 Hui Xu
Hui Xu Jessica R. Kenneson
Jessica R. Kenneson Douglas C. Goodwin
Douglas C. Goodwin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. Biol. , 10 July 2024
Sec. Molecular Sciences
Volume 3 - 2024 | https://doi.org/10.3389/fchbi.2024.1431412
This article is part of the Research Topic Structure-function relationship of enzymes through a chemical lens View all 6 articles
Many novel structural features impart a robust catalase activity to KatG that is absent from all other members of its superfamily. The conformationally dynamic “Arg switch” and oxidizable “proximal Trp” have both figured prominently in investigations of KatG structure and mechanism, but the full scope of their contributions to catalysis remains unclear. The switch (R418) appears to regulate active-site intramolecular electron transfer. The proximal Trp (W321) is a conspicuous site of radical formation, but W321•+ does not appear to participate directly in the KatG catalase cycle. To explore the extent to which these residues may cooperate in KatG’s catalase activity, we generated R418N and W321F/R418N KatG and compared their catalytic and spectroscopic properties to wt KatG. R418N KatG showed pH-independent susceptibility to H2O2-based inactivation, whereas wt KatG only showed this response under conditions where the Arg switch would be oriented away from the active site (i.e., low pH). Peroxidatic electron donors (PxEDs) prevented inactivation of wt and R418N KatG regardless of pH; however, protection of R418N KatG activity by this mechanism produced at least ten-fold greater extents of PxED oxidation. Elimination of the proximal Trp in addition to the Arg switch (i.e., W321F/R418N KatG) resulted in a near complete inability to sustain H2O2 degradation. Remarkably, W321F KatG showed resistance to H2O2-dependent inactivation indistinguishable from wt at pH 7 (i.e., when the Arg switch is oriented toward the active site) but sensitivity to H2O2-dependent inactivation indistinguishable from W321F/R418N KatG at pH 5 (i.e., when the Arg switch is oriented away from the active site). These data suggest loss of the Arg switch (either by mutagenesis or conformationally due to environmental pH) results in a KatG that is substantially compromised in the sustained degradation of H2O2. This can be overcome provided that KatG retains its ability to utilize the proximal Trp as a site of protein-based oxidation and has a PxED available to repair protein oxidation events. However, if both the Arg switch and the proximal Trp are absent, rapid H2O2-dependent inactivation is observed, and PxEDs are unable to effectively intervene to preserve KatG’s catalase activity.
Catalase-peroxidase (KatG) is a heme-dependent enzyme expressed in bacteria and lower-eukaryotes. Although KatG is broadly distributed among these organisms (Zámocký et al., 2010), it is prominently represented among pathogens (Krewall et al., 2020), ranging from the notorious agricultural culprit of rice blast disease, Magnaporthe grisea (Tanabe et al., 2011; Zámocký et al., 2012), to the prolific intracellular human pathogens Mycobacterium tuberculosis and Yersinia pestis (Garcia et al., 1999; Ng et al., 2004; Han et al., 2008; Eum et al., 2010; Corleis et al., 2012). The presence of KatG coincides with the copious production of H2O2 that accompanies host responses to infection. It is not unusual for such responses to be executed in cellular compartments (e.g., the neutrophil phagolysosome) that are also substantially acidic (i.e., ≤ pH 5.0).
Structurally, KatG has the scaffold of an enzyme from the non-animal peroxidase superfamily which also includes cytochrome c peroxidase (CcP) and horseradish peroxidase (HRP) as members. Naturally, KatG displays a classical peroxidase activity in the presence of H2O2 and well-known peroxidatic electron donors (PxEDs). However, in contrast to all other members of the superfamily, KatG also has a robust catalase activity that is comparable to typical (i.e., monofunctional) catalases (Njuma et al., 2014). This novel catalase activity depends on a protein-derived cofactor formed by the autocatalytic covalent linkage of an active-site Trp (W107 by M. tuberculosis KatG numbering), a Tyr (Y229), and a Met (M255) (Yamada et al., 2002; Carpena et al., 2003; Donald et al., 2003; Jakopitsch et al., 2003; Bertrand et al., 2004; Ghiladi et al., 2005a; Ghiladi et al., 2005b). This structure is referred to as the MYW cofactor (Figure 1A). An Arg residue positioned ∼20 Å from the heme center also plays a critical role. Replacement of this residue with an uncharged side chain diminishes catalase efficiency and increases peroxidase efficiency, both by nearly two orders of magnitude (Jakopitsch et al., 2004; Carpena et al., 2005; Zhao et al., 2012; Gasselhuber et al., 2016). This Arg (R418) is called the “arginine switch” because it is conformationally dynamic (Figure 1A); the dominant conformation of the switch is strongly dependent on pH (Carpena et al., 2005; Carpena et al., 2006; Gasselhuber et al., 2016). Together, these two structural features enable the peroxidatic structure of KatG to support a catalase activity which rivals that of typical monofunctional catalases (Switala and Loewen, 2002; Singh et al., 2008; Njuma et al., 2014). Though these features have been under investigation for years, many questions remain regarding the role of each structure in the unique catalytic abilities of KatG.
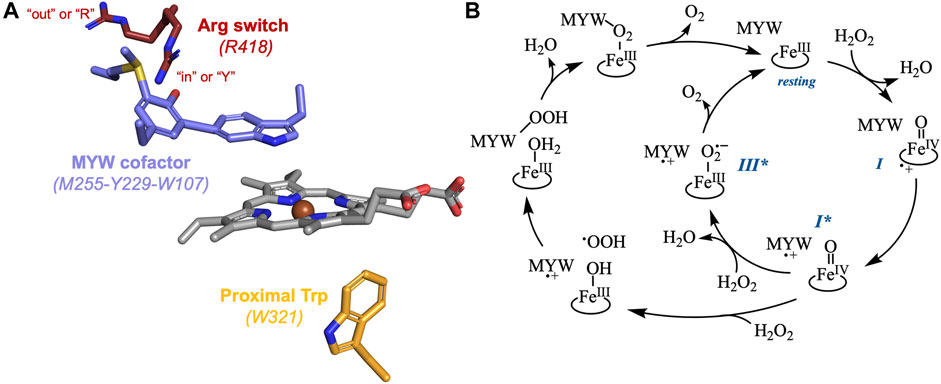
Figure 1. The active site structure (A) and two proposed catalytic cycles for KatG catalase activity (B). The KatG active site shows the relative positions of the arginine switch (maroon), the MYW cofactor (lilac), the heme (gray), and the proximal tryptophan (orange). The conformation of the switch where the guanidinium moiety is oriented toward the MYW phenolate oxygen is shown as “in” or “Y,” and the conformation where the side chain is oriented toward the enzyme’s solvent-accessible surface is shown as “out” or “R.” Both proposed KatG catalase mechanisms proceed through the FeIII (resting), FeIV=O[porphyrin]•+ (compound I [I]), and FeIV=O[MYW]•+ (compound I* [I*]) states. The mechanism in the inner circle posits formation of a FeIII-O2•-[MYW]•+ (compound III* [III*]) intermediate. The mechanism depicted by the outer circle progresses through a FeIII-OH2[MYW-OOH] state.
There is broad agreement that the MYW cofactor is a redox-active participant in a KatG-unique catalase mechanism. Specifically, it has been proposed that following an initial H2O2-dependent oxidation of the FeIII (i.e., resting) enzyme to a FeIV=O[porphyrin]•+ (i.e., compound I) state, an intramolecular electron transfer produces the FeIV=O[MYW]•+ form of the enzyme which has also been referred to as compound I* (Figure 1B) (Jakopitsch et al., 2007; Suarez et al., 2009; Zhao et al., 2010; Loewen et al., 2014; Njuma et al., 2014; Fita et al., 2015; Gasselhuber et al., 2016). The mechanism by which a second equivalent of H2O2 is oxidized to O2 by the FeIV=O[MYW]•+ intermediate, generating the FeIII state with a fully covalent MYW cofactor is still a matter of debate. Two mechanisms have been proposed for this KatG-dependent oxidation of H2O2 (Figure 1B). One hails back to a well-known reaction between peroxidase FeIV=O (i.e., compound II) intermediates and H2O2 to form FeIII-O2•- complexes referred to as compound III or oxyperoxidase (George, 1953; Nakajima and Yamazaki, 1987; Wariishi and Gold, 1989; Wariishi and Gold, 1990). Here, it is proposed that a FeIII-O2•-[MYW]•+ (i.e., compound III*) state enables intramolecular electron transfer to generate the resting enzyme (i.e., FeIII[MYW]) and free O2 (Jakopitsch et al., 2007; Suarez et al., 2009; Zhao et al., 2009; Vlasits et al., 2010). The second proposed mechanism invokes oxidation of H2O2 to OOH•, deprotonation of the indole nitrogen of the MYW•+ radical and formation of an MYW-OOH adduct (Loewen et al., 2014; Fita et al., 2015).
All KatGs, bacterial and fungal, also possess the conformationally dynamic and pH dependent Arg switch (Carpena et al., 2005; Carpena et al., 2006; Gasselhuber et al., 2016). In bacterial enzymes (e.g., R418 of MtKatG), the conformation of the guanidinium moeity is directed “in” toward the MYW phenolic oxygen at pH values above an apparent pKa of 6.5. Conversely, below the apparent pKa, the dominant conformation has the side chain pointed “out” away from the MYW cofactor (Figure 1A) (Carpena et al., 2005; Carpena et al., 2006). Though the Arg switch of fungal KatGs (e.g., R461 of Magneporthe grisea KatG) also exhibits such pH-dependent conformational changes, the Arg switch is more inclined to occupy the “in” conformation unless the pH values are substantially lower (≤ pH 5) (Gasselhuber et al., 2016).
The arginine switch plays a major role in directing KatG intramolecular electron transfers with far-reaching consequences for catalysis. In terms of facilitating catalatic O2 production, the Arg switch has been addressed in both proposed mechanisms for the catalase definitional H2O2 oxidation to O2. For the MYW• •OOH radical-coupling pathway, formation of the MYW-OOH intermediate reestablishes the cofactor’s tyrosine as a phenolate anion coinciding with the reorientation of the Arg switch guanidinium cation to its “in” conformation (Loewen et al., 2014; Fita et al., 2015). Alternatively, the formation of the MYW phenolate anion by FeIII-O2•- reduction of MYW•+ is also facilitated by the switch of the Arg guanidinium cation to its “in” conformation (Zhao et al., 2012; Kruft et al., 2015).
More broadly, a correspondence was first noted between the pH dependence of the Arg switch and the pH profiles for KatG’s peroxidase and catalase activities. The former was maximized at acidic pH (i.e., ≤5.0), conditions where the Arg switch would be oriented away from the active site (Carpena et al., 2006; Moore et al., 2008; Singh et al., 2008). The latter was maximized near pH 6.5 where the switch would roughly equally populate its in and out conformations. Accordingly, one of the hallmarks of variants lacking the switch (e.g., R418A MtKatG) was the diminished catalase and enhanced peroxidase activity mentioned above (Jakopitsch et al., 2004; Carpena et al., 2005; Carpena et al., 2006; Zhao et al., 2012). A more nuanced connection was revealed by calculations which suggested that pH would influence the oxidizability of the MYW cofactor, a phenomenon at least partially dependent on the conformational position of the Arg switch. Protonation of the MYW phenolate oxygen and displacement of the Arg switch to orient away from the active site (both of which would be expected at pH 5) made conversion of FeIV=O[porphyrin]•+ to FeIV=O[MYW]•+ a substantially less favorable possibility (Vidossich et al., 2007). This would be expected to diminish catalase activity and simultaneously enable formation of protein-based radicals at other sites that may support peroxidase activity but would not support catalase activity (Loewen et al., 2014). Such events are considered “off-catalase” intramolecular electron transfers. In this respect, the so-called proximal Trp (W321 in M. tuberculosis KatG) was identified as a prominent alternative site of oxidation (i.e., FeIV=O[W321]•+) (Vidossich et al., 2007). Consistent with this observation, reactions of KatG with peracetic acid (a peroxide that does not support catalase turnover), produce a proximal tryptophanyl radical state (Singh et al., 2007; Colin et al., 2009). Indeed, our own investigations have shown that the proximal Trp is a prominent site for off-catalase KatG protein oxidation (Njuma et al., 2017; Krewall et al., 2020). Here, W321 appears to progressively accumulate in its oxidized radical state as KatG catalase activity diminishes. This suggests that W321 acts as an electron donor to reduce the FeIV=O[porphyrin]•+ state, generating FeIV=O[W321]•+ instead of the catalase-essential FeIV=O[MYW]•+ intermediate. Clearly, the Arg switch plays a critical role in KatG intramolecular electron transfer whether toward or away from productive catalase turnover. Likewise, the proximal Trp is unmistakably a prominent site for off-catalase KatG protein oxidation. It is expected that subsequent intramolecular electron transfers occur, such that the protein-based radical migrates by hole-hopping toward the KatG protein surface via a series of Trp/Tyr residues. The remarkable abundance of Tyr and especially Trp residues in KatG has been noted previously (Fita et al., 2015; Njuma et al., 2017; Krewall et al., 2020). Indeed, the active-site bearing N-terminal domain of M. tuberculosis KatG contains 19 (4.32%) Trp and 16 (3.64%) Tyr.
We have observed that peroxidatic electron donors (PxEDs) dramatically stimulate KatG catalase activity despite being mutually exclusive with H2O2 for completing the peroxidase and catalase catalytic cycles (Ndontsa et al., 2012). Building on this counterintuitive observation, we have proposed that KatG’s peroxidase activity folds into a seamless and integrated whole where PxEDs are part of KatG’s mechanism for sustaining and maximizing its catalase-based H2O2 degradation across a wide pH range (Njuma et al., 2017; Krewall et al., 2020). The proximal Trp as a prominent site of off-catalase protein-based radical formation and the Arg switch as a regulator of intramolecular electron transfer suggests that these two residues may be indispensable to KatG’s integrated peroxidase-supported catalatic H2O2 degradation mechanism. However, this has not been directly examined. To investigate the mechanism and degree to which these residues cooperate to facilitate KatG’s capacity to rapidly degrade H2O2 over a broad range of conditions, we produced KatG variants targeting the Arg switch and proximal tryptophan (i.e., R418N and W321F/R418N) and evaluated their catalytic properties compared to the wild-type enzyme and a previously generated W321F KatG variant.
T4 DNA ligase, Pfu polymerase, and Escherichia coli XL-1 Blue cells were obtained from Agilent (La Jolla, CA). All oligonucleotide primers were purchased from Invitrogen (Carlsbad, CA). All restriction enzymes and Phusion high-fidelity PCR master mix with GC buffer were acquired from New England Biolabs (Beverly, MA). Benzonase nuclease was obtained from Milipore (Burlington, MA). Ampicillin and isopropyl β-D-thioglactopyranoside (IPTG) were purchased from Gold Biotechnology (St. Louis, MO). H2O2 (30%), hemin, sodium dithionite, chlorpromazine (CPZ), N,N,N’N’-tetramethyl-p-phenylenediamine dihydrochloride (TMPD), and L-ascorbic acid were obtained from Sigma (St. Louis, MO). Imidazole and nickel sulfate hexahydrate were purchased from Alfa Aesar (Haverhill, MA). Calcium chloride was acquired from Honeywell (Charlotte, NC). 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS), dibasic sodium phosphate, monobasic sodium phosphate, sodium chloride, and tetracycline hydrochloride were obtained from VWR (Radnor, PA). Sodium acetate trihydrate, Tris HCl, potassium chloride, and magnesium sulfate were acquired from Fisher (Hampton, NH). Magnesium chloride was obtained from Amresco (Dallas, TX). Ni-NTA resin was purchased from G-Biosciences (St. Louis, MO). Macro-Prep High Q resin and Econo-Pac 10DG desalting columns were purchased from Bio-Rad (Hercules, CA). Centrifugal filter units (30 kDa cutoff) were acquired from Pall Laboratory (Port Washington, NY). All buffers and media were prepared using water purified through a Barnstead EASYpure II UV ultrapure water system (18.2 megohms/cm resistivity). Quartz EPR tubes were purchased from Wilmad Lab Glass (Buena Vista, NJ).
Site-directed mutagenesis to produce the R418N and W321F/R418N variants of KatG was performed by the “Round-the-Horn” method (Moore, 2018) applied to the expression construct for wild-type M. tuberculosis KatG. This construct is pMRLB11, a pET23b-derived plasmid bearing the M. tuberculosis katG gene and was obtained from the TB vaccine Testing and Research Materials Contract at Colorado State University. The forward primer designed for the R418N substitution (5′-CACAACGATATGGGTCCCGTTGCGA-3′) included a site for codon replacement (bold) as well as a silent mutation designed to introduce a diagnostic restriction site for PshAI (underlined). The reverse primer (5′-GATCAGCTTGTACCAGGCCTTGGCGAA-3′) required no substitutions. Both primers were modified to include 5′-phosphoryl groups, allowing for blunt-end ligation following PCR. The W321F/R418N double variant was generated using the R418N primers with a previously-generated plasmid for expression of W321F KatG as a template (Njuma et al., 2017). All PCR reactions were carried out using Phusion High-Fidelity polymerase in GC-buffer-containing Master-Mix and 3% DMSO. All PCR products were initially treated with DpnI to degrade the starting template and then ligated using T4 DNA ligase. Ligation products were used to transform E. coli (XL-I Blue) by a standard heat shock protocol. Transformants were selected using ampicillin-containing media. Success of transformation was confirmed by PshAI digestion. Candidates that passed this screening were sent for full DNA sequence analysis (Laragen Sequencing, Culver City, CA).
E. coli (C41 [DE3]) containing the pHPEX3 plasmid (Varnado and Goodwin, 2004) were transformed using DpnI-treated/ligated PCR products containing either the single R418N or the double W321F/R418N codon substitutions. Transformants were then selected on the basis of ampicillin and tetracycline resistance. For expression, cultures were grown in Luria Bertani, Miller broth supplemented with 0.1 mg/mL ampicillin and 0.02 mg/mL tetracycline at 37°C until an OD600 of 0.30–0.45 was reached, at which time target protein expression was induced by the addition of IPTG to a final concentration of 1 mM. At this time, hemin prepared in 0.1 mM KOH was added to a final concentration of 8 μM in each flask. Post-induction cultures were grown for 4 h prior to harvesting by centrifugation at 4,100 × g for 20 min at 4-8°C. The supernatant was discarded and the pellet of harvested cells was frozen and stored at −20°C until purification. For purification, harvested cells were resuspended in 50 mM sodium phosphate, pH 7.0, 200 mM NaCl (5 mL for every L of expression culture). The resuspension buffer was supplemented with phenylmethylsulfonylfluoride (PMSF) (200 μL of 50 mM PMSF for every L of expression culture). The cell suspension was then homogenized using a glass tissue grinder, and the homogenized cell solution was then lysed by 8 sonication cycles (42 s on, 42 s off) using a Branson 250 Sonifier (Danbury, CT) equipped with a standard tip and set to 3.5 duty and constant output. Following sonication, benzonase nuclease (2 μL/L of expression culture volume) was added to the cell lysate, and the mixture was allowed to stir at 4°C for 4 h. The cell lysate was then centrifuged at 10,000 × g for 1 h (with slow deceleration) at 4°C to separate soluble protein from insoluble cell matter. Prior to its use, Ni-NTA was drained of its ethanol storage solution, washed with Barnstead-purified H2O, and then equilibrated with 50 mM Tris, pH 8.0. The supernatant from the centrifuged lysate was collected and then added to Ni-NTA resin in multiple 50 mL conical tubes and allowed to bind on rotator at 4°C for 12 h. Supernatant-incubated resin was collected in a column, and then washed according to the following steps: 1) 50 mM Tris, pH 8, and 2) 50 mM phosphate, pH 7.0, 200 mM NaCl. Target protein elution was monitored during sequential washes using 50 mM phosphate, pH 7, 200 mM NaCl, supplemented with 20, 50, 100, 200, and 500 mM imidazole. As confirmed by SDS-PAGE analyses of imidazole-containing fractions, KatG and its variants typically elute with 100 mM imidazole. Fraction(s) verified by SDS-PAGE to contain KatG were combined and concentrated using centrifugal filters with a 30 kDa cutoff limit. The concentrated protein was then added to Econo-Pac 10DG desalting columns for buffer exchange into 50 mM sodium phosphate, pH 7. After buffer exchange, a diagnostic spectrum was taken of the protein, and the protein was further concentrated in preparation for anion exchange chromatography. Macro-Prep High Q resin was used to purify the protein by anion exchange. KatG was purified from remaining contaminating proteins by elution using a linear gradient between 50 mM phosphate, pH 7, and 50 mM phosphate, pH 7, 500 mM NaCl. Heme content was estimated by evaluating the so-called Reinheitzahl (Rz) value which is the ratio of heme absorption at the Soret band λmax (in this case 407 nm) to that observed for protein absorption at 280 nm. Due to M. tuberculosis KatG’s extraordinarily high Tyr and especially Trp content, the Rz value is not expected to exceed 0.65. After analysis by SDS-PAGE and UV-visible spectroscopy, the fractions containing the highest-purity KatG (as judged by Rz evaluation of heme content) were combined, concentrated, and exchanged into 5 mM phosphate, pH 7.0. This was accomplished using centrifugal filtration (30 kD cutoff) by 3 – 5 repeated cycles of new buffer addition/concentration. The heme content and structural integrity of the purified protein was verified by UV-visible analyses of heme versus protein absorption, heme absorption features, and catalytic activity. The purified proteins were concentrated in 5 mM phosphate, pH 7.0, before being aliquoted and frozen (−80°C) for future investigation. The wt and R418N KatG were concentrated to 50–100 μM, and W321F/R418N KatG was concentrated to 20–30 μM.
Catalase activity was determined either by monitoring H2O2 degradation by UV-visible absorption (ε240 = 39.4 M-1cm-1) (Nelson and Kiesow, 1972) or O2 production by a Clark-type O2-sensitive electrode (Hansatech, Pentney, Norfolk, United Kingdom). Absorption-based catalase assays were carried out with a PC-driven UV-1601 spectrophotometer (Shimadzu, Kyoto, Japan), and assays contained 20 nM KatG (or one of its variants) in 100 mM phosphate, pH 7.0, at 23°C. Catalase assays where O2 production was monitored polarographically were carried out using 5 nM enzyme in 100 mM phosphate, pH 7, at 23°C.
Peroxidase activity was determined by monitoring the oxidation of ABTS to ABTS•+ spectrophotometrically at 417 nm (ε = 34,700 M-1cm-1) (Scott et al., 1993). To evaluate the effect of H2O2 concentration on KatG peroxidase activity, the concentration of ABTS was kept constant at 0.1 mM. Likewise, to determine the effect of ABTS concentration on the peroxidase activity, the concentration of H2O2 was held constant at 1.0 mM. All peroxidase activity was evaluated using 20 nM KatG in 50 mM acetate, pH 5, at ambient temperature. When evaluated, TMPD oxidation to TMPD•+ was monitored spectrophotometrically at 610 nm (ε = 11, 600 M-1cm-1) (Michaelis et al., 1939). Likewise, CPZ oxidation to CPZ•+ was measured at 525 nm (ε = 12,100 M-1cm-1). (Kelder et al., 1994). In all cases, these PxEDs were present in assays at a final concentration of 0.1 mM.
Steady-state kinetic data were evaluated to determine maximum turnover rate and catalytic efficiency (kcat and kon [i.e., kcat/KM], respectively) for each KatG variant. For nearly all measurements, a standard rectangular hyperbolic increase in rate as a function of substrate concentration was observed, and in these cases the data were fit using Eq. 1. Here and in our previous work, wild-type M. tuberculosis KatG shows a biphasic response of its catalase activity to H2O2 concentration at pH 5.0 (Ndontsa et al., 2012). To evaluate these kinetics an adapted Michaelis-Menten equation (Eq. 2) was used to account for an apparent second-order rate constant (kapp) which governs a less efficient catalase reaction. Initial rates obtained from experimental data were divided by total enzyme concentration in order to obtain a direct fit to kcat and kon.
The relative propensities of KatG and its variants to inactivation were evaluated by two methods. The first monitored the extent of O2 production as measured by O2-sensitive electrode in reactions containing 5 nM enzyme, 500 μM H2O2 and either 100 mM phosphate, pH 7, or 50 mM acetate, pH 5. The impact of peroxidatic electron donor (PxED) on inactivation was also evaluated. For these experiments, 0.1 mM ABTS, TMPD, or CPZ was also included. A second addition of H2O2 (0.5 mM) or enzyme (5 nM) was used to determine if O2 production had ceased because of substrate depletion or enzyme inactivation. If the former, a second addition of H2O2 but not fresh enzyme would induce more O2 production. If the latter, an addition of fresh enzyme but not H2O2 would induce more O2 production.
The second and complementary method of evaluating KatG inactivation recorded the remaining KatG catalase activity following a pre-incubation of the enzyme with H2O2. Pre-incubation reactions included 0.2 μM KatG or KatG variant and varying concentrations of H2O2 and were carried out in 100 mM phosphate, pH 7.0 or in 50 mM acetate, pH 5.0. When evaluating the impact of PxEDs on inactivation, these were included at a concentration of 0.1 mM (ABTS, TMPD, or CPZ as indicated). Following a 1-h incubation, an aliquot was withdrawn, and its activity measured by O2 production (O2-sensitive electrode) using a standard catalase assay which included 10 mM H2O2 and was carried out at ambient temperature in 100 mM phosphate, pH 7.0. All reactions were initiated by the addition of enzyme. The relative activity of H2O2-incubated enzyme was compared against the corresponding variant incubated for 1 h without added H2O2.
Transitions in heme spectra of wt KatG and its variants under steady-state conditions were monitored by photodiode array using a PC-upgraded SX18.MV rapid reaction analyzer from Applied Photophysics (Leatherhead, United Kingdom). In one syringe KatG (6 μM) in 5 mM phosphate, pH 7, was mixed against varying concentrations of H2O2 in either 100 mM phosphate, pH 7, or 50 mM acetate, pH 5. The final concentration of KatG monitored during reactions was 3 μM. When ascorbate and ABTS were present, they were included in the same syringe as H2O2 at concentrations of 1 mM and 0.2 mM, respectively. Accordingly, their final concentrations following mixing were 0.5 and 0.1 mM, respectively. To determine the extent oxidation of ABTS, ascorbate was left out and the accumulation of ABTS•+ was monitored at 645 nm (ε = 12,000 M-1cm-1). For these experiments, the final reactions contained 3 μM enzyme, 3 μM to 4 mM H2O2, and 0.1 mM ABTS.
Purified protein was concentrated to 300 μM using 30 kDa cutoff centrifugal filters for rapid freeze-quench sample preparation. Reactions were carried out and quenched using a RFQ-3 Quench-Flow Instrument (KinTek, Snow Shoe, PA). Enzyme (300 μM in 5 mM phosphate, pH 7.0) was placed in one syringe, and H2O2 (200 mM in 100 mM acetate, pH 5.0, or 200 mM phosphate, pH 7.0) was placed in the other. Accordingly, upon mixing reactions contained 150 μM KatG and 100 mM H2O2 (i.e., 667 molar equivalents). For W321F/R418N KatG, we observed high sensitivity to H2O2 inactivation at pH 5. Thus, the H2O2 concentration was decreased 10-fold to 20 mM (i.e., 66.7 molar equivalents upon mixing). Reactions designed to evaluate the impact of PxED included both ascorbate (10 mM) and ABTS (2 mM) along with H2O2. Reactions were quenched, frozen, and packed in liquid ethane (preparation described below). Samples of reaction time longer than 12 s were quenched manually in an ethanol-liquid nitrogen slurry. Prior to manual freeze-quenching, the enzyme and substrate were combined, gently mixed, and then added to the quartz EPR tube with a syringe. All samples were stored in liquid N2 until transferred to the EPR cavity for data collection.
Liquid ethane was produced by slowly bubbling ethane gas into a 50 mL conical tube prechilled and submerged in liquid nitrogen. The lid of the conical tube was altered to accommodate two tubes, one served to supply ethane gas to the chamber for freezing, and the second to outgas into mineral oil as a bubbler. Liquid ethane prepared for immediate use was kept in a isopentane-liquid nitrogen mixture (kept at 120–140 K). If necessary, liquid ethane could be frozen in pure liquid nitrogen to store for longer-term use (within a day). When needed, frozen ethane was thawed using a mixture of liquid isopentane-liquid nitrogen. The liquid ethane was used to fill the packing funnel and therefore for quenching the reactions and packing the frozen enzyme-substrate mixture into the EPR tube. The EPR tubes were packed in a KinTek Corporation sample packing unit maintained at 120–140 K with isopentane-liquid nitrogen mixture. All EPR spectra were collected at an X-band frequency using a Bruker EMX spectrometer in perpendicular mode. The spectrometer was equipped with an ESR900 liquid helium cryostat and an ITC temperature controller (Oxford Instruments). Spectra were recorded at 4.5 K. Unless otherwise indicated, instrument parameters were as follows: microwave frequency, 9.389 GHz; modulation amplitude, 1 G; modulation frequency, 100 kHz; microwave power, 1 mW; time constant, 163.84 ms; sweep time 335.54 s; number of scans, 1; conversion, 327.68 ms; resolution, 1024 point; harmonic 1st; receiver gain, 1.0 × 104; and phase 0 deg.
The sensitivity of select signals to microwave power saturation was evaluated by fitting normalized signal intensities versus microwave power using Eq. 3. Here, the parameter P½ is the microwave power at half saturation, and b is a parameter that accounts for contributions from inhomogeneous broadening. (Liu et al., 1998).
Reaction of wt KatG with 0.5 mM (100,000 molar equivalents) H2O2 at pH 7 yielded the full stoichiometric production of 0.25 mM O2 (Figure 2A) according to the 2:1 molar ratio of H2O2 consumed to O2 generated by the catalase reaction. The full depletion of the H2O2 substrate was confirmed when the production of new O2 could only be achieved upon addition of new H2O2, not the addition of fresh enzyme. The production of O2 by R418N KatG at pH 7 contrasted with wt KatG in multiple respects (Figure 2A). First, the initial rate of R418N KatG O2 production was only ∼11% of that observed for the wild-type enzyme. Second, only ∼60 μM O2 was produced, about 24% of that anticipated according to catalase stoichiometry. Third, only the addition of fresh R418N KatG and not H2O2 produced a new burst of O2 production. Taken together, these results indicate that substitution of the arginine switch with asparagine not only produces a KatG enzyme significantly less efficient in catalase-based H2O2 degradation, but also more prone to rapid inactivation and, therefore, unable to accomplish the full depletion of the substrate present in the reaction chamber.
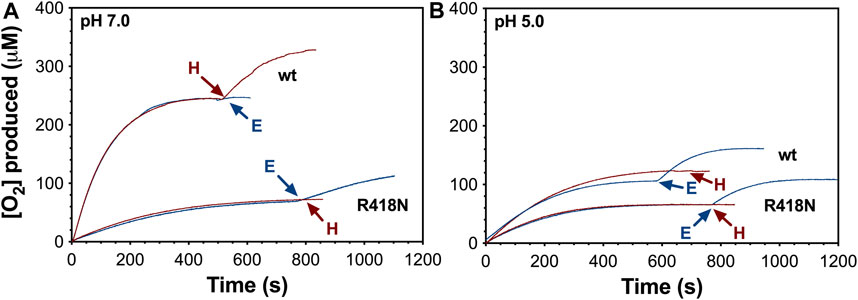
Figure 2. Catalatic O2 production by wt and R418N KatG at pH 7.0 (A) and pH 5.0 (B). All reactions were initiated by the addition of H2O2 (0.5 mM) to 5 nM enzyme. Second additions of 5.0 nM of the corresponding fresh enzyme (E, blue traces) or 0.5 mM new H2O2 (H, red traces) are indicated by the blue and red arrows, respectively. All reactions were carried out at 23°C. All traces are representative of 5 repeated measurements.
Notably, the O2 production profile of wt KatG much more closely resembled that of the R148N variant at pH 5 (Figure 2B). During reactions with 0.5 mM H2O2 (100,000 molar equivalents), both proteins were inactivated before fully consuming the H2O2 present. Indeed, only ∼40% (wt) and ∼25% (R418N) of the initially added H2O2 was accounted for in terms of O2 production. Consistent with this conclusion, only the addition of more enzyme promoted further O2 production by wt and R418N KatG. Though both proteins are prone to enzymatic inactivation at pH 5, wt KatG reproducibly yielded more O2 than R418N prior to inactivation (∼100 μM and ∼60 μM, respectively). Accordingly, wt KatG performed roughly 21,000 ± 3,000 catalase reaction cycles prior to irreversible inactivation, whereas R418N only achieved 12,000 ± 1,500 cycles before inactivation.
We evaluated the capacities of wt and R418N KatG to resist peroxide-dependent inactivation at pH 7 and 5 by evaluating the catalase activity of each enzyme following preincubation with increasing H2O2 concentrations (Figure 3). Under both pH conditions, each enzyme retained full activity following pretreatment with up to 2,000 molar equivalents H2O2. However, upon pre-reaction with not more than 30,000 molar equivalents H2O2, ∼50% loss of activity was detected for wt KatG at pH 5.0 and for R418N KatG under both pH conditions. Under all three reaction conditions, the complete loss of catalase activity was observed following pre-treatment with 70,000 equivalents H2O2. In stark contrast, at pH 7.0 wt KatG retained 70% of its catalase activity even following preincubation with 300,000 molar equivalents H2O2.
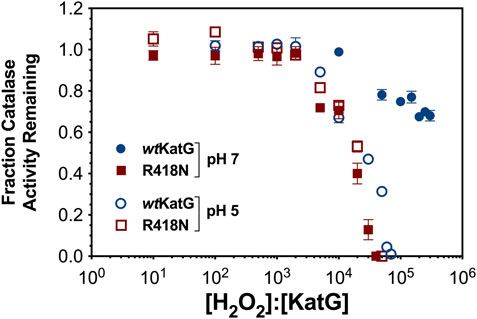
Figure 3. Resistance of wt and R418N KatG to H2O2-dependent inactivation at pH 7.0 and 5.0. Either wt KatG (blue circles) or R418N KatG (red squares) (0.2 μM) were preincubated for 1 hour with the number of molar equivalents H2O2 indicated on the x-axis at either pH 7 (solid symbols) or at pH 5.0 (open symbols). Preincubations carried out at pH 7.0 contained 100 mM phosphate, those carried out at pH 5.0 contained 50 mM acetate. An aliquot was withdrawn and catalase activity was determined by O2-sensitive electrode relative to that of the corresponding enzyme preincubated without H2O2. All activity assays were carried out in 100 mM phosphate, pH 7.0, and were initiated with 10 mM H2O2. All preincubations and activity assays were carried out at 23°C. Points represent the average of three replicates; error bars show standard deviations.
In light of the effect of pH on the position of KatG’s arginine switch (Carpena et al., 2006; Gasselhuber et al., 2016), the resilience of wt KatG at pH 7 indicates that the switch may assist KatG in resisting H2O2-dependent inactivation. Specifically, under conditions where the arginine switch is known to interact with the phenolate oxygen of the MYW cofactor (e.g., wt KatG at pH 7.0) (Carpena et al., 2006), we observe greater resistance to inactivation. Under conditions where the arginine switch is known to be pointed away from the MYW cofactor (e.g., wt KatG at pH 5.0) (Carpena et al., 2006) or the switch is absent entirely (e.g., R418N KatG under any conditions), we observe substantially diminished resistance to peroxide-dependent inactivation.
The inclusion of ABTS, a peroxidatic electron donor (PxED), served to protect the catalase activity of both wt and R418N KatG at pH 5 and pH 7. The effect of ABTS was most pronounced under conditions (pH and variant) that revealed the greatest propensity toward H2O2-dependent catalase inactivation in the first place (i.e., wt KatG at pH 5 and R418N KatG at pH 5 and 7). ABTS-enhanced R418N KatG-catalyzed O2 production rates were stimulated by 4-fold at pH 5 (Figure 4A) and 2-fold at pH 7 (data not shown); ABTS-enhanced, wt KatG-catalyzed O2 production rates increased by 9-fold at pH 5 (Figure 4A). In all three reactions (i.e., R418N KatG at pH 5 and 7; wt KatG at pH 5), the complete depletion of H2O2 was observed and catalase activity remained at the end of the reaction: 28% and 22% for R418N KatG at pH 7 and 5, respectively, and 73% for wt KatG at pH 5. For wt KatG at pH 7.0 the impact of ABTS was less prominent, but here as well, the PxED enabled the enzyme to retain more of its catalase activity than when it was withheld (data not shown). Consistent with these results, enhanced resistance to H2O2-dependent inactivation by wt and R418N KatG was imparted in the presence of ABTS. Indeed, an order of magnitude higher concentration of H2O2 could be tolerated by each enzyme when the PxED was included (Figure 4B).
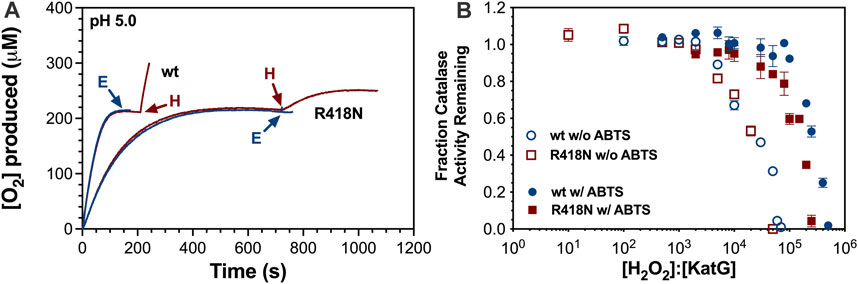
Figure 4. Effect of ABTS on catalatic O2 production and resistance to H2O2-dependent inactivation by wt and R418N KatG. O2 production (A) by wt or R418N KatG in the presence of 0.1 mM ABTS was initiated by the addition of 0.5 mM H2O2. Each reaction was supplemented by a second addition of H2O2 (H; red traces) or 5 nM fresh enzyme (E; blue traces) at the times indicated by the arrows. Resistance to H2O2 inactivation (B) of wt KatG (blue circles) or R418N KatG (red squares) was determined as described for Figure 3 in the absence (open symbols) or presence (closed symbols) of 0.1 mM ABTS. All O2 production traces (A) or preincubations with H2O2 (B) were carried out in 50 mM acetate, pH 5.0. Determination of remaining catalase activity was evaluated by a standard catalase activity assay out in 100 mM phosphate, pH 7.0, and initiated with 10 mM H2O2. All reactions were carried out at 23°C.
Not evident from the preservation of catalase activity (see Figure 4) was the extent to which the PxED was being oxidized over the course of the same reactions. Oxidation of ABTS to ABTS•+ by R418N KatG exceeded that observed for the wild-type enzyme (Figure 5). Certainly, in one respect this is reflected in the more efficient peroxidase activity displayed by R418N KatG compared to the wild-type enzyme (Table 1). However, considering the notion that PxED oxidation may reflect the correction/repair of off-catalase protein-based electron transfer, it is at least as important to quantify PxED oxidation events as it is to record rates. The most extensive ABTS•+ production was observed at lower pH conditions (i.e., pH 5.0), and strikingly, the utilization of ABTS was greatest on a cycle-by-cycle basis when H2O2 concentrations were low. Across all H2O2 concentrations and pH conditions evaluated, the extent of ABTS•+ generation by R418N KatG exceeded the wild-type enzyme. The same trend was observed for other peroxidatic electron donors (Supplementary Figure S1).
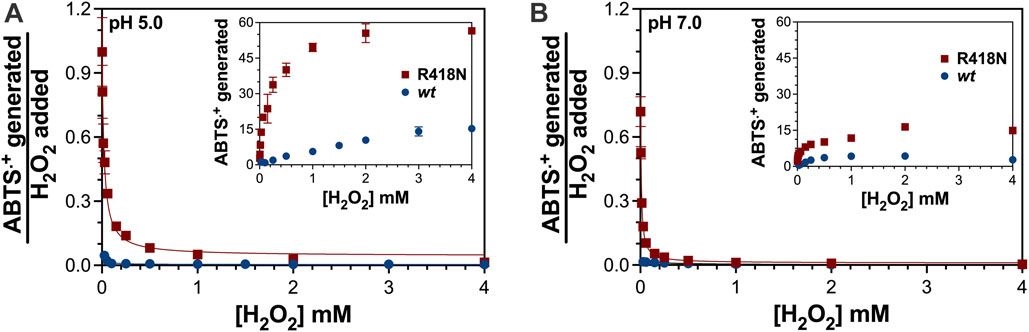
Figure 5. Oxidation of ABTS during catalatic consumption of H2O2 by wt and R418N KatG. The extent of ABTS oxidation by the two variants was evaluated at pH 5.0 (A) and pH 7.0 (B). The ABTS•+ generated was quantitated as described in Materials and Methods and the untransformed values as a function of H2O2 added to each reaction are shown in each inset. The stoichiometry of ABTS•+ generated was determined by dividing each [ABTS•+] generated by the corresponding [H2O2] used to generate it. All reactions contained 3 μM enzyme, 0.1 mM ABTS, and either 50 mM acetate, pH 5.0, or 100 mM phosphate, pH 7.0. Given the relative concentrations of H2O2 to enzyme (<1,400 molar equivalents), it should be noted that the full concentration of H2O2 was consumed over the course of each reaction.
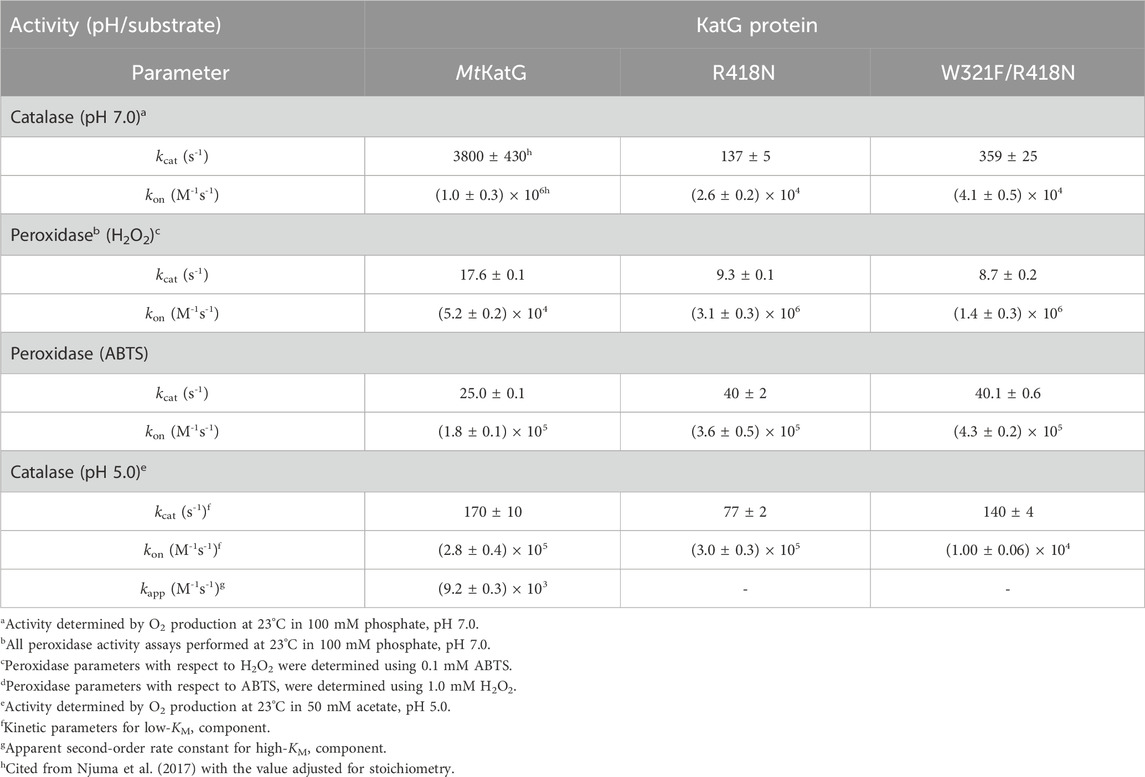
Table 1. Catalase and peroxidase kinetic parameters for MtKatG and variants.
In previous investigations, we observed that inclusion of PxEDs such as ABTS did not interfere with the accumulation or persistence of KatG intermediates connected with the enzyme’s catalase activity (i.e., the MYW•+ narrow-doublet radical and ferri-superoxo-like heme state). Simultaneously, inclusion of ABTS fully prevented the accumulation intermediates connected with enzyme inactivation (i.e., a series of singlet protein-based radical EPR spectra and late-reaction ferryl-like states) (Njuma et al., 2017; Krewall et al., 2020). Most prominently, we noted that an active-site Trp (the “proximal Trp”; W321 by M. tuberculosis KatG numbering) was a prominent site for off-catalase KatG protein oxidation, yielding an exchange-broadened protein-based radical (Njuma et al., 2017). By DFT QM/MM calculations, others have shown that there is a greater propensity towards oxidation of the proximal Trp observed when the arginine switch (R418) is pointed out and the MYW cofactor phenol is protonated (Vidossich et al., 2007). Consistent with this proposal, we observed the accumulation of a W321•+ intermediate that corresponded with progressively diminishing KatG catalase activity (Njuma et al., 2017). We have also observed that replacement of the proximal tryptophan with a non-oxidizable side chain (i.e., W321F KatG) produces an enzyme more susceptible to inactivation, particularly at low pH (Krewall et al., 2020).
To better understand the connection between the arginine switch, the proximal Trp, and H2O2-dependent inactivation of KatG, we prepared and characterized the W321F/R418N KatG variant. Even though initial-rate evaluation of the double variant’s catalase activity returned similar kinetic parameters at pH 7.0 and pH 5.0 as were observed for the R418N enzyme (Table 1), the ability of W321F/R418N KatG to sustain catalase turnover was substantially diminished compared to wild-type and even R418N KatG (Figure 6).
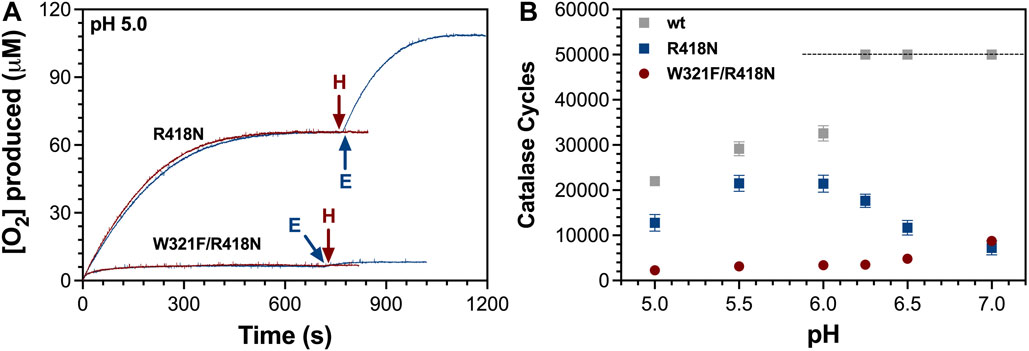
Figure 6. Catalatic O2 production by R418N and W321F/R418N KatG. The production of O2 over time by R418N and W321F/R418N KatG at pH 5.0 is shown (A). In neither instance was the addition of 0.5 mM more H2O2 (H) able to stimulate additional O2 production, but in both cases, addition of fresh enzyme (E) did stimulate new O2 production. The number of catalase cycles achieved as a function of pH (B) was calculated from the [O2] produced (initial reaction) at each pH divided by [E]T present in the reaction (5 nM) for wild-type (wt), R418N, and W321F/R418N KatG. Each reaction was initiated with 0.5 mM H2O2 allowing for a maximum of 50,000 catalase cycles (indicated by the dashed line). Accordingly, wt KatG was able to catalyze the full conversion of H2O2 to H2O and O2 at pH 6.25, 6.5, and 7.0. All reactions contained 5 nM KatG and were initiated by the addition of 0.5 mM H2O2. Reactions at pH 6.0, 6.25, 6.5, and 7 were buffered with 100 mM phosphate, and reactions at pH 5.0 and 5.5 were buffered with 50 mM acetate. All reactions were carried out at 23°C.
We also compared the ability of W321F and W321F/R418N variants to retain activity following preincubation with increasing concentrations of H2O2 (Figure 7). At pH 7.0, W321F KatG and the wild-type enzyme were indistinguishable in their robust ability to resist H2O2-dependent inactivation. However, at pH 5.0 the W321F variant showed much greater sensitivity to inactivation. More than 50% of the variant’s activity was lost following pretreatment with as few as 1,000 molar equivalents H2O2. To observe the same loss of activity with R418N KatG, 10-fold higher concentrations of H2O2 were required (Figure 7A). Clearly, when the proximal Trp is replaced by non-oxidizable Phe (i.e., W321F KatG) and the Arg switch is predominantly pointed away from the phenol moiety of the MYW cofactor at pH 5, KatG is significantly impaired in its ability to sustain catalase activity. We surmised that the effect at pH 5.0 was due to the combined absence of the proximal Trp (replaced by non-oxidizable Phe [i.e., W321F KatG]) and the Arg switch (predominantly pointed away from the MYW cofactor at pH 5.0). To evaluate this hypothesis further, we compared the resistance of wt, W321F, R418N, and W321F/R418N KatG to inactivation at pH 7.0 (Figure 7B). As anticipated, the variant lacking both the switch and the proximal Trp (i.e., W321F/R418N) showed very little resistance to H2O2-dependent inactivation.
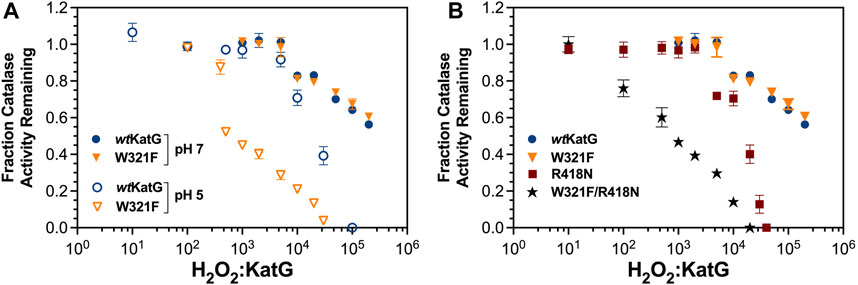
Figure 7. Resistance of W321F KatG and W321F/R418N KatG to H2O2-dependent inactivation. The ability of W321F KatG to retain its catalase activity following preincubation with increasing H2O2 concentrations (A) was monitored at pH 7.0 (filled triangles) and pH 5.0 (open triangles). These are compared against wt KatG at pH 7 (filled blue circles) and pH 5.0 (open blue circles). Resistance of wt (closed circles), W321F (closed triangles), R418N (closed squares), and W321F/R418N KatG (filled stars) to H2O2 inactivation at pH 7.0 is also shown (B). Preincubations and catalase activity measurements were carried out as described for Figure 3.
Our data suggest that the Arg switch and oxidation of the proximal Trp cooperate to preserve KatG catalysis. Previous investigations have pointed to a critical role for the Arg switch in the regulation of active-site intramolecular electron transfer (Vidossich et al., 2007; Loewen et al., 2014; Fita et al., 2015; Kruft et al., 2015). Our data indicate that a major consequence of disabling the arginine switch is the far greater propensity of KatG toward inactivation. Strikingly, the enzyme’s proximal Trp appears to be a failsafe against the consequences of displacement of the arginine switch from the active site. Accordingly, so long as the switch is oriented toward the MYW cofactor and the KatG active site, the presence or absence of the proximal Trp shows little fundamental contribution to KatG function. However, when conditions arise that alter the position (or presence) of the switch, the proximal Trp becomes an indispensable failsafe for preservation of KatG catalase activity. It is a failsafe which appears to be aided by the presence of a peroxidatic electron donor (PxED) to address (i.e., reduce) off-catalase protein-based radical intermediates. When the switch and the failsafe are absent, KatG becomes highly susceptible to H2O2-dependent inactivation.
To investigate the population of protein-based radicals generated during catalysis when the Arg switch is absent from its interaction with the MYW cofactor (i.e., to simulate wt KatG with the Arg switch positioned out), we performed RFQ-EPR on R418N KatG. Within 10 ms of mixing with H2O2 at either pH 5.0 or 7.0, R418N KatG formed an intense narrow doublet radical (10.6 G width as measured from peak-to-peak) (Figure 8A and Supplementary Figure S2A, respectively). This was concomitant with the near complete loss of signals between 800 and 1400 G corresponding to the high-spin ferric species of the resting enzyme (data not shown). In addition, the breadth of the signal, its g-value, and its sensitivity to power saturation (Supplementary Figures S3A, D) were consistent with the MYW•+ assigned by others as the cation radical of the MYW adduct (Suarez et al., 2009; Zhao et al., 2010). As has been observed with wt KatG (Njuma et al., 2017), the narrow doublet gave way to an exchange-broadened signal consistent with a radical centered on the enzyme’s proximal Trp (W321) residue (Figure 8B) (Ivancich et al., 2001; Singh et al., 2007; Colin et al., 2009; Njuma et al., 2017). The effect of microwave power on signal intensity for the spectrum observed 15 s after reaction with H2O2 (Supplementary Figure S3B) was not uniform. When evaluated at a magnetic field where exchange-broadening was clearly evident (e.g., 3300 G), a P½ value two orders of magnitude larger was observed when compared to that determined when the peak-to-trough intensity was evaluated instead (Supplementary Figures S3B, D). This differential in power saturation is consistent with our previous investigations of W321 oxidation during wt KatG reaction with H2O2, further supporting the idea that W321•+ is formed during R418N reaction with H2O2.
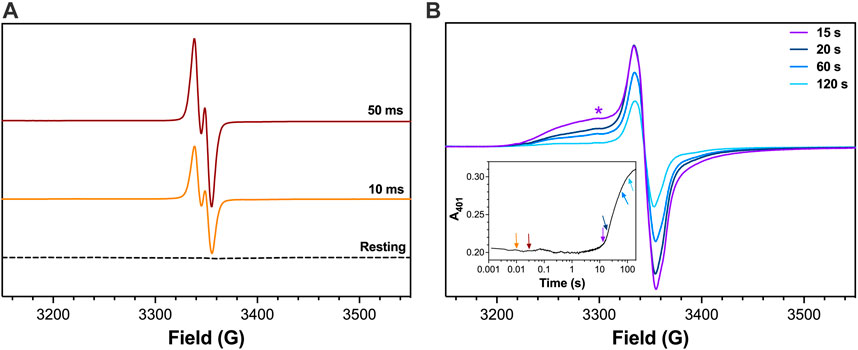
Figure 8. EPR spectra for R418N KatG freeze-quenched following reaction with H2O2. The R418N KatG variant (150 μM) was reacted with 667 molar equivalents H2O2 (i.e., 100 mM) at pH 5.0. Reactions were quenched at early (A) and late reaction times (B) as indicated. Features consistent with broadening due to exchange coupling (e.g., 3300G marked by *) were observed in the later reaction time-points. Reagents and protocols for RFQ-EPR are described in Materials and Methods. The spectrum for resting R418N KatG (dashed line, panel A) was frozen manually without the addition of any H2O2. A companion trace from optical stopped-flow monitoring the FeIII state at 401 nm for 3 μM R418N KatG reacted with 667 molar equivalents H2O2 is shown in the inset. The reaction times at which RFQ-EPR samples were collected and measured are indicated by the corresponding arrows. All reactions were carried out at 4°C. All EPR spectra were recorded at 4.5 K. EPR spectrometer settings are provided in Materials and Methods.
At pH 5.0 and 7.0, contribution of the exchange-broadened signal observed for KatG protein-based radicals was most prominent prior to the return of the FeIII state (Figure 8B, Supplementary Figure S2B, respectively). Following the re-emergence of the FeIII enzyme (e.g., 120 s post mixing with H2O2), the EPR spectrum was dominated by a singlet radical signal that lacked features of exchange broadening and showed minimal resistance to power saturation (Supplementary Figures S3C, D). In most respects, EPR signals observed for R418N KatG at pH 7.0 (Supplementary Figure S2) were highly similar to those detected at pH 5.0 (Figure 8) except that contributions from exchange-broadened radicals at pH 7.0 were generally diminished compared to those observed at pH 5.0.
To more definitively identify the exchange-broadened signal observed in R418N KatG EPR spectra, we carried out RFQ-EPR experiments with the W321F/R418N KatG variant. When the double variant was reacted with H2O2, the same narrow doublet radical was observed at early reaction times (Figure 9A). Consistent with the hypothesis that the exchange-broadened signal observed at late reaction times with R418N KatG belonged to a radical centered on W321, EPR spectra for the W321F/R418N variant recorded at similar reaction times lacked the exchange-broadened signal observed for R418N KatG (Figure 9B).
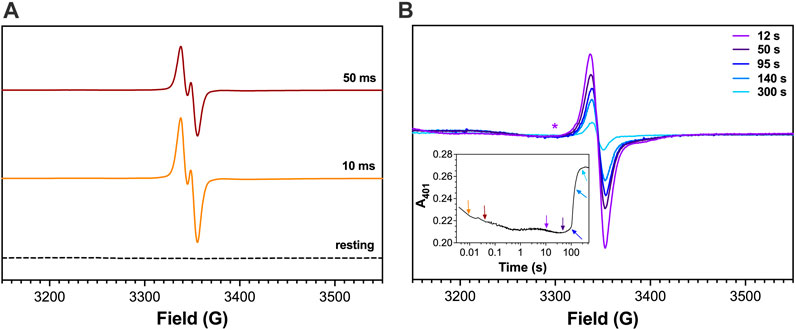
Figure 9. EPR spectra for W321F/R418N KatG freeze-quenched following reaction with H2O2. The W321F/R418N KatG double variant (150 μM) was reacted with 67 molar equivalents H2O2 (i.e., 10 mM) at pH 7.0. Reactions were quenched at early (A) and late reaction times (B) as indicated. The same magnetic field (3300 G) where exchange broadening is clearly present in Figure 8 is also marked here (*) for comparison. Reagents and protocols for RFQ-EPR are described in Materials and Methods. The spectrum for resting W321F/R418N KatG (dashed line, panel A) was frozen manually without the addition of any H2O2. A companion trace from optical stopped-flow monitoring the FeIII state at 401 nm reacted 3 μM W321F/R418N KatG with 67 molar equivalents H2O2 is shown in the inset. The reaction times at which RFQ-EPR samples were collected and measured are indicated by the corresponding arrows. All reactions were carried out at 4°C. All EPR spectra were recorded at 4.5 K. EPR spectrometer settings are provided in Materials and Methods.
Remarkably, W321F/R418N KatG showed nearly identical peroxidase activity to the R418N single variant (Table 2). Further, the overall extent of ABTS•+ produced as well as its generation on a cycle-by-cycle basis were nearly identical to R418N KatG at pH 5 and even favored the double variant at pH 7.0 (Supplementary Figure S4). Nevertheless, PxEDs showed no ability to preserve or extend catalase activity of W321F/R418N KatG (Figure 10). Across the pH range evaluated, after pre-reaction with 40,000 molar equivalents H2O2, wt KatG retained catalase activity even in the absence of an exogenous PxED, albeit to a lesser extent as pH decreased toward 5.0. Under the same conditions, neither R418N nor W321F/R418N KatG were able to retain any catalase activity (Figure 10A). Inclusion of a PxED enabled wild-type to retain 100% catalase activity across all pH conditions evaluated. This was observed not only for ABTS, but also for two other PxEDs, chlorpromazine (CPZ) and N,N,N′,N′-tetramethylphenylenediamine (TMPD) (Figures 10B–D, respectively). Nearly identical behavior was observed for R418N KatG. The sole exception was that some catalase activity was lost in the presence of ABTS, a phenomenon for which a buffer identity effect could not be fully ruled out. Nevertheless, in sharp contrast, pre-reaction of W321F/R418N KatG with 40,000 molar equivalents H2O2 resulted in the complete loss of catalase activity regardless of the absence, presence, or identity of the PxED.
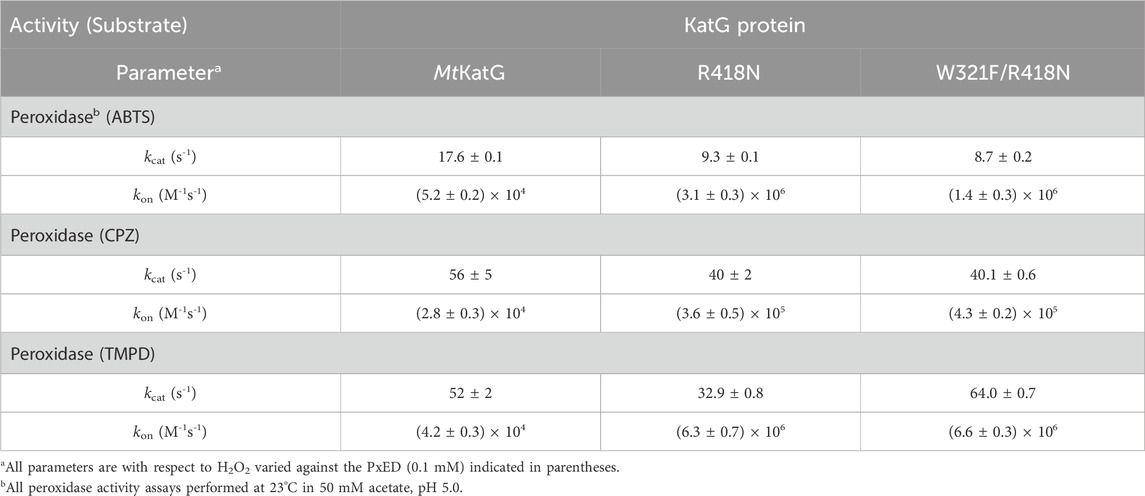
Table 2. Peroxidase kinetic parameters against three PxEDs for MtKatG and variants.
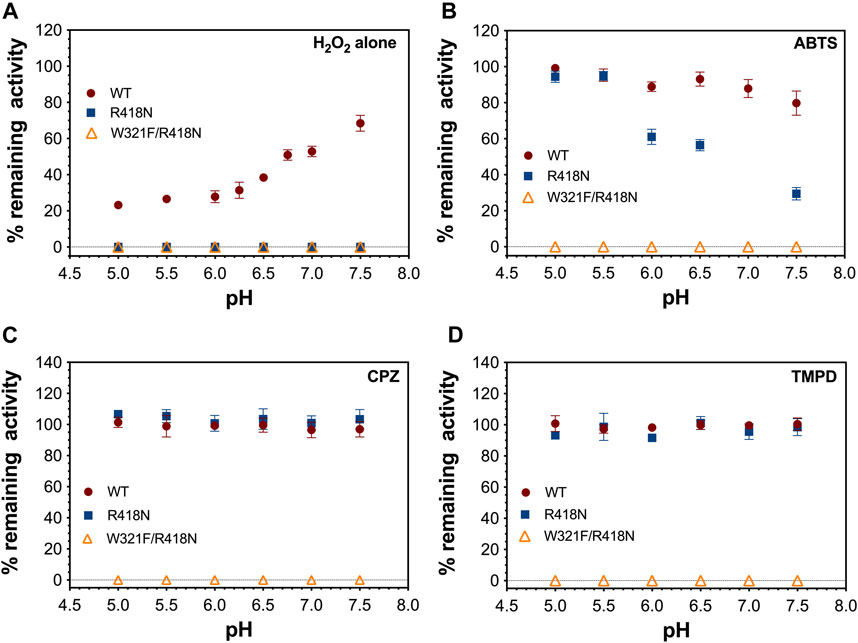
Figure 10. Effect of pH and peroxidatic electron donor on the preservation of catalase activity by KatG and its variants. Rates of catalatic O2 production by the indicated KatG variant (5 nM) were recorded upon addition of 0.2 mM H2O2 to reactions which contained no peroxidatic electron donor (A), 0.1 mM ABTS (B), 0.1 mM CPZ (C), or 0.1 mM TMPD (D). After cessation of O2 production, a second addition of H2O2 (0.2 mM) was made and a second rate of O2 production was recorded. The fraction of remaining activity was determined by dividing the second rate by the first. Reactions carried out at below 6.0 were buffered using 50 mM acetate; those carried out at pH 6.0 and above were buffered using 100 mM phosphate. All reactions were carried out at 23°C.
Taken together, our data indicate that the ability of KatG to maintain a robust catalase activity across a wide pH range depends on a unique mechanism for resisting peroxide-dependent inactivation. Our data indicate that this mechanism depends on the cooperation of the enzyme’s Arg switch, proximal Trp, and the availability of a PxED.
This suggests substantially expanded roles for these residues in KatG’s catalytic abilities. This is especially so for the proximal Trp. It has long been identified as a “hot spot” for protein-based radical formation (Ivancich et al., 2001; Singh et al., 2007; Vidossich et al., 2007; Colin et al., 2009), and it is strictly conserved across all KatGs. Nevertheless, a clear catalytic role for this residue has remained elusive. Such a role has been obscured by the fact that proximal Trp to Phe substitutions in KatG typically produce steady-state kinetic parameters for catalase and peroxidase activities that are similar to the wild-type enzyme (Jakopitsch et al., 2002; Yu et al., 2002; Wiseman et al., 2009; Njuma et al., 2017). A pivotal (albeit supporting) role for the Arg switch in KatG’s catalase activity has been clearer. Multiple studies, including this one, have observed that variants eliminating this conformationally dynamic, positively charged side chain (e.g., R418A or R418N KatG) diminishes catalase activity by two orders of magnitude at neutral pH (Jakopitsch et al., 2004; Carpena et al., 2005; Zhao et al., 2012). Nevertheless, these variants still show catalase activity that exceeds that of the other members of KatG’s superfamily by two orders of magnitude. Further, the catalase activities of wt and R418N KatG are appreciable and highly similar to one another at pH 5.0.
In the context of enzyme inactivation, an essential contribution for each residue and the seamless integration of KatG’s catalase and peroxidase activities come together to give a comprehensive view of KatG’s abilities to degrade H2O2 across a broad range of conditions (Figure 11). A sensitivity of wt KatG to peroxide-dependent inactivation emerges as pH decreases from 7 to 5. This is coincident with a shift in the dominant conformation of KatG’s Arg switch from its orientation toward the enzyme’s MYW cofactor and active site (i.e., “Y” or in; blue in Figure 11A) to its orientation toward the enzyme’s solvent accessible surface (i.e., “R” or out; green in Figure 11A). This, along with the protonation of the MYW phenolic oxygen, has been shown to increase the propensity toward protein oxidation by FeIV=O[porphyrin]•+ at the proximal Trp and away from the MYW cofactor (Vidossich et al., 2007). Our data suggest that this produces greater susceptibility to H2O2-dependent inactivation. Consistent with that model, removal of the switch entirely by mutagenesis (i.e., R418N KatG) produces a heightened sensitivity to inactivation across the pH range.

Figure 11. Proposed mechanism to connect Arg switch position and utilization of the proximal Trp failsafe. The critical branch point (A) concerns the relative frequency of MYW cofactor versus proximal Trp (PxW) oxidation. Above an apparent pKa of ∼6.5, the “Y” or in position of the switch (blue), MYW oxidation and the catalase cycle dominate. Below that apparent pKa, the “R” or out position of the switch is favored (green) and the propensity toward PxW oxidation substantially increases. (When the Arg switch is removed entirely by mutagenesis (e.g., R418N KatG), the effect is observed across the pH range.) The PxW•+ can be reduced at the expense of secondary protein-based radicals (PA). It is anticipated that these protein-based radicals migrate to nearby (likely surface-exposed) oxidizable amino acids (PB). The failsafe mechanism is completed (and active enzyme retained) when these protein-based radicals are reduced by an exogenous peroxidatic electron donor (PxED). In the absence of a PxED, protein-based radicals and subsequent advanced protein oxidation products accumulate, eventually resulting in irreversible enzymatic inactivation (B).
We have shown in our previous investigations that the proximal Trp is a prominent site of protein oxidation as KatG progressively loses activity over time during H2O2 degradation (Njuma et al., 2017), a phenomenon which is particularly noticeable at acidic pH. Taken together then, a decrease in pH necessarily produces an increase in the frequency of off-catalase intramolecular transfer, and the proximal Trp appears to be the principal conduit for channeling these oxidizing equivalents away from the active site. Here, the presence of exogenous PxEDs completes the mechanism for sustaining KatG’s catalase activity. So long as a suitable PxED is present, protein oxidation (starting at W321 and working toward the solvent accessible surface) can be resolved early before enzyme-inactivating modifications to the KatG protein can take place (Figure 11). To that end we have consistently observed by RFQ-EPR with wt and R418N KatG that inclusion of a PxED (e.g., ABTS) serves to quench all KatG protein-based radicals (including W321•+) with the notable exception of the narrow doublet MYW•+ species necessary for catalase activity. (Njuma et al., 2017). Accordingly, under all conditions where a greater propensity to H2O2-based inactivation is observed (i.e., R418N KatG regardless of pH and wt KatG at acidic pH), PxEDs show an ability to extend KatG’s catalase activity by way of their own sacrificial oxidation. Accordingly, the so-called peroxidase activity of KatG is more accurately understood as the manifestation of events which serve to repair KatG and sustain its catalase activity. As such, the dramatic increase in the accumulation of oxidized PxEDs (e.g., ABTS•+) generated by R418N KatG is reporting a corresponding increase in the frequency of off-catalase electron-transfer events.
Remarkably, our data show that utilizing this strategy depends on the proximal Trp. As conditions (i.e., low pH) necessitate an increased frequency of off-catalase electron transfer, this can be easily addressed by an exogenous PxED. Elimination of the Arg switch substantially expands the pH conditions and extent to which PxEDs are needed prevent inactivation and sustain KatG’s catalase activity, but electron-hole transfer via the proximal Trp and availability of a PxED are sufficient to address the issue. However, placing the proximal Trp out of reach as a point of KatG protein oxidation (i.e., W321F KatG), profoundly compromises the enzyme for handling the deleterious consequences of off-catalase electron transfer. This weakness is revealed when the frequency of off-pathway electron transfer is increased due to the withdrawal of the Arg switch (W321F KatG at low pH). Of course, removing the Arg switch altogether via mutagenesis (i.e., W321F/R418N KatG) makes the enzyme highly susceptible to H2O2-dependent inactivation irrespective of pH. Remarkably, exogenous PxEDs had no capacity to prevent the inactivation of this enzyme.
These data help explain the absolute conservation of the proximal Trp across all KatGs, even though repeated evaluation of variants replacing this Trp with Phe have shown only limited consequences in steady-state measurements of catalase and peroxidase activity (where initial rates are evaluated). These data also provide a mechanistic understanding of the seamless integration of KatG’s catalase and peroxidase activities. For many years, these had been supposed to be separate activities where the peroxidase was a limited activity observed under more extreme pH conditions and of little consequence to the physiological operation of KatG in catalatic H2O2 degradation. The integrated connection of these activities via the Arg switch, proximal Trp, and exogenous PxED enable robust H2O2 degradation, not only at pH 7 (a long-appreciated property of KatG), but also under highly acidic conditions as well. What is a beneficial property in H2O2 degradation catalysts for bacteria and lower eukaryotes writ large becomes indispensable for pathogens, particularly intracellular pathogens, who face the very large concentrations of H2O2 generated by host immune responses often within the confines of highly acidic compartments like the activated neutrophil phagolysosome.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
HX: Data curation, Formal Analysis, Investigation, Methodology, Visualization, Writing–original draft. JK: Conceptualization, Formal Analysis, Visualization, Writing–review and editing. LM: Data curation, Visualization, Writing–review and editing. DG: Conceptualization, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. National Science Foundation (NSF MCB 1616059). NSF funds supported the acquisition of materials, supplies, etc and graduate student effort for characterization of the proteins described.
We thank Dr. Evert Duin, Dr. Carly Engle, Dr. Robel Ghebreab, and Bryan Cronin for their assistance in RFQ sample preparation and the use of EPR spectrometer. The following reagent was obtained through BEI Resources, NIAID, NIH: Plasmid pMRLB.11 Containing Gene Rv 1908c (Protein KatG) from Mycobacterium tuberculosis, NR-13284. This work was supported in part by NSF (MCB 1616059).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchbi.2024.1431412/full#supplementary-material
Bertrand, T., Eady, N. A. J., Jones, J. N., Jesmin, , Nagy, J. M., Jamart-Grégoire, B., et al. (2004). Crystal structure of Mycobacterium tuberculosis catalase-peroxidase. J. Biol. Chem. 279 (37), 38991–38999. doi:10.1074/jbc.M402382200
Carpena, X., Loprasert, S., Mongkolsuk, S., Switala, J., Loewen, P. C., and Fita, I. (2003). Catalase-peroxidase KatG of burkholderia pseudomallei at 1.7Å resolution. J. Mol. Biol. 327 (2), 475–489. doi:10.1016/S0022-2836(03)00122-0
Carpena, X., Wiseman, B., Deemagarn, T., Herguedas, B., Ivancich, A., Singh, R., et al. (2006). Roles for Arg426 and Trp111 in the modulation of NADH oxidase activity of the catalase-peroxidase KatG from Burkholderia pseudomallei inferred from pH-induced structural changes. Biochemistry 45 (16), 5171–5179. doi:10.1021/bi060017f
Carpena, X., Wiseman, B., Deemagarn, T., Singh, R., Switala, J., Ivancich, A., et al. (2005). A molecular switch and electronic circuit modulate catalase activity in catalase-peroxidases. EMBO Rep. 6 (12), 1156–1162. doi:10.1038/sj.embor.7400550
Colin, J., Wiseman, B., Switala, J., Loewen, P. C., and Ivancich, A. (2009). Distinct role of specific tryptophans in facilitating electron transfer or as [Fe(IV)=O Trp⋅] intermediates in the peroxidase reaction of bulkholderia pseudomallei catalase-peroxidase: a multifrequency EPR spectroscopy investigation. J. Am. Chem. Soc. 131 (24), 8557–8563. doi:10.1021/ja901402v
Corleis, B., Korbel, D., Wilson, R., Bylund, J., Chee, R., and Schaible, U. E. (2012). Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell. Microbiol. 14 (7), 1109–1121. doi:10.1111/j.1462-5822.2012.01783.x
Donald, L. J., Krokhin, O. V., Duckworth, H. W., Wiseman, B., Deemagarn, T., Singh, R., et al. (2003). Characterization of the catalase-peroxidase KatG from Burkholderia pseudomallei by mass spectrometry. J. Biol. Chem. 278 (37), 35687–35692. doi:10.1074/jbc.M304053200
Eum, S.-Y., Kong, J.-H., Hong, M.-S., Lee, Y.-J., Kim, J.-H., Hwang, S.-H., et al. (2010). Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137 (1), 122–128. doi:10.1378/chest.09-0903
Fita, I., Carpena, X., and Loewen, P. C. (2015). “Catalase-peroxidase (KatG) structure and function,” in Heme Peroxidases. Editors E. Raven, and B. Dunford (Cambridge, United Kingdom: Royal Society of Chemistry), 135–155.
Garcia, E., Nedialkov, Y. A., Elliott, J., Motin, V. L., and Brubaker, R. R. (1999). Molecular characterization of katy (antigen 5), a thermoregulated chromosomally encoded catalase-peroxidase of yersinia pestis. J. Bacteriol. 181 (10), 3114–3122. doi:10.1128/jb.181.10.3114-3122.1999
Gasselhuber, B., Graf, M. M. H., Jakopitsch, C., Zamocky, M., Nicolussi, A., Furtmüller, P. G., et al. (2016). Interaction with the redox cofactor MYW and functional role of a mobile arginine in eukaryotic catalase-peroxidase. Biochemistry 55 (25), 3528–3541. doi:10.1021/acs.biochem.6b00436
George, P. (1953). The third intermediate compound of horseradish peroxidase and hydrogen peroxide. J. Biol. Chem. 201 (1), 427–434. doi:10.1016/s0021-9258(18)71385-9
Ghiladi, R. A., Knudsen, G. M., Medzihradszky, K. F., and de Montellano, P. R. O. (2005a). The met-tyr-trp cross-link in Mycobacterium tuberculosis catalase-peroxidase (KatG): autocatalytic formation and effect on enzyme catalysis and spectroscopic properties. J. Biol. Chem. 280 (24), 22651–22663. doi:10.1074/jbc.M502486200
Ghiladi, R. A., Medzihradszky, K. F., and Ortiz de Montellano, P. R. (2005b). Role of the Met−Tyr−Trp cross-link in Mycobacterium tuberculosis catalase-peroxidase (KatG) as revealed by KatG(M255I). Biochemistry 44 (46), 15093–15105. doi:10.1021/bi051463q
Han, Y., Geng, J., Qiu, Y., Guo, Z., Zhou, D., Bi, Y., et al. (2008). Physiological and regulatory characterization of KatA and KatY in Yersinia pestis. DNA Cell Biol. 27 (8), 453–462. doi:10.1089/dna.2007.0657
Ivancich, A., Dorlet, P., Goodin, D. B., and Un, S. (2001). Multifrequency high-field EPR study of the tryptophanyl and tyrosyl radical intermediates in wild-type and the W191G mutant of cytochrome c peroxidase. J. Am. Chem. Soc. 123 (21), 5050–5058. doi:10.1021/ja0036514
Jakopitsch, C., Ivancich, A., Schmuckenschlager, F., Wanasinghe, A., Pöltl, G., Furtmüller, P. G., et al. (2004). Influence of the unusual covalent adduct on the kinetics and formation of radical intermediates in synechocystis catalase peroxidase: a stopped-flow and EPR characterization of the MET275, TYR249, and ARG439 variants. J. Biol. Chem. 279 (44), 46082–46095. doi:10.1074/jbc.M408399200
Jakopitsch, C., Kolarich, D., Petutschnig, G., Furtmüller, P. G., and Obinger, C. (2003). Distal side tryptophan, tyrosine and methionine in catalase-peroxidases are covalently linked in solution. FEBS Lett. 552 (2-3), 135–140. doi:10.1016/s0014-5793(03)00901-3
Jakopitsch, C., Regelsberger, G., Georg Furtmüller, P., Rüker, F., Peschek, G. A., and Obinger, C. (2002). Engineering the proximal heme cavity of catalase-peroxidase. J. Inorg. Biochem. 91 (1), 78–86. doi:10.1016/s0162-0134(02)00374-4
Jakopitsch, C., Vlasits, J., Wiseman, B., Loewen, P. C., and Obinger, C. (2007). Redox intermediates in the catalase cycle of catalase-peroxidases from synechocystis PCC 6803, burkholderia pseudomallei, and Mycobacterium tuberculosis. Biochemistry 46 (5), 1183–1193. doi:10.1021/bi062266+
Kelder, P. P., de Mol, N. J., Fischer, M. J. E., and Janssen, L. H. M. (1994). Kinetic evaluation of the oxidation of phenothiazine derivatives by methemoglobin and horseradish peroxidase in the presence of hydrogen peroxide. Implications for the reaction mechanisms. Biochimica Biophysica Acta (BBA) - Protein Struct. Mol. Enzym. 1205 (2), 230–238. doi:10.1016/0167-4838(94)90238-0
Krewall, J. R., Minton, L. E., and Goodwin, D. C. (2020). “KatG structure and mechanism: using protein-based oxidation to confront the threats of reactive oxygen,” in Mechanistic enzymology: bridging structure and function Editor J. M. Miller (Washington, D.C: ACS Symposium Series), 1357, 83–120. doi:10.1021/bk-2020-1357.ch005
Kruft, B. I., Magliozzo, R. S., and Jarzęcki, A. A. (2015). Density functional theory insights into the role of the methionine-tyrosine-tryptophan adduct radical in the KatG catalase reaction: O2 release from the oxyheme intermediate. J. Phys. Chem. A 119 (26), 6850–6866. doi:10.1021/jp511358p
Liu, A., Pötsch, S., Davydov, A., Barra, A.-L., Rubin, H., and Gräslund, A. (1998). The tyrosyl free radical of recombinant ribonucleotide reductase from Mycobacterium tuberculosis is located in a rigid hydrophobic pocket. Biochemistry 37 (46), 16369–16377. doi:10.1021/bi981471p
Loewen, P. C., Carpena, X., Vidossich, P., Fita, I., and Rovira, C. (2014). An ionizable active-site tryptophan imparts catalase activity to a peroxidase core. J. Am. Chem. Soc. 136 (20), 7249–7252. doi:10.1021/ja502794e
Michaelis, L., Schubert, M. P., and Granick, S. (1939). The free radicals of the type of wurster's salts. J. Am. Chem. Soc. 61 (8), 1981–1992. doi:10.1021/ja01877a013
Moore, R. L., Powell, L. J., and Goodwin, D. C. (2008). The kinetic properties producing the perfunctory pH profiles of catalase-peroxidases. Biochimica Biophysica Acta (BBA) - Proteins Proteomics 1784 (6), 900–907. doi:10.1016/j.bbapap.2008.03.008
Moore, S. (2018). Round-the-horn site-directed mutagenesis. Available at: http://www.openwetware.org/wiki/'Round-the-horn_site-directed_mutagenesis.
Nakajima, R., and Yamazaki, I. (1987). The mechanism of oxyperoxidase formation from ferryl peroxidase and hydrogen peroxide. J. Biol. Chem. 262 (6), 2576–2581. doi:10.1016/S0021-9258(18)61544-3
Ndontsa, E. N., Moore, R. L., and Goodwin, D. C. (2012). Stimulation of KatG catalase activity by peroxidatic electron donors. Archives Biochem. Biophysics 525 (2), 215–222. doi:10.1016/j.abb.2012.06.003
Nelson, D. P., and Kiesow, L. A. (1972). Enthalpy of decomposition of hydrogen peroxide by catalase at 25° C (with molar extinction coefficients of H2O2 solutions in the UV). Anal. Biochem. 49 (2), 474–478. doi:10.1016/0003-2697(72)90451-4
Ng, V. H., Cox, J. S., Sousa, A. O., MacMicking, J. D., and McKinney, J. D. (2004). Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52 (5), 1291–1302. doi:10.1111/j.1365-2958.2004.04078.x
Njuma, O. J., Davis, I., Ndontsa, E. N., Krewall, J. R., Liu, A., and Goodwin, D. C. (2017). Mutual synergy between catalase and peroxidase activities of the bifunctional enzyme KatG is facilitated by electron-hole hopping within the enzyme. J. Biol. Chem. 292, 18408–18421. doi:10.1074/jbc.M117.791202
Njuma, O. J., Ndontsa, E. N., and Goodwin, D. C. (2014). Catalase in peroxidase clothing: interdependent cooperation of two cofactors in the catalytic versatility of KatG. Archives Biochem. Biophysics 544, 27–39. doi:10.1016/j.abb.2013.11.007
Scott, S. L., Chen, W. J., Bakac, A., and Espenson, J. H. (1993). Spectroscopic parameters, electrode potentials, acid ionization constants, and electron exchange rates of the 2,2'-azinobis(3-ethylbenzothiazoline-6-sulfonate) radicals and ions. J. Phys. Chem. 97 (25), 6710–6714. doi:10.1021/j100127a022
Singh, R., Switala, J., Loewen, P. C., and Ivancich, A. (2007). Two [Fe(IV)O Trp•] intermediates in M. tuberculosis catalase-peroxidase discriminated by multifrequency (9−285 GHz) EPR spectroscopy: reactivity toward isoniazid. J. Am. Chem. Soc. 129 (51), 15954–15963. doi:10.1021/ja075108u
Singh, R., Wiseman, B., Deemagarn, T., Jha, V., Switala, J., and Loewen, P. C. (2008). Comparative study of catalase-peroxidases (KatGs). Archives Biochem. Biophysics 471 (2), 207–214. doi:10.1016/j.abb.2007.12.008
Suarez, J., Ranguelova, K., Jarzecki, A. A., Manzerova, J., Krymov, V., Zhao, X., et al. (2009). An oxyferrous heme/protein-based radical intermediate is catalytically competent in the catalase reaction of Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 284 (11), 7017–7029. doi:10.1074/jbc.M808106200
Switala, J., and Loewen, P. C. (2002). Diversity of properties among catalases. Archives Biochem. Biophysics 401 (2), 145–154. doi:10.1016/S0003-9861(02)00049-8
Tanabe, S., Ishii-Minami, N., Saitoh, K. I., Otake, Y., Kaku, H., Shibuya, N., et al. (2011). The role of catalase-peroxidase secreted by Magnaporthe oryzae during early infection of rice cells. Mol. Plant-Microbe Interactions® 24 (2), 163–171. doi:10.1094/MPMI-07-10-0175
Varnado, C. L., and Goodwin, D. C. (2004). System for the expression of recombinant hemoproteins in Escherichia coli. Protein Expr. Purif. 35 (1), 76–83. doi:10.1016/j.pep.2003.12.001
Vidossich, P., Alfonso-Prieto, M., Carpena, X., Loewen, P. C., Fita, I., and Rovira, C. (2007). Versatility of the electronic structure of compound I in catalase-peroxidases. J. Am. Chem. Soc. 129 (44), 13436–13446. doi:10.1021/ja072245i
Vlasits, J., Jakopitsch, C., Bernroitner, M., Zamocky, M., Furtmüller, P. G., and Obinger, C. (2010). Mechanisms of catalase activity of heme peroxidases. Archives Biochem. Biophysics 500 (1), 74–81. doi:10.1016/j.abb.2010.04.018
Wariishi, H., and Gold, M. H. (1989). Lignin peroxidase compound III: formation, inactivation, and conversion to the native enzyme. FEBS Lett. 243 (2), 165–168. doi:10.1016/0014-5793(89)80122-X
Wariishi, H., and Gold, M. H. (1990). Lignin peroxidase compound III. Mechanism of formation and decomposition. J. Biol. Chem. 265 (4), 2070–2077. doi:10.1016/S0021-9258(19)39941-7
Wiseman, B., Colin, J., Smith, A. T., Ivancich, A., and Loewen, P. C. (2009). Mechanistic insight into the initiation step of the reaction of Burkholderia pseudomallei catalase-peroxidase with peroxyacetic acid. JBIC J. Biol. Inorg. Chem. 14 (5), 801–811. doi:10.1007/s00775-009-0493-9
Yamada, Y., Fujiwara, T., Sato, T., Igarashi, N., and Tanaka, N. (2002). The 2.0 Å crystal structure of catalase-peroxidase from Haloarcula marismortui. Nat. Struct. Biol. 9 (9), 691–695. doi:10.1038/nsb834
Yu, S., Chouchane, S., and Magliozzo, R. S. (2002). Characterization of the W321F mutant of Mycobacterium tuberculosis catalase-peroxidase KatG. Protein Sci. 11 (1), 58–64. doi:10.1110/ps.09902
Zámocký, M., Droghetti, E., Bellei, M., Gasselhuber, B., Pabst, M., Furtmüller, P. G., et al. (2012). Eukaryotic extracellular catalase-peroxidase from Magnaporthe grisea - biophysical/chemical characterization of the first representative from a novel phytopathogenic KatG group. Biochimie 94 (3), 673–683. doi:10.1016/j.biochi.2011.09.020
Zámocký, M., Furtmüller, P. G., and Obinger, C. (2010). Evolution of structure and function of Class I peroxidases. Archives Biochem. Biophysics 500, 45–57. doi:10.1016/j.abb.2010.03.024
Zhao, X., Khajo, A., Jarrett, S., Suarez, J., Levitsky, Y., Burger, R. M., et al. (2012). Specific function of the Met-Tyr-Trp adduct radical and residues Arg-418 and Asp-137 in the atypical catalase reaction of catalase-peroxidase KatG. J. Biol. Chem. 287 (44), 37057–37065. doi:10.1074/jbc.M112.401208
Zhao, X., Suarez, J., Khajo, A., Yu, S., Metlitsky, L., and Magliozzo, R. S. (2010). A radical on the met-tyr-trp modification required for catalase activity in catalase-peroxidase is established by isotopic labeling and site-directed mutagenesis. J. Am. Chem. Soc. 132 (24), 8268–8269. doi:10.1021/ja103311e
Zhao, X., Yu, S., Ranguelova, K., Suarez, J., Metlitsky, L., Schelvis, J. P. M., et al. (2009). Role of the oxyferrous heme intermediate and distal side adduct radical in the catalase activity of Mycobacterium tuberculosis KatG revealed by the W107F mutant. J. Biol. Chem. 284 (11), 7030–7037. doi:10.1074/jbc.M808107200
Keywords: protein-based radicals, peroxide-dependent inactivation, tryptophan, arginine, heme, enzyme inactivation, catalase-peroxidase, intramolecular electron transfer
Citation: Xu H, Kenneson JR, Minton LE and Goodwin DC (2024) A switch and a failsafe: KatG’s mechanism for preservation of catalase activity using a conformationally dynamic Arg and an active-site Trp. Front. Chem. Biol 3:1431412. doi: 10.3389/fchbi.2024.1431412
Received: 11 May 2024; Accepted: 18 June 2024;
Published: 10 July 2024.
Edited by:
Justin M. Miller, Middle Tennessee State University, United StatesReviewed by:
Walter Novak, Wabash College, United StatesCopyright © 2024 Xu, Kenneson, Minton and Goodwin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Douglas C. Goodwin, Z29vZHdkY0BhdWJ1cm4uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.