 Annie L. Miller
Annie L. Miller Rebekah E. James
Rebekah E. James Alan R. Harvey
Alan R. Harvey Dragana Trifunović
Dragana Trifunović Livia S. Carvalho
Livia S. Carvalho
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 04 August 2023
Sec. Molecular and Cellular Pathology
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1224078
This article is part of the Research Topic Inherited Retinal Dystrophies: A light at the end of the tunnel? View all 12 articles
Elucidation of the cellular changes that occur in degenerating photoreceptors of people with inherited retinal diseases (IRDs) has been a focus for many research teams, leading to numerous theories on how these changes affect the cell death process. What is clearly emerging from these studies is that there are common denominators across multiple models of IRD, regardless of the underlying genetic mutation. These common markers could open avenues for broad neuroprotective therapeutics to prevent photoreceptor loss and preserve functional vision. In recent years, the role of epigenetic modifications contributing to the pathology of IRDs has been a particular point of interest, due to many studies noting changes in these epigenetic modifications, which coincide with photoreceptor cell death. This review will discuss the two broad categories of epigenetic changes, DNA methylation and histone modifications, that have received particular attention in IRD models. We will review the altered epigenetic regulatory events that are believed to contribute to cell death in IRDs and discuss the therapeutic potential of targeting these alterations.
Inherited retinal diseases (IRDs) are a genetically and phenotypically diverse group of blinding diseases that can result in photoreceptor death, dysfunction, or developmental delay (Berger et al., 2010). Collectively, these diseases affect 1:2000 people worldwide and pose a significant socioeconomic problem due to healthcare costs, reduced workplace participation and an increased requirement for carer assistance (Berger et al., 2010; Galvin et al., 2020). However, treatments available for IRD are limited; only people with a mutation in one particular gene, RPE65, can receive the FDA-approved gene therapy drug Luxturna, leaving a critical gap in patient care (Maguire et al., 2021). Mutations in over 270 genes have been associated with IRD to date, and more are being discovered (Center DSTUoTHS, 2020). Due to this genetic heterogeneity, many researchers have investigated common targets that are independent of the underlying genetic mutations, with the aim of developing neuroprotective therapies that can treat a broader population of IRD patients. Such studies often focus on understanding the precise cell death mechanisms that lead to photoreceptor death. There is extensive debate in the field, with conflicting reports on whether apoptotic or non-apoptotic cell death mechanisms, or somewhere “in-between”, are the predominant cause of photoreceptor loss (Brunet et al., 2022). A seminal study by Arango-Gonzalez et al. (2014) identified a common non-apoptotic cell death pathway that was dysregulated in ten mouse models of IRD, with many of the components of this pathway linked to epigenetic regulation (Arango-Gonzalez et al., 2014). In recent years there has been increased research in this area, strengthening links between epigenomic modifications and cell death in IRD. This review will outline the current understanding of the association of two types of epigenetic modification, DNA methylation and histone modifications, with IRD pathology.
DNA methylation is a heritable genetic mark essential in multiple developmental processes such as genomic imprinting, X-chromosome inactivation and suppression of repetitive element transcription (Jin et al., 2011). DNA methylation functions by recruiting proteins involved in gene repression while also having a role in blocking DNA transcription factors (Moore et al., 2013). In eukaryotes, DNA methylation most often involves the addition of a methyl group to the C5 position of cytosine, forming 5-methylcytosine (5mC) (Moore et al., 2013). Other forms of DNA methylation exist, namely N6-methyladenine and N4-methylcytosine; however, their role in eukaryotes is far less clear, and thus they will not be a focus of this review (Xiao et al., 2018; Rodriguez et al., 2022). The level of methylation and demethylation of DNA is modulated by DNA methyltransferases (DNMTs) and ten-eleven translocase (TET) enzymes (Figure 1) (Moore et al., 2013; Rasmussen and Helin, 2016). DNMTs catalyse DNA methylation by transferring a methyl group to the fifth carbon of a cytosine to form 5mC (Moore et al., 2013). TET enzymes regulate the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), which can be further oxidised to form 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), leading to DNA demethylation (Moore et al., 2013). After oxidation to 5fC or 5caC, restoration of the molecule to a cytosine is modulated by thymine DNA glycosylase (TDG), which is an essential component of the base excision repair (BER) pathway (Moore et al., 2013). Changes to the proportion of oxidised cytosines and DNMTs are involved in multiple pathologies such as cancer and are thought to potentially contribute to photoreceptor degeneration in models of IRD (Wahlin et al., 2013; Farinelli et al., 2014; Locke et al., 2019). 5mC and 5hmC are the best understood of the cytosine derivatives and are thought to be the most biologically relevant thus far. This review will focus on studies that involve their dysregulation in IRDs.
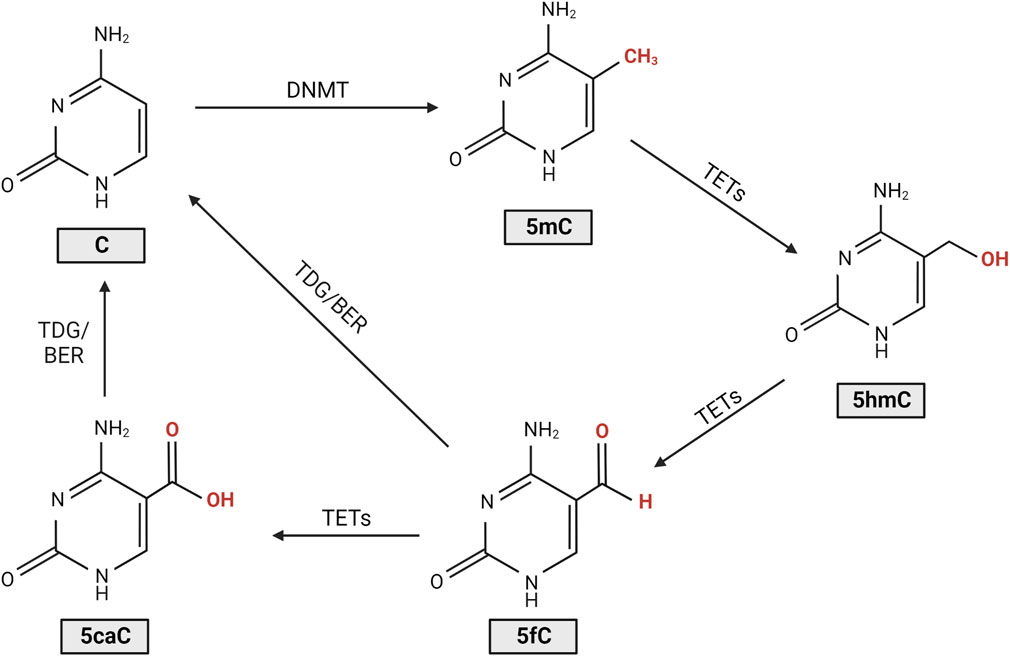
FIGURE 1. Cytosine DNA methylation and demethylation. DNA methyltransferases (DNMTs) introduce a methyl group to the cytosine (C), forming 5-methylcytosine (5mC). Ten-eleven translocase (TET) enzymes then regulate the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). Following oxidation to 5fC or 5caC, restoration of the molecule to cytosine is modulated by thymine DNA glycosylase (TDG), an essential component of the base excision repair (BER) pathway (Moore et al., 2013; Rasmussen and Helin, 2016).
Wahlin et al. (2013) first reported aberrant DNA methylation levels in the rd1 mouse model of retinitis pigmentosa (RP), a widely used model that displays rapid rod photoreceptor loss that peaks between postnatal days 12–14 (P12-14; for a summary of all preclinical models discussed in this review, refer to Supplementary Table S1) (Keeler, 1924; LaVail and Sidman, 1974; Portera-Cailliau et al., 1994; Wahlin et al., 2013). At timepoints corresponding to this peak of rod cell death, both rod and cone photoreceptors in the rd1 retina were found to possess increased immunoreactivity for 5mC and 5hmC compared to wildtype controls (Wahlin et al., 2013). This increase in 5hmC positive cells was seen as early as P9, and numbers were even greater at P10, prior to significant thinning of the outer nuclear layer (ONL) which occurs around 2 weeks postnatal (Wahlin et al., 2013). The authors noted that cells stained positively for either 5hmC or 5mC were also positive for the cell death terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) stain (Wahlin et al., 2013). The same group also investigated this phenomenon in homozygous rhodopsin-GFP knock-in mice carrying the P23H Rho mutation, which displayed 5mC positive cells in the early stages of degeneration. Similarly, in P23H adult retinal explants that were grown for 4 and 7 days, as the ONL degenerated there was an accumulation of 5mC positive cells, primarily in rods (Wahlin et al., 2013). These results were validated by a similar study that looked at 5mC expression in four models of RP, the rd1 and rd2 mouse models, which have mutations in the Pde6b and Prph2 genes, respectively, and the P23H and S334ter rat models, which harbor mutations in the Rho gene (Farinelli et al., 2014). It was shown that at the peak of cell death in each model there was an increase in 5mC expression in photoreceptors that was colocalised with TUNEL positivity (Farinelli et al., 2014). The rd1 mouse retina was further investigated at the ultrastructural level, revealing a severely altered chromatin structure which coincided with increased expression of the DNA methylating isozyme, DNMT3a. In microarray analysis, the rd1 mouse showed hypermethylation of genes involved in cell death and survival, cell morphology, and nervous system development, correlating with a transcriptional silencing action. Interestingly, rd1 retinal explants treated with the DNMT inhibitor decitabine showed a reduction in photoreceptor cell death after 4 days of treatment and a reduction in 5mC positive cells (Farinelli et al., 2014). These results suggest a potential role of DNA methylation in the pathological process of IRD and shows that DNA methylation may be a potential target for neuroprotection. However, research into this field is still in comparatively early stages, as only two studies currently have assessed the changes in DNA methylation in the context of IRD (Figure 2). As such, there is a need to understand the role of DNA methylation in the degenerative process, as well as the links between aberrant DNA methylation and photoreceptor loss in other models of IRD, and how to best translate any beneficial outcomes in preclinical research to the clinic.
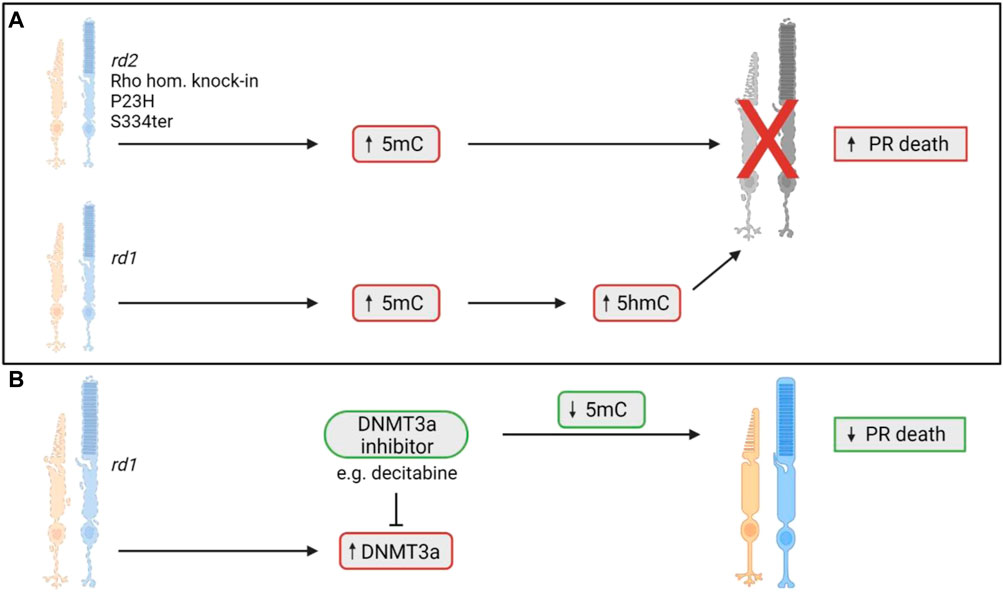
FIGURE 2. DNA methylation changes involved in photoreceptor death in models of IRD. (A) Previous studies have shown an upregulation of the demethylated cytosine molecule, 5-methylcytosine (5mC), in five models of IRD (Wahlin et al., 2013; Farinelli et al., 2014). The 5mC upregulation coincides with the photoreceptor degeneration found in each model (Wahlin et al., 2013; Farinelli et al., 2014). In the rd1 mouse model only, increased levels of the 5-hydroxymethylcytosine (5hmC) molecule was noted as well (Wahlin et al., 2013). 5hmC is an oxidised form of 5mC, with increased levels coinciding with photoreceptor death (Wahlin et al., 2013). (B) In the rd1 mouse, there was increased expression of DNMT3a, an enzyme responsible for the demethylation of cytosine to form 5mC. When rd1 retinal explants were treated with the DNMT3a inhibitor, decitabine, they noted a decrease in 5mC positive cells and a reduction in photoreceptor cell death (Farinelli et al., 2014). PR = photoreceptor.
Histone modifications permit significant changes in the regulation of DNA and play a major role in almost all fundamental biological processes. Modifications are complex, with many chemical groups that can be added to histones such as methyl, acetyl, and ADP-ribose units (Bannister and Kouzarides, 2011). Gene expression changes vary depending on the type and location of these modifications (Bannister and Kouzarides, 2011). Post-translational acetylation and deacetylation of histone proteins allow the bidirectional regulation of gene expression and chromatin architecture by opening (acetylation) or closing (deacetylation) the chromatin structure (Park and Kim, 2020). The dynamic process and balance of acetylation and deacetylation are modulated by histone acetyltransferases (HATs) and histone deacetylases (HDAC), respectively (Figure 3) (Bannister and Kouzarides, 2011). Over the years, many studies have reported an association between altered HDAC activity and the pathology of IRDs. HDACs counteract the acetylation process modulated by HATs by removing acetyl groups from histone proteins, deacetylating histones back to their basal state, thereby suppressing gene expression (Bannister and Kouzarides, 2011). HDACs can be separated into four broad categories: Class I (HDACs 1, 2, 3, 8), Class II (HDACs 4, 5, 6, 7, 9, 10), Class III (NAD-dependent sirtuins) and Class IV (HDAC11) (Bannister and Kouzarides, 2011; Balaiya et al., 2017). Each HDAC class and its isoforms have unique biological functions, tissue specificity, enzymatic activity and more (Balaiya et al., 2017; Park and Kim, 2020). Classical HDACs (Class I, II and IV) are distinct from sirtuins (Class III HDACs), so will be discussed separately.
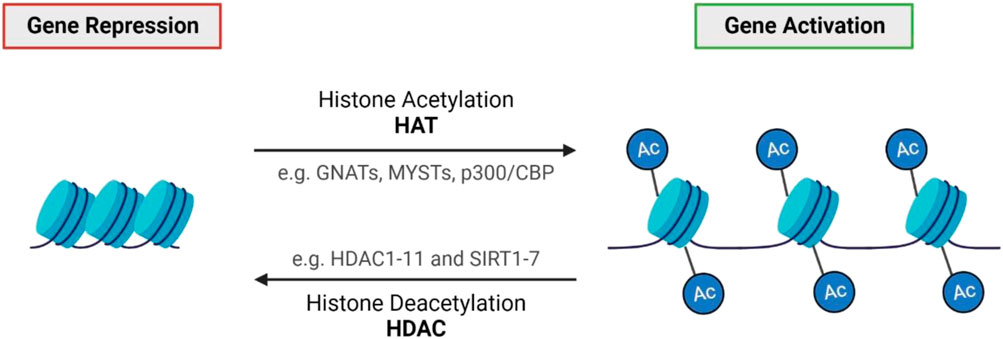
FIGURE 3. Histone acetylation and deacetylation. Histone acetylation generally results in gene activation through chromatin de-condensation, whereas deacetylation results in gene repression (Park and Kim, 2020). The balance of acetylation and deacetylation is modulated by two opposing classes of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Bannister and Kouzarides, 2011; Park and Kim, 2020).
In the field of IRDs, research has mainly focused on establishing the role of histone deacetylation in the context of photoreceptor degeneration. A substantial decrease in acetylation (hypoacetylation) was identified in the rd1 retina, thought to be due to an increase in HDAC class I, II, and IV activity (Sancho-Pelluz et al., 2010). Interestingly, approximately 94% of hypoacetylated cells were positive for TUNEL staining, while increased HDAC activity was detected 2 days before TUNEL positivity, suggesting that HDAC activity may precede the final stages of cell death (Sancho-Pelluz et al., 2010). This was further confirmed when rd1 explants were treated with the pan-HDAC inhibitor, trichostatin A (TSA), which caused a significant reduction in TUNEL-positive cells. However, when treated with the class I HDAC inhibitor, Scriptaid, no neuroprotective effects on photoreceptor survival were reported (Sancho-Pelluz et al., 2010). A later study went on to identify a potential causative role of HDAC in photoreceptor degeneration, highlighting that HDAC overactivity was a common feature in ten animal models of IRD: rd1, rd10, rd2, Cngb1−/−, Rho−/−, S334ter, P23H, Pde6ccpfl1, Cnga3−/−, and Rpe65−/− (Arango-Gonzalez et al., 2014).
Due to the identification of HDAC overactivity in multiple models of IRD, many studies have searched for neuroprotective effects of pharmacological inhibition of HDACs. In the Pde6ccpfl1 achromatopsia mouse model, treatment with TSA at P14, the time of onset of cone photoreceptor death in this model, resulted in cone rescue up to 10 days post-treatment (Trifunović et al., 2016). This study also showed improved localisation of cone-specific proteins, including opsins and cone transducin (GNAT2), and improved cone developmental migration patterns (Trifunović et al., 2016). When TSA was administered later in the disease stage at P18 the drug still displayed neuroprotective abilities, with a 10% increase in cone numbers and improved cone migration persisting as long as 12 days following a single intravitreal injection (Samardzija et al., 2019). TSA has also shown neuroprotective ability in rd10 retinal explants, with a five-fold increase in surviving photoreceptors (Trifunović et al., 2018). Administration of TSA in the rd1 and rd10 models at later stages of the disease, P19 and P42, respectively, was sufficient to preserve and support cone survival long-term while also allowing cones to remain light sensitive with preservation of visual function (Samardzija et al., 2020). Another pan-HDAC inhibitor, SAHA, was tested in 661W cells that were stressed with a non-specific phosphodiesterase inhibitor, resulting in improved cell survival, mitochondrial respiration and reduced mitochondrial fission in the 661W cells (Perron et al., 2021). When rd1 explants were treated with SAHA, the number of photoreceptors approximately doubled compared to controls (Perron et al., 2021; Dong et al., 2023).
Despite evidence that pharmacological HDAC inhibition is neuroprotective in several models of IRD, the molecular basis for this neuroprotection is poorly understood, mainly because HDAC inhibition drives concurrent transcriptional changes in numerous genes. For example, Samardzija et al. (2021) performed RNA sequencing analysis on rd1 cones treated with TSA, showing that TSA may have a multi-level protection mechanism via regulation of different pro-survival pathways including MAPK, PI3K-Akt and autophagy (Samardzija et al., 2020). These studies and others have highlighted the complexity of HDAC and the impact of its inhibition. As such, more broad transcriptional studies are required to help understand the mechanisms behind the neuroprotection that arises from HDAC inhibition.
Only one HDAC inhibitor has been tested in clinical trials for use in RP, valproic acid (VPA); however, it sparked much debate due to highly variable patient responses and concerns raised about the study design. VPA was already FDA-approved for use in epilepsy, bipolar and migraine disorders. As previous work in animal models of RP showed VPA could inhibit apoptosis, activate microglia and stimulate photoreceptor regeneration from glial cells, drug repurposing was suggested for its therapeutic use in RP (Clemson, 2010). Additionally, VPA was found to be a potent molecular chaperone with the ability to increase the yield of properly folded mutant rhodopsin in the RhoP23H/+ heterozygous knock-in mouse (Clemson, 2010; Kaushal et al., 2010). The initial human study reported that VPA had improved visual acuity in 9 of the 13 eyes from patients with RP; however, this study was criticised for a number of reasons, including a lack of controls and the failure to properly account for side effects from VPA use (Clemson et al., 2011; Sandberg et al., 2011). VPA was tested in a further three patients, but the trial was ended prematurely as the patients experienced a reduction in visual acuity and significant side effects including intolerable photophobia in one patient and torsional nystagmus in another, both of which were resolved upon cessation of VPA (Sisk, 2012). A subsequent non-randomised trial with ten patients showed an improved mean visual acuity after 3 months of daily VPA oral dosing, with average visual acuity progressing from 20/72 to 20/65 (Shanmugam et al., 2012). Similarly, a fourth study reported that 14 out of 15 RP patients treated with VPA had improved visual acuity (Kumar et al., 2014). Iraha et al. (2016) reported that after 6 months of VPA use, 16 out of 29 patients considered it “easier to see” when undergoing the Humphrey field analyser central 10–2 program. Patients showed improved best corrected visual acuity and visual field testing after treatment, but this improvement was lost once VPA administration was ceased (Iraha et al., 2016). Conversely, Bhalla et al. reported that in 31 patients with a range of different IRDs, there was, on average, a reduction in their visual field after VPA treatment, with most patients experiencing either no change or a slight decrease in visual acuity (Bhalla et al., 2013). Finally, a trial using VPA for 6–12 months on RP patients of unknown genotype, found no improvement in best corrected visual acuity measurements or visual field analyses, while noting potential decreases in some ERG measurement parameters (Totan et al., 2017).
Sisk (2012) suggested that genotype differences may be responsible for the variable patient outcomes (Sisk, 2012), a proposal validated by several studies conducted in animal models. A study conducted in four Xenopus laevis models, which expressed different RP-linked alleles of human rhodopsin, showed that administration of VPA in a Xenopus line with the P23H rhodopsin mutation was neuroprotective and led to an improvement in visual function (Vent-Schmidt et al., 2017). The other three Xenopus lines carrying the Q344ter, T17M, or T4K rhodopsin mutations did not demonstrate these same improvements (Vent-Schmidt et al., 2017). Similarly, a study carried out in two mouse models of autosomal recessive RP, the rd1 and rd10 mouse models, showed that daily injections for 12 days of VPA in rd1 mice resulted in a significant increase in photoreceptor rows, with several extra rows of rod nuclei compared to PBS injected controls (Mitton et al., 2014). On the other hand, when VPA was administered in the rd10 mouse model there was a failure of photoreceptor rescue and reduced visual function (Mitton et al., 2014). In 2018, the results of a randomised phase 2 multicentre placebo-controlled clinical trial of 90 patients with genetically characterised autosomal dominant RP revealed a small but significantly worse outcome for VPA-treated patients (Birch et al., 2018). Most adverse events reported were mild, but ultimately, the use of VPA in autosomal dominant RP was not supported (Birch et al., 2018). Future clinical translation and research of VPA or other HDAC inhibitors should consider the genotypes and clinical diagnosis of the patient and how that could affect their response to treatment with HDAC inhibitors. Importantly, when considering treatment regimes, different pan-HDAC inhibitors may have slightly different HDAC targets or have stronger affinities to certain isoforms, thus not all HDAC inhibitors will necessarily have the same effect in patients.
Isoform-specific HDAC inhibition has also been investigated, allowing for a deeper understanding of HDAC subtypes that may be associated with cell death and potentially reducing off-target toxicity sometimes associated with pan-HDAC inhibitors (Bieliauskas and Pflum, 2008; Vishwakarma et al., 2013). A study that used romidepsin, an HDAC1 and HDAC2 inhibitor, in the rd10 mouse, found that it caused significant neuroprotection and preservation of the rods, the ONL thickness increasing by approximately three-fold (Popova et al., 2021). Of concern, romidepsin also caused a reduction in weight gain throughout the treatment when compared to age-matched controls (Popova et al., 2021). In one study, no increase in cell survival was observed when the HDAC6 specific inhibitor, Tubastatin A, was applied to 661W cells that had been stressed with a non-specific phosphodiesterase inhibitor (Perron et al., 2021). Contrastingly, when 661W cells were stressed with hydrogen peroxide, treatment with Tubastatin A promoted cell survival, perhaps due to upregulation of heatshock proteins 25 and 70, heat shock transcription factor 1 and peroxiredoxin 1 (Leyk et al., 2017). Tubastatin A was then tested in the dyeucd6 zebrafish model of IRD, resulting in improved retinal morphology, as assessed by qualitative improvement of the photoreceptors, a slight improvement in outer segment length, and rescue of visual function (Leyk et al., 2017). The authors suggested that HDAC6 inhibition and the associated regulation of peroxiredoxin may play a role in protecting the photoreceptors in this model (Leyk et al., 2017). In the atp6v0e1−/− zebrafish model, HDAC6 inhibition with Tubastatin A led to improved visual function and cell morphology, the treated zebrafish showing an eight-fold improvement in vision and a 44.7% improvement in photoreceptor outer segment area (Sundaramurthi et al., 2020). Proteome sequencing after treatment revealed modulation of ubiquitin-proteasome, phototransduction, metabolism, and phagosome pathways. In addition, when using rd10 retinal explants, there was an increased number of cone arrestin-positive cells after treatment with Tubastatin A (Sundaramurthi et al., 2020). Another study used electroporation to overexpress HDAC4 to investigate its role in the degenerative process in newborn rd1 mice (Chen and Cepko, 2009). Retinae transfected to overexpress HDAC4 (but not HDAC5 or HDAC6) contained more rods at P50, at a time when these photoreceptors would usually have degenerated (Chen and Cepko, 2009). Furthermore, compared to the full-length HDAC4 protein, expression of a short N-terminal domain of HDAC4 resulted in a more extensive preservation of rd1 rods, greater cone survival and partial restoration of cone visual function (Guo et al., 2015). The authors speculated HDAC4s photoreceptor protection ability might be due to a restoration of altered gene expression of cell cycle progression genes Ccnb1 and Ccnd1, the transcription factors c-fos, c-jun, and p53, endoplasmic reticulum stress genes such as Atf4, Chop and Casp12 and apoptotic/cell death genes such as Bid and Parp1 (Guo et al., 2015).
In summary, HDAC overactivity seems to be a consistent feature in many preclinical models of IRD, with HDAC inhibition being neuroprotective. More recently, isoform-specific studies have highlighted that not all HDAC overactivity is necessarily deleterious, with evidence that HDAC4 can be neuroprotective. Further studies should validate if such results are consistent across different models of IRD, as well as looking at HDAC isoforms that have not been investigated yet. A summary of all studies that investigate HDAC changes and consequent HDAC modulation is shown in Figure 4.
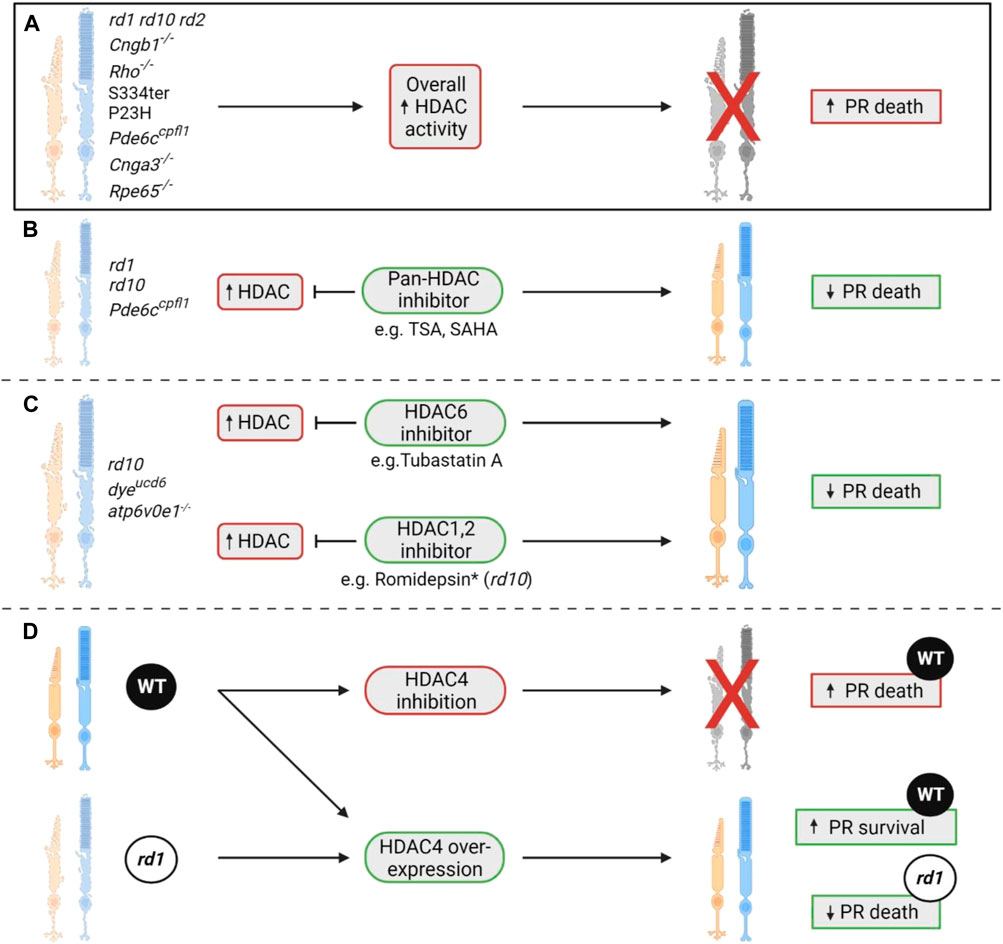
FIGURE 4. The role of HDACs in photoreceptor degeneration. (A) A seminal study showcased that histone deacetylase (HDAC) overactivity was a consistent phenomenon observed in ten different rodent models of IRD, namely the rd1, rd10, rd2, Cngb1−/−, Rho−/−, S334ter, P23H, Pde6ccpfl1, Cnga3−/−, and Rpe65−/− (Sancho-Pelluz et al., 2010; Arango-Gonzalez et al., 2014). This increase in overall HDAC activity coincided with the peak of cell death in each of these models (Sancho-Pelluz et al., 2010; Arango-Gonzalez et al., 2014). (B) Previous studies have shown that inhibition of HDAC with a pan-HDAC inhibitor such as trichostatin A (TSA) or SAHA can result in significant retention of photoreceptor numbers. These neuroprotective effects were displayed in two RP models (rd1 and rd10) and one achromatopsia model (Pde6ccpfl1) (Trifunović et al., 2016; Trifunović et al., 2018; Samardzija et al., 2019; Samardzija et al., 2020; Perron et al., 2021; Dong et al., 2023). (C) Isoform-specific HDAC inhibition has also proven beneficial in various models of IRD, with treatment with the HDAC1/2 inhibitor romidepsin allowing for preservation of rod numbers in the rd10 model of RP (Popova et al., 2021). *Despite romidepsin having neuroprotective effects in the retina, it caused a reduction in weight gain throughout treatment compared to age-matched wildtype controls, displaying a potential systemic toxicity (Popova et al., 2021). HDAC6 inhibition has also been shown to be neuroprotective in the dyeucd6, atp6v0e1−/− zebrafish models of inherited blindness and the rd10 mouse model of RP (Leyk et al., 2017; Sundaramurthi et al., 2020). Each model had HDAC6 inhibited via administration of the HDAC6 inhibitor Tubastatin A. In the two zebrafish models, improvements in retinal morphology and visual function were observed (Leyk et al., 2017; Sundaramurthi et al., 2020). In the rd10 model, an improvement in the number of cone arrestin positive cells was observed (Sundaramurthi et al., 2020). (D) HDAC4 inhibition in wildtype (WT) mice has been shown to cause photoreceptor death, implying that overexpression of HDAC4 is in fact neuroprotective (Chen and Cepko, 2009; Guo et al., 2015). This was validated when HDAC4 overexpression in WT and rd1 mice showed increased photoreceptor survival in both lines (Chen and Cepko, 2009; Guo et al., 2015). PR = photoreceptor.
The Class III HDACs, sirtuins, are a unique and highly conserved family of nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylases. They deacetylate both histone and non-histone proteins and are involved in cellular functions such as stress response, apoptosis, DNA repair, cell differentiation and much more (Balaiya et al., 2017). Seven sirtuins have been identified in mammals (SIRT1-7) (Balaiya et al., 2017). The role of sirtuins was investigated in the rd1 mouse, to ascertain if the overactive HDAC activity was derived from classical HDACs, sirtuins or both (Sancho-Pelluz et al., 2010). In the rd1 retina, while there was a small increase in overall sirtuin activity compared to wildtype controls, classic HDACs showed a much more substantial increase (Sancho-Pelluz et al., 2010). To further elucidate if sirtuins contributed to rd1 retinal pathology, the sirtuin inhibitor nicotinamide was administered to rd1 explants, but no improvement in photoreceptor survival was observed (Sancho-Pelluz et al., 2010). To elucidate which specific sirtuin isoforms might be important in photoreceptor degeneration, Sirt1 immunoreactivity was assessed in retinae from rd10 mice aged from P14 until 5 months of age (Jaliffa et al., 2009). There was strong Sirt1 staining at P15 in scattered cells throughout the ONL of the central retina (Jaliffa et al., 2009). Over time, Sirt1 immunoreactivity decreased as the rd10 retina degenerated, following an apparent central-to-periphery gradient (Jaliffa et al., 2009). This staining was seen mostly, if not exclusively, in the nucleus of the photoreceptors, and approximately 85% of the Sirt1-positive cells were also TUNEL-positive (Jaliffa et al., 2009). Additionally, of the Sirt1-positive cells, 82% were also positive for the apoptotic marker caspase-12, and 71% for mitochondrial apoptosis inducing factor, Aif (Jaliffa et al., 2009). In a different IRD model, the Nmnat1V9M/V9M mutant mouse, sirtuin expression changes were assessed indirectly by examining the sites they deacetylate, such as H3K9, H3K18, and H4K16 (Greenwald et al., 2021). H3K9ac is deacetylated by Sirt1 and potentially Sirt6, H3K18ac by Sirt7, and H4K16ac, by Sirt1, Sirt2, and possibly Sirt6 (Greenwald et al., 2021). All three sites showed no significant changes compared to wildtype, suggesting that these particular sirtuins were not dysregulated as a part of Nmnat1V9M/V9M disease progression (Greenwald et al., 2021). Overall, only one study by Jaliffa et al. (2009) has identified sirtuin expression changes might be relevant in the degeneration seen in models of IRD (Jaliffa et al., 2009). Clearly, more work needs to be done to understand the potential role of sirtuins in different IRDs, especially since sirtuin changes have been noted in several different neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease (Chandramowlishwaran et al., 2020). Increasing online access to single-cell sequencing data will permit more detailed and potentially revealing information about sirtuin expression in disease photoreceptors.
Histone methylation and demethylation are the processes whereby methyl groups are added or removed from histone proteins (Greer and Shi, 2012). The methylation process is dynamic and supported by various enzymes, which can add or remove methyl groups on different histone types, as well as specific residues on those histones (Figure 5).
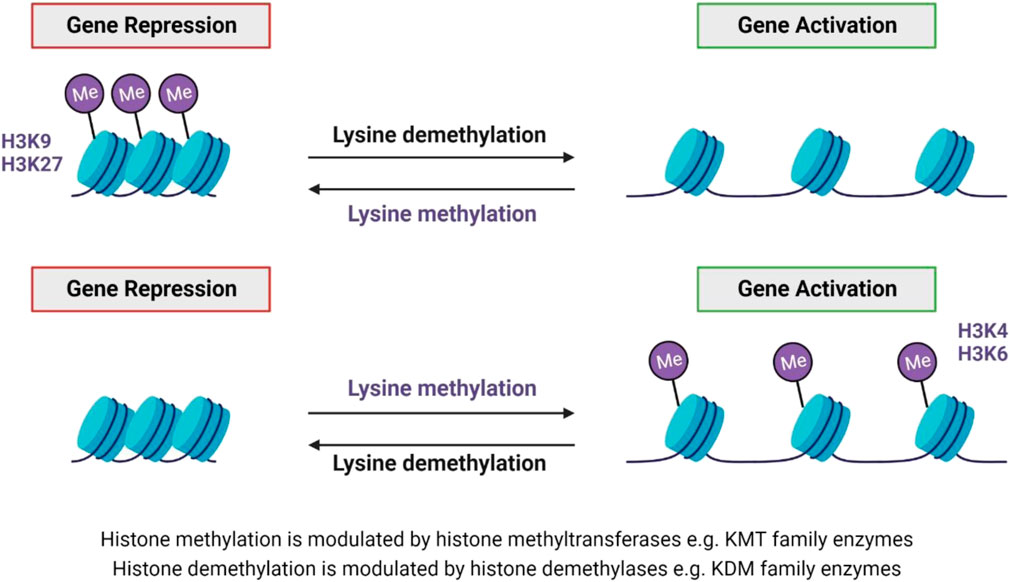
FIGURE 5. Lysine methylation and demethylation. Histone methytransferases modulate histone methylation, while demethylation is modulated by histone demethylases (Greer and Shi, 2012; Song et al., 2016). Both lysine methylation and demethylation can result in either gene repression or activation - dependent on the lysine residue that is methylated, e.g. H3K9 and H3K27 methylation results in gene repression, whereas H3K4 and H3K6 methylation result in gene activation (Basavarajappa and Subbanna, 2021).
Significantly, abnormal changes to these methylation marks have been associated with a multitude of diseases, including cancer and neurodegenerative disease (Song et al., 2016; Basavarajappa and Subbanna, 2021). Some forms of IRDs have also been associated with changes in methylation, but this field is still in its comparative infancy. In 2020, Zheng and colleagues made the first discovery of the involvement of histone methylation in IRDs, reporting increased expression of H3K27me3 in retinae from rd1 mice (Zheng et al., 2018). The global histone methylation inhibitor DZNep was administered subretinally at P0, resulting in the preservation of both the a- and b-wave in scotopic and photopic electroretinogram (ERG) recordings at P14 (Zheng et al., 2018). With the same treatment regime, ONL thickness was significantly retained by 70% compared to untreated controls (Zheng et al., 2018). Significant improvement in ONL thickness was also seen at P21 after treatment at P0; however, improvement in the ERGs was no longer present (Zheng et al., 2018). Another study in the rd10 model of RP reported that inhibition of the histone methylation eraser, lysine demethylase 1, LSD1, which specifically demethylates H3K4me1/2 and H3K9me1/2, resulted in reduced rod degeneration, preservation of vision, and influenced the expression of multiple genes including maintenance of rod-specific transcripts and downregulation of genes involved in inflammation, gliosis and cell death (Popova et al., 2021). The authors suggested that the neuroprotective activity of LSD1 inhibitors firstly targeted histone modifications, increasing accessibility of chromatin and upregulation of neuroprotective genes, then potentially inhibited transcription of inflammatory genes (Popova et al., 2021). Finally, in a recent study, we found that the ubiquitous H3K27me3 expression seen in wildtype cones was lost in the Pde6ccpfl1 mouse model of achromatopsia (Miller et al., 2022). Administration of GSK-J4, a histone demethylase inhibitor that targets H3K27me3, resulted in increased immunostaining of H3K27me3 in Pde6ccpfl1 cones, and increased cone survival in retinal explants. When GSK-J4 was administered to mice via a single intravitreal injection, there were significant transcriptional changes to pathways involved in mitochondrial dysfunction, endoplasmic reticulum stress and key epigenetic pathways (Miller et al., 2022). The role of histone methylation modifications and their contribution to IRD pathology has only recently been investigated, with current studies showing crucial differences in H3K27me3 status in cone and rod photoreceptors, where ubiquitous expression in rods is deleterious to survival, while it is beneficial in cones. A summary of all studies that have assessed changes in histone methylation in preclinical IRD models can be found in Figure 6. Future studies should investigate the differences between histone methylation patterns in rods versus cones and attempt to understand which changes to histone methylation sites are most relevant.
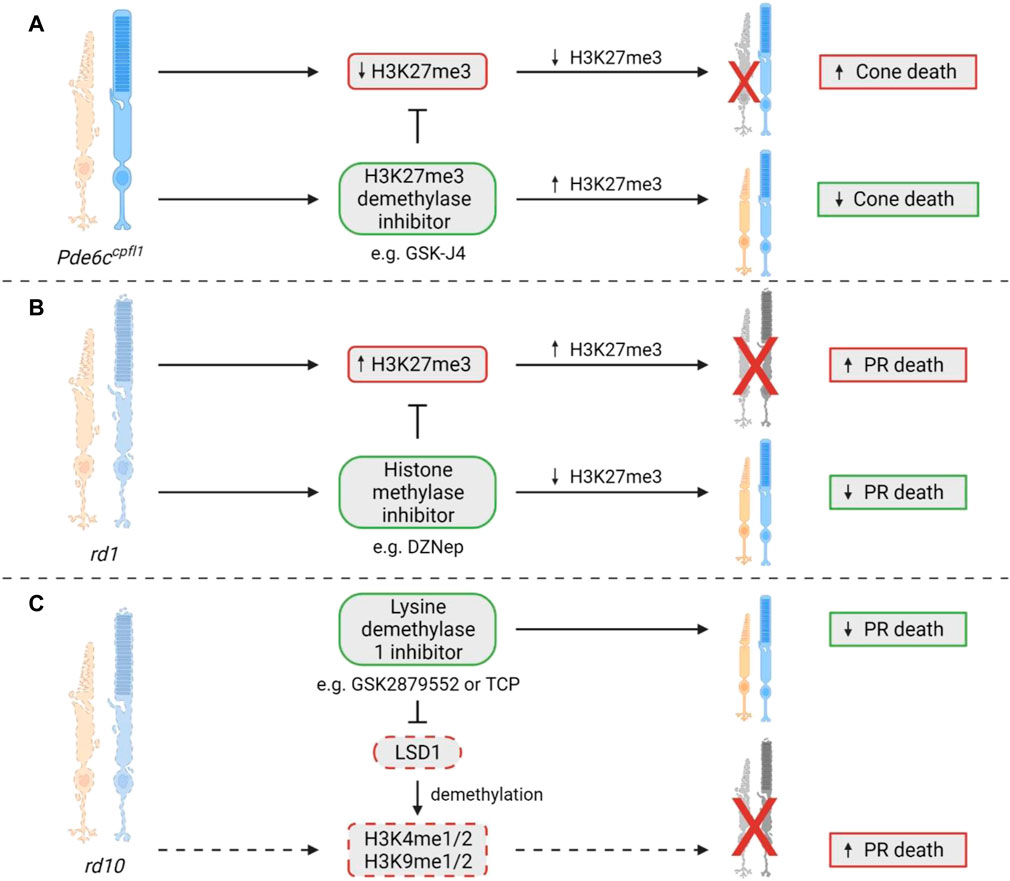
FIGURE 6. Histone methylation changes in IRDs. (A) In the Pde6ccpfl1 mouse model of achromatopsia, decreased expression of the usually ubiquitous H3K27me3 was noted in cone photoreceptors specifically. A significant cone photoreceptor survival was observed when H3K27me3 demethylation was inhibited via GSK-J4 administration in Pde6ccpfl1 retinal explants (Miller et al., 2022). (B) Interestingly, the rd1 mouse model, which shows degeneration of both rod and cone photoreceptors, showed an increase in H3K27me3 expression that coincides with photoreceptor cell death. After treatment with the histone methylase inhibitor DZNep, there was a significant improvement in photoreceptor survival (Zheng et al., 2018). (C) A study in the rd10 model of RP showed that treatment with a lysine demethylase 1 (LSD1) inhibitor, such as GSK2879552 or TCP, could cause a reduction in photoreceptor cell death. LSD1 is known to demethylate H3K4me1/2 and H3K9me1/2, which may contribute to cell death in this model, although the study did not directly assess this, so it remains to be confirmed (Popova et al., 2021). PR = photoreceptor.
Poly(ADP-ribosyl)ation is a post-translational modification involving the addition of ADP-ribose units on the glutamic or aspartic acid residues of histone and non-histone target proteins, catalysed by poly (ADP-ribose) polymerase (PARP; Figure 7) (Tong et al., 2001; Kraus and Lis, 2003; Quénet et al., 2009). Modifications can involve mono ADP-ribose additions or can involve chains of ADP-ribose polymers being added, called poly (ADP-ribose) (PAR) accumulation (Langelier et al., 2018). PAR accumulation generally causes transcriptional activation via chromatin de-condensation and the alteration of promoter and enhancer activity (Kraus and Lis, 2003; Quénet et al., 2009). This reaction is reversible due to the endo- and exo-glycosidic activity of poly (ADP-ribose) glycohydrolase (PARG) (Quénet et al., 2009). PARP is involved in various cellular roles, including cell proliferation, cell death, DNA repair, genomic stability, and epigenetic regulation (Tong et al., 2001; Quénet et al., 2009). The role of poly(ADP-ribosyl)ation, and the relevant molecules in this process have garnered attention for their potential role in neurodegeneration, including in IRDs.
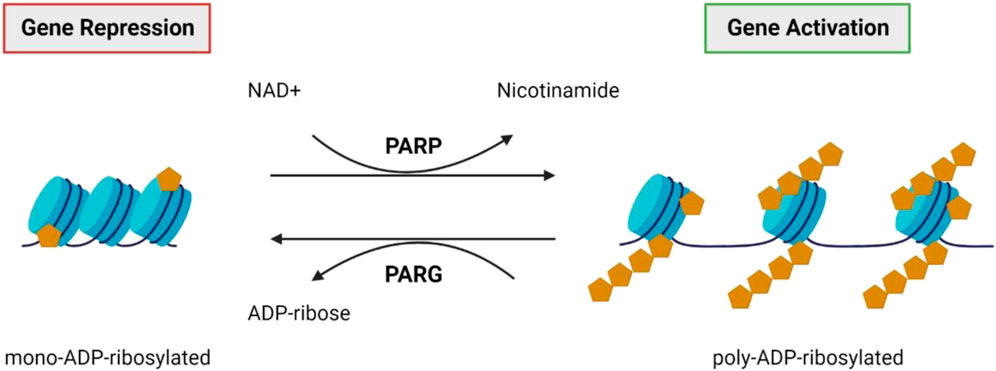
FIGURE 7. PARP and PARG. Poly (ADP-ribose) polymerases (PARPs) catalyse the attachment of poly-ADP-ribose units on the glutamic or aspartic acid residues of the target protein (Tong et al., 2001; Kraus and Lis, 2003; Quénet et al., 2009). Generally, this results in transcriptional activation via chromatin de-condensation and altered promoter and enhancer activity (Kraus and Lis, 2003; Quénet et al., 2009). This reaction is reversible by poly (ADP-ribose) glycohydrolase (PARG) activity (Quénet et al., 2009).
Paquet-Durand et al. (2007) first suggested that excessive activation of PARP may have a role in the photoreceptor death seen in rd1 mice (Paquet-Durand et al., 2007). As photoreceptors degenerated in rd1 retinae, there was a concomitant increase in PAR positive staining which was identified via immunohistochemistry, and increased PARP activity (Paquet-Durand et al., 2007). Interestingly, in P11 rd1 sections, 88% of PAR- or PARP-positive cells were also positive for the TUNEL cell death marker. Additionally, PAR- or PARP-positive cells were shown to co-localise with avidin and AIF, an oxidative damage marker and mitochondrial apoptosis-inducing factor, respectively (Paquet-Durand et al., 2007). The role of PARP activation in cell death was also established in two rat models of autosomal dominant RP with different mutations in the rhodopsin gene, P23H and S334ter. A significant activation of PARP was seen during cell death in these two models, which coincided with increased cellular oxidative stress, the activation of calpain, a protein linked to both apoptotic and necrotic cell death processes, and a reduction in its endogenous inhibitor calpastatin (Kaur et al., 2011). Another study looked at the impact of PARP in retinal degeneration using a Parp1 knockout (Parp1−/−) (Sahaboglu et al., 2010). The retina of the Parp1−/− mouse line was found to be morphologically similar to wildtype; however, there was a significant resistance to retinal degeneration when induced by blocking phosphodiesterase 6 (PDE6), an essential component of the phototransduction pathway. In contrast, application of the same PDE6 blocker caused rapid retinal degeneration in wildtype controls (Sahaboglu et al., 2010). The observed resistance to PDE6 induced retinal degeneration in Parp1−/− mutants suggests that PARP1 may be involved in photoreceptor degeneration via PARP-mediated cell death or a closely related mechanism (Sahaboglu et al., 2010). The role of PARP1 is largely opposed by its functional antagonist, poly-ADP-glycohydrolase (PARG), and another study by the same group investigated its effect in a Parg110 knockout (Parg110−/−) mouse. Parg110−/− mice were morphologically and functionally indistinguishable from wildtype mice, and when Parg110−/− mice were exposed to the PDE6 inhibitor there was a significant resistance to treatment, similar to that seen in Parp1−/− mice (Sahaboglu et al., 2014). The authors postulated that this resistance was due to low levels of PARP activity and reduced PAR accumulation, suggesting a positive regulation of PARP1 that must usually be present but is absent in the Parg110−/− retinae (Sahaboglu et al., 2014). Despite the initial assessment of PARG110 as a functional antagonist, this study revealed that there is, in fact, a positive feedback loop between PARP1 and PARG110, which is thought to be especially active in pathological conditions (Sahaboglu et al., 2014).
More broadly, PARP overactivity was consistently elevated compared to wildtype controls in ten models of IRD, namely the P23H and S334ter rat models of autosomal dominant RP, rd1, rd2, rd10, Cngb1−/− and Rho−/− mice models of autosomal recessive RP, the Rpe65−/− model of Leber’s congenital amaurosis and the Pde6ccpfl1 and Cnga3−/− models of achromatopsia (Arango-Gonzalez et al., 2014). This study also highlighted the consistent overactivity of other molecules involved in a non-apoptotic cell death pathway, including calpains, protein kinase G and HDAC. These observations suggest that similar mechanisms may modulate cell death in these ten models and may allow for generic neuroprotection using drugs that target these molecules across multiple models of IRD. Jiao et al. (2016) examined four additional models of RP, all with mutations in the Pde6a gene (three homozygous point mutations Pde6a R562W, D670G, V685M, and one compound heterozygous Pde6aV685M/R562W). In each of the four models there appeared to be PARP overactivation and PAR accumulation which correlated with the progression of photoreceptor degeneration (Jiao et al., 2016). Interestingly, the models that possessed the most rapid photoreceptor degeneration (V685M, Pde6aV685M/R562W) seemed to have lower levels of PARP activity. In contrast, in the slower degeneration models (R562W, D670G) there was a greater amount of PARP activity in dying cells. In the D670G mutant, the mildest form of all four models, almost 100% of PARP-positive cells were also TUNEL-positive (Jiao et al., 2016). This study also reported that pharmacological PARP inhibition using PJ34, was neuroprotective in all models to varying extents (Jiao et al., 2016). All models displayed a reduction in TUNEL-positive cells after treatment as well as an increase in the number of photoreceptor rows. There appeared to be an inverse correlation between the strength of the genetic insult and the efficacy of PJ34, with the D670G model, which has the slowest degeneration, having the best treatment effects (Jiao et al., 2016). Furthermore, addition of PJ34 to retinal explant cultures preserved the number of photoreceptor rows in all models except for the V685M at 10 days, but this effect was no longer evident by 16 days, an effect that could be due to the short-term viability of retinal explants or, perhaps, loss of treatment efficacy (Jiao et al., 2016). Similar effects of PJ34 were noted in two other mouse models of RP. There was a decrease in levels of poly(ADP-ribosyl)ation and photoreceptor cell death in rd1 retinal explants treated with PJ34 (Paquet-Durand et al., 2007), while rd2 explants had a reduction in photoreceptor death, decreased poly(ADP ribosyl)ation, and improved rhodopsin localisation in the outer segments of rods (Sahaboglu et al., 2017).
Interestingly, a study in the Nmnat1V9M/V9M mouse model of IRD, which harbours a mutation in a gene responsible for NAD+ biosynthesis, showed that PARP activity was elevated during disease progression, with increased PAR expression in the photoreceptors (Greenwald et al., 2021). As PARP is a consumer of nuclear NAD+, this finding may suggest the photoreceptors in the Nmnat1V9M/V9M mouse might be dying via PARthanatos. This unique cell death pathway occurs due to the overactivation of PARP and overproduction of PAR rather than through classic apoptotic pathways (Fatokun et al., 2014; Greenwald et al., 2021). This hypothesis was further validated in a subsequent study by the same group where RNA sequencing of Nmnat1V9M/V9M retinae at 3 weeks of age showed a significant upregulation in the expression of Parp1, Parp3, Tiparp (Parp7), Parp9, Parp12, Zchav1 (Parp13), Parp14 and Parp16 (Brown et al., 2022). By 4 weeks of age, PARP activity was significantly increased compared to wildtype controls (Brown et al., 2022). These increases in Parp expression appeared to coincide with reduced NAD+ activity, increased DNA damage, and increased immune reactivity in the retina (Brown et al., 2022). Furthermore, PARP upregulation has also been linked to endoplasmic reticulum (ER) stress-mediated cell death. In a model of achromatopsia caused by a mutation in the ATF6 gene, which is best known for its role in transducing signals related to ER stress, patient fibroblasts harbouring the ATF6Y567N/Y567N mutation were more sensitive to ER stress and PARP overexpression (Chiang et al., 2017; Hillary and FitzGerald, 2018). Lastly, use of a monoclonal antibody that targets TNF-alpha in rd10 RP mice resulted in a significant reduction in photoreceptor cell death, concurrently reducing PAR content, an indirect measurement of PARP activity (Martínez-Fernández de la Cámara et al., 2015).
Because dysregulated PARP activity seems to be a consistent feature during the death of photoreceptors in IRD, and the use of PJ34 to inhibit PARP appeared beneficial, multiple other PARP inhibitors have been tested to assess their effectiveness in preclinical models. These include inhibitors that are FDA-approved or in late stages of clinical trials, with the hope for easier drug repurposing in the future. R503, ABT-888 (in phase 3 clinical trials) and Olaparib (FDA-approved for use in ovarian cancer treatment) were all tested for their effectiveness in rd1 mice, with R503 and ABT-888 showing relative toxicity at low drug concentrations (Sahaboglu et al., 2016). Contrastingly, the FDA-approved Olaparib, which targets PARP1 and PARP2 isoforms, did not show toxicity and exhibited photoreceptor protection after treatment, in both short-term (treatment starting at P7 and finishing at P11) and long-term experiments (P7-P17). Olaparib reduced the number of TUNEL-positive cells and decreased PARylation while preserving ONL thickness (Sahaboglu et al., 2016). There was also a reduction in cGMP levels, thought to be an essential component of cell death in this model (Sahaboglu et al., 2016). However, this neuroprotective effect was lost by P24 (Sahaboglu et al., 2016). In a separate study, another two PARP inhibitors, BMN-673 (FDA-approved) and 3-aminobenzamine were utilised in the rd1 mouse, and both were able to reduce photoreceptor cell death by 25%–40%. The authors suggested this survival may be due to a relationship between PARP and the highly conserved kinase GSK and Wnt/catenin pathways, which are involved in various cellular processes such as differentiation, adult tissue homeostasis and apoptosis (Antolín and Mestres, 2014; Yang et al., 2016; Pai et al., 2017; Sahaboglu et al., 2020). Before treatment, there was a reduction in GSK-alpha immunoreactivity in rd1 retinae in the ganglion cell and inner cell layers, and a small but not significant reduction in the ONL. When treated with the PARP inhibitors, these expression levels were reversed towards wildtype levels. Beta-catenin showed a significantly lower expression in the RPE, but no significant reduction in the ganglion cell layer and inner nuclear layer. These changes were partially neutralised by BMN-673 in the ganglion cell layer and the RPE, and by 3-aminobenzamide in the ganglion cell layer, RPE and the inner nuclear layer (Sahaboglu et al., 2020).
Given the data suggesting the influence of PARP in multiple IRDs, and the fact that PARP inhibition generally enhances photoreceptor survival (summary in Figure 8), the next steps in this field should include developing a firm understanding of the mechanisms behind this protection. Analysis of PARP inhibition in clinical trials involving IRD patients should be undertaken to determine if PARP inhibitors can benefit all patients or only a small subset dependent on genotype or mutation, and determine the safety of long-term treatment and its effect on disease progression.
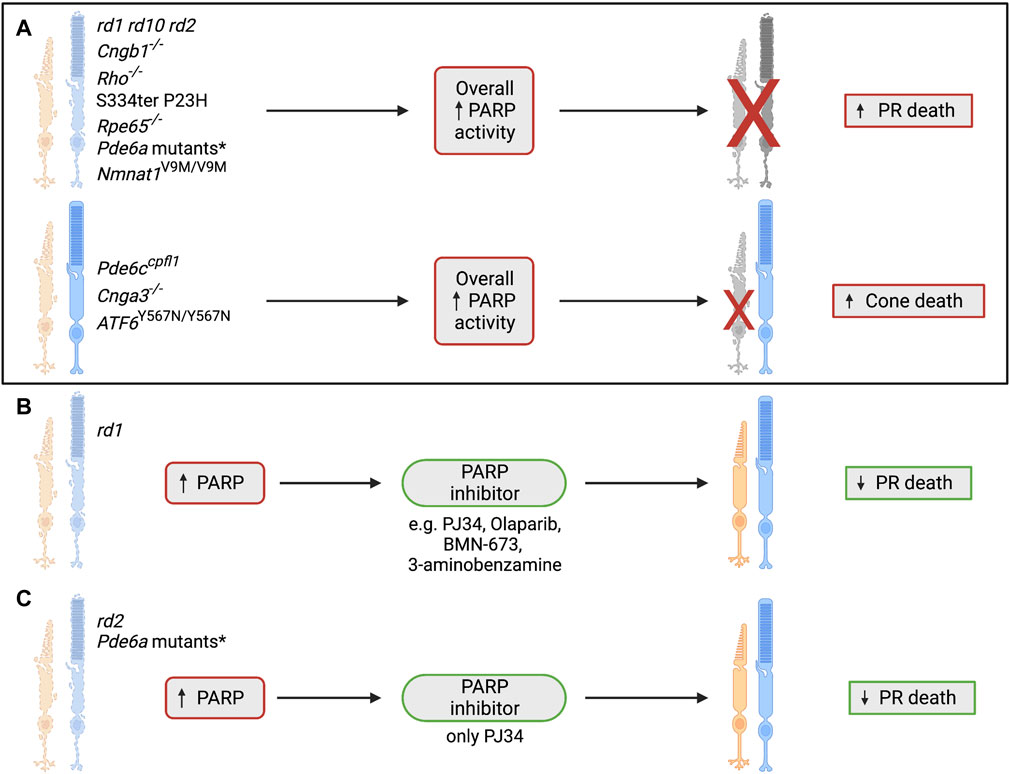
FIGURE 8. The role of poly(ADP-ribosyl)ation and PARP in IRDs. (A) PARP overactivity has previously been shown in many IRD models, including autosomal dominant RP, autosomal recessive RP, Leber’s congenital amaurosis, cone/rod dystrophy and achromatopsia. This consistent overactivity of PARP has been shown to coincide with photoreceptor cell death, suggesting a link between these two processes (Paquet-Durand et al., 2007; Kaur et al., 2011; Arango-Gonzalez et al., 2014; Jiao et al., 2016; Chiang et al., 2017; Greenwald et al., 2021). *The Pde6a mutant models that have shown an increase in overall PARP activity are Pde6a R562W, Pde6a D670G, Pde6a V685M and Pde6aV685M/R562W (Jiao et al., 2016). (B) Testing of several PARP inhibitors has taken place in the rd1 model and has indicated PARP inhibition as a strong candidate for neuroprotection of photoreceptors in IRD. Photoreceptor survival has been noted after administration of PJ34, Olaparib, BMN-673, and 3-aminobenzamine (Paquet-Durand et al., 2007; Sahaboglu et al., 2016; Sahaboglu et al., 2020). (C) Various IRD models have been used to test the PARP inhibitor PJ34, including the rd1, rd2, Pde6a R562W, Pde6a D670G, Pde6a V685M and Pde6aV685M/R562W models. PJ34 has shown neuroprotective benefits in all mentioned models and reduces photoreceptor death after administration (Paquet-Durand et al., 2007; Jiao et al., 2016; Sahaboglu et al., 2017). *The Pde6a mutant models that have shown a reduction in photoreceptor death after treatment with PJ34 are Pde6a R562W, Pde6a D670G, Pde6a V685M and Pde6aV685M/R562W (Jiao et al., 2016). PR = photoreceptor.
Post-translational modifications such as DNA methylation, histone acetylation, histone methylation and poly(ADP-ribosyl)ation all have complex interactions and functional interplay. Several studies in IRD have highlighted these complex relationships, emphasising that epigenetic modifications do not take place in isolation (Nakao, 2001; Lee et al., 2006; Ummarino et al., 2021; Khalid et al., 2022; Miller et al., 2022). In 2010, Sancho-Pelluz and colleagues discovered that both HDAC and PARP were overactive in rd1 mice (Sancho-Pelluz et al., 2010). Interestingly, they found that HDAC overactivity preceded PARP overactivity by approximately 2 days, with these findings later validated in a separate study that observed the same pattern of HDAC overactivity preceding PARP overactivity in ten different models of IRD (Sancho-Pelluz et al., 2010; Arango-Gonzalez et al., 2014). Additionally, it was found that PARP overactivity coincided with the peak of cell death, a determination based on positive TUNEL staining (Arango-Gonzalez et al., 2014). Notably, they found that calpain and PARP overactivity coincided with TUNEL staining, indicating that they may be involved in the final stages of cell death, as TUNEL labels DNA nick-ends which are associated with final stages of cell death, while HDAC overactivity and cGMP accumulation appeared to be found earlier in the cell death process (Arango-Gonzalez et al., 2014). Dong et al. (2023) also showed that treating rd1 explants with the PARP inhibitor Olaparib improved photoreceptor survival and reduced HDAC activity (Dong et al., 2023). In a similar line, overexpression of HDAC4 using electroporation significantly increased rod photoreceptor survival in rd1 mice retinae (Guo et al., 2015). HDAC4 overexpression led to a 50% decrease in Parp1 expression, along with other markers for cell death, cell cycle genes, and oxidative and endoplasmic reticulum stress, suggesting that Parp1, with other vital genes, may be involved in the protective effect seen with HDAC4 overexpression (Guo et al., 2015).
HDAC has also been shown to interact with other epigenetic modifications, such as histone methylation, where treatment with the pan-HDAC inhibitor TSA in the Pde6ccpfl1 mouse model of achromatopsia resulted in changes to histone methylation status. H3K27me3 levels which are severely reduced in Pde6ccpfl1 mice compared to wildtype, were partially restored to wildtype levels upon treatment, highlighting the effect that HDAC inhibition has on histone methylation (Miller et al., 2022). HDAC has also been shown to interact with DNA methylation via DMNT activity in rd1 and rd2 mice as well as in S334ter and P23H rat models of RP. Each model showed 5mC positive cells had very low or absent levels of acetylated lysine, suggesting a key interplay between HDAC and DNMT (Farinelli et al., 2014). Functional interplay between DNA methylation changes and poly(ADP-ribosyl)ation has been suggested; for instance, a study on rd1 retinae revealed that many cells in the ONL that were positive for 5mC staining were also positive for PAR staining (Wahlin et al., 2013; Ummarino et al., 2021). Contrastingly, another study showed that PARP inhibition during the peak of degeneration in rd1 retinae, did not cause any changes to 5mC and 5hmC levels, suggesting that DNA methylation may actually be unrelated or upstream to PARP activity (Sahaboglu et al., 2016).
The understanding of interactions between different epigenetic modifications in the context of IRD is still in relative infancy. In the future these types of studies may help us understand the neuroprotective effects of these drugs on a mechanistic level and may be used to leverage the use of multiple epigenetic modifying drugs for a synergistic and protective effect.
The potential role of epigenetic modifiers in IRD pathology has been gaining new insights in recent years. Roles for DNA methylation and histone modifications such as deacetylation, methylation, and poly(ADP-ribosyl)ation have been suggested, with modulation of each being a potential therapeutic target. The development of new cell-specific epigenetic techniques such as CUT&Tag, for example, will greatly assist in elucidating the role of histone modifications in IRD disease processes and its potential for therapeutic targeting. While understanding DNA methylation and histone methylation in IRD is still quite a new field, the influence of PARP and HDACs have been more extensively studied. PARP inhibition has been tested in multiple preclinical models and a better understanding of the mechanisms that underlie its neuroprotective action will only improve therapeutic options in the future. Both pan- and selective HDAC inhibition have shown promising potential in various preclinical models, although the HDAC inhibitor VPA remains the only drug that has so far proceeded to clinical trials. However, likely due to its different impact depending on the genetic basis of the IRD, its further use is currently discouraged due to inconsistent results in these clinical studies. A better understanding of how HDAC inhibitors affect people with different genotypes will facilitate future clinical translation of these types of drugs. There may also be sex differences in epigenetic regulation and drug metabolism that need to be considered (Gegenhuber and Tollkuhn, 2019; Li et al., 2019; Oliva et al., 2020; Saravanan et al., 2023). Clearly, interaction between each of these epigenetic regulators are very complex, with functional relationships via diverse molecules and intracellular pathways. In order to best understand these complex relationships, further omics studies are needed, ideally concurrently, which would allow for a better understanding of cell and mutation specific differences. Using multiple omics platforms in parallel would also allow for superior discernment of the changes underpinning the protective effects of epigenetic modulating drugs. This in-depth grasp of cellular mechanisms will be essential before successful translation of therapies to the clinic.
AM, AH, DT, and LC contributed to the conception of this review. AM wrote the first draft of the manuscript. RJ and AM created and edited the figures. All authors contributed to the article and approved the submitted version.
Figures in this manuscript were created using BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1224078/full#supplementary-material
5caC, 5-carboxylcytosine; 5fC, 5-formylcytosine; 5hmC, 5-hydroxymethylcytosine; 5mC, 5-methylcytosine; BER, base excision repair; DNMT, DNA methyltransferase; HAT, histone acetyltransferase; HDAC, histone deacetylase; IRD, inherited retinal disease; ONL, outer nuclear layer; PAR, poly (ADP-ribose); PARG, poly (ADP-ribose) glycohydrolase; PARP, poly (ADP-ribose) polymerase; rd, retinal degeneration [mouse model]; RP, retinitis pigmentosa; TDG, thymine DNA glycosylase; TET, ten-eleven translocase; TSA, trichostatin A; TUNEL, t erminal deoxynucleotidyl transferase dUTP nick end labelling; VPA, valproic acid.
Antolín, A. A., and Mestres, J. (2014). Linking off-target kinase pharmacology to the differential cellular effects observed among PARP inhibitors. Oncotarget 5 (10), 3023–3028. doi:10.18632/oncotarget.1814
Arango-Gonzalez, B., Trifunović, D., Sahaboglu, A., Kranz, K., Michalakis, S., Farinelli, P., et al. (2014). Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS One 9 (11), e112142. doi:10.1371/journal.pone.0112142
Balaiya, S., Abu-Amero, K. K., Kondkar, A. A., and Chalam, K. V. (2017). Sirtuins expression and their role in retinal diseases. Oxidative Med. Cell. Longev. 2017, 3187594. doi:10.1155/2017/3187594
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21 (3), 381–395. doi:10.1038/cr.2011.22
Basavarajappa, B. S., and Subbanna, S. (2021). Histone methylation regulation in neurodegenerative disorders. Int. J. Mol. Sci. 22 (9), 4654. doi:10.3390/ijms22094654
Berger, W., Kloeckener-Gruissem, B., and Neidhardt, J. (2010). The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin Eye Res. 29 (5), 335–375. doi:10.1016/j.preteyeres.2010.03.004
Bhalla, S., Joshi, D., Bhullar, S., Kasuga, D., Park, Y., and Kay, C. N. (2013). Long-term follow-up for efficacy and safety of treatment of retinitis pigmentosa with valproic acid. Br. J. Ophthalmol. 97 (7), 895–899. doi:10.1136/bjophthalmol-2013-303084
Bieliauskas, A. V., and Pflum, M. K. H. (2008). Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 37 (7), 1402–1413. doi:10.1039/b703830p
Birch, D. G., Bernstein, P. S., Iannacone, A., Pennesi, M. E., Lam, B. L., Heckenlively, J., et al. (2018). Effect of oral valproic acid vs placebo for vision loss in patients with autosomal dominant retinitis pigmentosa: A randomized phase 2 multicenter placebo-controlled clinical trial. JAMA Ophthalmol. 136 (8), 849–856. doi:10.1001/jamaophthalmol.2018.1171
Brown, E. E., Scandura, M. J., Mehrotra, S., Wang, Y., Du, J., and Pierce, E. A. (2022). Reduced nuclear NAD+ drives DNA damage and subsequent immune activation in the retina. Hum. Mol. Genet. 31 (9), 1370–1388. doi:10.1093/hmg/ddab324
Brunet, A. A., Harvey, A. R., and Carvalho, L. S. (2022). Primary and secondary cone cell death mechanisms in inherited retinal diseases and potential treatment options. Int. J. Mol. Sci. 23 (2), 726. doi:10.3390/ijms23020726
Center DSTUoTHS (2020). Center DSTUoTHS. RetNet 1996-2020. Available from: https://sph.uth.edu/retnet/.
Chandramowlishwaran, P., Vijay, A., Abraham, D., Li, G., Mwangi, S. M., and Srinivasan, S. (2020). Role of sirtuins in modulating neurodegeneration of the enteric nervous system and central nervous system. Front. Neurosci. 14, 614331. doi:10.3389/fnins.2020.614331
Chen, B., and Cepko, C. L. (2009). HDAC4 regulates neuronal survival in normal and diseased retinas. Sci. (New York, NY) 323 (5911), 256–259. doi:10.1126/science.1166226
Chiang, W. C., Chan, P., Wissinger, B., Vincent, A., Skorczyk-Werner, A., Krawczyński, M. R., et al. (2017). Achromatopsia mutations target sequential steps of ATF6 activation. Proc. Natl. Acad. Sci. U. S. A. 114 (2), 400–405. doi:10.1073/pnas.1606387114
Clemson, C. M. (2010). Development of a multi-site phase II clinical trial of valproic acid for retinitis pigmentosa.
Clemson, C. M., Tzekov, R., Krebs, M., Checchi, J. M., Bigelow, C., and Kaushal, S. (2011). Therapeutic potential of valproic acid for retinitis pigmentosa. Br. J. Ophthalmol. 95 (1), 89–93. doi:10.1136/bjo.2009.175356
Dong, Y., Yan, J., Yang, M., Xu, W., Hu, Z., Paquet-Durand, F., et al. (2023). Inherited retinal degeneration: Towards the development of a combination therapy targeting histone deacetylase, poly (ADP-Ribose) polymerase, and calpain. Biomolecules 13 (4), 581. doi:10.3390/biom13040581
Farinelli, P., Perera, A., Arango-Gonzalez, B., Trifunovic, D., Wagner, M., Carell, T., et al. (2014). DNA methylation and differential gene regulation in photoreceptor cell death. Cell Death Dis. 5 (12), e1558–e. doi:10.1038/cddis.2014.512
Fatokun, A. A., Dawson, V. L., and Dawson, T. M. (2014). Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171 (8), 2000–2016. doi:10.1111/bph.12416
Galvin, O., Chi, G., Brady, L., Hippert, C., Del Valle Rubido, M., Daly, A., et al. (2020). The impact of inherited retinal diseases in the republic of Ireland (ROI) and the United Kingdom (UK) from a cost-of-illness perspective. Clin. Ophthalmol. Auckl. NZ) 14, 707–719. doi:10.2147/OPTH.S241928
Gegenhuber, B., and Tollkuhn, J. (2019). Sex differences in the epigenome: A cause or consequence of sexual differentiation of the brain? Genes (Basel) 10 (6), 432. doi:10.3390/genes10060432
Greenwald, S. H., Brown, E. E., Scandura, M. J., Hennessey, E., Farmer, R., Du, J., et al. (2021). Mutant Nmnat1 leads to a retina-specific decrease of NAD+ accompanied by increased poly(ADP-ribose) in a mouse model of NMNAT1-associated retinal degeneration. Hum. Mol. Genet. 30 (8), 644–657. doi:10.1093/hmg/ddab070
Greer, E. L., and Shi, Y. (2012). Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13 (5), 343–357. doi:10.1038/nrg3173
Guo, X., Wang, S-B., Xu, H., Ribic, A., Mohns, E. J., Zhou, Y., et al. (2015). A short N-terminal domain of HDAC4 preserves photoreceptors and restores visual function in retinitis pigmentosa. Nat. Commun. 6, 8005. doi:10.1038/ncomms9005
Hillary, R. F., and FitzGerald, U. (2018). A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 25 (1), 48. doi:10.1186/s12929-018-0453-1
Iraha, S., Hirami, Y., Ota, S., Sunagawa, G. A., Mandai, M., Tanihara, H., et al. (2016). Efficacy of valproic acid for retinitis pigmentosa patients: A pilot study. Clin. Ophthalmol. 10, 1375–1384. doi:10.2147/OPTH.S109995
Jaliffa, C., Ameqrane, I., Dansault, A., Leemput, J., Vieira, V., Lacassagne, E., et al. (2009). Sirt1 involvement in rd10 mouse retinal degeneration. Invest. Ophthalmol. Vis. Sci. 50 (8), 3562–3572. doi:10.1167/iovs.08-2817
Jiao, K., Sahaboglu, A., Zrenner, E., Ueffing, M., Ekström, P. A. R., and Paquet-Durand, F. (2016). Efficacy of PARP inhibition in Pde6a mutant mouse models for retinitis pigmentosa depends on the quality and composition of individual human mutations. Cell Death Discov. 2 (1), 16040. doi:10.1038/cddiscovery.2016.40
Jin, B., Li, Y., and Robertson, K. D. (2011). DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2 (6), 607–617. doi:10.1177/1947601910393957
Kaur, J., Mencl, S., Sahaboglu, A., Farinelli, P., van Veen, T., Zrenner, E., et al. (2011). Calpain and PARP activation during photoreceptor cell death in P23H and S334ter rhodopsin mutant rats. PLoS One 6 (7), e22181. doi:10.1371/journal.pone.0022181
Kaushal, S., Noorwez, S. M., Tzekov, R., Huang, D., Li, Y., and Wen, R. (2010). The effect of valproic acid in mouse models of RP. Investigative Ophthalmol. Vis. Sci. 51 (13), 3735.
Keeler, C. E. (1924). The inheritance of a retinal abnormality in white mice. Proc. Natl. Acad. Sci. U. S. A. 10 (7), 329–333. doi:10.1073/pnas.10.7.329
Khalid, U., Simovic, M., Hammann, L. A., Iskar, M., Wong, J. K. L., Kumar, R., et al. (2022). A synergistic interaction between HDAC- and PARP inhibitors in childhood tumors with chromothripsis. Int. J. Cancer 151 (4), 590–606. doi:10.1002/ijc.34027
Kraus, W. L., and Lis, J. T. (2003). PARP goes transcription. Cell 113 (6), 677–683. doi:10.1016/s0092-8674(03)00433-1
Kumar, A., Midha, N., Gogia, V., Gupta, S., Sehra, S., and Chohan, A. (2014). Efficacy of oral valproic acid in patients with retinitis pigmentosa. J. Ocular Pharmacol. Ther. 30 (7), 580–586. doi:10.1089/jop.2013.0166
Langelier, M. F., Eisemann, T., Riccio, A. A., and Pascal, J. M. (2018). PARP family enzymes: Regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 53, 187–198. doi:10.1016/j.sbi.2018.11.002
LaVail, M. M., and Sidman, R. L. (1974). C57BL/6J mice with inherited retinal degeneration. Archives Ophthalmol. 91 (5), 394–400. doi:10.1001/archopht.1974.03900060406015
Lee, M. G., Wynder, C., Bochar, D. A., Hakimi, M. A., Cooch, N., and Shiekhattar, R. (2006). Functional interplay between histone demethylase and deacetylase enzymes. Mol. Cell Biol. 26 (17), 6395–6402. doi:10.1128/MCB.00723-06
Leyk, J., Daly, C., Janssen-Bienhold, U., Kennedy, B. N., and Richter-Landsberg, C. (2017). HDAC6 inhibition by tubastatin A is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis. 8 (8), e3028–e. doi:10.1038/cddis.2017.415
Li, B., Gografe, S., Munchow, A., Lopez-Toledano, M., Pan, Z. H., and Shen, W. (2019). Sex-related differences in the progressive retinal degeneration of the rd10 mouse. Exp. Eye Res. 187, 107773. doi:10.1016/j.exer.2019.107773
Locke, W. J., Guanzon, D., Ma, C., Liew, Y. J., Duesing, K. R., Fung, K. Y. C., et al. (2019). DNA methylation cancer biomarkers: Translation to the clinic. Front. Genet. 10, 1150. doi:10.3389/fgene.2019.01150
Maguire, A. M., Bennett, J., Aleman, E. M., Leroy, B. P., and Aleman, T. S. (2021). Clinical perspective: Treating RPE65-associated retinal dystrophy. Mol. Ther. 29 (2), 442–463. doi:10.1016/j.ymthe.2020.11.029
Martínez-Fernández de la Cámara, C., Hernández-Pinto, A. M., Olivares-González, L., Cuevas-Martín, C., Sánchez-Aragó, M., Hervás, D., et al. (2015). Adalimumab reduces photoreceptor cell death in A mouse model of retinal degeneration. Sci. Rep. 5, 11764. doi:10.1038/srep11764
Miller, A. L., Fuller-Carter, P. I., Masarini, K., Samardzija, M., Carter, K. W., Rashwan, R., et al. (2022). Increased H3K27 trimethylation contributes to cone survival in a mouse model of cone dystrophy. Cell Mol. Life Sci. 79 (8), 409. doi:10.1007/s00018-022-04436-6
Mitton, K. P., Guzman, A. E., Deshpande, M., Byrd, D., DeLooff, C., Mkoyan, K., et al. (2014). Different effects of valproic acid on photoreceptor loss in Rd1 and Rd10 retinal degeneration mice. Mol. Vis. 20, 1527–1544.
Moore, L. D., Le, T., and Fan, G. (2013). DNA methylation and its basic function. Neuropsychopharmacology 38 (1), 23–38. doi:10.1038/npp.2012.112
Nakao, M. (2001). Epigenetics: Interaction of DNA methylation and chromatin. Gene 278 (1), 25–31. doi:10.1016/s0378-1119(01)00721-1
Oliva, M., Munoz-Aguirre, M., Kim-Hellmuth, S., Wucher, V., Gewirtz, A. D. H., Cotter, D. J., et al. (2020). The impact of sex on gene expression across human tissues. Science 369 (6509), eaba3066. doi:10.1126/science.aba3066
Pai, S. G., Carneiro, B. A., Mota, J. M., Costa, R., Leite, C. A., Barroso-Sousa, R., et al. (2017). Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 10 (1), 101. doi:10.1186/s13045-017-0471-6
Paquet-Durand, F., Silva, J., Talukdar, T., Johnson, L. E., Azadi, S., van Veen, T., et al. (2007). Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J. Neurosci. 27 (38), 10311–10319. doi:10.1523/JNEUROSCI.1514-07.2007
Park, S-Y., and Kim, J-S. (2020). A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 52 (2), 204–212. doi:10.1038/s12276-020-0382-4
Perron, N. R., Nasarre, C., Bandyopadhyay, M., Beeson, C. C., and Rohrer, B. (2021). SAHA is neuroprotective in in vitro and in situ models of retinitis pigmentosa. Mol. Vis. 27, 151–160.
Popova, E. Y., Imamura Kawasawa, Y., Zhang, S. S-M., and Barnstable, C. J. (2021). Inhibition of epigenetic modifiers LSD1 and HDAC1 blocks rod photoreceptor death in mouse models of retinitis pigmentosa. J. Neurosci. 41 (31), 6775–6792. doi:10.1523/JNEUROSCI.3102-20.2021
Portera-Cailliau, C., Sung, C. H., Nathans, J., and Adler, R. (1994). Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 91 (3), 974–978. doi:10.1073/pnas.91.3.974
Quénet, D., El Ramy, R., Schreiber, V., and Dantzer, F. (2009). The role of poly(ADP-ribosyl)ation in epigenetic events. Int. J. Biochem. Cell Biol. 41 (1), 60–65. doi:10.1016/j.biocel.2008.07.023
Rasmussen, K. D., and Helin, K. (2016). Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 30 (7), 733–750. doi:10.1101/gad.276568.115
Rodriguez, F., Yushenova, I. A., DiCorpo, D., and Arkhipova, I. R. (2022). Bacterial N4-methylcytosine as an epigenetic mark in eukaryotic DNA. Nat. Commun. 13 (1), 1072. doi:10.1038/s41467-022-28471-w
Sahaboglu, A., Barth, M., Secer, E., Amo, E. M. D., Urtti, A., Arsenijevic, Y., et al. (2016). Olaparib significantly delays photoreceptor loss in a model for hereditary retinal degeneration. Sci. Rep. 6 (1), 39537. doi:10.1038/srep39537
Sahaboglu, A., Miranda, M., Canjuga, D., Avci-Adali, M., Savytska, N., Secer, E., et al. (2020). Drug repurposing studies of PARP inhibitors as a new therapy for inherited retinal degeneration. Cell. Mol. Life Sci. 77 (11), 2199–2216. doi:10.1007/s00018-019-03283-2
Sahaboglu, A., Sharif, A., Feng, L., Secer, E., Zrenner, E., and Paquet-Durand, F. (2017). Temporal progression of PARP activity in the Prph2 mutant rd2 mouse: Neuroprotective effects of the PARP inhibitor PJ34. PLoS One 12 (7), e0181374. doi:10.1371/journal.pone.0181374
Sahaboglu, A., Tanimoto, N., Bolz, S., Garrido, M. G., Ueffing, M., Seeliger, M. W., et al. (2014). Knockout of PARG110 confers resistance to cGMP-induced toxicity in mammalian photoreceptors. Cell Death Dis. 5 (5), e1234–e. doi:10.1038/cddis.2014.208
Sahaboglu, A., Tanimoto, N., Kaur, J., Sancho-Pelluz, J., Huber, G., Fahl, E., et al. (2010). PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function. PLOS ONE 5 (11), e15495. doi:10.1371/journal.pone.0015495
Samardzija, M., Corna, A., Gomez-Sintes, R., Jarboui, M. A., Armento, A., Roger, J. E., et al. (2020). HDAC inhibition ameliorates cone survival in retinitis pigmentosa mice. Cell Death & Differentiation.
Samardzija, M., Masarini, K., Ueffing, M., and Trifunović, D. (Editors) (2019). “HDAC inhibition prevents primary cone degeneration even after the onset of degeneration,” Retinal degenerative diseases (Cham: Springer International Publishing).
Sancho-Pelluz, J., Alavi, M. V., Sahaboglu, A., Kustermann, S., Farinelli, P., Azadi, S., et al. (2010). Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 1 (2), e24. doi:10.1038/cddis.2010.4
Sandberg, M. A., Rosner, B., Weigel-DiFranco, C., and Berson, E. L. (2011). Lack of scientific rationale for use of valproic acid for retinitis pigmentosa. Br. J. Ophthalmol. 95 (5), 744. doi:10.1136/bjo.2010.198176
Saravanan, M., Xu, R., Roby, O., Wang, Y., Zhu, S., Lu, A., et al. (2023). Tissue-specific sex difference in mouse eye and brain metabolome under fed and fasted states. Investigative Ophthalmol. Vis. Sci. 64 (3), 18. doi:10.1167/iovs.64.3.18
Shanmugam, P. M., Minija, C. K., Ramanjulu, R., Tekwani, P., and Saxena, M. (2012). Effect of short-term oral valproic acid on vision and visual field in retinitis pigmentosa. Ophthalmol. Ther. 1 (1), 6. doi:10.1007/s40123-012-0006-8
Sisk, R. A. (2012). Valproic acid treatment may be harmful in non-dominant forms of retinitis pigmentosa. Br. J. Ophthalmol. 96 (8), 1154–1155. doi:10.1136/bjophthalmol-2012-301950
Song, Y., Wu, F., and Wu, J. (2016). Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 9 (1), 49. doi:10.1186/s13045-016-0279-9
Sundaramurthi, H., Roche, S. L., Grice, G. L., Moran, A., Dillion, E. T., Campiani, G., et al. (2020). Selective histone deacetylase 6 inhibitors restore cone photoreceptor vision or outer segment morphology in zebrafish and mouse models of retinal blindness. Front. Cell Dev. Biol. 8, 689. doi:10.3389/fcell.2020.00689
Tong, W-M., Cortes, U., and Wang, Z-Q. (2001). Poly(ADP-ribose) polymerase: A guardian angel protecting the genome and suppressing tumorigenesis. Biochimica Biophysica Acta (BBA) - Rev. Cancer 1552 (1), 27–37. doi:10.1016/s0304-419x(01)00035-x
Totan, Y., Güler, E., Yüce, A., and Dervişogulları, M. S. (2017). The adverse effects of valproic acid on visual functions in the treatment of retinitis pigmentosa. Indian J. Ophthalmol. 65 (10), 984–988. doi:10.4103/ijo.IJO_978_16
Trifunović, D., Arango-Gonzalez, B., Comitato, A., Barth, M., Del Amo, E. M., Kulkarni, M., et al. (2016). HDAC inhibition in the cpfl1 mouse protects degenerating cone photoreceptors in vivo. Hum. Mol. Genet. 25 (20), 4462–4472. doi:10.1093/hmg/ddw275
Trifunović, D., Petridou, E., Comitato, A., Marigo, V., Ueffing, M., and Paquet-Durand, F. (Editors) (2018). “Primary rod and cone degeneration is prevented by HDAC inhibition,” Retinal degenerative diseases (Cham: Springer International Publishing).
Ummarino, S., Hausman, C., and Di Ruscio, A. (2021). The PARP way to epigenetic changes. Genes (Basel). 12 (3), 446. doi:10.3390/genes12030446
Vent-Schmidt, R. Y. J., Wen, R. H., Zong, Z., Chiu, C. N., Tam, B. M., May, C. G., et al. (2017). Opposing effects of valproic acid treatment mediated by histone deacetylase inhibitor activity in four transgenic X. laevis models of retinitis pigmentosa. J. Neurosci. 37 (4), 1039–1054. doi:10.1523/JNEUROSCI.1647-16.2016
Vishwakarma, S., Iyer, L. R., Muley, M., Singh, P. K., Shastry, A., Saxena, A., et al. (2013). Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. Int. Immunopharmacol. 16 (1), 72–78. doi:10.1016/j.intimp.2013.03.016
Wahlin, K. J., Enke, R. A., Fuller, J. A., Kalesnykas, G., Zack, D. J., and Merbs, S. L. (2013). Epigenetics and cell death: DNA hypermethylation in programmed retinal cell death. PLOS ONE 8 (11), e79140. doi:10.1371/journal.pone.0079140
Xiao, C. L., Zhu, S., He, M., Chen, D., Zhang, Q., Chen, Y., et al. (2018). N(6)-Methyladenine DNA modification in the human genome. Mol. Cell 71 (2), 306–318.e7. doi:10.1016/j.molcel.2018.06.015
Yang, E., Tacchelly-Benites, O., Wang, Z., Randall, M. P., Tian, A., Benchabane, H., et al. (2016). Wnt pathway activation by ADP-ribosylation. Nat. Commun. 7, 11430. doi:10.1038/ncomms11430
Keywords: inherited retinal disease, epigenetic changes, DNA methylation, histone methylation, histone acetylation, poly(ADP-ribosyl)ation
Citation: Miller AL, James RE, Harvey AR, Trifunović D and Carvalho LS (2023) The role of epigenetic changes in the pathology and treatment of inherited retinal diseases. Front. Cell Dev. Biol. 11:1224078. doi: 10.3389/fcell.2023.1224078
Received: 17 May 2023; Accepted: 20 July 2023;
Published: 04 August 2023.
Edited by:
Glenn Prazere Lobo, University of Minnesota Twin Cities, United StatesReviewed by:
Daniela Intartaglia, Università degli Studi di Napoli Federico II, ItalyCopyright © 2023 Miller, James, Harvey, Trifunović and Carvalho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Livia S. Carvalho, bGl2aWEuZG9zc2FudG9zY2FydmFsaG9AdXdhLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.