 Elsie Chit Yu Iu
Elsie Chit Yu Iu Ho So
Ho So Chi Bun Chan
Chi Bun Chan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 14 May 2024
Sec. Molecular and Cellular Pathology
Volume 12 - 2024 | https://doi.org/10.3389/fcell.2024.1403463
This article is part of the Research Topic Current Trends in Muscle Diseases and Their Treatment Strategies View all 8 articles
Sporadic inclusion body myositis (sIBM) is a distinct subcategory of Idiopathic Inflammatory Myopathies (IIM), characterized by unique pathological features such as muscle inflammation, rimmed vacuoles, and protein aggregation within the myofibers. Although hyperactivation of the immune system is widely believed as the primary cause of IIM, it is debated whether non-immune tissue dysfunction might contribute to the disease’s onset as patients with sIBM are refractory to conventional immunosuppressant treatment. Moreover, the findings that mitochondrial dysfunction can elicit non-apoptotic programmed cell death and the subsequent immune response further support this hypothesis. Notably, abnormal mitochondrial structure and activities are more prominent in the muscle of sIBM than in other types of IIM, suggesting the presence of defective mitochondria might represent an overlooked contributor to the disease onset. The large-scale mitochondrial DNA deletion, aberrant protein aggregation, and slowed organelle turnover have provided mechanistic insights into the genesis of impaired mitochondria in sIBM. This article reviews the disease hallmarks of sIBM, the plausible contributors of mitochondrial damage in the sIBM muscle, and the immunological responses associated with mitochondrial perturbations. Additionally, the potential application of mitochondrial-targeted chemicals as a new treatment strategy to sIBM is explored and discussed.
Idiopathic inflammatory myopathies (IIM), commonly known as myositis, is a heterogeneous group of muscle diseases with a global prevalence of 2–25 individuals per 100, 000 persons (Khoo et al., 2023). Based on the clinical, serological, and histological examination results, IIM can be classified into 5 major subtypes: dermatomyositis (DM), polymyositis (PM), immune-mediated necrotizing myopathy (IMNM), overlap myositis (OM), and sporadic inclusion body myositis (sIBM). Despite each IIM subtype has distinctive clinical and unique histopathological features, a great majority of IIM patients suffer from muscle weakness with muscle edema, fatty infiltration in muscle fibers, and elevated serum creatine kinase (CK) level (Tsamis et al., 2022). Additionally, most IIM patients demonstrate overactivation of cellular and humoral immunity as revealed by the presence of infiltrating granzyme and perforin-secreting CD8+ cytotoxic T cells and B cells in muscle and autoantibodies in their serum (Goebels et al., 1996; Greenberg et al., 2005; Gunawardena et al., 2009). The involvement of innate immunity in IIM is also evidenced by the muscle enrichment of dendritic cells and M2 macrophages for tissue repair (Rinnenthal et al., 2014).
Because the muscles of IIM patients exhibit mild-to-severe inflammation phenotypes, most treatments for IIM target the uncontrolled immune response. Glucocorticoids (GC) are the front-line treatment that is widely adopted in PM and DM (Greenberg, 2019). They are often administrated with other immunosuppressants like azathioprine and methotrexate to reduce steroid-related side effects like osteoporosis and cardiovascular diseases (Oddis, 2016). Second-line treatments such as cyclosporine, tacrolimus, and intravenous immunoglobin are used to alleviate the disease symptoms in refractory or severe PM and DM cases (Zeng et al., 2022; Skolka and Naddaf, 2023). Nonetheless, these immunosuppressive agents are not enduringly effective in treating some IIM patients, particularly those with sIBM (Skolka and Naddaf, 2023). A recent study using human sIBM xenografts demonstrated that T cell depletion does not alleviate the muscle degenerative features (Britson et al., 2022), further challenging the pathogenic role of inflammation in the disease. Therefore, it is suspected that the immune response might not be the fundamental cause of muscle damage in sIBM. Because abnormal mitochondrial changes are commonly found in the muscle of sIBM patients, which are more prevalent than other IIM subtypes (Oldfors et al., 2006), impaired mitochondrial function might be an underestimated factor in the pathogenesis of sIBM. Several excellent reviews have discussed how mitochondrial dysfunction may generate muscle weakness in sIBM (De Paepe, 2019; Chapa et al., 2023; Danieli et al., 2023), but the underlying mechanism of mitochondrial defects remains inconclusive. In this article, we review the recent findings of mitochondrial dysfunction in sIBM and discuss their possible linkage with various disease symptoms. We will also discuss some potential mitochondrial-based therapeutic strategies for the treatment of sIBM.
sIBM is a male-predominant disease that has a prevalence of 5–71 per million in the whole population (Tsamis et al., 2022). Typically occurring after the age of 50 (Snedden et al., 2022), sIBM is characterized by progressive and asymmetric weakness in the quadriceps and long finger flexors rather than an acute or subacute symmetrical proximal muscle weakness seen in other subtypes of IIM (Zubair et al., 2023). Individuals with sIBM experience a loss of muscle strength at a rate of 3.5%–28% per year (Zubair et al., 2023) and can become wheelchair-bound at a median time of 10.5 years after the disease onset (Skolka and Naddaf, 2023). Respiratory compromise and dysphagia are common risk factors for premature death in sIBM patients, which may account for their slight but significantly shortened lifespan when compared to healthy subjects (Shelly et al., 2021). Recently, an association between sIBM and malignancy has been proposed, although sIBM is not generally regarded as a risk factor for cancer (Damian et al., 2022). Due to the slowly progressive nature of symptom development, there is often a delay of 5–10 years between disease onset and confirmation (Huntley et al., 2019).
The diagnosis of sIBM is typically made in patients having experienced disease symptoms for more than 12 months with onset after 45-year-old, (Greenberg, 2019; Skolka and Naddaf, 2023), and most importantly exhibit histopathological hallmarks in their muscle, including endomysial lymphocyte infiltration, rimmed vacuoles, protein deposition, ragged red fibres, and the presence of cytochrome c oxidase (COX)-negative myofibers (Tanboon et al., 2020). The accumulation of proteins like amyloid beta (Aβ), ubiquitin, phosphor-tau, sequestosome 1 (p62), and TAR DNA binding protein 43 kDa (TDP-43) is also detected in the muscle of some sIBM patients (Skolka and Naddaf, 2023). Autoantibodies against cytosolic 50-nucleotidase (cN-1A), an enzyme highly expressed in the skeletal muscle that catalyses the hydrolysis of adenosine monophosphate (AMP) into adenosine and inorganic phosphate (Salam et al., 2022; Diederichsen et al., 2023) can be found in many sIBM patients but not in any other IIM subtypes (Yamashita et al., 2023), making it an important evidence to the disease diagnosis. Nevertheless, a substantial number of sIBM patients displayed no elevated anti-cN-1A antibody in their circulation, suggesting that the presence of antibody should not be considered a necessary criterion in the disease diagnosis (Mavroudis et al., 2021; Diederichsen et al., 2023). While histological analysis of muscle biopsy remains an essential procedure for sIBM diagnosis (Papadimas et al., 2019; Winkler et al., 2021), other methods such as multi-osmics profiling are being developed to screen for novel biomarkers with 100% sensitivity and specificity (Cantó Santos et al., 2023).
The presence of anti-cN-1A antibodies and the elevated level of inflammatory cytokines like interferon γ (IFN-γ), tumor necrosis factor α (TNF-α), interleukin 7 (IL-7), and interleukin 32 (IL-32) in the serum of sIBM patients indicate that both humoral and cellular immunities are provoked (Tsamis et al., 2022). The hyperactivation of cellular immunity in sIBM is demonstrated by the transformation of CD4+ helper T-cells into CD28− cytotoxic T-cells and the infiltration of CD8+ T cells in the patient’s muscle (Miller et al., 2018). These cytotoxic T-cells might upregulate the muscular expression of major histocompatibility complex I (MHC-I) genes and induce muscle cell death, possibly via perforin-granzymes or Fas ligand-mediated mechanisms (Fyhr and Oldfors, 1998; Snedden et al., 2022). Indeed, immunohistochemical analysis revealed stronger Fas immunoreactivity in the atrophic fibres of sIBM patients than that in other IIM subtypes (De Bleecker et al., 2001). Despite the clear engagement of the immune system in the pathogenesis of sIBM, disease alleviation via immune response inhibition is not satisfactory, as a significant number of patients are irresponsive to the classical immunosuppressive treatment (Askanas and Engel, 2008; Rygiel et al., 2015). For instance, some sIBM patients receiving long-term GC treatment for more than 5 years had a similar number of T-cells-invaded myofibers as those patients without treatment (Pruitt et al., 1996). Muscle atrophy of sIBM patients has also not ceased even though their CK levels have returned to normal after GC treatment (Miller et al., 2018). Moreover, the extent of immune cell infiltration in sIBM patients was found to correlate poorly with the severity of muscle weakness (Lightfoot et al., 2015). Hence, it is argued that prolonged immune system activation in sIBM is secondary to the intrinsic defects in the skeletal muscle.
A distinguishing characteristic of sIBM that differentiates it from other IIM subtypes is the presence of ragged red fibers in the patient’s muscle (Boncompagni et al., 2012; Tanboon et al., 2020), which appear as red rim in the speckled sarcoplasm after Gomori Trichrome staining. Because ragged red signal is caused by the accumulation of defective mitochondria below the plasma membrane, these unusual histological signals are not exclusive to sIBM but are also seen in the muscle of patients suffering from primary mitochondrial disorders, such as myoclonic epilepsy with ragged red fibers (MERRF) and mitochondrial thymidine kinase (T2K) deficiency (DiMauro et al., 1985; Jou et al., 2022). Indeed, some mitochondria in the muscles of sIBM patients are aberrant in structure and function. In contrast to DM, where single point mutations in mtDNA are frequently detected (Zhao et al., 2022), sIBM muscle features large-scale, single segment deletions (i.e., major rearrangement mutation) in the major arc of mtDNA molecules. It has been reported that 122 deletion breakpoints and 33 different single nucleotide deletions were detected in the mtDNA molecules of sIBM patients (Moslemi et al., 1997), which could lead to the elimination of up to 1/3 of the whole mtDNA. Another study confirmed an average of 67% lower mtDNA copy number in the quadriceps and tibialis anterior muscle of sIBM patients than in healthy subjects (Bhatt et al., 2019). Interestingly, heterogenous mtDNA molecules with differential deletion regions could be detected in a single myofiber (Rygiel et al., 2016), resulting in a high heteroplasmy in the muscle. Moreover, the mtDNA deletion might be present in continuous segments of the same muscle fiber, creating a spatially unique pattern of the mitochondrial protein COX staining within a single myofiber (Oldfors et al., 1995; Horvath et al., 1998).
Because the human mtDNA encodes genes for electron transfer complexes (ETC) subunits, mtDNA deletion might result in the loss of mitochondrial content (Joshi et al., 2014; Catalán-García et al., 2016a; Oikawa et al., 2020; Hedberg-Oldfors et al., 2021). In fact, the severity of mtDNA deletion is highly associated with the number of COX-deficient fibers, which are myofibers that contain abnormally low levels of the mitochondrial complex IV, in the sIBM patients’ muscles (Landfeldt et al., 2015). Using single-cell analysis, Rygiel reported that ∼85% of COX-deficient cells have major mtDNA deletion and rearrangement (Rygiel et al., 2016). Mitochondrion morphology is also disrupted in the myofiber of sIBM patients, with shortened and enlarged cristae and junction breaks, resulting in a reduced mitochondrial length/width ratio (Oikawa et al., 2020). As COX is an essential component of the ETC for ATP synthesis via OXPHOS, COX deficiency in the myoblasts of sIBM patients leads to a shift in ATP production from OXPHOS to glycolysis (Oikawa et al., 2020), which is a key indicator of mitochondrial dysfunction (Di Leo et al., 2023). The 31P-magnetic resonance spectroscopy assessments revealed that the muscle of sIBM patients has a low ability to synthesize ATP during resting, further supporting the notion of impaired mitochondrial respiration in sIBM (Lodi et al., 1998). A recent study also reported diminished mitochondrial enzymatic activities in cultured sIBM myofibers under low glucose availability, indicating compromised metabolic flexibility (Catalán-García et al., 2020). Consequently, this low mitochondrial activity might significantly impact the overall function of skeletal muscle, causing weakness in contraction strength and endurance (Gonzalez-Chapa et al., 2023).
The occurrence of mtDNA deletion in the muscle of sIBM patients is not limited to a consensus locus within a mtDNA molecule but is present in multiple regions. This suggests that the cause of mtDNA deletion and rearrangement might be more complex than previously assumed. The underlying mechanism that leads to the high frequency of mtDNA deletion in sIBM is still unknown. However, it has been hypothesized that most truncated mtDNA molecules in sIBM muscle are generated from the clonal expansion of a single defective molecular species (Moslemi et al., 1997). It has also been suggested that the mtDNA replication process in sIBM muscle is prone to errors, further contributing to the generation of multiple copies of defective mtDNA (Horvath et al., 1998). The high replication error in sIBM is not caused by any genetic defects in nuclear genes engaged in mitochondrial genome maintenance, as a recent study found no pathogenic variants in nuclear genes that contribute to the high levels of mtDNA deletions in sIBM (Hedberg-Oldfors et al., 2021). Instead, single nucleotide polymorphism of several key genes involved in mtDNA replication and maintenance, including the DNA helicase Twinkle, the DNA polymerase γ (POLG), and ribonucleotide-diphosphate reductase subunit M2B (RRM2B), has been identified in sIBM patients (Lindgren et al., 2015). Nevertheless, the functional consequence of these single mutations has not been elucidated.
A possible cause of high mtDNA damage in sIBM muscle is its unique localization and structure. Mitochondria are the organelle that produces ∼90% of the total ROS in the cells as the byproducts of, ETC complex I and III (Favaro et al., 2019). These short-lived yet highly reactive molecules are unstable and cause structural damage to mtDNA molecules (Urbina-Varela et al., 2020). The histone-free nature of mtDNA molecules further increases their risk of ROS-induced damage (Taylor and Turnbull, 2005). Histones protect DNA against hydroxyl radical-induced strand breaks, which is a critical defense mechanism against DNA damage (Ljungman and Hanawalt, 1992). Indeed, the high ROS level and mitochondrial deletion are closely associated with each other in many diseases, such as hepatocellular carcinoma (Moriya et al., 2001) and chronic periodontitis (Canakçi et al., 2006). It is important to note that this “ROS-induced mutation” model can only be valid if the ROS content in the muscle of sIBM patients is higher than that of the healthy subjects. Several studies have demonstrated that ROS level is augmented in the muscles of sIBM. First, myoblasts of sIBM patients displayed a higher ROS concentration when treated with a glutathione synthesis inhibitor (Oikawa et al., 2020). Second, a high level of deglycase DJ-1, an important mitochondrial protective protein against oxidative stress (Taira et al., 2004), has been found in the mitochondria of sIBM muscle (Terracciano et al., 2008), indicating the mitochondria are under high oxidative stress and require stronger protection. In support of this notion, fibroblasts cultured from sIBM patients displayed higher oxidative stress, concomitant with increased antioxidant defense (Cantó-Santos et al., 2023). Furthermore, the protein amount and gene expression of ROS scavengers, Cu, Zn- superoxide dismutase (Cu, Zn-SOD) and manganese superoxide dismutase (Mn-SOD), are augmented in vacuolated myofibers in sIBM (Askanas and Engel, 1998; Tsuruta et al., 2002). However, comparable muscular lipid peroxidation between sIBM patients and healthy subjects was also reported (Catalán-García et al., 2016b), making the ROS hypothesis inconclusive. Instead of having more mutation inducers in the muscle, it is possible that the DNA repair system is blemished in the patient’s tissue, hence facilitating the propagation of mutated mtDNA. Because no examination of the activity of DNA repairing machinery, such as apurinic/apyrimidinic endonuclease (APE) or DNA damage-binding protein (DDB) (Jang et al., 2019), in the sIBM sample, has been performed, it remains unknown if the system is involved in the accumulation of truncated mtDNA.
In addition to the occurrence of mtDNA deletion, which resulted in a loss of mitochondria in the muscle (Hedberg-Oldfors et al., 2021), the accumulation of Aβ in the tissue might further impair the function of the existing mitochondria in sIBM. Studies have shown that overproduction of β amyloid precursor protein (APP) in human muscle fibers resulted in decreased COX activity, enlarged mitochondria, and the formation of disrupted cristae that resemble the pathological features of sIBM (Askanas et al., 1996). Abnormal mitochondrial-related functions, including increased rate of ROS production, reduced TCA cycle activities, and a shift of fatty acid-to-glucose utilization, were also seen in the muscle of APP transgenic mice (Boncompagni et al., 2012). It is believed that the mislocation of Aβ to the mitochondrial membrane impedes the function of the mitochondrial transporter, hence hindering the imports of materials that are indispensable for mitochondrial functions (Askanas et al., 1996; Devi et al., 2006). Although this Aβ-mitochondrial interaction, as observed in neuronal tissues, is a logical linkage of Aβ overproduction to the dysregulated mitochondrial function, no colocalization of Aβ and mitochondria in the muscle of sIBM patients has been reported.
It is also possible that the mitochondrial defect in sIBM muscle is attributed to the delayed organelle turnover, which might result in the buildup of dysfunctional mitochondria. The “dysregulated myoproteostasis” model, which covers protein synthesis defect, improper folding, extensive post-translational modification, and impaired degradation of proteins, has been proposed by Askanas et al. to collectively explain the aggregation of abnormal proteins and organelles in sIBM muscle (Askanas et al., 2015). Several studies report that organelle degradation by macroautophagy is compromised in sIBM. First, there is a lack of p62 binding accuracy to LC3 in the patient’s muscle, although sIBM muscles contain a higher frequency of LC3-positive autophagosomes, indicating a stop of the autophagy process in its initial stages (Lünemann et al., 2007; Nogalska et al., 2010; Suzuki et al., 2019). Moreover, the activity of lysosomal enzymatic activity cathepsin D and B was lower than the healthy control in the sIBM muscle, which delays the clearance of LC3-associated autophagosomes and the ubiquitinated proteins (Nogalska et al., 2010). Indeed, the muscle biopsies of sIBM patients showed a high density of lipofuscin aggregates, a marker of lysosomal dysfunction (Lu et al., 2020). Nicot et al. (2014) further specified the mechanism by demonstrating that the autophagy cargo receptor NBR1-mediated removal of protein aggregates was inhibited in sIBM. This delayed autophagy provides a logical explanation for the accumulation of p62 and Aβ aggregates in the autophagic vacuoles in sIBM (Güttsches et al., 2017). Furthermore, several genes associated with autophagosome-lysosome processing have been identified as the risk alleles in sIBM (Weihl et al., 2015; Gang et al., 2016; Papadopoulos et al., 2017). Based on these findings, pharmacological inhibition of autophagy by chronic colchicine administration in mice is used as an animal model for sIBM research (Ching et al., 2013). Interestingly, chaperone-mediated autophagy is increased in the muscle of sIBM, as evidenced by elevated levels of lysosomal membrane protein LAMP2A and the chaperone Hsp70 co-aggregates (Cacciottolo et al., 2013). Because chaperone-mediated autophagy mainly targets specific proteins but not large organelles, this escalated chaperone-mediated autophagy might be a compensatory response to abnormal protein aggregation, like p62, in sIBM. Given that the removal of damaged mitochondria is autophagy-dependent, the dysregulated autophagy in sIBM might jeopardize its clearance. Although no comprehensive assessment of mitophagy, the specific pathway of autophagy to induce mitochondria degradation (Lu et al., 2023), has been performed in sIBM muscle, decreased expression of dynamin-related protein 1 (DRP1) has been detected in cultured myoblasts of sIBM patients (Oikawa et al., 2020). DRP1 is a critical factor that promotes the splitting of mitochondria for subsequent degradation (Picca et al., 2023); this low level of DRP1 indirectly supports the hypothesis that the clearance of mitochondria in sIBM muscle might be impaired. Moreover, abnormal accumulation of the mitochondrial fusion marker mitofusin 1 (MFN1) and the mitophagy receptor Bcl-2 adenovirus E1B19 19-kDa interacting protein (BNIP3) was observed in the ragged red fibers of the sIBM patients (Askanas et al., 2015). Because mitofusin accumulation triggers mitochondrial enlargement, which hinders the sequestration of damaged mitochondria (Joaquim and Escobar-Henriques, 2020), augmented mitofusin content in the sIBM muscle may result in defective clearance of Bnip3-tagged cargos.
Although the invasion of immune cells is commonly observed in all IIM, inflation of highly differentiated CD8+CD28− cytotoxic T cells is uniquely present in sIBM (Greenberg and Bourc’his, 2019). It is proposed that autoimmunity is the root cause of sIBM pathogenesis, as genetic mutation of TDP-43 and p62 in myotilinopathies and desminopathies do not result in the formation of protein aggregates (Olivé et al., 2008; Olivé et al., 2009). Moreover, stimulation of cultured muscle cells by inflammatory cytokines, such as IL-1β and IFNγ, or upregulation of much MHC class I alone is sufficient to trigger the formation of protein aggregates and rimmed vacuoles (Fréret et al., 2013; McCord and Day, 2023a; McCord and Day, 2023b). Furthermore, rhabdomyosarcoma cells challenged with the IgG from sIBM patients effectively induced the aggregation of p62 (Tawara et al., 2017). Finally, altering the immune system by virus infection like HIV can produce pathological features of sIBM like p62 aggregates and the formation of rimmed vacuoles in the muscle (Hiniker et al., 2016). While these findings support the causal relationship between autoimmunity and muscle degeneration, mitochondrial defects in the muscle of sIBM might have feed-forward activity to exaggerate tissue inflammation. Supporting this notion, Shelton et al. (2019) found that mutation of the mitochondrial transporter, aspartate glutamate carrier 1 (AGC1), produced a proinflammatory phenotype in the muscle biopsies of dogs. Indeed, recent studies in mitochondrial biology have confirmed that excessive mitochondrial dysfunction can trigger tissue damage and subsequent immune responses via regulated cell death mechanisms.
In principle, damaged mitochondria release their organelle contents, such as mtDNA, cardiolipins, and Ca2+, into the cytosol and the extracellular space (Picca et al., 2023). These mitochondrial-derived damage-associated molecular patterns (DAMPs) are regarded as foreign molecules by the pattern recognition receptors of the immune cells due to the bacterial ancestry of mitochondria. Induction of the innate immune response triggers the activation of pro-inflammatory pathways like toll-like receptor (TLR) signalling (Picca et al., 2023). Indeed, it has been reported that binding of oxidized cardiolipins to TLR4 in the cytosol initiates the NF-κB signalling, leading to increased myostatin (MSTN) expression in the sIBM muscle (Sachdev et al., 2018).
The presence of mtDNA in the cytosol is an intrinsic warning of pathogen infection or cellular dysfunction. These cytosolic mtDNAs are sensed by the cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS) and promote its dimerization, leading to the production of second messenger cyclic GMP-AMP (cGAMP). The binding of cGAMP to the endoplasmic reticulum protein STING (stimulator of interferon genes) induces its translocation to the Golgi apparatus, where it activates the TANK-binding kinase 1 (TBK1) to phosphorylate the transcription factors interferon regulatory factor 1 (IRF1) and the IκB kinase complex. Consequently, transcription of NFκB-targeted genes such as type I interferons will be enhanced, attracting the immune cells to the injured muscle (Zhang et al., 2022). Although no studies have been performed to evaluate the cGAS-STING pathway in the animal models of sIBM or patient samples, the mtDNA-induced cGAS-STING pathway activation is detected in cells when TDP-43 invades mitochondria (Yu et al., 2020). Moreover, a recent report by Irazoki et al. (2023) demonstrated that the release of mtDNA after mitochondrial dysfunction is sufficient to induce sterile inflammation in the skeletal muscle, further supporting the role of cGAS pathway in myositis development.
The release of mtDNA may activate the pyroptotic cell-death pathways via activating another intracellular DNA sensor NLRP (nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing) proteins to form inflammasomes (Marchi et al., 2023) and the subsequent pyroptosis signalling. Inflammasomes are multiprotein complex containing leucine-rich repeated containing proteins (e.g., NLRP2, AIM2, Pyrin), the adapter protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain CARD), and pro-caspase 1. Although the molecular details of mtDNA-induced NLRP activation are still unknown, the outcomes of inflammasome formation have been well-defined. Once the caspase 1 in the inflammasome is activated, it cleaves the membrane pore protein Gasdermin D and interleukins (IL-1 and IL-8). Consequently, the membrane permeability is increased, leading to cell swelling, leakage of cellular proteins, and the formation of functional IL-1β and IL-18, all of which are strong inflammation inducers (Sharma and Kanneganti, 2021). A recent study demonstrated that the formation of NLRP3 inflammasome and pyroptosis was upregulated in the muscle fibers of DM and PM (Liu et al., 2021), its activity in sIBM remains to be investigated. Nevertheless, it has been demonstrated that ketogenic diet could alleviate the clinical symptoms of sIBM patients, possibly through suppressing NLRP3 inflammation activation, suggesting that pyroptosis is involved in the pathogenesis of sIBM (Phillips et al., 2020).
Recently, Kamiya et al. (2022) reported that inhibiting the necroptosis signalling effectively ameliorated the invasion of CD8+ cytotoxic T lymphocytes and thus suppressed muscle injury in PM. Necroptosis is a caspase-independent form of programmed cell death that results in membrane rupture, cell swelling, and leakage of DAMPs to promote inflammation (Pasparakis and Vandenabeele, 2015). The canonical necroptosis pathway is typically initiated by the cytokine TNFα in caspase 8-inactive cells. Once TNFα binds to its cognate receptor TNFR1, the receptor forms a complex that contains TNFR1-associated death domain protein (TRADD) and receptor-interacting serine/threonine protein kinase 1 (RIPK1). In certain conditions such as growth factor deprivation, the ligand activated TNFR-TRADD-RIPK1 complex further recruits and activates caspase 8 to cleave the downstream apoptosis executors like BH3-interacting domain death agonist (BID) to induce mitochondrial member depolarization and release of cytochrome c for caspase 3 activation (Luo et al., 1998). In cells with the absence of caspase 8, activation of TNFR1 promotes the heterodimerization of RIPK1 and RIPK3, forming the complex necrosome. Because RIPK3 is a proteolytic substrate of caspase 8, the formation of necrosome will only be formed in the absence of caspase 8 (Kang et al., 2013). The necrosome further recruits the mixed-lineage kinase domain-like protein (MLKL), which is phosphorylated by RIPK3 to form active oligomers for plasma membrane translocation. Due to its porous nature, the insertion of MLKL oligomers causes leakage of cellular content to the extracellular environment or cell rupture to induce the tethering of immune cells. Interestingly, activation of RIPK1 and RIPK3 triggers mitochondrial dysfunction (Chen et al., 2018) and activates the pyruvate dehydrogenase complex to produce excessive ROS (Yang et al., 2018), respectively. The high ROS feedbacks to the necrosome complex formation, forming a positive feedback loop of the necroptosis pathway to further enhance the necroptosis signalling (Qiu et al., 2018). Peng et al. (2022) reported that the necroptosis machinery is highly expressed in several subtypes of IIM, including DM and IMNM. Moreover, overactivation of the necroptotic pathway is sufficient to cause cell death of healthy muscle cells (Peng et al., 2022). Although high expression of necroptosis inducer TNF-α is detected in the muscle of sIBM patients (Schmidt et al., 2008), the activity of caspase 8 in the muscle has never been studied, making it a mystery if necroptosis is also elevated in sIBM like other IIM subtypes.
Due to the immunosuppressant-resistant feature of sIBM, exercise remains the main-stay therapy for sIBM (Koo et al., 2019). In general, endurance exercise is known to benefit skeletal muscle by increasing the disposal of ROS-damaged proteins and the synthesis of new mitochondria to accelerate mitochondrial turnover, thereby maintaining a healthy mitochondria network with boosted OXPHOS capacity (Sorriento et al., 2021). Resistance exercise also enhances the cellular antioxidation capacity by activating FOXO3, a transcription factor that induces the expression of antioxidant enzymes like SOD, to improve muscle function in sIBM (Koo et al., 2019). Interestingly, Coudert et al. (2022) recently reported that testosterone supplementation and exercise training may exert additive effects in improving muscle performance and mitigating the overactivated immunity in sIBM patients. However, performing regular exercise relies heavily on self-motivation and is difficult for cane- or wheelchair-bound patients at the advanced stage of IBM. Therefore, there is an urge to develop agents that recapitulate the benefits of exercise, such as upregulating antioxidant activity, increasing mitochondrial biogenesis, or promoting mitochondrial removal in the muscle. Considering that AMPK activation is a critical event in initiating mitochondrial biogenesis and mitophagy during exercise, AMPK-activating agents are attractive candidates for this purpose. Indeed, the AMP analogue 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranosyl 5′-monophosphate (AICAR) downregulated the expression of atrophic marker Atrogin/MAFbx in the gastrocnemius muscle and relieved cancer-induced muscle atrophy in mice (Hall et al., 2018). Another Food and Drug Administration (FDA)-approved AMPK activator, metformin, is found to promote mitophagy in type 2 diabetic patients (Picca et al., 2023). In a cardiovascular disease mouse model, metformin treatment suppressed ROS production in the abnormal heart tissue in an AMPK-dependent manner, which prevented the NLRP3/IL-1β-mediated inflammation and cardiovascular lesions (Marek-Iannucci et al., 2021).
Antioxidant application may be helpful in rescuing cell death in the sIBM muscle by neutralizing ROS into less harmful products (Johnson et al., 2023). For example, the well-known antioxidant polyphenol resveratrol has been shown to suppress myostatin-mediated cell death by activating the NF-κB signalling in cultured sIBM myoblasts (Askanas et al., 2012). The use of mitochondrial-targeted antioxidants like MitoQ and MitoVitE, which are hundred folds more effective in rescuing mitochondrial oxidative stress-induced cell death in human fibroblasts than other non-specific, cellular antioxidants like idebenone and vitamin E (Jauslin et al., 2003), might also alleviate the symptoms of sIBM (Rostamzadeh et al., 2024). Indeed, studies in numerous experimental models have confirmed the protective effect of these mitochondrial-targeted antioxidants in treating ROS-associated diseases like Parkinson’s Disease and atherosclerosis (Jiang et al., 2020; Sulaimon et al., 2022), which provide a solid scientific basis to extend their translational potential in ameliorating sIBM. Nevertheless, caution must be exercised when antioxidants are used in the sIBM treatment because exogenous supplementation of NADH and GSH may disrupt the redox balance and impose reductive stress on the cell (Xiao and Loscalzo, 2019). In fact, multiple studies have correlated excessive reductive stress with the development of inflammatory-associated diseases like cardiomyopathy, muscular dystrophy, and Alzheimer’s disease (Pérez-Torres et al., 2017).
Target mitophagy, a specific form of autophagy that degrades mitochondria exclusively (Lu et al., 2023), may also be a viable approach to alleviate myopathies associated with excessive oxidative stress (Ito et al., 2022). Traditional mitophagy inducers like oligomycin and carbonyl cyanide m-chlorophenyl hydrazone (CCCP) have been widely adopted in the in vitro system but their high toxicity limits their clinical applicability (Lee et al., 2023). Therefore, considerable efforts have been devoted to screen for alternative inducers with low toxicity for clinical applications. In 2021, Luan et al. (2021) reported that Urolithin A (UroA) reversed the declined mitophagy in cultured myoblasts of Duchenne Muscular Dystrophy (DMD) patients and mdx mice, which led to a reduction in muscle damage. UroA is a metabolite of microflora produced from the polyphenols ellagic acid and ellagitannins in food (Faitg et al., 2024), which is considered safe for oral consumption in humans as a dietary supplement by the FDA (García-Villalba et al., 2022). In a randomized, double-blinded, placebo-controlled clinical trial, subjects consuming UroA for 4 months exhibited augmented expression of mitophagy markers in their muscle biopsy, which was associated with higher complex I and II-mediated respiration (Faitg et al., 2024). Another clinical trial demonstrated that UroA effectively improved mitochondrial function in the skeletal muscle of the elderly by upregulating mitochondrial gene expression (Andreux et al., 2019). The positive effect of UroA on mitochondrial respiration was also reflected by the 65% higher running capacity in the UroA-administrated Wistar rats (Nat Med, 2016). Mechanically, UroA triggers mitophagy via lowering the mitochondrial membrane potential, as short-term UroA treatment induced membrane depolarization followed by augmented mitophagy marker expression (Ryu et al., 2016). Based on the promising effect of UroA in improving muscle function of age-related atrophic fibers, which share some biochemical characteristics of sIBM myofibers (Romani et al., 2021), it is reasonable to assume that UroA could also be effective in ameliorating mitochondrial defects and reducing muscle damage in sIBM. In supporting the idea that maintaining mitochondria activity is important to sustain the survival of the sIBM muscle cells, Oikawa et al. (2020) reported that mitochonic acid 5 (MA-5), a plant derivative that facilitates mitochondrial activities, such as increasing ATP synthesis, reducing ROS production, and promoting OXPHOS, was effective in protecting the myoblasts of sIBM patients from the buthionine sulfoximine-induced cell death. Presumably, mitochondrial health-improving agents like UroA and MA-5 might represent safe and effective agents for sIBM patients to improve their muscle function.
Although pathological hallmarks of sIBM have been recognized for several decades, the precise molecular mechanism for these cellular abnormalities is still mysterious. Most studies on sIBM pathogenesis are associative in nature, and the lack of mechanistic studies hinders the development of effective treatment, making it still an incurable disease nowadays. The slow progress in the development of new sIBM therapy can be attributed, in part, to the uncertainty of the sequential relationship between muscle damage and overactivated immune response. Moreover, most studies of sIBM have primarily focused on the detrimental consequence of hyperactive immune cells and protein aggregation on skeletal muscle; the outcomes of other damaged organelles like mitochondria have received little attention. The conventional view of mitochondria solely as ATP-producing powerhouses biased our perception that defective mitochondria in IIM might only result in metabolic deficiency, thus underestimating its functional outcomes. Recent discoveries that highlight mitochondrial defects as inducers of immune response via pyroptosis and necroptosis in many different issues have provided new insights into the pathogenesis of IIM (Figure 1). Hence, it is imperative to recognize the etiological role of mitochondria and developing novel drugs that improve the mitochondrial health as a novel treatment strategy for sIBM.
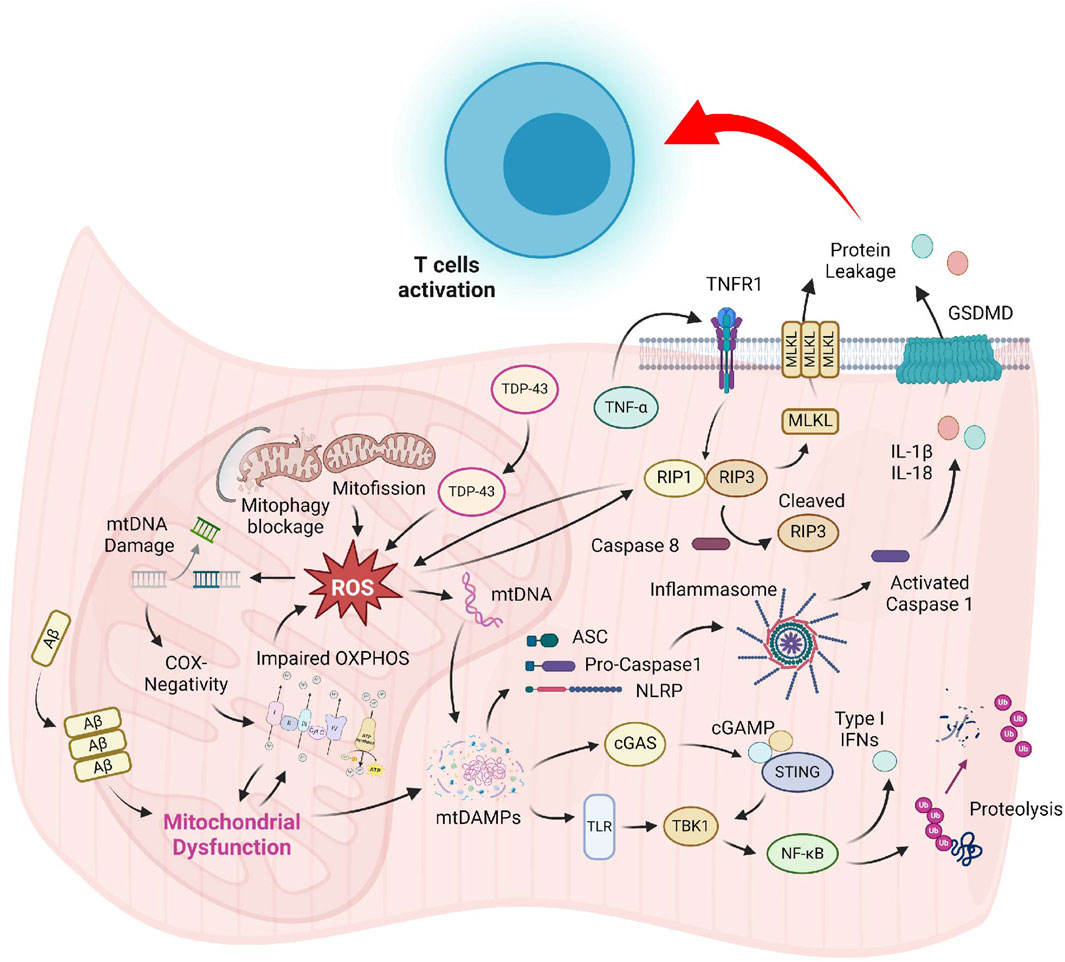
Figure 1. Prominent mitochondrial abnormalities in sIBM muscle include COX-negativity, OXPHOS suppression, delayed organelle clearance, and mislocalization of proteins. Augmented ROS produced by the dysfunctional mitochondrial might damage the organelle, resulting in the release of mitochondrial content into the sarcoplasm. These mtDAMPs are strong inducers of the TLR and cGAS-STING pathway, which promote inflammatory cytokines production and muscle breakdown via the NF-κB signaling. Accumulation of mtDAMPs and ROS might also induce the formation of NLRP-mediated inflammasome and TNF-α-triggered necrosome, leading to compromised sarcolemma integrity. Leakage of cellular content generates DAMPs that might serve as activation signals for T-cell recruitment in sIBM.
EI: Writing–original draft, Writing–review and editing. HS: Writing–review and editing. CC: Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work is supported by the HKU Seed Fund for Basic Research to CC and Hong Kong Health and Medical Research Fund (HMRF21222301) to CC and HS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Andreux, P. A., Blanco-Bose, W., Ryu, D., Burdet, F., Ibberson, M., Aebischer, P., et al. (2019). The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 1 (6), 595–603. doi:10.1038/s42255-019-0073-4
Askanas, V., and Engel, W. K. (1998). Sporadic inclusion-body myositis and hereditary inclusion-body myopathies: diseases of oxidative stress and aging? Archives Neurology 55 (7), 915–920. doi:10.1001/archneur.55.7.915
Askanas, V., and Engel, W. K. (2008). Inclusion-body myositis: muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer's and Parkinson's disease brains. Acta Neuropathol. 116 (6), 583–595. doi:10.1007/s00401-008-0449-0
Askanas, V., Engel, W. K., and Nogalska, A. (2012). Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J. Neuropathol. Exp. Neurol. 71 (8), 680–693. doi:10.1097/NEN.0b013e31826183c8
Askanas, V., Engel, W. K., and Nogalska, A. (2015). Sporadic inclusion-body myositis: a degenerative muscle disease associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochim. Biophys. Acta 1852 (4), 633–643. doi:10.1016/j.bbadis.2014.09.005
Askanas, V., McFerrin, J., Baqué, S., Alvarez, R. B., Sarkozi, E., and Engel, W. K. (1996). Transfer of beta-amyloid precursor protein gene using adenovirus vector causes mitochondrial abnormalities in cultured normal human muscle. Proc. Natl. Acad. Sci. U. S. A. 93 (3), 1314–1319. doi:10.1073/pnas.93.3.1314
Bhatt, P. S., Tzoulis, C., Balafkan, N., Miletic, H., Tran, G. T. T., Sanaker, P. S., et al. (2019). Mitochondrial DNA depletion in sporadic inclusion body myositis. Neuromuscul. Disord. 29 (3), 242–246. doi:10.1016/j.nmd.2019.02.001
Boncompagni, S., Moussa, C. E., Levy, E., Pezone, M. J., Lopez, J. R., Protasi, F., et al. (2012). Mitochondrial dysfunction in skeletal muscle of amyloid precursor protein-overexpressing mice. J. Biol. Chem. 287 (24), 20534–20544. doi:10.1074/jbc.M112.359588
Britson, K. A., Ling, J. P., Braunstein, K. E., Montagne, J. M., Kastenschmidt, J. M., Wilson, A., et al. (2022). Loss of TDP-43 function and rimmed vacuoles persist after T cell depletion in a xenograft model of sporadic inclusion body myositis. Sci Transl Med. 14 (628), eabi9196. doi:10.1126/scitranslmed.abi9196
Cacciottolo, M., Nogalska, A., D'Agostino, C., Engel, W. K., and Askanas, V. (2013). Chaperone-mediated autophagy components are upregulated in sporadic inclusion-body myositis muscle fibres. Neuropathol. Appl. Neurobiol. 39 (7), 750–761. doi:10.1111/nan.12038
Canakçi, C. F., Tatar, A., Canakçi, V., Cicek, Y., Oztas, S., and Orbak, R. (2006). New evidence of premature oxidative DNA damage: mitochondrial DNA deletion in gingival tissue of patients with periodontitis. J. Periodontol. 77 (11), 1894–1900. doi:10.1902/jop.2006.060108
Cantó-Santos, J., Valls-Roca, L., Tobías, E., García-García, F. J., Guitart-Mampel, M., Esteve-Codina, A., et al. (2023). Unravelling inclusion body myositis using a patient-derived fibroblast model. J. Cachexia Sarcopenia Muscle 14 (2), 964–977. doi:10.1002/jcsm.13178
Cantó Santos, J., Valls-Roca, L., Tobías, E., Oliva, C., García-García, F., Guitart-Mampel, M., et al. (2023). Integrated multi-omics analysis for inferring molecular players in inclusion body myositis. Antioxidants 12, 1639. doi:10.3390/antiox12081639
Catalán-García, M., García-García, F. J., Moreno-Lozano, P. J., Alcarraz-Vizán, G., Tort-Merino, A., Milisenda, J. C., et al. (2020). Mitochondrial dysfunction: a common hallmark underlying comorbidity between sIBM and other degenerative and age-related diseases. J. Clin. Med. 9 (5), 1446. doi:10.3390/jcm9051446
Catalán-García, M., Garrabou, G., Morén, C., Guitart-Mampel, M., Gonzalez-Casacuberta, I., Hernando, A., et al. (2016a). BACE-1, PS-1 and sAPPβ levels are increased in plasma from sporadic inclusion body myositis patients: surrogate biomarkers among inflammatory myopathies. Mol. Med. 21 (1), 817–823. doi:10.2119/molmed.2015.00168
Catalán-García, M., Garrabou, G., Morén, C., Guitart-Mampel, M., Hernando, A., Díaz-Ramos, À., et al. (2016b). Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin. Sci. (Lond) 130 (19), 1741–1751. doi:10.1042/cs20160080
Chapa, J., Macêdo, M., Naddaf, E., Saketkoo, L., and Lood, C. (2023). Mitochondrial transfer and implications for muscle function in idiopathic inflammatory myopathies. Clin. Exp. Rheumatology 42. doi:10.55563/clinexprheumatol/5lfq5x
Chen, S., Lv, X., Hu, B., Zhao, L., Li, S., Li, Z., et al. (2018). Critical contribution of RIPK1 mediated mitochondrial dysfunction and oxidative stress to compression-induced rat nucleus pulposus cells necroptosis and apoptosis. Apoptosis 23 (5-6), 299–313. doi:10.1007/s10495-018-1455-x
Ching, J. K., Ju, J. S., Pittman, S. K., Margeta, M., and Weihl, C. C. (2013). Increased autophagy accelerates colchicine-induced muscle toxicity. Autophagy 9 (12), 2115–2125. doi:10.4161/auto.26150
Coudert, J. D., Slater, N., Sooda, A., Beer, K., Lim, E. M., Boyder, C., et al. (2022). Immunoregulatory effects of testosterone supplementation combined with exercise training in men with Inclusion Body Myositis: a double-blind, placebo-controlled, cross-over trial. Clin. Transl. Immunol. 11 (9), e1416. doi:10.1002/cti2.1416
Damian, L., Login, C. C., Solomon, C., Belizna, C., Encica, S., Urian, L., et al. (2022). Inclusion body myositis and neoplasia: a narrative review. Int. J. Mol. Sci. 23 (13), 7358. doi:10.3390/ijms23137358
Danieli, M. G., Antonelli, E., Piga, M. A., Cozzi, M. F., Allegra, A., and Gangemi, S. (2023). Oxidative stress, mitochondrial dysfunction, and respiratory chain enzyme defects in inflammatory myopathies. Autoimmun. Rev. 22 (5), 103308. doi:10.1016/j.autrev.2023.103308
De Bleecker, J. L., Meire, V. I., Van Walleghem, I. E., Groessens, I. M., and Schröder, J. M. (2001). Immunolocalization of FAS and FAS ligand in inflammatory myopathies. Acta Neuropathol. 101 (6), 572–578. doi:10.1007/s004010000324
De Paepe, B. (2019). Sporadic inclusion body myositis: an acquired mitochondrial disease with extras. Biomolecules 9 (1), 15. doi:10.3390/biom9010015
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26 (35), 9057–9068. doi:10.1523/jneurosci.1469-06.2006
Diederichsen, L. P., Iversen, L. V., Nielsen, C. T., Jacobsen, S., Hermansen, M. L., Witting, N., et al. (2023). Myositis-related autoantibody profile and clinical characteristics stratified by anti-cytosolic 5'-nucleotidase 1A status in connective tissue diseases. Muscle Nerve 68 (1), 73–80. doi:10.1002/mus.27841
Di Leo, V., Bernardino Gomes, T. M., and Vincent, A. E. (2023). Interactions of mitochondrial and skeletal muscle biology in mitochondrial myopathy. Biochem. J. 480 (21), 1767–1789. doi:10.1042/bcj20220233
DiMauro, S., Bonilla, E., Zeviani, M., Nakagawa, M., and DeVivo, D. C. (1985). Mitochondrial myopathies. Ann. Neurol. 17 (6), 521–538. doi:10.1002/ana.410170602
Faitg, J., D'Amico, D., Rinsch, C., and Singh, A. (2024). Mitophagy activation by urolithin A to target muscle aging. Calcif. Tissue Int. 114 (1), 53–59. doi:10.1007/s00223-023-01145-5
Favaro, G., Romanello, V., Varanita, T., Andrea Desbats, M., Morbidoni, V., Tezze, C., et al. (2019). DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 10 (1), 2576. doi:10.1038/s41467-019-10226-9
Fréret, M., Drouot, L., Obry, A., Ahmed-Lacheheb, S., Dauly, C., Adriouch, S., et al. (2013). Overexpression of MHC class I in muscle of lymphocyte-deficient mice causes a severe myopathy with induction of the unfolded protein response. Am. J. Pathol. 183 (3), 893–904. doi:10.1016/j.ajpath.2013.06.003
Fyhr, I. M., and Oldfors, A. (1998). Upregulation of Fas/Fas ligand in inclusion body myositis. Ann. Neurol. 43 (1), 127–130. doi:10.1002/ana.410430123
Gang, Q., Bettencourt, C., Machado, P. M., Brady, S., Holton, J. L., Pittman, A. M., et al. (2016). Rare variants in SQSTM1 and VCP genes and risk of sporadic inclusion body myositis. Neurobiol. Aging 47, 218.e1–218.e9. doi:10.1016/j.neurobiolaging.2016.07.024
García-Villalba, R., Giménez-Bastida, J. A., Cortés-Martín, A., Ávila-Gálvez, M., Tomás-Barberán, F. A., Selma, M. V., et al. (2022). Urolithins: a comprehensive update on their metabolism, bioactivity, and associated gut microbiota. Mol. Nutr. Food Res. 66 (21), e2101019. doi:10.1002/mnfr.202101019
Goebels, N., Michaelis, D., Engelhardt, M., Huber, S., Bender, A., Pongratz, D., et al. (1996). Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J. Clin. Investig. 97 (12), 2905–2910. doi:10.1172/jci118749
Gonzalez-Chapa, J. A., Macêdo, M. B., and Lood, C. (2023). The emerging role of mitochondrial dysfunction in the pathogenesis of idiopathic inflammatory myopathies. Rambam Maimonides Med. J. 14 (2), e0006. doi:10.5041/rmmj.10493
Greenberg, M. V. C., and Bourc’his, D. (2019). The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol. 20 (10), 590–607. doi:10.1038/s41580-019-0159-6
Greenberg, S. A. (2019). Inclusion body myositis: clinical features and pathogenesis. Nat. Rev. Rheumatol. 15 (5), 257–272. doi:10.1038/s41584-019-0186-x
Greenberg, S. A., Bradshaw, E. M., Pinkus, J. L., Pinkus, G. S., Burleson, T., Due, B., et al. (2005). Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 65 (11), 1782–1787. doi:10.1212/01.wnl.0000187124.92826.20
Gunawardena, H., Betteridge, Z. E., and McHugh, N. J. (2009). Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatol. Oxf. 48 (6), 607–612. doi:10.1093/rheumatology/kep078
Güttsches, A. K., Brady, S., Krause, K., Maerkens, A., Uszkoreit, J., Eisenacher, M., et al. (2017). Proteomics of rimmed vacuoles define new risk allele in inclusion body myositis. Ann. Neurol. 81 (2), 227–239. doi:10.1002/ana.24847
Hall, D. T., Griss, T., Ma, J. F., Sanchez, B. J., Sadek, J., Tremblay, A. M. K., et al. (2018). The AMPK agonist 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), but not metformin, prevents inflammation-associated cachectic muscle wasting. EMBO Mol. Med. 10 (7), e8307. doi:10.15252/emmm.201708307
Hedberg-Oldfors, C., Lindgren, U., Basu, S., Visuttijai, K., Lindberg, C., Falkenberg, M., et al. (2021). Mitochondrial DNA variants in inclusion body myositis characterized by deep sequencing. Brain Pathol. 31 (3), e12931. doi:10.1111/bpa.12931
Hiniker, A., Daniels, B. H., and Margeta, M. (2016). T-cell-mediated inflammatory myopathies in HIV-positive individuals: a histologic study of 19 cases. J. Neuropathol. Exp. Neurol. 75 (3), 239–245. doi:10.1093/jnen/nlv023
Horvath, R., Fu, K., Johns, T., Genge, A., Karpati, G., and Shoubridge, E. A. (1998). Characterization of the mitochondrial DNA abnormalities in the skeletal muscle of patients with inclusion body myositis. J. Neuropathol. Exp. Neurol. 57 (5), 396–403. doi:10.1097/00005072-199805000-00003
Huntley, M. L., Gao, J., Termsarasab, P., Wang, L., Zeng, S., Thammongkolchai, T., et al. (2019). Association between TDP-43 and mitochondria in inclusion body myositis. Lab. Investig. 99 (7), 1041–1048. doi:10.1038/s41374-019-0233-x
Irazoki, A., Gordaliza-Alaguero, I., Frank, E., Giakoumakis, N. N., Seco, J., Palacín, M., et al. (2023). Disruption of mitochondrial dynamics triggers muscle inflammation through interorganellar contacts and mitochondrial DNA mislocation. Nat. Commun. 14 (1), 108. doi:10.1038/s41467-022-35732-1
Ito, A., Hashimoto, M., Tanihata, J., Matsubayashi, S., Sasaki, R., Fujimoto, S., et al. (2022). Involvement of Parkin-mediated mitophagy in the pathogenesis of chronic obstructive pulmonary disease-related sarcopenia. J. Cachexia Sarcopenia Muscle 13 (3), 1864–1882. doi:10.1002/jcsm.12988
Jang, S., Kumar, N., Beckwitt, E. C., Kong, M., Fouquerel, E., Rapić-Otrin, V., et al. (2019). Damage sensor role of UV-DDB during base excision repair. Nat. Struct. Mol. Biol. 26 (8), 695–703. doi:10.1038/s41594-019-0261-7
Jauslin, M. L., Meier, T., Smith, R. A., and Murphy, M. P. (2003). Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. Faseb J. 17 (13), 1972–1974. doi:10.1096/fj.03-0240fje
Jiang, Q., Yin, J., Chen, J., Ma, X., Wu, M., Liu, G., et al. (2020). Mitochondria-targeted antioxidants: a step towards disease treatment. Oxid. Med. Cell. Longev. 2020, 8837893. doi:10.1155/2020/8837893
Joaquim, M., and Escobar-Henriques, M. (2020). Role of mitofusins and mitophagy in life or death decisions. Front. Cell. Dev. Biol. 8, 572182. doi:10.3389/fcell.2020.572182
Johnson, G. A., Krishnamoorthy, R. R., and Stankowska, D. L. (2023). Modulating mitochondrial calcium channels (TRPM2/MCU/NCX) as a therapeutic strategy for neurodegenerative disorders. Front. Neurosci. 17, 1202167. doi:10.3389/fnins.2023.1202167
Joshi, P. R., Vetterke, M., Hauburger, A., Tacik, P., Stoltenburg, G., and Hanisch, F. (2014). Functional relevance of mitochondrial abnormalities in sporadic inclusion body myositis. J. Clin. Neurosci. 21 (11), 1959–1963. doi:10.1016/j.jocn.2014.05.051
Jou, C., Nascimento, A., Codina, A., Montoya, J., López-Gallardo, E., Emperador, S., et al. (2022). Pathological features in paediatric patients with TK2 deficiency. Int. J. Mol. Sci. 23 (19), 11002. doi:10.3390/ijms231911002
Kamiya, M., Mizoguchi, F., Kawahata, K., Wang, D., Nishibori, M., Day, J., et al. (2022). Targeting necroptosis in muscle fibers ameliorates inflammatory myopathies. Nat. Commun. 13 (1), 166. doi:10.1038/s41467-021-27875-4
Kang, T. B., Yang, S. H., Toth, B., Kovalenko, A., and Wallach, D. (2013). Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38 (1), 27–40. doi:10.1016/j.immuni.2012.09.015
Khoo, T., Lilleker, J. B., Thong, B. Y., Leclair, V., Lamb, J. A., and Chinoy, H. (2023). Epidemiology of the idiopathic inflammatory myopathies. Nat. Rev. Rheumatol. 19 (11), 695–712. doi:10.1038/s41584-023-01033-0
Koo, J. H., Kang, E. B., and Cho, J. Y. (2019). Resistance exercise improves mitochondrial quality control in a rat model of sporadic inclusion body myositis. Gerontology 65 (3), 240–252. doi:10.1159/000494723
Landfeldt, E., Lindgren, P., Bell, C. F., Schmitt, C., Guglieri, M., Straub, V., et al. (2015). Compliance to care guidelines for Duchenne muscular dystrophy. J. Neuromuscul. Dis. 2 (1), 63–72. doi:10.3233/jnd-140053
Lee, D. Y., Lee, K. M., Um, J. H., Kim, Y. Y., Kim, D. H., and Yun, J. (2023). The natural alkaloid palmatine selectively induces mitophagy and restores mitochondrial function in an alzheimer's disease mouse model. Int. J. Mol. Sci. 24 (22), 16542. doi:10.3390/ijms242216542
Lightfoot, A. P., McArdle, A., Jackson, M. J., and Cooper, R. G. (2015). In the idiopathic inflammatory myopathies (IIM), do reactive oxygen species (ROS) contribute to muscle weakness? Ann. Rheum. Dis. 74 (7), 1340–1346. doi:10.1136/annrheumdis-2014-207172
Lindgren, U., Roos, S., Hedberg Oldfors, C., Moslemi, A.-R., Lindberg, C., and Oldfors, A. (2015). Mitochondrial pathology in inclusion body myositis. Neuromuscul. Disord. 25 (4), 281–288. doi:10.1016/j.nmd.2014.12.010
Liu, D., Xiao, Y., Zhou, B., Gao, S., Li, L., Zhao, L., et al. (2021). PKM2-dependent glycolysis promotes skeletal muscle cell pyroptosis by activating the NLRP3 inflammasome in dermatomyositis/polymyositis. Rheumatol. Oxf. 60 (5), 2177–2189. doi:10.1093/rheumatology/keaa473
Ljungman, M., and Hanawalt, P. C. (1992). Efficient protection against oxidative DNA damage in chromatin. Mol. Carcinog. 5 (4), 264–269. doi:10.1002/mc.2940050406
Lodi, R., Taylor, D. J., Tabrizi, S. J., Hilton-Jones, D., Squier, M. V., Seller, A., et al. (1998). Normal in vivo skeletal muscle oxidative metabolism in sporadic inclusion body myositis assessed by 31P-magnetic resonance spectroscopy. Brain 121 (Pt 11), 2119–2126. doi:10.1093/brain/121.11.2119
Lu, J. Q., Monaco, C. M. F., Hawke, T. J., Yan, C., and Tarnopolsky, M. A. (2020). Increased intra-mitochondrial lipofuscin aggregates with spherical dense body formation in mitochondrial myopathy. J. Neurol. Sci. 413, 116816. doi:10.1016/j.jns.2020.116816
Lu, Y., Li, Z., Zhang, S., Zhang, T., Liu, Y., and Zhang, L. (2023). Cellular mitophagy: mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics 13 (2), 736–766. doi:10.7150/thno.79876
Luan, P., D'Amico, D., Andreux, P. A., Laurila, P. P., Wohlwend, M., Li, H., et al. (2021). Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci. Transl. Med. 13 (588), eabb0319. doi:10.1126/scitranslmed.abb0319
Lünemann, J. D., Schmidt, J., Schmid, D., Barthel, K., Wrede, A., Dalakas, M. C., et al. (2007). Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann. Neurol. 61 (5), 476–483. doi:10.1002/ana.21115
Luo, X., Budihardjo, I., Zou, H., Slaughter, C., and Wang, X. (1998). Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 94 (4), 481–490. doi:10.1016/s0092-8674(00)81589-5
Marchi, S., Guilbaud, E., Tait, S. W. G., Yamazaki, T., and Galluzzi, L. (2023). Mitochondrial control of inflammation. Nat. Rev. Immunol. 23 (3), 159–173. doi:10.1038/s41577-022-00760-x
Marek-Iannucci, S., Ozdemir, A. B., Moreira, D., Gomez, A. C., Lane, M., Porritt, R. A., et al. (2021). Autophagy-mitophagy induction attenuates cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight 6 (18), e151981. doi:10.1172/jci.insight.151981
Mavroudis, I., Knights, M., Petridis, F., Chatzikonstantinou, S., Karantali, E., and Kazis, D. (2021). Diagnostic accuracy of anti-cn1a on the diagnosis of inclusion body myositis. A hierarchical bivariate and bayesian meta-analysis. J. Clin. Neuromuscul. Dis. 23 (1), 31–38. doi:10.1097/CND.0000000000000353
McCord, B., and Day, R. M. (2023a). Cytotoxic immune cells do not affect TDP-43 and p62 sarcoplasmic aggregation but influence TDP-43 localisation. Sci. Rep. 13 (1), 15935. doi:10.1038/s41598-023-42824-5
McCord, B., and Day, R. M. (2023b). Influence of inflammatory cytokines IL-1β and IFNγ on sarcoplasmic aggregation of p62 and TDP-43 in myotubes. Mediat. Inflamm. 2023, 9018470. doi:10.1155/2023/9018470
Miller, F. W., Lamb, J. A., Schmidt, J., and Nagaraju, K. (2018). Risk factors and disease mechanisms in myositis. Nat. Rev. Rheumatol. 14 (5), 255–268. doi:10.1038/nrrheum.2018.48
Moriya, K., Nakagawa, K., Santa, T., Shintani, Y., Fujie, H., Miyoshi, H., et al. (2001). Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 61 (11), 4365–4370.
Moslemi, A. R., Lindberg, C., and Oldfors, A. (1997). Analysis of multiple mitochondrial DNA deletions in inclusion body myositis. Hum. Mutat. 10 (5), 381–386. doi:10.1002/(sici)1098-1004(1997)10:5<381::Aid-humu8>3.0.Co;2-i
Nicot, A. S., Lo Verso, F., Ratti, F., Pilot-Storck, F., Streichenberger, N., Sandri, M., et al. (2014). Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy 10 (6), 1036–1053. doi:10.4161/auto.28479
Nogalska, A., D'Agostino, C., Terracciano, C., Engel, W. K., and Askanas, V. (2010). Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am. J. Pathol. 177 (3), 1377–1387. doi:10.2353/ajpath.2010.100050
Oddis, C. V. (2016). Update on the pharmacological treatment of adult myositis. J. Intern. Med. 280 (1), 63–74. doi:10.1111/joim.12511
Oikawa, Y., Izumi, R., Koide, M., Hagiwara, Y., Kanzaki, M., Suzuki, N., et al. (2020). Mitochondrial dysfunction underlying sporadic inclusion body myositis is ameliorated by the mitochondrial homing drug MA-5. PLoS One 15 (12), e0231064. doi:10.1371/journal.pone.0231064
Oldfors, A., Moslemi, A. R., Fyhr, I. M., Holme, E., Larsson, N. G., and Lindberg, C. (1995). Mitochondrial DNA deletions in muscle fibers in inclusion body myositis. J. Neuropathol. Exp. Neurol. 54 (4), 581–587. doi:10.1097/00005072-199507000-00012
Oldfors, A., Moslemi, A. R., Jonasson, L., Ohlsson, M., Kollberg, G., and Lindberg, C. (2006). Mitochondrial abnormalities in inclusion-body myositis. Neurology 66 (2 Suppl. 1), S49–S55. doi:10.1212/01.wnl.0000192127.63013.8d
Olivé, M., Janué, A., Moreno, D., Gámez, J., Torrejón-Escribano, B., and Ferrer, I. (2009). TAR DNA-Binding protein 43 accumulation in protein aggregate myopathies. J. Neuropathol. Exp. Neurol. 68 (3), 262–273. doi:10.1097/NEN.0b013e3181996d8f
Olivé, M., van Leeuwen, F. W., Janué, A., Moreno, D., Torrejón-Escribano, B., and Ferrer, I. (2008). Expression of mutant ubiquitin (UBB+1) and p62 in myotilinopathies and desminopathies. Neuropathol. Appl. Neurobiol. 34 (1), 76–87. doi:10.1111/j.1365-2990.2007.00864.x
Papadimas, G. K., Kokkinis, C., Xirou, S., Chrysanthou, M., Kararizou, E., and Papadopoulos, C. (2019). Polymyositis with mitochondrial pathology or atypical form of sporadic inclusion body myositis: case series and review of the literature. Rheumatol. Int. 39 (8), 1459–1466. doi:10.1007/s00296-019-04314-8
Papadopoulos, C., Kirchner, P., Bug, M., Grum, D., Koerver, L., Schulze, N., et al. (2017). VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 36 (2), 135–150. doi:10.15252/embj.201695148
Pasparakis, M., and Vandenabeele, P. (2015). Necroptosis and its role in inflammation. Nature 517 (7534), 311–320. doi:10.1038/nature14191
Peng, Q. L., Zhang, Y. M., Liu, Y. C., Liang, L., Li, W. L., Tian, X. L., et al. (2022). Contribution of necroptosis to myofiber death in idiopathic inflammatory myopathies. Arthritis Rheumatol. 74 (6), 1048–1058. doi:10.1002/art.42071
Pérez-Torres, I., Guarner-Lans, V., and Rubio-Ruiz, M. E. (2017). Reductive stress in inflammation-associated diseases and the pro-oxidant effect of antioxidant agents. Int. J. Mol. Sci. 18 (10), 2098. doi:10.3390/ijms18102098
Phillips, M. C. L., Murtagh, D. K. J., Ziad, F., Johnston, S. E., and Moon, B. G. (2020). Impact of a ketogenic diet on sporadic inclusion body myositis: a case study. Front. Neurol. 11, 582402. doi:10.3389/fneur.2020.582402
Picca, A., Faitg, J., Auwerx, J., Ferrucci, L., and D’Amico, D. (2023). Mitophagy in human health, ageing and disease. Nat. Metab. 5 (12), 2047–2061. doi:10.1038/s42255-023-00930-8
Pruitt, J. N., Showalter, C. J., and Engel, A. G. (1996). Sporadic inclusion body myositis: counts of different types of abnormal fibers. Ann. Neurol. 39 (1), 139–143. doi:10.1002/ana.410390122
Qiu, X., Zhang, Y., and Han, J. (2018). RIP3 is an upregulator of aerobic metabolism and the enhanced respiration by necrosomal RIP3 feeds back on necrosome to promote necroptosis. Cell. Death Differ. 25 (5), 821–824. doi:10.1038/s41418-018-0075-x
Rinnenthal, J. L., Goebel, H. H., Preuße, C., Lebenheim, L., Schumann, M., Moos, V., et al. (2014). Inflammatory myopathy with abundant macrophages (IMAM): the immunology revisited. Neuromuscul. Disord. 24 (2), 151–155. doi:10.1016/j.nmd.2013.11.004
Romani, M., Sorrentino, V., Oh, C. M., Li, H., de Lima, T. I., Zhang, H., et al. (2021). NAD(+) boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell. Rep. 34 (3), 108660. doi:10.1016/j.celrep.2020.108660
Rostamzadeh, F., Najafipour, H., Aminizadeh, S., and Jafari, E. (2024). Therapeutic effects of the combination of moderate-intensity endurance training and MitoQ supplementation in rats with isoproterenol-induced myocardial injury: the role of mitochondrial fusion, fission, and mitophagy. Biomed. Pharmacother. 170, 116020. doi:10.1016/j.biopha.2023.116020
Rygiel, K. A., Miller, J., Grady, J. P., Rocha, M. C., Taylor, R. W., and Turnbull, D. M. (2015). Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol. Appl. Neurobiol. 41 (3), 288–303. doi:10.1111/nan.12149
Rygiel, K. A., Tuppen, H. A., Grady, J. P., Vincent, A., Blakely, E. L., Reeve, A. K., et al. (2016). Complex mitochondrial DNA rearrangements in individual cells from patients with sporadic inclusion body myositis. Nucleic Acids Res. 44 (11), 5313–5329. doi:10.1093/nar/gkw382
Ryu, D., Mouchiroud, L., Andreux, P. A., Katsyuba, E., Moullan, N., Nicolet-Dit-Félix, A. A., et al. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22 (8), 879–888. doi:10.1038/nm.4132
Sachdev, R., Kappes-Horn, K., Paulsen, L., Duernberger, Y., Pleschka, C., Denner, P., et al. (2018). Endoplasmic reticulum stress induces myostatin high molecular weight aggregates and impairs mature myostatin secretion. Mol. Neurobiol. 55 (11), 8355–8373. doi:10.1007/s12035-018-0997-9
Salam, S., Dimachkie, M. M., Hanna, M. G., and Machado, P. M. (2022). Diagnostic and prognostic value of anti-cN1A antibodies in inclusion body myositis. Clin. Exp. Rheumatol. 40 (2), 384–393. doi:10.55563/clinexprheumatol/r625rm
Schmidt, J., Barthel, K., Wrede, A., Salajegheh, M., Bähr, M., and Dalakas, M. C. (2008). Interrelation of inflammation and APP in sIBM: IL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain 131 (Pt 5), 1228–1240. doi:10.1093/brain/awn053
Sharma, B. R., and Kanneganti, T. D. (2021). NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 22 (5), 550–559. doi:10.1038/s41590-021-00886-5
Shelly, S., Mielke, M. M., Mandrekar, J., Milone, M., Ernste, F. C., Naddaf, E., et al. (2021). Epidemiology and natural history of inclusion body myositis: a 40-year population-based study. Neurology 96 (21), e2653–e2661. doi:10.1212/wnl.0000000000012004
Shelton, G. D., Minor, K. M., Li, K., Naviaux, J. C., Monk, J., Wang, L., et al. (2019). A mutation in the mitochondrial aspartate/glutamate carrier leads to a more oxidizing intramitochondrial environment and an inflammatory myopathy in Dutch shepherd dogs. J. Neuromuscul. Dis. 6 (4), 485–501. doi:10.3233/jnd-190421
Skolka, M. P., and Naddaf, E. (2023). Exploring challenges in the management and treatment of inclusion body myositis. Curr. Opin. Rheumatol. 35 (6), 404–413. doi:10.1097/bor.0000000000000958
Snedden, A. M., Kellett, K. A. B., Lilleker, J. B., Hooper, N. M., and Chinoy, H. (2022). The role of protein aggregation in the pathogenesis of inclusion body myositis. Clin. Exp. Rheumatol. 40 (2), 414–424. doi:10.55563/clinexprheumatol/pp0oso
Sorriento, D., Di Vaia, E., and Iaccarino, G. (2021). Physical exercise: a novel tool to protect mitochondrial health. Front. Physiol. 12, 660068. doi:10.3389/fphys.2021.660068
Sulaimon, L. A., Afolabi, L. O., Adisa, R. A., Ayankojo, A. G., Afolabi, M. O., Adewolu, A. M., et al. (2022). Pharmacological significance of MitoQ in ameliorating mitochondria-related diseases. Adv. Redox Res. 5, 100037. doi:10.1016/j.arres.2022.100037
Suzuki, N., Mori-Yoshimura, M., Yamashita, S., Nakano, S., Murata, K. Y., Mori, M., et al. (2019). The updated retrospective questionnaire study of sporadic inclusion body myositis in Japan. Orphanet J. Rare Dis. 14 (1), 155. doi:10.1186/s13023-019-1122-5
Taira, T., Saito, Y., Niki, T., Iguchi-Ariga, S. M., Takahashi, K., and Ariga, H. (2004). DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 5 (2), 213–218. doi:10.1038/sj.embor.7400074
Tanboon, J., Uruha, A., Stenzel, W., and Nishino, I. (2020). Where are we moving in the classification of idiopathic inflammatory myopathies? Curr. Opin. Neurol. 33 (5), 590–603. doi:10.1097/wco.0000000000000855
Tawara, N., Yamashita, S., Zhang, X., Korogi, M., Zhang, Z., Doki, T., et al. (2017). Pathomechanisms of anti-cytosolic 5'-nucleotidase 1A autoantibodies in sporadic inclusion body myositis. Ann. Neurol. 81 (4), 512–525. doi:10.1002/ana.24919
Taylor, R. W., and Turnbull, D. M. (2005). Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 6 (5), 389–402. doi:10.1038/nrg1606
Terracciano, C., Nogalska, A., Engel, W. K., Wojcik, S., and Askanas, V. (2008). In inclusion-body myositis muscle fibers Parkinson-associated DJ-1 is increased and oxidized. Free Radic. Biol. Med. 45 (6), 773–779. doi:10.1016/j.freeradbiomed.2008.05.030
Tsamis, K. I., Boutsoras, C., Kaltsonoudis, E., Pelechas, E., Nikas, I. P., Simos, Y. V., et al. (2022). Clinical features and diagnostic tools in idiopathic inflammatory myopathies. Crit. Rev. Clin. Lab. Sci. 59 (4), 219–240. doi:10.1080/10408363.2021.2000584
Tsuruta, Y., Furuta, A., Taniguchi, N., Yamada, T., Kira, J., and Iwaki, T. (2002). Increased expression of manganese superoxide dismutase is associated with that of nitrotyrosine in myopathies with rimmed vacuoles. Acta Neuropathol. 103 (1), 59–65. doi:10.1007/s004010100428
Urbina-Varela, R., Castillo, N., Videla, L. A., and Del Campo, A. (2020). Impact of mitophagy and mitochondrial unfolded protein response as new adaptive mechanisms underlying old pathologies: sarcopenia and non-alcoholic fatty liver disease. Int. J. Mol. Sci. 21 (20), 7704. doi:10.3390/ijms21207704
Weihl, C. C., Baloh, R. H., Lee, Y., Chou, T. F., Pittman, S. K., Lopate, G., et al. (2015). Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromuscul. Disord. 25 (4), 289–296. doi:10.1016/j.nmd.2014.12.009
Winkler, M., von Landenberg, C., Kappes-Horn, K., Neudecker, S., Kornblum, C., and Reimann, J. (2021). Diagnosis and clinical development of sporadic inclusion body myositis and polymyositis with mitochondrial pathology: a single-center retrospective analysis. J. Neuropathol. Exp. Neurol. 80 (11), 1060–1067–1067. doi:10.1093/jnen/nlab101
Xiao, W., and Loscalzo, J. (2019). Metabolic responses to reductive stress. Antioxidants Redox Signal. 32 (18), 1330–1347. doi:10.1089/ars.2019.7803
Yamashita, S., Tawara, N., Zhang, Z., Nakane, S., Sugie, K., Suzuki, N., et al. (2023). Pathogenic role of anti-cN1A autoantibodies in sporadic inclusion body myositis. J. Neurol. Neurosurg. Psychiatry 94 (12), 1018–1024. doi:10.1136/jnnp-2023-331474
Yang, Z., Wang, Y., Zhang, Y., He, X., Zhong, C. Q., Ni, H., et al. (2018). RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell. Biol. 20 (2), 186–197. doi:10.1038/s41556-017-0022-y
Yu, C. H., Davidson, S., Harapas, C. R., Hilton, J. B., Mlodzianoski, M. J., Laohamonthonkul, P., et al. (2020). TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 183 (3), 636–649. doi:10.1016/j.cell.2020.09.020
Zeng, R., Glaubitz, S., and Schmidt, J. (2022). Antibody therapies in autoimmune inflammatory myopathies: promising treatment options. Neurotherapeutics 19 (3), 911–921. doi:10.1007/s13311-022-01220-z
Zhang, Z., Zhou, H., Ouyang, X., Dong, Y., Sarapultsev, A., Luo, S., et al. (2022). Multifaceted functions of STING in human health and disease: from molecular mechanism to targeted strategy. Signal Transduct. Target Ther. 7 (1), 394. doi:10.1038/s41392-022-01252-z
Zhao, Y., Peng, C., Lai, R., Zhang, J., Zhang, X., and Guo, Z. (2022). The SNPs of mitochondrial DNA displacement loop region and mitochondrial DNA copy number associated with risk of polymyositis and dermatomyositis. Sci. Rep. 12 (1), 5903. doi:10.1038/s41598-022-09943-x
Keywords: mitochondria, myositis, muscle, pathogenesis, pyroptosis, necroptosis
Citation: Iu ECY, So H and Chan CB (2024) Mitochondrial defects in sporadic inclusion body myositis—causes and consequences. Front. Cell Dev. Biol. 12:1403463. doi: 10.3389/fcell.2024.1403463
Received: 19 March 2024; Accepted: 02 May 2024;
Published: 14 May 2024.
Edited by:
Shoulong Deng, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaCopyright © 2024 Iu, So and Chan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chi Bun Chan, Y2hhbmNiQGhrdS5oaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.