 Barnabas Poznansky
Barnabas Poznansky Sarah E. Cleary
Sarah E. Cleary Lisa A. Thompson
Lisa A. Thompson Holly A. Reeve*
Holly A. Reeve* Kylie A. Vincent
Kylie A. Vincent
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem. Eng., 06 August 2021
Sec. Microfluidic Engineering and Process Intensification
Volume 3 - 2021 | https://doi.org/10.3389/fceng.2021.718257
Translation of redox biocatalysis into a commercial hydrogenation flow reactor, with in-built electrolytic H2 generation, was achieved using immobilized enzyme systems. Carbon-supported biocatalysts were first tested in batch mode, and were then transferred into continuous flow columns for H2-driven, NADH-dependent asymmetric ketone reductions. The biocatalysts were thus handled comparably to heterogeneous metal catalysts, but operated at room temperature and 1–50 bar H2, highlighting that biocatalytic strategies enable implementation of hydrogenation reactions under mild–moderate conditions. Continuous flow reactions were demonstrated as a strategy for process intensification; high conversions were achieved in short residence times, with a high biocatalyst turnover frequency and productivity. These results show the prospect of using enzymes in reactor infrastructure designed for conventional heterogeneous hydrogenations.
As sustainability increases in importance in the chemical and pharmaceutical industries, new technologies that lower energy demands, use renewable resources and aid productivity and automation are critical (Jiménez-González et al., 2011; Bryan et al., 2018). Continuous processes and biocatalysis are key examples of technologies that can address these unmet needs. For hydrogenation reactions in particular, the confinement of a supported catalyst to a small-footprint packed-bed flow reactor minimizes energy demands and safety concerns associated with handling H2, and can improve contact between the H2 gas, heterogeneous catalyst and reaction solution. On the other hand, biocatalysis offers the possibility of replacing finite resource, toxic metal catalysts with renewable alternatives as well as enabling high productivities under mild reaction conditions. One particular area in which these benefits could align is the translation of redox biocatalysts into continuous processes for the asymmetric reduction of C = X bonds in the synthesis of chiral alcohols and amines (Devine et al., 2018; Tamborini et al., 2018; Adams et al., 2019; Santi et al., 2021).
The most well-established methods for asymmetric hydrogenations use precious-metal-based organometallic catalysts (such as Rh, Ru and Ir) (Noyori and Hashiguchi, 1997; Noyori, 2002; Zanotti-Gerosa et al., 2005). Although these homogeneous catalysts provide high enantioselectivity and total turnover numbers (TTN), they are expensive, toxic and difficult to recover and reuse. Attempts to mitigate this by catalyst immobilization (for example, onto mesoporous silica or organic polymers) are often associated with loss in catalyst stereoselectivity and efficiency (Wang and Ding, 2004; Mallat et al., 2007; Štefane and Požgan, 2016). This has hampered the heterogenization of organometallic complexes and their translation into flow reactors. By contrast, implementation of supported metal nanoparticles (such as Pd/C) in flow is commonplace for hydrogenation reactions; however, these catalysts are not inherently enantioselective (Irfan et al., 2011; Cossar et al., 2015; Hübner et al., 2016; Russell et al., 2017), and the selectivity can be difficult to tune, Figure 1A.
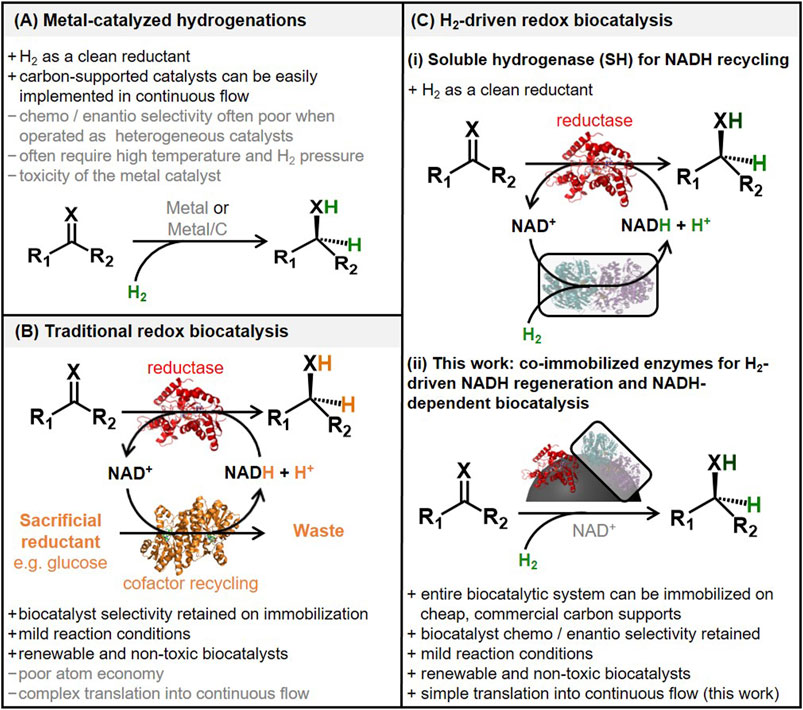
FIGURE 1. Comparison of asymmetric double bond reduction strategies. (A) Metal-catalyzed hydrogenation. (B) Double bond reduction by a reductase enzyme (red) with NADH recycling by glucose dehydrogenase, alcohol dehydrogenase or formate dehydrogenase (orange). (C) Double bond reduction by a reductase enzyme (red) with H2-driven NADH recycling performed [(C), i] in solution or [(C), ii] with enzymes co-immobilized onto carbon (gray). A list of pros (+) and cons (−) is shown.
One of the challenges in implementing biocatalysts for C = X bond reductions is that the relevant dehydrogenase enzymes rely on hydride transfer from expensive organic cofactors, commonly NADH or NADPH. A method for in situ regeneration of these cofactors is essential in commercial applications, and usually relies on carbon-based reductants, glucose, formate or isopropanol, at super-stoichiometric levels often with a second “cofactor recycling” enzyme, Figure 1B (Fernandez-Lafuente and Berenguer-Murcia, 2010). This leads to substantial generation of carbon-based waste, and also complicates the translation of redox biocatalysis into flow reactors. At present, nearly all biocatalytic C = X bond reductions are carried out as batch reactions with the enzymes in solution (Caron et al., 2006; Huisman et al., 2010; Tufvesson et al., 2010; Winkler et al., 2021).
Research has been carried out on the immobilization of enzymes onto a range of solid supports (Contente et al., 2017; Dall’Oglio et al., 2017; Döbber and Pohl, 2017; Peschke et al., 2017). In contrast to organometallic catalysts, enzymes typically retain excellent stereo-, regio- and chemo-selectivity once immobilized. Additional benefits such as increased lifetime (Liese and Hilterhaus, 2013; Rodrigues et al., 2013; Sheldon and van Pelt, 2013), improved reaction kinetics (for example, due to decreased product inhibition), higher catalytic efficiency and improved stability can also be achieved (Britton et al., 2018; Thompson et al., 2019; De Santis et al., 2020). However, the immobilization procedures are often lengthy and costly such that, at present, there are no clear standard methods. As a consequence, there is a desire for new strategies for implementing these highly selective biocatalysts in continuous processes.
We, and others, have previously described biocatalyst systems for H2-driven recycling of the NADH cofactor, coupled to NADH-dependent enzymes for selective C = X bond reductions, Figure 1C (Reeve et al., 2015; Al-Shameri et al., 2020; Thompson et al., 2020b; Rowbotham et al., 2020). These “biocatalytic hydrogenations” benefit from the use of H2 as a clean and often 100% atom-efficient reductant, as well as the inherent selectivity of biocatalysis for asymmetric reductions. We have demonstrated that these enzyme sequences can be entirely supported on carbon materials, meaning that the overall biocatalyst system can be handled similarly to carbon-supported metal nanoparticle catalysts (Reeve et al., 2015). Following the increasing trend in metal-catalyst hydrogenation being operated in flow reactors, we recently described the operation of our biocatalytic hydrogenation approaches in flow, using carbon nanotube based flow columns designed and prepared in-house (Zor et al., 2017; Thompson et al., 2020a). Although these proof-of-concept results were promising, the biocatalysts were clearly limited in operational stability and activity.
Here, we have explored whether established infrastructure for flow hydrogenations can be used to simplify translation of biocatalytic hydrogenations into continuous processes. Flow reactors such as the H-Cube (ThalesNano) are designed to achieve high availability of H2 with carbon-supported metal catalysts. In the H-cube, H2 is generated through water electrolysis, and this opens up future possibilities for using renewable electricity to drive reactions with sustainably produced H2. In this study, we first compare two biocatalyst systems for H2-driven NAD+ reduction and NADH recycling in batch, specifically focusing on activity and operational stability. We then implement these biocatalyst systems in the hydrogenation flow reactor, where H2 pressure, flow rate, temperature and reactant flow rate can be controlled with excellent precision. The results demonstrate a simple route to translating biocatalytic hydrogenations into a continuous process in a commercial flow reactor, with the best catalysts achieving high activity and operational stability as measured by the catalyst turnover frequency and total turnover number, respectively.
Activated charcoal (Darco, −100 mesh), sodium pyruvate, dimethyl sulfoxide (DMSO) and Trizma-base were purchased from Sigma-Aldrich and were used as received. Carbon black particles (Black Pearls 2000, BP 2000, Cabot Corporation), NAD+ and NADH (Prozomix), acetophenone (Acros) and GC standards [(S)-phenylethanol and (rac)-phenylethanol, Acros] were also used as supplied. All buffers were prepared with MilliQ water (Millipore, 18 MΩ cm). Commercially available lactate dehydrogenase (LDH, from rabbit muscle, Merck) was used as supplied.
Biocatalysts for H2-driven cofactor recycling (SH and Hyd1) were prepared in-house. Detailed protocols for expression and purification of the hydrogenases [Escherichia coli hydrogenase 1, Hyd1 (Joseph Srinivasan et al., 2021), and R. eutropha soluble hydrogenase, SH (Lauterbach and Lenz, 2013)] have been described previously. Alcohol dehydrogenase ADH-105 was provided by Johnson Matthey in the His-tagged and purified form.
All ultraviolet–visible (UV–Vis) spectra were acquired using an Agilent Cary 60 spectrophotometer. 1H NMR spectra were acquired using either a Bruker Avance III HD nanobay (400 MHz) or a Bruker Avance III HD (500 MHz) instrument. For samples in Tris-HCl (50 mM, pH 8.0), 10% v/v 2H2O was added to allow for field locking. Chiral-phase GC-FID data were obtained using a TRACE 1310 instrument (Thermo Fisher Scientific), with a CP-Chirasil-Dex CB column (Agilent), 25 m length, 0.25 mm diameter, 0.25 μm (film thickness), fitted with a guard of 10 m deactivated fused silica of the same diameter. Reactant and product peaks were determined by comparison against commercial standards. Enantiomeric purity was determined as the ratio of the area between the enantiomeric peaks.
In a glovebox (Glove Box Technology) under a protective N2 atmosphere (O2 < 10 ppm), carbon particles (BP2000) were suspended in deoxygenated Tris-HCl buffer (50 mM, pH 8.0) to make a 20 mg ml−1 slurry. Larger carbon agglomerates were dispersed, then sonicated twice for 30 min to give an ink-like slurry. The slurry was allowed to cool on ice. The designated amounts of Hyd1, SH and BP2000 slurry were combined in a centrifuge tube, mixed using a pipette and allowed to incubate at 4°C for 60 min. The catalyst slurry was centrifuged (12,000 × g), and the supernatant was then withdrawn using a pipette to remove any unadsorbed enzyme. The pellet was resuspended in deoxygenated Tris-HCl buffer (50 mM, pH 8.0) and separated into aliquot samples as needed. Samples were flash-frozen and stored at −80°C.
Activity assays were also performed in an anaerobic glovebox in deoxygenated Tris-HCl buffer (100 mM, pH 8.0) in a UV–Vis cuvette that was sealed with a rubber septum pierced with two needles to allow gas flow in and out. The concentration of NADH was monitored over time by the absorbance at 340 nm (ε = 6.22 mM−1cm−1) associated with the reduced cofactor, NADH.
A solution of 800 μl of 0.1 mM NAD+ in Tris-HCl (100 mM, pH 8.0) was saturated with H2 by bubbling for 10 min. The inlet needle was next adjusted such that it was streaming H2 through the cuvette headspace rather than through the solution. The H2-saturated solution (0.2 ml) was used to transfer the carbon-supported catalyst (see General Enzyme Immobilization Procedure for Batch Studies) into the cuvette by a fresh syringe and needle, and the resulting slurry was mixed once by gently syringing up and down. The reaction was monitored by UV–Vis, and the concentration of NADH (determined from A340 nm) was plotted against time to determine the specific activity from the linear phase of reaction.
Acetophenone reduction reactions were prepared on the benchtop in a N2-flushed Asynt OCTO mini parallel reactor, in which each reaction tube contained a magnetic stirrer bar. Solutions of NAD+ (0.5 mM) were prepared in Tris-HCl (40 mM, pH 8.0) and flushed with N2 followed by H2 before use. Stock solutions of acetophenone (2 M) in DMSO were added to the reaction mixtures to give the desired concentrations. The Asynt OCTO reactor was kept at room temperature under a steady flow (1 bar) of H2 throughout the reaction setup. H2-saturated NAD+ solution was added to each reaction tube, followed by SH/C or SH + Hyd1/C, ADH-105 and acetophenone in DMSO in the designated order of addition and amounts described below. The parallel reactor was left to stir at 350 r.p.m. at room temperature under a steady flow of H2 for the specified reaction time, during which aliquots (<50 μl) were collected and prepared for GC analysis by extraction into ethyl acetate (800 μl), after which 600 μl of the organic layer was dried over Na2SO4 and analysed by chiral-phase GC-FID. An additional 20 μL acetophenone (2 M in DMSO) was added at 19 h using a needle and syringe.
Biocatalytic cartridges (200 µl reactor volume) were prepared by two different methods. 1) A CatCart™ cartridge (30 × 4 mm) was packed and sealed with activated charcoal using a cartridge packer (ThalesNano). The required mixture of enzymes was manually pumped into the pre-sealed cartridge via syringe. The cartridge was then stored at 4°C for 1 h, then installed in the H-Cube and flushed with Tris-HCl buffer (50 mM, pH 8.0) to remove any unadsorbed enzyme. 2) A CatCart™ cartridge was filled with activated charcoal, and the required mixture of enzymes was then added into the cartridge before it was sealed. The cartridge was sealed using the cartridge packer, and was agitated on a vortex mixer and then stored at 4°C for 1 h. The column was then installed in the H-Cube and was flushed with Tris-HCl buffer (50 mM, pH 8.0) to remove any unadsorbed enzyme. Further experimental details are provided in the Supporting Information, Supplementary Table S1.
With the sealed CatCart™ biocatalyst cartridge installed in the H-Cube reactor, a liquid feed (Knauer H-Cube piston pump) containing NAD+ (1–5 mM) in Tris-HCl buffer (50 mM, pH 8.0) was pumped through the H-Cube with the H2 that was 1) generated by built-in water reservoir and electrolyzer (gas feed flow rate was set to 7–12 ml min−1); or 2) from a H2 cylinder pumped in by Vapourtec SF-10 peristaltic pumps (gas feed flow rate 0.6–1.2 ml min−1) using PTFE tubing (1/16″ OD, 1/32″ ID) and Tefzel™ fittings and mixed with H-Cube pump liquid solution at a Y-piece (PEEK, 1/4–28, 0.02″ thru-hole, IDEX). The liquid feed flow rate was set to 0.05–3 ml min−1 to give a 0.8–30 s residence time (tRes) through the biocatalyst cartridge. All reactions were performed at room temperature. Samples were collected using a fraction collector (BioRad 2110), and reactor volumes were combined to obtain a sufficient amount of material for analysis by UV–Vis spectroscopy. The ratio of absorbance at 260 and 340 nm was used to calculate the extent of conversion (using previously described calibration curves) (Reeve et al., 2015). Further experimental details are provided in the Supporting Information, Supplementary Table S2.
An appropriate sealed CatCart™ was prepared according to the procedures described in Preparation of Biocatalyst Cartridges for Hydrogenation Flow Reactor Studies. This was then installed in the H-Cube reactor and a liquid feed (Knauer H-Cube piston pump) containing sodium pyruvate (10–20 mM) and NAD+ (1 mM) in Tris-HCl buffer (50 mM, pH 8.0) was pumped through the H-Cube with the H2 that was 1) generated by built-in water reservoir and electrolyzer (gas feed flow rate was set to 7–12 ml min−1); or 2) obtained from a H2 cylinder pumped in by Vapourtec SF-10 peristaltic pumps (gas feed flow rate 0.6–1.2 ml min−1) using PTFE tubing (1/16″ OD, 1/32″ ID) and Tefzel™ fittings and mixed with H-Cube pump liquid solution at a Y-piece (PEEK, ¼-28, 0.02 thru-hole, IDEX).
The liquid feed flow rate was set to 0.05–1 ml min−1 to give a 0.8–18 s tRes through the biocatalytic cartridge. Samples were collected using a fraction collector (BioRad, 2110), and reactor volumes were combined to obtain a sufficient amount of material for analysis by 1H NMR spectroscopy. Reaction progress was determined by comparison to analytical standards and literature data. Further experimental details are provided in the Supporting Information, Supplementary Table S2.
An appropriate sealed CatCart™ was prepared according to the procedures described in Preparation of Biocatalyst Cartridges for Hydrogenation Flow Reactor Studies. This was then installed in the H-Cube reactor and a liquid feed (Knauer H-Cube piston pump) containing sodium pyruvate 92 mM) and NAD+ (1.7 mM) in Tris-HCl buffer (50 mM, pH 8.0) was pumped through the H-Cube with the H2 that was generated by the built-in water reservoir and electrolyzer (gas feed flow rate was set to 12 ml min−1); the liquid feed flow rate was set to 0.05 ml min−1 to give a 1 s tRes through the biocatalyst cartridge. The reaction solution was recirculated through the reactor and aliquots were removed periodically for analysis by 1H NMR spectroscopy. Reaction progress was determined by comparison to analytical standards and literature data. Further experimental details are provided in the Supporting Information, Supplementary Table S2.
In previous flow studies, we used the H2-oxidizing/NAD+-reducing soluble hydrogenase (SH) from Ralstonia eutropha immobilized on carbon nanotube lined flow columns (Thompson et al., 2020a). The hydrogenase module of the SH is natively electronically linked to the NAD+ reductase module by a series of electron-relay iron-sulfur clusters (see Figure 2A). However, the results were limited by the stability of the SH, achieving low total turnover number (TTN, mol product per mol SH). In our previous batch studies, we focused on co-immobilizing a separate NAD+ reductase (a protein possessing just the NAD+ reductase activity of the SH) with a more robust hydrogenase (E. coli hydrogenase 1, “Hyd1”) onto carbon particles; this system yielded much higher stability and cofactor regeneration activity compared with the SH. For reference, we have demonstrated that Hyd1 itself maintains activity after >5 days of continual H2-driven operation (FMN recycling in this case) at room temperature (Joseph Srinivasan et al., 2021). For this two-enzyme catalyst, the carbon plays dual roles, acting as an enzyme support and also electronically linking the enzymes by conducting electrons generated via H2-oxidation by the Hyd1 to the NAD+ reductase, thereby powering the NAD+ reductase to regenerate NADH. We wondered whether native SH activity and/or stability could be enhanced through co-immobilization with the robust Hyd1 (see Figure 2B). We considered this system under batch conditions, and then explored whether advantages could be attained in flow. Stability is highly desirable in flow as reactions can run continuously for as long as the catalysts are active, in contrast to batch conditions under which biocatalysis reactions are typically run for 24 h until high conversion is achieved. By analogy with supported metal nanoparticle catalysts, we refer to the SH supported on carbon as “SH/C”, and we refer to the co-immobilized SH and Hyd1 on carbon as “SH + Hyd1/C”.
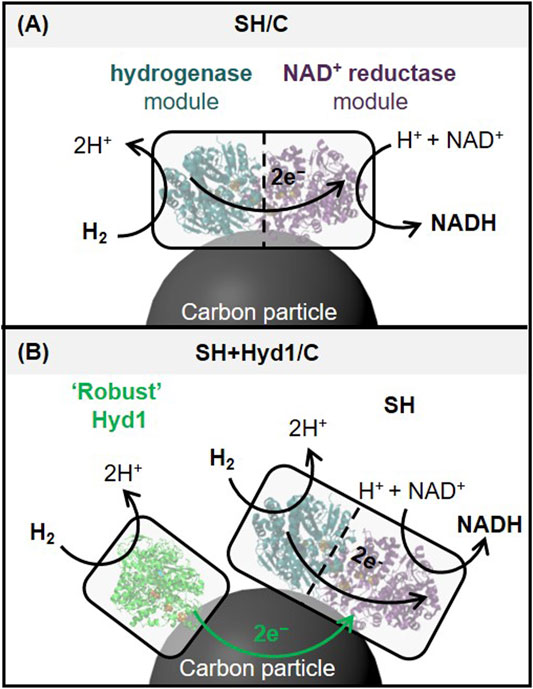
FIGURE 2. Illustration of the proposed boosting effect on NADH regeneration achieved by immobilizing a more active and robust hydrogenase (Hyd1) alongside SH on carbon. (A) Soluble hydrogenase consists of distinct modules for H2 oxidation (hydrogenase, dark green) and NAD+ reduction (NAD+ reductase, purple), with electron transfer between the modules facilitated by a chain of internal iron-sulfur electron relay centers. In this work, the SH is used as a heterogeneous biocatalyst by adsorption onto carbon particles. (B) Co-immobilization of an additional robust hydrogenase (Hyd1, light green) may provide additional H2-derived electrons through the carbon to the NAD+ reductase module, to supplement the relatively low activity and instability of the hydrogenase module of the SH.
First, the catalysts SH/C and SH + Hyd1/C were tested in H2-driven NADH-generating assays to determine the batch catalyst performance for the NADH generation step, Figure 3A. The enzymes were immobilized on carbon black (Black Pearls 2000, BP2000, ∼10 nm), which we have previously found to be a versatile enzyme support, according to the immobilization protocol described in General Enzyme Immobilization Procedure for Batch Studies(Poznansky et al., 2020). The enzyme-modified carbon particles were transferred into 800 μl of H2-saturated NAD+ (0.1 mM) in Tris-HCl (100 mM, pH 8.0) in a cuvette, then the reaction was monitored in situ by UV–Vis.
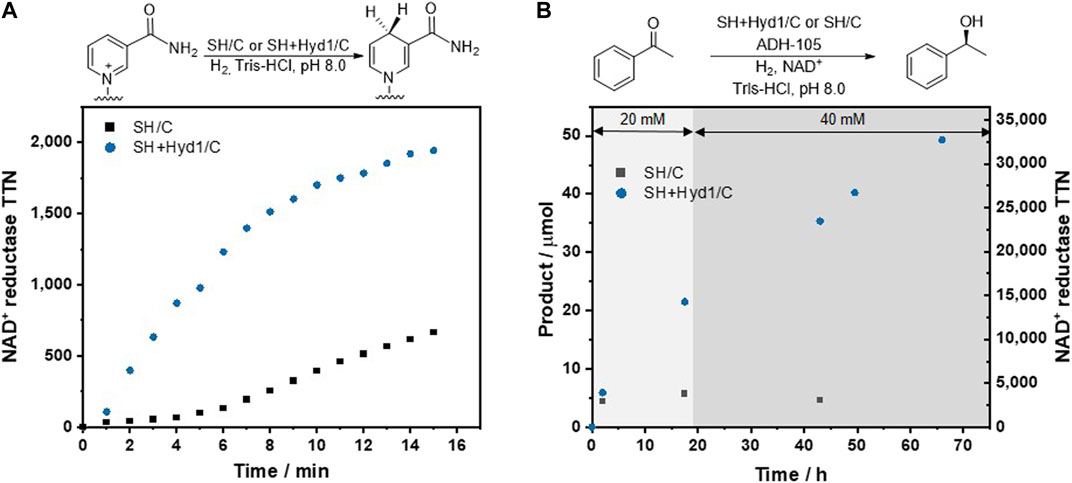
FIGURE 3. (A) Batch comparison of NAD+ reductase activity for H2-powered conversion of NAD+ to NADH using SH/C (12 μg SH, 30 μg BP 2,000) and SH + Hyd1/C [SH (4.8 μg), Hyd1 (3.4 μg), BP 2,000 (30 μg)]. Reactions conditions: H2-saturated NAD+ (0.1 mM) in Tris-HCl (100 mM, pH 8.0), 800 μl (see the Supporting Information Supplementary Figure S1 for full NAD+ and NADH structures). (B) Batch comparison between SH/C [SH (0.13 mg) and BP 2000 (0.40 mg)], and SH + Hyd1/C [SH (0.13 mg), Hyd1 (0.04 mg) and BP 2000 (0.40 mg)] catalysts using fed-batch conditions for the conversion of acetophenone to (S)-phenylethanol by H2-driven NADH recycling and ADH-105 in solution. Reaction conditions: acetophenone (20 mM plus a further 20 mM after 19 h), NAD+ (0.5 mM), Tris-HCl (100 mM, pH 8.0), 2 ml.
Results in Figure 3A are presented as TTN (mole of NADH produced per mole of the NAD+ reductase module of the SH) at each time point. Thus although the amount of SH is different between the two data sets in this figure, it is very clear that the activity of the NAD+ reductase module of the SH is much higher when the SH is co-immobilized with the additional hydrogenase, Hyd1. This is reproduced by a higher turnover frequency (TOF, mole NADH per mole NAD+ reductase module per minute) of 252 min−1 for SH + Hyd1/C compared to 65 min−1 for SH/C calculated from the linear phase of reaction. A brief lag phase observed for SH/C, likely due to activation of the hydrogenase module, is much less significant for the SH + Hyd1/C catalyst. These observations provide evidence that the hydrogenase module of the SH limits its overall activity, and that co-immobilization with a more active and robust hydrogenase (Hyd1) represents a more viable biocatalyst system.
The catalysts were next used for H2-driven NADH recycling coupled to a biocatalytic C = X bond reduction by an alcohol dehydrogenase (ADH-105): acetophenone reduction to (S)-phenylethanol. Batch reactions were performed in parallel using an H2-flushed Asynt OCTO mini parallel reactor. An H2-saturated NAD+ solution (2 ml of 0.5 mM solution in buffer) was added to each reaction tube, followed by SH/C or SH + Hyd1/C, ADH-105 (0.4 mg) and acetophenone (20 µl from a 2 M stock solution in DMSO) in this order. The parallel reactor was left to stir at 350 r.p.m. at room temperature under a steady flow of H2. A second dose of acetophenone (20 µl from a 2 M stock solution in DMSO) was added at 19 h. Reactions were analyzed for conversion and enantioselectivity by taking time-point aliquots for chiral-phase GC-FID, Figure 3B. Conversion to (S)-phenylethanol with >99% ee was determined at each point. Data in Figure 3B are presented on the left hand axis as conversion of the ketone to the product alcohol, and on the right hand axis as TTN for the NAD+ reductase module, based on the assumption that each equivalent of alcohol formed requires hydride transfer from one equivalent of NADH.
The 2 h time points show activity for both the SH/C and SH + Hyd1/C systems, with the conversion to (S)-phenylethanol being slightly lower for the SH/C system. By the subsequent time point at 17.5 h, the conversion achieved with the SH/C system had plateaued, reaching a TTN of just 3,800 for the NAD+ reductase module. The SH + Hyd1/C system was still active, however, having produced 21.5 μmol (S)-phenylethanol. Each reaction was supplied with an additional 20 mM acetophenone at 19 h. Although no further activity was exhibited by the SH/C system, the SH + Hyd1/C system continued to exhibit NADH-dependent acetophenone reduction, showing that the SH + Hyd1/C continued to recycle NADH. After 66 h, 49.0 μmol (S)-phenylethanol was produced by the SH + Hyd1/C system, equating to a TTN of 32,700 for the NAD+ reductase module. This represents a productivity of 47 g (S)-phenylethanol per gram SH. To evaluate the longevity of the SH + Hyd1/C further, we explored the possibility for re-use of the catalyst for further batch reactions. The SH + Hyd1/C displayed activity across three cycles of recovery and catalyst re-use, as outlined in the Supporting Information Supplementary Figure S2, retaining an SH productivity of 12.8 g(S)-phenylethanol gSH−1 on the 3rd reaction cycle.
Together, these batch results highlight that the SH + Hyd1/C displays improved activity and stability over the SH/C, making it a more appealing prospect for process intensification.
We next looked to determine whether these findings were also true in continuous flow. Previously, we found that the catalysts were limited not only by SH stability, but also by H2 availability in home-made flow reactors (Zor et al., 2017; Thompson et al., 2020a). We therefore also looked to pursue whether the inherently scalable H-Cube hydrogenation flow reactor would ease translation and improve process intensification options, as well as offering a more robust approach for catalyst and process comparisons.
To study the immobilized biocatalysts in continuous flow, enzyme-modified carbon particles were tested in the hydrogenation flow reactor within flow cartridges, prepared as described in Preparation of Biocatalyst Cartridges for Hydrogenation Flow Reactor Studies. Activated charcoal (>0.15 mm) was used as the support because a larger carbon particle diameter was required over BP2000 (∼10 nm) to avoid carbon leaching through the frits. For each of the flow results, parameters such as mass of enzyme and mass of carbon are specified in the relevant figure caption and experimental details are summarized in Supplementary Table S1 and Table S2 of the Supporting Information. An example of SH immobilization on carbon showing 100% immobilization yield and negligible leaching over 18 h is given in Supplementary Table S3 of the Supporting Information. First, we conducted a proof-of-concept experiment to determine the suitability of the reactor for H2-driven biocatalysis (Figure 4) by testing SH/C in a flow cartridge for the conversion of NAD+ to NADH.
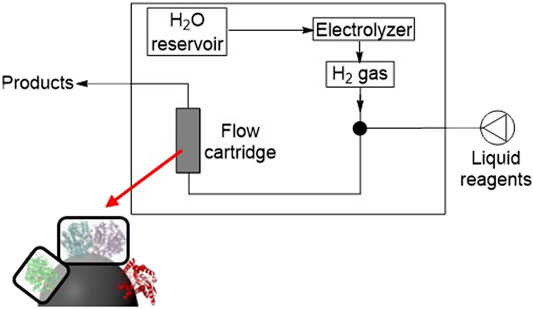
FIGURE 4. Schematic representation of the flow reactor, showing built-in electrolyzer for in situ H2 gas generation, with a flow cartridge containing carbon-supported biocatalysts.
A reaction solution containing NAD+ (1 mM) in buffer was pumped into the hydrogenation reactor at a range of flow rates (0.3, 1 or 3 ml min−1), where it was combined with H2 from the built-in water electrolysis H2 generator (approximately 12 ml min−1). As the gas flow rate made the major contribution to the total flow rate, this equated to tRes values through the reactor of 1, 0.9 or 0.8 s. The system pressure was set to 2, 10 or 50 bar. All of these parameters were tested across a single experiment on one biocatalytic cartridge, and the results are shown in Figure 5.
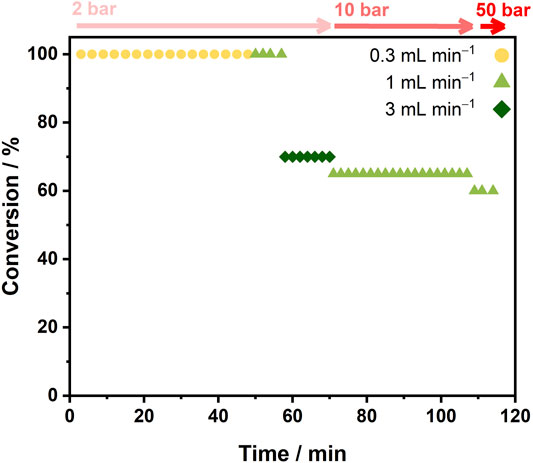
FIGURE 5. Continuous conversion of NAD+ to NADH using SH/C [SH (1.6 mg), activated charcoal (30 mg)] in the hydrogenation flow reactor over time under different liquid flow rates and pressures (top axis). Reaction conditions: NAD+ (1 mM), Tris-HCl buffer (50 mM, pH 8.0), liquid flow rate = 0.3, 1 or 3 ml min−1, H2 flow rate ≈12 ml min−1, tRes = 0.8, 0.9 or 1.0 s, reactor volume = 0.2 ml.
Quantitative conversion to NADH was achieved for liquid flow rates of 0.3 and 1 ml min−1 at 2 bar. At 3 ml min−1, the highest TOF of 223 min−1 was achieved (70% conversion), suggesting that this approach has overcome previous limitations of H2 availability in flow which were observed in earlier work (Thompson et al., 2020a). Increasing the pressure to 10 bar at 1 ml min−1 showed lower conversion (65%) compared with that at 2 bar; however, further increasing the system pressure to 50 bar had little effect on the conversion (60%). This demonstrated the stability of the biocatalytic cartridge at elevated pressure and, although the enzyme system does not require high pressure for operation, it shows the potential for use in processes where pressure is needed to keep up with H2 demand, or for multistep continuous processes in which higher pressures may be required for other reaction steps.
Having established the validity of the hydrogenation reactor for biocatalysis, we next examined intensifying the substrate throughput by performing experiments using higher concentrations of NAD+ over a longer period of time with the SH/C cartridge (see Supporting Information, Supplementary Figure S3). High conversion (>98%) of NAD+ (5 mM) to NADH is shown for 6.3 h and the system operated at an overall productivity of 108 gNADH gSH−1 an E factor of 0.9.
To determine whether addition of Hyd1 to the activated charcoal would display a boosting effect, as seen in batch conditions, we performed an activity lifetime comparison of SH/C against SH + Hyd1/C in continuous flow, Figure 6A. A cartridge containing SH/C and a cartridge containing SH + Hyd1/C were prepared with a decreased SH loading compared with the previous flow experiments, with the aim of putting the catalyst under strained conditions (<70% conversion). This allowed for differences between the two systems to be easily recognized. The cartridges were both tested under conditions of NAD+ (5 mM) in buffer, at 30°C, and at a liquid flow rate 0.05 ml min−1. Results in Figure 6A show that the SH/C is almost fully deactivated after 17 h (3,300 reactor volumes), displaying a TTN of 47,200 and an overall system productivity of 197 gNADH gSH−1. This deactivation is similar to that which was observed when SH was immobilized onto carbon nanotube-lined columns and used for NADH generation in continuous flow (Thompson et al., 2020a).
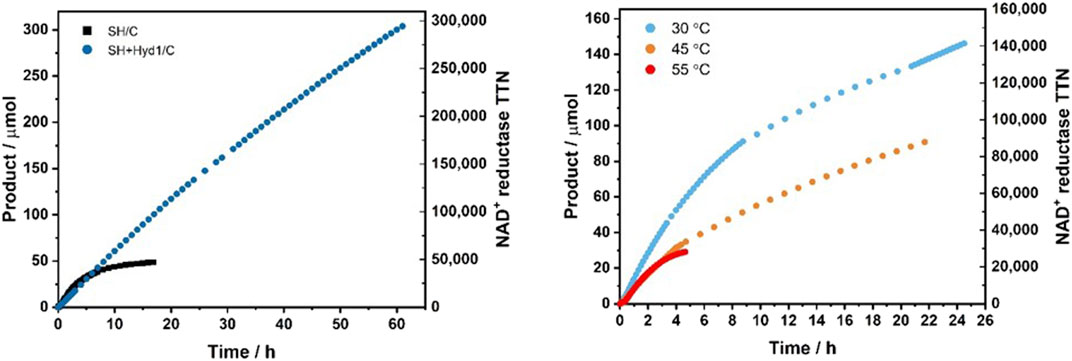
FIGURE 6. NADH generation by SH/C or SH + Hyd1/C under continuous flow hydrogenation conditions. The left y axis shows the cumulative amount of product formed, and the right y axis shows the TTN of the NAD+ reductase module of the SH. (A) Lifetime performance of SH/C [SH (0.18 mg), activated charcoal (20 mg)] vs. SH + Hyd1/C [SH (0.18 mg), Hyd1 (0.2 mg), activated charcoal (20 mg)] for the conversion of NAD+ to NADH in the hydrogenation flow reactor. Reaction conditions: NAD+ (5 mM), Tris-HCl buffer (50 mM, pH 8.0), 30°C, liquid flow rate = 0.05 ml min−1, H2 flow rate = 0.6 ml min−1, tRes = 18 s, reactor volume = 0.2 ml. (B) The effect of temperature on the lifetime performance of SH + Hyd1/C [SH (0.18 mg), Hyd1 (0.2 mg), activated charcoal (20 mg)] for the conversion of NAD+ to NADH in the flow reactor. Reaction conditions: NAD+ (5 mM), Tris-HCl buffer (50 mM, pH 8.0), liquid flow rate = 0.1 ml min−1, H2 flow rate = 0.6 ml min−1, tRes = 17 s, reactor volume = 0.2 ml).
Promisingly, after 61 h (11,895 reactor volumes) the SH + Hyd1/C was still displaying a TOF of 60 min−1 and had reached a TTN of 294,000 with an overall system productivity of 671 gNADH gSH−1. This clearly demonstrated that the addition of Hyd1 enhances the catalytic stability of the system. It is probable that the hydrogenase module of the SH denatures at a faster rate than the NAD+ reductase module, which leads to the decrease in activity displayed by the SH/C. The Hyd1 is able to provide supplementary electrons from H2 oxidation to the NAD+ reductase module by conduction through the activated charcoal, thus boosting the overall cofactor regeneration activity of the dual-enzyme system.
It was also found that the SH/C deactivates rapidly at >40°C in the hydrogenation flow reactor (see Supplementary Figure S4, Supporting Information). In contrast Hyd1 has excellent temperature stability (Joseph Srinivasan et al., 2021). To determine the activity lifetime of SH + Hyd1/C at elevated temperatures, three identical biocatalytic cartridges containing SH + Hyd1/C were prepared and were tested under conditions of NAD+ (5 mM) in buffer, at a liquid flow rate 0.1 ml min−1 in the flow reactor at 30°C, 45°C or 55°C, respectively. At 1 h, all cartridges showed comparable activity. However, at 4.6 h, the cartridge at 55°C had fully deactivated reaching an overall TTN of 34,600 for the NAD+ reductase module. At 21.7 h, the cartridge at 45°C had displayed an average TOF of 85 min−1, 83% of the average TOF displayed by the cartridge at 30°C at the same time point. The respectable performance of the system at 45°C suggests that, in the future, it could be combined with thermophilic NADH-dependent enzymes or work in cascade with other bio- and chemocatalysts that require elevated temperatures.
Enzymes for H2-driven cofactor recycling were next coupled to a model C=O bond reduction, the conversion of pyruvate to lactate by lactate dehydrogenase (LDH), Scheme 1. The biocatalyst cartridge was prepared by co-immobilizing LDH with SH on the activated carbon. The hydrogenation flow reactor was set up with a single liquid feed containing pyruvate (10 mM) and NAD+ (1 mM) in Tris-HCl buffer (50 mM, pH 8.0). The liquid flow rate was set to 0.1, 0.5 or 1 ml min−1, equating to a tRes of 1.7, 1.6 or 1.5 s, when combined with a H2 flow rate of 7 ml min−1. For each set of conditions, the reactor was allowed to reach steady-state and then multiple fractions were collected for analysis of pyruvate/lactate content by 1H NMR spectroscopy. Results of this experiment are presented in Figure 7. The conversion of pyruvate to lactate reached a steady-state conversion of >96% at 0.1 ml min−1 liquid flow rate, which was maintained for 100 min. The steady-state conversions achieved at 0.5 ml min−1 and 1 ml min−1 were 69 and 22%, respectively, resulting in a SH TTN of 47,200 across the whole experiment.
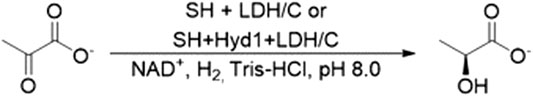
SCHEME 1. H2-powered conversion of pyruvate to lactate in a biocatalytic cartridge.
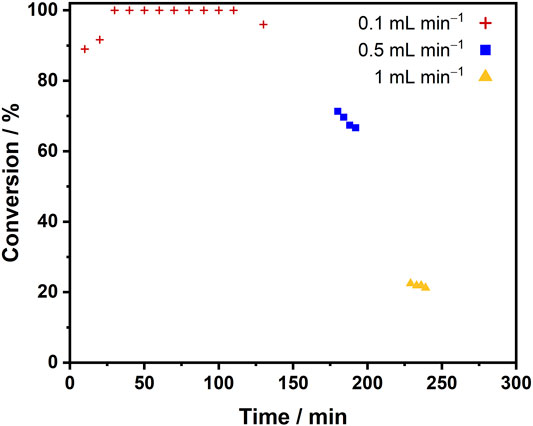
FIGURE 7. Conversion of pyruvate to lactate using SH + LDH/C [SH (1.6 mg), LDH (3 mg), activated charcoal (43 mg)] under liquid flow rates of 0.1 ml min−1, 0.5 ml min−1 and 1 ml min−1 in the hydrogenation flow reactor. Reaction conditions: pyruvate (10 mM), NAD+ (1 mM), Tris-HCl buffer (50 mM, pH 8.0), 25°C, liquid flow rate = 0.1, 0.5 or 1 ml min−1, gas flow rate ≈7 ml min−1, reactor volume = 0.2 ml.
Having established activity of the system for pyruvate to lactate conversion, the system was once again set up to determine whether Hyd1 would provide a similar boosting effect. To compare the activity lifetime of the two enzyme combinations, a cartridge containing SH + LDH/C and a cartridge containing SH + Hyd1+LDH/C were prepared. They were both tested under conditions of pyruvate (20 mM), NAD+ (1 mM) in buffer at 30°C, at a range of liquid flow rates (0.05 and 0.1 ml min−1) in the hydrogenation flow reactor, Figure 8.
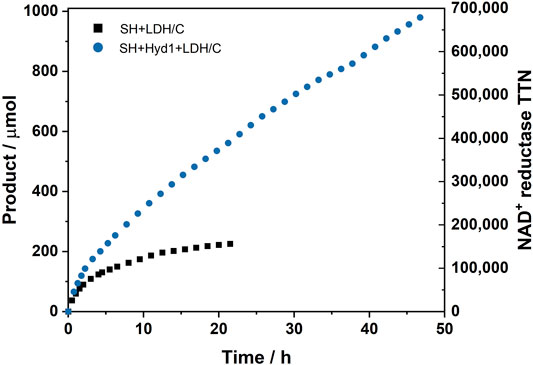
FIGURE 8. Lifetime performance of SH + LDH/C [SH (0.25 mg), LDH (3 mg), activated charcoal (30 mg)] vs. SH + Hyd1+LDH/C [SH (0.25 mg), Hyd1 (0.2 mg), LDH (3 mg), activated charcoal (30 mg)] for the conversion of pyruvate to lactate in the hydrogenation flow reactor. The cumulative amount of product formed is shown on the left y axis. The TTN of the NAD+ reductase module of the SH is shown on the right y axis. Reaction conditions: pyruvate (20 mM), NAD+ (1 mM), Tris-HCl buffer (50 mM, pH 8.0), 30°C, liquid flow rate = 0.05–0.1 ml min−1, H2 flow rate = 0.6–1.2 ml min−1, tRes = 9–18 s, reactor volume = 0.2 ml.
The results in Figure 8 show a similar trend to that observed in the NADH generation experiments shown in Figure 6A. The SH + LDH/C was almost entirely deactivated after 21.5 h, displaying a SH TTN of 156,400 (average TOF = 121 min−1) and an overall system productivity of 102 glactate gSH−1. The SH + Hyd1+LDH/C was still displaying catalytic TOF of 180 min−1 after 46.8 h of operation and reached a TTN of 679,800 (average TOF = 243 min−1), demonstrating an overall system productivity of 444 glactate gSH−1. The initial catalytic TOFs of the SH + LDH/C and the SH + Hyd1+LDH/C systems were 860 min−1and 1,013 min−1, respectively. The rapid consumption of the regenerated NADH by the LDH appears to drive an increase in SH catalytic performance for both of the H2-driven cofactor recycling systems; this is likely enhanced further by immobilizing the enzymes alongside each other on the carbon (Reeve et al., 2015).
Overall, the continuous flow system demonstrates an improvement in catalytic efficiency over the batch studies due to the intensification of conditions achievable in the flow reactor, namely increased H2 availability, high enzyme-to-substrate ratio, and consequently short tRes. To achieve higher conversions, one option would be to increase the catalyst bed volume, but this would have required more enzyme, and at present the SH and Hyd1 are both produced at laboratory scale although scale-up efforts are underway in separate research. Therefore a multi-pass set-up was explored in a closed loop reactor in which the reaction solution is recirculated through the hydrogenation flow reactor from a single, ambient pressure reactor vessel and combined constantly with the H2 stream from the electrolyzer. This was tested at gram scale for the conversion of pyruvate to lactate. A cartridge containing SH + Hyd1+LDH/C was prepared. A 100 ml solution of pyruvate (92 mM), NAD+ (1.7 mM) in buffer at 30°C was circulated through the hydrogenation flow reactor at a liquid flow rate 0.5 ml min−1 (complete pass = 200 min, tRes = 1 s). Time points were taken for analysis to measure the course of the reaction. The results in Figure 9 show that the closed loop, multi-pass flow system ran over a total of 67 h (20 complete reaction solution passes, giving overall tRes = 20 s), producing 0.525 g of lactate and demonstrating a SH TTN of 766,100 at an overall productivity of 500 glactate gSH−1. The cofactor recycling system displayed a maximum TOF of 1,060 min−1 (63,600 h−1), which is at the top end of typical rates achieved for the metal-catalyzed transfer hydrogenation of ketones (102–105 h−1) (Štefane and Požgan, 2016).
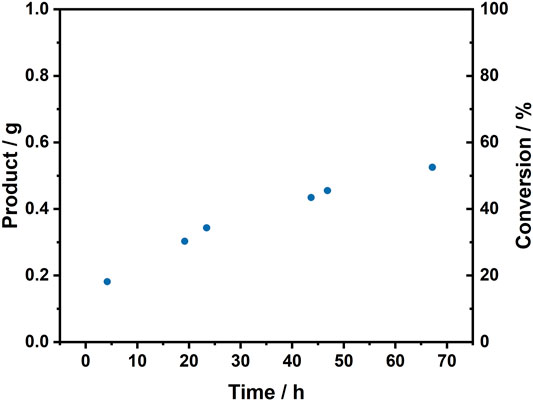
FIGURE 9. Closed loop, multi-pass flow reaction at gram scale. Increase in conversion of pyruvate to lactate with SH + Hyd1+LDH/C [SH (1.05 mg), Hyd1 (0.94 mg), LDH (5 mg), activated charcoal (60 mg)] in the hydrogenation flow reactor. A 100 ml solution of pyruvate (92 mM), NAD+ (1.7 mM) and Tris-HCl buffer (50 mM, pH 8.0) was recirculated through the system with time points taken periodically. Reaction conditions: liquid flow rate = 0.5 ml min−1, H2 flow rate ≈12 ml min−1, reactor volume = 0.2 ml, complete pass of reaction solution = 200 min, tRes = 1 s.
We have demonstrated the translation of carbon-supported biocatalytic hydrogenations from small-scale batch studies into a commercial hydrogenation flow reactor, where we were able to achieve full conversions in very short tRes (seconds) and high enzyme lifetime TTN (105) with reactions performed over 46.8 h. The immobilization of Hyd1 alongside the SH on carbon was shown to increase SH TOF and TTN in both batch and flow, with greater than six-fold improvement to TTN for the SH + Hyd1/C system in flow. Intensification in a closed loop, multi-pass flow set-up with SH + Hyd1+LDH/C gave 0.5 g product for the model pyruvate to lactate reaction. H2-powered biocatalysis carried out in the H-Cube hydrogenation flow reactor, using simply-prepared catalyst cartridges, is shown to give comparable catalyst TOF to supported metal transfer hydrogenation catalyst columns. The use of commercial carbon catalyst supports, together with the straightforward transfer of biocatalyst particles into this scalable commercial hydrogenation flow reactor highlights how this biocatalytic route could be slotted into existing reactor infrastructure. Numerous industrially-engineered dehydrogenase (keto-reductase) enzymes are available for catalysis of wide-ranging ketone reductions, and the methods we report here have the potential to simplify adoption of these NADH-dependent biocatalysts for asymmetric synthesis in flow.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
BP, SC, LT, and HR performed experiments; BP, SC, LT, and HR. analysed data. The manuscript was written with contributions from all of the authors. All of the authors have given approval to the final version of the manuscript.
SC, LT, HR, and KV are supported by Engineering and Physical Sciences Research Council (EPSRC) IB Catalyst award EP/N013514/1. BP is supported by iCASE funding from the Biotechnology and Biological Sciences Research Council (BBSRC, grant number BB/M011224/1), with support from Reddy’s Laboratories.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank O. Lenz (TU Berlin) for his gift of the Ralstonia eutropha strain HF904; staff at ThalesNano for access to a H-Cube Pro demo reactor; and B. Dominguez of Johnson Matthey for providing samples of ADH-105. BP thanks M. Bycroft [Dr. Reddy’s Laboratories (EU)] for useful discussions, project support and for use of their H-Cube Pro reactor. We acknowledge M. Ramirez Hernandez for preparing samples of SH and Hyd1.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fceng.2021.718257/full#supplementary-material
Adams, J. P., Brown, M. J. B., Diaz‐Rodriguez, A., Lloyd, R. C., and Roiban, G. D. (2019). Biocatalysis: A Pharma Perspective. Adv. Synth. Catal. 361, 2421–2432. doi:10.1002/adsc.201900424
Al-Shameri, A., Petrich, M. C., Junge Puring, K., Apfel, U. P., Nestl, B. M., and Lauterbach, L. (2020). Powering Artificial Enzymatic Cascades with Electrical Energy. Angew. Chem. Int. Ed. Engl. 59, 10929–10933. doi:10.1002/anie.202001302
Britton, J., Majumdar, S., and Weiss, G. A. (2018). Continuous Flow Biocatalysis. Chem. Soc. Rev. 47, 5891–5918. doi:10.1039/C7CS00906B
Bryan, M. C., Dunn, P. J., Entwistle, D., Gallou, F., Koenig, S. G., Hayler, J. D., et al. (2018). Key Green Chemistry Research Areas from a Pharmaceutical Manufacturers' Perspective Revisited. Green. Chem. 20, 5082–5103. doi:10.1039/C8GC01276H
Caron, S., Dugger, R. W., Ruggeri, S. G., Ragan, J. A., and Ripin, D. H. B. (2006). Large-Scale Oxidations in the Pharmaceutical Industry. Chem. Rev. 106, 2943–2989. doi:10.1021/cr040679f
Contente, M. L., Dall'Oglio, F., Tamborini, L., Molinari, F., and Paradisi, F. (2017). Highly Efficient Oxidation of Amines to Aldehydes with Flow-Based Biocatalysis. ChemCatChem 9, 3843–3848. doi:10.1002/cctc.201701147
Cossar, P. J., Hizartzidis, L., Simone, M. I., McCluskey, A., and Gordon, C. P. (2015). The Expanding Utility of Continuous Flow Hydrogenation. Org. Biomol. Chem. 13, 7119–7130. doi:10.1039/c5ob01067e
Dall’Oglio, F., Contente, M. L., Conti, P., Molinari, F., Monfredi, D., Pinto, A., et al. (2017). Flow-based Stereoselective Reduction of Ketones Using an Immobilized Ketoreductase/glucose Dehydrogenase Mixed Bed System. Catal. Commun. 93, 29–32. doi:10.1016/j.catcom.2017.01.025
De Santis, P., Meyer, L.-E., and Kara, S. (2020). The Rise of Continuous Flow Biocatalysis - Fundamentals, Very Recent Developments and Future Perspectives. React. Chem. Eng. 5, 2155–2184. doi:10.1039/D0RE00335B
Devine, P. N., Howard, R. M., Kumar, R., Thompson, M. P., Truppo, M. D., and Turner, N. J. (2018). Extending the Application of Biocatalysis to Meet the Challenges of Drug Development. Nat. Rev. Chem. 2, 409–421. doi:10.1038/s41570-018-0055-1
Döbber, J., and Pohl, M. (2017). HaloTag: Evaluation of a Covalent One-step Immobilization for Biocatalysis. J. Biotechnol. 241, 170–174. doi:10.1016/J.JBIOTEC.2016.12.004
Fernandez-Lafuente, R., and Berenguer-Murcia, A. (2010). New Trends in the Recycling of NAD(P)H for the Design of Sustainable Asymmetric Reductions Catalyzed by Dehydrogenases. Curr. Org. Chem. 14, 1000–1021. doi:10.2174/138527210791130514
Hübner, S., de Vries, J. G., and Farina, V. (2016). Why Does Industry Not Use Immobilized Transition Metal Complexes as Catalysts?. Adv. Synth. Catal. 358, 3–25. doi:10.1002/adsc.201500846
Huisman, G. W., Liang, J., and Krebber, A. (2010). Practical Chiral Alcohol Manufacture Using Ketoreductases. Curr. Opin. Chem. Biol. 14, 122–129. doi:10.1016/j.cbpa.2009.12.003
Irfan, M., Glasnov, T. N., and Kappe, C. O. (2011). Heterogeneous Catalytic Hydrogenation Reactions in Continuous-Flow Reactors. ChemSusChem 4, 300–316. doi:10.1002/cssc.201000354
Jiménez-González, C., Poechlauer, P., Broxterman, Q. B., Yang, B.-S., am Ende, D., Baird, J., et al. (2011). Key Green Engineering Research Areas for Sustainable Manufacturing: A Perspective from Pharmaceutical and Fine Chemicals Manufacturers. Org. Process. Res. Dev. 15, 900–911. doi:10.1021/op100327d
Joseph Srinivasan, S., Cleary, S. E., Ramirez, M. A., Reeve, H. A., Paul, C. E., and Vincent, K. A. (2021). E. coli Nickel‐Iron Hydrogenase 1 Catalyses Non‐native Reduction of Flavins: Demonstration for Alkene Hydrogenation by Old Yellow Enzyme Ene‐reductases**. Angew. Chem. Int. Ed. 60, 13824–13828. doi:10.1002/anie.202101186
Lauterbach, L., and Lenz, O. (2013). Catalytic Production of Hydrogen Peroxide and Water by Oxygen-Tolerant [NiFe]-Hydrogenase during H2Cycling in the Presence of O2. J. Am. Chem. Soc. 135, 17897–17905. doi:10.1021/ja408420d
Liese, A., and Hilterhaus, L. (2013). Evaluation of Immobilized Enzymes for Industrial Applications. Chem. Soc. Rev. 42, 6236–6249. doi:10.1039/C3CS35511J
Mallat, T., Orglmeister, E., and Baiker, A. (2007). Asymmetric Catalysis at Chiral Metal Surfaces. Chem. Rev. 107, 4863–4890. doi:10.1021/cr0683663
Noyori, R. (2002). Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 41, 2008–2022. doi:10.1002/1521-3773(20020617)41:12<2008::AID-ANIE2008>3.0.CO;2-4
Noyori, R., and Hashiguchi, S. (1997). Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Acc. Chem. Res. 30, 97–102. doi:10.1021/ar9502341
Peschke, T., Skoupi, M., Burgahn, T., Gallus, S., Ahmed, I., Rabe, K. S., et al. (2017). Self-Immobilizing Fusion Enzymes for Compartmentalized Biocatalysis. ACS Catal. 7, 7866–7872. doi:10.1021/acscatal.7b02230
Poznansky, B., Thompson, L. A., Warren, S. A., Reeve, H. A., and Vincent, K. A. (2020). Carbon as a Simple Support for Redox Biocatalysis in Continuous Flow. Org. Process. Res. Dev. 24, 2281–2287. doi:10.1021/acs.oprd.9b00410
Reeve, H. A., Lauterbach, L., Lenz, O., and Vincent, K. A. (2015). Enzyme-Modified Particles for Selective Biocatalytic Hydrogenation by Hydrogen-Driven NADH Recycling. ChemCatChem 7, 3480–3487. doi:10.1002/cctc.201500766
Rodrigues, R. C., Ortiz, C., Berenguer-Murcia, Á., Torres, R., and Fernández-Lafuente, R. (2013). Modifying Enzyme Activity and Selectivity by Immobilization. Chem. Soc. Rev. 42, 6290–6307. doi:10.1039/C2CS35231A
Rowbotham, J. S., Ramirez, M. A., Lenz, O., Reeve, H. A., and Vincent, K. A. (2020). Bringing Biocatalytic Deuteration into the Toolbox of Asymmetric Isotopic Labelling Techniques. Nat. Commun. 11, 1454. doi:10.1038/s41467-020-15310-z
Russell, C. C., Baker, J. R., Cossar, P. J., and McCluskey, A. (2017). “Recent Developments in the Use of Flow Hydrogenation in the Field of Medicinal Chemistry,” in New Advances In Hydrogenation Processes - Fundamentals And Applications. Editors M. T. Ravanchi. London: InTech. doi:10.5772/65518
Santi, M., Sancineto, L., Nascimento, V., Braun Azeredo, J., Orozco, E. V. M., Andrade, L. H., et al. (2021). Flow Biocatalysis: A Challenging Alternative for the Synthesis of APIs and Natural Compounds. Ijms 22, 990. doi:10.3390/ijms22030990
Sheldon, R. A., and van Pelt, S. (2013). Enzyme Immobilisation in Biocatalysis: Why, what and How. Chem. Soc. Rev. 42, 6223–6235. doi:10.1039/C3CS60075K
Štefane, B., and Požgan, F. (2016). Metal-Catalysed Transfer Hydrogenation of Ketones. Top. Curr. Chem. 374, 1–67. doi:10.1007/s41061-016-0015-5
Tamborini, L., Fernandes, P., Paradisi, F., and Molinari, F. (2018). Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification. Trends Biotechnol. 36, 73–88. doi:10.1016/j.tibtech.2017.09.005
Thompson, L. A., Rowbotham, J. S., Nicholson, J. H., Ramirez, M. A., Zor, C., Reeve, H. A., et al. (2020a). Rapid, Heterogeneous Biocatalytic Hydrogenation and Deuteration in a Continuous Flow Reactor. ChemCatChem 12, 3913–3918. doi:10.1002/cctc.202000161
Thompson, L. A., Rowbotham, J. S., Reeve, H. A., Zor, C., Grobert, N., and Vincent, K. A. (2020b). Biocatalytic Hydrogenations on Carbon Supports. Methods Enzymol. 630, 303–325. doi:10.1016/bs.mie.2019.10.017
Thompson, M. P., Peñafiel, I., Cosgrove, S. C., and Turner, N. J. (2019). Biocatalysis Using Immobilized Enzymes in Continuous Flow for the Synthesis of Fine Chemicals. Org. Process. Res. Dev. 23, 9–18. doi:10.1021/acs.oprd.8b00305
Tufvesson, P., Fu, W., Jensen, J. S., and Woodley, J. M. (2010). Process Considerations for the Scale-Up and Implementation of Biocatalysis. Food Bioproducts Process. 88, 3–11. doi:10.1016/j.fbp.2010.01.003
Wang, X., and Ding, K. (2004). Self-Supported Heterogeneous Catalysts for Enantioselective Hydrogenation. J. Am. Chem. Soc. 126, 10524–10525. doi:10.1021/ja047372i
Winkler, C. K., Schrittwieser, J. H., and Kroutil, W. (2021). Power of Biocatalysis for Organic Synthesis. ACS Cent. Sci. 7, 55–71. doi:10.1021/acscentsci.0c01496
Zanotti-Gerosa, A., Hems, W., Groarke, M., and Hancock, F. (2005). Ruthenium-catalysed Asymmetric Reduction of Ketones. Platinum Met. Rev. 49, 158–165. doi:10.1595/147106705X75421
Keywords: continuous flow, cofactor recycling, H-Cube, immobilized enzyme, hydrogenase
Citation: Poznansky B, Cleary SE, Thompson LA, Reeve HA and Vincent KA (2021) Boosting the Productivity of H2-Driven Biocatalysis in a Commercial Hydrogenation Flow Reactor Using H2 From Water Electrolysis. Front. Chem. Eng. 3:718257. doi: 10.3389/fceng.2021.718257
Received: 31 May 2021; Accepted: 27 July 2021;
Published: 06 August 2021.
Edited by:
Bartholomäus Pieber, Max Planck Institute of Colloids and Interfaces, GermanyReviewed by:
Florian Rudroff, Vienna University of Technology, AustriaCopyright © 2021 Poznansky, Cleary, Thompson, Reeve and Vincent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holly A. Reeve, aG9sbHlAaHlkcmVnZW5veGZvcmQuY29t; Kylie A. Vincent, a3lsaWUudmluY2VudEBjaGVtLm94LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.