 Zining Zhang
Zining Zhang Heng Zhang1,2†
Heng Zhang1,2† Xiang Liao
Xiang Liao Hsiang-i Tsai
Hsiang-i Tsai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 20 April 2023
Sec. Cancer Cell Biology
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1147676
This article is part of the Research TopicKinases for Cancer Prevention and TherapyView all 4 articles
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer. It has a poor response to conventional therapy and has an extremely poor 5-year survival rate. PDAC is driven by multiple oncogene mutations, with the highest mutation frequency being observed in KRAS. The KRAS protein, which binds to GTP, has phosphokinase activity, which further activates downstream effectors. KRAS mutation contributes to cancer cell proliferation, metabolic reprogramming, immune escape, and therapy resistance in PDAC, acting as a critical driver of the disease. Thus, KRAS mutation is positively associated with poorer prognosis in pancreatic cancer patients. This review focus on the KRAS mutation patterns in PDAC, and further emphases its role in signal transduction, metabolic reprogramming, therapy resistance and prognosis, hoping to provide KRAS target therapy strategies for PDAC.
The incidence of pancreatic cancer is increasing at a rate of 0.5%–1.0% per year, and pancreatic cancer is expected to be the second leading cause of cancer death by 2030. Pancreatic cancer includes pancreatic ductal adenocarcinoma (PDAC), which accounts for 90%, follicular carcinoma, pancreatic blastoma, and neuroendocrine tumors. The malignancy of pancreatic cancer is high and the 5-year survival rate of PDAC is as low as 10% (Park et al., 2021). Most PDAC patients fail to be cured by radical surgery due to diagnosis at late or advanced disease stages. Current chemotherapy avenues only prolong the survival of PDAC patients by a few months. Although advanced research tools, such as pancreatic duct-like organs (PDLOs) and pancreatic cancer patient-derived organoids (PDOs), provide unique advantages for developing precise medicines and novel drugs, targeted therapies, including immune therapies, for PDAC are still limited (Melzer et al., 2022). Thus, it is important to develop effective therapies for PDAC (Bear et al., 2020).
PDAC is characterized by a high (up to 95%) mutation rate in the Kirsten rat sarcoma virus oncogene homolog (KRAS) gene. KRAS signaling is essential for PDAC progression, as it regulates multiple downstream effectors. Moreover, KRAS signaling can remodel the PDAC tumor microenvironment (Eser et al., 2014; Hou et al., 2020; Ohara et al., 2022).Previously, KRAS signaling leading to PDAC was considered to be “non-druggable” (Ferguson et al., 2022). Strikingly, there have been several recent advances in the direct targeting of KRAS, particularly with KRASG12C inhibitors. Most existing KRAS inhibitors, such as AMG510 (sotorasib) and MRTX849 (adagrasib), have shown effect in non-small cell lung cancer treatment (Tang et al., 2021), with limited effect on PDAC. Although there are many therapeutic approaches targeting KRAS mutations, few are truly effective in suppressing the effects of KRAS mutations and related pathways in PDAC (Choi et al., 2019). Moreover, translation of the few promising results obtained through research to clinical application has been very disappointing.
PDAC is the most common pancreatic cancer. It involves the progressive differentiation of normal pancreatic ductal epithelium to precancerous lesions, such as pancreatic intraepithelial neoplasia (PanIN). The latest clinical statistics from Chinese PDAC patients revealed recurrent somatic mutations, including KRAS (83.2%), TP53 (70.6%), CDKN2A (28.8%), SMAD4 (23.0%), ARID1A (12.8%), and CDKN2B (8.9%) mutations (Zhang et al., 2022), with the highest percentage of mutations being observed in KRAS (Deramaudt and Rustgi, 2005). KRAS includes 150 structural domains, of which 144 can be resolved using X-ray crystallography and the remaining using NMR data-driven models. Among all known KRAS structures, eight are hypervariable region (HVR) peptides (flexible C-terminal structural elements), and the remaining 142 structures form a G domain (containing six β chains and five α helices) (Pantsar, 2020).
The RAS family of proteins, encoded by the highly homologous genes HRAS, NRAS, and KRAS, is the most common protein family, the genes of which (most importantly KRAS) are mutated in human cancer (Hamarsheh et al., 2020). Moreover, the KRAS mutation-activated signaling pathway could stimulate the expression of HRAS and NRAS proteins via the induction of eNOS and C118 expression to promote tumor growth (Counter, 2015). The most common mutation in KRAS occurs at codon 12 of the second exon, on the first or second nucleotide, resulting in a conformational change of the GTP-binding site and, thereby, reducing the intrinsic rate of GTP hydrolysis (Neuzillet et al., 2014). KRAS mutations mainly occur in GGT sequences, namely GAT (G12D, 40%), GTT (G12V, 33%), CGT (G12R, 15%), TGT (G12C), GCT (G12A) and AGT (G12S) (Zhang et al., 2022). Other KRAS mutations occur at codon 13 of the second exon (G13D, G13C, G13S and G13R) (7%), codon 61 of the third exon (Q61H, Q61R, Q61K and Q61L), codon 117 and 146 of the fourth exon (K117 and A146) (Buscail et al., 2020) (Figure 1). Additionally, point mutations in codons 12 and 13 of the second exon of KRAS have been identified in the sera of some pancreatic cancer patients (Nakano et al., 2018). Patients carrying KRASG12D have a worse prognosis (Valsangkar et al., 2015).
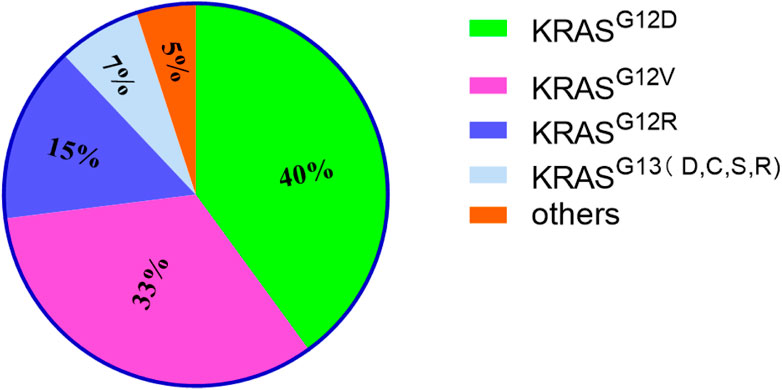
FIGURE 1. The proportion of KRAS mutations of different types. It mainly lists 4 types and others. Among them, KRASG12D accounts for the highest proportion.
KRAS mutation plays an important role in the initiation, development, proliferation, metastasis, metabolic reprogramming, and therapy resistance of PDAC. KRAS protein, which binds to GTP, having phosphokinase activity can activate various signaling pathways, such as the mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), RAL guanine nucleotide exchange factors (RAL-GEFs) and PLC pathways (Martins et al., 2014; Hamarsheh et al., 2020). The more classical signaling pathways activated by KRAS mutation include the RAF/MEK/ERK and PI3K/AKT/mTOR pathways (Mann et al., 2016; Ponz-Sarvise et al., 2019). KRAS/ERK signaling pathway mainly leads to the therapy resistance of PDAC, such as gemcitabine (Principe et al., 2022).In addition, the activation of ERK causes PanIN (Neuhöfer et al., 2021). KRAS/AKT signaling pathway can stimulate cellular nutrient uptake, energy production, regulation of cell cycle and apoptosis and synthesis of critical molecules (Ebrahimi et al., 2017; Dibble et al., 2022). KRAS/RAL-GEFs signaling pathway is crucial for the initiation and therapy resistance of PDAC (Seguin et al., 2014). KRAS/PLC signaling pathway can govern the survival of tumor cells (Buscail et al., 2020) (Figure 2).
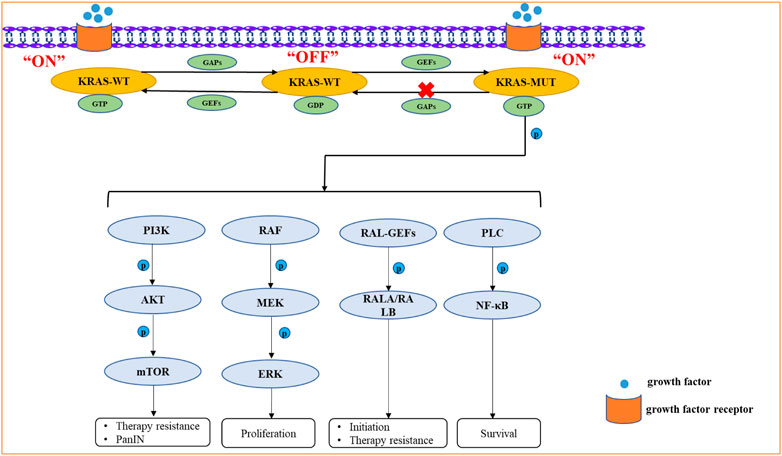
FIGURE 2. The signaling pathways activated by KRAS mutation include the RAF/MEK/ERK, PI3K/AKT/mTOR, RAL-GEFs, PLC signaling pathways. KRAS mutation can activate these signaling pathways by Phosphorylation. Due to the greatly reduced hydrolysis rate of GTP, it retains its active GTP binding state for an unusually long time. The KRAS protein, which binds to GTP, has phosphokinase activity and represents the “ON” state, which further activates downstream proteins.
KRAS mutation facilitates the transformation of acinar cells or ductal cells into pancreatic cancer cells (Flowers et al., 2021). Using a 3D cell culture system, the activation of RAF/MEK/ERK signaling, which is critical for acinus to duct metaplasia (ADM) progression, was studied in pancreatic acinar cells with KRASG12D mutation (Shi et al., 2013). In the SST22+/- pancreatic cancer mouse model, sustained PI3K-AKT signaling activation could cross with KRASG12D mice and further accelerate PDAC progression (Chalabi-Dchar et al., 2015). Using genetic and pharmacological methods, Ferro, R. & Falasca, M identified the KRASG12D/PI3K/PDK1 signaling pathway as a critical driver of ductal chemotaxis, PanIN transformation, and PDAC maintenance (Ferro and Falasca, 2014). Amplification of the CDK5 (cell cycle protein 5) gene or its major activators, p35 and p39, was observed in 67% of PDAC cases. Compared to the wild-type KRAS-mutation containing human pancreatic cancer cells, human pancreatic cancer cells containing the KRASG12D mutation displayed an increased CDK5 kinase activity and enhanced p35 cleavage, which contribute to early stage PDAC progression (Eggers et al., 2011). Both mTORC1 and mTORC2 were significantly activated in human and mouse ADM lesions and synergistically contributed to KRASG12D-driven ADM in vitro and in a pancreatic carcinoma mouse model (Zhao et al., 2021). The KRASG12D-activated RAF/MEK/ERK signaling pathway often acts synergistically with AKT, which is essential for PDAC initiation and progression (Collisson et al., 2012). In an observation study of pancreatic cancer initiation and progression among smokers, it was found that nicotine promoted the development of KRAS mutation-induced pancreatic cancer, which reduced the expression of GATA6 by activating the AKT-ERK-MYC signaling pathway, which is essential for accelerating PDAC development (Hermann et al., 2014). While assessing the relationship between KRAS mutation and inflammation, Zhang, W. et al. found that environmental factors can enhance KRAS mutation and, further, activate YAP-TAZ-JAK-STAT3 signaling, which is required for the development of ADM (Gruber et al., 2016). Pancreatitis could induce KRAS mutation synergistically with NOTCH signaling, and these mutations could further promote the conversion of PanIN into PDAC (De La and Murtaugh, 2009). While exploring potential therapeutic targets for PDAC, Gomes-Filho, S. M. et al. found that KRAS mutation could upregulate AURKA and TPX2 in PDAC cells to promote rapid PDAC cell proliferation (Gomes-Filho et al., 2020). KRAS mutation can lead to the spontaneous loss of GATA6, which may result in pancreatic acinar differentiation program GATA6 dysregulation and also upregulate epidermal growth factor (EGFR), an upstream activator of KRAS; this KRAS-GATA6-EGFR-KRAS positive loop could significantly boost PDAC initiation and progression (Martinelli et al., 2016). It has also been reported that KRASG12D-activated EGFR signaling enhanced Sox9 transcription and further promoted follicular-to-ductal transformation in mouse pancreatic tissue (Chen et al., 2015). KRAS mutation can inhibit the senescence of primary pancreatic ductal cells and maintain these cells in a permanent growth arrest state, which could lead to tumorigenesis under certain conditions (Lee and Bar-Sagi, 2010). By integrating genomics, single-cell chromatin analysis, and spatiotemporally controlled functional perturbations in an in situ mouse model, one study revealed that KRAS mutation may accelerate the malignant transformation of damaged pancreatic tissue (Alonso-Curbelo et al., 2021). Moreover, a study indicated that the Sox9-regulated formation of precancerous lesions and their development into tumors are related to KRAS mutation (Kopp et al., 2012). KRASG12D could also interact with GNASR201C, a key pancreatic cancer maintenance factor, to promote the initiation of intraductal papillary mucinous neoplasia (IPMN) and, thereby, IPMN-induced PDAC (Patra et al., 2018). SMAD4, a central mediator of TGF-β signaling, is specifically inactivated in more than half of PDAC cases, but the loss of SMAD4 expression in itself does not promote PDAC development and needs the presence of KRAS mutation (Zhao et al., 2018). Transcription activator factor 3 (ATF3) could activate the unfolded protein response (UPR) and promote the formation of ADM in pancreatitis in the presence of KRASG12D (Azizi et al., 2021). In addition, KRASG12D could activate enhancer networks in progenitor cells associated with pancreatitis to drive PDAC development (Li et al., 2021a). Histological analysis of human and mouse tumor tissues revealed that ALK4 drives PDAC formation through KRASG12D-induced ALK4 ligand activator A upregulation in transformed glandular follicle cells (Zhao et al., 2020). KRASG12D was also found to upregulate the ataxia capillaris group D complementary gene (ATDC) in PanIN subpopulations and accelerate PanIN progression in a mouse model (Wang et al., 2015). To determine whether the viral oncoprotein SV40 large T antigen (TAg) can promote PDAC in association with KRAS mutations, investigators used KRASG12D wide type and knockdown mouse models; they found that Tag could cause the development of highly invasive adenocarcinoma in the short term only in the presence of KRASG12D (Yamaguchi et al., 2014). Single Ink4a/Arf loss has no effect on tumor lesions but leads to their rapid progression to highly invasive and metastatic pancreatic adenocarcinoma in the presence of KRASG12D (Aguirre et al., 2003). In addition to the common precancerous lesions, PanIN, the co-existence of KRAS mutations with the SMAD4/DPC4 tumor suppressor gene deletion in mucinous cystic neoplasms (MCNs) may eventually lead to the development of PDAC (Izeradjene et al., 2007). KRAS mutation prevent the reversion of ADM development and accelerate pancreatic intraepithelial neoplasia (PanIN), eventually leading to pancreatic adenocarcinoma in both clinical samples as well as animal models (Liou et al., 2015) (Figure 3).
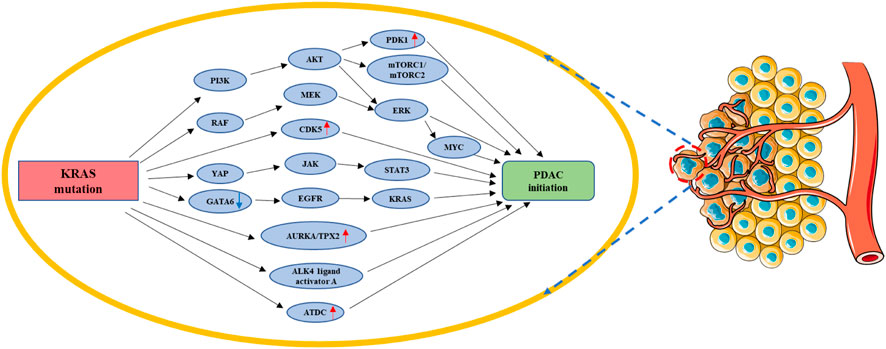
FIGURE 3. KRAS mutation in PDAC initiation. KRAS mutation could facilitate the transformation of acinar cells or ductal cells into pancreatic cancer cells by regulating PI3K/AKT/(PDK1&mTORC1/2&ERK-MYC), RAF/MEK/ERK, CDK5, YAP/JAK/STAT3, AURKA/TPX2, ALK4 ligand activator A, ATDC, and GA-TA6/EGFR/KRAS. Red arrow: The expression of related gene is upregulated. Blue arrow: The expression of related gene is downregulated.
KRAS mutation could accelerate PDAC progression depending on, not only the activation of signaling pathways, but also the induction of metabolic reprogramming (Hu et al., 2019). KRAS mutation could regulate glucose, amino acid, and lipid metabolism in PDAC cancer cells (Suzuki et al., 2022).
Pancreatic cancer cells facilitate glycolysis under adequate oxygen conditions; this is known as the “Warburg effect” (Chang et al., 2022). Glucose is transported into the cell through glucose transporters (GLUTs), which mainly include GLUT1, GLUT2, GLUT3 and GLUT4 (Reckzeh and Waldmann, 2020). The glucose transferred into the cytoplasm by the GLUT is catalyzed to glucose-6-phosphate(G-6-P) by hexokinase (HK) (De Jesus et al., 2022). Then, G-6-P can be converted to fructose-6-phosphate (F-6-P) by phosphohexose isomerase (PGI). Phosphofructokinase (PFK) can catalyze F-6-P to fructose-1,6-bisphosphate (F-1,6-BP) (Shi et al., 2017). F-1,6-BP can be catabolized to glyceraldehyde 3-phosphate (GA3P) and dihydroxyacetone phosphate (DHAP). Next, triose phosphate isomerase (TPI) converts DHAP to triose phosphate (TP). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) oxidizes GA3P to 1,3-diphosphoglyceric acid (1,3-DPG). 1,3-DPG is catalyzed by phosphoglycerate kinase (PGK) to produce 3-phosphoglyceric acid (3-PG). 3-PG is converted by phosphoglycerate mutase (PGAM) to glycero-2-phosphate (2-PG). 2-PG is dehydrated by enolase (ENO) to produce phosphate enol pyruvate (PEP). After this step, pyruvate kinase (PK) catalyzes the conversion of PEP to pyruvate and adenosine triphosphate (ATP). Then, the final step in glycolysis is that lactate dehydrogenase (LDH) catalyzes the conversion of pyruvate to L-lactate (Ganapathy-Kanniappan and Geschwind, 2013; Abdel-Wahab et al., 2019).
The fasting glucose level is a risk factor for pancreatic cancer occurrence in type 1 and type 2 diabetes patients, indicating that glucose metabolism is involved in pancreatic cancer development (Menini et al., 2020). A study of the United Kingdom Biobank found that people with diabetes were more than three times more likely to develop PDAC than those without diabetes (Sharma et al., 2022). Chronic type 2 diabetes is a risk factor for PDAC, which is also the cause of new onset diabetes. There is also a strong association with genetic mutations reported by PDAC patients (Andersen et al., 2017). In pancreatic cells, high glucose treatment favors the hexosamine pathway (HPB), which increases the level of O-glycosylation in pancreatic cells, leading to an increased level of O-glycosylation of PFK and a decrease in its activity, positive feedback to the HPB pathway. In diabetic patients, high glucose uses the enhanced HPB pathway to enhance O-glycosylation levels of RNM1, a subunit of nucleotide reductase (RNR), in pancreatic cells, leading to a reduction in the dNTP pool and causing DNA damage, which preferentially induces mutation in KRASG12D. The KRASG12D accelerates the conversion of pancreatic cells to pancreatic intraepithelial neoplasia and again mediates the reprogramming of glucose metabolism (Hu et al., 2019). KRASG12D promotes GLUT1 expression by upregulating SLC2A1, the gene encoding GLUT1. The expression of leukemia inhibitory factor receptor (LIFR) was significantly lower in pancreatic cancer compared to some cancers, but inhibition of KRAS mutation upregulated LIFR expression and was accompanied by a decrease in GLUT1 expression. It was found that LIFR can inhibit GLUT1 expression by activating STAT3, while KRAS mutation reversed this effect, allowing GLUT1 to be consistently expressed and promoting glycolysis (Liu et al., 2021).According to data from The Cancer Genome Atlas (TCGA) and the analysis of 89 informative PDAC tumor samples, KRAS mutation is involved in pancreatic cancer cell aerobic glycolysis (Zhu et al., 2021). PFKFB3/PFK2, a KRAS mutation-dependent glycolytic activator, was reported to contribute to PDAC aerobic glycolysis. KRASG12D/G12C can promote the expression of PFKFB3 and GLUT2 by activating p38γ MAPK signaling. p38γ MAPK can stabilize PFKFB3 by S467, which is the phosphorylation site of PFKFB3. Meanwhile, p-PFKFB3 can interact with GLUT2 and enhance the expression of GLUT2 (Wang et al., 2020).Among the clinical methods for tumor diagnosis, 2-[18F] fluoro-2-deoxy-D-glucose PET-CT is used as a relatively common means to assess tumors with standardized uptake values (SUVMAX). The tumor-specific “Warburg effect” increases the uptake of 2-[18F] fluoro-2-deoxy-D-glucose and the uptake is inversely correlated with the expression level of F-box and WD repeat domain-containing 7 (FBW7). FBW7 is an important molecule for targeting and destroying many cancer proteins, and its functional inhibition is an important driver of carcinogenesis. It was found that overexpression of FBW7 in pancreatic cancer cells activates the c-Myc signaling pathway to target thioredoxin-binding protein (TXNIP), a negative regulator of glucose metabolism, which in turn inhibits glycolysis-related genes, including GLUT1, GLUT4, HK2, LDHA and LDHB. However, KRAS mutation can destabilize FBW7 by activating ERK signaling, which ultimately promotes glycolysis and promotes proliferation of pancreatic cancer cells (Ji et al., 2016). The study found that cancer cells carrying KRAS mutation was more likely to survive when cultured under low-glucose condition and that this was closer to the in vivo microenvironment of PDAC (Yun et al., 2009; Sullivan et al., 2019). Moreover, the level of autophagy in KRAS-mutated cancer cells is increased under low-glucose condition. At the same time, the mitochondrial content and total membrane potential of PDAC cells are reduced. Therefore, the level of ROS in mitochondria is also decreased, which reduces the demand for NADPH and ensures that pancreatic cancer cells can continue to proliferate even under low-glucose condition. Both mitochondrial dysfunction and aerobic glycolysis are important features of cancer cells, and KRASG12V can promote mitochondrial dysfunction and facilitate the shift from oxidative phosphorylation to aerobic glycolysis (Hu et al., 2012).To explore the mechanism of the above phenomenon, Humpton, Tet al. found that KRASG12D can upregulate the mRNA and protein expression levels of Nix through activating MEK signaling pathway to induce mitochondrial autophagy, reduce mitochondrial glucose flux, and finally maintain redox homeostasis in PDAC cells (Humpton et al., 2019). The NADPH mentioned here is necessary for cellular maintenance of redox, and NAD(P)H oxidase (NOX) is an enzyme necessary to catalyze NAD(P)H oxidation, of which NAD(P)H oxidase 4 is one. In PDAC, KRAS mutation and p16 deletion are also required for its development, and both can collaborate to promote NOX4 expression, NAD(P)H oxidation and the production of the glycolytic substrate NAD + or NADP+, which can increase glycolysis allowing for sustained proliferation of PDAC cells. p16 deletion alone does not promote NOX4 expression, but also requires KRASG12V mutation to upregulate p22phox, a subunit required for NOX4 activation, through activation of NF-κB. The results indicate that NF-κB is the major transcription factor for p22phox promoter binding (Ju et al., 2017). In addition, NAD(P)H is also an important product of the pentose phosphate pathway (PPP) and is an essential molecule for maintaining the conversion of reduced and oxidized glutathione, which can be referred to as the oxidative branch of PPP. KRASG12D was found to increase glucose uptake to promote the production of NAD(P)H and ribose, and the oxidative and non-oxidative branches of PPP do not affect each other. Meanwhile, KRASG12D promotes the hexosamine pathway (HBP) to increase the level of protein glycosylation, and KRASG12D maintains pancreatic cancer by activating MAPK and Myc signaling pathways to upregulate glycolysis-related genes (GLUT1, HK1, ENO1, and LDHA), HBP-related genes (GFPT1), and PPP non-oxidative branch genes (RIA), respectively (Ying et al., 2012). KRASG12D also can promote glycolysis by using paracrine pathways. KRASG12D can promote the expression of type I cytokine receptor complexes (IL2rγ-IL4r⍺ and IL2rγ-IL13r⍺1) in pancreatic cancer cells, which can receive growth factor signals (IL4 and IL13) from invasive TH2 cells. Activated type I cytokine receptor complexes increase glycolytic flux by activating the JAK1-STAT6-Myc signaling pathway to upregulate glycolysis-related genes (HK2 and ENO1) (Dey et al., 2020). Recent study reveals that BZW1 (Basic leucine zipper and W2 domain–containing protein 1) can promote tumor growth by enhancing eIF2a phosphorylation to promote glycolysis in PDAC. However, the relationship between BZW1 and KRAS mutation has not been clearly elucidated (Li et al., 2022).
The use of nanoparticulate albumin bound paclitaxel (Nab-PTX) is one of the more commonly used treatments for pancreatic cancer, however, the RAS/RAF/MEK/ERK signaling pathway inhibits its targeted delivery and effectiveness. The drug efficacy of Nab-PTX was greatly enhanced when glucose deprivation experiments were simulated with an insulin-like growth factor 1 receptor inhibitor (IGF1R) (Li et al., 2021b). Studies have shown that glucose can be an effective, sensitive and inexpensive marker for precancerous lesions of pancreatic cancer. However, the standard threshold for detecting glucose has not been defined and, moreover, it has a low specificity. In contrast, KRAS is a highly specific but low-sensitivity biomarker. The combination of the two can greatly improve the detection of precancerous lesions in pancreatic cancer. Moreover, KRAS mutation can drive reprogramming of glucose metabolism, and a perfect combination of the two requires a lot of prospective studies before they can truly serve for clinical monitoring (Li et al., 2023) (Figure 4).
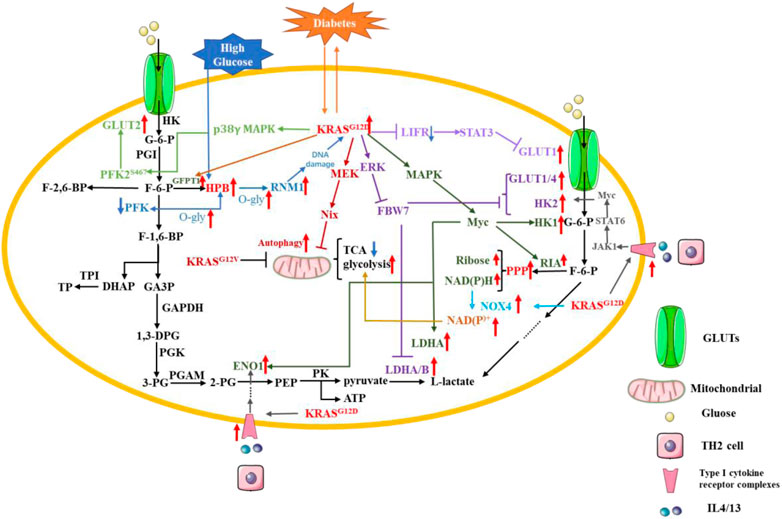
FIGURE 4. KRAS mutation in PDAC glucose metabolic reprogramming. Pancreatic cancer cells facilitate glycolysis under adequate oxygen conditions; this is known as the “Warburg effect. Red arrow: The expression of related gene is upregulated. Blue arrow: The expression of related gene is downregulated.
In addition to the characteristic “Warburg effect”, glutamine metabolism is another feature of tumor cell metabolism. Furthermore, glutamine is an important substrate for pancreatic cancer biomarkers, CA19-9, and glycosylation (Liu et al., 2023a). Although the “Warburg effect” reduces the availability of pyruvate for the tricarboxylic acid cycle (TCA), glutamine metabolism compensates for this. Glutamine is an alternative nutrient to glucose for PDAC cells as a source of energy and for biosynthesis, especially under glucose limitation conditions (Xu et al., 2022). KRASG12D-mutated pancreatic cancer cells were greatly killed when glycolysis-related genes PFKFB3 and glutamine-related GLS1 were simultaneously inhibited (Ozcan et al., 2021). Glutamine, one of the most abundant amino acids in the bloodstream, is a non-essential amino acid, but it provides precursors for the synthesis of a variety of substances, including proteins, lipids and nucleotides, and is an important source of carbon and nitrogen. It also provides essential precursors for the synthesis of NADPH and GSH and maintains redox homeostasis in cancer cells (Cluntun et al., 2017; Zhang et al., 2017; Delgir et al., 2021). Glutamine is transported into the cell via the transporters SLC38A2, SLC38A1, and SLC1A5, which allow the synthesis of hexosamine, aspartate, and nucleic acids in the cytoplasm (Scalise et al., 2017). Glutamine in the cytoplasm must pass through the glutamine transporter SLC1A5 variant to enter the mitochondria. After glutamine enters the mitochondria, glutaminases (GLSs) catalyze the production of glutamate and ammonia, which can be transported to the cytoplasm or remain within the mitochondria. Next, glutamate in the mitochondria is catalyzed by glutamate dehydrogenase 1 (GLUD1) or glutamate transaminase 2 (GPT2) and glutamic acid transaminase 2 (GOT2) to form α-ketoglutarate, which can be transported to the cytoplasm or involved in TCA (Wang et al., 2010; Yang et al., 2017; Stine and Dang, 2020).
The “Warburg effect” reduces the availability of pyruvate for the TCA, but glutamine can fuel the TCA cycle by KRAS mutation. KRAS mutation upregulated Protein interacting with never in mitosis A1 (PIN1) expression to maintain the normal function of mitochondria and redox homeostasis by synergistically activating c-Myc and NRF2, which is necessary for glutamine metabolism (Liang et al., 2019). However, the bloodstream system of pancreatic cancer is poor, and the cancer cells cannot take up enough glutamine. We call this part of cancer cells that do not get enough glutamine as glutamine-deficient cells. KRAS mutation-containing pancreatic cancer cells have great demand for glutamine, which is also termed as “glutamine addition”. α-ketoglutarate (α-KG) derived from the glutaminase-based catabolism of glutamine provides fuel for the tricarboxylic acid cycle (TCA) in pancreatic cancer cells. Supplementation with α-KG restored the growth and survival of glutamine-deficient pancreatic cancer cells with the help of glutamate-ammonia ligase (GLUL), which was elevated in the KRASG12D-induced mouse PDAC model (Bott et al., 2019). In most cancer cells, the GLUD1 catalyzed glutamate production of α-ketoglutarate is used as the main fuel for the TCA cycle. In contrast, pancreatic cancer cells can utilize α-ketoglutarate via a non-classical pathway that is mediated by KRASG12D inhibiting GLUD1 and promoting the expression of aspartate transaminase (GOT1). Glutamine-derived aspartate is translocated to the cytoplasm, subsequently, oxaloacetate is converted to malate and pyruvate, increasing the NADPH/NADP (+) ratio and thus maintaining the redox state of pancreatic cancer cells, which makes KRAS-mutated pancreatic cancer cells dependent on this pathway (Son et al., 2013). Thus, inhibition of the aspartate efflux-related protein, mitochondrial transporter uncoupling protein 2 (UCP2), resulted in reduced glutamine hydrolysis and NADPH/NADP+ and glutathione/glutathione disulfide ratios as well as increased levels of reactive oxygen species in KRAS-mutated pancreatic cancer cell lines. However, silencing UCP2 just reduced glutamine hydrolysis in wild-type KRAS PDAC cells, without affecting their redox homeostasis or proliferation rate (Raho et al., 2020). Moreover, KRASG12D in PDAC cells could also positively regulate the glutamine catabolic pathway by upregulating the O-GlcNAcylation of malate dehydrogenase 1 (MDH1), the expression of which was significantly elevated in clinical PDAC samples. This finding demonstrated that O-GlcNAcylation may also be related to signaling pathways involving KRAS mutation and glutamine metabolism in PDAC (Zhu et al., 2022) (Figure 5).
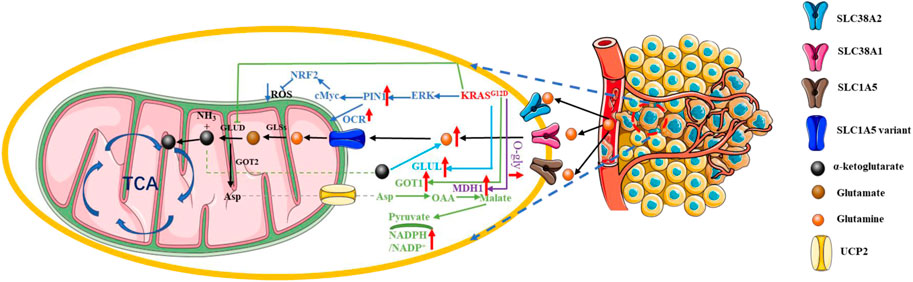
FIGURE 5. KRAS mutation in PDAC glutamine metabolic reprogramming. KRAS mutation-containing pancreatic cancer cells have great demand for glutamine; this is also termed as “glutamine addition”. Red arrow: The expression of related gene is upregulated. Blue arrow: The expression of related gene is downregulated.
Recent studies have identified PI3K-C2γ, a class II PI3K, as a negative regulator of PDAC, and its deletion leads to glutamine addiction in PDAC. PI3K-C2γ deletion leads to excessive activation of mTOR signaling, making PDAC glutamine-dependent. However, whether KRAS mutation have a regulatory role in this process has not been clearly elucidated (De Santis et al., 2023). In addition, M2 tumour-associated macrophages can secrete exosomes to promote glutamine metabolism in pancreatic cancer (Liu et al., 2023b). Pancreatic stellate cells can also promote glutamine metabolism in PDAC via the Wnt/β-catenin/TCF7 signaling pathway (Son et al., 2013). KRAS mutation promoting glutamine metabolism in pancreatic cancer can also be studied in relation to surrounding stromal cells or associated supporting cells. Glutamine addiction in pancreatic cancer is also a major cause of its chemoresistance (Ganguly et al., 2022; Park et al., 2023). A great deal of research is still needed on glutamine metabolism in pancreatic cancer, which is expected to provide efficient targets for the clinical treatment of pancreatic cancer.
Intraperitoneal fat can infiltrate the pancreas and negatively affect the normal physiological function of the pancreas. Obesity, like diabetes, increases the incidence of pancreatic cancer (Paternoster and Falasca, 2020). Most importantly, Wang, D et al. found that a high-fat diet (HFD), but not a high-carb diet (HCD), induced excessive activation of KRASG12D and further promoted metabolic reprogramming of PDAC (Wang et al., 2019). A high-fat, high-calorie diet (HFCD) increased pancreatic cancer incidence in P48+/Cre, LSL-KRASG12D (KC) mice, resulting in their exhibition of more extensive inflammation and fibrosis (Chang et al., 2017).A study showed that KRASG12D could upregulate FGL1 through the phosphorylation of STAT3, altering lipid metabolism and promoting the proliferation of PDAC cells (Chiu et al., 2021). As KRAS mutation can significantly reduce the expression of FGF21 by downregulating peroxisome proliferator-activated receptor gamma (PPARG), a lipid metabolism activator, injecting HFCD-fed KRASG12D/+ mice with recombinant human FGF21 (rhFGF21) could reduce abdominal fat accumulation, triglyceride level, and pancreatic tissue inflammation, ultimately resulting in longer survival and fewer metastases (Luo et al., 2019). KRAS mutation also downregulated hormone-sensitive lipase (HSL) by activating ERK signaling pathway to promote lipid storage and excess fatty acid storage in lipid droplets, which are required for pancreatic cancer cell proliferation and metastasis (Man et al., 2020). In addition, free mutant KRAS can also promote pancreatic cancer proliferation through fatty acid metabolism. Ferroptotic pancreatic cancer cells releasing KRASG12D can be taken up by macrophages through an autophagy-dependent mechanism, which polarizes macrophage to an M2-like phenotype through STAT3-dependent fatty acid oxidation (Dai et al., 2020) (Figure 6).
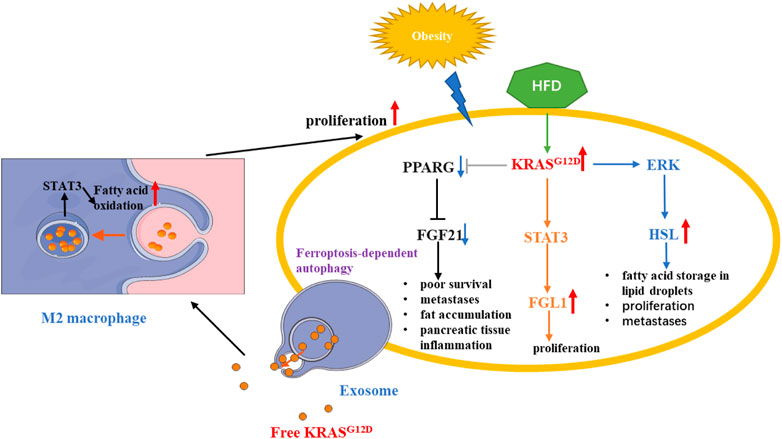
FIGURE 6. KRAS mutation in PDAC fatty acids metabolic reprogramming. KRAS mutation could alter lipid metabolism and promote the proliferation of PDAC cells by regulating STAT3/FGL1, FGF21, HSL. In addition, Autophagy-dependent PDAC cells releasing exosome, which contains KRASG12D, can promote fatty acid oxidation by STAT3 in M2 macrophage. Red arrow: The expression of related gene is upregulated. Blue arrow: The expression of related gene is downregulated.
Recent studies have shown that the lipid-rich lipid raft protein flotillin-1, which can be localized to the cell membrane, interacts with sevenless (SOS)-1 to facilitate the conversion of Ras-bound GDP to GTP. Tissue microarrays from multiple pancreatic cancer samples showed that flotillin-1 was highly expressed compared to normal pancreatic tissue and correlated with KRAS (Jin et al., 2023). In addition, the authors found that the SMARCD3, one of SWI/SNF (a nucleosome remodeling complex) subunits, is highly expressed in pancreatic cancer and can synergistically regulate lipid and fatty acid metabolism with FOXA1. Whether it is associated with KRAS mutation is also an unsolved mystery (Ferguson et al., 2023).
Histone H4 acetylation levels were elevated in pancreatic follicular cells containing KRASG12D prior to the appearance of precancerous lesions and this elevation was caused by the upregulation of acetyl coenzyme A by KRASG12D, which can promote ADM via the mevalonate pathway and thereby promote pancreatic carcinogenesis and epithelial modifications (Carrer et al., 2019). Acetyl coenzyme A synthase short chain family member 2 (ACSS2) is an acetyl coenzyme A synthase. ACSS2 promotes macropinocytosis and facilitates metabolic reprogramming of pancreatic cancer in pancreatic cancer (Zhou et al., 2022). In human PDAC cells, KRAS mutation has been shown to promote macropinocytosis, which is an essential strategy for nutrient uptake by cancer cells in the form of macromolecules and cellular debris (King et al., 2020). However, it is not known whether ACSS2 is regulated by KRAS mutation.
BCAA levels in booldstream are associated with an increased risk of PDAC in pancreatic cancer (Rossmeislová et al., 2021). Branched-chain amino acid (BCAA) metabolism has also been implicated in the development of PDAC. KRASG12V could stabilize BCAT2. Tissue-specific knockdown of BCAT2 in the pancreas can hinder the progression of PanIN in LSL-KRASG12D/+; Pdx1-Cre (KC) mice. A low-BCAA diet was also found to hinder the development of PDAC in mouse models. Thus, BCAA-mediated catabolism is critical for the development of KRAS-mutated PDAC (Li et al., 2020). Acetylation of BCAT2 promotes the degradation of BCAT2, which inhibits the cell proliferation of PDAC (Lei et al., 2020). In addition, BCAT1 promotes branched chain ketoacid addiction in mesenchymal-rich PDAC cells and pancreatic stellate cells can also promote pancreatic cancer progression by regulating BCAA metabolism (Zhu et al., 2020; Jiang et al., 2021). Whether KRAS mutation can affect BCAA metabolism in pancreatic cancer mesenchymal cells has not been studied in detail.
Early studies have confirmed that reactive oxygen species (ROS), especially hydrogen peroxide(H2O2), play an important role in maintaining cellular and tissue homeostasis (Park et al., 2021). However, some studies have highlighted that low levels of ROS are more able to promote the progression of pancreatic cancer with KRASG12D mutation (Melzer et al., 2022). Some studies have in turn emphasized that reducing ROS production delays the progression of pancreatic cancer with KRASG12D mutation (Eser et al., 2014; Bear et al., 2020; Ohara et al., 2022). The exact role of ROS in the cell is not clear (Hou et al., 2020). Recently, a mass spectrometry-based study found that the oncogenic KRASG12D mutation promotes the accumulation of H2O2, but not superoxide or hydroxyl radicals, in pancreatic cancer cells. The KRASG12D mutation inhibits the expression of the catalase gene (CAT) as a way to reduce the breakdown of H2O2. Because most oxygen radicals are unstable in the cell, being able to measure it precisely is difficult, so this finding is great. The findings suggest that excessive accumulation of H2O2 can modify intracellular metabolites, including lipids, deoxycholic acid and palmitoylcarnitine, to maintain the continued survival of pancreatic cancer cells (Ferguson et al., 2022). When exogenous H2O2 is added, the KRAS/AMPK signaling pathway is activated and promotes increased expression of glycolytic genes and lactate production. H2O2 can increase the flux of glycolysis (Tang et al., 2021). However, one question that while excess H2O2 promotes pancreatic cancer proliferation, its inevitable effects on unsaturated lipids, such as phospholipid-rich cell membranes (Choi et al., 2019), induce ferroptosis in pancreatic cancer cells (Zhang et al., 2022). However, an answer to this query was soon available, and recent studies have shown that KRASG12D can activate the MAPK signaling pathway, which in turn further activates NRF2, which in turn upregulates the expression of FSP1 (ferroptosis suppressor protein 1) and removes excess ROS, allowing pancreatic cancer cells to proliferate stably under safe conditions (Deramaudt and Rustgi, 2005). In summary, KRAS mutation can promote the proliferation of pancreatic cancer cells through a perfect collaborative regulatory pathway.
In addition to affecting pancreatic cancer cells biology in an intrinsic manner, KRAS mutation can also indirectly affect pancreatic cancer cells by regulating the tumor microenvironment (TME). KRAS mutation can suppress T-cell number and function in pancreatic cancer cells, while silencing KRAS can reverse this effect. KRASG12D was found to promote pancreatic cancer progression by upregulating the expression of the chemokine CXCR2 through the ERK pathway and thereby enhancing the infiltration of immunosuppressive cells into the TME (Purohit et al., 2016). KRASG12V-overexpressing pancreatic cancer cells were also found to enhance regulatory T cell (Treg) activity. In addition, the KRAS-MEK-ERK-AP1 signaling pathway induced PDAC cells to secrete abundant amounts of IL-10 and transforming growth factor-β1 (Zdanov et al., 2016), which are essential for maintaining Treg proliferation and function. KRAS mutation upregulated lipase H (LIPH) expression, enhancing the infiltration of tumor-associated macrophages (TAMs), Treg cells, and Th2/Th1 cells into the TME, while reducing the infiltration of CD8+ T cells and Th1 cells; this further contributed to tumor recurrence, worsening of histological grade, and poorer overall survival (Zhuang et al., 2022). The effectiveness of currently existing immune checkpoint inhibitors for treating KRAS mutation-containing pancreatic cancer is disappointing and indicates the need to explore additional pathways involved in immune evasion that should be targeted for therapy. Surprisingly, KRAS mutations were found to dysregulate cell death pathways, which are induced by the tumor necrosis factor (TNF) receptor. KRAS mutations in pancreatic cancer cells rely on both endogenous TNF-related apoptosis-inducing ligands (TRAILs) as well as their receptors to promote tumor growth and metastasis (von Karstedt and Walczak, 2020). Interestingly, KRAS mutations evaded immune therapy by upregulating programmed cell death 1 ligand 1 (PD-L1) transcription and protein expression (Coelho et al., 2017). KRAS mutation could also enhance the recycling of PD-L1 molecules in pancreatic cancer cells to the cell surface to promote the immune escape of pancreatic cancer cells (Hashimoto et al., 2019). High N6-methyladenosine (m6A) expression was associated with reduced PDAC immune infiltration and T-cell depletion, poor overall survival, and tumor recurrence. Moreover, KRAS mutation-containing PDAC tumors displayed high m6A scores, suggesting a correlation between KRAS, m6A, and immune escape (Zhou et al., 2021) (Figure 7).
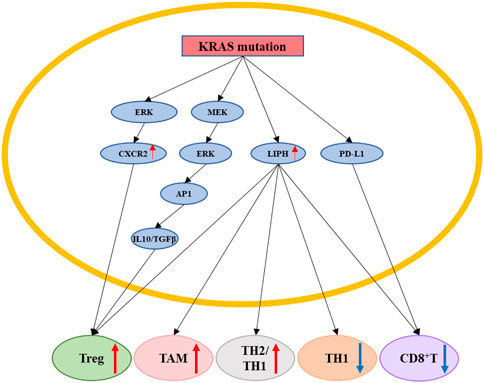
FIGURE 7. KRAS mutation promotes pancreatic cancer cells immune escape. KRAS mutation can indirectly affect pancreatic cancer cells by regulating the tumor immune microenvironment by regulating ERK/CXCR2, MEK/ERK/AP1/(IL10&TGFβ), LIPH, PD-L1. KRAS mutation could upregulate the proportion of Treg, TAM, TH2/TH1 cells, and downregulate the proportion of TH1, CD8+T cells. Red arrow: The expression of related gene is upregulated. Blue arrow: The expression of related gene is downregulated.
Targeting oncogenes, such as KRAS, NRG1, and NTRK, and their related molecules for therapy is always a hot topic in the tumor research field. However, most results of such research have been disappointing (Qian et al., 2020). KRAS is the most common oncogene in PDAC and targeting it has been very challenging, with KRAS mutation-containing PDAC long being considered undruggable. Most existing therapies for this cancer have focused on indirect targeting and have shown limited efficacy (Bannoura et al., 2021) (Table.1). In advanced stage PDAC, KRAS mutation upregulates the expression of the antioxidant factor NRF2 in response to higher oxidative stress levels in PDAC cells, which leads to chemoresistance (Mukhopadhyay et al., 2020). Several specific KRAS mutation inhibitors have been developed, most of which target KRASG12C. However, the KRASG12C mutation can only be detected in a small fraction of PDAC patients (Nagasaka et al., 2021). In addition, patients with KRASG12C mutation-containing PDAC respond poorly to KRASG12C inhibitors, mainly because: 1) KRASG12C co-exists with other KRAS point mutations or at a high copy number; 2) the presence of other mutant genes besides KRAS; 3) accelerated PDAC metastasis and novel genetic variations, such as MET amplification; NRAS, BRAF, MAP2K1, RET, ALK, RET, RAF1, and FGFR3 gene fusions; and loss of NF1 and PTEN tumor suppressor function (Nussinov et al., 2021). KRAS mutation not only drive the development of PDAC but also contribute to PDAC tumor heterogeneity during progression. Gemcitabine (GEM), a first-line PDAC chemotherapeutic drug, has limited efficacy in most patients, which may be due to KRAS mutation (Won et al., 2021). In one study, KRAS mutation-activated PI3K/AKT/mTOR and AXL signaling contributed to pancreatic cancer cell resistance to MEK (a KRAS effector) inhibitors (Pettazzoni et al., 2015). However, combining MEK inhibitors with a PI3K/mTOR inhibitor still failed to cure PDAC in the presence of KRASG13D (Neuzillet et al., 2014). Although the combination of MEK inhibitors with standard therapy (gemcitabine/Nab-paclitaxel) reduced tumor growth and metastasis in PDXs (patient-derived xenograft models), it may also elicit KRAS mutation, which can contribute to tumor heterogeneity. In vitro and in vivo analyses suggested that MEK inhibitor-induced PDAC heterogeneity facilitates the rapid proliferation of treatment-resistant cells (Pedersen et al., 2017). Related data show that CasRx can precisely and effectively silence the expression of mutant KRASG12D in PDAC cells and, further, eliminate KRASG12D-related downstream signaling, thereby, making PDAC cells sensitive to gemcitabine and inhibiting tumor growth (Jiang et al., 2020).
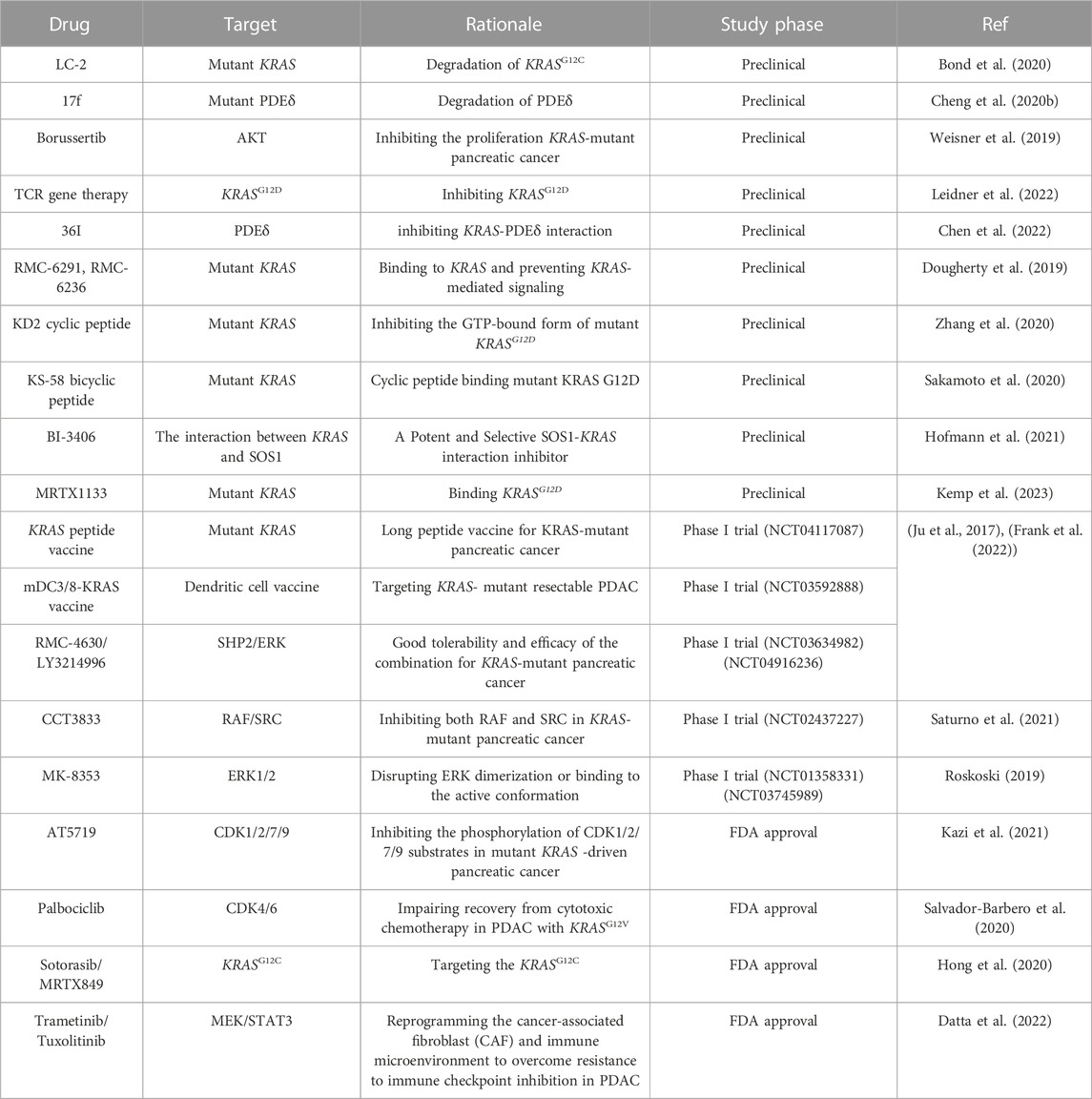
TABLE 1. Therapies for PDAC directly focused on KRAS mutation or indirectly on related effector.
As one of the most devastating digestive system cancers, PDAC is characterized by a lack of specific symptoms, fast progression, and poor prognosis (Chen et al., 2021a). Combining the identification of KRAS mutation and cytopathology increased the sensitivity, accuracy, and negative predictive value of pancreatic cancer diagnosis and molecular subtype classification. Compared to conventional serum biomarkers and medical imaging, examining the KRAS mutation level using genomic and transcriptomic techniques has significantly improved diagnostic sensitivity and specificity, indicating potential clinical application value (Lundy et al., 2021). In addition, the presence of KRAS mutations in the blood circulation was positively associated with poorer prognosis (Buscail et al., 2020). Moreover, KRASG12V mutations in the circulation of patients with PDAC was associated with a high percentage of circulating Tregs. Both, the presence of high KRASG12V mutations and Tregs in the circulation in PDAC, indicate advanced disease stage, extremely poor survival, and poor response to immunotherapy (Cheng et al., 2020a). Compared to clinical serum markers, circulating KRAS mutant DNA levels showed more sensitivity and specificity in predicting the effect of chemotherapy on tumor mass burden. Kruger et al. (2018) found that KRAS mutant DNA levels decreased immediately upon treatment of patients with PDAC with gemcitabine-based treatment, while there were no significant changes in the DNA levels of CA 19–9, CEA, and CYFRA 21–1 until 4 weeks after treatment; this indicated that KRAS mutant DNA is an early indicator of gemcitabine-based chemotherapy response. Repeated measurements of KRAS mutant DNA levels act as a superior marker for PDAC progression (sensitivity: 83%, specificity: 100%). In addition to chemotherapy, radiation therapy is also commonly used in pancreatic cancer treatment. Blocking KRAS mutations made pancreatic cancer tumors sensitive to radiation therapy both in vivo and in vitro (Brunner et al., 2005) (Allenson et al., 2017).
Local and distant metastasis indicate a poor prognosis of PDAC patients. The KRAS mutant DNA level in circulation was significantly consistent with metastasis to tissues in patients with advanced PDAC, indicating that the circulating KRAS mutant DNA level may be a potential biomarker to predict metastasis in PDAC (Patel et al., 2019). KRASG12D activated human heterogeneous cytosolic ribonucleoprotein A1 (HnRNPA1) in a ubiquitin-like protein (SUMO2)-dependent manner, promoting its export from cancer cells to the extracellular space in a vesicle-mediated (EV-mediated) manner as well as promoting lymphangiogenesis and lymph node metastasis. Mechanistically, a study revealed that KRASG12D induced HnRNPA1 hyperactivation, interacted with TSG101, and enhanced hnRNPA1 packaging and delivery via EVs, which were internalized by human endothelial lymphocytes in the tumor microenvironment and promoted lymph node metastasis (Luo et al., 2022). Through genomic, transcriptomic, methylomic, and molecular dynamics analyses, the KRAS mutant and the stability of the protein expressed by it could predict the poor prognosis of pancreatic cancer (Kaushik et al., 2021). In a mouse PDAC model, KRAS mutations promoted tumor-associated fibroblast heterogeneity, which resulted in pancreatic cancer cell metastasis and poor prognosis. Pan et al. (2021) found that tumor cells associated with metastasis-induced inflammatory CAFs (iCAFs) were with heterogeneity. CAFs from liver metastases exhibited a more homogeneous phenotype, while CAFs from lung metastases remained heterogeneous. Thus, it was difficult to implement a uniform approach to treat pancreatic cancer with a satisfactory survival rate. Compared to common PDAC, 73% adenosquamous carcinoma of the pancreas (ASCP) cases were found to involve KRAS mutations using whole-genome copy number variation (CNV), whole-exome sequencing, and transposase-accessible chromatin by sequencing (ATAC-seq), which may be the reason for higher malignancy and poorer prognosis in ASCP than in PDAC (Lenkiewicz et al., 2020).
The involvement of KRAS mutations has been widely researched in pancreatic cancer cell signaling activation, metabolic reprogramming, immune escape, therapy resistance, and prognosis. Although some KRAS mutation inhibitors have been used in clinical practice, they have shown few effects in the treatment of patients with PDAC. Due to the absence of specific symptoms, most KRAS mutations in PDAC are identified and studied in the advanced stage of the disease. Thus, the exact role of KRAS mutations in early stage PDAC is still unclear.
Review of existing literature in this area indicates that researchers should focus their attention on studying the role of KRAS mutations in early stage of pancreatic cancer to improve the early screening rate and, thereby, prolong the patient survival rate and improve the life quality in pancreatic cancer. This can be done through two ways. 1) The use of advanced research tools, such as organoids, patient-derived xenograft (PDX) models, and genetically engineered mouse models (GEMMs) for PDAC. However, due to the heterogeneous nature of pancreatic cancer cells, immune-deficient mice used to create PDX models lacked an effective immune microenvironment and pancreatic genetic diversity. Therefore, they could not completely simulate the pancreatic cancer TME and its response to treatments (Chen et al., 2021b). Although the establishment of an organoid requires an advanced organ culture model to form a pancreatic niche in vitro from the porcine bladder (PUB) (Melzer et al., 2022), it cannot acquire better experiment repeatability and confirm that every cell contained a genetic mutation, such as KRAS. Thus, to accurately simulate the pancreatic cancer microenvironment, a pancreatic model that contains stable KRAS mutations and can reveal the development of additional KRAS point mutations after the introduction of a certain treatment is required. Using such a model, one could better investigate the role of KRAS mutations in the early stages of pancreatic cancer. 2) The application of nanomaterials is another promising method for studying the role of KRAS mutations in early stage of pancreatic cancer. Traditional drug delivery methods have failed to distinguish cancer cells from normal cells (Conde et al., 2016). Nanomaterials could be an efficient medium for delivering siRNA to knock out KRAS (Mehta et al., 2019) from cells. However, these nanomaterials may elicit an immune response and, therefore, be hard to use in clinical practice. Thus, a nanomaterial that does not elicit an immune response from the human body is required. In addition, this nanomaterial should be able to modify various KRAS mutations and, therefore, act as a unified therapy.
ZZ wrote the manuscript and prepared the figures of the manuscript. HZ revised the manuscript. XL and HT finalized the manuscript together. All authors read and approved the final manuscript.
This study was supported by grants from the National Natural Science Foundation of China (82071984, 82001698, 82272066), Project of Social Development Guiding Science and Technology of Zhen-jiang City (FZ2021055), Infectious and Inflammatory Radiology Committee of Jiangsu Re-search Hospital Association (GY202205), Jinshan talents in the medical field of Zhenjiang (JSYCBS202101), and Postgraduate Innovation Project of Jiangsu Province (SJCX22_1878).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdel-Wahab, A. F., Mahmoud, W., and Al-Harizy, R. M. (2019). Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 150, 104511. doi:10.1016/j.phrs.2019.104511
Aguirre, A. J., Bardeesy, N., Sinha, M., Lopez, L., Tuveson, D. A., Horner, J., et al. (2003). Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes. Dev. 17, 3112–3126. doi:10.1101/gad.1158703
Allenson, K., Castillo, J., San Lucas, F. A., Scelo, G., Kim, D. U., Bernard, V., et al. (2017). High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann. Oncol. 28, 741–747. doi:10.1093/annonc/mdx004
Alonso-Curbelo, D., Ho, Y. J., Burdziak, C., Maag, J. L. V., Morris, J. P., Chandwani, R., et al. (2021). A gene-environment-induced epigenetic program initiates tumorigenesis. Nature 590, 642–648. doi:10.1038/s41586-020-03147-x
Andersen, D. K., Korc, M., Petersen, G. M., Eibl, G., Li, D., Rickels, M. R., et al. (2017). Diabetes, pancreatogenic diabetes, and pancreatic cancer. Diabetes 66, 1103–1110. doi:10.2337/db16-1477
Azizi, N., Toma, J., Martin, M., Khalid, M. F., Mousavi, F., Win, P. W., et al. (2021). Loss of activating transcription factor 3 prevents KRAS-mediated pancreatic cancer. Oncogene 40, 3118–3135. doi:10.1038/s41388-021-01771-z
Bannoura, S. F., Uddin, M. H., Nagasaka, M., Fazili, F., Al-Hallak, M. N., Philip, P. A., et al. (2021). Targeting KRAS in pancreatic cancer: New drugs on the horizon. Cancer Metastasis Rev. 40, 819–835. doi:10.1007/s10555-021-09990-2
Bear, A. S., Vonderheide, R. H., and O'Hara, M. H. (2020). Challenges and opportunities for pancreatic cancer immunotherapy. Cancer Cell. 38, 788–802. doi:10.1016/j.ccell.2020.08.004
Bond, M. J., Chu, L., Nalawansha, D. A., Li, K., and Crews, C. M. (2020). Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent. Sci. 6, 1367–1375. doi:10.1021/acscentsci.0c00411
Bott, A. J., Shen, J., Tonelli, C., Zhan, L., Sivaram, N., Jiang, Y. P., et al. (2019). Glutamine anabolism plays a critical role in pancreatic cancer by coupling carbon and nitrogen metabolism. Cell. Rep. 29, 1287–1298. doi:10.1016/j.celrep.2019.09.056
Brunner, T. B., Cengel, K. A., Hahn, S. M., Wu, J., Fraker, D. L., McKenna, W. G., et al. (2005). Pancreatic cancer cell radiation survival and prenyltransferase inhibition: The role of K-ras. Cancer Res. 65, 8433–8441. doi:10.1158/0008-5472.CAN-05-0158
Buscail, L., Bournet, B., and Cordelier, P. (2020). Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 17, 153–168. doi:10.1038/s41575-019-0245-4
Carrer, A., Trefely, S., Zhao, S., Campbell, S. L., Norgard, R. J., Schultz, K. C., et al. (2019). Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 9, 416–435. doi:10.1158/2159-8290.CD-18-0567
Chalabi-Dchar, M., Cassant-Sourdy, S., Duluc, C., Fanjul, M., Lulka, H., Samain, R., et al. (2015). Loss of somatostatin receptor subtype 2 promotes growth of KRAS-induced pancreatic tumors in mice by activating PI3K signaling and overexpression of CXCL16. Gastroenterology 148, 1452–1465. doi:10.1053/j.gastro.2015.02.009
Chang, H. H., Moro, A., Takakura, K., Su, H. Y., Mo, A., Nakanishi, M., et al. (2017). Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS One 12, e0184455. doi:10.1371/journal.pone.0184455
Chang, X., Liu, X., Wang, H., Yang, X., and Gu, Y. (2022). Glycolysis in the progression of pancreatic cancer. Am. J. Cancer Res. 12, 861–872.
Chen, N. M., Singh, G., Koenig, A., Liou, G. Y., Storz, P., Zhang, J. S., et al. (2015). NFATc1 links EGFR signaling to induction of Sox9 transcription and acinar-ductal transdifferentiation in the pancreas. Gastroenterology 148, 1024–1034. doi:10.1053/j.gastro.2015.01.033
Chen, X., Zeh, H. J., Kang, R., Kroemer, G., and Tang, D. (2021). Cell death in pancreatic cancer: From pathogenesis to therapy. Nat. Rev. Gastroenterol. Hepatol. 18, 804–823. doi:10.1038/s41575-021-00486-6
Chen, H., Zhuo, Q., Ye, Z., Xu, X., and Ji, S. (2021). Organoid model: A new hope for pancreatic cancer treatment? Biochim. Biophys. Acta Rev. Cancer 1875, 188466. doi:10.1016/j.bbcan.2020.188466
Chen, L., Zhang, J., Wang, X., Liu, G. S., Chen, Y., Ouyang, D., et al. (2022). Cell-modified plasmonic interface for the signal-amplified detection of Cucurbitacin E. Acta Pharm. Sin. B 12, 274–283. doi:10.1364/BOE.445679
Cheng, H., Luo, G., Jin, K., Fan, Z., Huang, Q., Gong, Y., et al. (2020). Kras mutation correlating with circulating regulatory T cells predicts the prognosis of advanced pancreatic cancer patients. Cancer Med. 9, 2153–2159. doi:10.1002/cam4.2895
Cheng, J., Li, Y., Wang, X., Dong, G., and Sheng, C. (2020b). Discovery of novel PDEδ degraders for the treatment of KRAS mutant colorectal cancer. J. Med. Chem. 63, 7892–7905. doi:10.1021/acs.jmedchem.0c00929
Chiu, C. F., Hsu, M. I., Yeh, H. Y., Park, J. M., Shen, Y. S., Tung, T. H., et al. (2021). Eicosapentaenoic acid inhibits KRAS mutant pancreatic cancer cell growth by suppressing hepassocin expression and STAT3 phosphorylation. Biomolecules 11, 370. doi:10.3390/biom11030370
Choi, M., Bien, H., Mofunanya, A., and Powers, S. (2019). Challenges in Ras therapeutics in pancreatic cancer. Semin. Cancer Biol. 54, 101–108. doi:10.1016/j.semcancer.2017.11.015
Cluntun, A. A., Lukey, M. J., Cerione, R. A., and Locasale, J. W. (2017). Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer 3, 169–180. doi:10.1016/j.trecan.2017.01.005
Coelho, M. A., de Carné Trécesson, S., Rana, S., Zecchin, D., Moore, C., Molina-Arcas, M., et al. (2017). Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 47, 1083–1099. doi:10.1016/j.immuni.2017.11.016
Collisson, E. A., Trejo, C. L., Silva, J. M., Gu, S., Korkola, J. E., Heiser, L. M., et al. (2012). A central role for RAF→MEK→ERK signaling in the Genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2, 685–693. doi:10.1158/2159-8290.CD-11-0347
Conde, J., Oliva, N., Zhang, Y., and Artzi, N. (2016). Local triple-combination therapy results in tumour regression and prevents recurrence in a colon cancer model. Nat. Mater 15, 1128–1138. doi:10.1038/nmat4707
Counter, C. (2015). Evaluating the role of nitric oxide synthase in oncogenic ras-driven tumorigenesis. Redox Biol. 5, 417. doi:10.1016/j.redox.2015.09.023
Dai, E., Han, L., Liu, J., Xie, Y., Kroemer, G., Klionsky, D. J., et al. (2020). Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 16, 2069–2083. doi:10.1080/15548627.2020.1714209
Datta, J., Dai, X., Bianchi, A., De Castro Silva, I., Mehra, S., Garrido, V. T., et al. (2022). Combined MEK and STAT3 inhibition uncovers stromal plasticity by enriching for cancer-associated fibroblasts with mesenchymal stem cell-like features to overcome immunotherapy resistance in pancreatic cancer. Gastroenterology 163, 1593–1612. doi:10.1053/j.gastro.2022.07.076
De Jesus, A., Keyhani-Nejad, F., Pusec, C. M., Goodman, L., Geier, J. A., Stoolman, J. S., et al. (2022). Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol. Cell. 82, 1261–1277.e9. doi:10.1016/j.molcel.2022.02.028
De La, O. J., and Murtaugh, L. C. (2009). Notch and kras in pancreatic cancer: At the crossroads of mutation, differentiation and signaling. Cell. Cycle 8, 1860–1864. doi:10.4161/cc.8.12.8744
De Santis, M. C., Gozzelino, L., Margaria, J. P., Costamagna, A., Ratto, E., Gulluni, F., et al. (2023). Lysosomal lipid switch sensitises to nutrient deprivation and mTOR targeting in pancreatic cancer. Gut 72, 360–371. doi:10.1136/gutjnl-2021-325117
Delgir, S., Bastami, M., Ilkhani, K., Safi, A., Seif, F., and Alivand, M. R. (2021). The pathways related to glutamine metabolism, glutamine inhibitors and their implication for improving the efficiency of chemotherapy in triple-negative breast cancer. Mutat. Res. Rev. Mutat. Res. 787, 108366. doi:10.1016/j.mrrev.2021.108366
Deramaudt, T., and Rustgi, A. K. (2005). Mutant KRAS in the initiation of pancreatic cancer. Biochim. Biophys. Acta 1756, 97–101. doi:10.1016/j.bbcan.2005.08.003
Dey, P., Li, J., Zhang, J., Chaurasiya, S., Strom, A., Wang, H., et al. (2020). Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov. 10, 608–625. doi:10.1158/2159-8290.CD-19-0297
Dibble, C. C., Barritt, S. A., Perry, G. E., Lien, E. C., Geck, R. C., DuBois-Coyne, S. E., et al. (2022). PI3K drives the de novo synthesis of coenzyme A from vitamin B5. Nature 608, 192–198. doi:10.1038/s41586-022-04984-8
Dougherty, P. G., Sahni, A., and Pei, D. (2019). Understanding cell penetration of cyclic peptides. Chem. Rev. 119, 10241–10287. doi:10.1021/acs.chemrev.9b00008
Ebrahimi, S., Hosseini, M., Shahidsales, S., Maftouh, M., Ferns, G. A., Ghayour-Mobarhan, M., et al. (2017). Targeting the akt/PI3K signaling pathway as a potential therapeutic strategy for the treatment of pancreatic cancer. Curr. Med. Chem. 24, 1321–1331. doi:10.2174/0929867324666170206142658
Eggers, J. P., Grandgenett, P. M., Collisson, E. C., Lewallen, M. E., Tremayne, J., Singh, P. K., et al. (2011). Cyclin-dependent kinase 5 is amplified and overexpressed in pancreatic cancer and activated by mutant K-Ras. Clin. Cancer Res. 17, 6140–6150. doi:10.1158/1078-0432.CCR-10-2288
Eser, S., Schnieke, A., Schneider, G., and Saur, D. (2014). Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 111, 817–822. doi:10.1038/bjc.2014.215
Ferguson, S., Yang, K. S., Zelga, P., Liss, A. S., Carlson, J. C. T., Del Castillo, C. F., et al. (2022). Single-EV analysis (sEVA) of mutated proteins allows detection of stage 1 pancreatic cancer. Sci. Adv. 8, eabm3453. doi:10.1126/sciadv.abm3453
Ferguson, L. P., Gatchalian, J., McDermott, M. L., Nakamura, M., Chambers, K., Rajbhandari, N., et al. (2023). Smarcd3 is an epigenetic modulator of the metabolic landscape in pancreatic ductal adenocarcinoma. Nat. Commun. 14, 292. doi:10.1038/s41467-023-35796-7
Ferro, R., and Falasca, M. (2014). Emerging role of the KRAS-PDK1 axis in pancreatic cancer. World J. Gastroenterol. 20, 10752–10757. doi:10.3748/wjg.v20.i31.10752
Flowers, B. M., Xu, H., Mulligan, A. S., Hanson, K. J., Seoane, J. A., Vogel, H., et al. (2021). Cell of origin influences pancreatic cancer subtype. Cancer Discov. 11, 660–677. doi:10.1158/2159-8290.CD-20-0633
Frank, K. J., Mulero-Sánchez, A., Berninger, A., Ruiz-Cañas, L., Bosma, A., Görgülü, K., et al. (2022). Extensive preclinical validation of combined RMC-4550 and LY3214996 supports clinical investigation for KRAS mutant pancreatic cancer. Cell. Rep. Med. 3, 100815. doi:10.1016/j.xcrm.2022.100815
Ganapathy-Kanniappan, S., and Geschwind, J. F. (2013). Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 12, 152. doi:10.1186/1476-4598-12-152
Ganguly, K., Bhatia, R., Rauth, S., Kisling, A., Atri, P., Thompson, C., et al. (2022). Mucin 5AC serves as the nexus for β-Catenin/c-Myc interplay to promote glutamine dependency during pancreatic cancer chemoresistance. Gastroenterol. 162, 253–268.e213.
Gomes-Filho, S. M., Dos Santos, E. O., Bertoldi, E. R. M., Scalabrini, L. C., Heidrich, V., Dazzani, B., et al. (2020). Aurora A kinase and its activator TPX2 are potential therapeutic targets in KRAS-induced pancreatic cancer. Cell. Oncol. (Dordr) 43, 445–460. doi:10.1007/s13402-020-00498-5
Gruber, R., Panayiotou, R., Nye, E., Spencer-Dene, B., Stamp, G., and Behrens, A. (2016). YAP1 and TAZ control pancreatic cancer initiation in mice by direct up-regulation of JAK-STAT3 signaling. Gastroenterology 151, 526–539. doi:10.1053/j.gastro.2016.05.006
Hamarsheh, S., Groß, O., Brummer, T., and Zeiser, R. (2020). Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 11, 5439. doi:10.1038/s41467-020-19288-6
Hashimoto, S., Furukawa, S., Hashimoto, A., Tsutaho, A., Fukao, A., Sakamura, Y., et al. (2019). ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc. Natl. Acad. Sci. U. S. A. 116, 17450–17459. doi:10.1073/pnas.1901765116
Hermann, P. C., Sancho, P., Cañamero, M., Martinelli, P., Madriles, F., Michl, P., et al. (2014). Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology 147, 1119–1133. doi:10.1053/j.gastro.2014.08.002
Hofmann, M. H., Gmachl, M., Ramharter, J., Savarese, F., Gerlach, D., Marszalek, J. R., et al. (2021). BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 11, 142–157. doi:10.1158/2159-8290.CD-20-0142
Hong, D. S., Fakih, M. G., Strickler, J. H., Desai, J., Durm, G. A., Shapiro, G. I., et al. (2020). KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 383, 1207–1217. doi:10.1056/NEJMoa1917239
Hou, P., Kapoor, A., Zhang, Q., Li, J., Wu, C. J., Li, J., et al. (2020). Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discov. 10, 1058–1077. doi:10.1158/2159-8290.CD-19-0597
Hu, Y., Lu, W., Chen, G., Wang, P., Chen, Z., Zhou, Y., et al. (2012). K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell. Res. 22, 399–412. doi:10.1038/cr.2011.145
Hu, C. M., Tien, S. C., Hsieh, P. K., Jeng, Y. M., Chang, M. C., Chang, Y. T., et al. (2019). High glucose triggers nucleotide imbalance through O-GlcNAcylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell. Metab. 29, 1334–1349. doi:10.1016/j.cmet.2019.02.005
Humpton, T. J., Alagesan, B., DeNicola, G. M., Lu, D., Yordanov, G. N., Leonhardt, C. S., et al. (2019). Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 9, 1268–1287. doi:10.1158/2159-8290.CD-18-1409
Izeradjene, K., Combs, C., Best, M., Gopinathan, A., Wagner, A., Grady, W. M., et al. (2007). Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 11, 229–243. doi:10.1016/j.ccr.2007.01.017
Ji, S., Qin, Y., Liang, C., Huang, R., Shi, S., Liu, J., et al. (2016). FBW7 (F-Box and WD repeat domain-containing 7) negatively regulates glucose metabolism by targeting the c-myc/TXNIP (Thioredoxin-Binding protein) Axis in pancreatic cancer. Clin. Cancer Res. 22, 3950–3960. doi:10.1158/1078-0432.CCR-15-2380
Jiang, W., Li, H., Liu, X., Zhang, J., Zhang, W., Li, T., et al. (2020). Precise and efficient silencing of mutant Kras(G12D) by CRISPR-CasRx controls pancreatic cancer progression. Theranostics 10, 11507–11519. doi:10.7150/thno.46642
Jiang, W., Qiao, L., Han, Y., Zhang, A., An, H., Xiao, J., et al. (2021). Pancreatic stellate cells regulate branched-chain amino acid metabolism in pancreatic cancer. Ann. Transl. Med. 9, 417. doi:10.21037/atm-21-761
Jin, H., Koh, M., Lim, H., Yong, H., Kim, E., Kim, S. Y., et al. (2023). Lipid raft protein flotillin-1 is important for the interaction between SOS1 and H-Ras/K-Ras, leading to Ras activation. Int. J. Cancer 152, 1933–1946. doi:10.1002/ijc.34443
Ju, H. Q., Ying, H., Tian, T., Ling, J., Fu, J., Lu, Y., et al. (2017). Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat. Commun. 8, 14437. doi:10.1038/ncomms14437
Kaushik, A. C., Wang, Y. J., Wang, X., and Wei, D. Q. (2021). Irinotecan and vandetanib create synergies for treatment of pancreatic cancer patients with concomitant TP53 and KRAS mutations. Brief. Bioinform 22, bbaa149. doi:10.1093/bib/bbaa149
Kazi, A., Chen, L., Xiang, S., Vangipurapu, R., Yang, H., Beato, F., et al. (2021). Global phosphoproteomics reveal CDK suppression as a vulnerability to KRas addiction in pancreatic cancer. Clin. Cancer Res. 27, 4012–4024. doi:10.1158/1078-0432.CCR-20-4781
Kemp, S. B., Cheng, N., Markosyan, N., Sor, R., Kim, I. K., Hallin, J., et al. (2023). Efficacy of a small-molecule inhibitor of KrasG12D in immunocompetent models of pancreatic cancer. Cancer Discov. 13, 298–311. doi:10.1158/2159-8290.CD-22-1066
King, B., Araki, J., Palm, W., and Thompson, C. B. (2020). Yap/Taz promote the scavenging of extracellular nutrients through macropinocytosis. Genes. Dev. 34, 1345–1358. doi:10.1101/gad.340661.120
Kopp, J. L., von Figura, G., Mayes, E., Liu, F. F., Dubois, C. L., Morris, J. P., et al. (2012). Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 22, 737–750. doi:10.1016/j.ccr.2012.10.025
Kruger, S., Heinemann, V., Ross, C., Diehl, F., Nagel, D., Ormanns, S., et al. (2018). Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann. Oncol. 29, 2348–2355. doi:10.1093/annonc/mdy417
Lee, K. E., and Bar-Sagi, D. (2010). Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell. 18, 448–458. doi:10.1016/j.ccr.2010.10.020
Lei, M. Z., Li, X. X., Zhang, Y., Li, J. T., Zhang, F., Wang, Y. P., et al. (2020). Acetylation promotes BCAT2 degradation to suppress BCAA catabolism and pancreatic cancer growth. Signal Transduct. Target Ther. 5, 70. doi:10.1038/s41392-020-0168-0
Leidner, R., Sanjuan Silva, N., Huang, H., Sprott, D., Zheng, C., Shih, Y. P., et al. (2022). Neoantigen T-cell receptor gene therapy in pancreatic cancer. N. Engl. J. Med. 386, 2112–2119. doi:10.1056/NEJMoa2119662
Lenkiewicz, E., Malasi, S., Hogenson, T. L., Flores, L. F., Barham, W., Phillips, W. J., et al. (2020). Genomic and epigenomic landscaping defines new therapeutic targets for adenosquamous carcinoma of the pancreas. Cancer Res. 80, 4324–4334. doi:10.1158/0008-5472.CAN-20-0078
Li, J. T., Yin, M., Wang, D., Wang, J., Lei, M. Z., Zhang, Y., et al. (2020). BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell. Biol. 22, 167–174. doi:10.1038/s41556-019-0455-6
Li, Y., He, Y., Peng, J., Su, Z., Li, Z., Zhang, B., et al. (2021). Mutant Kras co-opts a proto-oncogenic enhancer network in inflammation-induced metaplastic progenitor cells to initiate pancreatic cancer. Nat. Cancer 2, 49–65. doi:10.1038/s43018-020-00134-z
Li, R., Ng, T. S. C., Wang, S. J., Prytyskach, M., Rodell, C. B., Mikula, H., et al. (2021). Therapeutically reprogrammed nutrient signalling enhances nanoparticulate albumin bound drug uptake and efficacy in KRAS-mutant cancer. Nat. Nanotechnol. 16, 830–839. doi:10.1038/s41565-021-00897-1
Li, Z., Ge, Y., Dong, J., Wang, H., Zhao, T., Wang, X., et al. (2022). BZW1 facilitates glycolysis and promotes tumor growth in pancreatic ductal adenocarcinoma through potentiating eIF2α phosphorylation. Gastroenterology 162, 1256–1271.e14. doi:10.1053/j.gastro.2021.12.249
Li, S. Y., Wang, Z. J., Pan, C. Y., Wu, C., Li, Z. S., Jin, Z. D., et al. (2023). Comparative performance of endoscopic ultrasound-based techniques in patients with pancreatic cystic lesions: A network meta-analysis. Am. J. Gastroenterol. 118, 243–255. doi:10.14309/ajg.0000000000002088
Liang, C., Shi, S., Liu, M., Qin, Y., Meng, Q., Hua, J., et al. (2019). PIN1 maintains redox balance via the c-myc/NRF2 Axis to counteract kras-induced mitochondrial respiratory injury in pancreatic cancer cells. Cancer Res. 79, 133–145. doi:10.1158/0008-5472.CAN-18-1968
Liou, G. Y., Döppler, H., Braun, U. B., Panayiotou, R., Scotti Buzhardt, M., Radisky, D. C., et al. (2015). Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat. Commun. 6, 6200. doi:10.1038/ncomms7200
Liu, S., Gandler, H. I., Tošić, I., Ye, D. Q., Giaccone, Z. T., and Frank, D. A. (2021). Mutant KRAS downregulates the receptor for leukemia inhibitory factor (LIF) to enhance a signature of glycolysis in pancreatic cancer and lung cancer. Mol. Cancer Res. 19, 1283–1295. doi:10.1158/1541-7786.MCR-20-0633
Liu, C., Deng, S., Xiao, Z., Lu, R., Cheng, H., Feng, J., et al. (2023). Glutamine is a substrate for glycosylation and CA19-9 biosynthesis through hexosamine biosynthetic pathway in pancreatic cancer. Discov. Oncol. 14, 20. doi:10.1007/s12672-023-00628-z
Liu, H., Zhang, H., Liu, X., Guo, W., Liu, Q., Chen, L., et al. (2023). Pancreatic stellate cells exploit Wnt/β-catenin/TCF7-mediated glutamine metabolism to promote pancreatic cancer cells growth. Cancer Lett. 555, 216040. doi:10.1016/j.canlet.2022.216040
Lundy, J., Gao, H., Berry, W., Masoumi-Moghoddam, S., Jenkins, B. J., and Croagh, D. (2021). Targeted transcriptome and KRAS mutation analysis improve the diagnostic performance of EUS-FNA biopsies in pancreatic cancer. Clin. Cancer Res. 27, 5900–5911. doi:10.1158/1078-0432.CCR-21-1107
Luo, Y., Yang, Y., Liu, M., Wang, D., Wang, F., Bi, Y., et al. (2019). Oncogenic KRAS reduces expression of FGF21 in acinar cells to promote pancreatic tumorigenesis in mice on a high-fat diet. Gastroenterology 157, 1413–1428. doi:10.1053/j.gastro.2019.07.030
Luo, Y., Li, Z., Kong, Y., He, W., Zheng, H., An, M., et al. (2022). KRAS mutant-driven SUMOylation controls extracellular vesicle transmission to trigger lymphangiogenesis in pancreatic cancer. J. Clin. Invest. 132, e157644. doi:10.1172/JCI157644
Man, J., Pajic, M., and Joshua, A. M. (2020). Fats and mets, KRAS-driven lipid dysregulation affects metastatic potential in pancreatic cancer. Cancer Res. 80, 4886–4887. doi:10.1158/0008-5472.CAN-20-3082
Mann, K. M., Ying, H., Juan, J., Jenkins, N. A., and Copeland, N. G. (2016). KRAS-related proteins in pancreatic cancer. Pharmacol. Ther. 168, 29–42. doi:10.1016/j.pharmthera.2016.09.003
Martinelli, P., Madriles, F., Cañamero, M., Pau, E. C. d. S., Pozo, N. D., Guerra, C., et al. (2016). The acinar regulator Gata6 suppresses KrasG12V-driven pancreatic tumorigenesis in mice. Gut 65, 476–486. doi:10.1136/gutjnl-2014-308042
Martins, M., McCarthy, A., Baxendale, R., Guichard, S., Magno, L., Kessaris, N., et al. (2014). Tumor suppressor role of phospholipase C epsilon in Ras-triggered cancers. Proc. Natl. Acad. Sci. U. S. A. 111, 4239–4244. doi:10.1073/pnas.1311500111
Mehta, A., Dalle Vedove, E., Isert, L., and Merkel, O. M. (2019). Targeting KRAS mutant lung cancer cells with siRNA-loaded bovine serum albumin nanoparticles. Pharm. Res. 36, 133. doi:10.1007/s11095-019-2665-9
Melzer, M. K., Breunig, M., Arnold, F., Wezel, F., Azoitei, A., Roger, E., et al. (2022). Organoids at the PUB: The porcine urinary bladder serves as a pancreatic niche for advanced cancer modeling. Adv. Healthc. Mater 11, e2102345. doi:10.1002/adhm.202102345
Menini, S., Iacobini, C., de Latouliere, L., Manni, I., Vitale, M., Pilozzi, E., et al. (2020). Diabetes promotes invasive pancreatic cancer by increasing systemic and tumour carbonyl stress in Kras(G12D/+) mice. J. Exp. Clin. Cancer Res. 39, 152. doi:10.1186/s13046-020-01665-0
Mukhopadhyay, S., Goswami, D., Adiseshaiah, P. P., Burgan, W., Yi, M., Guerin, T. M., et al. (2020). Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res. 80, 1630–1643. doi:10.1158/0008-5472.CAN-19-1363
Nagasaka, M., Potugari, B., Nguyen, A., Sukari, A., Azmi, A. S., and Ou, S. H. I. (2021). KRAS inhibitors-yes but what next? Direct targeting of KRAS- vaccines, adoptive T cell therapy and beyond. Cancer Treat. Rev. 101, 102309. doi:10.1016/j.ctrv.2021.102309
Nakano, Y., Kitago, M., Matsuda, S., Nakamura, Y., Fujita, Y., Imai, S., et al. (2018). KRAS mutations in cell-free DNA from preoperative and postoperative sera as a pancreatic cancer marker: A retrospective study. Br. J. Cancer 118, 662–669. doi:10.1038/bjc.2017.479
Neuhöfer, P., Roake, C. M., Kim, S. J., Lu, R. J., West, R. B., Charville, G. W., et al. (2021). Acinar cell clonal expansion in pancreas homeostasis and carcinogenesis. Nature 597, 715–719. doi:10.1038/s41586-021-03916-2
Neuzillet, C., Tijeras-Raballand, A., de Mestier, L., Cros, J., Faivre, S., and Raymond, E. (2014). MEK in cancer and cancer therapy. Pharmacol. Ther. 141, 160–171. doi:10.1016/j.pharmthera.2013.10.001
Nussinov, R., Tsai, C. J., and Jang, H. (2021). Anticancer drug resistance: An update and perspective. Drug Resist Updat 59, 100796. doi:10.1016/j.drup.2021.100796
Ohara, Y., Valenzuela, P., and Hussain, S. P. (2022). The interactive role of inflammatory mediators and metabolic reprogramming in pancreatic cancer. Trends Cancer 8, 556–569. doi:10.1016/j.trecan.2022.03.004
Ozcan, S. C., Mutlu, A., Altunok, T. H., Gurpinar, Y., Sarioglu, A., Guler, S., et al. (2021). Simultaneous inhibition of PFKFB3 and GLS1 selectively kills KRAS-transformed pancreatic cells. Biochem. Biophys. Res. Commun. 571, 118–124. doi:10.1016/j.bbrc.2021.07.070
Pan, X., Zhou, J., Xiao, Q., Fujiwara, K., Zhang, M., Mo, G., et al. (2021). Cancer-associated fibroblast heterogeneity is associated with organ-specific metastasis in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 14, 184. doi:10.1186/s13045-021-01203-1
Pantsar, T. (2020). The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 18, 189–198. doi:10.1016/j.csbj.2019.12.004
Park, W., Chawla, A., and O'Reilly, E. M. (2021). Pancreatic cancer: A review. Jama 326, 851–862. doi:10.1001/jama.2021.13027
Park, S. J., Yoo, H. C., Ahn, E., Luo, E., Kim, Y., Sung, Y., et al. (2023). Enhanced glutaminolysis drives hypoxia-induced chemoresistance in pancreatic cancer. Cancer Res. 83, 735–752. doi:10.1158/0008-5472.CAN-22-2045
Patel, H., Okamura, R., Fanta, P., Patel, C., Lanman, R. B., Raymond, V. M., et al. (2019). Clinical correlates of blood-derived circulating tumor DNA in pancreatic cancer. J. Hematol. Oncol. 12, 130. doi:10.1186/s13045-019-0824-4
Paternoster, S., and Falasca, M. (20201873). The intricate relationship between diabetes, obesity and pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 1873, 188326. doi:10.1016/j.bbcan.2019.188326
Patra, K. C., Kato, Y., Mizukami, Y., Widholz, S., Boukhali, M., Revenco, I., et al. (2018). Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat. Cell. Biol. 20, 811–822. doi:10.1038/s41556-018-0122-3
Pedersen, K., Bilal, F., Bernadó Morales, C., Salcedo, M. T., Macarulla, T., Massó-Vallés, D., et al. (2017). Pancreatic cancer heterogeneity and response to Mek inhibition. Oncogene 36, 5639–5647. doi:10.1038/onc.2017.174
Pettazzoni, P., Viale, A., Shah, P., Carugo, A., Ying, H., Wang, H., et al. (2015). Genetic events that limit the efficacy of MEK and RTK inhibitor therapies in a mouse model of KRAS-driven pancreatic cancer. Cancer Res. 75, 1091–1101. doi:10.1158/0008-5472.CAN-14-1854
Ponz-Sarvise, M., Corbo, V., Tiriac, H., Engle, D. D., Frese, K. K., Oni, T. E., et al. (2019). Identification of resistance pathways specific to malignancy using organoid models of pancreatic cancer. Clin. Cancer Res. 25, 6742–6755. doi:10.1158/1078-0432.CCR-19-1398
Principe, D. R., Aissa, A. F., Kumar, S., Pham, T. N. D., Underwood, P. W., Nair, R., et al. (2022). Calcium channel blockers potentiate gemcitabine chemotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. U. S. A. 119, e2200143119. doi:10.1073/pnas.2200143119
Purohit, A., Varney, M., Rachagani, S., Ouellette, M. M., Batra, S. K., and Singh, R. K. (2016). CXCR2 signaling regulates KRAS(G12D)-induced autocrine growth of pancreatic cancer. Oncotarget 7, 7280–7296. doi:10.18632/oncotarget.6906
Qian, Y., Gong, Y., Fan, Z., Luo, G., Huang, Q., Deng, S., et al. (2020). Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 13, 130. doi:10.1186/s13045-020-00958-3
Raho, S., Capobianco, L., Malivindi, R., Vozza, A., Piazzolla, C., De Leonardis, F., et al. (2020). KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2, 1373–1381. doi:10.1038/s42255-020-00315-1
Reckzeh, E. S., and Waldmann, H. (2020). Small-molecule inhibition of glucose transporters GLUT-1-4. Chembiochem 21, 45–52. doi:10.1002/cbic.201900544
Roskoski, R. (2019). Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 142, 151–168. doi:10.1016/j.phrs.2019.01.039
Rossmeislová, L., Gojda, J., and Smolková, K. (2021). Pancreatic cancer: Branched-chain amino acids as putative key metabolic regulators? Cancer Metastasis Rev. 40, 1115–1139. doi:10.1007/s10555-021-10016-0
Sakamoto, K., Masutani, T., and Hirokawa, T. (2020). Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 10, 21671. doi:10.1038/s41598-020-78712-5
Salvador-Barbero, B., Álvarez-Fernández, M., Zapatero-Solana, E., El Bakkali, A., Menéndez, M. D. C., López-Casas, P. P., et al. (2020). CDK4/6 inhibitors impair recovery from cytotoxic chemotherapy in pancreatic adenocarcinoma. Cancer Cell. 37, 584–353.e346. doi:10.1016/j.ccell.2020.09.012
Saturno, G., Lopes, F., Niculescu-Duvaz, I., Niculescu-Duvaz, D., Zambon, A., Davies, L., et al. (2021). The paradox-breaking panRAF plus SRC family kinase inhibitor, CCT3833, is effective in mutant KRAS-driven cancers. Ann. Oncol. 32, 269–278. doi:10.1016/j.annonc.2020.10.483
Scalise, M., Pochini, L., Galluccio, M., Console, L., and Indiveri, C. (2017). Glutamine transport and mitochondrial metabolism in cancer cell growth. Front. Oncol. 7, 306. doi:10.3389/fonc.2017.00306
Seguin, L., Kato, S., Franovic, A., Camargo, M. F., Lesperance, J., Elliott, K. C., et al. (2014). An integrin β₃-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell. Biol. 16, 457–468. doi:10.1038/ncb2953
Sharma, S., Tapper, W. J., Collins, A., and Hamady, Z. Z. R. (2022). Predicting pancreatic cancer in the UK Biobank cohort using polygenic risk scores and diabetes mellitus. Gastroenterology 162, 1665–1674.e2. doi:10.1053/j.gastro.2022.01.016
Shi, G., DiRenzo, D., Qu, C., Barney, D., Miley, D., and Konieczny, S. F. (2013). Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 32, 1950–1958. doi:10.1038/onc.2012.210
Shi, L., Pan, H., Liu, Z., Xie, J., and Han, W. (2017). Roles of PFKFB3 in cancer. Signal Transduct. Target Ther. 2, 17044. doi:10.1038/sigtrans.2017.44
Son, J., Lyssiotis, C. A., Ying, H., Wang, X., Hua, S., Ligorio, M., et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. doi:10.1038/nature12040
Stine, Z. E., and Dang, C. V. (2020). Glutamine skipping the Q into mitochondria. Trends Mol. Med. 26, 6–7. doi:10.1016/j.molmed.2019.11.004
Sullivan, M. R., Danai, L. V., Lewis, C. A., Chan, S. H., Gui, D. Y., Kunchok, T., et al. (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, e44235. doi:10.7554/eLife.44235
Suzuki, T., Kishikawa, T., Sato, T., Takeda, N., Sugiura, Y., Seimiya, T., et al. (2022). Mutant KRAS drives metabolic reprogramming and autophagic flux in premalignant pancreatic cells. Cancer Gene Ther. 29, 505–518. doi:10.1038/s41417-021-00326-4
Tang, D., Kroemer, G., and Kang, R. (2021). Oncogenic KRAS blockade therapy: Renewed enthusiasm and persistent challenges. Mol. Cancer 20, 128. doi:10.1186/s12943-021-01422-7
Valsangkar, N. P., Ingkakul, T., Correa-Gallego, C., Mino-Kenudson, M., Masia, R., Lillemoe, K. D., et al. (2015). Survival in ampullary cancer: Potential role of different KRAS mutations. Surgery 157, 260–268. doi:10.1016/j.surg.2014.08.092
von Karstedt, S., and Walczak, H. (2020). An unexpected turn of fortune: Targeting TRAIL-rs in KRAS-driven cancer. Cell. Death Discov. 6, 14. doi:10.1038/s41420-020-0249-4
Wang, J. B., Erickson, J. W., Fuji, R., Ramachandran, S., Gao, P., Dinavahi, R., et al. (2010). Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 18, 207–219. doi:10.1016/j.ccr.2010.08.009
Wang, L., Yang, H., Abel, E. V., Ney, G. M., Palmbos, P. L., Bednar, F., et al. (2015). ATDC induces an invasive switch in KRAS-induced pancreatic tumorigenesis. Genes. Dev. 29, 171–183. doi:10.1101/gad.253591.114
Wang, D., Bi, Y., Hu, L., Luo, Y., Ji, J., Mao, A. Z., et al. (2019). Obesogenic high-fat diet heightens aerobic glycolysis through hyperactivation of oncogenic KRAS. Cell. Commun. Signal 17, 19. doi:10.1186/s12964-019-0333-7
Wang, F., Qi, X. M., Wertz, R., Mortensen, M., Hagen, C., Evans, J., et al. (2020). p38γ MAPK is essential for aerobic glycolysis and pancreatic tumorigenesis. Cancer Res. 80, 3251–3264. doi:10.1158/0008-5472.CAN-19-3281
Weisner, J., Landel, I., Reintjes, C., Uhlenbrock, N., Trajkovic-Arsic, M., Dienstbier, N., et al. (2019). Preclinical efficacy of covalent-allosteric AKT inhibitor borussertib in combination with trametinib in KRAS-mutant pancreatic and colorectal cancer. Cancer Res. 79, 2367–2378. doi:10.1158/0008-5472.CAN-18-2861
Won, E. J., Park, H., Chang, S. H., Kim, J. H., Kwon, H., Cho, Y. S., et al. (2021). One-shot dual gene editing for drug-resistant pancreatic cancer therapy. Biomaterials 279, 121252. doi:10.1016/j.biomaterials.2021.121252
Xu, Y., Yu, Z., Fu, H., Guo, Y., Hu, P., and Shi, J. (2022). Dual inhibitions on glucose/glutamine metabolisms for nontoxic pancreatic cancer therapy. ACS Appl. Mater Interfaces 14, 21836–21847. doi:10.1021/acsami.2c00111
Yamaguchi, T., Ikehara, S., Nakanishi, H., and Ikehara, Y. (2014). A genetically engineered mouse model developing rapid progressive pancreatic ductal adenocarcinoma. J. Pathol. 234, 228–238. doi:10.1002/path.4402
Yang, L., Venneti, S., and NagrathGlutaminolysis, D. (2017). Glutaminolysis: A hallmark of cancer metabolism. Annu. Rev. Biomed. Eng. 19, 163–194. doi:10.1146/annurev-bioeng-071516-044546
Ying, H., Kimmelman, A. C., Lyssiotis, C. A., Hua, S., Chu, G. C., Fletcher-Sananikone, E., et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 149, 656–670. doi:10.1016/j.cell.2012.01.058
Yun, J., Rago, C., Cheong, I., Pagliarini, R., Angenendt, P., Rajagopalan, H., et al. (2009). Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559. doi:10.1126/science.1174229
Zdanov, S., Mandapathil, M., Abu Eid, R., Adamson-Fadeyi, S., Wilson, W., Qian, J., et al. (2016). Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol. Res. 4, 354–365. doi:10.1158/2326-6066.CIR-15-0241
Zhang, J., Pavlova, N. N., and Thompson, C. B. (2017). Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. Embo J. 36, 1302–1315. doi:10.15252/embj.201696151
Zhang, Z., Gao, R., Hu, Q., Peacock, H., Peacock, D. M., Dai, S., et al. (2020). GTP-State-Selective cyclic peptide ligands of K-Ras(G12D) block its interaction with raf. ACS Cent. Sci. 6, 1753–1761. doi:10.1021/acscentsci.0c00514
Zhang, X., Mao, T., Zhang, B., Xu, H., Cui, J., Jiao, F., et al. (2022). Characterization of the genomic landscape in large-scale Chinese patients with pancreatic cancer. EBioMedicine 77, 103897. doi:10.1016/j.ebiom.2022.103897
Zhao, M., Mishra, L., and Deng, C. X. (2018). The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 14, 111–123. doi:10.7150/ijbs.23230
Zhao, Y., Wu, Z., Chanal, M., Guillaumond, F., Goehrig, D., Bachy, S., et al. (2020). Oncogene-induced senescence limits the progression of pancreatic neoplasia through production of activin A. Cancer Res. 80, 3359–3371. doi:10.1158/0008-5472.CAN-19-3763
Zhao, Y., Schoeps, B., Yao, D., Zhang, Z., Schuck, K., Tissen, V., et al. (2021). mTORC1 and mTORC2 converge on the arp2/3 complex to promote kras(g12d)-induced acinar-to-ductal metaplasia and early pancreatic carcinogenesis. Gastroenterology 160, 1755–1770.e17. doi:10.1053/j.gastro.2020.12.061
Zhou, Z., Zhang, J., Xu, C., Yang, J., Zhang, Y., Liu, M., et al. (2021). An integrated model of N6-methyladenosine regulators to predict tumor aggressiveness and immune evasion in pancreatic cancer. EBioMedicine 65, 103271. doi:10.1016/j.ebiom.2021.103271
Zhou, Z., Ren, Y., Yang, J., Liu, M., Shi, X., Luo, W., et al. (2022). Acetyl-coenzyme A synthetase 2 potentiates macropinocytosis and muscle wasting through metabolic reprogramming in pancreatic cancer. Gastroenterology 163, 1281–1293.e1. doi:10.1053/j.gastro.2022.06.058
Zhu, Z., Achreja, A., Meurs, N., Animasahun, O., Owen, S., Mittal, A., et al. (2020). Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat. Metab. 2, 775–792. doi:10.1038/s42255-020-0226-5
Zhu, L. L., Wu, Z., Li, R. K., Xing, X., Jiang, Y. S., Li, J., et al. (2021). Deciphering the genomic and lncRNA landscapes of aerobic glycolysis identifies potential therapeutic targets in pancreatic cancer. Int. J. Biol. Sci. 17, 107–118. doi:10.7150/ijbs.49243
Zhu, Q., Zhou, H., Wu, L., Lai, Z., Geng, D., Yang, W., et al. (2022). O-GlcNAcylation promotes pancreatic tumor growth by regulating malate dehydrogenase 1. Nat. Chem. Biol. 18, 1087–1095. doi:10.1038/s41589-022-01085-5
Zhuang, H., Chen, X., Wang, Y., Huang, S., Chen, B., Zhang, C., et al. (2022). Identification of LIPH as an unfavorable biomarkers correlated with immune suppression or evasion in pancreatic cancer based on RNA-seq. Cancer Immunol. Immunother. 71, 601–612. doi:10.1007/s00262-021-03019-x

Keywords: PDAC, KRAS mutation, phosphokinase, metabolic reprogramming, therapy resistance, poorer prognosis
Citation: Zhang Z, Zhang H, Liao X and Tsai H-i (2023) KRAS mutation: The booster of pancreatic ductal adenocarcinoma transformation and progression. Front. Cell Dev. Biol. 11:1147676. doi: 10.3389/fcell.2023.1147676
Received: 19 January 2023; Accepted: 10 April 2023;
Published: 20 April 2023.
Edited by:
Andy T. Y. Lau, Shantou University, ChinaReviewed by:
Rajeev Kumar Pandey, Johns Hopkins Medicine, United StatesCopyright © 2023 Zhang, Zhang, Liao and Tsai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Liao, bGlhb3hpYW5nMDI1QDEyNi5jb20=; Hsiang-i Tsai, VHNhaWhzaWFuZ2k4OEAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.