 Bipin Chakravarthy Paruchuri
Bipin Chakravarthy Paruchuri Sarah Smith
Sarah Smith Jessica Larsen
Jessica Larsen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. Eng., 12 October 2022
Sec. Biochemical Engineering
Volume 4 - 2022 | https://doi.org/10.3389/fceng.2022.997607
This article is part of the Research TopicWomen in Chemical EngineeringView all 8 articles
GM1 gangliosidosis is a lysosomal storage disorder caused by deficiency of β-galactosidase (βgal) and subsequent accumulation of GM1 ganglioside in lysosomes. One of the pathological aspects of GM1 gangliosidosis, and other storage disorders, is impaired autophagy, i.e., a reduced fusion of autophagosomes and lysosomes to degrade cellular waste. Enzyme replacement therapy (ERT) can effectively treat systemic deficiency but is limited by immunogenicity and shortened half-life of intravenously administered enzyme. In this paper, we report a hyaluronic acid-b-polylactic acid (HA-PLA) polymersome delivery system that can achieve an enzyme-responsive and sustained delivery of βgal to promote the cell’s self-healing process of autophagy. HA-PLA polymersomes have an average diameter of 138.0 ± 17.6 nm and encapsulate βgal with an efficiency of 77.7 ± 3.4%. In the presence of model enzyme Hyaluronidase, HA-PLA polymersomes demonstrate a two-fold higher release of encapsulant than without enzyme. We also identified reduced autophagy in a cellular model of GM1 Gangliosidosis (GM1SV3) compared to healthy cells, illustrated using immunofluorescence. Enhanced autophagy was reported in GM1SV3 cells treated with βgal-loaded polymersomes. Most notably, the fusion of lysosomes and autophagosomes in GM1SV3 cells returned to normal levels of healthy cells after 24 h of polymersome treatment. The HA-PLA polymersomes described here can provide a promising delivery system to treat GM1 Gangliosidosis.
With a prevalence of one in 5,000 to 8,000 live births, lysosomal storage diseases (LSDs) typically result from genetic mutations that lead to a lack of production or inappropriate folding of lysosomal enzymes, causing systemic pathology (Kelly and Bradbury, 2017). Linking these diseases together is the dysfunction of the lysosome, which leads to highly similar pathophysiology, especially in the LSDs that are prevalent in the central nervous system (CNS). These disorders are marked by failed autophagy (Figure 1), where the autophagosome and the lysosome are unable to fuse, causing the accumulation of both organelles. The autophagic failure causes a large burden on the affected cells, ultimately leading to premature cell death. This paper focuses on the autophagy-lysosomal pathway in an LSD, GM1 Gangliosidosis (GM1). In GM1, the lysosomal enzyme β-galactosidase (βgal) is missing or underproduced (Brunetti-Pierri and Scaglia, 2008; Johnson, 2015). βgal has the primary role of catabolizing down GM1 ganglioside to GM2 ganglioside (Sandhoff and Harzer, 2013). In the absence of this enzyme, GM1 ganglioside remains unmetabolized and accumulates in the cell along with other storage products, which leads to swollen lysosomes and ultimate cell death (Rha et al., 2021). The deficiency of βgal leads to the upregulation of other lysosomal enzymes, such as hexosaminidase A (De Maria et al., 1998; McCurdy et al., 2014). This is believed to be a compensatory mechanism, aiding the cell in the digestion of byproducts normally broken down by βgal. Although it is accepted that this upregulation occurs, the point within the disease progression where lysosomal enzyme upregulation occurs is unknown. This upregulation can inform decisions on treatment design, specifically utilizing upregulated enzyme activity to increase drug delivery through enzyme-responsive degradation (Paruchuri et al., 2021).
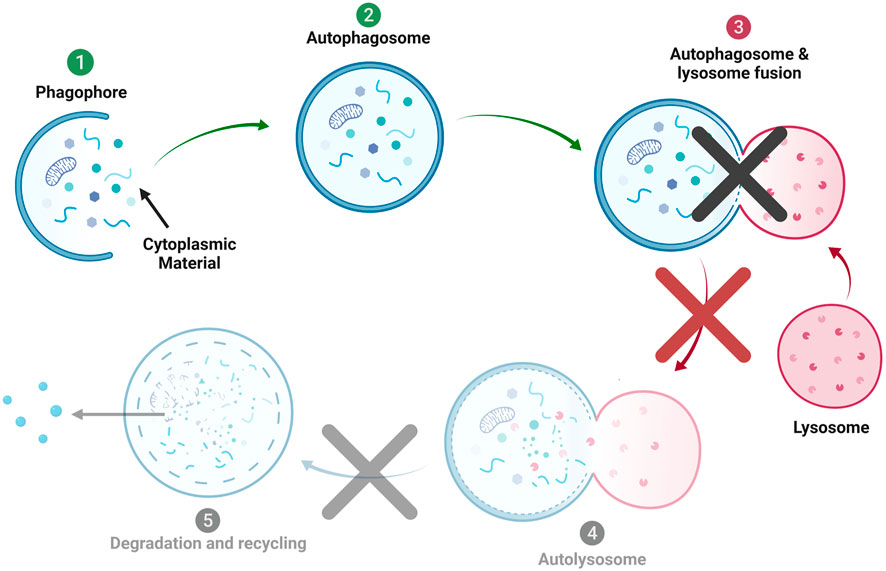
FIGURE 1. Autophagic Process in Lysosomal storage disorders (LSD). In many LSDs diseases, lack of autophagosome-lysosome fusion is believed to be the main cause of autophagic failure and apoptosis.
Among the approaches proposed to treat LSDs, enzyme replacement therapy (ERT) is the most often translated therapy and is clinically effective in reducing the lysosomal storage (Solomon and Muro, 2017). ERT involves periodic administration of the recombinant enzyme to be delivered to the cell and catabolizes the accumulated substrate. Even a partial restoration of the deficient enzyme activity can result in a clearance of the substrate and show a therapeutic effect (Muro, 2010; McCurdy et al., 2014). Currently, 15 ERTs have been approved to treat 11 different LSDs (Edelmann and Maegawa, 2020). Despite the advantages, ERT is limited by immunogenicity, short half-life in the circulation system, uneven biodistribution, and reduced activity of the enzyme (Kakkis et al., 1996; Ohashi et al., 2008; Wang et al., 2008; Banugaria et al., 2011; de la Fuente et al., 2021). Furthermore, when the CNS is involved, as it is in GM1 gangliosidosis, ERT is ineffective, as enzymes can not be transported through the blood-brain barrier (BBB). In case of severe immune response, systemic ERT may require immune modulation to maintain efficacy (Garman, Munroe and Richards, 2004; Dickson et al., 2008; Desai et al., 2020). One of the strategies to mitigate these limitations is the encapsulation of recombinant enzyme in nanoparticles (Mumtaz and Bachhawat, 1994; Muro, 2010; Blackman et al., 2018; Galliani et al., 2018; Grosso et al., 2019; Edelmann and Maegawa, 2020). Encapsulating enzymes in nanoparticles increases the therapeutic effect and reduces immune response by masking the enzyme molecule. Ultimately, although beyond the scope of this paper, targeting ligands can be added to the surface of these nanoparticles to facilitate the transport of enzyme through the BBB (Sánchez-Purrà et al., 2016; Kelly and Gross, 2017; Nabi et al., 2018; Iqbal et al., 2020).
Here we develop a nanoparticle tool capable of correcting autophagic failure using ERT and promoting healthy cellular digestion. We identify lysosomal hydrolase upregulation and impaired autophagy in a cellular model of GM1, isolated fibroblasts from GM1 affected felines (GM1SV3 cells), in comparison to isolated fibroblasts from healthy felines (NSV3 cells). A naturally occurring feline model of GM1 gangliosidosis was first identified in 1971 and closely replicates the human pathology of the juvenile form of disease (Baker et al., 1971; Baker and Lindsey, 1974; Martin et al., 2008). This model has been used extensively in pre-clinical studies to evaluate gene therapies targeting GM1 gangliosidosis (Rockwell et al., 2015; Gray-Edwards et al., 2017; Nicoli et al., 2021; Gross et al., 2022). These feline fibroblasts have previously been used to explore the efficacy of nanoparticle mediated enzyme replacement therapy (Reynolds et al., 1978; Kelly and Gross, 2017). Therefore, we use the feline cell model to evaluate our novel enzyme-responsive polymersomes that can respond to lysosomal enzymes and deliver βgal in a pathologically driven manner. We use hyaluronic acid (HA) and polylactic acid (PLA) as the hydrophilic and hydrophobic components, respectively, to create polymersomes that facilitate the entry of βgal into GM1-affected cells and restore normal autophagic function. These polymersomes could ultimately be used as a treatment for GM1 by helping the cells clear storage products through the promotion of autophagy.
Phenotypically, NSV3 (healthy) and GM1V3 (affected) cell lines are highly similar, with extended, spiny processes typical of fibroblast cell lines, as seen under bright field (BF) (Figure 2).
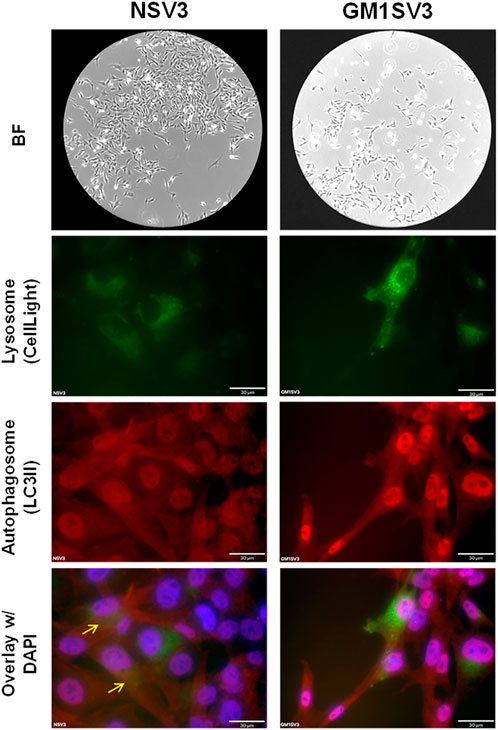
FIGURE 2. Phenotypic, Lysosomal, and Autophagic behavior of untreated NSV3 (healthy) and GM1SV3 (diseased) cell lines. (Scale bar = 30 μm).
GM1SV3 cells presented with a higher number of lysosomes (green) compared to healthy NSV3 cells. Similar behavior was observed with autophagosomes (red), where GM1SV3 cells presented more autophagosomes than NSV3 cells. When the green and red channels were overlaid, we observed that the autophagic flux in GM1SV3 cells was reduced in comparison to the healthy NSV3 cells (Figure 2).
The X-gal stained GM1SV3 cells qualitatively confirmed the deficiency of βgal as indicated by the negligible blue color compared to healthy cells (Figures 3A,B). Both results are consistent with the X-gal-stained tissue slices of GM1-affected and normal feline brain and spinal cord (McCurdy et al., 2014) (Figure 3C). Standard enzyme assay analysis indicated increased levels of lysosomal hydrolases hexosaminidase A and mannosidase in GM1SV3 cells compared to NSV3 cells. Fold normal enzyme activities presented in Figure 3D demonstrate the deviation between GM1SV3 and NSV3 cellular lysosomal enzyme activities, with larger numbers above a fold normal of one indicating upregulation.

FIGURE 3. Xgal staining results of (A) GM1SV3 and (B) NSV3 cell lines, compared to (C) brain (top) and spinal cord (bottom) of GM1-affected felines (From McCurdy et al., 2015. Reprinted with permission from AAAS). (D) Fold normal activities of β-galactosidase, hexosaminidase A, hexosaminidase total, and mannosidase in GM1SV3 cells. The dashed line represents activity in NSV3 cells. (n = 3).
The amphiphile HA-PLA was successfully synthesized using a two-step conjugation method (Supplementary Figure S1) (Deng et al., 2018). The absorbance at 3,284 cm−1 in the ATR-FITR spectrum of synthesized polymer was attributed to the stretching of -OH and -NH groups of HA, while the absorbance peaks for ester C=O and C-O stretching in PLA-NHS were observed at 1751 and 1,181 cm−1, respectively (Supplementary Figure S2). This confirmed the successful conjugation of the block polymers HA and PLA. The hydrophilic fraction (f) of the resultant copolymer was ∼0.17. HA-PLA polymersomes were self-assembled using the solvent injection method (Kelly and Bradbury, 2017). The hydrophobic membrane observed in TEM confirmed the vesicular structure of the self-assembled nanoparticles (Figure 4A). HA-PLA polymersomes have a monodisperse size distribution (Figure 4B) with a Z-average diameter of 138.0 ± 17.6 nm (PDI = 0.212 ± 0.06) and ζ-potential of -18.0 ± 3.8 mV, measured using DLS (Figure 4C).

FIGURE 4. Hyaluronic acid-polylactic acid (HA-PLA) polymersome characterization (A) The vesicular morphology of HA-PLA polymersomes observed by transmission electron microscope (scale bar: 1 μm, inset: 100 nm), (B) the size distribution of HA-PLA polymersomes observed by dynamic light scattering (n = 4), (C) the concentration of HA-PLA used in solvent injection method, Z-average diameter, polydispersity index and ζ-potential of HA-PLA polymersomes measured using dynamic light scattering (n = 4).
HA-PLA polymersomes encapsulated AF488-tagged βgal with an efficiency of 77.7 ± 3.4% and a loading capacity of 0.194 mg AF488 βgal/mL polymersomes. HA-PLA polymersomes were loaded with hydrophilic fluorescein isothiocyanate-dextran (FITC-D) to evaluate release with and without enzymatic degradation. In the presence of model cognate enzyme Hyaluronidase, a total release of 30.8 ± 4.4% was observed after 68 h, while incubation in buffer and with non-cognate βgal released only 14.4 ± 5.3% and 19.2 ± 4.8% of encapsulant (Figure 5).
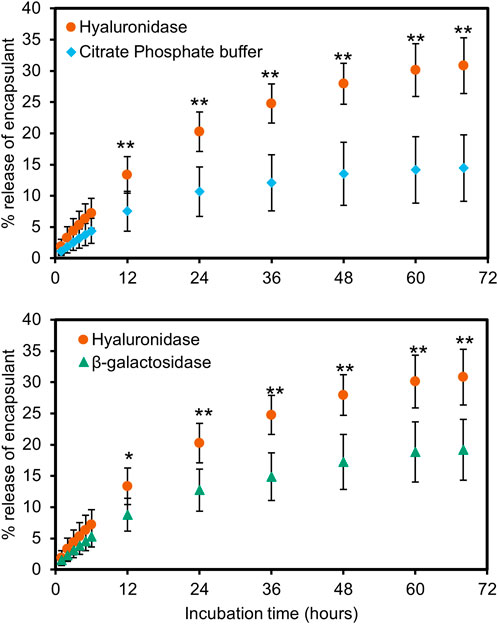
FIGURE 5. Release of encapsulated fluorescein isothiocyanate-dextran from Hyaluronic acid-polylactic acid (HA-PLA) polymersomes when incubated with hyaluronidase (●), citrate phosphate buffer (pH 4.8) (♦) and β-galactosidase (▲). These results show a higher release of encapsulant in the presence of hyaluronidase compared to buffer without enzyme (top) or in β-galactosidase, a non-cognate enzyme (bottom). (n = 4, * = p < 0.05, ** = p < 0.01).
The cellular uptake of HA-PLA polymersomes was studied using the fluorescent molecule FITC-bovine serum albumin (BSA) which was encapsulated with an efficiency of 93.3 ± 3.6%. GM1SV3 cells showed a similar fluorescence intensity for GM1SV3 cells incubated with free FITC-BSA and FITC-BSA loaded HA-PLA polymersomes (Figure 6A), indicating that cellular uptake of HA-PLA Ps was on par with the free drug. This observation was consistent with the quantified fluorescence results obtained using flow cytometry (Figure 6B).
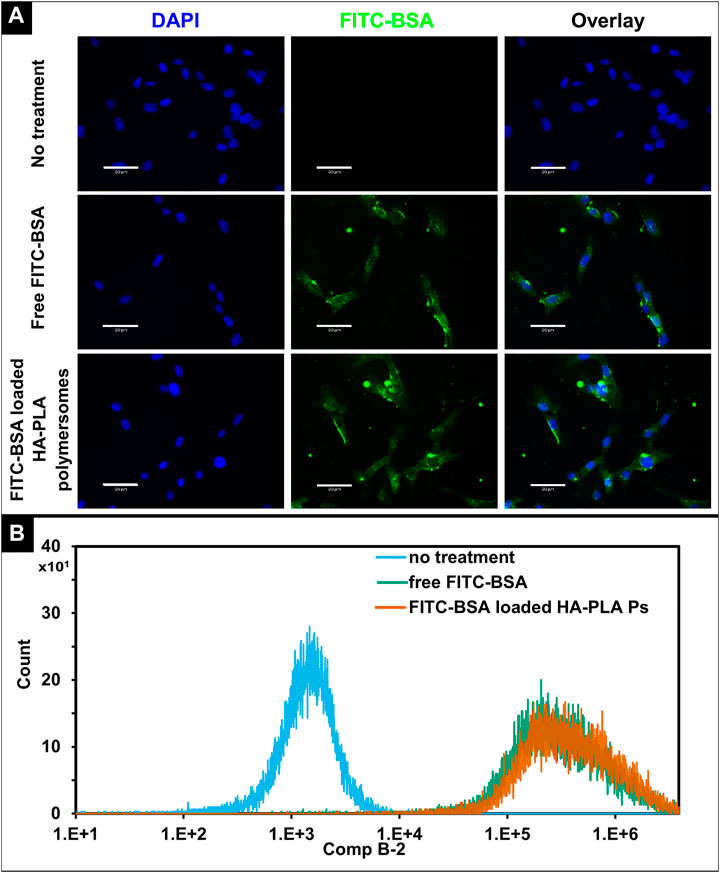
FIGURE 6. In vitro cellular uptake of Hyaluronic acid-polylactic acid (HA-PLA) polymersomes. (A) Fluorescent images of GM1SV3 cells without treatment, after treating with free albumin-fluorescein isothiocyanate conjugate (FITC-BSA) and FITC-BSA loaded HA-PLA polymersomes, (B) Flow cytometry histograms for unstained GM1SV3 cells, GM1SV3 cells treated with free FITC-BSA, and FITC-BSA loaded HA-PLA polymersomes (n = 3). (Scale bar = 60 μm).
We examined the therapeutic effect of current delivery system by treating the diseased cells βgal-loaded polymersomes at a dose of 0.35 mg βgal/cm2 to restore the enzyme levels (Kelly and Gross, 2017). After 24 h of polymersome treatment, GM1SV3 cells presented with reduced number of lysosomes and enhanced autophagy compared to untreated cells (Figure 7). Confocal imaging confirms findings, with observable co-localization after treatment with free βgal for 6 h (Supplementary Figure S3C) or βgal loaded HA-PLA polymersomes after 24 h (Supplementary Figure S3D). These observations are confirmed by image analysis quantifying the autophagy as co-localization percentage per cell (Figure 8). Importantly, after 24 h of polymersome treatment, with a co-localization percentage of 38.4 ± 11.1%, the autophagic behavior of GM1SV3 cells is no longer statistically different from NSV3 cells (38.4 ± 10.5%) or GM1SV3 cells treated with free βgal for 6 h (40.4 ± 12.7%).
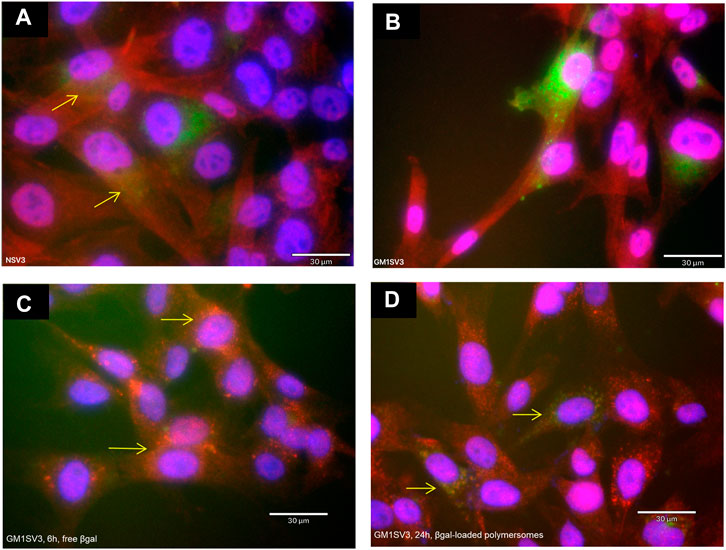
FIGURE 7. Autophagic behavior of (A) untreated healthy (NSV3) cells, (B) untreated diseased (GM1SV3) cells, after treating GM1SV3 cells with (C) free β-galactosidase (βgal) for 6 h and (D) βgal-loaded hyaluronic acid-polylactic acid (HA-PLA) polymersomes for 24 h (Scale bar = 30 μm). These results show that treatment with βgal-loaded HA-PLA polymersomes causes the autophagy of diseased cells to look more like healthy cells.
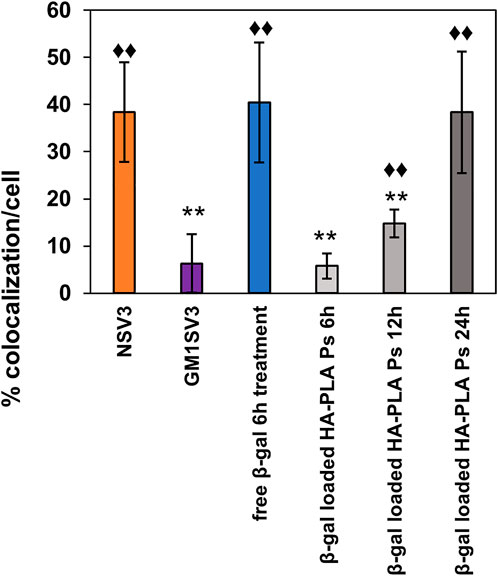
FIGURE 8. Co-localization (%) in untreated healthy (NSV3) and diseased (GM1SV3) cells, GM1SV3 cells treated with free βgal (6 h treatment), βgal-loaded hyaluronic acid-polylactic acid (HA-PLA) polymersomes for 6, 12 and 24 h. These results indicate that HA-PLA polymersomes achieved a therapeutic effect in 24 h, similar to 6 h of treatment with free βgal, due to the slow and controlled release of encapsulated βgal. Star (*) indicate a significant difference compared to NSV3 cells and diamond (♦) indicates a significant difference in colocalization compared to GM1SV3 cells (n = 3, * = p < 0.05, ** = p < 0.025).
Phenotypically, NSV3 and GM1SV3 cells presented similar morphological features, as characterized previously (Reynolds et al., 1978; Kelly and Bradbury, 2017; Chen et al., 2020; Gross et al., 2022). These cell lines also match with enzyme activity levels expected based on in vivo results from GM1 gangliosidosis-affected felines where the X-gal-stained tissues of diseased animals showed deficient enzyme levels compared to healthy controls (Figures 3A–C) (McCurdy et al., 2015). We also observed that NSV3 and GM1SV3 cells show differing levels of autophagic activity. Greater intensities of red and green indicate an increased presence of autophagosomes and lysosomes independently, while yellow in overlay images indicates the co-localization of autophagosomes and lysosomes. Therefore, the presence of yellow in NSV3 cells implies a normal fusion of these organelles to form autolysosomes, and the absence of yellow in GM1SV3 samples indicates an impaired fusion and disruption of normal autophagy. This was expected in in vitro GM1 model as similar results were seen in the GM1 mouse model (Liu et al., 2021) and other models of LSDs such as Multiple Sulfatase Deficiency (MSD) (Settembre et al., 2008), mucopolysaccharidosis type IIIA (Settembre et al., 2008), Pompe disease (Lim et al., 2018), and Gaucher disease (Kinghorn et al., 2016). In a mouse model of MSD, about a 40% decrease in co-localization compared to wild-type animals was reported. However, the results presented here were more dramatic than these, with an almost 90% decrease in co-localization in diseased cells. This difference could be explained by the differences occurring between diseases and disease models, with MSD co-localization studies using embryonic fibroblasts of MSD-affected mice and this study using skin fibroblasts of GM1-gangliosidosis-affected felines. Also, previously, an increase in expression of LC3-II, an autophagosome-specific marker, has been observed in GM1 (Takamura et al., 2008; Lieberman et al., 2012). The LC3-II fluorescence intensity was much greater in the βgal−/− mice samples compared to the wild type. This was also shown in our cell model when tagging autophagosome LC3-II with LC3 antibody, the red intensity tagging autophagosomes was greater in GM1SV3 than NSV3. This observed increase of LC3-II indicates that there is a buildup of autophagosomes that are not fusing with lysosomes to form the autophagosome complex. Because of this, GM1SV3 cells appear to be valid as a model of GM1 gangliosidosis, especially with respect to autophagic behavior.
Polymersomes are suitable for delivering the deficient enzyme to diseased cells (Iqbal et al., 2020), which can enhance the fusion of autophagosomes and lysosomes (Ivanova et al., 2019; Marques et al., 2020). Within the polymersome, an enzyme-responsive moiety can be incorporated in either 1) the hydrophilic corona, 2) the hydrophobic membrane or 3) the link between the two zones to offer better control over the release profile (Li et al., 2020; Paruchuri et al., 2021). In the current system, we synthesized an amphiphile with the enzyme-degradable HA as the hydrophilic block and acid-hydrolyzable PLA. As a result, HA is located in the hydrophilic corona of the self-assembled structures, where it is easily accessible to enzymes. The HA-PLA amphiphile self-assembled into vesicles with a membrane observed in the TEM imaging. HA-PLA polymersomes were within the biologically relevant size range of 10–200 nm: greater than 10 nm to avoid renal clearance (Choi et al., 2011) and smaller than 200 nm to avoid clearance through the reticuloendothelial system (Moghimi et al., 2001; Moghimi and Szebeni, 2003; Emerich and Thanos, 2007; Kulkarni and Feng, 2013). Drug loading of HA-PLA polymersomes was slightly lower than previously reported PEG1000-PLA5000 polymersomes (0.22 mg AF488 βgal/mL polymersomes) (Kelly and Bradbury, 2017). The high molecular weight of HA-PLA copolymer increases the thickness of hydrophobic membrane (Discher and Ahmed, 2006), which could limit the entry of AF488 βgal into polymersomes during the loading process.
When HA-PLA polymersomes are incubated with hyaluronidase, the enzymatic hydrolysis of HA in corona destabilizes the polymersome structure (Hu et al., 2017; Li et al., 2020; Bourgat et al., 2021; Paruchuri et al., 2021). As a result, a two-fold higher release of FITC-D was observed when treated with hyaluronidase compared to buffer. A similar phenomenon was previously observed with hyaluronic acid-g-deoxycholic acid (DOCA) micelles loaded with doxorubicin (Dox) targeting tumors (Kim et al., 2014). The release of encapsulated Dox was two-fold higher in the presence of hyaluronidase type II than without the enzyme. Other enzyme-responsive polymersomes and polymeric micelle systems also exhibited similar release behavior in the presence of esterase (Yan et al., 2020), hyaluronidase (Tücking et al., 2015) and matrix metalloproteinase-2 (Barve et al., 2020; Ramezani et al., 2020). Furthermore, in HA-PLA polymersomes, a significant difference in the amount released with and without hyaluronidase was observed starting at 12 h, indicating a slow degradation of polymersomes initially. Interestingly, a similar degradation behavior was reported previously in an HA-based in situ cross-linkable hydrogel. When exposed to hyaluronidase, the wet gel lost 50% of the original mass in 7 days but disappeared completely in 10 days (Bajaj et al., 2012). The release from HA-PLA polymersomes in buffer and with βgal were not significantly different, which is expected as βgal does not hydrolyze any component of polymersomes. This release behavior is translatable to other encapsulants as the drug release kinetics of enzyme-responsive polymersomes are determined by stimuli (rate of cleavage) and polymer degradation rate (Lee and Yeo, 2015; Hu et al., 2017). These results also suggest the utility of HA-PLA polymersomes in other diseases with upregulation of hexosaminidase A (Minami et al., 1980; Sferra et al., 2004) or hyaluronidase (Tan et al., 2011). HA is a primary component of the extracellular matrix and can bind with high affinity to the CD44 receptor, a transmembrane glycoprotein (Tiwari and Bahadur, 2019). The uptake mechanism of HA-grafted liposomes was previously reported to follow lipid raft-mediated pathways and the liposomes localized primarily in lysosomes (Syed et al., 2011). This native uptake mechanism HA nanoparticle and endocytosis of albumin (Frei, 2011) can explain the comparable fluorescence intensities of free FITC-BSA and FITC-BSA loaded polymersomes in GM1SV3 cells after 4 h treatment. Similarly, βgal-loaded HA-PLA polymersomes in our study could be taken up by cells and directed to lysosomes through the endosomal-lysosomal pathway. It is important to note that ultimately polymersomes could facilitate entry of payloads, like BSA, into areas they are unable to natively enter, like the BBB, through attachment of targeting ligands.
Within lysosomes, the HA in hydrophilic corona is degraded by hydrolases such as hexosaminidase A, β-glucuronidase, and lysosomal hyaluronidase (Gushulak et al., 2012) to induce the release of encapsulated βgal. Supplementing the deficient enzyme levels in LSD models can activate the autophagy pathway (Ivanova et al., 2019; Marques et al., 2020). In peripheral blood mononuclear cells (PBMCs) of patients with an LSD called Gaucher’s disease, treatment with recombinant human glucocerebrosidase resulted in normalized levels of lysosomal (LAMP1) and autophagosomes (LC3-1) markers closer to healthy controls during the first hour (Ivanova et al., 2019). This indicates the activation of autophagy, although the mechanism remains unclear. Similarly, in the current study, GM1SV3 cells treated with loaded polymersomes presented a higher intensity of yellow, suggesting a greater healthy autophagic activity compared to untreated cells. Figure 8 shows partial restoration of autophagic activity after 6 and 12 h of treatment with βgal-loaded polymersomes, and normalization of autophagic activity upon 24 h of treatment, indicated by no significant difference in colocalization compared to NSV3. In contrast, 6 h of treatment with free βgal was sufficient to increase the autophagic activity to normal levels. This result also indicates the slow and controlled release of the encapsulant from polymersomes compared to free βgal.
In this study, we developed an enzyme-responsive polymersome system made from HA and PLA that can degrade in the lysosomes and deliver the cargo to increase autophagic activity in lysosomal storage disorder GM1-affected cells. These polymersomes are capable of encapsulating active enzyme at high loading efficiencies of 77.7 ± 3.4%. Benchtop release studies demonstrate the role of enzyme-responsive degradation on the release of encapsulated payloads, with enzyme-responsive behavior apparent due to the two-fold increase in release when incubated with the cognate enzyme at the same time points in comparison to buffer or non-cognate enzyme incubations alone. When cultured with GM1-affected cells, GM1SV3, at low doses, autophagic activity is restored to healthy levels that are not statistically different from a healthy cell line, NSV3. Free βgal restores autophagic function as well, but is unable to cross the BBB (Przybilla et al., 2021), which could be facilitated through the addition of targeting ligands on the HA-PLA nanoparticle surface. Restoration of autophagic activity is likely to alleviate the burden of lysosomal storage in GM1-affected patients. To our knowledge, this study represents the first time that nanoparticles are used to alleviate the storage burden in lysosomal storage disease through targeted autophagic modulation, making this a promising study to promote further exploration towards a non-invasive treatment.
Dr. Douglas Martin of Auburn University donated skin fibroblasts from healthy cats, NSV3 cells, and GM1 gangliosidosis-affected cats, GM1SV3 cells. Media was Dulbecco’s Modified Eagle Media (DMEM) (ThermoFisher, Cat. Num. 12,430, Waltham, MA) with 10% Fetal Bovine Serum (FBS) and 1X Penicillin-Streptomycin (PS) (ThermoFisher, Cat. Num. 15140122, Waltham, MA). Passages were performed using 0.25% Trypsin (Corning, Cat. Num. 25-053-Cl, Manassas, VA). CellLight™ Lysosomes-GFP BacMam 2.0 (ThermoFisher, Cat. Num. C10507, Waltham, MA) and LC3 staining with an anti- LC3B Rabbit Polyclonal Antibody (ThermoFisher, Cat. Num. PA1-46286, Waltham, MA) (1:300) and Donkey anti-Rabbit IgG (H + L) ReadyProbes™ Secondary Antibody conjugated to Alexa Fluor 594 (ThermoFisher, Cat. Num R37119, Waltham, MA) were used for immunofluorescence. Fixation occurred with 4% paraformaldehyde (PFA) (Ted Pella, Cat. Num. 18,505, Redding, CA) or glutaraldehyde (Sigma Aldrich, Cat. No. G7651-10ML, St. Louis, MO) and growth/starvation media. Cells were permeabilized with 0.1% Triton 100X (Acros Organics, Cat. No. 215682500, Waltham, MA). When blocking was required, 5% donkey serum (Jackson ImmunoResearch, Cat. No. 017-000–121, West Grove, PA) was used. Mounting medium, Vectashield Antifade Mounting Medium with DAPI (Vector, Cat. Num. H-1200, Burlingame, CA) and cytoseal xyl (Fisher Scientific, Cat. No. 22050262, Waltham, PA), were also used.
Enzyme assays and x-gal staining require the use of the following materials: 4-methylumbelliferone (4-MU) (Sigma Aldrich, Cat. No. M1381, St. Louis, MO), 10N NaOH (BDH, Cat. No. BDH7247-1, Radnor, PA), citric acid (Fisher Scientific, Cat. No. A104-3, Hampton, NH), sodium phosphate (Sigma Aldrich, 7782-85–6, St. Louis, MO), sodium chloride (BDH, Cat. No. BDH9286, Radnor, PA), 0.1% Triton X-100 (Acros Organics, Cat. No. 215682500, Waltham, MA), magnesium chloride hexahydrate (Sigma Aldrich, Cat. No. M2670-100G, St. Louis, MO), sodium deoxycholate (Sigma Aldrich, Cat. No. D6750-10G, St. Louis, MO), potassium ferricyanide (Sigma Aldrich, Cat. No. 702587-50G, St. Louis, MO), potassium ferrocyanide (Fisher Scientific, Cat. No. P236500, Waltham, PA), and 0.05% bovine serum albumin (Fisher Scientific, Cat. No. BP1600-100, Hampton, NH). Substrates to measure enzyme activity were 4-methylumbelliferyl βgal (Acros Organics, Cat. No. 337210010, Waltham, MA), 4-methylumbelliferyl a-D-manopyranoside (Toronto Research Chemicals, Cat. No. M334745, Ontario Canada), 4-methylumbelliferyl 6-sulfa-2-acetoamido-2-deoxy-b-D-glucopyranosidase (Toronto Research Chemicals, Cat. No. M335000, Ontario Canada), and 4-methylumbelliferyl N-acetyl b-D-glucosaminide (Sigma Aldrich, Cat. No. M2133, St. Louis, MO). Lowry solution A consisted of copper sulfate (Fisher Scientific, Cat. No. C493-500, Waltham, MA), sodium potassium tartrate (BDH, Cat. No. BDH9272, Radnor, PA), and sodium carbonate (BDH, Cat. No. BDH9284, Radnor, PA). Solution B was made of Folin and Ciocalteu’s phenol reagent (Sigma Aldrich, Cat. No. F9252, St. Louis, MO) and MILLI-Q water.
Hyaluronic acid (molecular weight–5000 Da) (Lifecore Biomedical LLC, Cat No. HA5K, Chaska, MN), Poly (L-lactide)-N-hydroxysuccinimide endcap (Mn molecular weight ∼24,450 Da) (Akina, Inc., Cat. No. AI174, West Lafayette, IN), 1,4-diaminobutane (Fisher Scientific, Cat No. AC112120250, Hampton, NH), sodium cyanoborohydride (Chem-Impx Intl Inc., Cat. No. 4836, Wood Dale, IL), 10N NaOH (BDH, Cat. No. BDH7247-1, Radnor, PA), Sodium acetate trihydrate (BDH, Cat. No. BDH9278, Radnor, PA), N,N-diisopropyl- ethylamine (Acros Organics, Cat. No. 115220250, Waltham, MA) were used to synthesize HA-PLA. Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, Cat. No. 472301, St. Louis, MO) and D-mannitol (Fisher Scientific, Cat. No. M120, Hampton, NH) were used in polymersome synthesis. Fluorescently tagged βgal was prepared using βgal isolated from bovine liver (Sigma Aldrich, Cat. No. G1875, St. Louis, MO) and Alexa Fluor 488 (AF488) Protein Labeling Kit (Invitrogen, Cat. No. A10235, Waltham, MA). Fluorescein isothiocyanate–dextran (FITC-D) (Sigma Aldrich, Cat. No. 46944, St. Louis, MO), Albumin Fluorescein isothiocyanate Conjugate from bovine (FITC-BSA) (Sigma Aldrich, Cat. No. A9771, St. Louis, MO) were used for encapsulation and cellular uptake studies respectively. Hyaluronidase from bovine testes was used in release studies (Sigma Aldrich, Cat. No. H3506, St. Louis, MO).
NSV3 and GM1SV3 cells were passaged into chamber slides at a seeding density of 9.35 × 104 cells per well in normal growth media with 2 µL of CellLight per well. After overnight incubation, cells were fixed with ice-cold 4% PFA in the appropriate growth media (normal growth media or starvation media) for 15 min. Cells were then permeabilized with 0.1% Triton X-100 in PBS, 3 times for 5 min each. A blocking step was performed with 5% donkey serum in PBS for 1 h. Cells were then washed with PBS and incubated overnight at 4°C with 1:300 primary antibody dilution in PBS supplemented with 4% FBS for a total working volume of 500 μl. After a second overnight incubation, cells were washed 4 times with PBS for 10 min at a time. Secondary antibody was added to the appropriate wells ensuring untreated, primary-only, and secondary-only wells were run during each experiment as internal controls (Supplementary Figure S5) with 500 µl of appropriate growth media and allowed to incubate in the dark at room temperature for 30 min. Cells were washed with PBS, chambers removed, and one drop of Vectashield Antifade Mounting Medium with DAPI was added. Slides were then sealed and allowed to dry overnight at 4°C. The cells were imaged using both fluorescence microscopy (Echo Revolve) and confocal microscopy (Leica SPE Confocal). The fluorescence images were analyzed using ImageJ and the “Color Inspector 3D″ plugin. In order to quantify yellow and blue pixels, the LAB color code was used to isolate the color threshold. Scale bars were specified in the color range for quantification; the values used are found in Supplementary Table S2. After setting the color range, each cell body was isolated, and the frequency of yellow and blue pixels was determined. After obtaining this data, the percent co-localization was calculated as the number of yellow pixels per number of blue pixels.
After incubation of NSV3 and GM1SV3 cells in 6-well plates, the media was changed, and wells were manually scraped. After centrifugation, cells were re-suspended in Enzyme Isolation Buffer (0.1% Triton X-100 in 50 mM citrate phosphate buffer, pH 4.4 and 0.05% bovine serum albumin) and disrupted using 3 ml syringes with an 18-gauge needle. After enzyme isolation, enzyme activity of lysosomal hydrolases βgalactosidase, hexosaminidase A, hexosaminidase B, and mannosidase were measured using 4-methylumbelliferone enzyme assays described previously (Martin et al., 2005; Martin et al., 2008; Kelly and Gross, 2017).
X-gal staining method causes blue staining to indicate the activity of βgal, the deficient enzyme in GM1 gangliosidosis (Samoylova et al., 2008; McCurdy et al., 2014). NSV3 and GM1SV3 cells were passaged into chamber slides at a seeding density of 9.35 × 104 cells per well in normal growth media. After overnight incubation, X-gal staining was completed as previously described and compared to clinical feline data already published (McCurdy et al., 2015).
HA-PLA polymer was synthesized using a two-step conjugation method adapted from a previous report (Deng et al., 2018). Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra of the polymer blocks and the copolymer were obtained.
HA-PLA polymersomes were formed using the solvent injection method. Briefly, HA-PLA was dissolved in DMSO at a concentration of 0.8 μmol/ml. The polymer solution was injected into 8wt/v% mannitol in water at a rate of 20 μL/min under stirring, using a 20-gauge needle and a syringe pump. Polymersomes were subsequently lyophilized after slow freezing at -20°C, followed by overnight freezing at -80°C, and stored under vacuum conditions to be used later for enzyme encapsulation and in vitro experiments. The size distribution of HA-PLA polymersomes was obtained using Malvern Zetasizer Nano ZS (Malvern Ltd.) at 25°C after separation using 450 nm filters (n = 3). The morphology of polymersomes was observed using transmission electron microscope (TEM) imaging. The grid with polymersomes was stained using uranyl acetate and imaged with Hitachi HT7830 UHR 120 kV TEM.
Encapsulation efficiency and dye release were determined by UV-Vis analysis. The enzyme of interest βgal was tagged with fluorophore AF488 to allow the quantification of the encapsulated enzyme. AF488 tagged βgal was prepared at a concentration of 0.25 mg/ml and loaded into the polymersomes by dropwise addition of enzyme solution to lyophilized polymersomes. The unencapsulated enzyme was removed through dialysis of the loaded polymersomes for 4 h. The fluorescence of dialysate samples was measured in a UV spectrophotometer to calculate the amount of free enzyme by comparing it with a standard curve. Encapsulation efficiency (EE) and loading capacity (LC) of the polymersomes were calculated using the following equations.
Following the loading protocol mentioned above, FITC-D was encapsulated with an efficiency of 26.5 ± 6.6%. Loaded polymersomes were centrifuged and resuspended in 100 µL of 1 mg/ml hyaluronidase (cognate enzyme) solution. Polymersomes resuspended in 100 µL of 1 mg/ml βgal (non-cognate enzyme) solution and citrate phosphate buffer (pH 4.8) were used as controls. The samples were dialyzed against citrate phosphate buffer at 37°C for 68 h. The dialysate was analyzed periodically and compared against a standard curve of FITC-D to calculate release profiles in both enzymatic conditions.
FITC-bovine serum albumin (BSA) was used as a model encapsulant for βgal due to their similar molecular weights [∼66 kDa for FITC-BSA vs 68 kDa for βgal (DiCioccio et al., 1984)]. GM1SV3 cells were seeded in a 4-well chamber slide at a seeding density of 5 × 104 cells per cell. After overnight incubation, the cells were treated with free FITC-BSA and HA-PLA polymersomes loaded with FITC-BSA for 4 h to allow internalization. For fluorescent imaging, the cells were fixed with ice-cold 4% PFA 2 times for 10 min each. After fixation, cells were washed with PBS, the chambers were removed, and one drop of Vectashield Antifade Mounting Medium with DAPI was added to stain the nucleus. The slides were then sealed with a cover slip and dried overnight at 4°C. Fluorescence imaging was carried out with an ECHO Revolve microscope. For flow cytometry, after treating the cells with polymersomes, cells were harvested using trypsin and growth media, and the resultant cell pellet was resuspended in PBS for immediate flow cytometry.
GM1SV3 cells were passaged into chamber slides at a seeding density of 5 × 104 cells per well and incubated in growth media overnight, followed by in normal growth media with 2 µl of CellLight Lysosome-GFP per well for 16 h. Then the cells were treated with either free βgal for 6 h or βgal-loaded HA-PLA polymersomes at a dose of 0.35 mg/cm2 for 6, 12, or 24 h. After the polymersome treatment, the immunofluorescence labeling with anti-LC3B and image analysis were carried out, as mentioned above.
Statistical analysis between NSV3 co-localization and co-localization in GM1SV3 before and after treatment with free βgal or βgal-loaded polymersomes was completed by using a two-tailed student’s t-test with samples of unequal variance (p < 0.05, p < 0.025). Statistical analysis between the release of FITC-D with hyaluronidase and citrate phosphate buffer, between hyaluronidase and βgal was completed by using a student’s t-test with samples of unequal variance (p < 0.05 and p < 0.01).
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Conceptualization: BP, JL; methodology: BP and SS; writing “draft preparation by BP, sections added by SS; writing” review and editing: BP and JL; project administration and funding acquisition: JL. All authors have read and agreed to the published version of the manuscript.
This project was funded in part by the National Institutes of Health Project number 5P20GM103499-19 through the Developmental Research Project Program. This work was supported in part by the National Science Foundation EPSCoR Program under NSF Award # OIA-1655740 and the National Science Foundation CAREER program under NSF Award # 2047697. The article publishing charge was partially funded by the Clemson University Open Access Publishing Fund.
We thank Douglas Martin and Amanda Gross at Auburn University for the use of the NSV3 and GM1SV3 cell lines and Eric Davis for the use of ATR-FTIR spectrophotometer. We also thank Dorian Foster who collected zeta potential data for our HA-PLA polymersomes.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fceng.2022.997607/full#supplementary-material
Bajaj, G., Kim, M. R., Mohammed, S. I., and Yeo, Y. (2012). Hyaluronic acid-based hydrogel for regional delivery of paclitaxel to intraperitoneal tumors. J. Control. Release 158 (3), 386–392. Elsevier. doi:10.1016/j.jconrel.2011.12.001
Baker, H. J. J., Lindsey, J. R., McKhann, G. M., and Farrell, D. F. (1971). Neuronal GM1 gangliosidosis in a Siamese cat with beta-galactosidase deficiency. Science 174 (4011), 838–839. doi:10.1126/science.174.4011.838
Baker, H. J., and Lindsey, J. R. (1974). Animal model: feline GM1 gangliosidosis. Am. J. Pathol. 74 (3), 649–652.
Banugaria, S. G., Prater, S. N., Ng, Y. K., Kobori, J. A., Finkel, R. S., Ladda, R. L., et al. (2011). The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: Lessons learned from infantile Pompe disease. Genet. Med. 13 (8), 729–736. Elsevier. doi:10.1097/GIM.0b013e3182174703
Barve, A., Jain, A., Liu, H., Zhao, Z., and Cheng, K. (2020). Enzyme-responsive polymeric micelles of cabazitaxel for prostate cancer targeted therapy. Acta Biomater. 113, 501–511. Elsevier. doi:10.1016/j.actbio.2020.06.019
Blackman, L. D., Varlas, S., Arno, M. C., Houston, Z. H., Fletcher, N. L., Thurecht, K. J., et al. (2018). Confinement of therapeutic enzymes in selectively permeable polymer vesicles by polymerization-induced self-assembly (PISA) reduces antibody binding and proteolytic susceptibility’, ACS central science. ACS Cent. Sci. 4 (6), 718–723. doi:10.1021/acscentsci.8b00168
Bourgat, Y., Tiersch, B., Koetz, J., and Menzel, H. (2021). Enzyme degradable polymersomes from chitosan-g-[poly-l-lysine-block-ε-caprolactone] copolymer’. Macromol. Biosci. 21 (1), 2000259. Wiley-VCH Verlag. doi:10.1002/mabi.202000259
Brunetti-Pierri, N., and Scaglia, F. (2008). GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metabolism 94, 391–396. Academic Press. doi:10.1016/j.ymgme.2008.04.012
Chen, J. C., Luu, A. R., Wise, N., Angelis, R. D., Agrawal, V., Mangini, L., et al. (2020). Intracerebroventricular enzyme replacement therapy with β-galactosidase reverses brain pathologies due to GM1 gangliosidosis in mice. J. Biol. Chem. 295 (39), 13532–13555. American Society for Biochemistry and Molecular Biology Inc. doi:10.1074/jbc.RA119.009811
Choi, C. H. J., Zuckerman, J. E., Webster, P., and Davis, M. E. (2011). Targeting kidney mesangium by nanoparticles of defined size. Proc. Natl. Acad. Sci. U. S. A. 108 (16), 6656–6661. doi:10.1073/pnas.1103573108
de la Fuente, M., Lombardero, L., Gomez-Gonzalez, A., Solari, C., Angulo-Barturen, I., Acera, A., et al. (2021). Enzyme therapy: Current challenges and future perspectives. Int. J. Mol. Sci. 22 (17), 9181. doi:10.3390/ijms22179181
De Maria, R., Divari, S., Bo, S., Sonnino, S., Lotti, D., Capucchio, M. T., et al. (1998). β-galactosidase deficiency in a korat cat: A new form of feline G(m1)-gangliosidosis. Acta Neuropathol. 96 (3), 307–314. Springer. doi:10.1007/s004010050899
Deng, C., Xu, X., Tashi, D., Wu, Y., Su, B., and Zhang, Q. (2018). Co-administration of biocompatible self-assembled polylactic acid–hyaluronic acid block copolymer nanoparticles with tumor-penetrating peptide-iRGD for metastatic breast cancer therapy. J. Mat. Chem. B 6 (19), 3163–3180. The Royal Society of Chemistry. doi:10.1039/C8TB00319J
Desai, A. K., Baloh, C., Rosenberg, A., Kishnani, P., and Sleasman, J. (2020). Benefits of prophylactic short-course immune tolerance induction in patients with infantile Pompe disease: Demonstration of long-term safety and efficacy in an expanded cohort. Front. Immunol. 11, 1727. Frontiers Media S.A. doi:10.3389/fimmu.2020.01727
DiCioccio, R. A., Barlow, J. J., and Matta, K. L. (1984). Purification of a β-d-galactosidase from bovine liver by affinity chromatography. Carbohydr. Res. 127 (1), 109–120. Elsevier. doi:10.1016/0008-6215(84)85109-5
Dickson, P., Peinovich, M., McEntee, M., Lester, T., Le, S., Krieger, A., et al. (2008). Immune tolerance improves the efficacy of enzyme replacement therapy in canine mucopolysaccharidosis I’. J. Clin. Invest. 118 (8), 2868–2876. American Society for Clinical Investigation. doi:10.1172/JCI34676
Discher, D. E., and Ahmed, F. (2006). Polymersomes. Annu. Rev. Biomed. Eng. 8 (1), 323–341. doi:10.1146/annurev.bioeng.8.061505.095838
Edelmann, M. J., and Maegawa, G. H. B. (2020). CNS-targeting therapies for lysosomal storage diseases: Current advances and challenges. Front. Mol. Biosci. 7, 559804. Frontiers Media S.A. doi:10.3389/fmolb.2020.559804
Emerich, D. F., and Thanos, C. G. (2007). Targeted nanoparticle-based drug delivery and diagnosis. J. Drug Target. 15, 163–183. doi:10.1080/10611860701231810
Frei, E. (2011). Albumin binding ligands and albumin conjugate uptake by cancer cells. Diabetol. Metab. Syndr. 3, 11. BioMed Central. doi:10.1186/1758-5996-3-11
Galliani, M., Santi, M., Del Grosso, A., Cecchettini, A., Santorelli, F. M., Hofmann, S. L., et al. (2018). Cross-linked enzyme aggregates as versatile tool for enzyme delivery: Application to polymeric nanoparticles’. Bioconjug. Chem. 29 (7), 2225–2231. American Chemical Society. doi:10.1021/acs.bioconjchem.8b00206
Garman, R. D., Munroe, K., and Richards, S. M. (2004). Methotrexate reduces antibody responses to recombinant human α-galactosidase A therapy in a mouse model of Fabry disease’. Clin. Exp. Immunol. 137 (3), 496–502. Oxford Academic. doi:10.1111/j.1365-2249.2004.02567.x
Gray-Edwards, H. L., Regier, D. S., Shirley, J. L., Randle, A. N., Salibi, N., Thomas, S. E., et al. (2017). Novel biomarkers of human GM1 gangliosidosis reflect the clinical efficacy of gene therapy in a feline model. Mol. Ther. 25 (4), 892–903. Cell Press. doi:10.1016/J.YMTHE.2017.01.009
Gross, A. L., Gray-Edwards, H. L., Bebout, C. N., Ta, N. L., Nielsen, K., Brunson, B. L., et al. (2022). Intravenous delivery of adeno-associated viral gene therapy in feline GM1 gangliosidosis’. Brain 145 (2), 655–669. Oxford Academic. doi:10.1093/BRAIN/AWAB309
Grosso, A. D., Galliani, M., Angella, L., Santi, M., Tonazzini, I., Parlanti, G., et al. (2019). Brain-targeted enzyme-loaded nanoparticles: A breach through the blood-brain barrier for enzyme replacement therapy in krabbe disease’. Sci. Adv. 5 (11), eaax7462. American Association for the Advancement of Science. doi:10.1126/sciadv.aax7462
Gushulak, L., Hemming, R., Martin, D., Seyrantepe, V., Pshezhetsky, A., and Triggs-Raine, B. (2012). Hyaluronidase 1 and β-hexosaminidase have redundant functions in hyaluronan and chondroitin sulfate degradation.’. J. Biol. Chem. 287 (20), 16689–16697. American Society for Biochemistry and Molecular Biology. doi:10.1074/jbc.M112.350447
Hu, X., Zhang, Y., Xie, Z., Jing, X., Bellotti, A., and Gu, Z. (2017). ‘Stimuli-Responsive polymersomes for biomedical applications’. Biomacromolecules 18, 649–673. American Chemical Society. doi:10.1021/acs.biomac.6b01704
Iqbal, S., Blenner, M., Alexander-Bryant, A., and Larsen, J. (2020). Polymersomes for therapeutic delivery of protein and nucleic acid macromolecules: From design to therapeutic applications. Biomacromolecules 21 (4), 1327–1350. NLM (Medline). doi:10.1021/acs.biomac.9b01754
Ivanova, M. M., Changsila, E., laonou, C., and Ozlem, G. (2019). Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLOS ONE 14 (1), e0210617. Public Library of Science. doi:10.1371/journal.pone.0210617
Johnson, W. G. (2015). “β-Galactosidase deficiency: GM1 gangliosidosis, morquio B disease, and galactosialidosis,” in Rosenberg’s molecular and genetic basis of neurological and psychiatric disease (Netherland: Elsevier), 385–394. doi:10.1016/B978-0-12-410529-4.00034-6
Kakkis, E. D., McEntee, M. F., Schmidtchen, A., Neufeld, E. F., Ward, D. A., Gompf, R. E., et al. (1996). Long-term and high-dose trials of enzyme replacement therapy in the canine model of mucopolysaccharidosis I. Biochem. Mol. Med. 58 (2), 156–167. Academic Press Inc. doi:10.1006/bmme.1996.0044
Kelly, J. M., Bradbury, A., Martin, D. R., and Byrne, M. E. (2017). Emerging therapies for neuropathic lysosomal storage disorders. Prog. Neurobiol. 152, 166–180. Pergamon. doi:10.1016/j.pneurobio.2016.10.002
Kelly, J. M., Gross, A. L., Martin, D. R., and Byrne, M. E. (2017). Polyethylene glycol-b-poly(lactic acid) polymersomes as vehicles for enzyme replacement therapy. Nanomedicine 12 (23), 2591–2606. doi:10.2217/nnm-2017-0221
Kim, S. W., Oh, K. T., Youn, Y. S., and Lee, E. S. (2014). Hyaluronated nanoparticles with pH- and enzyme-responsive drug release properties. Colloids Surfaces B Biointerfaces 116, 359–364. Elsevier B.V. doi:10.1016/j.colsurfb.2014.01.017
Kinghorn, K. J., Gronke, S., Castillo-Quan, J. I., Woodling, N. S., Li, L., Sirka, E., et al. (2016). A Drosophila model of neuronopathic gaucher disease demonstrates lysosomal-autophagic defects and altered mTOR signalling and is functionally rescued by rapamycin’. J. Neurosci. 36 (46), 11654–11670. Society for Neuroscience. doi:10.1523/JNEUROSCI.4527-15.2016
Kulkarni, S. A., and Feng, S. S. (2013). Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm. Res. 30 (10), 2512–2522. Springer. doi:10.1007/s11095-012-0958-3
Lee, J. H., and Yeo, Y. (2015). ‘Controlled drug release from pharmaceutical nanocarriers’. Chem. Eng. Sci. 125, 75–84. Elsevier Ltd. doi:10.1016/j.ces.2014.08.046
Li, M., Zhao, G., Su, W. K., and Shuai, Q. (2020). Enzyme-responsive nanoparticles for anti-tumor drug delivery. Front. Chem. 8, 647. Frontiers Media S.A. doi:10.3389/fchem.2020.00647
Lieberman, A. P., Puertollano, R., Raben, N., Slaugenhaupt, S., Walkley, S. U., and Ballabio, A. (2012). Autophagy in lysosomal storage disorders. Autophagy 8, 719–730. doi:10.4161/auto.19469
Lim, J. A., Sun, B., Puertollano, R., and Raben, N. (2018). Therapeutic benefit of autophagy modulation in Pompe disease’, molecular therapy. Mol. Ther. 26 (7), 1783–1796. doi:10.1016/j.ymthe.2018.04.025
Liu, S., Feng, Y., Huang, Y., Jiang, X., Tang, C., Tang, F., et al. (2021). A GM1 gangliosidosis mutant mouse model exhibits activated microglia and disturbed autophagy’. Exp. Biol. Med. (Maywood). 246 (11), 1330–1341. SAGE Publications Inc. doi:10.1177/1535370221993052
Marques, A. R. A., Di Spiezio, A., ThieBen, N., Schmidt, L., Grotzinger, J., Lullmann-Rauch, R., et al. (2020). Enzyme replacement therapy with recombinant pro-CTSD (cathepsin D) corrects defective proteolysis and autophagy in neuronal ceroid lipofuscinosis’. Autophagy 16 (5), 811–825. Taylor and Francis Inc. doi:10.1080/15548627.2019.1637200
Martin, D. R., Cox, N. R., Morrison, N. E., Kennamer, D. M., Peck, S. L., Dodson, A. N., et al. (2005). Mutation of the GM2 activator protein in a feline model of GM2 gangliosidosis. Acta Neuropathol. 110 (5), 443–450. Springer-Verlag. doi:10.1007/s00401-005-1040-6
Martin, D. R., Rigat, B. A., Foureman, P., Varadarajan, G., Hwang, M., Krum, B. K., et al. (2008). Molecular consequences of the pathogenic mutation in feline GM1 gangliosidosis. Mol. Genet. Metabolism 94 (2), 212–221. Academic Press. doi:10.1016/j.ymgme.2008.02.004
McCurdy, V. J., Johnson, A. K., Gray-Edwards, H. L., Randle, A. N., Brunson, B. L., Morrison, N. E., et al. (2014). Sustained normalization of neurological disease after intracranial gene therapy in a feline model’. Sci. Transl. Med. 6 (231), 231ra48–279. NIH Public Access. doi:10.1126/scitranslmed.3007733
McCurdy, V. J., Rockwell, H. E., Arthur, J. R., Bradbury, A. M., Johnson, A. K., Randle, A. N., et al. (2015). Widespread correction of central nervous system disease after intracranial gene therapy in a feline model of Sandhoff disease. Gene Ther. 22 (2), 181–189. doi:10.1038/gt.2014.108
Minami, R., Nakamura, F., Kudoh, T., Oyanagi, K., and Nakao, T. (1980). An altered hexosaminidase A in the liver affected by hurler and hunter syndromes’. Tohoku J. Exp. Med. 132 (3), 329–335. doi:10.1620/tjem.132.329
Moghimi, S. M., Hunter, A. C., and Murray, J. C. (2001). Long-Circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev. 53 (2), 283–318.
Moghimi, S. M., and Szebeni, J. (2003). Stealth liposomes and long circulating nanoparticles: Critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 42, 463–478. Elsevier Ltd. doi:10.1016/S0163-7827(03)00033-X
Mumtaz, S., and Bachhawat, B. K. (1994). Enhanced intracellular stability and efficacy of PEG modified dextranase in the treatment of a model storage disorder. Biochimica Biophysica Acta - General Subj. 1199 (2), 175–182. Elsevier. doi:10.1016/0304-4165(94)90113-9
Muro, S. (2010). New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology, 189–204. doi:10.1002/wnan.73
Nabi, B., Rehman, S., Khan, S., Baboota, S., and Ali, J. (2018). Ligand conjugation: An emerging platform for enhanced brain drug delivery. Brain Res. Bull. 142, 384–393. Elsevier Inc. doi:10.1016/j.brainresbull.2018.08.003
Nicoli, E. R., Annunziata, I., d’Azzo, A., Platt, F. M., Tifft, C. J., and Stepien, K. M. (2021). GM1 gangliosidosis—a mini-review. Front. Genet. 12, 734878. Frontiers Media S.A. doi:10.3389/fgene.2021.734878
Ohashi, T., Iizuka, S., Ida, H., and Eto, Y. (2008). Reduced α-Gal A enzyme activity in Fabry fibroblast cells and Fabry mice tissues induced by serum from antibody positive patients with Fabry disease’. Mol. Genet. Metabolism 94 (3), 313–318. Academic Press. doi:10.1016/j.ymgme.2008.03.008
Paruchuri, B. C., Gopal, V., Sarupria, S., and Larsen, J. (2021). Toward enzyme-responsive polymersome drug delivery. Nanomedicine 16 (30), 2679–2693. doi:10.2217/nnm-2021-0194
Przybilla, M. J., Stewart, C., Carlson, T. W., Ou, L., Koniar, B. L., Sidhu, R., et al. (2021). Examination of a blood-brain barrier targeting β-galactosidase-monoclonal antibody fusion protein in a murine model of GM1-gangliosidosis. Mol. Genet. Metabolism Rep. 27, 100748. Elsevier Inc. doi:10.1016/j.ymgmr.2021.100748
Ramezani, P., Abnous, K., Taghdisi, S. M., Zahiri, M., Ramezani, M., and Alibolandi, M. (2020). Targeted MMP-2 responsive chimeric polymersomes for therapy against colorectal cancer. Colloids Surfaces B Biointerfaces 193, 111135. Elsevier B.V. doi:10.1016/j.colsurfb.2020.111135
Reynolds, G. D., Baker, H. J., and Reynolds, R. H. (1978). Enzyme replacement using liposome carriers in feline GM1 gangliosidosis fibroblasts’. Nature 275 (5682), 754–755. doi:10.1038/275754a0
Rha, A. K., Maguire, A. S., and Martin, D. R. (2021). ‘GM1 gangliosidosis: Mechanisms and management’. Appl. Clin. Genet. 14, 209–233. Dove Medical Press Ltd. doi:10.2147/TACG.S206076
Rockwell, H. E., McCurdy, V. J., Eaton, S. C., Wilson, D. U., Johnson, A. K., Randle, A. N., et al. (2015). AAV-mediated gene delivery in a feline model of sandhoff disease corrects lysosomal storage in the central nervous system. ASN Neuro 7 (2), 175909141556990. SAGE Publications Inc. doi:10.1177/1759091415569908
Samoylova, T. I., Martin, D. R., Morrison, N. E., Hwang, M., Cochran, A. M., Samoylov, A. M., et al. (2008). Generation and characterization of recombinant feline β-galactosidase for preclinical enzyme replacement therapy studies in GM1 gangliosidosis. Metab. Brain Dis. 23 (2), 161–173. Springer. doi:10.1007/s11011-008-9086-5
Sánchez-Purrà, M., Ramos, V., Petrenko, V., Torchilin, V., and Borros, S. (2016). Double-targeted polymersomes and liposomes for multiple barrier crossing. Int. J. Pharm. 511 (2), 946–956. doi:10.1016/j.ijpharm.2016.08.001
Sandhoff, K., and Harzer, K. (2013). Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 33 (25), 10195–10208. doi:10.1523/JNEUROSCI.0822-13.2013
Settembre, C., Fraldi, A., Jahreiss, L., Spampanato, C., Venturi, C., Medina, D., et al. (2008). A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 17 (1), 119–129. Oxford Academic. doi:10.1093/hmg/ddm289
Sferra, T. J., Backstrom, K., Wang, C., Rennard, R., Miller, M., and Hu, Y. (2004). Widespread correction of lysosomal storage following intrahepatic injection of a recombinant adeno-associated virus in the adult MPS VII mouse. Mol. Ther. 10 (3), 478–491. Cell Press. doi:10.1016/j.ymthe.2004.05.029
Solomon, M., and Muro, S. (2017). ‘Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives’. Adv. Drug Deliv. Rev. 118, 109–134. Elsevier. doi:10.1016/j.addr.2017.05.004
Syed, H., Qhattal, S., and Liu, X. (2011). Characterization of CD44-mediated cancer cell uptake and intracellular distribution of hyaluronan-grafted liposomes. Mol. Pharm. 8, 1233–1246. doi:10.1021/mp2000428
Takamura, A., Higaki, K., Kajimaki, K., Otsuka, S., Ninomiya, H., Matsuda, J., et al. (2008). Enhanced autophagy and mitochondrial aberrations in murine GM1-gangliosidosis. Biochem. Biophysical Res. Commun. 367 (3), 616–622. Academic Press. doi:10.1016/j.bbrc.2007.12.187
Tan, J.-X., Wang, X. Y., Li, H. Y., Su, X. L., Wang, L., Ran, L., et al. (2011). HYAL1 overexpression is correlated with the malignant behavior of human breast cancer. Int. J. Cancer 128 (6), 1303–1315. John Wiley & Sons, Ltd. doi:10.1002/ijc.25460
Tiwari, S., and Bahadur, P. (2019). Modified hyaluronic acid based materials for biomedical applications. Int. J. Biol. Macromol. 121, 556–571. Elsevier B.V. doi:10.1016/j.ijbiomac.2018.10.049
Tücking, K. S., Grutzner, V., Unger, R. E., and Schonherr, H. (2015). Dual enzyme-responsive capsules of hyaluronic acid-block-poly(lactic acid) for sensing bacterial enzymes. Macromol. Rapid Commun. 36 (13), 1248–1254. Wiley-Blackwell. doi:10.1002/marc.201500076
Wang, J., Lozier, J., Johnson, G., Kirshner, S., Verthelyi, D., Pariser, A., et al. (2008). ‘Neutralizing antibodies to therapeutic enzymes: Considerations for testing, prevention and treatment’. Nat. Biotechnol. 26, 901–908. Nature Publishing Group. doi:10.1038/nbt.1484
Yan, K., Zhang, S., Zhang, K., Miao, Y., Qiu, Y., Zhang, P., et al. (2020). Enzyme-responsive polymeric micelles with fluorescence fabricated through aggregation-induced copolymer self-assembly for anticancer drug delivery’. Polym. Chem. 11 (48), 7704–7713. The Royal Society of Chemistry. doi:10.1039/D0PY01328E
Keywords: enzyme-responsive polymersomes, hyaluronic acid, GM1 gangliosidosis, lysosomal storage disorder, enzyme replacement therapy, autophagy, drug delivery, nanomedicine
Citation: Paruchuri BC, Smith S and Larsen J (2022) Enzyme-responsive polymersomes ameliorate autophagic failure in a cellular model of GM1 gangliosidosis. Front. Chem. Eng. 4:997607. doi: 10.3389/fceng.2022.997607
Received: 19 July 2022; Accepted: 27 September 2022;
Published: 12 October 2022.
Edited by:
Lisa Volpatti, The University of Chicago, United StatesReviewed by:
Hailong Che, Shanghai University, ChinaCopyright © 2022 Paruchuri, Smith and Larsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica Larsen, bGFyc2VuakBjbGVtc29uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.