
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cell Dev. Biol., 24 May 2023
Sec. Cellular Biochemistry
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1205112
This article is part of the Research TopicLatest Developments on Transcription and Translation Regulation (Translation UK 2022)View all 7 articles
 Kleanthi Chalkiadaki1,2Elpida Statoulla1Maria Zafeiri1Nabila Haji3
Kleanthi Chalkiadaki1,2Elpida Statoulla1Maria Zafeiri1Nabila Haji3 Jean-Claude Lacaille3Craig M. Powell4
Jean-Claude Lacaille3Craig M. Powell4 Seyed Mehdi Jafarnejad5*
Seyed Mehdi Jafarnejad5* Arkady Khoutorsky6*
Arkady Khoutorsky6* Christos G. Gkogkas1*
Christos G. Gkogkas1*Tuberous sclerosis complex (TSC) is a rare monogenic disorder co-diagnosed with high rates of autism and is caused by loss of function mutations in the TSC1 or TSC2 genes. A key pathway hyperactivated in TSC is the mammalian/mechanistic target of rapamycin complex 1 (mTORC1), which regulates cap-dependent mRNA translation. We previously demonstrated that exaggerated cap-dependent translation leads to autism-related phenotypes and increased mRNA translation and protein expression of Neuroligin 1 (Nlgn1) in mice. Inhibition of Nlgn1 expression reversed social behavior deficits in mice with increased cap-dependent translation. Herein, we report elevated translation of Nlgn1 mRNA and an increase in its protein expression. Genetic or pharmacological inhibition of Nlgn1 expression in Tsc2+/− mice rescued impaired hippocampal mGluR-LTD, contextual discrimination and social behavior deficits in Tsc2+/− mice, without correcting mTORC1 hyperactivation. Thus, we demonstrate that reduction of Nlgn1 expression in Tsc2+/− mice is a new therapeutic strategy for TSC and potentially other neurodevelopmental disorders.
Loss of function mutations in the genes encoding tuberous sclerosis proteins TSC1 (hamartin) and TSC2 (tuberin) lead to the tuberous sclerosis complex (TSC), a rare autosomal dominant genetic disorder, with very high penetrance (Touraine et al., 2022). TSC manifests with multi-organ pathologies such as benign-tumor growth, skin abnormalities, lung lesions, and kidney problems. Central to TSC pathology are pervasive neurological symptoms such as seizures, cognitive difficulties, changes in behavior such as aggressive behavior, self-harming, attention deficit hyperactivity disorder and autism spectrum disorder (ASD) in ∼50% of children diagnosed with TSC.
ASD is a polygenic condition (Vorstman et al., 2017), affecting distinct domains of behavior and is accompanied by altered brain connectivity and excitation/inhibition dysregulation, which may engender a plethora of comorbid conditions such as epilepsy, sleep disturbance, anxiety, depression and peripheral changes in feeding and gastrointestinal function (Doernberg and Hollander, 2016). Several monogenic disorders, including TSC, are caused by mutations in genes (TSC1/TSC2, PTEN; phosphatase and tensin homolog, NF1; neurofibromin 1, FMR1; fragile X messenger ribonucleoprotein 1), which encode proteins that participate in signaling cascades converging to the mammalian/mechanistic target of rapamycin complex 1 (mTORC1) pathway (Kelleher and Bear, 2008), providing a possible unifying mechanistic explanation for several forms of syndromic and/or idiopathic ASD. TSC1/2 inhibit the small GTPase Rheb (RAS homolog-mTORC1 binding), which activates mTORC1 (Garami et al., 2003). Thus, loss of function of TSC1/2 engenders mTORC1 hyperactivation.
mTORC1 integrates extrinsic and intrinsic cues to elicit multilevel cellular responses ranging from cell proliferation and modulation of gene expression to autophagy and apoptosis, primarily by recruiting multiprotein signaling cascades (Liu and Sabatini, 2020). The best ascribed function of mTORC1 signaling is regulation of mRNA translation via phosphorylation of two major downstream effectors: 4E-BPs (eukaryotic initiation factor 4E-binding proteins 1, 2 and 3) and S6Ks (ribosomal protein S6 kinases 1 and 2), which regulate cap-dependent initiation of translation (Mamane et al., 2006). Hypo-phosphorylated 4E-BPs bind to the cap-binding protein, the eukaryotic translation initiation factor 4E (eIF4E), to preclude eIF4F complex formation, an early step in translation initiation (Sonenberg, 2008). Upon phosphorylation of 4E-BPs by mTORC1, eIF4F complex formation and cap-dependent translation are stimulated. Cap-dependent translation preferentially promotes the synthesis of a subset of mRNAs termed “eIF4E-senstitive” (Hinnebusch et al., 2016).
We previously showed that deletion of the predominant mouse brain isoform 4E-BP2, mimicking hyperactivation of the mTOR/4E-BP/eIF4E axis, engenders behaviors reminiscent of ASD (social behavior and repetitive/stereotypic behaviors), accompanied by hippocampal excitation/inhibition imbalance (Gkogkas et al., 2013). This phenotype was explained by preferential exaggerated cap-dependent translation of synaptic mRNAs such as neuroligin 1 (Nlgn1), by eIF4E, in the absence of 4E-BP2. Overexpression of eIF4E induces similar behavioral and molecular/cellular phenotypes in mice (Santini et al., 2013). Similarly, mTOR/4E-BP hyperactivation in Tsc2+/− mice is accompanied by a wide range of rapamycin-sensitive phenotypes: decreased hippocampal metabotropic glutamate receptor long-term depression (mGluR-LTD) (Auerbach et al., 2011), deficits in learning and memory (Ehninger et al., 2008) and autism-related behaviors such as changes in social interaction (Sato et al., 2012).
Nlgns are cell-adhesion molecules that regulate synaptic structure function, with known mutations linked to the pathogenesis of autism (Trobiani et al., 2020). Interestingly Nlgn1 knockout (KO) mice display modest changes in social behavior, but a profound increase in repetitive, stereotyped grooming and deficits in spatial learning and memory (Blundell et al., 2010). On the other hand, overexpression of Nlgn1 leads to learning and synaptic plasticity deficits by altering the E/I balance in the hippocampus (Dahlhaus et al., 2010).
Herein, we show that translation of Nlgn1 mRNA is significantly increased in the hippocampus of Tsc2+/− mice, leading to increased Nlgn1 protein expression. Pharmacological inhibition of mTORC1 or of cap-dependent translation restored translation of Nlgn1 mRNA in Tsc2+/− mice. Genetic deletion of one copy of Nlgn1 was sufficient to reverse the impairment of mGluR-LTD in Tsc2+/− mice. Furthermore, normalizing Nlgn1 expression in Tsc2+/− mice via genetic deletion of one Nlgn1 gene copy or via treatment with the cap-dependent translation inhibitor 4EGI-1, reversed hippocampal contextual memory impairment and social behavior deficits, without restoring mTORC1 signaling. Altogether, we reveal rescue of autism-related phenotypes in a mouse model of TSC by targeting Nlgn1, a key downstream effector molecule regulated by the mTORC1/4E-BP axis.
All procedures followed the UK Home Office and Canadian Council on Animal Care guidelines and were respectively approved by the University of Edinburgh, McGill University and Université de Montréal. Tsc2+/− mice (B6; 129S4-Tsc2tm1Djk/J, Jackson Laboratories) and Nlgn1+/− mice (Prof. C. Powell, UT Southwestern) were maintained on the C57Bl/6J background (backcrossed for more than 10 generations). For all experiments, littermates from heterozygote crossings were used both for Tsc2 and Nlgn1 models. Male and female mice were used where specified. Food and water were provided ad libitum, and mice were kept on a 12 h light/dark cycle. Pups were kept with their dams until weaning at postnatal day 21. After weaning, mice were group housed (maximum of four per cage) by sex. Cages were maintained in ventilated racks in temperature (20–21°C) and humidity (∼55%) controlled rooms, on a 12-h circadian cycle (7a.m.-7p.m. light period). For all behavioral assays, mice were handled/habituated for three consecutive days prior to experimental testing.
Polysome profile analysis was carried out as described previously (Gkogkas et al., 2013). Intact hippocampi from 2-month-old male mice were washed with ice-cold PBS containing 100 μg/mL cycloheximide, lysed in a hypotonic lysis buffer (5 mM Tris-HCl pH 7.5, 2.5 mM MgCl2, 1.5 mM KCl, 100 μg/mL cycloheximide, 2 mM DTT, 0.5% Triton X-100, and 0.5% sodium deoxycholate) and analyzed using polysome profiling. The Polysome to monosome ratio was calculated as the area under the A254 absorbance curve, using the function describing the absorbance values, processed with the definite integral command in MATLAB. Statistical analysis was carried out with a Student’s t-test.
Polysomal or total RNA was analyzed using a Biorad iQ SYBR Green Supermix kit as previously described (Gkogkas et al., 2013). Light polysomes correspond to fractions 11–13 (including monosomes, disomes, trisomes), while heavy polysomes (>3 ribosomes) correspond to fractions 14–18. For all experiments n = 4; results are presented in arbitrary units as relative amounts using serial dilutions of cortical or hippocampal RNA as RT-qPCR concentration standards. Statistical analysis was carried out with a Student’s t-test.
All tissues were dissociated in RIPA buffer (unless otherwise specified). Western blotting was previously described (Gkogkas et al., 2013). Antibodies against indicated proteins are summarized in Supplementary Table S1. Quantification of immunoblots was performed using ImageJ (NIH). Values were normalized to β-tubulin, HSC70, or another control (e.g., for phosphorylation) where specified and presented as percentage or fold change relative to control. Statistical analysis was carried out using a Student’s t-test or One-way ANOVA with a Bonferroni’s post-hoc test, where specified.
2-month-old mice were perfused intracardiacally with 4% paraformaldehyde. The brains were postfixed in PFA 4% for 24 h following extraction. The fixed brains were sectioned using a vibratome to acquire 40 µm-thick sections. Sections were permeabilized in 0.3% Triton X-100 in PBS for 30 min, blocked in 5% goat serum for 1 h, incubated with primary antibodies [1:129211, neuroligin (Synaptic Systems)] at 4°C overnight, washed in PBS, incubated with Alexa Fluor 488 or 594 (1:400, Molecular Probes), washed with PBS and mounted on microscope coverslips (Light Diagnostics). The specificity of the antibody was tested and confirmed using neuroligin 1 knockout hippocampal sections. Images were acquired with a Zeiss confocal microscope using a ×63 oil-immersion objective.
Transverse hippocampal slices (400 μm), prepared from 30 to 35-day-old male mice, were allowed to recover submerged for at least 2 h at 32°C in oxygenated artificial cerebrospinal fluid (ACSF) containing 124 mM NaCl, 5 mM KCl, 1.25 mM NaH2PO4, 2 mM MgSO4, 2 mM CaCl2, 26 mM NaHCO3 and 10 mM glucose, and for additional 30 min submerged in a recording chamber at 27°C–28°C, while continuously perfused with ACSF. CA1 and CA3 hippocampal regions were disconnected by a surgical cut. Field extracellular postsynaptic potentials (fEPSPs) were recorded in CA1 stratum radiatum with glass electrodes (2–3 MΩ) filled with ASCF. Schaffer collateral fEPSPs were evoked by stimulation with a concentric bipolar tungsten stimulating electrode placed in mid-stratum radiatum proximal to CA3 region. Baseline stimulation was applied at 0.033 Hz by delivering 0.1 ms pulses, with intensity adjusted to evoke 60% of maximal fEPSPs. Group I metabotropic glutamate receptor (mGluR) agonist (S)-3,5-dihydroxyphenylglycine (DHPG, 50 μM, Tocris) was added to ACSF for 10 min to induce mGluR-mediated LTD. fEPSP slope measurements were performed on digitized analogue recordings using the Clampfit analyze function and between 10% and 90% of maximal fEPSP amplitude. Statistical analyses were done on the 80–90 min period post-LTD induction, using One-way ANOVA, followed by Bonferroni’s post-hoc test.
This test was performed as in (Auerbach et al., 2011) and (Ehninger et al., 2008) with modifications. After handling, 2-month-old male mice were allowed to explore context A for 3 min, then conditioned to the training context A (metal floor, black walls, white lighting, no odor) with one 0.8 mA foot shock (2 s duration). Mice were removed 15 s after the shock was given and returned to home cage. Freezing was assessed 24 h later for 3 min as a proxy for contextual fear conditioning. To determine context specificity of the freezing response, mice trained in context A were separated into two groups: 50% of the mice were tested in context A (familiar context) and 50% in context B (novel context; plastic floor, black-white pattern walls, red light, no odor). Statistical analysis was carried out with One-way ANOVA with Bonferroni’s post-hoc test.
Pairs of 3–4 month-old male and female mice were tested for 15 min (testing only same sex and genotype pairs) in an open-field apparatus. Social behavior was analyzed for active interaction (sniffing, allo-grooming, mounting, or following) and rearing behavior. Statistical analysis was carried out using One-way ANOVA with Bonferroni’s post-hoc test.
Same-sex mouse dyads were introduced into an arena with Plexiglas walls that were opaque, measuring 39 cm in width, 26 cm in length, and 12 cm in height. The arena was placed 105 cm above ground level on a glass shelf to create a visual cliff, and it was illuminated with a 250-W LED light, providing an approximate of 3,000 lux. Within each open-field box, a single opaque PVC cylinder was present, measuring 7.5 cm in length and 3 cm in diameter, allowing only for joint proximity of the two animals (huddling). The time of tube co-occupancy, single occupancy or vacancy were measured over 180 min.
3–4-month-old mice were deeply anaesthetized with 1 mL kg-−1 of body weight of ketamine (55 mg per kg), xylazine (3 mg per kg) and medetomidine hydrochloride (0.3 mg per kg) injected intraperitoneally. Mice were placed in a Kopf stereotactic frame. A midline scalp incision was made to expose the skull, and bregma was used as the stereotaxic zero point. The guide cannulae, each 10 mm in length and 26-gauge in size, were implanted bilaterally into the hippocampi at AP -1.5 mm, ML ±1.65 mm, DV -1.8 mm (Paxinos Mouse Atlas). Cannulae were then fixed onto the skull with dental cement. Dummy cannulae were used to maintain clearance through each guide cannula. Mice were given a recovery period of at least 7 days following the surgery. During the infusion, a needle (33-gauge) was inserted into the guide cannula, 1.0 mm below the cannula tip. Using a peristaltic pump (connected to Hamilton 10 μL microsyringe via PE tubing), 0.5 μL per side of vehicle (50% DMSO in 100 mM Tris-Cl pH 7.2) rapamycin (Sigma, R0395, 30 nM per side) or 4EGI-1 (Sigma, 324519, 20 μg/μL) were infused into each hippocampus at a rate of 0.1 μL/min for 5 min. To prevent reflux, infusion needles remained in the guide cannulae for an additional 5 min following infusion.
Synaptosomes were prepared from freshly dissected hippocampus from 3–4-month-old mice as described in (Simbriger et al., 2020). Briefly, following rapid dissection, hippocampi were homogenized using a glass Dounce homogenizer in ice–cold sucrose buffer (320 mM sucrose, 5 mM Tris-HCl pH 7.4, 1 mM EDTA), and then centrifuged for 10 min at 1000 x g, 4°C. The pellet was resuspended in the same sucrose buffer and centrifuged again as before. The combined supernatant from both spins was further centrifuged for 10 min at 21,000 x g, 4°C, to pellet crude synaptosomes.
Experimenters were blinded to animal genotype during testing and scoring. All data are presented as mean ± S.E.M. (error bars). Statistical significance was set a priori at 0.05 (n.s.: non-significant). No nested data were collected in this study; n number corresponds to single observations (biological replicates). No randomization was performed in drug treatment. Sample size for behavioral, electrophysiological and biochemical analyses was determined with power calculations, based on previous data and effect size in refs (Gkogkas et al., 2013; Tuttle et al., 2017) using G*Power (a = 0.05, b: 0.2) (Faul et al., 2009). Normality test was performed using the Shapiro-Wilk test (α = 0.05). Details for statistical analysis are provided in Supplementary Table S2.
We previously demonstrated that Nlgn mRNAs are preferentially translated downstream of 4E-BP2/eIF4E and showed that knockdown of Nlgn1 in the hippocampus rescued behavioral deficits in Eif4ebp2−/− mice (Gkogkas et al., 2013). Since in Tuberous Sclerosis there is hyperactivation of mTORC1 and subsequently hyperphosphorylation of its downstream effectors, 4E-BPs and S6Ks, we reasoned that Nlgn1 mRNA translation might be increased in Tsc2+/− hippocampi. To examine translation of Nlgn1, we carried out polysome profiling on synaptosomes purified from dissected hippocampi of Tsc2+/− and wild type (Tsc2+/+; WT) mice. We did not detect any significant differences between Tsc2+/− and wild type groups in the overall sedimentation profiles of hippocampal tissue (Figure 1A), as evidenced by the unchanged Polysome/Monosome ratio (Figure 1A). RT-qPCR analysis of the RNA extracted from sucrose density gradient fractions revealed that Nlgn1 mRNA associated with heavier polysome fractions in Tsc2+/− synaptosomes compared with wild type (Figure 1B). In accordance with this finding, Nlgn1 protein expression was significantly increased (∼40%) in Tsc2+/− synaptosomes compared with wild type (Figure 1C), and in Nlgn1 immunofluorescence staining of the hippocampus stratum radiatum in Tsc2+/− mice (Figure 1D). We did not observe significant changes in total Nlgn1 mRNA amounts between the different genotypes (Figure 1E). Together, these findings indicate that the increased Nlgn1 expression in Tsc2+/− is due to translational and not transcriptional changes. To establish a direct link between translational control via mTORC1 hyperactivation and Nlgn1 protein expression in Tsc2+/− brain, we performed chronic in vivo treatment of Tsc2+/− mice with rapamycin (mTORC1 inhibitor) or 4EGI-1 (inhibitor of cap-dependent translation that disrupts the interaction between eIF4E and eIF4G). We previously established subthreshold dosage of rapamycin and 4EGI-1 that have no effect on Nlgn1 expression or autism-related and learning/memory phenotypes in wild type mice (Gkogkas et al., 2013). A regimen of 5 days of intrahippocampal rapamycin (30 nM) or 4EGI-1 (20 μg/μL) administration significantly reduced Nlgn1 protein amounts in Tsc2+/− but not in WT hippocampal synaptosomes (Figure 1F). Altogether, hyperactivation of mTORC1 in Tsc2+/− mice stimulates cap-dependent translation of Nlgn1 mRNA, resulting in increased Nlgn1 protein expression.
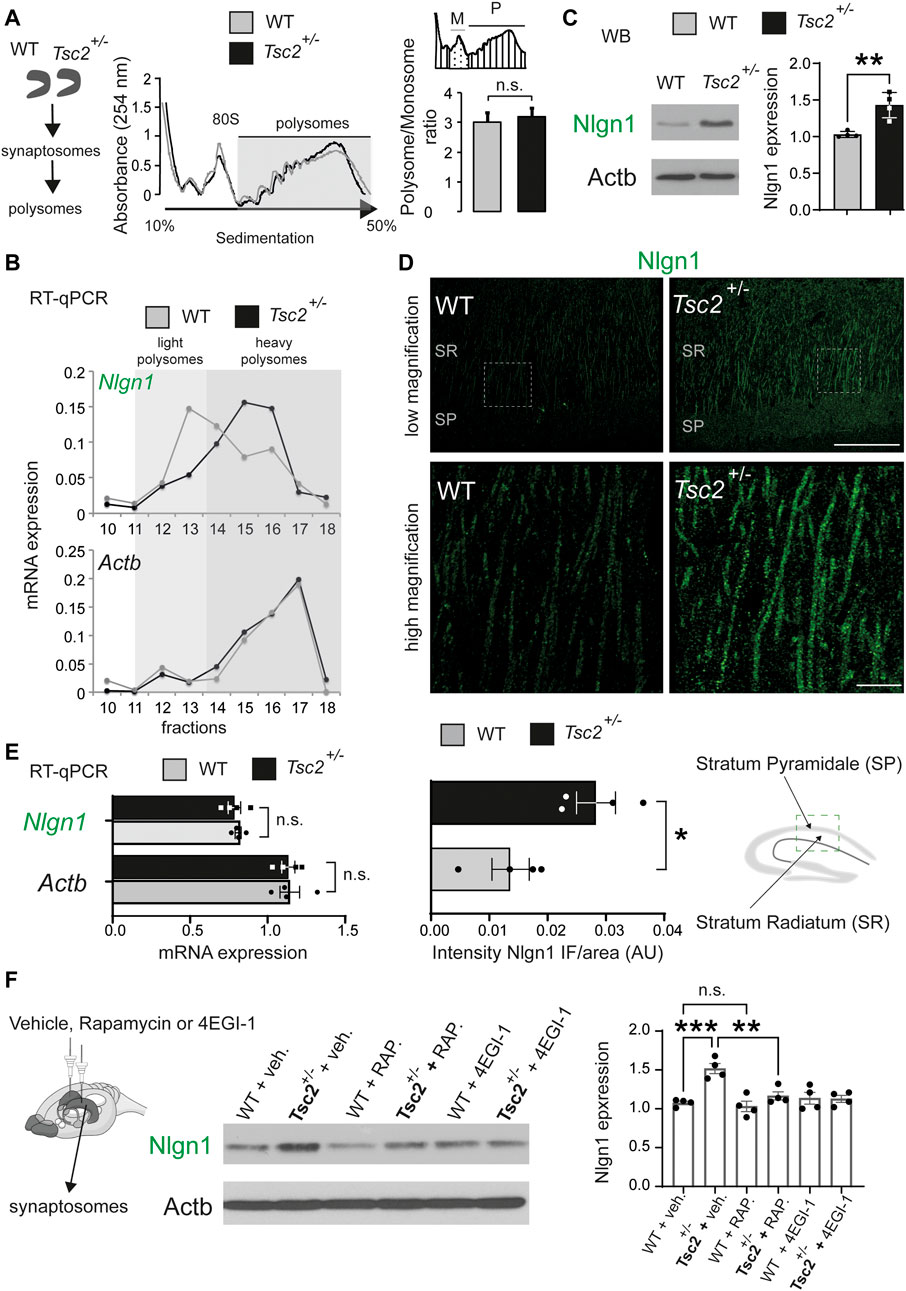
FIGURE 1. Exaggerated translation of Nlgn1 mRNA in Tsc2+/− mice. (A) Polysome fractionation in synaptosomes prepared from dissected hippocampi. Left: No difference in overall polysome sedimentation profiles between wild type (WT) and Tsc2+/− mice. Positions of the 80S ribosome peak and polysomes are indicated. Right: No difference in polysome (P)/monosome (M) ratio between WT and Tsc2+/− mice (n = 3, p = 0.15, Student’s t-test). (B) Increased Nlgn1 mRNA amounts associated with heavy polysome fractions in hippocampal synaptosomes from Tsc2+/− mice. RT-qPCR analysis of RNA extracted from polysomal fractions with specific primers for Nlgn1. No change in Actb mRNA distribution (n = 3; p = 0.003 light/heavy polysomes Student’s t-test). (C) Increased Nlgn1 protein amounts in synaptosomes from hippocampal lysates in Tsc2+/− mice. (n = 4, **p < 0.01, Student’s t-test). (D) Increased Nlgn1 immunostaining in CA1 pyramidal cells in Tsc2+/− hippocampus compared to WT shown with immunofluorescence. Top: low magnification; bottom: high magnification; SR, Stratum Radiatum; SP,: Stratum Pyramidale (n = 4, *p < 0.05, Student’s t-test). Scale bars: low magnification 80 μm, high magnification 20 μm. (E) No changes in Nlgn1 mRNA levels in Tsc2+/− mice. RT-qPCR analysis of total Nlgn1 or Actb mRNA in hippocampal synaptosomes from WT or Tsc2+/− mice (n = 4, Student’s t-test). (F) Chronic sub-threshold intrahippocampal infusion of rapamycin (30 nM) or 4EGI-1 (20 μg/μL) reduced Nlgn1 protein expression in synaptosomes from Tsc2+/− mice hippocampi. Left: Representative immunoblots of hippocampal synaptosome lysate are shown, probed with antisera against the indicated proteins; Actb: loading control. Right: Quantification of Nlgn1 protein expression in immunoblotting for the different groups (One-way ANOVA, with Bonferroni’s post-hoc, n = 4 per group, **p < 0.01, ***p < 0.001).
Previously, we showed that lentivirus-mediated knockdown of Nlgn1 reversed synaptic and behavioral deficits of Eif4ebp2−/− mice (Gkogkas et al., 2013). Given the increased Nlgn1 expression in the hippocampus of Tsc2+/− mice, we hypothesized that reducing Nlgn1 protein amounts could rescue the reduced mGluR-LTD in Tsc2+/− mice (Auerbach et al., 2011). To achieve a reduction of Nlgn1 amounts equivalent to our previous findings (Gkogkas et al., 2013), we first deleted one copy of Nlgn1 in Tsc2+/− mice. We crossed Nlgn1+/− to Tsc2+/− to obtain Tsc2+/−/Nlgn1+/− mice (Figure 2A) and confirmed that deletion of one Nlgn1 allele reduced Nlgn1 protein amounts in the hippocampus to WT levels (Figure 2B). To examine the effect of the normalization of Nlgn1 expression in Tsc2+/−/Nlgn1+/− mice on hippocampal mGluR-LTD, we induced LTD by stimulating group I metabotropic glutamate receptors in hippocampal slices with DHPG (Figure 2C). Tsc2+/− mice showed impaired mGluR-LTD relative to WT mice (Figure 2C). Deletion of one copy of Nlgn1 was sufficient to rescue the mGluR-LTD impairment in Tsc2+/−/Nlgn1+/− mice compared with WT (Figure 2C).
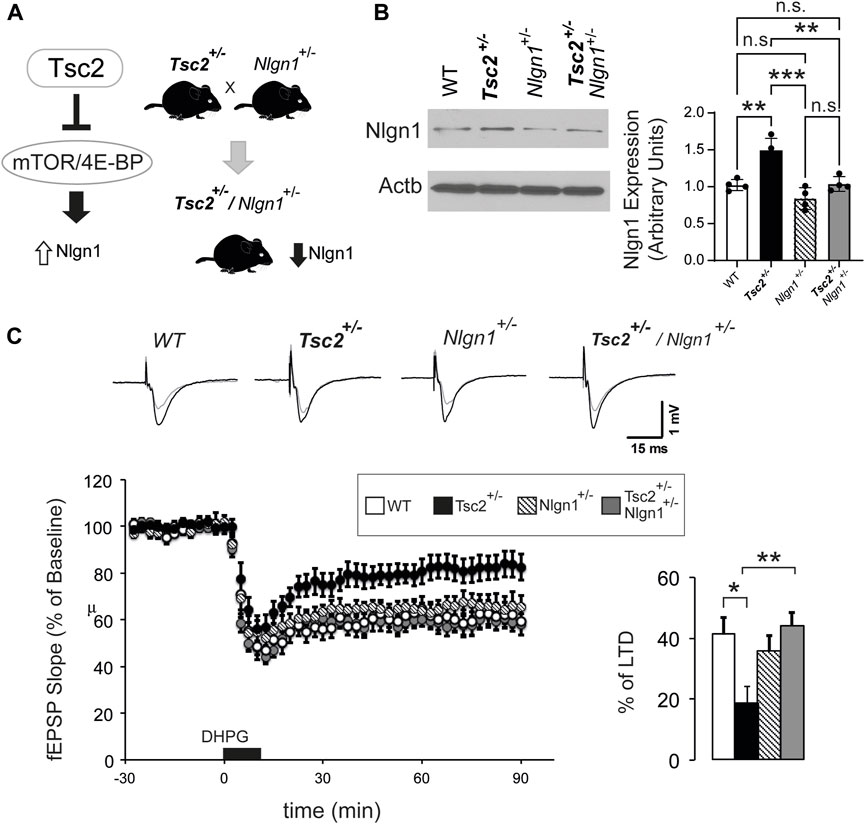
FIGURE 2. Nlgn1 haploinsufficiency rescues aberrant mGluR-LTD in Tsc2+/− mice. (A) Outline of breeding strategy to reduce Nlgn1 expression in Tsc2+/− mice. (B) Nlgn1 haploinsufficiency restores Nlgn1 expression in synaptosomes prepared from Tsc2+/− hippocampi. Representative immunoblots of hippocampal synaptosome lysates probed with antisera against the indicated proteins; Actb: loading control. Quantification of Nlgn1 protein expression is shown (One-way ANOVA, with Bonferroni’s post-hoc, n = 4 per group, **p < 0.01, ***p < 0.001). (C) Nlgn1 deletion rescued mGluR-LTD deficits in Tsc2+/− mice. Top: Representative traces from 4 groups (baseline in black and recording at 90 min in grey). Bottom left: LTD was induced by application of DHPG (50 µM for 10 min). Field excitatory postsynaptic potentials (fEPSP) were recorded over a 90-min period after DHPG-induced LTD. Bottom right: Quantification of the fEPSP slope (% decrease from baseline) during the last 10 min of the recording. One-way ANOVA with Bonferroni’s post-hoc n for groups: WT (13), Tsc2+/− (12), Nlgn1+/− (9) and Tsc2+/−/Nlgn1+/− (12), *p < 0.05, **p < 0.01.
We next examined whether the correction of synaptic deficits in Tsc2+/− via Nlgn1 deletion could reverse hippocampal-dependent context discrimination, which is impaired in Tsc2+/− mice (Ehninger et al., 2008). To this end, mice were trained in Context A (receiving a footshock) (Figure 3). Subsequently, 50% of the animals were tested in Context A (familiar) and the remaining 50% in Context B (novel). Tsc2+/− mice failed to discriminate between the two contexts (Figure 3A), as demonstrated by the non-significant difference in freezing percentage between familiar and novel contexts, in accordance with previous reports (Ehninger et al., 2008; Auerbach et al., 2011). Conversely, Tsc2+/−/Nlgn1+/− mice were able to discriminate between the two different contexts (Figure 3A), but without achieving complete rescue to wild type levels. Taken together, these data suggest that deletion of one copy of Nlgn1, restoring its protein expression to wild-type levels, is sufficient to reverse synaptic and behavioral deficits in a mouse model of TSC.
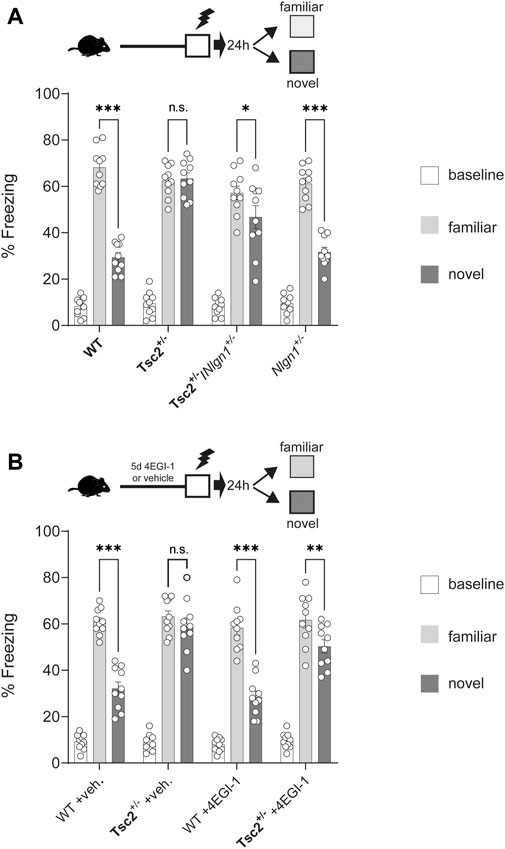
FIGURE 3. Rescue of hippocampal memory deficits in Tsc2+/− mice via Nlgn1 haploinsufficiency or inhibition of cap-dependent translation with 4EGI-1. Nlgn1 haploinsufficiency rescued context discrimination deficits in Tsc2+/− mice. (A,B) Chronic 4EGI-1 treatment rescued context discrimination deficits in Tsc2+/− mice. Percentage (%) Freezing shown at baseline, in familiar or novel context for the groups depicted; n = 10 for all groups; veh = vehicle. Two-way ANOVA with Bonferroni’s post-hoc *p < 0.05, **p < 0.01, ***p < 0.001.
To further validate the rescue effect of Nlgn1 deletion in Tsc2+/− mice, we used the 4EGI-1 inhibitor of cap-dependent translation, which we showed is effective in restoring Nlgn1 expression (Figure 1F). Chronic infusion (5 days) of 4EGI-1 (20 μg/μL, for 5 days) in the dorsal hippocampus led to a restoration of context discrimination in Tsc2+/− mice, but without achieving complete rescue to wild type levels (Figure 3B). Notably, the subthreshold regimen of 4EGI-1 that we used did not alter context discrimination behavior in WT mice (Figure 3B).
We finally examined behaviors reminiscent of autism (e.g., changes in social interaction) given their high prevalence in TSC patients. We subjected mice to social behavior analysis using a reciprocal social interaction test (Sato et al., 2012) and the tube co-occupancy test (TCOT) for social propinquity (Tuttle et al., 2017). Deletion of one copy of Nlgn1 or 4EGI-1 treatment (Figure 4A) in Tsc2+/− mice reversed deficits in reciprocal social interaction as evidenced by the active interaction time (Figures 4B, C) and also rescued the reduced co-occupancy time in the TCOT test (Figures 4E, F). Taken together, these data reveal that normalization of Nlgn1 protein expression rescues not only memory-related impairment, but also deficits in social behavior in Tsc2+/− mice.
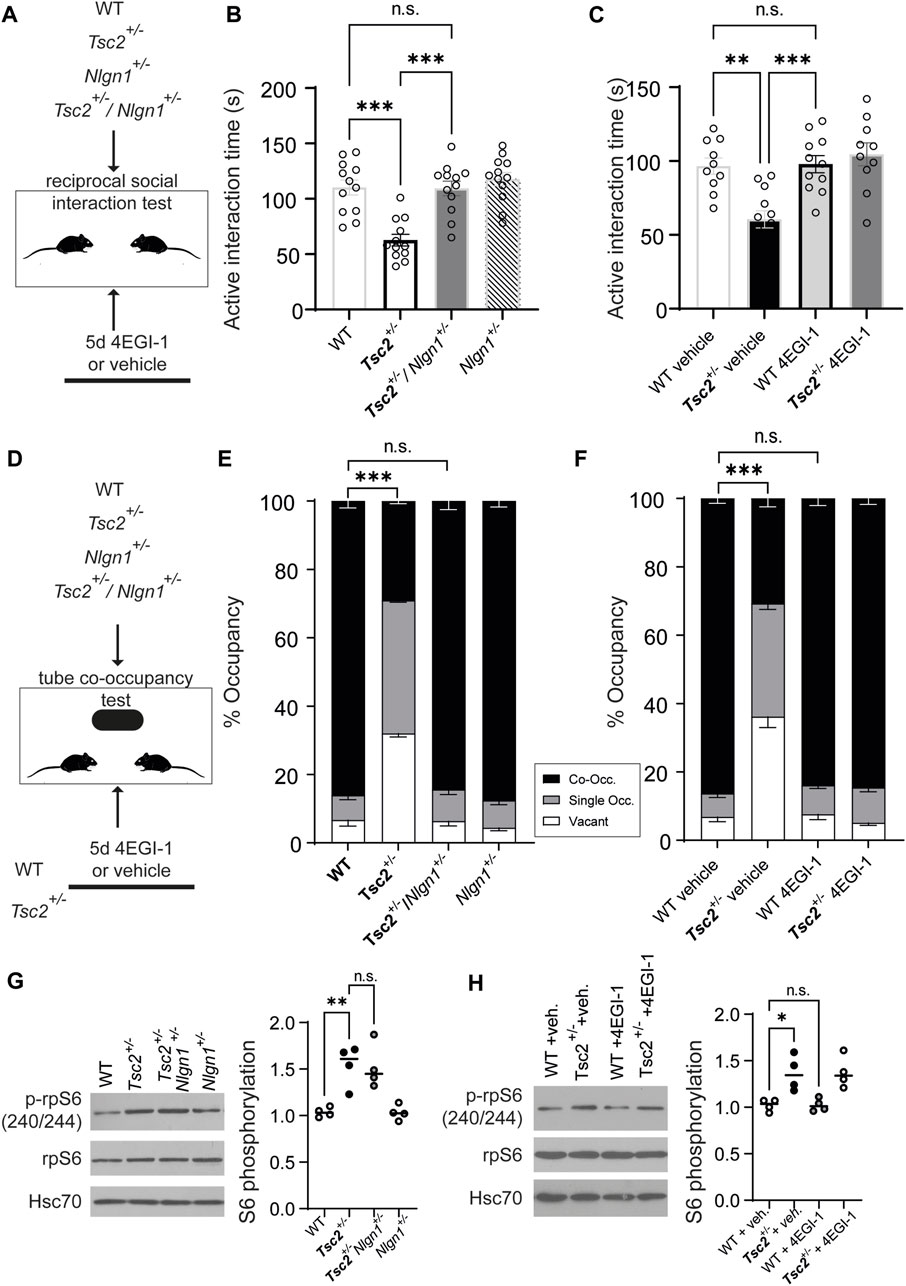
FIGURE 4. Rescue of social behavior deficits in Tsc2+/− mice via Nlgn1 haploinsufficiency or inhibition of cap-dependent translation with 4EGI-1 but no normalization of mTORC1 signaling. (A) Outline of the Reciprocal social interaction test for the indicated groups. (B,C) Active interaction during reciprocal social interaction is shown. For A, n = 10 for all groups. For (B) WT + veh (10), Tsc2+/− + veh. (12), WT +4EGI-1 (11), Tsc2+/− +4EGI-1 (10); veh = vehicle. (D) Outline of the Tube co-occupancy test for the indicated groups. (E,F) % Occupancy of the tube in the arena (Co-Occ: co-occupancy, Single Occ.: single occupancy or vacant). (G) Nlgn1 haploinsufficiency or (H) 4EGI-1 treatment do not restore mTORC1 signaling in hippocampal synaptosomes from Tsc2+/− mice. Left: Representative immunoblots of synaptosomes prepared from hippocampi, probed with antisera against the indicated proteins for the groups shown. Hsc70: loading control. Right: Quantification of phospho-rpS6 expression for the indicated groups. For (A–C): One-way ANOVA, with Bonferroni’s post-hoc and (D–F): Two-Way ANOVA with Bonferroni’s post-hoc (*p < 0.05, **p < 0.01, ***p < 0.001).
We reasoned that behavioral rescue following Nlgn1 expression normalization in Tsc2+/− mice is related to the hippocampal cellular phenotype observed (rescue of impaired hippocampal mGluR-LTD; Figure 2), yet it was still unclear whether Nlgn1 deletion affected mTORC1 hyperactivation in Tsc2+/−. To assess mTORC1 activity, we examined its common readout, the phosphorylation status of ribosomal protein S6 (p-rpS6, Figures 4G, H). Deletion of one copy of Nlgn1 or chronic 4EGI-1 treatment did not affect p-rpS6 in wild type or vehicle groups, and did not correct increased p-rpS6 in Tsc2+/−/Nlgn1+/− (Figure 4G) and Tsc2+/− - 4EGI-1 groups (Figure 4H). Thus, mGluR-LTD and behavioral phenotypes (context discrimination, social deficits) were corrected following normalization of Nlgn1 protein expression, while mTORC1 hyperactivation persisted.
ASD is a polygenic disorder and elucidating the contribution of converging signaling pathways will shed fresh light on its pathogenesis mechanisms and thus bolster potential new therapeutics. Dysregulated protein synthesis has emerged as a prominent process implicated in several monogenic forms of ASD and intellectual disability (Chen et al., 2019), as well as in sporadic ASD (Hooshmandi et al., 2020), and is often accompanied by changes in key signaling pathways upstream of translation such as mTOR (Rosina et al., 2019). Of these monogenic ASD forms, several conditions such as fragile X syndrome (FXS) and TSC are also linked to exaggerated global and mRNA-specific protein synthesis (Auerbach et al., 2011). It is evident that inhibitors of signaling upstream of protein synthesis (e.g., rapamycin and rapalogs) have the potential to be used as therapeutics, yet off-target and pervasive side effects cannot be excluded (Chu and Pelletier, 2018). We previously showed that the autism-related pathogenic effects of exaggerated cap-dependent translation on mouse brain physiology and behavior can be reversed by targeting “eIF4E-sensitive” mRNAs such as Nlgn1 (Gkogkas et al., 2013). Herein, we provide the first example of applying this strategy to rescue phenotypes in an adult mouse model of TSC, a rare genetic disorder, which is also a common monogenic form of ASD.
Synaptic protein synthesis was previously implicated in neurodevelopmental disorders such as ASD (Louros and Osterweil, 2016). The advent of genome-wide translational profiling (Ingolia et al., 2009) allowed to uncover potential new candidates for therapeutics, whose expression is regulated at the translational level, such as Nlgn1. Given the role of Nlgn1 in autism-related behaviors (Nguyen et al., 2020), we now offer genetic and pharmacological avenues to inhibit Nlgn1 for modulating TSC-related phenotypes (Figure 2; Figure 3; Figure 4). Neuroligins are cell-adhesion molecules critical for synaptic structure function and the regulation of the E/I balance; Nlgn1 is predominantly expressed in excitatory synapses (Liu et al., 2022). Thus, correcting Nlgn1 protein expression in Tsc2+/− mice may engender synaptic remodeling and correction of behaviors by normalizing excitatory transmission. Compared to our previous strategy employing lentiviruses expressing shRNA against Nlgn1, we now provide genetic evidence that we can achieve rescue by inducing Nlgn1 haploinsufficiency in Tsc2+/− mice (Figure 2; Figure 3; Figure 4), which further supports a TSC-Nlgn1 axis in TSC pathology. Furthermore, 4EGI-1, an inhibitor of cap-dependent translation was successfully used to achieve the same objective. 4EGI-1 and other similar acting compounds, alone or in synergy with other kinase inhibitors, are being explored as cancer therapeutics and thus could be repurposed for neurodevelopmental disorders such as TSC (Mahalingam et al., 2014).
By correcting Nlgn1 expression, we reversed the impairment of mGluR-LTD in Tsc2+/− mice. Previous work in mouse models of ASD (knockout models of Shank3, Pten, Fmr1, Tsc, Cntnap2, where mTORC1 signaling is dysregulated) has shown that mGluR-LTD is disrupted in various brain regions, including the prefrontal cortex and hippocampus (Lee et al., 2019). Moreover, mutations in genes related to mGluR signaling have been identified in individuals with ASD, further supporting a role for this pathway in the disorder (Hadley et al., 2014). mTORC1 inhibition with rapamycin rescued several behavioral phenotypes in Tsc2+/− mice (Ehninger and Silva, 2011) and in other models of neurodevelopmental disorders (Girodengo et al., 2022). Strikingly, we achieved rescue of TSC-related phenotypes in the Tsc2+/− mice, without correcting hyperactivated mTORC1 (Figure 4). Both Nlgn1 deletion and 4EGI-1 treatments did not alter rpS6 phosphorylation in animals where cellular (mGluR-LTD) and behavioral (context discrimination and social behavior) were normalized. This is an intriguing finding, suggesting that targeting mTORC1 downstream molecules, such as Nlgn1, in complex regulatory pathways is an appealing strategy, which may negate off-target effects and offer targeted therapeutics based on mechanistic understanding. In addition to mRNA translation, mTORC1 regulates several processes such as lipid metabolism and autophagy and thus our findings highlight the important role of translational control, downstream of mTORC1, in TSC.
We focused on the mTORC1/4E-BP/Nlgn1 axis and did not perform an exhaustive analysis of other candidate signaling pathways, such as ERK, which may be corrected in Tsc2+/− brains following normalization of Nlgn1 protein amounts. Moreover, we focused on hippocampus only and did not analyze other brain areas relevant to ASD/TSC such as forebrain or cerebellum. Further elucidation of the synaptic role of Nlgn1 and possibly other neuroligins in TSC and other forms of ASD is required. For example, examining other forms of synaptic plasticity, apart from LTD in ASD/TSC models may reveal new pharmacological avenues for treatment. Finally, our experiments were performed on adult mice. Potential clinical application of the mechanistic findings of this study in early life would require the study of younger animals.
Overall, our study revealed that reducing Nlgn1 expression is a possible new therapeutic avenue for TSC and plausibly for other neurodevelopmental disorders.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
All procedures followed the UK Home Office and Canadian Council on Animal Care guidelines and were respectively approved by the University of Edinburgh, McGill University and Université de Montréal.
CG, AK, and SJ conceived the project, designed experiments, and supervised the research. KC, ES, and MZ performed immunohistochemistry, biochemistry analyses and behavioral experiments. AK, NH, and J-CL performed electrophysiological studies. CP assisted with genetics of Nlgn1 transgenic animals and methodology. CG, AK, and SJ wrote the manuscript with input from all the authors. All authors contributed to the article and approved the submitted version.
This work was supported by grants to CG: Sir Henry Dale Fellowship from the Wellcome Trust and Royal Society (107687/Z/15/Z), a NARSAD Young Investigator grant from the Brain and Behavior Research Foundation (24968), a Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “2nd Call for H.F.R.I. Research Projects to support Faculty Members and Researchers” (Project Number: 2556) grant to CG and the RESEARCH-CREATE-INNOVATE grant T2EΔK-00852. It was also supported by grants from the Canadian Institutes of Health Research (CIHR) PJT-162412, and the Azrieli Centre for Autism Research (ACAR) to AK, and CIHR MOP-125985 and PJT-153311 to J-CL. J-CL is the recipient of the Canada Research Chair in Cellular and Molecular Neurophysiology (CRC 950-231066). SJ is supported by a Patrick G. Johnston Research Fellowship at Queen’s University Belfast.
The authors thank Annie Sylvestre and Nahum Sonenberg at McGill University for providing Tsc2 animals.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1205112/full#supplementary-material
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. doi:10.1038/nature10658
Blundell, J., Blaiss, C. A., Etherton, M. R., Espinosa, F., Tabuchi, K., Walz, C., et al. (2010). Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 30, 2115–2129. doi:10.1523/JNEUROSCI.4517-09.2010
Chen, Y. C., Chang, Y. W., and Huang, Y. S. (2019). Dysregulated translation in neurodevelopmental disorders: An overview of autism-risk genes involved in translation. Dev. Neurobiol. 79, 60–74. doi:10.1002/dneu.22653
Chu, J., and Pelletier, J. (2018). Therapeutic opportunities in eukaryotic translation. Cold Spring Harb. Perspect. Biol. 10, a032995. doi:10.1101/cshperspect.a032995
Dahlhaus, R., Hines, R. M., Eadie, B. D., Kannangara, T. S., Hines, D. J., Brown, C. E., et al. (2010). Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus 20, 305–322. doi:10.1002/hipo.20630
Doernberg, E., and Hollander, E. (2016). Neurodevelopmental disorders (ASD and ADHD): DSM-5, ICD-10, and ICD-11. CNS Spectr. 21, 295–299. doi:10.1017/S1092852916000262
Ehninger, D., Han, S., Shilyansky, C., Zhou, Y., Li, W., Kwiatkowski, D. J., et al. (2008). Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat. Med. 14, 843–848. doi:10.1038/nm1788
Ehninger, D., and Silva, A. J. (2011). Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends Mol. Med. 17, 78–87. doi:10.1016/j.molmed.2010.10.002
Faul, F., Erdfelder, E., Buchner, A., and Lang, A. G. (2009). Statistical power analyses using G*power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 41, 1149–1160. doi:10.3758/BRM.41.4.1149
Garami, A., Zwartkruis, F. J., Nobukuni, T., Joaquin, M., Roccio, M., Stocker, H., et al. (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466. doi:10.1016/s1097-2765(03)00220-x
Girodengo, M., Ultanir, S. K., and Bateman, J. M. (2022). Mechanistic target of rapamycin signaling in human nervous system development and disease. Front. Mol. Neurosci. 15, 1005631. doi:10.3389/fnmol.2022.1005631
Gkogkas, C. G., Khoutorsky, A., Ran, I., Rampakakis, E., Nevarko, T., Weatherill, D. B., et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. doi:10.1038/nature11628
Hadley, D., Wu, Z. L., Kao, C., Kini, A., Mohamed-Hadley, A., Thomas, K., et al. (2014). The impact of the metabotropic glutamate receptor and other gene family interaction networks on autism. Nat. Commun. 5, 4074. doi:10.1038/ncomms5074
Hinnebusch, A. G., Ivanov, I. P., and Sonenberg, N. (2016). Translational control by 5'-untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416. doi:10.1126/science.aad9868
Hooshmandi, M., Wong, C., and Khoutorsky, A. (2020). Dysregulation of translational control signaling in autism spectrum disorders. Cell Signal 75, 109746. doi:10.1016/j.cellsig.2020.109746
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., and Weissman, J. S. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. doi:10.1126/science.1168978
Kelleher, R. J., and Bear, M. F. (2008). The autistic neuron: Troubled translation? Cell 135, 401–406. doi:10.1016/j.cell.2008.10.017
Lee, K., Vyas, Y., Garner, C. C., and Montgomery, J. M. (2019). Autism-associated Shank3 mutations alter mGluR expression and mGluR-dependent but not NMDA receptor-dependent long-term depression. Synapse 73, e22097. doi:10.1002/syn.22097
Liu, G. Y., and Sabatini, D. M. (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203. doi:10.1038/s41580-019-0199-y
Liu, X., Hua, F., Yang, D., Lin, Y., Zhang, L., Ying, J., et al. (2022). Roles of neuroligins in central nervous system development: Focus on glial neuroligins and neuron neuroligins. J. Transl. Med. 20, 418. doi:10.1186/s12967-022-03625-y
Louros, S. R., and Osterweil, E. K. (2016). Perturbed proteostasis in autism spectrum disorders. J. Neurochem. 139, 1081–1092. doi:10.1111/jnc.13723
Mahalingam, P., Takrouri, K., Chen, T., Sahoo, R., Papadopoulos, E., Chen, L., et al. (2014). Synthesis of rigidified eIF4E/eIF4G inhibitor-1 (4EGI-1) mimetic and their in vitro characterization as inhibitors of protein-protein interaction. J. Med. Chem. 57, 5094–5111. doi:10.1021/jm401733v
Mamane, Y., Petroulakis, E., Lebacquer, O., and Sonenberg, N. (2006). mTOR, translation initiation and cancer. Oncogene 25, 6416–6422. doi:10.1038/sj.onc.1209888
Nguyen, T. A., Lehr, A. W., and Roche, K. W. (2020). Neuroligins and neurodevelopmental disorders: X-linked genetics. Front. Synaptic Neurosci. 12, 33. doi:10.3389/fnsyn.2020.00033
Rosina, E., Battan, B., Siracusano, M., Di Criscio, L., Hollis, F., Pacini, L., et al. (2019). Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl. Psychiatry 9, 50. doi:10.1038/s41398-018-0335-z
Santini, E., Huynh, T. N., Macaskill, A. F., Carter, A. G., Pierre, P., Ruggero, D., et al. (2013). Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493, 411–415. doi:10.1038/nature11782
Sato, A., Kasai, S., Kobayashi, T., Takamatsu, Y., Hino, O., Ikeda, K., et al. (2012). Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun. 3, 1292. doi:10.1038/ncomms2295
Simbriger, K., Amorim, I. S., Chalkiadaki, K., Lach, G., Jafarnejad, S. M., Khoutorsky, A., et al. (2020). Monitoring translation in synaptic fractions using a ribosome profiling strategy. J. Neurosci. Methods 329, 108456. doi:10.1016/j.jneumeth.2019.108456
Sonenberg, N. (2008). eIF4E, the mRNA cap-binding protein: from basic discovery to translational research. Biochem. Cell Biol. 86, 178–183. doi:10.1139/O08-034
Touraine, R., Hauet, Q., Harzallah, I., and Baruteau, A. E. (2022). Tuberous sclerosis complex: Genetic counselling and perinatal follow-up. Arch. Pediatr. 29, 5S3–5S7. doi:10.1016/S0929-693X(22)00283-4
Trobiani, L., Meringolo, M., Diamanti, T., Bourne, Y., Marchot, P., Martella, G., et al. (2020). The neuroligins and the synaptic pathway in Autism Spectrum Disorder. Neurosci. Biobehav Rev. 119, 37–51. doi:10.1016/j.neubiorev.2020.09.017
Tuttle, A. H., Tansley, S., Dossett, K., Tohyama, S., Khoutorsky, A., Maldonado-Bouchard, S., et al. (2017). Social propinquity in rodents as measured by tube cooccupancy differs between inbred and outbred genotypes. Proc. Natl. Acad. Sci. U. S. A. 114, 5515–5520. doi:10.1073/pnas.1703477114
Keywords: tuberous sclerosis, mTOR, translational control, autism, memory
Citation: Chalkiadaki K, Statoulla E, Zafeiri M, Haji N, Lacaille J-C, Powell CM, Jafarnejad SM, Khoutorsky A and Gkogkas CG (2023) Reversal of memory and autism-related phenotypes in Tsc2+/− mice via inhibition of Nlgn1. Front. Cell Dev. Biol. 11:1205112. doi: 10.3389/fcell.2023.1205112
Received: 13 April 2023; Accepted: 16 May 2023;
Published: 24 May 2023.
Edited by:
Cristina Montiel Duarte, Nottingham Trent University, United KingdomReviewed by:
Sidharth Mehan, Indo-Soviet Friendship College of Pharmacy, IndiaCopyright © 2023 Chalkiadaki, Statoulla, Zafeiri, Haji, Lacaille, Powell, Jafarnejad, Khoutorsky and Gkogkas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seyed Mehdi Jafarnejad, c20uamFmYXJuZWphZEBxdWIuYWMudWs=; Arkady Khoutorsky, YXJrYWR5Lmtob3V0b3Jza3lAbWNnaWxsLmNh; Christos G. Gkogkas, Y2drb2drYXNAYnJpLmZvcnRoLmdy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.