 Duo Zhang
Duo Zhang Murshid Imam
Murshid Imam Shunchao Yan
Shunchao Yan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 27 February 2024
Sec. Cancer Cell Biology
Volume 12 - 2024 | https://doi.org/10.3389/fcell.2024.1338448
This article is part of the Research TopicTherapeutic Potential Affected by Tumor Microenvironment in Triple-Negative Breast CancerView all 6 articles
Triple-negative breast cancer (TNBC) is a biologically and clinically heterogeneous disease. The G protein-coupled estrogen receptor (GPER) plays a crucial role in mediating the effect of estrogen and estrogen-like compounds in TNBC cells. Compared with other subtypes, GPER has a higher expression in TNBC. The GPER mechanisms have been thoroughly characterized and analyzed in estrogen receptor α (ERα) positive breast cancer, but not in TNBC. Our previous work revealed that a higher expression of GPER mRNA indicates a better prognosis for ERα-positive breast cancer; however, its effects in TNBC differ. Whether GPER could serve as a predictive prognostic marker or therapeutic target for TNBC remains unclear. In this review, we provide a detailed introduction to the subcellular localization of GPER, the different effects of various ligands, and the interactions between GPER and closely associated factors in TNBC. We focused on the internal molecular mechanisms specific to TNBC and thoroughly explored the role of GPER in promoting tumor development. We also discussed the interaction of GPER with specific cytokines and chemokines, and the relationship between GPER and immune evasion. Additionally, we discussed the feasibility of using GPER as a therapeutic target in the context of existing studies. This comprehensive review highlights the effects of GPER on TNBC, providing a framework and directions for future research.
According to the International Agency for Research on Cancers GLOBOCAN, global cancer burden data demonstrate that breast cancer has emerged as the most commonly diagnosed cancer, accounting for roughly 2.3 million (11.7%) new cases in 2020 (Sung et al., 2021; Chhikara and Parang, 2023). Breast cancer is the primary reason for cancer-related mortality among women worldwide (Cao et al., 2021). Triple-negative breast cancer (TNBC) accounts for approximately 15% of all breast cancer cases and >50% of breast cancer-related deaths (Morris et al., 2007). This subtype of breast cancer is particularly aggressive and lacks estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2).
TNBC demonstrates heterogeneity in both genetic expression and biological behavior, thereby contributing to its poor prognosis. Numerous studies have prioritized the accurate classification of TNBC to identify targeted treatments. Lehmann et al. (2011) identified seven subtypes of TNBC cell lines: luminal androgen receptor, immunomodulatory, mesenchymal stem-like, basal-like 1, basal-like 2, mesenchymal, and unstable. Moreover, Dai et al. (2017) proposed that TNBC cell lines can be divided into two distinct subtypes based on a diverse range of genetic expressions. The first subtype, triple-negative A, is characterized by an enrichment of basal markers, whereas triple-negative B is defined by a gene expression profile that indicates an increased potential for tumor invasiveness. This classification system assists in identifying high-risk individuals among patients with TNBC. Furthermore, Jiang et al. (2019) outlined a multi-omics landscape of TNBC, leading to the categorization of TNBC into four subtypes: immunomodulatory, basal-like immune-suppressed, luminal androgen receptor, and mesenchymal-like. This approach identified potential therapeutic targets for each subtype and optimized the precision treatment strategy for TNBC (Jiang et al., 2019). TNBC is resistant to specific hormone therapies and presents limited treatment options compared with those of other breast cancer types. To date, chemotherapy has been the primary approach for improving outcomes for patients with TNBC. Although new therapies such as poly (ADP-ribose) polymerase inhibitors and immunotherapy have been formulated in recent years, the TNBC prognosis remains poor due to heterogeneity (García Fernández et al., 2012; Li et al., 2017; Sharma, 2018; He et al., 2020; Yin L. et al., 2020). Therefore, identifying the molecular biological factors that impact or predict TNBC prognosis, exploring their mechanisms of action, and developing effective targeted therapeutic drugs with few side effects are crucial.
The lack of a response to hormones is regarded as an essential feature of TNBC. Nevertheless, researchers have observed the expression of certain non-classical steroid endocrine receptors in TNBC, in addition to classical ER. The G protein-coupled estrogen receptor (GPER), serving as a non-nuclear receptor, signifies the biological and clinical significance of steroid hormones in TNBC. Notably, GPER exhibits a higher expression level in TNBC than in other subtypes (Steiman et al., 2013). Existing research is controversial, with some studies indicating that GPER has a tumor-promoting effect in TNBC and is associated with increased recurrence rates, whereas others indicate the protective anti-tumor effect of GPER (Yu et al., 2014; Hernández-Silva et al., 2020). Therefore, a comprehensive perspective is required. In this review, we summarized the effects of GPER on TNBC from the perspective of signaling pathways and related mechanisms of action. Moreover, we have explored the potential of GPER as a prospective therapeutic target for TNBC based on existing original research.
Since the 1960s, researchers have observed that the uterine adenyl cyclase system and intracellular calcium have an instantaneous ability to react with estrogen in ovariectomized rats, which initiates cyclic adenosine monophosphate (cAMP) formation and induces calcium mobilization (Szego and Davis, 1967; Pietras and Szego, 1975). These effects are referred to as extranuclear, non-genomic effects (Luo and Liu, 2020). Thereafter, Aronica et al. (1994) demonstrated that estrogen increases adenylate cyclase activity, leading to increased cAMP production in human MCF-7 cells. At this time, researchers were not aware that GPER, but not just ERα played a key role in this effect (Aronica et al., 1994). Subsequently, the GPR30 gene was identified as a gene homologous to the G protein-coupled receptor (GPCR) family in breast cancer (Carmeci et al., 1997; Levoye et al., 2006). Moreover, Filardo et al. (2000) found that the rapid activation of extracellular signal-regulated kinases (ERK) by 17β-estradiol (E2) requires the participation of an orphan GPCR, with seven transmembrane domains. Maggiolini et al. (2004) demonstrated the ability of E2 to stimulate c-Fos gene expression in both ERα-positive MCF-7 and ERα-negative SKBR3 human breast cancer cells. This effect occurred through two different pathways: one involving the ERα and the other through the GPCR homolog GPER, independent of the ERα (Maggiolini et al., 2004). Revankar et al. (2005) eventually established GPER as a distinct ERα in SKBR3 cells, with a signaling pathway different from ERα-mediated signaling. Additionally, they observed E2 as the natural ligand for GPER and found that the effects of estrogen on this receptor were likely influenced by its intracellular localization. Subsequently, specific agonists (G-1) and antagonists (G-15 and G-36) targeting GPER were sequentially developed. With the deepening of our understanding of TNBC, studies on the involvement of GPER in TNBC have gradually increased. Since 2012, attention has been focused on the relationship between GPER activation and clinical pathological characteristics in TNBC. Numerous studies have focused on further investigating the mechanisms of GPER action in estrogen signaling and its implications for the development and progression of TNBC. The exploration of GPER in breast cancer is summarized in the form of a timeline in Figure 1A.
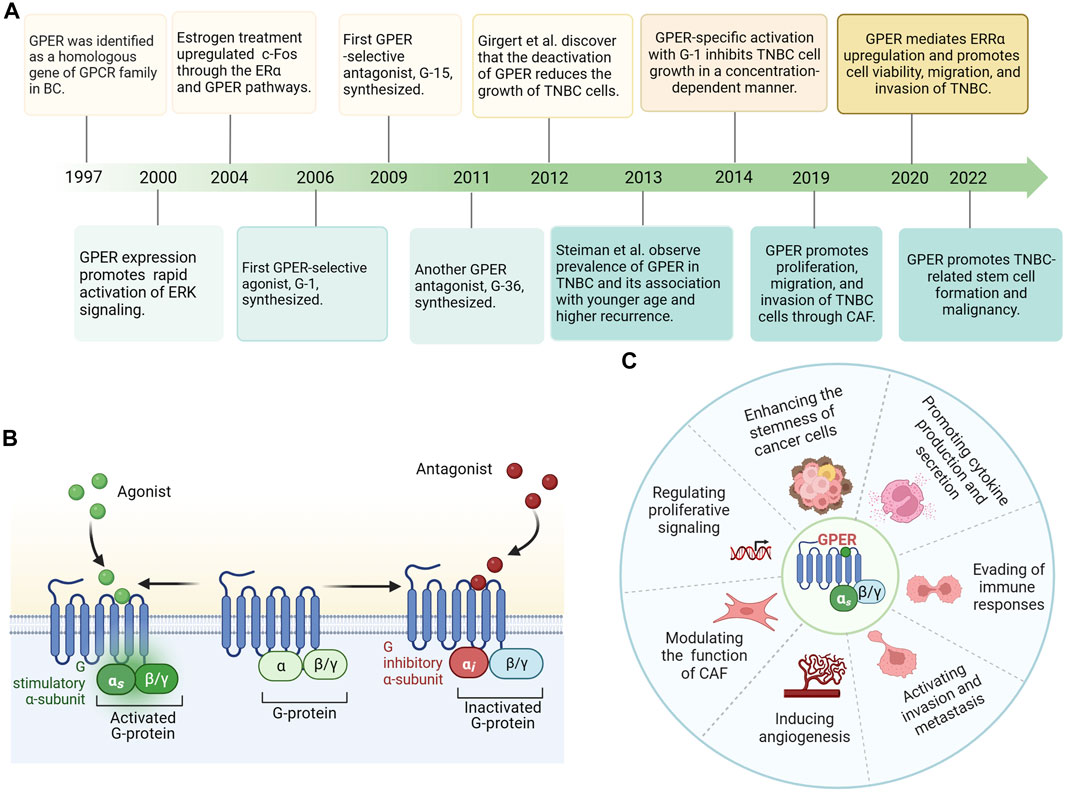
FIGURE 1. (A) Timeline of G protein-coupled estrogen receptor (GPER) exploration in breast cancer. (B) GPER protein is predominantly localized to the plasma membranes with seven transmembrane domains, ligand binding, and G protein binding pockets. Estrogen/agonist binding to GPER activates a stimulatory G protein α-subunit (Gαs) and results in GPER stimulation. Antagonist binding to GPER leads to an inhibitory G protein α-subunit (Gαi) and inactivated GPER. (C) GPER’s diverse functions include regulation of cell proliferation, invasion, metastasis, and angiogenesis, modulation of cancer-associated fibroblasts (CAFs), control of tumor stem cell functionality, cytokine production, and secretion, and evasion of immune responses. Abbreviations: GPER, G protein-coupled estrogen receptor; BC, breast cancer; TNBC, Triple-negative breast cancer; CAF, cancer-associated fibroblasts.
As a seven-transmembrane G protein-coupled receptor, GPER is typically assumed to be located on the plasma membrane, similar to most GPCRs (Thomas et al., 2005). While GPER is concentrated in the plasma membranes of certain tissues, its intracellular detection has also been reported (Filardo et al., 2007). Furthermore, the distribution of GPER varies across species, tissues, and cell types. Initially, Funakoshi et al. (2006) observed that GPER was localized in the plasma membrane of pyramidal neurons in the hippocampal CA2 area of the rat brain. Moreover, upon agonist stimulation, the GPER-transfected HeLa cells exhibited translocation of the receptor from the plasma membrane to the cytoplasm (Funakoshi et al., 2006). Additionally, GPER is primarily located in the plasma membrane of the uterine epithelia, myometrium, and renal epithelia (Cheng et al., 2011a; Gao et al., 2011; Lindsey et al., 2011; Maiti et al., 2011). However, a significant proportion of GPER is localized within the intracellular structure. Revankar et al. (2005) conducted a study using fluorescent E2 derivatives (E2-Alexas) to examine the localization features of GPER in monkey kidney fibroblasts. Interestingly, they observed that E2-Alexas did not effectively label the plasma membrane. Instead, it is primarily bound to the endoplasmic reticulum. Furthermore, staining with E2-Alexas or antibodies also revealed colocalization in the Golgi apparatus and nuclear membrane of GPER (Revankar et al., 2005). Similarly, in a study by Brailoiu et al. (2007), GPER was observed to be distributed within the neuronal cytoplasm using immunohistochemical analysis.
GPER primarily exhibits a cytoplasmic staining pattern in breast carcinoma tissues, with minimal presence on the cell surface (Thomas et al., 2005; Filardo et al., 2006; Luo et al., 2011). Interestingly, a fraction of tumor specimens exhibited both nuclear and cytoplasmic staining (Ignatov et al., 2011; Samartzis et al., 2014). The differential subcellular localization of GPER may be explained by a dynamic change mechanism (Cheng et al., 2011b). Under ligand-independent conditions, GPER employs an endocytic trafficking mechanism through clathrin-coated vesicles, accumulates in the perinuclear compartment, and is dispersed in the cytoplasm (Cheng et al., 2011b). Based on this evidence, we speculated that the different staining patterns may reflect the dynamic and time-dependent intracellular trafficking process of GPER. The action of GPER may be regulated by the amount of receptors present at different subcellular locations.
Differences in subcellular localization may have distinct biological implications for different breast carcinoma subtypes. Filardo et al. (2006) observed that the cytoplasmic staining of GPER in breast tumor tissue was nearly two-fold higher than that in tumors without GPER expression. Moreover, Sjöström et al. (2014) conducted a study to identify whether the subcellular localization of GPER could be an independent prognostic factor and revealed a positive correlation between the overexpression of plasma membrane GPER and a high histological grade. Therefore, plasma membrane localization of GPER may be a critical event that suggests a poor prognosis for breast cancer (Sjöström et al., 2014). Samartzis et al. (2014) observed a substantial correlation between the presence of cytoplasmic GPER and low tumor stage, luminal subtypes, and improved histological differentiation. In contrast, the expression of nuclear GPER is strongly associated with the triple-negative subtype and poorly differentiated tumors (Samartzis et al., 2014). Consistent results have been reported in our meta-analysis, where high GPER cytoplasmic expression, but not nuclear expression, is associated with improved outcomes in ERα-positive breast cancer (Zhang et al., 2022). As an understudied and important potential next frontier, whether the subcellular location of GPER in breast cancer has distinct prognostic implications needs further investigation.
Revankar et al. (2005) found that the natural hormone E2 is a paramount ligand of GPER (Thomas et al., 2005). Estriol (E3), an E2-based steroid, has been identified as a GPER antagonist, and estrone has been described as an agonist for GPER (Lappano et al., 2010). GPER exhibits a higher binding affinity to E2 than that of estrone and estriol (Revankar et al., 2005). The abundant cholesterol metabolite 27-hydroxycholesterol is a novel ligand for GPER, and its signaling axis plays a crucial role in ERα-negative breast cancer progression (Avena et al., 2022).
In 2006, researchers synthesized G-1, the first selective agonist for GPER (Bologa et al., 2006). Subsequent studies revealed a binding affinity of 10 nM between G-1 and GPER. In 2009, a selective antagonist of GPER, referred to as G-15, was shown to inhibit the calcium mobilizing effect of E2 in the SKBR3 cell line (Dennis et al., 2009). After 2 years, another GPER antagonist, G-36, was synthesized. This compound had a high affinity for GPER and a weak cross-reactivity with ERα (Dennis et al., 2011). Furthermore, Lappano et al. (2012) investigated two additional molecules, GPER-L1 and GPER-L2, which function as specific agonists of GPER, activating downstream signaling pathways.
Tamoxifen is the first accredited selective ERα modulator for breast cancer therapy and has demonstrated efficacy in reducing recurrence rates and improving the prognosis of ERα-positive breast tumors (Ariazi et al., 2006; Early Breast Cancer Trialists’ Collaborative Group, 2011). Tamoxifen acts as a GPER agonist, induces aromatase expression, and contributes to resistance to endocrine therapy in breast cancer (Meijer et al., 2006; van Agthoven et al., 2009; Ignatov et al., 2011; Catalano et al., 2014; Yin et al., 2017; Molina et al., 2020; Yu et al., 2020). Similarly, as a selective ERα downregulator, ICI182,780 (fulvestrant) displays significant binding to GPER and activates GPER in breast cancer (Osborne et al., 2004; Thomas et al., 2005).
Several plant-derived natural phytoestrogens have been shown to bind and/or activate ERα and other ERs (specifically GPER), such as daidzein (Kajta et al., 2013), genistein (Thomas and Dong, 2006; Vivacqua et al., 2006), resveratrol (Dong et al., 2013), and quercetin (Maggiolini et al., 2004). In addition, numerous xenoestrogens have been recognized as GPER ligands, including dichlorodiphenyltrichloroethane (Kajta et al., 2014), methoxychlor, atrazine (Thomas and Dong, 2006), bisphenol A (BPA) (Zhang et al., 2016), and bisphenol S (BPS) (Deng et al., 2018). The significance of BPA and BPS actions on GPER in the growth and migration of TNBC cells has been revealed. BPA is an industrially synthesized chemical compound with endocrine-disrupting effects that promote the growth of rat mammary tumor cells (Vandenberg et al., 2008; Wang et al., 2014; Zhang et al., 2016). In TNBC, BPA activates the focal adhesion kinase (FAK)/steroid receptor coactivator (SRC)/ERK signaling pathway and focal adhesion assembly by activating GPER and inducing epidermal growth factor receptor (EGFR) transactivation (Castillo Sanchez et al., 2016; Castillo-Sanchez et al., 2020). Tetrachlorobisphenol A (TCBPA) is a chlorinated derivative of BPA and exhibits estrogenic activity. Low TCBPA concentrations regulate the expression of GPER and its downstream target genes, resulting in the proliferation of TNBC cells (Lei et al., 2021). In comparison to BPA, BPS exhibits greater stability while exhibiting strong estrogenic responses. BPS was found to facilitate TNBC cell metastasis through the GPER/Hippo-Yes-associated protein 1 pathway (Zhao et al., 2008; Furth and Aylon, 2017), but it does not affect tumor proliferation (Deng et al., 2018). Deng et al. (2018) suggested that the reduced stimulation of proliferation by BPS in their study may be attributed to differences in cell lines or ERα/β expression status. Altogether, the positive and negative inhibitory effects of these GPER ligands provide a foundation for further exploration (Table 1).
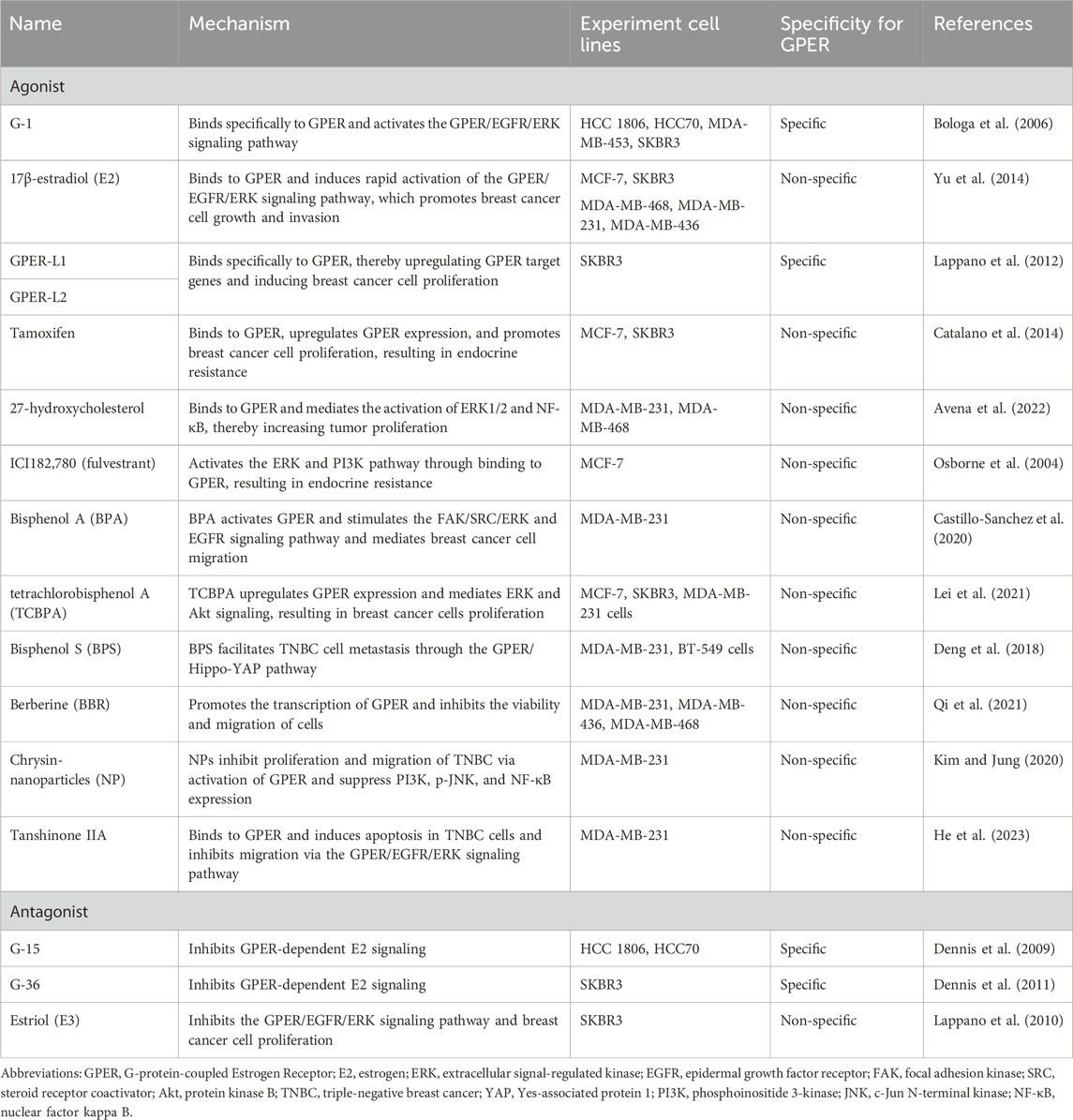
TABLE 1. Mechanisms of GPER agonists and antagonists in breast cancer.
In normal breast tissues, GPER is moderately expressed (Uhlén et al., 2015) and has been detected in diverse cancer cell lines and malignant tumors, such as endometrial carcinoma (He et al., 2009), ovarian cancer (Albanito et al., 2007), melanoma (Fábián et al., 2017), prostate cancer (Chan et al., 2010), thyroid cancer (Vivacqua et al., 2006), and testicular germ cell tumors (Franco et al., 2011). In invasive breast cancer, GPER exhibits a positivity rate estimated between 50% and 60%, as evidenced by immunohistochemistry (Filardo et al., 2006; Ignatov et al., 2011; Steiman et al., 2013). The expression of GPER varies among different ERα-positive breast cancer cell lines. In T47D cell lines, GPER is predominantly expressed at elevated levels in the cytoplasm, whereas in MCF-7 cells, GPER expression is primarily detected in the nucleus and is low (Samartzis et al., 2014). Moreover, within a cohort of ERα-negative breast cancer, the positivity rate of GPER exceeds 60% (Luo et al., 2011). Furthermore, in TNBC, GPER is frequently overexpressed, with rates reaching as high as 68.8% (Yu et al., 2014). Based on its expression levels, GPER holds promise as both a prognostic indicator and therapeutic target. Upon binding with an agonist, GPER is activated, playing a crucial role in the initiation and progression of tumors. Its diverse functions include the regulation of cell proliferation, invasion, metastasis, angiogenesis, modulation of cancer-associated fibroblasts (CAF), control of tumor stem cell functionality, cytokine production and secretion, and evasion of immune responses (Figures 1B,C). However, the function of GPER in TNBC and its potential effectiveness in combating highly invasive TNBC remain controversial. GPER overexpression is positively associated with metastatic capability, tumor size, HER2/neu and poor survival (Steiman et al., 2013). Using GPER-specific small interfering RNA (siRNA) knockdown, complete elimination of GPER expression was demonstrated, abolishing the stimulation of certain signaling pathways responsible for amplifying proliferation, and further substantiating the significant role of GPER in promoting the proliferation of breast cancer (Girgert et al., 2012). The potential involvement of GPER-associated signaling pathways in cell invasion and migration has also been demonstrated (Yu et al., 2014; Rigiracciolo et al., 2019; Ye et al., 2019; Yin et al., 2020). In vivo studies revealed that breast cancer in GPER-knockout mice resulted in diminished lung metastases compared with that in wild-type mice, indicating that animals with low or no GPER expression tend to develop less aggressive breast tumors (Marjon et al., 2014). Recent research has shown a significant correlation between GPER abundance and high-risk TNBC, characterized by G3 tumors, distant metastasis, stage III, and lymph node metastasis (Xu et al., 2022). Differing opinions suggest that GPER expression may suppress tumor initiation and progression (Weißenborn et al., 2014). GPER inhibits tumor cell growth by stimulating G2/M-phase cell cycle arrest, decreasing cyclin B expression, and promoting apoptosis in ERα-negative breast cancer cells (Wei et al., 2014). Weissenborn et al. (2017) suggested that TNBC with downregulated GPER expression has a poor outcome, which may be associated with promoter methylation changes in GPER. Additionally, TNBC models treated with GPER-specific agonists exhibited reduced distant migration ability and angiogenesis of tumor cells both in vivo and in vitro, leading to a significant reduction in lung metastasis (Chen et al., 2016).
ERs, such as ERα, belong to the nuclear receptor superfamily. E2 activates ERα in the cell nucleus, forming a dimer. The activated ERα causes gene expression in the nucleus by directly binding to estrogen response elements in the genome or by binding to other transcription factors and their response elements (Kato et al., 2005). This genomic effect is relatively slow but is an important pathway for estrogen to promote breast cancer growth (Watters et al., 1997). Additionally, a small fraction of ERα is localized to the cell membrane, enabling E2 to induce non-genomic effects through this membrane-bound ERα. Upon encountering E2 on the cell membrane, ERα rapidly activates the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway, which is a non-genomic effect that significantly contributes to promoting cell proliferation (Catalano et al., 2009; Puglisi et al., 2019). Furthermore, E2-ERα binding on the membrane activates the non-receptor tyrosine kinase SRC, which phosphorylates and activates aromatase, thereby promoting de novo synthesis of E2. This positive feedback effect also contributes to the genomic signaling of E2-ERα. Moreover, ERα signaling exhibits a ligand-independent activation pathway, exerting its effects through phosphorylation changes in ERα (Weigel and Zhang, 1998). Unlike the classic E2/ERα action pathway, E2-GPER primarily exerts its effects through fast, non-genomic signaling pathways. Distinguishing itself from ERα, which directly activates gene transcription by binding to transcriptional regulatory sites on DNA, GPER’s transcriptional effects are mediated indirectly through cAMP and EGFR. In breast cancer cell lines, E2-activated GPER couples to a trimeric G protein, and the Gα subunit dimer directly stimulates adenylyl cyclase, which in turn converts adenosine triphosphate into cAMP. In contrast, the Gβγ subunit dimer activates SRC tyrosine kinase, thereby activating α5β1 integrin and matrix metalloproteinase (MMP). Subsequently, the heparin-binding EGF-like growth factor is activated and stimulates EGFR transactivation (Filardo et al., 2000; Filardo and Thomas, 2005; Thomas et al., 2005; Filardo et al., 2008). This subsequently activates intracellular signaling events, specifically the PI3K/Akt and ERK1/2 pathways (Prossnitz et al., 2008). These two transcriptional pathways are regulated by cAMP-regulated enhancers and serum-regulated enhancers, respectively. After GPER transcriptional activation, genes for c-Fos, cyclins A, cyclins D1, and connective tissue growth factor (CTGF), which are involved in the biological processes of breast cancer, are upregulated. For example, CTGF is a cytokine that enhances the migration ability of the MDA-MB-231 cell line (Pupo et al., 2017). Moreover, in SKBR3 cells, which are ERα-negative, rapid transcriptional activation of c-Fos is induced by estradiol through activation of GPER/EGFR/mitogen-activated protein kinase (MAPK) signaling cascades (Maggiolini et al., 2004). The transcription factor c-Fos is an important invasion regulator in human mammary carcinomas, impacting cell migration, morphology, and proteolytic degradation of basal membranes (Milde-Langosch et al., 2004). The GPER mechanisms of action in TNBC and comprehensive molecular pathways are presented in Figure 2.
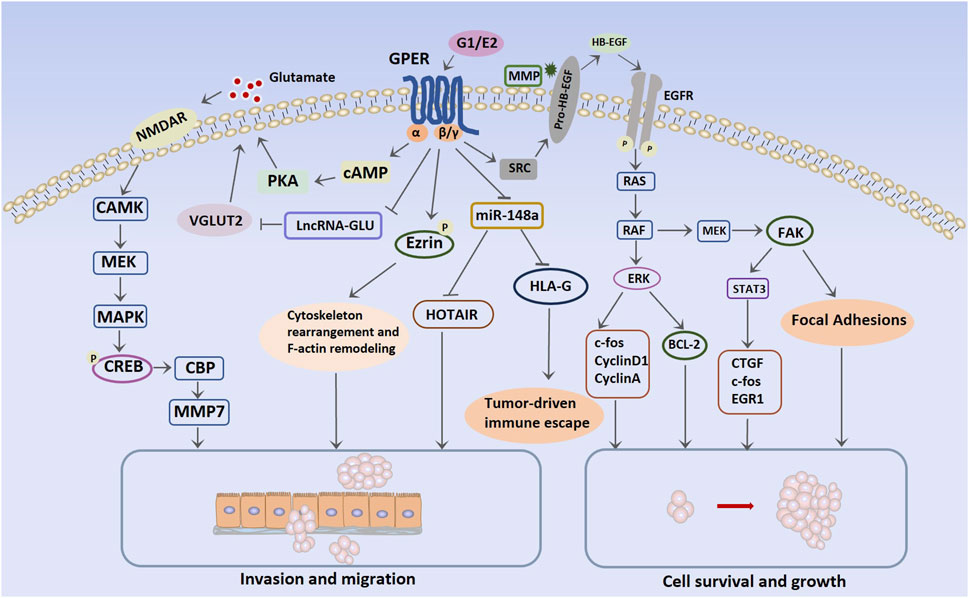
FIGURE 2. Molecular pathways mediated by G protein-coupled estrogen receptor in triple-negative breast cancer. Abbreviations: NMDAR, N-methyl-d-aspartate receptor; CAMK, Ca2+/calmodulin-dependent protein kinase; MEK, mitogen-activated protein kinase; MAPK, mitogen-activated protein kinase; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; CBP, CREB-binding protein; VGLUT2, vesicular glutamate transporter 2; HOTAIR, HOX transcript antisense intergenic RNA; HB-EGF, heparin-binding EGF-like growth factor; BCL-2, B-cell lymphoma 2; CTGF, connective tissue growth factor; EGR1: early growth response protein 1; HLA-G, human leukocyte antigen-G; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; SRC, steroid receptor coactivator.
Elevated GPER expression is directly related to a poor prognosis in TNBC (Steiman et al., 2013; Xu et al., 2022). Specifically, decreasing GPER expression with siRNA has been demonstrated to effectively inhibit the proliferation of TNBC cells (Girgert et al., 2012; 2014). In addition, G-1 has been shown to stimulate tissue proliferation by increasing mitosis (Scaling et al., 2014). Therefore, we summarized the relevant GPER mechanisms of action in TNBC cells.
GPER plays a pivotal role in numerous types of cancer, including endometrial cancer, thyroid cancer, melanoma, ovarian cancer, and breast cancer, via non-genomic signaling events (Revankar et al., 2005; Vivacqua et al., 2006; Albanito et al., 2007). Yu et al. (2014) observed that GPER was not only highly expressed in TNBC but was also involved in the activation of estrogen-mediated non-genomic ERK signaling. Based on clinicopathological evidence, p-ERK1/2 was detected in more than three-quarters of the GPER-positive TNBC specimens. Furthermore, high levels of GPER and p-ERK1/2 have been found to be prevalent in patients with a positive lymph nodes, large tumor size, and particularly an advanced clinical stage (Yu et al., 2014).
E2 or G-1 can induce Akt or ERK activation through GPER/EGFR signaling (Albanito et al., 2007; Fujiwara et al., 2012). Interestingly, Yu et al. (2014) found that in TNBC cells, E2, tamoxifen, and G-1 induced the rapid activation of p-ERK1/2 but not p-Akt signaling. Activation of GPER/EGFR/ERK upregulated proliferation-related genes, including genes for c-Fos, cyclin A, and cyclin D1, in MDA-MB-468 cells, thereby promoting cell cycle progression and proliferation. Additionally, E2/GPER/ERK significantly protected MDA-MB-468 cells when exposed to a serum-free medium through the upregulation of B-cell lymphoma (Bcl)-2 expression. This indicates that GPER/ERK signaling contributes to cell growth and survival by increasing Bcl-2 levels in TNBC cells (Yu et al., 2014). The Na+/H+ exchanger regulatory factor 1 (NHERF1) is a target actuator of estrogen signaling and is significantly downregulated in the early stages of TNBC. Wang et al. (2017) showed that NHERF1 co-localized with GPER in MDA-MB-231 cells, and overexpression of NHERF1 induced the inhibition of ERK1/2 and Akt signaling and proliferation of TNBC. Therefore, the activation of GPER/ERK non-genomic signaling is a crucial mechanism that triggers various downstream signaling pathways associated with cell proliferation in TNBC.
FAK is a cytoplasmatic protein tyrosine kinase and plays a crucial role in promoting tumor cell invasiveness, which is attributed to both kinase-independent and kinase-dependent scaffolding functions (Jean et al., 2014; Taliaferro-Smith et al., 2015; Shen et al., 2018; Rigiracciolo et al., 2021; Zhang et al., 2021). Notably, high expression of FAK has been observed in breast tumors. Increased levels of FAK in primary tumors are associated with a triple-negative phenotype, as well as invasive and metastatic breast cancer. Moreover, FAK amplification occurs during the early stages of breast tumorigenesis (Lark et al., 2005; Luo and Guan, 2010; Golubovskaya et al., 2014). Estrogen activates FAK via the GPER/c-SRC/mitogen-activated protein kinase kinase (MEK) transduction pathway, thereby mediating tumor cell proliferation and invasiveness (Rigiracciolo et al., 2019). Rigiracciolo et al. (2019) observed that GPER stimulated Y397 FAK phosphorylation and upregulated focal adhesions (FA) in invasive and metastatic TNBC. FAs are crucial subcellular structures that mediate cell adhesion to the extracellular matrix (ECM). Immunofluorescence studies have shown that GPER activation by E2 and G-1 leads to the involvement of FAK in signal transducer and activator of transcription (STAT)3 nuclear accumulation and changes in gene expression. As a critical factor in JAK/STAT signaling, STAT3 regulates tumor progression by controlling the cell cycle, apoptosis, angiogenesis, and immune evasion (Huang et al., 2010; Huynh et al., 2017). Additionally, both STAT3 and FAK participate in the regulation of GPER-mediated expression of multiple proliferation-associated genes, such as CTGF, c-Fos, and early growth response protein 1 (Joo et al., 2004; Pandey et al., 2009). Overall, estrogenic GPER signaling may promote the invasive ability of TNBC cells via FAK and STAT3.
Glutamate is a key compound in cell metabolism that stimulates certain downstream molecular mechanisms to promote the invasion and migration of cancer cells through the N-methyl-d-aspartate receptor (Rzeski et al., 2002; Li et al., 2018). Compared with HER2-positive and ERα-positive breast cancer cells, glutamate is heavily secreted in TNBC. Glutamate can induce the para secretion of hypoxia-inducible factor-1α (HIF-1α) in TNBC (Briggs et al., 2016). LncRNAs are the largest and most heterogeneous RNA family among the non-coding RNAs that participate in the normal biological behavior of cells. Dysregulation of lncRNA expression is conducive to the occurrence of malignant tumors. Notably, estrogen-induced lncRNAs are associated with the development of breast cancer (Volovat et al., 2020). Under the stimulation of G-1 or E2, GPER-regulated lncRNAs are downregulated, thereby increasing the transport activity of glutamate and the transcriptional activity of vesicular glutamate transporter 2 (VGLUT2). Upregulation of VGLUT2, in conjunction with GPER-cAMP/PKA signaling, leads to increased glutamate secretion through the lncRNA–Glutamate–VGLUT2 pathway. This facilitates the phosphorylation of N-methyl D-aspartate receptor subtype 2B and activates the glutamate N-methyl-D-aspartate receptor (Zeng et al., 2019). Furthermore, Ca2+/calmodulin-dependent protein kinase and MEK/MAPK signaling are activated, thereby enhancing cAMP response element-binding protein (CREB) phosphorylation, resulting in the recruitment of CREB-binding protein to the promoter regions of MMP7, which participates in TNBC invasion (Yin J. et al., 2020).
Furthermore, HOX transcript antisense intergenic RNA (HOTAIR), a crucial lncRNA, can promote tumor cell progression and the survival of breast cancer stem cells (Deng et al., 2017) and serves as a predictor of adverse prognostic events in patients with breast cancer. E2-GPER increases HOTAIR levels in TNBC cells by suppressing microRNA-148a (miR-148a) expression. Several studies have documented a positive association between the expression of HOTAIR and the metastatic potential of TNBC (Deng et al., 2017; Lu et al., 2018; Sun et al., 2018). G-15 blocks the E2-induced upregulation of HOTAIR and reverses cell migration (Tao et al., 2015).
Cancer stem cells are capable of self-renewal and differentiation, which enables them to initiate tumor growth in new areas and form new tumors (Valastyan and Weinberg, 2011). In the context of basal-like breast cancer, stem cells are often detected through the low expression of cluster of differentiation (CD) 24 and high expression of CD44 (Honeth et al., 2008; Li et al., 2013; O’Conor et al., 2018). These cell subpopulations remain viable during chemotherapy or neoadjuvant endocrine therapy, implying their significance in the development of drug resistance (Creighton et al., 2009; Croker and Allan, 2012). Furthermore, TNBC have a higher abundance of therapy-resistant cancer stem cells than that of other subtypes, leading to increased mortality, treatment failure, and recurrence (Honeth et al., 2008; Li et al., 2013; Ma et al., 2014). Zhu et al. (2022) used a spheroid culture method to induce sphere formation in MDA-MB-468 cells and compared the malignant characteristics, GPER levels, and stemness-related markers between spherical and adherent cells. The results revealed increased expression of GPER and a high percentage of the CD44+/CD24-subpopulation in spheroid MDA-MB-468 cells. Furthermore, enhanced tumor metastasis was observed. This indicated a close connection between GPER expression and the enhancement of stemness in malignant tumors. Additionally, both spheroid and adherent cells exhibited increased GPER expression after E2 treatment, whereas GPER expression decreased after G-15 treatment. These effects were most apparent in spheroid cells, indicating that GPER positively regulated the proliferation, invasiveness, and colony formation of TNBC cells in response to E2 signaling and that GPER expression is associated with the display of stemness characteristics (Zhu et al., 2022).
Ezrin-radixin-moesin (ERM) proteins, which belong to the actin-binding family, play a vital role in bridging the gap between the plasma membrane and actin cytoskeleton (Ponuwei, 2016). ERM proteins have a significant impact on cancer cell polarity and migration by regulating cell signaling, cytoactivity, and the cytoskeleton (Clucas and Valderrama, 2014). Clinical and pathological evidence has revealed that GPER and ezrin protein expression have a positive relationship with TNBC pathological tissues. Additionally, the high co-expression of GPER/ezrin is strongly correlated with a poor patient prognosis, and a high GPER expression is linked to a poor prognosis in young patients with TNBC with elevated estrogen levels but not in patients post menopause. E2 has been reported to rapidly promote the phosphorylation of the ezrin protein through GPER or ERβ, causing ezrin-dependent rearrangement of the cytoskeleton and F-actin remodeling, which stimulates the migration and invasion of ERα-negative cancer cells. Notably, the stimulation of GPER counteracts ezrin activation and migration induced by ERβ in co-expressed GPER and ERβ cells, such as MDA-MB-231 (Zhou et al., 2016). This could be attributed to the presence of a certain degree of mutual antagonism between ERs. Consequently, high GPER expression is a risk factor for TNBC in patients with high estrogen levels and is likely associated with the GPER-mediated activation of ezrin and its downstream pathways.
The malignant behavior of tumors is significantly correlated with the tumor microenvironment (TME) and CAFs, which are critical members of the TME and exhibit active migratory and invasive abilities in breast cancer (Luo et al., 2015; Lappano and Maggiolini, 2018). GPER is expressed in CAFs and acts as a transcriptional regulator of the CAF response to estrogen or G-1 (Luo et al., 2014). Additionally, GPER participates in the paracrine secretion of chemotactic, growth, and ECM-related factors by CAF, thereby triggering invasive behavior in breast tumor cells (Pupo et al., 2012), including F-actin reorganization, epithelial-mesenchymal transition (EMT), cancer cell migration, and angiogenesis (De Francesco et al., 2014). Moreover, activated GPER triggers a feed-forward loop, inducing the expression of interleukin (IL) 1β/IL1R1 target genes on CAF, thereby promoting the invasive characteristics of breast cancer cells (De Marco et al., 2016). Furthermore, both estrogen and G-1 can induce the upregulation of HIF-1α and vascular endothelial growth factor (VEGF) expression in CAFs, thereby promoting tumor angiogenesis (Roskoski, 2007; De Francesco et al., 2014). In TNBC, upregulated GPER in CAF may facilitate the expression of collagen type 1 and drive the crucial proliferation, invasion, and migration of cancer cells (Yang and Yao, 2019). The above evidence demonstrates that GPER exerts a pro-tumorigenic effect on tumor cell proliferation, invasion, and migration through CAF, regardless of hormone receptor status. These mechanisms are presented in Figure 3.
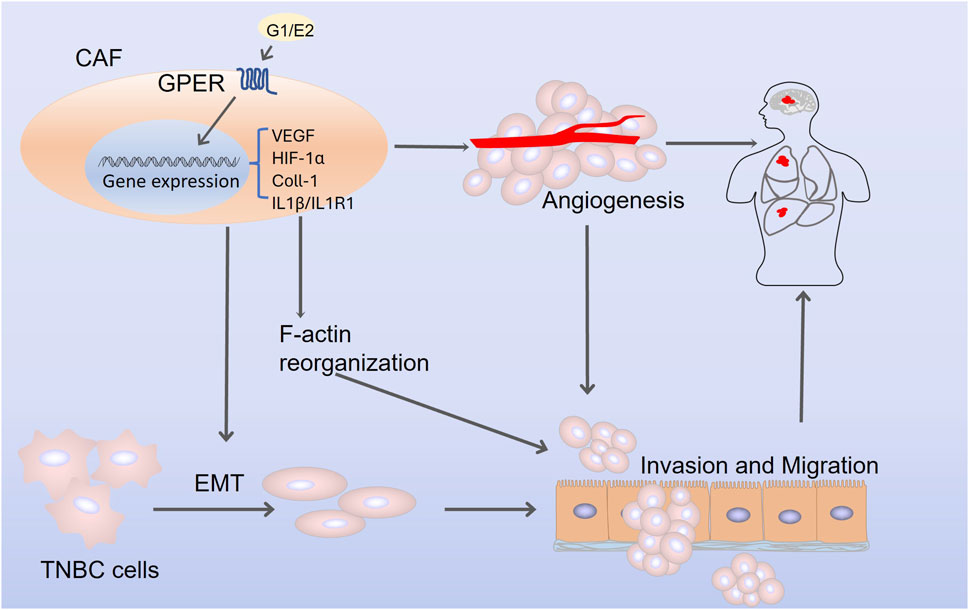
FIGURE 3. The mechanism by which G protein-coupled estrogen receptor promotes triple-negative breast cancer progression through cancer-associated fibroblasts. Abbreviations: CAF, cancer-associated fibroblasts; VEGF, vascular endothelial growth factor; EMT, epithelial-mesenchymal transition; HIF-1α, hypoxia-inducible factor-1α; Coll-1, collagen type 1; IL1β, interleukin 1β; IL1R1, interleukin 1 receptor 1.
The androgen receptor (AR) plays a vital role as a member of the nuclear receptor superfamily and participates in both physiological and pathological processes (Matsumoto et al., 2013; Anestis et al., 2020). Immunohistochemical staining of a TNBC tissue microarray containing 165 tumor specimens suggests that AR is positively correlated with tumor size, lymphatic metastasis, and high histological grade and negatively associated with GPER expression in TNBC (Shen et al., 2017). Dihydrotestosterone (DHT) enhances the proliferation of TNBC cells in a dose-dependent manner. Treatment with DHT leads to an upregulation of AR protein expression, whereas AR knockdown blocks the pro-proliferative effects induced by DHT. Intriguingly, DHT negatively regulates GPER expression, and AR deficiency significantly reduces the effectiveness of GPER suppression by DHT. Notably, the direct binding of AR to the GPER promoter, leading to negative regulation of GPER expression, is one of the contributing factors. The same conclusion was also observed in vivo, indicating that AR represses the pro-growth effects mediated by GPER in TNBC (Shen et al., 2017).
The prognostic influence of ERβ in TNBC is also disputable, with some studies suggesting that ERβ possesses anti-proliferative and -invasive functions in breast cancer cells, possibly by suppressing the PI3K/Akt/mTOR signaling pathway (Lei et al., 2020). In contrast, some studies have demonstrated that ERβ mediates TNBC invasion by regulating EGFR (Kyriakopoulou et al., 2022). In a recent study, silencing GPER expression using siRNA resulted in a weakening in the invasiveness of TNBC cells expressing ERβ while significantly enhancing the anti-invasive effects of ERβ (Schmitz et al., 2022). Activated ERβ suppresses tumor angiogenesis by downregulating VEGF protein levels (Salahuddin et al., 2022). However, GPER upregulates VEGF expression in breast cancer CAFs (De Marco et al., 2016). Additionally, in TNBC cells, the activation of GPER counteracts ezrin activation and migration induced by ERβ (Ye et al., 2019). Despite the apparent opposition in the effects of ERβ and GPER, their interaction mechanisms remain unclear and warrant further investigation.
As a classical ERα homologous orphan nuclear receptor, the overexpression of estrogen-related receptor α (ERRα) frequently correlates with unfavorable outcomes among individuals diagnosed with breast cancer (Cavallini et al., 2005; Chang et al., 2011). An in vitro study suggested that GPER-mediated estrogen-induced ERRα upregulation may promote TNBC cell viability, migration, and invasion. ERRα-GPER interactions lead to a malignant phenotype of TNBC, and this result was consistent with the conclusion drawn from the survival analysis (Ye et al., 2020). Additional research is necessary to determine the potential mechanisms that contribute to the interplay between GPER and ERRα in TNBC.
Activation of GPER has demonstrated effects on the synthesis of chemokines specific to white blood cells and intracellular signal transduction pathways, thereby governing the movement of immune cells and modulating inflammatory reactions. G-1 is an activator of GPER and promotes the synthesis of the anti-inflammatory cytokine IL-10 in CD4+ T lymphocytes (Brunsing and Prossnitz, 2011). G-1 suppresses the production of inflammatory molecules such as tumor necrosis factor (TNF), IL-6, and IL-12 in macrophages, which are triggered by lipopolysaccharides (Blasko et al., 2009). Following G-1-induced GPER activation, multiple genes, including GCSF, SOCS3, CXCL2, COX2, IL1RA, and IL1B, are upregulated through the p38 MAPK, ERK, and cAMP/PKA/CREB signaling pathways. Consequently, the production of CXCL8 protein is enhanced. Furthermore, GPER activation in neutrophils derived from healthy individuals enhanced the respiratory burst, cellular viability, and CD62L and CD11b activation marker levels (Rodenas et al., 2017).
Moreover, GPER interacts with specific chemokine receptors and cytokines, thereby impacting their signaling pathways and influencing physiological processes such as cell proliferation, migration, and tumor development. For example, the activation of GPER influences the synthesis of cytokines such as IL-6 and IL-8, which have important functions in promoting the growth and migration of cancer cells, particularly in low-oxygen environments (Bustos et al., 2017). In the case of breast cancer, the activation of GPER results in the suppression of nuclear factor kappa B (NF-κB) transcription, thereby decreasing the production of IL-6 induced by TNFα in the SKBR3 cell line. In addition, the activation of GPER is correlated with a reduction in IL-6 and VEGF-A levels in TNBC cellular models (Okamoto and Mizukami, 2016; Liang et al., 2017). Furthermore, GPER agonism by E2 inhibits TGF-β signaling in breast cancer (Kleuser et al., 2008). Both E2 and G-1 induce the release of IL-1β in CAFs, thereby resulting in an increase in the production of IL1R1. As a consequence of this molecular phenomenon, the invasive and migratory capabilities of breast cancer cells are heightened in the SKBR3 and MCF-7 cell lines. Notably, the absence of N-glycosylation in the N-terminal region of GPER causes the receptor to enter the nucleus, where it functions as a factor resembling transcription, thereby facilitating the upregulation of CTFG in CAFs and the SKBR3 cellular lineage (Pupo et al., 2017). Moreover, hypoxic conditions enhance the collaborative interaction between GPER and HIF1α in TNBC cells and CAF, thereby triggering the activation of the IL-β/IL1R1 signaling pathway and creating a feed-forward stimulatory loop. This causes changes in cytoskeleton structure and increased cellular activity, thus promoting TNBC invasiveness (Lappano et al., 2020).
GPER could mediate TNBC immune evasion and migration by regulating miR-148a in breast cancer. MiR-148a regulates tumor proliferation, invasion, and metastasis in various solid cancers by acting as an oncogene or cancer suppressor gene (Lujambio et al., 2008; Sakamoto et al., 2014). The miR-148a is specifically downregulated in TNBC (Chen et al., 2014). In their research, Tao et al. (2014) observed that estrogen-activated GPER inhibits miR-148a and stimulates the expression of human leukocyte antigen-G (HLA-G) in breast cancer (Tao et al., 2014). HLA-G is associated with the tumor-driven immune escape mechanism in the advanced stages of breast cancer (Jeong et al., 2014).
Furthermore, within the TME, the immune cells have significant contributions to tumor advancement. GPER expression is observed in diverse immune cells such as monocytes (Kvingedal and Smeland, 1997), natural killer cells (Wolfson et al., 2021), dendritic cells (Rodenas et al., 2017), macrophages (Rettew et al., 2010), polymorphonuclear cells (Blesson and Sahlin, 2012), as well as B and T lymphocytes. In breast cancer, the activation of GPER by various ligands elicits diverse responses within immune cells, influencing the constitution of the TME. Cytotoxic T lymphocytes (CTL) are the key effector cells in antitumor immunity and play an important role in immune responses against cancer. The activation of GPER leads to an elevation in IL-10 and IL-17, promoting the development of an immunosuppressive T cell phenotype. Furthermore, G-1 treatment leads to an upregulation of the programmed cell death 1 (PD-1) inhibitory receptor and CTL-associated protein 4 (CTLA-4) on isolated Foxp3+ regulatory T cells (Tregs) (Wang et al., 2009; Brunsing et al., 2013). The clinical significance of CTLA-4 and PD-1 expression in targeted cancer treatment has been acknowledged due to their ability to inhibit T cell proliferation (Zhao et al., 2018; Segovia-Mendoza et al., 2021). Thus, it is conceivable that GPER promotes the expression of PD-1 and CTLA-4 in Tregs derived from patients with breast cancer, thereby supporting the advancement of cancer. Furthermore, the use of GPER antagonists would favor the immune response against the tumor. In addition, GPER antagonists may also favor the clinical outcome of immunological checkpoints by reducing the expression of PD-1 and CTLA-4. The potential therapeutic approach of combining GPER antagonists with immunotherapy may serve as a strategy to boost the immune response and suppress the proliferation of breast cancer cells in patients.
Several studies have reported the antitumor functions of GPER. Our recent meta-analysis suggested that high GPER mRNA expression indicates improved overall survival in patients with ERα-positive breast cancer. Furthermore, subgroup analysis indicated that this may be related to the cytoplasmic expression of GPER (Zhang et al., 2022). Filardo et al. (2002) observed that the transfection of MDA-MB-231 cells with GPER stimulates adenylyl cyclase and inhibits EGFR-induced ERK1/2 activity (Filardo et al., 2002). Therefore, in TNBC, estrogen stimulation by GPER activation may have two different effects on the ERK1/2 pathway.
NF-κB regulates the main EMT transcription factors, including SNAIL1, SLUG, TWIST1, and SIP1, which are associated with the progression in breast cancer (Park et al., 2007; Pires et al., 2017). In TNBC, NK-κB plays an important role in the GPER-mediated inhibition of malignant tumor biological behaviors (Chen et al., 2016). Chen et al. (2016) reported that the activation of GPER by G-1 downregulates NF-κB activities and cytoskeletal protein fibronectin expression, and inhibits EMT in TNBC, resulting in a reduction in the aggressive characteristics of breast cancer. Wang et al. reported that E2-and G-1-activated GPER inhibit the expression and nuclear localization of NF-κB and STAT3 and suppress tumor angiogenesis and proliferation in MDA-MB-468 xenografts (Wang C. et al., 2018). The IL-6 cytokine family is widely expressed across vertebrates as a signaling molecule and contributes significantly to the malignant phenotype of cancer (Felcher et al., 2022). IL-6-activated STAT3 (Lee et al., 2009; Yu et al., 2009) elevates HIF-1α levels in MDA-MB-231 cells, which induces VEGF-A and MMP transcription (Xu et al., 2005; Eichten et al., 2016; Wang K. et al., 2018). Liang et al. (2017) found that the expression of IL-6 and VEGF can be suppressed by GPER-inhibiting NF-κB promoter activity in TNBC cells. Furthermore, the forceful inhibition of IL-6 expression in vitro reduces colony formation and cell survival and suppresses tumor engraftment in TNBC cell lines (Hanahan and Weinberg, 2011; Hartman et al., 2013; Felcher et al., 2022). Hence, activated GPER inhibits the expression of NF-κB and its downstream signaling, which are crucial in the suppression of TNBC progression. Notably, focusing on the interaction between NF-κB and GPER for the exploration of future treatments for TNBC is crucial.
Opposing evidence regarding the effect of GPER on TNBC is notable and may be associated with several factors. First, the role of G-1, as a specific ligand for GPER, in cancer cell survival and proliferation remains debatable. Further, G-1 exhibits a high affinity for GPER at a concentration of 10 nM and influences breast cancer cell proliferation in a dose-dependent manner (Table 2). Previous studies have suggested that G-1 promotes tumor cell proliferation within the range of 10 nM–1 μM, whereas high concentrations inhibit proliferation. Weißenborn et al. (2014) found that GPER treated with 1 μM G-1 inhibited TNBC cell growth. This inhibition was concentration-dependent and achieved by inducing cell cycle arrest in the G2/M phase, thereby enhancing the phosphorylation of histone h3, and promoting caspase-3-mediated apoptosis (Weißenborn et al., 2014). However, Lv et al. (2017) reported that the proliferation of TNBC cell lines was significantly inhibited by G-1 at relatively low concentrations (100 nM). G-1 inhibits microtubule assembly by occupying colchicine-binding sites within breast cancer cells, thereby inducing cell cycle arrest and triggering apoptosis. Interestingly, this inhibitory effect was not affected by GPER knockdown. Therefore, G-1 may act as a microtubule-targeting agent to inhibit breast cancer cell proliferation (Lv et al., 2017). This conclusion is similar to that of Wang et al. (2011) who demonstrated that G-1 inhibits the growth of breast and ovarian cancer cells independent of GPER at the micromolar level. In the studies mentioned above, a concentration of 1 µM was often used to assess the inhibitory effects of G-1 on breast cancer malignancy (Dennis et al., 2009; Chen et al., 2016; Liang et al., 2017). Consequently, based on this evidence, it is crucial for studies on the action of G-1 on breast cancer cells to consider both its effects on GPER and related signaling pathways and its non-GPER-dependent tumor inhibitory effect.
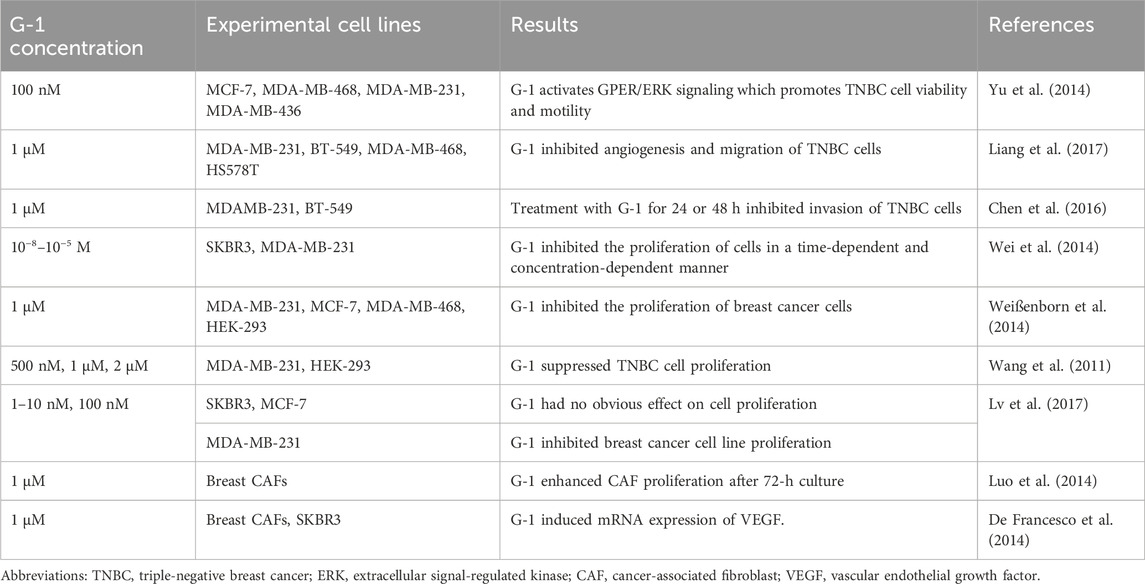
TABLE 2. Effects of different G-1 concentrations on breast cancer cell lines.
In addition, TNBC is an extremely heterogeneous cancer that can be classified into multiple subtypes, each with a distinct gene expression profile, resulting in significant differences in treatment response and prognosis. Moreover, differences in cell types, treatment conditions, and GPER subcellular localization may contribute to the previously discussed variations. Hence, further studies are required to fully elucidate the role of GPER in TNBC.
Ligand-activated GPER can promote TNBC progression. GPER ligands mainly include endogenous estrogen, exogenous estrogen, estrogen analogs, and specific ligands. Reducing biologically available estrogen may be an effective strategy to block GPER effects. There are various methods to treat patients with ERα-positive breast cancer through blocking estrogen. These methods include ovarian removal, inhibiting ovarian function by activating gonadotrophin-releasing hormone, or using aromatase inhibitors to suppress estrogen synthesis. Investigating the specific subgroup of TNBC that could potentially benefit from estrogen deprivation strategies represents a promising avenue for future research.
With regards to the dual role of GPER in TNBC, certain GPER-specific agonists, such as G-1 could also inhibit the progression of TNBC. Berberine (BBR) is a bioactive isoquinoline alkaloid that acts as an agonist of GPER to inhibit the nuclear translocation of the NF-κB subunit RELA/p65. BBR also significantly inhibits TNBC by modulating GPER protein levels (Qi et al., 2021). The small-molecule targeted agonist of GPER, LNS8801, has demonstrated impressive results in a multicenter Phase 1–2A clinical trial, exhibiting encouraging anti-tumor effects in patients with metastatic uveal melanoma, whether used alone or in combination with pembrolizumab. Additionally, LNS8801 alone or in combination with pembrolizumab was well tolerated and did not result in unexpected toxicity (Shoushtari et al., 2023). As shown in Table 1, a series of GPER agonists have been identified, showing different mechanisms of action. Notably, limited progress has been made in the development of effective GPER agonists for the treatment of breast cancer. Chrysin is a flavone that exhibits anti-cancer effects against various types of cancers (Kim et al., 2017) including TNBC (Yang et al., 2014). Chrysin nanoparticles (NP) are polymers with improved bioavailability and water solubility. In TNBC, chrysin NPs inhibit the expression of MMPs by activating GPER and suppressing factors such as NF-κB, phosphorylated-JNK, and PI3K. MMP regulation is crucial for tumor growth, proliferation, and angiogenesis. These studies elucidate the inhibitory effects of chrysin NPs on TNBC (Kim and Jung, 2020); hence, further clinical trials are warranted.
Small-molecule antagonists of GPER mainly include G-15 and G-36. In vitro and in vivo studies have demonstrated that both have high GPER affinity and can inhibit the activity of breast tumor cells. However, there is currently a lack of supporting clinical evidence (Dennis et al., 2009; Dennis et al., 2011). Recently, DeLeon et al. (2020) discovered a novel GPER antagonist, 2-cyclohexyl-4-isopropyl-N-(4-methoxybenzyl) aniline (CIMBA), which effectively reduced estrogen-induced cholesterol gallstone formation in female mice by inhibiting GPER signaling. CIMBA exhibits fewer off-target effects than G-15 and G-36 do when binding to the receptor, and is less likely than both to bind to ERα at high concentrations. This novel antagonist also presents potential for further research in the field of cancer (DeLeon et al., 2020). As shown in Table 1, several antagonists have been identified to inhibit GPER. However, additional experimental verification is crucial to determine their efficacy in TNBC.
GPER, like most GPCRs, relies on the action of heterotrimeric G proteins for its signaling mechanism (Nieto Gutierrez and McDonald, 2018). Directly targeting G proteins is an effective strategy (Campbell and Smrcka, 2018). G protein inhibitors can disrupt the conformational activation of the GPCR-Gαβγ pathway. For instance, the plant-derived depsipeptide FR900359 inhibits the constitutively active G protein α subunit Gαq in uveal melanoma, thereby halting tumor cell proliferation and inducing apoptosis (Onken et al., 2020). Additionally, β-adrenergic receptor kinase 1, as a Gβγ subunit sequestering peptide, blocks the activation of ERK1/2 in breast cancer cells (Filardo et al., 2000); an effect that requires further validation in animal models. Furthermore, somavert is a competitive growth hormone receptor inhibitor and is primarily used to treat acromegaly. However, treatment with somavert suppresses the expression of GPER in TNBC cells, inhibits EGFR activation, and downregulates proliferation-associated genes, such as genes for c-Fos and cyclin D (Girgert et al., 2018). Furthermore, gene therapy techniques, such as RNA interference or clustered regularly interspaced short palindromic repeats (CRISP)-CRISPR-associated protein 9, could be used to modulate GPER expression in vivo. However, these methods are predominantly experimental and are not yet widely used in clinical practice.
The GPER-dependent transactivation of EGFR, which plays a key role in initiating signaling pathways and influencing cellular activities, is crucial for the cellular signal transduction and function mediated by GPER (Quinn et al., 2009). Gefitinib is an inhibitor of the EGFR tyrosine kinase domain and effectively suppresses cancer cell proliferation by downregulating EGFR signaling, particularly within TNBC (Herbst et al., 2004; Bernsdorf et al., 2011). Although clinical trials using gefitinib as a monotherapy for breast cancer have exhibited unsatisfactory results, immunohistochemical analysis of post-treatment breast tumors has revealed complete inhibition of EGFR phosphorylation (Baselga et al., 2005). Girgert et al. (2017) demonstrated that gefitinib inhibits the development of TNBC cells by reducing GPER expression induced by E2. GPER antagonists efficiently impede the activation of both GPER and EGFR induced by EGF. The interplay between GPER and EGFR suggests that a therapeutic approach involving a combination of GPER and EGFR inhibitors holds promise for combating breast cancer. However, this potential requires comprehensive exploration through clinical trials.
Furthermore, the activation of the insulin-like growth factor (IGF)-1/IGF 1 receptor (IGF-1R) system induces stimulatory effects via GPER in highly malignant neoplasms such as mesothelioma and lung tumors (Avino et al., 2016). Moreover, De Marco et al. (2013) revealed a bidirectional interaction between the IGF system and GPER that promotes drug resistance and cancer progression. GPER is essential for the migratory effects induced by zinc via the IGF-1R pathway in ERα-negative breast cancer cells (Pisano et al., 2017). In addition, the fibroblast growth factor (FGF) and FGF receptor (FGFR) axis constitute a pivotal signal transduction pathway that mediates the intricate interaction between cancer cells and tumor stroma. The GPER-selective agonist G-1 induces the expression and release of FGF2 in CAFs through the activation of GPER. Subsequently, the secreted FGF2 activates the FGF2/FGFR1 paracrine signaling cascade, thereby promoting the invasion and migration of TNBC cells (Santolla et al., 2019). The interplay between GPER and these membrane growth factor receptors (GFRs) presents opportunities for the development of novel pharmacological approaches to target breast cancer progression. Therefore, combining GPER and GFR inhibitors emerges as a potential therapeutic strategy and warrants further study.
As a novel estrogen receptor, the biological role of GPER in TNBC has not yet been sufficiently elucidated compared with that in other breast cancer subtypes. This review presents a comprehensive overview of GPER, encompassing its subcellular structure, functional role, and internal molecular mechanisms. The analysis includes diverse ligand effects and the interactions between GPER and closely associated factors in TNBC. However, the impact of GPER on the malignant behavior of TNBC appears contradictory. Despite the existing studies, the feasibility of using GPER as a therapeutic target is insufficient, and further research is warranted.
DZ: Conceptualization, Visualization, Writing–original draft, Writing–review and editing. HC: Visualization, Writing–review and editing. JW: Writing–review and editing. JJ: Writing–review and editing. MI: Writing–review and editing. ZZ: Writing–review and editing. SY: Conceptualization, Funding acquisition, Supervision, Validation, Visualization, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Liaoning Province Science and Technology Project (Applied Basic Research Program) (grant number 2022JH2/101500066); Shenyang Science and Technology Bureau Project (grant number 21-173-9-69); and 345 Talent Project of Shengjing Hospital of China Medical University (to SY).
Figure 1 was created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Albanito, L., Madeo, A., Lappano, R., Vivacqua, A., Rago, V., Carpino, A., et al. (2007). G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 67, 1859–1866. doi:10.1158/0008-5472.CAN-06-2909
Anestis, A., Zoi, I., Papavassiliou, A. G., and Karamouzis, M. V. (2020). Androgen receptor in breast cancer-clinical and preclinical research insights. Molecules 25, 358. doi:10.3390/molecules25020358
Ariazi, E. A., Ariazi, J. L., Cordera, F., and Jordan, V. C. (2006). Estrogen receptors as therapeutic targets in breast cancer. Curr. Top. Med. Chem. 6, 195–213. doi:10.2174/156802606776173474
Aronica, S. M., Kraus, W. L., and Katzenellenbogen, B. S. (1994). Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc. Natl. Acad. Sci. U. S. A. 91, 8517–8521. doi:10.1073/pnas.91.18.8517
Avena, P., Casaburi, I., Zavaglia, L., Nocito, M. C., La Padula, D., Rago, V., et al. (2022). 27-Hydroxycholesterol binds GPER and induces progression of estrogen receptor-negative breast cancer. Cancers 14, 1521. doi:10.3390/cancers14061521
Avino, S., Marco, P. D., Cirillo, F., Santolla, M. F., Francesco, E. M. D., Perri, M. G., et al. (2016). Stimulatory actions of IGF-I are mediated by IGF-IR cross-talk with GPER and DDR1 in mesothelioma and lung cancer cells. Oncotarget 7, 52710–52728. doi:10.18632/oncotarget.10348
Baselga, J., Albanell, J., Ruiz, A., Lluch, A., Gascón, P., Guillém, V., et al. (2005). Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J. Clin. Oncol. 23, 5323–5333. doi:10.1200/JCO.2005.08.326
Bernsdorf, M., Ingvar, C., Jörgensen, L., Tuxen, M. K., Jakobsen, E. H., Saetersdal, A., et al. (2011). Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res. Treat. 126, 463–470. doi:10.1007/s10549-011-1352-2
Blasko, E., Haskell, C. A., Leung, S., Gualtieri, G., Halks-Miller, M., Mahmoudi, M., et al. (2009). Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J. Neuroimmunol. 214, 67–77. doi:10.1016/j.jneuroim.2009.06.023
Blesson, C. S., and Sahlin, L. (2012). Expression pattern and signalling pathways in neutrophil like HL-60 cells after treatment with estrogen receptor selective ligands. Mol. Cell Endocrinol. 361, 179–190. doi:10.1016/j.mce.2012.04.006
Bologa, C. G., Revankar, C. M., Young, S. M., Edwards, B. S., Arterburn, J. B., Kiselyov, A. S., et al. (2006). Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2, 207–212. doi:10.1038/nchembio775
Brailoiu, E., Dun, S. L., Brailoiu, G. C., Mizuo, K., Sklar, L. A., Oprea, T. I., et al. (2007). Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J. Endocrinol. 193, 311–321. doi:10.1677/JOE-07-0017
Briggs, K. J., Koivunen, P., Cao, S., Backus, K. M., Olenchock, B. A., Patel, H., et al. (2016). Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell 166, 126–139. doi:10.1016/j.cell.2016.05.042
Brunsing, R. L., Owens, K. S., and Prossnitz, E. R. (2013). The G protein-coupled estrogen receptor (GPER) agonist G-1 expands the regulatory T cell population under T(H)17 polarizing conditions. J. Immunother. 36, 190–196. doi:10.1097/CJI.0b013e31828d8e3b
Brunsing, R. L., and Prossnitz, E. R. (2011). Induction of interleukin-10 in the T helper type 17 effector population by the G protein coupled estrogen receptor (GPER) agonist G-1. Immunology 134, 93–106. doi:10.1111/j.1365-2567.2011.03471.x
Bustos, V., Nolan, Á. M., Nijhuis, A., Harvey, H., Parker, A., Poulsom, R., et al. (2017). GPER mediates differential effects of estrogen on colon cancer cell proliferation and migration under normoxic and hypoxic conditions. Oncotarget 8, 84258–84275. doi:10.18632/oncotarget.20653
Campbell, A. P., and Smrcka, A. V. (2018). Targeting G protein-coupled receptor signalling by blocking G proteins. Nat. Rev. Drug Discov. 17, 789–803. doi:10.1038/nrd.2018.135
Cao, W., Chen, H.-D., Yu, Y.-W., Li, N., and Chen, W.-Q. (2021). Changing profiles of cancer burden worldwide and in China: a secondary analysis of the global cancer statistics 2020. Chin. Med. J. Engl. 134, 783–791. doi:10.1097/CM9.0000000000001474
Carmeci, C., Thompson, D. A., Ring, H. Z., Francke, U., and Weigel, R. J. (1997). Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 45, 607–617. doi:10.1006/geno.1997.4972
Castillo Sanchez, R., Gomez, R., and Perez Salazar, E. (2016). Bisphenol A induces migration through a GPER-FAK-src-and ERK2-dependent pathway in MDA-MB-231 breast cancer cells. Chem. Res. Toxicol. 29, 285–295. doi:10.1021/acs.chemrestox.5b00457
Castillo-Sanchez, R., Ramirez-Ricardo, J., Martinez-Baeza, E., Cortes-Reynosa, P., Candanedo-Gonzales, F., Gomez, R., et al. (2020). Bisphenol A induces focal adhesions assembly and activation of FAK, Src and ERK2 via GPER in MDA-MB-231 breast cancer cells. Toxicol. Vitro 66, 104871. doi:10.1016/j.tiv.2020.104871
Catalano, S., Barone, I., Giordano, C., Rizza, P., Qi, H., Gu, G., et al. (2009). Rapid estradiol/ERalpha signaling enhances aromatase enzymatic activity in breast cancer cells. Mol. Endocrinol. 23, 1634–1645. doi:10.1210/me.2009-0039
Catalano, S., Giordano, C., Panza, S., Chemi, F., Bonofiglio, D., Lanzino, M., et al. (2014). Tamoxifen through GPER upregulates aromatase expression: a novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res. Treat. 146, 273–285. doi:10.1007/s10549-014-3017-4
Cavallini, A., Notarnicola, M., Giannini, R., Montemurro, S., Lorusso, D., Visconti, A., et al. (2005). Oestrogen receptor-related receptor alpha (ERRalpha) and oestrogen receptors (ERalpha and ERbeta) exhibit different gene expression in human colorectal tumour progression. Eur. J. Cancer 41, 1487–1494. doi:10.1016/j.ejca.2005.04.008
Chan, Q. K. Y., Lam, H.-M., Ng, C.-F., Lee, A. Y. Y., Chan, E. S. Y., Ng, H.-K., et al. (2010). Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G2 cell-cycle arrest. Cell Death Differ. 17, 1511–1523. doi:10.1038/cdd.2010.20
Chang, C., Kazmin, D., Jasper, J. S., Kunder, R., Zuercher, W. J., and McDonnell, D. P. (2011). The metabolic regulator ERRα, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell 20, 500–510. doi:10.1016/j.ccr.2011.08.023
Chen, Z., Ma, T., Huang, C., Zhang, L., Xu, T., Hu, T., et al. (2014). MicroRNA-148a: a potential therapeutic target for cancer. Gene 533, 456–457. doi:10.1016/j.gene.2013.09.067
Chen, Z.-J., Wei, W., Jiang, G.-M., Liu, H., Wei, W.-D., Yang, X., et al. (2016). Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-κB signals. Mol. Oncol. 10, 775–788. doi:10.1016/j.molonc.2016.01.002
Cheng, S.-B., Graeber, C. T., Quinn, J. A., and Filardo, E. J. (2011a). Retrograde transport of the transmembrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPER) from the plasma membrane towards the nucleus. Steroids 76, 892–896. doi:10.1016/j.steroids.2011.02.018
Cheng, S.-B., Quinn, J. A., Graeber, C. T., and Filardo, E. J. (2011b). Down-modulation of the G-protein-coupled estrogen receptor, GPER, from the cell surface occurs via a trans-golgi-proteasome pathway. J. Biol. Chem. 286, 22441–22455. doi:10.1074/jbc.M111.224071
Chhikara, B. S., and Parang, K. (2023). Global Cancer Statistics 2022: the trends projection analysis. Chem. Biol. Lett. 10, 451.
Clucas, J., and Valderrama, F. (2014). ERM proteins in cancer progression. J. Cell Sci. 127, 267–275. doi:10.1242/jcs.133108
Creighton, C. J., Li, X., Landis, M., Dixon, J. M., Neumeister, V. M., Sjolund, A., et al. (2009). Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. U. S. A. 106, 13820–13825. doi:10.1073/pnas.0905718106
Croker, A. K., and Allan, A. L. (2012). Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44⁺ human breast cancer cells. Breast Cancer Res. Treat. 133, 75–87. doi:10.1007/s10549-011-1692-y
Dai, X., Cheng, H., Bai, Z., and Li, J. (2017). Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 8, 3131–3141. doi:10.7150/jca.18457
De Francesco, E. M., Pellegrino, M., Santolla, M. F., Lappano, R., Ricchio, E., Abonante, S., et al. (2014). GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 74, 4053–4064. doi:10.1158/0008-5472.CAN-13-3590
DeLeon, C., Wang, H. H., Gunn, J., Wilhelm, M., Cole, A., Arnett, S., et al. (2020). A novel GPER antagonist protects against the formation of estrogen-induced cholesterol gallstones in female mice. J. Lipid Res. 61, 767–777. doi:10.1194/jlr.RA119000592
De Marco, P., Bartella, V., Vivacqua, A., Lappano, R., Santolla, M. F., Morcavallo, A., et al. (2013). Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene 32, 678–688. doi:10.1038/onc.2012.97
De Marco, P., Lappano, R., Francesco, E. M. D., Cirillo, F., Pupo, M., Avino, S., et al. (2016). GPER signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1β/IL1R1 response. Sci. Rep. 6, 24354. doi:10.1038/srep24354
Deng, J., Yang, M., Jiang, R., An, N., Wang, X., and Liu, B. (2017). Long non-coding RNA HOTAIR regulates the proliferation, self-renewal capacity, tumor formation and migration of the cancer stem-like cell (CSC) subpopulation enriched from breast cancer cells. PLoS ONE 12, e0170860. doi:10.1371/journal.pone.0170860
Deng, Q., Jiang, G., Wu, Y., Li, J., Liang, W., Chen, L., et al. (2018). GPER/Hippo-YAP signal is involved in Bisphenol S induced migration of triple negative breast cancer (TNBC) cells. J. Hazard Mater 355, 1–9. doi:10.1016/j.jhazmat.2018.05.013
Dennis, M. K., Burai, R., Ramesh, C., Petrie, W. K., Alcon, S. N., Nayak, T. K., et al. (2009). In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 5, 421–427. doi:10.1038/nchembio.168
Dennis, M. K., Field, A. S., Burai, R., Ramesh, C., Petrie, W. K., Bologa, C. G., et al. (2011). Identification of a GPER/GPR30 antagonist with improved estrogen receptor counter selectivity. J. Steroid Biochem. Mol. Biol. 127, 358–366. doi:10.1016/j.jsbmb.2011.07.002
Dong, W.-H., Chen, J.-C., He, Y.-L., Xu, J.-J., and Mei, Y.-A. (2013). Resveratrol inhibits K v 2.2 currents through the estrogen receptor GPR30-mediated PKC pathway. Am. J. Physiol. Cell Physiol. 305, C547–C557. doi:10.1152/ajpcell.00146.2013
Early Breast Cancer Trialists’ Collaborative Group (Ebctcg) Davies, C., Godwin, J., Gray, R., Clarke, M., Cutter, D., et al. (2011). Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 378, 771–784. doi:10.1016/S0140-6736(11)60993-8
Eichten, A., Su, J., Adler, A. P., Zhang, L., Ioffe, E., Parveen, A. A., et al. (2016). Resistance to anti-VEGF therapy mediated by autocrine IL6/STAT3 signaling and overcome by IL6 blockade. Cancer Res. 76, 2327–2339. doi:10.1158/0008-5472.CAN-15-1443
Fábián, M., Rencz, F., Krenács, T., Brodszky, V., Hársing, J., Németh, K., et al. (2017). Expression of G protein-coupled oestrogen receptor in melanoma and in pregnancy-associated melanoma. J. Eur. Acad. Dermatol Venereol. 31, 1453–1461. doi:10.1111/jdv.14304
Felcher, C. M., Bogni, E. S., and Kordon, E. C. (2022). IL-6 cytokine family: a putative target for breast cancer prevention and treatment. Int. J. Mol. Sci. 23, 1809. doi:10.3390/ijms23031809
Filardo, E., Quinn, J., Pang, Y., Graeber, C., Shaw, S., Dong, J., et al. (2007). Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 148, 3236–3245. doi:10.1210/en.2006-1605
Filardo, E. J., Graeber, C. T., Quinn, J. A., Resnick, M. B., Giri, D., DeLellis, R. A., et al. (2006). Distribution of GPR30, a seven membrane–spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 12, 6359–6366. doi:10.1158/1078-0432.CCR-06-0860
Filardo, E. J., Quinn, J. A., Bland, K. I., and Frackelton, A. R. (2000). Estrogen-induced activation of erk-1 and erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 14, 1649–1660. doi:10.1210/mend.14.10.0532
Filardo, E. J., Quinn, J. A., Frackelton, A. R., and Bland, K. I. (2002). Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling Axis. Mol. Endocrinol. 16, 70–84. doi:10.1210/mend.16.1.0758
Filardo, E. J., Quinn, J. A., and Sabo, E. (2008). Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids 73, 870–873. doi:10.1016/j.steroids.2007.12.025
Filardo, E. J., and Thomas, P. (2005). GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol. Metab. 16, 362–367. doi:10.1016/j.tem.2005.08.005
Franco, R., Boscia, F., Gigantino, V., Marra, L., Esposito, F., Ferrara, D., et al. (2011). GPR30 is overexpressed in post-puberal testicular germ cell tumors. Cancer Biol. Ther. 11, 609–613. doi:10.4161/cbt.11.6.14672
Fujiwara, S., Terai, Y., Kawaguchi, H., Takai, M., Yoo, S., Tanaka, Y., et al. (2012). GPR30 regulates the EGFR-Akt cascade and predicts lower survival in patients with ovarian cancer. J. Ovarian Res. 5, 35. doi:10.1186/1757-2215-5-35
Funakoshi, T., Yanai, A., Shinoda, K., Kawano, M. M., and Mizukami, Y. (2006). G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem. Biophys. Res. Commun. 346, 904–910. doi:10.1016/j.bbrc.2006.05.191
Furth, N., and Aylon, Y. (2017). The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death Differ. 24, 1488–1501. doi:10.1038/cdd.2017.99
Gao, F., Ma, X., Ostmann, A. B., and Das, S. K. (2011). GPR30 activation opposes estrogen-dependent uterine growth via inhibition of stromal ERK1/2 and estrogen receptor alpha (ERα) phosphorylation signals. Endocrinology 152, 1434–1447. doi:10.1210/en.2010-1368
García Fernández, A., Giménez, N., Fraile, M., González, S., Chabrera, C., Torras, M., et al. (2012). Survival and clinicopathological characteristics of breast cancer patient according to different tumour subtypes as determined by hormone receptor and Her2 immunohistochemistry. A single institution survey spanning 1998 to 2010. Breast 21, 366–373. doi:10.1016/j.breast.2012.03.004
Girgert, R., Emons, G., and Gründker, C. (2012). Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: possible application in targeted therapy. Breast Cancer Res. Treat. 134, 199–205. doi:10.1007/s10549-012-1968-x
Girgert, R., Emons, G., and Gründker, C. (2014). Inhibition of GPR30 by estriol prevents growth stimulation of triple-negative breast cancer cells by 17β-estradiol. BMC Cancer 14, 935. doi:10.1186/1471-2407-14-935
Girgert, R., Emons, G., and Gründker, C. (2017). 17β-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol. Rep. 37, 1212–1218. doi:10.3892/or.2016.5306
Girgert, R., Emons, G., and Gründker, C. (2018). Inhibition of growth hormone receptor by Somavert reduces expression of GPER and prevents growth stimulation of triple-negative breast cancer by 17β-estradiol. Oncol. Lett. 15, 9559–9566. doi:10.3892/ol.2018.8521
Golubovskaya, V. M., Ylagan, L., Miller, A., Hughes, M., Wilson, J., Wang, D., et al. (2014). High focal adhesion kinase expression in breast carcinoma is associated with lymphovascular invasion and triple-negative phenotype. BMC Cancer 14, 769. doi:10.1186/1471-2407-14-769
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Hartman, Z. C., Poage, G. M., den Hollander, P., Tsimelzon, A., Hill, J., Panupinthu, N., et al. (2013). Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 73, 3470–3480. doi:10.1158/0008-5472.CAN-12-4524-T
He, J., Tsang, J. Y., Xu, X., Li, J., Li, M., Chao, X., et al. (2020). AJCC 8th edition prognostic staging provides no better discriminatory ability in prognosis than anatomical staging in triple negative breast cancer. BMC Cancer 20, 18. doi:10.1186/s12885-019-6494-3
He, Y., Yang, K., Liu, J., Shi, D., Zhang, Z., Yang, J., et al. (2023). Tanshinone IIA inhibits triple-negative breast cancer cells MDA-MB-231 via G protein-coupled estrogen receptor- (GPER-) dependent signaling pathway. Dis. Markers 2023, 8371623. doi:10.1155/2023/8371623
He, Y.-Y., Cai, B., Yang, Y.-X., Liu, X.-L., and Wan, X.-P. (2009). Estrogenic G protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the MEK/ERK mitogen-activated protein kinase pathway. Cancer Sci. 100, 1051–1061. doi:10.1111/j.1349-7006.2009.01148.x
Herbst, R. S., Fukuoka, M., and Baselga, J. (2004). Gefitinib - a novel targeted approach to treating cancer. Nat. Rev. Cancer 4, 956–965. doi:10.1038/nrc1506
Hernández-Silva, C. D., Villegas-Pineda, J. C., and Pereira-Suárez, A. L. (2020). Expression and role of the G protein-coupled estrogen receptor (GPR30/GPER) in the development and immune response in female reproductive cancers. Front. Endocrinol. (Lausanne) 11, 544. doi:10.3389/fendo.2020.00544
Honeth, G., Bendahl, P.-O., Ringnér, M., Saal, L. H., Gruvberger-Saal, S. K., Lövgren, K., et al. (2008). The CD44+/CD24-phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 10, R53. doi:10.1186/bcr2108
Huang, C., Yang, G., Jiang, T., Huang, K., Cao, J., and Qiu, Z. (2010). Effects of IL-6 and AG490 on regulation of Stat3 signaling pathway and invasion of human pancreatic cancer cells in vitro. J. Exp. Clin. Cancer Res. 29, 51. doi:10.1186/1756-9966-29-51
Huynh, J., Etemadi, N., Hollande, F., Ernst, M., and Buchert, M. (2017). The JAK/STAT3 axis: a comprehensive drug target for solid malignancies. Semin. Cancer Biol. 45, 13–22. doi:10.1016/j.semcancer.2017.06.001
Ignatov, A., Ignatov, T., Weißenborn, C., Eggemann, H., Bischoff, J., Semczuk, A., et al. (2011). G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res. Treat. 128, 457–466. doi:10.1007/s10549-011-1584-1
Jean, C., Chen, X. L., Nam, J.-O., Tancioni, I., Uryu, S., Lawson, C., et al. (2014). Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J. Cell Biol. 204, 247–263. doi:10.1083/jcb.201307067
Jeong, S., Park, S., Park, B.-W., Park, Y., Kwon, O.-J., and Kim, H.-S. (2014). Human leukocyte antigen-G (HLA-G) polymorphism and expression in breast cancer patients. PLoS ONE 9, e98284. doi:10.1371/journal.pone.0098284
Jiang, Y.-Z., Ma, D., Suo, C., Shi, J., Xue, M., Hu, X., et al. (2019). Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell 35, 428–440. doi:10.1016/j.ccell.2019.02.001
Joo, A., Aburatani, H., Morii, E., Iba, H., and Yoshimura, A. (2004). STAT3 and MITF cooperatively induce cellular transformation through upregulation of c-fos expression. Oncogene 23, 726–734. doi:10.1038/sj.onc.1207174
Kajta, M., Litwa, E., Rzemieniec, J., Wnuk, A., Lason, W., Zelek-Molik, A., et al. (2014). Isomer-nonspecific action of dichlorodiphenyltrichloroethane on aryl hydrocarbon receptor and G-protein-coupled receptor 30 intracellular signaling in apoptotic neuronal cells. Mol. Cell Endocrinol. 392, 90–105. doi:10.1016/j.mce.2014.05.008
Kajta, M., Rzemieniec, J., Litwa, E., Lason, W., Lenartowicz, M., Krzeptowski, W., et al. (2013). The key involvement of estrogen receptor β and G-protein-coupled receptor 30 in the neuroprotective action of daidzein. Neuroscience 238, 345–360. doi:10.1016/j.neuroscience.2013.02.005
Kato, S., Sato, T., Watanabe, T., Takemasa, S., Masuhiro, Y., Ohtake, F., et al. (2005). Function of nuclear sex hormone receptors in gene regulation. Cancer Chemother. Pharmacol. 56, 4–9. doi:10.1007/s00280-005-0102-8
Kim, K. M., and Jung, J. (2020). Upregulation of G Protein-Coupled estrogen receptor by chrysin-nanoparticles inhibits tumor proliferation and metastasis in triple negative breast cancer xenograft model. Front. Endocrinol. (Lausanne) 11, 560605. doi:10.3389/fendo.2020.560605
Kim, K. M., Lim, H. K., Shim, S. H., and Jung, J. (2017). Improved chemotherapeutic efficacy of injectable chrysin encapsulated by copolymer nanoparticles. Int. J. Nanomedicine 12, 1917–1925. doi:10.2147/IJN.S132043
Kleuser, B., Malek, D., Gust, R., Pertz, H. H., and Potteck, H. (2008). 17-Beta-estradiol inhibits transforming growth factor-beta signaling and function in breast cancer cells via activation of extracellular signal-regulated kinase through the G protein-coupled receptor 30. Mol. Pharmacol. 74, 1533–1543. doi:10.1124/mol.108.046854
Kvingedal, A. M., and Smeland, E. B. (1997). A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. 407, 59–62. doi:10.1016/S0014-5793(97)00278-0
Kyriakopoulou, K., Kefali, E., Piperigkou, Z., Riethmüller, C., Greve, B., Franchi, M., et al. (2022). EGFR is a pivotal player of the E2/ERβ – mediated functional properties, aggressiveness, and stemness in triple-negative breast cancer cells. FEBS J. 289, 1552–1574. doi:10.1111/febs.16240
Lappano, R., and Maggiolini, M. (2018). GPER is involved in the functional liaison between breast tumor cells and cancer-associated fibroblasts (CAFs). J. Steroid Biochem. Mol. Biol. 176, 49–56. doi:10.1016/j.jsbmb.2017.02.019
Lappano, R., Rosano, C., De Marco, P., De Francesco, E. M., Pezzi, V., and Maggiolini, M. (2010). Estriol acts as a GPR30 antagonist in estrogen receptor-negative breast cancer cells. Mol. Cell Endocrinol. 320, 162–170. doi:10.1016/j.mce.2010.02.006
Lappano, R., Rosano, C., Santolla, M. F., Pupo, M., De Francesco, E. M., De Marco, P., et al. (2012). Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr. Cancer Drug Targets 12, 531–542. doi:10.2174/156800912800673284
Lappano, R., Talia, M., Cirillo, F., Rigiracciolo, D. C., Scordamaglia, D., Guzzi, R., et al. (2020). The IL1β-IL1R signaling is involved in the stimulatory effects triggered by hypoxia in breast cancer cells and cancer-associated fibroblasts (CAFs). J. Exp. Clin. Cancer Res. 39, 153. doi:10.1186/s13046-020-01667-y
Lark, A. L., Livasy, C. A., Dressler, L., Moore, D. T., Millikan, R. C., Geradts, J., et al. (2005). High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod. Pathol. 18, 1289–1294. doi:10.1038/modpathol.3800424
Lee, H., Herrmann, A., Deng, J.-H., Kujawski, M., Niu, G., Li, Z., et al. (2009). Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 15, 283–293. doi:10.1016/j.ccr.2009.02.015
Lehmann, B. D., Bauer, J. A., Chen, X., Sanders, M. E., Chakravarthy, A. B., Shyr, Y., et al. (2011). Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 121, 2750–2767. doi:10.1172/JCI45014
Lei, B., Tang, Q., Sun, S., Zhang, X., Huang, Y., and Xu, L. (2021). Insight into the mechanism of tetrachlorobisphenol A (TCBPA)-induced proliferation of breast cancer cells by GPER-mediated signaling pathways. Environ. Pollut. 275, 116636. doi:10.1016/j.envpol.2021.116636
Lei, S., Fan, P., Wang, M., Zhang, C., Jiang, Y., Huang, S., et al. (2020). Elevated estrogen receptor β expression in triple negative breast cancer cells is associated with sensitivity to doxorubicin by inhibiting the PI3K/AKT/mTOR signaling pathway. Exp. Ther. Med. 20, 1630–1636. doi:10.3892/etm.2020.8809
Levoye, A., Dam, J., Ayoub, M. A., Guillaume, J., and Jockers, R. (2006). Do orphan G-protein-coupled receptors have ligand-independent functions? new insights from receptor heterodimers. EMBO Rep. 7, 1094–1098. doi:10.1038/sj.embor.7400838
Li, H., Ma, F., Wang, H., Lin, C., Fan, Y., Zhang, X., et al. (2013). Stem cell marker aldehyde dehydrogenase 1 (ALDH1)-expressing cells are enriched in triple-negative breast cancer. Int. J. Biol. Markers 28, e357–e364. doi:10.5301/jbm.5000048
Li, L., Zeng, Q., Bhutkar, A., Galván, J. A., Karamitopoulou, E., Noordermeer, D., et al. (2018). GKAP acts as a genetic modulator of NMDAR signaling to govern invasive tumor growth. Cancer Cell 33, 736–751. doi:10.1016/j.ccell.2018.02.011
Li, X., Yang, J., Peng, L., Sahin, A. A., Huo, L., Ward, K. C., et al. (2017). Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res. Treat. 161, 279–287. doi:10.1007/s10549-016-4059-6
Liang, S., Chen, Z., Jiang, G., Zhou, Y., Liu, Q., Su, Q., et al. (2017). Activation of GPER suppresses migration and angiogenesis of triple negative breast cancer via inhibition of NF-κB/IL-6 signals. Cancer Lett. 386, 12–23. doi:10.1016/j.canlet.2016.11.003
Lindsey, S. H., Yamaleyeva, L. M., Brosnihan, K. B., Gallagher, P. E., and Chappell, M. C. (2011). Estrogen receptor GPR30 reduces oxidative stress and proteinuria in the salt-sensitive female mRen2.Lewis rat. Hypertension 58, 665–671. doi:10.1161/HYPERTENSIONAHA.111.175174
Lu, R., Zhang, J., Zhang, W., Huang, Y., Wang, N., Zhang, Q., et al. (2018). Circulating HOTAIR expression predicts the clinical response to neoadjuvant chemotherapy in patients with breast cancer. Cancer Biomark. 22, 249–256. doi:10.3233/CBM-170874
Lujambio, A., Calin, G. A., Villanueva, A., Ropero, S., Sánchez-Céspedes, M., Blanco, D., et al. (2008). A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. U. S. A. 105, 13556–13561. doi:10.1073/pnas.0803055105
Luo, H., Luo, P., Yang, G., Peng, Q., Liu, M., and Tu, G. (2011). G-Protein coupled estrogen receptor 1 expression in primary breast cancers and its correlation with clinicopathological variables. J. Breast Cancer 14, 185–190. doi:10.4048/jbc.2011.14.3.185
Luo, H., Tu, G., Liu, Z., and Liu, M. (2015). Cancer-associated fibroblasts: a multifaceted driver of breast cancer progression. Cancer Lett. 361, 155–163. doi:10.1016/j.canlet.2015.02.018
Luo, H., Yang, G., Yu, T., Luo, S., Wu, C., Sun, Y., et al. (2014). GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr. Relat. Cancer 21, 355–369. doi:10.1530/ERC-13-0237
Luo, J., and Liu, D. (2020). Does GPER really function as a G protein-coupled estrogen receptor in vivo? Front. Endocrinol. (Lausanne) 11, 148. doi:10.3389/fendo.2020.00148
Luo, M., and Guan, J.-L. (2010). Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 289, 127–139. doi:10.1016/j.canlet.2009.07.005
Lv, X., He, C., Huang, C., Hua, G., Wang, Z., Remmenga, S. W., et al. (2017). G-1 inhibits breast cancer cell growth via targeting colchicine-binding site of tubulin to interfere with microtubule assembly. Mol. Cancer Ther. 16, 1080–1091. doi:10.1158/1535-7163.MCT-16-0626
Ma, F., Li, H., Wang, H., Shi, X., Fan, Y., Ding, X., et al. (2014). Enriched CD44(+)/CD24(-) population drives the aggressive phenotypes presented in triple-negative breast cancer (TNBC). Cancer Lett. 353, 153–159. doi:10.1016/j.canlet.2014.06.022
Maggiolini, M., Vivacqua, A., Fasanella, G., Recchia, A. G., Sisci, D., Pezzi, V., et al. (2004). The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 279, 27008–27016. doi:10.1074/jbc.M403588200
Maiti, K., Paul, J. W., Read, M., Chan, E. C., Riley, S. C., Nahar, P., et al. (2011). G-1-Activated membrane estrogen receptors mediate increased contractility of the human myometrium. Endocrinology 152, 2448–2455. doi:10.1210/en.2010-0979
Marjon, N. A., Hu, C., Hathaway, H. J., and Prossnitz, E. R. (2014). G protein–coupled estrogen receptor regulates mammary tumorigenesis and metastasis. Mol. Cancer Res. 12, 1644–1654. doi:10.1158/1541-7786.MCR-14-0128-T
Matsumoto, T., Sakari, M., Okada, M., Yokoyama, A., Takahashi, S., Kouzmenko, A., et al. (2013). The androgen receptor in health and disease. Annu. Rev. Physiol. 75, 201–224. doi:10.1146/annurev-physiol-030212-183656
Meijer, D., van Agthoven, T., Bosma, P. T., Nooter, K., and Dorssers, L. C. J. (2006). Functional screen for genes responsible for tamoxifen resistance in human breast cancer cells. Mol. Cancer Res. 4, 379–386. doi:10.1158/1541-7786.MCR-05-0156
Milde-Langosch, K., Röder, H., Andritzky, B., Aslan, B., Hemminger, G., Brinkmann, A., et al. (2004). The role of the AP-1 transcription factors c-fos, FosB, fra-1 and fra-2 in the invasion process of mammary carcinomas. Breast Cancer Res. Treat. 86, 139–152. doi:10.1023/B:BREA.0000032982.49024.71
Molina, L., Bustamante, F., Ortloff, A., Ramos, I., Ehrenfeld, P., and Figueroa, C. D. (2020). Continuous exposure of breast cancer cells to tamoxifen upregulates GPER-1 and increases cell proliferation. Front. Endocrinol. (Lausanne) 11, 563165. doi:10.3389/fendo.2020.563165
Morris, G. J., Naidu, S., Topham, A. K., Guiles, F., Xu, Y., McCue, P., et al. (2007). Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and end results database. Cancer 110, 876–884. doi:10.1002/cncr.22836
Nieto Gutierrez, A., and McDonald, P. H. (2018). GPCRs: emerging anti-cancer drug targets. Cell Signal 41, 65–74. doi:10.1016/j.cellsig.2017.09.005
O’Conor, C. J., Chen, T., González, I., Cao, D., and Peng, Y. (2018). Cancer stem cells in triple-negative breast cancer: a potential target and prognostic marker. Biomark. Med. 12, 813–820. doi:10.2217/bmm-2017-0398
Okamoto, M., and Mizukami, Y. (2016). GPER negatively regulates TNFα-induced IL-6 production in human breast cancer cells via NF-κB pathway. Endocr. J. 63, 485–493. doi:10.1507/endocrj.EJ15-0571
Onken, M. D., Makepeace, C. M., Kaltenbronn, K. M., Kanai, S. M., Todd, T. D., Wang, S., et al. (2020). Abstract PR09: pharmacologic targeting of Gq reveals new pathways in uveal melanoma. Cancer Res. 80, PR09. doi:10.1158/1538-7445.MEL2019-PR09
Osborne, C. K., Wakeling, A., and Nicholson, R. I. (2004). Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 90, S2–S6. doi:10.1038/sj.bjc.6601629
Pandey, D. P., Lappano, R., Albanito, L., Madeo, A., Maggiolini, M., and Picard, D. (2009). Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 28, 523–532. doi:10.1038/emboj.2008.304
Park, B. K., Zhang, H., Zeng, Q., Dai, J., Keller, E. T., Giordano, T., et al. (2007). NF-kappaB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat. Med. 13, 62–69. doi:10.1038/nm1519
Pietras, R. J., and Szego, C. M. (1975). Endometrial cell calcium and oestrogen action. Nature 253, 357–359. doi:10.1038/253357a0
Pires, B. R. B., Mencalha, A. L., Ferreira, G. M., De Souza, W. F., Morgado-Díaz, J. A., Maia, A. M., et al. (2017). NF-kappaB is involved in the regulation of EMT genes in breast cancer cells. PLoS ONE 12, e0169622. doi:10.1371/journal.pone.0169622
Pisano, A., Santolla, M. F., De Francesco, E. M., De Marco, P., Rigiracciolo, D. C., Perri, M. G., et al. (2017). GPER, IGF-IR, and EGFR transduction signaling are involved in stimulatory effects of zinc in breast cancer cells and cancer-associated fibroblasts. Mol. Carcinog. 56, 580–593. doi:10.1002/mc.22518
Ponuwei, G. A. (2016). A glimpse of the ERM proteins. J. Biomed. Sci. 23, 35. doi:10.1186/s12929-016-0246-3
Prossnitz, E. R., Arterburn, J. B., Smith, H. O., Oprea, T. I., Sklar, L. A., and Hathaway, H. J. (2008). Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol. 70, 165–190. doi:10.1146/annurev.physiol.70.113006.100518
Puglisi, R., Mattia, G., Carè, A., Marano, G., Malorni, W., and Matarrese, P. (2019). Non-genomic effects of estrogen on cell homeostasis and remodeling with special focus on cardiac ischemia/reperfusion injury. Front. Endocrinol. (Lausanne) 10, 733. doi:10.3389/fendo.2019.00733
Pupo, M., Bodmer, A., Berto, M., Maggiolini, M., Dietrich, P.-Y., and Picard, D. (2017). A genetic polymorphism repurposes the G-protein coupled and membrane-associated estrogen receptor GPER to a transcription factor-like molecule promoting paracrine signaling between stroma and breast carcinoma cells. Oncotarget 8, 46728–46744. doi:10.18632/oncotarget.18156
Pupo, M., Pisano, A., Lappano, R., Santolla, M. F., De Francesco, E. M., Abonante, S., et al. (2012). Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Environ. Health Perspect. 120, 1177–1182. doi:10.1289/ehp.1104526
Qi, M., Liu, X., Zhou, Y., Wang, H., Zhao, Y., Ren, J., et al. (2021). Berberine inhibits MDA-MB-231 cells as an agonist of G protein-coupled estrogen receptor 1. Int. J. Mol. Sci. 22, 11466. doi:10.3390/ijms222111466
Quinn, J. A., Graeber, C. T., Frackelton, A. R., Kim, M., Schwarzbauer, J. E., and Filardo, E. J. (2009). Coordinate regulation of estrogen-mediated fibronectin matrix assembly and epidermal growth factor receptor transactivation by the G protein-coupled receptor, GPR30. Mol. Endocrinol. 23, 1052–1064. doi:10.1210/me.2008-0262
Rettew, J. A., McCall, S. H., and Marriott, I. (2010). GPR30/GPER-1 mediates rapid decreases in TLR4 expression on murine macrophages. Mol. Cell Endocrinol. 328, 87–92. doi:10.1016/j.mce.2010.07.017
Revankar, C. M., Cimino, D. F., Sklar, L. A., Arterburn, J. B., and Prossnitz, E. R. (2005). A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307, 1625–1630. doi:10.1126/science.1106943
Rigiracciolo, D. C., Cirillo, F., Talia, M., Muglia, L., Gutkind, J. S., Maggiolini, M., et al. (2021). Focal adhesion kinase fine tunes multifaced signals toward breast cancer progression. Cancers 13, 645. doi:10.3390/cancers13040645
Rigiracciolo, D. C., Santolla, M. F., Lappano, R., Vivacqua, A., Cirillo, F., Galli, G. R., et al. (2019). Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells. J. Exp. Clin. Cancer Res. 38, 58. doi:10.1186/s13046-019-1056-8
Rodenas, M. C., Tamassia, N., Cabas, I., Calzetti, F., Meseguer, J., Cassatella, M. A., et al. (2017). G protein-coupled estrogen receptor 1 regulates human neutrophil functions. Biomed. Hub. 2, 1–13. doi:10.1159/000454981
Roskoski, R. (2007). Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit. Rev. Oncol. Hematol. 62, 179–213. doi:10.1016/j.critrevonc.2007.01.006
Rzeski, W., Ikonomidou, C., and Turski, L. (2002). Glutamate antagonists limit tumor growth. Biochem. Pharmacol. 64, 1195–1200. doi:10.1016/S0006-2952(02)01218-2
Sakamoto, N., Naito, Y., Oue, N., Sentani, K., Uraoka, N., Oo, H. Z., et al. (2014). Micro RNA-148a is downregulated in gastric cancer, targets MMP 7, and indicates tumor invasiveness and poor prognosis. Cancer Sci. 105, 236–243. doi:10.1111/cas.12330
Salahuddin, A., Ghanem, H., Omran, G. A., and Helmy, M. W. (2022). Epigenetic restoration and activation of ERβ: an inspiring approach for treatment of triple-negative breast cancer. Med. Oncol. 39, 150. doi:10.1007/s12032-022-01765-1
Samartzis, E. P., Noske, A., Meisel, A., Varga, Z., Fink, D., and Imesch, P. (2014). The G protein-coupled estrogen receptor (GPER) is expressed in two different subcellular localizations reflecting distinct tumor properties in breast cancer. PLoS ONE 9, e83296. doi:10.1371/journal.pone.0083296
Santolla, M. F., Vivacqua, A., Lappano, R., Rigiracciolo, D. C., Cirillo, F., Galli, G. R., et al. (2019). GPER mediates a feedforward FGF2/FGFR1 paracrine activation coupling CAFs to cancer cells toward breast tumor progression. Cells 8, 223. doi:10.3390/cells8030223
Scaling, A. L., Prossnitz, E. R., and Hathaway, H. J. (2014). GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm. Cancer 5, 146–160. doi:10.1007/s12672-014-0174-1
Schmitz, V., Bauerschmitz, G., Gallwas, J., and Gründker, C. (2022). Suppression of G Protein-coupled estrogen receptor 1 (GPER1) enhances the anti-invasive efficacy of selective ERβ agonists. Anticancer Res. 42, 5187–5194. doi:10.21873/anticanres.16025
Segovia-Mendoza, M., Romero-Garcia, S., Lemini, C., and Prado-Garcia, H. (2021). Determining factors in the therapeutic success of checkpoint immunotherapies against PD-L1 in breast cancer: a focus on epithelial-mesenchymal transition activation. J. Immunol. Res. 2021, 6668573. doi:10.1155/2021/6668573
Sharma, P. (2018). Update on the treatment of early-stage triple-negative breast cancer. Curr Treat Options Oncol 19, 22. doi:10.1007/s11864-018-0539-8
Shen, J., Cao, B., Wang, Y., Ma, C., Zeng, Z., Liu, L., et al. (2018). Hippo component YAP promotes focal adhesion and tumour aggressiveness via transcriptionally activating THBS1/FAK signalling in breast cancer. J. Exp. Clin. Cancer Res. 37, 175. doi:10.1186/s13046-018-0850-z
Shen, Y., Yang, F., Zhang, W., Song, W., Liu, Y., and Guan, X. (2017). The androgen receptor promotes cellular proliferation by suppression of G-protein coupled estrogen receptor signaling in triple-negative breast cancer. Cell Physiol. Biochem. 43, 2047–2061. doi:10.1159/000484187
Shoushtari, A. N., Chaney, M. F., Cohen, J. V., Garyantes, T., Lin, J. J., Ishizuka, J. J., et al. (2023). The effect of LNS8801 alone and in combination with pembrolizumab in patients with metastatic uveal melanoma. J. Clin. Oncol. 41, 9543. doi:10.1200/JCO.2023.41.16_suppl.9543
Sjöström, M., Hartman, L., Grabau, D., Fornander, T., Malmström, P., Nordenskjöld, B., et al. (2014). Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res. Treat. 145, 61–71. doi:10.1007/s10549-014-2936-4
Steiman, J., Peralta, E. A., Louis, S., and Kamel, O. (2013). Biology of the estrogen receptor, GPR30, in triple negative breast cancer. Am. J. Surg. 206, 698–703. doi:10.1016/j.amjsurg.2013.07.014
Sun, Z., Wu, X. Y., and Wu, C. L. (2018). The association between LncRNA HOTAIR and cancer lymph node metastasis and distant metastasis: a meta-analysis. Neoplasma 65, 178–184. doi:10.4149/neo_2018_170114N34
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 71, 209–249. doi:10.3322/caac.21660
Szego, C. M., and Davis, J. S. (1967). Adenosine 3’,5’-monophosphate in rat uterus: acute elevation by estrogen. Proc. Natl. Acad. Sci. U. S. A. 58, 1711–1718. doi:10.1073/pnas.58.4.1711
Taliaferro-Smith, L., Oberlick, E., Liu, T., McGlothen, T., Alcaide, T., Tobin, R., et al. (2015). FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget 6, 4757–4772. doi:10.18632/oncotarget.3023
Tao, S., He, H., and Chen, Q. (2015). Estradiol induces HOTAIR levels via GPER-mediated miR-148a inhibition in breast cancer. J. Transl. Med. 13, 131. doi:10.1186/s12967-015-0489-x
Tao, S., He, H., Chen, Q., and Yue, W. (2014). GPER mediated estradiol reduces miR-148a to promote HLA-G expression in breast cancer. Biochem. Biophys. Res. Commun. 451, 74–78. doi:10.1016/j.bbrc.2014.07.073
Thomas, P., and Dong, J. (2006). Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 102, 175–179. doi:10.1016/j.jsbmb.2006.09.017
Thomas, P., Pang, Y., Filardo, E. J., and Dong, J. (2005). Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146, 624–632. doi:10.1210/en.2004-1064
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. doi:10.1126/science.1260419
Valastyan, S., and Weinberg, R. A. (2011). Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292. doi:10.1016/j.cell.2011.09.024
van Agthoven, T., Sieuwerts, A. M., Meijer-van Gelder, M. E., Look, M. P., Smid, M., Veldscholte, J., et al. (2009). Relevance of breast cancer antiestrogen resistance genes in human breast cancer progression and tamoxifen resistance. J. Clin. Oncol. 27, 542–549. doi:10.1200/JCO.2008.17.1462
Vandenberg, L. N., Maffini, M. V., Schaeberle, C. M., Ucci, A. A., Sonnenschein, C., Rubin, B. S., et al. (2008). Perinatal exposure to the xenoestrogen bisphenol-A induces mammary intraductal hyperplasias in adult CD-1 mice. Reprod. Toxicol. 26, 210–219. doi:10.1016/j.reprotox.2008.09.015
Vivacqua, A., Bonofiglio, D., Albanito, L., Madeo, A., Rago, V., Carpino, A., et al. (2006). 17beta-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the g protein-coupled receptor GPR30. Mol. Pharmacol. 70, 1414–1423. doi:10.1124/mol.106.026344
Volovat, S. R., Volovat, C., Hordila, I., Hordila, D.-A., Mirestean, C. C., Miron, O. T., et al. (2020). MiRNA and LncRNA as potential biomarkers in triple-negative breast cancer: a review. Front. Oncol. 10, 526850. doi:10.3389/fonc.2020.526850
Wang, C., Dehghani, B., Li, Y., Kaler, L. J., Proctor, T., Vandenbark, A. A., et al. (2009). Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J. Immunol. 182, 3294–3303. doi:10.4049/jimmunol.0803205
Wang, C., Li, J., Ye, S., Zhang, Y., Li, P., Wang, L., et al. (2018). Oestrogen inhibits VEGF expression and angiogenesis in triple-negative breast cancer by activating GPER-1. J. Cancer 9, 3802–3811. doi:10.7150/jca.29233
Wang, C., Lv, X., Jiang, C., and Davis, J. S. (2011). The putative G-protein coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian and breast cancer cells in a GPER-independent manner. Cell 144, 646–674.
Wang, J., Jenkins, S., and Lamartiniere, C. A. (2014). Cell proliferation and apoptosis in rat mammary glands following combinational exposure to bisphenol A and genistein. BMC Cancer 14, 379. doi:10.1186/1471-2407-14-379
Wang, K., Zhu, X., Zhang, K., Yin, Y., Chen, Y., and Zhang, T. (2018). Interleukin-6 contributes to chemoresistance in MDA-MB-231 cells via targeting HIF-1α. J. Biochem. Mol. Toxicol. 32, e22039. doi:10.1002/jbt.22039
Wang, Y., Peng, Z., Meng, R., Tao, T., Wang, Q., Zhao, C., et al. (2017). NHERF1 inhibits proliferation of triple-negative breast cancer cells by suppressing GPER signaling. Oncol. Rep. 38, 221–228. doi:10.3892/or.2017.5649
Watters, J. J., Campbell, J. S., Cunningham, M. J., Krebs, E. G., and Dorsa, D. M. (1997). Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology 138, 4030–4033. doi:10.1210/endo.138.9.5489
Wei, W., Chen, Z.-J., Zhang, K.-S., Yang, X.-L., Wu, Y.-M., Chen, X.-H., et al. (2014). The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Death Dis. 5, e1428. doi:10.1038/cddis.2014.398
Weigel, N. L., and Zhang, Y. (1998). Ligand-independent activation of steroid hormone receptors. J. Mol. Med. Berl. 76, 469–479. doi:10.1007/s001090050241
Weissenborn, C., Ignatov, T., Nass, N., Kalinski, T., Dan Costa, S., Zenclussen, A. C., et al. (2017). GPER promoter methylation controls GPER expression in breast cancer patients. Cancer Inves 35, 100–107. doi:10.1080/07357907.2016.1271886
Weißenborn, C., Ignatov, T., Ochel, H.-J., Costa, S. D., Zenclussen, A. C., Ignatova, Z., et al. (2014). GPER functions as a tumor suppressor in triple-negative breast cancer cells. J. Cancer Res. Clin. Oncol. 140, 713–723. doi:10.1007/s00432-014-1620-8
Wolfson, B., Padget, M. R., Schlom, J., and Hodge, J. W. (2021). Exploiting off-target effects of estrogen deprivation to sensitize estrogen receptor negative breast cancer to immune killing. J. Immunother. Cancer 9, e002258. doi:10.1136/jitc-2020-002258
Xu, Q., Briggs, J., Park, S., Niu, G., Kortylewski, M., Zhang, S., et al. (2005). Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 24, 5552–5560. doi:10.1038/sj.onc.1208719
Xu, T., Ma, D., Chen, S., Tang, R., Yang, J., Meng, C., et al. (2022). High GPER expression in triple-negative breast cancer is linked to pro-metastatic pathways and predicts poor patient outcomes. NPJ Breast Cancer 8, 100. doi:10.1038/s41523-022-00472-4
Yang, B., Huang, J., Xiang, T., Yin, X., Luo, X., Huang, J., et al. (2014). Chrysin inhibits metastatic potential of human triple-negative breast cancer cells by modulating matrix metalloproteinase-10, epithelial to mesenchymal transition, and PI3K/Akt signaling pathway: chrysin inhibits metastatic potential of breast cancer cells. J. Appl. Toxicol. 34, 105–112. doi:10.1002/jat.2941
Yang, K., and Yao, Y. (2019). Mechanism of GPER promoting proliferation, migration and invasion of triple-negative breast cancer cells through CAF. Am. J. Transl. Res. 11, 5858–5868.
Ye, S., Xu, Y., Li, J., Zheng, S., Sun, P., and Wang, T. (2019). Prognostic role of GPER/Ezrin in triple-negative breast cancer is associated with menopausal status. Endocr. Connect. 8, 661–671. doi:10.1530/EC-19-0164
Ye, S., Xu, Y., Wang, L., Zhou, K., He, J., Lu, J., et al. (2020). Estrogen-related receptor α (ERRα) and G protein-coupled estrogen receptor (GPER) synergistically indicate poor prognosis in patients with triple-negative breast cancer. Onco Targets Ther. 13, 8887–8899. doi:10.2147/OTT.S265372
Yin, H., Zhu, Q., Liu, M., Tu, G., Li, Q., Yuan, J., et al. (2017). GPER promotes tamoxifen-resistance in ER+ breast cancer cells by reduced Bim proteins through MAPK/Erk-TRIM2 signaling axis. Int. J. Oncol. 51, 1191–1198. doi:10.3892/ijo.2017.4117
Yin, J., Tu, G., Peng, M., Zeng, H., Wan, X., Qiao, Y., et al. (2020). GPER-regulated lncRNA-Glu promotes glutamate secretion to enhance cellular invasion and metastasis in triple-negative breast cancer. FASEB J. 34, 4557–4572. doi:10.1096/fj.201901384RR
Yin, L., Duan, J.-J., Bian, X.-W., and Yu, S. (2020). Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 22, 61. doi:10.1186/s13058-020-01296-5
Yu, H., Pardoll, D., and Jove, R. (2009). STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809. doi:10.1038/nrc2734
Yu, T., Cheng, H., Ding, Z., Wang, Z., Zhou, L., Zhao, P., et al. (2020). GPER mediates decreased chemosensitivity via regulation of ABCG2 expression and localization in tamoxifen-resistant breast cancer cells. Mol. Cell Endocrino 506, 110762. doi:10.1016/j.mce.2020.110762
Yu, T., Liu, M., Luo, H., Wu, C., Tang, X., Tang, S., et al. (2014). GPER mediates enhanced cell viability and motility via non-genomic signaling induced by 17β-estradiol in triple-negative breast cancer cells. J. Steroid Biochem. Mol. Biol. 143, 392–403. doi:10.1016/j.jsbmb.2014.05.003
Zeng, Q., Michael, I. P., Zhang, P., Saghafinia, S., Knott, G., Jiao, W., et al. (2019). Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–531. doi:10.1038/s41586-019-1576-6
Zhang, D., Wang, J., Chen, H., and Yan, S. (2022). Cytoplasmic G protein-coupled estrogen receptor 1 as a prognostic indicator of breast cancer: a meta-analysis. Technol. Cancer Res. Treat. 21, 15330338221131664. doi:10.1177/15330338221131664
Zhang, X.-L., Liu, N., Weng, S.-F., and Wang, H.-S. (2016). Bisphenol A increases the migration and invasion of triple-negative breast cancer cells via oestrogen-related receptor gamma. Basic Clin. Pharmacol. Toxicol. 119, 389–395. doi:10.1111/bcpt.12591
Zhang, Y., Liu, S., Zhou, S., Yu, D., Gu, J., Qin, Q., et al. (2021). Focal adhesion kinase: insight into its roles and therapeutic potential in oesophageal cancer. Cancer Lett. 496, 93–103. doi:10.1016/j.canlet.2020.10.005
Zhao, B., Ye, X., Yu, J., Li, L., Li, W., Li, S., et al. (2008). TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971. doi:10.1101/gad.1664408
Zhao, Y., Yang, W., Huang, Y., Cui, R., Li, X., and Li, B. (2018). Evolving roles for targeting CTLA-4 in cancer immunotherapy. Cell Physiol. Biochem. 47, 721–734. doi:10.1159/000490025
Zhou, K., Sun, P., Zhang, Y., You, X., Li, P., and Wang, T. (2016). Estrogen stimulated migration and invasion of estrogen receptor-negative breast cancer cells involves an ezrin-dependent crosstalk between G protein-coupled receptor 30 and estrogen receptor beta signaling. Steroids 111, 113–120. doi:10.1016/j.steroids.2016.01.021
Keywords: G protein-coupled estrogen receptor, triple-negative breast cancer, tumor development, mechanism, prognostic
Citation: Zhang D, Chen H, Wang J, Ji J, Imam M, Zhang Z and Yan S (2024) Current progress and prospects for G protein-coupled estrogen receptor in triple-negative breast cancer. Front. Cell Dev. Biol. 12:1338448. doi: 10.3389/fcell.2024.1338448
Received: 23 November 2023; Accepted: 08 February 2024;
Published: 27 February 2024.
Edited by:
Weipeng Zhao, Tianjin Medical University Cancer Institute and Hospital, ChinaReviewed by:
Jia Li, University of North Carolina at Charlotte, United StatesCopyright © 2024 Zhang, Chen, Wang, Ji, Imam, Zhang and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shunchao Yan, eWFuc2NAY211LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.