 Yoshitaka Furuta
Yoshitaka Furuta Zheng Zhou
Zheng Zhou- Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX, United States
Phosphatidylserine (PS) is a lipid component of the plasma membrane. It is asymmetrically distributed to the inner leaflet in live cells. In cells undergoing apoptosis, phosphatidylserine is exposed to the outer surfaces. The exposed phosphatidylserine acts as an evolutionarily conserved “eat-me” signal that attracts neighboring engulfing cells in metazoan organisms, including the nematode Caenorhabditis elegans, the fruit fly Drosophila melanogaster, and mammals. During apoptosis, the exposure of phosphatidylserine to the outer surface of a cell is driven by the membrane scramblases and flippases, the activities of which are regulated by caspases. Cells undergoing necrosis, a kind of cell death frequently associated with cellular injuries and morphologically distinct from apoptosis, were initially believed to allow passive exposure of phosphatidylserine through membrane rupture. Later studies revealed that necrotic cells actively expose phosphatidylserine before any rupture occurs. A recent study in C. elegans further reported that the calcium ion (Ca2+) plays an essential role in promoting the exposure of phosphatidylserine on the surfaces of necrotic cells. These findings indicate that necrotic and apoptotic cells, which die through different molecular mechanisms, use common and unique mechanisms for promoting the exposure of the same “eat me” signal. This article will review the mechanisms regulating the exposure of phosphatidylserine on the surfaces of necrotic and apoptotic cells and highlight their similarities and differences.
Introduction
The plasma membrane of eukaryotic cells is composed of the lipid bilayer and various membrane proteins. The lipid bilayer comprises glycerophospholipids, sphingomyelins, and cholesterol (Harayama and Riezman, 2018). Phosphatidylserine (PS), one type of glycerophospholipids, is a major component of the plasma membrane. In living cells, PS is almost exclusively localized to the inner leaflet of the plasma membrane (Balasubramanian and Schroit, 2003). PS exposure is observed when cells undergo apoptosis, a type of cell death that features cell shrinkage, chromatin condensation, nuclear DNA fragmentation, and a well-maintained plasma membrane integrity (Balasubramanian and Schroit, 2003; Kroemer et al., 2009). PS presented on the surfaces of apoptotic cells is an “eat-me” signal that attracts phagocytes to engulf apoptotic cells (Balasubramanian and Schroit, 2003; Segawa and Nagata, 2015). PS exposure also occurs in living cells under various physiological conditions. For example, mammalian platelets activated by vascular injuries expose PS, which recruits blood coagulation factors, and initiates blood clotting (Bevers et al., 1983; Dachary-Prigent et al., 1995). The exposure of PS on living cells and its biological significance have been extensively reviewed in another article (Shin and Takatsu, 2020) and is not covered here. Necrosis is another type of cell death that displays cell and organelle swelling, excessive intracellular membranes, and the subsequent rupture of intracellular and plasma membranes (Kroemer et al., 2009). Various conditions induce necrosis in cells, such as extreme temperature change, physical injury, hypoxia, hypo-osmotic shock, bacterial infection, ligands of transmembrane receptors such as the death receptors, and Ca2+ excitotoxicity (McCall, 2010; Moquin and Chan, 2010; Vlachos and Tavernarakis, 2010; Zhou and Yuan, 2014). Necrosis is closely associated with various diseases such as stroke, chronic inflammation, cancer, and neural and retinal degeneration (Yamashima, 2004; Noch and Khalili, 2009; Challa and Chan, 2010; Whelan et al., 2010; Nikoletopoulou and Tavernarakis, 2014; Shan et al., 2018; Zhang et al., 2023). For example, the hyperexcitation of neurons or glial cells induced by the constitutively active ion channels and related proteins causes excitotoxic necrosis (Noch and Khalili, 2009; Lai et al., 2014). Excitotoxic necrosis is a leading cause of neuronal damage in the brain ischemia (Tymianski, 2011; Lai et al., 2014). It also contributes to various aging-associated neurodegenerative disorders (Martin, 1999). Like apoptotic cells, necrotic cells are also engulfed and degraded by phagocytes (Hall et al., 1997; Krysko et al., 2006a; Poon et al., 2010b; Li et al., 2015; Schwegler et al., 2015). Efficient clearance of apoptotic and necrotic cells is essential for tissue homeostasis, tissue repair, and the suppression of harmful inflammatory and auto-immune responses caused by the contents of the dying cells during animal development and adulthood (Krysko et al., 2006a; Poon et al., 2010b; Poon et al., 2014). In addition, clearance of necrotic and apoptotic neurons helps to resolve the wounded area and facilitates tissue regeneration and the recovery from brain injury (Martin, 1999; Tymianski, 2011; Lai et al., 2014; Tovar-y-Romo et al., 2016; Tremblay et al., 2019). On the other hand, microglial cells, the professional phagocytes in the central nervous system, may also be activated by degenerating neurons and further enhance neural inflammation (Apolloni et al., 2014; Tremblay et al., 2019; Butler et al., 2021). Therefore, identifying cell clearance mechanisms is highly relevant to human health.
How necrotic cells are recognized and engulfed remained elusive for a long time. PS has been reported to serve as an “eat me” signal to recruit phagocytes for necrotic cells (Brouckaert et al., 2004; Schwegler et al., 2015; Budai et al., 2019). Whether the intracellular mechanisms that drive PS exposure are common or different between apoptotic and necrotic cells remains largely an unanswered question. Although besides PS, other “eat me” signals that attract phagocytes to necrotic cells were also identified (Gaipl et al., 2001; Böttcher et al., 2006; Poon et al., 2010a), this article will focus on PS-exposure mechanisms. Here we review the recent discoveries regarding the molecular mechanisms driving PS exposure on the surfaces of necrotic cells and how these mechanisms contribute to the phagocytosis of necrotic cells. Furthermore, we will compare these newly identified mechanisms to that driving PS exposure on the surfaces of apoptotic cells and discuss how these findings shed light on the roles of caspase 3 and Ca2+ in regulating PS exposure and dying cell-clearance.
PS on the surfaces of both apoptotic and necrotic cells acts as an “eat me” signal that attracts phagocytes
The molecular mechanisms of PS-dependent phagocytosis of apoptotic cells were extensively reviewed previously (Elliott and Ravichandran, 2016; Nagata, 2018; Atkin-Smith, 2021). In summary, in mammals, PS exposed on apoptotic cells is recognized by phagocytic receptors via two alternative mechanisms: phagocytic receptors such as Tim-1 and Tim-4, stabilin 1 and 2, and BAI1 on neighboring engulfing cells directly interact with the PS molecules on the surfaces of apoptotic cells to trigger phagocytosis (Miyanishi et al., 2007; Park et al., 2007; Ichimura et al., 2008; Park et al., 2008; Park et al., 2009). On the other hand, some phagocytotic receptors recognize PS indirectly via binding to the bridging molecules secreted into the extracellular space to interact with PS. For example, the αVβ3 integrin binds PS via the bridging protein MFG-E8 (Hanayama et al., 2002), and the Tryo3–Axl–Mer (TAM) family of receptors interact with PS via the bridging molecules Protein S or GAS6 in mammalian cells (Rothlin et al., 2015). PS on the surfaces of dying cells is directly recognized by the phagocytic receptor CED-1 in Caenorhabditis elegans (Zhou et al., 2001b; Li et al., 2015) and by Draper, the homolog of CED-1 in Drosophila (MacDonald et al., 2006; Tung et al., 2013). CED-1 was also proposed to interact with PS through TTR-52, a bridging molecule (Wang et al., 2010), indicating that it can use both mechanisms to recognize apoptotic cells. In the brain of mammals, microglia and astrocytes are known to engulf and degrade various extracellular materials (Uddin and Lim, 2022). MEGF10, the mammalian CED-1 ortholog, is expressed in astrocytes where it functions to phagocytose apoptotic cells (Scheib et al., 2012; Iram et al., 2016), synapses (Chung et al., 2013; Lee et al., 2021), and amyloid β aggregates (Singh et al., 2010; Fujita et al., 2020), demonstrating the functional conservation among the CED-1 family of proteins in phagocytosis.
In the nematode C. elegans, dominant (d) mutations in specific subunits of the DEG/ENaC super-family of sodium channels, Ca2+ channels, and trimeric G proteins induce specific neurons to undergo excitotoxic necrosis (Vlachos and Tavernarakis, 2010). In particular, dominant mutations in mec-4, which encodes a core subunit of a multimeric DEG/ENaC sodium channel expressed explicitly in the six mechanosensory (touch) neurons (Figure 1A), trigger the necrosis of these neurons during embryogenesis (Chalfie and Sulston, 1981; Driscoll and Chalfie, 1991). In mec-4(d) mutants, necrotic neurons swell to many times their original sizes and are easily distinguishable from living or apoptotic cells under Differential Interference Contrast (DIC) microscope (Figures 1B, C) (Chalfie and Sulston, 1981; Hall et al., 1997). Unlike apoptosis, mec-4(d)-induced necrosis does not require the CED-3 caspase activity (Ellis and Horvitz, 1986). Instead, the MEC-4(d) mutations render the mechanosensory Na+ channel permeable to Ca2+ by altering the channel conformation and in this manner, induce touch neurons to undergo excitotoxic necrosis (Driscoll and Chalfie, 1991; Bianchi et al., 2004). PS on the surfaces of necrotic cells can be detected by a secreted, fluorescently-tagged MFG-E8 reporter, a high-affinity and high-specificity PS-binding protein (Figure 1B) (Hanayama et al., 2002; Li et al., 2015). In addition to the touch neurons, other sensory neurons, motor neurons, and interneurons that undergo necrosis also expose PS, indicating that PS exposure is not a cell type-specific phenomenon (Furuta et al., 2021). That PS is detected on the surfaces of both the necrotic and apoptotic cells indicates that PS could serve as a common “eat me” signal for cells that die of different mechanisms to attract engulfing cells. Indeed, in C. elegans, the PS molecules on the necrotic and apoptotic cells' surfaces are recognized by the phagocytic receptor CED-1 localized on the plasma membrane of neighboring hypodermal cells, which subsequently engulf these cells (Li et al., 2015).
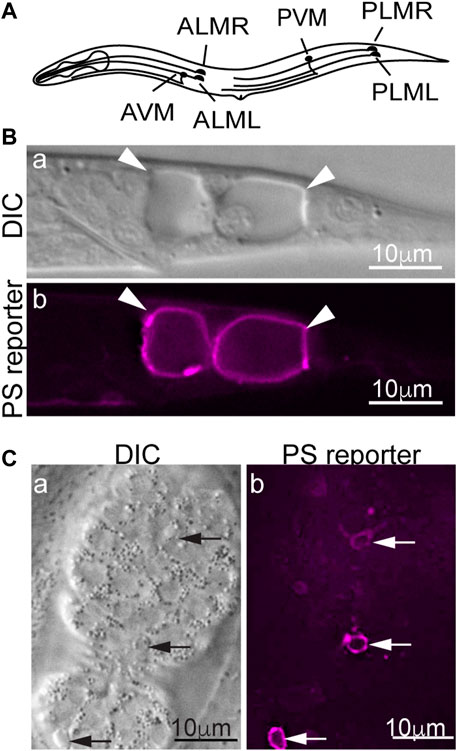
FIGURE 1. PS is detected on the surfaces of necrotic and apoptotic cells in Caenorhabditis elegans. MFG-E8 is a high-affinity PS-binding protein. Secreted MFG-E8mKate2 (the PS reporter) interacts explicitly with PS exposed on cell surfaces. (A) A diagram showing the positions and names of the six Caenorhabditis elegans touch neurons. (B) Differential Interference Contrast (DIC) and epifluorescence images of the tail of a mec-4(d) L1-stage larva carrying the MFG-E8::mKate2 reporter. Arrowheads mark the necrotic PLML and PLMR neurons. (C) DIC and epifluorescence images of a mid-stage embryo expressing MFG-E8::mKate2. Arrows indicate three apoptotic cells that expose PS.
Similarly, in mammals, PS has been reported to serve as an “eat me” signal for necrotic cells, although other “eat me” signals that attract phagocytes to necrotic cells, including the complement factors and histidine-rich glycoproteins, were also reported (Gaipl et al., 2001; Böttcher et al., 2006; Poon et al., 2010a). Please see (Krysko et al., 2006b; Westman et al., 2019), two excellent reviews for detailed descriptions of these other “eat me” signal molecules for necrotic cells. Reports have demonstrated that PS is externalized on the surfaces of necrotic mouse hybridoma cells induced by heat (Cocco and Ucker, 2001), necrotic human peripheral blood lymphocytes induced by heat (Böttcher et al., 2006), and necrotic mouse thymocytes induced by heat or H2O2 (Budai et al., 2019), all of which eventually attract macrophages. Various cells undergoing necroptosis, a death receptor-mediated programmed necrosis, including mouse fibrosarcoma cells, mouse embryonic fibroblast, human myelomonocyte, human keratinocyte, and mouse bone marrow-derived macrophage also externalize PS (Brouckaert et al., 2004; Gong et al., 2017; Zargarian et al., 2017). In addition, cells undergoing other types of regulated necrosis, such as pyroptosis and ferroptosis, also expose PS on their surfaces (de Vasconcelos et al., 2019; Klöditz and Fadeel, 2019). PS exposure promotes the engulfment of these cells (Brouckaert et al., 2004; Zargarian et al., 2017). Like in apoptotic cells, the PS receptors Tim-4, αVβ3 integrin, and the TAM receptors were reported to mediate the PS-dependent phagocytosis of necrotic mouse thymocytes (Budai et al., 2019).
The mechanisms of PS exposure on the surfaces of apoptotic cells
Molecular mechanisms of PS exposure of apoptotic cells have been extensively reviewed previously (Segawa and Nagata, 2015; Nagata et al., 2020). This article will briefly describe the findings reported in the literature. In mammalian living cells, PS is maintained almost exclusively in the inner leaflet of the plasma membrane, at least partly by the flippases (also called aminophospholipid translocases), the P4-type ATPases ATP11A and ATP11C, which flip PS from the outer to the inner leaflet in an ATP-dependent manner (Segawa et al., 2014; Segawa et al., 2016). In addition, CDC50A, a chaperon protein, plays an essential role in ensuring the plasma membrane localization and activities of ATP11A and ATP11C (Segawa et al., 2014; Segawa et al., 2016). CDC50A also acts as a chaperon for nine additional P4-type ATPases (Shin and Takatsu, 2020). ATP11C facilitates phospholipids translocation in all leukocytes (Yabas et al., 2016). ATP11C knockout mice display severe B-cell deficiency due to the remaining of PS on the surfaces of B cell precursors, which stimulates the engulfment of pre-B cells by phagocytes (Yabas et al., 2011; Segawa et al., 2018). This defect demonstrates that the asymmetrical enrichment of PS in the inner leaflet of the plasma membrane is essential for protecting cells from phagocytes.
In mouse W3 cells and human Jurkat cells induced to undergo apoptosis, activated caspase 3 has been reported to cleave and inactivate ATP11A and ATP11C (Segawa et al., 2014) (Figure 2A). However, mere inactivation of these flippases is not sufficient for maintaining a substantial level of PS exposure on the surfaces of apoptotic cells. Phospholipid scramblases, which bidirectionally translocate phospholipids between the two membrane leaflets (Bevers and Williamson, 2016), play active roles in exposing PS to the outer surfaces of apoptotic cells in both mammals and C. elegans. Suzuki et al. (2013) have identified the essential function of XK-related protein 8 (Xkr8), a phospholipid scramblase, in the exposure of PS on the surfaces of apoptotic cells from multiple human and mouse cell lines. Xkr8 has a caspase-recognition site that is cleaved by the activated mammalian caspase 3 in the cells undergoing apoptosis; this cleavage event activates the scramblase activity of Xkr8 (Suzuki et al., 2013) (Figure 2A). A recent study indicates that in addition to caspase cleavage, phosphorylation of XKr8 also promotes PS exposure (Sakuragi et al., 2019). Studies of Xkr8 knockout mice found that Xkr8 mediates the exposure of PS on the surfaces of apoptotic lymphocytes and aged neutrophils; furthermore, the lack of Xkr8 results in lupus-like autoimmune disease due to the defect of the clearance of apoptotic lymphocytes and aged neutrophils, indicating that the lack of PS exposure on apoptotic cells perturbed their removal (Kawano and Nagata, 2018). In mammalian apoptotic cells, in addition to Xkr8, the mammalian ATP Binding Cassette Subfamily A Members 1 and 7 (ABCA1 and ABCA7) transporters contribute another PS externalization activity (Alder-Baerens et al., 2005; Quazi and Molday, 2013). Both ABCA1 and ABCA7 facilitate the clearance of apoptotic cells (Hamon et al., 2000; Jehle et al., 2006), although controversial results have also been reported (Williamson et al., 2007). So far, whether ABCA1 and ABCA7 transporters are regulated by caspase activity remains elusive.
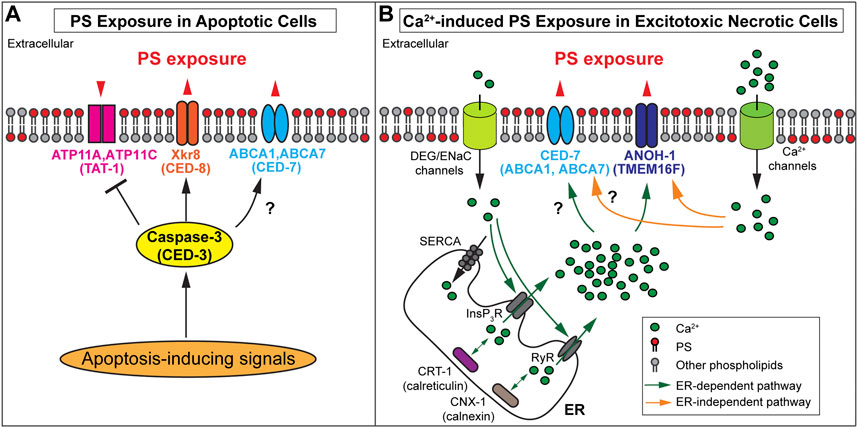
FIGURE 2. Mechanisms of PS exposure on apoptotic cells and necrotic cells (A) Protein names in paratheses are the Caenorhabditis elegans homologs of the mammalian proteins. In apoptotic cells, activated caspase-3 cleaves PS scramblases Xkr8 or CED-8 to trigger PS scrambling activity and cleaves the flippases ATP11A and ATP11C to inactivate the PS flipping activity. It is unclear whether the Caenorhabditis elegans homolog TAT-1 is inactivated by CED-3. Furthermore, ABCA1 and CED-7 are necessary for PS exposure on apoptotic cells. The black question mark indicates whether the caspase regulates these transporters remains elusive. (B) Protein names in paratheses are the mammalian homologs of the Caenorhabditis elegans proteins. In excitotoxic necrotic cells, Ca2+ influx through various ion channels elevates cytoplasmic Ca2+ levels in ER-dependent or ER-independent mechanisms. InsP3R (InsP3 receptor) and RyR (Ryanodine receptor) are ER Ca2+- release channels. SERCA (Sarco-Endoplasmic Reticulum Ca2+ ATPase) is a Ca2+ reuptake pump. Two ER chaperons, calreticulin, and calnexin are essential in establishing the Ca 2+ pool in the ER. The increased cytoplasmic Ca2+ is proposed to interact with ANOH-1 or TMEM16F to activate their PS scramblase activities. In addition, CED-7 is necessary to induce PS exposure in excitotoxic necrotic cells. However, it remains unknown whether its mammalian homolog(s) plays the same role and whether these transporters are regulated by cytoplasmic Ca2+. (A-B) Note that the biochemical activities of C. elegans TAT-1, CED-1, and ANOH-1 have not been examined.
PS exposure of apoptotic cells was also extensively studied in C. elegans, and most key molecular mechanisms are evolutionarily conserved. Caenorhabditis elegans have a close homolog for each of the mammalian ATP11A (TAT-1) (Darland-Ransom et al., 2008), Xkr8 (CED-8) (Stanfield and Horvitz, 2000), and ABCA1 (CED-7) (Wu and Horvitz, 1998a). These proteins were reported to function similarly to their mammalian homologs in regulating the exposure of PS on cell surfaces, that is, CED-8 and CED-7 promote PS exposure (Venegas and Zhou, 2007; Chen et al., 2013; Suzuki et al., 2013), whereas TAT-1 is proposed to maintain the asymmetric distribution of PS in the plasma membrane of living cells (Darland-Ransom et al., 2008). Both TAT-1 and CHAT-1, the C. elegans homolog of CDC50A, are required for the PS distribution to the cytoplasmic side of intracellular vesicles in the endocytosis pathway, yet the role of CHAT-1 in the PS exposure on plasma membrane has not been tested (Chen et al., 2010). The cleavage of CED-8 by CED-3, the C. elegans ortholog of caspase 3, is reported to activate the PS-exposure activity of CED-8 (Chen et al., 2013; Suzuki et al., 2013). Whether CED-3 activates CED-7 and inactivates TAT-1 through protease cleavage remains to be examined. In addition, an independent report indicates that knocking down tat-1 via RNA interference abrogates PS exposure instead of facilitating it (Zullig et al., 2007), casting some doubt on the proposed role of TAT-1 in suppressing PS exposure on living cells.
Cytoplasmic Ca2+ induces PS exposure on excitotoxic necrotic cells
PS is exposed on the surfaces of necrotic cells before cell rupture
Despite the common belief that PS is detected on the surfaces of necrotic cells due to the rupture of necrotic cell membranes (Atkin-Smith, 2021), the C. elegans necrotic neurons expose PS while maintaining the integrity of the plasma membrane (Li et al., 2015; Furuta et al., 2021). They are subsequently engulfed as intact cells (Hall et al., 1997; Li et al., 2015). These facts suggest that necrotic cells are able to expose PS actively. The PS exposure on necroptotic mammalian fibroblast cell lines (NIH 3T3 and L929) occurs preceding the loss of the plasma membrane integrity, again indicating that PS is actively exposed on cell surfaces before membrane disruption (Gong et al., 2017; Zargarian et al., 2017).
A mechanism that is dependent on the release of Ca2+ from the endoplasmic reticulum
Caspase activity is not necessary for inducing excitotoxic necrosis in C. elegans (Ellis and Horvitz, 1986; Chalfie and Wolinsky, 1990; Nagarajan et al., 2014); In excitotoxic necrosis, studies indicate that the elevation of intracellular Ca2+ levels plays a vital role in the induction of necrosis (Xu et al., 2001; Berliocchi et al., 2005; Celsi et al., 2009; Bano and Ankarcrona, 2018). Electrophysiological studies revealed that the MEC-4(d) mutations in C. elegans alter the conformation of the mechanosensory Na+ channel (of which MEC-4 is a subunit), allowing Ca2+ to enter the cytoplasm through this channel (Bianchi et al., 2004). By monitoring the signal intensity of an in vivo Ca2+ reporter specifically expressed in touch neurons in the developing mec-4(d) mutant embryos, it was observed that the cytoplasmic Ca2+ level increases in the touch neurons before cell swelling, a feature of necrosis, prior to PS exposure on necrotic cell surfaces Furuta et al., (2021); Furuta and Zhou (2021). Quantitative analysis found a strong correlation between the levels of cytoplasmic Ca2+ and the levels of PS exposure. The endoplasmic reticulum (ER) is a major intracellular Ca2+ storage pool (Sammels et al., 2010). The release of Ca2+ from the ER to the cytoplasm is essential for the induction of necrosis in mec-4(d) mutants (Xu et al., 2001). Inactivation of a major ER Ca2+-release channel, or an ER-located Ca2+-chaperon essential for the establishment of the Ca2+ pool inside the ER lumen, greatly reduces the level of PS exposed on the surfaces of necrotic cells in dominant mutant strains of mec-4, unc-8, and deg-1, each of which encodes a different subunit of the DEG/ENaC channel (Furuta et al., 2021). Furthermore, artificially increasing the cytoplasmic Ca2+ level through inactivating Sarco-Endoplasmic Reticulum Ca2+ ATPase (SERCA), the Ca2+ reuptake pump on the ER membrane (Treiman et al., 1998; Doan et al., 2015), induces both necrosis and PS exposure on necrotic cells (Furuta et al., 2021). In addition, inhibition of downstream necrotic events blocks the morphological changes of touch neurons but not the PS exposure (Furuta et al., 2021). Together, the above results indicate that 1) the increase of cytoplasmic Ca2+ induces both necrosis and PS exposure, 2) The Ca2+ leaked into touch neurons through the mutant DEG/ENaC channel in the mec-4(d), unc-8(d), or deg-1(d) mutant strains is necessary but not sufficient to induce PS exposure or necrosis; instead, the Ca2+-induced Ca2+ release from the ER, which further elevates the cytoplasmic Ca2+ level, is necessary for the induction of PS exposure and necrosis, and 3) the induction of PS exposure is independent of the induction of necrosis (Figure 2B). These results further implicate the existence of different proteins that act to induce various cellular events, such as PS exposure and cell swelling, in response to the elevated level of cytoplasmic Ca2+.
An ER-independent and Ca2+-dependent PS exposure mechanism
A dominant, constitutively active mutation of deg-3, which encodes a ligand-gated calcium channel belonging to the nicotinic acetylcholine receptor family (Treinin and Chalfie, 1995), like mec-4(d) mutations, results in the excitotoxic necrosis of neurons (Chalfie and Wolinsky, 1990). However, unlike mec-4(d) mutations, this mutation induces necrosis and PS exposure in a manner independent of the contribution of the ER Ca2+ pool (Xu et al., 2001; Furuta et al., 2021). The DEG-3 Ca2+ channel has high Ca2+ permeability, and the deg-3(u662) dominant mutation causes this channel to remain open constitutively (Treinin et al., 1998). In addition to deg-3(d), a dominant mutation in trp-4, which encodes a transient receptor potential (TRP) channel, another Ca2+ channel (Li et al., 2006), also induces the necrosis of neurons and PS exposure (Nagarajan et al., 2014). Interestingly, although trp-4(d)-induced necrosis is dependent on the ER Ca2+ release (Nagarajan et al., 2014), trp-4(d)-induced PS exposure is not (Furuta et al., 2021). TRP channels are highly permeable to Ca2+ (Gees et al., 2010). The TRP-4(d) mutation is likely to keep the Ca2+ channel in a constitutively open state. Together, the differential requirements for the ER contribution to PS exposure observed from the constitutively open DEG/ENaC channel and Ca2+ channels suggest that the critical factor that triggers PS exposure is the cytoplasmic Ca2+ level. When a mutant Ca2+ channel allows constitutive and high permeability influx of Ca2+ into neurons, the contribution of the Ca2+ from the ER pool is not necessary; on the other hand, when a mutant ion channel only allows a trickling amount of Ca2+ to enter a neuron, the “Ca2+-induced Ca2+ release” from the ER becomes essential for inducing PS exposure.
It is worth noting that a few previous studies indicated the role of Ca2+ in inducing PS exposure on apoptotic cells (Hampton et al., 1996; Verhoven et al., 1999; Zhivotovsky and Orrenius, 2011). On the other hand, lines of evidence demonstrate against the involvement of Ca2+ in PS exposure on the surfaces of apoptotic cells (Hampton et al., 1996; Schoenwaelder et al., 2009). In C. elegans, Furuta et al. (2021) demonstrated that impairing the release of ER Ca2+ into the cytoplasm did not affect the level of the PS on the surfaces of apoptotic cells. In conclusion, two different kinds of mechanisms regulate PS exposure: apoptotic cells utilize Ca2+-independent PS exposure driven by caspase activity, while necrotic cells utilize Ca2+-dependent mechanisms (Figure 2).
Scramblase and flippasses that play roles in Ca2+-dependent PS exposure on excitotoxic necrotic cells
Mammalian platelets activated during blood coagulation expose PS in response to Ca2+ influx but do not undergo cell death (Bevers et al., 1983; Dachary-Prigent et al., 1995). On the other hand, when platelets are induced to undergo apoptosis, they expose PS in a caspase-dependent yet Ca2+-independent manner (Schoenwaelder et al., 2009). These results again demonstrate the existence of both the Ca2+-dependent and -independent PS exposure mechanisms in platelets. Mammalian TMEM16F is a high-affinity Ca2+-binding protein and a Ca2+-dependent scramblase that is responsible for exposing PS when platelets are activated (Suzuki et al., 2010; Fujii et al., 2015). A mutation in TMEM16F was reported to cause Scott syndrome, a human bleeding disorder (Suzuki et al., 2010), demonstrating the essential role of human TMEM16F in blood clotting.
ANOH-1 is the C. elegans homolog of mammalian TMEM16F (Wang et al., 2013; Li et al., 2015). It is specifically expressed in neurons and presented on the plasma membrane of the excitotoxic necrotic touch neuron (Li et al., 2015). The amino acid sequences essential for the Ca2+-dependent PS scrambling activity identified in mammalian and the fungus Nectria haematococca TMEM16F (Suzuki et al., 2010; Brunner et al., 2014) are conserved in C. elegans ANOH-1 (Li et al., 2015). Deletion in the anoh-1 gene causes the reduction of PS exposure on neurons undergoing necrosis induced by the mec-4(d) and trp-4(d), suggesting that the Ca2+-induced PS exposure is dependent on ANOH-1 (Li et al., 2015; Furuta et al., 2021) (Figure 2B). In anoh-1(−); mec-4(d) double mutant larvae, necrotic touch neurons linger much longer before being engulfed, presumably as a result of the reduction of PS exposure activity (Li et al., 2015). On the other hand, anoh-1 deletion does not affect the exposure of PS on apoptotic cells (Li et al., 2015). Although its biochemical activity has not been determined, C. elegans ANOH-1 is proposed to act as a Ca2+-dependent phospholipid scramblase specific for necrotic cells (Li et al., 2015; Furuta et al., 2021).
Li et al. (2015) identified not only C. elegans ANOH-1 but also another C. elegans protein that promotes PS exposure on necrotic neurons. This protein is CED-7, the C. elegans homolog of ABCA1 transporter (Li et al., 2015). In ced-7 loss-of-function mutants, PS exposure on both apoptotic and necrotic cells are greatly reduced in C. elegans (Venegas and Zhou, 2007; Li et al., 2015). In ced-7; anoh-1 double mutants, the PS exposure defect is more severe than in each single mutant, and the necrotic touch neurons persist for much longer before being engulfed than in each of the anoh-1 or ced-7 single mutants, indicating that ANOH-1 and CED-7 act in parallel to promote PS exposure and the engulfment of necrotic cells (Li et al., 2015). ced-7 mutant phenotypes indicate that a common molecular mechanism that involved CED-7 is needed in both apoptotic and necrotic cells for efficient PS exposure. It is unclear whether C. elegans CED-7 is regulated by Ca2+ in necrotic cells (Figure 2B). Whether mammalian ABCA1 and ABCA7 play any role in the PS exposure on necrotic cells also awaits to be tested.
In addition to the scramblase activity, Ca2+ might also inactivate flippase(s) to “maintain” the exposed PS on the surface of necrotic cells. Segawa et al. (2018) reported that the PS exposed on the surfaces of lymphoma cells induced by Ca2+-ionophore is quickly internalized when the ionophore is removed. Yet, in ATP11A−/−ATP11C−/- double deletion cells, PS remains on the outer leaflet persistently. These observations indicate that cytoplasmic Ca2+ regulates flippase activities. Together, all the above information indicates that in necrotic cells, the increase of the cytoplasmic Ca2+ level might activate scramblase(s) and inactivate flippase(s) simultaneously to achieve continuous PS exposure. The mechanisms of how cytoplasmic Ca2+ induces PS exposure on necrotic cells need much further investigation.
Ca2+-independent PS-exposure on the surfaces of necrotic cells
Interestingly, a recent study conducted in TNF-induced necroptotic mouse embryonic fibroblast cells has found that although both Ca2+ influx and PS exposure occur when cells undergo necroptosis, PS exposure does not require either Ca2+ influx or caspase activity (Gong et al., 2017). This study further implicates that mixed lineage kinase-like (MLKL), which mediates membrane disruption in necroptosis, directly causes the exposure of PS. Another study showed that activated MLKL promotes PS exposure on interferon (IFN)-ɣ induced necroptotic mouse embryonic fibroblast cells and human colorectal adenocarcinoma cells (Chen et al., 2019), supporting the PS exposure-promoting role of MLKL in necroptotic cells.
In another example, a high dose of extracellular ATP induces PS exposure on mammalian T cells and other cell types and the eventual necrosis (Taylor et al., 2008; Ryoden et al., 2022). ATP causes these effects through binding to P2X7, an ATP-gated non-selective cation channel (Ryoden and Nagata, 2022). Activation of P2X7 was reported to cause Ca2+ influx (North, 2002; Ousingsawat et al., 2015). Furthermore, the Ca2+-dependent scramblase TMEM16F was reported to promote PS exposure in response to ATP in the HEK293 cells (Ousingsawat et al., 2015). However, a recent study reported that TMEM16F is dispensable for the P2X7-mediated PS exposure in a WR19L mouse lymphoblast-derived cell line; instead, Xk, a paralogue of the Xkr8 scramblase, and VPS13A, a cytoplasmic lipid transporter, are necessary for this event (Ryoden et al., 2022). Therefore, whether Ca2+ is required for this type of PS exposure is a matter of debate that needs further investigation. These studies nevertheless suggest that the requirement of Ca2+ for PS exposure might depend on how the necrosis is induced and the types of cells that undergo necrosis.
Summary
Here we summarize a few features behind the molecular mechanisms that regulate PS exposure on cell surfaces. First, cells die of apoptosis and necrosis, two different death mechanisms, expose the same “eat me” signal–PS and likely attract the same phagocytic receptor(s). For example, in C. elegans, the phagocytic receptor CED-1 recognizes both apoptotic and necrotic cells and initiates the engulfment of both kinds of dying cells. However, the upstream mechanisms that promote PS exposure are strikingly different between apoptotic and necrotic cells. Whereas apoptotic cells rely on caspase-mediated PS-exposure mechanisms, excitotoxic necrosis depends on a high level of cytoplasmic Ca2+ to induce PS exposure. The activation of different scramblases during apoptosis and necrosis discovered in C. elegans demonstrated the differential molecular mechanisms of PS exposure. On the other hand, C. elegans CED-7 facilitates PS exposure on both apoptotic and necrotic cells, suggesting that either CED-7 provides a basal-level, constitutive PS-exposure activity that is counteracted by the PS flippase(s) in living cells, or that CED-7 can be activated by multiple upstream signals. Nevertheless, both common and differential mechanisms are utilized to regulate PS exposure on dying cell surfaces. Interestingly, necroptotic cells and certain types of cells induced to undergo necrosis by ATP seem to apply specific Ca2+-independent mechanisms for PS exposure. Necrosis can be induced by various kinds of stimuli. The detailed mechanisms of PS exposure, which might be stimuli-specific and cell type-specific, await further investigation. Further dissecting the PS exposure mechanisms would help us understand the cell clearance mechanisms, especially in the disease context, and may serve as a critical step for future therapeutic intervention.
Author contributions
YF produced the first draft of the manuscript. YF and ZZ both revised the manuscript.
Funding
NIH R01GM104279: This grant supported the initial preparation of the submitted article. This grant is already completed. NIH R01NS129847: This grant support the completion of the submitted review article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alder-Baerens, N., Muller, P., Pohl, A., Korte, T., Hamon, Y., Chimini, G., et al. (2005). Headgroup-specific exposure of phospholipids in ABCA1-expressing cells. J. Biol. Chem. 280, 26321–26329. doi:10.1074/jbc.M413993200
Apolloni, S., Amadio, S., Parisi, C., Matteucci, A., Potenza, R. L., Armida, M., et al. (2014). Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis. models Mech. 7, 1101–1109. doi:10.1242/dmm.017038
Atkin-Smith, G. K. (2021). Phagocytic clearance of apoptotic, necrotic, necroptotic and pyroptotic cells. Biochem. Soc. Trans. 49, 793–804. doi:10.1042/BST20200696
Balasubramanian, K., and Schroit, A. J. (2003). Aminophospholipid asymmetry: A matter of life and death. Annu. Rev. Physiol. 65, 701–734. doi:10.1146/annurev.physiol.65.092101.142459
Bano, D., and Ankarcrona, M. (2018). Beyond the critical point: An overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci. Lett. 663, 79–85. doi:10.1016/j.neulet.2017.08.048
Berliocchi, L., Bano, D., and Nicotera, P. (2005). Ca2+ signals and death programmes in neurons. Philosophical Trans. R. Soc. Lond. Ser. B, Biol. Sci. 360, 2255–2258. doi:10.1098/rstb.2005.1765
Bevers, E. M., Comfurius, P., and Zwaal, R. F. (1983). Changes in membrane phospholipid distribution during platelet activation. Biochimica biophysica acta 736, 57–66. doi:10.1016/0005-2736(83)90169-4
Bevers, E. M., and Williamson, P. L. (2016). Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol. Rev. 96, 605–645. doi:10.1152/physrev.00020.2015
Bianchi, L., Gerstbrein, B., Frokjaer-Jensen, C., Royal, D. C., Mukherjee, G., Royal, M. A., et al. (2004). The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: Implications for necrosis initiation. Nat. Neurosci. 7, 1337–1344. doi:10.1038/nn1347
Böttcher, A., Gaipl, U. S., Fürnrohr, B. G., Herrmann, M., Girkontaite, I., Kalden, J. R., et al. (2006). Involvement of phosphatidylserine, alphavbeta3, CD14, CD36, and complement C1q in the phagocytosis of primary necrotic lymphocytes by macrophages. Arthritis rheumatism 54, 927–938. doi:10.1002/art.21660
Brouckaert, G., Kalai, M., Krysko, D. V., Saelens, X., Vercammen, D., Ndlovu, M., et al. (2004). Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol. Biol. Cell 15, 1089–1100. doi:10.1091/mbc.e03-09-0668
Brunner, J. D., Lim, N. K., Schenck, S., Duerst, A., and Dutzler, R. (2014). X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516, 207–212. doi:10.1038/nature13984
Budai, Z., Ujlaky-Nagy, L., Kis, G. N., Antal, M., Bankó, C., Bacsó, Z., et al. (2019). Macrophages engulf apoptotic and primary necrotic thymocytes through similar phosphatidylserine-dependent mechanisms. FEBS open bio 9, 446–456. doi:10.1002/2211-5463.12584
Butler, C. A., Popescu, A. S., Kitchener, E. J. A., Allendorf, D. H., Puigdellívol, M., and Brown, G. C. (2021). Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 158, 621–639. doi:10.1111/jnc.15327
Celsi, F., Pizzo, P., Brini, M., Leo, S., Fotino, C., Pinton, P., et al. (2009). Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. Biochimica biophysica acta 1787, 335–344. doi:10.1016/j.bbabio.2009.02.021
Chalfie, M., and Sulston, J. (1981). Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev. Biol. 82, 358–370. doi:10.1016/0012-1606(81)90459-0
Chalfie, M., and Wolinsky, E. (1990). The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature 345, 410–416. doi:10.1038/345410a0
Challa, S., and Chan, F. K. (2010). Going up in flames: Necrotic cell injury and inflammatory diseases. Cell. Mol. life Sci. CMLS 67, 3241–3253. doi:10.1007/s00018-010-0413-8
Chen, B., Jiang, Y., Zeng, S., Yan, J., Li, X., Zhang, Y., et al. (2010). Endocytic sorting and recycling require membrane phosphatidylserine asymmetry maintained by TAT-1/CHAT-1. PLoS Genet. 6, e1001235. doi:10.1371/journal.pgen.1001235
Chen, J., Kuroki, S., Someda, M., and Yonehara, S. (2019). Interferon-γ induces the cell surface exposure of phosphatidylserine by activating the protein MLKL in the absence of caspase-8 activity. J. Biol. Chem. 294, 11994–12006. doi:10.1074/jbc.RA118.007161
Chen, Y. Z., Mapes, J., Lee, E. S., Skeen-Gaar, R. R., and Xue, D. (2013). Caspase-mediated activation of Caenorhabditis elegans CED-8 promotes apoptosis and phosphatidylserine externalization. Nat. Commun. 4, 2726. doi:10.1038/ncomms3726
Chung, W. S., Clarke, L. E., Wang, G. X., Stafford, B. K., Sher, A., Chakraborty, C., et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. doi:10.1038/nature12776
Cocco, R. E., and Ucker, D. S. (2001). Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol. Biol. Cell 12, 919–930. doi:10.1091/mbc.12.4.919
Dachary-Prigent, J., Pasquet, J. M., Freyssinet, J. M., and Nurden, A. T. (1995). Calcium involvement in aminophospholipid exposure and microparticle formation during platelet activation: A study using Ca2+-ATPase inhibitors. Biochemistry 34, 11625–11634. doi:10.1021/bi00036a039
Darland-Ransom, M., Wang, X., Sun, C. L., Mapes, J., Gengyo-Ando, K., Mitani, S., et al. (2008). Role of C. elegans TAT-1 protein in maintaining plasma membrane phosphatidylserine asymmetry. Science 320, 528–531. doi:10.1126/science.1155847
de Vasconcelos, N. M., Van Opdenbosch, N., Van Gorp, H., Parthoens, E., and Lamkanfi, M. (2019). Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell death Differ. 26, 146–161. doi:10.1038/s41418-018-0106-7
Doan, N. T., Paulsen, E. S., Sehgal, P., Moller, J. V., Nissen, P., Denmeade, S. R., et al. (2015). Targeting thapsigargin towards tumors. Steroids 97, 2–7. doi:10.1016/j.steroids.2014.07.009
Driscoll, M., and Chalfie, M. (1991). The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 349, 588–593. doi:10.1038/349588a0
Elliott, M. R., and Ravichandran, K. S. (2016). The dynamics of apoptotic cell clearance. Dev. Cell 38, 147–160. doi:10.1016/j.devcel.2016.06.029
Ellis, H. M., and Horvitz, H. R. (1986). Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829. doi:10.1016/0092-8674(86)90004-8
Fujii, T., Sakata, A., Nishimura, S., Eto, K., and Nagata, S. (2015). TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. U. S. A. 112, 12800–12805. doi:10.1073/pnas.1516594112
Fujita, Y., Maeda, T., Sato, C., Sato, M., Hatakeyama, H., Ota, Y., et al. (2020). Engulfment of toxic amyloid β-protein in neurons and astrocytes mediated by MEGF10. Neuroscience 443, 1–7. doi:10.1016/j.neuroscience.2020.07.016
Furuta, Y., Pena-Ramos, O., Li, Z., Chiao, L., and Zhou, Z. (2021). Calcium ions trigger the exposure of phosphatidylserine on the surface of necrotic cells. PLoS Genet. 17, e1009066. doi:10.1371/journal.pgen.1009066
Furuta, Y., and Zhou, Z. (2021). Simultaneous monitoring cytoplasmic calcium ion and cell surface phosphatidylserine in the necrotic touch neurons of Caenorhabditis elegans. Bio-protocol 11, e4187. doi:10.21769/BioProtoc.4187
Gaipl, U. S., Kuenkele, S., Voll, R. E., Beyer, T. D., Kolowos, W., Heyder, P., et al. (2001). Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell death Differ. 8, 327–334. doi:10.1038/sj.cdd.4400826
Gees, M., Colsoul, B., and Nilius, B. (2010). The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2, a003962. doi:10.1101/cshperspect.a003962
Gong, Y. N., Guy, C., Olauson, H., Becker, J. U., Yang, M., Fitzgerald, P., et al. (2017). ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169, 286–300.e16. e216. doi:10.1016/j.cell.2017.03.020
Hall, D. H., Gu, G., Garcia-Anoveros, J., Gong, L., Chalfie, M., and Driscoll, M. (1997). Neuropathology of degenerative cell death in Caenorhabditis elegans. J. Neurosci. official J. Soc. Neurosci. 17, 1033–1045. doi:10.1523/JNEUROSCI.17-03-01033.1997
Hamon, Y., Broccardo, C., Chambenoit, O., Luciani, M. F., Toti, F., Chaslin, S., et al. (2000). ABC1 promotes engulfment of apoptotic cells and transbilayer redistribution of phosphatidylserine. Nat. Cell Biol. 2, 399–406. doi:10.1038/35017029
Hampton, M. B., Vanags, D. M., Porn-Ares, M. I., and Orrenius, S. (1996). Involvement of extracellular calcium in phosphatidylserine exposure during apoptosis. FEBS Lett. 399, 277–282. doi:10.1016/s0014-5793(96)01341-5
Hanayama, R., Tanaka, M., Miwa, K., Shinohara, A., Iwamatsu, A., and Nagata, S. (2002). Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187. doi:10.1038/417182a
Harayama, T., and Riezman, H. (2018). Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 19, 281–296. doi:10.1038/nrm.2017.138
Ichimura, T., Asseldonk, E. J., Humphreys, B. D., Gunaratnam, L., Duffield, J. S., and Bonventre, J. V. (2008). Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J. Clin. investigation 118, 1657–1668. doi:10.1172/JCI34487
Iram, T., Ramirez-Ortiz, Z., Byrne, M. H., Coleman, U. A., Kingery, N. D., Means, T. K., et al. (2016). Megf10 is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J. Neurosci. official J. Soc. Neurosci. 36, 5185–5192. doi:10.1523/JNEUROSCI.3850-15.2016
Jehle, A. W., Gardai, S. J., Li, S., Linsel-Nitschke, P., Morimoto, K., Janssen, W. J., et al. (2006). ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J. Cell Biol. 174, 547–556. doi:10.1083/jcb.200601030
Kawano, M., and Nagata, S. (2018). Lupus-like autoimmune disease caused by a lack of Xkr8, a caspase-dependent phospholipid scramblase. Proc. Natl. Acad. Sci. U. S. A. 115, 2132–2137. doi:10.1073/pnas.1720732115
Klöditz, K., and Fadeel, B. (2019). Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell death Discov. 5, 65. doi:10.1038/s41420-019-0146-x
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., et al. (2009). Classification of cell death: Recommendations of the nomenclature committee on cell death 2009. Cell death Differ. 16, 3–11. doi:10.1038/cdd.2008.150
Krysko, D. V., D'Herde, K., and Vandenabeele, P. (2006a). Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 11, 1709–1726. doi:10.1007/s10495-006-9527-8
Krysko, D. V., Denecker, G., Festjens, N., Gabriels, S., Parthoens, E., D'Herde, K., et al. (2006b). Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell death Differ. 13, 2011–2022. doi:10.1038/sj.cdd.4401900
Lai, T. W., Zhang, S., and Wang, Y. T. (2014). Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 115, 157–188. doi:10.1016/j.pneurobio.2013.11.006
Lee, J. H., Kim, J. Y., Noh, S., Lee, H., Lee, S. Y., Mun, J. Y., et al. (2021). Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 590, 612–617. doi:10.1038/s41586-020-03060-3
Li, W., Feng, Z., Sternberg, P. W., and Xu, X. Z. (2006). A C. elegans stretch receptor neuron revealed by a mechanosensitive TRP channel homologue. Nature 440, 684–687. doi:10.1038/nature04538
Li, Z., Venegas, V., Nagaoka, Y., Morino, E., Raghavan, P., Audhya, A., et al. (2015). Necrotic cells actively attract phagocytes through the collaborative action of two distinct PS-exposure mechanisms. PLoS Genet. 11, e1005285. doi:10.1371/journal.pgen.1005285
MacDonald, J. M., Beach, M. G., Porpiglia, E., Sheehan, A. E., Watts, R. J., and Freeman, M. R. (2006). The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50, 869–881. doi:10.1016/j.neuron.2006.04.028
Martin, J. B. (1999). Molecular basis of the neurodegenerative disorders. N. Engl. J. Med. 340, 1970–1980. doi:10.1056/NEJM199906243402507
McCall, K. (2010). Genetic control of necrosis - another type of programmed cell death. Curr. Opin. Cell Biol. 22, 882–888. doi:10.1016/j.ceb.2010.09.002
Miyanishi, M., Tada, K., Koike, M., Uchiyama, Y., Kitamura, T., and Nagata, S. (2007). Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439. doi:10.1038/nature06307
Moquin, D., and Chan, F. K. (2010). The molecular regulation of programmed necrotic cell injury. Trends Biochem. Sci. 35, 434–441. doi:10.1016/j.tibs.2010.03.001
Nagarajan, A., Ning, Y., Reisner, K., Buraei, Z., Larsen, J. P., Hobert, O., et al. (2014). Progressive degeneration of dopaminergic neurons through TRP channel-induced cell death. J. Neurosci. official J. Soc. Neurosci. 34, 5738–5746. doi:10.1523/JNEUROSCI.4540-13.2014
Nagata, S. (2018). Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol. 36, 489. doi:10.1146/annurev-immunol-042617-053010
Nagata, S., Sakuragi, T., and Segawa, K. (2020). Flippase and scramblase for phosphatidylserine exposure. Curr. Opin. Immunol. 62, 31–38. doi:10.1016/j.coi.2019.11.009
Nikoletopoulou, V., and Tavernarakis, N. (2014). Necrotic cell death in Caenorhabditis elegans. Methods Enzym. 545, 127–155. doi:10.1016/B978-0-12-801430-1.00006-8
Noch, E., and Khalili, K. (2009). Molecular mechanisms of necrosis in glioblastoma: The role of glutamate excitotoxicity. Cancer Biol. Ther. 8, 1791–1797. doi:10.4161/cbt.8.19.9762
North, R. A. (2002). Molecular physiology of P2X receptors. Physiol. Rev. 82, 1013–1067. doi:10.1152/physrev.00015.2002
Ousingsawat, J., Wanitchakool, P., Kmit, A., Romao, A. M., Jantarajit, W., Schreiber, R., et al. (2015). Anoctamin 6 mediates effects essential for innate immunity downstream of P2X7 receptors in macrophages. Nat. Commun. 6, 6245. doi:10.1038/ncomms7245
Park, D., Tosello-Trampont, A. C., Elliott, M. R., Lu, M., Haney, L. B., Ma, Z., et al. (2007). Bai1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430–434. doi:10.1038/nature06329
Park, S. Y., Jung, M. Y., Kim, H. J., Lee, S. J., Kim, S. Y., Lee, B. H., et al. (2008). Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell death Differ. 15, 192–201. doi:10.1038/sj.cdd.4402242
Park, S. Y., Jung, M. Y., Lee, S. J., Kang, K. B., Gratchev, A., Riabov, V., et al. (2009). Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J. Cell Sci. 122, 3365–3373. doi:10.1242/jcs.049569
Poon, I. K., Hulett, M. D., and Parish, C. R. (2010a). Histidine-rich glycoprotein is a novel plasma pattern recognition molecule that recruits IgG to facilitate necrotic cell clearance via FcgammaRI on phagocytes. Blood 115, 2473–2482. doi:10.1182/blood-2009-07-234013
Poon, I. K., Hulett, M. D., and Parish, C. R. (2010b). Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell death Differ. 17, 381–397. doi:10.1038/cdd.2009.195
Poon, I. K., Lucas, C. D., Rossi, A. G., and Ravichandran, K. S. (2014). Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 14, 166–180. doi:10.1038/nri3607
Quazi, F., and Molday, R. S. (2013). Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J. Biol. Chem. 288, 34414–34426. doi:10.1074/jbc.M113.508812
Rothlin, C. V., Carrera-Silva, E. A., Bosurgi, L., and Ghosh, S. (2015). TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 33, 355–391. doi:10.1146/annurev-immunol-032414-112103
Ryoden, Y., and Nagata, S. (2022). The XK plasma membrane scramblase and the VPS13A cytosolic lipid transporter for ATP-induced cell death. BioEssays news Rev. Mol. Cell. Dev. Biol. 44, e2200106. doi:10.1002/bies.202200106
Ryoden, Y., Segawa, K., and Nagata, S. (2022). Requirement of Xk and Vps13a for the P2X7-mediated phospholipid scrambling and cell lysis in mouse T cells. Proc. Natl. Acad. Sci. U. S. A. 119, e2119286119. doi:10.1073/pnas.2119286119
Sakuragi, T., Kosako, H., and Nagata, S. (2019). Phosphorylation-mediated activation of mouse Xkr8 scramblase for phosphatidylserine exposure. Proc. Natl. Acad. Sci. U. S. A. 116, 2907–2912. doi:10.1073/pnas.1820499116
Sammels, E., Parys, J. B., Missiaen, L., De Smedt, H., and Bultynck, G. (2010). Intracellular Ca2+ storage in health and disease: A dynamic equilibrium. Cell calcium 47, 297–314. doi:10.1016/j.ceca.2010.02.001
Scheib, J. L., Sullivan, C. S., and Carter, B. D. (2012). Jedi-1 and MEGF10 signal engulfment of apoptotic neurons through the tyrosine kinase Syk. J. Neurosci. official J. Soc. Neurosci. 32, 13022–13031. doi:10.1523/JNEUROSCI.6350-11.2012
Schoenwaelder, S. M., Yuan, Y., Josefsson, E. C., White, M. J., Yao, Y., Mason, K. D., et al. (2009). Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood 114, 663–666. doi:10.1182/blood-2009-01-200345
Schwegler, M., Wirsing, A. M., Dollinger, A. J., Abendroth, B., Putz, F., Fietkau, R., et al. (2015). Clearance of primary necrotic cells by non-professional phagocytes. Biol. Cell 107, 372–387. doi:10.1111/boc.201400090
Segawa, K., Kurata, S., and Nagata, S. (2016). Human type IV P-type ATPases that work as plasma membrane phospholipid flippases and their regulation by caspase and calcium. J. Biol. Chem. 291, 762–772. doi:10.1074/jbc.M115.690727
Segawa, K., Kurata, S., Yanagihashi, Y., Brummelkamp, T. R., Matsuda, F., and Nagata, S. (2014). Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168. doi:10.1126/science.1252809
Segawa, K., and Nagata, S. (2015). An apoptotic 'eat me' signal: Phosphatidylserine exposure. Trends Cell Biol. 25, 639–650. doi:10.1016/j.tcb.2015.08.003
Segawa, K., Yanagihashi, Y., Yamada, K., Suzuki, C., Uchiyama, Y., and Nagata, S. (2018). Phospholipid flippases enable precursor B cells to flee engulfment by macrophages. Proc. Natl. Acad. Sci. U. S. A. 115, 12212–12217. doi:10.1073/pnas.1814323115
Shan, B., Pan, H., Najafov, A., and Yuan, J. (2018). Necroptosis in development and diseases. Genes & Dev. 32, 327–340. doi:10.1101/gad.312561.118
Shin, H. W., and Takatsu, H. (2020). Phosphatidylserine exposure in living cells. Crit. Rev. Biochem. Mol. Biol. 55, 166–178. doi:10.1080/10409238.2020.1758624
Singh, T. D., Park, S. Y., Bae, J. S., Yun, Y., Bae, Y. C., Park, R. W., et al. (2010). MEGF10 functions as a receptor for the uptake of amyloid-β. FEBS Lett. 584, 3936–3942. doi:10.1016/j.febslet.2010.08.050
Stanfield, G. M., and Horvitz, H. R. (2000). The ced-8 gene controls the timing of programmed cell deaths in C. elegans. Mol. Cell 5, 423–433. doi:10.1016/s1097-2765(00)80437-2
Suzuki, J., Denning, D. P., Imanishi, E., Horvitz, H. R., and Nagata, S. (2013). Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406. doi:10.1126/science.1236758
Suzuki, J., Umeda, M., Sims, P. J., and Nagata, S. (2010). Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838. doi:10.1038/nature09583
Taylor, S. R., Gonzalez-Begne, M., Dewhurst, S., Chimini, G., Higgins, C. F., Melvin, J. E., et al. (2008). Sequential shrinkage and swelling underlie P2X7-stimulated lymphocyte phosphatidylserine exposure and death. J. Immunol. 180, 300–308. doi:10.4049/jimmunol.180.1.300
Tovar-y-Romo, L. B., Penagos-Puig, A., and Ramírez-Jarquín, J. O. (2016). Endogenous recovery after brain damage: Molecular mechanisms that balance neuronal life/death fate. J. Neurochem. 136, 13–27. doi:10.1111/jnc.13362
Treiman, M., Caspersen, C., and Christensen, S. B. (1998). A tool coming of age: Thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol. Sci. 19, 131–135. doi:10.1016/s0165-6147(98)01184-5
Treinin, M., and Chalfie, M. (1995). A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron 14, 871–877. doi:10.1016/0896-6273(95)90231-7
Treinin, M., Gillo, B., Liebman, L., and Chalfie, M. (1998). Two functionally dependent acetylcholine subunits are encoded in a single Caenorhabditis elegans operon. Proc. Natl. Acad. Sci. U. S. A. 95, 15492–15495. doi:10.1073/pnas.95.26.15492
Tremblay, M. E., Cookson, M. R., and Civiero, L. (2019). Glial phagocytic clearance in Parkinson's disease. Mol. Neurodegener. 14, 16. doi:10.1186/s13024-019-0314-8
Tung, T. T., Nagaosa, K., Fujita, Y., Kita, A., Mori, H., Okada, R., et al. (2013). Phosphatidylserine recognition and induction of apoptotic cell clearance by Drosophila engulfment receptor Draper. J. Biochem. 153, 483–491. doi:10.1093/jb/mvt014
Tymianski, M. (2011). Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat. Neurosci. 14, 1369–1373. doi:10.1038/nn.2951
Uddin, M. S., and Lim, L. W. (2022). Glial cells in Alzheimer's disease: From neuropathological changes to therapeutic implications. Ageing Res. Rev. 78, 101622. doi:10.1016/j.arr.2022.101622
Venegas, V., and Zhou, Z. (2007). Two alternative mechanisms that regulate the presentation of apoptotic cell engulfment signal in Caenorhabditis elegans. Mol. Biol. Cell 18, 3180–3192. doi:10.1091/mbc.e07-02-0138
Verhoven, B., Krahling, S., Schlegel, R. A., and Williamson, P. (1999). Regulation of phosphatidylserine exposure and phagocytosis of apoptotic T lymphocytes. Cell death Differ. 6, 262–270. doi:10.1038/sj.cdd.4400491
Vlachos, M., and Tavernarakis, N. (2010). Non-apoptotic cell death in Caenorhabditis elegans. Dev. Dyn. 239, 1337–1351. doi:10.1002/dvdy.22230
Wang, X., Li, W., Zhao, D., Liu, B., Shi, Y., Chen, B., et al. (2010). Caenorhabditis elegans transthyretin-like protein TTR-52 mediates recognition of apoptotic cells by the CED-1 phagocyte receptor. Nat. Cell Biol. 12, 655–664. doi:10.1038/ncb2068
Wang, Y., Alam, T., Hill-Harfe, K., Lopez, A. J., Leung, C. K., Iribarne, D., et al. (2013). Phylogenetic, expression, and functional analyses of anoctamin homologs in Caenorhabditis elegans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R1376–R1389. doi:10.1152/ajpregu.00303.2012
Westman, J., Grinstein, S., and Marques, P. E. (2019). Phagocytosis of necrotic debris at sites of injury and inflammation. Front. Immunol. 10, 3030. doi:10.3389/fimmu.2019.03030
Whelan, R. S., Kaplinskiy, V., and Kitsis, R. N. (2010). Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu. Rev. Physiol. 72, 19–44. doi:10.1146/annurev.physiol.010908.163111
Williamson, P., Halleck, M. S., Malowitz, J., Ng, S., Fan, X., Krahling, S., et al. (2007). Transbilayer phospholipid movements in ABCA1-deficient cells. PloS one 2, e729. doi:10.1371/journal.pone.0000729
Wu, Y., and Horvitz, H. R. (1998a). The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell 93, 951–960. doi:10.1016/s0092-8674(00)81201-5
Xu, K., Tavernarakis, N., and Driscoll, M. (2001). Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron 31, 957–971. doi:10.1016/s0896-6273(01)00432-9
Yabas, M., Jing, W., Shafik, S., Bröer, S., and Enders, A. (2016). ATP11C facilitates phospholipid translocation across the plasma membrane of all leukocytes. PloS one 11, e0146774. doi:10.1371/journal.pone.0146774
Yabas, M., Teh, C. E., Frankenreiter, S., Lal, D., Roots, C. M., Whittle, B., et al. (2011). ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat. Immunol. 12, 441–449. doi:10.1038/ni.2011
Yamashima, T. (2004). Ca2+-dependent proteases in ischemic neuronal death: A conserved 'calpain-cathepsin cascade' from nematodes to primates. Cell calcium 36, 285–293. doi:10.1016/j.ceca.2004.03.001
Zargarian, S., Shlomovitz, I., Erlich, Z., Hourizadeh, A., Ofir-Birin, Y., Croker, B. A., et al. (2017). Phosphatidylserine externalization, "necroptotic bodies" release, and phagocytosis during necroptosis. PLoS Biol. 15, e2002711. doi:10.1371/journal.pbio.2002711
Zhang, Q., Hu, X. M., Zhao, W. J., Ban, X. X., Li, Y., Huang, Y. X., et al. (2023). Targeting necroptosis: A novel therapeutic option for retinal degenerative diseases. Int. J. Biol. Sci. 19, 658–674. doi:10.7150/ijbs.77994
Zhivotovsky, B., and Orrenius, S. (2011). Calcium and cell death mechanisms: A perspective from the cell death community. Cell calcium 50, 211–221. doi:10.1016/j.ceca.2011.03.003
Zhou, W., and Yuan, J. (2014). Necroptosis in health and diseases. Semin. Cell Dev. Biol. 35, 14–23. doi:10.1016/j.semcdb.2014.07.013
Zhou, Z., Hartwieg, E., and Horvitz, H. R. (2001b). CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 104, 43–56. doi:10.1016/s0092-8674(01)00190-8
Keywords: apoptotic cells, necrotic cells, phagocytosis, phosphatidylserine, flippases, phospholipid scramblases, Ca2+, caspase
Citation: Furuta Y and Zhou Z (2023) How do necrotic cells expose phosphatidylserine to attract their predators—What’s unique and what’s in common with apoptotic cells. Front. Cell Dev. Biol. 11:1170551. doi: 10.3389/fcell.2023.1170551
Received: 21 February 2023; Accepted: 27 March 2023;
Published: 05 April 2023.
Edited by:
Nu Zhang, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Magnus Olsson, Karolinska Institutet (KI), SwedenDavid Ucker, University of Illinois at Chicago, United States
Copyright © 2023 Furuta and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zheng Zhou, emhlbmd6QGJjbS50bWMuZWR1
†Present address: Yoshitaka Furuta, Memory and Brain Research Center, Department of Neuroscience, Baylor College of Medicine, Houston, TX, United States