 Alejandra Navarro-Martínez
Alejandra Navarro-Martínez Cristina Vicente-García
Cristina Vicente-García Jaime J. Carvajal
Jaime J. Carvajal
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 04 August 2023
Sec. Molecular and Cellular Pathology
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1216726
This article is part of the Research Topic Personalized Medicine for Neuromuscular Disorders View all 9 articles
Neuromuscular junctions (NMJs) are a special type of chemical synapse that transmits electrical stimuli from motor neurons (MNs) to their innervating skeletal muscle to induce a motor response. They are an ideal model for the study of synapses, given their manageable size and easy accessibility. Alterations in their morphology or function lead to neuromuscular disorders, such as the congenital myasthenic syndromes, which are caused by mutations in proteins located in the NMJ. In this review, we highlight novel potential candidate genes that may cause or modify NMJs-related pathologies in humans by exploring the phenotypes of hundreds of mouse models available in the literature. We also underscore the fact that NMJs may differ between species, muscles or even sexes. Hence the importance of choosing a good model organism for the study of NMJ-related diseases: only taking into account the specific features of the mammalian NMJ, experimental results would be efficiently translated to the clinic.
Neuromuscular junctions (NMJs) have been the subject of study since the early 19th century and are the ideal model for understanding synapses. Their main function is the transmission of signals between the motor neuron (MN) and the skeletal muscle fiber to induce muscle contraction and movement (Kuhne, 1863; Dale, 1914; Couteaux, 1947; Fatt and Katz, 1951; Fatt and Katz, 1952; Birks et al., 1960).
The position of the NMJ in the muscle fiber, its morphology, or degree of development of the postsynaptic region may vary between muscles or species; but despite these particularities, all NMJs contain these main components: 1) a non-myelinating terminal Schwann cell (tSC) that forms a “cap” over the part of the nerve terminal that does not face the postsynaptic region and that is important for synaptogenesis, synaptic transmission, and NMJ maintenance and repair (Santosa et al., 2018); 2) a nerve terminal filled with neurotransmitter vesicles; 3) the synaptic cleft in between the MN and its innervating muscle; 4) the postsynaptic membrane containing neurotransmitter receptors; and 5) the muscle sarcoplasm that provides structural and metabolic support (Engel, 2008). Acetylcholine (ACh) is the neurotransmitter involved in synaptic transmission in vertebrate NMJs. After ACh is released from the presynaptic nerve terminal into the synaptic cleft, it binds to nicotinic ACh receptors (AChR) in the postsynaptic muscle region triggering a response in the muscle (Engel, 2008).
Mutations in proteins located at the NMJ cause the so-called congenital myasthenic syndromes, inherited neuromuscular disorders characterized by abnormal NMJ morphology and/or signal transmission at the motor endplate (Engel, 2020). In this review we aim at highlighting potential novel candidate genes causative or modifiers of NMJ-related disorders based on data from already-published mouse models. Further, we seek to draw attention on very important factors that need to be taken into account in the diagnosis and treatment of NMJ-related disorders. Indeed, while the mouse is the model organism of choice in the study of human diseases, it may not be the most informative in the case of NMJ-related diseases. Further, only a few muscle types, and normally only in males, are usually analyzed. Thus, caution should be taken when translating to the clinic findings derived from studies that disregard these factors.
In 1991, the first printed version of the Gene Table of Neuromuscular Disorders was published (Dubowitz, 1991). It contained a total of 7 nuclear genes and 16 mapped loci, together with their associated neuromuscular disease. This table has been annually updated ever since, and besides de printed version, a computerized online version that allows interactivity is also available at www.musclegenetable.fr. This database currently hosts 641 unique genes and 1,129 diseases grouped into 16 distinctive categories (Cohen et al., 2021). Group 11 contains the congenital myasthenic syndromes, with a total of 42 such syndromes caused by mutations in 34 unique genes; most of them being AChR subunits or genes involved in AChR clustering or ACh metabolism (Engel, 2020). Remarkably, genes associated with other diseases also affect the NMJ, especially genes related with MN diseases, but also with “pure” muscular dystrophies or myopathies. That is the case of STAC3, a component of the excitation-contraction coupling machinery (Polster et al., 2016) whose mutations can be found in patients with Bailey-Block congenital myopathy (Horstick et al., 2013; Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 255995: 10/03/2023. World Wide Web URL: https://omim.org/). Also known as Native American myopathy because it was first described in the Lumbee Indian tribe in North Carolina (Bailey and Bloch, 1987). This autosomal recessive disorder is characterized by congenital weakness, arthrogryposis and myopathic facies, among multiple other symptoms. KO mice for the gene show perinatal lethality due to their inability to breathe, as well as a wide array of muscular defects (Nelson et al., 2013; Reinholt et al., 2013). While the NMJs of these mutants are correctly formed, they occupy a broader space, presenting an increase in the frequency of miniature endplate potentials, likely as a secondary consequence of the muscle defects (Nelson et al., 2013), but underscoring the interdependence of MN and muscle development for correct morphology and synaptic transmission at the NMJ. However, only in a few neuromuscular disorders, potential phenotypes of the NMJ have been directly addressed in humans. This can be probably explained by the limited availability of samples, the difficulty in accurately assessing NMJ function in humans or simply due to the fact that the multitude of other neural or muscle-specific phenotypes are of higher priority because of prevalence or clinical significance. In any case, there exists the possibility that genes involved in already-described diseases, also alter the NMJ, potentially impacting on the pathophysiological mechanisms or disease treatments.
This observation prompted us to review the literature in search of mouse gene models that have already shown any abnormality related with NMJ morphology or function. All these genes could be potential modifiers of some diseases or even the cause of the few for which the gene has not been mapped yet, as is the case of autosomal recessive distal spinal muscular atrophy motor neuron disease (Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 607088: 16/08/2019. World Wide Web URL: https://omim.org/).
The Mouse Genome Informatics MGI database (http://www.informatics.jax.org) currently harbors 258 single or multigenic mouse models in different genetic backgrounds, with annotations associated with abnormal NMJ structure or function. Specifically, the terms used to retrieve these results were: “abnormal neuromuscular synapse morphology”, “failure of neuromuscular synapse presynaptic differentiation”, “failure of neuromuscular synapse postsynaptic differentiation”, “abnormal endplate potential” and “abnormal miniature endplate potential”. These 258 models (Supplementary Table S1) are composed of 132 unique loci, 64 of which have not been associated with any human neuromuscular disease registered at the full gene table database (Supplementary Table S2), even if some of them are well-known players in NMJ formation, maintenance and function (Tables 1–9 and Supplementary Table S3). Possible explanations include the fact that most of these models are constitutive knock-outs (cKOs) that lead to embryonic or early postnatal lethality, usually related with the impossibility of inflating the lungs due to a lack of neural input to the diaphragm muscle (Tables 1–9 and Supplementary Table S3). Deficiencies in these genes would probably cause abortions or still-births in humans as well. However, they can still be candidates to take into account for potential single nucleotide polymorphisms (SNPs) as causative or modifiers of specific diseases.
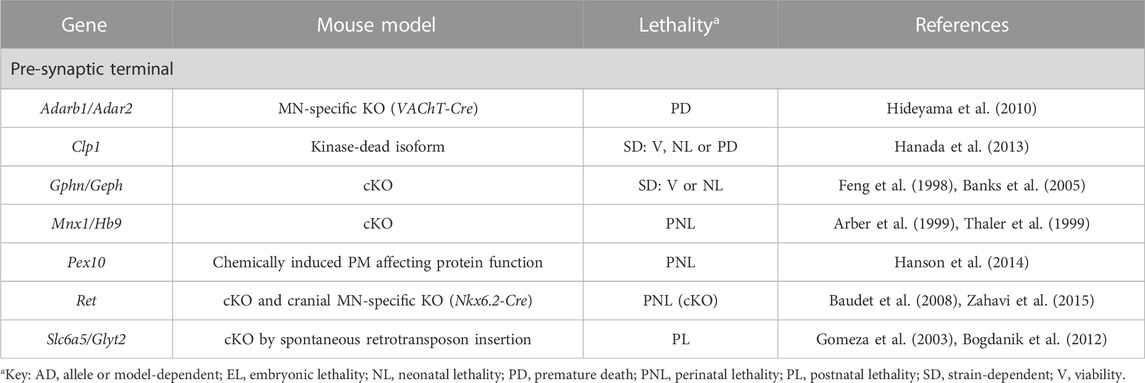
TABLE 1. Novel candidate genes for NMJ-related diseases in humans associated with motor neuron identity, function and survival. All models are constitutive unless otherwise stated. Lethal phenotypes can have complete or incomplete penetrance. cKO, constitutive KO; MN, motor neuron; NMJ, neuromuscular junction; PM, point-mutation.
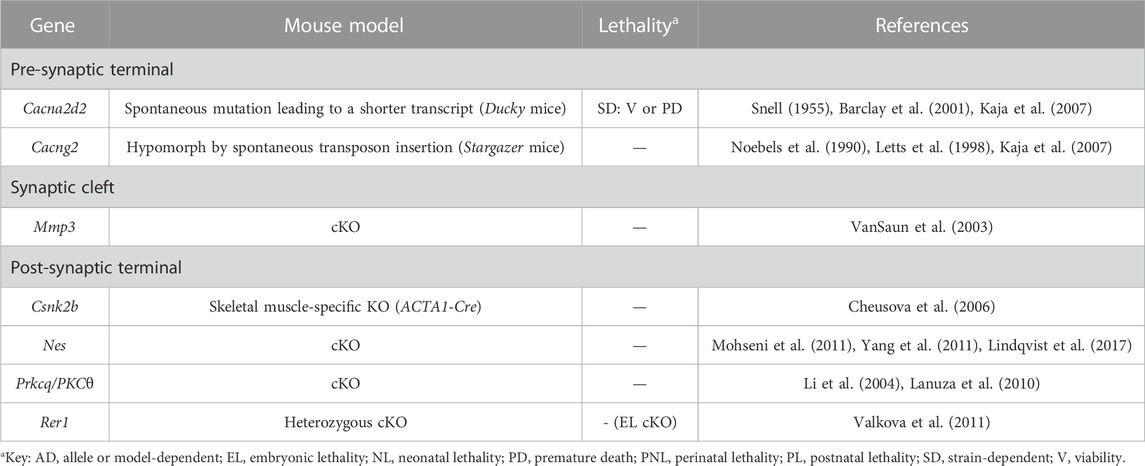
TABLE 2. Novel candidate genes for NMJ-related diseases in humans associated with channels. All models are constitutive unless otherwise stated. Lethal phenotypes can have complete or incomplete penetrance. cKO, constitutive KO.
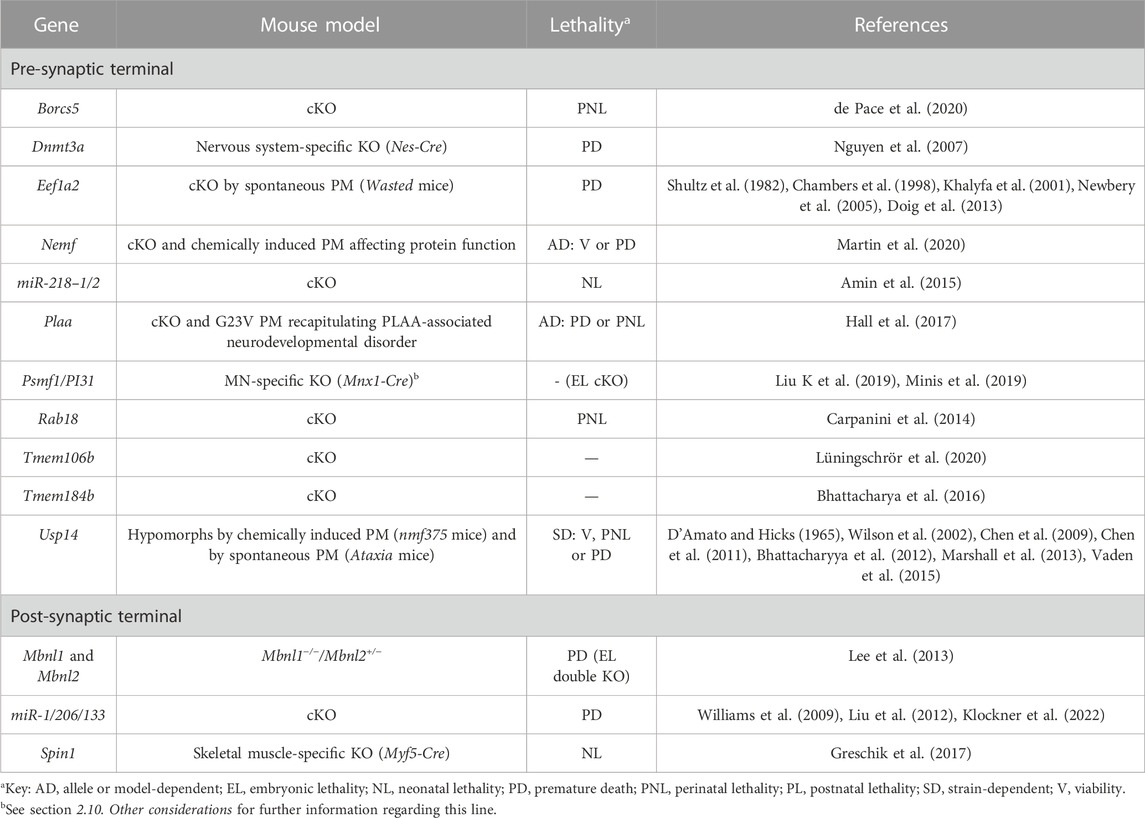
TABLE 3. Novel candidate genes for NMJ-related diseases in humans associated with the regulation of gene expression, as well as with the processes of proteostasis and autophagy. All models are constitutive unless otherwise stated. Lethal phenotypes can have complete or incomplete penetrance. cKO, constitutive KO; MN, motor neuron; PM, point-mutation.
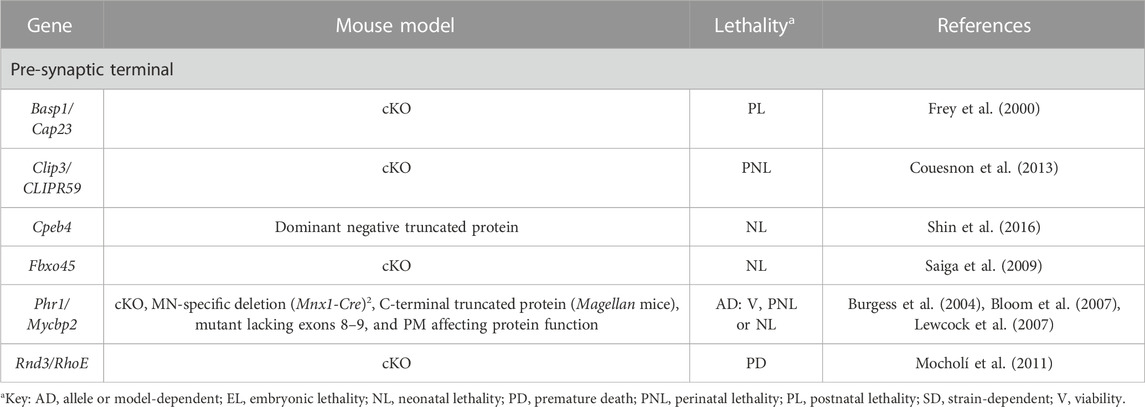
TABLE 4. Novel candidate genes for NMJ-related diseases in humans associated with cytoskeleton dynamics. All models are constitutive unless otherwise stated. Lethal phenotypes can have complete or incomplete penetrance. cKO, constitutive KO; MN, motor neuron; PM, point-mutation.
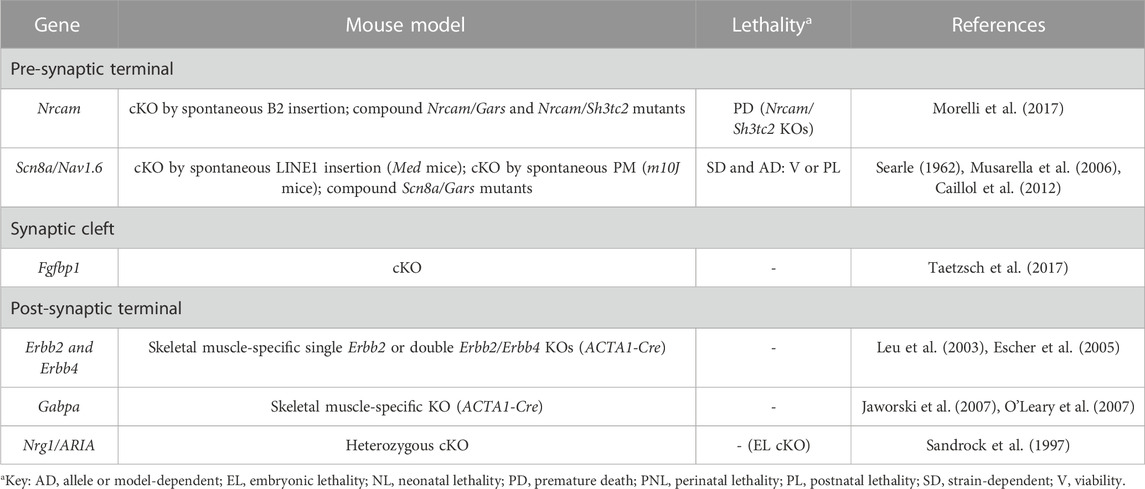
TABLE 5. Novel candidate genes for NMJ-related diseases in humans associated with defective signalling and synaptic transmission. All models are constitutive unless otherwise stated. Lethal phenotypes can have complete or incomplete penetrance. cKO, constitutive KO; PM, point-mutation.
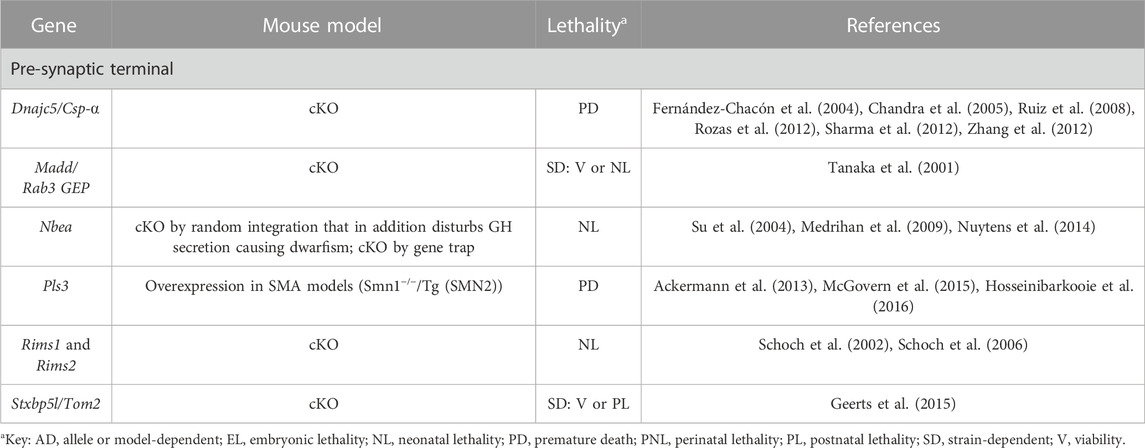
TABLE 6. Novel candidate genes for NMJ-related diseases in humans associated with endocytosis, synaptic vesicle formation and neurotransmitter release. All models are constitutive unless otherwise stated. Lethal phenotypes can have complete or incomplete penetrance. cKO, constitutive KO; SMA, spinal muscle atrophy.
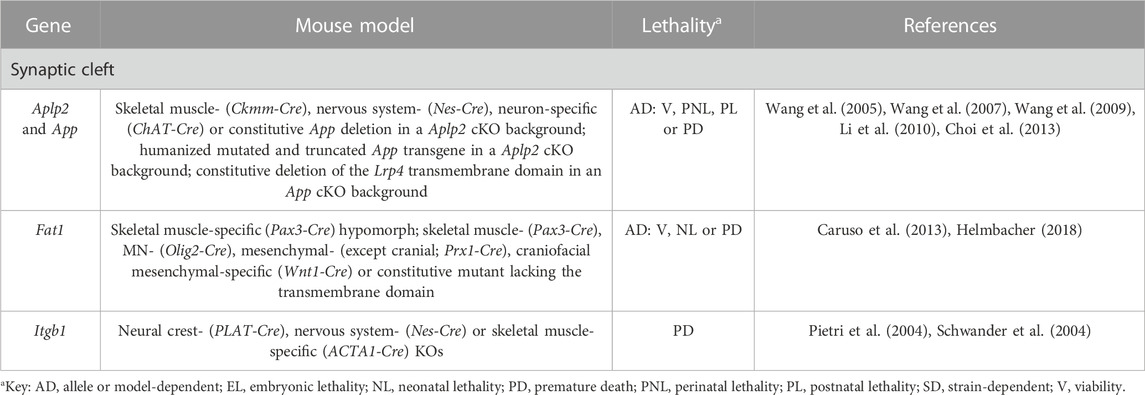
TABLE 7. Novel candidate genes for NMJ-related diseases in humans associated with adhesion. All models are constitutive unless otherwise stated. Lethal phenotypes can have complete or incomplete penetrance. cKO, constitutive KO; MN, motor neuron; SMA, spinal muscle atrophy.
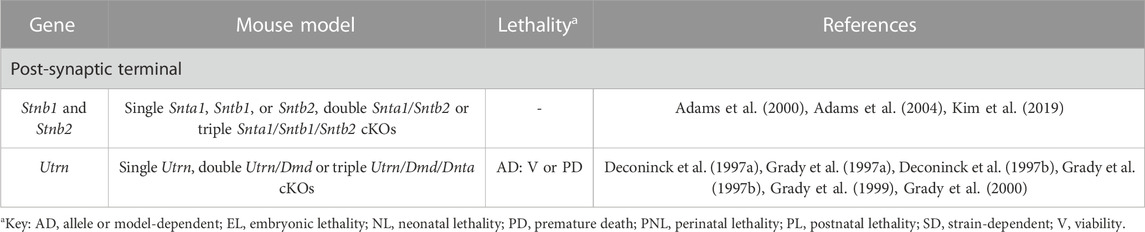
TABLE 8. Novel candidate genes for NMJ-related diseases in humans associated with the DGC complex. All models are constitutive unless otherwise stated. Lethal phenotypes can have complete or incomplete penetrance. cKO, constitutive KO.
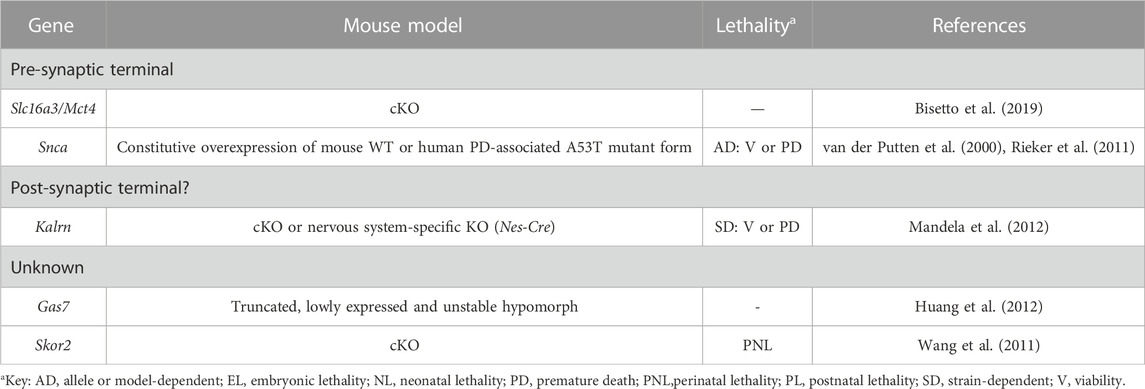
TABLE 9. Novel candidate genes for NMJ-related diseases in humans with unknown function. All models are constitutive unless otherwise stated. Lethal phenotypes can have complete or incomplete penetrance. cKO, constitutive KO.
We sought to characterize these 64 loci following the same classification used in the congenital myasthenic syndromes, which have been classically subdivided according to the specific location of the mutant protein as presynaptic, synaptic or postsynaptic (Tables 1–9, Supplementary Table S3 and Figure 1). In the next sections, we will briefly discuss the molecular mechanisms affected when these genes are not normally expressed. Of note, even if we originally aimed at finding genes that have a direct impact on NMJ morphology or function, our search has also uncovered genes whose malfunction lead to indirect defects in the NMJ, particularly when they affect motor neuron survival. In any case, the results obtained here help to expand the repertoire of genes to take into consideration when studying human pathologies.
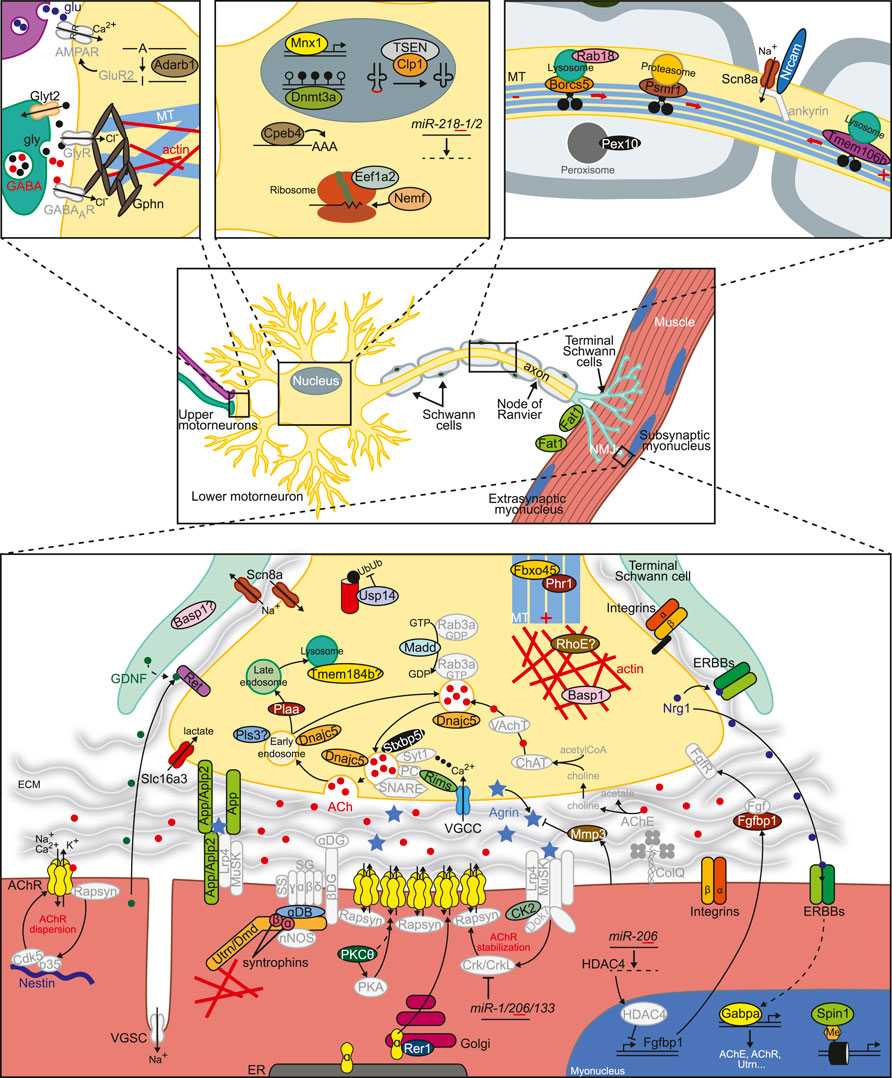
FIGURE 1. Factors involved in the regulation of NMJ formation, maintenance or function. Novel proteins or miRNAs whose dysfunction disturbs NMJ structure or function as described in this work appear in color. See Tables 1–9, Supplementary Table S3 and text for further details. Additional important factors need for normal NMJ biology are also included in grey. The basic NMJ template was obtained from Motifolio drawing toolkits (Motifolio Inc, Ellicott City, MD, USA). ECM, extracellular matrix; ER, endoplasmic reticulum; DG, dystroglycan; NMJ, neuromuscular junction; PC, priming complex (Munc13, Rab3/27); SG, sarcoglycan; SS, sarcospan; VGSC/VGCC, voltage-gated sodium/calcium channel.
Many of the novel candidates that could be indirectly associated with NMJ diseases are essential for MN development, function or viability (Table 1, Supplementary Table S3 and Figure 1). If MNs fail to reach their target muscles, NMJs obviously fail to form, while if innervation is correctly established but MNs later die, NMJs degenerate. Therefore, defects in the NMJ in the models described in this section are just secondary to defects in MNs. For example, in mice lacking the transcription factor Mnx1, MNs are generated, but their identity is not correctly specified because the interneuron identity program is not efficiently repressed (Arber et al., 1999; Thaler et al., 1999). This causes problems in axon projections and pathfinding, ultimately leading to the lack of innervation, and thus of NMJs, of certain muscles like the diaphragm (Arber et al., 1999). Thus, Mnx1 KO mice are perinatal lethal due to their inability to breath. MNs are also born in Slc6a5/Glyt2 mutant mice (Gomeza et al., 2003; Bogdanik et al., 2012). However, in the absence of this glycine transporter, essential to sustain prolonged glycinergic inhibitory transmission in the spinal cord, MNs exhibit increased excitability, that is, uncoordinated and excessive firing, together with an acceleration in postnatal NMJ maturation (Bogdanik et al., 2012). These molecular events result in a neuromotor deficiency that resembles human hyperekplexia, but unlike in humans, who can compensate the impairment in glycinergic transmission with increased GABAergic inhibition, Slc6a5 mutant mice only survive for 2 weeks (Gomeza et al., 2003; Bogdanik et al., 2012). Glycinergic synaptic activity is also disturbed in Gphn/Geph mutant animals (Feng et al., 1998) given that this tubulin-binding protein anchors all glycine and several GABAA receptor types to the microtubule network in the presynaptic MN. In these animals, MN survival and innervation patterns are affected during development, albeit differentially depending on the target muscle (Banks et al., 2005).
Reduced MN integrity and survival is additionally observed in mutant mice for Adarb1/Adar2, Ret, Clp1 and Pex10 for different reasons. In the absence Adarb1/Adar2, the mRNA editing needed for the expression of the correct isoform of the AMPA receptor subunit GluR2 does not take place, thus disturbing the properties of this channel and, ultimately, MN viability (Hideyama et al., 2010). Similarly, if the GDNF receptor Ret is missing, MNs cannot integrate GDNF inputs, which interferes with the proper organization and maturation of the presynaptic terminals and NMJs during development and regeneration, resulting in increased MN death (Baudet et al., 2008). The exact molecular mechanism is unknown, but it has been hypothesized that GDNF could activate Ret eliciting a signaling cascade within MNs, or it could be incorporated in the MN in a Ret-dependent manner and exert specific actions there (Baudet et al., 2008; Zahavi et al., 2015). In parallel, impaired function of Clp1, a component of the tSEN complex involved in RNA maturation, causes the accumulation of small aberrant RNA species that trigger p53-dependent apoptosis of MNs (Hanada et al., 2013). Finally, when the activity of the peroxisome-associated Pex10 protein is disturbed in SCs, these cells die (Hanson et al., 2014); not only do they accumulate toxic molecules, but they fail to synthesize essential molecules like plasmalogens, which are very abundant in myelin. Eventually, the lack of support from SCs has a negative impact in MN integrity and synaptic transmission (Hanson et al., 2014). Indeed, the role of SCs in NMJ development has proven fundamental. In the absence of SCs, even if axons appear defasciculated, MNs reach the target muscles and NMJs are initially formed. However, nerve terminals eventually retract and NMJs thus fail to be maintained (Lin et al., 2000). Curiously, NMJs can be preserved if muscle activity is blocked (Liu Y et al., 2019). This finding suggests that SCs counteract the presumable destabilizing activity muscle exerts in the developing NMJ, and which causes MN retraction (Liu K et al., 2019).
Ducky (Snell, 1955) and Stargazer (Noebels et al., 1990) mice are spontaneous mutants for the Cacna2d2 (Barclay et al., 2001) and Cacng2 (Letts et al., 1998) subunits of the voltage-gated calcium channels (VGCCs) required to conduct the presynaptic calcium influx along MN axons necessary for neurotransmitter release at the terminals (Table 2, Supplementary Table S3 and Figure 1). Both are epilepsy models that show spike-wave seizures and ataxia, among other features (Noebels et al., 1990; Barclay et al., 2001). In addition, mutations in these genes cause very mild phenotypes observed at the NMJ level, probably reflecting the existence of redundancy with other accessory channel subunits (Kaja et al., 2007).
The rest of the novel candidate genes causative or modifiers of NMJ-related disorders in this section are associated to various extents to postsynaptic AChRs (Table 2, Supplementary Table S3 and Figure 1). Here, we will briefly describe NMJ formation [recently reviewed in (Rodríguez Cruz et al., 2020)], highlighting the role of the candidate genes found in our search. During NMJ formation, small AChR clusters appear in the center of the muscle fibers in a nerve-independent process but that is mediated by Lrp4/MuSK/Dok7 and rapsyn at the muscle terminal (Lin et al., 2001; Yang et al., 2001; Okada et al., 2006; Weatherbee et al., 2006). Then, MNs innervate some of the prepatterned clusters, that get stabilized via neural-derived agrin signaling on Lrp4/MuSK/Dok7 at the postsynaptic terminal (McMahan, 1990; Glass et al., 1996; Okada et al., 2006; Kim et al., 2008; Zhang et al., 2008). Of note, MNs can organize de novo AChR clusters in the absence of prepatterned clusters (Lin et al., 2008; Liu et al., 2008). After agrin-induced stimulation, a series of events take place before AChR cluster stabilization, including phosphorylation of multiple MuSK residues, which function as docking sites for downstream signaling. Casein kinase II (CK2) holoenzyme is responsible for phosphorylating MuSK at its kinase inert domain (Cheusova et al., 2006). In the absence of the CK2 regulatory subunit Csnk2b, MuSK phosphorylation is disturbed, not only causing a negative impact on AChR cluster stabilization and postsynaptic structure, but also on synaptic transmission (Cheusova et al., 2006), leading to embryonic lethality in constitutive null mice (Buchou et al., 2003). AChR clustering and stability is controlled by phosphorylation of its different subunits as well. In this regard, agrin-induced tyrosine phosphorylation of the AChRβ subunit contributes to rapsyn association and receptor clustering (Borges et al., 2008). In parallel, PKCθ would be involved in the regulation of PKA-mediated phosphorylation of the AChRε subunit, and in the direct phosphorylation of AChRδ, two events that produce opposing effects: AChR stability or loss, respectively (Lanuza et al., 2010). This would explain the delay in postsynaptic maturation present in PKCθ deficient mice (Lanuza et al., 2010), which also show a delay in the early postnatal synapse elimination process to achieve monoinnervation by an unknown mechanism (Li et al., 2004). Whether the absence of PKCθ activity at the presynaptic terminal contributes to any of these two phenotypes cannot be discarded (Lanuza et al., 2010).
Agrin signaling terminates with its removal from the synaptic cleft. Mmp3 is a metalloprotease secreted by muscle that degrades the major components of the extracellular matrix and other proteins including agrin (VanSaun and Werle, 2000). In Mmp3 deficient mice, agrin accumulates at the synaptic basal lamina, and further, it also appears abnormally located outside the central endplate area where AChR should only cluster (VanSaun et al., 2003). As a consequence, the structure of the postsynaptic membrane is greatly disturbed (VanSaun et al., 2003).
Not all prepatterned AChR clusters get stabilized. Those receptors that had integrated in the muscle membrane extrasynaptically do not receive agrin input and are dispersed through a Cdk5-dependent mechanism triggered by ACh (Lin et al., 2005). The intermediate filament protein nestin has been shown to play a role in this process (Mohseni et al., 2011; Yang et al., 2011). The model poses that unphosphorylated nestin and Cdk5 constitutively interact at the postsynaptic terminal. Upon ACh stimulation, p35 is recruited and activates Cdk5, which in turn phosphorylates nestin. This results in destabilization of the intermediate filament network, the release of active Cdk5 and AChR cluster dispersal (Yang et al., 2011). Thus, in nestin KO mice, Cdk5 activity is impaired, hindering extrasynaptic AChR dispersal (Mohseni et al., 2011; Yang et al., 2011), as well as disturbing proliferation in muscle, causing frequent spontaneous regeneration (Lindqvist et al., 2017). Of note, Cdk5 can be activated by a second mechanism that involves rapsyn and calpain (Chen et al., 2007).
Finally, the correct assembly of the AChR complexes at the membrane is undoubtedly essential for proper function. Rer1 is a Golgi-associated protein part of the quality control mechanism that retains unassembled or immature membrane protein complexes in the endoplasmic reticulum, ensuring only proper mature complexes are discharged [e.g., (Kaether et al., 2007)]. When Rer1 activity is compromised, unassembled AChR⍺ subunits -and no other subunit type-escape this quality control mechanism and accumulate in the postsynaptic membrane, or are bound for lysosome-mediated degradation, reducing the number of fully assembled AChRs at the muscle membrane (Valkova et al., 2011).
Dnmt3a is required for de novo DNA methylation, and is essential for the establishment of correct DNA methylation patterns during development. When this gene is removed specifically in the nervous system by means of Nestin-Cre-mediated recombination, NMJs are highly fragmented. Mutant mice exhibit problems in neuromuscular function and motor coordination in the absence of gross anatomical alterations in the cerebellum (Nguyen et al., 2007). The exact molecular mechanism is unknown but probably related with abnormal gene expression in MNs during development through disturbed methylation of specific targets (Nguyen et al., 2007). Indeed, even if spinal cord MNs appear normal in the mutants, in vitro it has been shown that Dnmt3a is required for MN differentiation (Ziller et al., 2018) (Table 3, Supplementary Table S3 and Figure 1). The potential contribution of skeletal muscle, considering nestin is also expressed in this tissue, cannot be discarded [(Mohseni et al., 2011; Yang et al., 2011; Lindqvist et al., 2017), see section 2.10 below].
In mice harboring a skeletal muscle-specific deletion of the chromatin reader Spin1, the expression of bHLH transcription factors involved in myogenesis, and by extension of their target genes, is altered during development and after birth (Greschik et al., 2017). Many of these targets are associated with the NMJ, such as Dok7, AChE, Colq and several AChR subunits. This not only results in severe morphological alterations of the postsynaptic muscle terminal, but also in defects in the presynaptic MN, including a reduced number of synaptic vesicles and the presence of vacuoles (Greschik et al., 2017) (Table 3, Supplementary Table S3 and Figure 1).
At the post-transcriptional regulation level, at least two micro-RNA (miRNA) families are associated with NMJ development (Table 3, Supplementary Table S3 and Figure 1). Being the most abundant MN miRNAs (Amin et al., 2015), the miR-218 family is particularly important. In the absence of the miR-218-1 and miR-218-2 paralogs, hundreds of neuronal enriched genes from multiple molecular pathways are deregulated (Amin et al., 2015). This massive alteration in the transcriptome does not impact on MN development, but rather on MN maintenance and survival, as well as on NMJ formation (Amin et al., 2015). Of note, miR-218 gene variants have recently been associated with amyotrophic lateral sclerosis (ALS) susceptibility (Reichenstein et al., 2019). The second miRNA family, miR-1/206/133, is involved in NMJ formation from the muscle side. The lack of these species alleviates the repression of their target gene CRX, which acts downstream the Lrp4/MuSK/Dok7 cascade and is involved in agrin-induced stabilization of forming AChR clusters (Rodríguez Cruz et al., 2020) through the recruitment of, at least, the Rho GTPase RAC1 (Klockner et al., 2022). Intriguingly, miR-1/206/133 are dispensable for skeletal muscle development since only NMJ formation is altered in the triple KOs (Klockner et al., 2022). In contrast, if only miR-206 is removed, NMJ development remains unperturbed, presumably due to compensation by other members of the family (Williams et al., 2009). Still, its target Hdac4 is overexpressed, causing a negative effect on retrograde FGF signaling from muscle to its innervating neuron, where abnormalities are evident (Williams et al., 2009).
Further, Mbnl1 and Mbnl2 are RNA binding proteins involved in multiple aspects of RNA biology (Nakka et al., 2018). In double Mbnl1−/−/Mbnl2+/− mutants, NMJs are greatly compromised presumably as a secondary effect to the severe defects observed in muscles, which develop a myotonic dystrophy (DM)-like phenotype (Lee et al., 2013). The mechanism by which NMJ deteriorate is probably related with a defect in the processing of one or multiple transcripts involved in NMJ development, maintenance and/or function. In particular, changes in alternative splicing and polyadenylation events are observed in the compound mutants (Lee et al., 2013) (Table 3, Supplementary Table S3 and Figure 1). In this sense, it is important to mention that alternative splicing has been shown to be a crucial layer in the regulation of NMJ development. For example, only neurons produce an alternatively spliced isoform of agrin called Z+ agrin in mammals, which is up to 1,000-fold more potent in AChR clustering than the Z− isoforms (Ferns et al., 1993). Nova1 and Nova2 neuron-specific splicing factors regulate Z+ agrin splicing and thus, double mutant mice show defective morphology and function of the NMJ, and are paralyzed (Ruggiu et al., 2008). Remarkably, rescuing Z+ agrin expression restores some morphological defects, but physiologic MN activity remains impaired (Ruggiu et al., 2008). This would indicate that Nova factors target additional undefined transcripts that are essential for synaptic transmission, highlighting the importance of post-transcriptional regulatory mechanisms during NMJ development and function.
At the protein level, impaired translation in MNs seems to lie at the root of the denervation and neurodegeneration defects observed in Eef1a2 (Shultz et al., 1982; Chambers et al., 1998; Khalyfa et al., 2001; Newbery et al., 2005; Doig et al., 2013) and Nemf (Martin et al., 2020) deficient animals, presumably due to the inability of these cells to maintain proper homeostasis (Table 3, Supplementary Table S3 and Figure 1). Remarkably, the core translation machinery component Eef1a2 is spontaneously mutated in the so-called Wasted mice (Shultz et al., 1982; Chambers et al., 1998), which develop an aggressive, early-onset form of neurodegeneration that can be prevented in constitutive heterozygotes (Griffiths et al., 2012), but not if reintroducing the gene specifically in muscle (Doig et al., 2013). In parallel, a KO and two-point mutation alleles that interfere with protein function have been described for Nemf (Martin et al., 2020). This rescue factor is part of the Ribosome-associated Quality Control RQC complex that resolves stalled ribosomes during translation (Martin et al., 2020). All alleles exhibit marked motor defects, with the KO being the most severe and showing complete preweaning lethality (Martin et al., 2020).
A number of novel candidates are involved in the degradation and recycling of proteins and organelles in the presynaptic MN terminal, either mediated by the endosomal-lysosomal system or the proteasome (Table 3, Supplementary Table S3 and Figure 1). Some of these genes play a role in the axonal transport of lysosomes and proteasomes through the microtubule network. That is the case of Borcs5 (de Pace et al., 2020; Lüningschrör et al., 2020) and Tmem106b (Lüningschrör et al., 2020), in charge of the anterograde and retrograde transport of lysosomes (or lysosome-like particles), respectively. Mice defective for either gene show a reduction in the number of lysosomes in their axon terminals, causing impaired proteolysis and autophagy, and thus affecting the structure and function of axons (de Pace et al., 2020; Lüningschrör et al., 2020). In this same line, in vitro studies suggest that the absence of the small GTPase Rab18 could be associated with impaired lysosomal trafficking and autophagy in neurons (Nian et al., 2019). This would explain the in vivo phenotypes of Rab18 KO animals, which accumulate neurofilament and microtubule proteins at presynaptic terminals (Carpanini et al., 2014). In parallel, when the adapter protein Psmf1 is absent, the anterograde transport of proteasomes to MN terminals is impaired, eliciting proteotoxic stress followed by axonal degeneration and motor dysfunction (Liu Y et al., 2019; Minis et al., 2019).
When the process of endosomal-lysosomal-mediated degradation itself is altered in the MN terminal, problems in the NMJ arise as well (Table 3, Supplementary Table S3 and Figure 1). For example, the phospholipase A2 activator Plaa contributes to the sorting of ubiquitinated cargos from early to late endosomes or multivesicular bodies for their eventual degradation in lysosomes (Hall et al., 2017). When Plaa function is compromised, waste products accumulate at the NMJs and synaptic vesicle recycling -and neurotransmission by extension-is impaired (Hall et al., 2017). Presumably, Tmem184b would also participate in autophagosomal structure or clearance of cellular debris (Bhattacharya et al., 2016). Therefore, Tmem184b KO animals exhibit neurodegeneration due to the aberrant accumulation of recycled cargos at terminal synapses (Bhattacharya et al., 2016). Interestingly, via the same impaired mechanism, the absence of this protein has a neuroprotective role after injury since axon degeneration is delayed (Bhattacharya et al., 2016).
Ataxia mice or axJ mice, characterized by severe tremor and hindlimb paralysis, arose in 1965 (D’Amato and Hicks, 1965) as a spontaneous mutation in the Usp14 gene (Wilson et al., 2002). Phenotypic analyses of this and other Usp14 mutant models indicate that the lack of this deubiquitinating enzyme causes a defect in ubiquitin homeostasis at the presynaptic terminal with two immediate consequences (Table 3, Supplementary Table S3 and Figure 1). Firstly, proteasomal-mediated degradation of synaptic proteins involved in neurotransmitter release and NMJ shaping during postnatal development is impaired. Secondly, MNs are unable to recycle ubiquitin for additional protein targeting, on the other (Wilson et al., 2002; Chen et al., 2009; Chen et al., 2011; Bhattacharyya et al., 2012; Marshall et al., 2013). Proteasome-independent ubiquitin signaling through JNK (Vaden et al., 2015) is also affected in the mutants. All in all, these molecular malfunctions result in a wide array of phenotypic alterations that depend on the mouse strain and specific mutant allele. In all cases, though, NMJs are damaged.
The neuronal RNA binding protein Cpeb4 is involved in mRNA polyadenylation, central in cellular metabolism. Cpeb4 null mice are mostly normal (Shin et al., 2016). However, a truncated version of the protein that only contains an unstructured low complexity domain has a dominant toxic effect in mice, triggering abnormal ribosomal biogenesis and function, as well as elevated levels of stress response genes, including Drr1 (Shin et al., 2016). This gene encodes for an actin-bundling protein that interferes with neurite outgrowth (Schmidt M et al., 2011) and as a consequence, with the proper formation of NMJs due to the disruption of actin organization (Shin et al., 2016) (Table 4, Supplementary Table S3 and Figure 1).
Similarly, the calmodulin binding protein Basp1 would regulate actin dynamics at the presynaptic terminal, presumably by promoting subplasmalemmal actin accumulation (Frey et al., 2000). It would also control tSC-mediated synaptic growth and strength by an unknown mechanism (Frey et al., 2000). In any case, its function is very important for the organism since null mice exhibit a high postnatal mortality rate (Frey et al., 2000) (Table 4, Supplementary Table S3 and Figure 1).
Rnd proteins like Rnd3/RhoE are constitutively active members of the Rho GTPase family, and participate in multiple cellular functions, such as in the control of actin cytoskeleton dynamics (Guasch et al., 1998). In the absence of Rnd3/RhoE, mice display severe growth retardation, abnormal posture, motor impairments, and convulsions among other phenotypes (Mocholí et al., 2011). It has been proposed that Rnd3/RhoE could activate the RhoA/ROCK pathway in MNs, leading to the inactivation of cofilin, involved in actin depolymerization (Bamburg et al., 2021). This would generate MN axon growth or migration problems, as in the case of RhoE-defective hippocampal neurons (Peris et al., 2012). Alternatively, RhoE has been associated with skeletal muscle maturation, so a delay in myogenesis could have a negative impact on MN survival and/or establishment due to the lack of trophic factors (Mocholí et al., 2011) (Table 4, Supplementary Table S3 and Figure 1).
Phr1 is an atypical E3 ubiquitin-protein ligase that mediates ubiquitination of non-lysine residues on target proteins (Mabbitt et al., 2020). Its function is essential to stabilize the microtubule network in developing MNs, which in Phr1 null mice present an abnormal organization with disorganized microtubules not oriented in the direction of axon growth and extending past the actin-rich growth cone at the tips (Lewcock et al., 2007). Molecularly, it has been hypothesized that Phr1 might function via regulation of MAPK signaling (Lewcock et al., 2007) and/or impaired ubiquitination of specific protein targets (Mabbitt et al., 2020), possibly in complex with Fbxo45 (Saiga et al., 2009) (Table 4, Supplementary Table S3 and Figure 1).
The last candidate in this section is Clip3/CLIPR-59, which is specifically expressed in MNs, where it localizes to the trans-Golgi network. When Clip3/CLIPR-59 activity is compromised, mice die perinatally due to altered innervation of the diaphragm, and consequently, to the inability to breath (Couesnon et al., 2013). While different muscle types are differentially affected, in general the number of nerve terminals is reduced in all of them, while no defects in axon guidance are observed in the mutants. This points to a specific role for Clip3/CLIPR-59 in NMJ maintenance and stability, probably related with protein/membrane trafficking or cytoskeleton remodeling (Couesnon et al., 2013). Given that this protein is involved in myoblast fusion in vitro, a potential role of the postsynaptic terminal in NMJ stabilization cannot be discarded (Sun et al., 2015) (Table 4, Supplementary Table S3 and Figure 1).
Throughout development and after birth, multiple signaling pathways are established between the different elements that compose the NMJ to ensure its proper formation, maintenance and function (Rodríguez Cruz et al., 2020) (Table 5, Supplementary Table S3 and Figure 1). One such pathway comprises the MN-secreted neuregulin1/Nrg1 signal, and its Erbb receptors located in skeletal muscle and tSCs (Belotti and Schaeffer, 2020). Not surprisingly, when this pathway is disturbed either under Nrg1 haploinsufficiency (Sandrock et al., 1997) or by muscle-specific deletion of the Erbb2/Erbb4 receptors (Leu et al., 2003; Escher et al., 2005), postsynaptic integrity is compromised. Although controversial (Belotti and Schaeffer, 2020), defective Nrg1/Erbb signaling may affect the expression of subsynaptic genes like some AChR subunits via the GABP⍺ transcription factor, whose role in the process has also been questioned (Jaworski et al., 2007; O’Leary et al., 2007). Other possibilities include the destabilization of AChR clusters through impaired phosphorylation of the dystrophin-associated glycoprotein complex (DGC) component ⍺-dystrobrevin (Schmidt N et al., 2011), and the negative impact that impaired Nrg1/Erbb signaling would have on tSCs survival (Lin et al., 2000; Newbern and Birchmeier, 2010).
Additionally, FGF signaling from skeletal muscle to the innervating MN is essential for the formation and maintenance of NMJs (Rodríguez Cruz et al., 2020). For this reason, when the FGF carrier Fgfbp1 is not secreted to the synaptic cleft, NMJs mature slowly and deteriorate fast (Taetzsch et al., 2017). The exact FGF molecule and the presynaptic receptor that mediate in this process are unknown (Taetzsch et al., 2017). There is also the possibility that the lack of Fgfbp1 in mice has an indirect effect by eliciting other cellular changes that ultimately affect NMJ structural integrity (Taetzsch et al., 2017).
Scn8a/Nav1.6 is a voltage-gated sodium channel localized at presynaptic MNs. Several mutants have been described for this gene, with Med (Motor Endplate Disease) mice arising spontaneously in the 60s (Searle, 1962; Duchen et al., 1967; Burgess et al., 1995). These animals are characterized by a fatal form of progressive muscular weakness, and as early as in the 70s, it was already proposed that the disease was caused by progressive failure of neuromuscular transmission leading to muscle denervation (Duchen, 1970; Duchen and Stefani, 1971). Indeed, defective sodium currents in tSCs have been proposed to negatively affect the synthesis of growth factors necessary to support the development and function of NMJs (Musarella et al., 2006; Caillol et al., 2012). Defects in action potential generation in the presynaptic neuron can also contribute to the observed phenotypes, especially in the case of the m10J allele (Morelli et al., 2017). Mice carrying this mutation in heterozygosis do not show NMJ defects. However, they exhibit exacerbation of the phenotypes of Gars heterozygotes (Morelli et al., 2017), which constitute axonal models of the dominant axonal neuropathy Charcot-Marie-Tooth (CMT) disease and display defects in NMJ morphology and synaptic transmission on their own (Seburn et al., 2006; Achilli et al., 2009; Morelli et al., 2017). In these double Gars/Scn8a heterozygotes, reduced axon diameter and impaired sodium currents, presumably causing depolarization problems, synergize to produce more severe reductions in nerve conduction velocity and NMJ defects (Morelli et al., 2017). Similarly, mutants for the cell adhesion molecule Nrcam present normal NMJs, but the phenotypes of either demyelinating (Sh3tc2 KO) or axonal (Gars heterozygotes) models of CMT are also exacerbated, possibly due to impaired generation of sodium currents at the presynaptic terminal (Morelli et al., 2017). It is important to mention that Nrcam binds ankyrins intracellularly, which in turn bind sodium channels like Scn8a, which appears mislocalized and functionally compromised in Nrcam mutants (Morelli et al., 2017).
The release of ACh at the NMJ involves multiple steps such as ACh production, and synaptic vesicle formation, exocytosis and recycling. Defects in any of these steps could impair neurotransmission and cause motor problems (Table 6, Supplementary Table S3 and Figure 1). For example, the guanine nucleotide exchange factor Madd regulates the recycling between the GDP/GTP-bound forms of Rab3A, implicated in calcium-dependent neurotransmitter release (Tanaka et al., 2001). It is also probably required for the formation and trafficking of synaptic vesicles (Tanaka et al., 2001). Thus, in the absence of Madd, neurotransmission is severely impaired (Tanaka et al., 2001), as in the case of Rims1/2 mutants (Schoch et al., 2006). Of note, these proteins act as scaffolds, interacting with several molecules at the active zone, including Rab3A, being essential for synaptic vesicle exocytosis (Schoch et al., 2002; Schoch et al., 2006). If Rims interact with the same molecules in central synapses and NMJs, then they would participate in the tethering of VGCCs to the active zones, enabling high calcium concentrations in the vicinity of synaptic vesicles to trigger exocytosis (Kiyonaka et al., 2007; Kaeser et al., 2011). Nbea is another scaffolding protein required for vesicle trafficking (Su et al., 2004). While synaptic transmission at the NMJ is blocked in Nbea null mice causing lethal paralysis in newborns (Su et al., 2004), the exact mechanism of action remains unknown: either the action potential that travels along MN axons fails to reach the presynaptic terminal, or it fails to trigger neurotransmitter release (Su et al., 2004). A possible contribution of impaired trafficking of specific proteins to the presynaptic terminal, as observed in neurons in the central nervous system, cannot be discarded (Medrihan et al., 2009). The role of Stxbp5l/Tom2 in NMJ biology is also not clear, especially in comparison with its paralog Stxbp5/Tom1, a well-known vesicle fusion inhibitor (Sakisaka et al., 2008). Due to its similarity, Stxbp5l/Tom2 may contribute to this function as well. In any case, mice deficient for this protein suggest that Stxbp5l/Tom2 may inhibit spontaneous ACh release to favor sustained secretion upon repetitive stimulation (Geerts et al., 2015).
In contrast, multiple studies have explored the function of Dnajc5/CSP-⍺ revealing that this chaperone impinges on many aspects of the neurotransmission process. Mice deficient for Dnajc5/CSP-⍺ develop a progressive sensorimotor disorder with progressive muscle weakness that leads to premature death (Fernández-Chacón et al., 2004). At the molecular level, these animals show an impairment in the maintenance of the synaptic release sites (Rozas et al., 2012), in the recycling process of the different components of the neurotransmitter release machinery (Fernández-Chacón et al., 2004; Rozas et al., 2012; Sharma et al., 2012; Zhang et al., 2012), as well as in the assembly of the SNARE complex (Chandra et al., 2005; Zhang et al., 2012), involved in vesicle release, proving Dnajc5/CSP-⍺ essential to maintain continued synaptic function (Fernández-Chacón et al., 2004; Chandra et al., 2005; Zhang et al., 2012). All these defects eventually cause activity-dependent neurodegeneration of the NMJ and defects in synaptic transmission (Fernández-Chacón et al., 2004; Ruiz et al., 2008). Reduced sensitivity to calcium-mediated neurotransmitter release may also contribute to the observed phenotypes (Ruiz et al., 2008).
Finally, whether the actin-bundling protein Pls3 participates significantly in neurotransmission remains a subject of debate. It was initially proposed that Pls3 overexpression had a significant beneficial effect upon the spinal muscle atrophy (SMA) phenotype (Oprea et al., 2008). However, findings in this regard from different studies are contradictory, surely because they are not homogeneous, the various laboratories using different overexpression models (tagged vs non-tagged Pls3 protein, random genomic insertion vs. targeted insertion at the ROSA26 locus) and mouse backgrounds (different strains; SMA model or not) (Ackermann et al., 2013; McGovern et al., 2015). All in all, it seems that if Pls3 does indeed have any beneficial role, the extent of protection would be related with the severity of the SMA model, with the most severe forms being irrecoverable due to multi-organ deficits (Ackermann et al., 2013). Protection would be through the rescue of endocytic processes that affect synaptic vesicle recycling in these models (Hosseinibarkooie et al., 2016).
The functions of the amyloid precursor protein APP go beyond its widely recognized association with Alzheimer’s disease. Indeed, the adhesion activities of APP and APP-like protein 2 (Aplp2) ensure the correct apposition between the pre- and postsynaptic terminals of the NMJ in the so-called transsynaptic APP interaction model (Wang et al., 2009). When this complex is disturbed in double App/Aplp2 KO mice, the percentage of apposition is reduced and the presynaptic terminal morphology disturbed (Wang et al., 2005) (Table 7, Supplementary Table S3 and Figure 1). Also, APP regulates the presynaptic expression and activity of the high-affinity choline transporter CHT that mediates choline uptake from the synaptic cleft for ACh synthesis (Wang et al., 2007). These abnormalities are responsible for the defective synaptic transmission observed in the mutants (Wang et al., 2005). Further, the APP-Aplp2 complex interacts with Lrp4, likely to integrate agrin-induced AChR stabilization signals (Choi et al., 2013). The highly conserved APP C-terminal domain is indispensable for these functions (Li et al., 2010).
Adhesion plays a fundamental role during development as well. It has been shown that when integrin β1 is absent from tSCs, their migration along MN axons to the terminals is affected because this collagen receptor is required for the interaction between both cell types (Pietri et al., 2004) (Table 7, Supplementary Table S3 and Figure 1). Thus, tSC-specific functions during NMJ development are impeded (Pietri et al., 2004). If the expression is abolished in skeletal muscle, the interaction between the pre- and post-synaptic terminals is perturbed, impacting upon presynaptic differentiation and NMJ formation (Schwander et al., 2004). In contrast, no gross defects are observed in MN-specific mutants (Schwander et al., 2004).
Similarly, the concerted action of the protocadherin Fat1 in skeletal muscle, connective tissue and MNs during development, directly or indirectly controls the coordinated collective migration of myogenic precursors, as well as MN axonal growth and specification through gene expression regulation of MN-specific genes (Caruso et al., 2013; Helmbacher, 2018). When Fat1 activity is missing, the coupling of muscular and neuronal morphogenesis is altered, resulting in a wide array of abnormalities such as the misshaping and mispositioning of some muscle groups, poor motor performance, and defects in NMJ integrity (Caruso et al., 2013; Helmbacher, 2018) (Table 7, Supplementary Table S3 and Figure 1).
The DGC is a multiprotein complex in charge of anchoring the cytoskeleton of muscle fibers to the extracellular matrix, thus being required for muscle stability (Ibraghimov-Beskrovnaya et al., 1992) (Table 8, Supplementary Table S3 and Figure 1). Utrophin (Utrn) and dystrophin (Dmd) are among its main components. While Utrn is located at the crests of the junctional folds where AChRs cluster, Dmd localizes at the base together with VGSCs (Flucher and Daniels, 1989; Sealock et al., 1991; Bewick et al., 1992). Not surprisingly, double Utrn/Dmd KOs present largely disassembled DGCs (Deconinck et al., 1997b; Grady et al., 1997b), unlike single mutants (Deconinck et al., 1997a; Grady et al., 1997a; Grady et al., 1997b) presumably due to the compensation between both proteins. As a consequence, AChR distribution is altered and postsynaptic folding almost inexistent, albeit with minimal effects in neurotransmission (Deconinck et al., 1997b; Grady et al., 1997b). ⍺-dystrobrevin (Dnta) is a soluble DGC component required to establish interactions with other proteins like the AChRs, so its absence in triple Utrn/Dmd/Dnta KOs worsens the phenotype (Grady et al., 1999; Grady et al., 2000). Syntrophins are also adapter proteins for the DGC, so in line with the previous models, single ⍺1-syntrophin (Snta1) (Adams et al., 2000) and, especially, double ⍺1-syntrophin/β2-syntrophin (Snta1/Sntb2) KOs (Adams et al., 2004) exhibit disturbed organization and stability of postsynaptic folds due to the disruption of the DGC. In contrast, single Sntb1 or Sntb2 (Adams et al., 2004) mutants have no overt phenotype, possibly because of compensation by ⍺1-syntrophin.
The pathogenic mechanisms that cause NMJ abnormalities in the last set of candidate genes are largely unknown and can only be hypothesized (Table 9, Supplementary Table S3 and Figure 1). For example, it has been shown that aged Gas7-deficient mice have fewer MNs than expected, which, in addition, show reduced terminal sprouting at NMJs (Huang et al., 2012). They also present changes in fiber type composition and size, but only in slow skeletal muscles (Huang et al., 2012). These abnormalities result in motor activity defects. Given its role in actin and tubulin polymerization in vitro (She et al., 2002; Akiyama et al., 2009), the lack of Gas7 could compromise the organization of cytoskeletal network of axon terminals. Alternatively, since it is also expressed in tSCs and skeletal muscle, where it localizes to the sarcomeres, NMJ abnormalities could result from a defect in tSCs supportive role on MNs, and/or to a muscle-autonomous problem in the contraction machinery (Huang et al., 2012).
Kalrn is a Rho GDP/GTP exchange factor that is expressed in multiple tissue-specific isoforms from the same gene, which complicates studies on its function. Null mice for most isoforms show marked abnormalities in the pre- and postsynaptic terminals of the NMJ, concomitant with reduced neuromuscular function (Mandela et al., 2012). These phenotypes are probably mostly related with the functions of the Kalrn9 and Kalrn12 isoforms in skeletal muscle given that defects are more pronounced in constitutive than in neuron-specific mutants (Mandela et al., 2012), where the Kalrn7 isoform is predominant (Penzes et al., 2000). Impaired clustering of receptors, channels and signaling proteins at the NMJ, or impaired sarcomeric formation in the muscle fibers may underlie the observed phenotypes (Mandela et al., 2012). Altered basal secretion of peptides from unspecified tissues could also contribute, while a role for Kalrn7 at the presynaptic terminal can also not be discarded (Mandela et al., 2012).
In one of the earliest publications regarding the function of the transcriptional co-repressor Skor2 in mammals (Wang et al., 2011), the authors mentioned that 90% of Skor2 KO pups “die within 48 h of birth, probably owing to defective NMJ formation and respiration failure” but they did not elaborate further. This and follow-up publications revealed a role for Skor2 in the development of GABAergic neurons in the cerebellum (Wang et al., 2011; Nakatani et al., 2014) and midbrain (Kirjavainen et al., 2022), but the phenotypes in NMJs remain unexplored to date.
In contrast, a metabolic problem could explain the phenotypes of Slc16a3/Mct4 deficient mice. This proton-linked transporter mediates the transport of metabolites like lactate or pyruvate across the plasma membrane. If its activity is disrupted in the presynaptic terminal, the motor unit is destabilized, and NMJs degenerate among other phenotypes (Bisetto et al., 2019). If metabolic pathways responsible for ATP production are disturbed in the absence of Slc16a3/Mct4, there would not be enough energy for neurotransmitter release and NMJ would eventually deteriorate (Bisetto et al., 2019). Surprisingly, even if mutant muscles accumulate high amounts of lactate, they are morphologically and functionally unperturbed (Bisetto et al., 2019).
Finally, the most predominant hallmark of Parkinson’s Disease (PD) is the presence of the so-called Lewy bodies, protein aggregates that contain ⍺-synuclein (Snca), among other proteins, and that accumulate within neurons disturbing cellular homeostasis and eventually causing cell death (Srinivasan et al., 2021). When trying to model PD in mice, several authors have overexpressed mouse Snca (Rieker et al., 2011) or a mutant form of human SNCA described in PD patients (van der Putten et al., 2000). In these models, NMJs degenerate and mice display motor abnormalities, being the human mutant overexpression model the most severe (van der Putten et al., 2000). They also manifest ubiquitin immunopathology, especially when overexpressing the mouse form Rieker et al. (2011). Since Snca has been shown to be involved in the regulation of synaptic vesicle trafficking [e.g., (Burré et al., 2010)], these defects could be related with disturbed recycling of synaptic vesicles and/or with the inhibition of the proteasome catalytic activity due to ubiquitination overload, thus causing a problem in synapse maintenance (van der Putten et al., 2000; Rieker et al., 2011).
Lgals1 encodes for galectin-1, which binds numerous complex carbohydrates and, among other functions, is involved in the removal of nerve terminals at the NMJ after denervation (Plachta et al., 2007) directly via the Sema3a pathway (Quintá et al., 2014), and indirectly through the recruitment of macrophages (Gaudet et al., 2009). However, Lgals1 constitutive KO mice do not have a defect in NMJ morphology despite having such annotation in the MGI database. Similarly, even if Utp14b, essential for spermatogenesis (Beamer et al., 1988) is associated with “failure in neuromuscular synapse postsynaptic differentiation” at MGI, the referenced paper is unrelated (Beamer et al., 1988), and we failed to find any other relationship with the NMJ in the literature.
Interestingly, Nmf67 mice present increased fragmentation of the postsynaptic terminal and premature death, among other defects in the heart and eyes. However, the gene altered in this chemically induced model is yet to be identified https://www.jax.org/strain/004472.
Caution should be taken when using compound models that involve previously generated mutant alleles even if they had extensively been used in the literature before. For example, the Mnx1-Cre line is a very common resource to drive Cre-mediated recombination in motor neurons (Yang et al., 2001). Mnx1-Cre heterozygotes are fully viable and fertile, but may have reduced endplate and muscle fiber sizes (Ackermann et al., 2013). Thus, Mnx1 happloinsufficiency may have a phenotype on its own that should be taken into account especially if studying NMJs. Further, Cre-mediated recombination in lines driving tissue-specific Cre expression might not be very efficient or even inaccurate, as in the case of the Nes-Cre line used for generating neural-specific Dnmt3a KOs, in which recombination is observed in non-neural tissues as well (Nguyen et al., 2007). Indeed, nestin is also expressed in skeletal muscle where it would participate in the dispersal of extrasynaptic AChRs (Mohseni et al., 2011; Yang et al., 2011; Lindqvist et al., 2017), as mentioned above. In parallel, when random integration is used to generate transgenic animals, only in few occasions the exact genomic location where the transgene has integrated is determined. However, this could be an essential factor since the transgene might have inactivated an endogenous gene influencing the phenotypic outcome of the model, as shown for some of the most highly used transgenic mouse lines (Goodwin et al., 2019). For example, a profilin transgene containing a mutation associated with ALS disrupts the Mdga2 gene, involved in cell-cell interactions, according to The Jackson Laboratory (https://www.jax.org/strain/030568). However, the authors of the model, which leads to abnormal NMJ morphology among other phenotypes, never stated this in the original publication (Fil et al., 2017) and thus, the potential contribution of Mdga2 to the observed phenotypes is currently unknown. Even if homologous recombination is used to target a specific locus, the potential scars or exogenous sequences introduced in the process could have unexpected consequences. For example, the three different mutant models available for the myogenic regulatory factor Mrf4, still harbor the pgk-neomicine resistance cassette that was used for homologous recombination in all cases (Olson et al., 1996). Surprisingly, these models exhibit diverse phenotypes that range from complete viability to lethality at birth. This variability is explained by the fact that the resistance cassette affects the expression of the neighboring Myf5 gene (Vicente-García et al., unpublished results) to various extents, as hypothesized (Olson et al., 1996), making these models compound Mrf4:Myf5 mutants. All in all, the above-mentioned factors should therefore always be taken into account when drawing conclusions from our experiments.
Finally, the list of novel candidate genes for NMJ-associated diseases described here is not exhaustive, as it relies on the manual curation of the MGI database that needs to continuously scan the increasingly vast literature. Also, the fact that we use very specific terms to retrieve models with deficient NMJs could also contribute to some essential genes being overlooked: the approach described here is thus highly selective at the cost of a potential high number of false negatives. For example, key genes known to have a direct impact in NMJ development, maintenance or function, like some genes of the Wnt signaling pathway (e.g., Vangl2: Boëx et al., 2022; Messéant et al., 2017; Wls; Shen et al., 2018), are missing from the search results. Further, null mice for the MuSK binding protein Biglycan present defects in synapse stability, which leads to morphological abnormalities (Amenta et al., 2012). However, this phenotype is still not included in the database. Likewise, while Sam68 KO mice are included in the database, their only phenotypic annotations are associated with the role of this RNA-binding protein in bone marrow mesenchymal cell differentiation (Richard et al., 2005), while its function in maintaining NMJ integrity (de Paola et al., 2020) still awaits incorporation. In any case, our approach has indeed enabled the unveiling of mostly understudied genes in the context of NMJ biology in the expectation that they would be recognized in the field.
The mouse is the most commonly used model organism for the study of NMJ-related diseases. However, it must be taken into account that NMJs exhibit species-specific characteristics, with many differences existing between mice and humans. Thus, the mouse may not the most suitable model organism in all cases. Further, differences also exist between muscle types or even sexes, which are factors that need to be considered in the study of NMJ disorders as well.
The physiology of NMJs has been extensively studied in vertebrates and invertebrates (e.g., Couteaux, 1955 and reviewed in Clarac and Pearlstein, 2007). Although the invertebrate nervous system is considered “simple”, many of the findings obtained in invertebrates have been extrapolated to vertebrates. For example, studies on the crayfish (Wojtowicz et al., 1994) established the basis and principles for analyses in other invertebrates, such as Drosophila (Lnenicka, 2020), and later, in more complex organisms (Ackermann et al., 2015).
Even if mammalian NMJs have been more deeply characterized, little is known about the differences between the NMJs of different species even if these differences were already apparent in the 1950s (Couteaux, 1955), especially in comparison to humans. To date, the mouse is the main model organism used both for the study of NMJs and for the development of disease models that affect them. However, in many cases, the results obtained in these models have not been successfully translated to humans, as in ALS models (Clerc et al., 2016). This may be due to differences in the morphology or functionality of NMJs between both species. Indeed, human NMJs are much smaller than in the mouse, and they also have much thinner pre-synaptic axons with more rudimentary nerve endings and a different post-synaptic conformation (Jones et al., 2017). In addition, human NMJs vary from those in mice and other species in the so-called safety factor, originally defined as the excess of released ACh to ensure that neuromuscular transmission takes place (Wood and Slater, 2001). Precisely, morphological diversity may underlie differences in the safety factor between species. In general, it seems that the safety factor in humans is lower than in mice (Wood and Slater, 2001). Of note, variation or reduction in the safety factor is associated with different pathological conditions (Wood and Slater, 2001), such as congenital myasthenic syndromes (Engel et al., 2015), or myasthenia gravis (Plomp et al., 2017).
Thus, to better understand the form and function of the NMJ in human health and disease, it is important to identify animal models that more closely resemble human NMJs. Given the clear differences identified between humans and mice (Jones et al., 2017), larger animal models could provide more suitable, albeit probably more impractical, alternatives.
Boehm and others have recently compared the morphology of the NMJs of four different hindlimb muscles in six mammalian species (mouse, cat, dog, pig, sheep, and human) (Boehm et al., 2020). Using the NMJ-morph tool (Jones et al., 2016), they performed a deep qualitative and quantitative characterization. In the qualitative analysis, they observed that mouse NMJs have a characteristic, so-called “pretzel” morphology, while human NMJs were surprisingly small and nummular (coin-shaped) in morphology (Jones et al., 2017). Strikingly, the morphology of pig and sheep NMJs was very similar to each other, and the closest to that of humans.
In the quantitative analysis, a total of 21 pre- and post-synaptic variables were measured, including nerve terminal areas, the number terminal branches, AChR and endplate areas, and the degree of fragmentation of the endplate, among others (Table 10). Most of these variables were significantly larger in mice and dogs compared to humans. In contrast, these parameters attained the lowest values in cats. When considering overall size, cats and humans had strikingly small NMJs, while dogs presented the largest ones. As in the qualitative analysis, sheep and pig NMJs showed the highest similarity to human NMJs, especially sheep, with 19 of the 21 parameters analyzed showing no statistically significant differences (18/21 in the case of pigs) (Boehm et al., 2020).
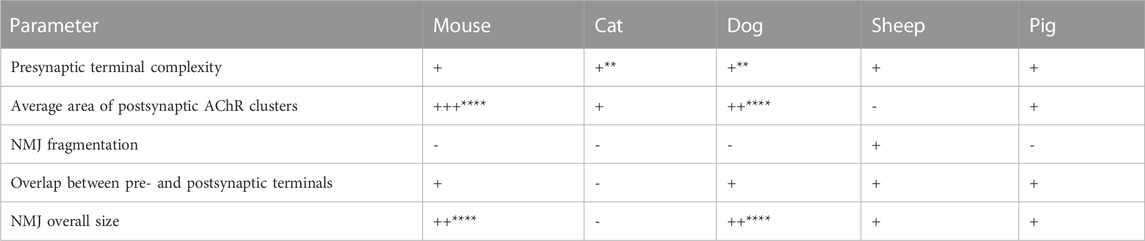
TABLE 10. Characteristics of NMJs in different mammalian species taking the human as reference. Key: (−) lower, (+) greater, (++) two times larger, (+++) three or more times larger values than in humans. Significance levels: **p < 0.01, ****p < 0.0001. Original data from (Boehm et al., 2020).
This study disproves the theory that NMJ size is inversely correlated with body mass, a theory proposed over 3 decades ago (Balice-Gordon and Lichtman, 1990). Indeed, it was hypothesized that the small size of human NMJs compared to mouse ones was due to the difference in size between both species. However, now according to Boehm et al. (2020), this is not the case, as rats or cats, larger animals than mice, have equal and smaller NMJs relative to humans, respectively.
To date, this is the only study that has carried out in-depth comparative analyses of NMJs in different mammalian species, highlighting the limitations of using the mouse as a model. Its main drawback, though, is that it only considers hindlimb muscles, so it could be possible that for other muscles, the animals with the greatest similarity to humans are not sheep. It would be therefore important to extend this work to more muscle types of diverse developmental origin.
In addition to differences in morphology and size of the pre- and post-synaptic components, differences can also be observed in tSCs (Santosa et al., 2018). In comparison with mice, despite having a similar number of tSCs per NMJ, human tSCs are significantly smaller, covering less AChR synaptic area, but extending extrasynaptically (Alhindi et al., 2021), at least in the peroneus brevis hindlimb muscle. These features, whereas being normal in human tSCs, have been considered as “pathological” in mice, as they have been described in mouse models of ALS and in denervation experiments (Kang et al., 2014; Carrasco et al., 2016). This highlights the importance of understanding the structure and function of all components of the NMJ in each species, in order to be able to efficiently translate scientific findings to humans.
Furthermore, the above-mentioned morphological differences among the NMJs of the diverse species are accompanied by distinct functionalities, as exemplified by their differential response to aging. Numerous studies based on work with mice suggest that mammalian NMJs are unstable with age (Valdez et al., 2010; Willadt et al., 2016), but little is known about what happens in humans. Indeed, during aging, mouse NMJs suffer post-synaptic fragmentation, pre-synaptic axon diameter decreases, partially innervated NMJs appear, and a reduction of AChR density takes place (Valdez et al., 2010). In humans, however, it has been shown that these events do not occur: there is only a slight increase in the diameter of pre-synaptic axons, with no signs of remodeling or fragmentation of the NMJs, which remain fairly stable throughout (Jones et al., 2017), in contrast to other mammalian species.
For years it has been hypothesized that there was a positive correlation between NMJ size and muscle fiber diameter (Balice-Gordon and Lichtman, 1990). However, a recent study has failed to find any relationship between the morphology of the NMJ and the diameter of the fiber it innervates (Boehm et al., 2020), suggesting that fiber size is not a determining factor in NMJ morphology.
In contrast, fiber type does play a role: in general, NMJs innervating fast type II (a, x, and b) muscle fibers appear to be more elaborate, with a greater number of branches and a larger area than those innervating slow type I fibers (Sieck and Prakash, 1997; Mejia Maza et al., 2021). Further, NMJs innervating type II fibers show less overlap between the pre- and post-synaptic terminals, leading to increased susceptibility to neuromuscular failure and subsequent fatigue, while NMJs innervating type I fibers are more resistant to failure and less prone to fatigue (Sieck and Prakash, 1997). However, in the mouse this is not always the case. For example, in the hindlimb soleus muscle, slow type I fibers have larger NMJs than their fast Ia counterparts. This could be due to the fact that the soleus is a major postural muscle, thus subject to constant physical activity and therefore more neurotransmission, which could lead to the expansion of both pre- and post-synaptic structures. This adaptation would allow the NMJ to store more ACh and express a greater number of receptors, which would help delay the onset of neuromuscular failure and fatigue (Deschenes et al., 2013).
In addition to the differences associated with the type of fiber that make up the muscles, it appears that the physiological state of the muscle may influence the morphology of the NMJ that innervates it. Numerous studies have shown that aging produces morphological changes in NMJs (Deschenes et al., 2013; Willadt et al., 2016; Burke et al., 2021; Dobrowolny et al., 2021). For example, in mouse extensor digitorum longus, gastrocnemius and adductor longus limb muscles, there is an increase in endplate area as well as an increased number of AChR clusters at 34 months of age (Burke et al., 2021). Exercise and caloric restriction are additional factors that produce alterations in NMJs and they can even prevent their deterioration with aging (Valdez et al., 2010).
Furthermore, the function that a muscle performs could also influence NMJ morphology. For example, the splenius capitis cranial muscle in the rat has higher ratios of endplate size to muscle fiber volume than the quadriceps limb muscle, with which it has important functional and structural differences, even if both are considered fast muscles (Pfister and Zenker, 1984). Indeed, cranial muscles perform very different functions to other muscles in the body, such as the laryngeal muscles, which regulate the shape and tension of the vocal cords that mediate the control of phonation, vocalization, breathing, and swallowing. They have a fast shortening speed, high active tension, and resistance to fatigue. Their functional and structural differences with the muscles of the limbs is such that they remain intact or only slightly affected in some degenerative neuromuscular diseases: their specific characteristics may thus confer resistance to certain pathologies (Feng et al., 2012). Feng and others compared the structure and morphology of NMJs of laryngeal muscles with those of hindlimb muscles in rats (Feng et al., 2012). They analyzed 3 laryngeal muscles (laryngeal thyroarytenoid (TA), cricothyroid (CT), and posterior cricoarytenoid (PCA)) and 2 hindlimb muscles (soleus (SOL) and extensor digitorum longus (EDL)) and found significant differences between them. In particular, in laryngeal muscles, NMJs were more fragmented the higher the proportion of fast fibers in the muscle, with the PCA and TA showing the highest degree of fragmentation. This relationship does not occur in hindlimb muscles, which, in contrast, present the largest NMJs (Feng et al., 2012). Furthermore, they confirmed that, as in humans, polyinnervation is also present in rat laryngeal muscles, a finding that had only been previously observed in cranial muscles (Feng et al., 2012).
Given the remarkable differences among the NMJs of diverse muscles within the same organism, it stands to reason that there may also be differences between the NMJs of females and males of the same species. This would not be surprising considering the numerous studies that have shown sex-specific characteristics of skeletal muscle, including differential fiber type composition or size, and contraction strength (Staron et al., 2000; Davegårdh et al., 2019; O’Reilly et al., 2021). However, little is known about the possible morphological differences between NMJs of both sexes, since there are few works that directly addresses this issue, to the best of our knowledge. One such study found that in the case of rat EDL, SOL, and PL hindlimb muscles, there are no significant differences in the NMJs of females and males, although in all three muscles fiber sizes differ, being male fibers larger than those in females (Deschenes et al., 2013). This is in contrast to one of the laryngeal muscles, the TA, in which females have more fragmented NMJs than males (Feng et al., 2012), which might contribute to sexual dimorphism in the voice (Lenell and Johnson, 2017). These variations in NMJs could be the reason why certain muscle diseases affect both sexes differently, as in the case of spasmodic dysphonia, characterized by involuntary movements and inconsistent contraction of the laryngeal muscles, and mainly affecting females (Feng et al., 2012). However, further comparative studies are needed to determine whether differences in the NMJs of other muscles exist, which may allow us to understand the differential involvement of both sexes in some pathologies.
NMJs are complex structures responsible for the translation of nervous inputs into functional motor outcomes. Malfunction in any of its components -MNs, skeletal muscle or tSCs-result in defects in the development, maintenance, activity and integrity of the NMJ. In humans, 42 NMJ-specific disorders, the congenital myasthenic syndromes, have been described as indicated in the Gene Table of Neuromuscular Disorders (www.musclegenetable.fr), a specialized database that collects all known neuromuscular diseases and their causative genes, if known (Dubowitz, 1991). However, “pure” MN or skeletal muscle dystrophies can also lead to NMJ deficiencies. In addition, the multitude of mouse mutant lines generated by the scientific community has taught us that there are probably many more genes essential for NMJ biology. Several reasons could explain the fact that they have not been associated with a disease in humans. First, their function could be so essential that their absence is incompatible with life and thus, it is not possible to find patients carrying mutations in these genes. Second, the function of these genes may not be as essential in humans due to the existence of redundant or compensatory mechanisms, or simply because their functions in mice and humans have evolved divergently. Therefore, their malfunction would not cause significant phenotypes in humans. Third, many genes are pleiotropic and therefore control several traits even in different organs or at different developmental time points. Deficiencies in one such genes could cause earlier or more serious phenotypes than defects in the NMJ, especially if they do not affect gross motor skills. In this scenario, abnormalities in the NMJ would probably be missed unless specifically addressed. Or fourth, they have not been associated with a human disease to date. In any case, we provide here a list of novel candidate genes that should be considered in the study of the pathophysiological mechanisms of diseases that affect the NMJ and their potential treatments. This list is not exhaustive since it relies on manually curated data available at the MGI database. Thus, attention should be paid in the future for novel annotations and references that could expand the repertoire of potential candidate genes associated with NMJ-related diseases.
The mouse is the most commonly used model for the study of NMJ-related diseases, however, as described in this review, it may not always be the best choice. Numerous studies have shown that there are important differences between mouse and human NMJs, which may explain the failure to translate the results obtained with this animal to the clinic. In addition, we have seen how larger mammalian models, such as sheep or pigs, could provide a better model, closer to humans, for the study of these pathologies. Nevertheless, more studies are needed to gain a detailed understanding of the features of the NMJs of small and large mammals so that we have a model as close as possible to humans. It is also important to bear in mind that muscle properties and even biological sex can influence NMJ morphology and function. For example, it has been shown that the type of fiber that innervates the NMJ affects its size. However, comparative analyses that take into account these factors are very limited, especially in the case of sex-specific differences. Thus, the field would greatly benefit from more studies in this regard in order to develop efficient treatments for NMJ-related diseases in human.
AN-M, CV-G, and JC contributed to conception and design of the study. AN-M, CV-G, and JC wrote the first draft of the manuscript. AN-M, CV-G, and JC wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by the Spanish Ministry of Education, Culture and Sports through fellowship PRE2019-091097 to AN-M, an Institutional Grant CEX2020-001088-M (Maria de Maeztu Ecellence Unit, Department of Gene Regulation and Morphogenesis at CABD), an AFM research grant #22447, and PID2020-117058GB-I00 from the Spanish Ministry of Science and Innovation to JC, and ProyExcel_00153 from the Andalusian Government Junta de Andalucia (PAIDI 2020; 2021 call) to CV-G.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1216726/full#supplementary-material
Achilli, F., Bros-Facer, V., Williams, H. P., Banks, G. T., AlQatari, M., Chia, R., et al. (2009). An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Dis. Model Mech. 2, 359–373. doi:10.1242/dmm.002527
Ackermann, B., Kröber, S., Torres-Benito, L., Borgmann, A., Peters, M., Hosseini Barkooie, S. M., et al. (2013). Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 22, 1328–1347. doi:10.1093/hmg/dds540
Ackermann, F., Waites, C. L., and Garner, C. C. (2015). Presynaptic active zones in invertebrates and vertebrates. EMBO Rep. 16, 923–938. doi:10.15252/embr.201540434
Adams, M. E., Kramarcy, N., Fukuda, T., Engel, A. G., Sealock, R., and Froehner, S. C. (2004). Structural abnormalities at neuromuscular synapses lacking multiple syntrophin isoforms. J. Neurosci. 24, 10302–10309. doi:10.1523/JNEUROSCI.3408-04.2004
Adams, M. E., Kramarcy, N., Krall, S. P., Rossi, S. G., Rotundo, R. L., Sealock, R., et al. (2000). Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 150, 1385–1398. doi:10.1083/jcb.150.6.1385
Akiyama, H., Gotoh, A., Shin, R.-W., Koga, T., Ohashi, T., Sakamoto, W., et al. (2009). A novel role for hGas7b in microtubular maintenance: Possible implication in tau-associated pathology in alzheimer disease. J. Biol. Chem. 284, 32695–32699. doi:10.1074/jbc.M109.035998
Alhindi, A., Boehm, I., Forsythe, R. O., Miller, J., Skipworth, R. J. E., Simpson, H., et al. (2021). Terminal Schwann cells at the human neuromuscular junction. Brain Commun. 3, fcab081. doi:10.1093/braincomms/fcab081
Amenta, A. R., Creely, H. E., Mercado, M. L., Hagiwara, H., McKechnie, B. A., Lechner, B. E., et al. (2012). Biglycan is an extracellular MuSK binding protein important for synapse stability. J. Neurosci. 32, 2324–2334. doi:10.1523/JNEUROSCI.4610-11.2012
Amin, N. D., Bai, G., Klug, J. R., Bonanomi, D., Pankratz, M. T., Gifford, W. D., et al. (2015). Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 350, 1525–1529. doi:10.1126/science.aad2509
Arber, S., Han, B., Mendelsohn, M., Smith, M., Jessell, T. M., and Sockanathan, S. (1999). Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron 23, 659–674. doi:10.1016/s0896-6273(01)80026-x
Bailey, A. G., and Bloch, E. C. (1987). Malignant hyperthermia in a three-month-old American Indian infant. Anesth. Analg. 66, 1043–1045. doi:10.1213/00000539-198710000-00028
Balice-Gordon, R. J., and Lichtman, J. W. (1990). In vivo visualization of the growth of pre- and postsynaptic elements of neuromuscular junctions in the mouse. J. Neurosci. 10, 894–908. doi:10.1523/JNEUROSCI.10-03-00894.1990
Bamburg, J. R., Minamide, L. S., Wiggan, O., Tahtamouni, L. H., and Kuhn, T. B. (2021). Cofilin and actin dynamics: Multiple modes of regulation and their impacts in neuronal development and degeneration. Cells 10, 2726. doi:10.3390/cells10102726
Banks, G. B., Kanjhan, R., Wiese, S., Kneussel, M., Wong, L. M., O’Sullivan, G., et al. (2005). Glycinergic and GABAergic synaptic activity differentially regulate motoneuron survival and skeletal muscle innervation. J. Neurosci. 25, 1249–1259. doi:10.1523/JNEUROSCI.1786-04.2005
Barclay, J., Balaguero, N., Mione, M., Ackerman, S. L., Letts, V. A., Brodbeck, J., et al. (2001). Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J. Neurosci. 21, 6095–6104. doi:10.1523/JNEUROSCI.21-16-06095.2001
Baudet, C., Pozas, E., Adameyko, I., Andersson, E., Ericson, J., and Ernfors, P. (2008). Retrograde signaling onto Ret during motor nerve terminal maturation. J. Neurosci. 28, 963–975. doi:10.1523/JNEUROSCI.4489-07.2008
Beamer, W. G., Cunliffe-Beamer, T. L., Shultz, K. L., Langley, S. H., and Roderick, T. H. (1988). Juvenile spermatogonial depletion (jsd): A genetic defect of germ cell proliferation of male mice. Biol. Reprod. 38, 899–908. doi:10.1095/biolreprod38.4.899
Belotti, E., and Schaeffer, L. (2020). Regulation of gene expression at the neuromuscular junction. Neurosci. Lett. 735, 135163. doi:10.1016/j.neulet.2020.135163
Bewick, G. S., Nicholson, L., Young, C., O’Donnell, E., and Slater, C. R. (1992). Different distributions of dystrophin and related proteins at nerve-muscle junctions. Neuroreport 3, 857–860. doi:10.1097/00001756-199210000-00009
Bhattacharya, M. R. C., Geisler, S., Pittman, S. K., Doan, R. A., Weihl, C. C., Milbrandt, J., et al. (2016). TMEM184b promotes axon degeneration and neuromuscular junction maintenance. J. Neurosci. 36, 4681–4689. doi:10.1523/JNEUROSCI.2893-15.2016
Bhattacharyya, B. J., Wilson, S. M., Jung, H., and Miller, R. J. (2012). Altered neurotransmitter release machinery in mice deficient for the deubiquitinating enzyme Usp14. Am. J. Physiol. Cell Physiol. 302, C698–C708. doi:10.1152/ajpcell.00326.2010
Birks, R., Huxley, H. E., and Katz, B. (1960). The fine structure of the neuromuscular junction of the frog. J. Physiol. 150, 134–144. doi:10.1113/jphysiol.1960.sp006378
Bisetto, S., Wright, M. C., Nowak, R. A., Lepore, A. C., Khurana, T. S., Loro, E., et al. (2019). New insights into the lactate shuttle: Role of MCT4 in the modulation of the exercise capacity. iScience 22, 507–518. doi:10.1016/j.isci.2019.11.041
Bloom, A. J., Miller, B. R., Sanes, J. R., and DiAntonio, A. (2007). The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 21, 2593–2606. doi:10.1101/gad.1592107
Boehm, I., Alhindi, A., Leite, A. S., Logie, C., Gibbs, A., Murray, O., et al. (2020). Comparative anatomy of the mammalian neuromuscular junction. J. Anat. 237, 827–836. doi:10.1111/joa.13260
Boëx, M., Cottin, S., Halliez, M., Bauché, S., Buon, C., Sans, N., et al. (2022). The cell polarity protein Vangl2 in the muscle shapes the neuromuscular synapse by binding to and regulating the tyrosine kinase MuSK. Sci. Signal 15, eabg4982. doi:10.1126/scisignal.abg4982
Bogdanik, L. P., Chapman, H. D., Miers, K. E., Serreze, D. v., and Burgess, R. W. (2012). A MusD retrotransposon insertion in the mouse Slc6a5 gene causes alterations in neuromuscular junction maturation and behavioral phenotypes. PLoS One 7, e30217. doi:10.1371/journal.pone.0030217
Borges, L. S., Yechikhov, S., Lee, Y. I., Rudell, J. B., Friese, M. B., Burden, S. J., et al. (2008). Identification of a motif in the acetylcholine receptor β subunit whose phosphorylation regulates rapsyn association and postsynaptic receptor localization. J. Neurosci. 28, 11468–11476. doi:10.1523/JNEUROSCI.2508-08.2008
Buchou, T., Vernet, M., Blond, O., Jensen, H. H., Pointu, H., Olsen, B. B., et al. (2003). Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol. Cell Biol. 23, 908–915. doi:10.1128/MCB.23.3.908-915.2003
Burgess, D. L., Kohrman, D. C., Galt, J., Plummer, N. W., Jones, J. M., Spear, B., et al. (1995). Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease”. Nat. Genet. 10, 461–465. –5. doi:10.1038/ng0895-461
Burgess, R. W., Peterson, K. A., Johnson, M. J., Roix, J. J., Welsh, I. C., and O’Brien, T. P. (2004). Evidence for a conserved function in synapse formation reveals Phr1 as a candidate gene for respiratory failure in newborn mice. Mol. Cell Biol. 24, 1096–1105. doi:10.1128/MCB.24.3.1096-1105.2004
Burke, S. K., Fenton, A. I., Konokhova, Y., and Hepple, R. T. (2021). Variation in muscle and neuromuscular junction morphology between atrophy-resistant and atrophy-prone muscles supports failed re-innervation in aging muscle atrophy. Exp. Gerontol. 156, 111613. doi:10.1016/j.exger.2021.111613
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Südhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi:10.1126/science.1195227
Caillol, G., Vacher, H., Musarella, M., Bellouze, S., Dargent, B., and Autillo-Touati, A. (2012). Motor endplate disease affects neuromuscular junction maturation. Eur. J. Neurosci. 36, 2400–2408. doi:10.1111/j.1460-9568.2012.08164.x
Carpanini, S. M., McKie, L., Thomson, D., Wright, A. K., Gordon, S. L., Roche, S. L., et al. (2014). A novel mouse model of Warburg Micro syndrome reveals roles for RAB18 in eye development and organisation of the neuronal cytoskeleton. Dis. Model Mech. 7, 711–722. doi:10.1242/dmm.015222
Carrasco, D. I., Seburn, K. L., and Pinter, M. J. (2016). Altered terminal Schwann cell morphology precedes denervation in SOD1 mice. Exp. Neurol. 275 (1), 172–181. doi:10.1016/j.expneurol.2015.09.014
Caruso, N., Herberth, B., Bartoli, M., Puppo, F., Dumonceaux, J., Zimmermann, A., et al. (2013). Deregulation of the protocadherin gene FAT1 alters muscle shapes: Implications for the pathogenesis of facioscapulohumeral dystrophy. PLoS Genet. 9, e1003550. doi:10.1371/journal.pgen.1003550
Chambers, D. M., Peters, J., and Abbott, C. M. (1998). The lethal mutation of the mouse wasted (wst) is a deletion that abolishes expression of a tissue-specific isoform of translation elongation factor 1alpha, encoded by the Eef1a2 gene. Proc. Natl. Acad. Sci. U. S. A. 95, 4463–4468. doi:10.1073/pnas.95.8.4463
Chandra, S., Gallardo, G., Fernández-Chacón, R., Schlüter, O. M., and Südhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. doi:10.1016/j.cell.2005.09.028
Chen, F., Qian, L., Yang, Z.-H., Huang, Y., Ngo, S. T., Ruan, N.-J., et al. (2007). Rapsyn interaction with calpain stabilizes AChR clusters at the neuromuscular junction. Neuron 55, 247–260. doi:10.1016/j.neuron.2007.06.031
Chen, P.-C., Bhattacharyya, B. J., Hanna, J., Minkel, H., Wilson, J. A., Finley, D., et al. (2011). Ubiquitin homeostasis is critical for synaptic development and function. J. Neurosci. 31, 17505–17513. doi:10.1523/JNEUROSCI.2922-11.2011
Chen, P.-C., Qin, L.-N., Li, X.-M., Walters, B. J., Wilson, J. A., Mei, L., et al. (2009). The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J. Neurosci. 29, 10909–10919. doi:10.1523/JNEUROSCI.2635-09.2009
Cheusova, T., Khan, M. A., Schubert, S. W., Gavin, A.-C., Buchou, T., Jacob, G., et al. (2006). Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction. Genes Dev. 20, 1800–1816. doi:10.1101/gad.375206
Choi, H. Y., Liu, Y., Tennert, C., Sugiura, Y., Karakatsani, A., Kröger, S., et al. (2013). APP interacts with LRP4 and agrin to coordinate the development of the neuromuscular junction in mice. Elife 2, e00220. doi:10.7554/eLife.00220
Clarac, F., and Pearlstein, E. (2007). Invertebrate preparations and their contribution to neurobiology in the second half of the 20th century. Brain Res. Rev. 54, 113–161. doi:10.1016/j.brainresrev.2006.12.007
Clerc, P., Lipnick, S., and Willett, C. (2016). A look into the future of ALS research. Drug Discov. Today 21, 939–949. doi:10.1016/j.drudis.2016.02.002
Cohen, E., Bonne, G., Rivier, F., and Hamroun, D. (2021). The 2022 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 31, 1313–1357. doi:10.1016/j.nmd.2021.11.004
Couesnon, A., Offner, N., Bernard, V., Chaverot, N., Backer, S., Dimitrov, A., et al. (2013). CLIPR-59: A protein essential for neuromuscular junction stability during mouse late embryonic development. Development 140, 1583–1593. doi:10.1242/dev.087106
Couteaux, R. (1955). Localization of cholinesterases at neuromuscular junctions. Int. Rev. Cytol. 4, 335–375. doi:10.1016/S0074-7696(08)60463-5
Dale, H. H. (1914). The action of certain esters and ethers of choline and their relation to muscarine. J. Pharmacol. Exp. Ther. 6, 147–190.
D’Amato, C. J., and Hicks, S. P. (1965). Neuropathologic alterations in the ataxia (paralytic) mouse. Arch. Pathol. (Chic) 80, 604–612.
Davegårdh, C., Hall Wedin, E., Broholm, C., Henriksen, T. I., Pedersen, M., Pedersen, B. K., et al. (2019). Sex influences DNA methylation and gene expression in human skeletal muscle myoblasts and myotubes. Stem Cell Res. Ther. 10, 26. doi:10.1186/s13287-018-1118-4
de Pace, R., Britt, D. J., Mercurio, J., Foster, A. M., Djavaherian, L., Hoffmann, V., et al. (2020). Synaptic vesicle precursors and lysosomes are transported by different mechanisms in the axon of mammalian neurons. Cell Rep. 31, 107775. doi:10.1016/j.celrep.2020.107775
de Paola, E., Forcina, L., Pelosi, L., Pisu, S., la Rosa, P., Cesari, E., et al. (2020). Sam68 splicing regulation contributes to motor unit establishment in the postnatal skeletal muscle. Life Sci. Alliance 3, e201900637. doi:10.26508/lsa.201900637
Deconinck, A. E., Potter, A. C., Tinsley, J. M., Wood, S. J., Vater, R., Young, C., et al. (1997a). Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J. Cell Biol. 136, 883–894. doi:10.1083/jcb.136.4.883
Deconinck, A. E., Rafael, J. A., Skinner, J. A., Brown, S. C., Potter, A. C., Metzinger, L., et al. (1997b). Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90, 717–727. doi:10.1016/s0092-8674(00)80532-2
Deschenes, M. R., Hurst, T. E., Ramser, A. E., and Sherman, E. G. (2013). Presynaptic to postsynaptic relationships of the neuromuscular junction are held constant across age and muscle fiber type. Dev. Neurobiol. 73, 744–753. doi:10.1002/dneu.22095
Dobrowolny, G., Barbiera, A., Sica, G., and Scicchitano, B. M. (2021). Age-related alterations at neuromuscular junction: Role of oxidative stress and epigenetic modifications. Cells 10, 1307. doi:10.3390/cells10061307
Doig, J., Griffiths, L. A., Peberdy, D., Dharmasaroja, P., Vera, M., Davies, F. J. C., et al. (2013). In vivo characterization of the role of tissue-specific translation elongation factor 1A2 in protein synthesis reveals insights into muscle atrophy. FEBS J. 280, 6528–6540. doi:10.1111/febs.12554
Dubowitz, V. (1991). Neuromuscular disorders: Gene location. Neuromuscul. Disord. 1, 75–76. doi:10.1016/0960-8966(91)90050-3
Duchen, L. W. (1970). Hereditary motor end-plate disease in the mouse: Light and electron microscopic studies. J. Neurol. Neurosurg. Psychiatry 33, 238–250. doi:10.1136/jnnp.33.2.238
Duchen, L. W., Searle, A. G., and Strich, A. J. (1967). An hereditary motor endplate disease in the mouse. J. Physiol. (Lond.) 189, 4–6P.
Duchen, L. W., and Stefani, E. (1971). Electrophysiological studies of neuromuscular transmission in hereditary “motor end-plate disease” of the mouse. J. Physiol. 212, 535–548. doi:10.1113/jphysiol.1971.sp009340
Engel, A. G. (2020). “Congenital myasthenic syndromes,” in Rosenberg’s molecular and genetic basis of neurological and psychiatric disease. Editors P RA, and E Al. (Amsterdam, Netherlands: Elsevier), 539–558. doi:10.1016/B978-0-12-813866-3.00032-1
Engel, A. G., Shen, X.-M., Selcen, D., and Sine, S. M. (2015). Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 14, 420–434. doi:10.1016/S1474-4422(14)70201-7
Engel, A. G. (2008). The neuromuscular junction. Handb. Clin. Neurology 91, 103–148. Elsevier. doi:10.1016/S0072-9752(07)01503-5
Escher, P., Lacazette, E., Courtet, M., Blindenbacher, A., Landmann, L., Bezakova, G., et al. (2005). Synapses form in skeletal muscles lacking neuregulin receptors. Science 308, 1920–1923. doi:10.1126/science.1108258
Fatt, P., and Katz, B. (1951). An analysis of the end-plate potential recorded with an intracellular electrode. J. Physiol. 115, 320–370. doi:10.1113/JPHYSIOL.1951.SP004675
Fatt, P., and Katz, B. (1952). Spontaneous subthreshold activity at motor nerve endings. J. Physiol. 117, 109–128. doi:10.1113/jphysiol.1952.sp004735
Feng, G., Tintrup, H., Kirsch, J., Nichol, M. C., Kuhse, J., Betz, H., et al. (1998). Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science 282, 1321–1324. doi:10.1126/science.282.5392.1321
Feng, X., Zhang, T., Ralston, E., and Ludlow, C. L. (2012). Differences in neuromuscular junctions of laryngeal and limb muscles in rats. Laryngoscope 122, 1093–1098. doi:10.1002/lary.23218
Fernández-Chacón, R., Wölfel, M., Nishimune, H., Tabares, L., Schmitz, F., Castellano-Muñoz, M., et al. (2004). The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron 42, 237–251. doi:10.1016/s0896-6273(04)00190-4
Ferns, M., Campanelli, J. T., Hoch, W., Scheller, R. H., and Hall, Z. M. (1993). The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron 11, 491–502. doi:10.1016/0896-6273(93)90153-i
Fil, D., DeLoach, A., Yadav, S., Alkam, D., MacNicol, M., Singh, A., et al. (2017). Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 26, 686–701. doi:10.1093/hmg/ddw429
Flucher, B. E., and Daniels, M. P. (1989). Distribution of Na+ channels and ankyrin in neuromuscular junctions is complementary to that of acetylcholine receptors and the 43 kd protein. Neuron 3, 163–175. doi:10.1016/0896-6273(89)90029-9
Frey, D., Laux, T., Xu, L., Schneider, C., and Caroni, P. (2000). Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J. Cell Biol. 149, 1443–1454. doi:10.1083/jcb.149.7.1443
Gaudet, A. D., Leung, M., Poirier, F., Kadoya, T., Horie, H., and Ramer, M. S. (2009). A role for galectin-1 in the immune response to peripheral nerve injury. Exp. Neurol. 220, 320–327. doi:10.1016/j.expneurol.2009.09.007
Geerts, C. J., Plomp, J. J., Koopmans, B., Loos, M., van der Pijl, E. M., van der Valk, M. A., et al. (2015). Tomosyn-2 is required for normal motor performance in mice and sustains neurotransmission at motor endplates. Brain Struct. Funct. 220, 1971–1982. doi:10.1007/s00429-014-0766-0
Glass, D. J., Bowen, D. C., Stitt, T. N., Radziejewski, C., Bruno, J., Ryan, T. E., et al. (1996). Agrin acts via a MuSK receptor complex. Cell 85, 513–523. doi:10.1016/s0092-8674(00)81252-0
Gomeza, J., Ohno, K., Hülsmann, S., Armsen, W., Eulenburg, V., Richter, D. W., et al. (2003). Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron 40, 797–806. doi:10.1016/s0896-6273(03)00673-1
Goodwin, L. O., Splinter, E., Davis, T. L., Urban, R., He, H., Braun, R. E., et al. (2019). Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. Genome Res. 29, 494–505. doi:10.1101/gr.233866.117
Grady, R. M., Grange, R. W., Lau, K. S., Maimone, M. M., Nichol, M. C., Stull, J. T., et al. (1999). Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat. Cell Biol. 1, 215–220. doi:10.1038/12034
Grady, R. M., Merlie, J. P., and Sanes, J. R. (1997a). Subtle neuromuscular defects in utrophin-deficient mice. J. Cell Biol. 136, 871–882. doi:10.1083/jcb.136.4.871
Grady, R. M., Teng, H., Nichol, M. C., Cunningham, J. C., Wilkinson, R. S., and Sanes, J. R. (1997b). Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for duchenne muscular dystrophy. Cell 90, 729–738. doi:10.1016/s0092-8674(00)80533-4
Grady, R. M., Zhou, H., Cunningham, J. M., Henry, M. D., Campbell, K. P., and Sanes, J. R. (2000). Maturation and maintenance of the neuromuscular synapse: Genetic evidence for roles of the dystrophin-glycoprotein complex. Neuron 25, 279–293. doi:10.1016/s0896-6273(00)80894-6
Greschik, H., Duteil, D., Messaddeq, N., Willmann, D., Arrigoni, L., Sum, M., et al. (2017). The histone code reader Spin1 controls skeletal muscle development. Cell Death Dis. 8, e3173, e3173. doi:10.1038/cddis.2017.468
Griffiths, L. A., Doig, J., Churchhouse, A. M. D., Davies, F. C. J., Squires, C. E., Newbery, H. J., et al. (2012). Haploinsufficiency for translation elongation factor eEF1A2 in aged mouse muscle and neurons is compatible with normal function. PLoS One 7, e41917. doi:10.1371/journal.pone.0041917
Guasch, R. M., Scambler, P., Jones, G. E., and Ridley, A. J. (1998). RhoE regulates actin cytoskeleton organization and cell migration. Mol. Cell Biol. 18, 4761–4771. doi:10.1128/MCB.18.8.4761
Hall, E. A., Nahorski, M. S., Murray, L. M., Shaheen, R., Perkins, E., Dissanayake, K. N., et al. (2017). PLAA mutations cause a lethal infantile epileptic encephalopathy by disrupting ubiquitin-mediated endolysosomal degradation of synaptic proteins. Am. J. Hum. Genet. 100, 706–724. doi:10.1016/j.ajhg.2017.03.008
Hanada, T., Weitzer, S., Mair, B., Bernreuther, C., Wainger, B. J., Ichida, J., et al. (2013). CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature 495, 474–480. doi:10.1038/nature11923
Hanson, M. G., Fregoso, V. L., Vrana, J. D., Tucker, C. L., and Niswander, L. A. (2014). Peripheral nervous system defects in a mouse model for peroxisomal biogenesis disorders. Dev. Biol. 395, 84–95. doi:10.1016/j.ydbio.2014.08.026
Helmbacher, F. (2018). Tissue-specific activities of the Fat1 cadherin cooperate to control neuromuscular morphogenesis. PLoS Biol. 16, e2004734. doi:10.1371/journal.pbio.2004734
Hideyama, T., Yamashita, T., Suzuki, T., Tsuji, S., Higuchi, M., Seeburg, P. H., et al. (2010). Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J. Neurosci. 30, 11917–11925. doi:10.1523/JNEUROSCI.2021-10.2010
Horstick, E. J., Linsley, J. W., Dowling, J. J., Hauser, M. A., McDonald, K. K., Ashley-Koch, A., et al. (2013). Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat. Commun. 4, 1952. doi:10.1038/ncomms2952
Hosseinibarkooie, S., Peters, M., Torres-Benito, L., Rastetter, R. H., Hupperich, K., Hoffmann, A., et al. (2016). The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am. J. Hum. Genet. 99, 647–665. doi:10.1016/j.ajhg.2016.07.014
Huang, B.-T., Chang, P.-Y., Su, C.-H., Chao, C. C.-K., and Lin-Chao, S. (2012). Gas7-deficient mouse reveals roles in motor function and muscle fiber composition during aging. PLoS One 7, e37702. doi:10.1371/journal.pone.0037702
Ibraghimov-Beskrovnaya, O., Ervasti, J. M., Leveille, C. J., Slaughter, C. A., Sernett, S. W., and Campbell, K. P. (1992). Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355, 696–702. doi:10.1038/355696a0
Jaworski, A., Smith, C. L., and Burden, S. J. (2007). GA-binding protein is dispensable for neuromuscular synapse formation and synapse-specific gene expression. Mol. Cell Biol. 27, 5040–5046. doi:10.1128/MCB.02228-06
Jones, R. A., Harrison, C., Eaton, S. L., Llavero Hurtado, M., Graham, L. C., Alkhammash, L., et al. (2017). Cellular and molecular anatomy of the human neuromuscular junction. Cell Rep. 21, 2348–2356. doi:10.1016/j.celrep.2017.11.008
Jones, R. A., Reich, C. D., Dissanayake, K. N., Kristmundsdottir, F., Findlater, G. S., Ribchester, R. R., et al. (2016). NMJ-morph reveals principal components of synaptic morphology influencing structure-function relationships at the neuromuscular junction. Open Biol. 6, 160240. doi:10.1098/rsob.160240
Kaeser, P. S., Deng, L., Wang, Y., Dulubova, I., Liu, X., Rizo, J., et al. (2011). RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144, 282–295. doi:10.1016/j.cell.2010.12.029
Kaether, C., Scheuermann, J., Fassler, M., Zilow, S., Shirotani, K., Valkova, C., et al. (2007). Endoplasmic reticulum retention of the gamma-secretase complex component Pen2 by Rer1. EMBO Rep. 8, 743–748. doi:10.1038/sj.embor.7401027
Kaja, S., Todorov, B., van de Ven, R. C. G., Ferrari, M. D., Frants, R. R., van den Maagdenberg, A. M. J. M., et al. (2007). Redundancy of Cav2.1 channel accessory subunits in transmitter release at the mouse neuromuscular junction. Brain Res. 1143, 92–101. doi:10.1016/j.brainres.2007.01.063
Kang, H., Tian, L., Mikesh, M., Lichtman, J. W., and Thompson, W. J. (2014). Terminal Schwann cells participate in neuromuscular synapse remodeling during reinnervation following nerve injury. J. Neurosci. 34, 6323–6333. doi:10.1523/JNEUROSCI.4673-13.2014
Khalyfa, A., Bourbeau, D., Chen, E., Petroulakis, E., Pan, J., Xu, S., et al. (2001). Characterization of elongation factor-1A (eEF1A-1) and eEF1A-2/S1 protein expression in normal and wasted mice. J. Biol. Chem. 276, 22915–22922. doi:10.1074/jbc.M101011200
Kim, M. J., Whitehead, N. P., Bible, K. L., Adams, M. E., and Froehner, S. C. (2019). Mice lacking α-β1- and β2-syntrophins exhibit diminished function and reduced dystrophin expression in both cardiac and skeletal muscle. Hum. Mol. Genet. 28, 386–395. doi:10.1093/hmg/ddy341
Kim, N., Stiegler, A. L., Cameron, T. O., Hallock, P. T., Gomez, A. M., Huang, J. H., et al. (2008). Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 135, 334–342. doi:10.1016/j.cell.2008.10.002
Kirjavainen, A., Singh, P., Lahti, L., Seja, P., Lelkes, Z., Makkonen, A., et al. (2022). Gata2, Nkx2-2 and Skor2 form a transcription factor network regulating development of a midbrain GABAergic neuron subtype with characteristics of REM-sleep regulatory neurons. Development 149, dev200937. doi:10.1242/dev.200937
Kiyonaka, S., Wakamori, M., Miki, T., Uriu, Y., Nonaka, M., Bito, H., et al. (2007). RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat. Neurosci. 10, 691–701. doi:10.1038/nn1904
Klockner, I., Schutt, C., Gerhardt, T., Boettger, T., and Braun, T. (2022). Control of CRK-RAC1 activity by the miR-1/206/133 miRNA family is essential for neuromuscular junction function. Nat. Commun. 13, 3180. doi:10.1038/s41467-022-30778-7
Kuhne, W. (1863). Die muskelspindeln. Arch. fur Pathol. Anat. Physiol. fur Klin. Med. 28, 528e538. doi:10.1007/BF01942820
Lanuza, M. A., Besalduch, N., González, C., Santafé, M. M., Garcia, N., Tomàs, M., et al. (2010). Decreased phosphorylation of δ and ε subunits of the acetylcholine receptor coincides with delayed postsynaptic maturation in PKC θ deficient mouse. Exp. Neurol. 225, 183–195. doi:10.1016/j.expneurol.2010.06.014
Lee, K.-Y., Li, M., Manchanda, M., Batra, R., Charizanis, K., Mohan, A., et al. (2013). Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 5, 1887–1900. doi:10.1002/emmm.201303275
Lenell, C., and Johnson, A. M. (2017). Sexual dimorphism in laryngeal muscle fibers and ultrasonic vocalizations in the adult rat. Laryngoscope 127, E270–E276. doi:10.1002/lary.26561
Letts, V. A., Felix, R., Biddlecome, G. H., Arikkath, J., Mahaffey, C. L., Valenzuela, A., et al. (1998). The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nat. Genet. 19, 340–347. doi:10.1038/1228
Leu, M., Bellmunt, E., Schwander, M., Fariñas, I., Brenner, H. R., and Müller, U. (2003). Erbb2 regulates neuromuscular synapse formation and is essential for muscle spindle development. Development 130, 2291–2301. doi:10.1242/dev.00447
Lewcock, J. W., Genoud, N., Lettieri, K., and Pfaff, S. L. (2007). The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56, 604–620. doi:10.1016/j.neuron.2007.09.009
Li, H., Wang, Z., Wang, B., Guo, Q., Dolios, G., Tabuchi, K., et al. (2010). Genetic dissection of the amyloid precursor protein in developmental function and amyloid pathogenesis. J. Biol. Chem. 285, 30598–30605. doi:10.1074/jbc.M110.137729
Li, M.-X., Jia, M., Yang, L.-X., Jiang, H., Lanuza, M. A., Gonzalez, C. M., et al. (2004). The role of the theta isoform of protein kinase C (PKC) in activity-dependent synapse elimination: Evidence from the PKC theta knock-out mouse in vivo and in vitro. J. Neurosci. 24, 3762–3769. doi:10.1523/JNEUROSCI.3930-03.2004
Lin, S., Landmann, L., Ruegg, M. A., and Brenner, H. R. (2008). The role of nerve-versus muscle-derived factors in mammalian neuromuscular junction formation. J. Neurosci. 28, 3333–3340. doi:10.1523/JNEUROSCI.5590-07.2008
Lin, W., Burgess, R. W., Dominguez, B., Pfaff, S. L., Sanes, J. R., and Lee, K. F. (2001). Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 410, 1057–1064. doi:10.1038/35074025
Lin, W., Dominguez, B., Yang, J., Aryal, P., Brandon, E. P., Gage, F. H., et al. (2005). Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron 46, 569–579. doi:10.1016/j.neuron.2005.04.002
Lin, W., Sanchez, H. B., Deerinck, T., Morris, J. K., Ellisman, M., and Lee, K. F. (2000). Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 97, 1299–1304. doi:10.1073/pnas.97.3.1299
Lindqvist, J., Torvaldson, E., Gullmets, J., Karvonen, H., Nagy, A., Taimen, P., et al. (2017). Nestin contributes to skeletal muscle homeostasis and regeneration. J. Cell Sci. 130, 2833–2842. doi:10.1242/jcs.202226
Liu, K., Jones, S., Minis, A., Rodriguez, J., Molina, H., and Steller, H. (2019). PI31 is an adaptor protein for proteasome transport in axons and required for synaptic development. Dev. Cell 50, 509–524.e10. doi:10.1016/j.devcel.2019.06.009
Liu, N., Williams, A. H., Maxeiner, J. M., Bezprozvannaya, S., Shelton, J. M., Richardson, J. A., et al. (2012). microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J. Clin. Invest. 122, 2054–2065. doi:10.1172/JCI62656
Liu Y, Y., Sugiura, Y., Chen, F., Lee, K. F., Ye, Q., and Lin, W. (2019). Blocking skeletal muscle DHPRs/Ryr1 prevents neuromuscular synapse loss in mutant mice deficient in type III Neuregulin 1 (CRD-Nrg1). PLoS Genet. 15, e1007857. doi:10.1371/journal.pgen.1007857
Liu, Y., Padgett, D., Takahashi, M., Li, H., Sayeed, A., Teichert, R. W., et al. (2008). Essential roles of the acetylcholine receptor ɣ-subunit in neuromuscular synaptic patterning. Development 135, 1957–1967. doi:10.1242/dev.018119
Lnenicka, G. A. (2020). Crayfish and Drosophila NMJs. Neurosci. Lett. 732, 135110. doi:10.1016/j.neulet.2020.135110
Lüningschrör, P., Werner, G., Stroobants, S., Kakuta, S., Dombert, B., Sinske, D., et al. (2020). The FTLD risk factor TMEM106B regulates the transport of lysosomes at the axon initial segment of motoneurons. Cell Rep. 30, 3506–3519.e6. doi:10.1016/j.celrep.2020.02.060
Mabbitt, P. D., Loreto, A., Déry, M.-A., Fletcher, A. J., Stanley, M., Pao, K.-C., et al. (2020). Structural basis for RING-Cys-Relay E3 ligase activity and its role in axon integrity. Nat. Chem. Biol. 16, 1227–1236. doi:10.1038/s41589-020-0598-6
Mandela, P., Yankova, M., Conti, L. H., Ma, X.-M., Grady, J., Eipper, B. A., et al. (2012). Kalrn plays key roles within and outside of the nervous system. BMC Neurosci. 13, 136. doi:10.1186/1471-2202-13-136
Marshall, A. G., Watson, J. A., Hallengren, J. J., Walters, B. J., Dobrunz, L. E., Francillon, L., et al. (2013). Genetic background alters the severity and onset of neuromuscular disease caused by the loss of ubiquitin-specific protease 14 (usp14). PLoS One 8, e84042. doi:10.1371/journal.pone.0084042
Martin, P. B., Kigoshi-Tansho, Y., Sher, R. B., Ravenscroft, G., Stauffer, J. E., Kumar, R., et al. (2020). NEMF mutations that impair ribosome-associated quality control are associated with neuromuscular disease. Nat. Commun. 11, 4625. doi:10.1038/s41467-020-18327-6
McGovern, V. L., Massoni-Laporte, A., Wang, X., Le, T. T., Le, H. T., Beattie, C. E., et al. (2015). Plastin 3 expression does not modify spinal muscular atrophy severity in the ∆7 SMA mouse. PLoS One 10, e0132364. doi:10.1371/journal.pone.0132364
McMahan, U. J. (1990). The agrin hypothesis. Cold Spring Harb. Symp. Quant. Biol. 55, 407–418. doi:10.1101/sqb.1990.055.01.041
Medrihan, L., Rohlmann, A., Fairless, R., Andrae, J., Döring, M., Missler, M., et al. (2009). Neurobeachin, a protein implicated in membrane protein traffic and autism, is required for the formation and functioning of central synapses. J. Physiol. 587, 5095–5106. doi:10.1113/jphysiol.2009.178236
Mejia Maza, A., Jarvis, S., Lee, W. C., Cunningham, T. J., Schiavo, G., Secrier, M., et al. (2021). NMJ-Analyser identifies subtle early changes in mouse models of neuromuscular disease. Sci. Rep. 11, 12251. doi:10.1038/s41598-021-91094-6
Messéant, J., Ezan, J., Delers, P., Glebov, K., Marchiol, C., Lager, F., et al. (2017). Wnt proteins contribute to neuromuscular junction formation through distinct signaling pathways. Development 144, 1712–1724. doi:10.1242/dev.146167
Minis, A., Rodriguez, J. A., Levin, A., Liu, K., Govek, E.-E., Hatten, M. E., et al. (2019). The proteasome regulator PI31 is required for protein homeostasis, synapse maintenance, and neuronal survival in mice. Proc. Natl. Acad. Sci. U. S. A. 116, 24639–24650. doi:10.1073/pnas.1911921116
Mocholí, E., Ballester-Lurbe, B., Arqué, G., Poch, E., Peris, B., Guerri, C., et al. (2011). RhoE deficiency produces postnatal lethality, profound motor deficits and neurodevelopmental delay in mice. PLoS One 6, e19236. doi:10.1371/journal.pone.0019236
Mohseni, P., Sung, H.-K., Murphy, A. J., Laliberte, C. L., Pallari, H.-M., Henkelman, M., et al. (2011). Nestin is not essential for development of the CNS but required for dispersion of acetylcholine receptor clusters at the area of neuromuscular junctions. J. Neurosci. 31, 11547–11552. doi:10.1523/JNEUROSCI.4396-10.2011
Morelli, K. H., Seburn, K. L., Schroeder, D. G., Spaulding, E. L., Dionne, L. A., Cox, G. A., et al. (2017). Severity of demyelinating and axonal neuropathy mouse models is modified by genes affecting structure and function of peripheral nodes. Cell Rep. 18, 3178–3191. doi:10.1016/j.celrep.2017.03.009
Musarella, M., Alcaraz, G., Caillol, G., Boudier, J.-L., Couraud, F., and Autillo-Touati, A. (2006). Expression of Nav1.6 sodium channels by Schwann cells at neuromuscular junctions: Role in the motor endplate disease phenotype. Glia 53, 13–23. doi:10.1002/glia.20252
Nakatani, T., Minaki, Y., Kumai, M., Nitta, C., and Ono, Y. (2014). The c-Ski family member and transcriptional regulator Corl2/Skor2 promotes early differentiation of cerebellar Purkinje cells. Dev. Biol. 388, 68–80. doi:10.1016/j.ydbio.2014.01.016
Nakka, K., Ghigna, C., Gabellini, D., and Dilworth, F. J. (2018). Diversification of the muscle proteome through alternative splicing. Skelet. Muscle 8, 8. doi:10.1186/s13395-018-0152-3
Nelson, B. R., Wu, F., Liu, Y., Anderson, D. M., McAnally, J., Lin, W., et al. (2013). Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc. Natl. Acad. Sci. U. S. A. 110, 11881–11886. doi:10.1073/pnas.1310571110
Newbern, J., and Birchmeier, C. (2010). Nrg1/ErbB signaling networks in Schwann cell development and myelination. Semin. Cell Dev. Biol. 21, 922–928. doi:10.1016/j.semcdb.2010.08.008
Newbery, H. J., Gillingwater, T. H., Dharmasaroja, P., Peters, J., Wharton, S. B., Thomson, D., et al. (2005). Progressive loss of motor neuron function in wasted mice: Effects of a spontaneous null mutation in the gene for the eEF1 A2 translation factor. J. Neuropathol. Exp. Neurol. 64, 295–303. doi:10.1093/jnen/64.4.295
Nguyen, S., Meletis, K., Fu, D., Jhaveri, S., and Jaenisch, R. (2007). Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev. Dyn. 236, 1663–1676. doi:10.1002/dvdy.21176
Nian, F.-S., Li, L.-L., Cheng, C.-Y., Wu, P.-C., Lin, Y.-T., Tang, C.-Y., et al. (2019). Rab18 collaborates with Rab7 to modulate lysosomal and autophagy activities in the nervous system: An overlapping mechanism for warburg micro syndrome and charcot-marie-tooth neuropathy type 2B. Mol. Neurobiol. 56, 6095–6105. doi:10.1007/s12035-019-1471-z
Noebels, J. L., Qiao, X., Bronson, R. T., Spencer, C., and Davisson, M. T. (1990). Stargazer: A new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res. 7, 129–135. doi:10.1016/0920-1211(90)90098-g
Nuytens, K., Tuand, K., Fu, Q., Stijnen, P., Pruniau, V., Meulemans, S., et al. (2014). The dwarf phenotype in GH240B mice, haploinsufficient for the autism candidate gene Neurobeachin, is caused by ectopic expression of recombinant human growth hormone. PLoS One 9, e109598. doi:10.1371/journal.pone.0109598
Okada, K., Inoue, A., Okada, M., Murata, Y., Kakuta, S., Jigami, T., et al. (2006). The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 312, 1802–1805. doi:10.1126/science.1127142
O’Leary, D. A., Noakes, P. G., Lavidis, N. A., Kola, I., Hertzog, P. J., and Ristevski, S. (2007). Targeting of the ETS factor GABPalpha disrupts neuromuscular junction synaptic function. Mol. Cell Biol. 27, 3470–3480. doi:10.1128/MCB.00659-06
Olson, E. N., Arnold, H. H., Rigby, P. W., and Wold, B. J. (1996). Know your neighbors: Three phenotypes in null mutants of the myogenic bHLH gene MRF4. Cell 85, 1–4. doi:10.1016/s0092-8674(00)81073-9
Oprea, G. E., Kröber, S., McWhorter, M. L., Rossoll, W., Müller, S., Krawczak, M., et al. (2008). Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320, 524–527. doi:10.1126/science.1155085
O’Reilly, J., Ono-Moore, K. D., Chintapalli, S. v., Rutkowsky, J. M., Tolentino, T., Lloyd, K. C. K., et al. (2021). Sex differences in skeletal muscle revealed through fiber type, capillarity, and transcriptomics profiling in mice. Physiol. Rep. 9, e15031. doi:10.14814/phy2.15031
Penzes, P., Johnson, R. C., Alam, M. R., Kambampati, V., Mains, R. E., and Eipper, B. A. (2000). An isoform of kalirin, a brain-specific GDP/GTP exchange factor, is enriched in the postsynaptic density fraction. J. Biol. Chem. 275, 6395–6403. doi:10.1074/jbc.275.9.6395
Peris, B., Gonzalez-Granero, S., Ballester-Lurbe, B., García-Verdugo, J.-M., Pérez-Roger, I., Guerri, C., et al. (2012). Neuronal polarization is impaired in mice lacking RhoE expression. J. Neurochem. 121, 903–914. doi:10.1111/j.1471-4159.2012.07733.x
Pfister, J., and Zenker, W. (1984). The splenius capitis muscle of the rat, architecture and histochemistry, afferent and efferent innervation as compared with that of the quadriceps muscle. Anat. Embryol. Berl. 169, 79–89. doi:10.1007/BF00300589
Pietri, T., Eder, O., Breau, M. A., Topilko, P., Blanche, M., Brakebusch, C., et al. (2004). Conditional beta1-integrin gene deletion in neural crest cells causes severe developmental alterations of the peripheral nervous system. Development 131, 3871–3883. doi:10.1242/dev.01264
Plachta, N., Annaheim, C., Bissière, S., Lin, S., Rüegg, M., Hoving, S., et al. (2007). Identification of a lectin causing the degeneration of neuronal processes using engineered embryonic stem cells. Nat. Neurosci. 10, 712–719. doi:10.1038/nn1897
Plomp, J. J., Huijbers, M. G. M., and Verschuuren, J. J. G. M. (2017). Neuromuscular synapse electrophysiology in myasthenia gravis animal models. Ann. N. Y. Acad. Sci. 1412, 146–153. doi:10.1111/nyas.13507
Polster, A., Nelson, B. R., Olson, E. N., and Beam, K. G. (2016). Stac3 has a direct role in skeletal muscle-type excitation-contraction coupling that is disrupted by a myopathy-causing mutation. Proc. Natl. Acad. Sci. U. S. A. 113, 10986–10991. doi:10.1073/pnas.1612441113
Quintá, H. R., Pasquini, J. M., Rabinovich, G. A., and Pasquini, L. A. (2014). Glycan-dependent binding of galectin-1 to neuropilin-1 promotes axonal regeneration after spinal cord injury. Cell Death Differ. 21, 941–955. doi:10.1038/cdd.2014.14
Reichenstein, I., Eitan, C., Diaz-Garcia, S., Haim, G., Magen, I., Siany, A., et al. (2019). Human genetics and neuropathology suggest a link between miR-218 and amyotrophic lateral sclerosis pathophysiology. Sci. Transl. Med. 11, eaav5264. doi:10.1126/scitranslmed.aav5264
Reinholt, B. M., Ge, X., Cong, X., Gerrard, D. E., and Jiang, H. (2013). Stac3 is a novel regulator of skeletal muscle development in mice. PLoS One 8, e62760. doi:10.1371/journal.pone.0062760
Richard, S., Torabi, N., Franco, G. V., Tremblay, G. A., Chen, T., Vogel, G., et al. (2005). Ablation of the Sam68 RNA binding protein protects mice from age-related bone loss. PLoS Genet. 1, e74. doi:10.1371/journal.pgen.0010074
Rieker, C., Dev, K. K., Lehnhoff, K., Barbieri, S., Ksiazek, I., Kauffmann, S., et al. (2011). Neuropathology in mice expressing mouse alpha-synuclein. PLoS One 6, e24834. doi:10.1371/journal.pone.0024834
Rodríguez Cruz, P. M., Cossins, J., Beeson, D., and Vincent, A. (2020). The neuromuscular junction in health and disease: Molecular mechanisms governing synaptic formation and homeostasis. Front. Mol. Neurosci. 13, 610964. doi:10.3389/fnmol.2020.610964
Rozas, J. L., Gómez-Sánchez, L., Mircheski, J., Linares-Clemente, P., Nieto-González, J. L., Vázquez, M. E., et al. (2012). Motorneurons require cysteine string protein-α to maintain the readily releasable vesicular pool and synaptic vesicle recycling. Neuron 74, 151–165. doi:10.1016/j.neuron.2012.02.019
Ruggiu, M., Herbst, R., Kim, N., Jevsek, M., Fak, J. J., Mann, M. A., et al. (2008). Rescuing Z+ agrin splicing in Nova null mice restores synapse formation and unmasks a physiologic defect in motor neuron firing. Proc. Natl. Acad. Sci. U. S. A. 106, 3513–3518. doi:10.1073/pnas.0813112106
Ruiz, R., Casañas, J. J., Südhof, T. C., and Tabares, L. (2008). Cysteine string protein-alpha is essential for the high calcium sensitivity of exocytosis in a vertebrate synapse. Eur. J. Neurosci. 27, 3118–3131. doi:10.1111/j.1460-9568.2008.06301.x
Saiga, T., Fukuda, T., Matsumoto, M., Tada, H., Okano, H. J., Okano, H., et al. (2009). Fbxo45 forms a novel ubiquitin ligase complex and is required for neuronal development. Mol. Cell Biol. 29, 3529–3543. doi:10.1128/MCB.00364-09
Sakisaka, T., Yamamoto, Y., Mochida, S., Nakamura, M., Nishikawa, K., Ishizaki, H., et al. (2008). Dual inhibition of SNARE complex formation by tomosyn ensures controlled neurotransmitter release. J. Cell Biol. 183, 323–337. doi:10.1083/jcb.200805150
Sandrock, A. W., Dryer, S. E., Rosen, K. M., Gozani, S. N., Kramer, R., Theill, L. E., et al. (1997). Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science 276, 599–603. doi:10.1126/science.276.5312.599
Santosa, K. B., Keane, A. M., Jablonka-Shariff, A., Vannucci, B., and Snyder-Warwick, A. K. (2018). Clinical relevance of terminal Schwann cells: An overlooked component of the neuromuscular junction. J. Neurosci. Res. 96, 1125–1135. doi:10.1002/jnr.24231
Schmidt, M., Schülke, J.-P., Liebl, C., Stiess, M., Avrabos, C., Bock, J., et al. (2011). Tumor suppressor down-regulated in renal cell carcinoma 1 (DRR1) is a stress-induced actin bundling factor that modulates synaptic efficacy and cognition. Proc. Natl. Acad. Sci. U. S. A. 108, 17213–17218. doi:10.1073/pnas.1103318108
Schmidt, N., Akaaboune, M., Gajendran, N., Martinez-Pena y Valenzuela, I., Wakefield, S., Thurnheer, R., et al. (2011). Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of α-dystrobrevin. J. Cell Biol. 195, 1171–1184. doi:10.1083/jcb.201107083
Schoch, S., Castillo, P. E., Jo, T., Mukherjee, K., Geppert, M., Wang, Y., et al. (2002). RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415, 321–326. doi:10.1038/415321a
Schoch, S., Mittelstaedt, T., Kaeser, P. S., Padgett, D., Feldmann, N., Chevaleyre, V., et al. (2006). Redundant functions of RIM1alpha and RIM2alpha in Ca(2+)-triggered neurotransmitter release. EMBO J. 25, 5852–5863. doi:10.1038/sj.emboj.7601425
Schwander, M., Shirasaki, R., Pfaff, S. L., and Müller, U. (2004). Beta1 integrins in muscle, but not in motor neurons, are required for skeletal muscle innervation. J. Neurosci. 24, 8181–8191. doi:10.1523/JNEUROSCI.1345-04.2004
Sealock, R., Butler, M. H., Kramarcy, N. R., Gao, K. X., Murnane, A. A., Douville, K., et al. (1991). Localization of dystrophin relative to acetylcholine receptor domains in electric tissue and adult and cultured skeletal muscle. J. Cell Biol. 113, 1133–1144. doi:10.1083/jcb.113.5.1133
Seburn, K. L., Nangle, L. A., Cox, G. A., Schimmel, P., and Burgess, R. W. (2006). An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron 51, 715–726. doi:10.1016/j.neuron.2006.08.027
Sharma, M., Burré, J., Bronk, P., Zhang, Y., Xu, W., and Südhof, T. C. (2012). CSPα knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 31, 829–841. doi:10.1038/emboj.2011.467
She, B.-R., Liou, G.-G., and Lin-Chao, S. (2002). Association of the growth-arrest-specific protein Gas7 with F-actin induces reorganization of microfilaments and promotes membrane outgrowth. Exp. Cell Res. 273, 34–44. doi:10.1006/excr.2001.5435
Shen, C., Li, L., Zhao, K., Bai, L., Wang, A., Shu, X., et al. (2018). Motoneuron Wnts regulate neuromuscular junction development. eLife 7, e34625. doi:10.7554/eLife.34625
Shin, J., Salameh, J. S., and Richter, J. D. (2016). Impaired neurodevelopment by the low complexity domain of CPEB4 reveals a convergent pathway with neurodegeneration. Sci. Rep. 6, 29395. doi:10.1038/srep29395
Shultz, L. D., Sweet, H. O., Davisson, M. T., and Coman, D. R. (1982). Wasted, a new mutant of the mouse with abnormalities characteristic to ataxia telangiectasia. Nature 297, 402–404. doi:10.1038/297402a0
Sieck, G. C., and Prakash, Y. S. (1997). Morphological adaptations of neuromuscular junctions depend on fiber type. Can. J. Appl. Physiol. 22, 197–230. doi:10.1139/h97-014
Snell, G. D. (1955). Ducky, a new second chromosome mutation in the mouse. J. Hered. 46, 27–29. doi:10.1093/oxfordjournals.jhered.a106505
Srinivasan, E., Chandrasekhar, G., Chandrasekar, P., Anbarasu, K., Vickram, A. S., Karunakaran, R., et al. (2021). Alpha-synuclein aggregation in Parkinson’s disease. Front. Med. (Lausanne) 8, 736978. doi:10.3389/fmed.2021.736978
Staron, R. S., Hagerman, F. C., Hikida, R. S., Murray, T. F., Hostler, D. P., Crill, M. T., et al. (2000). Fiber type composition of the vastus lateralis muscle of young men and women. J. Histochem Cytochem 48, 623–629. doi:10.1177/002215540004800506
Su, Y., Balice-Gordon, R. J., Hess, D. M., Landsman, D. S., Minarcik, J., Golden, J., et al. (2004). Neurobeachin is essential for neuromuscular synaptic transmission. J. Neurosci. 24, 3627–3636. doi:10.1523/JNEUROSCI.4644-03.2004
Sun, Y., Ren, W., Côté, J.-F., Hinds, P. W., Hu, X., and Du, K. (2015). ClipR-59 interacts with Elmo2 and modulates myoblast fusion. J. Biol. Chem. 290, 6130–6140. doi:10.1074/jbc.M114.616680
Taetzsch, T., Tenga, M. J., and Valdez, G. (2017). Muscle fibers secrete FGFBP1 to slow degeneration of neuromuscular synapses during aging and progression of ALS. J. Neurosci. 37, 70–82. doi:10.1523/JNEUROSCI.2992-16.2016
Tanaka, M., Miyoshi, J., Ishizaki, H., Togawa, A., Ohnishi, K., Endo, K., et al. (2001). Role of Rab3 GDP/GTP exchange protein in synaptic vesicle trafficking at the mouse neuromuscular junction. Mol. Biol. Cell 12, 1421–1430. doi:10.1091/mbc.12.5.1421
Thaler, J., Harrison, K., Sharma, K., Lettieri, K., Kehrl, J., and Pfaff, S. L. (1999). Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron 23, 675–687. doi:10.1016/s0896-6273(01)80027-1
Vaden, J. H., Bhattacharyya, B. J., Chen, P.-C., Watson, J. A., Marshall, A. G., Phillips, S. E., et al. (2015). Ubiquitin-specific protease 14 regulates c-Jun N-terminal kinase signaling at the neuromuscular junction. Mol. Neurodegener. 10, 3. doi:10.1186/1750-1326-10-3
Valdez, G., Tapia, J. C., Kang, H., Clemenson, G. D., Gage, F. H., Lichtman, J. W., et al. (2010). Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. U. S. A. 107, 14863–14868. doi:10.1073/pnas.1002220107
Valkova, C., Albrizio, M., Röder, I., Schwake, M., Betto, R., Rudolf, R., et al. (2011). Sorting receptor Rer1 controls surface expression of muscle acetylcholine receptors by ER retention of unassembled alpha-subunits. Proc. Natl. Acad. Sci. U. S. A. 108, 621–625. doi:10.1073/pnas.1001624108
van der Putten, H., Wiederhold, K. H., Probst, A., Barbieri, S., Mistl, C., Danner, S., et al. (2000). Neuropathology in mice expressing human alpha-synuclein. J. Neurosci. 20, 6021–6029. doi:10.1523/JNEUROSCI.20-16-06021.2000
VanSaun, M., Herrera, A. A., and Werle, M. J. (2003). Structural alterations at the neuromuscular junctions of matrix metalloproteinase 3 null mutant mice. J. Neurocytol. 32, 1129–1142. doi:10.1023/B:NEUR.0000021907.68461.9c
VanSaun, M., and Werle, M. J. (2000). Matrix metalloproteinase-3 removes agrin from synaptic basal lamina. J. Neurobiol. 43, 369–9. doi:10.1002/1097-4695(20000905)44:3<369:aid-neu7>3.0.co;2-z
Wang, B., Harrison, W., Overbeek, P. A., and Zheng, H. (2011). Transposon mutagenesis with coat color genotyping identifies an essential role for Skor2 in sonic hedgehog signaling and cerebellum development. Development 138, 4487–4497. doi:10.1242/dev.067264
Wang, B., Yang, L., Wang, Z., and Zheng, H. (2007). Amyolid precursor protein mediates presynaptic localization and activity of the high-affinity choline transporter. Proc. Natl. Acad. Sci. U. S. A. 104, 14140–14145. doi:10.1073/pnas.0704070104
Wang, P., Yang, G., Mosier, D. R., Chang, P., Zaidi, T., Gong, Y.-D., et al. (2005). Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. J. Neurosci. 25, 1219–1225. doi:10.1523/JNEUROSCI.4660-04.2005
Wang, Z., Wang, B., Yang, L., Guo, Q., Aithmitti, N., Songyang, Z., et al. (2009). Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J. Neurosci. 29, 10788–10801. doi:10.1523/JNEUROSCI.2132-09.2009
Weatherbee, S. D., Anderson, K. V., and Niswander, L. A. (2006). LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development 133, 4993–5000. doi:10.1242/dev.02696
Willadt, S., Nash, M., and Slater, C. R. (2016). Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci. Rep. 6, 24849. doi:10.1038/srep24849
Williams, A. H., Valdez, G., Moresi, V., Qi, X., McAnally, J., Elliott, J. L., et al. (2009). MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 326, 1549–1554. doi:10.1126/science.1181046
Wilson, S. M., Bhattacharyya, B., Rachel, R. A., Coppola, V., Tessarollo, L., Householder, D. B., et al. (2002). Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat. Genet. 32, 420–425. doi:10.1038/ng1006
Wojtowicz, J. M., Marin, L., and Atwood, H. L. (1994). Activity-induced changes in synaptic release sites at the crayfish neuromuscular junction. J. Neurosci. 14, 3688–3703. doi:10.1523/JNEUROSCI.14-06-03688.1994
Wood, S. J., and Slater, C. R. (2001). Safety factor at the neuromuscular junction. Prog. Neurobiol. 64, 393–429. doi:10.1016/s0301-0082(00)00055-1
Yang, J., Dominguez, B., de Winter, F., Gould, T. W., Eriksson, J. E., and Lee, K.-F. (2011). Nestin negatively regulates postsynaptic differentiation of the neuromuscular synapse. Nat. Neurosci. 14, 324–330. doi:10.1038/nn.2747
Yang, X., Arber, S., William, C., Li, L., Tanabe, Y., Jessell, T. M., et al. (2001). Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 30, 399–410. doi:10.1016/s0896-6273(01)00287-2
Zahavi, E. E., Ionescu, A., Gluska, S., Gradus, T., Ben-Yaakov, K., and Perlson, E. (2015). A compartmentalized microfluidic neuromuscular co-culture system reveals spatial aspects of GDNF functions. J. Cell Sci. 128, 1241–1252. doi:10.1242/jcs.167544
Zhang, B., Luo, S., Wang, Q., Suzuki, T., Xiong, W. C., and Mei, L. (2008). LRP4 serves as a coreceptor of agrin. Neuron 60, 285–297. doi:10.1016/j.neuron.2008.10.006
Zhang, Y.-Q., Henderson, M. X., Colangelo, C. M., Ginsberg, S. D., Bruce, C., Wu, T., et al. (2012). Identification of CSPα clients reveals a role in dynamin 1 regulation. Neuron 74, 136–150. doi:10.1016/j.neuron.2012.01.029
Keywords: neuromuscular junction, synaptic transmission, human disease, mouse models, comparative analysis
Citation: Navarro-Martínez A, Vicente-García C and Carvajal JJ (2023) NMJ-related diseases beyond the congenital myasthenic syndromes. Front. Cell Dev. Biol. 11:1216726. doi: 10.3389/fcell.2023.1216726
Received: 04 May 2023; Accepted: 21 July 2023;
Published: 04 August 2023.
Edited by:
Kathrin Meyer, Nationwide Children’s Hospital, United StatesReviewed by:
Matilde Cescon, University of Padua, ItalyCopyright © 2023 Navarro-Martínez, Vicente-García and Carvajal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Vicente-García, Y3ZpY2dhckB1cG8uZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.