 Diego Benítez
Diego Benítez Cecilia Ortíz1†
Cecilia Ortíz1† Marcelo A. Comini
Marcelo A. Comini- 1Group Redox Biology of Trypanosomes, Institut Pasteur de Montevideo, Montevideo, Uruguay
- 2Laboratorio de Síntesis Orgánica, Departamento de Química Orgánica, Facultad de Química, Universidad de la República, Montevideo, Uruguay
Introduction: In vivo imaging technology based on bioluminescence has contributed to the study of different pathophysiological conditions involving inherited or transmissible diseases. Here, we aimed to establish a bioluminescent model of acute African trypanosomiasis for a manifold of applications. African trypanosomiasis is a neglected tropical disease that threatens human and animal health, mainly in sub-Saharan countries, for which new chemotherapies are needed.
Methods: The model relies on a hypervirulent bloodstream form of Trypanosoma brucei brucei, which constitutively expresses red-shifted luciferase, and an infection-susceptible murine host, Balb/cJ mouse. In vivo and ex vivo imaging techniques were applied to obtain a spatial, temporal, and quantitative (parasite load) resolution of the infection process and to refine the animal endpoint criterion.
Results: The model proved suitable for validating the essentiality of the parasite enzyme glucose 6-phosphate dehydrogenase by reverse genetics (tetracycline-inducible double-strand RNA interference). The efficacy of drugs (monotherapy or a new combination) for the treatment of the acute stage of the disease was successfully explored by in vivo imaging.
Discussion: The new bioluminescent model developed here may represent a valuable tool for speeding up the drug discovery process and the investigation of host-pathogen interactions in the acute stage of African sleeping sickness.
1 Introduction
Zoonotic infectious diseases continue to be one of the major leading causes of human and animal death worldwide (World Health Organization, 2019; World Health Organization, 2022). Among them, human African trypanosomiasis (HAT), or sleeping sickness, is a neglected disease caused by Trypanosoma brucei gambiense and T. brucei rhodesiense. Other related Trypanosoma (sub)species (e.g., T. brucei brucei) are the etiological agents of animal African trypanosomiasis, which represents a major obstacle to the nutritional and economic development of the endemic regions (Shaw et al., 2014). In humans, the acute or first stage of the disease compromises the hemolymphatic system and, if uncontrolled, may progress into a lethal meningoencephalitis (or second-stage). HAT threatens ∼70 million people and, similar to other zoonotic diseases, pathogen eradication is unfeasible due to the complex life cycle of the microorganism that involves an invertebrate and different wildlife hosts.
The development of immunoprophylactic approaches against HAT is almost impossible because the pathogens have evolved very efficient mechanisms to evade the host immune response (Stijlemans et al., 2016). Thus, chemotherapy, along with disease control and prevention are the only choice to fight HAT (Aksoy et al., 2017).
After several decades without new pharmacological discoveries, between 2009 and 2018 the nitroheterocycles Nifurtimox (in combination with Eflornitine; Priotto et al., 2009) and Fexinidazole (Mesu et al., 2018) were approved for the treatment of second stage HAT. Although these two drugs fully replaced the use of the highly toxic Melarsoprol (an arsenic-based drug), they share the same core scaffold, consequently, the possibility of developing cross-resistance is yet feasible (Sokolova et al., 2010). The remaining drugs in clinical use for first-stage HAT (pentamidine and suramin) require a complex administration regime, have limited efficacy, face pathogen resistance, and/or important side effects (Franco et al., 2018). Therefore, better drugs are needed for combating this disease.
In this regard, the availability of the genome sequence for each major pathogenic species of African trypanosomes (Shanmugasundram et al., 2023) offers the opportunity to identify indispensable and parasite-specific drug target candidates. Although the function and essentiality of several trypanosomal genes have been studied in vitro by reverse genetic approaches (e.g., Alsford et al., 2011), the physiological relevance of a few of them has been validated in vivo (Mutomba et al., 1999; Abdulla et al., 2008; Wyllie et al., 2009; Price et al., 2010; Manta et al., 2013; Loureiro et al., 2015; Currier et al., 2019).
Drug target validation, the preclinical assessment of efficacy for a hit/lead compound, and the study of the pathophysiological phenomena occurring during infection and treatment are research areas that largely benefit from state-of-the-art models and techniques that preserve pathogen and host integrity. In this regard, bioluminescence models applied to infectious diseases have proven to be powerful tools for quantifying and localizing pathogens or host cells (e.g., infected, altered, damaged, or responsive) as well as the associated signalling or response events (Suff and Waddington, 2017). The development of bioluminescent reporter cell lines of African trypanosomes dates back nearly 30 years ago. The different reporter cell lines were generated on laboratory strains (originally isolated from animals or patients) with capacity (pleomorphic cell line) or not (monomorphic cell line) to complete the full differentiation cycle of the parasite. Based on the fact that the luciferase (LUC) gene from the beetle Photinus pyralis (PpyLUC) contains a peroxisomal targeting sequence, the first use of PpyLUC in African trypanosomes was to investigate protein import into the glycosomes (a parasite-specific organelle closely related to peroxisomes that plays a key energetic role in bloodstream parasites; Sommer et al., 1992). PpyLUC was also widely employed as a reporter to study the regulation of gene expression (Hug et al., 1993), and to improve expression systems for these parasites (Wirtz and Clayton, 1995). Further developments and applications were driven by the discovery and sequence optimization of novel LUC-encoding genes and by the development of in vivo imaging technology (Promega, 2005; Badr and Tannous, 2011; Xu et al., 2016). The first in vivo studies with bioluminescent African trypanosomes were performed using transgenic T. brucei spp. that expressed a stably integrated copy of the sea pansy Renilla reniformis (RrLUC) in the β-tubulin locus. These reporter cell lines contributed to disclosing parasite tropism for the extravascular space of testis in male mice, a niche where the pathogen escapes drug action (Claes et al., 2009), and the, unexpected, early colonization of the brain during the acute stage of the disease (Giroud et al., 2009).
PpyLUC (Emission wavelength 560 nm) is expected to have better in vivo performance than RrLUC (Emission wavelength 480 nm) because it uses a substrate (D-luciferin) with more favorable in vivo kinetics than coelenterazine (substrate of RrLUC), and emits light with higher tissue penetration. However, the authors above (Claes et al., 2009) preferred not to use PpyLUC as a reporter on the basis that a glycosomal localization of this LUC (Sommer et al., 1992) may impair energy metabolism and thereby lower bioluminescence signal.
Further optimization of the original sequence encoding for PpyLUC led to a LUC variant (PpyLUC2) with cytosolic localization (e.g., by the removal of the peroxisomal targeting sequence) and faster response rates (Promega, 2005). A comparative study performed with different strains of bloodstream T. b. brucei expressing RrLUC or PpyLUC2 (Myburgh et al., 2013) demonstrated that despite the higher in vitro activity of RrLUC, PpyLUC2 was far more sensitive for in vivo detection of the pathogen. This result is in line with the predicted better tissue penetration of the greenish-yellow light emitted by PpyLUC (Badr and Tannous, 2011). An additional contribution of this study was to generate a laboratory model of the second stage of African trypanosomiasis that reduced from 180 to 60 days the time required to assess drug efficacy.
In a parallel study, McLatchie et al. (2013) reported the generation and characterization of bioluminescent T. b. brucei cell lines with improved performance for in vivo applications. For this purpose, they selected PpyRE9H-LUC as a reporter gene and tested discrete expression regulatory elements (promoter, 5′- and 3′-UTR sequences) and a loci-specific integration site. PpyRE9H-LUC is a codon-optimized LUC that emits red-shifted light (Emission wavelength 617 nm) and is more thermostable at 37°C than PpyLUC (Branchini et al., 2010). The best reporter clones obtained for mono and pleomorphic T. b. brucei yielded, under in vitro conditions, a robust and stable (at least 2- and 3-month, respectively) expression of PpyRE9H-LUC in the absence of selective pressure. More importantly, the level of imaging sensitivity obtained in different mouse genetic backgrounds with this system surpassed that reported previously for African trypanosomes expressing other LUC variants. Independent studies further confirmed the higher in vivo sensitivity of the PpyRE9H-LUC pleomorphic T. b. brucei cell line (McLatchie et al., 2013), which contributed to reducing significantly the time required for the assessment of drug preclinical efficacy in a murine chronic model of sleeping sickness (Burrell-Saward et al., 2015; Burrell-Saward et al., 2017).
Conclusive evidence about the in vitro and in vivo performance of LUC variants was provided by Van Reet et al., 2014. They expressed RrLUC and two red-shifted LUC, namely, PpyRE9H-LUC and the click beetle LUC (CBR-LUC), in different T. brucei spp. (i.e., T. b. brucei, T. b. gambiense, and T. b. rodhesiense). To allow comparisons, each reporter gene was cloned into the same promoterless expression vector that integrates into the β-tubulin locus (Claes et al., 2009). Independently of the genetic and phenotypic background of the trypanosoma subspecies, the red-shifted LUC proved superior to RrLUC in revealing parasites’ distribution across the animal body. Furthermore, PpyRE9H-LUC displayed an in vitro and in vivo sensitivity 10- to 20-fold higher than CBR-LUC.
More recently, we have reported the generation and in vitro characterization of a PpyRE9H-LUC expressing cell line of monomorphic T. b. brucei (Benítez et al., 2020) that is being used for drug screening purposes (Barreiro-Costa et al., 2022; Dibello et al., 2022; Mesa et al., 2022; Pazos et al., 2022; Rivas et al., 2022; Ballesteros-Casallas et al., 2023; Ihnatenko et al., 2023; Lindhof et al., 2023). In this cell line, the expression cassette previously shown to yield the best expression levels of PpyRE9H-LUC in T. brucei (i.e., rDNA promoter, 5′-UTR of the Variable Surface Glycoprotein, and the 3′-UTR from the β-tubulin; McLatchie et al., 2013), was stably integrated into the non-transcribed rRNA spacer sequence, which is a multicopy locus of the Trypanosoma genome (Quijada et al., 2002). However, at variance with all previously reported T. brucei LUC-reporters, we selected a host cell line harboring one copy of the T7-RNA polymerase gene and two copies of the sequence coding for the tetracycline (Tet) repressor protein (Alibu et al., 2005). This genetic background allows performing reverse genetic studies with Tet-inducible expression systems with the added value of monitoring pathogen viability by bioluminescence, a goal not yet addressed in African trypanosomes. Another issue not yet explored by previous studies using bioluminescent trypanosomes is the (re)definition of euthanasia criterion based on the quantitative analysis of in vivo bioluminescence.
Bearing all this in mind, here we present a detailed characterization of a bioluminescent acute murine model of sleeping sickness based on the bloodstream form of a hypervirulent and PpyRE9H-LUC reporter T. b. brucei cell line that is amenable to genetic manipulation. The application of the model for the genetic validation of drug target candidates and therapeutic efficacy assays was validated. Furthermore, to minimize animal pain and distress, the criterion for assay end-point was refined based on quantitative image analysis of parasite load.
2 Material and methods
2.1 Reagents
Unless otherwise stated all chemical reagents were of analytical grade and purchased from Sigma-Aldrich.
2.2 Parasite culture
This study employed a clone of the bloodstream form of monomorphic T. b. brucei (strain 427, cell line 514-1313; Alibu et al., 2005) that has been genetically engineered to express PpyRE9H-LUC stably and constitutively (Benítez et al., 2020). Hereon, the cell line is named T7TetR-PpyRE9H-LUC. The parasites were axenically grown in HMI-9 medium (Hirumi and Hirumi, 1989) supplemented with 10% fetal bovine serum, 100 U/mL penicillin (Gibco), 100 μg/mL streptomycin (Gibco), 0.2 μg/mL bleomycin (Jena Bioscience), 4 μg/mL G418 disulfate salt and 0.8 μg/mL puromycin (Invitrogen), inside a humidified incubator (Thermo Fisher SCIENTIFIC) with controlled temperature (37°C) and CO2 (5%) in 25 cm2 vented cap culture-flask (Corning).
Whenever required, blood samples from infected animals (treated or not with Diminazene aceturate, DAC) were diluted 1:100 in fresh culture medium devoid of selection antibiotics, and the presence/number of parasites was determined by microscopy up to 2 weeks of culture time. For technical details on blood sampling/processing and parasite counting refer to section 2.11.
To obtain a homogenous population of cells in a similar metabolic status and growth stage (logarithmic phase), parasite cultures were synchronized by at least two consecutive passages every 24 h at an initial cell density ≤ 3–5×104 parasites/mL in 10 mL fresh culture medium.
2.3 Generation and phenotypic characterization of bioluminescent RNAi cell line for glucose-6-phosphate dehydrogenase (G6PDH)
The T7TetR-PpyRE9H-LUC cell line was transfected with the linearized pHD-677_G6PDHARNi plasmid that allows the Tet-inducible posttranscriptional gene silencing of T. brucei G6PDH (Cordeiro et al., 2009). The transfection and selection conditions were exactly as those described for the generation of T7TetR-PpyRE9H-LUC (Benítez et al., 2020) except that hygromycin (5 μg/mL; Jena Bioscience) was used as selection antibiotic. The new transgenic cell line was called T7TetR-PpyRE9H-LUC RNAi-G6PDH.
The growth phenotype of the clones was studied by cultivating the cells for 5 days in 10 mL culture medium lacking all selection antibiotics but in the presence or absence of 1 μg/mL oxytetracycline (Oxytet, Oximic, Laboratorios Microsules; 5 mg/mL stock solution in water, sterilized by filtration). Every 24 h, LUC activity was determined as described in the next section, parasites’ number was assessed by counting on a Neubauer chamber (Precicolor HBG, Germany), and the cultures were diluted back to the initial cell density of 5×104 cells/mL.
G6PDH activity was determined in cell extracts of two RNAi clones as described next. Two million parasites from 48 h Oxytet-induced or non-induced cultures corresponding to two T7TetR-PpyRE9H-LUC RNAi-G6PDH clones (clon A2 and A6) that showed a deleterious growth phenotype were harvested by centrifugation (2000 g, 10 min at 4 °C). The cell pellet was washed twice with 2 mL of cold PBS 1% w/v glucose and then resuspended in 100 μL of G6PDH reaction buffer (Tris 50 mM, 5 mM MgCl2, and 250 mM NaCl, pH 7.5). Cells were lysed by performing five freezing-thawing cycles in liquid nitrogen. Finally, 5 μL of 0.1% v/v Triton 100X was added to the samples and the supernatant was separated from cell debris by centrifugation (10,000 g, 10 min at 4°C). In a quartz cuvette (final reaction volume of 500 μL), the G6PDH activity of the clarified parasites lysates was measured spectrophotometrically (Absorbance 340 nm) at saturating concentrations of substrates (NADP 700 μM and glucose 6-phosphate 6 mM) and room temperature, using a Cary 50 Bio spectrophotometer (Agilent Technologies). G6PDH activity was calculated from initial velocities using GraphPad Prism software (version 8.0.1).
2.4 In vitro LUC activity
LUC activity was determined using the Firefly, Luciferase Assay System from Promega (catalog number E4530). One million parasites from synchronized cultures or lysed blood samples (section 2.11), were pelleted by centrifugation (2000 g for 10 min at room temperature) and gently washed twice in Phosphate Buffer Saline (PBS)-glucose 1% (w/v). The samples were further processed according to the protocol of the commercial supplier, and the bioluminescent signal was read in a LUMIstar OPTIMA Microplate (BMG LABTECH) luminometer, according to the protocol described in Benítez et al. (2020).
If the number of parasites obtained from blood was between 0.2 and <1 million, then the signal was corrected to 1 million applying the linear regression equation published by Benítez et al. (2020).
2.5 Animal housing, infection, and ethics statement
All animal experiments employed six to 8 weeks old Balb/cJ female mice bred at the animal facility of the Laboratory Animals Biotechnology (Institut Pasteur de Montevideo). After infection, they were housed in individual airtight, HEPA filter cages with negative pressure (IsoCage N - Biocontainment System, TECNIPLAST) in a controlled environment at 20 ± 1°C with a relative humidity of 40–60% in a 14/10 light-dark cycle. Food and water were administered ad libitum.
All infections were performed with T7TetR-PpyRE9H-LUC or T7TetR-PpyRE9H-LUC RNAi-G6PDH parasites in the logarithmic growth phase showing optimal levels of LUC activity, which was verified as described in Section 2.4. Next, the parasites were resuspended in 0.3 mL of fresh culture medium at different cell densities, depending on the experiment, and inoculated to the animals in a single intraperitoneal (ip) injection. Individual animals numbered with codes # 0, 2, 20, 5, or 50 (marks made by ear punching or cutting method) were used for each group and experiment. The animals from each study group were hosted in individual cages.
The experimentation protocols were approved by the Animal Use and Ethic Committee (CEUA) of the Institut Pasteur de Montevideo (Protocol 017-2023) and adhered to the National Law for Laboratory Animal Experimentation (Law nr. 18.611) as well as the international FELASA guidelines governing the use of laboratory animals. Mice showing an impaired health status and/or a parasite load of ≥ 108 cells/mL blood were euthanized (standard end-point criterion).
2.6 Acute mouse infection model of African trypanosomiasis
Three groups of mice (n=3/group) were infected with 102, 103, or 104 T7TetR-PpyRE9H-LUC parasites as described in section 2.5. One non-infected mouse was used as a control. From day 4 post-infection onwards, bioluminescence was assessed at different time points in vivo and ex vivo in blood samples by the methods described in sections 2.7 and 2.4, respectively.
For eliminating systemic infection, 3 mice in an advanced stage of the disease were treated with a single dose of 40 mg/kg DAC (Santa Cruz Biotechnology) administered via ip, and imaged at day 60 post-infection (imaging procedure described in section 2.8). At this time point, these animals were euthanized under terminal anesthesia [165 mg/kg Ketamine (Ketona 50, Richmond Vet Pharma) and 21 mg/kg Xylazine (SETON2%, GALIER)], and blood samples obtained by submandibular puncture with a 21G needle were cultured according with section 2.2 to verify DAC efficacy.
2.7 In vivo imaging and data analysis
For imaging, mice were injected via ip with D-luciferin (Perkin Elmer) 150 mg/kg, and, 2–4 min later, anesthetized with 2.5% isoflurane: 95% oxygen. Next, light emission was recorded at room temperature using the In Vivo Xtreme II equipment (Bruker), back-illuminated with a four megapixels charge-coupled device detector, and Molecular Imaging Software “MI” 7.5 wins for image acquisition in two modalities: luminescence and X-ray or reflectance. During image acquisition, the animals were always placed in a ventral position and anesthetized with a constant flux of 2.5% isoflurane in oxygen.
The settings used for the luminescence modality are: High sensitivity mode, Binning 4 x 4 pixels, Time-lapse 30 s exposure time/Interval 60 s, Field of view 190 mm, f-Stop 1.1, Focal plane 0, Tray Focal reference, and Stop After 20 or 10 min. This setting yields a real-time imaging of 28.5 or 14.25 min, and 20 or 10 luminescence’s pictures, respectively.
The settings used for the X-ray modality are: High-speed sensitivity mode, No Binning, 4 s exposure time, Field of view 190 mm, f-Stop 5.60, Focal plane 0, X-ray Focal reference.
The settings used for the reflectance modality are: High sensitivity mode, No Binning, 0.1 s exposure time, Field of view 190 mm, f-Stop 1.1, Focal plane 0, and Tray Focal reference.
The luminescence and X-ray or reflectance images were analyzed employing the free software from Fiji (Schindelin et al., 2012). The luminescence images (a 16-bit deep image that corresponds to a maximum of 65,535 gray values or levels of signal intensity) were converted to a stack in TIF format and pseudocolored (“Fire” style). The bioluminescence signal is expressed in arbitrary units denominated “counts”. X-ray or reflectance images were overlapped with the corresponding luminescence images of interest.
For analyses of whole-body luminescence, an ROI (Region Of Interest) of 95 pixels width x 270 pixels height, which covers the animal contour according to the X-ray images, was defined. This ROI was further subdivided into three rectangular areas of 95 pixels in width and heights of 50, 75, and 145 pixels that correspond to regions embracing the head, thorax, and abdomen, respectively. For time course analysis of in vivo bioluminescence, the data (counts per sec or day for each ROI) were plotted using the program GraphPad Prism software (version 8.0.1).
2.8 Multimodal Animal Rotation System (MARS) and ex-vivo imaging
For these experiments, five mice were infected with 103 parasites from the cell line T7TetR-PpyRE9H-LUC as described in section 2.5, and one non-infected was used as control. During the infection, the animals were in vivo imaged using the luminescent modality and the settings described above (section 2.7).
Two animals coursing the early and one the late stage of the disease were subjected to MARS and ex-vivo imaging analysis.
For MARS imaging, the animals were injected with D-luciferin and anesthetized before and during image acquisition as indicated in section 2.7. The settings used for luminescence modality were: High sensitivity mode, Binning 4 x 4 pixels, 30 s exposure time, Field of view 190 mm, f-Stop 1.1, Focal plane 0, Tray Focal reference.
For X-ray modality, the settings consisted of: High-speed sensitivity mode, No Binning, 3 s exposure time, Field of view 190 mm, f-Stop 4, Focal plane 0, and X-ray Focal reference. Images were acquired at different animal positions using the following rotation settings: start at 0°, range 360°, and increment of 30°, which delivers a total of 13 luminescence and X-ray images overlapped (note: the first and the last image correspond to the same animal position).
The images were processed (smoothed and pseudocolored) and exported as a video file (AVI format, at one frame per second) using the Bruker Rotation Software.
For ex-vivo imaging, the mice were injected via ip with D-luciferin 150 mg/kg and, 7 minutes later, with phenobarbital 100 mg/kg to achieve terminal anesthesia. A blood sample (200 µL) was collected in a 1.5 mL tube (Corning) containing 20 µL of K3EDTA anticoagulant (Wiener) and parasitemia was determined. Next, the animals were bled and intracardially perfused with 40 mL of PBS-glucose 1% w/v followed by 10 mL of 0.3 mg/mL D-luciferin in PBS-glucose 1% w/v. After perfusion, the organs and tissues were individually dissected, transferred to a 15 mm diameter Petri dish, soaked in 0.3 mg/mL D-luciferin in PBS-glucose 1% w/v pre-heated 5 min at 37°C and then imaged. The settings for the luminescence modality were: High sensitivity mode, Binning 4 x 4 pixels, 5 min exposure time, Field of view 190 mm, f-Stop 1.1, Focal plane 0, Tray Focal reference. The settings for reflectance modality were: High sensitivity mode, No Binning, 0.1 s exposure time, Field of view 190 mm, f-Stop 1.1, Focal plane 0, Tray Focal reference. The luminescence and reflectance images were pseudocolored (“Fire” and “Grays” style, respectively) and overlapped. A picture of the preparations was taken from the top with an Apple iPad, version 12.1.1 (16C50), model MD788LL/B.
For each organ of interest, the mean (RLU: Relative Luminescence Units/ROI) and maximum signal intensity were determined using the Fiji software.
2.9 In vivo double strand RNA interference assay in T. b. brucei-infected mice
For this experiment, the animals were divided into two groups (n=3/group). One of them was fed, 2 days before infection and along the course of the experiment, with water containing 5 mg/mL Oxytet (prepared fresh every 48 h), and the other group was always fed with tap water. Mice from both groups were infected with 103 parasites from the cell line T7TetR-PpyRE9H-LUC RNAi-G6PDH as described in section 2.5.
At different time points of the infection, all animals were imaged for luminescence for 14.25 min (or single image for X-ray modality) applying the method described in section 2.7. In end-point situations, blood samples were taken (section 2.11) and LUC activity was determined (section 2.4) ex vivo or in culture samples from parasites isolated from animals.
2.10 Therapeutic efficacy assay
Twelve mice were infected with 103 parasites from the cell line T7TetR-PpyRE9H-LUC as described in section 2.5 and divided into four groups (n=3/group). From day 2–5 post-infection, the animals were treated daily with 250 mg/kg Deferoxamine Mesylate dissolved in PBS (Dfx 1 x), 250 mg/kg Dfx plus 50 mg/kg Nfx (group: Dfx 1 x + Nfx 0.5 x) or 2,750 mg/kg DMSO dissolved in PBS (group: vehicles). The drug control group (group: Nfx 1 x) was treated on day 2 with a single dose of 100 mg/kg Nifurtimox dissolved in DMSO 100% v/v. The compounds and the vehicles were given via ip in volumes of 50 µL (for different Nfx doses or vehicles) or 300 µL (for Dfx). At different time points during the infection, the following procedures were applied: in vivo imaging of animals (luminescence for 14.25 min and single X-ray image) as described in section 2.7, determination of parasitemia (section 2.11) and blood sample culture to confirm infection or cure (section 2.2).
2.11 Parasitemia determination
Blood samples (≤50 µL), taken from the submandibular vein employing a needle of 27G, were collected in a tube containing 5 µL of tri-potassium ethylenediamine tetra-acetic acid anticoagulant (K3EDTA). After thorough homogenization, an aliquot was diluted 1:20 in a hypotonic solution (BD Pharm Lyse) to lyse red blood cells, incubated for 2 min at room temperature, and diluted 1:1 in PBS-glucose 1% w/v. The cell suspension is placed in a Neubauer Chamber and parasites are counted using light microscopy. The minimum parasite density detectable by this technique is 5×104 cells/mL.
2.12 Statistics
Analysis of survival curves was performed employing the log-rank (Mantel-Cox) test and the t-test was applied for the comparison of two median (unpaired samples, one-tailed and alfa 0.05), using the GraphPad Prism software (version 8.0.1). The log-rank test allowed us to estimate the death hazard functions of the groups under comparison (study vs. control).
3 Results
3.1 In vivo characterization of reporter cell line and acute infection model
3.1.1 Infectivity
We have previously reported the generation and phenotypic characterization of the cell line T7TetR-PpyRE9H-LUC (Benítez et al., 2020). Under in vitro culture conditions, the reporter parasites expressed LUC stably and constitutively in the absence of selective pressure and presented a growth pattern and sensitivity to drugs indistinguishable from the parental cell line.
Here we extended the phenotypic profiling of the reporter cell line to the more challenging in vivo conditions. First, we determined the infectivity of the cell line and monitored parasitemia and LUC activity along the course of mice infection, the latter by in vivo and ex vivo imaging or analysis of different biological samples. Mice from the Balb/cJ strain were used as hosts because they represent an extended laboratory research model of African trypanosomiasis wherein the disease resembles well that occurring in humans (Brun and Kaminsky, 1999; Antoine-Moussiaux et al., 2008; Claes et al., 2009; Giroud et al., 2009; McLatchie et al., 2013).
Injected via ip at inoculums spanning three orders of magnitude: 102, 103 and 104 trypanosomes/mouse, the reporter parasites were able to establish and develop the infection in all mice (Figure 1), with unnoticeable differences concerning infections reported for wildtype strains of T. brucei brucei (Manta et al., 2013; Hiller et al., 2014; Musunda et al., 2015; Currier et al., 2019). For instance, independently of the inoculum, the acute infection is characterized by two phases, an early stage (days 1–7) where the level of parasites in the bloodstream remains controlled, albeit transiently, upon some initial ascending trend, and a late stage (days 8 and on) where this condition is no longer sustained and parasitemia raises uncontrollably reaching highest values. Nonetheless, during the early stage, the trends in parasitemia (i.e., ascending or descending) differed according to the amount of parasites injected: at higher inoculums high parasitemia levels were achieved a few days later upon primo-infection. Similarly, animal survival (Figure 1A) and parasitemia (Figure 1B) correlated well with the inoculum. Overall, in mice infected with as few as 100 trypanosomes, the parasitemia showed a slower kinetics increase during the first and second week upon infection (Figure 1B) that resulted in better animal health scores and a mean survival time of 15 days (Figure 1A). In contrast, animals infected with 100-fold more parasites (104) presented a significantly shorter mean survival time (10 days) as a consequence of the more rapid and sustained onset of higher parasitemia levels (Figure 1B) and impaired health status. The animal group infected with 103 trypanosomes displayed an intermediate behavior.
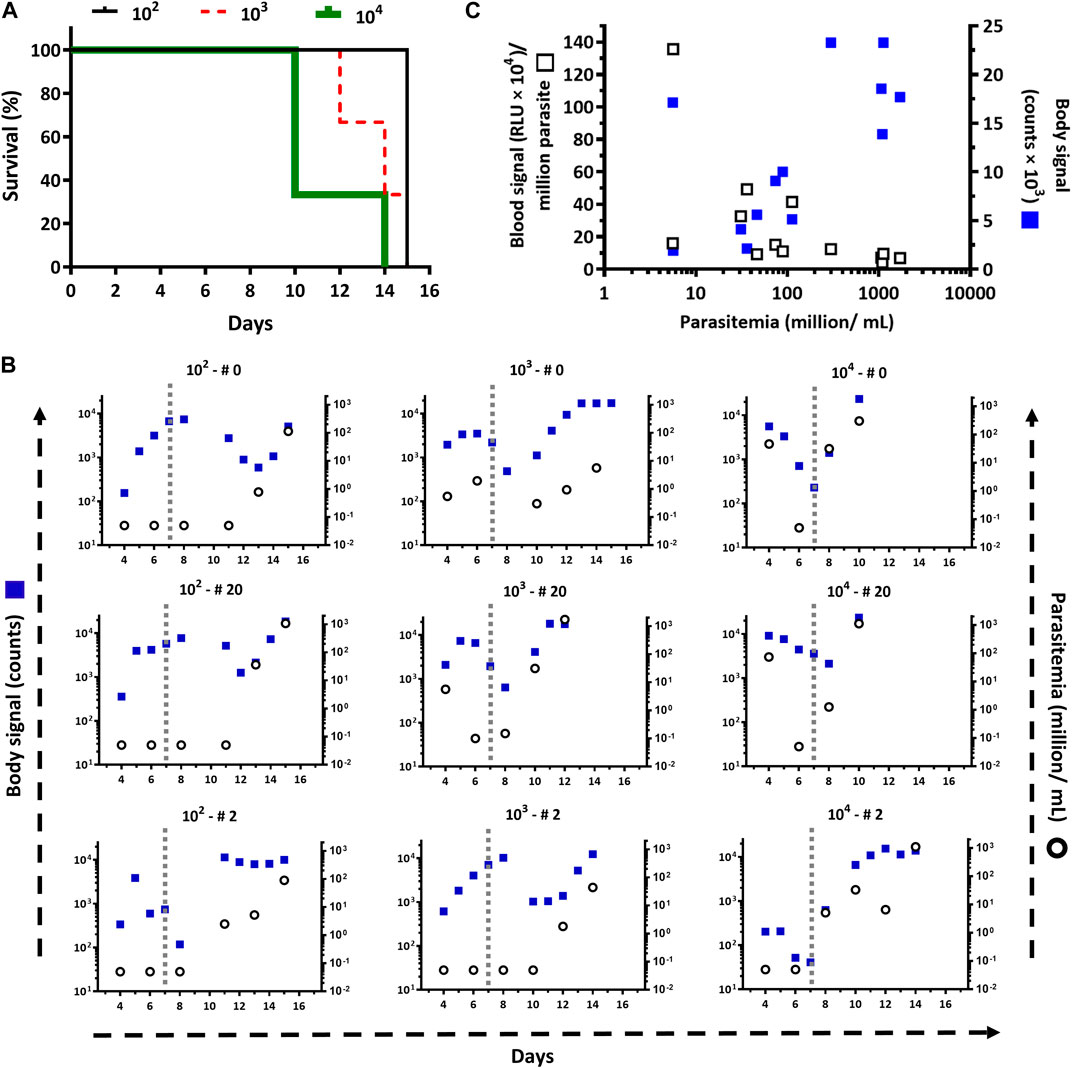
Figure 1. Infectivity of the T. b. brucei reporter cell line. (A) Survival plots, based on a standard end-point criterion, for mice (n=3/group) infected intraperitoneally with 104, 103, and 102 bloodstream trypanosomes of the cell line T7TetR-PpyRE9H-LUC. The mean survival times are 10, 14, and 15 days, respectively. According to the Log Rank (Mantel-Cox), the differences are statistically significant for groups 102 and 104 (p = 0.0224). (B) Parasite burden in the blood (Parasitemia) and whole body (Body signal) determined for each animal by cell counting and by in vivo imaging, respectively. The vertical dotted line demarks the boundary between the early and late phases of the acute infection. The parasite inoculum used for infection (102, 103, and 104) and the animal numbers (# 0, 20, and 2) are shown at the top of each plot. (C) Correlation of parasitemia vs. in vivo (Body signal) or ex vivo (Blood signal) bioluminescence signal.
3.1.2 Infection dynamics monitored by bioimaging
While blood samples were taken at specific time points, from day 4 post-infection and on, all mice were subjected to almost daily non-invasive analysis of bioluminescence to obtain more detailed information about the optimal conditions for in vivo imaging and on the development of the infection/disease. First, the bioluminescence signal generated upon substrate administration was monitored for 30 min (Supplementary Figure S1). Although the temporal development of bioluminescence and its intensity differed between animals (from the same or different groups) and days, overall, the maximum body signal was achieved around 15 min post-administration of D-luciferin. After this time point, in most animals, the signal reached a plateau or presented a slightly descending trend. The exceptions to these rules were animals from the group infected with 102 parasites that at the very early stage of the infection (only at day 4) showed a steady increase of signal during the full acquisition time (Supplementary Figure S1A) and one animal (#0) from the group infected with 104 parasites that at a late stage of the infection (day 10) showed a significant drop in signal 15 min after the acquisition began (Supplementary Figure S1C). We do have not a clear explanation for this rapid in vivo consumption of substrate (e.g., parasites with higher LUC expression or energetic metabolism, inadequate bioavailability of substrate) because such a phenomenon was not observed in animals having similar or higher parasitemia (day 15 in Supplementary Figure S1A, day 12 in Supplementary Figure S1B, and day 10 and 14 in Supplementary Figure S1C). In contrast, the slow kinetics of signal development observed at day 4 in the animals infected with 102 parasites can be ascribed to the lower parasitemia they presented (i.e., trypanosomes undetectable in blood by light microscopy).
Bioimaging during the infection (Supplementary Figure S2) allowed us to visualize the dynamics of the process and to recognize with higher accuracy the different stages of the acute infection mentioned above. In general, for the first 2 days of imaging (days 4 and 5), there was a good correlation between body bioluminescence and inoculum (Figure 1B). Although differing in magnitude and amplitude, the signal showed a similar dynamic among the animal groups. In animals infected with 102 trypanosomes, the signal increased up to days 7–8, then dropped remarkably between days 8–12 to finally raise to almost maximum values on days 12–15. For animals infected with 103 trypanosomes, the signal exhibited a bell-like pattern from day 4 to 8-10 and then ascended to saturation levels. At variance with the behavior observed in the other groups, the signal in mice inoculated with 104 trypanosomes started from a maximum or lower level and decreased steadily up to day 7–8, and then raised to maximum values until the end-point of the experiment. Most likely, a sudden increase in signal occurred during days 0–3 post-infection, which was missed by our imaging schedule that started on day 4.
A valley point between the bioluminescence peaks is observed in all mice from the different groups (Figure 1B) and coincides well with the recrudescence of the infection, which is evidenced by the increase in parasitemia and the impaired animal health status. Expectedly, the time point at which the signal drops (valley) correlates negatively with the infection inoculum: days 10–13, days 8–9, and days 7–8 for infections performed with 102, 103, and 104 trypanosomes, respectively.
Qualitative image analysis was performed to assess the body distribution of the parasites during the infection. For animals infected with a low number of parasites (102 or 103 trypanosomes), the first 4–6 days post-infection (early stage), the bioluminescence signal is mostly confined to the abdominal region (Figure 2). This agrees with the administration route (ip) used to infect the mice. While the infection progresses, in most of these animals, the abdominal signal increases in intensity and extends to the thoracic region (days 7–8). The next days (valley point: days 8–10 for group 103 and days 12–13 for group 102), the signal at the thorax and abdomen drops significantly, both in intensity and area covered, but remains detectable at these sites. When the infection enters the late stage, there is a massive expansion and increase of the signal in the whole body of mice from both groups, particularly in the upper region: thorax and head. As discussed above, in animals infected with a large number of parasites (104), the infection advanced more rapidly. Thus, at variance with animals from groups 102 and 103, which showed a focused abdominal signal, on the first day of imaging (day 4), two out of three mice from the 104 group presented an intense signal encompassing the abdomen and thorax. Nonetheless, after this initial bioluminescence peak, the dynamics of body signal distribution and intensity were fairly similar to that described above for groups 102 and 103. As will be discussed later (section 3.3), the signal from different regions of interest was quantified to identify infection hallmarks that may help to refine the end-point criteria.
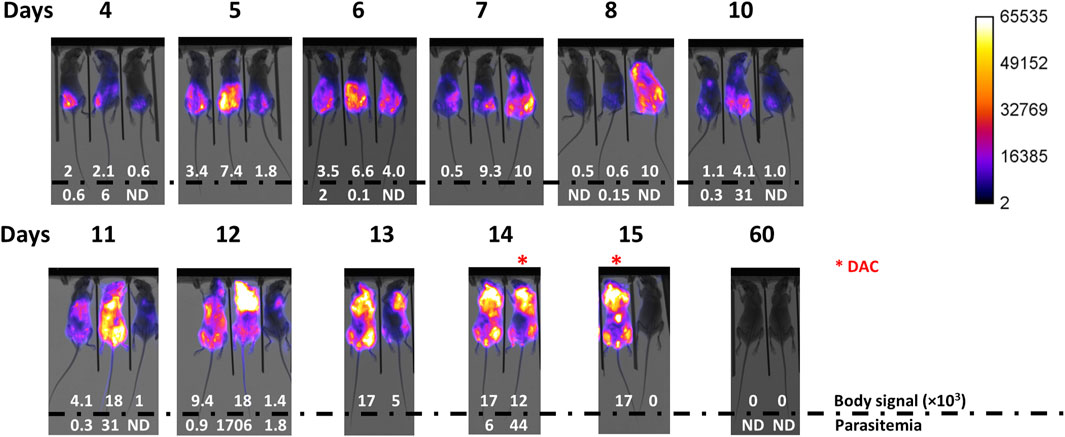
Figure 2. Infection development monitored by body signal and parasitemia. Mice (n = 3) were infected via ip with 103 T7TetR-PpyRE9H-LUC parasites and subjected, from day 4 post-infection on, to almost daily in vivo imaging and parasitemia assessment. Merge images of X-ray and luminescence are shown for each animal (# 0, #20, and #2 from left to right). Max body signal is expressed in counts units and a value = 0 indicates <0.1 counts. Parasitemia is expressed in units of million parasites/mL blood and ND indicates <5 × 104 parasites/mL blood (detection limit of the microscopy counting method). *, indicates mice treated with Diminazene aceturate (DAC). Animal # 20 was sacrificed on day 12 according to the conventional end-point criterion.
3.1.3 Contribution of bloodstream parasites to bioluminescence
The distribution of bioluminescence along the animal body agrees well with the biology of African trypanosomes, which are extracellular parasites that dwell in different internal body fluids (e.g., blood, lymph, cerebrospinal, and interstitial fluids). Therefore, not surprisingly, the measurement of in vivo bioluminescence proved more sensitive than estimating parasite load in blood by microscopy counting, particularly during the early stage of the infection. For instance, in all animals where parasites remained undetected in blood samples, the bioluminescence signal was always detected in vivo (Figure 1B). This apparent higher sensitivity of the in vivo bioluminescence may stem from the fact that: 1) in vivo imaging measures light emitted by parasites located in different parts of the animal body, and not in a restricted compartment of it, and 2) the blood volume analyzed (5–50 µL) represents a minor fraction of the mouse volemia (0.2–2%). However, we noticed that in many cases, the magnitude of the increase in parasitemia did not match the changes in the bioluminescence signal (Figure 1B). A possible reason for that is insufficient bioavailability or rapid consumption/clearance of D-luciferin in infected animals. However, this hypothesis was ruled out based on data from kinetics measurements of in vivo bioluminescence that showed increasing or stable signal levels within 30 min (Supplementary Figure S1). To get some insights into the contribution of bloodstream parasites to in vivo bioluminescence, LUC activity was measured in blood samples from infected animals (Figure 1C). To our surprise, this analysis revealed a lack of correlation between the signal produced by bloodstream parasites and parasitemia (R2 for linear regression = 0.1729), which contrasted significantly with the positive linear correlation observed for whole-body bioluminescence vs. parasitemia (R2 for linear regression = 0.4003) (Supplementary Figure S3). This result led us to speculate that either the reporter gene was not stably integrated into the genome, or that its expression and/or the energetic metabolism of bloodstream parasites is downregulated during parasitemia peaks. To test this hypothesis, bioluminescence was assessed in blood samples of representative animals coursing the parasitemia peak (i.e., group 102: animal 0, day 15; group 103: animal 20, day 12; group 104: animal 2, day 14) and in parasites, from these samples, that were grown in vitro and synchronized in the absence of antibiotic pressure. As shown in the Supplementary Table S1, the signal emitted by bloodstream trypanosomes was negligible (0.6%–8.7%) but fully recovered upon in vitro culture (94–101%), reaching levels comparable with that produced by parasites used for animal infection. An identical result was obtained for bloodstream parasites isolated at longer times upon infection (from 18 up to 36 days) and from animals corresponding to different independent experiments (Supplementary Table S1; see sections 3.2.1 and 3.2.2). Taking together, this data demonstrates that the reporter gene is stably integrated into the parasite genome and that a downregulated expression of the reporter gene and/or limited ATP availability may account for the lower LUC activity of parasites colonizing the bloodstream at the parasitemia peaks.
3.1.4 Acute infection by a monomorphic T. brucei does not compromise CNS
As quoted earlier, during the chronic stage of the infection, African trypanosomes are capable of crossing the BBB to colonize the central nervous system. In the laboratory, this is typically observed in infections performed with pleomorphic cell lines of T. brucei spp., which produce a disease characterized by regular (every 1–2 weeks) peaks of parasitemia of minor magnitude than those observed in infections with monomorphic parasites (Claes et al., 2009; Giroud et al., 2009; McLatchie et al., 2013; Burrell-Saward et al., 2015). It has been shown that pleomorphic trypanosomes invade the brain meninges as early as 5 days post-infection, whereas penetration of the BBB takes about 21 days (Myburgh et al., 2013). Treatment with DAC, a drug unable to cross the BBB (Jennings et al., 1977; Gichuki and Brun, 1999), cleared meningeal (and systemic) parasites but not those localized in the brain, which were responsible for the relapse of the infection (Myburgh et al., 2013). At the late stage of the acute infection by monomorphic trypanosomes, we observed a significant increase of bioluminescence in the mice head (Figure 2 and Supplementary Figure S2) suggesting a potential compromise of the tissues/organs therein located. To confirm this, three animals at the terminal stage of the infection (group 103: animal 2 at day 14 and animal 0 at day 15; group 102: animal 2 at day 15) received a single dose of DAC 40 mg/kg and were monitored by in vivo imaging, parasitemia, and clinical state. In 24 hs, DAC was able to fully clear the bioluminescence signal from the whole animal body and to turn undetectable the presence of parasites in the bloodstream (Figure 2, group 102: animal 2 at day 14 and group 102: animal 2 at day 15). The treated animals remained alive and displayed an excellent clinical status ∼45 days upon DAC administration and 60 days upon infection, with undetectable signals of bioluminescence and of parasites in blood (Figure 2, Supplementary Figure S2, day 60). This result suggests that the monomorphic cell line does not invade the central nervous system (CNS).
3.2 3D and ex vivo imaging of mice at different stages of the acute infection
To further characterize the model, an independent infection experiment was performed, which included animal imaging (ventral position and full animal rotation: MARS) and ex vivo imaging of different organs/tissues from three mice where body signal distribution and intensity differed during the infection (Figure 3). In mouse 5, infection developed with the typical first peak and valley at week one, and with a minor compromise of the thoracic region (Figure 3A). In mouse 0, the body signal increased up to day 5 post-infection, and remained stable for the next 3 days, albeit on day 8 the thorax signal retracted to the abdominal cavity (Figure 3A). During the first week of infection, mouse 20 exhibited signal dynamics and intensity similar to that of mouse 5, however, at day 8 bioluminescence dropped markedly and within the next 3 days raised to maximum levels in the full body, particularly in the thorax and head (day 11; Figure 3A).
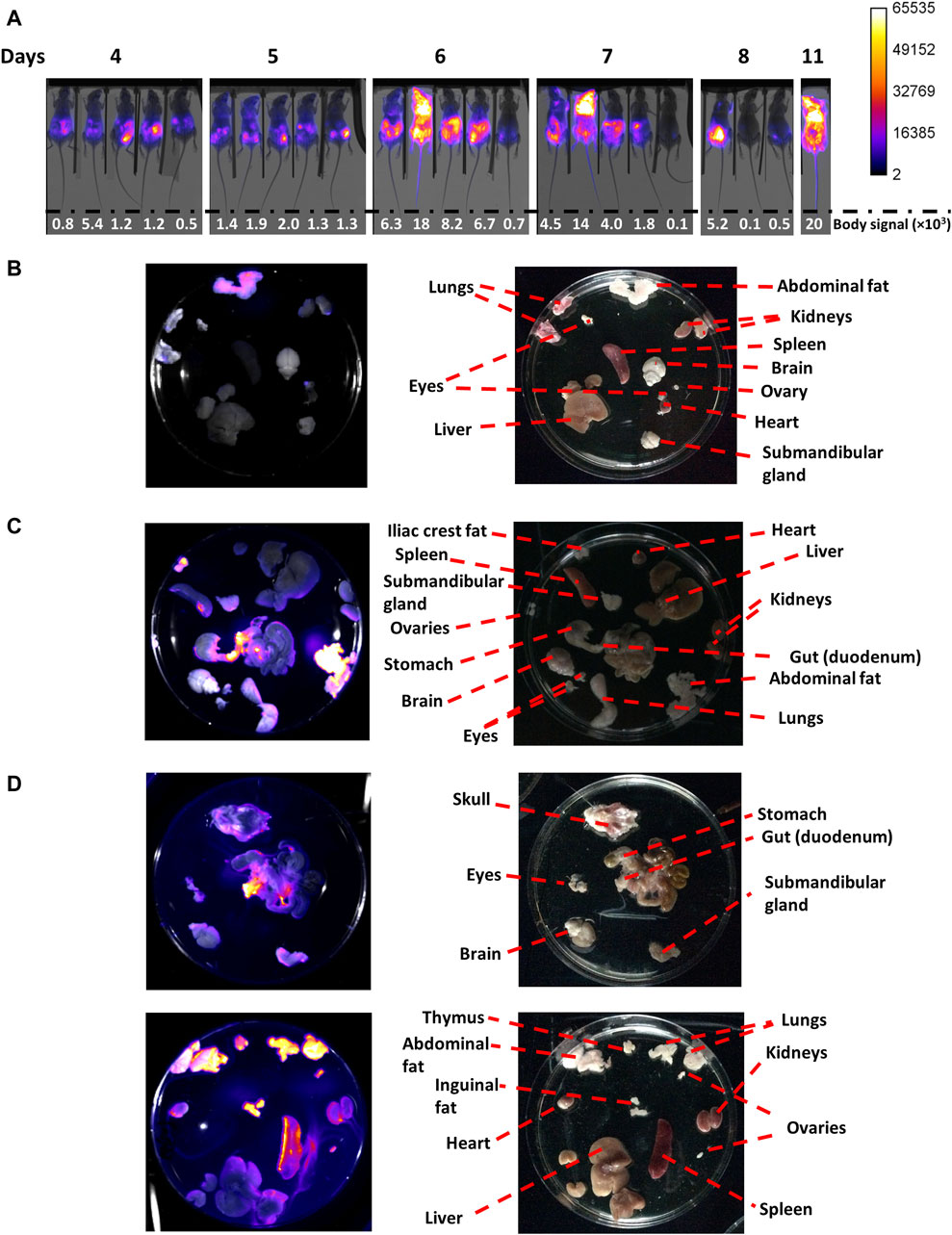
Figure 3. In vivo and ex vivo imaging of mice coursing acute African trypanosomiasis. Mice (n = 5) were infected via ip with 103 T7TetR-PpyRE9H-LUC parasites and subjected to (A) in vivo imaging, where the merge images of X-ray and luminescence are shown for each animal (#0, #2, #20, #5 and #50 from left to right) and body signal is expressed in counts units, and (B) ex vivo imaging for mouse #5 at day 7 (0.8 × 106 parasites/mL), (C) mouse #0 at day 8 (0.4 × 106 parasites/mL, early phase of the acute infection), and (D) mouse #20 at day 11 (950 × 106 parasites/mL, late phase of the acute infection). For the ex vivo images, the left panel contains luminescence images overlapped to the corresponding reflectance image acquired with the In vivo Xtreme II equipment (Bruker), and the right panel shows photographs taken with an Apple iPad, version 12.1.1 (16C50), model MD788LL/B.
Considering the analysis above, different organs and tissues of extensively perfused animals were imaged at day 7 (mouse 5, parasitemia = 0.8 x 106 parasites/mL), 8 (mouse 0, parasitemia = 0.4 x 106 parasites/mL) and 11 (mouse 20, parasitemia = 9.5 x 108 parasites/mL; Figures 3B–D, respectively).
In mouse 5, at day 7, bioluminescence was located in the abdomen with a more intense signal in its upper region (Figure 3A). MARS analysis showed two light sources in the abdomen, better observed from the ventral position, one transversal and below the diaphragm, and the second one centered at the pelvis (Supplementary Video S1). In line with this, ex vivo imaging revealed a notorious signal in the abdominal fat (mean ∼10,300, max 29,530 counts) and, slightly above background level, in the apical regions of the kidneys (mean ∼900, max 8920 counts) and ovary (mean ∼1,200, max 9530 counts), and lungs (mean ∼1,190, max 7735 counts) (Figure 3B). Taking into account the prominent signal observed for abdominal fat, and that the kidneys and ovary are located in the abdominal cavity and are surrounded by a fat layer, we speculate that the signal in these organs stems from their corresponding fat layers. Interestingly, at this time point no signal was detected in the spleen, which is a primary target organ during the first stage of African trypanosomiasis (Blum et al., 2006).
For mouse 0, at day 8, the bioluminescence signal was high in the abdominal region and significantly lower, in intensity and area, in the thoracic region (Figure 3A). MARS analysis showed three infectious sites, one in the thorax (at the left side of the ventral position image), and the other two in similar localizations as those described previously for animal 5 (Supplementary Video S2). Once again, the abdominal fat displayed the highest signal (mean ∼35,700, max 65,535 counts), followed by fat from the iliac crest (mean ∼17,000, max 47,001 counts), and the duodenum (mean ∼23,300, max 65,535 counts), and a localized region of an enlarged spleen (likely the anchorage site of the splenic artery) (Figure 3C).
In contrast, the mouse at the terminal stage (day 11, mouse 20) presented strong bioluminescence in the whole body, albeit the saturation levels were located in the thorax and head (Figure 3A). In agreement with this, MARS analysis revealed a distribution of the signal across the body but especially focused on the thorax (observed from a ventral position) and head (better observed from a dorsal position) (Supplementary Video S3). Ex vivo imaging confirmed the compromise of multiple organs and tissues, with the strongest bioluminescence observed in the abdominal (mean ∼22,700, max 65,535 counts), and inguinal fat (mean ∼28,000, max 65,314 counts), the lungs (mean ∼32,300, max 65,535 counts), the thymus (mean ∼21,900, max 45,412 counts), the ovaries (mean ∼19,000, max 3812 counts), the duodenum (mean ∼28,500, max 65,535 counts), the enlarged spleen (mean ∼20,000, max 65,324 counts), the kidneys (mean ∼12,600, max 38,512 counts), and the heart (mean ∼10,300, max 31,661 counts) (Figure 3D). Minor levels of bioluminescence were observed in the submandibular gland and the skulls. No significant levels of bioluminescence were detected in the liver, which is another organ infected by T. brucei spp. (Blum et al., 2006). Although an intense signal was observed in the head of this animal, there was no bioluminescence detected in the brain, which agrees well with the absence of infection relapse after DAC treatment in the late stage (Figure 2, day 60). Therefore, and supported by other studies (Myburgh et al., 2013), it is tempting to speculate that the head´s signal observed in vivo stems from the colonization of the meningeal spaces. Taken together, these results demonstrate that under our experimental conditions, and despite causing a severe acute infection, monomorphic T. b. brucei does not colonize the CNS.
3.3 Refining end-point criterion based on quantitative in vivo image analysis
Based on the characterization of the acute murine infection presented above for monomorphic T. b. brucei, we next aimed to establish a quantitative parametrization of in vivo bioluminescence to redefine the end-point criterion based on a non-invasive method.
With this purpose, four major ROIs were considered for the analysis of in vivo bioluminescence: the full mouse body, the abdomen, the thorax, and the head (Figure 4A). Taking into account the spatial dynamics of the infection and to determine which of them is more sensitive in highlighting a compromise of vital organs, the following signal ratios were calculated: abdomen/body, [head+thorax]/body, and [head+thorax]/abdomen (Figure 4A). Furthermore, to cover a large spectrum of possible infection scenarios, the analysis was performed in all the animals that have been infected with inoculum encompassing three different orders of magnitude (Figure 1).
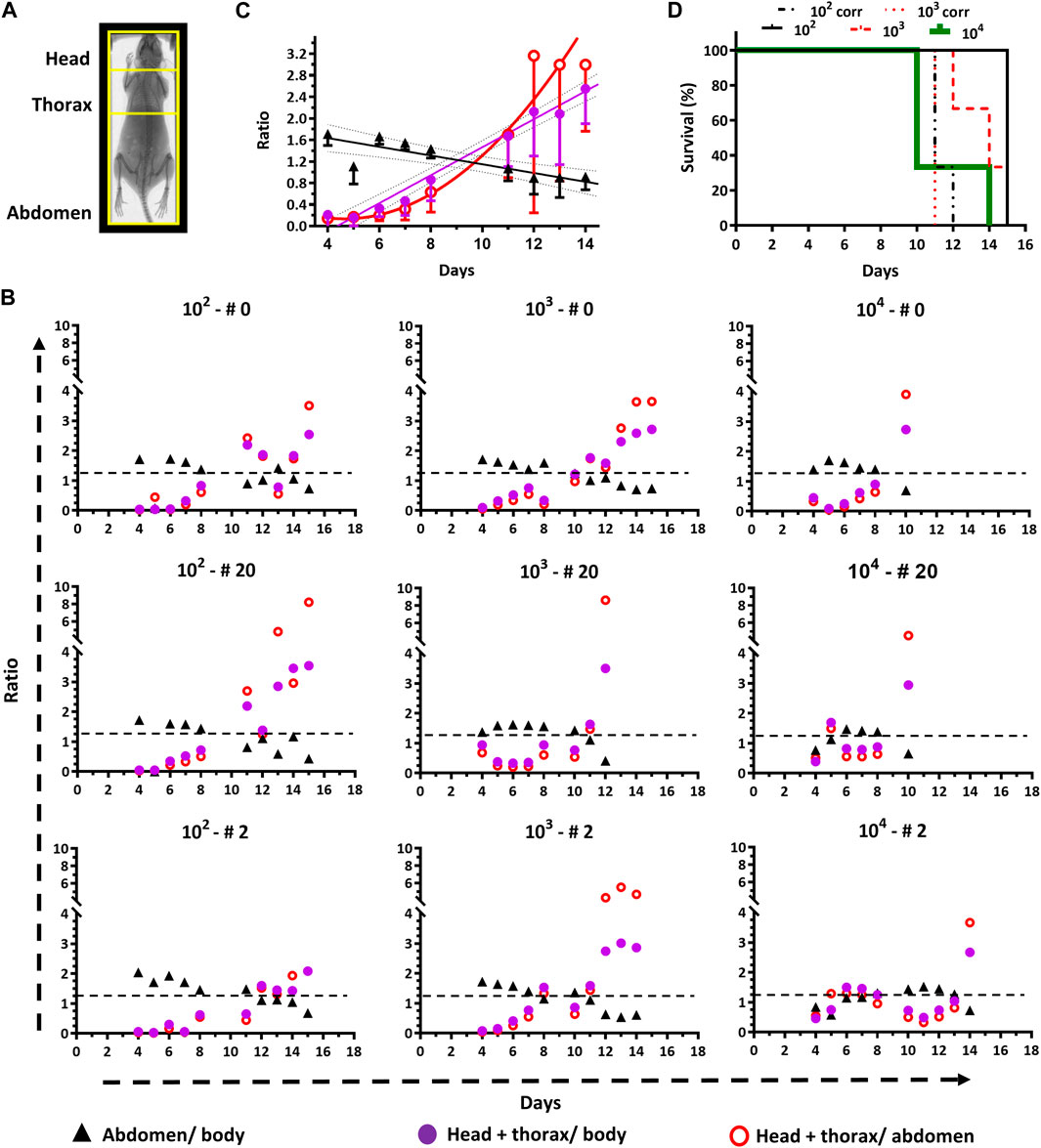
Figure 4. End-point criteria refined by quantitative in vivo imaging. (A) The Regions of Interest (ROI) that were selected for image analysis are framed in yellow on a representative mouse X-ray photograph. (B) Time course plots for signal ratios of abdomen/body, [head + thorax]/body, and [head + thorax]/abdomen for animals (n = 3) infected with different inoculums (102, 103 or 104 parasites). The black dashed line indicates the cut-off value (1.2) suggested as end-point criterion (i.e., <1.2 for abdomen/body and >1.2 for [head + thorax]/body). (C) Time course plots of signal ratios (mean ± SD) from different ROIs of animals infected with 102 and 103 parasites. The data fitted to linear or non-linear equations are shown with solid lines. The cut-off value of 1.2 corresponds to the interception of the linear regression plots for the abdomen/body and the [head + thorax]/body ratios. (D) Survival plot analyzed by standard (for groups 102, 103, and 104) and bioluminescence (groups 102 and 103) end-point criteria. For the first criterion, the mean survival times are 10, 14, and 15 days, for groups 102, 103 and 104, respectively, and the Long Rank (Mantel-Cox) test indicates a significant difference between groups 102 and 104 (p-value 0.0224). For the second criterion, the mean survival time for both groups (102 and 103) is 11 days. The Long Rank (Mantel-Cox) test yielded significant differences for the mean survival time of the group 102 (p-value 0.0224) and 103 (p-value 0.0253) when the conventional and bioluminescence end-point criteria were compared.
Independently of the inoculum, during the first week of infection, the signal ratio abdomen/body appears overall stable by fluctuating between values of 1–2, but shows an overall decreasing trend (values <1) while the infection progresses (Figure 4B).
For instance, at day 4 post-infection the ratio is 1.47 ± 0.40 and reaches a value of 0.63 ± 0.13, which is statistically significant (p-value <0.0001), at the corresponding end-point conditions of each animal. In contrast, the ratios estimated for [head+thorax]/body, and [head+thorax]/abdomen at day 4 (0.38 ± 0.34 and 0.26 ± 0.26, respectively) and the end-point (2.92 ± 0.36 and 4.99 ± 1.99, respectively) show a clear increase that is 8-fold and 19-fold higher than the decrease observed for the ratio abdomen/body. Taking together, the ratiometric analyses agree well with the abdominal cavity being the site of inoculation from which then the pathogen spread to other tissues and organs. Furthermore, these data demonstrate that bioluminescence signals from the upper body of the animal are more sensitive markers of infection burden and progression.
Interestingly, for most animals, the plots for the ratios [head + thorax]/body, and [head + thorax]/abdomen can be decomposed into at least three phases that match the previous description of the acute infection (section 3.1). During the first week and in all mice, these ratios remain stable (group 104) or show a slight increase trending in the groups infected with the low and medium inoculum. In all mice (except for #0 from group 104), and matching the body signal valley shown in Figure 1B, the ratios reach a transient plateau or decrease (time window from day 7–12). The third phase shows, in most cases, a sudden and remarkable increase in the ratios (>2-folds for the [head + thorax]/body ratio and >3-folds for the [head + thorax]/abdomen ratio).
To define an experimental end-point based on quantitative image analysis, the average for the different signal ratios (abdomen/body, [head + thorax]/body, and [head + thorax]/abdomen) along time from mice belonging to groups 102 and 103 were plotted and analyzed (Figure 4C). The group infected with 104 parasites was excluded from the analysis because of the rapid disease onset. The data corresponding to the signal from the abdomen, and head + thorax normalized to the body signal fitted well to a linear regression equation (R2 = 0.775 and R2 = 0.977, respectively) while that from [head+thorax]/abdomen could be fitted to a non-linear regression function (R2 = 0.701). This representation of the data shows clearly the opposite decreasing and increasing trend in ratio signal for the lower (ratio abdomen/body) and upper (ratio [head+thorax] vs. body or abdomen) regions of the animal body. Considering this quantitative analysis we propose as bioluminescence-based end-point criterion the interception of the linear plots, which has a value of 1.2. Thus, animals displaying a ratio of abdomen/body <1.2 and a ratio of [head+thorax]/body >1.2 should be excluded from the study. Applying this end-point criterion to the survival analysis of the experiment involving these mice revealed that the infection performed with 102 or 103 trypanosomes (Figure 4D), contributed to lowering the mean survival time from 15 to 11 days (Long Rank (Mantel-Cox) test p-value = 0.0224) and from 14 to 11 days (Long Rank (Mantel-Cox) test p-value = 0.0253), respectively. This significant shortening of the survival assay contributes to reducing unnecessary animal pain and distress caused by the trypanosomal infection, without compromising the robustness of the experimental data. In contrast, for animals infected with a larger amount of trypanosomes (104), there was no difference in the survival time of the animals subjected to the signal end-point criterion compared to the clinical- and parasitemia-based parametrization of the end-point, likely because of the rapid development of the infection.
3.4 Applications
3.4.1 Validation of a molecular target candidate by in vivo reverse genetic and imaging
The reporter LUC cell line was generated from a parental cell line that encodes for the T7-RNA polymerase and the Tet-repressor protein (cell line 514-1313; Alibu et al., 2005), which allows the Tet-regulated expression of trans-sequences (full gene or fragments) either dependent on the heterologous T7-promoter or on trypanosomatids´ promoter sequences. The potential of our reporter cell line to interrogate gene function in vitro and in vivo was verified by co-transfecting it with an integrative DNA vector that enabled the Tet-inducible downregulation of the T. brucei G6PDH by double-strand RNA interference (dsRNAi; Gupta et al., 2011). G6PDH is the first enzyme from the pentose phosphate pathway (Comini et al., 2013) and was selected as a molecular target because its essentiality for bloodstream T. b. brucei has been demonstrated in vitro but not in vivo (Gupta et al., 2011).
Although the efficiency of the RNAi construct used to downregulate G6PDH has been previously assessed (Gupta et al., 2011), here we verified its functionality on the LUC cell line by measuring G6PDH activity in cell extracts from two clones (A2 and A6) grown for 48 h in the presence of Oxytet. Under steady-state conditions, G6PDH activity ranged from 0.7 to 3.3 μmol/min in samples from non-induced cultures and was undetectable on Oxytet-induced samples from both RNAi clones. One representative clone (A6) was selected for further in vitro and in vivo phenotypic analysis. By the cell counting technique, a marked and transient (24 h) growth arrest was observed on day 3 of sustained dsRNAi induction (Figure 5A). Thereafter, cell growth was resumed though at a lower rate than non-induced cells. Worth noting, the cumulative LUC activity reported in Figure 5A stems from determinations performed with an identical amount of parasites. The bioluminescence emitted by the Oxytet-induced parasites displayed a variation pattern similar but not identical to that recorded by cell counting. For instance, already by day 2 the signal produced by Oxytet-induced parasites was 65% lower compared to untreated cells and remained stable for the next 24 h. Furthermore, in Oxytet-induced parasites, the signal slope from day 3 on was 0.3-fold lower than that determined by counting or than that corresponding to the non-induced cells. These factors determine that bioluminescence is more sensitive than cell counting for assessing parasite viability. Interestingly, in Oxytet-induced parasites, the decline in LUC activity anticipated by 24 h the subsequent growth arrest, which reinforces the important role of G6PDH in the parasite´s energetic and biosynthetic metabolism.
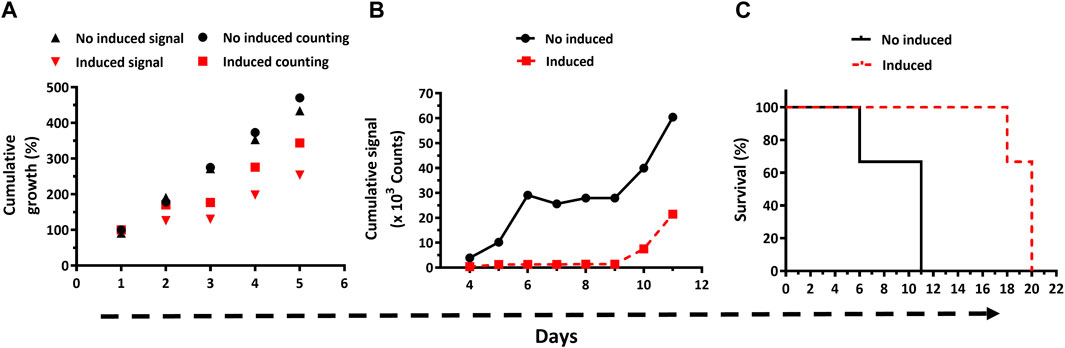
Figure 5. In vitro and in vivo bioluminescence-based phenotypic analysis of bloodstream Trypanosoma brucei with downregulated expression of Glucose-6-phosphate dehydrogenase. (A) Parasite growth (expressed as % cumulative growth) in cultures lacking (black symbols) or containing Oxytet (red symbols) was monitored daily by cell counting (counting) and bioluminescent (signal) methods. (B) In vivo cumulative bioluminescent signal for animals (n = 3/group) infected with T7TetR-PpyRE9H-LUC RNAi-G6PDH parasites and feed with water without (no induced) or containing Oxytet (induced). (C) Survival plots based on the standard (─ and ---) and bioluminescence (-.-. and …) end-point criteria are depicted for infected mice feed (induced) or not with Oxytet (no induced). Statistical analysis (Long Rank, Mantel-Cox test) indicates that differences in mean survival times for the non- and Oxytet-induced groups obtained by the standard and bioluminescence end-point criteria are significant (p-values of 0.0295 and 0.0246, respectively).
Having verified in vitro the integrity of the reporter parasites and of the RNAi system that silences the expression of the indispensable G6PDH, mice were infected with this transgenic cell line, and the infection was monitored by classical methods and in vivo imaging (Supplementary Figure S4). During the first 8 days post-infection, the infection was notoriously controlled by animals fed with water containing Oxytet, which presented infection foci of low to almost null signal intensity in the abdominal region. On day 10, an intense signal appeared in the abdomen of these animals, indicating infection relapse that in the subsequent days (11–15) extended to the thoracic region and reached the head at the end-point days (18–20). In contrast, in mice fed with water lacking Oxytet the bioluminescence signal was higher and broadly distributed across the body during the first week. Only one animal from this group (#50) and between days 6–8 did not obey this rule. On days 10 and 11, an intense signal covered the full body and, mainly, the head/thorax of the remaining living animals from this group (#5 and 50). Such a level of signal intensity in the head/thorax was not observed in mice fed with Oxytet, not even at the end-point day. Quantification of the cumulative bioluminescence in the animals’ bodies revealed a notorious difference in signal development for both groups (Figure 5B). In mice receiving plain water, the signal increased almost exponentially during the early stage of the acute infection (days 4–6), then remained stable for the next 3 days (infection control window), and from day 10 and on resumed the initial increasing tendency. For animals fed with Oxytet, the level of cumulative signal remained low and stable until day 9, from there on (days 10 and 11), it displayed a remarkable increase, albeit lower than that occurring in the control group. Another important difference is that at day 9 the maximum cumulative signal in the control group was 30-fold higher than in the Oxytet-fed group.
Strikingly, at day 18 the intensity of the body signal in the animals from the Oxytet group remained stable (#0) or decreased ∼3-folds (#5 and 50; Supplementary Figure S4). To verify is such phenomena can be linked to a decimated parasite population or a lower expression/activity of the reporter gene, blood samples were collected at this time point to estimate parasitemia and LUC activity. In two (mouse #0 and #50 at day 18 and 20, respectively) of the animals analyzed, the parasitemia was high (7.6 and 33 x 108 parasites/mL blood, respectively) yet bioluminescence signal ex vivo was negligible (<1% compared to parasites grown in vitro; Supplementary Table S1). However, upon a short in vitro growth in the culture medium, the parasites isolated from animals recovered full LUC activity. This result demonstrates the stable integration of the LUC cassette and supports our previous assumption (section 3.1): at a terminal stage of the infection, the parasites undergo nutrient starvation, which affects LUC expression and/or ATP availability.
In line with the development of the infection described above, animals infected with parasites subjected to in vivo induction of G6PDH dsRNAi showed a mean survival time of 20 days, which is 9 days longer and statistically significant (p = 0.0295) compared to the mean survival of the mice from the control group (i.e., infected with the same parasite cell line but not subjected to dsRNAi induction; Figure 5C).
Applying the bioluminescence-based end-point criterion, the mean survival time was 10 for the no induced and 18 days for the induced dsRNAi condition (Figure 5C), and yet statistically significant (p = 0.0246).
In summary, these results confirm the suitability of the LUC reporter bloodstream T. brucei for addressing in vitro and in vivo gene function in a non-invasive manner.
3.4.2 Therapeutic efficacy studies
Next, we were interested in assessing the potential of the bioluminescent acute infection model of African trypanosomiasis for performing the preclinical evaluation of drugs (or candidates). For this pilot experiment (mice = 3/group), we selected the following drugs: Nfx, which is currently used for the treatment of HAT (Sokolova et al., 2010) and Dfx, an iron chelator with cytostatic action against bloodstream African trypanosomes (Breidbach et al., 2002; Merschjohann and Steverding, 2006) that is clinically approved for the treatment of iron-related diseases and -poisoning (American Society of Health-System Pharmacists, 2023). Dfx mainly distributes in blood (Miller and Morgan, 2014), therefore, we expect that it will be effective against systemic parasites. So far, this drug has never been tested in vivo against African trypanosomes. For Nfx, we tested a reported curative dose (100 mg/kg x day; Sokolova et al., 2010), and for Dfx a dose (250 mg/kg x day) that is below the reported LD50 for mice (1,760 mg/kg via ip) and did not cause acute toxicity in mice upon a daily administration during 9 days (Bosque et al., 1995). In addition, a combinatory treatment between Nfx and Dfx was assayed and consisted of administering the first at half its curative dose (50 mg/kg x day). Drugs and vehicle alone (2,750 mg/kg DMSO in PBS) were administered ip daily for 4 days, starting 48 h post-infection.
Compared to the group treated with the drug vehicles (mean survival = 18 days), only mice treated with 100 mg/kg Nfx survived for the full extension of the experiment (60 days; p = 0.0224), whereas those receiving 250 mg/kg Dfx alone (mean survival = 20 days or 19 days when bioluminescence-based end-point criterion is applied) or in combination with 50 mg/kg Nfx (mean survival = 23 days) showed an extended life-span, though not statistically significant (p> 0.15; Figure 6A).
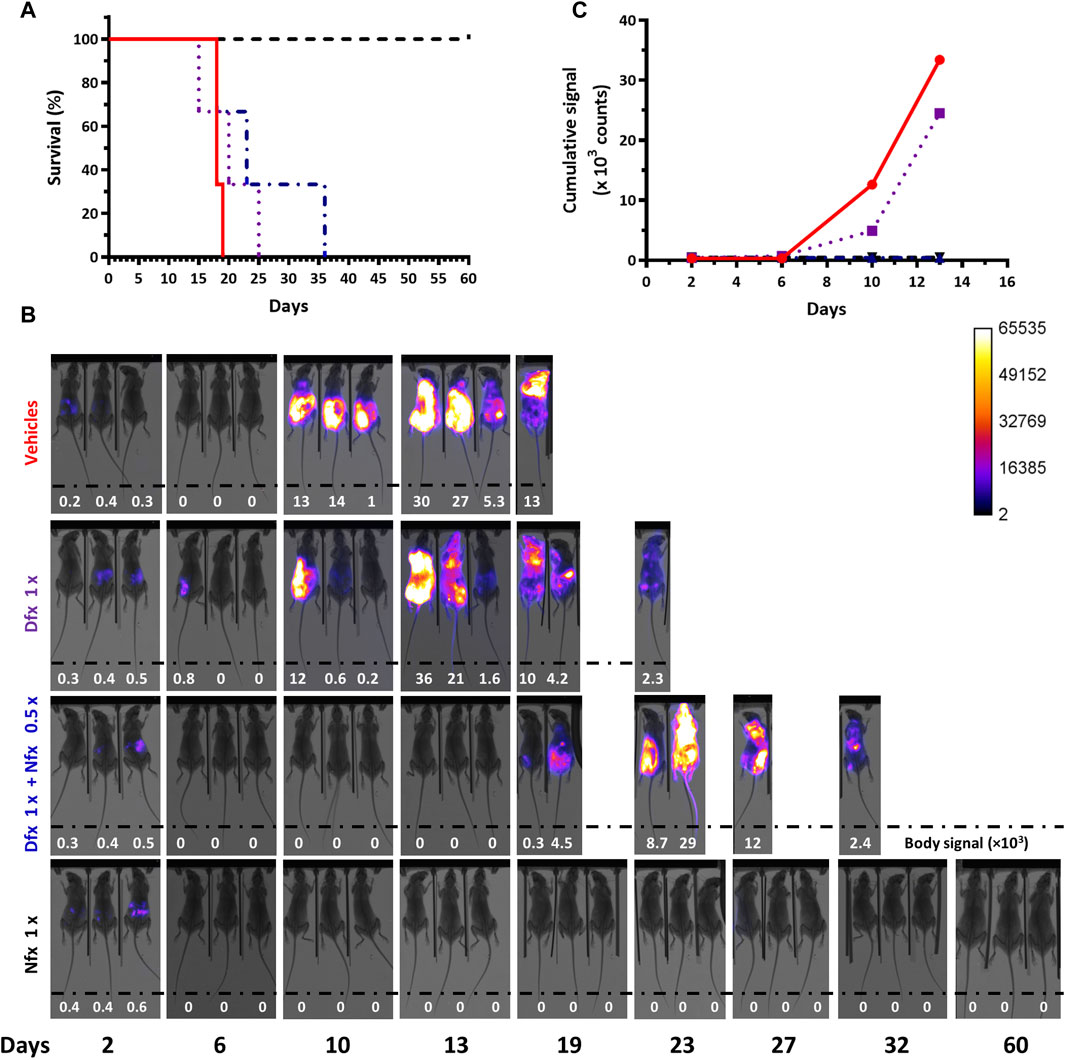
Figure 6. Therapeutic efficacy assays in acute African trypanosomiasis monitored by in vivo imaging. This pilot experiment (n=3 mice/group) evaluated the therapeutic efficacy of Deferoxamine 250 mg/kg x day for 4 days (Dfx 1x), a combination of Dfx 250 mg/kg x day + Nifurtimox 50 mg/kg x day for 4 days (Dfx 1x + Nfx 0.5x), a single dose of Nifurtimox 100 mg/kg (Nfx 1x) and the vehicles PBS and DMSO. (A) Survival plots. The mean survival times are 18, 20, and 23 days, for the vehicles, Dfx 1x and Dfx1x + Nfx 0.5x groups, respectively. The Long Rank (Mantel-Cox) test indicates a significant difference in survival time only for the treatment with Nfx 1x (p-value 0.0224). (B) In vivo imaging of the infected mice (#0, #2, and #20 from left to right) from each group. The merge of X-ray and luminescence images is shown. The max body signal values (in counts units) are shown as inset at the bottom of each image and a value = 0 indicates <0.1 counts. (C) Cumulative bioluminescent signal (counts) vs. day from day 2 until 13 post-infection for the different mice groups.
In vivo imaging confirmed the successful infection of all 12 animals (Figure 6B). In mice receiving the vehicles, the evolvement and body distribution of the signal were similar to that observed in previous experiments (section 3.1). Compared to animals from the control group, 2 out of 3 animals treated with Dfx presented low or null signals at day 10 post-infection, which suggests certain residual action of the chelating agent on trypanosomes. However, this effect was transient since 3 days later bioluminescence reappeared. Interestingly, at this time point (day 13), the signal was undetectable in mice treated with the drug combination, which, though not curative, indicates an improvement in infection control when compared to the single Dfx treatment. Similar to the behavior observed in some animals coursing the terminal phase of the infection (Supplementary Figure S2C and Supplementary Figure S4), the last survivors from the group treated with Dfx (day 23) or the drug combination (day 36) showed a decrease in body signal (Figure 6B). LUC activity in a blood sample collected at this stage for one of these mice (drug combination) was almost null but returned to basal levels (97%) when the bloodstream parasites were cultivated in vitro for a short time (Supplementary Table S1). This confirms once again the stable integration of the reporter gene under a challenging condition (drug treatment). In the animals treated with Nfx alone, the signal was detected in their abdomen on day 2 and then remained undetectable until the end of the assay (day 60), in agreement with the curative effect of this drug dosis.
Quantification of the temporal cumulative body signal for all animals from each group until the end-point day of the vehicle control group (day 13, Figure 6C) supported the qualitative descriptions above. Compared to this group, at days 10 and 13, bioluminescence remained significantly low in mice treated with Nfx alone or combined with Dfx (>27- and >71-fold lower, for days 10 and 13, respectively).
Altogether, the results obtained confirmed that the bioluminescent infection model of acute trypanosomiasis can be used to monitor drug efficacy.
4 Discussion
Our original premise was to develop a bioluminescent cell line of T. brucei suitable for addressing all stages of the early phase drug discovery process: from target validation in vitro and in vivo to compound screening and drug efficacy tests in animal models. To meet this goal we considered that genetic reverse approaches for target validation may require inducible expression systems (i.e., dsRNAi, conditional knockout, dominant negative). Therefore, we selected a T. b. brucei cell line that constitutively expresses the Tet-repressor protein and the T7-RNA Polymerase (cell line 514-1313; Alibu et al., 2005). This cell line has been successfully used to genetically validate several molecular targets in vitro and in vivo (Comini et al., 2004; Manta et al., 2013; Loureiro et al., 2015; Schichler et al., 2023). Another important feature of the selected cell line (bloodstream and monomorphic form of strain 427) is its lack of pathogenicity for humans and ease of in vitro culture and genetic manipulation. Unlike the pleomorphic variant, monomorphic parasites have lost the capacity to undergo differentiation to non-replicative or insect stages (Silvester et al., 2017), resulting in a high proliferative/infective rate that leads to acute and fulminant infections in susceptible mouse strains (i.e., Balb/cJ, Duleu et al., 2004). Therefore, the monomorphic bloodstream form can be considered a hypervirulent parasite variant that, when applied to drug discovery and drug target validation, biases the identification of potent growth inhibitors and molecular targets essential for parasite proliferation and virulence in vitro and in vivo. Regarding the LUC variant, we chose a sequence from P. pyralis luciferase optimized for thermal stability and emission of red-shifted light (PpyRE9H-LUC, Branchini et al., 2010) that has been previously validated for deep-tissue imaging in trypanosomal infections (McLatchie et al., 2013; Lewis et al., 2014; Lewis et al., 2015). Furthermore, the activity of PpyRE9H-LUC is ATP-dependent, allowing us to correlate the metabolic status/viability of the parasites with the signal.
Recently, we reported the generation of the T7TetR-PpyRE9H-LUC T. brucei cell line and its use for compound screening (Benítez et al., 2020). Here we extended its use to different in vivo applications, some of which have not been addressed by other studies (i.e., refinement of animal end-point criterion based on a thorough quantitative analysis of infection images and gene-function studies).
First, we demonstrated that T7TetR-PpyRE9H-LUC was fully infective to Balb/cJ mice, as few as 100 parasites injected via ip led to the development of fulminant disease where the survival times showed an inverse correlation with the inoculum (e.g., the higher the inoculum, the lower the animal survival). The clinical signs and anatomopathological features of the infected animals were similar to those observed or reported for infections performed with the wild-type monomorphic strain (Brun and Kaminsky, 1999; Claes et al., 2009). These data confirmed that the in vivo phenotype of the transgenic parasites was not altered by the expression of the reporter gene and the genetic manipulation performed.
In laboratory models of African trypanosomiasis, the measurement of parasitemia along with the monitoring of animal health status are considered the gold standard parameters to monitor an active infection in the organism (Brun and Kaminsky, 1999). However, as shown here, the microscopy counting method is far less sensitive than whole-body in vivo imaging, particularly in the early stage of infection (first-week post-infection), when parasites disappear from the bloodstream due to the strong innate immune response of the host. In this regard, ex vivo imaging of different organs revealed that during this phase monomorphic parasites are preferentially located in abdominal fat and, to a minor extent, in adipose tissue surrounding lungs and kidneys. Our data complement prior studies conducted with pleomorphic T. b. brucei on mice from a different sex and strain (C57BL/6J male), which highlighted the early colonization of the interstitium of adipose tissues by the parasites (Trindade et al., 2016). Although adipose tissue is considered to be a proper immune organ (Barthelemy et al., 2023), clearly, therein, the parasites are hidden from the systemic immune response. In agreement with previous studies (Claes et al., 2009; McLatchie et al., 2013), in the late stage of the disease (second week post-infection), the presence of the parasite is detected in multiple organs: lungs, spleen, and thymus. In contrast, the determination of bioluminescence in blood samples proved poorly reliable to quantitatively assess parasitemia (e.g., lack of correlation between parasite load and luminescence signal). We could link this phenomenon to an impaired energetic metabolism of the parasites or a low expression of the reporter protein, and not to an unstable integration of the transgene since parasites isolated from mice bloodstream and grown in fresh culture medium recovered maximum LUC activity.
As shown in our study, the estimation of infection burden by measuring the dynamic changes in bioluminescence signal across different regions of the animal body supersedes in sensitivity and resolution the quantification of parasitemia (either by microscopy counting or by luminescence in blood samples). Pretend to define an in vivo “detection limit” for our system, as established for a pleomorphic reporter cell line (McLatchie et al., 2013), seems difficult to address because it will depend on many dynamic variables such as the metabolic status of the pathogen and the pharmacokinetics of D-luciferin at the target organ or tissue, both being influenced by the pathophysiological status of each animal. Instead, we found that the quantitative analysis of segmented images from the upper (head + thorax) and lower (abdomen) regions of the animal normalized to whole body signal provides more accurate information about the progress of the infection. Worth noting, the normalization against the body´s signal contributes to canceling out differences in instrumental settings. As shown in our study, the relative signal from these body regions changes in a sustained and opposite manner (signal ratio decrease for abdomen and increase for head + thorax) that matches the severity of the infection and allows to set an unbiased end-point criterion. On this basis, we propose that the crossing point of the linear regressions of these ratios determines the cutoff value for interrupting animal experimentation. Under our experimental and instrumental conditions, this value was 1.2, and according to this, animals presenting ratios of [head + thorax]/body and abdomen/body higher and lower, respectively, than that value should be excluded from the assay. The application of this rule to our experimental data revealed a significant shortening (3–4 days) of the assay end-point compared to the standard parameters. We noted that the new end-point criterion may also prevent cases of sudden death in animals asymptomatic or with mild signs of disease (e.g., null parasitemia). This phenomenon has been previously reported in the literature (Brun and Kaminsky, 1999; Magez et al., 2002).
Previous studies conducted with different strains of T. brucei spp. and rodents have shown that, independently of infection route and inoculum, the pathogen invades the brain parenchyma within hours or a few days upon primo-infection and before microvascular inflammation is detectable and disruption of BBB happens, or meningoencephalitis is fully established (Wirtz et al., 1999; Frevert et al., 2012; Laperchia et al., 2016). In our model, an intense bioluminescence signal was observed at the animal head, which was suggestive of brain colonization. However, ex vivo imaging and treatment with DAC (a drug unable to cross the BBB) ruled out this hypothesis since no bioluminescence signal was detected in the dissected brain, and mice coursing the late stage of the acute infection were cured upon administration of a single drug dose. In laboratory models of chronic African trypanosomiasis, DAC treatment has been employed for eliminating systemic parasites and studying infection relapse from CNS (Jennings et al., 1977; McLatchie et al., 2013). Further studies with our reporter cell line are needed to verify if the signal observed in the head corresponds to a meningeal infection. Thus, we can conclude that the mouse infection model with the monomorphic T7TetR-PpyRE9H-LUC cell line represents an acute or hematolymphatic model of animal African trypanosomiasis.
As initially expected, T7TetR-PpyRE9H-LUC allowed the Tet-inducible downregulation of an endogenous gene, namely, G6PDH, when stably transfected with the proper RNAi vector. G6PDH is the first enzyme of the pentose phosphate pathway that produces reducing power in the form of NADPH upon oxidation of glucose 6-phosphate into 6-phospho-gluconolactone. NADPH is a key redox cofactor that fuels several biosynthetic reactions (i.e., lipid and nucleotide biosynthesis) and the antioxidant system of the parasite (Comini et al., 2013). The indispensability of G6PDH for the in vitro growth of T. brucei had been previously addressed using the conventional counting method (Gupta et al., 2011) but, so far, not been investigated in vivo for any trypanosomatid. Here we confirmed and showed for the first time that post-transcriptional silencing of G6PDH expression impairs the in vitro and in vivo growth of African trypanosomes, respectively, as revealed by the low bioluminescence levels (single point or cumulative) recorded in parasite cultures or infected animals that were treated with the G6PDH-dsRNAi inducer: Oxytet. The survival time of mice fed with Oxytet (18–20 days) almost doubled that of those receiving plain water (10–11 days). However, the anti-proliferative effect exerted by G6PDH silencing was transitory and upon day 10 post-infection this deleterious phenotype reverted and led to mice death in the subsequent days. This behavior is not surprising, it has been previously reported for in vivo studies using genetically modified T. brucei with Tet-inducible systems (Lecordier et al., 2005; Manta et al., 2013), and has been ascribed to the appearance of Tet-insensitive mutants due to the impossibility to administer mice the corresponding selective pressure for the inducible expression systems (e.g., RNAi or dominant negative) all along the course of the experiment.
In conclusion, at the in vitro level, the use of LUC as a viability reporter conferred the following advantages: it allows a rapid phenotypic screening of a larger number of transfected clones, and drops in bioluminescence signal anticipate those occurring on cell number (counting method), which may shorten the screening time or provide information about the metabolic role of the molecular target. At the in vivo level, this experiment demonstrates that the T7TetR-PpyRE9H-LUC cell line can be used for the validation and study of molecular targets in a non-invasive fashion.
Thanks to intensive monitoring programs, which include early diagnosis and treatment, HAT cases dropped dramatically in the last decades (Gao et al., 2020). Under such a scenario, drugs active against the acute stage of the infection or with prophylactic potential (i.e., to control epidemics in villages or protect travelers visiting these regions) may be needed shortly. In this respect, we turn our attention to a drug repurposing approach based on a clinically approved iron chelator, Dfx. The exploration of chelating agents for the treatment of HAT was originally conducted by Clarkson and collaborators (Clarkson and Brohn, 1976; Clarkson et al., 1981), who tested a combination of the iron chelator salicylhydroxamic acid and glycerol to simultaneously block the aerobic- and anaerobic-dependent energetic metabolism of the pathogen. Almost concurrently, several other chelators were successfully assayed in vitro and in vivo against African trypanosomes (Shapiro et al., 1982). Despite the promising results obtained, further pre-clinical research on chelating agents for HAT was discontinued. The investigation on iron chelators was resumed almost 2 decades later with several studies reporting low μM EC50 (half maximal effective concentration) against the infective stage of T. brucei spp. for Dfx (Breidbach et al., 2002), several commercial iron chelators (Merschjohann and Steverding, 2006), and the antimalarial drugs ferroquine (Pomel et al., 2013), deferiprone and a chloroquine-deferiprone conjugate (Gehrke et al., 2013). For the related pathogen Trypanosoma cruzi, it has been shown that Dfx has a trypanostatic effect in vivo (Arantes et al., 2011) and potentiates the effect of the nitroheterocyle drug benznidazole when applied in a combined therapy (Francisco et al., 2008). Iron chelators display a cytostatic effect and the mode of action of Dfx (or desferal) involves the depletion of cellular iron by chelating the non-protein bound form of the transition metal (also called “free-iron”) and is safely used to treat acute iron poisoning or, in a long-term fashion, chronic iron overload caused by repeated blood transfusions (e.g., in anemia or thalassemia; Aydinok, 2023).
Based on this evidence, here we investigated the therapeutic potential of Dfx, administered alone or in combination with half the curative dose of the cytotoxic drug Nfx, in our in vivo model of acute African trypanosomiasis. The rationale behind the combination of cytostatic (Dfx) and cytotoxic (Nfx) drugs was to potentiate the trypanocidal activity of the last while reducing its side effects. According to the data obtained, Dfx was not curative at the dose and regimen tested but delayed parasite proliferation and slightly extended animal survival. When combined with Nfx (a drug that is used in combination with Eflornithine to treat the chronic stage of HAT; (Priotto et al., 2009), the animal life span was further extended but the combination lacked the curative effect observed for the nitroheterocycle drug administered at its full dose. The quantitative analysis of in vivo bioluminescence provided a clearer picture of the drug´s therapeutic effect during the first 2 weeks of infection. Body signal, which is synonymous with parasite burden, raised almost exponentially in untreated animals while it remained at very low levels in mice receiving Nfx alone or combined with Dfx. In animals receiving Dfx alone, bioluminescence also showed a time-dependent increase but of a minor magnitude than that recorded for the untreated group.
The in vivo inhibition of parasite growth exerted by a short treatment (4 days) with this drug combination is a promising result that prompts to explore and optimize the dose/regimen and/or other drug combinations with iron chelators. In this regard, current and future medicinal chemistry and target validation initiatives for HAT should strongly consider the localization of T. brucei in fat depots. Highly hydrophilic drugs, such as Dfx (logP −2.2) or Oxytet (logP −1.1, the drug used to induce G6PDH RNAi in our study), will have a low bioavailability in fatty tissues, which may account for the relapse or lower efficacy observed in the RNAi and therapeutic assays here presented. It should also be noted that recent studies demonstrated that (monomorphic and pleomorphic) trypanosomes residing in this tissue display metabolic adaptations and a slow growth rate, which altogether may contribute to treatment failure (Trindade et al., 2016; Trindade et al., 2022).
In conclusion, the bioluminescent in vivo model of Acute African trypanosomiasis fully met our initial expectancies as it proved useful for monitoring parasite burden, understanding the pathophysiology of the acute infection, validating a molecular target, testing the drug’s efficacy, and refining animal experimentation. From a more translational perspective and to accelerate the bench-to-bedside drug discovery for HAT, the results of the therapeutic efficacy experiment here presented should encourage researchers to conduct preclinical studies based on drug repositioning and combinations.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by Animal Use and Ethic Committee (CEUA) of the Institut Pasteur de Montevideo. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
DB: Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing–original draft, Writing–review and editing. CO: Investigation, Methodology, Writing–review and editing. ED: Investigation, Methodology, Writing–review and editing. MC: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors belong to the Uruguayan National System of Researchers from ANII (Agencia Nacional de Investigación e Innovación, Uruguay) and thank the support of PEDECIBA (Programa de Desarrollo de las Ciencias Básicas) from Ministerio de Educación y Cultura, Uruguay, and FOCEM (Fondo para la Convergencia Estructural del Mercosur, COF 03/11).
Acknowledgments
DB and ED acknowledge Institut Pasteur de Montevideo and Comisión Académica de Posgrado (Universidad de la República, Uruguay), respectively, for postdoctoral funding. Paul A. M. Michels (Centre for Immunity, Infection, and Evolution, and Centre for Translational and Chemical Biology, The University of Edinburgh, United Kingdom) is acknowledged for providing the plasmid phD677G6PDHARNi. Hugo Cerecetto and Elena Aguilera (Grupo de Química Orgánica Medicinal, Instituto de Química Biológica, Facultad de Ciencias, Universidad de la República, Montevideo, Uruguay) are acknowledged for the gift of purified Nifurtimox. Staff from the Institut Pasteur de Montevideo, Uruguay, is gratefully acknowledged for technical support in the use/maintenance of the Bruker Xtreme II equipment (MSc. Ana Paula Arévalo from the Laboratory Animals Biotechnology, and Luis Inchausti from the Maintenance and Service Unit), animal manipulation (Natalia Lago from the former Neuroinflammation and Gene Therapy Laboratory, and Tatiana Basika from the Cellular Biology Unit) and scripts for image analysis (Federico Lecumberry, Alfredo Solari, and Tabaré De los Campos from the former Signal Processing Laboratory).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchbi.2024.1433511/full#supplementary-material
Abbreviations
BBB, Blood-brain barrier; Dfx, Deferoxamine; DAC, Diminazene aceturate; G6PDH, Glucose-6-phosphate dehydrogenase; HAT, Human African trypanosomiasis; ip, Intraperitoneal; LUC, Luciferase; MARS, Multimodal Animal Rotation System; Nfx, Nifurtimox; Oxytet, Oxytetracycline; PpyLUC, Photinus pyralis luciferase; PpyRE9H or RE9H-LUC, Photinus pyralis red-shifted luciferase; ROI, Region Of Interest; RLU, Relative Luminescence Units; RrLUC, Renilla reniformis luciferase; Tet, Tetracycline; T7TetR-PpyRE9H-LUC, Trypanosoma brucei brucei, bloodstream form, strain 449, cell line 514-1313 expressing RE9H-LUC; T7TetR-PpyRE9H-LUC RNAi-G6PDH, T7TetR-PpyRE9H-LUC stably transfected with phD677G6PDHARNi (RNAi vector for Glucose-6-phosphate dehydrogenase from T. b. brucei).
References
Abdulla, M. H., O’Brien, T., Mackey, Z. B., Sajid, M., Grab, D. J., and McKerrow, J. H. (2008). RNA interference of Trypanosoma brucei cathepsin B and L affects disease progression in a mouse model. PLoS Negl. Trop. Dis. 2 (9), e298. doi:10.1371/journal.pntd.0000298
Aksoy, S., Buscher, P., Lehane, M., Solano, P., and Van Den Abbeele, J. (2017). Human African trypanosomiasis control: achievements and challenges. PLoS Negl. Trop. Dis. 11 (4), e0005454. doi:10.1371/journal.pntd.0005454
Alibu, V. P., Storm, L., Haile, S., Clayton, C., and Horn, D. (2005). A doubly inducible system for RNA interference and rapid RNAi plasmid construction in Trypanosoma brucei. Mol. Biochem. Parasitol. 139 (1), 75–82. doi:10.1016/j.molbiopara.2004.10.002
Alsford, S., Turner, D. J., Obado, S. O., Sanchez-Flores, A., Glover, L., Berriman, M., et al. (2011). High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21 (6), 915–924. doi:10.1101/gr.115089.110
American Society of Health-System Pharmacists (2023). “Deferoxamine mesylate” monograph. Available at: https://www.drugs.com/monograph/deferoxamine-mesylate.html (Accessed December 26, 2023).
Antoine-Moussiaux, N., Magez, S., and Desmecht, D. (2008). Contributions of experimental mouse models to the understanding of African trypanosomiasis. Trends Parasitol. 24 (9), 411–418. doi:10.1016/j.pt.2008.05.010
Arantes, J. M., Francisco, A. F., de Abreu Vieira, P. M., Silva, M., Araújo, M. S. S., de Carvalho, A. T., et al. (2011). Trypanosoma cruzi: desferrioxamine decreases mortality and parasitemia in infected mice through a trypanostatic effect. Exp. Parasitol. 128 (4), 401–408. doi:10.1016/j.exppara.2011.05.011
Aydinok, Y. (2023). Combination chelation therapy. Ann. N. Y. Acad. Sci. 1529 (1), 33–41. doi:10.1111/nyas.15052
Badr, C. E., and Tannous, B. A. (2011). Bioluminescence imaging: progress and applications. Trends Biotechnol. 29 (12), 624–633. doi:10.1016/j.tibtech.2011.06.010
Ballesteros-Casallas, A., Quiroga, C., Ortíz, C., Benítez, D., Denis, P. A., Figueroa, D., et al. (2023). Mode of action of p-quinone derivatives with trypanocidal activity studied by experimental and in silico models. Eur. J. Med. Chem. 246, 114926. doi:10.1016/j.ejmech.2022.114926
Barreiro-Costa, O., Quiroga Lozano, C., Muñoz, E., Rojas-Silva, P., Medeiros, A., Comini, M. A., et al. (2022). Evaluation of the anti-Leishmania mexicana and Trypanosoma brucei activity, and mode of action of 4,4′-(arylmethylene)bis(3-methyl-1-phenyl-1H-pyrazol-5-ol). Biomedicines 10 (8), 1913. doi:10.3390/biomedicines10081913
Barthelemy, J., Bogard, G., and Wolowczuk, I. (2023). Beyond energy balance regulation: the underestimated role of adipose tissues in host defense against pathogens. Front. Immunol. 14, 1083191. doi:10.3389/fimmu.2023.1083191
Benítez, D., Dibello, E., Bonilla, M., and Comini, M. A. (2020). A simple, robust, and affordable bioluminescent assay for drug discovery against infective African trypanosomes. Drug Dev. Res. 83 (2), 253–263. doi:10.1002/ddr.21634
Blum, J., Schmid, C., and Burri, C. (2006). Clinical aspects of 2541 patients with second stage human African trypanosomiasis. Acta Trop. 97 (1), 55–64. doi:10.1016/j.actatropica.2005.08.001
Bosque, M. A., Domingo, J. L., and Corbella, J. (1995). Assessment of the developmental toxicity of deferoxamine in mice. Arch. Toxicol. 69 (7), 467–471. doi:10.1007/s002040050200
Branchini, B. R., Ablamsky, D. M., Davis, A. L., Southworth, T. L., Butler, B., Fan, F., et al. (2010). Red-emitting luciferases for bioluminescence reporter and imaging applications. Anal. Biochem. 396 (2), 290–297. doi:10.1016/j.ab.2009.09.009
Breidbach, T., Scory, S., Krauth-Siegel, R. L., and Steverding, D. (2002). Growth inhibition of bloodstream forms of Trypanosoma brucei by the iron chelator deferoxamine. Int. J. Parasitol. 32 (4), 473–479. doi:10.1016/s0020-7519(01)00310-1
Brun, R., and Kaminsky, R. (1999). “Animal models of acute (first-stage) sleeping sickness,” in Handbook of animal models of infection. Editors O. Zak, and M. Sande (New York: Academic Press), 789–793.
Burrell-Saward, H., Harris, A. J., de LaFlor, R., Sallam, H., Alavijeh, M. S., Ward, T. H., et al. (2017). Dose-dependent effect and pharmacokinetics of fexinidazole and its metabolites in a mouse model of human African trypanosomiasis. Int. J. Antimicrob. Agents 50 (2), 203–209. doi:10.1016/j.ijantimicag.2017.01.038
Burrell-Saward, H., Rodgers, J., Bradley, B., Croft, S. L., and Ward, T. H. (2015). A sensitive and reproducible in vivo imaging mouse model for evaluation of drugs against late-stage human African trypanosomiasis. J. Antimicrob. Chemother. 70 (2), 510–517. doi:10.1093/jac/dku393
Claes, F., Vodnala, S. K., Van Reet, N., Boucher, N., Lunden-Miguel, H., Baltz, T., et al. (2009). Bioluminescent imaging of Trypanosoma brucei shows preferential testis dissemination which may hamper drug efficacy in sleeping sickness. PLoS Negl. Trop. Dis. 3 (7), e486. doi:10.1371/journal.pntd.0000486
Clarkson, A. B., and Brohn, F. H. (1976). Trypanosomiasis: an approach to chemotherapy by the inhibition of carbohydrate catabolism. Science 194 (4261), 204–206. doi:10.1126/science.986688
Clarkson, A. B., Grady, R. W., Grossman, S. A., McCallum, R. J., and Brohn, F. H. (1981). Trypanosoma brucei brucei: a systematic screening for alternatives to the salicylhydroxamic acid-glycerol combination. Mol. Biochem. Parasitol. 3 (5), 271–291. doi:10.1016/0166-6851(81)90002-5
Comini, M. A., Guerrero, S. A., Haile, S., Menge, U., Lünsdorf, H., and Flohé, L. (2004). Validation of Trypanosoma brucei trypanothione synthetase as drug target. Free Radic. Biol. Med. 36 (10), 1289–1302. doi:10.1016/j.freeradbiomed.2004.02.008
Comini, M. A., Ortíz, C., and Cazzulo, J. J. (2013). “Drug targets in trypanosomal and leishmanial pentose phosphate pathway,” in Trypanosomatid diseases: molecular routes to drug discovery (drug discovery in infectious diseases). Editors T. Jäger, O. Koch, L. Flohé, and P. M. Selzer (Oxford, UK: Wiley-Blackwell), 297–313.
Cordeiro, A. T., Thiemann, O. H., and Michels, P. A. M. (2009). Inhibition of Trypanosoma brucei glucose-6-phosphate dehydrogenase by human steroids and their effects on the viability of cultured parasites. Bioorg. Med. Chem. 17 (6), 2483–2489. doi:10.1016/j.bmc.2009.01.068
Currier, R. B., Ulrich, K., Leroux, A. E., Dirdjaja, N., Deambrosi, M., Bonilla, M., et al. (2019). An essential thioredoxin-type protein of Trypanosoma brucei acts as redox-regulated mitochondrial chaperone. PLoS Pathog. 15 (9), e1008065. doi:10.1371/journal.ppat.1008065
Dibello, E., Comini, M. A., and Benítez, D. (2022). “A simple, robust and affordable bioluminescent assay for drug screening against infective African trypanosomes,” in Bioluminescence: methods and protocols, 4th edition, series methods in molecular biology. Editor S.-B. Kim (New York: Humana Press), 253–263. doi:10.1007/978-1-0716-2453-1_11
Duleu, S., Vincendeau, P., Courtois, P., Semballa, S., Lagroye, I., Daulouede, S., et al. (2004). Mouse strain susceptibility to trypanosome infection: an arginase-dependent effect. J. Immunol. 172 (10), 6298–6303. doi:10.4049/jimmunol.172.10.6298
Francisco, A. F., de Abreu Vieira, P. M., Arantes, J. M., Pedrosa, M. L., Martins, H. R., Silva, M., et al. (2008). Trypanosoma cruzi: effect of benznidazole therapy combined with the iron chelator desferrioxamine in infected mice. Exp. Parasitol. 120 (4), 314–319. doi:10.1016/j.exppara.2008.08.002
Franco, J., Scarone, L., and Comini, M. A. (2018). “Drugs and drug resistance in african and American trypanosomiasis,” in Annual reports in medicinal chemistry: neglected diseases: extensive space for modern drug discovery. Editor M. Botta (Academic Press, Elsevier Inc), 97–133.
Frevert, U., Movila, A., Nikolskaia, O. V., Raper, J., Mackey, Z. B., Abdulla, M., et al. (2012). Early invasion of brain parenchyma by african trypanosomes. PLoS ONE 7 (8), e43913. doi:10.1371/journal.pone.0043913
Gao, J. M., Qian, Z. Y., Hide, G., Lai, D. H., Lun, Z. R., and Wu, Z. D. (2020). Human African trypanosomiasis: the current situation in endemic regions and the risks for non-endemic regions from imported cases. Parasitology 147 (9), 922–931. doi:10.1017/S0031182020000645
Gehrke, S. S., Pinto, E. G., Steverding, D., Pleban, K., Tempone, A. G., Hider, R. C., et al. (2013). Conjugation to 4-aminoquinoline improves the anti-trypanosomal activity of Deferiprone-type iron chelators. Bioorg. Med. Chem. 21 (3), 805–813. doi:10.1016/j.bmc.2012.11.009
Gichuki, C., and Brun, R. (1999). “Animal models of CNS (second stage) sleeping sickness,” in Handbook of animal models of infection. Editors O. Zak, and M. Sande (New York: Academic Press), 795–800.
Giroud, C., Ottones, F., Coustou, V., Dacheux, D., Biteau, N., Miezan, B., et al. (2009). Murine models for Trypanosoma brucei gambiense disease progression - from silent to chronic infections and early brain tropism. PLoS Negl. Trop. Dis. 3 (9), e509. doi:10.1371/journal.pntd.0000509
Gupta, S., Igoillo-Esteve, M., Michels, P. A., and Cordeiro, A. T. (2011). Glucose-6-phosphate dehydrogenase of trypanosomatids: characterization, target validation, and drug discovery. Mol. Biol. Int. 2011, 135701–135710. doi:10.4061/2011/135701
Hiller, C., Nissen, A., Benítez, D., Comini, M. A., and Krauth-Siegel, R. L. (2014). Cytosolic peroxidases protect the lysosome of bloodstream African trypanosomes from iron-mediated membrane damage. PLoS Pathog. 10 (4), e1004075. doi:10.1371/journal.ppat.1004075
Hirumi, H., and Hirumi, K. (1989). Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75 (6), 985–989. doi:10.2307/3282883
Hug, M., Carruthers, V. B., Hartmann, C., Sherman, D. S., Cross, G. A. M., and Clayton, C. (1993). A possible role for the 3’-untranslated region in developmental regulation in Trypanosoma brucei. Mol. Biochem. Parasitol. 61 (1), 87–95. doi:10.1016/0166-6851(93)90161-p
Ihnatenko, I., Müller, M. J., Orban, O. C. F., Lindhof, J. C., Benítez, D., Ortíz, C., et al. (2023). The indole motif is essential for the antitrypanosomal activity of N5-substituted paullones. PLoS One 18 (11), e0292946. doi:10.1371/journal.pone.0292946
Jennings, F. W., Whitelaw, D. D., and Urquhart, G. M. (1977). The relationship between duration of infection with Trypanosoma brucei in mice and the efficacy of chemotherapy. Parasitology 75 (2), 143–153. doi:10.1017/s0031182000062284
Laperchia, C., Palomba, M., Seke Etet, P. F., Rodgers, J., Bradley, B., Montague, P., et al. (2016). Trypanosoma brucei invasion and T-cell infiltration of the brain parenchyma in experimental sleeping sickness: timing and correlation with functional changes. PLoS Negl. Trop. Dis. 10 (12), e0005242. doi:10.1371/journal.pntd.0005242
Lecordier, L., Walgraffe, D., Devaux, S., Poelvoorde, P., Pays, E., and Vanhamme, L. (2005). Trypanosoma brucei RNA interference in the mammalian host. Mol. Biochem. Parasitol. 140 (1), 127–131. doi:10.1016/j.molbiopara.2004.12.009
Lewis, M. D., Fortes Francisco, A., Taylor, M. C., Burrell-Saward, H., Mclatchie, A. P., Miles, M. A., et al. (2014). Bioluminescence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cell. Microbiol. 16 (9), 1285–1300. doi:10.1111/cmi.12297
Lewis, M. D., Francisco, A. F., Taylor, M. C., and Kelly, J. M. (2015). A new experimental model for assessing drug efficacy against Trypanosoma cruzi infection based on highly sensitive in vivo imaging. SLAS Discov. 20 (1), 36–43. doi:10.1177/1087057114552623
Lindhof, J. C., Ihnatenko, I., Müller, M. J., Orban, O. C. F., Ortíz, C., Benítez, D., et al. (2023). Discovery of antitrypanosomal indolylacetamides by a deconstruction-optimization strategy applied to paullones. ChemMedChem 18 (10), e202300036. doi:10.1002/cmdc.202300036
Loureiro, I., Faria, J., Clayton, C., Macedo-Ribeiro, S., Santarém, N., Roy, N., et al. (2015). Ribose 5-phosphate isomerase B knockdown compromises Trypanosoma brucei bloodstream form infectivity. PLoS Negl. Trop. Dis. 9 (1), e3430. doi:10.1371/journal.pntd.0003430
Magez, S., Stijlemans, B., Caljon, G., Eugster, H. P., and De Baetselier, P. (2002). Control of experimental Trypanosoma brucei infections occurs independently of lymphotoxin-alpha induction. Infect. Immun. 70 (3), 1342–1351. doi:10.1128/IAI.70.3.1342-1351.2002
Manta, B., Pavan, C., Sturlese, M., Medeiros, A., Crispo, M., Berndt, C., et al. (2013). Iron-sulfur cluster binding by mitochondrial monothiol glutaredoxin-1 of Trypanosoma brucei: molecular basis of iron-sulfur cluster coordination and relevance for parasite infectivity. Antioxidants Redox Signal. 19 (7), 665–682. doi:10.1089/ars.2012.4859
McLatchie, A. P., Burrell-Saward, H., Myburgh, E., Lewis, M. D., Ward, T. H., Mottram, J. C., et al. (2013). Highly sensitive in vivo imaging of Trypanosoma brucei expressing “Red-Shifted” luciferase. PLoS Negl. Trop. Dis. 7, e2571. doi:10.1371/journal.pntd.0002571
Merschjohann, K., and Steverding, D. (2006). In vitro growth inhibition of bloodstream forms of Trypanosoma brucei and Trypanosoma congolense by iron chelators. Kinetoplastid Biol. Dis. 5, 3. doi:10.1186/1475-9292-5-3
Mesa, J. M., Comini, M. A., Dibello, E., and Gamenara, D. (2022). Organocatalytic synthesis and anti-trypanosomal activity evaluation of L-pentofuranose-mimetic iminosugars. Eur. J. Org. Chem. 2022 (34), e202200636. doi:10.1002/ejoc.202200636
Mesu, V. K. B. K., Kalonji, W. M., Bardonneau, C., Mordt, O. V., Blesson, S., Simon, F., et al. (2018). Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: a pivotal multicentre, randomised, non-inferiority trial. Lancet 391 (10116), 144–154. doi:10.1016/S0140-6736(17)32758-7
Miller, S. J., and Morgan, B. W. (2014). “Deferoxamine,” in Encyclopedia of toxicology (third edition). Editor P. Wexler (Academic Press), 1154–1156.
Musunda, B., Benítez, D., Dirdjaja, N., Comini, M. A., and Krauth-Siegel, R. L. (2015). Glutaredoxin-deficiency confers bloodstream Trypanosoma brucei with improved thermotolerance. Mol. Biochem. Parasitol. 204 (2), 93–105. doi:10.1016/j.molbiopara.2016.02.001
Mutomba, M. C., Li, F., Gottesdiener, K. M., and Wang, C. C. (1999). A Trypanosoma brucei bloodstream form mutant deficient in ornithine decarboxylase can protect against wild-type infection in mice. Exp. Parasitol. 91 (2), 176–184. doi:10.1006/expr.1998.4363
Myburgh, E., Coles, J. A., Ritchie, R., Kennedy, P. G. E., McLatchie, A. P., Rodgers, J., et al. (2013). In vivo imaging of trypanosome-brain interactions and development of a rapid screening test for drugs against CNS stage trypanosomiasis. PLoS Negl. Trop. Dis. 7 (8), e2384. doi:10.1371/journal.pntd.0002384
Pazos, M., Dibello, E., Mesa, J. M., Sames, D., Comini, M. A., Seoane, G., et al. (2022). Iboga inspired N-Indolylethyl-Substituted isoquinuclidines as a bioactive scaffold: chemoenzymatic synthesis and characterization as GDNF releasers and antitrypanosoma agents. Molecules 27 (3), 829. doi:10.3390/molecules27030829
Pomel, S., Biot, C., Bories, C., and Loiseau, P. M. (2013). Antiprotozoal activity of ferroquine. Parasitol. Res. 112 (2), 665–669. doi:10.1007/s00436-012-3183-4
Price, H. P., Güther, M. L., Ferguson, M. A., and Smith, D. F. (2010). Myristoyl-CoA: protein N-myristoyltransferase depletion in trypanosomes causes avirulence and endocytic defects. Mol. Biochem. Parasitol. 169 (1), 55–58. doi:10.1016/j.molbiopara.2009.09.006
Priotto, G., Kasparian, S., Mutombo, W., Ngouama, D., Ghorashian, S., Arnold, U., et al. (2009). Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet 374 (9683), 56–64. doi:10.1016/S0140-6736(09)61117-X
Promega (2005). Luciferase reporters: less is more. pGL4 vectors: a new generation of luciferase reporter vectors. Available at: https://www.promega.com/-/media/files/resources/promega-notes/89/pgl4-vectors-a-new-generation-of-luciferase-reporter-vectors.pdf?rev=b8d3f2a0978a4bfa8b54a8ae33ca3ce4&sc_lang=en.
Quijada, L., Guerra-Giraldez, C., Drozdz, M., Hartmann, C., Irmer, H., and Ben-Dov, C. (2002). Expression of the human RNA-binding protein HuR in Trypanosoma brucei increases the abundance of mRNAs containing AU-rich regulatory elements. Nucleic Acids Res. 30 (20), 4414–4424. doi:10.1093/nar/gkf577
Rivas, F., Del Mármol, C., Scalese, G., Pérez-Díaz, L., Machado, I., Blacque, O., et al. (2022). New multifunctional Ru(II) organometallic compounds show activity against Trypanosoma brucei and Leishmania infantum. J. Inorg. Biochem. 237, 112016. doi:10.1016/j.jinorgbio.2022.112016
Schichler, D., Konle, A., Spath, E. M., Riegler, S., Klein, A., Seleznev, A., et al. (2023). Characterisation of TbSmee1 suggests endocytosis allows surface-bound cargo to enter the trypanosome flagellar pocket. J. Cell Sci. 136 (20), jcs261548. doi:10.1242/jcs.261548
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9 (7), 676–682. doi:10.1038/nmeth.2019
Shanmugasundram, A., Starns, D., Böhme, U., Amos, B., Wilkinson, P. A., Harb, O. S., et al. (2023). TriTrypDB: an integrated functional genomics resource for kinetoplastida. PLoS Negl. Trop. Dis. 17 (1), e0011058. doi:10.1371/journal.pntd.0011058
Shapiro, A., Nathan, H. C., Hutner, S. H., Garofalo, J., McLaughlin, S. D., Rescigno, D., et al. (1982). In vivo and in vitro activity by diverse chelators against Trypanosoma brucei brucei. J. Protozoology 29 (1), 85–90. doi:10.1111/j.1550-7408.1982.tb02885.x
Shaw, A. P. M., Cecchi, G., Wint, G. R. W., Mattioli, R. C., and Robinson, T. P. (2014). Mapping the economic benefits to livestock keepers from intervening against bovine trypanosomosis in Eastern Africa. Prev. Veterinary Med. 113 (2), 197–210. doi:10.1016/j.prevetmed.2013.10.024
Silvester, E., McWilliam, K. R., and Matthews, K. R. (2017). The cytological events and molecular control of life cycle development of Trypanosoma brucei in the mammalian bloodstream. Pathogens 6 (3), 29. doi:10.3390/pathogens6030029
Sokolova, A. Y., Wyllie, S., Patterson, S., Oza, S. L., Read, K. D., and Fairlamb, A. H. (2010). Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 54 (7), 2893–2900. doi:10.1128/AAC.00332-10
Sommer, J. M., Cheng, Q. L., Keller, G. A., and Wang, C. C. (1992). In vivo import of firefly luciferase into the glycosomes of Trypanosoma brucei and mutational analysis of the C-terminal targeting signal. Mol. Biol. Cell 3 (7), 749–759. doi:10.1091/mbc.3.7.749
Stijlemans, B., Caljon, G., Van Den Abbeele, J., Van Ginderachter, J. A., Magez, S., and De Trez, C. (2016). Immune evasion strategies of Trypanosoma brucei within the mammalian host: progression to pathogenicity. Front. Immunol. 7, 233. doi:10.3389/fimmu.2016.00233
Suff, N., and Waddington, S. N. (2017). The power of bioluminescence imaging in understanding host-pathogen interactions. Methods 127, 69–78. doi:10.1016/j.ymeth.2017.07.001
Trindade, S., De Niz, M., Costa-Sequeira, M., Bizarra-Rebelo, T., Bento, F., Dejung, M., et al. (2022). Slow growing behavior in African trypanosomes during adipose tissue colonization. Nat. Commun. 13 (1), 7548. doi:10.1038/s41467-022-34622-w
Trindade, S., Rijo-Ferreira, F., Carvalho, T., Pinto-Neves, D., Guegan, F., Aresta-Branco, F., et al. (2016). Trypanosoma brucei parasites occupy and functionally adapt to the adipose tissue in mice. Cell Host Microbe 19 (6), 837–848. doi:10.1016/j.chom.2016.05.002
Van Reet, N., Van de Vyver, H., Pyana, P. P., Van der Linden, A. M., and Büscher, P. (2014). A panel of trypanosoma brucei strains tagged with blue and red-shifted luciferases for bioluminescent imaging in murine infection models bioluminescent imaging in murine infection models. PLoS Negl. Trop. Dis. 8 (8), e3054. doi:10.1371/journal.pntd.0003054
Wirtz, E., and Clayton, C. (1995). Inducible gene expression in trypanosomes mediated by a prokaryotic repressor. Science 268 (5214), 1179–1183. doi:10.1126/science.7761835
Wirtz, E., Leal, S., Ochatt, C., and Cross, G. A. M. (1999). A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99 (1), 89–101. doi:10.1016/s0166-6851(99)00002-x
World Health Organization (2019). The tripartite zoonoses guide (TZG). Available at: https://www.who.int/initiatives/tripartite-zoonosis-guide (Accessed February 12, 2024).
World Health Organization (2022). Collaborating for better global health: new tripartite operational tools. Available at: https://www.who.int/news/item/21-09-2022-collaborating-for-better-global-health--new-tripartite-operational-tools (Accessed February 12, 2024).
Wyllie, S., Oza, S. L., Patterson, S., Spinks, D., Thompson, S., and Fairlamb, A. H. (2009). Dissecting the essentiality of the bifunctional trypanothione synthetase-amidase in Trypanosoma brucei using chemical and genetic methods. Mol. Microbiol. 74 (3), 529–540. doi:10.1111/j.1365-2958.2009.06761.x
Keywords: bioluminescence, drug efficacy, genetic validation, glucose 6-phosphate dehydrogenase, dsRNAi, red-shifted luciferase, Trypanosoma brucei, 3Rs principle
Citation: Benítez D, Ortíz C, Dibello E and Comini MA (2024) Expanding the applications of a bioluminescent mouse infection model of acute African trypanosomiasis. Front. Chem. Biol 3:1433511. doi: 10.3389/fchbi.2024.1433511
Received: 16 May 2024; Accepted: 19 July 2024;
Published: 13 August 2024.
Edited by:
Sung Bae Kim, National Institute of Advanced Industrial Science and Technology (AIST), JapanReviewed by:
Brian C. Schaefer, Uniformed Services University of the Health Sciences, United StatesAlicia Ponte-Sucre, Central University of Venezuela, Venezuela
Copyright © 2024 Benítez, Ortíz, Dibello and Comini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo A. Comini, bWNvbWluaUBwYXN0ZXVyLmVkdS51eQ==; Diego Benítez, ZGJlbml0ZXpAcGFzdGV1ci5lZHUudXk=
†Present address: Cecilia Ortíz, Laboratorio de Biotecnología, Universidad ORT, Montevideo, Uruguay.