 Asli E. Atici
Asli E. Atici Timothy R. Crother
Timothy R. Crother Magali Noval Rivas
Magali Noval Rivas
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 06 November 2023
Sec. Cellular Biochemistry
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1290046
This article is part of the Research Topic Mitochondrial Plasticity and Quality Control in Health and Disease View all 12 articles
Cardiovascular diseases (CVDs) are one of the primary causes of mortality worldwide. An optimal mitochondrial function is central to supplying tissues with high energy demand, such as the cardiovascular system. In addition to producing ATP as a power source, mitochondria are also heavily involved in adaptation to environmental stress and fine-tuning tissue functions. Mitochondrial quality control (MQC) through fission, fusion, mitophagy, and biogenesis ensures the clearance of dysfunctional mitochondria and preserves mitochondrial homeostasis in cardiovascular tissues. Furthermore, mitochondria generate reactive oxygen species (ROS), which trigger the production of pro-inflammatory cytokines and regulate cell survival. Mitochondrial dysfunction has been implicated in multiple CVDs, including ischemia-reperfusion (I/R), atherosclerosis, heart failure, cardiac hypertrophy, hypertension, diabetic and genetic cardiomyopathies, and Kawasaki Disease (KD). Thus, MQC is pivotal in promoting cardiovascular health. Here, we outline the mechanisms of MQC and discuss the current literature on mitochondrial adaptation in CVDs.
Mitochondria are central signaling hubs for eukaryotic cells involved in metabolism, inflammation, calcium regulation, and cell death (Figure 1) (Galluzzi et al., 2012; Lopez-Crisosto et al., 2017; Mehta et al., 2017; Galluzzi et al., 2018). Mitochondria are double membrane organelles consisting of an outer (OMM) and an inner mitochondrial membrane (IMM), both delimiting an intermembrane space (Figure 1). The IMM folds inwards, forming crests (cristae), which increase the surface area available for mitochondrial reactions (Protasoni and Zeviani, 2021) (Figure 1). Through the breakdown of carbohydrates and fatty acids and the production of adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS), mitochondria are the primary source of energy for eukaryotic cells (Madeira, 2018). This process is mediated by products from the tricarboxylic acid cycle (TCA cycle) entering the mitochondrial electron transport chain (ETC), a group of proteins located in the IMM (Figure 1) (Martínez-Reyes and Chandel, 2020). Eleven ETC subunits are encoded in mitochondria’s circular DNA (mtDNA) and are crucial to correctly assembling these complexes (Vercellino and Sazanov, 2022). The remaining ETC subunits are encoded by the nuclear DNA and imported into the mitochondria (Yan et al., 2019) (Figure 1). Mitochondria are also involved in ROS production and communicate with other organelles, such as the endoplasmic reticulum (ER), via channels and pores, allowing metabolites and protein transport (Figure 1). Mitochondria are a highly dynamic network of organelles, strictly regulated by mitochondrial quality control mechanisms (MQC) (Ni et al., 2015). Due to cardiac tissues’ elevated energy requirement, maintenance of cardiovascular homeostasis is heavily dependent on mitochondrial quality. Indeed, cardiomyocytes utilize the ATP that mitochondria produce from carbohydrates and fatty acid-driven oxidative phosphorylation. Therefore, mitochondrial dysfunction affects cardiomyocytes’ function and contractility. Calcium levels are also central to cardiac activity. Calcium storage and regulation by mitochondria and the ER modulate cardiac function and impact electrical conduction (Lai and Qiu, 2020). Damaged or dysfunctional mitochondria are constantly eliminated via a mitochondria-specific degradation machinery, known as mitophagy (Sica et al., 2015; Harper et al., 2018). When mitophagy is dysregulated, damaged mitochondria accumulate inside cardiomyocytes, vascular smooth muscle cells (VSMCs), and endothelial cells, which can trigger inflammatory responses. Severe mitochondrial dysfunction and lack of clearance of damaged mitochondria can also initiate cell death and lead to tissue damage or loss (Hu et al., 2015; Amgalan et al., 2017; Gisterå and Hansson, 2017). Therefore, perturbations in mitochondrial quality promote the pathogenesis of CVDs, including ischemia/reperfusion, hypertension, atherosclerosis, heart failure, diabetic and genetic cardiomyopathies, and Kawasaki Disease (KD) (Bravo-San Pedro et al., 2017; Suomalainen and Battersby, 2018). Herein, we discuss several pathways related to mitochondrial metabolism and dynamics, how aged and dysfunctional mitochondria are eliminated, how new mitochondria are produced, and the importance of MQC to several CVDs.
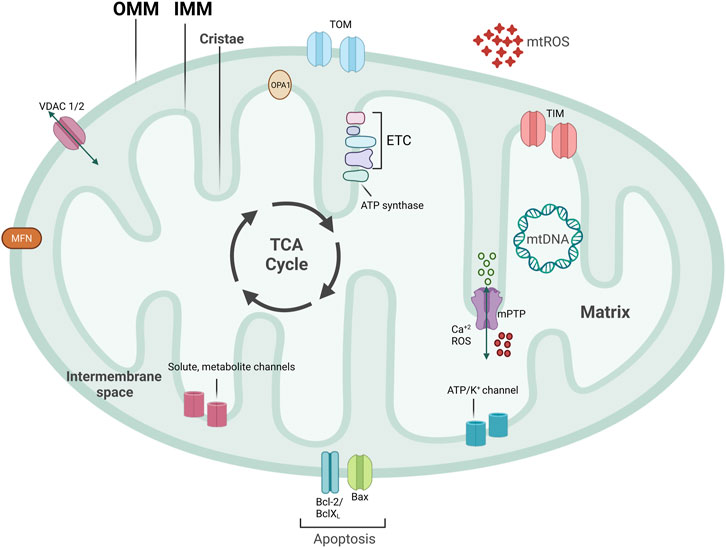
FIGURE 1. Mitochondrial structure. Mitochondria are double-membraned organelles with cristae formation. Mitochondria functioning is central for bioenergetic activities, producing ATP, an important source of ROS production. Mitochondria have several channels and pores to allow for metabolite and protein import, pivotal for inter-organelle communication. Part of ETC subunits are encoded in mitochondria’s circular DNA, named mtDNA. TCA cycle that produces important electron carriers for ETC, as well as ATP, and resides on the IMM. mtROS, mitochondrial reactive oxygen species; ETC, electron transport chain; mtDNA, mitochondrial DNA; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; mPTP, mitochondrial permeability transition pore; VDAC, voltage-dependent anion channel.
The Tricarboxylic acid (TCA) cycle (citric acid cycle or Krebs cycle) comprises a series of reactions occurring in the mitochondrial matrix, involving numerous substrates crucial to cellular metabolism (Figure 1). Metabolites of the TCA cycle regulate cellular function. They are necessary for the biosynthesis of lipids, nucleotides, and proteins (McDonnell et al., 2016), as well as chromatin modifications, DNA methylation, and post-translational protein modifications (Wellen et al., 2009; Moussaieff et al., 2015). The TCA cycle is initiated by a reaction that produces six-carbon citrate from two-carbon acetyl-CoA. Acetyl-CoA is generated from the oxidation of amino acids, glucose, fatty acids, or pyruvate (Figure 2). Acetyl-CoA combines with a four-carbon oxaloacetate molecule to yield citrate, which initiates the TCA cycle (Figure 2). Then, the oxidative decarboxylation of citrate isomer, isocitrate, forms α-ketoglutarate, a five-carbon molecule. Conversion of α-ketoglutarate to succinyl-CoA, a four-carbon molecule, yields two carbon dioxide (CO2) and two nicotinamide adenine dinucleotide (NADH or NAD+) molecules. Succinyl-CoA is then converted into succinate-producing GTP, which can later be converted into ATP (Shi and Tu, 2015; Martínez-Reyes et al., 2016). Oxidation of succinate to four-carbon fumarate transfers two hydrogens to flavin adenine dinucleotide (FAD), forming FADH2 (hydroquinone form). Lastly, fumarate is converted to malate and oxaloacetate, forming acetyl-CoA to proceed with the cycle (Martínez-Reyes and Chandel, 2020) (Figure 2). As detailed in the next section, byproducts NADH and FADH2 from the TCA cycle are essential electron donors for mitochondrial OXPHOS (Figure 2).
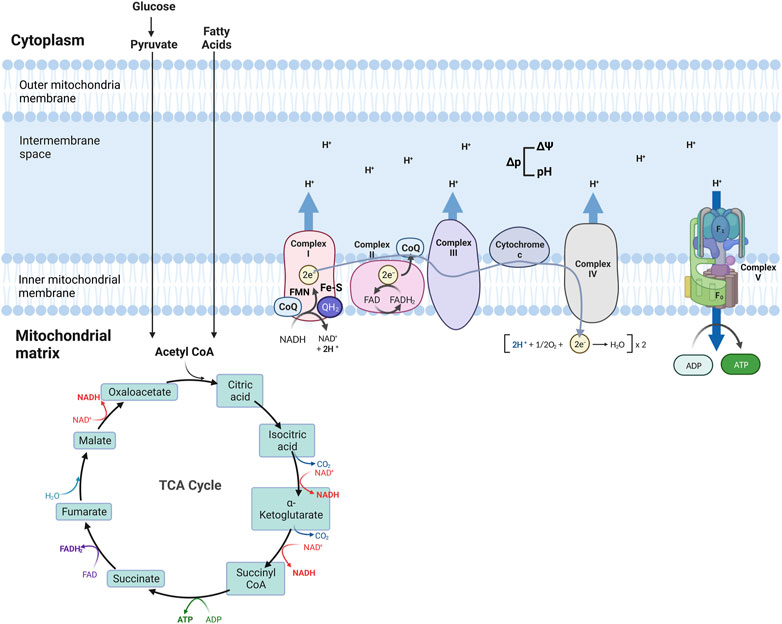
FIGURE 2. Schematic of mitochondria electron transport chain (ETC) and Oxidative phosphorylation (OXPHOS). ETC comprises four complexes that reside in the inner mitochondrial membrane that leads to the production of ATP. The TCA cycle is the source of NADH and FADH2, which are required for electron transport between the complexes, finalized by the production of ATP at the last step.
Four transmembrane complexes form the ETC. These complexes use cytochrome c and ubiquinone to transfer electrons through the IMM, where the ETC is located (Figure 2). ETC complexes form hetero-, or homodimers called super-complexes to optimize their functioning (Iwata et al., 1998; Guo et al., 2017). Together with ATP synthase, these complexes form the OXPHOS system in the mitochondrial inner membrane (Figure 2).
The ETC is fueled by electrons supplied via by-products from the TCA cycle that are transferred to oxygen (O2), a process that also results in the formation of a proton gradient in the mitochondrial intermembrane space and the production of ATP (Martínez-Reyes et al., 2016). More specifically, byproducts of the TCA cycle, NADH and FADH2, are electron sources for Complex I (NADH: ubiquinone oxidoreductase) or Complex II (succinate dehydrogenase), respectively (Figure 2). Transfer of electrons from NADH to Complex I is achieved by ubiquinone (CoQ) and requires co-factors flavin-mononucleotide (FMN) and iron-sulfur (FeS) clusters, where CoQ is reduced to ubiquinol (QH2). Transfer of electrons to Complex I promotes the pumping of four protons into the mitochondria intermembrane space by Complex I. Electrons from FADH2 are conveyed to CoQ, with the co-factor FeS, and ultimately transferred to Complex II. Electrons entering the ETC are transferred to Complex III (coenzyme Q: cytochrome c reductase) via the oxidation of QH2 and the activity of the 2Fe-2S cluster. Electrons are then transferred to Cytochrome c, which releases two more protons into the mitochondrial intermembrane space. Cytochrome c is a mobile electron carrier that cargoes electrons from Complex III to Complex IV (cytochrome c oxidase). Complex IV comprises three subunits: I, II, and III. Subunit II interacts with reduced cytochrome c, transferring electrons to subunit I. This transfer also causes O2 to bind to Complex IV, which is reduced to water. Complex IV pumps protons from the mitochondrial matrix; some of them go into the intermembrane space and contribute to the proton gradient, while the remaining protons are used to form water molecules. The use of O2 for this process is known as the mitochondrial respiration (Fernie et al., 2004; Guo et al., 2018; Zhao et al., 2019; Vercellino and Sazanov, 2022).
As a result of the electron transport process, ten protons are pumped from the mitochondrial matrix into the intermembrane space where a proton gradient is built, which is the basis for the mitochondrial membrane potential (ΔΨ) (Figure 2) (Zorova et al., 2018). As the last step of oxidative phosphorylation, ATP synthase uses the proton motive force (Δp), the combination of ΔΨ and proton concentration, to produce ATP. More specifically, Δp couples electron transport from the other complexes to the activity of ATP synthase. ATP synthase consists of an extra-membranous (F1) and a transmembrane (F0) domain, which functions with a rotational mechanism to produce ATP. At the last step of OXPHOS, protons pumped to the intermembrane space return to the mitochondrial matrix through F0, dispersing the proton gradient. This allows for the rotation of ATP synthase, which induces the addition of phosphate to ADP to form ATP (Figure 2) (Reid et al., 1966; Capaldi and Aggeler, 2002; Harrington et al., 2023).
The process of electron transfer between the ETC complexes could be more efficient. Protons may leak and migrate into the mitochondrial matrix, which results in incomplete coupling of O2 with no ATP production (Cheng et al., 2017). Electrons may also leak and exit the ETC before being converted into water from Complex I and III, resulting in reduced O2 and increased production of reactive oxygen species (ROS) (Cheng et al., 2017). Mitochondria are a significant source of ROS, which can damage mitochondria and result in cell death when produced at high concentrations or act as a signaling molecule when made at low concentrations (Murphy et al., 2016). ROS comprises superoxide anion (O2•-), hydroxyl radical (•OH), and hydrogen peroxide (H2O2). Reduction of O2 by one electron causes the formation of O2•- and can be dismutated to H2O2 (Murphy, 2009; Cadenas, 2018). Complex II has also been shown to be involved in ROS production as it performs a reverse transfer of electrons from succinate to ubiquinone and back to Complex I, a ROS production site (Liu et al., 2002; Yankovskaya et al., 2003). Various other mitochondrial enzymes also contribute to mitochondrial ROS production. Oxidation of lipids by acyl-CoA dehydrogenase or glycerol-α-phosphate dehydrogenase yields mitochondrial ROS in the tissues (St-Pierre et al., 2002; Lambertucci et al., 2008). In addition, the TCA cycle enzymes pyruvate and α-ketoglutarate dehydrogenase also increase mitochondrial ROS production (Starkov et al., 2004).
Voltage-dependent anion channels (VDACs) are OMM proteins that form aqueous pores to exchange metabolites (Colombini et al., 1996). There are three VDAC isoforms (VDAC1, 2, and 3), which are very similar in sequence and are expressed in cardiac tissue cells but have diverse functions (Zinghirino et al., 2021). VDAC1 and 2 play a role in mitochondrial bioenergetics and mitochondrial apoptotic pathways (Ravi et al., 2021). Nuclear-encoded proteins are responsible for modulating the redox state, and VDACs can sense the imbalance in the redox state in the mitochondria (Storz et al., 2005). It has been suggested that VDAC3 is a sensor of mtROS and VDAC1 is involved in ROS-induced apoptotic pathways (Brahimi-Horn et al., 2015; Reina et al., 2016). VDAC1 also partakes in the translocation of O2•- to the cytosol from the mitochondria (Han et al., 2003). Furthermore, VDAC3 has also been shown as a target of ROS produced by Complex III of the ETC in mitochondria isolated from rat hearts (Bleier et al., 2015). In support of this, mice lacking VDAC3 have elevated mtROS production after being fed a high-salt diet (Zou et al., 2018). Similarly, a recent study showed that the absence of VDAC3 in human cells results in elevated ROS production and an increase in the ROS scavenging system to cope with oxidative stress, hinting at the protective effects of VDAC3 during oxidative damage. Additionally, the same study showed that VDAC3 deletion suppresses mitochondrial biogenesis (Reina et al., 2022).
O2•- can react with nitric oxide (NO) that produces reactive nitrogen species (RNS). NO is critical during inflammation and acts as a pro-inflammatory mediator by promoting neutrophil accumulation, downregulation of adhesion molecules, and upregulation of apoptosis at high concentrations (Albina et al., 1991; Lu et al., 1996; Shin et al., 1996). NO also boosts vasodilatation in the cardiovascular system and activates macrophages (Coleman, 2001).
Numerous enzymes manage ROS detoxification and convert these byproducts into less reactive forms (Dubois-Deruy et al., 2020). O2•- is very reactive and exhibits a short half-life, as it is constantly converted to H2O2 by mitochondrial manganese superoxide dismutase (MnSOD). H2O2 is less reactive and can diffuse out of the mitochondria as a cytosolic messenger (Giorgio et al., 2007; Candas and Li, 2014). Catalases detoxify H2O2 (Sepasi Tehrani and Moosavi-Movahedi, 2018). These Fe-heme-containing enzymes catalyze hydrogen peroxide into water and oxygen, mainly in the cytoplasmic peroxisomes (Kirkman and Gaetani, 2007). Glutathione peroxidases (GPx) and peroxiredoxins (Prx) are also involved in the breakdown of hydrogen peroxide. Glutathione is a co-factor and an electron donor for GPx. Prx is mainly located in the mitochondria, and like GPx, it converts H2O2 to water (Forman et al., 2009; Murphy, 2009; Forkink et al., 2010; Hossain et al., 2015).
ROS production can affect mtDNA integrity and lead to mtDNA damage. ROS production site is close to mtDNA in the IMM (Sampath, 2014). Importantly, mtDNA replication occurs in an asymmetric route, where heavy strands are left single-stranded, causing spontaneous deamination on the exposed nucleotides (Tanaka and Ozawa, 1994; Reyes et al., 1998). ROS-induced mtDNA damage causes missense mutations, deletions, or base substitutions, jeopardizing mtDNA integrity and mitochondrial functioning (Richter et al., 1988; Yakes and Van Houten, 1997). Numerous mtDNA repair pathways are used by the mitochondria, which take advantage of genomic DNA repair mechanisms (Kazak et al., 2012; Stein and Sia, 2017). The base excision repair (BER) pathway is the primary mechanism for mtDNA repair. Overall, BER eliminates oxidized, deaminated, or methylated bases, single-strand breaks, and alkylation damage (Sykora et al., 2012; Krokan and Bjørås, 2013). BER is a multi-step process that involves 1) recognition and removal of the target bases, 2) removal of abasic site, 3) end processing/gap filling, and finally, 4) DNA ligation (Jacobs and Schär, 2012). DNA glycosylases recognize and remove damaged DNA, and mitochondrial glycosylases include Uracil DNA glycosylase (UNG), 8-Oxoguanine DNA glycosylase (OGG1), and Nei-like DNA glycosylase 1 (NEIL1) (Hu et al., 2005). When damaged mtDNA accumulates, this interferes with the OXPHOS functioning (Ma et al., 2020). Consequently, mitophagy pathways are activated to clear defective mitochondria.
Mutations of mtDNA itself or nuclear genes that encode for mitochondrial proteins can lead to perturbations in mitochondrial functioning and cause mitochondrial diseases (Russell et al., 2020). These mutations can include point mutations and rearrangements in mtDNA. Point mutations can alter genes that encode proteins, transfer RNA (tRNA) or ribosomal RNA (rRNA) and are inherited maternally (Pereira et al., 2021). Rearrangements of mtDNA are usually deletions or duplications and can either be inherited maternally or occur de novo (Chinnery and Hudson, 2013). Mutations that arise on nuclear genes that encode mitochondrial proteins can be inherited as X-linked, autosomal dominant, or autosomal recessive. These mutations usually affect ETC-related nuclear genes, mitochondrial import machinery, mitochondrial translational factors, or CoQ10 biosynthesis (Coenen et al., 2004; MacKenzie and Payne, 2007; Potgieter et al., 2013). Various nuclear-encoded proteins are responsible for maintaining mtDNA, which controls the synthesis of mitochondria deoxyribonucleoside triphosphates (dNTPs) and mtDNA replication. Any mutations in these genes may lead to the depletion of dNTPs or deficiency in mtDNA replication, inducing a decrease in the overall levels of mtDNA (El-Hattab and Scaglia, 2013). These mutations result in disturbances in ETC assembly and functioning, ultimately leading to a deficiency in organ ATP production, especially in high-energy-demanding cardiac tissues (Chinnery, 1993). Furthermore, as the OXPHOS system is defective due to mutations of mtDNA or nuclear DNA, NADH accumulates, blocking the TCA cycle and favoring the production of lactate from pyruvate, resulting in elevated levels of lactate that interferes with proper cardiac tissue functioning (El-Hattab and Scaglia, 2013). Cells can have identical mtDNA (homoplasmy), as well as a combination of different types (heteroplasmy) (Pereira et al., 2021). Mutations that alter all copies of mtDNA are homoplasmic, whereas mutations that affect certain copies of mtDNA and cause co-existence of normal and mutant mtDNA are called heteroplasmic mutations (Ballana et al., 2008; Stefano et al., 2017). Mitochondria are randomly distributed to the daughter cells during cell division, causing heteroplasmic mtDNA mutations to be inherited by chance. Therefore, mutant mtDNAs can accumulate at different rates in different tissues. Hence, as the mutated mtDNA content increases, energy production decreases, enabling CVDs susceptibility (Saneto and Sedensky, 2013; Gonçalves, 2019).
MQC mechanisms are essential to maintain cellular mitochondrial homeostasis. MQC consists of a spectrum of various coordinated pathways. The primary function of MQC mechanisms is to ensure mitochondrial quality and maintain mitochondrial functions by limiting oxidative damage to mitochondria (Picca et al., 2018). ROS scavenging is a crucial first response to oxidative damage, and under physiological conditions, ROS production and detoxification are efficiently maintained (Lennicke and Cochemé, 2021). However, organelle damage can occur if ROS scavenging is insufficient to prevent a proper defense (Richter et al., 1995). ROS generation can also damage mtDNA due to the mitochondria’s inadequate DNA repair system (Druzhyna et al., 2008). At this point, a secondary line of defense is necessary for the mitochondria to continue normal functioning. These MQC mechanisms can be divided into two categories: molecular or organelle-level quality control mechanisms (Ng et al., 2021). Molecular level quality control includes the activation of mitochondrial chaperones, precisely heat shock protein 60 (HSP60), HSP70, and HSP90 (Bahr et al., 2022). These chaperones ensure proper folding of newly synthesized proteins and refolding of damaged ones before they are imported into the mitochondria (Becker and Craig, 1994). When misfolded or damaged proteins accumulate, they must be degraded via the ubiquitin-proteasome system (UPS). However, if the mitochondrial damage cannot be repaired via chaperones or UPS, organelle-level quality control mechanisms come into play, as summarized below (Kwon and Ciechanover, 2017; Jin et al., 2018) (Figure 3).
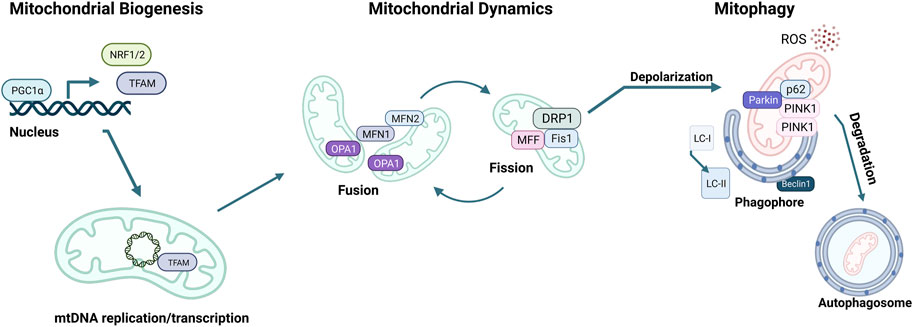
FIGURE 3. Mitochondrial quality control (MQC) mechanisms. The master regulator of mitochondrial biogenesis is PGC-1α, which induces NRF1, NRF2, and TFAM expression in the nucleus. Mitochondria life cycle includes fusion, which forms elongated mitochondrial networks, and fission, which creates fragmented mitochondria. MFN1, MFN2, and OPA1 are responsible for the membrane fusion of mitochondria, whereas DRP1, FIS1, and MFF mediate fission. Mitochondria accumulate oxidative damage during their expected lifespan or in the case of CVDs. Fission enables the fragmentation of damaged mitochondria to be separated and degraded. The elimination of damaged mitochondria is achieved via mitophagy, initiated by the accumulation of PINK1 kinase in the mitochondrial membrane, followed by the recruitment of Parkin, which targets the mitochondria to the autophagosome. p62 targets the mitochondria to the autophagosome and is eliminated during active autophagy. Assembly of the autophagosome requires Beclin1 and attachment of LC3-I onto phosphatidylethanolamine to form LC3-II.
The transcription and replication of the nuclear and mitochondrial genome are essential to mitochondrial biogenesis (mitobiogenesis), the process of self-replication of mitochondria. mtDNA encodes for 11 polypeptides of the ETC, as well as 22 tRNA and 2 rRNA (Fernández-Silva et al., 2003). Nonetheless, most of the mitochondrial proteins are encoded in the nuclear genome and transported into the mitochondria. For this reason, replication of mitochondria relies on mtDNA and nuclear DNA transcription to promote protein and lipid synthesis. Lipids, such as phosphatidylethanolamine (PE) or cardiolipin, are necessary for mitochondrial functioning and are made in the mitochondria but require ER-derived lipids (Tatsuta et al., 2014). Therefore, importing essential proteins and lipids from the nucleus or ER is critical for mitobiogenesis.
The master regulator of mitobiogenesis is the transcriptional coregulator peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC1α), which was first identified in brown adipose tissue as a coactivator of PPARγ (Puigserver et al., 1998). Numerous transcription factors are induced by PGC1α during external stimuli, such as exercise, cold, or fasting (Wu et al., 1999; Zong et al., 2002; Nisoli et al., 2005). Nuclear respiratory factors 1 and 2 (NRF1, 2) are two of such transcription factors, increasing the expression of mitochondrial transcription factor A (TFAM), which in turn translocates to mitochondria and induces mtDNA transcription (Virbasius and Scarpulla, 1994). Mitochondria resident micro-RNAs (miRs) also fine-tune mitobiogenesis by targeting TFAM or other mitobiogenesis factors, as well as relaying signals from the nucleus to the mitochondria to replenish the mitochondrial pool when necessary (Das et al., 2012; Das et al., 2014; Roman et al., 2020; Dogan et al., 2022). During mitobiogenesis, nuclear-encoded genes necessary for mitochondrial function are transcribed in the nucleus and translated into the cytosol with a mitochondrial localization signal. These proteins are transported to the mitochondria and are folded by mitochondrial chaperones (Wiedemann et al., 2004). Similarly, lipids are synthesized in the ER and transferred to the mitochondria during mitobiogenesis. This process involves the mitochondria-associated membranes (MAMs), which are mitochondria-ER contact sites (Gaigg et al., 1995).
Numerous external stimuli regulate PGC1α activity. Cold exposure and exercise induce PGC1α expression through β-adrenergic stimulation and cAMP response element-binding protein (cAMP/CREB) signaling (Wu et al., 1999; Baar et al., 2002). Moreover, AMPK activation in response to exercise also boosts PGC1α expression (Little et al., 2010). Posttranslational modifications are essential for the PGC1α activity (Miller et al., 2019). It has been shown that AMPK phosphorylates PGC1α on T177 and S538 in skeletal muscle (Jäger et al., 2007). Another crucial posttranslational modification on PGC1α is done by the NAD+-dependent deacetylase Sirtuin 1 (SIRT1). Deacetylation and activation of PGC1α by SIRT1 are essential, as NAD+ levels are highly dependent on AMPK activity. Therefore, in addition to its role in mitobiogenesis, PGC1α conjugates cellular energy metabolism, redox state, and mitochondrial activity (Gerhart-Hines et al., 2007; Coste et al., 2008; Cantó et al., 2009).
The mitochondrial network is highly dynamic, constantly remodeling through fission and fusion to decrease stress and replace damaged components (Parra et al., 2011; Chan, 2020). Fusion of two mitochondria is driven by dynamin-related GTPases mitofusin1 and 2 (MFN1, MFN2) on the OMM surface and optic atrophy protein 1 (OPA1) and cardiolipin on the IMM (Chen et al., 2003; Olichon et al., 2007; Paradies et al., 2019) (Figure 3). MFN1 also participates in ER-associated degradation (ERAD) through the degradation of damaged mitochondria via polyubiquitination by the E3 ubiquitin ligase Parkin (Picca et al., 2018). Mitochondrial fusion allows for the fusion of two mitochondria that are in close proximity to share mtDNA, metabolites, and enzymes. Fusion of the OMM requires homo- and heterodimerization of MFN1 and MFN2 (Figure 3) (Ishihara et al., 2004; Chen and Chan, 2009; Hall et al., 2014). OPA1 plays a role in the fusion of the IMM while preserving the cristae structure (Malka et al., 2005; Song et al., 2007). This fusion process sustains the mitochondrial membrane and its permeability to enhance protection from oxidative damage.
Damaged mitochondria can also undergo fission, where one mitochondrion divides into two mitochondria (Chen and Chan, 2009; Ong and Hausenloy, 2010; Elgass et al., 2013). Fission is orchestrated by GTPase dynamin-related protein 1 (DRP1), mitochondrial fission one protein (FIS1), and mitochondrial fission factor (MFF). FIS1 inhibits mitochondrial fusion and acts as a receptor for the binding of DRP1 to the OMM. Once recruited, DRP1 triggers cleavage of mitochondria via interacting with FIS1 and MFF (Palmer et al., 2011; Onoue et al., 2013; Otera et al., 2013; Atkins et al., 2016). Splitting off damaged mitochondria through fission preserves the healthy pool of mitochondria, optimal OXPHOS functioning, and efficient allocation of mitochondrial materials (Quiles and Gustafsson Å, 2022).
A balance between mitochondrial fission and fusion is critical for maintaining optimal mitochondria functioning at homeostasis or during pathological conditions, especially in CVDs. Clearance of damaged mitochondria is central to the control of mitochondrial quality. It is achieved through mitophagy, a process linked to fission since fragmented mitochondria are degraded via the autophagolysosomes (Zaffagnini and Martens, 2016). Mitochondrial fission occurs on the MAMs, where DRP1 also localizes and promotes nucleation of mitochondrial fission. ER tubules can also wrap around the mitochondria and cause constriction of mitochondrial membranes during fission (Friedman et al., 2011; Yang and Yang, 2013).
A pivotal mechanism of MQC is a mitochondria-specific autophagy process named mitophagy (Figure 4). Clearance of mitochondria by mitophagy is indispensable during cellular differentiation, as well as for tissues that require high energy, such as the cardiovascular system (McWilliams et al., 2018). Mitophagy is needed to overcome stress in pathological conditions, and evidence from multiple studies suggests that a deficit in mitophagy mechanisms contributes to the progression of CVDs (Tong et al., 2019; Tan et al., 2021). Briefly, the elimination of mitochondria by mitophagy requires the autophagy receptors to load mitochondrion to the autophagosome through light chain-3 (LC3) (Figures 3, 4). Mitochondria are then engulfed by the autophagosome, which merges with a lysosome to degrade its contents through the lysosomal enzymatic activity (Rüb et al., 2017). Mitophagy can be ubiquitin-dependent or receptor-dependent. Ubiquitin-dependent mitophagy mainly includes PINK1/Parkin-dependent pathways (Figure 4A). Receptor-dependent mitophagy includes Bcl-2 and adenovirus E1B19kDa-interacting protein 3 (BNIP3), FUNDC1, and lipid-dependent mitophagy pathways (Figure 4B).
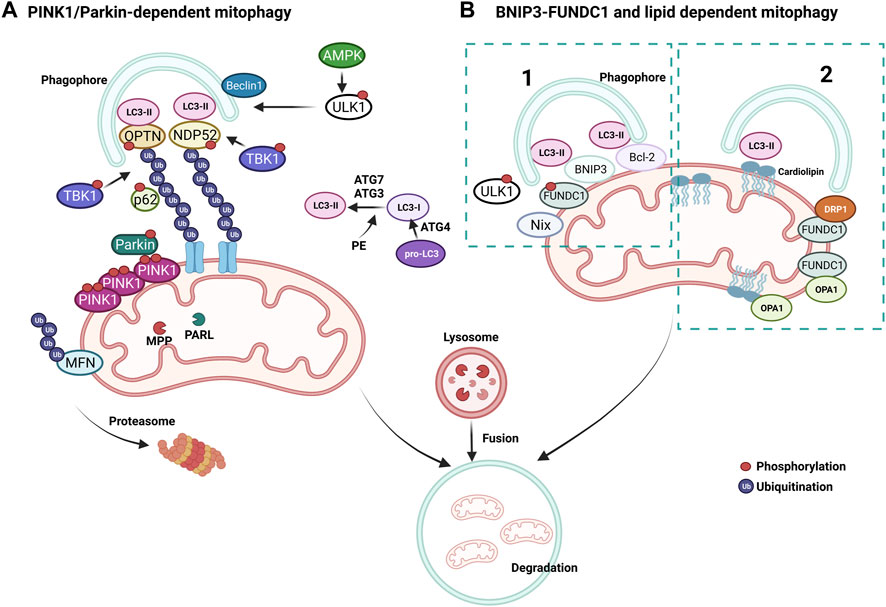
FIGURE 4. Mechanisms of mitophagy. (A) PINK1/Parkin-dependent mitophagy. PINK1 accumulates on damaged mitochondria and recruits Parkin to promote the ubiquitination of OMM proteins. Poly-ubiquitin chains provide an eat me signal to initiate autophagy. TBK1 phosphorylates OPTN and NDP52 to induce clearance of damaged mitochondria. (B) Parkin-independent mitophagy pathways, (1) BNIP3/FUNDC1 mediated and (2) lipid-mediated mitophagy. Mitophagy receptors BNIP3 and FUNDC1 favor mitochondrial fission via DRP1 interaction and OPA1 release.
During basal conditions, serine/threonine kinase PTEN-induced kinase 1 (PINK1) is constantly imported into the mitochondria as it bears a mitochondrial target signal (Lazarou et al., 2012). Once imported, PINK1 is cleaved by matrix processing peptidases, mitochondrial processing peptidase (MPP), and presenilins-associated rhomboid-like protein (PARL) to eliminate the mitochondrial target signal and its hydrophobic transmembrane helix within the transmembrane domain. After processing, PINK1 is released into the cytoplasm, where the UPS constantly degrades it. When mitochondrial damage occurs, PINK1 cannot be transported back to the IMM and accumulates on the OMM, unable to be cleaved by PARL, forming homodimers. Phosphorylation of PINK1 on S228 and S402 is required for its activation and engages Parkin to the OMM, inducing mitophagy (Lazarou et al., 2012; Okatsu et al., 2013; Yamano and Youle, 2013). Interaction of Parkin with PINK1 causes conformational changes that allow for its opening and phosphorylation on S65 by PINK1, simultaneously activating its E3 ubiquitin ligase activity (Ordureau et al., 2014; Pickrell and Youle, 2015) (Figure 4A). As a mechanism that further augments mitophagy, Parkin-mediated ubiquitination of OMM proteins serves as substrates for PINK1, which consequently recruits more Parkin (Nguyen et al., 2016). MFN2 can also be phosphorylated by PINK1 on the OMM and ubiquitinated by Parkin to prevent the fusion of damaged mitochondria with healthy ones (Chen et al., 2013) (Figure 4A). Mitochondria undergoing mitophagy are isolated through the interaction of autophagy-related gene (ATG) and LC3 proteins with a phagophore formation. First, the carboxyl-terminal region of pro-LC3 is processed by ATG4, a cysteine protease (Figure 4A). This results in glycine residues being exposed and the formation of LC3-I. Then, phosphatidylethanolamine (PE) binds to LC3-I via ATG7 and ATG3, forming LC-II as the first ubiquitin-like reaction. Following this reaction, together with ATG10, ATG7 deposits ATG12 to ATG5, which assembles the ATG12-5 complex. This complex later binds to ATG16, which endorses the recruitment of ATG8 with PE on the autophagosomes. Damaged mitochondria are identified through their LC3-interacting region (LIR), which ultimately leads to their engulfment by the autophagosome (Tanida et al., 2004; Nakatogawa, 2013; Dooley et al., 2014). Adaptor proteins sequestosome 1 (p62/SQSTM1), nuclear dot protein 52kDa (NDP52), optineurin (OPTN), and Tax-binding protein-1 (TAX1BP1) are crucial during the tethering of damaged mitochondria to the autophagosomal membranes via recognition of ubiquitinated proteins (Villa et al., 2018). The OMM proteins are eliminated when mitochondrial adaptor proteins contact LC3, which binds to the autophagosomal membranes. OPTN and NDP52 are found on the OMM of damaged mitochondria (Figure 4A). TANK-binding kinase 1 (TBK1) is responsible for the phosphorylation of OPTN and p62 at S177 and S403, respectively. These phosphorylations induce their tethering with ubiquitinated sites and LC3 (Figure 4A). Consecutively, OPTN and NDP52 induce autophagy via recruiting autophagy-related unc-51-like autophagy-activating kinase 1 (ULK1), which is phosphorylated and activated by AMP-activated protein kinase (AMPK) on S555 (Egan et al., 2011). Next, ULK1 recruits the Beclin-1 complex, which promotes the nucleation of phagophores, making autophagosome formation more efficient around the damaged mitochondria (Russell et al., 2013). This final step fully isolates targeted mitochondria inside the autophagosomes and triggers their fusion with lysosomes to form autolysosomes, leading to the degradation of the targeted mitochondria (Narendra et al., 2010; Weil et al., 2018; Yamano et al., 2020). Activation of mitophagy is not strictly dependent on AMPK activation, whereas ULK1 deficiency impairs PINK1/Parkin-dependent mitophagy (Vargas et al., 2019).
Bcl-2, BNIP3, and BNIP3-like (Nix) are all part of the Bcl-2 family of proteins and are constitutively expressed on the OMM (Ma et al., 2020) (Figure 4B). They have a prominent role in apoptosis but are also essential mitophagy receptors. These receptors are bound to the OMM and mainly interact with LC3 via the LIR motif to facilitate mitophagy, independent of the ubiquitin (Gustafsson Å and Dorn, 2019). It has been shown that BNIP3 and Nix are involved in the elimination of mitochondria in adult heart tissues (Lampert et al., 2019) and that hypoxic conditions can upregulate BNIP3 (Gálvez et al., 2006). Another mitophagy receptor, the FUN14 domain containing 1 (FUNDC1), mediates mitophagy (Figure 4B). This receptor is also relevant to hypoxia and ischemia, as hypoxic stimuli are linked with FUNDC1 dephosphorylation on its LIR via Phosphoglycerate mutase family Member 5 (PGAM5) to promote its binding with LC3 (Ren et al., 2020; Xiao et al., 2020). ULK1 is also known to phosphorylate FUNDC1 on its LIR motif to favor interaction with LC3 during mitophagy (Chen et al., 2014; Wu et al., 2014). Intriguingly, FUNDC1 has also been shown to interact with OPA1 and DRP1 to regulate mitochondrial dynamics during mitochondrial stress (Chen M. et al., 2016).
Lipids, such as ceramide or cardiolipin, which can prompt mitophagy with their LIR motif, are present in the OMM. Ceramide is a sphingolipid synthesized by ceramide synthase (CeS). During ER stress, ceramides are translocated from the ER and accumulate on the OMM, triggering mitophagy (Chu et al., 2013). Accumulated ceramides on the OMM can bind LC3 through its ceramide-binding domain and promote the binding of Beclin1. Ceramide-mediated mitophagy also relies on DRP1, which initiates the recognition of ceramide in the OMM by the autophagolysosomes (Sentelle et al., 2012; Antón et al., 2016) (Figure 4B). Importantly, ceramides can build a channel with active BCL2-associated X protein (BAX) that allows the release of cytochrome c to initiate apoptosis. Another important lipid in mitophagy, cardiolipin, is a phospholipid synthesized by cardiolipin synthase (CRD1) in the mitochondria. Cardiolipin (CL) is present on mitochondrial membranes and cristae structures and is primarily found in the IMM (Daum, 1985). CL binds to OPA1 in physiological conditions to regulate OPA1 assembly during mitochondrial fusion and interacts with LC3 during mitochondrial stress (Liu et al., 2003). During mitochondrial damage, CL is transported from the IMM to the OMM (Chu et al., 2013). This movement allows CL to be exposed to the cytosol, bind to LC3, and serve as a recruitment site for adaptor proteins during mitophagy (Chu et al., 2013). Exposed CL preferentially associates with Beclin1 on the OMM during mitophagy and is crucial for phagophore formation (Huang et al., 2012). Notably, pro-IL-1α directly binds to CL in lipopolysaccharide (LPS)-treated macrophages through an LC3 binding domain while blocking mitophagy and activating NLRP3 inflammasome, further supporting the importance of CL in mitochondrial homeostasis (Dagvadorj et al., 2021).
Mitochondrial networks cannot function separately from other subcellular compartments and rely heavily on inter-organelle communication. Mitochondria continuously coordinate with the nucleus, the ER, and other vesicular organelles (Lin et al., 2020). Importantly, inter-organelle communication of mitochondria and cytosolic organelles is crucial in the development and pathogenesis of several diseases. Most of the mitochondrial proteins are encoded in the nucleus and must be translocated into the mitochondria via transmembrane complexes (Pfanner et al., 2019). As mitochondria are self-monitoring organelles, any perturbations trigger retrograde signaling from mitochondria to the nucleus or other organelles to cope with the stress and reestablish proper mitochondrial functioning (Chowdhury et al., 2020; Vizioli et al., 2020; Booth et al., 2021).
When mitochondrial protein import is dysfunctional, a signal from the mitochondria, called UPRmt, is initiated and relayed to the nucleus. This signal activates the required transcription factors to cope with stress and induces a repair mechanism to normalize the mitochondrial protein import (Shpilka and Haynes, 2018). UPRmt aims to optimize metabolism by blocking oxidative phosphorylation and the TCA cycle, decreasing the mitochondrial load and ultimately alleviating mitochondrial stress. Simultaneously, the expression of genes responsible for glycolysis and amino acid metabolism is augmented to provide alternative energy resources (Anzell et al., 2018). Aside from switching the reliance on specific energy resources, UPRmt also promotes the expression of mitochondrial chaperones, anti-oxidative proteins, and proteases to cope with the accumulation of dysfunctional proteins (Zhu L. et al., 2021). This decreases the mitochondrial burden while enabling the proper processing of damaged mitochondrial proteins (Quirós et al., 2016). Impairment of OXPHOS machinery, mtDNA damage, disruption of mitochondrial protein synthesis, accumulation of misfolded or unfolded proteins in the mitochondria, or the buildup of ROS causes mitochondrial dysfunction. These events trigger UPRmt (Qureshi et al., 2017). It has been shown that during mammalian UPRmt, transcription factors C/EBP homologous protein (CHOP), activating transcription factor 4 (ATF4), and ATF5 are activated, which are also involved in the unfolded protein response of the ER to alleviate ER stress caused by the accumulation of unfolded or misfolded proteins (Shpilka and Haynes, 2018; Yildirim et al., 2022). Activation of the kinases general control non-derepressible-2 kinase (GCN2), protein kinase RNA (PKR), PKR-like endoplasmic reticulum kinase (PERK), and heme-regulated inhibitor kinase (HRI) controls the phosphorylation of eukaryotic translation initiation factor 2 subunit 1 (eIF2α), which is the major player during organelle stress (Teske et al., 2013). Activated eIF2α inhibits global protein translation to reduce organelle stress, enhancing the expression of specific transcription factors like CHOP, ATF4, and ATF5. These transcription factors promote the expression of chaperones and proteases to cope with the UPRmt. Notably, the expression of ROS detoxifying enzymes, mitobiogenesis machinery, and mitochondrial import proteins are also enhanced to favor mitochondrial remodeling during stress (Qureshi et al., 2017).
Mitochondrial stress, which stems from the loss of mitochondrial membrane potential, releases calcium (Ca2+) from the mitochondria to the cytoplasm through voltage-dependent L-type Ca2+ channels. This increase in cytosolic Ca2+ triggers more Ca2+ to be released from the sarcoplasmic reticulum through the ryanodine receptor 2 (RYR2). Consequently, Ca2+ is cleared away from the cytoplasm via sarcoplasmic or ER resident ATPase (SERCA) family proteins, as well as solute carrier family 8 member A1 (SLC8A1) (Kho et al., 2012). During basal conditions, Ca2+ enters the mitochondria via the calcium uniporter protein (MCU) (Baughman et al., 2011). In addition, Ca2+ release occurs through the Ca2+ antiporter SLC8B1. Transient increases in Ca2+ levels can aid OXPHOS machinery. However, during mitochondrial membrane potential loss, free cytosolic Ca2+ activates calcineurin. In turn, the nuclear factor-κB (NF-κB) and nuclear factor of activated T cells (NFATC) are activated and translocated to the nucleus. This translocation stimulates the expression of genes responsible for Ca2+ storage or transport (Liu et al., 2017). Increased Ca2+ levels can activate various kinases like calcium/calmodulin-dependent protein kinase IV (CAMKIV), Ca2+-dependent protein kinase C, c-Jun N-terminal kinases (JNK), and p38 MAPK. These kinases fine-tune the mitochondrial adaptation to Ca2+ level fluctuations and metabolism, as well as cell proliferation and glucose metabolism (Giorgi et al., 2018).
During severe unresolved stress, like calcium overload, excessive ROS, or mtDNA damage, intrinsic apoptosis is initiated as mitochondrial homeostasis is jeopardized (Brumatti et al., 2010). Programmed cell death is orchestrated by pro-apoptotic proteins of the B cell lymphoma (BCL) 2 family. When apoptosis is triggered, BCL-associated X (BAX) and Bcl2 homologous antagonist/killer (BAK) are incorporated into the OMM and oligomerize, resulting in a pore formation on the mitochondria. This pore increases membrane permeability and promotes the release of pro-apoptotic factors (Lomonosova and Chinnadurai, 2008). Under physiological conditions, BAX is inactive and shuttles between the cytosol and the OMM, whereas BAK resides on the OMM and interacts with VDAC2 (Cheng et al., 2003; Schellenberg et al., 2013). Under apoptotic stimuli, BAX is no longer shuttled to the cytosol and together with BAK, gets activated via the pro-apoptotic Bcl2 homology-3 (BH3) domain-containing factors, BCL2 binding component 3 (also known as p53-upregulated modulator of apoptosis, PUMA), BCL2-interacting mediator of cell death (BIM), phorbol-12-myristate-13-acetate-induced protein 1 (NOXA) and BH3-interacting domain death agonist (BID) (Kuwana et al., 2005). Multiple pro-survival proteins counteract with the pro-apoptotic factors to reduce the permeabilization of the OMM, such as BCL2, BCL2 like 1 (BCL2L1 also known as BCL-XL), myeloid cell leukemia 1 (MCL1), BCL2 like 2 (BCL2L2, also known as BCL-W), and BCL2 related protein A1 (BCL2A1) (Moldoveanu et al., 2014). These factors reside on the OMM or ER and inhibit the binding of pro-apoptotic proteins (Czabotar et al., 2014). If the permeabilization of OMM is maintained, cytochrome c is released to initiate caspase-independent cell death. Notably, the release of cytochrome c causes apoptosome formation, a multi-protein structure composed of apoptotic protease-activating factor-1 (APAF-1), deoxyATP (dATP), and pro-caspase-9, where pro-caspase-9 gets activated (Chen et al., 2011). Consequently, caspases 3 and 7 are also cleaved and activated to promote cell death (Li et al., 1997; McArthur et al., 2018).
Mitochondrial apoptosis can produce type I interferon when caspases are inhibited (Rongvaux et al., 2014; White et al., 2014). In this context, mtDNA is released to the cytosol as a damage-associated molecular pattern (DAMP) during BAX/BAK-mediated pore formation and apoptosis (Cosentino et al., 2022). The presence of any DNA in the cytoplasm (e.g., pathogenic, nuclear, or mitochondrial) triggers a signal to activate 2′3′-cyclic GMP-AMP (cGAMP) synthase (cGAS), which detects cytosolic DNA. This detection allows cGAS dimerization and production of second messenger cGAMP to relay the message to and activate the stimulator of interferon genes (STING) (Diner et al., 2013; Sun et al., 2013). ER resident STING then shuttles to Golgi, activates TBK1, and gets phosphorylated by TBK1 (Zhang C. et al., 2019). Interferon regulatory factor 3 (IRF3) phosphorylation by active TBK1 promotes its dimerization and transport to the nucleus, which activates interferons and other cytokines (Tanaka and Chen, 2012). Oxidized mtDNA can exit the mitochondria through mPTP and VDAC, where oxidation promotes its fragmentation (Xian et al., 2022). In the cytosol, mtDNA fragments interact with NLRP3 to induce inflammasome activation and STING phosphorylation on S365, a pre-requisite for IRF3 activation (Chen Q. et al., 2016). Similarly, when released to the cytosol during programmed cell death, oxidized mtDNA binds to the NLRP3 inflammasome and activates it (Shimada et al., 2012). It has been recently shown that glycolytic enzyme Hexokinase 2 dissociation from VDAC on the OMM is essential for NLRP3 inflammasome activation. This dissociation results in Ca2+ uptake by mitochondria released from the ER and pore formation by VDAC oligomerization, allowing protein and mtDNA to exit from the mitochondria (Baik et al., 2023). Consequently, NLRP3 assembly is promoted and crucial during inflammatory responses.
Pathogenic or mitochondrial dsRNA in the cytoplasm is detected by the retinoic acid-inducible gene I (RIG-I) as well as melanoma differentiation-associated protein 5 (MDA5), which enables activation and aggregation of mitochondrial antiviral signaling protein (MAVS) on the OMM (Reikine et al., 2014). Accumulation of MAVS promotes IRF3 activation and stimulates the expression of antiviral response genes in the nucleus (Hou et al., 2011). Furthermore, bacterial infection can also activate an innate immune response that involves mitochondria. LPS or glycans on the bacterial wall can induce immune responses via ubiquitylation. This allows for the formation of pro-inflammatory ubiquitin linkages with the mitochondria and causes OMM permeabilization, enabling the formation of endolysosomes (Hamacher-Brady et al., 2014).
Mitochondrial dysfunction has been involved in the development of several CVDs (Killackey et al., 2020). CVDs are often associated with defective ETC machinery, excessive ROS production, impaired energetics, and MQC, as well as abnormal Ca2+ homeostasis (Forte et al., 2021). Notably, the dysregulation of proteins responsible for mitochondrial dynamics, mitophagy, or mitobiogenesis is closely linked to the advancement of CVDs (Chehaitly et al., 2022). Hence, understanding the contribution of mitochondrial health and normal functions to CVDs is crucial to develop novel and more efficient therapeutic approaches for these conditions. Here, we discuss various CVDs and the implications of mitochondrial dysfunction in this context (Table 1).
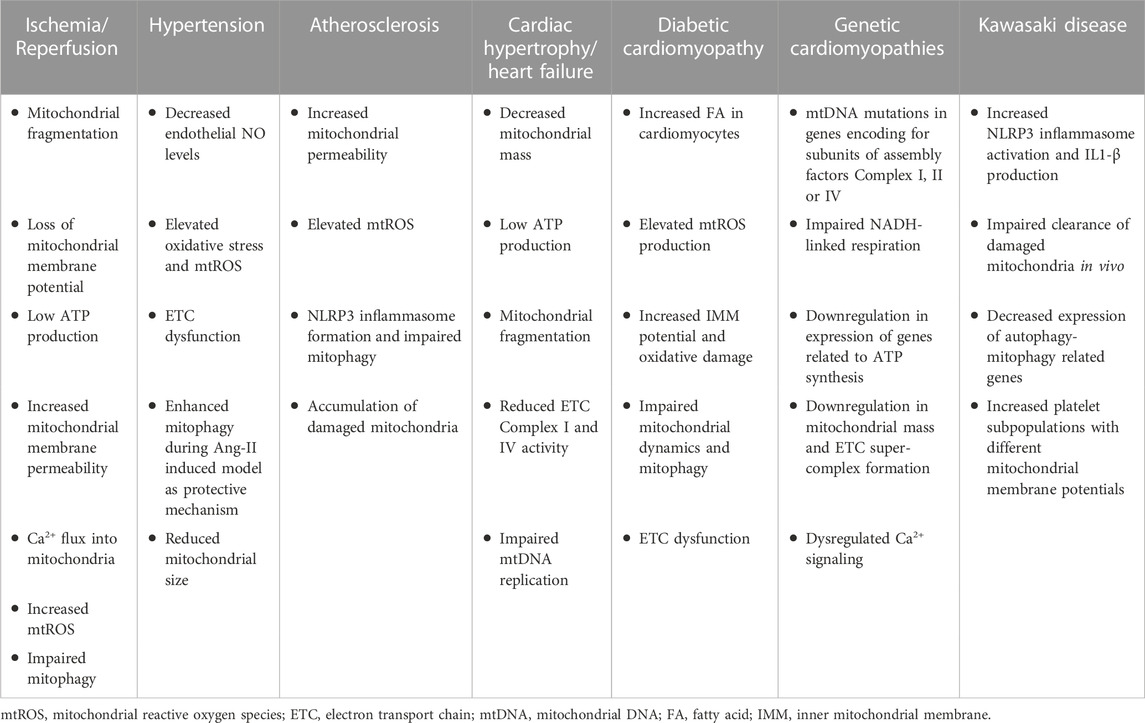
TABLE 1. Mitochondrial phenotype in CVDs. Overview of mitochondrial impairment during multiple CVDs.
Blood flow restoration of ischemic tissues or areas results in functional and structural tissue alterations, known as ischemia-reperfusion (I/R) injury. I/R injury usually stems from occlusion of coronary vessels, leading to tissue hypoxia and a deficiency in ATP levels (Peoples et al., 2019).
Mitochondria respond to cardiac tissue I/R injury, and a cascade of mitochondrial fragmentation and cell death, associated with elevated oxidative stress and loss of mitochondrial membrane potential is initiated upon reperfusion (Murphy and Steenbergen, 2008). Oxygen deprivation immediately results in mitochondrial dysfunction since heart tissues rely heavily on OXPHOS for their energy demand. In vitro, hypoxia/reoxygenation of cardiomyocytes revealed that these cells exhibit less mitochondrial fusion, lower ATP production, and higher membrane permeability (Liu and Hajnóczky, 2011). Furthermore, overexpression of mitochondrial fusion protein MFN2 or DRP1-dependent blockade of mitochondrial fission increased cell survival (Jahani-Asl et al., 2007). Mice overexpressing OPA1 display less severe I/R injury with better mitochondrial cristae remodeling (Varanita et al., 2015). Similarly, reduced OPA1 levels indicating a decrease in mitochondrial fusion during I/R injury in mice were rescued by melatonin treatment, which promotes AMPK signaling and diminishes caspase-9-dependent apoptosis (Zhang Y. et al., 2019). Supporting these findings, melatonin treatment also increased the expression of SIRT3 and the activity of the ROS-metabolizing enzyme SOD2 in cardiomyocytes during I/R by blocking the mitochondrial fission (Bai et al., 2021). Reduced infarct size is also observed in rats when mitochondrial fusion is promoted pharmacologically (Maneechote et al., 2019). Supporting these notions, cardiac-specific MFN1 knock-out mice are protected against excessive ROS production as these mice have a delayed mitochondrial permeability transition pore (mPTP) opening, resulting in increased cell survival (Papanicolaou et al., 2012). I/R injury also causes an increase in oxidative stress and promotes Ca2+ flux into the mitochondria, allowing a dysfunctional electrochemical gradient of IMM and impairing the ETC activity (Halestrap, 1999). Excess Ca2+ inside the mitochondria and elevated ROS levels cause the opening of the mPTP and render permeability of the IMM, resulting in depolarization, membrane potential loss, and cell death (Kwong and Molkentin, 2015). Similarly, overflow of Ca2+ into mitochondria due to mitochondrial calcium uniporter (MCU) activation is fundamental to I/R injury. Overexpression of Histidine triad nucleotide-binding 2 (HINT2) blocks MCU activity and alleviates Ca+2 burden and fragmentation of mitochondria in mice during I/R injury (Li et al., 2021).
Clearance of defective mitochondria by mitophagy is beneficial during I/R injury. Evidence indicates defective mitophagy, dysfunctional mitochondria, and myocardial impairment after I/R injury in DRP1 knock-out mice (Ikeda et al., 2015). Similarly, Parkin knock-out mice have exacerbated infarct size and dysfunctional mitochondria with impaired mitophagy, suggesting that by clearing defective mitochondria, mitophagy is beneficial during I/R injury (Kubli et al., 2013). Also, autophagy proteins such as Beclin 1, BNIP3, and FUNDC1 are implicated in the I/R injury (Georgakopoulos et al., 2017). FUNDC1 protects the heart by promoting mitophagy to eliminate damaged or non-functional mitochondria (Zhang et al., 2017). Furthermore, in a hypoxia model, the overexpression of necroptosis modulator receptor-interacting protein 3 (RIPK3) blocks the AMPK pathway and inhibits Parkin-dependent mitophagy in cardiomyocytes, leading to cardiomyocyte necroptosis and exacerbated cardiac damage (Zhu P. et al., 2021).
Data from FUNDC1 knock-out mice showed that this mitophagy factor regulates MQC, is essential for platelet activation, and ultimately decreases heart I/R injury (Zhang et al., 2016). In addition, an anti-diabetes drug, empagliflozin, has been reported to promote AMPK1/ULK1/FUNDC1-dependent mitophagy and attenuated mitochondrial dysfunction in both in vitro and in vivo I/R injury models (Cai et al., 2022). Upregulation of mitophagy and mitobiogenesis is observed in human cardiac tissues after cardiopulmonary bypass surgery, indicating that a balance between these two arms of MQC is crucial during surgery-induced I/R injury (Andres et al., 2017). These findings provide insight into how mechanisms of MQC regulate tissue homeostasis during I/R injury. However, therapeutic strategies to fine-tune mitochondrial dynamics during I/R injury need further investigation.
Hypertension, characterized by increased blood pressure and a decline in vascular function, is associated with inflammation, endothelial impairment, and mitochondrial dysfunction (Barki-Harrington et al., 2004; Lahera et al., 2017). During hypertension, oxidative damage in mitochondria is triggered by the activation of Angiotensin (Ang) II, which decreases endothelial NO levels and exacerbates vascular oxidative stress. ROS production increases during hypertension due to Ang II-mediated protein kinase-C activity and excess production of O2•- and H2O2 (Doughan et al., 2008). Excessive ROS production may lead to mtDNA mutations and ETC machinery dysfunction, both contributing to hypertension progression (Griendling et al., 2021).
MQC also plays a pivotal role in the development of angiogenic function. It has been shown that the knock-down of MFN1, a protein involved in mitochondrial fusion, results in a less angiogenic response of endothelial cells to vascular endothelial growth factor (VEGF), induces endothelial NO synthase (eNOS) and decreases mitochondrial membrane potential (Lugus et al., 2011). Moreover, inhibition of mitochondrial fission by blocking DRP1 activity results in a blockade of apoptosis in an Ang II hypertensive rat model. In this context, SIRT1 is degraded by Ang II, resulting in p53 acetylation, which promotes DRP1 expression and cell apoptosis (Qi et al., 2018). Furthermore, genetic deletion of SIRT3 in mice leads to hypertension by inducing endothelial dysfunction, vascular inflammation, and hypertrophy. In contrast, global SIRT3 overexpression blocks Ang II-induced hypertension (Dikalova et al., 2020). Elevated FUNDC1-mediated mitophagy during hypoxic pulmonary hypertension induces hypoxia-inducible factor 1α (HIF1α) and pulmonary artery smooth muscle cell proliferation, leading to pulmonary vascular remodeling (Liu et al., 2022).
Mitophagy acts as a protective mechanism during hypertension and enhanced ATG-5-mediated mitophagy during Ang II-induced hypertension results in reduced levels of ROS and inflammation (Pan et al., 2012). Mitochondrial mass and structure are also affected during hypertension. Reduced mitochondrial size and osmotic swelling disrupt the mitochondrial OXPHOS efficiency (Ritz and Berrut, 2005). Cristae structure and ETC stability of mitochondria are jeopardized during hypertension, as it has been shown that there is a loss of CL, a crucial phospholipid for the membrane structure (Cogliati et al., 2013).
Atherosclerosis is a progressive cardiovascular disease resulting in the formation of plaques containing inflammatory cells, the narrowing of arteries, and decreased blood flow, increasing the risks of cardiovascular complications, such as myocardial infarction and stroke. Plaque formation occurs via low-density lipoprotein (LDL) uptake in the arterial wall, leading to immune cell infiltration, inflammation, calcification, and vascular smooth muscle (VSMC) migration into the plaque (Gimbrone and García-Cardeña, 2016). Accumulation of oxidized LDL in the arterial wall can interfere with the proper functioning of mitochondrial respiration dynamics, cause the opening of mPTP and increase mitochondrial permeabilization, and promote ROS production (Khwaja et al., 2021). This eventually results in endothelial cell apoptosis and accelerates atherosclerosis. In addition, excess ROS production and mitochondrial dysfunction during atherosclerosis have been shown to activate NLRP3 inflammasome formation and boosts mitophagy (Jin Y. et al., 2022). Of note, loss of OGG1 induces oxidative mtDNA damage and subsequent NLRP3 inflammasome activation, exacerbating atherosclerosis in mice (Tumurkhuu et al., 2016). Importantly, depletion of an abundant cholesterol biosynthetic intermediate in coronary lesions, desmosterol, results in mtROS accumulation and NLRP3 inflammasome activation in macrophages, favoring atherosclerotic plaque formation (Zhang X. et al., 2021).
VSMCs can be activated by the platelet-derived growth factor (PDGF) during atherosclerotic plaque formation. MFN2 is less expressed in the atherosclerotic plaques, which favors mitochondrial fission. Inhibition of mitochondrial fission decreases VSMCs proliferation (Salabei and Hill, 2013). Similarly, inhibition of DRP1, a mitochondrial fission regulator, decreased endothelial impairment and atherosclerosis progression in apolipoprotein E (ApoE) knock-out mice, as well as VSMCs calcification (Rogers et al., 2017; Wang et al., 2017). Accumulation of p62 and LC3-II has been reported in cells from atherosclerotic plaques, which indicates impaired autophagy/mitophagy (Sergin et al., 2016). Decreased LC3-II levels are detected in patients with unstable atherosclerotic plaques, leading to reduced autophagy/mitophagy and defective clearance of damaged mitochondria and cell death in the arterial wall (Swaminathan et al., 2014). Supporting these findings, deleting the ATG-5 gene in mice, which is involved in autophagy, results in increased production of IL-1β, impaired cholesterol clearance, and increased atherosclerotic plaque formation (Razani et al., 2012). SIRT1 inhibition accelerates mouse atherosclerosis progression through the increased acetylation of ATG5 (Yang et al., 2017). Similarly, VSMC-specific deletion of ATG7 in ApoE−/− mice leads to impaired autophagic flux, accumulation of fragmented mitochondria, and inefficient OXPHOS activity with unstable atherosclerotic plaque formation susceptible to rupture (Nahapetyan et al., 2019). Furthermore, the importance of ER-mitochondria signaling in atherosclerosis progression has been shown through the modulation of the ER-resident kinase PERK. Lipid-activated PERK can activate Lon protease-1, resulting in mitochondrial PINK1 degradation and blockade of mitophagy. Inhibition of mitophagy increases ROS production due to the accumulation of damaged mitochondria and boosts inflammasome activation in macrophages, contributing further to atherosclerotic plaque formation (Onat et al., 2019). In addition, in the absence of a secreted protein known to participate in atherosclerosis, apolipoprotein A-I binding protein (AIBP), PINK1 is cleaved, and mitophagy is blocked, which also promotes atherosclerosis progression and plaque formation (Duan et al., 2022).
Cardiac hypertrophy, a condition that results in the thickening of heart muscles, is an acute response to enhanced hemodynamic overload and mechanical stress on cardiac tissues. When prolonged, cardiac hypertrophy becomes pathological and can result in heart failure (Levy et al., 1990). Cardiac hypertrophy is associated with increased sarcomere production, thickening of the ventricular wall and induces changes in cardiac tissue gene expression, metabolism, and contractility (Schiattarella and Hill, 2015; Nakamura and Sadoshima, 2018). After hypertrophy, cardiomyocytes need more energy and increase mitochondrial mass at an early stage. However, this is followed by a decrease in mitochondrial mass and an impairment of cardiac contractility as the disease progresses (Zak et al., 1980; Goffart et al., 2004). Ultimately, the decline in mitochondrial mass reduces the production of ATP and diastolic dysfunction, causing heart failure (Flarsheim et al., 1996). BNIP3 expression increases in stressed cardiomyocytes, favoring mitochondrial fragmentation (Chaanine et al., 2016). Blocking mitochondrial fission through the deletion of DRP1 in mice leads to hypertrophy caused by norepinephrine signaling. On the other hand, inhibition of mitochondrial fusion by blocking MFN2 stimulates the hypertrophic response (Pennanen et al., 2014). Mitochondrial fusion is affected during cardiac hypertrophy, as reduced expression of MFN2 and OPA1 has been reported in cardiomyocytes in the context of heart failure (Chen et al., 2009). Furthermore, the activity of Complex I and IV from the mitochondrial ETC is also reduced during heart failure (Arbustini et al., 1998). mtDNA replication is also impaired in heart failure, causing a decrease in mtDNA-encoded proteins and mitochondrial biogenesis (Sheeran and Pepe, 2017). Interestingly, the deletion of OMA1, an inner mitochondrial membrane protease that determines cristae structure for optimal ETC functioning, alleviates myocardial damage by preventing cardiomyocyte death (Acin-Perez et al., 2018). The expression of dual-specificity tyrosine-regulated kinase 1B (DYRK1B) is increased in failing hearts of humans and mice and overexpression of this kinase impairs ejection fraction as well as cardiac fibrosis in mice by downregulating the mitobiogenesis regulator PGC1α (Zhuang et al., 2022). An important inflammatory response during pathological conditions is pyroptosis, a form of inflammatory cell death (Shi et al., 2015). During this process, NLRP3 inflammasome is induced, activating caspase-1, which cleaves gasdermin D (GSDMD) to permeabilize the cells, resulting in IL-1β and IL-18 secretion from the rupturing cells (Kayagaki et al., 2015; Shi et al., 2015). In mouse and human hypertrophic myocardia, it has been shown that there is an amplification of GSDMD that activates the mitochondrial STING axis, causing mitochondrial dysfunction and contributing to cardiomyocyte pyroptosis (Han et al., 2022). Notably, choline supplementation in rats alleviated cardiac hypertrophy by promoting UPRmt through activating the SIRT3-AMPK axis as an adaptive response to cardiac dysfunction (Xu et al., 2019). Mitochondrial ROS scavenging is crucial to protect the cardiac tissues from oxidative damage (Xin et al., 2021). Recently, the deletion of mitochondrial peroxiredoxin 3 (Prdx3) in mice has been shown to cause cardiac hypertrophy and heart failure. In addition, the same study showed that Prdx3 interacts with PINK1 and blocks its proteolytic cleavage during mitophagy, playing a protective role during hypertrophy-induced mitochondrial dysfunction (Sonn et al., 2022). Of note, Parkin levels decline in hypertrophic cardiomyocytes, as well as mouse hearts, and overexpression of Parkin in mice blocked Ang-II-induced hypertrophy and improved heart function via boosting mitophagy, highlighting the importance of clearance of damaged mitochondria during cardiac hypertrophy (Sun et al., 2022).
Diabetic cardiomyopathy is a pathological process leading to changes in myocardial structure and function observed in patients with diabetes mellitus in the absence of other cardiac risk factors, such as coronary artery diseases or hypertension (Jia et al., 2018). Elevated levels of fatty acids in cardiomyocytes from diabetic patients can interfere with mitochondrial metabolism. In a healthy heart, ATP is produced by fatty acid oxidation and, to some extent, from carbohydrates (Duncan, 2011). Mitochondria and peroxisomes are responsible for the β-oxidation of fatty acids (FAO), a process that depends on oxygen availability. This process is not optimal when cardiac tissue needs high levels of ATP. In the context of diabetes, increased levels of fatty acids and ATP produced from the oxidation of FAO result in a loss of flexibility as to mitochondrial fuel sources (Duncan, 2011). Consequently, heart contraction efficiency starts to decline, causing a lack of blood flow and cardiomyopathy (Liesa and Shirihai, 2013). Nuclear receptor family PPARs are heavily involved in the pathogenesis of diabetic cardiomyopathy. Elevated levels of fatty acids inside cardiomyocytes interfere with FAO and lead to increased PPAR-γ expression and decreased PPAR-α expression (Wang L. et al., 2021). In line with this, an increase in fatty acids boosts ROS production by increasing IMM potential, resulting in oxidative damage and mitochondrial dysfunction (Schilling, 2015). Prolonged ROS levels increase and induce cardiomyocyte apoptosis, which is compensated by fibroblast recruitment. This changes the cellular composition of cardiac tissue, which can lead to heart failure (Flarsheim et al., 1996; Dabkowski et al., 2010).
Type 2 diabetic cardiomyopathy results in perturbations in the mitochondrial Ca2+ signaling (Dillmann, 2019). In a murine model of diabetic cardiomyopathy, mice fed with a high-fat and high-sucrose diet showed cardiac hypertrophy, dysfunction, and insulin resistance (Dia et al., 2020). This phenotype was associated with decreased interactions between IP3R and VDAC1, causing a decline in Ca2+ flux into the mitochondria (Dia et al., 2020). In a rat model of type 2 diabetes, the expression of secreted frizzled-related protein 2 (SFRP2), a protein involved in Wnt signaling and angiogenesis, is downregulated in rat cardiomyocytes and cardiac tissues. This was associated with a reduction in mitochondrial membrane potential and increased oxidative stress via the regulation of mitobiogenesis through the AMPK/PGC1α axis (Ma et al., 2021). While exercise in mice increased the expression of the mitochondrial deacetylase SIRT3 during diabetes-induced cardiac impairment and preserved cardiac functions by promoting the AMPK/SIRT3 pathway (Jin L. et al., 2022), SIRT3 deficiency exacerbates diabetic cardiomyopathy by activating NLRP3 and promoting the accumulation of mtROS (Song et al., 2021). The anti-diabetic drug empagliflozin has also been shown to inhibit cardiac dysfunction during diabetic cardiomyopathy by increasing the expression of Nrf2 in type 2 diabetic mice. Additionally, in mice treated with empagliflozin, the expression of mitochondrial fission proteins was decreased with an upregulation of the fusion machinery, contributing to the recovery of cardiac tissues during this pathogenic condition (Wang J. et al., 2022). Notably, a decline in NAD+ to NADH ratio in diabetic mice is linked to the downregulation of SOD2, deficit in mitochondrial bioenergetics, and a concomitant increase in oxidative damage. Increasing NAD+ levels via nicotinamide phosphoribosyltransferase (NAMPT), the enzyme producing the intermediate for NAD+, relieved cardiac dysfunction in diabetic mice, showing the possible therapeutic potential of NAD+ supplementation in this disease (Chiao et al., 2021).
Mitochondrial dynamics are also impaired in type 2 diabetic cardiomyopathy. The decline in MFN1,2, OPA1, and DRP1 function has been implicated in the development of diabetic cardiomyopathy (Williams and Caino, 2018). A decrease in MFN2 expression, followed by impaired mitochondrial fusion, mitochondrial fragmentation, and related OXPHOS dysfunction, has been reported in diabetic cardiomyopathy (Montaigne et al., 2014). Perturbations in mitochondrial dynamics activate mitophagy and autophagy, the mechanisms of MQC. Previous data suggest that autophagy is blocked in type 1 diabetes, as shown by decreased levels of LC3, ATG5, and 12 in cardiomyocytes (Xu et al., 2013). Lower levels of PINK1 and Parkin are also reported in the same study, hinting at the suppression of mitophagy in the hearts of type 1 diabetic mice (Xu et al., 2013). In mouse models of type 1 diabetes, autophagy and mitophagy are augmented in cardiac tissues (Tao and Xu, 2020; Zhang M. et al., 2021). Furthermore, feeding mice with a high saturated fatty acid diet induces autophagy and cardiomyopathy (Russo et al., 2012). In addition, mice fed a high-fat diet developing diabetic cardiomyopathy show an initial peak of cardiac autophagy, which declines later. However, in this study, mitophagy activation was sustained for 2 months, leading to Parkin deficiency and impaired mitophagy, worsening the disease and suggesting a protective effect of mitophagy during the early stages of diabetic cardiomyopathy (Tong et al., 2019). Interestingly, the expression of TOM70, a mitochondrial outer membrane protein, is decreased in the heart tissues of diabetic mice (Wang et al., 2020). Overexpressing TOM70 rescues mitochondrial dysfunction, oxidative damage, and apoptosis in cardiac tissues, indicating that TOM70 could be a therapeutic target in treating diabetic cardiomyopathy (Wang et al., 2020).
Genetic cardiomyopathies arise from chromosomal abnormalities interfering with the normal function of the heart (McKenna et al., 2017). Hypertrophic cardiomyopathy is the most prevailing, affecting 1 in 500 people (Tuohy et al., 2020). It is usually caused by autosomal dominant mutations of genes that code for sarcomere proteins and mutations of mitochondria-related genes (Teekakirikul et al., 2019). Arrhythmogenic right ventricular cardiomyopathy is a familial heart disease that manifests by mutations of desmosomal proteins, which are intermediate muscle tissue filaments (Krahn et al., 2022). Consequently, ventricular wall thinning and ballooning occur in affected patients (Shah et al., 2023). Another congenital cardiomyopathy is left ventricular non-compaction, which is initiated during the embryonic stage and perturbs the normal development of the heart muscle (Singh and Patel, 2023). In addition, restrictive cardiomyopathy is characterized by the restriction of the left ventricle, causing a significant increase in ventricular pressure and myocardial stiffness (Rapezzi et al., 2022). Finally, dilated cardiomyopathy is manifested with enlarged ventricles, causes systolic dysfunction, and is inherited in an autosomally dominant manner (De Paris et al., 2019).
Genetic cardiomyopathies are mitochondrial diseases related to either disruption in ETC complex subunits or factors involved in their assembly, mitochondria encoded tRNAs, rRNAs, and proteins responsible for mtDNA maintenance and CoQ10 synthesis (Yamada and Nomura, 2021). These cardiomyopathies are often associated with mutations in mtDNA encoded Complex I subunit genes NADH-ubiquinone oxidoreductase chain 1 and 5 (MT-ND1 and 5) and nuclear-encoded Complex I subunit genes such as NADH: ubiquinone oxidoreductase core subunits S2 (NDUFS2), V2 (NDUFV2) or A2 (NDUFA2). Similarly, mutations in nuclear factors involved in Complex I assembly such as acyl-CoA dehydrogenase family member 9 (ACAD9) and NADH: ubiquinone oxidoreductase complex assembly factor 1 (NDUFAF1), are also associated with the development of genetic cardiomyopathies (Fassone and Rahman, 2012; Brecht et al., 2015; Dubucs et al., 2023). All subunits of Complex II are nuclear-encoded, and mutations of subunits A, D (SDHA and SDHD), as well as Complex II assembly factor 4, have been reported in patients with hypertrophic, dilated, and non-compaction cardiomyopathies (Jain-Ghai et al., 2013; Alston et al., 2015; Wang X. et al., 2022). Likewise, mutations in mtDNA encoded cytochrome b gene (MTCYB) have also been identified in patients with hypertrophic and dilated cardiomyopathy (Hagen et al., 2013; Carossa et al., 2014). Supporting these findings, mutations in Complex IV subunit genes COX6B1, MTCO2, and MTCO3 have been associated with dilated and hypertrophic cardiomyopathies (Abdulhag et al., 2015; Yang et al., 2022). Moreover, a possibly pathogenic m.9856T>C (Ile217Thr) mutation in MTCO3 has been identified in a left ventricular non-compaction cardiomyopathy patient, together with a significant decrease in mtDNA copy number in the patient group compared with controls (Liu et al., 2013). Interestingly, hypertrophic cardiomyopathy has been associated with m.2336T>C homoplasmic mutation of mitochondrial 16S rRNA gene (MTRNR2), and restrictive cardiomyopathy has been linked to m.1555A>G mutation of the mitochondrial 12S rRNA gene (MTRNR1) (Liu et al., 2014; Li et al., 2019). Furthermore, c.467T>G and c.950C>G mutations in mitochondrial ribosomal proteins L44 and L3 (MRPL44 and 3) have also been reported in hypertrophic cardiomyopathy, respectively (Galmiche et al., 2011; Distelmaier et al., 2015). Mutation in genes involved in CoQ10 biosynthesis, specifically COQ2, 4, and 9, have been reported in patients with hypertrophic cardiomyopathy (Doimo et al., 2014; Desbats et al., 2015; Mantle et al., 2023).
Mitochondrial dysfunction in hypertrophic cardiomyopathy has recently been associated with impaired NADH-linked respiration, as well as fatty acid oxidation in septal myectomy tissues from patients, due to mitochondrial fragmentation, which was rescued by NAD+ supplementation (Nollet et al., 2023). Interestingly, cardiac-specific deletion of cytochrome c assembly factor Cox10 resulted in mitochondrial cardiomyopathy in mice, leading to OXPHOS defects. Furthermore, the activation of mitochondrial peptidase OMA1 appears protective during mitochondrial cardiomyopathy (Ahola et al., 2022). A multi-omics study investigating healthy controls and patients with hypertrophic cardiomyopathy indicates a global downregulation in the expression of mitochondrial genes involved in ATP synthesis, coupled with mitochondrial damage manifested by low cristae density and less oxidative phosphorylation (Ranjbarvaziri et al., 2021).
During dilated cardiomyopathy, mitochondria numbers first increase to compensate for the energy deficit and then decrease over time, which results in less ATP production, deficiencies in cardiac contractility, and more ROS production (Goffart et al., 2004; Ramaccini et al., 2020). It has been reported that cardiomyocytes lacking the circadian rhythm activation factor basic helix-loop-helix ARNT like 1 (BMAL1) gene exhibit a dilated cardiomyopathy-like phenotype with reduced expression of mitochondrial BCL2 interacting protein 3 (BNIP3) protein, resulting in defective mitophagy and cardiomyocyte function (Li et al., 2020). Additionally, in a doxorubicin-induced mice model of dilated cardiomyopathy, increased expression of full-length SIRT3 prevented cardiac dysfunction by blocking superoxide generation (Tomczyk et al., 2022).
Mitochondrial dysfunction also contributes heavily to arrhythmogenic cardiomyopathy, as there are deficits in intracellular Ca2+ dynamics and ion channel functioning (Kim et al., 2019). Connexin 43 (Cx43), found on IMM of subsarcolemmal cardiomyocytes for regulation of ATP synthesis and respiration, is shown to decrease in arrhythmogenic cardiomyopathy patients, resulting in dysfunctional cardiomyocyte excitability (Noorman et al., 2013).
Pathogenic variants of desmin, a crucial component of the intermediate filaments of cardiac or skeletal muscle cells, can cause desminopathy, characterized by cardiomyopathy and skeletal myopathy (van Spaendonck-Zwarts et al., 2011). Dilated, arrhythmogenic, and restricted cardiomyopathy is often linked to desminopathy (Taylor et al., 2007; Otten et al., 2010; Lorenzon et al., 2013). A recent study identified a p.(S57L) mutation in desmin, which was associated with a striking downregulation of mitochondrial proteins and bioenergetics, coupled with a decrease in ETC Complex I + III + IV super-complex formation (Kubánek et al., 2020). In addition, VDAC1 has been shown to aggregate on muscle fibers together with apoptotic proteins, both in patients with desminopathy and in the rat model of this disease (Li et al., 2016).
KD is an acute febrile systemic vasculitis of unknown etiology, affecting infants and young children, and is the leading cause of acquired heart disease in developed countries (Kawasaki et al., 1974; Newburger et al., 2016; McCrindle et al., 2017). Coronary arteritis and coronary artery aneurysms (CAAs) occur in approximately 25% of untreated children, which is reduced to 3%–5% with high-dose intravenous immunoglobulin (IVIG) treatment (Burns et al., 1998; Tremoulet et al., 2008). Transcriptome analysis of whole blood from KD patients as well as cardiovascular tissues collected from Lactobacillus casei cell wall extract (LCWE)-injected mice, an experimental mouse model mimicking KD vasculitis, showed increased expression of genes associated with IL-1β and the NLRP3 inflammasome pathway (Hoang et al., 2014; Lee et al., 2015; Wakita et al., 2016; Porritt et al., 2021). Supporting NLRP3 activation, high levels of serum concentration of the pro-inflammatory cytokines IL-1β and IL-18 have been reported in the plasma of children with KD during the acute phase of the disease (Alphonse et al., 2016). Blocking the IL-1 pathway in two different experimental mouse models of KD, the LCWE- and the Candida albicans water-soluble fraction (CAWS), either pharmacologically or using knock-out mice, significantly decreases vasculitis severity (Lee et al., 2012; Lee et al., 2015; Anzai et al., 2020; Porritt et al., 2020). Several case studies have also demonstrated the efficacy of Anakinra, an IL-1 receptor antagonist, in treating IVIG-resistant KD patients (Cohen et al., 2012; Shafferman et al., 2014; Guillaume et al., 2018; Blonz et al., 2020; Koné-Paut et al., 2021).
Mitochondrial dysfunction and impaired autophagy and mitophagy can trigger NLRP3 inflammasome activation (Zhang N. P. et al., 2019). Impaired clearance of damaged mitochondria has been reported in the LCWE-induced murine model of KD vasculitis, shown by a decreased autophagic flux and blockade of autophagy by mTOR pathway activity, contributing to NLRP3 inflammasome activation and ROS accumulation (Marek-Iannucci et al., 2021). The expression of LC3B, BECN1, and ATG16L1 mRNA is reduced in whole blood of KD patients compared with febrile and healthy controls, while IVIG treatment increases the expression of these autophagy-related genes (Huang et al., 2020). However, low expression of ATG16L1 mRNA persists in patients developing coronary artery lesions, further indicating that the autophagy/mitophagy pathway is disrupted during KD and could be a possible therapeutic target (Huang et al., 2020). Statins, specifically atorvastatin, have been used to treat KD patients developing giant coronary aneurysms to improve endothelial cell homeostasis (Niedra et al., 2014). Studies have shown that statins also block NLRP3 inflammasome activation by promoting autophagy/mitophagy (Andres et al., 2014; Peng et al., 2018; Tremoulet et al., 2019). In addition, alleviating ER stress in mice by deletion or pharmacological inhibition of ER resident endoribonuclease IRE1 also reduces LCWE-induced KD vasculitis as well as caspase-1 activity and IL-1β production, highlighting the importance of ER stress pathways in KD progression (Marek-Iannucci et al., 2022). Serum levels of β-hydroxy-γ-trimethylammonium butyrate (carnitine), an amine that transfers long-chain fatty acids across the IMM for β-oxidation, is found to be higher in IVIG non-responsive KD patients, hinting towards altered mitochondrial fatty acid metabolism and mitochondrial fuel dependency (Muto et al., 2022).
Increased platelet count is commonly reported in KD patients in the second to third week of disease onset (Levin et al., 1985; Arora et al., 2020), as well as in the LCWE-induced mouse model of KD (Kocatürk et al., 2023). Two separate platelet sub-populations co-exist in the peripheral blood of KD patients (Pietraforte et al., 2014). The first one is characterized by mitochondrial membrane potential loss, whereas the second one exhibits mitochondrial membrane potential hyperpolarization. These platelet sub-populations may potentially influence inflammasome responses, the redox state, and calcium balance in the development of the KD vasculitis (Pietraforte et al., 2014). Therefore, further studies are warranted to investigate the potential role of mitophagy and autophagy on the development of KD cardiovascular lesions.
Modulating mitochondrial function and reducing mitochondrial oxidative stress could be a therapeutic target to prevent and/or ameliorate CVDs. Mitochondria-targeted antioxidant MitoQ, is a synthetic ROS-scavenger that mimics the activity of mitochondrial CoQ10 (Murphy and Smith, 2007). MitoQ treatment is available orally, and safe (Rossman et al., 2018; Oliver and Reddy, 2019), and its uptake by the mitochondria is dependent on membrane potential (James et al., 2007). MitoQ alleviates doxorubicin-induced cardiomyopathy in rats (Chandran et al., 2009) and promotes vascular endothelial function in older patients by reducing age-related ROS accumulation (Rossman et al., 2018). Similarly, another mitochondria-targeted ROS scavenger mimetic, mitoTEMPO, decreases mPTP opening to prevent mitochondrial apoptosis (Liang et al., 2010), and reduces diabetic cardiomyopathy in mice (Ni et al., 2016).
Mitochondria generate ATP through OXPHOS, and byproducts of the TCA cycle, such as NAD+ in its reduced form, NADH, contribute as a primary source of electrons to this process (Figure 2). NAD+ is a cofactor involved in cellular metabolism and mitochondrial fitness, and reduced NAD+ levels are associated with dysfunctional mitochondria (Srivastava, 2016). Therefore, targeting and boosting NAD+ could improve mitochondrial function in CVDs. Dietary supplementation with NAD+ precursor nicotinamide mononucleotide (NMN) can restore mitochondrial function in murine models of heart failure (HF) (Wang Y. J. et al., 2021), dilated cardiomyopathy (Martin et al., 2017) and I/R injury (Yamamoto et al., 2014). NMN has also been shown to promote autophagy and to decrease hypertension-mediated stroke in rats (Forte et al., 2020). Oral supplementation of mice with nicotinamide riboside (NR), another NAD+ intermediate, also boosts NAD+ and attenuates heart failure and dilated cardiomyopathy (Diguet et al., 2018). Chronic supplementation with NR is well tolerated and effectively boosts NAD+ levels in healthy middle-aged and older adults (Martens et al., 2018).
Another important regulator of autophagy/mitophagy, SIRT1, an NAD-dependent deacetylase, is non-specifically activated by resveratrol, a natural polyphenol produced in plants (Baur and Sinclair, 2006), as well as synthetic sirtuin-activating compounds such as SRT1720 (Bonkowski and Sinclair, 2016). These molecules have been shown to limit disease severity in multiple rodent CVD models, including Ang-II-induced atherosclerosis (Chen et al., 2015), cardiac hypertrophy (Chan et al., 2008), and diabetic cardiomyopathy (Sulaiman et al., 2010). Boosting NAD+ levels activates sirtuins and improves mitochondrial function (Kane and Sinclair, 2018).
The AMPK pathway is another crucial regulator of mitochondrial biogenesis and mitophagy. As mentioned above, AMPK activates PGC1α to promote mitochondrial biogenesis (Kukidome et al., 2006; Jäger et al., 2007). Metformin activates AMPK and promotes survival during heart failure in mice (Gundewar et al., 2009). Resveratrol activates mitochondrial biogenesis via AMPK and downstream PGC1α and TFAM (Lagouge et al., 2006). Notably, resveratrol improves cardiac function in mice and rats with hypertension (Rimbaud et al., 2011; Magyar et al., 2012). Caloric restriction and exercise can also induce the AMPK pathway, promote mitophagy, and prevent inflammatory response (Laker et al., 2017; Chen et al., 2020). Furthermore, inhibition of mTOR with rapamycin induces mitophagy in mice cardiomyocytes and increases lifespan (Wei et al., 2015; Yang et al., 2019). Promising developments in approaches/drugs improving mitochondrial homeostasis in cardiac tissues are crucial for discovering novel therapeutic agents, and future studies are needed to explore better targets in the context of CVDs.
Proper mitochondrial functioning is essential for tissue homeostasis, particularly in cardiac tissues with high demand for energy, such as the heart. Therefore, MQC mechanisms are central to maintaining cardiac homeostasis via the clearance of damaged mitochondria. Adapting the mitochondrial network to stress allows the heart to preserve its function during disease. The pathology of CVDs is highly connected to mitochondrial structure, membrane potential, dynamics, and clearance. Maintaining the health of the mitochondrial network via MQC is necessary for the outcome of CVDs. Therefore, mitochondria-targeting agents are critical therapeutic options to prevent or alleviate mitochondrial dysfunction during cardiac pathology. Current studies have shown that cardiac tissue protection can be achieved through mitophagy, ROS detoxification mechanisms, replenishing mitochondria pool, and inter-organelle communication. Targeting these MQC pathways is pivotal in discovering new treatments for CVDs in the future.
AA: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing–original draft, Writing–review and editing. TC: Conceptualization, Formal analysis, Investigation, Methodology, Supervision, Writing–review and editing. MN: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing–review and editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institutes of Health (grant number: R01 HL159297).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdulhag, U. N., Soiferman, D., Schueler-Furman, O., Miller, C., Shaag, A., Elpeleg, O., et al. (2015). Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 23 (2), 159–164. doi:10.1038/ejhg.2014.85
Acin-Perez, R., Lechuga-Vieco, A. V., Del Mar Muñoz, M., Nieto-Arellano, R., Torroja, C., Sánchez-Cabo, F., et al. (2018). Ablation of the stress protease OMA1 protects against heart failure in mice. Sci. Transl. Med. 10 (434), eaan4935. doi:10.1126/scitranslmed.aan4935
Ahola, S., Rivera Mejías, P., Hermans, S., Chandragiri, S., Giavalisco, P., Nolte, H., et al. (2022). OMA1-mediated integrated stress response protects against ferroptosis in mitochondrial cardiomyopathy. Cell. Metab. 34 (11), 1875–1891.e7. doi:10.1016/j.cmet.2022.08.017
Albina, J. E., Abate, J. A., and Henry, W. L. (1991). Nitric oxide production is required for murine resident peritoneal macrophages to suppress mitogen-stimulated T cell proliferation. Role of IFN-gamma in the induction of the nitric oxide-synthesizing pathway. J. Immunol. 147 (1), 144–148. doi:10.4049/jimmunol.147.1.144
Alphonse, M. P., Duong, T. T., Shumitzu, C., Hoang, T. L., McCrindle, B. W., Franco, A., et al. (2016). Inositol-triphosphate 3-kinase C mediates inflammasome activation and treatment response in Kawasaki disease. J. Immunol. 197 (9), 3481–3489. doi:10.4049/jimmunol.1600388
Alston, C. L., Ceccatelli Berti, C., Blakely, E. L., Oláhová, M., He, L., McMahon, C. J., et al. (2015). A recessive homozygous p.Asp92Gly SDHD mutation causes prenatal cardiomyopathy and a severe mitochondrial complex II deficiency. Hum. Genet. 134 (8), 869–879. doi:10.1007/s00439-015-1568-z
Amgalan, D., Chen, Y., and Kitsis, R. N. (2017). Death receptor signaling in the heart: Cell survival, apoptosis, and necroptosis. Circulation 136 (8), 743–746. doi:10.1161/circulationaha.117.029566
Andres, A. M., Hernandez, G., Lee, P., Huang, C., Ratliff, E. P., Sin, J., et al. (2014). Mitophagy is required for acute cardioprotection by simvastatin. Antioxid. Redox Signal 21 (14), 1960–1973. doi:10.1089/ars.2013.5416
Andres, A. M., Tucker, K. C., Thomas, A., Taylor, D. J., Sengstock, D., Jahania, S. M., et al. (2017). Mitophagy and mitochondrial biogenesis in atrial tissue of patients undergoing heart surgery with cardiopulmonary bypass. JCI Insight 2 (4), e89303. doi:10.1172/jci.insight.89303
Antón, Z., Landajuela, A., Hervás, J. H., Montes, L. R., Hernández-Tiedra, S., Velasco, G., et al. (2016). Human Atg8-cardiolipin interactions in mitophagy: Specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy 12 (12), 2386–2403. doi:10.1080/15548627.2016.1240856
Anzai, F., Watanabe, S., Kimura, H., Kamata, R., Karasawa, T., Komada, T., et al. (2020). Crucial role of NLRP3 inflammasome in a murine model of Kawasaki disease. J. Mol. Cell. Cardiol. 138, 185–196. doi:10.1016/j.yjmcc.2019.11.158
Anzell, A. R., Maizy, R., Przyklenk, K., and Sanderson, T. H. (2018). Mitochondrial quality control and disease: Insights into ischemia-reperfusion injury. Mol. Neurobiol. 55 (3), 2547–2564. doi:10.1007/s12035-017-0503-9
Arbustini, E., Diegoli, M., Fasani, R., Grasso, M., Morbini, P., Banchieri, N., et al. (1998). Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am. J. Pathol. 153 (5), 1501–1510. doi:10.1016/s0002-9440(10)65738-0
Arora, K., Guleria, S., Jindal, A. K., Rawat, A., and Singh, S. (2020). Platelets in Kawasaki disease: Is this only a numbers game or something beyond? Genes. Dis. 7 (1), 62–66. doi:10.1016/j.gendis.2019.09.003
Atkins, K., Dasgupta, A., Chen, K. H., Mewburn, J., and Archer, S. L. (2016). The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: Implications for human disease. Clin. Sci. (Lond) 130 (21), 1861–1874. doi:10.1042/cs20160030
Baar, K., Wende, A. R., Jones, T. E., Marison, M., Nolte, L. A., Chen, M., et al. (2002). Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. Faseb J. 16 (14), 1879–1886. doi:10.1096/fj.02-0367com
Bahr, T., Katuri, J., Liang, T., and Bai, Y. (2022). Mitochondrial chaperones in human health and disease. Free Radic. Biol. Med. 179, 363–374. doi:10.1016/j.freeradbiomed.2021.11.015
Bai, Y., Yang, Y., Gao, Y., Lin, D., Wang, Z., and Ma, J. (2021). Melatonin postconditioning ameliorates anoxia/reoxygenation injury by regulating mitophagy and mitochondrial dynamics in a SIRT3-dependent manner. Eur. J. Pharmacol. 904, 174157. doi:10.1016/j.ejphar.2021.174157
Baik, S. H., Ramanujan, V. K., Becker, C., Fett, S., Underhill, D. M., and Wolf, A. J. (2023). Hexokinase dissociation from mitochondria promotes oligomerization of VDAC that facilitates NLRP3 inflammasome assembly and activation. Sci. Immunol. 8 (84), eade7652. doi:10.1126/sciimmunol.ade7652
Ballana, E., Govea, N., de Cid, R., Garcia, C., Arribas, C., Rosell, J., et al. (2008). Detection of unrecognized low-level mtDNA heteroplasmy may explain the variable phenotypic expressivity of apparently homoplasmic mtDNA mutations. Hum. Mutat. 29 (2), 248–257. doi:10.1002/humu.20639
Barki-Harrington, L., Perrino, C., and Rockman, H. A. (2004). Network integration of the adrenergic system in cardiac hypertrophy. Cardiovasc Res. 63 (3), 391–402. doi:10.1016/j.cardiores.2004.03.011
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476 (7360), 341–345. doi:10.1038/nature10234
Baur, J. A., and Sinclair, D. A. (2006). Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 5 (6), 493–506. doi:10.1038/nrd2060
Becker, J., and Craig, E. A. (1994). Heat-shock proteins as molecular chaperones. Eur. J. Biochem. 219 (1-2), 11–23. doi:10.1007/978-3-642-79502-2_2
Bleier, L., Wittig, I., Heide, H., Steger, M., Brandt, U., and Dröse, S. (2015). Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 78, 1–10. doi:10.1016/j.freeradbiomed.2014.10.511
Blonz, G., Lacroix, S., Benbrik, N., Warin-Fresse, K., Masseau, A., Trewick, D., et al. (2020). Severe late-onset Kawasaki disease successfully treated with anakinra. J. Clin. Rheumatol. 26 (2), e42–e43. doi:10.1097/rhu.0000000000000814
Bonkowski, M. S., and Sinclair, D. A. (2016). Slowing ageing by design: The rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell. Biol. 17 (11), 679–690. doi:10.1038/nrm.2016.93
Booth, D. M., Várnai, P., Joseph, S. K., and Hajnóczky, G. (2021). Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Mol. Cell. 81 (18), 3866–3876.e2. doi:10.1016/j.molcel.2021.07.014
Brahimi-Horn, M. C., Giuliano, S., Saland, E., Lacas-Gervais, S., Sheiko, T., Pelletier, J., et al. (2015). Knockout of Vdac1 activates hypoxia-inducible factor through reactive oxygen species generation and induces tumor growth by promoting metabolic reprogramming and inflammation. Cancer Metab. 3, 8. doi:10.1186/s40170-015-0133-5
Bravo-San Pedro, J. M., Kroemer, G., and Galluzzi, L. (2017). Autophagy and mitophagy in cardiovascular disease. Circ. Res. 120 (11), 1812–1824. doi:10.1161/circresaha.117.311082
Brecht, M., Richardson, M., Taranath, A., Grist, S., Thorburn, D., and Bratkovic, D. (2015). Leigh syndrome caused by the MT-ND5 m.13513G>A mutation: A case presenting with WPW-like conduction defect, cardiomyopathy, hypertension and hyponatraemia. JIMD Rep. 19, 95–100. doi:10.1007/8904_2014_375
Brumatti, G., Salmanidis, M., and Ekert, P. G. (2010). Crossing paths: Interactions between the cell death machinery and growth factor survival signals. Cell. Mol. Life Sci. 67 (10), 1619–1630. doi:10.1007/s00018-010-0288-8
Burns, J. C., Capparelli, E. V., Brown, J. A., Newburger, J. W., and Glode, M. P. (1998). Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. US/Canadian Kawasaki syndrome study group. Pediatr. Infect. Dis. J. 17 (12), 1144–1148. doi:10.1097/00006454-199812000-00009
Cadenas, S. (2018). Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 1859 (9), 940–950. doi:10.1016/j.bbabio.2018.05.019
Cai, C., Guo, Z., Chang, X., Li, Z., Wu, F., He, J., et al. (2022). Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 52, 102288. doi:10.1016/j.redox.2022.102288
Candas, D., and Li, J. J. (2014). MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid. Redox Signal 20 (10), 1599–1617. doi:10.1089/ars.2013.5305
Cantó, C., Gerhart-Hines, Z., Feige, J. N., Lagouge, M., Noriega, L., Milne, J. C., et al. (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458 (7241), 1056–1060. doi:10.1038/nature07813
Capaldi, R. A., and Aggeler, R. (2002). Mechanism of the F(1)F(0)-type ATP synthase, a biological rotary motor. Trends Biochem. Sci. 27 (3), 154–160. doi:10.1016/s0968-0004(01)02051-5
Carossa, V., Ghelli, A., Tropeano, C. V., Valentino, M. L., Iommarini, L., Maresca, A., et al. (2014). A novel in-frame 18-bp microdeletion in MT-CYB causes a multisystem disorder with prominent exercise intolerance. Hum. Mutat. 35 (8), 954–958. doi:10.1002/humu.22596
Chaanine, A. H., Kohlbrenner, E., Gamb, S. I., Guenzel, A. J., Klaus, K., Fayyaz, A. U., et al. (2016). FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Heart Circ. Physiol. 311 (6), H1540-H1559–h1559. doi:10.1152/ajpheart.00549.2016
Chan, A. Y., Dolinsky, V. W., Soltys, C. L., Viollet, B., Baksh, S., Light, P. E., et al. (2008). Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J. Biol. Chem. 283 (35), 24194–24201. doi:10.1074/jbc.M802869200
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 15, 235–259. doi:10.1146/annurev-pathmechdis-012419-032711
Chandran, K., Aggarwal, D., Migrino, R. Q., Joseph, J., McAllister, D., Konorev, E. A., et al. (2009). Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by mito-Q. Biophys. J. 96 (4), 1388–1398. doi:10.1016/j.bpj.2008.10.042
Chehaitly, A., Guihot, A. L., Proux, C., Grimaud, L., Aurrière, J., Legouriellec, B., et al. (2022). Altered mitochondrial opa1-related fusion in mouse promotes endothelial cell dysfunction and atherosclerosis. Antioxidants (Basel) 11 (6), 1078. doi:10.3390/antiox11061078
Chen, G., Han, Z., Feng, D., Chen, Y., Chen, L., Wu, H., et al. (2014). A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell. 54 (3), 362–377. doi:10.1016/j.molcel.2014.02.034
Chen, G., Kroemer, G., and Kepp, O. (2020). Mitophagy: An emerging role in aging and age-associated diseases. Front. Cell. Dev. Biol. 8, 200. doi:10.3389/fcell.2020.00200
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum. Mol. Genet. 18 (R2), R169–R176. doi:10.1093/hmg/ddp326
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell. Biol. 160 (2), 189–200. doi:10.1083/jcb.200211046
Chen, L., Gong, Q., Stice, J. P., and Knowlton, A. A. (2009). Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 84 (1), 91–99. doi:10.1093/cvr/cvp181
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016a). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12 (4), 689–702. doi:10.1080/15548627.2016.1151580
Chen, Q., Sun, L., and Chen, Z. J. (2016b). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17 (10), 1142–1149. doi:10.1038/ni.3558
Chen, Y. B., Aon, M. A., Hsu, Y. T., Soane, L., Teng, X., McCaffery, J. M., et al. (2011). Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell. Biol. 195 (2), 263–276. doi:10.1083/jcb.201108059
Chen, Y., Dorn, G. W., and 2nd, (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340 (6131), 471–475. doi:10.1126/science.1231031
Chen, Y. X., Zhang, M., Cai, Y., Zhao, Q., and Dai, W. (2015). The Sirt1 activator SRT1720 attenuates angiotensin II-induced atherosclerosis in apoE⁻/⁻ mice through inhibiting vascular inflammatory response. Biochem. Biophys. Res. Commun. 465 (4), 732–738. doi:10.1016/j.bbrc.2015.08.066
Cheng, E. H., Sheiko, T. V., Fisher, J. K., Craigen, W. J., and Korsmeyer, S. J. (2003). VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301 (5632), 513–517. doi:10.1126/science.1083995
Cheng, J., Nanayakkara, G., Shao, Y., Cueto, R., Wang, L., Yang, W. Y., et al. (2017). Mitochondrial proton leak plays a critical role in pathogenesis of cardiovascular diseases. Adv. Exp. Med. Biol. 982, 359–370. doi:10.1007/978-3-319-55330-6_20
Chiao, Y. A., Chakraborty, A. D., Light, C. M., Tian, R., Sadoshima, J., Shi, X., et al. (2021). NAD(+) redox imbalance in the heart exacerbates diabetic cardiomyopathy. Circ. Heart Fail 14 (8), e008170. doi:10.1161/circheartfailure.120.008170
Chinnery, P. F. (1993). "Primary mitochondrial disorders overview," in GeneReviews(®), eds M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle (WA): University of Washington, Seattle
Chinnery, P. F., and Hudson, G. (2013). Mitochondrial genetics. Br. Med. Bull. 106 (1), 135–159. doi:10.1093/bmb/ldt017
Chowdhury, A. R., Zielonka, J., Kalyanaraman, B., Hartley, R. C., Murphy, M. P., and Avadhani, N. G. (2020). Mitochondria-targeted paraquat and metformin mediate ROS production to induce multiple pathways of retrograde signaling: A dose-dependent phenomenon. Redox Biol. 36, 101606. doi:10.1016/j.redox.2020.101606
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell. Biol. 15 (10), 1197–1205. doi:10.1038/ncb2837
Coenen, M. J., Antonicka, H., Ugalde, C., Sasarman, F., Rossi, R., Heister, J. G., et al. (2004). Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N. Engl. J. Med. 351 (20), 2080–2086. doi:10.1056/NEJMoa041878
Cogliati, S., Frezza, C., Soriano, M. E., Varanita, T., Quintana-Cabrera, R., Corrado, M., et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 155 (1), 160–171. doi:10.1016/j.cell.2013.08.032
Cohen, S., Tacke, C. E., Straver, B., Meijer, N., Kuipers, I. M., and Kuijpers, T. W. (2012). A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann. Rheum. Dis. 71 (12), 2059–2061. doi:10.1136/annrheumdis-2012-201658
Coleman, J. W. (2001). Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 1 (8), 1397–1406. doi:10.1016/s1567-5769(01)00086-8
Colombini, M., Blachly-Dyson, E., and Forte, M. (1996). VDAC, a channel in the outer mitochondrial membrane. Ion. Channels 4, 169–202. doi:10.1007/978-1-4899-1775-1_5
Cosentino, K., Hertlein, V., Jenner, A., Dellmann, T., Gojkovic, M., Peña-Blanco, A., et al. (2022). The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell. 82 (5), 933–949.e9. doi:10.1016/j.molcel.2022.01.008
Coste, A., Louet, J. F., Lagouge, M., Lerin, C., Antal, M. C., Meziane, H., et al. (2008). The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1{alpha}. Proc. Natl. Acad. Sci. U. S. A. 105 (44), 17187–17192. doi:10.1073/pnas.0808207105
Czabotar, P. E., Lessene, G., Strasser, A., and Adams, J. M. (2014). Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell. Biol. 15 (1), 49–63. doi:10.1038/nrm3722
Dabkowski, E. R., Baseler, W. A., Williamson, C. L., Powell, M., Razunguzwa, T. T., Frisbee, J. C., et al. (2010). Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am. J. Physiol. Heart Circ. Physiol. 299 (2), H529–H540. doi:10.1152/ajpheart.00267.2010
Dagvadorj, J., Mikulska-Ruminska, K., Tumurkhuu, G., Ratsimandresy, R. A., Carriere, J., Andres, A. M., et al. (2021). Recruitment of pro-IL-1α to mitochondrial cardiolipin, via shared LC3 binding domain, inhibits mitophagy and drives maximal NLRP3 activation. Proc. Natl. Acad. Sci. U. S. A. 118 (1), e2015632118. doi:10.1073/pnas.2015632118
Das, S., Bedja, D., Campbell, N., Dunkerly, B., Chenna, V., Maitra, A., et al. (2014). miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One 9 (5), e96820. doi:10.1371/journal.pone.0096820
Das, S., Ferlito, M., Kent, O. A., Fox-Talbot, K., Wang, R., Liu, D., et al. (2012). Nuclear miRNA regulates the mitochondrial genome in the heart. Circ. Res. 110 (12), 1596–1603. doi:10.1161/circresaha.112.267732
Daum, G. (1985). Lipids of mitochondria. Biochim. Biophys. Acta 822 (1), 1–42. doi:10.1016/0304-4157(85)90002-4
De Paris, V., Biondi, F., Stolfo, D., Merlo, M., and Sinagra, G. (2019). “Pathophysiology,” in Dilated cardiomyopathy: From genetics to clinical management. Editors G. Sinagra, M. Merlo, and B. Pinamonti (Cham (CH): Springer).
Desbats, M. A., Lunardi, G., Doimo, M., Trevisson, E., and Salviati, L. (2015). Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J. Inherit. Metab. Dis. 38 (1), 145–156. doi:10.1007/s10545-014-9749-9
Dia, M., Gomez, L., Thibault, H., Tessier, N., Leon, C., Chouabe, C., et al. (2020). Reduced reticulum-mitochondria Ca(2+) transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res. Cardiol. 115 (6), 74. doi:10.1007/s00395-020-00835-7
Diguet, N., Trammell, S. A. J., Tannous, C., Deloux, R., Piquereau, J., Mougenot, N., et al. (2018). Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation 137 (21), 2256–2273. doi:10.1161/circulationaha.116.026099
Dikalova, A. E., Pandey, A., Xiao, L., Arslanbaeva, L., Sidorova, T., Lopez, M. G., et al. (2020). Mitochondrial deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ. Res. 126 (4), 439–452. doi:10.1161/circresaha.119.315767
Dillmann, W. H. (2019). Diabetic cardiomyopathy. Circ. Res. 124 (8), 1160–1162. doi:10.1161/circresaha.118.314665
Diner, E. J., Burdette, D. L., Wilson, S. C., Monroe, K. M., Kellenberger, C. A., Hyodo, M., et al. (2013). The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell. Rep. 3 (5), 1355–1361. doi:10.1016/j.celrep.2013.05.009
Distelmaier, F., Haack, T. B., Catarino, C. B., Gallenmüller, C., Rodenburg, R. J., Strom, T. M., et al. (2015). MRPL44 mutations cause a slowly progressive multisystem disease with childhood-onset hypertrophic cardiomyopathy. Neurogenetics 16 (4), 319–323. doi:10.1007/s10048-015-0444-2
Dogan, A. E., Hamid, S. M., Yildirim, A. D., Yildirim, Z., Sen, G., Riera, C. E., et al. (2022). PACT establishes a posttranscriptional brake on mitochondrial biogenesis by promoting the maturation of miR-181c. J. Biol. Chem. 298(7), 102050. doi:10.1016/j.jbc.2022.102050
Doimo, M., Desbats, M. A., Cerqua, C., Cassina, M., Trevisson, E., and Salviati, L. (2014). Genetics of coenzyme q10 deficiency. Mol. Syndromol. 5(3-4), 156–162. doi:10.1159/000362826
Dooley, H. C., Razi, M., Polson, H. E., Girardin, S. E., Wilson, M. I., and Tooze, S. A. (2014). WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell. 55 (2), 238–252. doi:10.1016/j.molcel.2014.05.021
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102 (4), 488–496. doi:10.1161/circresaha.107.162800
Druzhyna, N. M., Wilson, G. L., and LeDoux, S. P. (2008). Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 129 (7-8), 383–390. doi:10.1016/j.mad.2008.03.002
Duan, M., Chen, H., Yin, L., Zhu, X., Novák, P., Lv, Y., et al. (2022). Mitochondrial apolipoprotein A-I binding protein alleviates atherosclerosis by regulating mitophagy and macrophage polarization. Cell. Commun. Signal 20 (1), 60. doi:10.1186/s12964-022-00858-8
Dubois-Deruy, E., Peugnet, V., Turkieh, A., and Pinet, F. (2020). Oxidative stress in cardiovascular diseases. Antioxidants (Basel) 9 (9), 864. doi:10.3390/antiox9090864
Dubucs, C., Aziza, J., Sartor, A., Heitz, F., Sevely, A., Sternberg, D., et al. (2023). Severe antenatal hypertrophic cardiomyopathy secondary to ACAD9-related mitochondrial complex I deficiency. Mol. Syndromol. 14 (2), 101–108. doi:10.1159/000526022
Duncan, J. G. (2011). Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim. Biophys. Acta 1813 (7), 1351–1359. doi:10.1016/j.bbamcr.2011.01.014
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331 (6016), 456–461. doi:10.1126/science.1196371
El-Hattab, A. W., and Scaglia, F. (2013). Mitochondrial DNA depletion syndromes: Review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 10 (2), 186–198. doi:10.1007/s13311-013-0177-6
Elgass, K., Pakay, J., Ryan, M. T., and Palmer, C. S. (2013). Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 1833 (1), 150–161. doi:10.1016/j.bbamcr.2012.05.002
Fassone, E., and Rahman, S. (2012). Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 49 (9), 578–590. doi:10.1136/jmedgenet-2012-101159
Fernández-Silva, P., Enriquez, J. A., and Montoya, J. (2003). Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 88 (1), 41–56. doi:10.1113/eph8802514
Fernie, A. R., Carrari, F., and Sweetlove, L. J. (2004). Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 7 (3), 254–261. doi:10.1016/j.pbi.2004.03.007
Flarsheim, C. E., Grupp, I. L., and Matlib, M. A. (1996). Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am. J. Physiol. 271 (2), H192–H202. doi:10.1152/ajpheart.1996.271.1.H192
Forkink, M., Smeitink, J. A., Brock, R., Willems, P. H., and Koopman, W. J. (2010). Detection and manipulation of mitochondrial reactive oxygen species in mammalian cells. Biochim. Biophys. Acta 1797 (6-7), 1034–1044. doi:10.1016/j.bbabio.2010.01.022
Forman, H. J., Zhang, H., and Rinna, A. (2009). Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 30 (1-2), 1–12. doi:10.1016/j.mam.2008.08.006
Forte, M., Bianchi, F., Cotugno, M., Marchitti, S., De Falco, E., Raffa, S., et al. (2020). Pharmacological restoration of autophagy reduces hypertension-related stroke occurrence. Autophagy 16 (8), 1468–1481. doi:10.1080/15548627.2019.1687215
Forte, M., Schirone, L., Ameri, P., Basso, C., Catalucci, D., Modica, J., et al. (2021). The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 178 (10), 2060–2076. doi:10.1111/bph.15068
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334 (6054), 358–362. doi:10.1126/science.1207385
Gaigg, B., Simbeni, R., Hrastnik, C., Paltauf, F., and Daum, G. (1995). Characterization of a microsomal subfraction associated with mitochondria of the yeast, Saccharomyces cerevisiae. Involvement in synthesis and import of phospholipids into mitochondria. Biochim. Biophys. Acta 1234 (2), 214–220. doi:10.1016/0005-2736(94)00287-y
Galluzzi, L., Kepp, O., Trojel-Hansen, C., and Kroemer, G. (2012). Mitochondrial control of cellular life, stress, and death. Circ. Res. 111 (9), 1198–1207. doi:10.1161/circresaha.112.268946
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell. Death Differ. 25 (3), 486–541. doi:10.1038/s41418-017-0012-4
Galmiche, L., Serre, V., Beinat, M., Assouline, Z., Lebre, A. S., Chretien, D., et al. (2011). Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum. Mutat. 32 (11), 1225–1231. doi:10.1002/humu.21562
Gálvez, A. S., Brunskill, E. W., Marreez, Y., Benner, B. J., Regula, K. M., Kirschenbaum, L. A., et al. (2006). Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J. Biol. Chem. 281 (3), 1442–1448. doi:10.1074/jbc.M509056200
Georgakopoulos, N. D., Wells, G., and Campanella, M. (2017). The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 13 (2), 136–146. doi:10.1038/nchembio.2287
Gerhart-Hines, Z., Rodgers, J. T., Bare, O., Lerin, C., Kim, S. H., Mostoslavsky, R., et al. (2007). Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. Embo J. 26 (7), 1913–1923. doi:10.1038/sj.emboj.7601633
Gimbrone, M. A., and García-Cardeña, G. (2016). Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 118 (4), 620–636. doi:10.1161/circresaha.115.306301
Giorgi, C., Marchi, S., and Pinton, P. (2018). The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell. Biol. 19 (11), 713–730. doi:10.1038/s41580-018-0052-8
Giorgio, M., Trinei, M., Migliaccio, E., and Pelicci, P. G. (2007). Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell. Biol. 8 (9), 722–728. doi:10.1038/nrm2240
Gisterå, A., and Hansson, G. K. (2017). The immunology of atherosclerosis. Nat. Rev. Nephrol. 13 (6), 368–380. doi:10.1038/nrneph.2017.51
Goffart, S., von Kleist-Retzow, J. C., and Wiesner, R. J. (2004). Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res. 64 (2), 198–207. doi:10.1016/j.cardiores.2004.06.030
Gonçalves, V. F. (2019). Mitochondrial genetics. Adv. Exp. Med. Biol. 1158, 247–255. doi:10.1007/978-981-13-8367-0_13
Griendling, K. K., Camargo, L. L., Rios, F. J., Alves-Lopes, R., Montezano, A. C., and Touyz, R. M. (2021). Oxidative stress and hypertension. Circ. Res. 128 (7), 993–1020. doi:10.1161/circresaha.121.318063
Guillaume, M. P., Reumaux, H., and Dubos, F. (2018). Usefulness and safety of anakinra in refractory Kawasaki disease complicated by coronary artery aneurysm. Cardiol. Young 28 (5), 739–742. doi:10.1017/s1047951117002864
Gundewar, S., Calvert, J. W., Jha, S., Toedt-Pingel, I., Ji, S. Y., Nunez, D., et al. (2009). Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 104 (3), 403–411. doi:10.1161/circresaha.108.190918
Guo, R., Gu, J., Zong, S., Wu, M., and Yang, M. (2018). Structure and mechanism of mitochondrial electron transport chain. Biomed. J. 41 (1), 9–20. doi:10.1016/j.bj.2017.12.001
Guo, R., Zong, S., Wu, M., Gu, J., and Yang, M. (2017). Architecture of human mitochondrial respiratory megacomplex I(2)III(2)IV(2). Cell. 170 (6), 1247–1257. doi:10.1016/j.cell.2017.07.050
Gustafsson Å, B., and Dorn, G. W. (2019). Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol. Rev. 99 (1), 853–892. doi:10.1152/physrev.00005.2018
Hagen, C. M., Aidt, F. H., Havndrup, O., Hedley, P. L., Jespersgaard, C., Jensen, M., et al. (2013). MT-CYB mutations in hypertrophic cardiomyopathy. Mol. Genet. Genomic Med. 1 (1), 54–65. doi:10.1002/mgg3.5
Halestrap, A. P. (1999). The mitochondrial permeability transition: Its molecular mechanism and role in reperfusion injury. Biochem. Soc. Symp. 66, 181–203. doi:10.1042/bss0660181
Hall, A. R., Burke, N., Dongworth, R. K., and Hausenloy, D. J. (2014). Mitochondrial fusion and fission proteins: Novel therapeutic targets for combating cardiovascular disease. Br. J. Pharmacol. 171 (8), 1890–1906. doi:10.1111/bph.12516
Hamacher-Brady, A., Choe, S. C., Krijnse-Locker, J., and Brady, N. R. (2014). Intramitochondrial recruitment of endolysosomes mediates Smac degradation and constitutes a novel intrinsic apoptosis antagonizing function of XIAP E3 ligase. Cell. Death Differ. 21 (12), 1862–1876. doi:10.1038/cdd.2014.101
Han, D., Antunes, F., Canali, R., Rettori, D., and Cadenas, E. (2003). Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278 (8), 5557–5563. doi:10.1074/jbc.M210269200
Han, J., Dai, S., Zhong, L., Shi, X., Fan, X., Zhong, X., et al. (2022). GSDMD (gasdermin D) mediates pathological cardiac hypertrophy and generates a feed-forward amplification cascade via mitochondria-STING (stimulator of interferon genes) Axis. Hypertension 79 (11), 2505–2518. doi:10.1161/hypertensionaha.122.20004
Harper, J. W., Ordureau, A., and Heo, J. M. (2018). Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell. Biol. 19 (2), 93–108. doi:10.1038/nrm.2017.129
Harrington, J. S., Ryter, S. W., Plataki, M., Price, D. R., and Choi, A. M. K. (2023). Mitochondria in health, disease, and aging. Physiol. Rev. 103, 2349–2422. doi:10.1152/physrev.00058.2021
Hoang, L. T., Shimizu, C., Ling, L., Naim, A. N., Khor, C. C., Tremoulet, A. H., et al. (2014). Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med. 6 (11), 541. doi:10.1186/s13073-014-0102-6
Hossain, M. A., Bhattacharjee, S., Armin, S. M., Qian, P., Xin, W., Li, H. Y., et al. (2015). Hydrogen peroxide priming modulates abiotic oxidative stress tolerance: Insights from ROS detoxification and scavenging. Front. Plant Sci. 6, 420. doi:10.3389/fpls.2015.00420
Hou, F., Sun, L., Zheng, H., Skaug, B., Jiang, Q. X., and Chen, Z. J. (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 146 (3), 448–461. doi:10.1016/j.cell.2011.06.041
Hu, J., de Souza-Pinto, N. C., Haraguchi, K., Hogue, B. A., Jaruga, P., Greenberg, M. M., et al. (2005). Repair of formamidopyrimidines in DNA involves different glycosylases: Role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 280 (49), 40544–40551. doi:10.1074/jbc.M508772200
Hu, Y. F., Chen, Y. J., Lin, Y. J., and Chen, S. A. (2015). Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 12 (4), 230–243. doi:10.1038/nrcardio.2015.2
Huang, F. C., Huang, Y. H., Kuo, H. C., and Li, S. C. (2020). Identifying downregulation of autophagy markers in Kawasaki disease. Child. (Basel) 7 (10), E166. doi:10.3390/children7100166
Huang, W., Choi, W., Hu, W., Mi, N., Guo, Q., Ma, M., et al. (2012). Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell. Res. 22 (3), 473–489. doi:10.1038/cr.2012.24
Ikeda, Y., Shirakabe, A., Maejima, Y., Zhai, P., Sciarretta, S., Toli, J., et al. (2015). Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 116 (2), 264–278. doi:10.1161/circresaha.116.303356
Ishihara, N., Eura, Y., and Mihara, K. (2004). Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell. Sci. 117 (26), 6535–6546. doi:10.1242/jcs.01565
Iwata, S., Lee, J. W., Okada, K., Lee, J. K., Iwata, M., Rasmussen, B., et al. (1998). Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 281 (5373), 64–71. doi:10.1126/science.281.5373.64
Jacobs, A. L., and Schär, P. (2012). DNA glycosylases: In DNA repair and beyond. Chromosoma 121 (1), 1–20. doi:10.1007/s00412-011-0347-4
Jäger, S., Handschin, C., St-Pierre, J., and Spiegelman, B. M. (2007). AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U. S. A. 104 (29), 12017–12022. doi:10.1073/pnas.0705070104
Jahani-Asl, A., Cheung, E. C., Neuspiel, M., MacLaurin, J. G., Fortin, A., Park, D. S., et al. (2007). Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J. Biol. Chem. 282 (33), 23788–23798. doi:10.1074/jbc.M703812200
Jain-Ghai, S., Cameron, J. M., Al Maawali, A., Blaser, S., MacKay, N., Robinson, B., et al. (2013). Complex II deficiency--a case report and review of the literature. Am. J. Med. Genet. A 161a (2), 285–294. doi:10.1002/ajmg.a.35714
James, A. M., Sharpley, M. S., Manas, A. R., Frerman, F. E., Hirst, J., Smith, R. A., et al. (2007). Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 282 (20), 14708–14718. doi:10.1074/jbc.M611463200
Jia, G., Hill, M. A., and Sowers, J. R. (2018). Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ. Res. 122 (4), 624–638. doi:10.1161/circresaha.117.311586
Jin, L., Geng, L., Ying, L., Shu, L., Ye, K., Yang, R., et al. (2022a). FGF21-Sirtuin 3 Axis confers the protective effects of exercise against diabetic cardiomyopathy by governing mitochondrial integrity. Circulation 146 (20), 1537–1557. doi:10.1161/circulationaha.122.059631
Jin, Q., Li, R., Hu, N., Xin, T., Zhu, P., Hu, S., et al. (2018). DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 14, 576–587. doi:10.1016/j.redox.2017.11.004
Jin, Y., Liu, Y., Xu, L., Xu, J., Xiong, Y., Peng, Y., et al. (2022b). Novel role for caspase 1 inhibitor VX765 in suppressing NLRP3 inflammasome assembly and atherosclerosis via promoting mitophagy and efferocytosis. Cell. Death Dis. 13 (5), 512. doi:10.1038/s41419-022-04966-8
Kane, A. E., and Sinclair, D. A. (2018). Sirtuins and NAD(+) in the development and treatment of metabolic and cardiovascular diseases. Circ. Res. 123 (7), 868–885. doi:10.1161/circresaha.118.312498
Kawasaki, T., Kosaki, F., Okawa, S., Shigematsu, I., and Yanagawa, H. (1974). A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics 54 (3), 271–276. doi:10.1542/peds.54.3.271
Kayagaki, N., Stowe, I. B., Lee, B. L., O'Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526 (7575), 666–671. doi:10.1038/nature15541
Kazak, L., Reyes, A., and Holt, I. J. (2012). Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell. Biol. 13 (10), 659–671. doi:10.1038/nrm3439
Kho, C., Lee, A., and Hajjar, R. J. (2012). Altered sarcoplasmic reticulum calcium cycling--targets for heart failure therapy. Nat. Rev. Cardiol. 9 (12), 717–733. doi:10.1038/nrcardio.2012.145
Khwaja, B., Thankam, F. G., and Agrawal, D. K. (2021). Mitochondrial DAMPs and altered mitochondrial dynamics in OxLDL burden in atherosclerosis. Mol. Cell. Biochem. 476 (4), 1915–1928. doi:10.1007/s11010-021-04061-0
Killackey, S. A., Philpott, D. J., and Girardin, S. E. (2020). Mitophagy pathways in health and disease. J. Cell. Biol. 219 (11), e202004029. doi:10.1083/jcb.202004029
Kim, J. C., Pérez-Hernández, M., Alvarado, F. J., Maurya, S. R., Montnach, J., Yin, Y., et al. (2019). Disruption of Ca(2+)(i) homeostasis and connexin 43 hemichannel function in the right ventricle precedes overt arrhythmogenic cardiomyopathy in plakophilin-2-deficient mice. Circulation 140 (12), 1015–1030. doi:10.1161/circulationaha.119.039710
Kirkman, H. N., and Gaetani, G. F. (2007). Mammalian catalase: A venerable enzyme with new mysteries. Trends Biochem. Sci. 32 (1), 44–50. doi:10.1016/j.tibs.2006.11.003
Kocatürk, B., Lee, Y., Nosaka, N., Abe, M., Martinon, D., Lane, M. E., et al. (2023). Platelets exacerbate cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight 8 (14), e169855. doi:10.1172/jci.insight.169855
Koné-Paut, I., Tellier, S., Belot, A., Brochard, K., Guitton, C., Marie, I., et al. (2021). Phase II open label study of anakinra in intravenous immunoglobulin-resistant Kawasaki disease. Arthritis Rheumatol. 73 (1), 151–161. doi:10.1002/art.41481
Krahn, A. D., Wilde, A. A. M., Calkins, H., La Gerche, A., Cadrin-Tourigny, J., Roberts, J. D., et al. (2022). Arrhythmogenic right ventricular cardiomyopathy. JACC Clin. Electrophysiol. 8 (4), 533–553. doi:10.1016/j.jacep.2021.12.002
Krokan, H. E., and Bjørås, M. (2013). Base excision repair. Cold Spring Harb. Perspect. Biol. 5 (4), a012583. doi:10.1101/cshperspect.a012583
Kubánek, M., Schimerová, T., Piherová, L., Brodehl, A., Krebsová, A., Ratnavadivel, S., et al. (2020). Desminopathy: Novel desmin variants, a new cardiac phenotype, and further evidence for secondary mitochondrial dysfunction. J. Clin. Med. 9 (4), 937. doi:10.3390/jcm9040937
Kubli, D. A., Zhang, X., Lee, Y., Hanna, R. A., Quinsay, M. N., Nguyen, C. K., et al. (2013). Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 288 (2), 915–926. doi:10.1074/jbc.M112.411363
Kukidome, D., Nishikawa, T., Sonoda, K., Imoto, K., Fujisawa, K., Yano, M., et al. (2006). Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 55 (1), 120–127. doi:10.2337/diabetes.55.01.06.db05-0943
Kuwana, T., Bouchier-Hayes, L., Chipuk, J. E., Bonzon, C., Sullivan, B. A., Green, D. R., et al. (2005). BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell. 17 (4), 525–535. doi:10.1016/j.molcel.2005.02.003
Kwon, Y. T., and Ciechanover, A. (2017). The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 42 (11), 873–886. doi:10.1016/j.tibs.2017.09.002
Kwong, J. Q., and Molkentin, J. D. (2015). Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell. Metab. 21 (2), 206–214. doi:10.1016/j.cmet.2014.12.001
Lagouge, M., Argmann, C., Gerhart-Hines, Z., Meziane, H., Lerin, C., Daussin, F., et al. (2006). Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 127 (6), 1109–1122. doi:10.1016/j.cell.2006.11.013
Lahera, V., de Las Heras, N., López-Farré, A., Manucha, W., and Ferder, L. (2017). Role of mitochondrial dysfunction in hypertension and obesity. Curr. Hypertens. Rep. 19 (2), 11. doi:10.1007/s11906-017-0710-9
Lai, L., and Qiu, H. (2020). The physiological and pathological roles of mitochondrial calcium uptake in heart. Int. J. Mol. Sci. 21 (20), 7689. doi:10.3390/ijms21207689
Laker, R. C., Drake, J. C., Wilson, R. J., Lira, V. A., Lewellen, B. M., Ryall, K. A., et al. (2017). Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 8 (1), 548. doi:10.1038/s41467-017-00520-9
Lambertucci, R. H., Hirabara, S. M., Silveira Ldos, R., Levada-Pires, A. C., Curi, R., and Pithon-Curi, T. C. (2008). Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J. Cell. Physiol. 216 (3), 796–804. doi:10.1002/jcp.21463
Lampert, M. A., Orogo, A. M., Najor, R. H., Hammerling, B. C., Leon, L. J., Wang, B. J., et al. (2019). BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15 (7), 1182–1198. doi:10.1080/15548627.2019.1580095
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell. 22 (2), 320–333. doi:10.1016/j.devcel.2011.12.014
Lee, Y., Schulte, D. J., Shimada, K., Chen, S., Crother, T. R., Chiba, N., et al. (2012). Interleukin-1β is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation 125 (12), 1542–1550. doi:10.1161/circulationaha.111.072769
Lee, Y., Wakita, D., Dagvadorj, J., Shimada, K., Chen, S., Huang, G., et al. (2015). IL-1 signaling is critically required in stromal cells in Kawasaki disease vasculitis mouse model: Role of both IL-1α and IL-1β. Arterioscler. Thromb. Vasc. Biol. 35 (12), 2605–2616. doi:10.1161/atvbaha.115.306475
Lennicke, C., and Cochemé, H. M. (2021). Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell. 81 (18), 3691–3707. doi:10.1016/j.molcel.2021.08.018
Levin, M., Holland, P. C., Nokes, T. J., Novelli, V., Mola, M., Levinsky, R. J., et al. (1985). Platelet immune complex interaction in pathogenesis of Kawasaki disease and childhood polyarteritis. Br. Med. J. Clin. Res. Ed. 290 (6480), 1456–1460. doi:10.1136/bmj.290.6480.1456
Levy, D., Garrison, R. J., Savage, D. D., Kannel, W. B., and Castelli, W. P. (1990). Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 322 (22), 1561–1566. doi:10.1056/nejm199005313222203
Li, D., Sun, Y., Zhuang, Q., Song, Y., Wu, B., Jia, Z., et al. (2019). Mitochondrial dysfunction caused by m.2336T>C mutation with hypertrophic cardiomyopathy in cybrid cell lines. Mitochondrion 46, 313–320. doi:10.1016/j.mito.2018.08.005
Li, E., Li, X., Huang, J., Xu, C., Liang, Q., Ren, K., et al. (2020). BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell. 11 (9), 661–679. doi:10.1007/s13238-020-00713-x
Li, H., Zheng, L., Mo, Y., Gong, Q., Jiang, A., and Zhao, J. (2016). Voltage-dependent anion channel 1(VDAC1) participates the apoptosis of the mitochondrial dysfunction in desminopathy. PLoS One 11 (12), e0167908. doi:10.1371/journal.pone.0167908
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S., et al. (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91 (4), 479–489. doi:10.1016/s0092-8674(00)80434-1
Li, S., Chen, J., Liu, M., Chen, Y., Wu, Y., Li, Q., et al. (2021). Protective effect of HINT2 on mitochondrial function via repressing MCU complex activation attenuates cardiac microvascular ischemia-reperfusion injury. Basic Res. Cardiol. 116 (1), 65. doi:10.1007/s00395-021-00905-4
Liang, H. L., Sedlic, F., Bosnjak, Z., and Nilakantan, V. (2010). SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. Free Radic. Biol. Med. 49 (10), 1550–1560. doi:10.1016/j.freeradbiomed.2010.08.018
Liesa, M., and Shirihai, O. S. (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell. Metab. 17 (4), 491–506. doi:10.1016/j.cmet.2013.03.002
Lin, T. K., Lin, K. J., Lin, K. L., Liou, C. W., Chen, S. D., Chuang, Y. C., et al. (2020). When friendship turns sour: Effective communication between mitochondria and intracellular organelles in Parkinson's disease. Front. Cell. Dev. Biol. 8, 607392. doi:10.3389/fcell.2020.607392
Little, J. P., Safdar, A., Cermak, N., Tarnopolsky, M. A., and Gibala, M. J. (2010). Acute endurance exercise increases the nuclear abundance of PGC-1alpha in trained human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298 (4), R912–R917. doi:10.1152/ajpregu.00409.2009
Liu, J., Dai, Q., Chen, J., Durrant, D., Freeman, A., Liu, T., et al. (2003). Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 1 (12), 892–902.
Liu, R., Xu, C., Zhang, W., Cao, Y., Ye, J., Li, B., et al. (2022). FUNDC1-mediated mitophagy and HIF1α activation drives pulmonary hypertension during hypoxia. Cell. Death Dis. 13 (7), 634. doi:10.1038/s41419-022-05091-2
Liu, S., Bai, Y., Huang, J., Zhao, H., Zhang, X., Hu, S., et al. (2013). Do mitochondria contribute to left ventricular non-compaction cardiomyopathy? New findings from myocardium of patients with left ventricular non-compaction cardiomyopathy. Mol. Genet. Metab. 109 (1), 100–106. doi:10.1016/j.ymgme.2013.02.004
Liu, T., Zhang, L., Joo, D., and Sun, S. C. (2017). NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2, 17023-. doi:10.1038/sigtrans.2017.23
Liu, X., and Hajnóczky, G. (2011). Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell. Death Differ. 18 (10), 1561–1572. doi:10.1038/cdd.2011.13
Liu, Y., Fiskum, G., and Schubert, D. (2002). Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 80 (5), 780–787. doi:10.1046/j.0022-3042.2002.00744.x
Liu, Z., Song, Y., Li, D., He, X., Li, S., Wu, B., et al. (2014). The novel mitochondrial 16S rRNA 2336T>C mutation is associated with hypertrophic cardiomyopathy. J. Med. Genet. 51 (3), 176–184. doi:10.1136/jmedgenet-2013-101818
Lomonosova, E., and Chinnadurai, G. (2008). BH3-only proteins in apoptosis and beyond: An overview. Oncogene 27(1), S2–S19. doi:10.1038/onc.2009.39
Lopez-Crisosto, C., Pennanen, C., Vasquez-Trincado, C., Morales, P. E., Bravo-Sagua, R., Quest, A. F. G., et al. (2017). Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 14 (6), 342–360. doi:10.1038/nrcardio.2017.23
Lorenzon, A., Beffagna, G., Bauce, B., De Bortoli, M., Li Mura, I. E., Calore, M., et al. (2013). Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am. J. Cardiol. 111 (3), 400–405. doi:10.1016/j.amjcard.2012.10.017
Lu, L., Bonham, C. A., Chambers, F. G., Watkins, S. C., Hoffman, R. A., Simmons, R. L., et al. (1996). Induction of nitric oxide synthase in mouse dendritic cells by IFN-gamma, endotoxin, and interaction with allogeneic T cells: Nitric oxide production is associated with dendritic cell apoptosis. J. Immunol. 157 (8), 3577–3586. doi:10.4049/jimmunol.157.8.3577
Lugus, J. J., Ngoh, G. A., Bachschmid, M. M., and Walsh, K. (2011). Mitofusins are required for angiogenic function and modulate different signaling pathways in cultured endothelial cells. J. Mol. Cell. Cardiol. 51 (6), 885–893. doi:10.1016/j.yjmcc.2011.07.023
Ma, K., Chen, G., Li, W., Kepp, O., Zhu, Y., and Chen, Q. (2020). Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell. Dev. Biol. 8, 467. doi:10.3389/fcell.2020.00467
Ma, T., Huang, X., Zheng, H., Huang, G., Li, W., Liu, X., et al. (2021). SFRP2 improves mitochondrial dynamics and mitochondrial biogenesis, oxidative stress, and apoptosis in diabetic cardiomyopathy. Oxid. Med. Cell. Longev. 2021, 9265016. doi:10.1155/2021/9265016
MacKenzie, J. A., and Payne, R. M. (2007). Mitochondrial protein import and human health and disease. Biochim. Biophys. Acta 1772 (5), 509–523. doi:10.1016/j.bbadis.2006.12.002
Madeira, V. M. C. (2018). Overview of mitochondrial bioenergetics. Methods Mol. Biol. 1782, 1–6. doi:10.1007/978-1-4939-7831-1_1
Magyar, K., Halmosi, R., Palfi, A., Feher, G., Czopf, L., Fulop, A., et al. (2012). Cardioprotection by resveratrol: A human clinical trial in patients with stable coronary artery disease. Clin. Hemorheol. Microcirc. 50 (3), 179–187. doi:10.3233/ch-2011-1424
Malka, F., Guillery, O., Cifuentes-Diaz, C., Guillou, E., Belenguer, P., Lombès, A., et al. (2005). Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 6 (9), 853–859. doi:10.1038/sj.embor.7400488
Maneechote, C., Palee, S., Kerdphoo, S., Jaiwongkam, T., Chattipakorn, S. C., and Chattipakorn, N. (2019). Balancing mitochondrial dynamics via increasing mitochondrial fusion attenuates infarct size and left ventricular dysfunction in rats with cardiac ischemia/reperfusion injury. Clin. Sci. (Lond) 133 (3), 497–513. doi:10.1042/cs20190014
Mantle, D., Millichap, L., Castro-Marrero, J., and Hargreaves, I. P. (2023). Primary coenzyme Q10 deficiency: An update. Antioxidants (Basel) 12 (8), 1652. doi:10.3390/antiox12081652
Marek-Iannucci, S., Ozdemir, A. B., Moreira, D., Gomez, A. C., Lane, M., Porritt, R. A., et al. (2021). Autophagy-mitophagy induction attenuates cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight 6 (18), e151981. doi:10.1172/jci.insight.151981
Marek-Iannucci, S., Yildirim, A. D., Hamid, S. M., Ozdemir, A. B., Gomez, A. C., Kocatürk, B., et al. (2022). Targeting IRE1 endoribonuclease activity alleviates cardiovascular lesions in a murine model of Kawasaki disease vasculitis. JCI Insight 7 (6), e157203. doi:10.1172/jci.insight.157203
Martens, C. R., Denman, B. A., Mazzo, M. R., Armstrong, M. L., Reisdorph, N., McQueen, M. B., et al. (2018). Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun. 9 (1), 1286. doi:10.1038/s41467-018-03421-7
Martin, A. S., Abraham, D. M., Hershberger, K. A., Bhatt, D. P., Mao, L., Cui, H., et al. (2017). Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich's ataxia cardiomyopathy model. JCI Insight 2 (14), e93885. doi:10.1172/jci.insight.93885
Martínez-Reyes, I., and Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11 (1), 102. doi:10.1038/s41467-019-13668-3
Martínez-Reyes, I., Diebold, L. P., Kong, H., Schieber, M., Huang, H., Hensley, C. T., et al. (2016). TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell. 61 (2), 199–209. doi:10.1016/j.molcel.2015.12.002
McArthur, K., Whitehead, L. W., Heddleston, J. M., Li, L., Padman, B. S., Oorschot, V., et al. (2018). BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359 (6378), eaao6047. doi:10.1126/science.aao6047
McCrindle, B. W., Rowley, A. H., Newburger, J. W., Burns, J. C., Bolger, A. F., Gewitz, M., et al. (2017). Diagnosis, treatment, and long-term management of Kawasaki disease: A scientific statement for health professionals from the American heart association. Circulation 135 (17), e927–e999. doi:10.1161/cir.0000000000000484
McDonnell, E., Crown, S. B., Fox, D. B., Kitir, B., Ilkayeva, O. R., Olsen, C. A., et al. (2016). Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell. Rep. 17 (6), 1463–1472. doi:10.1016/j.celrep.2016.10.012
McKenna, W. J., Maron, B. J., and Thiene, G. (2017). Classification, epidemiology, and global burden of cardiomyopathies. Circ. Res. 121 (7), 722–730. doi:10.1161/circresaha.117.309711
McWilliams, T. G., Prescott, A. R., Montava-Garriga, L., Ball, G., Singh, F., Barini, E., et al. (2018). Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell. Metab. 27 (2), 439–449. doi:10.1016/j.cmet.2017.12.008
Mehta, M. M., Weinberg, S. E., and Chandel, N. S. (2017). Mitochondrial control of immunity: Beyond ATP. Nat. Rev. Immunol. 17 (10), 608–620. doi:10.1038/nri.2017.66
Miller, K. N., Clark, J. P., and Anderson, R. M. (2019). Mitochondrial regulator PGC-1a-Modulating the modulator. Curr. Opin. Endocr. Metab. Res. 5, 37–44. doi:10.1016/j.coemr.2019.02.002
Moldoveanu, T., Follis, A. V., Kriwacki, R. W., and Green, D. R. (2014). Many players in BCL-2 family affairs. Trends Biochem. Sci. 39 (3), 101–111. doi:10.1016/j.tibs.2013.12.006
Montaigne, D., Marechal, X., Coisne, A., Debry, N., Modine, T., Fayad, G., et al. (2014). Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130 (7), 554–564. doi:10.1161/circulationaha.113.008476
Moussaieff, A., Rouleau, M., Kitsberg, D., Cohen, M., Levy, G., Barasch, D., et al. (2015). Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell. Metab. 21 (3), 392–402. doi:10.1016/j.cmet.2015.02.002
Murphy, E., Ardehali, H., Balaban, R. S., DiLisa, F., Dorn, G. W., Kitsis, R. N., et al. (2016). Mitochondrial function, Biology, and role in disease: A scientific statement from the American heart association. Circ. Res. 118 (12), 1960–1991. doi:10.1161/res.0000000000000104
Murphy, E., and Steenbergen, C. (2008). Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 88 (2), 581–609. doi:10.1152/physrev.00024.2007
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417 (1), 1–13. doi:10.1042/bj20081386
Murphy, M. P., and Smith, R. A. (2007). Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 47, 629–656. doi:10.1146/annurev.pharmtox.47.120505.105110
Muto, T., Nakamura, N., Masuda, Y., Numoto, S., Kodama, S., Miyamoto, R., et al. (2022). Serum free carnitine levels in children with Kawasaki disease. Pediatr. Int. 64 (1), e14849. doi:10.1111/ped.14849
Nahapetyan, H., Moulis, M., Grousset, E., Faccini, J., Grazide, M. H., Mucher, E., et al. (2019). Altered mitochondrial quality control in Atg7-deficient VSMCs promotes enhanced apoptosis and is linked to unstable atherosclerotic plaque phenotype. Cell. Death Dis. 10 (2), 119. doi:10.1038/s41419-019-1400-0
Nakamura, M., and Sadoshima, J. (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15 (7), 387–407. doi:10.1038/s41569-018-0007-y
Nakatogawa, H. (2013). Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 55, 39–50. doi:10.1042/bse0550039
Narendra, D., Kane, L. A., Hauser, D. N., Fearnley, I. M., and Youle, R. J. (2010). p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6 (8), 1090–1106. doi:10.4161/auto.6.8.13426
Newburger, J. W., Takahashi, M., and Burns, J. C. (2016). Kawasaki disease. J. Am. Coll. Cardiol. 67 (14), 1738–1749. doi:10.1016/j.jacc.2015.12.073
Ng, M. Y. W., Wai, T., and Simonsen, A. (2021). Quality control of the mitochondrion. Dev. Cell. 56 (7), 881–905. doi:10.1016/j.devcel.2021.02.009
Nguyen, T. N., Padman, B. S., and Lazarou, M. (2016). Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell. Biol. 26 (10), 733–744. doi:10.1016/j.tcb.2016.05.008
Ni, H. M., Williams, J. A., and Ding, W. X. (2015). Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13. doi:10.1016/j.redox.2014.11.006
Ni, R., Cao, T., Xiong, S., Ma, J., Fan, G. C., Lacefield, J. C., et al. (2016). Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 90, 12–23. doi:10.1016/j.freeradbiomed.2015.11.013
Niedra, E., Chahal, N., Manlhiot, C., Yeung, R. S., and McCrindle, B. W. (2014). Atorvastatin safety in Kawasaki disease patients with coronary artery aneurysms. Pediatr. Cardiol. 35 (1), 89–92. doi:10.1007/s00246-013-0746-9
Nisoli, E., Tonello, C., Cardile, A., Cozzi, V., Bracale, R., Tedesco, L., et al. (2005). Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310 (5746), 314–317. doi:10.1126/science.1117728
Nollet, E. E., Duursma, I., Rozenbaum, A., Eggelbusch, M., Wüst, R. C. I., Schoonvelde, S. A. C., et al. (2023). Mitochondrial dysfunction in human hypertrophic cardiomyopathy is linked to cardiomyocyte architecture disruption and corrected by improving NADH-driven mitochondrial respiration. Eur. Heart J. 44 (13), 1170–1185. doi:10.1093/eurheartj/ehad028
Noorman, M., Hakim, S., Kessler, E., Groeneweg, J. A., Cox, M. G., Asimaki, A., et al. (2013). Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart rhythm. 10 (3), 412–419. doi:10.1016/j.hrthm.2012.11.018
Okatsu, K., Uno, M., Koyano, F., Go, E., Kimura, M., Oka, T., et al. (2013). A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J. Biol. Chem. 288 (51), 36372–36384. doi:10.1074/jbc.M113.509653
Olichon, A., Elachouri, G., Baricault, L., Delettre, C., Belenguer, P., and Lenaers, G. (2007). OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell. Death Differ. 14 (4), 682–692. doi:10.1038/sj.cdd.4402048
Oliver, D. M. A., and Reddy, P. H. (2019). Small molecules as therapeutic drugs for Alzheimer's disease. Mol. Cell. Neurosci. 96, 47–62. doi:10.1016/j.mcn.2019.03.001
Onat, U. I., Yildirim, A. D., Tufanli, Ö., Çimen, I., Kocatürk, B., Veli, Z., et al. (2019). Intercepting the lipid-induced integrated stress response reduces atherosclerosis. J. Am. Coll. Cardiol. 73 (10), 1149–1169. doi:10.1016/j.jacc.2018.12.055
Ong, S. B., and Hausenloy, D. J. (2010). Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. 88 (1), 16–29. doi:10.1093/cvr/cvq237
Onoue, K., Jofuku, A., Ban-Ishihara, R., Ishihara, T., Maeda, M., Koshiba, T., et al. (2013). Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J. Cell. Sci. 126 (1), 176–185. doi:10.1242/jcs.111211
Ordureau, A., Sarraf, S. A., Duda, D. M., Heo, J. M., Jedrychowski, M. P., Sviderskiy, V. O., et al. (2014). Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell. 56 (3), 360–375. doi:10.1016/j.molcel.2014.09.007
Otera, H., Ishihara, N., and Mihara, K. (2013). New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 1833 (5), 1256–1268. doi:10.1016/j.bbamcr.2013.02.002
Otten, E., Asimaki, A., Maass, A., van Langen, I. M., van der Wal, A., de Jonge, N., et al. (2010). Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart rhythm. 7 (8), 1058–1064. doi:10.1016/j.hrthm.2010.04.023
Palmer, C. S., Osellame, L. D., Laine, D., Koutsopoulos, O. S., Frazier, A. E., and Ryan, M. T. (2011). MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12 (6), 565–573. doi:10.1038/embor.2011.54
Pan, L., Li, Y., Jia, L., Qin, Y., Qi, G., Cheng, J., et al. (2012). Cathepsin S deficiency results in abnormal accumulation of autophagosomes in macrophages and enhances Ang II-induced cardiac inflammation. PLoS One 7 (4), e35315. doi:10.1371/journal.pone.0035315
Papanicolaou, K. N., Ngoh, G. A., Dabkowski, E. R., O'Connell, K. A., Ribeiro, R. F., Stanley, W. C., et al. (2012). Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am. J. Physiol. Heart Circ. Physiol. 302 (1), H167–H179. doi:10.1152/ajpheart.00833.2011
Paradies, G., Paradies, V., Ruggiero, F. M., and Petrosillo, G. (2019). Role of cardiolipin in mitochondrial function and dynamics in health and disease: Molecular and pharmacological aspects. Cells 8 (7), 728. doi:10.3390/cells8070728
Parra, V., Verdejo, H., del Campo, A., Pennanen, C., Kuzmicic, J., Iglewski, M., et al. (2011). The complex interplay between mitochondrial dynamics and cardiac metabolism. J. Bioenerg. Biomembr. 43 (1), 47–51. doi:10.1007/s10863-011-9332-0
Peng, S., Xu, L. W., Che, X. Y., Xiao, Q. Q., Pu, J., Shao, Q., et al. (2018). Atorvastatin inhibits inflammatory response, attenuates lipid deposition, and improves the stability of vulnerable atherosclerotic plaques by modulating autophagy. Front. Pharmacol. 9, 438. doi:10.3389/fphar.2018.00438
Pennanen, C., Parra, V., López-Crisosto, C., Morales, P. E., Del Campo, A., Gutierrez, T., et al. (2014). Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell. Sci. 127 (12), 2659–2671. doi:10.1242/jcs.139394
Peoples, J. N., Saraf, A., Ghazal, N., Pham, T. T., and Kwong, J. Q. (2019). Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 51 (12), 1–13. doi:10.1038/s12276-019-0355-7
Pereira, C. V., Gitschlag, B. L., and Patel, M. R. (2021). Cellular mechanisms of mtDNA heteroplasmy dynamics. Crit. Rev. Biochem. Mol. Biol. 56 (5), 510–525. doi:10.1080/10409238.2021.1934812
Pfanner, N., Warscheid, B., and Wiedemann, N. (2019). Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell. Biol. 20 (5), 267–284. doi:10.1038/s41580-018-0092-0
Picca, A., Mankowski, R. T., Burman, J. L., Donisi, L., Kim, J. S., Marzetti, E., et al. (2018). Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 15 (9), 543–554. doi:10.1038/s41569-018-0059-z
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85 (2), 257–273. doi:10.1016/j.neuron.2014.12.007
Pietraforte, D., Gambardella, L., Marchesi, A., Tarissi de Jacobis, I., Villani, A., Del Principe, D., et al. (2014). Platelets in Kawasaki patients: Two different populations with different mitochondrial functions. Int. J. Cardiol. 172 (2), 526–528. doi:10.1016/j.ijcard.2014.01.022
Porritt, R. A., Markman, J. L., Maruyama, D., Kocaturk, B., Chen, S., Lehman, T. J. A., et al. (2020). Interleukin-1 beta-mediated sex differences in Kawasaki disease vasculitis development and response to treatment. Arterioscler. Thromb. Vasc. Biol. 40 (3), 802–818. doi:10.1161/ATVBAHA.119.313863
Porritt, R. A., Zemmour, D., Abe, M., Lee, Y., Narayanan, M., Carvalho, T. T., et al. (2021). NLRP3 inflammasome mediates immune-stromal interactions in vasculitis. Circ. Res. 129 (9), e183–e200. doi:10.1161/circresaha.121.319153
Potgieter, M., Pretorius, E., and Pepper, M. S. (2013). Primary and secondary coenzyme Q10 deficiency: The role of therapeutic supplementation. Nutr. Rev. 71 (3), 180–188. doi:10.1111/nure.12011
Protasoni, M., and Zeviani, M. (2021). Mitochondrial structure and bioenergetics in normal and disease conditions. Int. J. Mol. Sci. 22 (2), 586. doi:10.3390/ijms22020586
Puigserver, P., Wu, Z., Park, C. W., Graves, R., Wright, M., and Spiegelman, B. M. (1998). A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 92 (6), 829–839. doi:10.1016/s0092-8674(00)81410-5
Qi, J., Wang, F., Yang, P., Wang, X., Xu, R., Chen, J., et al. (2018). Mitochondrial fission is required for angiotensin II-induced cardiomyocyte apoptosis mediated by a sirt1-p53 signaling pathway. Front. Pharmacol. 9, 176. doi:10.3389/fphar.2018.00176
Quiles, J. M., and Gustafsson Å, B. (2022). The role of mitochondrial fission in cardiovascular health and disease. Nat. Rev. Cardiol. 19 (11), 723–736. doi:10.1038/s41569-022-00703-y
Quirós, P. M., Mottis, A., and Auwerx, J. (2016). Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell. Biol. 17 (4), 213–226. doi:10.1038/nrm.2016.23
Qureshi, M. A., Haynes, C. M., and Pellegrino, M. W. (2017). The mitochondrial unfolded protein response: Signaling from the powerhouse. J. Biol. Chem. 292 (33), 13500–13506. doi:10.1074/jbc.R117.791061
Ramaccini, D., Montoya-Uribe, V., Aan, F. J., Modesti, L., Potes, Y., Wieckowski, M. R., et al. (2020). Mitochondrial function and dysfunction in dilated cardiomyopathy. Front. Cell. Dev. Biol. 8, 624216. doi:10.3389/fcell.2020.624216
Ranjbarvaziri, S., Kooiker, K. B., Ellenberger, M., Fajardo, G., Zhao, M., Vander Roest, A. S., et al. (2021). Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation 144 (21), 1714–1731. doi:10.1161/circulationaha.121.053575
Rapezzi, C., Aimo, A., Barison, A., Emdin, M., Porcari, A., Linhart, A., et al. (2022). Restrictive cardiomyopathy: Definition and diagnosis. Eur. Heart J. 43 (45), 4679–4693. doi:10.1093/eurheartj/ehac543
Ravi, B., Kanwar, P., Sanyal, S. K., Bheri, M., and Pandey, G. K. (2021). VDACs: An outlook on biochemical regulation and function in animal and plant systems. Front. Physiol. 12, 683920. doi:10.3389/fphys.2021.683920
Razani, B., Feng, C., Coleman, T., Emanuel, R., Wen, H., Hwang, S., et al. (2012). Autophagy links inflammasomes to atherosclerotic progression. Cell. Metab. 15 (4), 534–544. doi:10.1016/j.cmet.2012.02.011
Reid, R. A., Moyle, J., and Mitchell, P. (1966). Synthesis of adenosine triphosphate by a protonmotive force in rat liver mitochondria. Nature 212 (5059), 257–258. doi:10.1038/212257a0
Reikine, S., Nguyen, J. B., and Modis, Y. (2014). Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front. Immunol. 5, 342. doi:10.3389/fimmu.2014.00342
Reina, S., Checchetto, V., Saletti, R., Gupta, A., Chaturvedi, D., Guardiani, C., et al. (2016). VDAC3 as a sensor of oxidative state of the intermembrane space of mitochondria: The putative role of cysteine residue modifications. Oncotarget 7 (3), 2249–2268. doi:10.18632/oncotarget.6850
Reina, S., Nibali, S. C., Tomasello, M. F., Magrì, A., Messina, A., and De Pinto, V. (2022). Voltage Dependent Anion Channel 3 (VDAC3) protects mitochondria from oxidative stress. Redox Biol. 51, 102264. doi:10.1016/j.redox.2022.102264
Ren, J., Sun, M., Zhou, H., Ajoolabady, A., Zhou, Y., Tao, J., et al. (2020). FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci. Adv. 6 (38), eabc8561. doi:10.1126/sciadv.abc8561
Reyes, A., Gissi, C., Pesole, G., and Saccone, C. (1998). Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol. Biol. Evol. 15 (8), 957–966. doi:10.1093/oxfordjournals.molbev.a026011
Richter, C., Gogvadze, V., Laffranchi, R., Schlapbach, R., Schweizer, M., Suter, M., et al. (1995). Oxidants in mitochondria: From physiology to diseases. Biochim. Biophys. Acta 1271 (1), 67–74. doi:10.1016/0925-4439(95)00012-s
Richter, C., Park, J. W., and Ames, B. N. (1988). Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. U. S. A. 85 (17), 6465–6467. doi:10.1073/pnas.85.17.6465
Rimbaud, S., Ruiz, M., Piquereau, J., Mateo, P., Fortin, D., Veksler, V., et al. (2011). Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS One 6 (10), e26391. doi:10.1371/journal.pone.0026391
Ritz, P., and Berrut, G. (2005). Mitochondrial function, energy expenditure, aging and insulin resistance. Diabetes Metab. 31(2), 5s67–5S73. doi:10.1016/s1262-3636(05)73654-5
Rogers, M. A., Maldonado, N., Hutcheson, J. D., Goettsch, C., Goto, S., Yamada, I., et al. (2017). Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ. Res. 121 (3), 220–233. doi:10.1161/circresaha.116.310293
Roman, B., Kaur, P., Ashok, D., Kohr, M., Biswas, R., O'Rourke, B., et al. (2020). Nuclear-mitochondrial communication involving miR-181c plays an important role in cardiac dysfunction during obesity. J. Mol. Cell. Cardiol. 144, 87–96. doi:10.1016/j.yjmcc.2020.05.009
Rongvaux, A., Jackson, R., Harman, C. C., Li, T., West, A. P., de Zoete, M. R., et al. (2014). Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 159 (7), 1563–1577. doi:10.1016/j.cell.2014.11.037
Rossman, M. J., Santos-Parker, J. R., Steward, C. A. C., Bispham, N. Z., Cuevas, L. M., Rosenberg, H. L., et al. (2018). Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 71 (6), 1056–1063. doi:10.1161/hypertensionaha.117.10787
Rüb, C., Wilkening, A., and Voos, W. (2017). Mitochondrial quality control by the Pink1/Parkin system. Cell. Tissue Res. 367 (1), 111–123. doi:10.1007/s00441-016-2485-8
Russell, O. M., Gorman, G. S., Lightowlers, R. N., and Turnbull, D. M. (2020). Mitochondrial diseases: Hope for the future. Cell. 181 (1), 168–188. doi:10.1016/j.cell.2020.02.051
Russell, R. C., Tian, Y., Yuan, H., Park, H. W., Chang, Y. Y., Kim, J., et al. (2013). ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell. Biol. 15 (7), 741–750. doi:10.1038/ncb2757
Russo, S. B., Baicu, C. F., Van Laer, A., Geng, T., Kasiganesan, H., Zile, M. R., et al. (2012). Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J. Clin. Investig. 122 (11), 3919–3930. doi:10.1172/jci63888
Salabei, J. K., and Hill, B. G. (2013). Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. 1 (1), 542–551. doi:10.1016/j.redox.2013.10.011
Sampath, H. (2014). Oxidative DNA damage in disease--insights gained from base excision repair glycosylase-deficient mouse models. Environ. Mol. Mutagen 55 (9), 689–703. doi:10.1002/em.21886
Saneto, R. P., and Sedensky, M. M. (2013). Mitochondrial disease in childhood: mtDNA encoded. Neurotherapeutics 10 (2), 199–211. doi:10.1007/s13311-012-0167-0
Schellenberg, B., Wang, P., Keeble, J. A., Rodriguez-Enriquez, R., Walker, S., Owens, T. W., et al. (2013). Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol. Cell. 49 (5), 959–971. doi:10.1016/j.molcel.2012.12.022
Schiattarella, G. G., and Hill, J. A. (2015). Inhibition of hypertrophy is a good therapeutic strategy in ventricular pressure overload. Circulation 131 (16), 1435–1447. doi:10.1161/circulationaha.115.013894
Schilling, J. D. (2015). The mitochondria in diabetic heart failure: From pathogenesis to therapeutic promise. Antioxid. Redox Signal 22 (17), 1515–1526. doi:10.1089/ars.2015.6294
Sentelle, R. D., Senkal, C. E., Jiang, W., Ponnusamy, S., Gencer, S., Selvam, S. P., et al. (2012). Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 8 (10), 831–838. doi:10.1038/nchembio.1059
Sepasi Tehrani, H., and Moosavi-Movahedi, A. A. (2018). Catalase and its mysteries. Prog. Biophys. Mol. Biol. 140, 5–12. doi:10.1016/j.pbiomolbio.2018.03.001
Sergin, I., Bhattacharya, S., Emanuel, R., Esen, E., Stokes, C. J., Evans, T. D., et al. (2016). Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci. Signal 9 (409), ra2. doi:10.1126/scisignal.aad5614
Shafferman, A., Birmingham, J. D., and Cron, R. Q. (2014). High dose anakinra for treatment of severe neonatal Kawasaki disease: A case report. Pediatr. Rheumatol. Online J. 12, 26. doi:10.1186/1546-0096-12-26
Shah, S. N., Umapathi, K. K., and Oliver, T. I. (2023). “Arrhythmogenic right ventricular cardiomyopathy,” in StatPearls (Treasure Island (FL): StatPearls Publishing).
Sheeran, F. L., and Pepe, S. (2017). Mitochondrial bioenergetics and dysfunction in failing heart. Adv. Exp. Med. Biol. 982, 65–80. doi:10.1007/978-3-319-55330-6_4
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526 (7575), 660–665. doi:10.1038/nature15514
Shi, L., and Tu, B. P. (2015). Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell. Biol. 33, 125–131. doi:10.1016/j.ceb.2015.02.003
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36 (3), 401–414. doi:10.1016/j.immuni.2012.01.009
Shin, W. S., Hong, Y. H., Peng, H. B., De Caterina, R., Libby, P., and Liao, J. K. (1996). Nitric oxide attenuates vascular smooth muscle cell activation by interferon-gamma. The role of constitutive NF-kappa B activity. J. Biol. Chem. 271 (19), 11317–11324. doi:10.1074/jbc.271.19.11317
Shpilka, T., and Haynes, C. M. (2018). The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell. Biol. 19 (2), 109–120. doi:10.1038/nrm.2017.110
Sica, V., Galluzzi, L., Bravo-San Pedro, J. M., Izzo, V., Maiuri, M. C., and Kroemer, G. (2015). Organelle-specific initiation of autophagy. Mol. Cell. 59 (4), 522–539. doi:10.1016/j.molcel.2015.07.021
Singh, D. P., and Patel, H. (2023). “Left ventricular noncompaction cardiomyopathy,” in StatPearls (Treasure Island (FL): StatPearls Publishing).
Song, S., Ding, Y., Dai, G. L., Zhang, Y., Xu, M. T., Shen, J. R., et al. (2021). Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 42 (2), 230–241. doi:10.1038/s41401-020-0490-7
Song, Z., Chen, H., Fiket, M., Alexander, C., and Chan, D. C. (2007). OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell. Biol. 178 (5), 749–755. doi:10.1083/jcb.200704110
Sonn, S. K., Song, E. J., Seo, S., Kim, Y. Y., Um, J. H., Yeo, F. J., et al. (2022). Peroxiredoxin 3 deficiency induces cardiac hypertrophy and dysfunction by impaired mitochondrial quality control. Redox Biol. 51, 102275. doi:10.1016/j.redox.2022.102275
Srivastava, S. (2016). Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin. Transl. Med. 5 (1), 25. doi:10.1186/s40169-016-0104-7
St-Pierre, J., Buckingham, J. A., Roebuck, S. J., and Brand, M. D. (2002). Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 277 (47), 44784–44790. doi:10.1074/jbc.M207217200
Starkov, A. A., Fiskum, G., Chinopoulos, C., Lorenzo, B. J., Browne, S. E., Patel, M. S., et al. (2004). Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 24 (36), 7779–7788. doi:10.1523/jneurosci.1899-04.2004
Stefano, G. B., Bjenning, C., Wang, F., Wang, N., and Kream, R. M. (2017). Mitochondrial heteroplasmy. Adv. Exp. Med. Biol. 982, 577–594. doi:10.1007/978-3-319-55330-6_30
Stein, A., and Sia, E. A. (2017). Mitochondrial DNA repair and damage tolerance. Front. Biosci. (Landmark Ed. 22 (5), 920–943. doi:10.2741/4525
Storz, P., Döppler, H., and Toker, A. (2005). Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol. Cell. Biol. 25 (19), 8520–8530. doi:10.1128/mcb.25.19.8520-8530.2005
Sulaiman, M., Matta, M. J., Sunderesan, N. R., Gupta, M. P., Periasamy, M., and Gupta, M. (2010). Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 298 (3), H833–H843. doi:10.1152/ajpheart.00418.2009
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339 (6121), 786–791. doi:10.1126/science.1232458
Sun, T., Han, Y., Li, J. L., Jiao, X. Y., Zuo, L., Wang, J., et al. (2022). FOXO3a-dependent PARKIN negatively regulates cardiac hypertrophy by restoring mitophagy. Cell. Biosci. 12 (1), 204. doi:10.1186/s13578-022-00935-y
Suomalainen, A., and Battersby, B. J. (2018). Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell. Biol. 19 (2), 77–92. doi:10.1038/nrm.2017.66
Swaminathan, B., Goikuria, H., Vega, R., Rodríguez-Antigüedad, A., López Medina, A., Freijo Mdel, M., et al. (2014). Autophagic marker MAP1LC3B expression levels are associated with carotid atherosclerosis symptomatology. PLoS One 9 (12), e115176. doi:10.1371/journal.pone.0115176
Sykora, P., Wilson, D. M., and Bohr, V. A. (2012). Repair of persistent strand breaks in the mitochondrial genome. Mech. Ageing Dev. 133 (4), 169–175. doi:10.1016/j.mad.2011.11.003
Tan, Y., Li, M., Wu, G., Lou, J., Feng, M., Xu, J., et al. (2021). Short-term but not long-term high fat diet feeding protects against pressure overload-induced heart failure through activation of mitophagy. Life Sci. 272, 119242. doi:10.1016/j.lfs.2021.119242
Tanaka, M., and Ozawa, T. (1994). Strand asymmetry in human mitochondrial DNA mutations. Genomics 22 (2), 327–335. doi:10.1006/geno.1994.1391
Tanaka, Y., and Chen, Z. J. (2012). STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 5 (214), ra20. doi:10.1126/scisignal.2002521
Tanida, I., Ueno, T., and Kominami, E. (2004). LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell. Biol. 36 (12), 2503–2518. doi:10.1016/j.biocel.2004.05.009
Tao, T., and Xu, H. (2020). Autophagy and obesity and diabetes. Adv. Exp. Med. Biol. 1207, 445–461. doi:10.1007/978-981-15-4272-5_32
Tatsuta, T., Scharwey, M., and Langer, T. (2014). Mitochondrial lipid trafficking. Trends Cell. Biol. 24 (1), 44–52. doi:10.1016/j.tcb.2013.07.011
Taylor, M. R., Slavov, D., Ku, L., Di Lenarda, A., Sinagra, G., Carniel, E., et al. (2007). Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 115 (10), 1244–1251. doi:10.1161/circulationaha.106.646778
Teekakirikul, P., Zhu, W., Huang, H. C., and Fung, E. (2019). Hypertrophic cardiomyopathy: An overview of genetics and management. Biomolecules 9 (12), 878. doi:10.3390/biom9120878
Teske, B. F., Fusakio, M. E., Zhou, D., Shan, J., McClintick, J. N., Kilberg, M. S., et al. (2013). CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell. 24 (15), 2477–2490. doi:10.1091/mbc.E13-01-0067
Tomczyk, M. M., Cheung, K. G., Xiang, B., Tamanna, N., Fonseca Teixeira, A. L., Agarwal, P., et al. (2022). Mitochondrial sirtuin-3 (SIRT3) prevents doxorubicin-induced dilated cardiomyopathy by modulating protein acetylation and oxidative stress. Circ. Heart Fail 15 (5), e008547. doi:10.1161/circheartfailure.121.008547
Tong, M., Saito, T., Zhai, P., Oka, S. I., Mizushima, W., Nakamura, M., et al. (2019). Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ. Res. 124 (9), 1360–1371. doi:10.1161/circresaha.118.314607
Tremoulet, A. H., Best, B. M., Song, S., Wang, S., Corinaldesi, E., Eichenfield, J. R., et al. (2008). Resistance to intravenous immunoglobulin in children with Kawasaki disease. J. Pediatr. 153 (1), 117–121. doi:10.1016/j.jpeds.2007.12.021
Tremoulet, A. H., Jain, S., Jone, P. N., Best, B. M., Duxbury, E. H., Franco, A., et al. (2019). Phase I/IIa trial of atorvastatin in patients with acute Kawasaki disease with coronary artery aneurysm. J. Pediatr. 215, 107–117. doi:10.1016/j.jpeds.2019.07.064
Tumurkhuu, G., Shimada, K., Dagvadorj, J., Crother, T. R., Zhang, W., Luthringer, D., et al. (2016). Ogg1-Dependent DNA repair regulates NLRP3 inflammasome and prevents atherosclerosis. Circ. Res. 119 (6), e76–e90. doi:10.1161/circresaha.116.308362
Tuohy, C. V., Kaul, S., Song, H. K., Nazer, B., and Heitner, S. B. (2020). Hypertrophic cardiomyopathy: The future of treatment. Eur. J. Heart Fail 22 (2), 228–240. doi:10.1002/ejhf.1715
University of Washington (2023). Seattle. GeneReviews is a registered trademark of the University of Washington. Seattle. All rights reserved.
van Spaendonck-Zwarts, K. Y., van Hessem, L., Jongbloed, J. D., de Walle, H. E., Capetanaki, Y., van der Kooi, A. J., et al. (2011). Desmin-related myopathy. Clin. Genet. 80 (4), 354–366. doi:10.1111/j.1399-0004.2010.01512.x
Varanita, T., Soriano, M. E., Romanello, V., Zaglia, T., Quintana-Cabrera, R., Semenzato, M., et al. (2015). The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell. Metab. 21 (6), 834–844. doi:10.1016/j.cmet.2015.05.007
Vargas, J. N. S., Wang, C., Bunker, E., Hao, L., Maric, D., Schiavo, G., et al. (2019). Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol. Cell. 74 (2), 347–362. doi:10.1016/j.molcel.2019.02.010
Vercellino, I., and Sazanov, L. A. (2022). The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell. Biol. 23 (2), 141–161. doi:10.1038/s41580-021-00415-0
Villa, E., Marchetti, S., and Ricci, J. E. (2018). No parkin zone: Mitophagy without parkin. Trends Cell. Biol. 28 (11), 882–895. doi:10.1016/j.tcb.2018.07.004
Virbasius, J. V., and Scarpulla, R. C. (1994). Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. U. S. A. 91 (4), 1309–1313. doi:10.1073/pnas.91.4.1309
Vizioli, M. G., Liu, T., Miller, K. N., Robertson, N. A., Gilroy, K., Lagnado, A. B., et al. (2020). Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes. Dev. 34 (5-6), 428–445. doi:10.1101/gad.331272.119
Wakita, D., Kurashima, Y., Crother, T. R., Noval Rivas, M., Lee, Y., Chen, S., et al. (2016). Role of interleukin-1 signaling in a mouse model of Kawasaki disease-associated abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 36 (5), 886–897. doi:10.1161/atvbaha.115.307072
Wang, J., Huang, X., Liu, H., Chen, Y., Li, P., Liu, L., et al. (2022a). Empagliflozin ameliorates diabetic cardiomyopathy via attenuating oxidative stress and improving mitochondrial function. Oxid. Med. Cell. Longev. 2022, 1122494. doi:10.1155/2022/1122494
Wang, L., Cai, Y., Jian, L., Cheung, C. W., Zhang, L., and Xia, Z. (2021a). Impact of peroxisome proliferator-activated receptor-α on diabetic cardiomyopathy. Cardiovasc Diabetol. 20 (1), 2. doi:10.1186/s12933-020-01188-0
Wang, P., Wang, D., Yang, Y., Hou, J., Wan, J., Ran, F., et al. (2020). Tom70 protects against diabetic cardiomyopathy through its antioxidant and antiapoptotic properties. Hypertens. Res. 43 (10), 1047–1056. doi:10.1038/s41440-020-0518-x
Wang, Q., Zhang, M., Torres, G., Wu, S., Ouyang, C., Xie, Z., et al. (2017). Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of drp1-mediated mitochondrial fission. Diabetes 66 (1), 193–205. doi:10.2337/db16-0915
Wang, X., Zhang, X., Cao, K., Zeng, M., Fu, X., Zheng, A., et al. (2022b). Cardiac disruption of SDHAF4-mediated mitochondrial complex II assembly promotes dilated cardiomyopathy. Nat. Commun. 13 (1), 3947. doi:10.1038/s41467-022-31548-1
Wang, Y. J., Paneni, F., Stein, S., and Matter, C. M. (2021b). Modulating sirtuin Biology and nicotinamide adenine diphosphate metabolism in cardiovascular disease-from bench to bedside. Front. Physiol. 12, 755060. doi:10.3389/fphys.2021.755060
Wei, Y., Zhang, Y. J., Cai, Y., and Xu, M. H. (2015). The role of mitochondria in mTOR-regulated longevity. Biol. Rev. Camb Philos. Soc. 90 (1), 167–181. doi:10.1111/brv.12103
Weil, R., Laplantine, E., Curic, S., and Génin, P. (2018). Role of optineurin in the mitochondrial dysfunction: Potential implications in neurodegenerative diseases and cancer. Front. Immunol. 9, 1243. doi:10.3389/fimmu.2018.01243
Wellen, K. E., Hatzivassiliou, G., Sachdeva, U. M., Bui, T. V., Cross, J. R., and Thompson, C. B. (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324 (5930), 1076–1080. doi:10.1126/science.1164097
White, M. J., McArthur, K., Metcalf, D., Lane, R. M., Cambier, J. C., Herold, M. J., et al. (2014). Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 159 (7), 1549–1562. doi:10.1016/j.cell.2014.11.036
Wiedemann, N., Frazier, A. E., and Pfanner, N. (2004). The protein import machinery of mitochondria. J. Biol. Chem. 279 (15), 14473–14476. doi:10.1074/jbc.R400003200
Williams, M., and Caino, M. C. (2018). Mitochondrial dynamics in type 2 diabetes and cancer. Front. Endocrinol. (Lausanne) 9, 211. doi:10.3389/fendo.2018.00211
Wu, W., Tian, W., Hu, Z., Chen, G., Huang, L., Li, W., et al. (2014). ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 15 (5), 566–575. doi:10.1002/embr.201438501
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 98 (1), 115–124. doi:10.1016/s0092-8674(00)80611-x
Xian, H., Watari, K., Sanchez-Lopez, E., Offenberger, J., Onyuru, J., Sampath, H., et al. (2022). Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 55 (8), 1370–1385.e8. doi:10.1016/j.immuni.2022.06.007
Xiao, Y., Chen, W., Zhong, Z., Ding, L., Bai, H., Chen, H., et al. (2020). Electroacupuncture preconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting mitophagy mediated by the mTORC1-ULK1-FUNDC1 pathway. Biomed. Pharmacother. 127, 110148. doi:10.1016/j.biopha.2020.110148
Xin, Y., Zhang, X., Li, J., Gao, H., Li, J., Li, J., et al. (2021). New insights into the role of mitochondria quality control in ischemic heart disease. Front. Cardiovasc Med. 8, 774619. doi:10.3389/fcvm.2021.774619
Xu, M., Xue, R. Q., Lu, Y., Yong, S. Y., Wu, Q., Cui, Y. L., et al. (2019). Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc Res. 115 (3), 530–545. doi:10.1093/cvr/cvy217
Xu, X., Kobayashi, S., Chen, K., Timm, D., Volden, P., Huang, Y., et al. (2013). Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J. Biol. Chem. 288 (25), 18077–18092. doi:10.1074/jbc.M113.474650
Yakes, F. M., and Van Houten, B. (1997). Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 94 (2), 514–519. doi:10.1073/pnas.94.2.514
Yamada, T., and Nomura, S. (2021). Recent findings related to cardiomyopathy and genetics. Int. J. Mol. Sci. 22 (22), 12522. doi:10.3390/ijms222212522
Yamamoto, T., Byun, J., Zhai, P., Ikeda, Y., Oka, S., and Sadoshima, J. (2014). Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS One 9 (6), e98972. doi:10.1371/journal.pone.0098972
Yamano, K., Kikuchi, R., Kojima, W., Hayashida, R., Koyano, F., Kawawaki, J., et al. (2020). Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J. Cell. Biol. 219 (9), e201912144. doi:10.1083/jcb.201912144
Yamano, K., and Youle, R. J. (2013). PINK1 is degraded through the N-end rule pathway. Autophagy 9 (11), 1758–1769. doi:10.4161/auto.24633
Yan, C., Duanmu, X., Zeng, L., Liu, B., and Song, Z. (2019). Mitochondrial DNA: Distribution, mutations, and elimination. Cells 8 (4), 379. doi:10.3390/cells8040379
Yang, J., Chen, S., Duan, F., Wang, X., Zhang, X., Lian, B., et al. (2022). Mitochondrial cardiomyopathy: Molecular epidemiology, diagnosis, models, and therapeutic management. Cells 11 (21), 3511. doi:10.3390/cells11213511
Yang, J. Y., and Yang, W. Y. (2013). Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 4, 2428. doi:10.1038/ncomms3428
Yang, M., Linn, B. S., Zhang, Y., and Ren, J. (2019). Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (9), 2293–2302. doi:10.1016/j.bbadis.2019.05.007
Yang, X., Wei, J., He, Y., Jing, T., Li, Y., Xiao, Y., et al. (2017). SIRT1 inhibition promotes atherosclerosis through impaired autophagy. Oncotarget 8 (31), 51447–51461. doi:10.18632/oncotarget.17691
Yankovskaya, V., Horsefield, R., Törnroth, S., Luna-Chavez, C., Miyoshi, H., Léger, C., et al. (2003). Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299 (5607), 700–704. doi:10.1126/science.1079605
Yildirim, A. D., Citir, M., Dogan, A. E., Veli, Z., Yildirim, Z., Tufanli, O., et al. (2022). ER stress-induced sphingosine-1-phosphate lyase phosphorylation potentiates the mitochondrial unfolded protein response. J. Lipid Res. 63 (10), 100279. doi:10.1016/j.jlr.2022.100279
Zaffagnini, G., and Martens, S. (2016). Mechanisms of selective autophagy. J. Mol. Biol. 428 (9), 1714–1724. doi:10.1016/j.jmb.2016.02.004
Zak, R., Rabinowitz, M., Rajamanickam, C., Merten, S., and Kwiatkowska-Patzer, B. (1980). Mitochondrial proliferation in cardiac hypertrophy. Basic Res. Cardiol. 75 (1), 171–178. doi:10.1007/bf02001410
Zhang, C., Shang, G., Gui, X., Zhang, X., Bai, X. C., and Chen, Z. J. (2019a). Structural basis of STING binding with and phosphorylation by TBK1. Nature 567 (7748), 394–398. doi:10.1038/s41586-019-1000-2
Zhang, M., Sui, W., Xing, Y., Cheng, J., Cheng, C., Xue, F., et al. (2021a). Angiotensin IV attenuates diabetic cardiomyopathy via suppressing FoxO1-induced excessive autophagy, apoptosis and fibrosis. Theranostics 11 (18), 8624–8639. doi:10.7150/thno.48561
Zhang, N. P., Liu, X. J., Xie, L., Shen, X. Z., and Wu, J. (2019b). Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. 99 (6), 749–763. doi:10.1038/s41374-018-0177-6
Zhang, W., Ren, H., Xu, C., Zhu, C., Wu, H., Liu, D., et al. (2016). Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 5, e21407. doi:10.7554/eLife.21407
Zhang, W., Siraj, S., Zhang, R., and Chen, Q. (2017). Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy 13 (6), 1080–1081. doi:10.1080/15548627.2017.1300224
Zhang, X., McDonald, J. G., Aryal, B., Canfrán-Duque, A., Goldberg, E. L., Araldi, E., et al. (2021b). Desmosterol suppresses macrophage inflammasome activation and protects against vascular inflammation and atherosclerosis. Proc. Natl. Acad. Sci. U. S. A. 118 (47), e2107682118. doi:10.1073/pnas.2107682118
Zhang, Y., Wang, Y., Xu, J., Tian, F., Hu, S., Chen, Y., et al. (2019c). Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal Res. 66 (2), e12542. doi:10.1111/jpi.12542
Zhao, R. Z., Jiang, S., Zhang, L., and Yu, Z. B. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 44 (1), 3–15. doi:10.3892/ijmm.2019.4188
Zhu, L., Zhou, Q., He, L., and Chen, L. (2021a). Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free Radic. Biol. Med. 163, 125–134. doi:10.1016/j.freeradbiomed.2020.12.013
Zhu, P., Wan, K., Yin, M., Hu, P., Que, Y., Zhou, X., et al. (2021b). RIPK3 induces cardiomyocyte necroptosis via inhibition of AMPK-parkin-mitophagy in cardiac remodelling after myocardial infarction. Oxid. Med. Cell. Longev. 2021, 6635955. doi:10.1155/2021/6635955
Zhuang, L., Jia, K., Chen, C., Li, Z., Zhao, J., Hu, J., et al. (2022). DYRK1B-STAT3 drives cardiac hypertrophy and heart failure by impairing mitochondrial bioenergetics. Circulation 145 (11), 829–846. doi:10.1161/circulationaha.121.055727
Zinghirino, F., Pappalardo, X. G., Messina, A., Nicosia, G., De Pinto, V., and Guarino, F. (2021). VDAC genes expression and regulation in mammals. Front. Physiol. 12, 708695. doi:10.3389/fphys.2021.708695
Zong, H., Ren, J. M., Young, L. H., Pypaert, M., Mu, J., Birnbaum, M. J., et al. (2002). AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. U. S. A. 99 (25), 15983–15987. doi:10.1073/pnas.252625599
Zorova, L. D., Popkov, V. A., Plotnikov, E. Y., Silachev, D. N., Pevzner, I. B., Jankauskas, S. S., et al. (2018). Mitochondrial membrane potential. Anal. Biochem. 552, 50–59. doi:10.1016/j.ab.2017.07.009
Keywords: mitochondria, cardiovascular disease, mitochondrial dynamics, mitophagy, mitobiogenesis, ROS
Citation: Atici AE, Crother TR and Noval Rivas M (2023) Mitochondrial quality control in health and cardiovascular diseases. Front. Cell Dev. Biol. 11:1290046. doi: 10.3389/fcell.2023.1290046
Received: 06 September 2023; Accepted: 16 October 2023;
Published: 06 November 2023.
Edited by:
Azhar Ali, National University of Singapore, SingaporeReviewed by:
Andreas Brodehl, Heart and Diabetes Center North Rhine-Westphalia, GermanyCopyright © 2023 Atici, Crother and Noval Rivas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magali Noval Rivas, bWFnYWxpLm5vdmFscml2YXNAY3NtYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.