 Xue Du
Xue Du Anna G. Butler
Anna G. Butler Holly Y. Chen
Holly Y. Chen- Department of Cell, Developmental and Integrative Biology, Heersink School of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
The retina is part of the central nervous system specialized for vision. Inherited retinal diseases (IRD) are a group of clinically and genetically heterogenous disorders that lead to progressive vision impairment or blindness. Although each disorder is rare, IRD accumulatively cause blindness in up to 5.5 million individuals worldwide. Currently, the pathophysiological mechanisms of IRD are not fully understood and there are limited treatment options available. Most IRD are caused by degeneration of light-sensitive photoreceptors. Genetic mutations that abrogate the structure and/or function of photoreceptors lead to visual impairment followed by blindness caused by loss of photoreceptors. In healthy retina, photoreceptors structurally and functionally interact with retinal pigment epithelium (RPE) and Müller glia (MG) to maintain retinal homeostasis. Multiple IRD with photoreceptor degeneration as a major phenotype are caused by mutations of RPE- and/or MG-associated genes. Recent studies also reveal compromised MG and RPE caused by mutations in ubiquitously expressed ciliary genes. Therefore, photoreceptor degeneration could be a direct consequence of gene mutations and/or could be secondary to the dysfunction of their interaction partners in the retina. This review summarizes the mechanisms of photoreceptor-RPE/MG interaction in supporting retinal functions and discusses how the disruption of these processes could lead to photoreceptor degeneration, with an aim to provide a unique perspective of IRD pathogenesis and treatment paradigm. We will first describe the biology of retina and IRD and then discuss the interaction between photoreceptors and MG/RPE as well as their implications in disease pathogenesis. Finally, we will summarize the recent advances in IRD therapeutics targeting MG and/or RPE.
1 Introduction
Among the five senses of humans (vision, taste, touch, smell, hearing), vision is considered as the most important one for most people (Hutmacher, 2019). Approximately a hundred years ago, research on perception and perceptual memory was mainly focused on the vision, through which we perceive most of information (Ripley and Politzer, 2010; Hutmacher, 2019). Vision starts with light entering the eye, being filtered through the cornea, and focused onto the retina by the lens. The retina is a multi-layered structure specialized for vision. The light-sensitive photoreceptors in the retina capture photons and convert them into electrical impulses, which are integrated and processed by interneurons, and transmitted to the lateral geniculate nucleus, pretectal nuclei, and superior colliculus in the brain by retinal ganglion cells (Smith and Czyz, 2023).
Up to 75% of people consider losing the sense of vision the scariest compared to the other four senses (Hutmacher, 2019). Vision loss stems from the inability of the retina to detect the light and/or transmit visual signals to the brain (Wright et al., 2010; Veleri et al., 2015; Chen et al., 2021b). Amongst the plethora of blinding disorders, inherited retinal diseases (IRD) have particular significance. IRD are a group of clinically and genetically heterogeneous disorders characterized by progressive vision impairment or loss. Although individually rare, IRD have an accumulative prevalence of up to 5.5 million cases globally (Ben-Yosef, 2022). As a major cause of childhood blindness (John et al., 2022), IRD frequently destine children to a lifetime of severe vision impairment and/or blindness and cause a considerable burden on family and societies (Leroy et al., 2021). In 2019, the total costs spent on IRD were estimated between $13.4 and $31.8 billion in the United States and between $1.6 and $6.7 billion in Canada (Gong et al., 2021). Currently, there is only one FDA-approved gene therapy drug voretigene neparvovec (Luxturna®) to treat IRD caused by RPE65 mutations (Pierce and Bennett, 2015), and the long-term effect seems variable (Gardiner et al., 2020; Wang et al., 2020; Leroy et al., 2022). In addition, with over 280 disease-causing genes of IRD (RetNet, https://sph.uth.edu/retnet/), the development of individualized gene therapy protocols would not be an optimal option particularly for an individually rare disease (Tambuyzer et al., 2020). Therefore, gene-agnostic paradigms are being developed as a more desirable therapeutic approach (Scholl et al., 2016) and a comprehensive understanding of the cellular and molecular mechanisms of IRD is a premise for designing effective and long-lasting therapeutics.
A majority of IRD are due to dysfunction and/or degeneration of the light-sensitive photoreceptors (Wright et al., 2010; Verbakel et al., 2018), with disruption of photoreceptor outer segment biogenesis/function, phototransduction, synapses, metabolism the most frequent causes (Veleri et al., 2015; Zelinger and Swaroop, 2018). We note that development and homeostasis of photoreceptors heavily rely on the interaction with the retinal pigment epithelium (RPE) and Müller glia (MG). Consistently, an increasing number of studies suggest that pathologies of RPE or MG may compromise photoreceptor survival (Wright et al., 2010; Mysore et al., 2014; Pellissier et al., 2015; Veleri et al., 2015; Duncan et al., 2018; Amato et al., 2021; Chen et al., 2023a). Therefore, degeneration of photoreceptors in IRD could be a direct consequence of genetic mutations and/or secondary to compromised RPE/MG. This review aims to summarize our current understanding in the interaction among these cell types and explore how dysfunction of one cell type could compromise the other one, a process that is particularly important in designing long-lasting and efficacious therapeutics. As some of these mechanisms could be common among various mutations, a comprehensive understanding of the mechanisms underlying the cell-cell interaction in the outer retina should hold the promise to identify therapeutic targets for gene-agonistic therapies.
2 Retinal structure and function
When light strikes the eye, the cornea and the lens bend and invert the light to focus it on the retina, a receptive inner layer lining the posterior part of the eyes (Masland, 2012; Erskine and Herrera, 2014). The retina is comprised of neurons (photoreceptors, bipolar cells, horizontal cells, amacrine cells, and retinal ganglion cells) and glial cells (MG) forming three distinct cellular layers [the outer nuclear layer (ONL), inner nuclear layer (INL), and ganglion cell layer (GCL)] and two plexiform layers (the outer plexiform layer and inner plexiform layer) enriched in cellular processes and synapses (Figure 1) (Hoon et al., 2014).
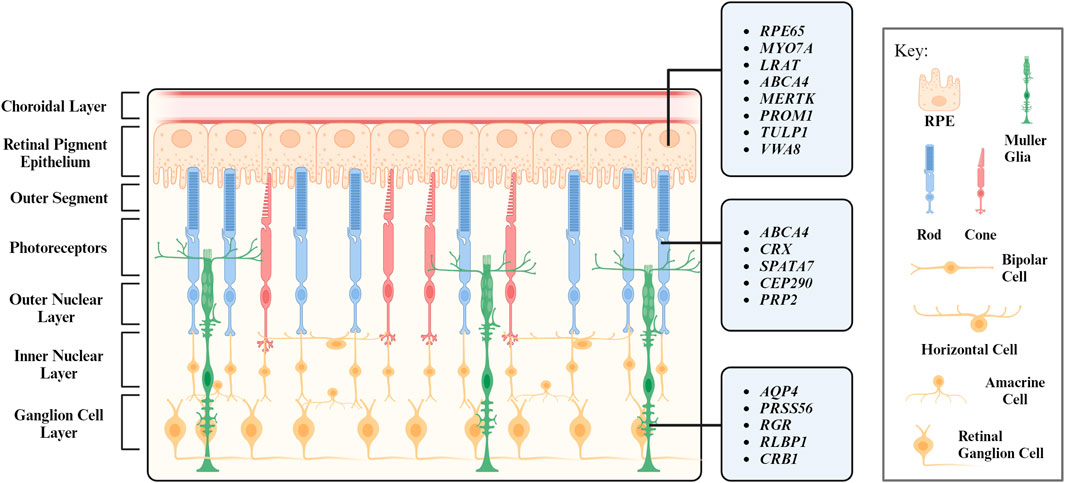
FIGURE 1. Layers of the retina and their respective cell types. Genes associated with inherited retinal degeneration expressed in retinal pigment epithelium (RPE), photoreceptors (PR), and Müller Glia (MG) are listed.
Every retinal cell type has its unique and vital role in maintaining the retinal function. Photoreceptors are light-sensitive neurons located at the ONL and can be further categorized into rod and cone photoreceptors based on their morphology and function. Rod photoreceptors were historically considered to function under dim light for night vision. Recent studies reveal they also support cone survival and function (Leveillard et al., 2004; Pahlberg et al., 2017; Tikidji-Hamburyan et al., 2017). Cone photoreceptors operate at bright illumination conditions and are responsible for color and high acuity vision as well as non-image forming responses (Gooley et al., 2010; Lall et al., 2010; Molday and Moritz, 2015; Walmsley et al., 2015). Retinal interneuron bipolar, horizontal, and amacrine cells are mainly located at the INL. Bipolar cells process visual signals from photoreceptors and transmit them to retinal ganglion cells, a process modulated by horizontal cells and amacrine cells with both excitatory and inhibitory properties (Diamond, 2017). Visual signals are transmitted to the brain via the optic nerves, which are axons of the retinal ganglion cells (Mead and Tomarev, 2016).
The structure and function of the retina is maintained by MG, which are the most abundant glial cells in the retina. They play a crucial role in maintaining the homeostasis of retina and provide structural, metabolic, and functional support to retinal neurons (Reichenbach and Bringmann, 2013; Tworig and Feller, 2021). RPE is another type of supporting cell for the retina. Located juxtaposed to photoreceptors (Figure 1), RPE form the outer retina-blood barrier, with their apical and basal side interfacing with photoreceptors and the Bruch’s membrane, respectively (Lakkaraju et al., 2020). RPE tightly interact with photoreceptors to coordinate metabolism and visual cycle (Palczewski and Kiser, 2020; Hurley, 2021; Nolan et al., 2021). We will further discuss the interaction between photoreceptors and their interaction partners MG and RPE in the following sections.
3 Genetics and biology of inherited retinal degenerative diseases
The genesis and health of the retina are maintained by numerous structural and functional components. Mutations in genes encoding for these components could lead to progressive, visually debilitating diseases collectively termed as IRD. The discovery of the first two IRD-causing genes (RHO and CHM) can be traced back to 1990 (Cremers et al., 1990; Dryja et al., 1990). With the advances in sequencing technologies and genetic mapping, more than 300 IRD-associated loci are mapped and over 280 disease-causing genes with diverse roles in the retina have been identified (RetNet; https://web.sph.uth.edu/RetNet/).
IRD are highly heterogeneous both genetically and phenotypically. They can be monogenic, digenic, or even more complex, and inherited as autosomal recessive, autosomal dominant, or X-linked. Recently, a comprehensive analysis on 1243 proband-parent trios in 22 subgroups of inherited eye disorders by targeted exome sequencing reveals de novo mutations contributes to approximately 7% of pathogenicity (Li et al., 2023). De novo mutations could arise from patients with a simplex disease and cause autosomal dominant phenotypes, such as in the case of the Arg677Ter mutation of RP1 (Schwartz et al., 2003). The presence of de novo mutations supports the hypothesis of mutational hotspots (Schwartz et al., 2003; Daiger et al., 2007). Besides RP1, a recent study on a large cohort indicates the high prevalence of ABCA4 and USH2A mutations (Karali et al., 2022). The pleiotropic effect of numerous IRD-causing genes, in which mutations in one gene could cause diverse phenotypes, adds more complexity to the clinical manifestations (den Hollander et al., 2010; Valente et al., 2006; Siemiatkowska et al., 2014; Daiger et al., 2007). More than 50 major types of IRD have been documented globally, with retinitis pigmentosa (RP), Leber congenital amaurosis (LCA), Stargardt disease as the most common forms (Duncan et al., 2018; Schneider et al., 2022). RP, which is the most prevalent IRD and contributes to approximately half of the IRD cases (Daiger et al., 2013), is characterized by initial rod photoreceptor degeneration followed by gradual loss of cone cells (Verbakel et al., 2018). LCA is a group of severe retinal dystrophy and the leading cause of inherited childhood blindness (Kumaran et al., 2017). Visual impairment or blindness caused by dysfunction or degeneration of photoreceptors is congenital or manifested within the first few months after birth in LCA patients (Cideciyan and Jacobson, 2019). Different from RP and LCA, in which compromised peripheral vision caused by dysfunctional rods is the earliest and most common phenotype (Shintani et al., 2009; Chacon-Camacho and Zenteno, 2015), Stargardt disease is the most prevalent inherited macular dystrophy characterized by gradual loss of central vision (Gelisken and De Laey, 1985; Weleber, 1994). Most Stargardt patients experience significant reduction in visual acuity in their first or second decade of life, which is associated with loss of photoreceptors and/or RPE (Molday, 2015).
While dysfunction or death of photoreceptors is a common phenotype shared by almost every type of IRD, disease-causing genes encode for proteins involved in diverse cell types, signaling pathways, and cellular functions. Notably, numerous IRD with photoreceptor degeneration are caused by mutations in genes associated with RPE and MG structure/function (Table 1, 2). Therefore, photoreceptor degeneration in IRD could be a direct cause of genetic mutations and/or secondary to pathologies of its interaction partner such as MG and RPE. In favor of the latter, therapeutic approaches targeting MG and RPE show promising outcome in maintaining photoreceptor survival and function in preclinical models and clinical trials (Cideciyan et al., 2009; Jacobson et al., 2012; LaVail et al., 2016; Buck et al., 2021). In the following sections, we will focus on the function of RPE and MG as well as their interaction with photoreceptors, with an aim to unravel their potential role in IRD pathogenesis.
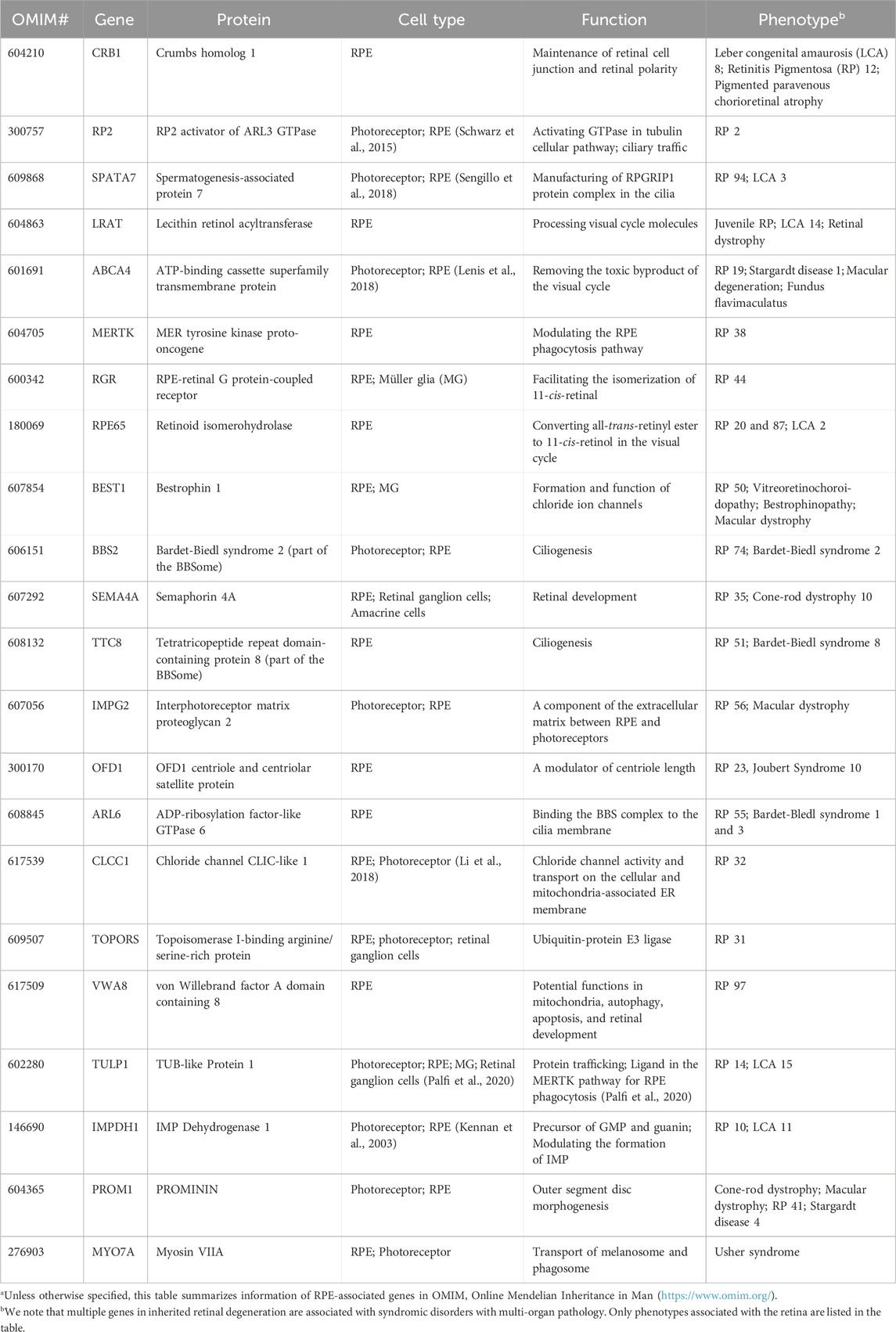
TABLE 1. Retinal pigment epithelium (RPE) genes associated with inherited retinal degenerationa.
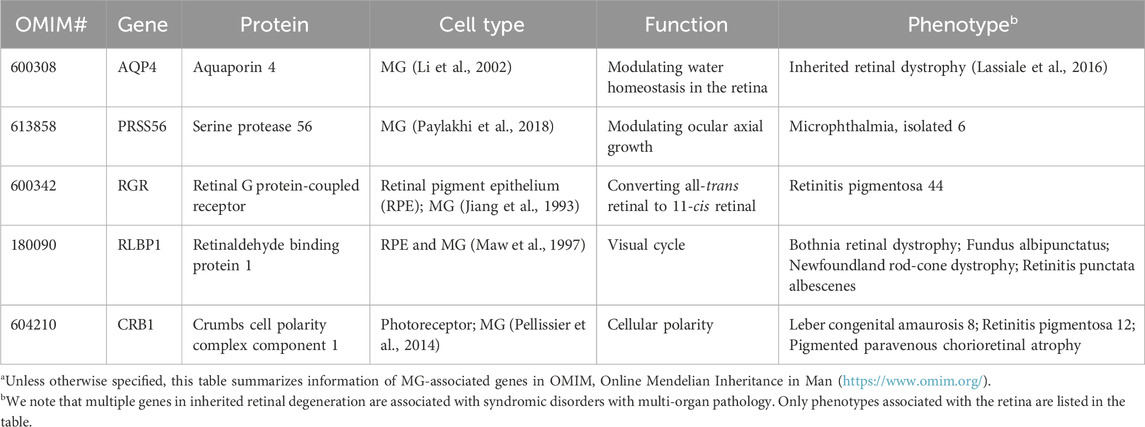
TABLE 2. Müller glia (MG) genes associated with inherited retinal degenerationa.
4 Photoreceptor-RPE interaction in IRD
RPE form a monolayer between the outer retina and the choroidal layer (Figure 1). The basolateral side of RPE faces the Bruch’s membrane, an elastin- and collagen-rich extracellular matrix between the RPE and the fenestrated choroidal capillaries of the eye, and the apical side is juxtaposed to photoreceptors (Strauss, 2005; Booij et al., 2010). The tight junction of RPE enables the layer to act as a semipermeable membrane to provide structural and functional support for the retina. Due to the close relationship between photoreceptors and RPE, mutations in RPE genes are often associated with photoreceptor degeneration in IRD (Table 1). In this section, we will discuss how mutations in RPE-associated disease-causing genes abrogate RPE function and lead to photoreceptor degeneration in IRD (Figure 2).
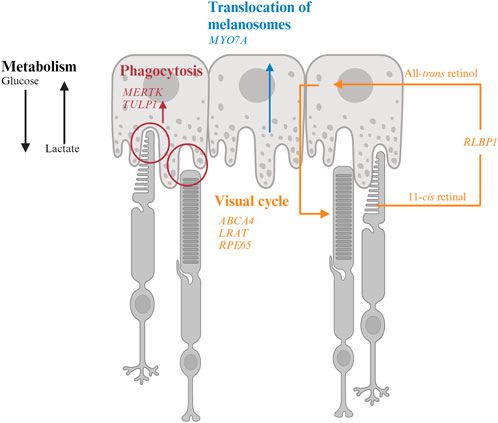
FIGURE 2. Interaction of retinal pigment epithelium (RPE) and photoreceptor and genes associated with inherited retinal degeneration. RPE harbor melanosomes that absorb light. The distribution of melanosomes is modulated by MYO7A. MERTK and TULP1 facilitate phagocytosis of photoreceptor outer segments and their mutations could disrupt the metabolic homeostasis between photoreceptors and RPE. LRAT, RPE65, and ABCA4 encode for key molecules in visual processing. The transport of 11-cis retinal from RPE to photoreceptors and all-trans retinol from photoreceptors to RPE are facilitated by CRALBP encoded by RLBP1.
4.1 Absorption of light and reduction of oxidative stress
While light is essential for vision, exposure to bright illumination could cause permanent photic damage to the retina (Youssef et al., 2011). Human RPE contain three types of pigment granules at various stages, which absorb and filter approximately 60% of light with various wavelengths to protect the retina. Melanin-containing melanosomes are formed between early embryogenesis and up to 2 years in humans. Lipofuscin granules accumulate with increasing age and melanolipofuscin granules are a feature of aged RPE (Feeney, 1978; Boulton, 2014). When the filtered light reaches the retina, it initiates the visual process, which requires tremendous amount of energy. Reactive oxygen species (ROS) is thus generated as a by-product of active metabolism by mitochondria in photoreceptors (Wong-Riley, 2010). ROS triggers oxidative stress and subsequent retinal damage and is strongly implicated in retinal degeneration (Ray et al., 2012; Bellezza, 2018; Ozawa, 2020). RPE harbor a high concentration of cellular enzymatic antioxidants (Newsome et al., 1990; Blanks et al., 1992; Oliver and Newsome, 1992; Miceli et al., 1994; Tate et al., 1995; Feng et al., 2010; Murthy et al., 2014; Biswal et al., 2018; Sun et al., 2018; Zheng et al., 2022b), which should facilitate the alleviation of oxidative stress in RPE themselves as well as the outer retina. Melanosomes also have been demonstrated to have a potential antioxidant role in RPE (Burke et al., 2011).
4.1.1 MYO7A-associated Usher syndrome 1B
MYO7A encodes myosin VIIA in photoreceptors and RPE. Mutations in MYO7A are associated with the most common Usher Syndrome type 1B characterized by congenital deafness and progressive retinal degeneration (Smith et al., 1994). Myo7a-null mice reveals the function of Myo7a in the apical localization of melanosomes and phagosomes via actin-based motor activity in RPE as well as the selective transport of opsins and other phototransduction proteins to the outer segments in photoreceptors (Lopes et al., 2011). Consequently, mutations in MYO7A disrupt the proper function of melanosomes for light absorption to protect the retina as well as the localization of phototransduction machineries to initiate vision, which lead to visual impairment and retinal damage in patients.
4.2 Visual cycle
Phototransduction is a visual process that converts light into electrical signals in photoreceptors. Photoreceptors contain a high concentration of visual pigment (i.e., opsin) at the outer segments (Ebrey and Koutalos, 2001). The type of opsin defines the photoreceptor subtype. Rod photoreceptors harbor rhodopsin. Cone photoreceptors can be further categorized into L- (long, 564 nm), M- (medium, 533 nm) or S- (short, 437 nm) type depending on the maximal spectral sensitivity of their opsins. Rod and cone opsins are present in the membranous discs of outer segments. Each opsin molecule is covalently bound to chromophore 11-cis retinal to become light-sensitive. Upon photon capture, 11-cis retinal is isomerized to all-trans form, which triggers a conformational change in opsins and initiates a cascade of biochemical events to initiate the phototransduction cascade. The photobleached pigment releases all-trans retinal into the disc bilayer. All-trans retinal cannot be processed by photoreceptors and thus is transported back to RPE for recycling and then returned to photoreceptors in the cis form as part of the visual cycle (Strauss, 2005; Palczewski and Kiser, 2020).
4.2.1 ABCA4-associated Stargardt disease
ABCA4 is a member of the superfamily of ATP-binding cassette transporters primarily localized along the rim region of photoreceptor outer segment disc membranes (Allikmets et al., 1997; Azarian and Travis, 1997; Illing et al., 1997) but is also expressed by RPE (Lenis et al., 2018). Mutations in ABCA4 are the major cause of Stargardt disease and a subset of cone-rod dystrophy with progressive blindness in children and young adults (Allikmets et al., 1997; Maugeri et al., 2000; Burke et al., 2014).
In photoreceptors, the free all-trans retinaldehyde combines rapidly and reversibly with phosphatidylethanolamine (PE) in the disc membrane to form N-retinylidene-phosphatidylethanolamine (N-ret-PE). While the retinylidene-bearing head group facing the outer segment cytoplasm is reduced to all-trans-retinol by retinol dehydrogenase 8 in the first step to regenerate visual chromophore (Rattner et al., 2000), the one located on the disc luminal surface is flipped to cytoplasmic side by ABCA4 (Sun and Nathans, 1997; Quazi et al., 2012). As retinaldehyde is toxic to photoreceptors (Getter et al., 2019), its efficient clearance and recycle not only facilitate the continuation of visual cycle but also maintain photoreceptor survival. Therefore, mutations in ABCA4 lead to accumulation of retinaldehyde and delay the visual cycle, which contribute to visual impairment and photoreceptor degeneration in IRD. A key pathologic feature of Stargardt disease is the accumulation of fluorescent lipofuscin granules in RPE. As ABCA4 has long been considered to be a photoreceptor-specific gene, the RPE phenotype is thought to be the major lipofuscin fluorophore A2E converted from bisretinoids from the outer segments with accumulation of retinaldehyde (Mata et al., 2000). However, ABCA4 is found to be expressed by RPE in a recent study (Lenis et al., 2018). The RPE of dark-adapted Abca4 mice accumulate lipofuscin the same rate as the ones under normal diurnal cycle, suggesting the lipofuscin is not contributed by the phagocytosed outer segments with accumulated retinaldehyde. Further investigation reveals that ABCA4 recycles the retinaldehyde released from the phagocytosed photoreceptor outer segments in RPE endolysosomes (Lenis et al., 2018). RPE-specific expression of ABCA4 show partial rescue of both the lipofuscin accumulation and photoreceptor degeneration (Lenis et al., 2018), suggesting that the phenotypes in ABCA4-Stargardt are contributed by both RPE and photoreceptor pathologies.
4.2.2 LRAT-, RPE65-, RLBP1-associated RP
LRAT, RPE6, and RLBP1 encode for enzymes involved in the visual cycle. Lecithin retinol acyltransferase encoded by LRAT catalyzes the first critical step to esterify all-trans retinol from photoreceptors into all-trans retinyl ester. Retinoid isomerohydrolase encoded by RPE65 then converts the all-trans retinyl ester to 11-cis retinol, which is transported to the photoreceptors (4). Mutations in either of these two RPE-specific genes limit the availability of 11-cis retinal to photoreceptors, leading to early-onset visual impairment and subsequent photoreceptor degeneration. The transport of all-trans retinol from photoreceptors to RPE as well as 11-cis retinol from RPE to photoreceptor are carried by Retinaldehyde-binding protein 1 (RLBP1) (Xue et al., 2015; Napoli, 2016). Therefore, RLBP1 prevents the accumulation of toxic retinoid compounds in photoreceptors and RPE and facilitate the completion of visual cycle. Mutations of RLBP1 could lead to early-onset visual impairment and retinal degeneration.
4.3 Metabolism
Photoreceptors and RPE share a unique symbiotic relationship in their co-dependent metabolic pathways. Each day, approximately 10% of photoreceptor outer segments are phagocytosed by RPE for daily renewal (Kevany and Palczewski, 2010; Viegas and Neuhauss, 2021). Phagocytosis of the outer segments also facilitates nutrient supply to photoreceptors. Glucose is transported from the choroidal circulation and supplied to photoreceptors by RPE through glucose transporter 1 (GLUT1), which is maintained at the apical side of RPE by phagocytosis of photoreceptor outer segments (Figure 2). Glucose is preferentially supplied to photoreceptors to maintain their high metabolic demand (Hurley, 2021). In photoreceptors, glucose is converted to ATP as energy source and, to lactate, which is shuttled to RPE for energy. Lactate is then converted to pyruvate by lactate dehydrogenase (LDH) in RPE to produce ATP through the Krebs or tricarboxylic acid (TCA) cycle and reduce NAD+ to NADH to inhibit glycolysis (Kanow et al., 2017; Hurley, 2021). Another approach to inhibit glycolysis in RPE is to activate the Akt pathway by phosphatidylserine on the outer segments (Viegas and Neuhauss, 2021). As photoreceptors are rich in lipids, the remaining products from phagocytosed outer segments contain sufficient phospholipids, fatty acids, cholesterol, and proteins to support the energy demand of RPE (Nolan et al., 2021; Ramachandra Rao and Fliesler, 2021; Viegas and Neuhauss, 2021). These lipids are broken down into ketone bodies by hydroxymethylglutaryl-coenzyme A (CoA) synthase 2 in RPE through the mitochondrial β-oxidation pathways (Nolan et al., 2021). The ketone bodies are released to the apical side of the RPE probably to be taken up by photoreceptors as another source of energy supply (Nolan et al., 2021). Approximately 80% of materials in the phagocytosed outer segments are recycled back to photoreceptors or removed to the blood stream as waste, a process regulated by ATP-driven Na+/K+ pumps (Mazzoni et al., 2014; Country, 2017; Kwon and Freeman, 2020).
4.3.1 MERTK- and TULP1-associated RP
MERTK encodes a widely expressed receptor tyrosine kinase Mer, which is involved in numerous cellular processes and signal transduction pathways. The onset of visual impairment in MERTK-RP patients is within the second decade of life, with progressive decline of visual acuity (Gal et al., 2000; Tschernutter et al., 2006).
In the retina, MERTK is expressed in RPE and involved in the phagocytosis of outer segments of rod photoreceptors. Disruption of this process caused by MERTK mutations could impede the energy supply to RPE. Due to lack of lactate and reduced Akt activity, RPE starts to uptake glycolysis and starve photoreceptors.
Tubby like 1 (TULP1) binds to MERTK to stimulate RPE phagocytosis. Mutations of TULP1 are also associated with severe early-onset IRD (Table 1). TULP1 is also expressed in photoreceptors, in which it is localized in the inner segments and engaged in the trafficking of photoreceptor opsins to the outer segments (Grossman et al., 2011; Palfi et al., 2020). The essential roles of TULP1 in both RPE and photoreceptors explain the more severe phenotypes in RP carrying TULP1 mutations the MERTK ones.
4.3.2 PROM1-and VWA8-associated IRD
Autophagy is a surveillance mechanism to degrade nucleic acids, lipids, and proteins to maintain cellular homeostasis. The autophagy pathway is responsible to breakdown the phagocytosed outer segments. Dysregulation of autophagy has been associated with various ocular disorder (Frost et al., 2014). Mutations in PROM1 and VWA1, both of which are implicated in autophagy, could interfere with RPE metabolism and cause various types of IRD (Bhattacharya et al., 2017; Kong et al., 2023). PROM1 encodes for Prominin-1 and is located to the open rims of photoreceptor outer segments to regulate disc morphogenesis in Xenopus laevis (Han et al., 2012; Carr et al., 2021). PROM1-IRD could be contributed by impaired disc formation. RPE-specific von Willebrand factor A domain containing 8 encoded by VWA8 is well known for the regulation of mitophagy (i.e., autophagy of the mitochondria). Mutations in VWA8 aberrantly activate the degradation of mitochondria and lead to defective retinal development and subsequent retinal degeneration in autosomal dominant RP (Kong et al., 2023). Surprisingly, treatment of malaria drug chloroquine or hydroxychloroquine, both of which act as autophagy inhibitor, could lead to damage to the macular cones outside of the fovea due to reduced lysosomal activity and outer segment phagocytosis (Stokkermans et al., 2023). Therefore, photoreceptor degeneration in VWA8-RP could be caused by compromised RPE metabolisms and/or retinal developmental defects.
4.3.3 Bietti’s Crystalline Dystrophy
First described by Italian Ophthalmologist Dr. G.B. Bietti in 1937, Bietti’s Crystalline Dystrophy (BCD) is a rare autosomal recessive ocular disease characterized by yellow-white crystalline lipid deposits in the retina and sometimes cornea, degeneration of RPE, and sclerosis of the choroidal vessels (Saatci et al., 2023). The typical onset of BCD is between the second and third decades of life, and patients gradually lose peripheral and/or central visual acuity till legal blindness (Sayadi and Mekni, 2022). Although the pathophysiology of BCD is not yet fully understood, it is mainly caused by biallelic mutations in CYP4V2 (Lin et al., 2005). CYP4V2 encodes for a member of the cytochrome P450 hemethiolate protein superfamily which is involved in oxidizing fatty acid precursors. Dysfunctional lipid metabolism in RPE may disrupt the metabolic homeostasis between photoreceptors and RPE. Loss of fatty acid metabolism reduces the ketone bodies supplied to photoreceptors. RPE may consume glucose as energy supply, which disrupts photoreceptor function and leads to their starvation.
4.4 Ion channels
RPE express voltage- and ligand-gated potassium ion (K+), chloride ion (Cl−), and calcium ion (Ca2+)-conducting channels. These ion channels are crucial not only for the normal physiology of RPE but also for the interaction with photoreceptors. In the darkness, the K+ ions enter RPE through their Na+/K+-ATPases at the apical side and exit by the basolateral membrane to control the K+ concentration in the subretinal space and maintain the Na+-K+ equilibrium in photoreceptors (Baylor, 1996). When exposed to light, the hyperpolarization of photoreceptors reduces the release of K+ and leads to hyperpolarization of the apical membrane of RPE (Oakley, 1977), which subsequently inhibits the Na+/K+/2Cl− co-transporters and results in an increase of Na+ and a decrease of the intracellular Cl− concentration. Recent studies demonstrate that Na+ channels are strongly implicated in phagocytosis of photoreceptor outer segments and the lateral spread of voltage spikes via gap junctions in RPE (Johansson et al., 2019; Ignatova et al., 2023). Besides the Na+ channels, Cl− and Ca2+ channels are also implicated in the phagocytosis of photoreceptor outer segments and their subsequent degradation by the autophagy pathway (Busschaert et al., 2017; Zheng et al., 2022a). Genetic mutations of chloride intracellular channel 4 (CLIC4) have been shown to lead to dry age-related macular degeneration potentially via dysregulation of the autophagy pathway, although the precise mechanisms require further investigation (Chuang et al., 2022). Cl− channels, together with the K+ and Ca2+ ones, also have an important function in transepithelial transport of ions and water (Wollmann et al., 2006; Wimmers et al., 2007). The retina generates a large amount of water due to the high metabolic turnover. This water is eliminated by both RPE and MG. Transepithelial water transport from the apical to the basolateral side of the RPE is achieved by Ca2+-dependent modulation of K+ or Cl− channels and aquaporin-1 channels (Stamer et al., 2003; Dvoriashyna et al., 2020).
4.4.1 KCNJ13-LCA
KCNJ13 encodes for potassium inwardly rectifying channel subfamily J member 13 in RPE. Mutations in KCNJ13 lead to LCA16 characterized by significant central and peripheral vision loss in young children. Although the underlying mechanisms are not yet fully understood, mutations in KCNJ13 lead to compromised cell alignment and phagocytosis in human induced pluripotent stem cell-derived RPE (Kanzaki et al., 2020), which was consistent with the function of K+ and relevant ions (e.g., Cl−, Ca2+) in the regulation of phagocytosis.
4.4.2 Bestrophinopathy
Bestrophinopathy is the collective term of a phenotypically heterogeneous group of degenerative ocular diseases caused by mutations in the Bestrophin (BEST) genes, specifically the BEST1 gene (Pasquay et al., 2015; Guziewicz et al., 2017). Initially BEST mutations were identified in IRD including Best vitelliform macular dystrophy (VMD), which is the most common form, autosomal dominant vitreoretinochoroidopathy (ADVIRC), and autosomal recessive bestrophinopathy (ARB). BEST1 mutations are subsequently found to be implicated in more complex ocular disorders with the involvement of the anterior segment such as autosomal dominant microcornea, early-onset cataract, and posterior staphyloma (MRCS) syndrome (Johnson et al., 2017; Pfister et al., 2021). BEST1 is a Ca2+-activated Cl− channel localized to the basolateral membrane of RPE (Marmorstein et al., 2000). Although the exact role of BEST1 in RPE is unclear, its mutations cause a spectrum of phenotypes associated with compromised ion channels including altered permeability to large anions, dysregulated intracellular Ca2+ signaling, impaired anion channel activity, and mistrafficking of protein to the basalaterol membrane of RPE.
4.5 Immaturity of RPE in retinal ciliopathy
The primary cilium is a ubiquitous, microtubule-based organelle for modulating diverse signaling pathways and sensing external environment (Chen et al., 2021a). Mutations in genes associated with primary cilia are a major caused of IRD (Zelinger and Swaroop, 2018). Although primary cilia present in various retinal cell types (Lepanto et al., 2016; Ning et al., 2021), how defects of the primary cilia impact the function of every cell type is largely unexplored. CEP290 encodes for a centrosomal/ciliary protein located at the transition zone and is responsible for initiating its formation by tethering the microtubules to the ciliary membrane (Craige et al., 2010; Wu et al., 2020). Mutations in CEP290 compromise the formation of the transition zone and thus disrupt the biogenesis photoreceptor outer segments (Rachel et al., 2012; Rachel et al., 2015; Parfitt et al., 2016; Shimada et al., 2017). CEP290-LCA patients suffer from visual impairment at birth or infancy, with rod photoreceptors degenerating within the first decade of life followed by cone cell death (Cideciyan and Jacobson, 2019). A recent study indicates patient-derived RPE harbor defective apical processes, compromised phagocytosis, and reduced adult-specific gene expression (May-Simera et al., 2018). As the primary cilia is a regulator of various signaling pathway, the immaturity of RPE is caused by simultaneously suppressing canonical WNT and activating PKCδ pathways due to compromised primary cilia. Notably, such RPE defects precedes photoreceptor degeneration in a ciliopathy animal model (May-Simera et al., 2018). Ablation of primary cilia specifically in RPE also leads to photoreceptor degeneration in another animal model (Kretschmer et al., 2023). Therefore, although photoreceptor degeneration the major phenotypes in IRD caused by defective primary cilia, compromised RPE function could accelerate this process.
5 MG-photoreceptor interaction in IRD
Following injury or photoreceptor degeneration at the late stages of IRD, MG in the retina of fish and many non-mammalian vertebrates are able to dedifferentiate into neural progenitors which have the capacity to differentiate into all retinal neurons to replace the lost ones (Raymond et al., 2006; Bernardos et al., 2007; Thummel et al., 2008). However, MG in mammals have only limited capacity to regenerate the retina. Recent studies start to reveal growth factors and transcriptional machineries involved in the reprogramming of MG (Lahne et al., 2020; Todd et al., 2021; Todd et al., 2022). On the other hand, mammalian MG have adapted morphological, biochemical, physiological, and genetical machineries to activate reactive gliosis in response to the loss of retinal cells (Dyer and Cepko, 2000; Bringmann et al., 2009; Goldman, 2014; Vecino et al., 2016). Upon photoreceptor cell death, MG form scars to prevent further expansion of the injuries. Although this process is beneficial for photoreceptor survival, it may accelerate the degeneration process upon prolonged activation (Bringmann et al., 2006).
Indeed, MG are among the first cell types to respond to photoreceptor stress by secretion of antioxidants and neurotrophic factors (Jones et al., 2016; Leinonen et al., 2023). A recent study reveals MG can uptake damaged mitochondria from cone photoreceptors in zebrafish (Hutto et al., 2023). Therefore, compromised MG could contribute to photoreceptor degeneration during IRD progression. Consistently, MG-associated genes were found to be dysregulated at the early stages of IRD, which precedes photoreceptor dysfunction (Deng et al., 2018; Chen et al., 2023a). In this section, we will describe the role of MG in the retina and discuss how disease-causing genes impact MG function and lead to photoreceptor degeneration.
5.1 Structural support
MG are the predominant glial cells in the retina. The somata of MG reside in the INL and the two stem processes radiate in opposite directions, spanning the entire thickness of the retina (Figure 1). MG keep close contact with all types of retinal neurons to provide essential structural and functional support for their development and survival (Figure 3). In vertebrate retina, the apical processes of MG are attached to each other and to the inner segments of photoreceptors to form the outer limiting membrane (OLM) by adherens junctions and desmosomes. The adherens junctions interact with the actin cytoskeleton and intermediate filaments through zonula occludens (ZO)-1 and desmosomes respectively (Hartsock and Nelson, 2008). They also contain transmembrane proteins such as cadherins to interact with various cytoplasmic proteins (Chifflet et al., 2004). Adherens junctions perform multiple cellular functions including initiation and stabilization of cell–cell adhesion, regulation of the actin cytoskeleton, intracellular signaling, and transcriptional regulation (Omri et al., 2010). Mutations in OLM-associated genes interfere with photoreceptor maturation, function, and vision, and are implicated in multiple IRD including LCA, RP, and childhood cone-rod dystrophy as well as syndromic disorders (e.g., Usher syndrome) (Table 2) (Figure 3) (Khan et al., 2011; Bujakowska et al., 2012; Ratnam et al., 2013; Quinn et al., 2019b).
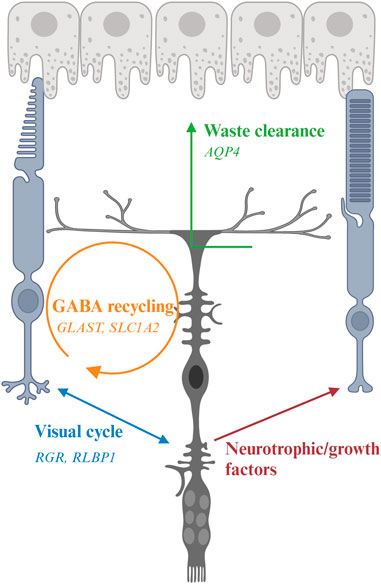
FIGURE 3. Interaction of Müller glia (MG) and photoreceptor and genes associated with inherited retinal degeneration. MG maintain osmotic homeostasis of the retina by AQP4 that facilitate the transport of ions and water. MG support PR by recycling glutamate and GABA neurotransmitters with the aid of other retinal cells, a process modulated by multiple genes including GLAST, and releasing neurotrophic factors. MG also facilitate the visual process of cone photoreceptors by expressing RGR and RLBP1.
5.1.1 CRB1-associated IRD
Mutations in the CRB1 gene are associated with variable phenotypes in various IRD including LCA and RP (Bujakowska et al., 2012). Some patients also develop macular dystrophy (Bujakowska et al., 2012). The Crumbs (CRB) protein was first identified in Drosophila as a key regulator of apical polarity (Tepass et al., 1990). It is expressed in the retina and the brain. Among the three genes of the family in humans, CRB1 and CRB2 are expressed in the photoreceptor and MG (Pellissier et al., 2014; Quinn et al., 2019a). CRB1 contains transmembrane and cytoplasmic domains. It constitutes the adherens junctions and interacts with the actin cytoskeleton through the cytoplasmic domain (Gosens et al., 2008). Consistent with the function of adherens junctions, CRB1 has an evolutionarily conserved function to regulate cellular polarity. Animal models carrying Crb1 mutations display disruption of the ONL, disorganization of the retinal layers, and loss of photoreceptor cell polarization (Mehalow et al., 2003; van de Pavert et al., 2004). The phenotypes of the animal models are consistent with the clinical features of patients carrying CRB1 mutations, whose retinas are thickened and show an altered lamination (Jacobson et al., 2003), suggesting an important function of CRB1 in the formation of the ONL and the regulation of retinal morphogenesis. Although CRB2 mutations in patients do not display phenotypes associated with the retina, CRB2 has been shown to be a modifier of CRB1 in diseases (Pellissier et al., 2014; Quinn et al., 2019b). MG-specific knockout of CRB1 and knockdown of CRB2 in mice and patient retinal organoids reveal disorganization of retinal structure and visual defects (Buck et al., 2021; Boon et al., 2023), suggesting that photoreceptor degeneration in CRB1-associated IRD is at least partially contributed by MG.
5.2 Clearance of ions, water, and cellular debris
The phototransduction cascade triggers hyperpolarization of photoreceptors by modulation of ion channels in their cell membranes. The visual signals are transmitted to second-order interneurons such as bipolar cells in the INL through neurotransmitters at the synaptic terminals. These processes are modulated by ions such as K+ and Na+, which affect conductance and permeability of the channels (Oakley, 1977; Mao et al., 2003). However, excessive K+ accumulated at the extracellular space has long been shown to lead to cell apoptosis (Bortner et al., 1997; Hughes and Cidlowski, 1999). MG mediate the transportation of excessive extracellular K+ to extraretinal fluid-filled space (blood vessels, vitreous, and subretinal space) via passive currents through Kir channels (Bringmann et al., 2006; Reichenbach and Bringmann, 2013; Reichenbach and Bringmann, 2020). Excessive water is also removed by MG via the aquaporin-4 (AQP4) channel (Reichenbach and Bringmann, 2013). Clearance of excessive water is critical to protect retinal neurons since water accumulation has been proposed as a pathogenic factor for retinal degeneration (Vecino et al., 2016). MG also phagocytose outer segments shed from cone photoreceptors as well as other cell debris to maintain retinal homeostasis (Bejarano-Escobar et al., 2017).
5.2.1 AQP4-associated IRD
Aquaporin 4 encoded by AQP4 is a membrane transport protein expressed in multiple epithelial and neurosupportive cells (Li et al., 2014). In the retina, AQP4 is highly expressed in MG and astrocytes. The expression and localization of AQP4 are dependent on syntrophins, and the elimination of α1-and β1-syntrophins induce an almost complete loss of AQP4 (Neely et al., 2001; Katoozi et al., 2020). Studies have shown that reduced AQP4 level causes altered MG cell volume (Netti et al., 2021). Depletion of AQP4 increases the susceptibility of MG toward osmotic stress and renders a higher risk of retinal degeneration upon light damage (Pannicke et al., 2010; Li et al., 2014). In Aqp4−/− mice, retinal hyperfusion and upregulated GFAP are observed, which consequently associated with the loss of retinal ganglion cells in congenital glaucoma (Maisam Afzali et al., 2022).
5.3 Regulation of synaptic transmission
Another well-studied function of MG is the regulation of synaptic transmission via recycling of neurotransmitter glutamate and gamma-aminobutyric acid (GABA). Glutamate is the most abundant excitatory neurotransmitter in the central nervous system including the retina (Zhou and Danbolt, 2014). MG uptake the released glutamate from excitatory retinal neurons by glutamate-aspartate transporter [GLAST; also known as excitatory amino acid transporter 1 (EAAT1) or solute carrier family 1, member 3 (SLC1A3)] to prevent ion toxicity and to maintain a fine visual resolution (Bringmann et al., 2006; Bringmann et al., 2013; Vandenberg and Ryan, 2013). The glutamate in MG is converted to glutamine by glutamine synthetase, an enzyme exclusively expressed by glia cells in retina (Pfeiffer et al., 2020). The glutamine is then recycled by retinal neurons for synthesis of glutamate and GABA. GABA is the main inhibitory neurotransmitter in retina (Yang, 2004). By recycling GABA via corresponding receptors and transporters, MG act as important modulators of visual processes through a fast termination of GABAergic signaling via their highly efficient GABA uptake (Biedermann et al., 2002).
5.3.1 Animal models and patient data associated with GLAST
Although no IRD-causing mutations in glutamate-aspartate transporters have been reported, animal models and glaucoma patient samples reveal the role of GLAST in retinal degeneration. As glutamate transporters play a critical role in the recycling of glutamate, impaired function of glutamate transporters could cause glutamate accumulation in the extracellular matrix and contribute to excitotoxicity to retinal neuronal cells. Downregulation of GLAST expression has been reported in human glaucomatous eyes (Naskar et al., 2000) and mutations in GLAST are also found in glaucoma patients (Yanagisawa et al., 2020). Consistently, overexpression of Glast by AAV transduction protects retinal ganglion cells from degeneration in experimental autoimmune encephalomyelitis rats (Boccuni et al., 2023), highlighting a protective role of MG on photoreceptors.
5.3.2 CEP290-LCA
Although MG harbor primary cilia, their function in MG remains largely unexplored. A recent study reveals dysregulation of gene associated with MG development and function in CEP290-LCA patient-derived retinal organoids (Chen et al., 2023a). Notably, the expression of GLU1, which encodes for glutamine synthetase to convert glutamine from glutamate, is downregulated in patient retinal organoids. However, how it impacts synaptic transmission is not investigated in this study. Whether the dysregulation of MG-associated genes is due to defects of the primary cilia caused by CEP290 mutations or caused by the response of MG to photoreceptor dysfunction requires further investigation.
5.4 Secretion of neurotrophic and growth factors
MG secrete a variety of trophic and growth factors to regulate neuronal survival and neuritogenesis and to protect retinal neurons against excitotoxicity (Vecino et al., 2016; Tworig and Feller, 2021). Some well-studied examples include pigment epithelium-derived factor (PEDF), vascular endothelial growth factor (VEGF), glial cell-derived neurotrophic factor (GDNF), interleukin-6 (IL-6), ciliary neurotrophic factor (CNTF), brain-derived neurotrophic factor (BDNF), and nerve growth factor (NGF). Retinal ganglion cells and photoreceptors together with MG itself have receptors for these neurotrophins and growth factors.
5.4.1 VEGF in IRD
Although VEGF is mainly associated with age-related macular degeneration (AMD), diabetic retinopathy (DR), and retinopathy of prematurity (ROP) (Hu et al., 2021a), VEGF levels have been found to be dysregulated in IRD (Salom et al., 2008). Intravitreal injection of VEGF in rd1 mice show enhanced proliferation of retinal progenitor cells that have the potential to differentiate into retinal neurons (Nishiguchi et al., 2007). However, as there are no retinal progenitor cells even in newborn, the therapeutic potential of this approach remains to be determined.
5.5 Lipid metabolism
Photoreceptor outer segments are enriched in fatty acids and cholesterol, which are essential for maintaining metabolic homeostasis in the outer retina (see Section 4.3). These lipid components are channeled to photoreceptors by MG via their fatty acid-binding and transferring proteins. Docosahexaenoic acid (DHA), a trophic factor implicated in photoreceptor development and function, is taken up and processed by MG before supplying to photoreceptors (Politi et al., 2001; Shindou et al., 2017). MG also possess low-density lipoproteins (LDL) receptors that facilitate transport of circulating lipids to retinal neurons. This process is particularly crucial to maintaining photoreceptor outer segments and retinal ganglion cell axons as well as synapse formation (Mauch et al., 2001).
5.5.1 APOE in IRD
Apolipoprotein E (APOE) is a plasma lipid transport protein that is mainly expressed in RPE but also expressed in MG and photoreceptors (Shanmugaratnam et al., 1997; Wickremasinghe et al., 2011; Hu et al., 2021b). APOE has been linked to the pathogenesis of AMD due to its function and potential role in drusen formation (Hu et al., 2021b). A recent study revealed dysregulation of APOE in CEP290-LCA patient-derived retinal organoids (Chen et al., 2023a). As these organoids harbor minimal RPE, the APOE is expressed by MG and/or photoreceptors, yet it is unclear which cell type(s) contribute to this phenotype. Other studies also indicate an association between APOE and glaucoma, but the association is controversial (Wang et al., 2014; Liuska et al., 2023). The role of APOE in IRD pathogenesis requires further elucidation.
5.6 Regulation of visual processes
Conversion of 11-cis retinal to the all-trans form in phototransduction requires the continuous recycling of the chromophore. Although RPE is the major site of this process in the visual cycle, the recycling rate is slow and the number of rod photoreceptors in human retina outweighs the cone ones, which may pose challenges for cone photoreceptors to obtain sufficient chromophore (Wang and Kefalov, 2011). Therefore, the presence of cone-specific visual cycle has long been proposed (Wang and Kefalov, 2011). MG express multiple retinoid-processing proteins such as CRALBP and retinol dehydrogenase-10 (RDH10), suggesting their implication in cone-specific visual cycle. Retinal G protein-coupled receptor (RGR) is a non-visual opsin in intracellular membranes of RPE and MG. It covalently binds to all-trans retinaldehyde and converts it to the 11-cis form (Hao and Fong, 1999). RGR lacks the motif to interact with G protein coupled receptor (Fritze et al., 2003), consistent with their role as a photoisomerase instead of a signaling molecule to activate the phototransduction cascade. Recent studies show that RGR couples with RDH10 to convert all-trans retinol to 11-cis retinol in a light-dependent manner (Morshedian et al., 2019; Tworak et al., 2023). Besides the role in cone-specific visual cycle, MG have also been reported to serve as optic fibers and direct the light to photoreceptors in guinea pigs (Agte et al., 2011), yet whether this function preserves in human retina remains further investigation.
5.6.1 RGR- and RLBP1-associated IRD
Mutations in RGR is a pathogenic factor for RP (Table 2). Under continuous light treatment, cone photoreceptors of Rgr−/− mice lose their sensitivity sooner compared to the wild type ones (Morshedian et al., 2019). A recent study using an innovative cell type-specific gene reactivation approach confirms RGR is critical for cone photoreceptor function and such supporting function is contributed by both RPE and a subset of MG (Tworak et al., 2023). This finding raises an interesting yet challenging question on the targeted cell types for therapies of RGR-RP. Further investigation is also needed to identify the molecular signature of the MG subset responsible for the cone-specific visual cycle.
Likewise, mutations in RLBP1 can cause various IRD including Bothnia dystrophy, retinitis pigmentosa, retinitis punctata albescens, fundus albipunctatus, and Newfoundland rod–cone dystrophy (Hipp et al., 2015) (Table 2). Compromised visual cycle and dysfunction of photoreceptors especially the cones are the primary phenotypes in patients (Kolesnikov et al., 2021). In Rlbp1−/− mice, reduced M-cone dark adaptation, mislocalization of opsin, and loss of photoreceptors are observed (Xue et al., 2015). These defects are demonstrated to be contributed by MG, and restoration CRALBP expression in MG improves M-cone sensitivity (Xue et al., 2015), suggesting an impact of MG pathology in visual defects of photoreceptors.
6 Recent advances in therapies
The advances in genetic approaches to identify targets, model systems to test therapeutics, and retinal imaging and molecular biology to evaluate therapeutic outcomes have established a promising environment for developing treatments for IRD. The first FDA approved gene therapy drug Luxturna further inspires the burgeon of numerous therapeutic approaches to maintain or restore vision. These proof-of-concept evidence not only demonstrates the clinical values but also provides new insights on the molecular mechanisms of disease pathogenesis. In this section, we will explore current therapeutic modalities to maintain photoreceptor survival and discuss how the role of RPE and MG in IRD pathogenesis could potentially impact the outcomes of IRD treatments. For therapeutic approaches to restore vision for late-stage IRD patients with little or no photoreceptors viable in the retina, we direct our readers to other excellent reviews discussing cell replacement therapy, retinal prosthetics, and direct brain stimulation (Yue et al., 2016; Bosking et al., 2017; Uyama et al., 2021; Chen et al., 2023b; Chew and Iannaccone, 2023).
6.1 Gene therapy
Gene-based therapy involves the delivery of genetic materials as an intervention to modify the expression of disease-associated proteins in target cells/tissues. The main goal of gene therapy is to restore the protein function compromised by the mutated genes permanently to reduce the need for long-term medication dependence (Mendell et al., 2021). Dating back to over seven decades to the first observation of viral gene transfer, gene therapy has been demonstrated to be a relatively safe and long-term efficacious therapeutic approach for treatment of various genetic disorders that once considered incurable (Wirth et al., 2013; Tamura and Toda, 2020). Although adverse side effects were reported in two human clinical trials in late 1990s (Wirth et al., 2013; Tamura and Toda, 2020), with the development of new technologies and viral vectors, gene-based therapies have been applied in over 200 human clinical trials involving various tissues and cell types without any incidents of deaths or cancers (Ginn et al., 2018).
As an enclosed, immune-privileged site protected by the blood-retina barrier (Chen et al., 2019), the retina offers a unique opportunity for gene therapy. Only low doses of gene therapy vectors are needed to treat the retina due to its small size and a lack of cellular proliferation in adulthood (Trapani and Auricchio, 2018), and thus the risk of systemic dissemination of the vectors and immune responses is generally negligible (Amato et al., 2021). In addition, surgical procedures or clinical practices have been well established to deliver the gene therapy machineries into the retina, and the therapeutic outcome can be easily monitored by ocular imaging technologies including optical coherence tomography and fundus imaging (Drag et al., 2023). Therefore, gene therapy has been extensively evaluated in the retina in the past two decades. Currently, 73 clinical trials for IRD are recruiting, 39 starting soon, and 154 have been completed (https://clinicaltrials.gov/). Disorders being targeted for genetic therapy include RP, LCA, choroideremia, achromatopsia, Leber’s hereditary optic neuropathy, usher syndrome, X-linked retinoschisis, and Stargardt disease (Nuzbrokh et al., 2021). Besides IRD, gene therapy has also been applied for treatment of age-related macular degeneration by inhibiting VEGF (Heier et al., 2017; Grishanin et al., 2019) or the complement cascade (Cashman et al., 2011).
6.1.1 Current progress and clinical success
6.1.1.1 Strategies of gene-based therapy
Depending on the molecular mechanisms of disease pathogenesis, different strategies are applied in IRD treatment. When a disease-causing mutation disrupts the normal gene function, such as in the case of autosomal recessive or X-linked recessive IRD, gene augmentation (also called gene replacement) could be a promising approach to restore the normal function of the mutated gene. As one of the most commonly used gene therapy strategies, the feasibility and success of gene augmentation therapy in IRD treatments have been demonstrated by numerous clinical and preclinical studies. Besides RPE65 expression by Luxturna for treatment of LCA, delivery of ND4, REP1, ABCA4, RPGR, MERTK, RS1, and CNGA are currently at various stages of clinical trials targeting Leber hereditary optic neuropathy (Yang et al., 2016; Guy et al., 2017; Zhang et al., 2019; Yuan et al., 2020), choroideremia (Dimopoulos et al., 2018; Lam et al., 2019; Morgan et al., 2022), Stargardt disease (Parker et al., 2022), X-linked RP (Cehajic-Kapetanovic et al., 2020), autosomal recessive RP (Ghazi et al., 2016), X-linked retinoschisis (Mishra et al., 2021), and achromatopsia (Fischer et al., 2020), respectively. Despite signs of inflammation and adverse outcomes in some cases, majority of the clinical studies demonstrate safety and/or efficacy of this approach in functional improvement of vision in patients. Besides delivery of normal genes, for IRD caused by splicing defects, antisense oligonucleotides (AON) have been shown to be a long-lasting and effective approach to restore normal splicing and consequently protein function, such as in the case of LCA caused by CEP290 mutations (Cideciyan et al., 2019; Russell et al., 2022). A preclinical study of the AON approach also showed promising results for correcting the splicing defects in Usher syndrome caused by USH2A mutations (Slijkerman et al., 2016). CRISPR/Cas9-mediated genome editing has recently been successfully applied for treatment of CEP290-LCA in degenerative models, demonstrating safety and efficacy in the retina (Ruan et al., 2017). The newly developed self-limiting CRISPR/Cas9 system minimizes the duration of Cas9 expression to futher reduce the immune response (Ruan et al., 2017).
In contrast to loss-of-function mutations in recessive IRD, gain-of-function mutations in autosomal dominant diseases lead to the formation of aberrant proteins that disrupt normal cellular or tissue functions. In this case, the therapeutic goal is to prevent the expression of the altered genes. Downregulation of gene expression by ribozyme, small interfering RNA, and AON has been developed. Although these techniques have been well optimized and extensively applied in in vitro models, their application for therapeutics in vivo is still challenging due to the specificity, off-target effect, and degradation of RNA molecules. Currently, these transcriptional silencing strategies have been successfully applied in animal models of RP caused by RHO mutations (Lewin et al., 1998; Chadderton et al., 2009; Hernan et al., 2011; O'Reilly et al., 2008; Murray et al., 2015). A CRISPR/Cas9-mediated transcript degradation approach has also been successfully applied in vitro and preclinical models of RHO-RP (Bakondi et al., 2016; Li et al., 2018; Tsai et al., 2018). Besides autosomal dominant diseases, the genome-editing approaches, either by CRISPR/Cas9-system or by transcription activator-like effector nuclease (TALEN), can also be applied to correct the mutations underlying recessive disorders. One successful example is the correction of Crb1 mutation in rd8 degenerative model by TALEN-Mediated homology-directed repair. Even the heterozygous mice restore the morphology of the ONL and show a normal retinal phenotype (Low et al., 2014), suggesting the efficiency of the correction could meet the therapeutic needs.
6.1.1.2 Delivery approach
Recombinant adeno-associated virus (AAV) vectors are currently widely used in ocular gene therapy due to therapeutic benefits including non-integrating nature, low immune response, and long duration of transgene expression (Wu et al., 2006; Amato et al., 2021). Recombinant AAV (rAAV) vectors offer a high number of tissue-specific serotypes including AAV 1, 2, 4, 5, 6, 7, 8, and 9 for retinal cells, and thus improve the specificity of viral transduction. The safety and efficacy of AAV-mediated gene therapy have been demonstrated by most of the completed and ongoing clinical studies including Luxturna. One notable limitation of AAV is the small packaging limit, and thus the genetic materials carried by AAV cannot exceed 4.7 kb (Wu et al., 2010), and therefore it is not applicable for diseases caused by mutations of large genes. Lentiviruses, a retrovirus with a larger packing capacity of up to 8kb, become a more compelling alternative to AAV vectors in this case. Lentiviruses pose risk of mutagenesis due to their nature to integrate into the host genome, yet such risk could be justified for treatment of the post-mitotic retina (Drag et al., 2023). The initial trial of ABCA4 delivered by lentiviral vectors for treatment of Stargardt disease has showed promising safety data and the evaluation of efficacy is currently ongoing (Parker et al., 2022).
Despite the positive data from lentiviral vectors, they still pose risks for insertional mutagenesis, germline transmission, and adverse immune response (Drag et al., 2023). Non-viral delivery of genetic materials is being developed as an alternative for IRD caused by mutations of large genes. Although non-viral delivery by physical (e.g., direct injection of genetic materials) or chemical (e.g., nanoparticles) approaches are not as popular as the viral ones due to the low efficiency, immune response, and duration of the effect, recent advances have started to overcome these issues (Sharma and Paschalis, 2022). Various bioengineering materials such as synthetic polymers, lipid cations, liposome-based nanoparticles, and polysaccharides show good transfection efficiency. The use of polyethylene glycol (PEG) combined with cell type-specific promoter or ligand could largely improve the specificity and efficacy of therapies and reduce immunogenicity. The feasibility of such approach has been demonstrated by the relatively efficient and durable therapeutic outcomes in cell cultures and an ABCA4-associated animal model in a recent study (Sun et al., 2020).
6.1.1.3 Administration route
Gene therapies are typically delivered to target retinal cells by subretinal or intravitreal injection in clinical studies. Subretinal injection is a commonly used approach to deliver gene therapy machineries to the subretinal area for treatment of RPE and/or photoreceptors (Planul and Dalkara, 2017). In addition, the subretinal area is a closed immune-privileged compartment and thus further reduce the immune response (Xian and Huang, 2015; Chen et al., 2019). However, subretinal injection could potentially trigger several complications including retinal tears, cataract progression, retinal detachment, or retinal/choroidal hemorrhages (Tripepi et al., 2023). Such a delicate procedure also requires skilled and experienced surgeons for successful administration (Huang et al., 2022).
Intravitreal injection offers several advantages over subretinal injection. It is less invasive and less technically challenging, thus can be performed in a clinic setting and offer the opportunity of gene therapy to larger populations (Amato et al., 2021). However, although efficiently transducing inner retinal cells, intravitreal injection is less effective on outer retinal cells due to dilution of vectors in vitreous cavity and the thick inner limiting membrane in primates (Planul and Dalkara, 2017). A higher therapeutic dose of vectors is thus needed to achieve a desirable outcome, which could pose a significant risk of immunogenicity (Drag et al., 2023).
Suprachoroidal delivery in the space between the sclera and the choroid has also been shown to be a safe approach in preclinical and clinical studies yet it poses risk to spread viral vectors into the systemic circulation and could lead to adverse physical and immune response (Kansara et al., 2020; Naftali Ben Haim and Moisseiev, 2021; Tan et al., 2021).
6.1.2 Strength and limitation
The success of gene therapy has been well demonstrated by the approval of FDA as the first therapy to treat IRD. Numerous of completed and ongoing clinical trials targeting IRD caused by various disease-causing genes show promising safety and efficacy data so far. Approvals of more gene-based therapies are expected in the near future. The advances in engineered viral vectors and cell type-specific promoters further improve the specificity and safety of this approach.
However, gene therapy still faces several problems waiting to be addressed. One of the biggest challenges of traditional gene therapy is their dependence on the targeted disease-causing genes. With over 280 disease-causing genes of IRD (RetNet, https://sph.uth.edu/retnet/) currently identified, it could be time-consuming, expensive, and labor-intensive to design gene therapy for individual gene. In addition, the gene(s) responsible for disease phenotypes are not always identified in patients. Next-generation whole exome sequencing in combination with genetic linkage or homozygosity mapping approaches should facilitate the identification of the causal genes and the design of effective gene therapy (Siemiatkowska et al., 2014). Different mutations in the same gene could lead to various clinical phenotypes, and mutations in different genes may show comparable clinical manifestations. Such complexity poses challenges to gene therapy targeting a single disease-causing gene. Besides, as a rare disease, developing treatments for each IRD may not be favorable for pharmaceutical companies. Therefore, gene-agnostic gene therapy independent of the mutated genes are being developed. This approach targets the converging pathway(s) in multiple IRD and thus has the potential to significantly reduce the cost of development and commercialization of IRD treatments. OCU400, a nuclear hormone receptor-based gene therapy, is currently under clinical trials for treatment of RP caused by NR2E3 or RHO mutations (NCT05203939). AAV-mediated expression of Txnip, which modulates the nutrient supply, has been shown to prolong the survival of cone photoreceptors and improve visual acuity in multiple RP models (Xue et al., 2021), providing promising proof-of-concept evidence for the feasibility of this approach.
Another limitation of AAV-mediated gene therapy is the incapacity to delivery large genes. Although lentiviral vectors and non-viral approaches are being developed to carry large genes into the retina, they may lead to adverse immune response and safety issues and thus require further improvement. Concurrently, AAV-mediated dual vector approach, in which the disease-causing gene is split into two parts and packaged into separate AAV vectors, are being developed. Although the reconstitution efficiency was relatively low initially (McClements and MacLaren, 2017), a recent study improves the technology and demonstrates promising results in animal models and human pluripotent stem cell-derived retinal organoids (Riedmayr et al., 2023), suggesting the feasibility of this approach.
Besides, several clinical studies demonstrated confounding therapeutic outcomes, which may due to the treatment window and patient variability (Ghazi et al., 2016; Fischer et al., 2020; Mishra et al., 2021; Hahn et al., 2022). To maximize the therapeutic potential of gene therapy, a better understanding of disease pathogenesis and careful selection and characterization of patients during the recruitment period could be essential.
6.1.3 RPE and MG as therapeutic targets
The clinical phenotypes of IRD caused by RPE-associated genes reveal a role of RPE in photoreceptor degeneration. Gene augmentation therapies by AAV vector carrying these genes driven by RPE-associated or CMV promoters have shown positive therapeutic outcome in preclinical and clinical studies (Choi et al., 2015; Ghazi et al., 2016; MacLachlan et al., 2018; Dyka et al., 2019; Sun et al., 2020). In the treatment of CEP290-LCA, although AON drug QR110 has shown favorable therapeutic outcome (Jacobson et al., 2017; Cideciyan et al., 2019), the AON is driven by CMV promoter that is able to drive gene expression in RPE and delivered by AAV2, which can readily transduce both photoreceptors and RPE. Therefore, whether the therapeutic outcome of QR110 is due to correction of CEP290 splicing defects in photoreceptor only or both photoreceptor and RPE requires further investigation. Mutations in genes associated with primary cilia are a major caused of IRD (Zelinger and Swaroop, 2018). Further investigation on RPE structure and function in IRD caused by mutations of ciliary genes is needed to determine whether RPE should be included as a therapeutic target in gene therapy.
Mutations in MG-associated genes including RLBP1 and CRB1 could lead to photoreceptor degeneration in IRD. Although gene augmentation therapies of RLBP1 reveal visual improvement in preclinical models (Choi et al., 2015; MacLachlan et al., 2018), CMV promoter is applied to drive the expression of RLBP1 and thus it is difficult to dissect whether the therapeutic outcome is resulted from the improvement of RPE, MG, or both. Studies on CRB1- and CRB2- associated RP and LCA illustrate the role of MG in disease pathogenesis. Novel MG-specific RP mouse and human pluripotent stem cell-derived retinal organoid models, which harbor completely loss of CRB1 and reduced levels of CRB2 specifically in MG, reveal disorganization of retinal structure and visual defects (Buck et al., 2021; Boon et al., 2023). Delivery of CMV-CRB2 is more potent than CMV-CRB1 in protection of vision in animal models (Buck et al., 2021). Gene augmentation therapy with CRB2 also improves the outer retinal phenotypes in CRB1 knockout retinal organoids (Boon et al., 2023). These results suggest that compromised CRB1 and CRB2 functions in MG are the major cause of photoreceptor degeneration. In addition, a recent study indicates dysregulation of MG-associated genes involved in development and function in retinal organoids derived from CEP290-LCA patients (Chen et al., 2023a). Although the primary cilia are a ubiquitous organelle, their roles are not heavily investigated except in photoreceptors. Therefore, it is unclear whether the dysregulated MG genes are caused by defects of the primary cilia or responses to compromised photoreceptors.
Understanding the cellular mechanisms of IRD pathogenesis is crucial for the success of a safe, long-lasting, and efficacious gene therapy. Current gene therapies have careful selection of AAV serotypes and cell type-specific promoters to limit the non-specific expression of the delivered genes. However, if mutations of the disease-associated genes compromise the function of RPE and/or MG, which contribute to pathogenesis, RPE and/or MG should be included as the targeted cell types. Transduction of RPE and MG in treatments will impact not only the selection of AAV serotype and promoter but also the route of administration as intravitreal injection is more efficient than subretinal injection for transducing inner retinal cells. Such as in the case of RGR-RP, the RGR-dependent cone visual cycle is mediated by both RPE and a subtype of MG (Tworak et al., 2023). It could be challenging to deliver viral vectors to both cell types efficiently. In addition, the dose of AAV should also be taken into account as RPE and MG are different cell types from photoreceptors and thus they may have distinct tolerance of AAV transduction and ectopic expression of genetic materials.
6.2 Pharmaceutical therapy
Although drug screening using cell lines have successfully identified therapeutics for various diseases, its application for IRD treatment remains slow due to the limited cell number in the retina and the technical challenge to maintain retinal primary cultures. Recent advances in synthetic chemistry, AI-based screening, structural biology, and the advances in pluripotent stem cell differentiation and high-throughput screening technologies should spur the design of novel bioactive molecules and drug repurposing for IRD treatment.
6.2.1 Current progress
Antioxidants such as vitamin A, vitamin B3, DHA, and lutein have been shown to delay or inhibit the apoptosis of photoreceptors and preserve patient vision (Guadagni et al., 2015). Likewise, applications of neurotrophic agents including CNTF, BDNF, and anti-apoptotic drugs (e.g., tauroursodeoxycholic acid, rasagiline, norgestrel, and myriocin) have shown positive therapeutic outcomes in cell cultures and animal models of RP (Dias et al., 2018).
Besides inhibiting photoreceptor apoptosis to preserve vision, another approach is to target the compromised function and/or restore the dysregulated pathways associated with the mutated genes. A repurposed drug metformin has been shown to facilitate the clearance of lipid deposits caused by ABCA4 mutations in human pluripotent stem cell-derived and in vivo RPE (Amin et al., 2022; Farnoodian et al., 2022). A selective estrogen receptor modulator raloxifene is also shown to inhibit photoreceptor apoptosis and mitigate toxicity of retinaldehyde in ABCA4-associated models (Getter et al., 2019). As Metformin is an FDA-approved treatment for type 2 diabetes, National Eye Institute is launching a Phase I/II trial to evaluate the therapeutic effect of Metformin on Stargardt disease patients. Lumacaftor, another FDA-approved drug for the treatment of cystic fibrosis, has been shown to rescue the ABCA4 trafficking mutants by restoration of Hsp27 in HEK cells (Liu et al., 2019). Yet the therapeutic value of Lumacaftor for preserving vision requires more tests on degenerative models and/or human pluripotent stem cell-derived RPE. A flavonoid drug eupatilin is able to partially restore photoreceptor outer segments and visual function in degenerative models caused by CEP290 mutations (Kim et al., 2018). Disruption of protein homeostasis is another common cause of photoreceptor degeneration in IRD. Modulations of this process, either by regulation of the autophagy pathway and the ubiquitin-proteasome system, or by the use of small molecule chaperon, is able to reduce photoreceptor apoptosis and preserve light detection ability in multiple degenerative models (Mockel et al., 2012; Chen et al., 2018; Yao et al., 2018; Qiu et al., 2019; Intartaglia et al., 2022; Chen et al., 2023a).
6.2.2 Strength and limitation
Pharmaceutical drugs can be produced at reasonable costs and their manufacturing is scalable (Tambuyzer et al., 2020), which are desirable particularly for rare diseases like IRD. As pharmaceutical drugs act by targeting dysregulated cellular function and/or signaling pathways, which could be shared by multiple IRD caused by different mutations, they hold the promise to be translated into cost-effective gene-agonistic therapies. Various drugs currently identified for treatment of IRD act as neurotrophic factors or apoptotic inhibitors, and thus they could be applied together with gene therapy drugs to achieve better therapeutic outcomes. The establishment of high-throughput screening platform using retina of degenerative models (Campbell et al., 2018; Chen et al., 2018), cell lines (Kim et al., 2018), or pluripotent stem cell-derived retinal organoids (Chen et al., 2023a) should accelerate drug discovery for treatment of IRD.
However, pharmaceutical drugs, particularly the small molecules, are highly penetrant and thus could lead to off-target effects. Therefore, it is crucial yet challenging to identify the right molecule with an excellent pharmacological effect and pharmacokinetics and few off-target impact, which sometimes requires extensive optimization of the lead compound (Tambuyzer et al., 2020). A deeper understanding of the key signaling pathways involved in the therapeutic processes should facilitate the refinement of the lead compound. Furthermore, over 90% of drug candidates fail in Phase I clinical trials due to the differences in pathophysiology and pharmacokinetics between humans and animal models (Horvath et al., 2016). Human pluripotent stem cell-derived retinal organoids structurally and functionally recapitulate in vivo retina in various important aspect (Cowan et al., 2020; Saha et al., 2022) and thus offers a promising platform to test drugs in a human context. Recent studies have begun to employ the organoid platform to evaluate the dose and therapeutic outcome of drugs (Chen et al., 2023a; Corral-Serrano et al., 2023).
6.2.3 Targeting RPE and MG by pharmaceutical approaches
Due to the penetrant property of pharmaceutical drugs, all retinal cell types including RPE and MG are targeted in the treatment. A recent study revealed partial restoration of dysregulated genes in patient-derived retinal organoids upon treatment of IRD drug reserpine (Chen et al., 2023a). However, it is unclear whether such improvement in MG is a therapeutic outcome of the drug treatment or the secondary effect of photoreceptor restoration. Furthermore, the therapeutic and toxic doses may differ among retinal cell types. Evaluation of all retinal cell types in the drug treatment should facilitate the selection of optimal doses to achieve desirable therapeutic outcomes without interfering the homeostasis of the retina. Besides, all targeted cell types should be included in the initial screening to avoid toxic side effects due to undesirable interaction of the drug with cell type-specific molecules. One typical example is the interaction of chloroquine and hydroxychloroquine with melanin, which abrogates the lysosomal activity of RPE and leads to subsequent macular degeneration (Stokkermans et al., 2023).
7 Conclusion and outlook
IRD is a common cause of childhood visual impairment and blindness, yet the disease mechanisms have not been comprehensively investigated and limited treatment options are currently available. A vast majority of IRD are caused by dysfunction and/or degeneration of photoreceptors. In healthy retina, photoreceptor homeostasis is maintained by MG and RPE, which have been shown to remove cellular debris, damaged mitochondria, and excessive ions and release trophic and growth factors. Photoreceptors on the other hand provide metabolites to MG and RPE as energy source. Therefore, photoreceptors tightly interact with MG and RPE and they share a symbiotic relationship to maintain homeostasis of the outer retina. Consequently, genetic mutations that compromise the one could disrupt the others. In favor of this postulation, recent studies reveal MG and RPE pathologies in IRD caused by mutations in photoreceptor-specific or ubiquitously expressed genes, suggesting photoreceptor degeneration could be contributed by cellular dysfunction resulted from genetic mutations and MG/RPE pathologies. MG and RPE should therefore be taken into account in designing treatments for IRD.
With over 280 IRD-causing genes, it is time-consuming and labor-intensive to develop gene therapy targeting individual disease-causing gene. Gene therapeutic approaches targeting shared dysregulated functions (e.g., immune response, glucose metabolism, oxidative stress) among IRD caused by different mutations are being developed (Venkatesh et al., 2015; Xiong et al., 2015; Wang et al., 2019; Xue et al., 2021). MG/RPE pathologies in IRD could be at least partially contributed by their response to photoreceptor dysfunction such as phagocytosis of cell debris and damaged mitochondria. Therefore, designing therapeutics targeting relevant compromised MG/RPE function should hold the promise to be developed into mutation/gene-independent treatments targeting multiple IRD.
Author contributions
XD: Conceptualization, Writing–original draft, Writing–review and editing. AB: Conceptualization, Writing–original draft, Writing–review and editing. HC: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was supported by the Start-up package from Department of Cell, Developmental and Integrative Biology at the University of Alabama of Birmingham and Individual Investigator Research Award of Foundation Fighting Blindness (BR-CMM-0623-0855-UAB) to HC.
Acknowledgments
The figures were created with BioRender.com. We apologize to those whose work could not be included due to space limitation.
Conflict of interest
HC is listed as inventor on a patent application by National Institutes of Health (PCT/US 2021/040157).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agte, S., Junek, S., Matthias, S., Ulbricht, E., Erdmann, I., Wurm, A., et al. (2011). Muller glial cell-provided cellular light guidance through the vital Guinea-pig retina. Biophys. J. 101, 2611–2619. doi:10.1016/j.bpj.2011.09.062
Allikmets, R., Singh, N., Sun, H., Shroyer, N. F., Hutchinson, A., Chidambaram, A., et al. (1997). A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 15, 236–246. doi:10.1038/ng0397-236
Amato, A., Arrigo, A., Aragona, E., Manitto, M. P., Saladino, A., Bandello, F., et al. (2021). Gene therapy in inherited retinal diseases: an update on current state of the art. Front. Med. (Lausanne) 8, 750586. doi:10.3389/fmed.2021.750586
Amin, S. V., Khanna, S., Parvar, S. P., Shaw, L. T., Dao, D., Hariprasad, S. M., et al. (2022). Metformin and retinal diseases in preclinical and clinical studies: insights and review of literature. Exp. Biol. Med. (Maywood) 247, 317–329. doi:10.1177/15353702211069986
Azarian, S. M., and Travis, G. H. (1997). The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt's disease (ABCR). FEBS Lett. 409, 247–252. doi:10.1016/s0014-5793(97)00517-6
Bakondi, B., Lv, W., Lu, B., Jones, M. K., Tsai, Y., Kim, K. J., et al. (2016). In vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol. Ther. 24, 556–563. doi:10.1038/mt.2015.220
Baylor, D. (1996). How photons start vision. Proc. Natl. Acad. Sci. U. S. A. 93, 560–565. doi:10.1073/pnas.93.2.560
Bejarano-Escobar, R., Sanchez-Calderon, H., Otero-Arenas, J., Martin-Partido, G., and Francisco-Morcillo, J. (2017). Muller glia and phagocytosis of cell debris in retinal tissue. J. Anat. 231, 471–483. doi:10.1111/joa.12653
Bellezza, I. (2018). Oxidative stress in age-related macular degeneration: nrf2 as therapeutic target. Front. Pharmacol. 9, 1280. doi:10.3389/fphar.2018.01280
Ben-Yosef, T. (2022). Inherited retinal diseases. Int. J. Mol. Sci. 23, 13467. doi:10.3390/ijms232113467
Bernardos, R. L., Barthel, L. K., Meyers, J. R., and Raymond, P. A. (2007). Late-stage neuronal progenitors in the retina are radial Muller glia that function as retinal stem cells. J. Neurosci. 27, 7028–7040. doi:10.1523/JNEUROSCI.1624-07.2007
Bhattacharya, S., Yin, J., Winborn, C. S., Zhang, Q., Yue, J., and Chaum, E. (2017). Prominin-1 is a novel regulator of autophagy in the human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 58, 2366–2387. doi:10.1167/iovs.16-21162
Biedermann, B., Bringmann, A., and Reichenbach, A. (2002). High-affinity GABA uptake in retinal glial (Muller) cells of the Guinea pig: electrophysiological characterization, immunohistochemical localization, and modeling of efficiency. Glia 39, 217–228. doi:10.1002/glia.10097
Biswal, M. R., Justis, B. D., Han, P., Li, H., Gierhart, D., Dorey, C. K., et al. (2018). Daily zeaxanthin supplementation prevents atrophy of the retinal pigment epithelium (RPE) in a mouse model of mitochondrial oxidative stress. PLoS One 13, e0203816. doi:10.1371/journal.pone.0203816
Blanks, J. C., Pickford, M. S., and Organisciak, D. T. (1992). Ascorbate treatment prevents accumulation of phagosomes in RPE in light damage. Invest. Ophthalmol. Vis. Sci. 33, 2814–2821.
Boccuni, I., Bas-Orth, C., Bruehl, C., Draguhn, A., and Fairless, R. (2023). Glutamate transporter contribution to retinal ganglion cell vulnerability in a rat model of multiple sclerosis. Neurobiol. Dis. 187, 106306. doi:10.1016/j.nbd.2023.106306
Booij, J. C., Baas, D. C., Beisekeeva, J., Gorgels, T. G., and Bergen, A. A. (2010). The dynamic nature of Bruch's membrane. Prog. Retin Eye Res. 29, 1–18. doi:10.1016/j.preteyeres.2009.08.003
Boon, N., Lu, X., Andriessen, C. A., Orlova, M., Quinn, P. M. J., Boon, C. J. F., et al. (2023). Characterization and AAV-mediated CRB gene augmentation in human-derived CRB1(KO) and CRB1(KO)CRB2(+/-) retinal organoids. Mol. Ther. Methods Clin. Dev. 31, 101128. doi:10.1016/j.omtm.2023.101128
Bortner, C. D., Hughes, F. M., and Cidlowski, J. A. (1997). A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 272, 32436–32442. doi:10.1074/jbc.272.51.32436
Bosking, W. H., Beauchamp, M. S., and Yoshor, D. (2017). Electrical stimulation of visual cortex: relevance for the development of visual cortical prosthetics. Annu. Rev. Vis. Sci. 3, 141–166. doi:10.1146/annurev-vision-111815-114525
Boulton, M. E. (2014). Studying melanin and lipofuscin in RPE cell culture models. Exp. Eye Res. 126, 61–67. doi:10.1016/j.exer.2014.01.016
Bringmann, A., Grosche, A., Pannicke, T., and Reichenbach, A. (2013). GABA and glutamate uptake and metabolism in retinal glial (muller) cells. Front. Endocrinol. (Lausanne) 4, 48. doi:10.3389/fendo.2013.00048
Bringmann, A., Iandiev, I., Pannicke, T., Wurm, A., Hollborn, M., Wiedemann, P., et al. (2009). Cellular signaling and factors involved in Muller cell gliosis: neuroprotective and detrimental effects. Prog. Retin Eye Res. 28, 423–451. doi:10.1016/j.preteyeres.2009.07.001
Bringmann, A., Pannicke, T., Grosche, J., Francke, M., Wiedemann, P., Skatchkov, S. N., et al. (2006). Muller cells in the healthy and diseased retina. Prog. Retin Eye Res. 25, 397–424. doi:10.1016/j.preteyeres.2006.05.003
Buck, T. M., Vos, R. M., Alves, C. H., and Wijnholds, J. (2021). AAV-CRB2 protects against vision loss in an inducible CRB1 retinitis pigmentosa mouse model. Mol. Ther. Methods Clin. Dev. 20, 423–441. doi:10.1016/j.omtm.2020.12.012
Bujakowska, K., Audo, I., Mohand-Said, S., Lancelot, M. E., Antonio, A., Germain, A., et al. (2012). CRB1 mutations in inherited retinal dystrophies. Hum. Mutat. 33, 306–315. doi:10.1002/humu.21653
Burke, J. M., Kaczara, P., Skumatz, C. M., Zareba, M., Raciti, M. W., and Sarna, T. (2011). Dynamic analyses reveal cytoprotection by RPE melanosomes against non-photic stress. Mol. Vis. 17, 2864–2877.
Burke, T. R., Duncker, T., Woods, R. L., Greenberg, J. P., Zernant, J., Tsang, S. H., et al. (2014). Quantitative fundus autofluorescence in recessive Stargardt disease. Invest. Ophthalmol. Vis. Sci. 55, 2841–2852. doi:10.1167/iovs.13-13624
Busschaert, N., Park, S. H., Baek, K. H., Choi, Y. P., Park, J., Howe, E. N. W., et al. (2017). A synthetic ion transporter that disrupts autophagy and induces apoptosis by perturbing cellular chloride concentrations. Nat. Chem. 9, 667–675. doi:10.1038/nchem.2706
Campbell, L. J., West, M. C., and Jensen, A. M. (2018). A high content, small molecule screen identifies candidate molecular pathways that regulate rod photoreceptor outer segment renewal. Sci. Rep. 8, 14017. doi:10.1038/s41598-018-32336-y
Carr, B. J., Stanar, P., and Moritz, O. L. (2021). Distinct roles for prominin-1 and photoreceptor cadherin in outer segment disc morphogenesis in CRISPR-altered X. laevis. J. Cell Sci. 134, jcs253906. doi:10.1242/jcs.253906
Cashman, S. M., Ramo, K., and Kumar-Singh, R. (2011). A non membrane-targeted human soluble CD59 attenuates choroidal neovascularization in a model of age related macular degeneration. PLoS One 6, e19078. doi:10.1371/journal.pone.0019078
Cehajic-Kapetanovic, J., Xue, K., Martinez-Fernandez De La Camara, C., Nanda, A., Davies, A., Wood, L. J., et al. (2020). Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 26, 354–359. doi:10.1038/s41591-020-0763-1
Chacon-Camacho, O. F., and Zenteno, J. C. (2015). Review and update on the molecular basis of Leber congenital amaurosis. World J. Clin. Cases 3, 112–124. doi:10.12998/wjcc.v3.i2.112
Chadderton, N., Millington-Ward, S., Palfi, A., O'Reilly, M., Tuohy, G., Humphries, M. M., et al. (2009). Improved retinal function in a mouse model of dominant retinitis pigmentosa following AAV-delivered gene therapy. Mol. Ther. 17, 593–599. doi:10.1038/mt.2008.301
Chen, H. Y., Kelley, R. A., Li, T., and Swaroop, A. (2021a). Primary cilia biogenesis and associated retinal ciliopathies. Semin. Cell Dev. Biol. 110, 70–88. doi:10.1016/j.semcdb.2020.07.013
Chen, H. Y., Lehmann, O. J., and Swaroop, A. (2021b). Genetics and therapy for pediatric eye diseases. EBioMedicine 67, 103360. doi:10.1016/j.ebiom.2021.103360
Chen, H. Y., Swaroop, M., Papal, S., Mondal, A. K., Song, H. B., Campello, L., et al. (2023a). Reserpine maintains photoreceptor survival in retinal ciliopathy by resolving proteostasis imbalance and ciliogenesis defects. Elife 12, e83205. doi:10.7554/eLife.83205
Chen, M., Luo, C., Zhao, J., Devarajan, G., and Xu, H. (2019). Immune regulation in the aging retina. Prog. Retin Eye Res. 69, 159–172. doi:10.1016/j.preteyeres.2018.10.003
Chen, X., Xu, N., Li, J., Zhao, M., and Huang, L. (2023b). Stem cell therapy for inherited retinal diseases: a systematic review and meta-analysis. Stem Cell Res. Ther. 14, 286. doi:10.1186/s13287-023-03526-x
Chen, Y., Chen, Y., Jastrzebska, B., Golczak, M., Gulati, S., Tang, H., et al. (2018). A novel small molecule chaperone of rod opsin and its potential therapy for retinal degeneration. Nat. Commun. 9, 1976. doi:10.1038/s41467-018-04261-1
Chew, L. A., and Iannaccone, A. (2023). Gene-agnostic approaches to treating inherited retinal degenerations. Front. Cell Dev. Biol. 11, 1177838. doi:10.3389/fcell.2023.1177838
Chifflet, S., Correa, V., Nin, V., Justet, C., and Hernandez, J. A. (2004). Effect of membrane potential depolarization on the organization of the actin cytoskeleton of eye epithelia. The role of adherens junctions. Exp. Eye Res. 79, 769–777. doi:10.1016/j.exer.2004.08.031
Choi, V. W., Bigelow, C. E., Mcgee, T. L., Gujar, A. N., Li, H., Hanks, S. M., et al. (2015). AAV-mediated RLBP1 gene therapy improves the rate of dark adaptation in Rlbp1 knockout mice. Mol. Ther. Methods Clin. Dev. 2, 15022. doi:10.1038/mtm.2015.22
Chuang, J. Z., Yang, N., Nakajima, N., Otsu, W., Fu, C., Yang, H. H., et al. (2022). Retinal pigment epithelium-specific CLIC4 mutant is a mouse model of dry age-related macular degeneration. Nat. Commun. 13, 374. doi:10.1038/s41467-021-27935-9
Cideciyan, A. V., and Jacobson, S. G. (2019). Leber congenital amaurosis (LCA): potential for improvement of vision. Invest. Ophthalmol. Vis. Sci. 60, 1680–1695. doi:10.1167/iovs.19-26672
Cideciyan, A. V., Jacobson, S. G., Drack, A. V., Ho, A. C., Charng, J., Garafalo, A. V., et al. (2019). Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 25, 225–228. doi:10.1038/s41591-018-0295-0
Cideciyan, A. V., Swider, M., Aleman, T. S., Tsybovsky, Y., Schwartz, S. B., Windsor, E. A., et al. (2009). ABCA4 disease progression and a proposed strategy for gene therapy. Hum. Mol. Genet. 18, 931–941. doi:10.1093/hmg/ddn421
Corral-Serrano, J. C., Sladen, P. E., Ottaviani, D., Rezek, O. F., Athanasiou, D., Jovanovic, K., et al. (2023). Eupatilin improves cilia defects in human CEP290 ciliopathy models. Cells 12, 1575. doi:10.3390/cells12121575
Country, M. W. (2017). Retinal metabolism: a comparative look at energetics in the retina. Brain Res. 1672, 50–57. doi:10.1016/j.brainres.2017.07.025
Cowan, C. S., Renner, M., De Gennaro, M., Gross-Scherf, B., Goldblum, D., Hou, Y., et al. (2020). Cell types of the human retina and its organoids at single-cell resolution. Cell 182, 1623–1640. doi:10.1016/j.cell.2020.08.013
Craige, B., Tsao, C. C., Diener, D. R., Hou, Y., Lechtreck, K. F., Rosenbaum, J. L., et al. (2010). CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 190, 927–940. doi:10.1083/jcb.201006105
Cremers, F. P., Van De Pol, D. J., Van Kerkhoff, L. P., Wieringa, B., and Ropers, H. H. (1990). Cloning of a gene that is rearranged in patients with choroideraemia. Nature 347, 674–677. doi:10.1038/347674a0
Daiger, S. P., Bowne, S. J., and Sullivan, L. S. (2007). Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 125, 151–158. doi:10.1001/archopht.125.2.151
Daiger, S. P., Sullivan, L. S., and Bowne, S. J. (2013). Genes and mutations causing retinitis pigmentosa. Clin. Genet. 84, 132–141. doi:10.1111/cge.12203
Deng, W. L., Gao, M. L., Lei, X. L., Lv, J. N., Zhao, H., He, K. W., et al. (2018). Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Rep. 10, 2005–1281. doi:10.1016/j.stemcr.2018.05.012
Den Hollander, A. I., Black, A., Bennett, J., and Cremers, F. P. (2010). Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J. Clin. Invest. 120, 3042–3053. doi:10.1172/JCI42258
Diamond, J. S. (2017). Inhibitory interneurons in the retina: types, circuitry, and function. Annu. Rev. Vis. Sci. 3, 1–24. doi:10.1146/annurev-vision-102016-061345
Dias, M. F., Joo, K., Kemp, J. A., Fialho, S. L., Da Silva Cunha, A., Woo, S. J., et al. (2018). Molecular genetics and emerging therapies for retinitis pigmentosa: basic research and clinical perspectives. Prog. Retin Eye Res. 63, 107–131. doi:10.1016/j.preteyeres.2017.10.004
Dimopoulos, I. S., Hoang, S. C., Radziwon, A., Binczyk, N. M., Seabra, M. C., Maclaren, R. E., et al. (2018). Two-year results after AAV2-mediated gene therapy for choroideremia: the alberta experience. Am. J. Ophthalmol. 193, 130–142. doi:10.1016/j.ajo.2018.06.011
Drag, S., Dotiwala, F., and Upadhyay, A. K. (2023). Gene therapy for retinal degenerative diseases: progress, challenges, and future directions. Invest. Ophthalmol. Vis. Sci. 64, 39. doi:10.1167/iovs.64.7.39
Dryja, T. P., Mcgee, T. L., Hahn, L. B., Cowley, G. S., Olsson, J. E., Reichel, E., et al. (1990). Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 323, 1302–1307. doi:10.1056/NEJM199011083231903
Duncan, J. L., Pierce, E. A., Laster, A. M., Daiger, S. P., Birch, D. G., Ash, J. D., et al. (2018). Inherited retinal degenerations: current landscape and knowledge gaps. Transl. Vis. Sci. Technol. 7, 6. doi:10.1167/tvst.7.4.6
Dvoriashyna, M., Foss, A. J. E., Gaffney, E. A., and Repetto, R. (2020). Fluid and solute transport across the retinal pigment epithelium: a theoretical model. J. R. Soc. Interface 17, 20190735. doi:10.1098/rsif.2019.0735
Dyer, M. A., and Cepko, C. L. (2000). Control of Muller glial cell proliferation and activation following retinal injury. Nat. Neurosci. 3, 873–880. doi:10.1038/78774
Dyka, F. M., Molday, L. L., Chiodo, V. A., Molday, R. S., and Hauswirth, W. W. (2019). Dual ABCA4-AAV vector treatment reduces pathogenic retinal A2E accumulation in a mouse model of autosomal recessive Stargardt disease. Hum. Gene Ther. 30, 1361–1370. doi:10.1089/hum.2019.132
Ebrey, T., and Koutalos, Y. (2001). Vertebrate photoreceptors. Prog. Retin Eye Res. 20, 49–94. doi:10.1016/s1350-9462(00)00014-8
Erskine, L., and Herrera, E. (2014). Connecting the retina to the brain. ASN Neuro 6, 1759091414562107. doi:10.1177/1759091414562107
Farnoodian, M., Bose, D., Khristov, V., Susaimanickam, P. J., Maddileti, S., Mariappan, I., et al. (2022). Cell-autonomous lipid-handling defects in Stargardt iPSC-derived retinal pigment epithelium cells. Stem Cell Rep. 17, 2438–2450. doi:10.1016/j.stemcr.2022.10.001
Feeney, L. (1978). Lipofuscin and melanin of human retinal pigment epithelium. Fluorescence, enzyme cytochemical, and ultrastructural studies. Invest. Ophthalmol. Vis. Sci. 17, 583–600.
Feng, Z., Liu, Z., Li, X., Jia, H., Sun, L., Tian, C., et al. (2010). α-Tocopherol is an effective Phase II enzyme inducer: protective effects on acrolein-induced oxidative stress and mitochondrial dysfunction in human retinal pigment epithelial cells. J. Nutr. Biochem. 21, 1222–1231. doi:10.1016/j.jnutbio.2009.10.010
Fischer, M. D., Michalakis, S., Wilhelm, B., Zobor, D., Muehlfriedel, R., Kohl, S., et al. (2020). Safety and vision outcomes of subretinal gene therapy targeting cone photoreceptors in achromatopsia: a nonrandomized controlled trial. JAMA Ophthalmol. 138, 643–651. doi:10.1001/jamaophthalmol.2020.1032
Fishman, G. A., Roberts, M. F., Derlacki, D. J., Grimsby, J. L., Yamamoto, H., Sharon, D., et al. (2004). Novel mutations in the cellular retinaldehyde-binding protein gene (RLBP1) associated with retinitis punctata albescens: evidence of interfamilial genetic heterogeneity and fundus changes in heterozygotes. Arch. Ophthalmol. 122, 70–75. doi:10.1001/archopht.122.1.70
Fritze, O., Filipek, S., Kuksa, V., Palczewski, K., Hofmann, K. P., and Ernst, O. P. (2003). Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. Proc. Natl. Acad. Sci. U. S. A. 100, 2290–2295. doi:10.1073/pnas.0435715100
Frost, L. S., Mitchell, C. H., and Boesze-Battaglia, K. (2014). Autophagy in the eye: implications for ocular cell health. Exp. Eye Res. 124, 56–66. doi:10.1016/j.exer.2014.04.010
Gal, A., Li, Y., Thompson, D. A., Weir, J., Orth, U., Jacobson, S. G., et al. (2000). Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat. Genet. 26, 270–271. doi:10.1038/81555
Gardiner, K. L., Cideciyan, A. V., Swider, M., Dufour, V. L., Sumaroka, A., Komaromy, A. M., et al. (2020). Long-term structural outcomes of late-stage RPE65 gene therapy. Mol. Ther. 28, 266–278. doi:10.1016/j.ymthe.2019.08.013
Gelisken, O., and De Laey, J. J. (1985). A clinical review of Stargardt's disease and/or fundus flavimaculatus with follow-up. Int. Ophthalmol. 8, 225–235. doi:10.1007/BF00137651
Getter, T., Suh, S., Hoang, T., Handa, J. T., Dong, Z., Ma, X., et al. (2019). The selective estrogen receptor modulator raloxifene mitigates the effect of all-trans-retinal toxicity in photoreceptor degeneration. J. Biol. Chem. 294, 9461–9475. doi:10.1074/jbc.RA119.008697
Ghazi, N. G., Abboud, E. B., Nowilaty, S. R., Alkuraya, H., Alhommadi, A., Cai, H., et al. (2016). Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum. Genet. 135, 327–343. doi:10.1007/s00439-016-1637-y
Ginn, S. L., Amaya, A. K., Alexander, I. E., Edelstein, M., and Abedi, M. R. (2018). Gene therapy clinical trials worldwide to 2017: an update. J. Gene Med. 20, e3015. doi:10.1002/jgm.3015
Goldman, D. (2014). Muller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci. 15, 431–442. doi:10.1038/nrn3723
Gong, J., Cheung, S., Fasso-Opie, A., Galvin, O., Moniz, L. S., Earle, D., et al. (2021). The impact of inherited retinal diseases in the United States of America (US) and Canada from a cost-of-illness perspective. Clin. Ophthalmol. 15, 2855–2866. doi:10.2147/OPTH.S313719
Gooley, J. J., Rajaratnam, S. M., Brainard, G. C., Kronauer, R. E., Czeisler, C. A., and Lockley, S. W. (2010). Spectral responses of the human circadian system depend on the irradiance and duration of exposure to light. Sci. Transl. Med. 2, 31ra33. doi:10.1126/scitranslmed.3000741
Gosens, I., Den Hollander, A. I., Cremers, F. P., and Roepman, R. (2008). Composition and function of the Crumbs protein complex in the mammalian retina. Exp. Eye Res. 86, 713–726. doi:10.1016/j.exer.2008.02.005
Grishanin, R., Vuillemenot, B., Sharma, P., Keravala, A., Greengard, J., Gelfman, C., et al. (2019). Preclinical evaluation of ADVM-022, a novel gene therapy approach to treating wet age-related macular degeneration. Mol. Ther. 27, 118–129. doi:10.1016/j.ymthe.2018.11.003
Grossman, G. H., Watson, R. F., Pauer, G. J., Bollinger, K., and Hagstrom, S. A. (2011). Immunocytochemical evidence of Tulp1-dependent outer segment protein transport pathways in photoreceptor cells. Exp. Eye Res. 93, 658–668. doi:10.1016/j.exer.2011.08.005
Guadagni, V., Novelli, E., Piano, I., Gargini, C., and Strettoi, E. (2015). Pharmacological approaches to retinitis pigmentosa: a laboratory perspective. Prog. Retin Eye Res. 48, 62–81. doi:10.1016/j.preteyeres.2015.06.005
Guy, J., Feuer, W. J., Davis, J. L., Porciatti, V., Gonzalez, P. J., Koilkonda, R. D., et al. (2017). Gene therapy for leber hereditary optic neuropathy: low- and medium-dose visual results. Ophthalmology 124, 1621–1634. doi:10.1016/j.ophtha.2017.05.016
Guziewicz, K. E., Sinha, D., Gomez, N. M., Zorych, K., Dutrow, E. V., Dhingra, A., et al. (2017). Bestrophinopathy: an RPE-photoreceptor interface disease. Prog. Retin Eye Res. 58, 70–88. doi:10.1016/j.preteyeres.2017.01.005
Hahn, L. C., Van Schooneveld, M. J., Wesseling, N. L., Florijn, R. J., Ten Brink, J. B., Lissenberg-Witte, B. I., et al. (2022). X-linked retinoschisis: novel clinical observations and genetic spectrum in 340 patients. Ophthalmology 129, 191–202. doi:10.1016/j.ophtha.2021.09.021
Han, Z., Anderson, D. W., and Papermaster, D. S. (2012). Prominin-1 localizes to the open rims of outer segment lamellae in Xenopus laevis rod and cone photoreceptors. Invest. Ophthalmol. Vis. Sci. 53, 361–373. doi:10.1167/iovs.11-8635
Hao, W., and Fong, H. K. (1999). The endogenous chromophore of retinal G protein-coupled receptor opsin from the pigment epithelium. J. Biol. Chem. 274, 6085–6090. doi:10.1074/jbc.274.10.6085
Hartsock, A., and Nelson, W. J. (2008). Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 1778, 660–669. doi:10.1016/j.bbamem.2007.07.012
Heier, J. S., Kherani, S., Desai, S., Dugel, P., Kaushal, S., Cheng, S. H., et al. (2017). Intravitreous injection of AAV2-sFLT01 in patients with advanced neovascular age-related macular degeneration: a phase 1, open-label trial. Lancet 390, 50–61. doi:10.1016/S0140-6736(17)30979-0
Hernan, I., Gamundi, M. J., Planas, E., Borras, E., Maseras, M., and Carballo, M. (2011). Cellular expression and siRNA-mediated interference of rhodopsin cis-acting splicing mutants associated with autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 52, 3723–3729. doi:10.1167/iovs.10-6933
Hipp, S., Zobor, G., Glockle, N., Mohr, J., Kohl, S., Zrenner, E., et al. (2015). Phenotype variations of retinal dystrophies caused by mutations in the RLBP1 gene. Acta Ophthalmol. 93, E281–E286. doi:10.1111/aos.12573
Hoon, M., Okawa, H., Della Santina, L., and Wong, R. O. (2014). Functional architecture of the retina: development and disease. Prog. Retin Eye Res. 42, 44–84. doi:10.1016/j.preteyeres.2014.06.003
Horvath, P., Aulner, N., Bickle, M., Davies, A. M., Nery, E. D., Ebner, D., et al. (2016). Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 15, 751–769. doi:10.1038/nrd.2016.175
Hu, J. Y., Zhu, M. L., Li, D., Wu, Q., and Le, Y. Z. (2021a). VEGF as a direct functional regulator of photoreceptors and contributing factor to diabetes-induced alteration of photoreceptor function. Biomolecules 11, 988. doi:10.3390/biom11070988
Hu, M. L., Quinn, J., and Xue, K. M. (2021b). Interactions between apolipoprotein E metabolism and retinal inflammation in age-related macular degeneration. Life-Basel 11, 635. doi:10.3390/life11070635
Huang, P., Narendran, S., Pereira, F., Fukuda, S., Nagasaka, Y., Apicella, I., et al. (2022). The learning curve of murine subretinal injection among clinically trained ophthalmic surgeons. Transl. Vis. Sci. Technol. 11, 13. doi:10.1167/tvst.11.3.13
Hughes, F. M., and Cidlowski, J. A. (1999). Potassium is a critical regulator of apoptotic enzymes in vitro and in vivo. Adv. Enzyme Regul. 39, 157–171. doi:10.1016/s0065-2571(98)00010-7
Hurley, J. B. (2021). Retina metabolism and metabolism in the pigmented epithelium: a busy intersection. Annu. Rev. Vis. Sci. 7, 665–692. doi:10.1146/annurev-vision-100419-115156
Hutmacher, F. (2019). Why is there so much more research on vision than on any other sensory modality? Front. Psychol. 10, 2246. doi:10.3389/fpsyg.2019.02246
Hutto, R. A., Rutter, K. M., Giarmarco, M. M., Parker, E. D., Chambers, Z. S., and Brockerhoff, S. E. (2023). Cone photoreceptors transfer damaged mitochondria to Muller glia. Cell Rep. 42, 112115. doi:10.1016/j.celrep.2023.112115
Ignatova, I., Frolov, R., and Nymark, S. (2023). The retinal pigment epithelium displays electrical excitability and lateral signal spreading. BMC Biol. 21, 84. doi:10.1186/s12915-023-01559-5
Illing, M., Molday, L. L., and Molday, R. S. (1997). The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J. Biol. Chem. 272, 10303–10310. doi:10.1074/jbc.272.15.10303
Intartaglia, D., Giamundo, G., Naso, F., Nusco, E., Di Giulio, S., Salierno, F. G., et al. (2022). Induction of autophagy promotes clearance of RHO(P23H) aggregates and protects from retinal degeneration. Front. Aging Neurosci. 14, 878958. doi:10.3389/fnagi.2022.878958
Jacobson, S. G., Cideciyan, A. V., Aleman, T. S., Pianta, M. J., Sumaroka, A., Schwartz, S. B., et al. (2003). Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum. Mol. Genet. 12, 1073–1078. doi:10.1093/hmg/ddg117
Jacobson, S. G., Cideciyan, A. V., Ratnakaram, R., Heon, E., Schwartz, S. B., Roman, A. J., et al. (2012). Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch. Ophthalmol. 130, 9–24. doi:10.1001/archophthalmol.2011.298
Jacobson, S. G., Cideciyan, A. V., Sumaroka, A., Roman, A. J., Charng, J., Lu, M., et al. (2017). Outcome measures for clinical trials of leber congenital amaurosis caused by the intronic mutation in the CEP290 gene. Invest. Ophthalmol. Vis. Sci. 58, 2609–2622. doi:10.1167/iovs.17-21560
Jiang, M., Pandey, S., and Fong, H. K. W. (1993). An opsin homologue in the retina and pigment epithelium. Investigative Ophthalmol. Vis. Sci. 34, 3669–3678.
Johansson, J. K., Karema-Jokinen, V. I., Hakanen, S., Jylha, A., Uusitalo, H., Vihinen-Ranta, M., et al. (2019). Sodium channels enable fast electrical signaling and regulate phagocytosis in the retinal pigment epithelium. BMC Biol. 17, 63. doi:10.1186/s12915-019-0681-1
John, M. C., Quinn, J., Hu, M. L., Cehajic-Kapetanovic, J., and Xue, K. (2022). Gene-agnostic therapeutic approaches for inherited retinal degenerations. Front. Mol. Neurosci. 15, 1068185. doi:10.3389/fnmol.2022.1068185
Johnson, A. A., Guziewicz, K. E., Lee, C. J., Kalathur, R. C., Pulido, J. S., Marmorstein, L. Y., et al. (2017). Bestrophin 1 and retinal disease. Prog. Retin Eye Res. 58, 45–69. doi:10.1016/j.preteyeres.2017.01.006
Jones, B. W., Pfeiffer, R. L., Ferrell, W. D., Watt, C. B., Marmor, M., and Marc, R. E. (2016). Retinal remodeling in human retinitis pigmentosa. Exp. Eye Res. 150, 149–165. doi:10.1016/j.exer.2016.03.018
Kanow, M. A., Giarmarco, M. M., Jankowski, C. S., Tsantilas, K., Engel, A. L., Du, J., et al. (2017). Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife 6, e28899. doi:10.7554/eLife.28899
Kansara, V., Muya, L., Wan, C. R., and Ciulla, T. A. (2020). Suprachoroidal delivery of viral and nonviral gene therapy for retinal diseases. J. Ocul. Pharmacol. Ther. 36, 384–392. doi:10.1089/jop.2019.0126
Kanzaki, Y., Fujita, H., Sato, K., Hosokawa, M., Matsumae, H., Shiraga, F., et al. (2020). KCNJ13 gene deletion impairs cell alignment and phagocytosis in retinal pigment epithelium derived from human-induced pluripotent stem cells. Invest. Ophthalmol. Vis. Sci. 61, 38. doi:10.1167/iovs.61.5.38
Karali, M., Testa, F., Di Iorio, V., Torella, A., Zeuli, R., Scarpato, M., et al. (2022). Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Sci. Rep. 12, 20815. doi:10.1038/s41598-022-24636-1
Katoozi, S., Rao, S. B., Skauli, N., Froehner, S. C., Ottersen, O. P., Adams, M. E., et al. (2020). Functional specialization of retinal Muller cell endfeet depends on an interplay between two syntrophin isoforms. Mol. Brain 13, 40. doi:10.1186/s13041-020-00581-w
Kennan, A., Aherne, A., Bowne, S. J., Daiger, S. P., Farrar, G. J., Kenna, P. F., et al. (2003). On the role of IMPDH1 in retinal degeneration. Adv. Exp. Med. Biol. 533, 13–18. doi:10.1007/978-1-4615-0067-4_2
Kevany, B. M., and Palczewski, K. (2010). Phagocytosis of retinal rod and cone photoreceptors. Physiol. (Bethesda) 25, 8–15. doi:10.1152/physiol.00038.2009
Khan, M. I., Kersten, F. F., Azam, M., Collin, R. W., Hussain, A., Shah, S. T., et al. (2011). CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology 118, 1444–1448. doi:10.1016/j.ophtha.2010.10.047
Kim, Y. J., Kim, S., Jung, Y., Jung, E., Kwon, H. J., and Kim, J. (2018). Eupatilin rescues ciliary transition zone defects to ameliorate ciliopathy-related phenotypes. J. Clin. Invest. 128, 3642–3648. doi:10.1172/JCI99232
Kolesnikov, A. V., Kiser, P. D., Palczewski, K., and Kefalov, V. J. (2021). Function of mammalian M-cones depends on the level of CRALBP in Muller cells. J. General Physiology 153, e202012675. doi:10.1085/jgp.202012675
Kong, L., Chu, G., Ma, W., Liang, J., Liu, D., Liu, Q., et al. (2023). Mutations in VWA8 cause autosomal-dominant retinitis pigmentosa via aberrant mitophagy activation. J. Med. Genet. 60, 939–950. doi:10.1136/jmg-2022-108888
Kretschmer, V., Schneider, S., Matthiessen, P. A., Reichert, D., Hotaling, N., Glassser, G., et al. (2023). Deletion of IFT20 exclusively in the RPE ablates primary cilia and leads to retinal degeneration. PLoS Biol. 21, e3002402. doi:10.1371/journal.pbio.3002402
Kumaran, N., Moore, A. T., Weleber, R. G., and Michaelides, M. (2017). Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 101, 1147–1154. doi:10.1136/bjophthalmol-2016-309975
Kwon, W., and Freeman, S. A. (2020). Phagocytosis by the retinal pigment epithelium: recognition, resolution, recycling. Front. Immunol. 11, 604205. doi:10.3389/fimmu.2020.604205
Lahne, M., Nagashima, M., Hyde, D. R., and Hitchcock, P. F. (2020). Reprogramming muller glia to regenerate retinal neurons. Annu. Rev. Vis. Sci. 6, 171–193. doi:10.1146/annurev-vision-121219-081808
Lakkaraju, A., Umapathy, A., Tan, L. X., Daniele, L., Philp, N. J., Boesze-Battaglia, K., et al. (2020). The cell biology of the retinal pigment epithelium. Prog. Retin Eye Res. 78, 100846. doi:10.1016/j.preteyeres.2020.100846
Lall, G. S., Revell, V. L., Momiji, H., Al Enezi, J., Altimus, C. M., Guler, A. D., et al. (2010). Distinct contributions of rod, cone, and melanopsin photoreceptors to encoding irradiance. Neuron 66, 417–428. doi:10.1016/j.neuron.2010.04.037
Lam, B. L., Davis, J. L., Gregori, N. Z., Maclaren, R. E., Girach, A., Verriotto, J. D., et al. (2019). Choroideremia gene therapy phase 2 clinical trial: 24-month results. Am. J. Ophthalmol. 197, 65–73. doi:10.1016/j.ajo.2018.09.012
Lassiale, S., Valamanesh, F., Klein, C., Hicks, D., Abitbol, M., Versaux-Botteri, C., et al. (2016). Changes in aquaporin-4 and Kir4.1 expression in rats with inherited retinal dystrophy. Exp. Eye Res. 148, 33–44. doi:10.1016/j.exer.2016.05.010
Lavail, M. M., Yasumura, D., Matthes, M. T., Yang, H., Hauswirth, W. W., Deng, W. T., et al. (2016). Gene therapy for MERTK-associated retinal degenerations. Adv. Exp. Med. Biol. 854, 487–493. doi:10.1007/978-3-319-17121-0_65
Leinonen, H. O., Bull, E., and Fu, Z. (2023). Neural and Muller glial adaptation of the retina to photoreceptor degeneration. Neural Regen. Res. 18, 701–707. doi:10.4103/1673-5374.354511
Lenis, T. L., Hu, J., Ng, S. Y., Jiang, Z., Sarfare, S., Lloyd, M. B., et al. (2018). Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 115, E11120–E11127. doi:10.1073/pnas.1802519115
Lepanto, P., Davison, C., Casanova, G., Badano, J. L., and Zolessi, F. R. (2016). Characterization of primary cilia during the differentiation of retinal ganglion cells in the zebrafish. Neural Dev. 11, 10. doi:10.1186/s13064-016-0064-z
Leroy, B. P., Birch, D. G., Duncan, J. L., Lam, B. L., Koenekoop, R. K., Porto, F. B. O., et al. (2021). LEBER congenital amaurosis due to CEP290 MUTATIONS-SEVERE VISION impairment with A high unmet medical need: a review. Retina 41, 898–907. doi:10.1097/IAE.0000000000003133
Leroy, B. P., Fischer, M. D., Flannery, J. G., Maclaren, R. E., Dalkara, D., Scholl, H. P. N., et al. (2022). Gene therapy for inherited retinal disease: long-term durability of effect. Ophthalmic Res. 66, 179–196. doi:10.1159/000526317
Leveillard, T., Mohand-Said, S., Lorentz, O., Hicks, D., Fintz, A. C., Clerin, E., et al. (2004). Identification and characterization of rod-derived cone viability factor. Nat. Genet. 36, 755–759. doi:10.1038/ng1386
Lewin, A. S., Drenser, K. A., Hauswirth, W. W., Nishikawa, S., Yasumura, D., Flannery, J. G., et al. (1998). Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat. Med. 4, 967–971. doi:10.1038/nm0898-967
Li, X. M., Wendu, R. L., Yao, J., Ren, Y., Zhao, Y. X., Cao, G. F., et al. (2014). Abnormal glutamate metabolism in the retina of aquaporin 4 (AQP4) knockout mice upon light damage. Neurol. Sci. 35, 847–853. doi:10.1007/s10072-013-1610-7
Li, J., Patil, R. V., and Verkman, A. S. (2002). Mildly abnormal retinal function in transgenic mice without Muller cell aquaporin-4 water channels. Investigative Ophthalmol. Vis. Sci. 43, 573–579.
Lin, J., Nishiguchi, K. M., Nakamura, M., Dryja, T. P., Berson, E. L., and Miyake, Y. (2005). Recessive mutations in the CYP4V2 gene in East Asian and Middle Eastern patients with Bietti crystalline corneoretinal dystrophy. J. Med. Genet. 42, e38. doi:10.1136/jmg.2004.029066
Li, P., Kleinstiver, B. P., Leon, M. Y., Prew, M. S., Navarro-Gomez, D., Greenwald, S. H., et al. (2018). Allele-specific CRISPR-cas9 genome editing of the single-base P23H mutation for rhodopsin-associated dominant retinitis pigmentosa. CRISPR J. 1, 55–64. doi:10.1089/crispr.2017.0009
Liu, Q., Sabirzhanova, I., Bergbower, E. A. S., Yanda, M., Guggino, W. G., and Cebotaru, L. (2019). The CFTR corrector, VX-809 (lumacaftor), rescues ABCA4 trafficking mutants: a potential treatment for Stargardt disease. Cell Physiol. Biochem. 53, 400–412. doi:10.33594/000000146
Liuska, P. J., Ramo, J. T., Lemmela, S., Kaarniranta, K., Uusitalo, H., Lahtela, E., et al. (2023). Association of APOE haplotypes with common age-related ocular diseases in 412,171 individuals. Invest. Ophthalmol. Vis. Sci. 64, 33. doi:10.1167/iovs.64.14.33
Li, W., He, X. D., Yang, Z. T., Han, D. M., Sun, Y., Chen, Y. X., et al. (2023). De novo mutations contributes approximately 7% of pathogenicity in inherited eye diseases. Invest. Ophthalmol. Vis. Sci. 64, 5. doi:10.1167/iovs.64.2.5
Lopes, V. S., Gibbs, D., Libby, R. T., Aleman, T. S., Welch, D. L., Lillo, C., et al. (2011). The Usher 1B protein, MYO7A, is required for normal localization and function of the visual retinoid cycle enzyme, RPE65. Hum. Mol. Genet. 20, 2560–2570. doi:10.1093/hmg/ddr155
Low, B. E., Krebs, M. P., Joung, J. K., Tsai, S. Q., Nishina, P. M., and Wiles, M. V. (2014). Correction of the Crb1rd8 allele and retinal phenotype in C57BL/6N mice via TALEN-mediated homology-directed repair. Invest. Ophthalmol. Vis. Sci. 55, 387–395. doi:10.1167/iovs.13-13278
Maclachlan, T. K., Milton, M. N., Turner, O., Tukov, F., Choi, V. W., Penraat, J., et al. (2018). Nonclinical safety evaluation of scAAV8-RLBP1 for treatment of RLBP1 retinitis pigmentosa. Mol. Ther. Methods Clin. Dev. 8, 105–120. doi:10.1016/j.omtm.2017.12.001
Maisam Afzali, A., Stuve, L., Pfaller, M., Aly, L., Steiger, K., Knier, B., et al. (2022). Aquaporin-4 prevents exaggerated astrocytosis and structural damage in retinal inflammation. J. Mol. Med. Berl. 100, 933–946. doi:10.1007/s00109-022-02202-6
Mao, B. Q., Macleish, P. R., and Victor, J. D. (2003). Role of hyperpolarization-activated currents for the intrinsic dynamics of isolated retinal neurons. Biophys. J. 84, 2756–2767. doi:10.1016/S0006-3495(03)75080-2
Marmorstein, A. D., Marmorstein, L. Y., Rayborn, M., Wang, X., Hollyfield, J. G., and Petrukhin, K. (2000). Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. U. S. A. 97, 12758–12763. doi:10.1073/pnas.220402097
Masland, R. H. (2012). The neuronal organization of the retina. Neuron 76, 266–280. doi:10.1016/j.neuron.2012.10.002
Mata, N. L., Weng, J., and Travis, G. H. (2000). Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 97, 7154–7159. doi:10.1073/pnas.130110497
Mauch, D. H., Nagler, K., Schumacher, S., Goritz, C., Muller, E. C., Otto, A., et al. (2001). CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357. doi:10.1126/science.294.5545.1354
Maugeri, A., Klevering, B. J., Rohrschneider, K., Blankenagel, A., Brunner, H. G., Deutman, A. F., et al. (2000). Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 67, 960–966. doi:10.1086/303079
Maw, M. A., Kennedy, B., Knight, A., Bridges, R., Roth, K. E., Mani, E. J., et al. (1997). Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat. Genet. 17, 198–200. doi:10.1038/ng1097-198
May-Simera, H. L., Wan, Q., Jha, B. S., Hartford, J., Khristov, V., Dejene, R., et al. (2018). Primary cilium-mediated retinal pigment epithelium maturation is disrupted in ciliopathy patient cells. Cell Rep. 22, 189–205. doi:10.1016/j.celrep.2017.12.038
Mazzoni, F., Safa, H., and Finnemann, S. C. (2014). Understanding photoreceptor outer segment phagocytosis: use and utility of RPE cells in culture. Exp. Eye Res. 126, 51–60. doi:10.1016/j.exer.2014.01.010
Mcclements, M. E., and Maclaren, R. E. (2017). Adeno-associated virus (AAV) dual vector strategies for gene therapy encoding large transgenes. Yale J. Biol. Med. 90, 611–623.
Mead, B., and Tomarev, S. (2016). Evaluating retinal ganglion cell loss and dysfunction. Exp. Eye Res. 151, 96–106. doi:10.1016/j.exer.2016.08.006
Mehalow, A. K., Kameya, S., Smith, R. S., Hawes, N. L., Denegre, J. M., Young, J. A., et al. (2003). CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum. Mol. Genet. 12, 2179–2189. doi:10.1093/hmg/ddg232
Mendell, J. R., Al-Zaidy, S. A., Rodino-Klapac, L. R., Goodspeed, K., Gray, S. J., Kay, C. N., et al. (2021). Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther. 29, 464–488. doi:10.1016/j.ymthe.2020.12.007
Miceli, M. V., Liles, M. R., and Newsome, D. A. (1994). Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp. Cell Res. 214, 242–249. doi:10.1006/excr.1994.1254
Mishra, A., Vijayasarathy, C., Cukras, C. A., Wiley, H. E., Sen, H. N., Zeng, Y., et al. (2021). Immune function in X-linked retinoschisis subjects in an AAV8-RS1 phase I/IIa gene therapy trial. Mol. Ther. 29, 2030–2040. doi:10.1016/j.ymthe.2021.02.013
Mockel, A., Obringer, C., Hakvoort, T. B., Seeliger, M., Lamers, W. H., Stoetzel, C., et al. (2012). Pharmacological modulation of the retinal unfolded protein response in Bardet-Biedl syndrome reduces apoptosis and preserves light detection ability. J. Biol. Chem. 287, 37483–37494. doi:10.1074/jbc.M112.386821
Molday, R. S. (2015). Insights into the molecular properties of ABCA4 and its role in the visual cycle and Stargardt disease. Prog. Mol. Biol. Transl. Sci. 134, 415–431. doi:10.1016/bs.pmbts.2015.06.008
Molday, R. S., and Moritz, O. L. (2015). Photoreceptors at a glance. J. Cell Sci. 128, 4039–4045. doi:10.1242/jcs.175687
Morgan, J. I. W., Jiang, Y. Y., Vergilio, G. K., Serrano, L. W., Pearson, D. J., Bennett, J., et al. (2022). Short-term assessment of subfoveal injection of adeno-associated virus-mediated hCHM gene augmentation in choroideremia using adaptive optics ophthalmoscopy. JAMA Ophthalmol. 140, 411–420. doi:10.1001/jamaophthalmol.2022.0158
Morshedian, A., Kaylor, J. J., Ng, S. Y., Tsan, A., Frederiksen, R., Xu, T., et al. (2019). Light-driven regeneration of cone visual pigments through a mechanism involving RGR opsin in muller glial cells. Neuron 102, 1172–1183. doi:10.1016/j.neuron.2019.04.004
Murray, S. F., Jazayeri, A., Matthes, M. T., Yasumura, D., Yang, H., Peralta, R., et al. (2015). Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest. Ophthalmol. Vis. Sci. 56, 6362–6375. doi:10.1167/iovs.15-16400
Murthy, R. K., Ravi, K., Balaiya, S., Brar, V. S., and Chalam, K. V. (2014). Lutein protects retinal pigment epithelium from cytotoxic oxidative stress. Cutan. Ocul. Toxicol. 33, 132–137. doi:10.3109/15569527.2013.812108
Mysore, N., Koenekoop, J., Li, S., Ren, H., Keser, V., Lopez-Solache, I., et al. (2014). A review of secondary photoreceptor degenerations in systemic disease. Cold Spring Harb. Perspect. Med. 5, a025825. doi:10.1101/cshperspect.a025825
Naftali Ben Haim, L., and Moisseiev, E. (2021). Drug delivery via the suprachoroidal space for the treatment of retinal diseases. Pharmaceutics 13, 967. doi:10.3390/pharmaceutics13070967
Napoli, J. L. (2016). Functions of intracellular retinoid binding-proteins. Subcell. Biochem. 81, 21–76. doi:10.1007/978-94-024-0945-1_2
Naskar, R., Vorwerk, C. K., and Dreyer, E. B. (2000). Concurrent downregulation of a glutamate transporter and receptor in glaucoma. Investigative Ophthalmol. Vis. Sci. 41, 1940–1944.
Neely, J. D., Amiry-Moghaddam, M., Ottersen, O. P., Froehner, S. C., Agre, P., and Adams, M. E. (2001). Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. U. S. A. 98, 14108–14113. doi:10.1073/pnas.241508198
Netti, V., Fernández, J., Melamud, L., Garcia-Miranda, P., Di Giusto, G., Ford, P., et al. (2021). Aquaporin-4 removal from the plasma membrane of human muller cells by AQP4-IgG from patients with neuromyelitis optica induces changes in cell volume homeostasis: the first step of retinal injury? Mol. Neurobiol. 58, 5178–5193. doi:10.1007/s12035-021-02491-x
Newsome, D. A., Dobard, E. P., Liles, M. R., and Oliver, P. D. (1990). Human retinal pigment epithelium contains two distinct species of superoxide dismutase. Invest. Ophthalmol. Vis. Sci. 31, 2508–2513.
Ning, K., Sendayen, B. E., Kowal, T. J., Wang, B., Jones, B. W., Hu, Y., et al. (2021). Primary cilia in amacrine cells in retinal development. Invest. Ophthalmol. Vis. Sci. 62, 15. doi:10.1167/iovs.62.9.15
Nishiguchi, K. M., Nakamura, M., Kaneko, H., Kachi, S., and Terasaki, H. (2007). The role of VEGF and VEGFR2/Flk1 in proliferation of retinal progenitor cells in murine retinal degeneration. Investigative Ophthalmol. Vis. Sci. 48, 4315–4320. doi:10.1167/iovs.07-0354
Nolan, N. D., Jenny, L. A., Wang, N. K., and Tsang, S. H. (2021). Retinal pigment epithelium lipid metabolic demands and therapeutic restoration. Taiwan J. Ophthalmol. 11, 216–220. doi:10.4103/tjo.tjo_31_21
Nuzbrokh, Y., Ragi, S. D., and Tsang, S. H. (2021). Gene therapy for inherited retinal diseases. Ann. Transl. Med. 9, 1278. doi:10.21037/atm-20-4726
Oakley, B. (1977). Potassium and the photoreceptor-dependent pigment epithelial hyperpolarization. J. Gen. Physiol. 70, 405–425. doi:10.1085/jgp.70.4.405
Oliver, P. D., and Newsome, D. A. (1992). Mitochondrial superoxide dismutase in mature and developing human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 33, 1909–1918.
Omri, S., Omri, B., Savoldelli, M., Jonet, L., Thillaye-Goldenberg, B., Thuret, G., et al. (2010). The outer limiting membrane (OLM) revisited: clinical implications. Clin. Ophthalmol. 4, 183–195. doi:10.2147/opth.s5901
O'Reilly, M., Millington-Ward, S., Palfi, A., Chadderton, N., Cronin, T., Mcnally, N., et al. (2008). A transgenic mouse model for gene therapy of rhodopsin-linked Retinitis Pigmentosa. Vis. Res. 48, 386–391. doi:10.1016/j.visres.2007.08.014
Ozawa, Y. (2020). Oxidative stress in the light-exposed retina and its implication in age-related macular degeneration. Redox Biol. 37, 101779. doi:10.1016/j.redox.2020.101779
Pahlberg, J., Frederiksen, R., Pollock, G. E., Miyagishima, K. J., Sampath, A. P., and Cornwall, M. C. (2017). Voltage-sensitive conductances increase the sensitivity of rod photoresponses following pigment bleaching. J. Physiol. 595, 3459–3469. doi:10.1113/JP273398
Palczewski, K., and Kiser, P. D. (2020). Shedding new light on the generation of the visual chromophore. Proc. Natl. Acad. Sci. U. S. A. 117, 19629–19638. doi:10.1073/pnas.2008211117
Palfi, A., Yesmambetov, A., Humphries, P., Hokamp, K., and Farrar, G. J. (2020). Non-photoreceptor expression of Tulp1 may contribute to extensive retinal degeneration in Tulp1-/- mice. Front. Neurosci. 14, 656. doi:10.3389/fnins.2020.00656
Pannicke, T., Wurm, A., Iandiev, I., Hollborn, M., Linnertz, R., Binder, D. K., et al. (2010). Deletion of aquaporin-4 renders retinal glial cells more susceptible to osmotic stress. J. Neurosci. Res. 88, 2877–2888. doi:10.1002/jnr.22437
Parfitt, D. A., Lane, A., Ramsden, C. M., Carr, A. F., Munro, P. M., Jovanovic, K., et al. (2016). Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell Stem Cell 18, 769–781. doi:10.1016/j.stem.2016.03.021
Parker, M. A., Erker, L. R., Audo, I., Choi, D., Mohand-Said, S., Sestakauskas, K., et al. (2022). Three-year safety results of SAR422459 (EIAV-ABCA4) gene therapy in patients with ABCA4-associated Stargardt disease: an open-label dose-escalation phase I/IIa clinical trial, cohorts 1-5. Am. J. Ophthalmol. 240, 285–301. doi:10.1016/j.ajo.2022.02.013
Pasquay, C., Wang, L. F., Lorenz, B., and Preising, M. N. (2015). Bestrophin 1--phenotypes and functional aspects in bestrophinopathies. Ophthalmic Genet. 36, 193–212. doi:10.3109/13816810.2013.863945
Paylakhi, S., Labelle-Dumais, C., Tolman, N. G., Sellarole, M. A., Seymens, Y., Saunders, J., et al. (2018). Muller glia-derived PRSS56 is required to sustain ocular axial growth and prevent refractive error. Plos Genet. 14, e1007244. doi:10.1371/journal.pgen.1007244
Pellissier, L. P., Lundvig, D. M., Tanimoto, N., Klooster, J., Vos, R. M., Richard, F., et al. (2014). CRB2 acts as a modifying factor of CRB1-related retinal dystrophies in mice. Hum. Mol. Genet. 23, 3759–3771. doi:10.1093/hmg/ddu089
Pellissier, L. P., Quinn, P. M., Alves, C. H., Vos, R. M., Klooster, J., Flannery, J. G., et al. (2015). Gene therapy into photoreceptors and Muller glial cells restores retinal structure and function in CRB1 retinitis pigmentosa mouse models. Hum. Mol. Genet. 24, 3104–3118. doi:10.1093/hmg/ddv062
Pfeiffer, R. L., Marc, R. E., and Jones, B. W. (2020). Muller cell metabolic signatures: evolutionary conservation and disruption in disease. Trends Endocrinol. Metab. 31, 320–329. doi:10.1016/j.tem.2020.01.005
Pfister, T. A., Zein, W. M., Cukras, C. A., Sen, H. N., Maldonado, R. S., Huryn, L. A., et al. (2021). Phenotypic and genetic spectrum of autosomal recessive bestrophinopathy and best vitelliform macular dystrophy. Invest. Ophthalmol. Vis. Sci. 62, 22. doi:10.1167/iovs.62.6.22
Pierce, E. A., and Bennett, J. (2015). The status of RPE65 gene therapy trials: safety and efficacy. Cold Spring Harb. Perspect. Med. 5, a017285. doi:10.1101/cshperspect.a017285
Planul, A., and Dalkara, D. (2017). Vectors and gene delivery to the retina. Annu. Rev. Vis. Sci. 3, 121–140. doi:10.1146/annurev-vision-102016-061413
Politi, L., Rotstein, N., and Carri, N. (2001). Effects of docosahexaenoic acid on retinal development: cellular and molecular aspects. Lipids 36, 927–935. doi:10.1007/s11745-001-0803-8
Qiu, Y., Yao, J., Jia, L., Thompson, D. A., and Zacks, D. N. (2019). Shifting the balance of autophagy and proteasome activation reduces proteotoxic cell death: a novel therapeutic approach for restoring photoreceptor homeostasis. Cell Death Dis. 10, 547. doi:10.1038/s41419-019-1780-1
Quazi, F., Lenevich, S., and Molday, R. S. (2012). ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 3, 925. doi:10.1038/ncomms1927
Quinn, P. M., Buck, T. M., Mulder, A. A., Ohonin, C., Alves, C. H., Vos, R. M., et al. (2019a). Human iPSC-derived retinas recapitulate the fetal CRB1 CRB2 complex formation and demonstrate that photoreceptors and muller glia are targets of AAV5. Stem Cell Rep. 12, 906–919. doi:10.1016/j.stemcr.2019.03.002
Quinn, P. M., Mulder, A. A., Henrique Alves, C., Desrosiers, M., De Vries, S. I., Klooster, J., et al. (2019b). Loss of CRB2 in Muller glial cells modifies a CRB1-associated retinitis pigmentosa phenotype into a Leber congenital amaurosis phenotype. Hum. Mol. Genet. 28, 105–123. doi:10.1093/hmg/ddy337
Rachel, R. A., May-Simera, H. L., Veleri, S., Gotoh, N., Choi, B. Y., Murga-Zamalloa, C., et al. (2012). Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J. Clin. Invest. 122, 1233–1245. doi:10.1172/JCI60981
Rachel, R. A., Yamamoto, E. A., Dewanjee, M. K., May-Simera, H. L., Sergeev, Y. V., Hackett, A. N., et al. (2015). CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies. Hum. Mol. Genet. 24, 3775–3791. doi:10.1093/hmg/ddv123
Ramachandra Rao, S., and Fliesler, S. J. (2021). Cholesterol homeostasis in the vertebrate retina: biology and pathobiology. J. Lipid Res. 62, 100057. doi:10.1194/jlr.TR120000979
Ratnam, K., Vastinsalo, H., Roorda, A., Sankila, E. M., and Duncan, J. L. (2013). Cone structure in patients with usher syndrome type III and mutations in the Clarin 1 gene. JAMA Ophthalmol. 131, 67–74. doi:10.1001/2013.jamaophthalmol.2
Rattner, A., Smallwood, P. M., and Nathans, J. (2000). Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J. Biol. Chem. 275, 11034–11043. doi:10.1074/jbc.275.15.11034
Ray, P. D., Huang, B. W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24, 981–990. doi:10.1016/j.cellsig.2012.01.008
Raymond, P. A., Barthel, L. K., Bernardos, R. L., and Perkowski, J. J. (2006). Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Dev. Biol. 6, 36. doi:10.1186/1471-213X-6-36
Reichenbach, A., and Bringmann, A. (2013). New functions of Muller cells. Glia 61, 651–678. doi:10.1002/glia.22477
Reichenbach, A., and Bringmann, A. (2020). Glia of the human retina. Glia 68, 768–796. doi:10.1002/glia.23727
Riedmayr, L. M., Hinrichsmeyer, K. S., Thalhammer, S. B., Mittas, D. M., Karguth, N., Otify, D. Y., et al. (2023). mRNA trans-splicing dual AAV vectors for (epi)genome editing and gene therapy. Nat. Commun. 14, 6578. doi:10.1038/s41467-023-42386-0
Ripley, D. L., and Politzer, T. (2010). Vision disturbance after TBI. NeuroRehabilitation 27, 215–216. doi:10.3233/NRE-2010-0599
Ruan, G. X., Barry, E., Yu, D., Lukason, M., Cheng, S. H., and Scaria, A. (2017). CRISPR/Cas9-Mediated genome editing as a therapeutic approach for leber congenital amaurosis 10. Mol. Ther. 25, 331–341. doi:10.1016/j.ymthe.2016.12.006
Russell, S. R., Drack, A. V., Cideciyan, A. V., Jacobson, S. G., Leroy, B. P., Van Cauwenbergh, C., et al. (2022). Intravitreal antisense oligonucleotide sepofarsen in Leber congenital amaurosis type 10: a phase 1b/2 trial. Nat. Med. 28, 1014–1021. doi:10.1038/s41591-022-01755-w
Saatci, A. O., Atas, F., Cetin, G. O., and Kayabasi, M. (2023). Diagnostic and management strategies of Bietti crystalline dystrophy: current perspectives. Clin. Ophthalmol. 17, 953–967. doi:10.2147/OPTH.S388292
Saha, A., Capowski, E., Fernandez Zepeda, M. A., Nelson, E. C., Gamm, D. M., and Sinha, R. (2022). Cone photoreceptors in human stem cell-derived retinal organoids demonstrate intrinsic light responses that mimic those of primate fovea. Cell Stem Cell 29, 487–489. doi:10.1016/j.stem.2022.02.003
Salom, D., Diaz-Llopis, M., Garcia-Delpech, S., Udaondo, P., Sancho-Tello, M., and Romero, F. J. (2008). Aqueous humor levels of vascular endothelial growth factor in retinitis pigmentosa. Investigative Ophthalmol. Vis. Sci. 49, 3499–3502. doi:10.1167/iovs.07-1168
Sayadi, J., and Mekni, M. (2022). Crystals lens deposits: a rare occurrence in Bietti corneo-retinal dystrophy. Pan Afr. Med. J. 43, 7. doi:10.11604/pamj.2022.43.7.34356
Schneider, N., Sundaresan, Y., Gopalakrishnan, P., Beryozkin, A., Hanany, M., Levanon, E. Y., et al. (2022). Inherited retinal diseases: linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin Eye Res. 89, 101029. doi:10.1016/j.preteyeres.2021.101029
Scholl, H. P., Strauss, R. W., Singh, M. S., Dalkara, D., Roska, B., Picaud, S., et al. (2016). Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 8, 368rv6. doi:10.1126/scitranslmed.aaf2838
Schwartz, S. B., Aleman, T. S., Cideciyan, A. V., Swaroop, A., Jacobson, S. G., and Stone, E. M. (2003). De novo mutation in the RP1 gene (Arg677ter) associated with retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 44, 3593–3597. doi:10.1167/iovs.03-0155
Schwarz, N., Carr, A. J., Lane, A., Moeller, F., Chen, L. L., Aguila, M., et al. (2015). Translational read-through of the RP2 Arg120stop mutation in patient iPSC-derived retinal pigment epithelium cells. Hum. Mol. Genet. 24 (4), 972–986. doi:10.1093/hmg/ddu509
Sengillo, J. D., Lee, W., Bilancia, C. G., Jobanputra, V., and Tsang, S. H. (2018). Phenotypic expansion and progression of SPATA7-associated retinitis pigmentosa. Doc. Ophthalmol. 136 (2), 125–133. doi:10.1007/s10633-018-9626-1
Shanmugaratnam, J., Berg, E., Kimerer, L., Johnson, R. J., Amaratunga, A., Schreiber, B. M., et al. (1997). Retinal Muller glia secrete apolipoproteins E and J which are efficiently assembled into lipoprotein particles. Brain Res. Mol. Brain Res. 50, 113–120. doi:10.1016/s0169-328x(97)00176-9
Sharma, J., and Paschalis, E. I. (2022). The future of non-viral gene delivery for the treatment of inherited retinal diseases. Mol. Ther. Nucleic Acids 30, 354. doi:10.1016/j.omtn.2022.10.011
Shimada, H., Lu, Q., Insinna-Kettenhofen, C., Nagashima, K., English, M. A., Semler, E. M., et al. (2017). In vitro modeling using ciliopathy-patient-derived cells reveals distinct cilia dysfunctions caused by CEP290 mutations. Cell Rep. 20, 384–396. doi:10.1016/j.celrep.2017.06.045
Shindou, H., Koso, H., Sasaki, J., Nakanishi, H., Sagara, H., Nakagawa, K. M., et al. (2017). Docosahexaenoic acid preserves visual function by maintaining correct disc morphology in retinal photoreceptor cells. J. Biol. Chem. 292, 12054–12064. doi:10.1074/jbc.M117.790568
Shintani, K., Shechtman, D. L., and Gurwood, A. S. (2009). Review and update: current treatment trends for patients with retinitis pigmentosa. Optometry 80, 384–401. doi:10.1016/j.optm.2008.01.026
Siemiatkowska, A. M., Collin, R. W., Den Hollander, A. I., and Cremers, F. P. (2014). Genomic approaches for the discovery of genes mutated in inherited retinal degeneration. Cold Spring Harb. Perspect. Med. 4, a017137. doi:10.1101/cshperspect.a017137
Slijkerman, R. W., Vache, C., Dona, M., Garcia-Garcia, G., Claustres, M., Hetterschijt, L., et al. (2016). Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. Mol. Ther. Nucleic Acids 5, e381. doi:10.1038/mtna.2016.89
Smith, A. M., and Czyz, C. N. (2023). Neuroanatomy, cranial nerve 2 (optic). Treasure Island (FL): StatPearls.
Smith, R. J., Berlin, C. I., Hejtmancik, J. F., Keats, B. J., Kimberling, W. J., Lewis, R. A., et al. (1994). Clinical diagnosis of the usher syndromes. Usher syndrome consortium. Am. J. Med. Genet. 50, 32–38. doi:10.1002/ajmg.1320500107
Stamer, W. D., Bok, D., Hu, J., Jaffe, G. J., and Mckay, B. S. (2003). Aquaporin-1 channels in human retinal pigment epithelium: role in transepithelial water movement. Invest. Ophthalmol. Vis. Sci. 44, 2803–2808. doi:10.1167/iovs.03-0001
Stokkermans, T. J., Falkowitz, D. M., and Trichonas, G. (2023). Chloroquine and hydroxychloroquine toxicity. Treasure Island (FL): StatPearls.
Strauss, O. (2005). The retinal pigment epithelium in visual function. Physiol. Rev. 85, 845–881. doi:10.1152/physrev.00021.2004
Sun, D., Schur, R. M., Sears, A. E., Gao, S. Q., Vaidya, A., Sun, W., et al. (2020). Non-viral gene therapy for Stargardt disease with ECO/pRHO-ABCA4 self-assembled nanoparticles. Mol. Ther. 28, 293–303. doi:10.1016/j.ymthe.2019.09.010
Sun, H., and Nathans, J. (1997). Stargardt's ABCR is localized to the disc membrane of retinal rod outer segments. Nat. Genet. 17, 15–16. doi:10.1038/ng0997-15
Sun, Y., Zheng, Y., Wang, C., and Liu, Y. (2018). Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 9, 753. doi:10.1038/s41419-018-0794-4
Tambuyzer, E., Vandendriessche, B., Austin, C. P., Brooks, P. J., Larsson, K., Miller Needleman, K. I., et al. (2020). Therapies for rare diseases: therapeutic modalities, progress and challenges ahead. Nat. Rev. Drug Discov. 19, 93–111. doi:10.1038/s41573-019-0049-9
Tamura, R., and Toda, M. (2020). Historic overview of genetic engineering technologies for human gene therapy. Neurol. Med. Chir. (Tokyo) 60, 483–491. doi:10.2176/nmc.ra.2020-0049
Tan, T. E., Fenner, B. J., Barathi, V. A., Tun, S. B. B., Wey, Y. S., Tsai, A. S. H., et al. (2021). Gene-based therapeutics for acquired retinal disease: opportunities and progress. Front. Genet. 12, 795010. doi:10.3389/fgene.2021.795010
Tate, D. J., Miceli, M. V., and Newsome, D. A. (1995). Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 36, 1271–1279.
Tepass, U., Theres, C., and Knust, E. (1990). Crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelial cells and required for organization of epithelia. Cell 61, 787–799. doi:10.1016/0092-8674(90)90189-l
Thummel, R., Kassen, S. C., Enright, J. M., Nelson, C. M., Montgomery, J. E., and Hyde, D. R. (2008). Characterization of Muller glia and neuronal progenitors during adult zebrafish retinal regeneration. Exp. Eye Res. 87, 433–444. doi:10.1016/j.exer.2008.07.009
Tikidji-Hamburyan, A., Reinhard, K., Storchi, R., Dietter, J., Seitter, H., Davis, K. E., et al. (2017). Rods progressively escape saturation to drive visual responses in daylight conditions. Nat. Commun. 8, 1813. doi:10.1038/s41467-017-01816-6
Todd, L., Hooper, M. J., Haugan, A. K., Finkbeiner, C., Jorstad, N., Radulovich, N., et al. (2021). Efficient stimulation of retinal regeneration from Muller glia in adult mice using combinations of proneural bHLH transcription factors. Cell Rep. 37, 109857. doi:10.1016/j.celrep.2021.109857
Todd, L., Jenkins, W., Finkbeiner, C., Hooper, M. J., Donaldson, P. C., Pavlou, M., et al. (2022). Reprogramming Muller glia to regenerate ganglion-like cells in adult mouse retina with developmental transcription factors. Sci. Adv. 8, eabq7219. doi:10.1126/sciadv.abq7219
Trapani, I., and Auricchio, A. (2018). Seeing the light after 25 Years of retinal gene therapy. Trends Mol Med. 24, 669–681. doi:10.1016/j.molmed.2018.06.006
Tripepi, D., Jalil, A., Ally, N., Buzzi, M., Moussa, G., Rothschild, P. R., et al. (2023). The role of subretinal injection in ophthalmic surgery: therapeutic agent delivery and other indications. Int. J. Mol. Sci. 24, 10535. doi:10.3390/ijms241310535
Tsai, Y. T., Wu, W. H., Lee, T. T., Wu, W. P., Xu, C. L., Park, K. S., et al. (2018). Clustered regularly interspaced short palindromic repeats-based genome surgery for the treatment of autosomal dominant retinitis pigmentosa. Ophthalmology 125, 1421–1430. doi:10.1016/j.ophtha.2018.04.001
Tschernutter, M., Jenkins, S. A., Waseem, N. H., Saihan, Z., Holder, G. E., Bird, A. C., et al. (2006). Clinical characterisation of a family with retinal dystrophy caused by mutation in the Mertk gene. Br. J. Ophthalmol. 90, 718–723. doi:10.1136/bjo.2005.084897
Tworak, A., Kolesnikov, A. V., Hong, J. D., Choi, E. H., Luu, J. C., Palczewska, G., et al. (2023). Rapid RGR-dependent visual pigment recycling is mediated by the RPE and specialized Muller glia. Cell Rep. 42, 112982. doi:10.1016/j.celrep.2023.112982
Tworig, J. M., and Feller, M. B. (2021). Muller glia in retinal development: from specification to circuit integration. Front. Neural Circuits 15, 815923. doi:10.3389/fncir.2021.815923
Uyama, H., Mandai, M., and Takahashi, M. (2021). Stem-cell-based therapies for retinal degenerative diseases: current challenges in the establishment of new treatment strategies. Dev. Growth Differ. 63, 59–71. doi:10.1111/dgd.12704
Valente, E. M., Silhavy, J. L., Brancati, F., Barrano, G., Krishnaswami, S. R., Castori, M., et al. (2006). Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 38, 623–625. doi:10.1038/ng1805
Vandenberg, R. J., and Ryan, R. M. (2013). Mechanisms of glutamate transport. Physiol. Rev. 93, 1621–1657. doi:10.1152/physrev.00007.2013
Van De Pavert, S. A., Kantardzhieva, A., Malysheva, A., Meuleman, J., Versteeg, I., Levelt, C., et al. (2004). Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J. Cell Sci. 117, 4169–4177. doi:10.1242/jcs.01301
Vecino, E., Rodriguez, F. D., Ruzafa, N., Pereiro, X., and Sharma, S. C. (2016). Glia-neuron interactions in the mammalian retina. Prog. Retin Eye Res. 51, 1–40. doi:10.1016/j.preteyeres.2015.06.003
Veleri, S., Lazar, C. H., Chang, B., Sieving, P. A., Banin, E., and Swaroop, A. (2015). Biology and therapy of inherited retinal degenerative disease: insights from mouse models. Dis. Model Mech. 8, 109–129. doi:10.1242/dmm.017913
Venkatesh, A., Ma, S., Le, Y. Z., Hall, M. N., Ruegg, M. A., and Punzo, C. (2015). Activated mTORC1 promotes long-term cone survival in retinitis pigmentosa mice. J. Clin. Invest. 125, 1446–1458. doi:10.1172/JCI79766
Verbakel, S. K., Van Huet, R. A. C., Boon, C. J. F., Den Hollander, A. I., Collin, R. W. J., Klaver, C. C. W., et al. (2018). Non-syndromic retinitis pigmentosa. Prog. Retin Eye Res. 66, 157–186. doi:10.1016/j.preteyeres.2018.03.005
Viegas, F. O., and Neuhauss, S. C. F. (2021). A metabolic landscape for maintaining retina integrity and function. Front. Mol. Neurosci. 14, 656000. doi:10.3389/fnmol.2021.656000
Walmsley, L., Hanna, L., Mouland, J., Martial, F., West, A., Smedley, A. R., et al. (2015). Colour as a signal for entraining the mammalian circadian clock. PLoS Biol. 13, e1002127. doi:10.1371/journal.pbio.1002127
Wang, J. S., and Kefalov, V. J. (2011). The cone-specific visual cycle. Prog. Retin Eye Res. 30, 115–128. doi:10.1016/j.preteyeres.2010.11.001
Wang, S. K., Xue, Y., Rana, P., Hong, C. M., and Cepko, C. L. (2019). Soluble CX3CL1 gene therapy improves cone survival and function in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 116, 10140–10149. doi:10.1073/pnas.1901787116
Wang, X., Yu, C., Tzekov, R. T., Zhu, Y., and Li, W. (2020). The effect of human gene therapy for RPE65-associated Leber's congenital amaurosis on visual function: a systematic review and meta-analysis. Orphanet J. Rare Dis. 15, 49. doi:10.1186/s13023-020-1304-1
Wang, Y., Zhou, Y. F., Zhao, B. Y., Gu, Z. Y., and Li, S. L. (2014). Apolipoprotein E gene ε4ε4 is associated with elevated risk of primary open angle glaucoma in Asians: a meta-analysis. Bmc Med. Genet. 15, 60. doi:10.1186/1471-2350-15-60
Weleber, R. G. (1994). Stargardt's macular dystrophy. Arch. Ophthalmol. 112, 752–754. doi:10.1001/archopht.1994.01090180050033
Wickremasinghe, S. S., Xie, J., Lim, J., Chauhan, D. S., Robman, L., Richardson, A. J., et al. (2011). Variants in the APOE gene are associated with improved outcome after anti-VEGF treatment for neovascular AMD. Invest. Ophthalmol. Vis. Sci. 52, 4072–4079. doi:10.1167/iovs.10-6550
Wimmers, S., Karl, M. O., and Strauss, O. (2007). Ion channels in the RPE. Prog. Retin Eye Res. 26, 263–301. doi:10.1016/j.preteyeres.2006.12.002
Wirth, T., Parker, N., and Yla-Herttuala, S. (2013). History of gene therapy. Gene 525, 162–169. doi:10.1016/j.gene.2013.03.137
Wollmann, G., Lenzner, S., Berger, W., Rosenthal, R., Karl, M. O., and Strauss, O. (2006). Voltage-dependent ion channels in the mouse RPE: comparison with Norrie disease mice. Vis. Res. 46, 688–698. doi:10.1016/j.visres.2005.08.030
Wong-Riley, M. T. (2010). Energy metabolism of the visual system. Eye Brain 2, 99–116. doi:10.2147/EB.S9078
Wright, A. F., Chakarova, C. F., Abd El-Aziz, M. M., and Bhattacharya, S. S. (2010). Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 11, 273–284. doi:10.1038/nrg2717
Wu, Z., Asokan, A., and Samulski, R. J. (2006). Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol. Ther. 14, 316–327. doi:10.1016/j.ymthe.2006.05.009
Wu, Z., Pang, N., Zhang, Y., Chen, H., Peng, Y., Fu, J., et al. (2020). CEP290 is essential for the initiation of ciliary transition zone assembly. PLoS Biol. 18, e3001034. doi:10.1371/journal.pbio.3001034
Wu, Z., Yang, H., and Colosi, P. (2010). Effect of genome size on AAV vector packaging. Mol. Ther. 18, 80–86. doi:10.1038/mt.2009.255
Xian, B., and Huang, B. (2015). The immune response of stem cells in subretinal transplantation. Stem Cell Res. Ther. 6, 161. doi:10.1186/s13287-015-0167-1
Xiong, W., Maccoll Garfinkel, A. E., Li, Y., Benowitz, L. I., and Cepko, C. L. (2015). NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Invest. 125, 1433–1445. doi:10.1172/JCI79735
Xue, Y., Shen, S. Q., Jui, J., Rupp, A. C., Byrne, L. C., Hattar, S., et al. (2015). CRALBP supports the mammalian retinal visual cycle and cone vision. J. Clin. Invest. 125, 727–738. doi:10.1172/JCI79651
Xue, Y., Wang, S. K., Rana, P., West, E. R., Hong, C. M., Feng, H., et al. (2021). AAV-Txnip prolongs cone survival and vision in mouse models of retinitis pigmentosa. Elife 10, e66240. doi:10.7554/eLife.66240
Yanagisawa, M., Namekata, K., Aida, T., Katou, S., Takeda, T., Harada, T., et al. (2020). EAAT1 variants associated with glaucoma. Biochem. Biophys. Res. Commun. 529, 943–949. doi:10.1016/j.bbrc.2020.06.099
Yang, X. L. (2004). Characterization of receptors for glutamate and GABA in retinal neurons. Prog. Neurobiol. 73, 127–150. doi:10.1016/j.pneurobio.2004.04.002
Yang, S., Ma, S. Q., Wan, X., He, H., Pei, H., Zhao, M. J., et al. (2016). Long-term outcomes of gene therapy for the treatment of Leber's hereditary optic neuropathy. EBioMedicine 10, 258–268. doi:10.1016/j.ebiom.2016.07.002
Yao, J., Qiu, Y., Frontera, E., Jia, L., Khan, N. W., Klionsky, D. J., et al. (2018). Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 14, 1226–1238. doi:10.1080/15548627.2018.1463121
Youssef, P. N., Sheibani, N., and Albert, D. M. (2011). Retinal light toxicity. Eye (Lond) 25, 1–14. doi:10.1038/eye.2010.149
Yuan, J., Zhang, Y., Liu, H., Wang, D., Du, Y., Tian, Z., et al. (2020). Seven-year follow-up of gene therapy for leber's hereditary optic neuropathy. Ophthalmology 127, 1125–1127. doi:10.1016/j.ophtha.2020.02.023
Yue, L., Weiland, J. D., Roska, B., and Humayun, M. S. (2016). Retinal stimulation strategies to restore vision: fundamentals and systems. Prog. Retin Eye Res. 53, 21–47. doi:10.1016/j.preteyeres.2016.05.002
Zelinger, L., and Swaroop, A. (2018). RNA biology in retinal development and disease. Trends Genet. 34, 341–351. doi:10.1016/j.tig.2018.01.002
Zhang, Y., Li, X., Yuan, J., Tian, Z., Liu, H., Wang, D., et al. (2019). Prognostic factors for visual acuity in patients with Leber's hereditary optic neuropathy after rAAV2-ND4 gene therapy. Clin. Exp. Ophthalmol. 47, 774–778. doi:10.1111/ceo.13515
Zheng, W. V., Xu, W., Li, Y., Qin, J., Zhou, T., Li, D., et al. (2022b). Anti-aging effect of β-carotene through regulating the KAT7-P15 signaling axis, inflammation and oxidative stress process. Cell Mol. Biol. Lett. 27, 86. doi:10.1186/s11658-022-00389-7
Zheng, Q., Chen, Y., Chen, D., Zhao, H., Feng, Y., Meng, Q., et al. (2022a). Calcium transients on the ER surface trigger liquid-liquid phase separation of FIP200 to specify autophagosome initiation sites. Cell 185, 4082–4098. doi:10.1016/j.cell.2022.09.001
Keywords: cell-cell interaction, inherited retinal disease, photoreceptor, Müller glia, retinal pigment epithelium
Citation: Du X, Butler AG and Chen HY (2024) Cell-cell interaction in the pathogenesis of inherited retinal diseases. Front. Cell Dev. Biol. 12:1332944. doi: 10.3389/fcell.2024.1332944
Received: 03 November 2023; Accepted: 06 February 2024;
Published: 04 March 2024.
Edited by:
Steven J. Fliesler, University at Buffalo, United StatesReviewed by:
Brian Perkins, Cole Eye Institute, United StatesSriganesh Ramachandra Rao, University at Buffalo, United States
Stephen Tsang, Columbia University, United States
Copyright © 2024 Du, Butler and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holly Y. Chen, Y2hlbmhAdWFiLmVkdQ==
†These authors have contributed equally to this work and share first authorship