 Farah Kotaich†
Farah Kotaich† Damien Caillol
Damien Caillol Pascale Bomont
Pascale Bomont
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 18 December 2023
Sec. Cell Growth and Division
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1275155
This article is part of the Research Topic Cytoskeletal Alterations in Aging and Disease View all 9 articles
Neurofilaments (NFs) are the most abundant component of mature neurons, that interconnect with actin and microtubules to form the cytoskeleton. Specifically expressed in the nervous system, NFs present the particularity within the Intermediate Filament family of being formed by four subunits, the neurofilament light (NF-L), medium (NF-M), heavy (NF-H) proteins and α-internexin or peripherin. Here, we review the current knowledge on NF proteins and neurofilaments, from their domain structures and their model of assembly to the dynamics of their transport and degradation along the axon. The formation of the filament and its behaviour are regulated by various determinants, including post-transcriptional (miRNA and RBP proteins) and post-translational (phosphorylation and ubiquitination) modifiers. Altogether, the complex set of modifications enable the neuron to establish a stable but elastic NF array constituting the structural scaffold of the axon, while permitting the local expression of NF proteins and providing the dynamics necessary to fulfil local demands and respond to stimuli and injury. Thus, in addition to their roles in mechano-resistance, radial axonal outgrowth and nerve conduction, NFs control microtubule dynamics, organelle distribution and neurotransmission at the synapse. We discuss how the studies of neurodegenerative diseases with NF aggregation shed light on the biology of NFs. In particular, the NEFL and NEFH genes are mutated in Charcot-Marie-Tooth (CMT) disease, the most common inherited neurological disorder of the peripheral nervous system. The clinical features of the CMT forms (axonal CMT2E, CMT2CC; demyelinating CMT1F; intermediate I-CMT) with symptoms affecting the central nervous system (CNS) will allow us to further investigate the physiological roles of NFs in the brain. Thus, NF-CMT mouse models exhibit various degrees of sensory-motor deficits associated with CNS symptoms. Cellular systems brought findings regarding the dominant effect of NF-L mutants on NF aggregation and transport, although these have been recently challenged. Neurofilament detection without NF-L in recessive CMT is puzzling, calling for a re-examination of the current model in which NF-L is indispensable for NF assembly. Overall, we discuss how the fundamental and translational fields are feeding each-other to increase but also challenge our knowledge of NF biology, and to develop therapeutic avenues for CMT and neurodegenerative diseases with NF aggregation.
Neurofilaments (NFs) belong to a family of proteins called Intermediate Filaments (IFs), that form the cytoskeleton of the cell together with the actin filaments and microtubules (MTs) (Eriksson et al., 2009; Herrmann et al., 2009). The name refers to the 10 nm diameter of the filaments that is categorised as “intermediate” between the 7 nm actin filaments (Grazi et al., 1993) and 25 nm MT (Wade, 2009). IF proteins are encoded by one of the largest gene families present in the human genome that comprises ∼70 genes and phylogenetically derives from a common lamin-like ancestor (Peter and Stick, 2015). Historically, the classification of IFs was divided into 6 major groups according to their amino acid sequences and their structural and physico-chemical properties: types I and II are constituted by acidic and neutral-basic keratin proteins, respectively; type III includes vimentin, desmin, GFAP and peripherin; type IV comprises nestin, synemin, ⍺-internexin and NF proteins; type V is composed of the nuclear lamins and type VI by the lens-specific Bfsp1/Bfsp2 (Eriksson et al., 2009; Omary, 2009). Gene expression of IFs has been shown to be tissue-specific (Omary, 2009) to provide a customizable cytoplasmic meshwork adapted to various tissues and cell types. Thus, mutations in IF genes have been shown to cause a wide range of >80 tissue-specific diseases, from skin diseases (keratin), myopathies (desmin), cataract (Bfsp) to neuropathies (NFs, GFAP) (Omary, 2009). The crucial roles of IFs in physiology is further supported by the discovery of a wide aggregation of IF proteins in numerous pathologies, including desmin bodies in myopathies and NF accumulation in most neurodegenerative diseases (Omary, 2009). Within the IF family, NFs are selectively expressed in the nervous system, where they constitute the most abundant cytoskeletal component of mature neurons and represent as much as 15% of the total protein content of peripheral nerves (Yuan et al., 2017). NFs form a backbone structure composed of three subunits - the neurofilament light protein (NF-L), the neurofilament medium protein (NF-M) and the neurofilament heavy protein (NF-H). This structure can further incorporate ⍺-internexin in the central nervous system (CNS) and peripherin in the peripheral nervous system (Liem and Messing, 2009). The manipulation of NF stoichiometry in mice has revealed the fundamental role of NFs in the development, maintenance, survival and regeneration of the nervous system (Lariviere and Julien, 2004; Liem and Messing, 2009). In the biomedical field, NFs have received much attention as their aggregation constitutes a pathological hallmark for most neurodegenerative diseases (Liem and Messing, 2009; Perrot and Eyer, 2009; Didonna and Opal, 2019) and they represent a universal biomarker for neurodegeneration (Gafson et al., 2020; Bomont, 2021), as a result of their detection in cerebrospinal fluid and blood. While the exact contribution of NF aggregation in disease still remains unclear, the discovery of disease-causing mutations in genes controlling the dynamics of NFs (GAN, TRIM2, KIF5A, SACS, HSPB1) further evidence their importance for neuronal homeostasis and functions (Bomont, 2021). More directly, the NEFL and NEFH genes have been shown to be mutated in several forms of the Charcot-Marie-Tooth (CMT) disease (Laurá et al., 2019), the most common inherited neurological disorder of the peripheral nervous system.
This review aims to present the achievements of both fields of fundamental research and study of diseases and to discuss how these findings have enriched our knowledge of NF biology and roles in physiology. First, we will present NFs, from their assembly to their dynamics and roles in neuronal physiology. Secondly, we will focus on diseases to detail the genetics of NF-CMTs, the cellular and physiological effects of NF mutants in vitro and in animal models, to reach conclusions on the ongoing therapeutic perspectives towards NFs.
Like all members of the IF family, NFs have a tripartite structural organisation (Figure 1A). A central conserved segment consisting of an ⍺-helical rod domain is flanked by a short N-terminal segment named the head domain and a C-terminal segment of variable size named the tail domain (Herrmann and Aebi, 2016). In the IF family, NFs have the specificity of being heteropolymers formed by four proteins (Yuan et al., 2017): the NF triplet constituted by the NF-L (for Light), NF-M (for Medium) and NF-H (for Heavy) proteins, and a fourth subunit α-internexin or peripherin that is specifically expressed within the central and peripheral nervous system, respectively (Yuan et al., 2006; 2012). The rod domain contains long stretches of hydrophobic heptad repeats and promotes the assembly of the filament through the formation of ⍺-helical coiled-coil dimers (Herrmann and Aebi, 2016). Both extremities of the NF proteins can be subjected to various post-translational modifications to modulate the stability and organisation of the filament, in particular the long C-terminal domains of NF-M and NF-H which are highly phosphorylated.
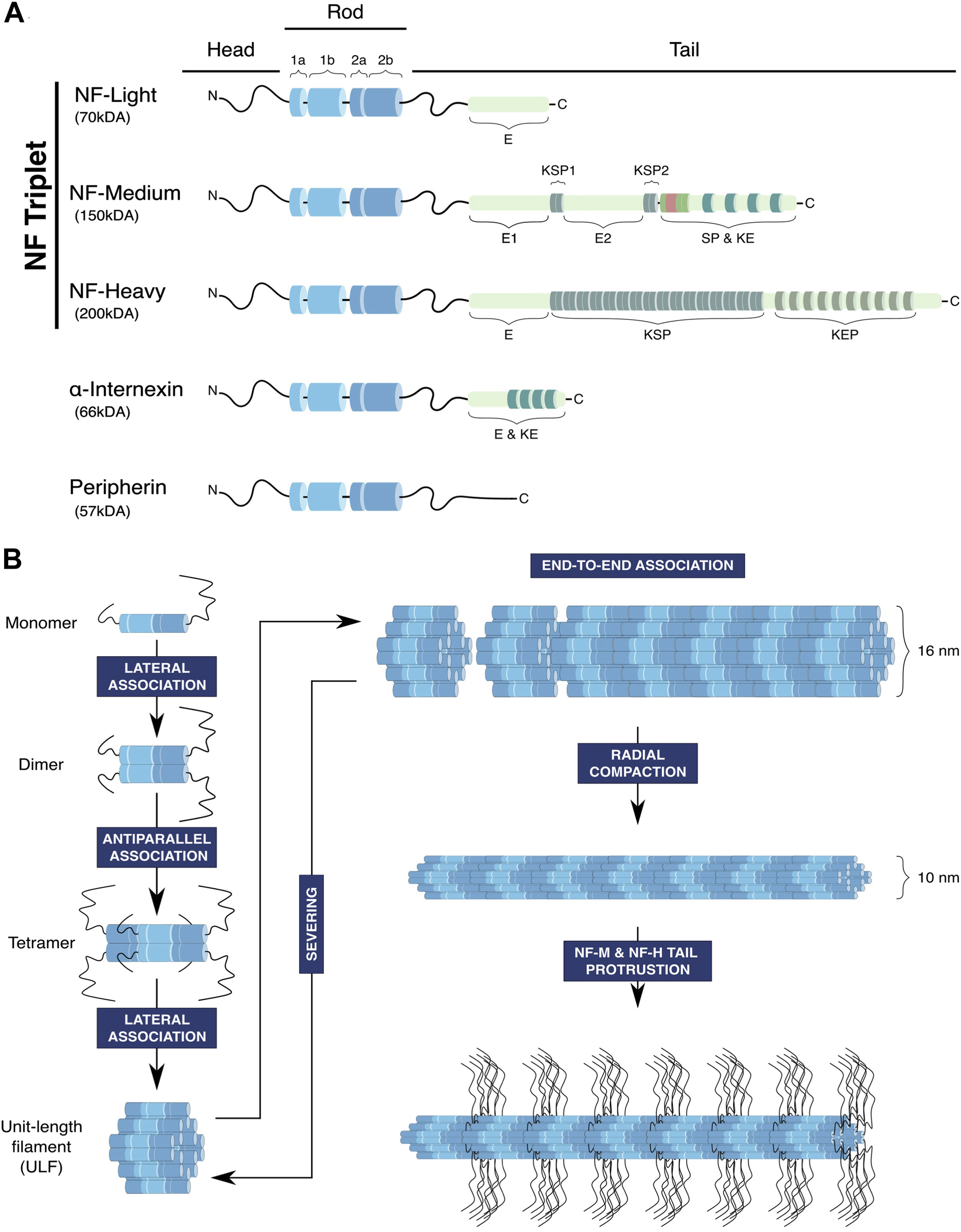
FIGURE 1. Structure and assembly of neurofilaments. (A) Neurofilaments are composed of the core NF triplet: NF-Light (NF-L), NF-Medium (NF-M) and NF-Heavy (NF-H) subunits, which integrates a fourth subunit, peripherin in the peripheral nervous system and ⍺-internexin in the central nervous system. NF subunits have a tripartite structure composed of a head domain, a conserved rod domain containing α-helical coils ensuring coiled-coil interactions necessary for NF assembly and a tail domain of variable length that is subjected to post-translational modifications, in particular phosphorylation of lysine-serin-proline repeats (KSP). (E: glutamic-acid-rich segment, KE: lysine-glutamic acid, KEP: lysine-glutamic acid-proline, SP: serine-proline, KSP: lysine-serine-proline). (B) NFs follow an assembly process common to other IFs, starting with a sequence of successive association of polypeptides to form the basic unit of NFs, the Unit-Length Filament (ULF). Firstly, the rod domains of NF monomers interact to form parallel coiled-coil heterodimers. Secondly, dimers associate and generate antiparallel tetramers, which consequently lose their polarity. Then, the lateral association of eight tetramers forms a cylindrical structure called the unit-length filament (ULF), which can expand through end-to-end association (annealing) to generate the neurofilament. In principle, fragmentation may occur through the severing of the filament, but whether this mechanism acts on the immature 16 nm filament is not determined. Finally, the filament matures through radial compaction to reach a diameter of 10 nm, and phosphorylation of the long tails of NF-M and NF-H induces their projection outward from the filament.
With their specific composition, NFs are considered to assemble through a sequential mode similarly to other IF filaments (Figure 1B). The first step begins with the association of monomers to form parallel coiled-coil dimers though the central rod domain. Then, dimers associate in antiparallel tetramers, hence generating non-polar polymers. The subsequent step involves the lateral association of eight tetramers to form a ring structure called the Unit-Length Filaments (ULFs), which measures 16 nm in diameter (Herrmann et al., 1996). The ULF is thought to constitute the basic unit of NFs, whose longitudinal association through end-to-end annealing (and shortening through severing) will define the length of the filament (Colakoğlu and Brown, 2009; Uchida et al., 2013). Finally, NFs are subjected to radial compaction followed by the protrusion of the tail domains of the NF-M and NF-H subunits promoted by their phosphorylation, generating mature NFs of 10 nm in diameter able to interact with other components of the cytoskeleton and organelles. Indeed, ultrastructural studies have revealed that the regular projections radiating outward from the filament core (Hisanaga and Hirokawa, 1988) form cross-bridges between NFs, organelles and other cytoskeletal elements (Hirokawa, 1982).
Regarding the mode of assembly of the filament, in vitro studies revealed that NF assembly does not require nucleotide binding or hydrolysis but depends on the physicochemical properties of the subunits and the environment (temperature, pH, ions … ). While this supports the idea that the intrinsic nature of NF polypeptides can drive assembly, this does not exclude that cofactors can regulate their assembly in vivo (Chernyatina et al., 2015; Herrmann and Aebi, 2016). As examples, Sacsin (mutated in the ataxia ARSACs presenting NF aggregation), NUDEL and the 14-3-3 protein have been shown to bind to NF-L and regulate the assembly/disassembly state of NFs (Nguyen et al., 2004; Miao et al., 2013; Dabbaghizadeh et al., 2022). Investigation of the intrinsic capacities of single NF subunits to polymerise revealed that only NF-L, peripherin and α-internexin are able to form homopolymeric filaments in vitro (Ching and Liem, 1993; Cui et al., 1995; Carter et al., 1998). Thus, NF-M and NF-H incorporation require the presence of NF-L, supporting the notion that NF-L (together with peripherin or α-internexin) constitutes the backbone of the filament, exposing NF-M and NF-H (and their long tail domains) at the periphery. The assembly model would be that NF-L copolymerises with NF-M and NF-H, thus forming NF-L/NF-M and NF-L/NF-H hetero-tetramers, which are then able to associate together through NF-L to form the ULFs (Yuan et al., 2006; 2012).
NFs are the most abundant cytoskeletal component of mature neurons and the expression of individual NF subunits follows a temporal sequence synchronised with neuronal development. There are discrepancies between different neuronal types, localisation in the brain and organism species, and consequently the exact expression profile of the different NF proteins is not fully established. Still, a general picture proposes that immature neurons do not express NF proteins but other IF types, such as vimentin, nestin and synemin (Bott and Winckler, 2020; Bomont, 2021). Upon neuronal differentiation, the expression of these IF types is downregulated and progressively replaced by NF proteins in a sequential manner (Figure 2). Thus, NF-L is first expressed (together with peripherin or α-internexin) upon neuronal differentiation, followed by NF-M during axonal elongation, and NF-H concomitantly to radial outgrowth and myelination (Kirkcaldie and Dwyer, 2017; Bomont, 2021). The subsequent phosphorylation of the NF-H and NF-M tails triggers the formation of cross-bridges between NF polymers, membranes and other cytoskeletal components, therefore stabilising the network and negatively regulating axonal elongation. The change in IF expression during neuronal maturation demonstrates the crucial role of NFs in neuronal maturation and physiology. Still, the concomitant and residual expression of several IF types in mature neurons suggests a cooperation of these different elements in sustaining neuronal functions, such as the presence of nestin in the distal region of growing axons (Bott and Winckler, 2020).
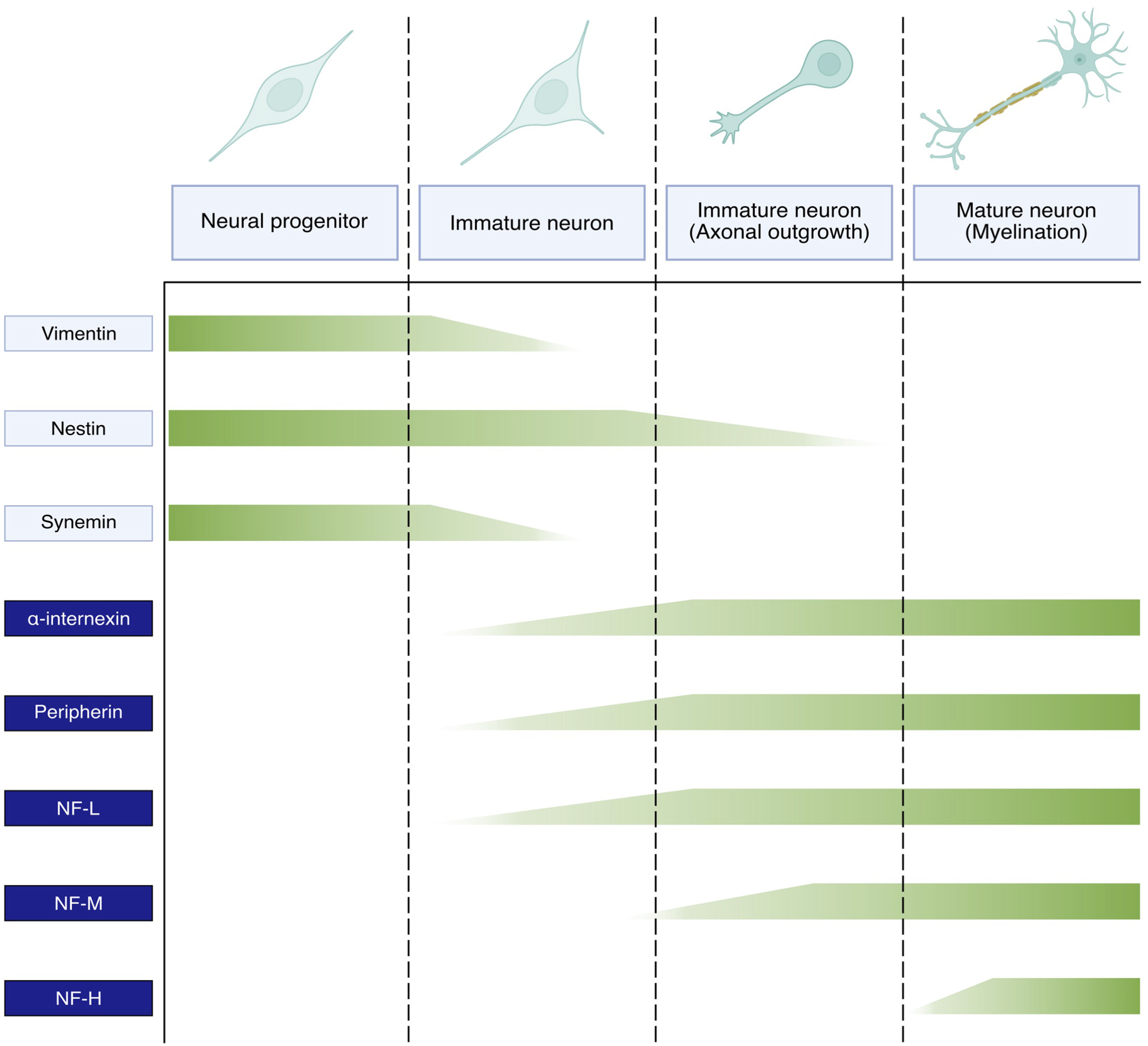
FIGURE 2. Expression pattern of neuronal intermediate filaments during neuronal maturation. Vimentin, nestin, synemin present in the ectodermal cells committed to neuronal differentiation are gradually downregulated in favour of NF subunits in immature neurons. The expression of NF proteins is sequential and coupled with neuronal maturation. Thus, NF-L alongside either ⍺-internexin in the CNS or peripherin in the PNS initially appear in immature neurons, followed by NF-M during axonal outgrowth and NF-H during axon myelination and radial outgrowth.
The ability of the cell to control gene expression and its proteome is essential for sustaining cell viability and physiological functions. These regulations can occur at the genomic, transcriptomic and protein levels. For NFs, both post-transcriptional and post-translational modifications (PTMs) have been identified.
Post-transcriptional regulations are RNA-mediated mechanisms that control many aspects of RNA life, including biogenesis, alternative splicing, stability, transport and translation efficiency. For NFs, post-transcriptional modifications have been found to be triggered by microRNAs (miRNAs) and RNA-binding proteins (RBPs) (Yuan and Nixon, 2023). Collectively, these regulatory mechanisms ensure a spatio-temporal expression of isoforms in specific neuronal compartments and are therefore essential for neuronal physiology. While miRNAs represent a common mechanism that is deregulated in neurodegenerative diseases (Juźwik et al., 2019), increasing evidence shows the direct role of RNA-mediated mechanisms in the pathophysiology of various neurodegenerative diseases, with dozens of RBP identified as causal genes for amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) alone (Yuan and Nixon, 2023). Regarding NF mRNAs, the conserved 3′UTR seems to serve as a hub for modifications, as many RBPs (TDP43 (Strong et al., 2007), 14-3–3 (Miao et al., 2013), Aldolase A/C (Cañete-Soler et al., 2005), FUS (Lagier-Tourenne et al., 2012) bind to this region to either stabilise, destabilise or modulate the mRNA of NF-L, NF-M and NF-H. MiRNAs (miR-b1336 (Ishtiaq et al., 2014), MiR-146a (Campos-Melo et al., 2013), miR-233-3p (Campos-Melo et al., 2018), whose biogenesis can be controlled by RBPs, also bind to the 3′UTR regions of the NF mRNAs. Other RBPs (hnRNP K (Thyagarajan and Szaro, 2004; 2008), ELAVL2 (Antic et al., 1999)) are capable of modulating mRNA transport and translation, hence directing the local synthesis of NF proteins in neuronal sub-compartments. Less is known for α-internexin and peripherin, with the RBP hnRNPK modulating only α-internexin mRNA translation. Overall, studies on post-transcriptional modifications revealed that protein synthesis does not occur exclusively in the soma, but that NF mRNAs can be protected/destabilised along their journey, delivered and translated in different compartments, therefore providing the neuron with a high level of dynamics in fulfilling local demands and responding to stimuli.
Post-translational modifications (PTM) of proteins play an important role in physiology by modulating various properties including protein activity, interaction, subcellular localisation, stability and turn-over. Proteomic analyses have identified or predicted specific sites on NF proteins that can be modified by PTMs (MacTaggart and Kashina, 2021). NFs can be the targets of various PTMs, including glycosylation, acetylation, SUMOylation, methylation, but the most studied are phosphorylation and to a lesser extent ubiquitination (Figure 3).
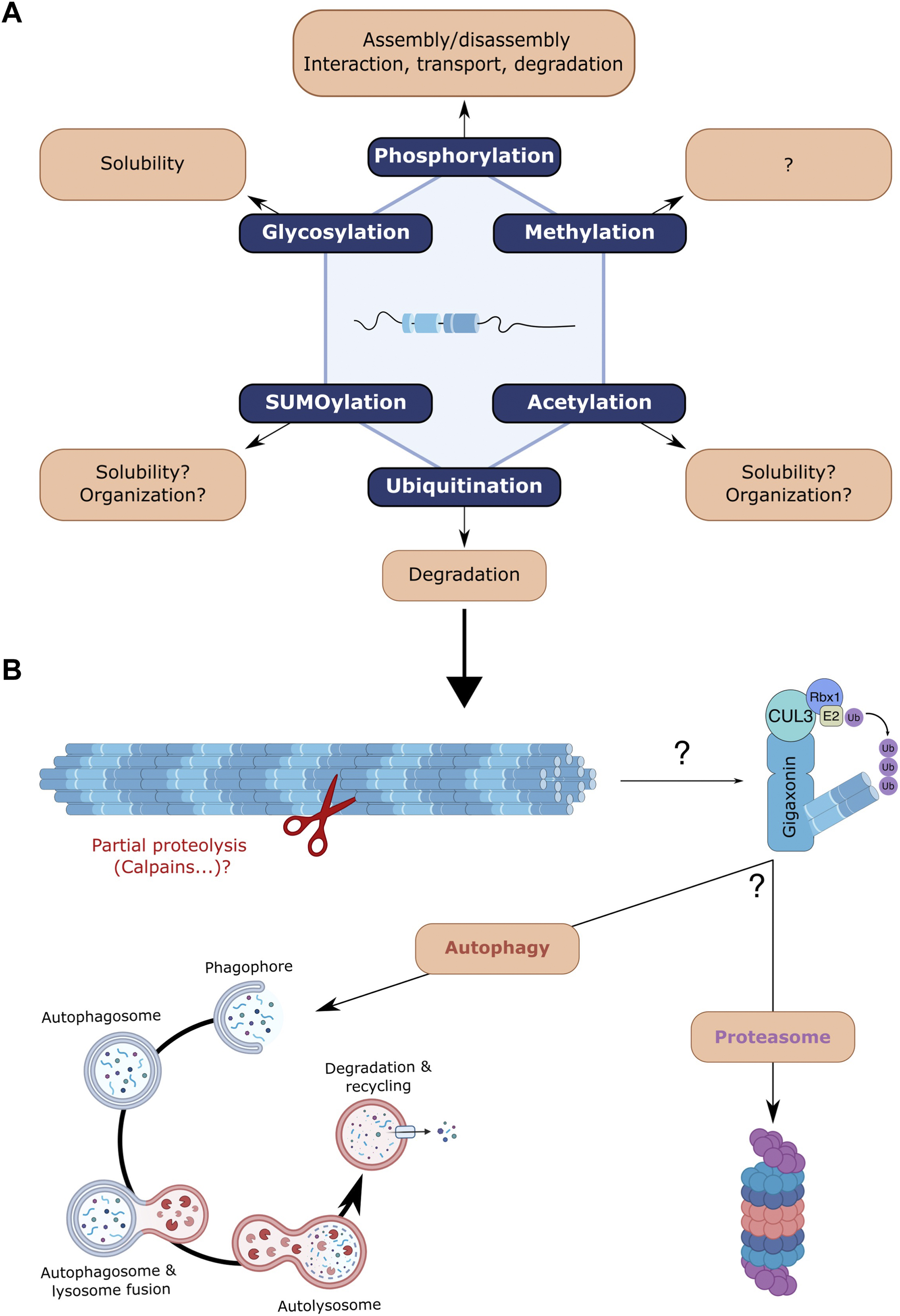
FIGURE 3. Post-translational modifications of neurofilaments and focus on ubiquitin-dependent degradation. (A) NFs undergo various post-translational modifications (PMTs) that can regulate their behaviour: methylation, acetylation, SUMOylation, glycosylation, phosphorylation and ubiquitination. Very little is known regarding the effects of most NF-specific PTMs but findings from other IFs may suggest a role in regulating filament solubility and organisation (acetylation, SUMOylation). Glycosylation was also shown to modulate the solubility of NFs. Phosphorylation, the most studied PTM has revealed various roles, including filament assembly/disassembly, interaction with NFs and other cytoskeletal components, NF transport and protection from proteolytic degradation. (B) Focus on the ubiquitination and proteolysis of NFs. While the Trim2-E3 ligase was shown to ubiquitinate NF-L, gigaxonin (which at least induces peripherin ubiquitination) is the only E3 ligase capable of degrading all NF subunits. Whether the gigaxonin-mediated clearance is dependent on the proteasome and/or autophagy pathways remains to be determined, but autophagy and proteasome have been identified as degradative routes for NF proteins in vivo. Various unsolved questions concern the early requirement of partial proteolysis by proteases, the NF forms that are substrates for proteolysis, the cooperation of both degradative routes and the spatio-temporal signalling that activates and regulates them.
The three polypeptides (NF-L, NF-M and NF-H) were the first IFs identified as being subjected to phosphorylation (Pant et al., 1978) (Figure 3A). The level of phosphorylation of NF-M and NF-H is found to be particularly high when compared to the basal phosphorylation level of other IFs. This is related to their particularly long tail domains rich in lysine-serine-proline (KSP) motifs. All NF subunits can be phosphorylated in their head domain, but only NF-M and NF-H are highly phosphorylated in their tail domains (Dale and Garcia, 2012; Holmgren et al., 2012). Interestingly, this phosphorylation in different portions of NF proteins is modulated in distinct subcellular compartments. Thus, the head domain is more phosphorylated in the soma, while the tail domain is more phosphorylated in the axons (Sihag et al., 2007). This implies a tight spatio-temporal regulation of kinase/phosphatase during development, when newly synthetised NF proteins in the cell body enter the axons. Phosphorylation in the head of NF subunits (mediated by PKA, PKC and CAMK II, cdk5 … ) has been mainly shown to regulate the assembly-disassembly of the filament, but it is also exerting negative control of tail domain phosphorylation, presumably to avoid abnormal stabilisation and aggregation of NFs in the soma (Yuan et al., 2017). NF tail domain phosphorylation (mediated by CDKs, MAPKs … ) occurs mostly at the KSP sites and preferentially upon their entry into the axons. Interestingly, the degree of phosphorylation increases as the NFs progress distally into the axons, therefore contributing to the maturation of the filament and the interaction with other cytoskeleton components. Accordingly, myelination and synaptogenesis regulate the phosphorylation of NF tail domains, inducing NF spacing and promoting axonal radial outgrowth (Lariviere and Julien, 2004; Holmgren et al., 2012).
Collectively, NF phosphorylation plays a crucial role in the formation, maintenance and remodelling of the NF network (this will be expanded in the following sections), and alterations of this balance are associated with the settings of neurodegenerative diseases (Dale and Garcia, 2012; Binukumar et al., 2013).
Ubiquitination is a PTM involved in the turn-over of NF proteins (Figure 3B). With an estimated half-life of >8 months in sciatic nerves, the NF array was initially shown to be extremely stable in vivo. Surprisingly, genetic alterations of NFs evidenced a certain dynamic in the NF array. Thus, NFs were found to be degraded in a synchronous manner along the nerve and much faster (estimated 3 weeks half-life) in the absence of a preexisting NF array (Millecamps et al., 2007). Interestingly, the tail domains of the NF-M and NF-H proteins are critical as they protect the filament in vivo from proteolytic degradation (Rao et al., 2012). Also, NFs, when incorporated in the array, exhibit a slower decay than the surrounding less-phosphorylated NF particles (Boumil et al., 2018). Therefore, the NF array itself, whose density varies between neuronal subtypes and along individual axons, is a key determinant of NF stability within neurons. The mechanisms leading to the degradation of the NF array are not fully elucidated but the gigaxonin-E3 ubiquitin ligase (Bomont et al., 2000) has been found to be a universal regulator of IF steady state (Bomont, 2016). While the absence of gigaxonin in patients suffering from a neurodegenerative disease called GAN induces an abnormal accumulation of several IF types (Lescouzères and Bomont, 2020), its overexpression has been shown to induce the clearance of several IF types, including the NF-L, NF-M, NF-M, peripherin and α-internexin (Israeli et al., 2016). Extremely difficult to evidence, ubiquitination has so far only been revealed for peripherin (Israeli et al., 2016). Another E3 ligase (Trim2) has been shown to ubiquitinate NF-L (Balastik et al., 2008), although it does not regulate NF abundance (Li et al., 2020). The generalised action of the gigaxonin relies on its interaction with the central rod domain, which is conserved between all IF types (Mahammad et al., 2013). Many areas require further investigation, such as the forms that are degraded (filament, ULF, tetramers), the prerequisite action of calpain, the calcium-activated protease necessary for NF degradation in Wallerian degeneration (Glass et al., 1994; Park et al., 2013a; 2013b; Ma et al., 2013). Regarding NF proteins, very little is known about the implication of the proteasome but recent evidence points towards the autophagy pathway. Firstly, the gigaxonin-E3 ligase was shown to regulate the flux of the autophagy pathway (Scrivo et al., 2019), by promoting the elongation of the autophagosomes (Bomont, 2019; Lescouzères and Bomont, 2020). Secondly, the abundance of the NF triplet (NF-L, NF-M and NF-H) has been shown to be regulated by autophagy activity in vitro, to a greater extent than the proteasome (Rao et al., 2023). In mouse brains, the levels of NF-M and NF-H (but not NF-L) could be increased by < 2 fold upon inhibition of the autophagic flux, while proteasome inhibition has less effect and only on NF-M. Furthermore, NF particles have been detected in the lumen of double-membrane autophagosomes but with no filamentous structure, suggesting that shorter processed NFs (oligomers, ULF, monomers … ) are addressed to the autophagic pathway (Rao et al., 2023). Altogether, these findings reveal that NF proteins are substrates for both the proteasome and the autophagy pathways, with a specificity towards subunits that remains to be scrutinised. In the future, it would be important to determine the spatio-temporal variations of intrinsic (structure, PTM of subunit, assembly state … ) and extrinsic (composition of degradative machineries, cooperation between systems, inducers … ) properties of these crucial regulations sustaining NF steady state.
In contrast to the microtubule and actin networks, which are remodelled by rapid depolymerisation and polymerisation waves, NFs have long been considered as an extremely stable and non-dynamic network. Still, NF particles were found to incorporate and leave the NF array (Nixon and Logvinenko, 1986; Lewis and Nixon, 1988). The apolar structure of NFs represents a major hurdle to investigate the dynamic aspects of the network, but the development of methodologies adapted to IFs has allowed us to examine the behaviour of NFs.
As discussed earlier, NF subunits exhibit a sequential expression pattern accompanying key stages of neuronal maturation (Figure 2). Synthetised in the cell body, NF proteins are then transported to the axonal compartment (Yuan et al., 2009). Over the last decades, two critical aspects of NF maturation have been the subject of intense debate, with conflicting findings obtained as a result of the different model systems and methodologies used to visualise the NF array: the identity of the NF structures transported along the axon and the characteristics of transport (Figure 4). In both cases, the emergence of optical methodologies adapted to IFs and the manipulation of individual subunits permitted to evidence that multiple assembly forms of NFs are transported along the axon, and that the apparent slow axonal transport of NFs is driven by a net fast movement but with intermittent prolonged pausing.
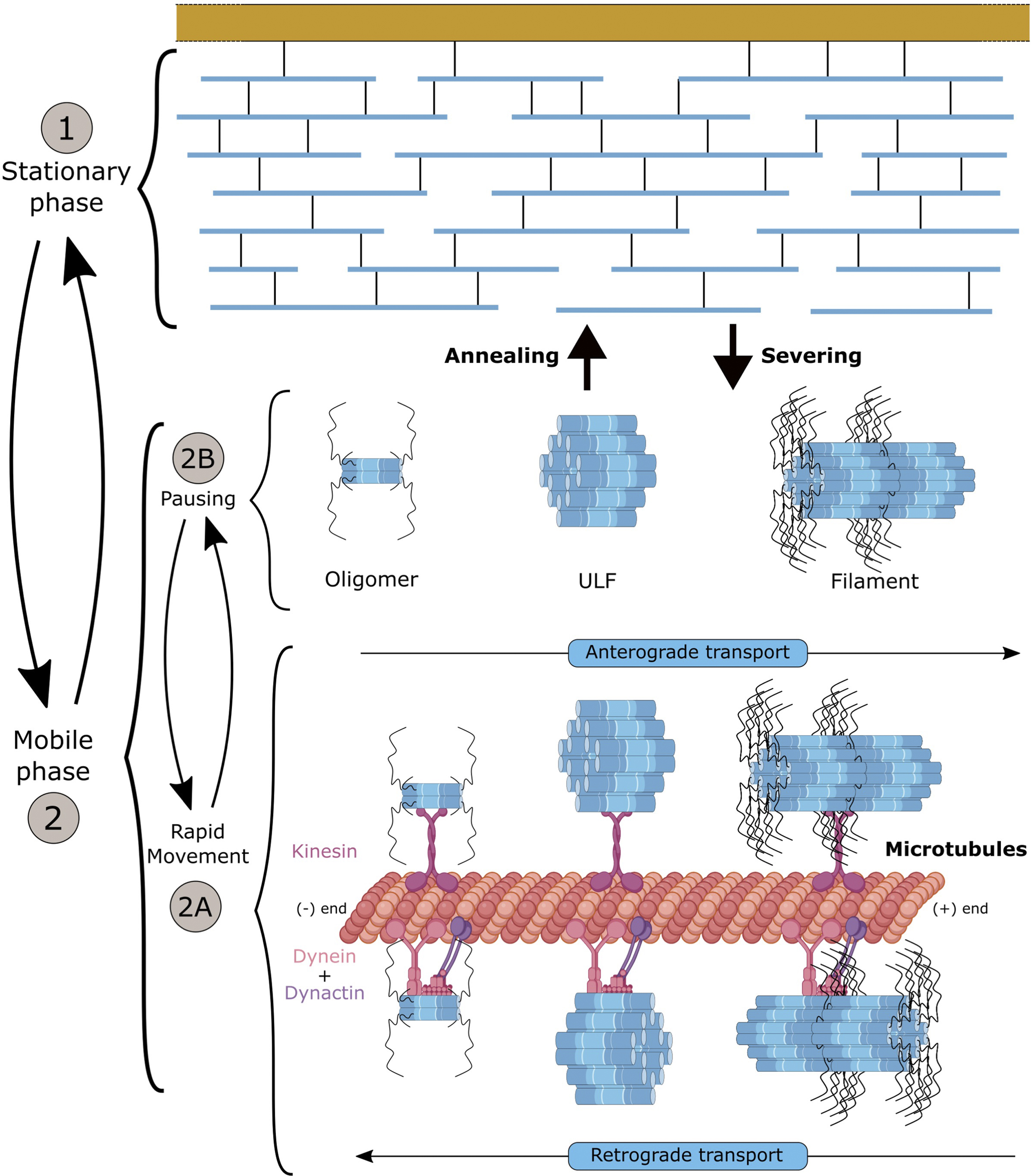
FIGURE 4. Axonal transport of neurofilaments and formation of the network. NFs can be distinguished into two phases: (1) the stationary phase and (2) the mobile phase into which NFs can enter cyclically. The mobile phase appears as a “slow” transport but results from bidirectional and net “fast” movements powered by the dynein and kinesin motors onto microtubules (2A), interrupted by short pauses (2B). Distinct NF structures (heterooligomers, ULF or short filament) that coexist in the same axon can be further incorporated into the stationary network for prolonged pausing. Exchange of NF portions has been shown to occur along the filament through annealing and severing. Overall, NFs spend 97% of their time in an immobile phase (short and long pauses), but it is assumed that NFs can cycle between the different phases, to provide to the abundant NF network the dynamics necessary to adjust NF content and respond to developmental stimuli and insults. The mechanisms controlling the switch from movement to pausing (2A-2B) and integration into the filament (1) are not fully understood, but they may involve many factors (detachment of NFs from molecular motors, access to MTs, etc.). Likewise, the structure of the NFs, and the regulatory elements enabling local annealing and severing without compromising the entire filament are not known.
It is worth noting that the outcomes obtained in studies performed in vitro and in vivo should take into account the great variability of NF content between both systems and may not be comparable. Thus, adult mice axons express NF levels that can exceed 100–1,000 folds of what is present in embryonic primary neurons.
Regarding the first aspect, NFs were shown to be transported either in the form of non-filamentous NF structures, oligomers or short polymers (Yuan et al., 2017). Interestingly, these forms can co-exist in the same axon, with a spatial localisation that would suggest an assembly into filament as the NF structures are transported along the axon (Yuan et al., 2009). Additional findings in vivo showed that the formation of a mature NF network is not a prerequisite for the transports of NF forms (Millecamps et al., 2007), and that NF content and variation along the axon does not regulate the rate of NF transport (Rao et al., 2012; Yuan et al., 2015a), even-though NF removal from axons induces a faster NF transport rate (Millecamps et al., 2007). The life cycle of NFs was originally proposed to begin by expression of NF subunits in the soma, followed by the transport of NF structures along the axon and network formation, and finally the degradation of the entire pool of NFs as they progress through the synaptic terminals. It is now shown that as in degradation, NF transport can occur all along the axon, but only a small population of NF structures is transported at any given time, while the majority of the array is stationary (Figure 4). This would enable the neuron to maintain an abundant NF network at a minimal energy cost along the entire length of the long axon, while locally adjusting NF content in response to myelination and various environmental signals.
Initial studies performed in vivo using radioisotope pulse labelling established that NFs move in the slow compartment (0.25 mm/day) (Black and Lasek, 1980), but in two distinct pools with one being in continuous motion and the second being more stationary (Nixon and Logvinenko, 1986). In vitro studies in neuron types presenting a scarcity of NFs, combined with the development of live-cell imaging (photo-bleaching/activating/conversion) and computational methodologies enabled to scrutinise the characteristics of NF transport. Collectively, in cellular system, the global slow transport of NFs was shown to result of a net fast transport (50–400 mm/day) that is interrupted by prolonged pauses (Roy et al., 2000; Wang et al., 2000). With an average velocity of 1 ym s-1 (Fenn et al., 2018), NF transport is bidirectional and powered by molecular motors. More specifically, interactions of specific NF subunits with MT-based and actin filament-based motors were identified: KIF5A with NF-M and NF-H (Yabe et al., 2000; Jung et al., 2005), dynein with NF-M (Wagner et al., 2004) and myosinVa with NF-L (Rao et al., 2002a). Consistent with the abnormal accumulation of NFs in the cell body in KIF5A knockout mice (Xia et al., 2003) and altered NF transport in KIF5A KO neurons (Uchida et al., 2009), overexpression of KIF5A mutants (as identified in the SPG10 disease, a form of human hereditary spastic paraplegia) evidenced a defect in the frequency of NF transport along the axon (Wang and Brown, 2010). Disrupting dynein was shown to reduce the (retrograde) transport of NFs (He et al., 2005; Uchida et al., 2009). Myosin-depleted neurons revealed a prolonged stay of NFs in the stationary network, suggesting a role in engaging NFs onto the MT network (Alami et al., 2009). While interactions with the molecular motors and their cooperativity in promoting the bidirectional motility of different forms of NF structures are not fully characterised, a general model for NF transport has been proposed. In the current model (Figure 4), the mobile phase is characterised by the bidirectional transport of various NF forms along MTs, with a fast net transport powered by molecular motors that is intermittent and interrupted by short pauses (estimated at 30 s). NFs can switch to a stationary phase, in which NFs undergo prolonged pauses (estimated at 60 min), presumably as a result of their incorporation into the stable interconnected filament. Overall, NFs pause 97% of the time, which would explain their global slow transport of 0.5 mm/day (Trivedi et al., 2007). While many aspects of this model remain to be investigated, the model puts forward the argument that the short pausing behaviour may be due to the detachment of molecular motors from NFs, and that the incorporation into the stationary network may result from the unavailability or prevention of reattachment to motors or weak MT density (Shea, 2000). This model has been essentially built in cellular systems conveniently presenting a scarcity of NFs. Therefore, it may present different characteristic in vivo, in nerves presenting significantly higher NF density.
The second subject of debate concerns the capacity of all NFs, including those present in the stationary network, to switch to a mobile state. The first view, based on computational analysis, proposes a single NF population model in which transported NFs form the entire NF content of the axon (Brown and Jung, 2013), that would suggest a high dynamic of the network and imply weak and reversible interactions between filaments. Accordingly, in vivo assessment of NF motility in large myelinated axons revealed a bidirectional spread from the photoactivated area (Boyer et al., 2022) that involved >80% of the network, following 3 h of activation (Fenn et al., 2023). The second view, supported by various anterior in vivo studies, proposes a two-NF population model in which transported NFs represent a small fraction maintaining a long-lived stationary network (Yuan et al., 2015a). In this situation, the local variation of NF content along the axon would not be determined by NF transport, but instead by the rate of incorporation of NFs into the filament, and by the half-life of the stationary NFs. The presence of the stationary NF network would permit the neuron to maintain axonal integrity at low energy cost while permitting local NF density regulation in response to various stimuli. Solving the issue of the population undergoing motility in axons is important but complex, and discrepancies cannot simply be attributed to the biological system, as respective studies have been performed in vivo.
Regarding the possible mode of integration of moving NF particles into the preexisting filaments, in vitro studies revealed that subunit exchange and severing occur along the length of the filament (Colakoğlu and Brown, 2009; Uchida et al., 2013). Presumably, the filament would have a topologically open architecture enabling portions of the filament to dissociate or associate without compromising the integrity of the network. While little is known regarding the pathways that control NF behaviour, myelination (Monsma et al., 2014, 201; Walker et al., 2019) and phosphorylation of the NF-H (or NF-M) tail domain (Jung et al., 2000a; 2000b; Ackerley et al., 2003) that are argued to detach NFs from motors (Shea and Flanagan, 2001; Holmgren et al., 2013) have been identified as negative regulators of NF motility. Surprisingly, depletion of the heavily phosphorylated tail of either NF-H (Rao et al., 2002a; Yuan et al., 2006), NF-M (Rao et al., 2003) or both (Rao et al., 2012) does not alter NF transport in optic nerve axons. While this does not exclude a pathological role attributed to tail phosphorylation in modulating NF transport (i.e., hyperphosphorylation), these findings may suggest relevant phosphorylation sites outside long tail domains, as suggested by the partial NF transport defect upon phosphorylation manipulation in the NF-L head domain (Yates et al., 2009). Equally puzzling is the finding that the gigaxonin-E3 ligase, responsible for the degradation of IFs was recently shown to modulate NF transport in dorsal root ganglion (DRG) sensory neurons (Renganathan et al., 2023).
Gene manipulations in mice, along with the identification of causal genes in neurodegenerative diseases, demonstrated the importance of NFs in neuronal physiology and shed light into to the molecular mechanisms sustaining various functions (Figure 5) (Yuan et al., 2017; Bomont, 2021).
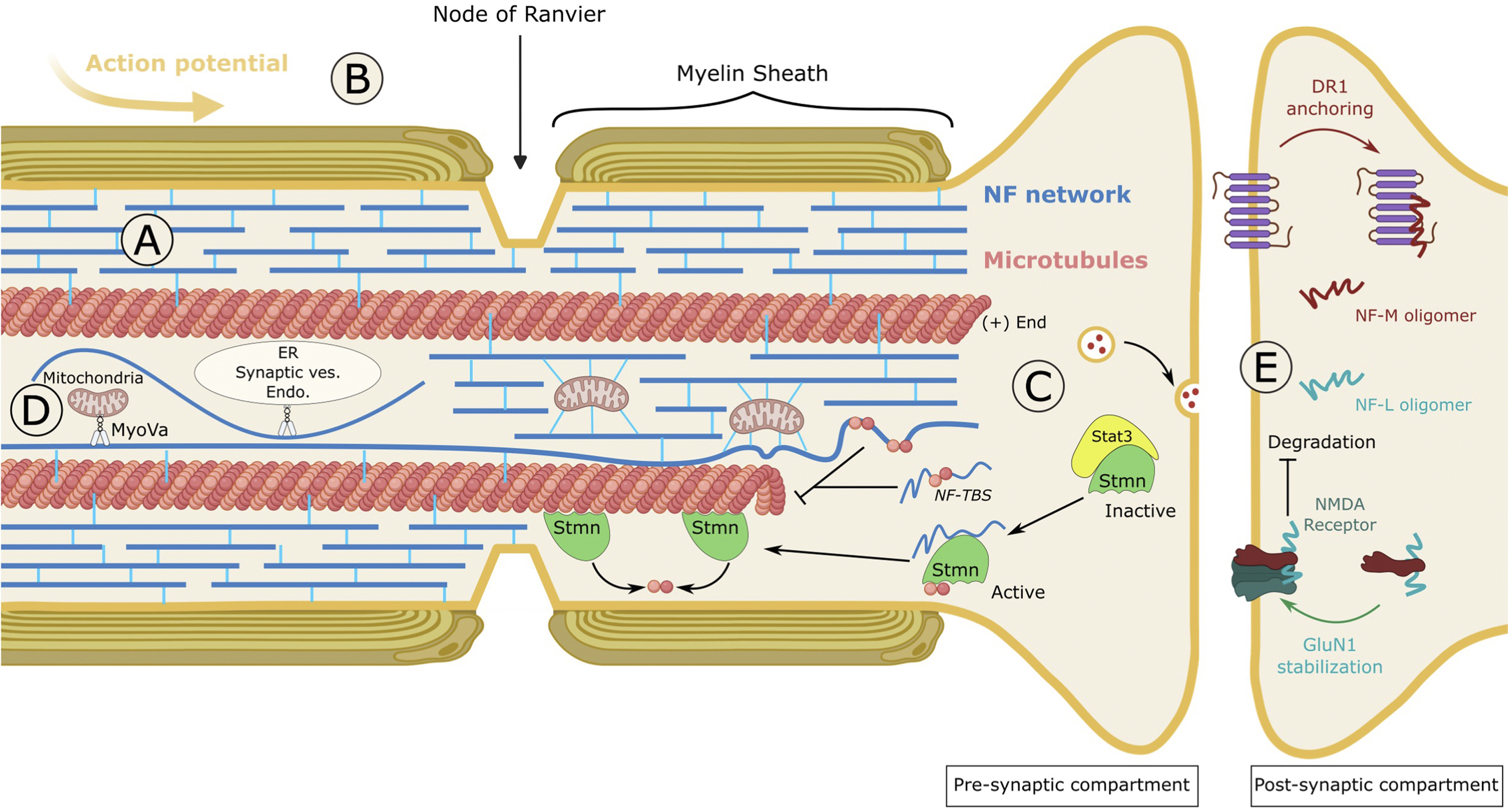
FIGURE 5. Cellular and physiological roles of neurofilaments. (A) Structural support of the axon. NFs form an abundant interconnected network that exhibits exceptional viscoelastic properties. Resisting an average stretching of 260%, NFs confer a mechano-resistance to long axons. (B) Axon diameter and nerve conduction. NF density and spacing within the axon are key determinants of radial axonal outgrowth, myelination and conductivity. Large, myelinated axons are filled with interspaced NFs. Many conditions decreasing the axonal content of NFs or the protrusion of their tails alter axonal caliber and nerve conduction. One puzzling finding is the greater role of the NF-M tail, which is shorter than the NF-H tail. (C) Regulator of microtubule dynamics. NFs act as a negative regulator of MT dynamics through two mechanisms. On the one hand, a tubulin-binding site (TBS), present in the head domain of NF proteins binds to and sequesters unassembled tubulin, therefore inhibiting MT polymerisation. On the other hand, NFs disrupt the inactive Stat3-Stmn complex and release Stmn which is a microtubule-destabilising factor. In conditions where MTs are destabilised, NF depletion can promote the formation of Stat3-Stmn complex, hence stimulating the regrowth of MTs. (D) Distribution of organelles. Most abundant scaffold of the cell, NFs play a role in the positioning of organelles. The direct interaction of NFs with mitochondria anchors them to the network, hence decreasing their motility on MTs. Alternatively, the binding of NFs to the actin-molecular motor Myosin Va (MyoVa) regulates the distribution of the ER, mitochondria and endosomes within the axon, possibly by permitting exchange between actin, NFs and MTs. (E) Neurotransmission. Exhibiting different assemblies than those found in the axon (short oligomers), synaptic resident NFs regulate receptor trafficking and stability at the membrane of the post-synaptic compartment. NF-M associates with the D1R to anchor the internalised receptors on endosomes, to promote desensitisation to D1R stimulants such as cocaine. NF-L associates with GluN1, one of the subunits of the NMDA receptor, and stabilises it at the plasma membrane by protecting it from ubiquitin-dependent degradation. Thus, NF-L regulates the density and length of dendritic spines and stimulates various NMDAR-dependent responses such as spatial and social memory. NMDAR: N-methyl-D-aspartate receptor.
As the most abundant constituent of mature neurons, NFs have long been known for the protection they provide to long axons from mechanical insults (Figure 5A). Intensively inter-connected through the tail domains of NF-M/H, the NF array presents structural properties that can resist an average stretching of 260% before rupture (Kreplak et al., 2005; Sapra and Medalia, 2021). The exceptional viscoelasticity of this scaffold provides at the same time the mechano-resistance necessary to protect axons during the lifetime of an individual and adaptation to environmental stress.
Many studies have established that NFs are essential for the radial growth of large myelinated axons and for nerve conduction (Figure 5B). NF content has been shown to correlate with axonal diameter (and is reduced at the node of Ranvier), and the protrusion of NF tails outwards the highly dense filament was evidenced as a key determinant of NF spacing and radial axonal outgrowth (Lariviere and Julien, 2004). Thus, genetic manipulations to decrease NF density or spacing result in the reduction of axon caliber and nerve conduction. Amongst NFs, NF-L was shown to be a major determinant of NF content in axons. Indeed, depletion of NF-L through genetic manipulation (Zhu et al., 1997), in a spontaneous quail line (Sakaguchi et al., 1993) and in CMT patients with recessive mutations in the NEFL gene (Sainio et al., 2021) substantially inhibits radial growth and nerve conduction. Interfering with NF spacing has a similar effect on decreasing axonal caliber and conductivity but, surprisingly, the NF-M tail domain (as seen in the NFM-tailless (Rao et al., 2003), NFM/H tailless mice (Garcia et al., 2003)) and not the NF-H tail was shown to be the key determinant (Rao et al., 2002b). While this would suggest that the specific phosphorylation of KSP repeats within NF-M is the driver of radial axonal outgrowth, the systematic inactivation of all known KSP sites in the NF-M tail does not alter axonal caliber (Garcia et al., 2009). Another unexpected finding is the regulation of internode length (the distance between the nodes of Ranvier) by the phosphorylation of the tail domain of NF-M (Villalón et al., 2018).
Interconnected with the actin filaments and microtubules, NFs have been shown to regulate MT dynamics (Figure 5C). This line of research emerged from the observation (carried out in NF content-lowering conditions as seen in NF mouse models and the quail line) that MT and NF densities are inversely correlated, sometimes without changing the level of soluble tubulin (Eyer and Peterson, 1994; Zhao and Szaro, 1995; Zhu et al., 1997; Elder et al., 1998; Jacomy et al., 1999; Bocquet et al., 2009). Locally, MTs have also been shown to be reduced or excluded from densely packed NFs in various diseases, such as GAN (Donaghy et al., 1988; Ganay et al., 2011), suggesting a decreased ability of MTs to polymerise in NF rich regions. In NFHLacZ transgenic mice, NFs aggregate in the soma, preventing their export to the axon which are therefore depleted in NFs. Examination of the distribution of MTs and tubulin levels revealed that while axons lacking NFs are filled with MTs, the soma packed with NFs are devoid of MTs, even with normal tubulin levels (Bocquet et al., 2009). The negative effect that NFs exert on MT dynamics is mediated by two mechanisms. On one hand, a tubulin-binding site (TBS) identified in the head domain of NF proteins has been shown to bind soluble tubulin and to be sufficient to inhibit MT polymerisation (Bocquet et al., 2009). Therefore, NFs would act as a reservoir for unassembled tubulin, providing a source throughout the axon for rapid local polymerisation. On the other hand, NFs have been shown to destabilise MTs through disruption of the stathmin complex. This has been revealed in mice with progressive motor neuronopathy (pmn), characterised by a drastic destabilisation of MTs due to TBCE mutations and an increase in NF content. Depletion of NFs in the pmn model results in the regrowth of MTs, due to the inactivation of the MT-destabiliser stathmin, through increased interaction with Stat3 (Yadav et al., 2016).
Considering the key role of MTs for axonal transport of organelles, the regulatory action that NFs exert on MTs dynamics is likely to impact the distribution of organelles. More directly, NFs also constitute a cellular scaffold for the docking and organisation of organelles (Figure 5D). NFs have been shown to interact with organelles either directly (i.e., phosphorylated NFs binds to mitochondria (Wagner et al., 2003)), or through the actin-molecular motor MyosinVa (MyoVa) (Rao et al., 2002a). Thus, the motility of mitochondria is regulated by NF density. It is increased in the absence of NFs as seen in the NF-L KO mice and recessive NF-L CMT (Perrot and Julien, 2009; Gentil et al., 2012; Sainio et al., 2021) and decreased upon NF aggregation as seen in dominant NF-L CMT and GAN (Gentil et al., 2012; Saporta et al., 2015; Israeli et al., 2016; Nath et al., 2023). While these data suggest that NFs serve as a docking site, other NF-independent mechanisms susceptible of redistributing mitochondria to cope with insult may be considered (Van Lent et al., 2021). Surprisingly, NF-L interacts with the motor domain of MyoVa (Rao et al., 2002a; 2011), an actin-based molecular motor which transports many cargoes, including synaptic vesicles, endosomes, mitochondria, and endoplasmic reticulum (ER). NF-L depletion was shown to induce a redistribution of the ER, mitochondria, and endosomes to the periphery, hence revealing that NFs are crucial for MyoVa-mediated distribution of organelles within the axon (Rao et al., 2011). It is hypothesised that by binding to the motor domain of MyoVa, NFs may anchor organelles along the axon and facilitate their distribution across actin and MTs that are highly interconnected.
NFs are also critical regulators of neurotransmission (Figure 5E). Synapses have long been considered as the degradative site for NFs and, as a result, NF presence in this compartment was considered as an axonal contaminant (Yuan et al., 2017). More recent data has proven this to be wrong, with synaptic NF assemblies being very specific and playing crucial roles in synapse functions. NFs isolated from synapses present differences from axonal NFs, with a higher proportion of α-internexin and reduced phosphorylation of NF-M (Yuan et al., 2015b). This distinct population of NFs is more abundant in the post-synaptic compartment and can only occasionally form 10 nm short polymers. These characteristics may suggest an immature-like state of NFs (such as during development) that would confer greater local plasticity to the cytoskeleton. Considering that monomers are inefficiently transported along the axons, synaptic NFs would be at the very least transported in an oligomeric assembly form or be alternatively synthetised from mRNA at the synapse (Yuan et al., 2015b). Gene depletion studies in mice revealed the crucial roles of NFs in regulating receptor targeting and stability at the post-synaptic membrane, as well as substantial alterations in neurotransmission and behaviour.
NF-M, which was previously shown to interact with the dopamine D1 receptor (D1R) (Kim et al., 2002), plays a role in its recycling to the membrane by anchoring internalised D1R on endosomes at the synapse lumen (Yuan et al., 2015b). Thus, NF-M deletion increases the redistribution of post-synaptic D1R from endosomes to the membrane and amplifies dopamine-D1R-mediated motor responses to cocaine. NF-L was reported to interact with the glutamatergic GluN1, one of the four subunits of the N-methyl-D-aspartic acid receptor (NMDAR) (Ehlers et al., 1998). A study revealed that NF-L stabilises GluN1 at the membrane, by protecting it from ubiquitin-dependent degradation. In agreement with the known role of the NMDAR in regulating dendritic spine morphology, NF-L depletion, which decreases GluN1 level in vivo, also decreases the density and length of dendritic spines (Yuan et al., 2018). Moreover, NF-L depletion was shown to alter neurotransmission and cause various behavioural defects as seen in NMDAR hypofunction, included but not restricted to spatial and social memory. Altogether, these findings show that a unique population of NFs in the synapses plays key roles in neurotransmission in vivo and may suggest a potential implication in addiction, memory, and psychiatric and neurological diseases (Yuan and Nixon, 2016).
Neurofilaments sustain critical functions from neurodevelopment to adulthood, and alterations of this network are the direct or contributing causes of various neurodegenerative diseases in humans (Didonna and Opal, 2019; Bomont, 2021). Thus, abnormal aggregation of NFs represents one of the earliest pre-symptomatic hallmarks for most, if not all, neurodegenerative diseases. Interestingly, rare NF variants (possibly polymorphisms or gene modifiers) have been associated with amyotrophic lateral sclerosis, Alzheimer’s and Parkinson’s disease (Yuan et al., 2017), but their role in pathogenicity is still uncertain. In addition, several NF-controlling genes have been identified as the genetic cause of numerous neurological diseases. These include NF assembly (Sacsin, HSPB1), NF transport (KIF5A) and NF degradation regulators (gigaxonin) (Bomont, 2021). In these pathologies, determining the contribution of NFs to disease is challenged by the fact that the respective proteins regulate multiples targets and functions. In this review, we will focus on Charcot-Marie-Tooth (CMT) disease, for which NEFL and NEFH are causal genes. It is still important to highlight that NF mutations can also cause hereditary spastic paraplegia (NEFL), spinal muscular atrophy (NEFH) but also myopathy (NEFL), which evidence a broader phenotypic spectrum of NF mutations (Wang and Brown, 2010; Agrawal et al., 2014; Ando et al., 2022).
Charcot-Marie-Tooth disease, also known as hereditary motor and sensory neuropathy (HMSN) is the most common inherited neurological disorder of the peripheral nervous system, with a worldwide prevalence rate of 1 in 2,500 (Laurá et al., 2019; Rudnik-Schöneborn et al., 2020). CMT is a motor and sensory neuropathy caused by the progressive deterioration of the peripheral nerves, composed of the lower motor axons projecting from the spinal cord and the sensory axons from the dorsal root ganglia. Thus, CMT is most commonly characterised by muscle weakness, amyotrophy and sensory loss that start in the lower limbs and progress slowly in a length-dependent manner. A high incidence of foot deformities (pes cavus) is also seen in CMT. There is considerable variability concerning the age of onset, the severity of the symptoms and the progression of the disease between different forms of CMT, even in the same family. Most patients present symptoms in the first or second decade of life, although it may also start in infancy or at an advanced age. The clinical presentation of the disease can be highly variable. Indeed, more severely affected patients may become non-ambulant by the third decade of life, whereas other individuals can only present minimal symptoms such as pes cavus or minor distal muscle atrophy. Some mutation carriers may even age without experiencing any clinical signs. Historically, CMT was described as a group of disorders primarily affecting the peripheral nervous system, but in recent years many forms of other non-neurological and neurological symptoms within the central nervous system have been found (Pipis et al., 2019), including dementia, cerebellar syndrome, hearing impairment and optic atrophy.
CMT is classified into two main types, according to neurophysiology and histopathological features at the nerve biopsy: the demyelinating (CMT1) and the axonal (CMT2) forms (Laurá et al., 2019; Rudnik-Schöneborn et al., 2020). CMT1 is characterised by a reduction of the upper limb motor nerve conduction velocities (MNCV) (below 38 m/s) and a predominant alteration of the Schwann cells, the glial cells of the peripheral nervous system that form the myelin sheath around the nerve fibers necessary for saltatory conduction. CMT2 has a MNCV above 38 m/s and is primarily caused by axonal degeneration. Another form, the intermediate form (I-CMT), has emerged, with MNCV ranging between 25 m/s and 45 m/s (Berciano et al., 2017). Considering the invasiveness of sural nerve biopsy, the associated pain and poor diagnostic value in pinpointing specific genes, this procedure is no longer conducted in regular practice. Confusingly, recessive forms of CMT1 have been named CMT4. CMT are further classified according to the mode of inheritance (mostly dominant but also recessive and X-linked) and the genetic cause. The most common form of CMT (60% of cases) is CMT1A, which is caused by a genomic duplication comprising the peripheral myelin protein 22 gene (PMP22). In CMT2 (which represents about 30% of all CMTs), the mitofusin gene (MFN2) is the most common form (CMT2A), which accounts for 20% of axonal CMT. The development of next-generation sequencing (NGS) technologies (gene panels, whole-exome/whole-genome/mitochondrial/high-throughput transcriptome sequencing) revolutionised genetic diagnosis and allowed the number of genes to be considerably expanded to >100 for CMT (Rossor et al., 2013; Pipis et al., 2019). This revealed the high genetic heterogeneity, which is further complicated by the fact that several genes can cause different forms of CMT. For example, the NEFL gene, originally discovered in a Russian family suffering from axonal CMT2E, has been further implicated in CMT1F and I-CMT.
The NEFL gene was the first causal gene to be identified in a dominant form of axonal CMT2 (CMT2E) (Mersiyanova et al., 2000). Since then, various mutations have been identified throughout the NEFL gene and more recently in the NEFH gene (Figure 6 with associated bibliography). Overall, NF-CMT accounts for 1% of diagnosed CMT cases and the majority of mutations are inherited through a dominant mode, although recessive forms and sporadic cases have been identified. As for many other CMT forms, NF-CMT have revealed a broader clinical picture than initially described. Thus, patients with NF mutations have shown to develop several defects within the central nervous system, including ataxia, hearing loss, dementia, nystagmus, tremor, schizophrenia, dysarthria, spasticity, and pain in addition to sensory-motor defects (Fabrizi et al., 2004; 2007; Züchner et al., 2004; Miltenberger-Miltenyi et al., 2007; Hashiguchi et al., 2014; Berciano et al., 2015; 2016; Doppler et al., 2017; Horga et al., 2017; Lerat et al., 2019; Machado et al., 2019; Kim et al., 2021; Chao et al., 2023).
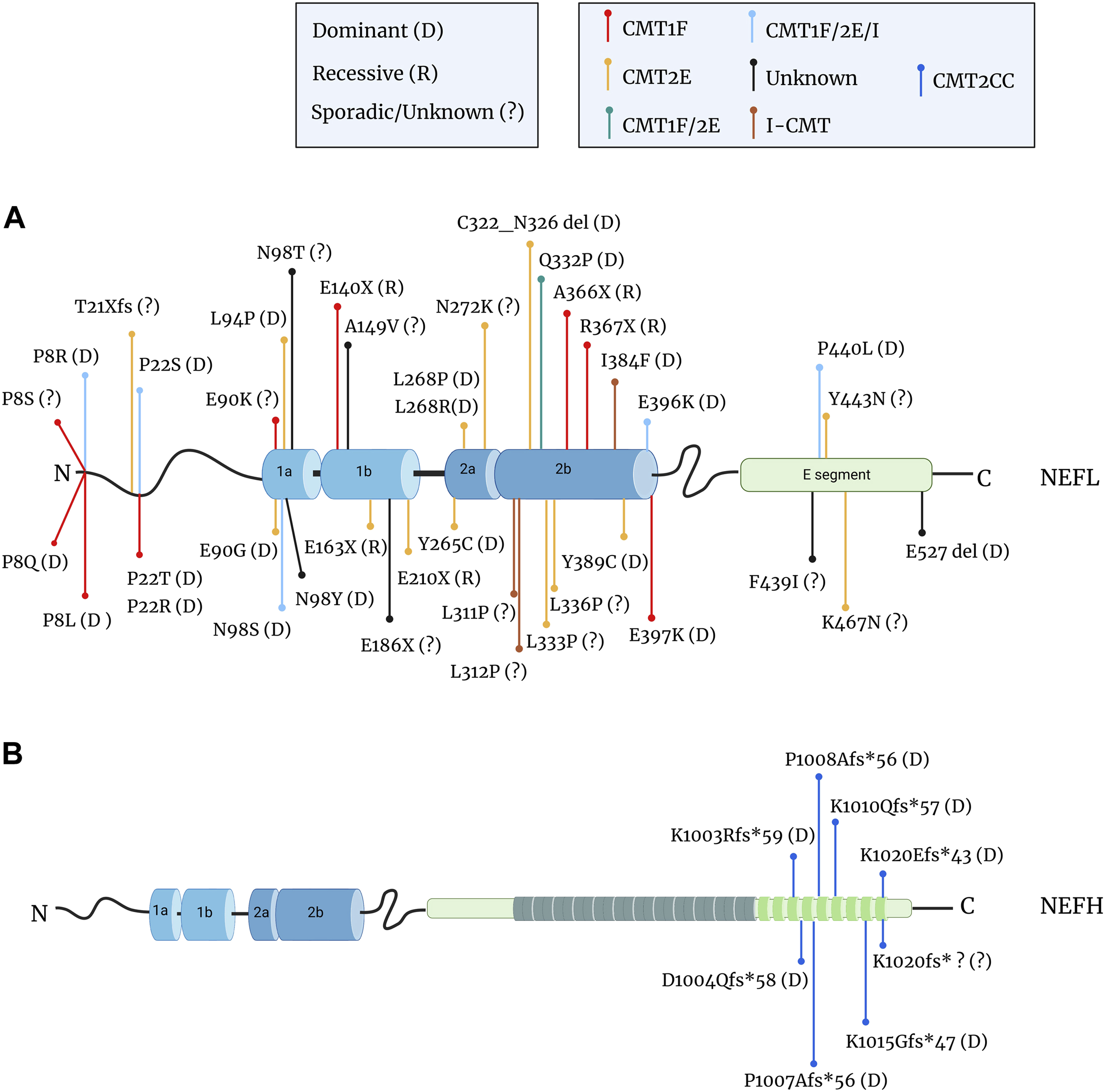
FIGURE 6. Mutations in the NEFL and NEFH genes in CMT patients. (A) Localisation of the mutations in the NF-L protein that cause demyelinating CMT1F (red dot), axonal CMT2E (yellow dot), intermediate I-CMT (brown dot), unknown CMT (black dot) or mixed forms: CMT1F or CMT2E (green dot); CMT1F, CMT2E, or I-CMT (light blue dot). Missense mutations are the most common, but frameshift (fs), deletion (del), and nonsense mutations (X) have been reported. Mutations are often inherited through a dominant mode (D) but recessive transmission (R) also occurs and several cases are undetermined or sporadic (?). The nonsense mutations are located within the rod domain and are all recessive, while missense and in-frame deletion mutations are dominantly inherited. (B) Mutations in the tail domain of the NF-H protein cause axonal CMT2CC (dark blue dot). Nucleotide changes (e.g., deletion, insertion) create frameshift mutations that are inherited through an dominant mode or de novo. Data from: (Mersiyanova et al., 2000; De Jonghe et al., 2001; Georgiou et al., 2002; Yoshihara et al., 2002; Jordanova et al., 2003; Choi et al., 2004; 2012; Fabrizi et al., 2004; 2007; Züchner et al., 2004; Leung et al., 2006; Miltenberger-Miltenyi et al., 2007; Shin et al., 2008; Abe et al., 2009; Bhagavati et al., 2009; Yum et al., 2009; Benedetti et al., 2010; Lin et al., 2011; Sivera et al., 2013; Elbracht et al., 2014; Hashiguchi et al., 2014; Manganelli et al., 2014; Berciano et al., 2015; 2016; Drew et al., 2015; Rebelo et al., 2016; Yang et al., 2016; Doppler et al., 2017; Horga et al., 2017; Jacquier et al., 2017; Nam et al., 2017; Bian et al., 2018; Fu and Yuan, 2018; Sainio et al., 2018; Ikenberg et al., 2019; Lerat et al., 2019; Machado et al., 2019; Aruta et al., 2021; Ando et al., 2022; Kim et al., 2022; Pipis et al., 2022; Chao et al., 2023; Petrucci et al., 2023).
As seen in Figure 6, the mutation types and positions differ greatly between the NEFL and NEFH genes.
Mutations in the NEFL gene have been identified in all domains of the protein: in the head, the ⍺-helical rod and in the tail (Figure 6A), domains which regulate various NF functions (see previous paragraph). Interestingly, the severity of the symptoms does not seem to correlate with the position of mutations (Stone et al., 2021), hence revealing an equal importance of all domains for NF functions.
So far, missense mutations are mostly inherited through a dominant mode and represent the most common CMT-causing mutations in the NEFL gene. Less frequently frameshift, in-frame deletion and nonsense mutations were identified. Interestingly, the latter mutations are only found in the rod domain where they segregate exclusively through a recessive mode of inheritance. Studies on the NFLA367X mutant revealed that the premature termination of NEFL translation in both alleles abolishes NF-L expression in IPSC-MNs, due to the specific instability of its mRNA through a mechanism called non-sense mediated mRNA decay (Sainio et al., 2018). Astonishingly, a filamentous network positive for all other NF subunits could be seen in these cells (Sainio et al., 2018; 2021), findings which are surprising considering the current model presenting the necessity of the NF-L subunit for proper filament assembly. One could assume that in the IPSC-MN, α–internexin and peripherin may substitute NF-L in forming heteropolymers with NF-M and NF-H. Still, in vivo, this compensation does not seem to occur, as rare electron micrographs of patient biopsies evidence axons filled with MTs but with no filaments (NEFLE210X and NEFLE163X) (Yum et al., 2009; Fu and Yuan, 2018)). A comprehensive analysis of all recessive nonsense mutations regarding the stability of the mRNA, the expression of NF-L in patient-derived cells, as well as the visualisation of the NF network in biopsies will be essential to further examine the requirement of NF-L in adult stages.
Another intriguing aspect is the clinical variability of the disease. Since the first mutation in the NEFL gene in an axonal form of CMT (CMT2E), others have been revealed in cases of demyelinating neuropathy (CMT1F) with reduced MNCV (Figure 6A). To make the picture even more complex, a variable clinical presentation (CMT1F and/or CMT2E and/or I-CMT) can result from distinct mutations on the same amino acid, or even from the same mutation (positions P8, P22, E90, N98, E396, P440). This is puzzling as NFs are confined to neuronal cells and not expressed in myelinating-Schwann cells. To explain this, several hypotheses could be proposed. An assumption would be that the demyelination (and reduced MNCV) may result from a general decrease in axonal caliber rather than axonal degeneration, as shown in the NEFLN98S/+ knock-in model (Lancaster et al., 2018). In this context, all NF cases may correspond to axonal CMTs and would indicate a clinical variability due to gene modifiers or environmental factors. Alternatively, one could conceive that a crosstalk exists between the neuron and the Schwann cell, whereby dysfunctional NFs within the axons could signal to the glial cells. Finally, rare in vivo data have evidenced a transient induction of NF proteins in both early Schwann cells differentiation and dedifferentiation during Wallerian degeneration, which may reveal critical roles in myelin formation and repair after damage (Kelly et al., 1992; Roberson et al., 1992; Fabrizi et al., 1997; Yin et al., 2001). In patients, one could hypothesise that NF mutations may affect the functionality of the myelinating-Schwann cell, acting at the early stage of their maturation.
Mutations within the NEFH gene form an homogeneous group of dominant axonal CMT (CMT2CC) (Figure 6B), which differ from classical CMT due to their early and prominent involvement in proximal muscles and rapid decline of motor capacities (Aruta et al., 2021). The first cases (D1004Qfs*58 and P1008Afs*56) identified a novel type of mutation in NF-CMT (Rebelo et al., 2016): a frameshift variation that induces a loss of the terminating codon and the translation of short fragments in the 3′UTR of the NEFH mRNA. Predicted to be amyloidogenic, these extra fragments induce aggregation of the NF-H protein in neuronal cell lines. The identification of other mutations confirmed the homogeneity of the genetic alteration in CMT2CC and identifies a hot spot in the C-terminal portion of the tail domain of NF-H, that extends its length by < 60 amino acids.
Several laboratories have studied the effects of the NEFL/H mutations on filament assembly and cellular functions. Studies were mostly conducted in vitro, with only three mouse models generated for the NF-CMTs. Here, we will distinguish the findings evidenced in vitro and in vivo, and will discuss their relevance for NF biology and disease.
In cells, studies on NF mutants were systematically conducted using overexpression, which makes sense for dominant forms. Accordingly, we will not discuss results obtained for the recessively inherited NEFLE210X mutation (with no NFL mutant in patient biopsy) and the NEFLT21Xfs found at heterozygous state (unclear if haplo-insufficient or dominant form). Various cellular systems have been used to determine the ability of single NF proteins (wild-type or mutants) to form a filament. To avoid bias of interpretation due to the presence of endogenous NF subunits (or IF types), the preferred cellular system is the SW13vim- human adrenal adenocarcinoma cells, which are deprived of IFs due to the spontaneous silencing of vimentin. An alternative yet rarely used IF-free system is the Sf9 insect cells. Once the ability of NF subunit to homopolymerise is established, research groups may combine other neuronal cell lines (CAD, SH-EP, NSC34, Neuro2a) and/or primary neurons with long neurites (cortical neuron, motor neuron, DRG sensory neuron).
Thus, the use of SW13- cells has helped demonstrate that only the human NF-L protein is able to homopolymerise, while the other cell types confirm the ability of wild-type NF-L to form filament in various conditions (Table 1 and associated bibliography). Indeed, when overexpressed, neither mouse nor rat NF-L can form a filamentous structure in the absence of other NF subunits (SW13vim- and Sf9 cells). Rodent NF-L was shown to form filament only as heteropolymer with another overexpressed NF subunit: NF-M and/or NF-H or ⍺-internexin in SW13vim- cells, or NF-M in Sf9 cells. Similarly, when IF proteins are endogenously expressed in SW13vim+ cells (vimentin), neuronal cell line and primary neurons (all NF subunits), the rodent NF-L protein is able to incorporate and colocalise with the endogenous network. Beautiful electron micrographs carried out using a quick-freeze deep-etching method in Sf9 cells revealed that co-expressed mouse NF-M and NF-L proteins co-assemble in parallelly arranged 10 nm filaments that form cross-bridges, similarly to what is seen in rat peripheral nerves (Nakagawa et al., 1995).

TABLE 1. Summary of assembly capacity of wild-type and CMT-mutated NF-L and NF-H proteins in vitro. In this table, we find the NF subunits overexpressed in different cellular systems. The capacity of wild type (WT) and mutant NF proteins to homopolymerise is determined in IF-free systems (SW13vim- or Sf9). Their effects on other IFs (exogenously or endogenously expressed) is shown in non-neuronal and neuronal cell types. Note that the study performed on the recessively inherited NEFLE210X mutation is not presented, as the patient has no detectable NF-L. Abbreviations: h, human; m, mouse; r, rat. Cell types used in the studies: SW13, a human adrenocarcinoma cell line that does not express vimentin (vim-) or expresses vimentin (vim+); Sf9, an insect clonal isolate of Spodoptera frugiperda Sf21 cells; Neuronal cell lines are: CAD (Cath. -a-differentiated), a mouse catecholaminergic neuronal tumour cell line; NSC34, a mouse motor neuron-like cell line; Neuro2a, a mouse neural crest-derived cell line; SH-EP, a human neuroblastoma cell line; primary neuronal cells are MNs, mouse primary motor neurons; DRG, mouse dorsal root ganglia sensory neurons.
The effects of NF-L mutants (from human or rodent origin) have been extensively evaluated, mostly in SW13vim- cells, alone or in combination with one or two other NF subunits (Table 1). The detailed picture is complex but we can extract a few major points from these studies. Firstly, various NF-L mutants were shown to aggregate when overexpressed in SW13vim- cells, in the soma and proximal neurites in neurons. Secondly, the form of aggregates is highly variable, ranging from puncta, inclusions, small aggregates and large aggregate of various shapes. At this point, correlating a type of aggregate with specific mutations would be very difficult, considering the variability between studies and experimental conditions. Third, NF-L mutants seem to exert a dominant effect on other ectopically expressed wild-type subunits (alone or in combination) in SW13vim- cells and various neuronal types. Interestingly, this also applies to the endogenous network, as vimentin in SW13vim+ cells and endogenous NF-M in neurons were shown to colocalise with mutant NF-L. Nevertheless, effects vary greatly. Indeed, while in some cases other subunits co-aggregate with the mutant NF-L proteins, several reports show on the contrary that the other NF proteins (and vimentin) can protect to some degree the mutant NF-L from severe aggregation. For example, NFLP8R, NFLP8Q and NFLP22S form dense aggregates in SW13vim- cells, but are incorporated in an “apparent normal” network or bundle upon co-expression of NF-M, or together with endogenous vimentin filament in SW13vim+ cells (Perez-Olle et al., 2002; 2004; 2005; Tradewell et al., 2009). Other evidence shows that the mouse NFLP8R and NFLQ332P do not form aggregate in primary motor neurons at an early stage and have the capacity to be incorporated into the endogenous NF array (Gentil et al., 2012). Interestingly, a recent study has re-examined the capacity of human NF-L mutants to form aggregates in different cellular contexts expressing increasing NF content: the SW13vim- cells (no IFs), SW13vim- co-transfected with NF-M, SW13vim- co-transfected with NF-M + peripherin, primary cortical neurons co-transfected with NF-M (and containing endogenous NFs). Unexpectedly, none of the 7 human NF-L mutants aggregate in SW13vim- cells (Stone et al., 2019), as previously reported. Instead, they are able to form heteropolymers, with variable dependency on NF content (NF-M or NF-M + peripherin). In rat cortical neurons, rat NF-L mutants integrate into the endogenous NF array without forming any aggregate. Thus, with the examples cited above, these findings present the notion that while NF-L mutants may aggregate in certain contexts (possibly due to excessive overexpression or imbalance with other NF subunits), they are able to be incorporated into the filament as the wild-type protein. This different appreciation of the effects of NEFL mutations in dominant forms of CMT may reconcile with the rare data available from patients, which show more of an increased density and disorganised network than a defect in filament formation (Lus et al., 2003; Fabrizi et al., 2004; 2007; Doppler et al., 2017). Obtaining ultrastructural analysis of the NF array in patients and standardised biological systems to control protein (over)expression would be essential in order to compare and determine the effects of dominant mutations. Alternatively, a time-course study in motor neurons demonstrated that NFLP8R and NFLQ332P mutants could initially integrate into a pre-existing NF network and aggregate 7 days after overexpression, which may also indicate a long-term deleterious effect on NF assembly (Tradewell et al., 2009). Using the same mutants, another study reported that the mutants affect the oligomerisation process, possibly compromising filament formation (Gentil et al., 2013). Along with this, purified NF-LP22S and NF-LP22T are unable to form filament in vitro, even in the presence of wild-type NF-L (Sasaki et al., 2006), hence suggesting that NF-L mutants disrupt proper oligomer formation in cell-free systems.
Trying to conciliate data obtained from overexpressing systems in vitro and from patients is quite challenging. Still, the fact that the IPSC-MNs endogenously expressing the NFLP8R mutant form distinct inclusions in the soma and neurites (Van Lent et al., 2021) clearly demonstrates that NEFL mutations can induce “aggregate” formation in a physiological context. Moreover, the detection of NFs as “aggregates” (using immunofluorescence) in the soma of DRG neurons from the knock-in NEFLN98S/+ mouse and as “packed/disorganised filaments” (using electron microscopy) (Zhao et al., 2017) demonstrates that endogenous NF-L mutants are seen differently depending on the level of analysis. Thus, we may suggest that NF mutants alter filament properties in various ways, and that what we visualise as “aggregates” in cells with low magnification methods may results from an abnormal arrangement of the filament.
Unlike the human NF-L protein, human NF-H itself is unable to form filament, as seen in SW13vim- cells, even in the presence of ectopic NF-L (Bian et al., 2018) (Table 1). In some studies, human NF-H was shown to be evenly distributed in the cytoplasm (Neuro2a, SH-EP), while others showed a filamentous network (SH-EP, Neuro2a) upon co-expression of NF-L in primary motor neurons. While these discrepancies may be explained by different times of analysis or expression levels, this clearly indicates that the human NF-H is self-assembly incompetent but can be incorporated to a pre-existing NF network. Rodent NF-H was shown to be unable to form filaments in any context, except in cortical neurons co-expressing NF-L (Table 1). Considering that purified NF-H is incapable of forming filament in assembly in vitro tests but does so in presence of purified NF-L (Geisler and Weber, 1981), more extensive analysis may be required to firmly document the capacity of NF-H to form heteropolymers in cells. Nevertheless, overexpression of several frameshift mutants of NF-H consistently revealed perinuclear aggregates, which trap ectopically expressed WT-NF-L, hence demonstrating the dominant effect conferred by the extra C-terminal fragment of NF-H.
The findings that NF mutants form aggregates in the soma or proximal segments in primary neurons (Brownlees et al., 2002; Perez-Olle et al., 2004; 2005; Zhai et al., 2007; Tradewell et al., 2009; Jacquier et al., 2017) or in IPSC-MNs (Van Lent et al., 2021) have led to the conclusion that their transport is reduced in disease. We could only find two studies directly addressing the effect of NF-L mutants on NF transport in rat cortical neurons. In the first study, the transport of EGFP-NF-M was reduced by about 50% in the context of aggregate-prone NFLP8R and NFLQ332P (Brownlees et al., 2002), while the other revealed an overall normal movement of various filament competent EGFP-NF-L mutants (Stone et al., 2019). While further studies will be needed to solve this issue, these findings suggest that the NF transport impairment in the context of aggregates may be caused by the alteration of NF integrity.
More literature is available on axonal transport, particularly of mitochondria. Thus, aberrant localisation, reduced length, reduced fusion, abnormal trafficking (reduced or increased), and decreased basal respiration were reported in neuronal cells expressing NFLP8R, NFLP8L, NFLN98S and NFLQ332P (Brownlees et al., 2002; Perez-Olle et al., 2004; 2005; Tradewell et al., 2009; Gentil et al., 2012; Saporta et al., 2015), and also in IPSC-MN with NEFLP8R mutation (Van Lent et al., 2021). However, no alteration of mitochondria dynamics was evidenced in human motor neuron spheroids with the NFLN98S mutant (Maciel et al., 2020). Other dysfunctions, such as Golgi fragmentation and lysosomal motility were occasionally reported for NF-L mutants (Perez-Olle et al., 2005; Van Lent et al., 2021). Rare studies have evidenced electrophysiological and neuronal dysfunctions for NEFL mutations. Thus, reduced action potential threshold was reported in IPSC-MN with NEFLN98S mutation (Saporta et al., 2015); reduced neurite length and hyperconnectivity were evidenced in IPSC-MN with NEFLP8R mutation (Van Lent et al., 2021), as well as apoptosis in SH-EP and spinal cord motor neurons overexpressing NF-H mutant (Jacquier et al., 2017) and progressive neurotoxicity in motor neurons overexpressing NFLP8R and NFLQ332P (Zhai et al., 2007). In apparent normal NF networks, neurons deprived of NF-L (as seen in the recessive forms of NF-CMT) also showed interesting abnormalities. Thus, IPSC-MN with NEFLA366X (or CRISPR-KO cells) exhibit increased mitochondria trafficking and reduced amplitude of miniature excitatory postsynaptic currents (Sainio et al., 2021).
Overall, studies predominantly investigated mitochondria dynamics, but the current knowledge regarding the physiological mechanisms underlying the NF-CMT pathology is rather limited. Efforts initiated to investigate neuronal (electro)physiology in cellular systems would be of great benefit to the field, in combination with the study of NF-CMT in animal models.
To date, only three animal models have been generated for NF-L CMT disease. Here we will not discuss the transient expression of NF-H mutant in animals, which only confirmed aggregation and caspase-3 activation in chick embryos (Jacquier et al., 2017) and revealed shorter motor axons in zebrafish (Rebelo et al., 2016). It is worth noting that one transgenic mouse model overexpressing a mouse NEFLL394P mutation not identified in CMT disease showed specific alterations of motor neurons, with abnormal NF accumulation in the soma and axonal degeneration (Lee et al., 1994).
Two transgenic models were created, overexpressing the human NEFL E397K (hNEFLE397K (Shen et al., 2011)) or the P22S transgene (hNEFLP22S (Dequen et al., 2010)), the latter in an inducible manner using the Tet-off tetracycline-regulated system.
From 4 months of age, hNEFLE397K mice exhibit abnormal gait (Dale et al., 2012), sensory defects (reduced sensitivity to mechanical stimuli) (Villalón et al., 2017), motor deficits (reduced locomotion, reduced motor coordination), aberrant limb posture, reduced MNCV and muscle atrophy of the lower limbs, without denervation (Shen et al., 2011). The total number of motor and sensory axons is not affected but a shift towards a decrease in large caliber axons is observed. Regarding NFs, there was no sign of NF aggregation or impaired disorganisation/density in hNEFLE397K mice, only increased NF phosphorylation in the soma of spinal motor neuron (Shen et al., 2011). Detailed analysis of muscle spindle morphology revealed a reduced area at pre-symptomatic stage, suggesting proprioceptive sensory deficits in the hNEFLE397K model (Villalón et al., 2017).
The hNEFLP22S mice exhibit from 6 months of age aberrant limb posture, abnormal gait, learning defects, motor dysfunctions (reduced coordination), but in contrast to the hNEFLE397K mice, the model presents no sensory deficits (normal mechanical response) and present muscle hypertrophy with signs of denervation (Dequen et al., 2010; Filali et al., 2011). As for the other model, the hNEFLP22S mice do not present NF alterations (except for rare NF-H positive inclusions in spinal motor neurons) nor neuronal degeneration. Interestingly, the removal of the mutant NF-L transgene (following 3 months of treatment with doxycycline) ameliorates several motor abnormalities, attenuates muscle hypertrophy and restores muscle innervation (Dequen et al., 2010).
Overall, although both CMT models present symptoms of the sensory-motor system, none of the two transgenic mouse models exhibit overt neurodegeneration and NF alterations, and only mild signs of nerve pathology were reported.
A third NEFLN98S/+ knock-in mouse model was generated by the introduction of the CMT mutation at the endogenous locus. This mouse model presents abnormal hindlimb posture, develops tremor from 4 weeks of age (Adebola et al., 2015), and subsequently exhibits altered balance/gait from 6 weeks of age (Lancaster et al., 2018). Mobility and sensitivity to thermal stimuli remain unaltered. Interestingly, an abnormal distribution of NF-L was observed in different parts of the nervous system (spinal cord, DRG neurons, cerebellum) at pre-symptomatic stage (post-natal day 7) and persisted during aging (18 months of age). Thus, accumulation of NF-L was detected in the soma of neurons, while a decrease was observed in the axonal tracts. In the sciatic nerves, only a few NFs could be detected upon ultrastructural examination (12 months of age) and, as expected from the literature, increased MT density and decreased axonal caliber were detected (Adebola et al., 2015; Lancaster et al., 2018). Early analysis (from 8 weeks of age) of various regions of the nervous system (optic nerves, spinal cord, femoral/sciatic/caudal nerves) evidenced fewer NFs, smaller myelinated axons and reported a reduction of both the amplitude and NCV in the caudal nerves (Lancaster et al., 2018). In contrary, cell bodies and proximal axons of the sensory-motor system exhibit NF accumulation and are disorganised. Another study revealed NF aggregation in the soma of DRG neurons in adult NEFLN98S/+ mice and showed that the NF-L mutant switches the soluble/insoluble balance towards insolubility in tissue (Zhao et al., 2017). Overall, these findings suggest that the NEFLN98S mutant has a dominant effect on NF architecture, most likely trapping and disorganising NFs within the soma. While the phenotype is milder compared to human CMT disease, the NEFLN98S/+ mouse is the first model to present NF abnormalities and signs of axonal degeneration and regeneration.
Interestingly, several in vivo studies have revealed the essential role of NFs in promoting axonal regeneration. Indeed, the hNEFLE397K mice show reduced recovery from nerve crush injury on sciatic nerves, which exacerbates the neuropathy: increased mechanical allodynia, premature gait dysfunction and disrupted limb coordination (Villalón et al., 2015). Reduced regeneration was also reported in two mouse models depleted of axonal NFs, NEFL−/− (Zhu et al., 1997) and NEFHLACZ mice (Cassereau et al., 2013), in which NFs were shown to accumulate in the soma.
To date, there is no treatment for any form of the NFs-CMT disease. However, several therapeutic strategies aimed at enhancing expression of wild-type NF-L in recessive forms, inactivating mutated NF-L and restoring normal NF organisation have been investigated. Other approaches targeting the downstream cellular alterations are also undergoing.
In recessive forms of NF-CMT, heterozygous carriers were reported to be asymptomatic, suggesting that a single functional NEFL allele is sufficient for normal neurological function (Abe et al., 2009; Yum et al., 2009; Fu and Yuan, 2018). Thus, drugs that induce translational read-through or inhibit non-sense mediated mRNA decay were tested in IPSC-MNs carrying the NEFLA367X homozygous mutation. While this first attempt failed to show an increase of NF-L level (due to neurotoxicity) (Sainio et al., 2021), this avenue is certainly worth further investigation. In another IPSC-MN model for the dominant NEFLN98S mutation, authors used the CRISPR/cas9 technology to correct the mutation (NEFLN98S−cor) or specifically silence the N98S allele (NEFLN98S−fs) by introducing a frameshift in the sequence (Feliciano et al., 2021). This strategy induces a 42% reduction of NF-LN98S−fs expression in the IPSC-MN, due to the specific decrease of the NEFL mutant transcript. Interestingly, gene editing restored normal intensity of NFs in the soma and electrical activity in both the NEFLN98S−cor and NEFLN98S−fs MNs, hence representing a promising therapeutic avenue for dominant missense mutations in the NEFL gene. Another interesting approach, initiated on IPSC-MNs is based on the amelioration of the mitochondrial-associated defects in CMT2, as shown by the benefits induced by MAP3K12 inhibitors on mitochondrial morphology and basal respiration in NEFLLP8R human MNs (Van Lent et al., 2021).
Three dimensional spheroids were recently developed from IPSC-MNs of NF-CMT2 patients. NEFLN98S spheroids revealed abnormal NF distribution along the axons (10-fold increase of NF deposits without change in abundance), phenotype which is partially reversed by two kinase inhibitors (PLKi and CK2) known to phosphorylate NFs (Maciel et al., 2020). The role of phosphorylation in regulating various aspects of NF dynamics has been exploited as a possible therapeutic strategy for NF-CMT. In particular, the phosphorylation of the head domain of NFs is known to induce polymer disassembly. The findings demonstrating that purified NF-LP22S and NF-LP22T proteins are less phosphorylated by ERK and cdk5 kinases (Sasaki et al., 2006) led to the proposal that their aggregation may result from an excessive stability of the proteins. Thus, increasing PKA-dependent phosphorylation (acting on other residues in the head) was shown to increase the solubility of NF-L mutants and to partially dissolve NF aggregates in cortical neurons (Sasaki et al., 2006). Other alternative approaches aiming at destabilising NFs would be to modulate regulatory factors in disease. In this direction, the 14-3-3 protein, which was shown to interact with the NF-L head domain in a phospho-dependent manner induces NF disassembly in vitro and reduces aggregation caused by NF-LP8L, NF-LP22S and NF-LP22T mutants in SW13vim-cells NFs (Miao et al., 2013).
Amongst therapeutic strategies initiated for NF-CMT, there is a particular focus on molecular chaperones which protect cells from misfolded proteins. This interest merged from the findings that mutations in the small heat-shock protein 27 (HSP27 also called HSPB1) are causal for the dominant axonal CMT2F (Evgrafov et al., 2004) and induce aggregation when overexpressed in SW13vim-cells, NSC34 cells and cortical neurons (Evgrafov et al., 2004; Ackerley et al., 2006; Zhai et al., 2007). Studies showed that the wild-type HSPB1 associates with NF proteins preferentially in a polymerising state to interfere with it and increase the pool of soluble NF particles (Nefedova et al., 2017). This study indicates that HSPB1 modulates the transition between tetramers and filament and suggests that its mutation in CMT2F would cause an excessive stabilisation of the wild-type NF (leading to NF aggregation). It also suggests that HSPB1 could be used to destabilise NF aggregates in CMT2E. Thus, overexpression of HSPB1 was shown to decrease aggregation induced by the NF-LP8R and NF-LQ332P mutants in SW13vim- cells (Zhai et al., 2007). Importantly, similar results were obtained in primary motor neurons transfected with the NF-LP8R mutant, resulting in an improvement of cell viability, hence representing a substantial beneficial outcome for therapeutic efficacy. In addition, the fact that the neurotoxicity of mutant HSPB1 on motor neurons is almost eradicated in NEFL−/− motor neurons (Zhai et al., 2007) indicates that NFs represent a major target for therapeutic development for various forms of CMTs. Intriguingly, heat shock proteins exhibit a specificity toward NF-L mutants, with HSPB1 being preferentially effective on NF-LP8R but not NF-LQ332P and HSPA1 only on NF-LQ332P in MNs (Tradewell et al., 2009). This implies that the efficacy of therapy using chaperone or chaperone inducers must carefully examine the type of chaperone that would be effective on given NF-L mutants. Treatment with celastrol, an inducer of HSPA1 in motor neuron is only able to decrease NF-LQ332P inclusions and not NF-LP8R ones (Gentil et al., 2013). As celastrol exhibits a cell type specific mode of action (induction of HSPA1 specifically in motor neurons, HSPB1 specifically in sensory neurons), its benefit in treating a sensory motor neuropathy appears to be rather limited. Nevertheless, these studies have opened the avenue for the identification of chaperone inducers as a therapeutic strategy for NF-CMT.
Noteworthy, HSPB1 has also been shown to control NF transport (through regulation of NF binding to kinesin via Cdk5-mediated phosphorylation of NFs) (Holmgren et al., 2013), MT stability (Almeida-Souza et al., 2011) (by binding to tubulin) and autophagy flux (through interaction with the autophagy receptor p62) (Haidar et al., 2019), which may suggest multiple functions of HSPB1 on NFs and may therefore reveal potential downstream targets for therapeutic intervention in NF-CMTs (Vendredy et al., 2020).
Neurofilaments are composed of four subunits, whose stoichiometry varies between neuronal types during development and aging. With a general mode of assembly akin to IFs, NFs are expected to exhibit a high heterogeneity, with NF-L (and to a lesser extent ⍺-internexin/peripherin) as a core protein. One challenge ahead will be to scrutinise the molecular and structural diversity of NFs and to determine how this contributes to cell signalling. NFs have been shown to play key physiological roles in protecting axonal architecture, controlling nerve conduction and neurotransmission and regulating cytoskeletal dynamics. Considering the broad clinical picture of NF-CMT forms in the CNS and the identification of NF mutations in other neurological diseases, the field will certainly uncover additional unknown fundamental roles of NFs in various parts of the nervous system. Another interesting aspect to consider in the future is the mechanisms whereby NFs respond to neuronal needs within the nervous system. With a basal stationary NF network that provides the structural scaffold of the axon, the neuron has established a sophisticated set of NF regulators to provide, at a minimal energy cost, the dynamics necessary to fulfil local demands and respond to normal stimuli and injury. While our knowledge on NFs has considerably increased in the last few decades, the transport and turn-over of NFs are key determinants of NF dynamics that require further investigation. A major challenge ahead is to apprehend with a spatial and temporal resolution the various elements that regulate NF dynamics and signalling. While manipulating NF stoichiometry continues to be an essential approach in tackling these key questions, a deep investigation of the various NF diseases using IPSC, organoids and animal models is absolutely required to expand our knowledge on NF biology and its physiological relevance. Thus, the study of recessive NF-CMT forms may suggest a temporal control for the requirement of specific core proteins in filament assembly. Conflicting outcomes obtained on dominant CMT forms are challenging our view on what is an aggregate and how NF mutants affect filament assembly and structure. In the future, understanding the yet unknown mechanisms underlying neurodegeneration in disease is a main biomedical challenge that will fuel discoveries on the physiological roles of NFs, therapeutic innovation in NF-CMTs, and more widely in most neurodegenerative diseases.
FK: Writing–original draft. DC: Writing–original draft. PB: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Resources, Supervision, Validation, Visualization, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. PB is supported by the Institut National de la Santé et de la Recherche Médicale (INSERM). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement n° 864893). FK and DC are the recipients of a PhD fellowship from the Association Française contre les Myopathies (AFM) and the ERC program, respectively. Figures have been designed with the free and open-source Inkscape software and partly with Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abe, A., Numakura, C., Saito, K., Koide, H., Oka, N., Honma, A., et al. (2009). Neurofilament light chain polypeptide gene mutations in Charcot-Marie-Tooth disease: nonsense mutation probably causes a recessive phenotype. J. Hum. Genet. 54, 94–97. doi:10.1038/jhg.2008.13
Ackerley, S., James, P. A., Kalli, A., French, S., Davies, K. E., and Talbot, K. (2006). A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum. Mol. Genet. 15, 347–354. doi:10.1093/hmg/ddi452
Ackerley, S., Thornhill, P., Grierson, A. J., Brownlees, J., Anderton, B. H., Leigh, P. N., et al. (2003). Neurofilament heavy chain side arm phosphorylation regulates axonal transport of neurofilaments. J. Cell Biol. 161, 489–495. doi:10.1083/jcb.200303138
Adebola, A. A., Di Castri, T., He, C.-Z., Salvatierra, L. A., Zhao, J., Brown, K., et al. (2015). Neurofilament light polypeptide gene N98S mutation in mice leads to neurofilament network abnormalities and a Charcot-Marie-Tooth Type 2E phenotype. Hum. Mol. Genet. 24, 2163–2174. doi:10.1093/hmg/ddu736
Agrawal, P. B., Joshi, M., Marinakis, N. S., Schmitz-Abe, K., Ciarlini, P. D. S. C., Sargent, J. C., et al. (2014). Expanding the phenotype associated with the NEFL mutation: neuromuscular disease in a family with overlapping myopathic and neurogenic findings. JAMA Neurol. 71, 1413. doi:10.1001/jamaneurol.2014.1432
Alami, N. H., Jung, P., and Brown, A. (2009). Myosin Va increases the efficiency of neurofilament transport by decreasing the duration of long-term pauses. J. Neurosci. 29, 6625–6634. doi:10.1523/JNEUROSCI.3829-08.2009
Almeida-Souza, L., Asselbergh, B., d’Ydewalle, C., Moonens, K., Goethals, S., de Winter, V., et al. (2011). Small heat-shock protein HSPB1 mutants stabilize microtubules in Charcot-Marie-Tooth neuropathy. J. Neurosci. 31, 15320–15328. doi:10.1523/JNEUROSCI.3266-11.2011
Ando, M., Higuchi, Y., Okamoto, Y., Yuan, J., Yoshimura, A., Takei, J., et al. (2022). An NEFH founder mutation causes broad phenotypic spectrum in multiple Japanese families. J. Hum. Genet. 67, 399. doi:10.1038/s10038-022-01019-y
Antic, D., Lu, N., and Keene, J. D. (1999). ELAV tumor antigen, Hel-N1, increases translation of neurofilament M mRNA and induces formation of neurites in human teratocarcinoma cells. Genes Dev. 13, 449–461. doi:10.1101/gad.13.4.449
Aruta, F., Severi, D., Iovino, A., Spina, E., Barghigiani, M., Ruggiero, L., et al. (2021). Proximal weakness involvement in the first Italian case of Charcot-Marie-Tooth 2CC harboring a novel frameshift variant in NEFH. J. Peripher Nerv. Syst. 26, 231–234. doi:10.1111/jns.12454
Balastik, M., Ferraguti, F., Pires-da Silva, A., Lee, T. H., Alvarez-Bolado, G., Lu, K. P., et al. (2008). Deficiency in ubiquitin ligase TRIM2 causes accumulation of neurofilament light chain and neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 105, 12016–12021. doi:10.1073/pnas.0802261105
Benedetti, S., Previtali, S. C., Coviello, S., Scarlato, M., Cerri, F., Di Pierri, E., et al. (2010). Analyzing histopathological features of rare charcot-marie-tooth neuropathies to unravel their pathogenesis. Arch. Neurol. 67. doi:10.1001/archneurol.2010.303
Berciano, J., García, A., Gallardo, E., Peeters, K., Pelayo-Negro, A. L., Álvarez-Paradelo, S., et al. (2017). Intermediate Charcot–Marie–Tooth disease: an electrophysiological reappraisal and systematic review. J. Neurol. 264, 1655–1677. doi:10.1007/s00415-017-8474-3
Berciano, J., García, A., Peeters, K., Gallardo, E., De Vriendt, E., Pelayo-Negro, A. L., et al. (2015). NEFL E396K mutation is associated with a novel dominant intermediate Charcot-Marie-Tooth disease phenotype. J. Neurol. 262, 1289–1300. doi:10.1007/s00415-015-7709-4
Berciano, J., Peeters, K., García, A., López-Alburquerque, T., Gallardo, E., Hernández-Fabián, A., et al. (2016). NEFL N98S mutation: another cause of dominant intermediate Charcot-Marie-Tooth disease with heterogeneous early-onset phenotype. J. Neurol. 263, 361–369. doi:10.1007/s00415-015-7985-z
Bhagavati, S., Maccabee, P. J., and Xu, W. (2009). The neurofilament light chain gene (NEFL) mutation Pro22Ser can be associated with mixed axonal and demyelinating neuropathy. J. Clin. Neurosci. 16, 830–831. doi:10.1016/j.jocn.2008.08.030
Bian, X., Lin, P., Li, J., Long, F., Duan, R., Yuan, Q., et al. (2018). Whole-genome linkage analysis with whole-exome sequencing identifies a novel frameshift variant in NEFH in a Chinese family with charcot-marie-tooth 2: a novel variant in NEFH for charcot-marie-tooth 2. Neurodegener. Dis. 18, 74–83. doi:10.1159/000487754
Binukumar, B. K., Shukla, V., Amin, N. D., Reddy, P., Skuntz, S., Grant, P., et al. (2013). Topographic regulation of neuronal intermediate filaments by phosphorylation, role of peptidyl-prolyl isomerase 1: significance in neurodegeneration. Histochem Cell Biol. 140, 23–32. doi:10.1007/s00418-013-1108-7
Black, M. M., and Lasek, R. J. (1980). Slow components of axonal transport: two cytoskeletal networks. J. Cell Biol. 86, 616–623. doi:10.1083/jcb.86.2.616
Bocquet, A., Berges, R., Frank, R., Robert, P., Peterson, A. C., and Eyer, J. (2009). Neurofilaments bind tubulin and modulate its polymerization. J. Neurosci. 29, 11043–11054. doi:10.1523/JNEUROSCI.1924-09.2009
Bomont, P., Cavalier, L., Blondeau, F., Ben Hamida, C., Belal, S., Tazir, M., et al. (2000). The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Genet. 26, 370–374. doi:10.1038/81701
Bomont, P. (2016). Degradation of the intermediate filament family by gigaxonin. Methods Enzymol. 569, 215–231. doi:10.1016/bs.mie.2015.07.009
Bomont, P. (2019). GAN (gigaxonin) E3 ligase and ATG16L1: master and commander of autophagosome production. Autophagy 15, 1650–1652. doi:10.1080/15548627.2019.1628546
Bomont, P. (2021). The dazzling rise of neurofilaments: physiological functions and roles as biomarkers. Curr. Opin. Cell Biol. 68, 181–191. doi:10.1016/j.ceb.2020.10.011
Bott, C. J., and Winckler, B. (2020). Intermediate filaments in developing neurons: beyond structure. Cytoskelet. Hob. 77, 110–128. doi:10.1002/cm.21597
Boumil, E. F., Vohnoutka, R., Lee, S., Pant, H., and Shea, T. B. (2018). Assembly and turnover of neurofilaments in growing axonal neurites. Biol. Open 7, bio028795. doi:10.1242/bio.028795
Boyer, N. P., Julien, J.-P., Jung, P., and Brown, A. (2022). Neurofilament transport is bidirectional in vivo. eNeuro 9. doi:10.1523/ENEURO.0138-22.2022
Brown, A., and Jung, P. (2013). A critical reevaluation of the stationary axonal cytoskeleton hypothesis. Cytoskelet. Hob. 70, 1–11. doi:10.1002/cm.21083
Brownlees, J., Ackerley, S., Grierson, A. J., Jacobsen, N. J. O., Shea, K., Anderton, B. H., et al. (2002). Charcot-Marie-Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport. Hum. Mol. Genet. 11, 2837–2844. doi:10.1093/hmg/11.23.2837
Campos-Melo, D., Droppelmann, C. A., He, Z., Volkening, K., and Strong, M. J. (2013). Altered microRNA expression profile in amyotrophic lateral sclerosis: a role in the regulation of NFL mRNA levels. Mol. Brain 6, 26. doi:10.1186/1756-6606-6-26
Campos-Melo, D., Hawley, Z. C. E., and Strong, M. J. (2018). Dysregulation of human NEFM and NEFH mRNA stability by ALS-linked miRNAs. Mol. Brain 11, 43. doi:10.1186/s13041-018-0386-3
Cañete-Soler, R., Reddy, K. S., Tolan, D. R., and Zhai, J. (2005). Aldolases A and C are ribonucleolytic components of a neuronal complex that regulates the stability of the light-neurofilament mRNA. J. Neurosci. 25, 4353–4364. doi:10.1523/JNEUROSCI.0885-05.2005
Carter, J., Gragerov, A., Konvicka, K., Elder, G., Weinstein, H., and Lazzarini, R. A. (1998). Neurofilament (NF) assembly; divergent characteristics of human and rodent NF-L subunits. J. Biol. Chem. 273, 5101–5108. doi:10.1074/jbc.273.9.5101
Cassereau, J., Nicolas, G., Lonchampt, P., Pinier, M., Barthelaix, A., Eyer, J., et al. (2013). Axonal regeneration is compromised in NFH-LacZ transgenic mice but not in NFH-GFP mice. Neuroscience 228, 101–108. doi:10.1016/j.neuroscience.2012.10.011
Chao, H.-C., Hsiao, C.-T., Lai, K.-L., Tsai, Y.-S., Lin, K.-P., Liao, Y.-C., et al. (2023). Clinical and genetic characterization of NEFL-related neuropathy in Taiwan. J. Formos. Med. Assoc. 122, 132–138. doi:10.1016/j.jfma.2022.08.008
Chernyatina, A. A., Guzenko, D., and Strelkov, S. V. (2015). Intermediate filament structure: the bottom-up approach. Curr. Opin. Cell Biol. 32, 65–72. doi:10.1016/j.ceb.2014.12.007
Ching, G. Y., and Liem, R. K. (1993). Assembly of type IV neuronal intermediate filaments in nonneuronal cells in the absence of preexisting cytoplasmic intermediate filaments. J. Cell Biol. 122, 1323–1335. doi:10.1083/jcb.122.6.1323
Choi, B.-O., Koo, S. K., Park, M.-H., Rhee, H., Yang, S.-J., Choi, K.-G., et al. (2012). Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth disease. Hum. Mutat. 33, 1610–1615. doi:10.1002/humu.22143
Choi, B.-O., Lee, M. S., Shin, S. H., Hwang, J. H., Choi, K.-G., Kim, W.-K., et al. (2004). Mutational analysis of PMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot-Marie-Tooth neuropathy patients. Hum. Mutat. 24, 185–186. doi:10.1002/humu.9261
Colakoğlu, G., and Brown, A. (2009). Intermediate filaments exchange subunits along their length and elongate by end-to-end annealing. J. Cell Biol. 185, 769–777. doi:10.1083/jcb.200809166
Cui, C., Stambrook, P. J., and Parysek, L. M. (1995). Peripherin assembles into homopolymers in SW13 cells. J. Cell Sci. 108, 3279–3284. doi:10.1242/jcs.108.10.3279
Dabbaghizadeh, A., Paré, A., Cheng-Boivin, Z., Dagher, R., Minotti, S., Dicaire, M.-J., et al. (2022). The J domain of Sacsin disrupts intermediate filament assembly. Int. J. Mol. Sci. 23, 15742. doi:10.3390/ijms232415742
Dale, J. M., and Garcia, M. L. (2012). Neurofilament phosphorylation during development and disease: which came first, the phosphorylation or the accumulation? J. Amino Acids, 1–10. doi:10.1155/2012/382107
Dale, J. M., Villalon, E., Shannon, S. G., Barry, D. M., Markey, R. M., Garcia, V. B., et al. (2012). Expressing hNF-LE397K results in abnormal gaiting in a transgenic model of CMT2E. Genes Brain Behav. 11, 360–365. doi:10.1111/j.1601-183X.2012.00771.x
De Jonghe, P., Mersivanova, I., Nelis, E., Del Favero, J., Martin, J. J., Van Broeckhoven, C., et al. (2001). Further evidence that neurofilament light chain gene mutations can cause Charcot-Marie-Tooth disease type 2E. Ann. Neurol. 49, 245–249. doi:10.1002/1531-8249(20010201)49:2<245::aid-ana45>3.0.co;2-a
Dequen, F., Filali, M., Larivière, R. C., Perrot, R., Hisanaga, S.-I., and Julien, J.-P. (2010). Reversal of neuropathy phenotypes in conditional mouse model of Charcot-Marie-Tooth disease type 2E. Hum. Mol. Genet. 19, 2616–2629. doi:10.1093/hmg/ddq149
Didonna, A., and Opal, P. (2019). The role of neurofilament aggregation in neurodegeneration: lessons from rare inherited neurological disorders. Mol. Neurodegener. 14, 19. doi:10.1186/s13024-019-0318-4
Donaghy, M., King, R. H., Thomas, P. K., and Workman, J. M. (1988). Abnormalities of the axonal cytoskeleton in giant axonal neuropathy. J. Neurocytol. 17, 197–208. doi:10.1007/BF01674207
Doppler, K., Kunstmann, E., Krüger, S., and Sommer, C. (2017). Painful Charcot-Marie-Tooth neuropathy type 2E/1F due to a novel NEFL mutation. Muscle Nerve 55, 752–755. doi:10.1002/mus.25410
Drew, A. P., Zhu, D., Kidambi, A., Ly, C., Tey, S., Brewer, M. H., et al. (2015). Improved inherited peripheral neuropathy genetic diagnosis by whole-exome sequencing. Mol. Genet. Genomic Med. 3, 143–154. doi:10.1002/mgg3.126
Ehlers, M. D., Fung, E. T., O’Brien, R. J., and Huganir, R. L. (1998). Splice variant-specific interaction of the NMDA receptor subunit NR1 with neuronal intermediate filaments. J. Neurosci. 18, 720–730. doi:10.1523/JNEUROSCI.18-02-00720.1998
Elbracht, M., Senderek, J., Schara, U., Nolte, K., Klopstock, T., Roos, A., et al. (2014). Clinical and morphological variability of the E396K mutation in the neurofilament light chain gene in patients with Charcot-Marie- Tooth disease type 2E. Clin. Neuropathol. 33, 335–343. doi:10.5414/NP300742
Elder, G. A., Friedrich, V. L., Bosco, P., Kang, C., Gourov, A., Tu, P. H., et al. (1998). Absence of the mid-sized neurofilament subunit decreases axonal calibers, levels of light neurofilament (NF-L), and neurofilament content. J. Cell Biol. 141, 727–739. doi:10.1083/jcb.141.3.727
Eriksson, J. E., Dechat, T., Grin, B., Helfand, B., Mendez, M., Pallari, H.-M., et al. (2009). Introducing intermediate filaments: from discovery to disease. J. Clin. Invest. 119, 1763–1771. doi:10.1172/JCI38339
Evgrafov, O. V., Mersiyanova, I., Irobi, J., Van Den Bosch, L., Dierick, I., Leung, C. L., et al. (2004). Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 36, 602–606. doi:10.1038/ng1354
Eyer, J., and Peterson, A. (1994). Neurofilament-deficient axons and perikaryal aggregates in viable transgenic mice expressing a neurofilament-beta-galactosidase fusion protein. Neuron 12, 389–405. doi:10.1016/0896-6273(94)90280-1
Fabrizi, C., Kelly, B. M., Gillespie, C. S., Schlaepfer, W. W., Scherer, S. S., and Brophy, P. J. (1997). Transient expression of the neurofilament proteins NF-L and NF-M by Schwann cells is regulated by axonal contact. J. Neurosci. Res. 50, 291–299. doi:10.1002/(SICI)1097-4547(19971015)50:2<291::AID-JNR17>3.0.CO;2-B
Fabrizi, G. M., Cavallaro, T., Angiari, C., Bertolasi, L., Cabrini, I., Ferrarini, M., et al. (2004). Giant axon and neurofilament accumulation in Charcot-Marie-Tooth disease type 2E. Neurology 62, 1429–1431. doi:10.1212/01.wnl.0000120664.07186.3c
Fabrizi, G. M., Cavallaro, T., Angiari, C., Cabrini, I., Taioli, F., Malerba, G., et al. (2007). Charcot-Marie-Tooth disease type 2E, a disorder of the cytoskeleton. Brain 130, 394–403. doi:10.1093/brain/awl284
Feliciano, C. M., Wu, K., Watry, H. L., Marley, C. B. E., Ramadoss, G. N., Ghanim, H. Y., et al. (2021). Allele-specific gene editing rescues pathology in a human model of charcot-marie-tooth disease type 2E. Front. Cell Dev. Biol. 9, 723023. doi:10.3389/fcell.2021.723023
Fenn, J. D., Johnson, C. M., Peng, J., Jung, P., and Brown, A. (2018). Kymograph analysis with high temporal resolution reveals new features of neurofilament transport kinetics. Cytoskelet. Hob. 75, 22–41. doi:10.1002/cm.21411
Fenn, J. D., Li, Y., Julien, J.-P., Jung, P., and Brown, A. (2023). The mobility of neurofilaments in mature myelinated axons of adult mice. eNeuro 10. doi:10.1523/ENEURO.0029-23.2023
Filali, M., Dequen, F., Lalonde, R., and Julien, J.-P. (2011). Sensorimotor and cognitive function of a NEFL(P22S) mutant model of Charcot-Marie-Tooth disease type 2E. Behav. Brain Res. 219, 175–180. doi:10.1016/j.bbr.2010.12.022
Fu, J., and Yuan, Y. (2018). A novel homozygous nonsense mutation in NEFL causes autosomal recessive Charcot–Marie–Tooth disease. Neuromuscul. Disord. 28, 44–47. doi:10.1016/j.nmd.2017.09.018
Gafson, A. R., Barthélemy, N. R., Bomont, P., Carare, R. O., Durham, H. D., Julien, J.-P., et al. (2020). Neurofilaments: neurobiological foundations for biomarker applications. Brain 143, 1975. doi:10.1093/brain/awaa098
Ganay, T., Boizot, A., Burrer, R., Chauvin, J. P., and Bomont, P. (2011). Sensory-motor deficits and neurofilament disorganization in gigaxonin-null mice. Mol. Neurodegener. 6, 25. doi:10.1186/1750-1326-6-25
Garcia, M. L., Lobsiger, C. S., Shah, S. B., Deerinck, T. J., Crum, J., Young, D., et al. (2003). NF-M is an essential target for the myelin-directed “outside-in” signaling cascade that mediates radial axonal growth. J. Cell Biol. 163, 1011–1020. doi:10.1083/jcb.200308159
Garcia, M. L., Rao, M. V., Fujimoto, J., Garcia, V. B., Shah, S. B., Crum, J., et al. (2009). Phosphorylation of highly conserved neurofilament medium KSP repeats is not required for myelin-dependent radial axonal growth. J. Neurosci. 29, 1277–1284. doi:10.1523/JNEUROSCI.3765-08.2009
Geisler, N., and Weber, K. (1981). Self-assembly in vitro of the 68,000 molecular weight component of the mammalian neurofilament triplet proteins into intermediate-sized filaments. J. Mol. Biol. 151, 565–571. doi:10.1016/0022-2836(81)90011-5
Gentil, B. J., Minotti, S., Beange, M., Baloh, R. H., Julien, J.-P., and Durham, H. D. (2012). Normal role of the low-molecular-weight neurofilament protein in mitochondrial dynamics and disruption in Charcot-Marie-Tooth disease. FASEB J. 26, 1194–1203. doi:10.1096/fj.11-196345
Gentil, B. J., Mushynski, W. E., and Durham, H. D. (2013). Heterogeneity in the properties of NEFL mutants causing Charcot-Marie-Tooth disease results in differential effects on neurofilament assembly and susceptibility to intervention by the chaperone-inducer, celastrol. Int. J. Biochem. Cell Biol. 45, 1499–1508. doi:10.1016/j.biocel.2013.04.009
Georgiou, D.-M., Zidar, J., Korosec, M., Middleton, L. T., Kyriakides, T., and Christodoulou, K. (2002). A novel NF-L mutation Pro22Ser is associated with CMT2 in a large Slovenian family. Neurogenetics 4, 93–96. doi:10.1007/s10048-002-0138-4
Glass, J. D., Schryer, B. L., and Griffin, J. W. (1994). Calcium-mediated degeneration of the axonal cytoskeleton in the Ola mouse. J. Neurochem. 62, 2472–2475. doi:10.1046/j.1471-4159.1994.62062472.x
Grazi, E., Schwienbacher, C., and Magri, E. (1993). Osmotic stress is the main determinant of the diameter of the actin filament. Biochem. Biophys. Res. Commun. 197, 1377–1381. doi:10.1006/bbrc.1993.2629
Haidar, M., Asselbergh, B., Adriaenssens, E., De Winter, V., Timmermans, J.-P., Auer-Grumbach, M., et al. (2019). Neuropathy-causing mutations in HSPB1 impair autophagy by disturbing the formation of SQSTM1/p62 bodies. Autophagy 15, 1051–1068. doi:10.1080/15548627.2019.1569930
Hashiguchi, A., Higuchi, Y., Nomura, M., Nakamura, T., Arata, H., Yuan, J., et al. (2014). Neurofilament light mutation causes hereditary motor and sensory neuropathy with pyramidal signs. J. Peripher Nerv. Syst. 19, 311–316. doi:10.1111/jns.12102
He, Y., Francis, F., Myers, K. A., Yu, W., Black, M. M., and Baas, P. W. (2005). Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J. Cell Biol. 168, 697–703. doi:10.1083/jcb.200407191
Herrmann, H., and Aebi, U. (2016). Intermediate filaments: structure and assembly. Cold Spring Harb. Perspect. Biol. 8, a018242. doi:10.1101/cshperspect.a018242
Herrmann, H., Häner, M., Brettel, M., Müller, S. A., Goldie, K. N., Fedtke, B., et al. (1996). Structure and assembly properties of the intermediate filament protein vimentin: the role of its head, rod and tail domains. J. Mol. Biol. 264, 933–953. doi:10.1006/jmbi.1996.0688
Herrmann, H., Strelkov, S. V., Burkhard, P., and Aebi, U. (2009). Intermediate filaments: primary determinants of cell architecture and plasticity. J. Clin. Invest. 119, 1772–1783. doi:10.1172/JCI38214
Hirokawa, N. (1982). Cross-linker system between neurofilaments, microtubules, and membranous organelles in frog axons revealed by the quick-freeze, deep-etching method. J. Cell Biol. 94, 129–142. doi:10.1083/jcb.94.1.129
Hisanaga, S., and Hirokawa, N. (1988). Structure of the peripheral domains of neurofilaments revealed by low angle rotary shadowing. J. Mol. Biol. 202, 297–305. doi:10.1016/0022-2836(88)90459-7
Holmgren, A., Bouhy, D., De Winter, V., Asselbergh, B., Timmermans, J.-P., Irobi, J., et al. (2013). Charcot-Marie-Tooth causing HSPB1 mutations increase Cdk5-mediated phosphorylation of neurofilaments. Acta Neuropathol. 126, 93–108. doi:10.1007/s00401-013-1133-6
Holmgren, A., Bouhy, D., and Timmerman, V. (2012). Neurofilament phosphorylation and their proline-directed kinases in health and disease. J. Peripher Nerv. Syst. 17, 365–376. doi:10.1111/j.1529-8027.2012.00434.x
Horga, A., Laurà, M., Jaunmuktane, Z., Jerath, N. U., Gonzalez, M. A., Polke, J. M., et al. (2017). Genetic and clinical characteristics of NEFL-related Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 88, 575–585. doi:10.1136/jnnp-2016-315077
Ikenberg, E., Reilich, P., Abicht, A., Heller, C., Schoser, B., and Walter, M. C. (2019). Charcot-Marie-Tooth disease type 2CC due to a frameshift mutation of the neurofilament heavy polypeptide gene in an Austrian family. Neuromuscul. Disord. 29, 392–397. doi:10.1016/j.nmd.2019.02.007
Ishtiaq, M., Campos-Melo, D., Volkening, K., and Strong, M. J. (2014). Analysis of novel NEFL mRNA targeting microRNAs in amyotrophic lateral sclerosis. PLoS ONE 9, e85653. doi:10.1371/journal.pone.0085653
Israeli, E., Dryanovski, D. I., Schumacker, P. T., Chandel, N. S., Singer, J. D., Julien, J. P., et al. (2016). Intermediate filament aggregates cause mitochondrial dysmotility and increase energy demands in giant axonal neuropathy. Hum. Mol. Genet. 25, 2143–2157. doi:10.1093/hmg/ddw081
Jacomy, H., Zhu, Q., Couillard-Després, S., Beaulieu, J. M., and Julien, J. P. (1999). Disruption of type IV intermediate filament network in mice lacking the neurofilament medium and heavy subunits. J. Neurochem. 73, 972–984. doi:10.1046/j.1471-4159.1999.0730972.x
Jacquier, A., Delorme, C., Belotti, E., Juntas-Morales, R., Solé, G., Dubourg, O., et al. (2017). Cryptic amyloidogenic elements in mutant NEFH causing Charcot-Marie-Tooth 2 trigger aggresome formation and neuronal death. Acta Neuropathol. Commun. 5, 55. doi:10.1186/s40478-017-0457-1
Jordanova, A., De Jonghe, P., Boerkoel, C. F., Takashima, H., De Vriendt, E., Ceuterick, C., et al. (2003). Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain 126, 590–597. doi:10.1093/brain/awg059
Jung, C., Lee, S., Ortiz, D., Zhu, Q., Julien, J.-P., and Shea, T. B. (2005). The high and middle molecular weight neurofilament subunits regulate the association of neurofilaments with kinesin: inhibition by phosphorylation of the high molecular weight subunit. Brain Res. Mol. Brain Res. 141, 151–155. doi:10.1016/j.molbrainres.2005.08.009
Jung, C., Yabe, J. T., Lee, S., and Shea, T. B. (2000a). Hypophosphorylated neurofilament subunits undergo axonal transport more rapidly than more extensively phosphorylated subunits in situ. Cell Motil. Cytoskelet. 47, 120–129. doi:10.1002/1097-0169(200010)47:2<120::AID-CM3>3.0.CO;2-6
Jung, C., Yabe, J. T., and Shea, T. B. (2000b). C-terminal phosphorylation of the high molecular weight neurofilament subunit correlates with decreased neurofilament axonal transport velocity. Brain Res. 856, 12–19. doi:10.1016/s0006-8993(99)02314-8
Juźwik, C. A., S Drake, S., Zhang, Y., Paradis-Isler, N., Sylvester, A., Amar-Zifkin, A., et al. (2019). microRNA dysregulation in neurodegenerative diseases: a systematic review. Prog. Neurobiol. 182, 101664. doi:10.1016/j.pneurobio.2019.101664
Kelly, B. M., Gillespie, C. S., Sherman, D. L., and Brophy, P. J. (1992). Schwann cells of the myelin-forming phenotype express neurofilament protein NF-M. J. Cell Biol. 118, 397–410. doi:10.1083/jcb.118.2.397
Kim, H. J., Kim, S. B., Kim, H. S., Kwon, H. M., Park, J. H., Lee, A. J., et al. (2022). Phenotypic heterogeneity in patients with NEFL-related Charcot-Marie-Tooth disease. Mol. Genet. Genomic Med. 10, e1870. doi:10.1002/mgg3.1870
Kim, H. S., Kim, H. J., Nam, S. H., Kim, S. B., Choi, Y. J., Lee, K. S., et al. (2021). Clinical and neuroimaging features in charcot-marie-tooth patients with GDAP1 mutations. J. Clin. Neurol. 17, 52–62. doi:10.3988/jcn.2021.17.1.52
Kim, O.-J., Ariano, M. A., Lazzarini, R. A., Levine, M. S., and Sibley, D. R. (2002). Neurofilament-M interacts with the D1 dopamine receptor to regulate cell surface expression and desensitization. J. Neurosci. 22, 5920–5930. doi:10.1523/JNEUROSCI.22-14-05920.2002
Kirkcaldie, M. T. K., and Dwyer, S. T. (2017). The third wave: intermediate filaments in the maturing nervous system. Mol. Cell. Neurosci. 84, 68–76. doi:10.1016/j.mcn.2017.05.010
Kreplak, L., Bär, H., Leterrier, J. F., Herrmann, H., and Aebi, U. (2005). Exploring the mechanical behavior of single intermediate filaments. J. Mol. Biol. 354, 569–577. doi:10.1016/j.jmb.2005.09.092
Lagier-Tourenne, C., Polymenidou, M., Hutt, K. R., Vu, A. Q., Baughn, M., Huelga, S. C., et al. (2012). Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15, 1488–1497. doi:10.1038/nn.3230
Lancaster, E., Li, J., Hanania, T., Liem, R., Scheideler, M. A., and Scherer, S. S. (2018). Myelinated axons fail to develop properly in a genetically authentic mouse model of Charcot-Marie-Tooth disease type 2E. Exp. Neurol. 308, 13–25. doi:10.1016/j.expneurol.2018.06.010
Lariviere, R. C., and Julien, J.-P. (2004). Functions of intermediate filaments in neuronal development and disease. J. Neurobiol. 58, 131–148. doi:10.1002/neu.10270
Laurá, M., Pipis, M., Rossor, A. M., and Reilly, M. M. (2019). Charcot–Marie–Tooth disease and related disorders: an evolving landscape. Curr. Opin. Neurology 32, 641–650. doi:10.1097/WCO.0000000000000735
Lee, I. B., Kim, S. K., Chung, S. H., Kim, H., Kwon, T. K., Min, D. S., et al. (2008). The effect of rod domain A148V mutation of neurofilament light chain on filament formation. BMB Reports 41 (12), 868–874. doi:10.5483/bmbrep.2008.41.12.868
Lee, M. K., Marszalek, J. R., and Cleveland, D. W. (1994). A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron 13, 975–988. doi:10.1016/0896-6273(94)90263-1
Lee, M. K., Xu, Z., Wong, P. C., and Cleveland, D. W. (1993). Neurofilaments are obligate heteropolymers in vivo. J. Cell Biol. 122 (6), 1337–1350. doi:10.1083/jcb.122.6.1337
Lerat, J., Magdelaine, C., Beauvais-Dzugan, H., Espil, C., Ghorab, K., Latour, P., et al. (2019). A novel pathogenic variant of NEFL responsible for deafness associated with peripheral neuropathy discovered through next-generation sequencing and review of the literature. J. Peripher Nerv. Syst. 24, 139–144. doi:10.1111/jns.12310
Lescouzères, L., and Bomont, P. (2020). E3 ubiquitin ligases in neurological diseases: focus on gigaxonin and autophagy. Front. Physiol. 11, 1022. doi:10.3389/fphys.2020.01022
Leung, C. L., Nagan, N., Graham, T. H., and Liem, R. K. H. (2006). A novel duplication/insertion mutation ofNEFL in a patient with Charcot-Marie-Tooth disease. Am. J. Med. Genet. 140A, 1021–1025. doi:10.1002/ajmg.a.31242
Lewis, S. E., and Nixon, R. A. (1988). Multiple phosphorylated variants of the high molecular mass subunit of neurofilaments in axons of retinal cell neurons: characterization and evidence for their differential association with stationary and moving neurofilaments. J. Cell Biol. 107, 2689–2701. doi:10.1083/jcb.107.6.2689
Li, J. J., Sarute, N., Lancaster, E., Otkiran-Clare, G., Fagla, B. M., Ross, S. R., et al. (2020). A recessive Trim2 mutation causes an axonal neuropathy in mice. Neurobiol. Dis. 140, 104845. doi:10.1016/j.nbd.2020.104845
Liem, R. K. H., and Messing, A. (2009). Dysfunctions of neuronal and glial intermediate filaments in disease. J. Clin. Invest. 119, 1814–1824. doi:10.1172/JCI38003
Lin, K.-P., Soong, B.-W., Yang, C.-C., Huang, L.-W., Chang, M.-H., Lee, I.-H., et al. (2011). The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One 6, e29393. doi:10.1371/journal.pone.0029393
Lus, G., Nelis, E., Jordanova, A., Löfgren, A., Cavallaro, T., Ammendola, A., et al. (2003). Charcot-Marie-Tooth disease with giant axons: a clinicopathological and genetic entity. Neurology 61, 988–990. doi:10.1212/wnl.61.7.988
Ma, M., Ferguson, T. A., Schoch, K. M., Li, J., Qian, Y., Shofer, F. S., et al. (2013). Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol. Dis. 56, 34–46. doi:10.1016/j.nbd.2013.03.009
Machado, R., Pinto-Basto, J., and Negrão, L. (2019). The first Portuguese family with NEFL-related Charcot-Marie-Tooth type 2 disease. Acta Myol. 38, 180–183.
Maciel, R., Correa, R., Bosso Taniguchi, J., Prufer Araujo, I., and Saporta, M. A. (2020). Human tridimensional neuronal cultures for phenotypic drug screening in inherited peripheral neuropathies. Clin. Pharmacol. Ther. 107, 1231–1239. doi:10.1002/cpt.1718
MacTaggart, B., and Kashina, A. (2021). Posttranslational modifications of the cytoskeleton. Cytoskelet. Hob. 78, 142–173. doi:10.1002/cm.21679
Mahammad, S., Murthy, S. N. P., Didonna, A., Grin, B., Israeli, E., Perrot, R., et al. (2013). Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J. Clin. Invest. 123, 1964–1975. doi:10.1172/JCI66387
Manganelli, F., Tozza, S., Pisciotta, C., Bellone, E., Iodice, R., Nolano, M., et al. (2014). Charcot-Marie-Tooth disease: frequency of genetic subtypes in a Southern Italy population. J. Peripher Nerv. Syst. 19, 292–298. doi:10.1111/jns.12092
Mersiyanova, I. V., Perepelov, A. V., Polyakov, A. V., Sitnikov, V. F., Dadali, E. L., Oparin, R. B., et al. (2000). A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am. J. Hum. Genet. 67, 37–46. doi:10.1086/302962
Miao, L., Teng, J., Lin, J., Liao, X., and Chen, J. (2013). 14-3-3 proteins interact with neurofilament protein-L and regulate dynamic assembly of neurofilaments. J. Cell Sci. 126, 427–436. doi:10.1242/jcs.105817
Millecamps, S., Gowing, G., Corti, O., Mallet, J., and Julien, J.-P. (2007). Conditional NF-L transgene expression in mice for in vivo analysis of turnover and transport rate of neurofilaments. J. Neurosci. 27, 4947–4956. doi:10.1523/JNEUROSCI.5299-06.2007
Miltenberger-Miltenyi, G., Janecke, A. R., Wanschitz, J. V., Timmerman, V., Windpassinger, C., Auer-Grumbach, M., et al. (2007). Clinical and electrophysiological features in Charcot-Marie-Tooth disease with mutations in the NEFL gene. Arch. Neurol. 64, 966–970. doi:10.1001/archneur.64.7.966
Monsma, P. C., Li, Y., Fenn, J. D., Jung, P., and Brown, A. (2014). Local regulation of neurofilament transport by myelinating cells. J. Neurosci. 34, 2979–2988. doi:10.1523/JNEUROSCI.4502-13.2014
Nakagawa, T., Chen, J., Zhang, Z., Kanai, Y., and Hirokawa, N. (1995). Two distinct functions of the carboxyl-terminal tail domain of NF-M upon neurofilament assembly: cross-bridge formation and longitudinal elongation of filaments. J. Cell Biol. 129, 411–429. doi:10.1083/jcb.129.2.411
Nam, D. E., Jung, S.-C., Yoo, D. H., Choi, S. S., Seo, S.-Y., Kim, G. H., et al. (2017). Axonal Charcot-Marie-Tooth neuropathy concurrent with distal and proximal weakness by translational elongation of the 3′ UTR in NEFH: nam et al. J. Peripher Nerv. Syst. 22, 200–207. doi:10.1111/jns.12223
Nath, B., Phaneuf, D., and Julien, J.-P. (2023). Axonal transport defect in gigaxonin deficiency rescued by tubastatin A. Neurotherapeutics. doi:10.1007/s13311-023-01393-1
Nefedova, V. V., Sudnitsyna, M. V., and Gusev, N. B. (2017). Interaction of small heat shock proteins with light component of neurofilaments (NFL). Cell Stress Chaperones 22, 467–479. doi:10.1007/s12192-016-0757-6
Nguyen, M. D., Shu, T., Sanada, K., Larivière, R. C., Tseng, H.-C., Park, S. K., et al. (2004). A NUDEL-dependent mechanism of neurofilament assembly regulates the integrity of CNS neurons. Nat. Cell Biol. 6, 595–608. doi:10.1038/ncb1139
Nixon, R. A., and Logvinenko, K. B. (1986). Multiple fates of newly synthesized neurofilament proteins: evidence for a stationary neurofilament network distributed nonuniformly along axons of retinal ganglion cell neurons. J. Cell Biol. 102, 647–659. doi:10.1083/jcb.102.2.647
Omary, M. B. (2009). IF-pathies: a broad spectrum of intermediate filament-associated diseases. J. Clin. Invest. 119, 1756–1762. doi:10.1172/JCI39894
Pant, H. C., Shecket, G., Gainer, H., and Lasek, R. J. (1978). Neurofilament protein is phosphorylated in the squid giant axon. J. Cell Biol. 78, R23–R27. doi:10.1083/jcb.78.2.r23
Park, J. Y., Jang, S. Y., Shin, Y. K., Koh, H., Suh, D. J., Shinji, T., et al. (2013a). Mitochondrial swelling and microtubule depolymerization are associated with energy depletion in axon degeneration. Neuroscience 238, 258–269. doi:10.1016/j.neuroscience.2013.02.033
Park, J. Y., Jang, S. Y., Shin, Y. K., Suh, D. J., and Park, H. T. (2013b). Calcium-dependent proteasome activation is required for axonal neurofilament degradation. Neural Regen. Res. 8, 3401–3409. doi:10.3969/j.issn.1673-5374.2013.36.005
Perez-Olle, R., Jones, S. T., and Liem, R. K. H. (2004). Phenotypic analysis of neurofilament light gene mutations linked to Charcot-Marie-Tooth disease in cell culture models. Hum. Mol. Genet. 13, 2207–2220. doi:10.1093/hmg/ddh236
Perez-Olle, R., Leung, C. L., and Liem, R. K. H. (2002). Effects of Charcot-Marie-Tooth-linked mutations of the neurofilament light subunit on intermediate filament formation. J. Cell Sci. 115, 4937–4946. doi:10.1242/jcs.00148
Perez-Olle, R., Lopez-Toledano, M. A., Goryunov, D., Cabrera-Poch, N., Stefanis, L., Brown, K., et al. (2005). Mutations in the neurofilament light gene linked to Charcot-Marie-Tooth disease cause defects in transport. J. Neurochem. 93, 861–874. doi:10.1111/j.1471-4159.2005.03095.x
Perrot, R., and Eyer, J. (2009). Neuronal intermediate filaments and neurodegenerative disorders. Brain Res. Bull. 80, 282–295. doi:10.1016/j.brainresbull.2009.06.004
Perrot, R., and Julien, J.-P. (2009). Real-time imaging reveals defects of fast axonal transport induced by disorganization of intermediate filaments. FASEB J. 23, 3213–3225. doi:10.1096/fj.09-129585
Peter, A., and Stick, R. (2015). Evolutionary aspects in intermediate filament proteins. Curr. Opin. Cell Biol. 32, 48–55. doi:10.1016/j.ceb.2014.12.009
Petrucci, A., Lispi, L., Garibaldi, M., Frezza, E., Moro, F., Massa, R., et al. (2023). NEFL-related charcot-marie tooth disease due to P440L mutation in two Italian families: expanding the phenotype and defining modulating factors. Eur. Neurol. 86, 185–192. doi:10.1159/000529706
Pipis, M., Cortese, A., Polke, J. M., Poh, R., Vandrovcova, J., Laura, M., et al. (2022). Charcot-Marie-Tooth disease type 2CC due to NEFH variants causes a progressive, non-length-dependent, motor-predominant phenotype. J. Neurol. Neurosurg. Psychiatry 93, 48–56. doi:10.1136/jnnp-2021-327186
Pipis, M., Rossor, A. M., Laura, M., and Reilly, M. M. (2019). Next-generation sequencing in Charcot-Marie-Tooth disease: opportunities and challenges. Nat. Rev. Neurol. 15, 644–656. doi:10.1038/s41582-019-0254-5
Rao, M. V., Campbell, J., Yuan, A., Kumar, A., Gotow, T., Uchiyama, Y., et al. (2003). The neurofilament middle molecular mass subunit carboxyl-terminal tail domains is essential for the radial growth and cytoskeletal architecture of axons but not for regulating neurofilament transport rate. J. Cell Biol. 163, 1021–1031. doi:10.1083/jcb.200308076
Rao, M. V., Darji, S., Stavrides, P. H., Goulbourne, C. N., Kumar, A., Yang, D.-S., et al. (2023). Autophagy is a novel pathway for neurofilament protein degradation in vivo. Autophagy 19 (4), 1277–1292. doi:10.1080/15548627.2022.2124500
Rao, M. V., Engle, L. J., Mohan, P. S., Yuan, A., Qiu, D., Cataldo, A., et al. (2002a). Myosin Va binding to neurofilaments is essential for correct myosin Va distribution and transport and neurofilament density. J. Cell Biol. 159, 279–290. doi:10.1083/jcb.200205062
Rao, M. V., Garcia, M. L., Miyazaki, Y., Gotow, T., Yuan, A., Mattina, S., et al. (2002b). Gene replacement in mice reveals that the heavily phosphorylated tail of neurofilament heavy subunit does not affect axonal caliber or the transit of cargoes in slow axonal transport. J. Cell Biol. 158, 681–693. doi:10.1083/jcb.200202037
Rao, M. V., Mohan, P. S., Kumar, A., Yuan, A., Montagna, L., Campbell, J., et al. (2011). The myosin Va head domain binds to the neurofilament-L rod and modulates endoplasmic reticulum (ER) content and distribution within axons. PLoS One 6, e17087. doi:10.1371/journal.pone.0017087
Rao, M. V., Yuan, A., Campbell, J., Kumar, A., and Nixon, R. A. (2012). The C-terminal domains of NF-H and NF-M subunits maintain axonal neurofilament content by blocking turnover of the stationary neurofilament network. PLoS One 7, e44320. doi:10.1371/journal.pone.0044320
Rebelo, A. P., Abrams, A. J., Cottenie, E., Horga, A., Gonzalez, M., Bis, D. M., et al. (2016). Cryptic amyloidogenic elements in the 3′ UTRs of neurofilament genes trigger axonal neuropathy. Am. J. Hum. Genet. 98, 597–614. doi:10.1016/j.ajhg.2016.02.022
Renganathan, B., Zewe, J. P., Cheng, Y., Paumier, J., Kittisopikul, M., Ridge, K. M., et al. (2023). Gigaxonin is required for intermediate filament transport. FASEB J. 37, e22886. doi:10.1096/fj.202202119R
Roberson, M. D., Toews, A. D., Goodrum, J. F., and Morell, P. (1992). Neurofilament and tubulin mRNA expression in Schwann cells. J. Neurosci. Res. 33, 156–162. doi:10.1002/jnr.490330120
Rossor, A. M., Polke, J. M., Houlden, H., and Reilly, M. M. (2013). Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat. Rev. Neurol. 9, 562–571. doi:10.1038/nrneurol.2013.179
Roy, S., Coffee, P., Smith, G., Liem, R. K., Brady, S. T., and Black, M. M. (2000). Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J. Neurosci. 20, 6849–6861. doi:10.1523/JNEUROSCI.20-18-06849.2000
Rudnik-Schöneborn, S., Auer-Grumbach, M., and Senderek, J. (2020). Charcot-Marie-Tooth disease and hereditary motor neuropathies – update 2020. Med. Genet. 32, 207–219. doi:10.1515/medgen-2020-2038
Sainio, M. T., Rasila, T., Molchanova, S. M., Järvilehto, J., Torregrosa-Muñumer, R., Harjuhaahto, S., et al. (2021). Neurofilament light regulates axon caliber, synaptic activity, and organelle trafficking in cultured human motor neurons. Front. Cell Dev. Biol. 9, 820105. doi:10.3389/fcell.2021.820105
Sainio, M. T., Ylikallio, E., Mäenpää, L., Lahtela, J., Mattila, P., Auranen, M., et al. (2018). Absence of NEFL in patient-specific neurons in early-onset Charcot-Marie-Tooth neuropathy. Neurol. Genet. 4, e244. doi:10.1212/NXG.0000000000000244
Sakaguchi, T., Okada, M., Kitamura, T., and Kawasaki, K. (1993). Reduced diameter and conduction velocity of myelinated fibers in the sciatic nerve of a neurofilament-deficient mutant quail. Neurosci. Lett. 153, 65–68. doi:10.1016/0304-3940(93)90078-y
Saporta, M. A., Dang, V., Volfson, D., Zou, B., Xie, X. S., Adebola, A., et al. (2015). Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp. Neurol. 263, 190–199. doi:10.1016/j.expneurol.2014.10.005
Sapra, K. T., and Medalia, O. (2021). Bend, push, stretch: remarkable structure and mechanics of single intermediate filaments and meshworks. Cells 10, 1960. doi:10.3390/cells10081960
Sasaki, T., Gotow, T., Shiozaki, M., Sakaue, F., Saito, T., Julien, J.-P., et al. (2006). Aggregate formation and phosphorylation of neurofilament-L Pro22 Charcot-Marie-Tooth disease mutants. Hum. Mol. Genet. 15, 943–952. doi:10.1093/hmg/ddl011
Scrivo, A., Codogno, P., and Bomont, P. (2019). Gigaxonin E3 ligase governs ATG16L1 turnover to control autophagosome production. Nat. Commun. 10, 780. doi:10.1038/s41467-019-08331-w
Shea, T. B., and Flanagan, L. A. (2001). Kinesin, dynein and neurofilament transport. Trends Neurosci. 24, 644–648. doi:10.1016/s0166-2236(00)01919-6
Shea, T. B. (2000). Microtubule motors, phosphorylation and axonal transport of neurofilaments. J. Neurocytol. 29, 873–887. doi:10.1023/a:1010951626090
Shen, H., Barry, D. M., Dale, J. M., Garcia, V. B., Calcutt, N. A., and Garcia, M. L. (2011). Muscle pathology without severe nerve pathology in a new mouse model of Charcot–Marie–Tooth disease type 2E. Hum. Mol. Genet. 20, 2535–2548. doi:10.1093/hmg/ddr152
Shin, J. S., Chung, K. W., Cho, S. Y., Yun, J., Hwang, S. J., Kang, S. H., et al. (2008). NEFL Pro22Arg mutation in Charcot-Marie-Tooth disease type 1. J. Hum. Genet. 53, 936–940. doi:10.1007/s10038-008-0333-8
Sihag, R. K., Inagaki, M., Yamaguchi, T., Shea, T. B., and Pant, H. C. (2007). Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp. Cell Res. 313, 2098–2109. doi:10.1016/j.yexcr.2007.04.010
Sivera, R., Sevilla, T., Vílchez, J. J., Martínez-Rubio, D., Chumillas, M. J., Vázquez, J. F., et al. (2013). Charcot-Marie-Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology 81, 1617–1625. doi:10.1212/WNL.0b013e3182a9f56a
Stone, E. J., Kolb, S. J., and Brown, A. (2021). A review and analysis of the clinical literature on Charcot–Marie–Tooth disease caused by mutations in neurofilament protein L. Cytoskeleton 78, 97–110. doi:10.1002/cm.21676
Stone, E. J., Uchida, A., and Brown, A. (2019). Charcot–Marie–Tooth disease Type 2E/1F mutant neurofilament proteins assemble into neurofilaments. Cytoskeleton 76, 423–439. doi:10.1002/cm.21566
Strong, M. J., Volkening, K., Hammond, R., Yang, W., Strong, W., Leystra-Lantz, C., et al. (2007). TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci. 35, 320–327. doi:10.1016/j.mcn.2007.03.007
Thyagarajan, A., and Szaro, B. G. (2008). Dynamic endogenous association of neurofilament mRNAs with K-homology domain ribonucleoproteins in developing cerebral cortex. Brain Res. 1189, 33–42. doi:10.1016/j.brainres.2007.11.012
Thyagarajan, A., and Szaro, B. G. (2004). Phylogenetically conserved binding of specific K homology domain proteins to the 3′-untranslated region of the vertebrate middle neurofilament mRNA. J. Biol. Chem. 279, 49680–49688. doi:10.1074/jbc.M408915200
Tradewell, M. L., Durham, H. D., Mushynski, W. E., and Gentil, B. J. (2009). Mitochondrial and axonal abnormalities precede disruption of the neurofilament network in a model of charcot-marie-tooth disease type 2E and are prevented by heat shock proteins in a mutant-specific fashion. J. Neuropathol. Exp. Neurol. 68, 642–652. doi:10.1097/NEN.0b013e3181a5deeb
Trivedi, N., Jung, P., and Brown, A. (2007). Neurofilaments switch between distinct mobile and stationary states during their transport along axons. J. Neurosci. 27, 507–516. doi:10.1523/JNEUROSCI.4227-06.2007
Uchida, A., Alami, N. H., and Brown, A. (2009). Tight functional coupling of kinesin-1A and dynein motors in the bidirectional transport of neurofilaments. Mol. Biol. Cell 20, 4997–5006. doi:10.1091/mbc.e09-04-0304
Uchida, A., Çolakoğlu, G., Wang, L., Monsma, P. C., and Brown, A. (2013). Severing and end-to-end annealing of neurofilaments in neurons. Proc. Natl. Acad. Sci. U. S. A. 110, E2696–E2705. doi:10.1073/pnas.1221835110
Van Lent, J., Verstraelen, P., Asselbergh, B., Adriaenssens, E., Mateiu, L., Verbist, C., et al. (2021). Induced pluripotent stem cell-derived motor neurons of CMT type 2 patients reveal progressive mitochondrial dysfunction. Brain 144, 2471–2485. doi:10.1093/brain/awab226
Vendredy, L., Adriaenssens, E., and Timmerman, V. (2020). Small heat shock proteins in neurodegenerative diseases. Cell Stress Chaperones 25, 679–699. doi:10.1007/s12192-020-01101-4
Villalón, E., Barry, D. M., Byers, N., Frizzi, K., Jones, M. R., Landayan, D. S., et al. (2018). Internode length is reduced during myelination and remyelination by neurofilament medium phosphorylation in motor axons. Exp. Neurol. 306, 158–168. doi:10.1016/j.expneurol.2018.05.009
Villalón, E., Dale, J. M., Jones, M., Shen, H., and Garcia, M. L. (2015). Exacerbation of Charcot-Marie-Tooth type 2E neuropathy following traumatic nerve injury. Brain Res. 1627, 143–153. doi:10.1016/j.brainres.2015.09.024
Villalón, E., Jones, M. R., Sibigtroth, C., Zino, S. J., Dale, J. M., Landayan, D. S., et al. (2017). Muscle spindle alterations precede onset of sensorimotor deficits in Charcot-Marie-Tooth type 2E. Genes Brain Behav. 16, 260–270. doi:10.1111/gbb.12341
Wade, R. H. (2009). On and around microtubules: an overview. Mol. Biotechnol. 43, 177–191. doi:10.1007/s12033-009-9193-5
Wagner, O. I., Ascaño, J., Tokito, M., Leterrier, J.-F., Janmey, P. A., and Holzbaur, E. L. F. (2004). The interaction of neurofilaments with the microtubule motor cytoplasmic dynein. Mol. Biol. Cell 15, 5092–5100. doi:10.1091/mbc.e04-05-0401
Wagner, O. I., Lifshitz, J., Janmey, P. A., Linden, M., McIntosh, T. K., and Leterrier, J.-F. (2003). Mechanisms of mitochondria-neurofilament interactions. J. Neurosci. 23, 9046–9058. doi:10.1523/JNEUROSCI.23-27-09046.2003
Walker, C. L., Uchida, A., Li, Y., Trivedi, N., Fenn, J. D., Monsma, P. C., et al. (2019). Local acceleration of neurofilament transport at nodes of ranvier. J. Neurosci. 39, 663–677. doi:10.1523/JNEUROSCI.2272-18.2018
Wang, L., and Brown, A. (2010). A hereditary spastic paraplegia mutation in kinesin-1A/KIF5A disrupts neurofilament transport. Mol. Neurodegener. 5, 52. doi:10.1186/1750-1326-5-52
Wang, L., Ho, C. L., Sun, D., Liem, R. K., and Brown, A. (2000). Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat. Cell Biol. 2, 137–141. doi:10.1038/35004008
Xia, C.-H., Roberts, E. A., Her, L.-S., Liu, X., Williams, D. S., Cleveland, D. W., et al. (2003). Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J. Cell Biol. 161, 55–66. doi:10.1083/jcb.200301026
Yabe, J. T., Jung, C., Chan, W. K., and Shea, T. B. (2000). Phospho-dependent association of neurofilament proteins with kinesin in situ. Cell Motil. Cytoskelet. 45, 249–262. doi:10.1002/(SICI)1097-0169(200004)45:4<249::AID-CM1>3.0.CO;2-M
Yadav, P., Selvaraj, B. T., Bender, F. L. P., Behringer, M., Moradi, M., Sivadasan, R., et al. (2016). Neurofilament depletion improves microtubule dynamics via modulation of Stat3/stathmin signaling. Acta Neuropathol. 132, 93–110. doi:10.1007/s00401-016-1564-y
Yang, Y., Gu, L.-Q., Burnette, W. B., and Li, J. (2016). N98S mutation in NEFL gene is dominantly inherited with a phenotype of polyneuropathy and cerebellar atrophy. J. Neurol. Sci. 365, 46–47. doi:10.1016/j.jns.2016.04.007
Yates, D. M., Manser, C., De Vos, K. J., Shaw, C. E., McLoughlin, D. M., and Miller, C. C. J. (2009). Neurofilament subunit (NFL) head domain phosphorylation regulates axonal transport of neurofilaments. Eur. J. Cell Biol. 88, 193–202. doi:10.1016/j.ejcb.2008.11.004
Yin, Q., Kemp, G. J., Yu, L. G., Wagstaff, S. C., and Frostick, S. P. (2001). Expression of Schwann cell-specific proteins and low-molecular-weight neurofilament protein during regeneration of sciatic nerve treated with neurotrophin-4. Neuroscience 105, 779–783. doi:10.1016/s0306-4522(01)00216-0
Yoshihara, T., Yamamoto, M., Hattori, N., Misu, K., Mori, K., Koike, H., et al. (2002). Identification of novel sequence variants in the neurofilament-light gene in a Japanese population: analysis of Charcot-Marie-Tooth disease patients and normal individuals. J. Peripher Nerv. Syst. 7, 221–224. doi:10.1046/j.1529-8027.2002.02028.x
Yuan, A., Hassinger, L., Rao, M. V., Julien, J.-P., Miller, C. C. J., and Nixon, R. A. (2015a). Dissociation of axonal neurofilament content from its transport rate. PLoS One 10, e0133848. doi:10.1371/journal.pone.0133848
Yuan, A., and Nixon, R. A. (2023). Posttranscriptional regulation of neurofilament proteins and tau in health and disease. Brain Res. Bull. 192, 115–127. doi:10.1016/j.brainresbull.2022.10.017
Yuan, A., and Nixon, R. A. (2016). Specialized roles of neurofilament proteins in synapses: relevance to neuropsychiatric disorders. Brain Res. Bull. 126, 334–346. doi:10.1016/j.brainresbull.2016.09.002
Yuan, A., Rao, M. V., Sasaki, T., Chen, Y., Kumar, A., Veeranna, N., et al. (2006). Alpha-internexin is structurally and functionally associated with the neurofilament triplet proteins in the mature CNS. J. Neurosci. 26, 10006–10019. doi:10.1523/JNEUROSCI.2580-06.2006
Yuan, A., Rao, M. V., Veeranna, N., and Nixon, R. A. (2017). Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb. Perspect. Biol. 9, a018309. doi:10.1101/cshperspect.a018309
Yuan, A., Sasaki, T., Kumar, A., Peterhoff, C. M., Rao, M. V., Liem, R. K., et al. (2012). Peripherin is a subunit of peripheral nerve neurofilaments: implications for differential vulnerability of CNS and peripheral nervous system axons. J. Neurosci. 32, 8501–8508. doi:10.1523/JNEUROSCI.1081-12.2012
Yuan, A., Sasaki, T., Rao, M. V., Kumar, A., Kanumuri, V., Dunlop, D. S., et al. (2009). Neurofilaments form a highly stable stationary cytoskeleton after reaching a critical level in axons. J. Neurosci. 29, 11316–11329. doi:10.1523/JNEUROSCI.1942-09.2009
Yuan, A., Sershen, H., Veeranna, N., Basavarajappa, B. S., Kumar, A., Hashim, A., et al. (2015b). Neurofilament subunits are integral components of synapses and modulate neurotransmission and behavior in vivo. Mol. Psychiatry 20, 986–994. doi:10.1038/mp.2015.45
Yuan, A., Veeranna, N., Sershen, H., Basavarajappa, B. S., Smiley, J. F., Hashim, A., et al. (2018). Neurofilament light interaction with GluN1 modulates neurotransmission and schizophrenia-associated behaviors. Transl. Psychiatry 8, 167. doi:10.1038/s41398-018-0194-7
Yum, S. W., Zhang, J., Mo, K., Li, J., and Scherer, S. S. (2009). A novel recessive Nefl mutation causes a severe, early-onset axonal neuropathy. Ann. Neurol. 66, 759–770. doi:10.1002/ana.21728
Zhai, J., Lin, H., Julien, J.-P., and Schlaepfer, W. W. (2007). Disruption of neurofilament network with aggregation of light neurofilament protein: a common pathway leading to motor neuron degeneration due to Charcot-Marie-Tooth disease-linked mutations in NFL and HSPB1. Hum. Mol. Genet. 16, 3103–3116. doi:10.1093/hmg/ddm272
Zhao, J., Brown, K., and Liem, R. K. H. (2017). Abnormal neurofilament inclusions and segregations in dorsal root ganglia of a Charcot-Marie-Tooth type 2E mouse model. PLoS One 12, e0180038. doi:10.1371/journal.pone.0180038
Zhao, Y., and Szaro, B. G. (1995). The optic tract and tectal ablation influence the composition of neurofilaments in regenerating optic axons of Xenopus laevis. J. Neurosci. 15, 4629–4640. doi:10.1523/JNEUROSCI.15-06-04629.1995
Zhu, Q., Couillard-Després, S., and Julien, J.-P. (1997). Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp. Neurol. 148, 299–316. doi:10.1006/exnr.1997.6654
Keywords: neurofilament, cytoskeleton, intermediate filaments, Charcot-Marie-Tooth disease, CMT, neurodegenerative diseases, mutations, dynamics
Citation: Kotaich F, Caillol D and Bomont P (2023) Neurofilaments in health and Charcot-Marie-Tooth disease. Front. Cell Dev. Biol. 11:1275155. doi: 10.3389/fcell.2023.1275155
Received: 09 August 2023; Accepted: 02 October 2023;
Published: 18 December 2023.
Edited by:
Coralie Fassier, INSERM U968 Institut de la Vision, FranceReviewed by:
Mala V. Rao, New York University, United StatesCopyright © 2023 Kotaich, Caillol and Bomont. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pascale Bomont, cGFzY2FsZS5ib21vbnRAaW5zZXJtLmZy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.