 Masahiro Takumi
Masahiro Takumi Aiichiro Nagaki
Aiichiro Nagaki- Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Kyoto, Japan
Electrochemistry provides a green and atom-efficient route to synthesize pharmaceutical and useful functional molecules, as it eliminates the need for the harsh chemical oxidants and reductants commonly used in traditional chemical reactions. To promote the implementation of electrochemical processes in the industry, there is a strong demand for the development of technologies that would allow for scale-up and a shortened reaction process time. Herein, we report that electrolysis was successfully accomplished using a flow-divided-electrochemical reactor within a few seconds, enabling the desired chemical conversion in a short period of time. Moreover, the narrow electrode gap of the flow reactor, which offers greener conditions than the conventional batch reactor, resulted in the continuous flash synthesis of C-arylglycosides.
Introduction
Electrochemistry has received considerable attention in recent years from the academic and the industrial communities because it offers an efficient and benign route to synthesize useful compounds (Frontana-Uribe et al., 2010; Horn et al., 2016; Wiebe et al., 2018; Pollok and Waldvogel, 2020). Various electrochemical transformations have been developed to produce valuable compounds, including pharmaceuticals and agrochemicals (Yoshida et al., 2008; Yan et al., 2017; Yuan and Lei, 2019; Röckl et al., 2020; Shi et al., 2021). The remarkable advantage of this methodology is that electrons can work as traceless redox reagents, reducing hazardous chemicals and enabling a green synthetic process. This development of scalable electrochemical technologies has been necessitated in recent years in order to promote the implementation of this environmentally friendly process in the industrial field (Wills et al., 2021). However, traditional batch electrochemical processes encounter many challenges in increasing productivity from the laboratory scale to the industrial scale. As the electrochemical reactions proceed under heterogeneous conditions, the simple strategy involving an enlargement of the cell volume is not appropriate, because the cell voltage drastically increases when the electrode gap increases. Furthermore, the drawbacks of batch reactors such as insufficient heat transfer, mixing and, long electrolysis duration due to the low electrode surface-to-volume ratio make scaling-up difficult. To address these issues, the incorporation of electrochemistry into flow chemistry is believed to constitute a promising solution (Atobe et al., 2018; Pletcher et al., 2018; Nöel at al., 2019; Tanbouza et al., 2020). Flow technology not only enhances the reproducibility and selectivity of electrochemical reactions due to its excellent temperature and mass transfer abilities, but also improves the productivity through continuous operation. In recent years, Baran et al. developed numerous elegant electrochemical transformations and extended them to flow conditions (Peter at al., 2019; Hu et al., 2020; Gnaim et al., 2021). In their reports, the developed batch conditions were smoothly applied to a flow reactor in which electrodes were stacked in parallel, achieving 100 g-scale production. Similarly, many researchers have developed unique flow reactors, and new reactors continue to be developed (Horii et al., 2007; Green et al., 2015; Folgueiras-Amador et al., 2017; Gütz at al., 2017; Laudadio et al., 2019a; Laudadio et al., 2019b; Cao and Nöel, 2019; Wang et al., 2019; Mo et al., 2020; Jud et al., 2021; Ošeka et al., 2021; Sato et al., 2021; Zhong et al., 2021). We, on the other hand, recently developed a novel divided-type flow electrochemical reactor that allows shortening of the electrolysis process, and reported its application in short-lived carbocations (Takumi et al., 2022). A general feature of electrochemistry is that forced electron transfer between the electrode and substrate can irreversibly generate highly reactive intermediates, such as cations, anions, and radicals, leading to unique and rapid reactions (Yoshida, 2005; Yoshida et al., 2018). However, the long electrolysis duration due to the low surface-to-volume ratio of the electrode in the conventional batch reactor causes decomposition of the reactive intermediates, and its usefulness cannot be fully exploited. To address this long-standing issue, a flow electrochemical reactor newly developed in our laboratory has provided a solution. The proposed flow reactor can accomplish electrolysis within a few seconds, enabling the flash generation of short-lived carbocations and subsequent reactions with nucleophiles before the cations decompose. Notably, this reactor allows for significant shortening of the electrolysis process and rapid subsequent chemical transformation by utilizing highly reactive carbocations which contribute to a significant increase in the productivity.
To demonstrate further synthetic utility, we report a simple and rapid electrochemical production method for C-arylglycosides using the developed flow reactor.
Results and Discussion
To validate the performance of our flow reactor, we focused on the electrochemical synthesis of C-arylglycosides as a model reaction. C-Arylglycosides represent key structural motifs found in various biologically active natural compounds that are relevant for medicinal chemistry (Bililign et al., 2005). They exhibit higher stability to hydrolytic enzymes in vivo than O-glycosides due to the strong C-C bond formed between sugar and aromatic moieties, and are therefore considered to be promising therapeutic agents. This has attracted the attention of many researchers, and accordingly, various chemical synthetic methodologies have been developed (Yang and Yu, 2017; Kitamura et al., 2018; Liao et al., 2018; Xu et al., 2020). Moreover, studies on the bioactivity of compounds that mimic and/or simplify natural scaffolds have been reported (Matos et al., 2020). Generally, these synthetic methodologies are carried out by adding external chemical reagents, such as Lewis acids and organometallic reagents; however, there are few reports on the use of electrochemical methods (Xu and Moeller, 2010; Smith and Moeller, 2013; Okamoto et al., 2021). We commenced our investigation of the electrochemical reaction of thioglycoside 1 with 2-naphthol 2 using a divided-type batch electrochemical reactor (Table 1). A solution of 1 containing a supporting electrolyte in CH2Cl2 was anodically oxidized at −75°C under a constant current until 1.25 F/mol of electricity was consumed. After electrolysis, a solution of 2 in CH2Cl2 was added, following which the mixture was stirred at 0°C for 30 min and then treated with Et3N to obtain the corresponding products. Target compound 3 is a structurally simplified analogue of 8-β-D-glucosylgenistein, and its therapeutic potential has been studied (Matos et al., 2020). In the initial trial with Bu4NOTf, the corresponding O-glycoside was obtained in 91% yield (α/β = 80/20) instead of the desired C-glycoside 3 (Table 1, entry 1). In contrast, replacing the supporting electrolyte with Bu4NB(C6F5)4 produced a moderate yield of the desired C-arylglycoside (Table 1, entry 2). Finally, we found that employing the Bu4NBF4/CH2Cl2 system provided the target product with an even better yield (Table 1, entry 3); therefore, we selected this supporting electrolyte, which is widely used in electrochemistry for further studies. In addition, electrolysis in the presence of nucleophile 2 gave a lower conversion and yield, presumably because of the undesired oxidation of 2 (Table 1, entry 4).
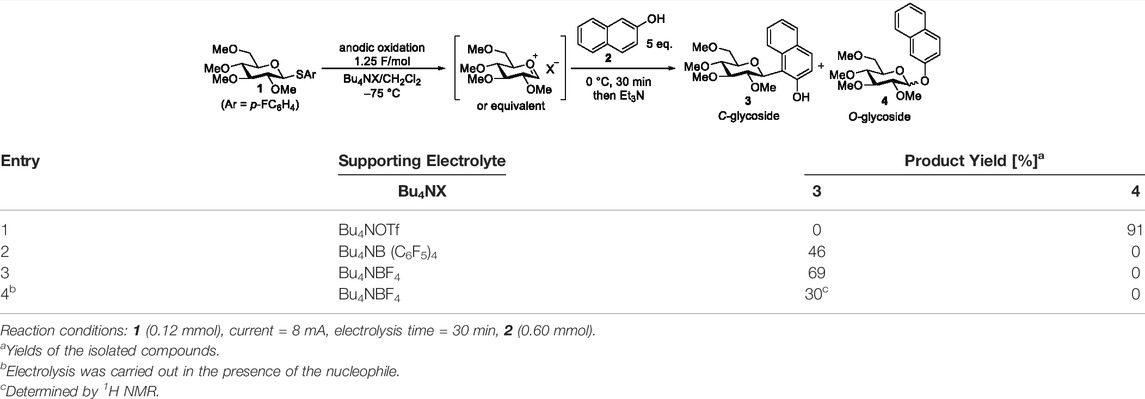
TABLE 1. Screening of supporting electrolyte for electrochemical C-glycoside synthesis in the batch reactor.
Having developed a simple electrochemical method, we next optimized the electrolysis temperature on a larger reaction scale (Table 2). Initially, the reaction on a larger scale at −75°C resulted in a slight decrease in the yield (Table 2, entry 1). This led us to increase the electrolysis temperature (Table 2, entries 1–4). By applying a higher temperature, the product yields increased, and as shown in entries 1–4, we found that temperatures of approximately 0°C to −25°C are suitable for this transformation, furnishing the desired C-arylglycoside in 82% yield at −25°C. However, electrolysis at 20°C resulted in low yields, presumably because the intermediate decomposed during electrolysis.

TABLE 2. Optimization of electrolysis temperature in the batch reactor.
With the obtained benchmark conditions, we attempted to reduce the reaction process time in a batch reactor. As electrolysis was carried out under constant-current conditions, increasing the current should decrease the electrolysis time and improve the productivity (Table 3). The increase in the current resulted in a significant increase in the cell voltage and a decrease in the yield (Table 3, entries 1–4), suggesting that the heat generated at the anode by the electrical resistance was not sufficiently removed, causing decomposition of the intermediate. To conduct the transformation in a greener manner, a lower concentration of the supporting electrolyte was applied (Table 3, entries 5–8). This, however, caused a significant increase in the cell voltage and a further decrease in the yield. These experiments suggest that there are limitations of batch reactors in terms of increasing the productivity.
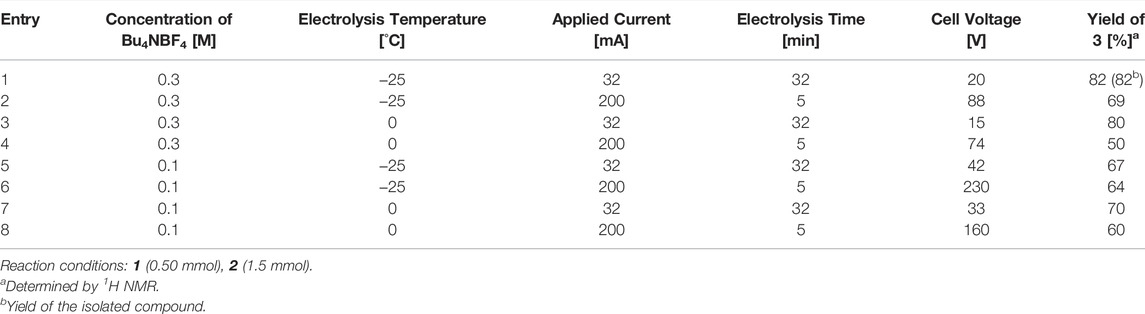
TABLE 3. Effect of applied current and concentration of supporting electrolyte.
To address these issues observed in the batch reactor, the reaction was applied to our flow electrochemical reactor system (Figure 1). The flow electrochemical reactor was equipped with a carbon felt anode and platinum-plated cathode divided by a diaphragm composed of glass filters and PTFE membranes, resulting in an interelectrode gap of 1 mm (see Supplementary Material for more details). The electrolysis temperature was controlled using a Peltier cooling system. For the flow system, a solution of the substrate (0.02 M) containing a supporting electrolyte (Bu4NBF4, 0.1 M) in CH2Cl2 was introduced by syringe pumping into the anodic chamber with a carbon felt anode. A trifluoromethanesulfonic acid (TfOH) solution (0.05 M) containing the supporting electrolyte (Bu4NBF4, 0.1 M) in CH2Cl2 was also introduced into a cathodic chamber equipped with platinum-plated cathodes to promote electrolysis. Current was applied to consume 1.25 F/mol, and constant-current electrolysis was performed at 0°C. The intermediate generated in the anodic chamber was quickly transferred to a microtube reactor (R1), mixed at speed with nucleophile 2 in a micromixer (M1), and reacted in R2 and R3 to yield C-arylglycoside 3.
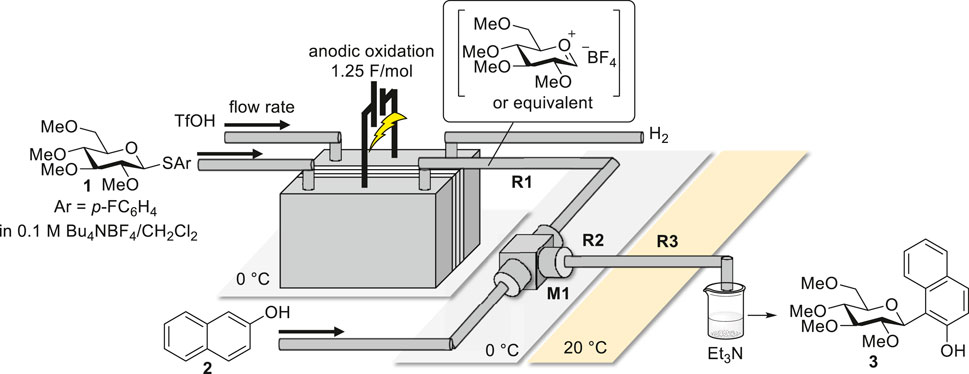
FIGURE 1. Schematic of C-glycoside synthesis using a flow electrochemical reactor system.
To evaluate the performance of the flow system, the reaction was performed by varying the flow rate, and the results are summarized in Table 4. In the first trial, with a flow rate of 1.0 ml/min, the desired product 3 was afforded in moderate yield (Table 4, entry 1). Next, we carried out the reactions under higher-flow-rate conditions. When the flow rate was gradually increased to 5.0 ml/min with the same electrochemical setup, the yield also increased (Table 4, entries 2–5). Notably, the quantitative electrochemical conversion of 1 was maintained even at 5.0 ml/min, implying that electrolysis could be accomplished in 5 s. Furthermore, the increase in the flow rate not only reduced the electrolysis time but also enabled the quick transfer of the intermediate generated in the anodic chamber and the immediate reaction with the nucleophile, which contributed to the improved yields and productivity. In addition, the high-temperature controllability of this flow system allowed for steady production under high-current conditions, e.g., 200 mA, where a drop in the yield was observed in the batch reactor. Moreover, the cell voltage could be suppressed to a much lower level even at a reduced supporting electrolyte concentration owing to the narrow interelectrode gap. In addition, electrochemical access to a 5-membered ring C-arylglycoside 5 was successfully accomplished, suggesting the high generality of the present methodology (Figure 2).

TABLE 4. Productivity and cell voltage using a flow electrochemical reactor system.
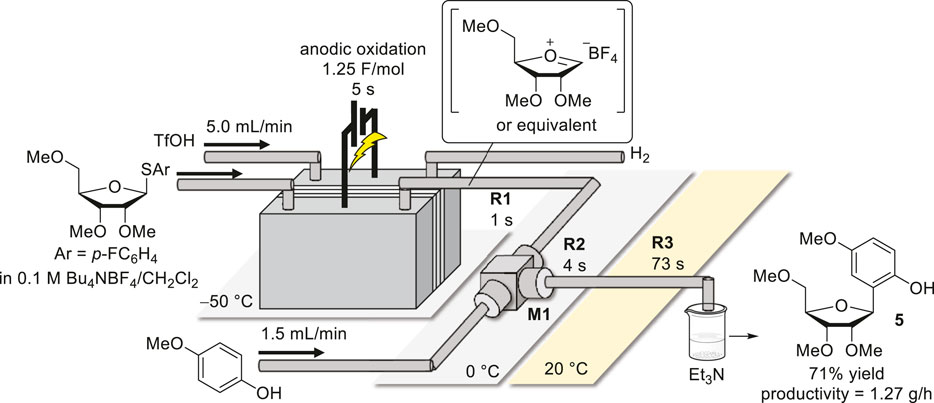
FIGURE 2. Schematic of 5-membered ring C-arylglycoside synthesis using a flow electrochemical reactor system.
Following these successes, we proceeded to further improve the productivity. To achieve this, we tested the flow reaction at a higher concentration of 1. To our delight, the increase in concentration and applied current resulted in a further increase in the yield and productivity without a decrease in the electrolysis efficiency (Figure 3).
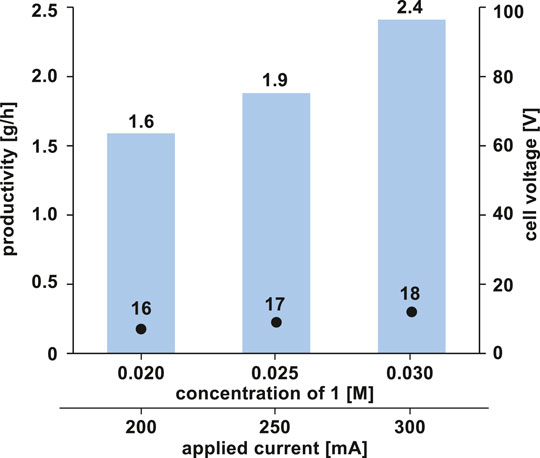
FIGURE 3. Improving productivity by increasing substrate concentration. Bars and plots represent the productivity and cell voltage, respectively.
During this series of investigations, general strategies for improving the productivity, i.e., increasing the flow rate and concentration, were confirmed to be applicable. Furthermore, the narrow electrode gap and precise temperature controllability enabled mild and green reactions to be carried out in a more practical manner compared to the batch. Finally, continuous production of the target compound was demonstrated (Figure 4). Plunger pumps were used instead of syringe pumps for long-term operation. After 60 min of operation in a single passage, 2.5 g of the desired product was obtained (77% yield). During the operation, the electrolysis temperature was precisely controlled at 0°C (Supplementary Figure S8), suggesting the robustness of the system. The total reaction time, including electrolysis and reaction with the nucleophile, was within 75 s, enabling on-demand access to the product.
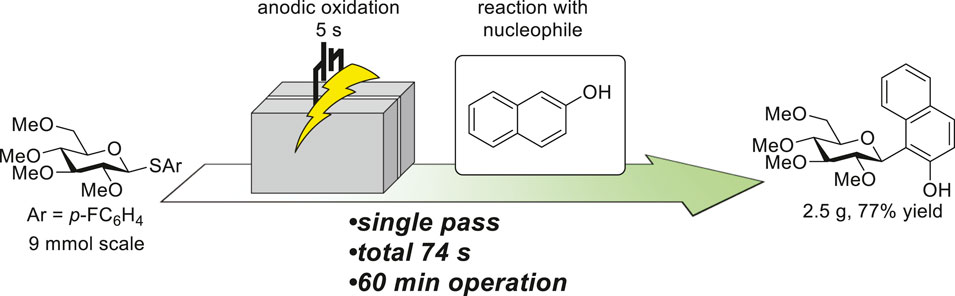
FIGURE 4. Flash and continuous production of C-arylglycoside. See Supplementary Information for detailed conditions.
A simple and highly productive electrochemical C-arylglycosylation reaction was performed under flow. The highly efficient electrolysis ability of the flow reactor, which can accomplish electrolysis within a few seconds in a single passage through the reactor, drastically shortened the electrolysis time, enabling rapid production. Furthermore, owing to the narrow interelectrode gap and the high-temperature controllability of the reactor, continuous manufacturing for 60 min was steadily achieved, demonstrating the practicality of the flow system. The research presented herein will facilitate the introduction of electrochemistry into the manufacturing process.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
MT and AN conceived the project, directed the research, and prepared the manuscript. MT performed all experiment.
Funding
This work was supported by JSPS KAKENHI Grant Numbers JP15H05849, JP17K06910, JP20KK0121, and JP21H01936. This work was also partially supported by AMED (Nos. JP20ak0101090 and JP21ak0101156), JST A-step program (No. 18067420), CREST (No. JPMJCR18R1), and the Ogasawara Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fceng.2022.862766/full#supplementary-material
References
Atobe, M., Tateno, H., and Matsumura, Y. (2018). Applications of Flow Microreactors in Electrosynthetic Processes. Chem. Rev. 118, 4541–4572. doi:10.1021/acs.chemrev.7b00353
Bililign, T., Griffith, B. R., and Thorson, J. S. (2005). Structure, Activity, Synthesis and Biosynthesis of Aryl-C-Glycosides. Nat. Prod. Rep. 22, 742–760. doi:10.1039/B407364A
Cao, Y., and Noël, T. (2019). Efficient Electrocatalytic Reduction of Furfural to Furfuryl Alcohol in a Microchannel Flow Reactor. Org. Process. Res. Dev. 23, 403–408. doi:10.1021/acs.oprd.8b00428
de Matos, A. M., Blázquez-Sánchez, M. T., Bento-Oliveira, A., de Almeida, R. F. M., Nunes, R., Lopes, P. E. M., et al. (2020). Glucosylpolyphenols as Inhibitors of Aβ-Induced Fyn Kinase Activation and Tau Phosphorylation: Synthesis, Membrane Permeability, and Exploratory Target Assessment within the Scope of Type 2 Diabetes and Alzheimer's Disease. J. Med. Chem. 63, 11663–11690. doi:10.1021/acs.jmedchem.0c00841
Folgueiras-Amador, A. A., Philipps, K., Guilbaud, S., Poelakker, J., and Wirth, T. (2017). An Easy-To-Machine Electrochemical Flow Microreactor: Efficient Synthesis of Isoindolinone and Flow Functionalization. Angew. Chem. Int. Ed. 56, 15446–15450. doi:10.1002/anie.201709717
Frontana-Uribe, B. A., Little, R. D., Ibanez, J. G., Palma, A., and Vasquez-Medrano, R. (2010). Organic Electrosynthesis: a Promising green Methodology in Organic Chemistry. Green. Chem. 12, 2099–2119. doi:10.1039/C0GC00382D
Gnaim, S., Takahira, Y., Wilke, H. R., Yao, Z., Li, J., Delbrayelle, D., et al. (2021). Electrochemically Driven Desaturation of Carbonyl Compounds. Nat. Chem. 13, 367–372. doi:10.1038/s41557-021-00640-2
Green, R. A., Brown, R. C. D., Pletcher, D., and Harji, B. (2015). A Microflow Electrolysis Cell for Laboratory Synthesis on the Multigram Scale. Org. Process. Res. Dev. 19, 1424–1427. doi:10.1021/acs.oprd.5b00260
Gütz, C., Stenglein, A., and Waldvogel, S. R. (2017). Highly Modular Flow Cell for Electroorganic Synthesis. Org. Process. Res. Dev. 21, 771–778. doi:10.1021/acs.oprd.7b00123
Horii, D., Fuchigami, T., and Atobe, M. (2007). A New Approach to Anodic Substitution Reaction Using Parallel Laminar Flow in a Micro-flow Reactor. J. Am. Chem. Soc. 129, 11692–11693. doi:10.1021/ja075180s
Horn, E. J., Rosen, B. R., and Baran, P. S. (2016). Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method. ACS Cent. Sci. 2, 302–308. doi:10.1021/acscentsci.6b00091
Hu, P., Peters, B. K., Malapit, C. A., Vantourout, J. C., Wang, P., Li, J., et al. (2020). Electroreductive Olefin-Ketone Coupling. J. Am. Chem. Soc. 142, 20979–20986. doi:10.1021/jacs.0c11214
Jud, W., Kappe, C. O., and Cantillo, D. (2021). Development and Assembly of a Flow Cell for Single‐Pass Continuous Electroorganic Synthesis Using Laser‐Cut Components. Chem. Methods 1, 36–41. doi:10.1002/cmtd.202000042
Kitamura, K., Ando, Y., Matsumoto, T., and Suzuki, K. (2018). Total Synthesis of Aryl C-Glycoside Natural Products: Strategies and Tactics. Chem. Rev. 118, 1495–1598. doi:10.1021/acs.chemrev.7b00380
Laudadio, G., Barmpoutsis, E., Schotten, C., Struik, L., Govaerts, S., Browne, D. L., et al. (2019a). Sulfonamide Synthesis through Electrochemical Oxidative Coupling of Amines and Thiols. J. Am. Chem. Soc. 141, 5664–5668. doi:10.1021/jacs.9b02266
Laudadio, G., Bartolomeu, A. d. A., Verwijlen, L. M. H. M., Cao, Y., de Oliveira, K. T., and Noël, T. (2019b). Sulfonyl Fluoride Synthesis through Electrochemical Oxidative Coupling of Thiols and Potassium Fluoride. J. Am. Chem. Soc. 141, 11832–11836. doi:10.1021/jacs.9b06126
Liao, H., Ma, J., Yao, H., and Liu, X.-W. (2018). Recent Progress of C-Glycosylation Methods in the Total Synthesis of Natural Products and Pharmaceuticals. Org. Biomol. Chem. 16, 1791–1806. doi:10.1039/c8ob00032h
Mo, Y., Lu, Z., Rughoobur, G., Patil, P., Gershenfeld, N., Akinwande, A. I., et al. (2020). Microfluidic Electrochemistry for Single-Electron Transfer Redox-Neutral Reactions. Science 368, 1352–1357. doi:10.1126/science.aba3823
Noël, T., Cao, Y., and Laudadio, G. (2019). The Fundamentals behind the Use of Flow Reactors in Electrochemistry. Acc. Chem. Res. 52, 2858–2869. doi:10.1021/acs.accounts.9b00412
Okamoto, K., Tsutsui, M., Morizumi, H., Kitano, Y., and Chiba, K. (2021). Electrochemical Synthesis of Imino‐ C ‐Nucleosides by "Reactivity Switching" Methodology for In Situ Generated Glycoside Donors. Eur. J. Org. Chem. 2021, 2479–2484. doi:10.1002/ejoc.202100106
Ošeka, M., Laudadio, G., van Leest, N. P., Dyga, M., Bartolomeu, A. d. A., Gooßen, L. J., et al. (2021). Electrochemical Aziridination of Internal Alkenes with Primary Amines. Chem 7, 255–266. doi:10.1016/j.chempr.2020.12.002
Peters, B. K., Rodriguez, K. X., Reisberg, S. H., Beil, S. B., Hickey, D. P., Kawamata, Y., et al. (2019). Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-Ion Battery Chemistry. Science 363, 838–845. doi:10.1126/science.aav5606
Pletcher, D., Green, R. A., and Brown, R. C. D. (2018). Flow Electrolysis Cells for the Synthetic Organic Chemistry Laboratory. Chem. Rev. 118, 4573–4591. doi:10.1021/acs.chemrev.7b00360
Pollok, D., and Waldvogel, S. R. (2020). Electro-organic Synthesis - a 21st century Technique. Chem. Sci. 11, 12386–12400. doi:10.1039/D0SC01848A
Röckl, J. L., Pollok, D., Franke, R., and Waldvogel, S. R. (2020). A Decade of Electrochemical Dehydrogenative C,C-Coupling of Aryls. Acc. Chem. Res. 53, 45–61. doi:10.1021/acs.accounts.9b00511
Sato, E., Fujii, M., Tanaka, H., Mitsudo, K., Kondo, M., Takizawa, S., et al. (2021). Application of an Electrochemical Microflow Reactor for Cyanosilylation: Machine Learning-Assisted Exploration of Suitable Reaction Conditions for Semi-large-scale Synthesis. J. Org. Chem. 86, 16035–16044. doi:10.1021/acs.joc.1c01242
Shi, S.-H., Liang, Y., and Jiao, N. (2021). Electrochemical Oxidation Induced Selective C-C Bond Cleavage. Chem. Rev. 121, 485–505. doi:10.1021/acs.chemrev.0c00335
Smith, J. A., and Moeller, K. D. (2013). Oxidative Cyclizations, the Synthesis of Aryl-Substituted C-Glycosides, and the Role of the Second Electron Transfer Step. Org. Lett. 15, 5818–5821. doi:10.1021/ol402826z
Takumi, M., Sakaue, H., and Nagaki, A. (2022). Flash Electrochemical Approach to Carbocations. Angew. Chem. Int. Ed. 61, e202116177. doi:10.1002/anie.202116177
Tanbouza, N., Ollevier, T., and Lam, K. (2020). Bridging Lab and Industry with Flow Electrochemistry. iScience 23, 101720. doi:10.1016/j.isci.2020.101720
Wang, D., Wang, P., Wang, S., Chen, Y.-H., Zhang, H., and Lei, A. (2019). Direct Electrochemical Oxidation of Alcohols with Hydrogen Evolution in Continuous-Flow Reactor. Nat. Commun. 10, 2796–2803. doi:10.1038/s41467-019-10928-0
Wiebe, A., Gieshoff, T., Möhle, S., Rodrigo, E., Zirbes, M., and Waldvogel, S. R. (2018). Electrifying Organic Synthesis. Angew. Chem. Int. Ed. 57, 5594–5619. doi:10.1002/anie.201711060
Wills, A. G., Charvet, S., Battilocchio, C., Scarborough, C. C., Wheelhouse, K. M. P., Poole, D. L., et al. (2021). High-Throughput Electrochemistry: State of the Art, Challenges, and Perspective. Org. Process. Res. Dev. 25, 2587–2600. doi:10.1021/acs.oprd.1c00167
Xu, G., and Moeller, K. D. (2010). Anodic Coupling Reactions and the Synthesis of C-Glycosides. Org. Lett. 12, 2590–2593. doi:10.1021/ol100800u
Xu, L.-Y., Fan, N.-L., and Hu, X.-G. (2020). Recent Development in the Synthesis of C-Glycosides Involving Glycosyl Radicals. Org. Biomol. Chem. 18, 5095–5109. doi:10.1039/d0ob00711k
Yan, M., Kawamata, Y., and Baran, P. S. (2017). Synthetic Organic Electrochemical Methods since 2000: On the Verge of a Renaissance. Chem. Rev. 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Yang, Y., and Yu, B. (2017). Recent Advances in the Chemical Synthesis of C-Glycosides. Chem. Rev. 117, 12281–12356. doi:10.1021/acs.chemrev.7b00234
Yoshida, J.-i. (2005). Flash Chemistry Using Electrochemical Method and Microsystems. Chem. Commun. 36, 4509–4516. doi:10.1039/B508341A
Yoshida, J.-i., Kataoka, K., Horcajada, R., and Nagaki, A. (2008). Modern Strategies in Electroorganic Synthesis. Chem. Rev. 108, 2265–2299. doi:10.1021/cr0680843
Yoshida, J.-i., Shimizu, A., and Hayashi, R. (2018). Electrogenerated Cationic Reactive Intermediates: The Pool Method and Further Advances. Chem. Rev. 118, 4702–4730. doi:10.1021/acs.chemrev.7b00475
Yuan, Y., and Lei, A. (2019). Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions. Acc. Chem. Res. 52, 3309–3324. doi:10.1021/acs.accounts.9b00512
Keywords: electrochemistry, flow chemistry, flow electrochemical reactor, C-arylglycosides, microreactor
Citation: Takumi M and Nagaki A (2022) Flash Synthesis and Continuous Production of C-Arylglycosides in a Flow Electrochemical Reactor. Front. Chem. Eng. 4:862766. doi: 10.3389/fceng.2022.862766
Received: 26 January 2022; Accepted: 07 March 2022;
Published: 12 April 2022.
Edited by:
Giancarlo Cravotto, University of Turin, ItalyReviewed by:
Alessandro Massi, University of Ferrara, ItalyGianvito Vilé, Politecnico di Milano, Italy
Copyright © 2022 Takumi and Nagaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aiichiro Nagaki, YW5hZ2FraUBzYmNoZW0ua3lvdG8tdS5hYy5qcA==