for humans and wildlife. Environ. Health Perspect.103 (Suppl. 7), 135–136.
23
CortesC.VapnikV. (1995). Support-Vector Networks. Mach. Learn.20, 273–297. 10.1007/BF00994018
24
Cortes-CirianoI. (2016). Bioalerts: a python library for the derivation of structural alerts from bioactivity and toxicity data sets. J. Cheminform.8:13. 10.1186/s13321-016-0125-7
25
CoverT.HartP. (1967). Nearest neighbor pattern classification. IEEE Trans. Inform. Theory13, 21–27. 10.1109/TIT.1967.1053964
26
DavisA. P.GrondinC. J.JohnsonR. J.SciakyD.KingB. L.McMorranR.et al. (2017). The comparative toxicogenomics database: update 2017. Nucleic Acids Res.45, D972–D978. 10.1093/nar/gkw838
27
DemsarJ.CurkT.ErjavecA.GorupC.HocevarT.MilutinovicM.et al. (2013). Orange: data mining toolbox in python. J. Mach. Learn. Res.14, 2349–2353. Available online at: https://orange.biolab.si/citation/
28
DengL.HintonG.KingsburyB. (2013). New types of deep neural network learning for speech recognition and related applications: an overview, in IEEE International Conference on Acoustics, Speech and Signal Processing (Vancouver, BC), 8599–8603.
29
DevillersJ.MombelliE. (2010). Evaluation of the OECD QSAR Application Toolbox and Toxtree for estimating the mutagenicity of chemicals. Part 1. Aromatic amines. SAR QSAR Environ. Res.21, 753–769. 10.1080/1062936X.2010.528959
30
DikS.PenningsJ. L.van LoverenH.EzendamJ. (2015). Development of an in vitro test to identify respiratory sensitizers in bronchial epithelial cells using gene expression profiling. Toxicol. In Vitro30(1 Pt B), 274–280. 10.1016/j.tiv.2015.10.010
31
DiplarisS.TsoumakasG.MitkasP. A.VlahavasI. (2005). Protein classification with multiple algorithms. Adv. Inform. Proc.3746, 448–456. 10.1007/11573036_42
32
DoddareddyM. R.KlaasseE. C.Shagufta IjzermanA. P.BenderA. (2010). Prospective validation of a comprehensive in silico hERG model and its applications to commercial compound and drug databases. Chem. Med. Chem.5, 716–729. 10.1002/cmdc.201000024
33
DongJ.CaoD. S.MiaoH. Y.LiuS.DengB. C.YunY. H.et al. (2015). ChemDes: an integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform.7:60. 10.1186/s13321-015-0109-z
34
DongJ.WangN.-N.LiuK.-Y.ZhuM.-F.YunY.-H.ZengW.-B.et al. (2017a). ChemBCPP: a freely available web server for calculating commonly used physicochemical properties. Chemometr. Intell. Lab. Syst.171, 65–73. 10.1016/j.chemolab.2017.10.006
35
DongJ.YaoZ. J.ZhuM. F.WangN. N.LuB.ChenA. F.et al. (2017b). ChemSAR: an online pipelining platform for molecular SAR modeling. J. Cheminform.9:27. 10.1186/s13321-017-0215-1
36
DuH.CaiY.YangH.ZhangH.XueY.LiuG.et al. (2017). In silico prediction of chemicals binding to aromatase with machine learning methods. Chem. Res. Toxicol.30, 1209–1218. 10.1021/acs.chemrestox.7b00037
37
DuvenaudD.MaclaurinD.Aguilera-IparraguirreJ.Gmez-BombarelliR.HirzelT.Aspuru-GuzikA. N.et al. (2015). Convolutional Networks on Graphs for Learning Molecular Fingerprints. ArXiv e-prints [Online], (1509). Available online at: http://adsabs.harvard.edu/abs/2015arXiv150909292D (Accessed Sept 1, 2015).
38
ElithJ.LeathwickJ. R.HastieT. (2008). A working guide to boosted regression trees. J. Anim. Ecol.77, 802–813. 10.1111/j.1365-2656.2008.01390.x
39
FanD.YangH.LiF.SunL.DiP.LiW.et al. (in press). In silico prediction of chemical genotoxicity using machine learning methods structural alerts. Toxicol. Res.10.1039/C7TX00259A
40
FerrariT.CattaneoD.GiniG.Golbamaki BakhtyariN.ManganaroA.BenfenatiE. (2013). Automatic knowledge extraction from chemical structures: the case of mutagenicity prediction. SAR QSAR Environ. Res.24, 631–649. 10.1080/1062936X.2013.773376
41
FlorisM.RaitanoG.MeddaR.BenfenatiE. (2017). Fragment prioritization on a large mutagenicity dataset. Mol. Inform.36:1600133. 10.1002/minf.201600133
42
FongerG. C.HakkinenP.JordanS.PublickerS. (2014). The National Library of Medicine's (NLM) Hazardous Substances Data Bank (HSDB): background, recent enhancements and future plans. Toxicology325, 209–216. 10.1016/j.tox.2014.09.003
43
FowlerS.SchnallJ. G. (2014). TOXNET: information on toxicology and environmental health. Am. J. Nurs.114, 61–63. 10.1097/01.NAJ.0000443783.75162.79
44
FrankE.HallM.TriggL.HolmesG.WittenI. H. (2004). Data mining in bioinformatics using Weka. Bioinformatics20, 2479–2481. 10.1093/bioinformatics/bth261
45
GaultonA.HerseyA.NowotkaM.BentoA. P.ChambersJ.MendezD.et al. (2017). The ChEMBL database in 2017. Nucleic Acids Res.45, D945–D954. 10.1093/nar/gkw1074
46
GibajaE.VenturaS. (2015). A tutorial on multilabel learning. Acm Comput. Surveys47, 1–38. 10.1145/2716262
47
GilsonM. K.LiuT.BaitalukM.NicolaG.HwangL.ChongJ. (2016). BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res.44, D1045–D1053. 10.1093/nar/gkv1072
48
GintantG.SagerP. T.StockbridgeN. (2016). Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov.15, 457–471. 10.1038/nrd.2015.34
49
GiriS.BaderA. (2015). A low-cost, high-quality new drug discovery process using patient-derived induced pluripotent stem cells. Drug Discov. Today20, 37–49. 10.1016/j.drudis.2014.10.011
50
GodboleS.SarawagiS. (2004). Discriminative methods for multi-labeled classification. Adv. Knowl. Discov. Data Min. Proc.3056, 22–30. 10.1007/978-3-540-24775-3_5
51
GohG. B.HodasN. O.VishnuA. (2017). Deep learning for computational chemistry. J. Comput. Chem.38, 1291–1307. 10.1002/jcc.24764
52
GolbamakiA.BenfenatiE. (2016). In silico methods for carcinogenicity assessment. Methods Mol. Biol.1425, 107–119. 10.1007/978-1-4939-3609-0_6
53
GolbamakiA.BenfenatiE.GolbamakiN.ManganaroA.MerdivanE.RoncaglioniA.et al. (2016). New clues on carcinogenicity-related substructures derived from mining two large datasets of chemical compounds. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev.34, 97–113. 10.1080/10590501.2016.1166879
54
GoldL. S.ManleyN. B.SloneT. H.RohrbachL.GarfinkelG. B. (2005). Supplement to the Carcinogenic Potency Database (CPDB): results of animal bioassays published in the general literature through 1997 and by the National Toxicology Program in 1997-1998. Toxicol. Sci.85, 747–808. 10.1093/toxsci/kfi161
55
GrünF.BlumbergB. (2007). Perturbed nuclear receptor signaling by environmental obesogens as emerging factors in the obesity crisis. Rev. Endocr. Metab. Disord.8, 161–171. 10.1007/s11154-007-9049-x
56
Halling-SørensenB.Nors NielsenS.LanzkyP. F.IngerslevF.Holten LützhøftH. C.JørgensenS. E. (1998). Occurrence, fate and effects of pharmaceutical substances in the environment–a review. Chemosphere36, 357–393. 10.1016/S0045-6535(97)00354-8
57
HansenK.MikaS.SchroeterT.SutterA.ter LaakA.Steger-HartmannT.et al. (2009). Benchmark data set for in Silico prediction of ames mutagenicity. J. Chem. Inf. Model.49, 2077–2081. 10.1021/ci900161g
58
HeJ.PengT.YangX.LiuH. (2017). Development of QSAR models for predicting the binding affinity of endocrine disrupting chemicals to eight fish estrogen receptor. Ecotoxicol. Environ. Saf.148, 211–219. 10.1016/j.ecoenv.2017.10.023
59
HewittM.PrzybylakK. (2016). In silico models for hepatotoxicity. Methods Mol. Biol.1425, 201–236. 10.1007/978-1-4939-3609-0_11
60
HsiehJ. H.SedykhA.HuangR.XiaM.TiceR. R. (2015). A data analysis pipeline accounting for artifacts in Tox21 quantitative high-throughput screening assays. J. Biomol. Screen.20, 887–897. 10.1177/1087057115581317
61
HuangR.XiaM.SakamuruS.ZhaoJ.ShahaneS. A.Attene-RamosM.et al. (2016). Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat. Commun.7:10425. 10.1038/ncomms10425
62
HuhD.HamiltonG. A.IngberD. E. (2011). From 3D cell culture to organs-on-chips. Trends Cell Biol.21, 745–754. 10.1016/j.tcb.2011.09.005
63
HuhD.MatthewsB. D.MammotoA.Montoya-ZavalaM.HsinH. Y.IngberD. E. (2010). Reconstituting organ-level lung functions on a chip. Science328, 1662–1668. 10.1126/science.1188302
64
JackJ.WambaughJ.ShahI. (2013). Systems toxicology from genes to organs. Methods Mol. Biol.930, 375–397. 10.1007/978-1-62703-059-5_17
65
JarvisJ.SeedM. J.StocksS. J.AgiusR. M. (2015). A refined QSAR model for prediction of chemical asthma hazard. Occup. Med. (Lond).65, 659–666. 10.1093/occmed/kqv105
66
JudsonR.RichardA.DixD.HouckK.ElloumiF.MartinM.et al. (2008). ACToR–Aggregated computational toxicology resource. Toxicol. Appl. Pharmacol.233, 7–13. 10.1016/j.taap.2007.12.037
67
KaziusJ.McGuireR.BursiR. (2005). Derivation and validation of toxicophores for mutagenicity prediction. J. Med. Chem.48, 312–320. 10.1021/jm040835a
68
KaziusJ.NijssenS.KokJ.BäckT.IjzermanA. P. (2006). Substructure mining using elaborate chemical representation. J. Chem. Inf. Model.46, 597–605. 10.1021/ci0503715
69
KlekotaJ.RothF. P. (2008). Chemical substructures that enrich for biological activity. Bioinformatics24, 2518–2525. 10.1093/bioinformatics/btn479
70
KolleS. N.van RavenzwaayB.LandsiedelR. (2017). Regulatory accepted but out of domain: in vitro skin irritation tests for agrochemical formulations. Regul. Toxicol. Pharmacol.89, 125–130. 10.1016/j.yrtph.2017.07.016
71
KuhnM.CampillosM.LetunicI.JensenL. J.BorkP. (2010). A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol.6:343. 10.1038/msb.2009.98
72
LandrumG. (2017). RDKit. Available online at: http://www.rdkit.org.
73
LeCunY.BengioY.HintonG. (2015). Deep learning. Nature521, 436–444. 10.1038/nature14539
74
LeiT.ChenF.LiuH.SunH.KangY.LiD.et al. (2017). ADMET Evaluation in drug discovery. part 17: development of quantitative and qualitative prediction models for chemical-induced respiratory toxicity. Mol. Pharm.14, 2407–2421. 10.1021/acs.molpharmaceut.7b00317
75
LeiT.LiY.SongY.LiD.SunH.HouT. (2016). ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling. J. Cheminformat.8:6. 10.1186/S13321-016-0117-7
76
LepailleurA.PoezevaraG.BureauR. (2013). Automated detection of structural alerts (chemical fragments) in (eco)toxicology. Comput. Struct. Biotechnol. J.5:e201302013. 10.5936/csbj.201302013
77
LiF.FanD.WangH.YangH.LiW.TangY.et al. (2017). In silico prediction of pesticide aquatic toxicity with chemical category approaches. Toxicol. Res.6, 831–842. 10.1039/C7TX00144D
78
LiX.ChenL.ChengF.WuZ.BianH.XuC.et al. (2014). In silico prediction of chemical acute oral toxicity using multi-classification methods. J. Chem. Inf. Model.54, 1061–1069. 10.1021/ci5000467
79
LiX.DuZ.WangJ.WuZ. R.LiW. H.LiuG. X.et al. (2015). In silico estimation of chemical carcinogenicity with binary and ternary classification methods. Mol. Inform.34, 228–235. 10.1002/minf.201400127
80
MatsunoO. (2012). Drug-induced interstitial lung disease: mechanisms and best diagnostic approaches. Respir. Res.13:39. 10.1186/1465-9921-13-39
81
MaunzA.GütleinM.RautenbergM.VorgrimmlerD.GebeleD.HelmaC. (2013). lazar: a modular predictive toxicology framework. Front. Pharmacol.4:38. 10.3389/fphar.2013.00038
82
MayrA.KlambauerG.UnterthinerT.HochreiterS. (2016). DeepTox: toxicity prediction using deep learning. Front. Environ. Sci.3:80. 10.3389/fenvs.2015.00080
83
MazzatortaP.CroninM. T. D.BenfenatiE. (2006). A QSAR study of avian oral toxicity using support vector machines and genetic algorithms. QSAR Comb. Sci.25, 616–628. 10.1002/qsar.200530189
84
MekenyanO.PatlewiczG.KusevaC.PopovaI.MehmedA.KotovS.et al. (2014). A mechanistic approach to modeling respiratory sensitization. Chem. Res. Toxicol.27, 219–239. 10.1021/tx400345b
85
MombelliE.DevillersJ. (2010). Evaluation of the OECD (Q)SAR Application Toolbox and Toxtree for predicting and profiling the carcinogenic potential of chemicals. SAR QSAR Environ. Res.21, 731–752. 10.1080/1062936X.2010.528598
86
MullinerD.SchmidtF.StolteM.SpirklH. P.CzichA.AmbergA. (2016). Computational models for human and animal hepatotoxicity with a global application scope. Chem. Res. Toxicol.29, 757–767. 10.1021/acs.chemrestox.5b00465
87
O'BoyleN. M.BanckM.JamesC. A.MorleyC.VandermeerschT.HutchisonG. R. (2011). Open Babel: an open chemical toolbox. J. Cheminform.3:33. 10.1186/1758-2946-3-33
88
PatlewiczG.JeliazkovaN.SaffordR. J.WorthA. P.AleksievB. (2008). An evaluation of the implementation of the cramer classification scheme in the toxtree software. SAR QSAR Environ. Res.19, 495–524. 10.1080/10629360802083871
89
PaulS. M.MytelkaD. S.DunwiddieC. T.PersingerC. C.MunosB. H.LindborgS. R.et al. (2010). How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov.9, 203–214. 10.1038/nrd3078
90
PedregosaF.VaroquauxG.GramfortA.MichelV.WeissR.DubourgV.et al. (2011). Scikit-learn: machine learning in python. J. Mach. Learn. Res.12, 2825–2830. Available online at: http://scikit-learn.org/stable/about.html#citing-scikit-learn
91
PhRMA (2015). 2015 Biopharmaceutical Research Industry Profle.Washington, DC: Pharmaceutical Research and Manufacturers of America.
92
PizzoF.GadaletaD.LombardoA.NicolottiO.BenfenatiE. (2015). Identification of structural alerts for liver and kidney toxicity using repeated dose toxicity data. Chem. Cent. J.9:62. 10.1186/s13065-015-0139-7
93
RaymondJ. W.RogersT. N.ShonnardD. R.KlineA. A. (2001). A review of structure-based biodegradation estimation methods. J. Hazard. Mater.84, 189–215. 10.1016/S0304-3894(01)00207-2
94
ReadJ.PfahringerB.HolmesG. (2008). Multi-label classification using ensembles of pruned sets, ICDM 2008: Eighth IEEE International Conference on Data Mining, Proceedings (Pisa), 995–1000.
95
ReadJ.PfahringerB.HolmesG.FrankE. (2011). Classifier chains for multi-label classification. Mach. Learn.85, 333–359. 10.1007/s10994-011-5256-5
96
SauerJ. M.HartungT.LeistM.KnudsenT. B.HoengJ.HayesA. W. (2015). Systems toxicology: the future of risk assessment. Int. J. Toxicol.34, 346–348. 10.1177/1091581815576551
97
SauvantM. P.PepinD.PiccinniE. (1999). Tetrahymena pyriformis: a tool for toxicological studies. A review. Chemosphere38, 1631–1669. 10.1016/S0045-6535(98)00381-6
98
SchmidtU.StruckS.GrueningB.HossbachJ.JaegerI. S.ParolR.et al. (2009). SuperToxic: a comprehensive database of toxic compounds. Nucleic Acids Res.37, D295-D299. 10.1093/nar/gkn850
99
SchusterD.LaggnerC.LangerT. (2005). Why drugs fail - A study on side effects in new chemical entities. Curr. Pharm. Des.11, 3545–3559. 10.2174/138161205774414510
100
SegallM. D.BarberC. (2014). Addressing toxicity risk when designing and selecting compounds in early drug discovery. Drug Discov. Today19, 688–693. 10.1016/j.drudis.2014.01.006
101
ShahI.LiuJ.JudsonR. S.ThomasR. S.PatlewiczG. (2016). Systematically evaluating read-across prediction and performance using a local validity approach characterized by chemical structure and bioactivity information. Regul. Toxicol. Pharmacol.79, 12–24. 10.1016/j.yrtph.2016.05.008
102
ShenJ.ChengF.XuY.LiW.TangY. (2010). Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model50, 1034–1041. 10.1021/ci100104j
103
ShenQ.JiangJ. H.JiaoC. X.LinW. Q.ShenG. L.YuR. Q. (2004). Hybridized particle swarm algorithm for adaptive structure training of multilayer feed-forward neural network: QSAR studies of bioactivity of organic compounds. J. Comput. Chem.25, 1726–1735. 10.1002/jcc.20094
104
SinghK. P.GuptaS.RaiP. (2013). Predicting carcinogenicity of diverse chemicals using probabilistic neural network modeling approaches. Toxicol. Appl. Pharmacol.272, 465–475. 10.1016/j.taap.2013.06.029
105
SokolovaM.SzpakowiczS. (2010). In Handbook of Research on Machine Learning Applications and Trends: Algorithms, Methods, and Techniques. Hershey, PA: IGI Global.
106
SonnetP.GuillonJ.EnguehardC.DallemagneP.BureauR.RaultS.AuvrayP.et al. (1998). Design and synthesis of a new type of non steroidal human aromatase inhibitors. Bioorg. Med. Chem. Lett.8, 1041–1044. 10.1016/S0960-894X(98)00157-7
107
SteinbeckC.HanY. Q.KuhnS.HorlacherO.LuttmannE.WillighagenE. (2003). The Chemistry Development Kit (CDK): an open-source Java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci.43, 493–500. 10.1021/ci025584y
108
SunL.YangH.LiJ.WangT.LiW.LiuG.et al. (2017). In silico prediction of compounds binding to human plasma proteins by QSAR models. ChemMedChem. [Epub ahead of print]. 10.1002/cmdc.201700582
109
SunL.ZhangC.ChenY. J.LiX.ZhuangS. L.LiW. H.et al. (2015). In silico prediction of chemical aquatic toxicity with chemical category approaches and substructural alerts. Toxicol. Res.4, 452–463. 10.1039/C4TX00174E
110
SushkoI.SalminaE.PotemkinV. A.PodaG.TetkoI. V. (2012). ToxAlerts: a web server of structural alerts for toxfic chemicals and compounds with potential adverse reactions. J. Chem. Inf. Model.52, 2310–2316. 10.1021/ci300245q
111
TanabeK.KuritaT.NishidaK.LucićB.AmićD.SuzukiT. (2013). Improvement of carcinogenicity prediction performances based on sensitivity analysis in variable selection of SVM models. SAR QSAR Environ. Res.24, 565–580. 10.1080/1062936X.2012.762425
112
TatonettiN. P.YeP. P.DaneshjouR.AltmanR. B. (2012). Data-driven prediction of drug effects and interactions. Sci. Transl. Med.4:125ra131. 10.1126/scitranslmed.3003377
113
TetkoI. V.GasteigerJ.TodeschiniR.MauriA.LivingstoneD.ErtlP.et al. (2005). Virtual computational chemistry laboratory–design and description. J. Comput. Aided Mol. Des.19, 453–463. 10.1007/s10822-005-8694-y
114
ToropovA. A.BenfenatiE. (2006). QSAR models of quail dietary toxicity based on the graph of atomic orbitals. Bioorg. Med. Chem. Lett.16, 1941–1943. 10.1016/j.bmcl.2005.12.085
115
TsoumakasG.KatakisI.TaniarD. (2007). Multi-label classification: an overview. Int. J. Data Warehousing Min.3, 1–13. 10.4018/jdwm.2007070101
116
TsoumakasG.KatakisI.VlahavasI. (2008). Effective and efficient multilabel classification in domains with large number of labels, in Ecml/pkdd Workshop on Mining Multidimensional Data (Ho Chi Minh City).
117
TsoumakasG.KatakisI.VlahavasI. (2010). Mining Multi-label Data, in Data Mining and Knowledge Discovery Handbook, eds MaimonO.RokachL. (Boston, MA: Springer), 667–685.
118
TsoumakasG.VlahavasI. (2007). Random k-labelsets: an ensemble method for multilabel classification, in Proceedings of Machine Learning. ECML 2007 (Warsaw).
119
VerheyenG. R.BraekenE.Van DeunK.Van MiertS. (2017). Evaluation of existing (Q)SAR models for skin and eye irritation and corrosion to use for REACH registration. Toxicol. Lett.265, 47–52. 10.1016/j.toxlet.2016.11.007
120
VermaR. P.MatthewsE. J. (2015a). Estimation of the chemical-induced eye injury using a weight-of-evidence (WoE) battery of 21 artificial neural network (ANN) c-QSAR models (QSAR-21): part I: irritation potential. Regul. Toxicol. Pharmacol.71, 318–330. 10.1016/j.yrtph.2014.11.011
121
VermaR. P.MatthewsE. J. (2015b). Estimation of the chemical-induced eye injury using a Weight-of-Evidence (WoE) battery of 21 artificial neural network (ANN) c-QSAR models (QSAR-21): part II: corrosion potential. Regul. Toxicol. Pharmacol.71, 331–336. 10.1016/j.yrtph.2014.12.004
122
WangJ. R.FengJ.SunX.ChenS. S.ChenB. (2014). Simplified Constraints Rank-SVM for Multi-label Classification. Pattern Recogn.483(Pt I), 229–236. 10.1007/978-3-662-45646-0_23
123
WangQ.LiX.YangH.CaiY.WangY.WangZ.et al. (2017). In silico prediction of serious eye irritation or corrosion potential of chemicals. RSC Adv.7, 6697–6703. 10.1039/C6RA25267B
124
WangS.LiY.WangJ.ChenL.ZhangL.YuH.et al. (2012). ADMET evaluation in drug discovery. 12. Development of binary classification models for prediction of hERG potassium channel blockage. Mol. Pharm.9, 996–1010. 10.1021/mp300023x
125
WangS.SunH.LiuH.LiD.LiY.HouT. (2016). ADMET evaluation in drug discovery. 16. Predicting hERG Blockers by combining multiple pharmacophores and machine learning approaches. Mol. Pharm. 13, 2855–2866. 10.1021/acs.molpharmaceut.6b00471
126
WangY.XiaoJ.SuzekT. O.ZhangJ.WangJ.BryantS. H. (2009). PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res.37(Suppl. 2), W623–W633. 10.1093/nar/gkp456
127
WilhelmusK. R. (2001). The Draize eye test. Surv. Ophthalmol.45, 493–515. 10.1016/S0039-6257(01)00211-9
128
Williams-DeVaneC. R.WolfM. A.RichardA. M. (2009). DSSTox chemical-index files for exposure-related experiments in ArrayExpress and Gene Expression Omnibus: enabling toxico-chemogenomics data linkages. Bioinformatics25, 692–694. 10.1093/bioinformatics/btp042
129
WishartD.ArndtD.PonA.SajedT.GuoA. C.DjoumbouY.et al. (2015). T3DB: the toxic exposome database. Nucleic Acids Res.43, D928-D934. 10.1093/nar/gku1004
130
XuC.ChengF.ChenL.DuZ.LiW.LiuG.et al. (2012). In silico prediction of chemical Ames mutagenicity. J. Chem. Inf. Model.52, 2840–2847. 10.1021/ci300400a
131
XuY.DaiZ.ChenF.GaoS.PeiJ.LaiL. (2015). Deep learning for drug-induced liver injury. J. Chem. Inf. Model.55, 2085–2093. 10.1021/acs.jcim.5b00238
132
XuY.PeiJ.LaiL. (2017). Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J. Chem. Inf. Model.57, 2672–2685. 10.1021/acs.jcim.7b00244
133
XueY.LiZ. R.YapC. W.SunL. Z.ChenX.ChenY. Z. (2004). Effect of molecular descriptor feature selection in support vector machine classification of pharmacokinetic and toxicological properties of chemical agents. J. Chem. Inf. Comput. Sci.44, 1630–1638. 10.1021/ci049869h
134
YangH.LiJ.WuZ.LiW.LiuG.TangY. (2017a). Evaluation of different methods for identification of structural alerts using chemical ames mutagenicity data set as a benchmark. Chem. Res. Toxicol.30, 1355–1364. 10.1021/acs.chemrestox.7b00083
135
YangH.LiX.CaiY.WangQ.LiW.LiuG.et al. (2017b). In silico prediction of chemical subcellular localization via multi-classification methods. Medchemcomm. 8, 1225–123410.1039/C7MD00074J
136
YapC. W. (2011). PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem.32, 1466–1474. 10.1002/jcc.21707
137
ZhangC.ChengF.LiW.LiuG.LeeP. W.TangY. (2016a). In silico prediction of drug induced liver toxicity using substructure pattern recognition method. Mol. Inform.35, 136–144. 10.1002/minf.201500055
138
ZhangC.ChengF.SunL.ZhuangS.LiW.LiuG.et al. (2015). In silico prediction of chemical toxicity on avian species using chemical category approaches. Chemosphere122, 280–287. 10.1016/j.chemosphere.2014.12.001
139
ZhangC.ZhouY.GuS.WuZ.WuW.LiuC.et al. (2016b). In silico prediction of hERG potassium channel blockage by chemical category approaches. Toxicol. Res.5, 570–582. 10.1039/C5TX00294J
140
ZhangH.CaoZ.LiM.LiY.PengC. (2016). Novel naive Bayes classification models for predicting the carcinogenicity of chemicals. Food Chem. Toxicol.97, 141–149. 10.1016/j.fct.2016.09.005
141
ZhangL.AiH.ChenW.YinZ.HuH.ZhuJ.et al. (2017). CarcinoPred-EL: novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci. Rep.7:2118. 10.1038/s41598-017-02365-0
142
ZhangM. L.ZhouZ. H. (2006). Multilabel neural networks with applications to functional genomics and text categorization. IEEE Trans. Knowl. Data Eng.18, 1338–1351. 10.1109/TKDE.2006.162
143
ZhangM. L.ZhouZ. H. (2007). ML-KNN: a lazy learning approach to multi-label leaming. Pattern Recognit.40, 2038–2048. 10.1016/j.patcog.2006.12.019
144
ZhangM. L.ZhouZ. H. (2014). A review on multi-label learning algorithms. IEEE Trans. Knowl. Data Eng.26, 1819–1837. 10.1109/TKDE.2013.39
145
ZhuH.MartinT. M.YeL.SedykhA.YoungD. M.TropshaA. (2009). Quantitative structure-activity relationship modeling of rat acute toxicity by oral exposure. Chem. Res. Toxicol.22, 1913–1921. 10.1021/tx900189p
146
ZhuX.KruhlakN. L. (2014). Construction and analysis of a human hepatotoxicity database suitable for QSAR modeling using post-market safety data. Toxicology321, 62–72. 10.1016/j.tox.2014.03.009
Summary
Keywords
drug safety, chemical toxicity, drug design, machine learning, structural alerts
Citation
Yang H, Sun L, Li W, Liu G and Tang Y (2018) In Silico Prediction of Chemical Toxicity for Drug Design Using Machine Learning Methods and Structural Alerts. Front. Chem. 6:30. doi: 10.3389/fchem.2018.00030
Received
11 January 2018
Accepted
05 February 2018
Published
20 February 2018
Volume
6 - 2018
Edited by
Daniela Schuster, Paracelsus Private Medical University of Salzburg, Austria
Reviewed by
Huixiao Hong, United States Food and Drug Administration, United States; Heebeom Koo, Catholic University of Korea, South Korea
Updates
Copyright
© 2018 Yang, Sun, Li, Liu and Tang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Tang ytang234@ecust.edu.cn
This article was submitted to Medicinal and Pharmaceutical Chemistry, a section of the journal Frontiers in Chemistry
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.