 Sarah Pickles1,2†Desiree Zanetti Alepuz1†
Sarah Pickles1,2†Desiree Zanetti Alepuz1† Yuka Koike1Mei Yue1Jimei Tong1Pinghu Liu3Yugui Zhou3Karen Jansen-West1Lillian M. Daughrity1Yuping Song1
Yuka Koike1Mei Yue1Jimei Tong1Pinghu Liu3Yugui Zhou3Karen Jansen-West1Lillian M. Daughrity1Yuping Song1 Michael DeTure1
Michael DeTure1 Björn Oskarsson4Neill R. Graff-Radford4
Björn Oskarsson4Neill R. Graff-Radford4 Bradley F. Boeve5
Bradley F. Boeve5 Ronald C. Petersen5Keith A. Josephs5
Ronald C. Petersen5Keith A. Josephs5 Dennis W. Dickson1,2,4
Dennis W. Dickson1,2,4 Michael E. Ward6
Michael E. Ward6 Lijin Dong3
Lijin Dong3 Mercedes Prudencio1,2Casey N. Cook1,2
Mercedes Prudencio1,2Casey N. Cook1,2 Leonard Petrucelli1,2*
Leonard Petrucelli1,2*- 1Department of Neuroscience, Mayo Clinic, Jacksonville, FL, United States
- 2Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic, Jacksonville, FL, United States
- 3Genetic Engineering Core, National Eye Institute, National Institutes of Health, Bethesda, MD, United States
- 4Department of Neurology, Mayo Clinic, Jacksonville, FL, United States
- 5Department of Neurology, Mayo Clinic, Rochester, MN, United States
- 6National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, United States
Treatments for neurodegenerative disease, including Frontotemporal dementia (FTD) and Amyotrophic lateral sclerosis (ALS), remain rather limited, underscoring the need for greater mechanistic insight and disease-relevant models. Our ability to develop novel disease models of genetic risk factors, disease modifiers, and other FTD/ALS-relevant targets is impeded by the significant amount of time and capital required to develop conventional knockout and transgenic mice. To overcome these limitations, we have generated a novel CRISPRi interference (CRISPRi) knockin mouse. CRISPRi uses a catalytically dead form of Cas9, fused to a transcriptional repressor to knockdown protein expression, following the introduction of single guide RNA against the gene of interest. To validate the utility of this model we have selected the TAR DNA binding protein (TDP-43) splicing target, stathmin-2 (STMN2). STMN2 RNA is downregulated in FTD/ALS due to loss of TDP-43 activity and STMN2 loss is suggested to play a role in ALS pathogenesis. The involvement of STMN2 loss of function in FTD has yet to be determined. We find that STMN2 protein levels in familial FTD cases are significantly reduced compared to controls, supporting that STMN2 depletion may be involved in the pathogenesis of FTD. Here, we provide proof-of-concept that we can simultaneously knock down Stmn2 and express the expanded repeat in the Chromosome 9 open reading frame 72 (C9ORF72) gene, successfully replicating features of C9-associated pathology. Of interest, depletion of Stmn2 had no effect on expression or deposition of dipeptide repeat proteins (DPRs), but significantly decreased the number of phosphorylated Tdp-43 (pTdp-43) inclusions. We submit that our novel CRISPRi mouse provides a versatile and rapid method to silence gene expression in vivo and propose this model will be useful to understand gene function in isolation or in the context of other neurodegenerative disease models.
Introduction
Individuals diagnosed with FTD experience impaired behavioral and language function due to neuronal loss in the frontal and temporal lobes of the brain, whereas those with ALS will suffer a progressive loss of motor neurons in the cortex, brain stem, and spinal cord, resulting in paralysis and eventually death. At autopsy, almost all ALS cases and roughly half of FTD cases show a mislocalization of TDP-43 from a diffuse nuclear distribution to nuclear and cytoplasmic inclusions in affected brain regions (Ling et al., 2013). Individuals with an expanded G4C2 repeat in the C9ORF72 gene, the most common genetic cause of both FTD and ALS (C9FTD/ALS), have TDP-43 positive inclusions, as well as C9FTD/ALS specific pathology related to the expanded repeat. In particular, repeat-containing sense (G4C2) and antisense (C4G2) RNA transcripts lead to the formation of nuclear RNA foci and accumulation of five dipeptide repeat proteins produced through unconventional repeat-associated non-AUG (RAN) translation, including poly(GA), poly(GP), poly(GR), poly(PR) and poly(PA) (Balendra and Isaacs, 2018). Along with the C9ORF72 repeat expansion, pathogenic mutations in the Progranulin (GRN) gene are also causative for FTD with TDP-43 pathology, leading to haploinsufficiency of the encoded progranulin protein (Rhinn et al., 2022). At present, those diagnosed with either FTD or ALS have limited treatment options, strongly underscoring the need for better insight into disease mechanisms to facilitate the discovery of more effective therapeutic strategies (Andrews et al., 2020).
Currently, the field’s ability to evaluate the role of newly identified genetic risk factors, disease modifiers, and therapeutic targets in pre-clinical animals is hampered by the significant time and costs associated with developing conventional knockout or transgenic mouse models. Genome editing in the mouse central nervous system (CNS) has been achieved by developing CRISPR/Cas9 knockin mice and delivery of single guide RNA (sgRNA) via adeno-associated virus (AAV) (Platt et al., 2014). These mice provide a model to probe the loss of function of virtually any gene. Epigenetic modulation of gene expression via CRISPR activation (CRISPRa) or interference (CRISPRi) has recently been described, whereby a catalytically dead form of Cas9 is fused to transcriptional enhancer or repressor and targeted to the transcription start site of a gene of interest by sgRNA, leading to histone acetylation or methylation and corresponding changes in gene expression (Kampmann, 2018). CRISPRa/i has been successfully used to alter gene expression in the mouse brain (Zheng et al., 2018; Carpenter et al., 2020; Gemberling et al., 2021). Compared to Cas9, CRISPRi avoids tissue mosaicism due to incomplete loss of function and gain of function phenotypes within cell populations (Smits et al., 2019) and does not induce double-strand breaks, mitigating DNA-damage associated toxicity and large genomic deletions or translocations (Haapaniemi et al., 2018; Ihry et al., 2018; Kosicki et al., 2018). CRISPRi mice may be particularly advantageous when combined with established mouse models of FTD/ALS to avoid complex breeding strategies which may be labor-intensive, costly, and require multiple crosses over several generations.
STMN2 is a microtubule-binding protein that is highly expressed in neurons and plays an important role in axonal growth and maintenance (Shin et al., 2012; Shin et al., 2014). STMN2 splicing is regulated by TDP-43, such that in conditions of TDP-43 deficiency, a cryptic exon is included within the RNA, leading to the production of a non-functional truncated STMN2 variant and the concomitant loss of full-length STMN2 in ALS and FTD cases (Klim et al., 2019; Melamed et al., 2019; Prudencio et al., 2020). Flies lacking the STMN2 ortholog have defective synapse stability at the neuromuscular junction (NMJ) (Graf et al., 2011), accompanied by paralysis and reduced life span (Duncan et al., 2013). Knockdown of Stmn2 in the midbrain of mice results in the selective loss of dopaminergic neurons and impaired motor function, pointing towards a role for Stmn2 in neuronal survival (Wang et al., 2019). Collectively, Stmn2 knockout mice exhibit deficits in motor behavior due to the denervation of the NMJ (Guerra San Juan et al., 2022; Krus et al., 2022; Lopez-Erauskin et al., 2022). Moreover, motor deficits were rescued by crossing Stmn2 knockout mice to mice expressing human STMN2, suggesting that human STMN2 performs a similar role in NMJ maintenance (Guerra San Juan et al., 2022). Similarly, restoring STMN2 levels in induced pluripotent stem cell (iPSC)-derived motor neurons depleted of either STMN2 or TDP-43, rescued axonal regeneration and growth defects (Klim et al., 2019; Melamed et al., 2019). Taken together, these data strongly support that loss of STMN2 protein contributes to ALS pathogenesis. However, as STMN2 RNA levels are also reduced in the frontal cortex of FTD patients (Prudencio et al., 2020), it is important to assess the contribution of STMN2 deficiency to FTD-associated phenotypes, in particular given the recent development of therapeutic strategies to rescue STMN2 splicing and restore STMN2 expression and function. In addition, given that the TDP-43 binding site within intron 1 of the human STMN2 gene is not conserved in mice (Melamed et al., 2019; Klim et al., 2021), previous mouse models with impaired Tdp-43 function lacked Stmn2 misprocessing and loss of function. To this end, the current goals of the study include: 1) Assess whether STMN2 loss of function is a feature of FTD, 2) generate a CRISPRi mouse model using Stmn2 depletion as a proof-of concept and 3) combine CRISPRi of Stmn2 with a C9ORF72 expanded repeat. The creation of this dual model serves to demonstrate the ease with which knockdown can be combined with established neurodegenerative disease models and will allow us to evaluate if Stmn2 deficiency in the presence of the expanded C9ORF72 repeat and endogenous Tdp-43 pathology exacerbates pathology, thereby providing a more complete model of FTD.
Materials and methods
Human post-mortem tissues
The Mayo Clinic Florida Brain Bank provided post-mortem brain tissue from individuals with confirmed FTD with TDP-43 inclusions and individuals without neuropathological features. We obtained written informed consent from all participants or their family members. The Mayo Clinic Institutional Review Board and Ethics Committee approved all protocols associated with the collection of human tissues. The sample size was determined based on the availability of tissue. A description of patient demographic information for both control individuals and FTD cases can be found in Supplementary Table S1.
Animal studies
All animal procedures were conducted in accordance with the National Institutes of Health Guide for Care and Use of Experimental Animals and were approved by both the Mayo Clinic Institutional Animal Care and Use Committee (IACUC Protocol number A00005405-20) and the National Eye Institute ACUS committee (ASP number NEI-626).
Generation of CRISPRi knockin mouse model
The CRISPRi knockin mouse line used in this study was created at the Genetic Engineering Core, National Eye Institute, National Institutes of Health. The system uses a human codon-optimized dCas9 from Streptococcus pyogenes, fused with a nuclear localization sequence (NLS, 2X), an HA tag, and the coding sequence of blue fluorescent protein (BFP). The modified dCas9 is fused with the KRAB (Krüppel-associated box) domain of the human KOX1 gene at its C-terminus, which facilitates the recruitment of chromatin-modifying factors to the targeted gene locus (Margolin et al., 1994).
To create the CRISPRi knockin mouse line, a vector was designed to target the Rosa26 locus of the previously constructed mouse genome (Liu et al., 2019). The CRISPRi cassette containing the essential elements of the system was obtained by PCR amplification from pHF-SFFV-dCas9-BFP-KRAB, a gift from Stanley Qi and Jonathan Weissman (Addgene plasmid #46911) (Gilbert et al., 2014). The cassette was then inserted between the Cytomegalovirus early enhancer/chicken β actin (CAG) promoter and the rabbit beta-globin polyadenylation (RBG-PA) signal sequence in the mouse Rosa26 targeting vector, which contains a phosphoglycerate kinase-neomycin (PGK-Neo) cassette flanked by two FRT sites. To ensure insulation of the transgene, the CAG-CRISPRi-RBG-PA cassette was flanked by two copies of the 5′-HS4 chicken β-globin (cHS4) insulator (Chung et al., 1993).
To introduce the CRISPRi knockin allele at the Rosa26 locus, the targeting vector was linearized using AscI and then electroporated into mouse W4 embryonic stem (ES) cells as described (Bradley et al., 1984). Homologous recombination (HR) events were selected using neomycin (neo) selection followed by HR screening using a long genomic PCR assay. Two PCR assays were performed: one with forward primer 5′-CGGCACTACTGTGTTGGCGGACTGGCGGGAC-3′ and reverse primer 5′-GAACATACGTCATTATTGACGTCAATG-3′ to detect the left arm HR (2.85 kb PCR product) and another with forward primer 5′-GGTGTGGCGGACCGCTATCAGGACATAGC-3′ and reverse primer 5′-CATGTTTAGTAATGGCTGCAGACT-3″ to detect the right arm HR (2.145 kb PCR product). ES clones carrying the CRISPRi knockin allele at the Rosa26 locus were then expanded and microinjected into blastocyst embryos obtained from C57BL6/J female donors. Male chimeric F0 pups with 90% or higher agouti coat color were crossed with wildtype C57Bl6/J females to achieve germline transmission of the knockin allele. F1 pups carrying the germline-transmitted knockin allele were bred with wild-type C57Bl6/J mice for several generations until they became congenic with the C57Bl6/J background. Finally, the neo cassette was removed by crossing the CRISPRi knockin mice with a germline flippase (FLP) recombination strain (actin-FLP, Jackson lab).
Generation of AAV vectors and P0 injections
To produce AAV expressing sgRNA and sgRNA plus a C9ORF72 G4G2 repeat, the coding sequence for the mouse U6 promoter, including modified stem loop 1, 2 and guide sequence site from pU6-sgRNA EF1Alpha-puro-T2A-BFP, a gift from Jonathan Weismann and Martin Kampmann (Addgene plasmid #60955) (Gilbert et al., 2014), were cloned into the pAAV CBA EGFP backbone and pAAV CBA C9ORF72 with 2 or 149 G4C2 repeats (referred to as 2R and 149R, respectively) (Chew et al., 2015). To generate AAV sgStmn2 we selected four sgRNA sequences against Stmn2, as described previously (Horlbeck et al., 2016). Top and bottom oligonucleotides were annealed and ligated into BstXI and BlpI restriction sites within the AAV sgRNA or sgRNA-2R/149R backbone. Sequences for sgRNA are as follow sgStmn2#1: 5′-GGCCGCGGAGAGAGCGAGAG-3′, sgStmn2#2: 5′-GGGAGTGGTGGCGAAGGCAA-3′, sgStmn2#3: 5′-GGGGATGTGCACGCACGGAG-3′, sgStmn2#4: 5′-GCGAGAGAGGAGTCTGAGTA-3′ and sgCtrl: 5′-GGACTAAGCGCAAGCACCTA-3′.
To produce AAV9 virus we followed a previously described protocol (Chew et al., 2015). Briefly, AAV vectors were co-transfected with helper plasmids in HEK293T cells. After 72 h, cells were harvested, treated with 50 Units/mL Benzonase (Sigma-Aldrich), and lysed by several cycles of freeze-thaw. The virus was purified using a discontinuous iodixanol gradient and diluted in PBS.
Intracerebroventricular injections (ICV) of AAV were performed as previously described with some minor modifications (Chew et al., 2015). In a few words, a 30-gauge needle attached to a 10 μL syringe (Hamilton Company) was inserted into the lateral ventricle of cryo anesthetized CRISPRi pups at postnatal day 0 (P0). Two microliters (1.5E10 genomes/μL) of AAV9 virus were manually injected into each ventricle. Following injection, pups were placed on a heated pad until they recovered from anesthesia, at which time they were placed back into their home cage. Approximately three-to-four-week later pups were weaned, DNA was extracted from tails and genotyped for the presence of CRISPRi transgene using the following primer set: dCas9 forward: 5′- ATCAACCGGCTGTCCGACTA-3′, dCas9 reverse: 5′-CCTCTGGGTAATCAGCTTGG-3′.
Mouse tissue processing
Mice were euthanized by CO2 and brains were harvested and cut sagittally across the midline. Sagittal half brains were fixed in 10% formalin, embedded in paraffin, sectioned, and mounted on glass slides. The other half brain was dissected and frozen on dry ice. Frozen mouse forebrain tissues were homogenized in ice-cold Tris-EDTA (TE; 50 mM Tris, pH 8.0, 50 mM NaCl, 1 mM EDTA), with 2× protease and phosphatase inhibitors.
RNA extraction, cDNA synthesis, and qRT-PCR
To extract total RNA, brain homogenates were mixed with Trizol LS (Thermo Fisher Scientific) and frozen. The following day, RNA was extracted using the Direct-zol RNA MiniPrep kit (Zymo Research). cDNA was synthesized from 250 ng of RNA using the High-Capacity cDNA transcription kit (Applied Biosystems). Quantitative reverse transcription-PCR (qRT-PCR) was performed using the SYBR GreenER qPCR SuperMix (Invitrogen, Thermo Fisher Scientific). Each sample was run in triplicate and processed on a Prism 7900HT Fast Real-Time system (Applied Biosystems). Samples were analyzed using the ΔΔCt method and normalized to the endogenous controls (Rplp0 and Gapdh). The following primers and their sequences were used:
Stmn1 forward: 5′-TCTGTCCCCGATTTCCCCC-3′, Stmn1 reverse: 5′-AGCTGCTTCAAGACTTCCGC-3’.
Stmn2 forward: 5′-GCAATGGCCTACAAGGAAAA-3′, Stmn2 reverse: 5′-GGTGGCTTCAAGATCAGCTC-3′.
Stmn3 forward: 5′-CCAGCACCGTATCTGCCTAC-3′, Stmn3 reverse 5′- CAGAGGCCCGCTTATCCAG-3’.
Stmn4 forward: 5′-GCGTCATCTCTGATATGGAAGTC-3′, Stmn4 reverse: 5′- CTCTTCTAATGATGGGTCTCGC-3’.
Gapdh forward: 5′-CATGGCCTTCCGTGTTCCTA-3′, Gapdh reverse: 5′-CCTGCTTCACCACCTTCTTGAT-3′.
Rplp0 forward: 5′-ACTGGTCTAGGACCCGAGAAG-3′, Rplp0 reverse: 5′-CTCCCACCTTGTCTCCAGTC-3′.
Iba1/Aif1 forward: 5′-GGATTTGCAGGGAGGAAAAG-3′, Iba1/Aif1 reverse: 5′-TGGGATCATCGAGGAATTG-3′.
Cd68 forward: 5′-GACACTTCGGGCCATGTT-3′, Cd68 reverse: 5′-GAGGAGGACCAGGCCAAT-3.’
Gfap forward: 5′-GGAGAGGGACAACTTTGCAC-3′, Gfap reverse: 5′-AGCCTCAGGTTGGTTTCATC-3′.
C9orf72 G4C2 forward: 5′-TAGTACTCGCTGAGGGTGAAC-3′, C9orf72 G4C2 reverse: 5′-CTACAGGCTGCGGTTGTTTC-3′.
Tardbp forward: 5′-GGGGCAATCTGGTATATGTTG-3′, Tardbp reverse: 5′-TGGACTGCTCTTTTCACTTTCA-3’.
Protein extraction and immunoblotting
To prepare protein lysates from mouse brains, Triton X-100 and Sodium dodecyl sulfate (SDS) were added to brain homogenates at final concentrations of 1% and 2%, respectively. Homogenates were sonicated on ice and centrifuged at ×16,000 g for 20 min. The supernatants were collected, and protein concentrations were determined by a bicinchoninic acid (BCA) assay (ThermoFisher Scientific). For human post-mortem frontal cortex samples, 50 mg of tissue was homogenized in Radioimmunoprecipitation buffer (RIPA, 25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% sodium deoxycholate, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, protease, and phosphatase inhibitors). Tissue homogenates were sonicated on ice and centrifuged at ×100,000 g for 30 min at 4°C. The supernatant was collected and is referred to as the RIPA-soluble fraction, with protein concentration determined by BCA assay. The RIPA-insoluble pellet was extracted using 7 M urea buffer, sonicated, and then centrifuged for 30 min at ×100, 000 g at room temperature. Protein concentrations of the urea-soluble fractions were determined by Bradford assay.
Protein lysates were diluted in Tris-Glycine SDS sample buffer (ThermoFisher Scientific) and 5% beta-mercaptoethanol (Sigma Aldrich). Protein samples were heat denatured for 5 min at 95°C and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 4%–20% Tris-Glycine gels (Life Technologies). Protein was subsequently transferred to Polyvinylidene (PVDF) membrane (Millipore). Membranes were first blocked with 5% non-fat dry milk in Tris-buffered saline with Triton (TBS-T, 100 mM Tris-HCl pH 7.5, 140 mM NaCl, 0.1% Triton X-100) and incubated overnight at 4°C with mouse anti-STMN2 (1:1000 MAB6930, R&D Systems), rabbit anti-TDP-43 (1:5,000 12892-AP ProteinTech) and mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (1:30,000, H86504M, Meridian Life Sciences), diluted in TBS-T plus 5% nonfat dry milk. Membranes were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:5,000; Jackson ImmunoResearch) and signal was detected by Enhanced chemiluminescence (ECL, PerkinElmer). Quantitative densitometry was performed using ImageJ, and Stmn2 levels were normalized to Gapdh expression.
Immunohistochemistry
Paraffin-embedded brains were deparaffinized in xylene and rehydrated through a series of ethanol solutions. Antigen retrieval was performed by steaming in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6), distilled water, or pH 9 Tris-EDTA for 30 min. Tissues were immunostained with polyclonal antibodies against poly(GP) [1:10,000, Rb5823 (Gendron et al., 2013)] and Gfap (1:2,500, BioGenex). For phosphorylated Tdp-43 (pTdp-43) staining, tissue sections were incubated with Dual Endogenous Enzyme Block (DAKO) and blocked with 2% normal goat serum. Slides were incubated with our novel MA1290/1291AP in-house antibody (1:100), an anti-rabbit antibody for both mouse and human TDP-43 phosphorylated at serine 409/410, overnight at 4°C. The recognized antigen is amino acid residues 404 to 414 of full-length TDP-43 with phosphorylation of S409/410 (CSMDSK[pS][pS]GWGM), and equivalent to our previously described Rb3655 antibody (Todd et al., 2020; Wu et al., 2021). The following day, slides were washed and incubated with biotinylated goat anti-rabbit secondary (1:200, Vectastain® ABC-HRP Kit, Peroxidase (Rabbit IgG) PK-4001) for 2 h at room temperature. Slides were incubated with avidin-biotin complex solution for 30 min. Slides were then incubated with 3,3′-diaminobenzidine (DAB, Acros Organics) activated with hydrogen peroxide, and the reaction was stopped by rinsing slides in distilled water. Sections were counterstained with hematoxylin, dehydrated through a series of ethanol and xylene washes, and coverslipped with Cytoseal mounting media (Thermo Fisher Scientific).
Quantification of neuropathology
The burden of GP inclusions and Gfap immunoreactivity was quantified using digital pathology technology by Leica Biosystems. We used a ScanScope® AT2 (Leica Biosystems) to obtain high-resolution digitized images of immunostained slides. ImageScope® software (v12.1; Leica Biosystems) was used to annotate the cortex on mid-sagittal serial sections stained for GP of Gfap for each mouse. A custom-designed Positive Pixel Count algorithm was used to quantify either poly-GP burden or Gfap immunoreactivity in the cortex. pTdp-43 inclusions in the cortex of each mouse sample were counted manually in a blinded fashion, with the number of pTdp-43 inclusions per mm2 determined for each slide.
Immunoassays for dipeptide repeat proteins and phosphorylated TDP-43
Previously characterized Meso Scale Discovery (MSD) sandwich immunoassays were used to quantify poly (GP) and poly (GA) (Gendron et al., 2015) from mouse brains. The abundance of pTDP-43 in the urea-soluble fraction from the frontal cortex of human cases and controls had been measured in a previous study by sandwich Meso Scale Discovery (MSD) immunoassay (Prudencio et al., 2020). Lysates were diluted in Tris buffered saline (TBS) and an equal amount of protein was loaded in duplicate wells. Response values corresponding to the intensity of emitted light upon electrochemical stimulation of the assay plate using the MSD QUICKPLEX SQ120 were acquired.
Statistics
GraphPad Prism 9 (GraphPad software) was used for all statistical analyses. The type of statistical analysis used, the number of subjects, and the p values used are indicated in the figure and/or legend. To compare STMN2 protein abundance in FTD and controls, the data was analyzed with a single-variable (unadjusted) and a multivariable linear regression model (adjusted), considering age at death and sex. p-values less than 0.0125 were considered statistically significant, after comparing the four groups, all FTD combined and each genotype separately. To compare pTDP-43 abundance between FTD groups, that data was analyzed using both single-variable (unadjusted) and multivariable linear regression (adjusted) models, factoring in age at death and sex. p-values less than 0.025 were considered statistically significant after multiple comparisons. Associations between STMN2 protein and TDP-43 or pTDP-43 protein levels in FTD cases were determined using a single-variable and multivariable linear regression model, the latter was adjusted for age at death and sex. p-values less than 0.025 were considered statistically significant after multiple comparisons. Due to their skewed distribution, STMN2, TDP-43 and pTDP-43 protein levels were plotted on a base 2 logarithmic scale.
Results
STMN2 protein is reduced in the frontal cortex of familial FTD cases
Previous studies demonstrated a reduction of STMN2 RNA in FTD and ALS cases (Klim et al., 2019; Melamed et al., 2019; Prudencio et al., 2020) and a decrease in STMN2 immunoreactivity in motor neurons from ALS cases compared to controls (Klim et al., 2019). However, STMN2 protein levels in FTD cases have yet to be evaluated. To do so, we assembled an FTD cohort with a neuropathological diagnosis of frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP), with an approximately equal number of cases having an expanded G4C2 repeat in the C9ORF72 gene, GRN mutations or no known FTD-causative mutation (Supplementary Table S1). We then measured detergent-soluble STMN2 protein in the frontal cortex by Western blot, which revealed a significant or nominally significant reduction of STMN2 protein in all FTLD-TDP cases compared to controls in unadjusted analysis and adjusted analysis when controlling for age and sex, respectively (Figure 1A; Supplementary Table S2). Lower levels of STMN2 protein in the FTLD-TDP group were largely driven by carriers of a C9ORF72 expanded repeat and GRN mutations, as individuals without a mutation had similar levels of STMN2 protein compared to controls. We speculated that cases harboring mutations may have greater reductions in STMN2 protein due to lower levels of soluble TDP-43 protein. To that end, we measured soluble TDP-43 levels in our cohort of controls and FTLD-TDP cases. Compared to controls, we observed a significant or nominally significant reduction in TDP-43 levels in unadjusted and analysis adjusted for age and sex, respectively (Figure 1B; Supplementary Table S2). Like STMN2 levels, the reduction in soluble TDP-43 protein was more pronounced in mutation carriers. Additionally, lower levels of TDP-43 protein were significantly associated with reduced levels of soluble STMN2 in FTLD-TDP cases in both unadjusted and adjusted analyses controlled for age and sex (Supplementary Table S3). We further suspected that levels of pTDP-43 pathology may also be greater in mutation carriers. To evaluate this possibility, we used pTDP-43 protein levels we had measured in these same cases from a previous study (Prudencio et al., 2020) and compared the pTDP-43 burden in sporadic and familial FTLD-TDP cases. Indeed, mutation carriers had significantly elevated levels of pTDP-43 than those without, in both unadjusted and adjusted analysis (Figure 1C; Supplementary Table S4), in line with our previous findings (Prudencio et al., 2017). Higher levels of pTDP-43 were found to have a significant or nominally significant association with lower levels of STMN2 protein in FTLD-TDP cases (Supplementary Table S3). Taken together, we provide evidence of decreased STMN2 and TDP-43 abundance in FTLD-TDP cases harboring C9ORF72 and GRN mutations. These findings suggest that loss of STMN2 protein is a feature of familial FTD and may contribute to pathogenesis.
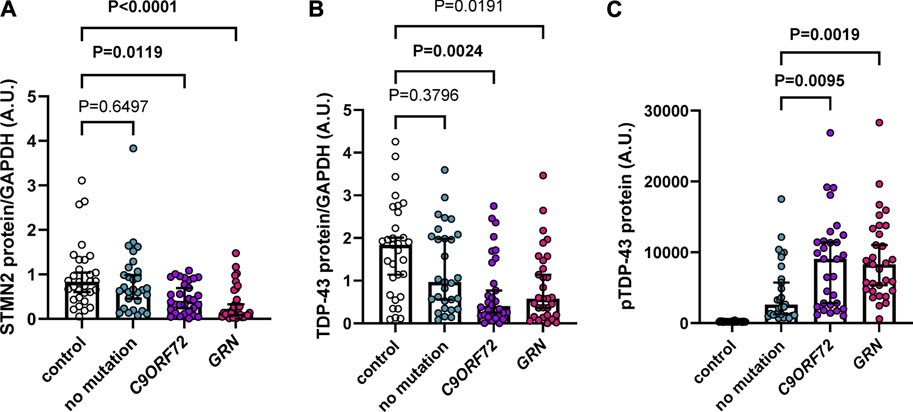
FIGURE 1. Soluble STMN2 and TDP-43 protein are reduced in the frontal cortex of familial FTLD-TDP cases. (A,B) Soluble STMN2 (A), TDP-43 (B) protein levels were measured by Western blot in the frontal cortex of FTLD-TDP cases (n = 90) and non-neurological disease controls (n = 31). Following quantification of STMN2 (A) or TDP-43 (B) immunoreactivity, levels were normalized to GAPDH to control for protein loading. (C) pTDP-43 protein levels, as measured by immunoassay, are significantly elevated in cases with a C9ORF72 repeat expansion or GRN mutations compared to those without mutations. Horizontal bars represent median ± 95% confidence intervals. p-values from the linear regression model adjusting for age and sex are shown. Values for both unadjusted and adjusted linear regression models are reported in Supplementary Tables S2, S4.
Creation of a Stmn2 loss of function model using CRISPRi
To model Stmn2 loss of function in vivo and assess its relevance in FTD, we first generated a novel knockin CRISPRi mouse model using a system that previously demonstrated effective silencing of target genes in vitro (Gilbert et al., 2014). The CRISPRi machinery was integrated into the Rosa26 locus and expresses a catalytically dead form of Cas9 (dCas9) fused to a transcriptional repressor domain, KRAB (Figure 2A). AAV was used to deliver sgRNA to direct CRISPRi machinery to the Stmn2 transcription start site (Figure 2B). We tested four sgRNAs targeted against Stmn2 from a CRISPRi library of the whole mouse genome (Horlbeck et al., 2016). To deliver guides to the mouse brain, ICV injections of AAV sgStmn2 (#1–4) were performed at postnatal day zero, and mice were harvested at 2 months of age (Figure 2B). Notably, all four guides were able to significantly reduce Stmn2 RNA (Figure 2C) and protein (Figures 2D, E) compared to CRISPRi mice injected with enhanced Green Fluorescent Protein (eGFP) or non-transgenic mice (NT, lacking the CRISPRi machinery). While Stmn2 protein knockdown ranged from 40% to 75%, mice injected with sgStmn2#1 displayed the largest reduction in Stmn2 protein (Figures 2D, E). We therefore selected sgStmn2#1 for further experiments and will refer to it as sgStmn2 hereafter. We were also able to modulate the levels of Stmn2 knockdown by titrating the amount of virus delivered, with the highest levels of virus showing the largest decrease in both Stmn2 RNA (Supplementary Figure S1A) and protein levels (Supplementary Figures S1B, C). Taken together, we were able to achieve robust knockdown of Stmn2 in the brain of CRISPRi mice.
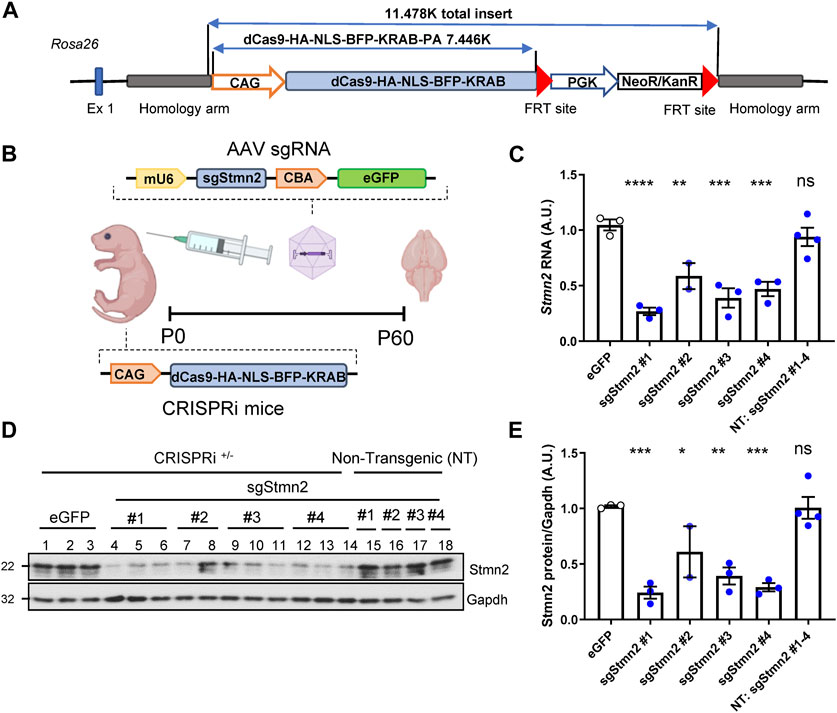
FIGURE 2. Generation of Stmn2 knockdown mouse model using CRISPRi. (A) Schematic of CRISPRi machinery used to create CRISPRi knockin mice. (B) Schematic of CRISPRi mice, AAV sgRNA construct, and delivery of virus by ICV injection to create an in vivo model of Stmn2 loss of function. (C–E) Stmn2 RNA and protein levels were assessed in the brain tissue of 2-month-old CRISPRi mice and/or non-transgenic littermate controls injected with AAV encoding eGFP, or four guides targeting Stmn2 (sgStmn2#1-4). (C) Stmn2 RNA levels, normalized to endogenous controls Rplp0 and Gapdh, as measured by qRT-PCR, are significantly reduced in CRISPRi mice injected with AAV sgStmn2. Immunoblot (D) and quantification of Stmn2 protein normalized to Gapdh levels (E) reveal a reduction of Stmn2 protein in CRISPRi mice injected with sgStmn2. Data presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, non-significant (ns), as analyzed using one-way ANOVA, followed by Dunnett’s post hoc analysis, n = 2–4 animals per group (C,E). Schematic (B) created with BioRender.com.
Establishing a dual CRISPRi Stmn2 loss of function and C9ORF72 expanded repeat model
Our group has previously shown that in comparison to mice expressing a non-pathological number of 2 G4C2 repeats (2R), mice expressing 149 G4C2 repeats (149R) recapitulate C9FTD relevant disease features including pTdp-43 inclusions, accumulation of RNA foci and DPR proteins from both sense and antisense transcripts, neurodegeneration and behavioral deficits (Chew et al., 2015; Chew et al., 2019). To understand if Stmn2 deficiency would impact C9ORF72-associated pathology we used AAV to simultaneously express either sgStmn2 or a non-targeting guide RNA (sgCtrl) along with either 2R or 149R.
Each AAV (sgCtrl-2R, sgCtrl-149R, sgStmn2-2R, and sgStmn2-149R) was introduced to the mouse CNS via ICV injection at postnatal day 0 (Figure 3A). Animals were harvested at 6 months of age, a time point when we previously observed robust pathology and gliosis in the 149R-AAV model (Chew et al., 2019). We first confirmed the expression of the G4C2 repeat transcript, with similar levels observed between all four groups (Figure 3B). Moreover, as anticipated, Stmn2 RNA levels were significantly reduced in mice injected with sgStmn2 compared to mice injected with the non-targeting sgCtrl (Figure 3C). Stmn2 protein levels were also significantly reduced in sgStmn2-2R and sgStmn2-149R mice (Figures 3D, E). Of note, we did not observe any differences in Stmn2 knockdown efficiency as a function of the G4C2 repeat number. We also measured transcripts of other stathmin family genes to both verify the specificity of the guide RNA for Stmn2 and exclude any possible compensatory upregulation mechanisms (Gagliardi et al., 2022). Analysis of Stmn1, 3, or 4 transcript levels revealed no significant differences among any of the four groups (Supplementary Figures S2A–C).
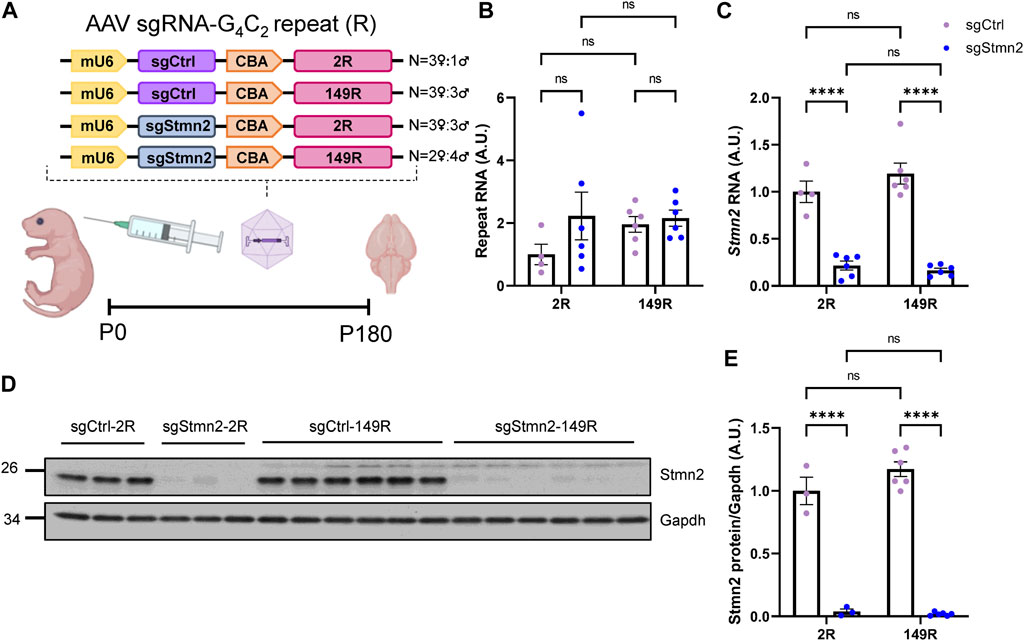
FIGURE 3. Generation of dual C9ORF72 expanded repeat and Stmn2 loss of function model in CRISPRi mice. (A) Schematic of AAV sgRNA-G4C2 repeat construct and delivery of virus to create a dual model. (B–E) The levels of the C9ORF72 G4C2 repeat (B), Stmn2 RNA (C), and Stmn2 protein (D,E) were measured in brain tissue of 6-month-old CRISPRi mice injected with AAV encoding sgCtrl or sgStmn2, with either 2 (2R) or 149 G4C2 repeats (149R). (B) Levels of G4C2 repeat RNA are similar regardless of sgRNA or repeat expression. (C) Stmn2 RNA levels, normalized to endogenous controls Rplp0 and Gapdh as measured by qRT-PCR, are significantly reduced in CRISPRi mice receiving sgStmn2 compared to sgCtrl, regardless of repeat number. Immunoblot (D) and quantification of Stmn2 protein normalized to Gapdh levels (E) reveal a significant reduction of Stmn2 protein in CRISPRi mice injected with sgStmn2 compared to sgCtrl, regardless of repeat number. Data presented as mean ± SEM. ****p < 0.0001, non-significant (ns), as analyzed using two-way ANOVA, followed by Tukey’s post hoc analysis, n = 3–6 animals per group (B–E). Schematic (A) created with BioRender.com.
Dual CRISPRi Stmn2 loss of function and C9ORF72 expanded repeat model recapitulates C9-associated pathology
To validate whether our CRISPRi dual model replicated C9ORF72 expanded repeat-associated pathology, we assessed DPR protein expression and pTdp-43 accumulation. First, we measured the expression of DPR proteins in the cortex using established immunoassays (Chew et al., 2019). As anticipated, we detected robust expression of poly(GP) (Figure 4A) and poly(GA) (Figure 4B) in sgCtrl-149R and sgStmn2-149R mice compared to animals not expressing the expanded repeat. Reduction of Stmn2 protein levels did not impact either poly(GA) or poly (GP) protein levels. Immunohistochemical analysis using a poly(GP)-specific antibody revealed both diffuse nuclear staining in the cortex as well as cytoplasmic inclusions that were restricted to animals expressing the expanded repeat (149R) (Figures 4C, D). The number of GP inclusions was similar between sgCtrl-149R and sgStmn2-149R animals consistent with our immunoassay results.
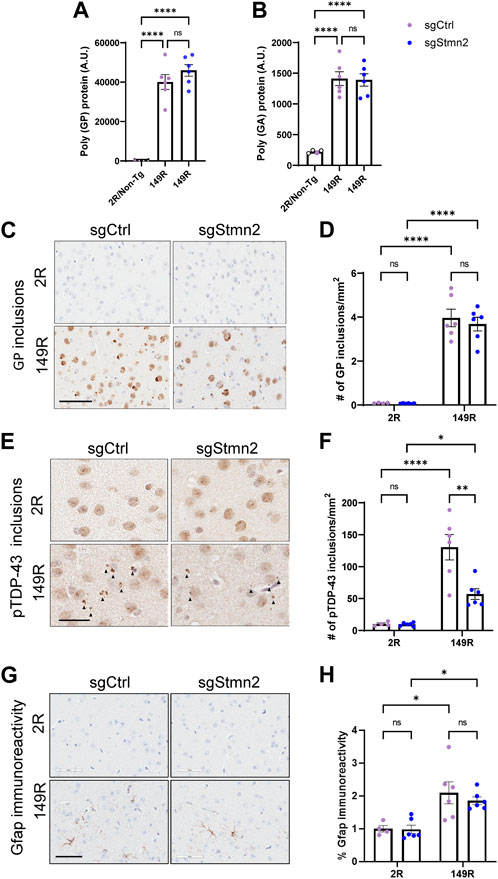
FIGURE 4. Dual C9ORF72 expanded repeat and Stmn2 loss of function model recapitulates C9-associated pathology. (A–H) DPR accumulation, pTdp-43 pathology, and gliosis were analyzed in the brains of 6-month-old mice. (A,B) Immunoassay measuring soluble poly-GP (A) and poly-GA (B) show a significant accumulation of DPRs in sgCtrl-149R and sgStmn2-149R mice. (C) Representative images of immunohistochemical analysis of poly(GP) with quantitative analysis of the number of GP-positive inclusions in the cortex (D). (E) Representative images of pTdp-43 immunoreactivity and quantitative analysis of the number of pTdp-43 inclusions observed in the cortex (F). (G) Representative images of immunohistochemical analysis of Gfap levels with quantitative analysis of percent Gfap immunoreactivity (H). Data presented as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001, non-significant (ns), as analyzed using two-way ANOVA, followed by Tukey’s post hoc analysis, n = 4–6 animals per group (A–H). Scale bar is 100 uM (C) and 60 uM (E,G). Solid arrows indicate pTdp-43 inclusions (E).
We next evaluated pTdp-43 inclusions and observed small cytoplasmic inclusions in the cortex of sgCtrl-149R mice that were absent in sgCtrl-2R mice (Figures 4E, F). Unexpectedly, we found fewer pTdp-43 inclusions in sgStmn2-149R mice compared to sgCtrl-149R mice (Figure 4F) in the absence of changes in Tardbp RNA levels (Supplementary Figure S3A).
We also assessed gliosis in the cortex and observed a significant increase in Gfap immunoreactivity, a marker of activated astrocytes, (Figures 4G, H), in mice expressing the C9ORF72 expanded repeat, in line with our previous mouse model (Chew et al., 2019). Levels of the Gfap transcript were also significantly elevated in the 149R groups (Supplementary Figure S4A). Additionally, RNA levels of markers of activated microglia, (Iba1/Aif1, Supplementary Figure S4B), and phagocytic microglia (Cd68, Supplementary Figure S4C) were significantly increased in groups expressing the expanded repeat, suggestive of microgliosis, although this was not confirmed at the protein level. Reduction of Stmn2 protein did not consistently induce or exacerbate gliosis. However, we did observe a significant increase in Cd68 transcript levels in the sgStmn2-2R group compared to sgCtrl-2R (Supplementary Figure S4C). A significant increase of Gfap RNA levels was observed in Stmn2 depleted mice (sgStmn2-149R) compared to controls (sgCtrl-149R) in animals expressing the expanded repeat (Supplementary Figure S4A), although this was not preserved at the protein level (Figure 4H). Together, these data provide proof-of-concept that we were able to successfully deliver guide RNA to knock down a target of interest in CRISPRi mice in a disease model (in this case mice expressing the expanded C9ORF72 repeat). Overall, loss of Stmn2 in the presence of the C9ORF72 expanded repeat does not exacerbate C9-mediated pathology at 6 months of age in the current model.
Discussion
Previous studies had suggested STMN2 is integral for axon growth, maintenance, and stability (Shin et al., 2012; Shin et al., 2014). Loss of Stmn2 expression in fly and mouse models have identified a role for Stmn2 in motor coordination, innervation of the NMJ, and neuronal survival of dopaminergic neurons in the substantia nigra (Graf et al., 2011; Duncan et al., 2013; Wang et al., 2019; Guerra San Juan et al., 2022; Krus et al., 2022; Lopez-Erauskin et al., 2022). The discovery that TDP-43 regulates STMN2 processing and that STMN2 RNA levels are reduced in ALS/FTD cases have fueled speculation of its involvement in the pathogenesis of these diseases. Recent evidence from three Stmn2 loss of function mouse models strongly support that the loss of STMN2 contributes to ALS (Guerra San Juan et al., 2022; Krus et al., 2022; Lopez-Erauskin et al., 2022). However, the role of STMN2 in the central nervous system beyond the spinal cord and its relevance to FTD remain unresolved. We find that soluble STMN2 and TDP-43 protein levels are significantly reduced in the frontal cortex of FTD cases with GRN mutations and a C9ORF72 repeat expansion, these findings support the notion that STMN2 deficiency is a feature of familial FTD. However, the specific contribution of STMN2 loss in the pathogenesis of FTD has yet to be delineated. Regardless, these data support the development of therapies aimed at restoring STMN2 levels in FTD with TDP-43 pathology. Indeed, antisense oligonucleotides (ASO) are attractive therapeutics for the treatment of neurodegenerative diseases following the promising clinical trial results and Federal Drug Administration approval of nusinersen for the treatment of Spinal Muscular Atrophy (Schoch and Miller, 2017; Mercuri et al., 2018). ASOs to correct STMN2 splicing and augment protein levels were shown effective in TDP-43 depleted iPSC-derived motor neurons, TDP-43 depleted primary cortical neurons from mice with a humanized STMN2 gene and in mice with constitutive misprocessing of the human STMN2 gene (Baughn et al., 2023). Given these compelling pre-clinical results, phase 1 trials using ASOs to restore STMN2 splicing levels in ALS patients are currently underway (Corporation, 2022). ASOs to restore STMN2 levels in FTD are also in the preclinical phase.
To provide a tool to rapidly interrogate the contribution of loss of function of a target of interest we generated a novel CRISPRi knockin mouse model and used knockdown of Stmn2 as a test-case. We achieved a robust knockdown of Stmn2 RNA and protein, which can be titrated either by the selection of guide RNA or the amount of virus. As multiple human diseases are caused by haploinsufficiency (Dang et al., 2008), including FTD with GRN mutations (Rhinn et al., 2022), the ability to fine-tune the level of knockdown by either guide design or the amount of AAV delivered will be of great value. Further, AAV delivery of guide RNA offers long-term gene silencing compared to ASOs (Murlidharan et al., 2014). In addition, knockdown is achieved postnatally, circumventing embryonic or perinatal lethality if the target of interest is required during embryogenesis or development. Indeed, Stmn2 likely plays an unresolved role in early development, as two groups reported perinatal lethality in homozygous Stmn2 knockouts (Krus et al., 2022; Lopez-Erauskin et al., 2022) or mice with constitutive human STMN2 processing defects (Baughn et al., 2023).
Genetic screens in fly and yeast have identified the nuclear pore complex and nuclear export/import factors as possible modifiers of C9ORF72-mediated toxicity (Freibaum et al., 2015; Jovicic et al., 2015; Zhang et al., 2015; Boeynaems et al., 2016), however these candidates have yet to be evaluated in vivo or in C9FTD/ALS mouse models. Likewise, CRISPR/Cas9 screens examining modifiers of DPR toxicity (Kramer et al., 2018) or RAN-translation (Cheng et al., 2019) have also produced promising candidate genes requiring validation in preclinical animal models. Evaluation of promising candidates using the CRISPRi mouse could serve as a natural extension of CRISPR screens in human cell lines. Taken together, the CRISPRi model offers high flexibility in target selection, can be rapidly generated, and is more economical compared to conventional knockouts.
We also created a dual model where Stmn2 protein deficiency was combined with the expression of a C9ORF72 expanded repeat. DPR accumulation was similar between sgCtrl-149R and sgStmn2-149R animals as evidenced by immunoassay and immunohistochemical analysis. Unexpectedly, we did observe a significant decrease in the number of pTdp-43 inclusions in the cortex of sgStmn2-149R mice. Modulating Stmn2 expression has been reported to alter levels of other neurogenerative disease proteins, although the mechanisms are not defined. Knockdown of Stmn2 in the midbrain has been shown to lead to increased levels of phosphorylated alpha-synuclein (Wang et al., 2019). In addition, overexpression of STMN2 in the hippocampus of an Alzheimer’s disease mouse model resulted in decreased amyloid β (Aβ) staining and plaque burden, presumably through STMN2-dependent regulation of the intracellular trafficking and processing of amyloid precursor protein (APP) (Wang et al., 2013). TDP-43 regulates STMN2 expression, but in turn, it is possible that Stmn2 affects pTdp-43 levels either directly or indirectly. Alternatively, the accumulation of pTdp-43, which correlates with neurodegeneration in C9FTD (Mackenzie et al., 2013), and loss of Stmn2 may synergize and lead to neuronal loss. At present, we did not test this speculation directly, as reports on the requirement for Stmn2 for neuronal survival have been mixed. Depletion of Stmn2 in the substantia nigra led to a marked decrease of dopaminergic hydroxylase positive (TH+) neurons, but not overall neuron counts (Wang et al., 2019). Despite motor impairments and denervation at the NMJ, Stmn2 knockouts did not display overt neurodegeneration of motor neurons in the spinal cord (Guerra San Juan et al., 2022; Krus et al., 2022; Lopez-Erauskin et al., 2022). These findings may indicate that STMN2 expression may be required for specific neuronal subpopulations. Further study is required to clarify the link between Stmn2 and levels of TDP-43 and other disease-associated proteins. Suppressing expression of the splicing factor SYF2 in a mouse model overexpressing human TDP-43 can rescue motor deficits and TDP-43 pathology (Linares et al., 2023). Depletion of SYF2 in the context of the CRISPRi C9ORF72 model could extend these observations to FTD. Beyond C9ORF72-mediated FTD, we anticipate this model may be useful to interrogate newly identified risk factors and disease modifiers in other neurodegenerative diseases. Indeed, our group has reported Spinocerebellar Ataxia (SCA3) (Jansen-West et al., 2022) and Tau (Cook et al., 2015) models which could be combined with knockdown of disease modifiers in our CRISPRi mouse. This and other CRISPRi mouse models could be a first step toward the development of more complex gene silencing models, such as cell-type specific expression of the dCas9 machinery to rapidly determine cell-autonomous and non-cell autonomous gene function, or inducible expression of repression machinery to compare gene silencing early and late in disease course.
Despite the advantages of the CRISPRi model our study did have limitations. We were able to achieve a robust knockdown of Stmn2 RNA and protein in the dual CRISPRi model, but we did not eliminate expression. Therefore, it is possible that the remaining Stmn2 protein levels were sufficient to carry out its function in the cortex. Stmn2+/− heterozygous mice also exhibit deficits in motor behavior and neuromuscular innervation that only became evident with advanced age (12 months compared to 3 months in homozygous mice) (Krus et al., 2022). Denervation in the heterozygote was also less severe than the Stmn2 knockout (Krus et al., 2022), suggesting that even low levels of Stmn2 can maintain synaptic integrity. We assessed pathology in the cortex at 6 months of age, which may not be sufficient if the effects of Stmn2 depletion are age dependent. We did not characterize FTD-related behavioral deficits nor neurodegeneration in this proof-of-concept study. These would be evident next steps, which may shed light on the role of Stmn2 in brain regions beyond the spinal cord.
Our findings of reduced STMN2 protein levels in C9ORF72 and GRN-linked FTD support efforts focused on determining the contribution of STMN2 deficiency in FTD pathogenesis. Additionally, our results provide a rationale for the development and use of ASOs to correct STMN2 splicing for individuals with FTD, particularly those harboring genetic mutations. Further, we provide proof-of-concept that CRISPRi-mediated knockdown in a disease model is feasible, demonstrating the ability to suppress Stmn2 expression alone and in combination with the C9ORF72 expanded repeat. Further, our dual model replicates disease features previously reported in a C9ORF72 mouse. We propose that CRISPRi mice provide a versatile and rapid method to silence the expression of novel targets of interest and will be useful to interrogate gene function alone or in the context of other disease models in vivo. We anticipate this model will accelerate the translation of newly identified targets to preclinical disease models and ultimately to the clinic.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the Mayo Clinic Institutional Review Board and Ethics Committee approved all protocols associated with the collection of human tissues. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal study was approved by the Mayo Clinic Institutional Animal Care and Use Committee and National Eye Institute ACUS committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SP, DZA, MW, LD, MP, CC, and LP conceived and designed the study. PL, YZ, MW, and LD created CRISPRi knock in mice. SP, DZA, YK, MY, JT, KJ-W, LMD, YS, MP, and CC performed experiments. MD, BO, NG-R, BB, RP, KJ, and DD provided human tissue samples and information regarding pathology, genetics and clinical diagnosis pertaining to those cases. SP and DZA analyzed the data. SP, DZA, MP, CC, and LP wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants R35NS097273 (LP), U54NS123743 (LP and MP), P01NS084974 (LP), AG62077-02-03 (LP) RF1NS120992 (MP), R01AG37491 (KJ), R01AG063780 (CC), Target ALS (MP) and the Robert Packard Center for ALS Research at Johns Hopkins (LP). YK is supported by Milton Safenowitz Postdoctoral Fellowship Program from the Amyotrophic Lateral Sclerosis Association (21-PDF-582). SP is supported by a BrightFocus Alzheimer’s Disease Research Grant (A2020279F).
Acknowledgments
We would like to thank the patients and their families for their donation to the Mayo Clinic Brain Bank, Monica Castanedes-Casey for assistance with immunohistochemistry, and all members of the Petrucelli laboratory for their helpful discussions and comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1251551/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Knockdown of Stmn2 can be modulated in CRISPRi mice. (A) Stmn2 RNA levels were measured by qRT-PCR in 2-month-old CRISPRi mice injected with AAV encoding sgCtrl or sgStmn2 at various titers. (B,C) Stmn2 protein levels were examined by immunoblot (B), followed by quantification and normalization to Gapdh to control for protein loading (C). Data presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, non-significant (ns), with analysis performed by one-way ANOVA followed by Tukey’s post hoc analysis, n = 2–3 animals per group (A,C).
SUPPLEMENTARY FIGURE S2 | Knockdown of Stmn2 does not lead to upregulation of Stathmin family genes. (A-C) The levels of Stmn1 (A), Stmn3 (B), and Stmn4 RNA (C) were measured by qRT-PCR and normalized to endogenous controls Rplp0 and Gapdh, in brain tissue of 6-month old CRISPRi mice injected with AAV encoding sgCtrl or sgStmn2 with either 2 or 149 G4C2 repeats. Data presented as mean ± SEM. non-significant (ns), as analyzed using two-way ANOVA, followed by Tukey’s post hoc analysis, n = 4–6 animals per group.
SUPPLEMENTARY FIGURE S3 | Knockdown of Stmn2 does not impact Tardbp levels. (A) The levels of Tardbp were measured by qRT-PCR and normalized to endogenous controls Rplp0 and Gapdh in brain tissue of 6-month-old CRISPRi mice injected with AAV encoding sgCtrl or sgStmn2 with either 2 or 149 G4C2 repeats. Data presented as mean ± SEM. non-significant (ns), as analyzed using two-way ANOVA, followed by Tukey’s post hoc analysis, n = 4–6 animals per group.
SUPPLEMENTARY FIGURE S4 | Transcript levels of glial markers are increased in CRISPRi mice expressing a C9ORF72-expanded repeat. RNA levels of glial markers were assessed by qRT PCR and normalized to endogenous controls Rplp0 and Gapdh, revealing a significant increase in GFAP levels (A), Iba1/Aif1 levels (B), and Cd68 levels (C) in mice expressing the expanded repeat. Data presented as mean ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001, non-significant (ns), as analyzed using two-way ANOVA, followed by Tukey’s post hoc analysis, n = 4–6 animals per group.
SUPPLEMENTARY TABLE S1 | Characteristics of controls and FTLD-TDP cases.
SUPPLEMENTARY TABLE S2 | Comparison of STMN2 and TDP-43 protein levels in the frontal cortex of controls and FTLD-TDP cases.
SUPPLEMENTARY TABLE S3 | Association of STMN2 protein with TDP-43 and pTDP-43 protein levels in FTLD-TDP cases.
SUPPLEMENTARY TABLE S4 | Comparison of pTDP-43 protein levels between FTLD-TDP cases.
References
Andrews, J. A., Jackson, C. E., Heiman-Patterson, T. D., Bettica, P., Brooks, B. R., and Pioro, E. P. (2020). Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 21, 509–518. doi:10.1080/21678421.2020.1771734
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi:10.1038/s41582-018-0047-2
Baughn, M. W., Melamed, Z., López-Erauskin, J., Beccari, M. S., Ling, K., Zuberi, A., et al. (2023). Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science 379, 1140–1149. doi:10.1126/science.abq5622
Boeynaems, S., Bogaert, E., Michiels, E., Gijselinck, I., Sieben, A., Jovicic, A., et al. (2016). Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 6, 20877. doi:10.1038/srep20877
Bradley, A., Evans, M., Kaufman, M. H., and Robertson, E. (1984). Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature 309, 255–256. doi:10.1038/309255a0
Carpenter, M. D., Hu, Q., Bond, A. M., Lombroso, S. I., Czarnecki, K. S., Lim, C. J., et al. (2020). Nr4a1 suppresses cocaine-induced behavior via epigenetic regulation of homeostatic target genes. Nat. Commun. 11, 504. doi:10.1038/s41467-020-14331-y
Cheng, W., Wang, S., Zhang, Z., Morgens, D. W., Hayes, L. R., Lee, S., et al. (2019). CRISPR-Cas9 screens identify the RNA helicase DDX3X as a repressor of C9ORF72 (GGGGCC)n repeat-associated non-AUG translation. Neuron 104, 885–898. doi:10.1016/j.neuron.2019.09.003
Chew, J., Cook, C., Gendron, T. F., Jansen-West, K., Del Rosso, G., Daughrity, L. M., et al. (2019). Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol. Neurodegener. 14, 9. doi:10.1186/s13024-019-0310-z
Chew, J., Gendron, T. F., Prudencio, M., Sasaguri, H., Zhang, Y. J., Castanedes-Casey, M., et al. (2015). Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154. doi:10.1126/science.aaa9344
Chung, J. H., Whiteley, M., and Felsenfeld, G. (1993). A 5' element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 74, 505–514. doi:10.1016/0092-8674(93)80052-g
Cook, C., Kang, S. S., Carlomagno, Y., Lin, W. L., Yue, M., Kurti, A., et al. (2015). Tau deposition drives neuropathological, inflammatory and behavioral abnormalities independently of neuronal loss in a novel mouse model. Hum. Mol. Genet. 24, 6198–6212. doi:10.1093/hmg/ddv336
Corporation, Q. (2022). “QurAlis receives Health Canada clinical trial application authorization for QRL-201, a first-in-class STATHMIN-2 precision therapy for ALS,” in First ever study to evaluate a therapy that rescues STATHMIN-2 expression in ALS patients expected to initiate in Canada in early 2023 with other countries to follow. Editor K. Vincent (Cambridge, Massachusetts: QurAlis).
Dang, V. T., Kassahn, K. S., Marcos, A. E., and Ragan, M. A. (2008). Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur. J. Hum. Genet. 16, 1350–1357. doi:10.1038/ejhg.2008.111
Duncan, J. E., Lytle, N. K., Zuniga, A., and Goldstein, L. S. (2013). The microtubule regulatory protein stathmin is required to maintain the integrity of axonal microtubules in Drosophila. PLoS One 8, e68324. doi:10.1371/journal.pone.0068324
Freibaum, B. D., Lu, Y., Lopez-Gonzalez, R., Kim, N. C., Almeida, S., Lee, K. H., et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. doi:10.1038/nature14974
Gagliardi, D., Pagliari, E., Meneri, M., Melzi, V., Rizzo, F., Comi, G. P., et al. (2022). Stathmins and motor neuron diseases: Pathophysiology and therapeutic targets. Biomedicines 10, 711. doi:10.3390/biomedicines10030711
Gemberling, M., Siklenka, K., Rodriguez, E., Tonn-Eisinger, K. R., Barrera, A., Liu, F., et al. (2021). Transgenic mice for in vivo epigenome editing with CRISPR-based systems. bioRxiv 2003, 434430.doi:10.1038/s41592-021-01207-2
Gendron, T. F., Bieniek, K. F., Zhang, Y. J., Jansen-West, K., Ash, P. E., Caulfield, T., et al. (2013). Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844. doi:10.1007/s00401-013-1192-8
Gendron, T. F., Van Blitterswijk, M., Bieniek, K. F., Daughrity, L. M., Jiang, J., Rush, B. K., et al. (2015). Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol. 130, 559–573. doi:10.1007/s00401-015-1474-4
Gilbert, L. A., Horlbeck, M. A., Adamson, B., Villalta, J. E., Chen, Y., Whitehead, E. H., et al. (2014). Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 159, 647–661. doi:10.1016/j.cell.2014.09.029
Graf, E. R., Heerssen, H. M., Wright, C. M., Davis, G. W., and Diantonio, A. (2011). Stathmin is required for stability of the Drosophila neuromuscular junction. J. Neurosci. 31, 15026–15034. doi:10.1523/JNEUROSCI.2024-11.2011
Guerra San Juan, I., Nash, L. A., Smith, K. S., Leyton-Jaimes, M. F., Qian, M., Klim, J. R., et al. (2022). Loss of mouse Stmn2 function causes motor neuropathy. Neuron 110, 4031. doi:10.1016/j.neuron.2022.11.003
Haapaniemi, E., Botla, S., Persson, J., Schmierer, B., and Taipale, J. (2018). CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 24, 927–930. doi:10.1038/s41591-018-0049-z
Horlbeck, M. A., Gilbert, L. A., Villalta, J. E., Adamson, B., Pak, R. A., Chen, Y., et al. (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5, e19760. doi:10.7554/eLife.19760
Ihry, R. J., Worringer, K. A., Salick, M. R., Frias, E., Ho, D., Theriault, K., et al. (2018). p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 24, 939–946. doi:10.1038/s41591-018-0050-6
Jansen-West, K., Todd, T. W., Daughrity, L. M., Yue, M., Tong, J., Carlomagno, Y., et al. (2022). Plasma PolyQ-ATXN3 levels associate with cerebellar degeneration and behavioral abnormalities in a new AAV-based SCA3 mouse model. Front. Cell. Dev. Biol. 10, 863089. doi:10.3389/fcell.2022.863089
Jovicic, A., Mertens, J., Boeynaems, S., Bogaert, E., Chai, N., Yamada, S. B., et al. (2015). Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 18, 1226–1229. doi:10.1038/nn.4085
Kampmann, M. (2018). CRISPRi and CRISPRa screens in mammalian cells for precision Biology and medicine. ACS Chem. Biol. 13, 406–416. doi:10.1021/acschembio.7b00657
Klim, J. R., Pintacuda, G., Nash, L. A., Guerra San Juan, I., and Eggan, K. (2021). Connecting TDP-43 pathology with neuropathy. Trends Neurosci. 44, 424–440. doi:10.1016/j.tins.2021.02.008
Klim, J. R., Williams, L. A., Limone, F., Guerra San Juan, I., Davis-Dusenbery, B. N., Mordes, D. A., et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 22, 167–179. doi:10.1038/s41593-018-0300-4
Kosicki, M., Tomberg, K., and Bradley, A. (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771. doi:10.1038/nbt.4192
Kramer, N. J., Haney, M. S., Morgens, D. W., Jovicic, A., Couthouis, J., Li, A., et al. (2018). CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 50, 603–612. doi:10.1038/s41588-018-0070-7
Krus, K. L., Strickland, A., Yamada, Y., Devault, L., Schmidt, R. E., Bloom, A. J., et al. (2022). Loss of Stathmin-2, a hallmark of TDP-43-associated ALS, causes motor neuropathy. Cell. Rep. 39, 111001. doi:10.1016/j.celrep.2022.111001
Linares, G. R., Li, Y., Chang, W. H., Rubin-Sigler, J., Mendonca, S., Hong, S., et al. (2023). SYF2 suppression mitigates neurodegeneration in models of diverse forms of ALS. Cell. Stem Cell. 30, 171–187.e14. doi:10.1016/j.stem.2023.01.005
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi:10.1016/j.neuron.2013.07.033
Liu, P., Li, Y., Lei, J., and Dong, L. (2019). Creating knockin alleles in mouse embryonic stem cells by CRISPR/Cas9-Mediated homologous recombination without Drug selection. Methods Mol. Biol. 1874, 115–137. doi:10.1007/978-1-4939-8831-0_7
Lopez-Erauskin, J., Bravo-Hernandez, M., Presa, M., Baughn, M. W., Melamed, Z. E., Beccari, M. S., et al. (2022). Stathmin-2 loss leads to neurofilament-dependent axonal collapse driving motor and sensory denervation. bioRxiv.
Mackenzie, I. R., Arzberger, T., Kremmer, E., Troost, D., Lorenzl, S., Mori, K., et al. (2013). Dipeptide repeat protein pathology in C9ORF72 mutation cases: Clinico-pathological correlations. Acta Neuropathol. 126, 859–879. doi:10.1007/s00401-013-1181-y
Margolin, J. F., Friedman, J. R., Meyer, W. K., Vissing, H., Thiesen, H. J., and Rauscher, F. J. (1994). Krüppel-associated boxes are potent transcriptional repression domains. Proc. Natl. Acad. Sci. U. S. A. 91, 4509–4513. doi:10.1073/pnas.91.10.4509
Melamed, Z., Lopez-Erauskin, J., Baughn, M. W., Zhang, O., Drenner, K., Sun, Y., et al. (2019). Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 22, 180–190. doi:10.1038/s41593-018-0293-z
Mercuri, E., Darras, B. T., Chiriboga, C. A., Day, J. W., Campbell, C., Connolly, A. M., et al. (2018). Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 378, 625–635. doi:10.1056/NEJMoa1710504
Murlidharan, G., Samulski, R. J., and Asokan, A. (2014). Biology of adeno-associated viral vectors in the central nervous system. Front. Mol. Neurosci. 7, 76. doi:10.3389/fnmol.2014.00076
Platt, R. J., Chen, S., Zhou, Y., Yim, M. J., Swiech, L., Kempton, H. R., et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 159, 440–455. doi:10.1016/j.cell.2014.09.014
Prudencio, M., Gonzales, P. K., Cook, C. N., Gendron, T. F., Daughrity, L. M., Song, Y., et al. (2017). Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet. 26, 3421–3431. doi:10.1093/hmg/ddx233
Prudencio, M., Humphrey, J., Pickles, S., Brown, A. L., Hill, S. E., Kachergus, J. M., et al. (2020). Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Investig. 130, 6080–6092. doi:10.1172/JCI139741
Rhinn, H., Tatton, N., Mccaughey, S., Kurnellas, M., and Rosenthal, A. (2022). Progranulin as a therapeutic target in neurodegenerative diseases. Trends Pharmacol. Sci. 43, 641–652. doi:10.1016/j.tips.2021.11.015
Schoch, K. M., and Miller, T. M. (2017). Antisense oligonucleotides: Translation from mouse models to human neurodegenerative diseases. Neuron 94, 1056–1070. doi:10.1016/j.neuron.2017.04.010
Shin, J. E., Geisler, S., and Diantonio, A. (2014). Dynamic regulation of SCG10 in regenerating axons after injury. Exp. Neurol. 252, 1–11. doi:10.1016/j.expneurol.2013.11.007
Shin, J. E., Miller, B. R., Babetto, E., Cho, Y., Sasaki, Y., Qayum, S., et al. (2012). SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. U. S. A. 109, E3696–E3705. doi:10.1073/pnas.1216204109
Smits, A. H., Ziebell, F., Joberty, G., Zinn, N., Mueller, W. F., Clauder-Münster, S., et al. (2019). Biological plasticity rescues target activity in CRISPR knock outs. Nat. Methods 16, 1087–1093. doi:10.1038/s41592-019-0614-5
Todd, T. W., Mceachin, Z. T., Chew, J., Burch, A. R., Jansen-West, K., Tong, J., et al. (2020). Hexanucleotide repeat expansions in c9FTD/ALS and SCA36 confer selective patterns of neurodegeneration in vivo. Cell. Rep. 31, 107616. doi:10.1016/j.celrep.2020.107616
Wang, J., Shan, C., Cao, W., Zhang, C., Teng, J., and Chen, J. (2013). SCG10 promotes non-amyloidogenic processing of amyloid precursor protein by facilitating its trafficking to the cell surface. Hum. Mol. Genet. 22, 4888–4900. doi:10.1093/hmg/ddt339
Wang, Q., Zhang, Y., Wang, M., Song, W. M., Shen, Q., Mckenzie, A., et al. (2019). The landscape of multiscale transcriptomic networks and key regulators in Parkinson's disease. Nat. Commun. 10, 5234. doi:10.1038/s41467-019-13144-y
Wu, Y., Shao, W., Todd, T. W., Tong, J., Yue, M., Koga, S., et al. (2021). Microglial lysosome dysfunction contributes to white matter pathology and TDP-43 proteinopathy in GRN-associated FTD. Cell. Rep. 36, 109581. doi:10.1016/j.celrep.2021.109581
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi:10.1038/nature14973
Keywords: frontotemporal dementia, amyotrophic lateral sclerosis, STMN2, C9orf72, CRISPR interference, TDP-43
Citation: Pickles S, Zanetti Alepuz D, Koike Y, Yue M, Tong J, Liu P, Zhou Y, Jansen-West K, Daughrity LM, Song Y, DeTure M, Oskarsson B, Graff-Radford NR, Boeve BF, Petersen RC, Josephs KA, Dickson DW, Ward ME, Dong L, Prudencio M, Cook CN and Petrucelli L (2023) CRISPR interference to evaluate modifiers of C9ORF72-mediated toxicity in FTD. Front. Cell Dev. Biol. 11:1251551. doi: 10.3389/fcell.2023.1251551
Received: 01 July 2023; Accepted: 26 July 2023;
Published: 07 August 2023.
Edited by:
Yuzuru Imai, Juntendo University, JapanReviewed by:
Claudia Fallini, University of Rhode Island, United StatesJoseph W. Lewcock, Genentech Inc., United States
Copyright © 2023 Pickles, Zanetti Alepuz, Koike, Yue, Tong, Liu, Zhou, Jansen-West, Daughrity, Song, DeTure, Oskarsson, Graff-Radford, Boeve, Petersen, Josephs, Dickson, Ward, Dong, Prudencio, Cook and Petrucelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonard Petrucelli, cGV0cnVjZWxsaS5sZW9uYXJkQG1heW8uZWR1
†These authors have contributed equally to this work