 Peipei Li1,2
Peipei Li1,2 Shen Li3Le Wang4Hongmin Li4Yang Wang4Hongbing Liu3
Shen Li3Le Wang4Hongmin Li4Yang Wang4Hongbing Liu3 Xin Wang3Xiaodan Zhu4Zhangsuo Liu1,2*†
Xin Wang3Xiaodan Zhu4Zhangsuo Liu1,2*† Fanglei Ye4*†Yuan Zhang4*†
Fanglei Ye4*†Yuan Zhang4*†- 1Department of Integrated Traditional and Western Nephrology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Henan Province Research Center for Kidney Disease, Zhengzhou, China
- 3Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 4Department of Otology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Sensorineural deafness becomes an inevitable worldwide healthy problem, yet the current curative therapy is limited. Emerging evidences demonstrate mitochondrial dysfunction plays a vital role of in the pathogenesis of deafness. Reactive oxygen species (ROS)-induced mitochondrial dysfunction combined with NLRP3 inflammasome activation is involved in cochlear damage. Autophagy not only clears up undesired proteins and damaged mitochondria (mitophagy), but also eliminate excessive ROS. Appropriate enhancement of autophagy can reduce oxidative stress, inhibit cell apoptosis, and protect auditory cells. In addition, we further discuss the interplays linking ROS generation, NLRP3 inflammasome activation, and autophagy underlying the pathogenesis of deafness, including ototoxic drugs-, noise- and aging-related hearing loss.
1 Introduction
Hearing loss affects almost 5% of the world’s population and impacts people ranges of all ages (Sheffield and Smith, 2019). Hearing loss causes communication barriers between people, leading to isolation, depression, dementia, and other psychological problems. This primary effect not only impairs the quality life of patients themselves but also causes indirect economic losses to society due to the reduced productivity caused by communication difficulties (Cunningham and Tucci, 2017). Therefore, a comprehensive understanding mechanism of hearing loss is extremely important for disease prevention and treatment.
Hearing loss is mainly considered a sensory disorder in humans. Multiple factors contribute to the pathogenesis of sensorineural hearing loss (SNHL), such as noise exposure, ototoxic drugs (aminoglycoside antibiotics, platin-based anticancer drugs, and loop diuretics), genetic mutations, aging, and chronic conditions. Histopathological changes of SNHL are characterized by mechanosensory hair cell damage, spiral ganglion neuron (SGN) loss, and stria vascularis atrophy (Keithley, 2020; Rousset et al., 2020). Emerging studies have suggested that mitochondrial DNA damage, reactive oxygen species (ROS) overproduction, and inflammatory mediators activation are associated with subsequent cochlear damage. Mitochondria ROS could induce inflammasome activation that promotes various disease progression (Martinon, 2010; Sorbara and Girardin, 2011). Moreover, ROS could also induce cellular defense process such as autophagy, a cytoprotective manner that deliver damaged organelles to lysosomes for degrader (Wang and Klionsky, 2003; Vernon and Tang, 2013). Current studies reveal autophagy exhibits an antioxidative capacity to protect against hair cell damage and possesses the potential to alleviate noise-induced hearing loss (NIHL) (Ye et al., 2019). This review mainly discusses the underlying mechanism affecting cochlear damage and hearing loss, including ROS-induced oxidative stress, autophagy, and NLRP3 inflammasome (Figure 1).
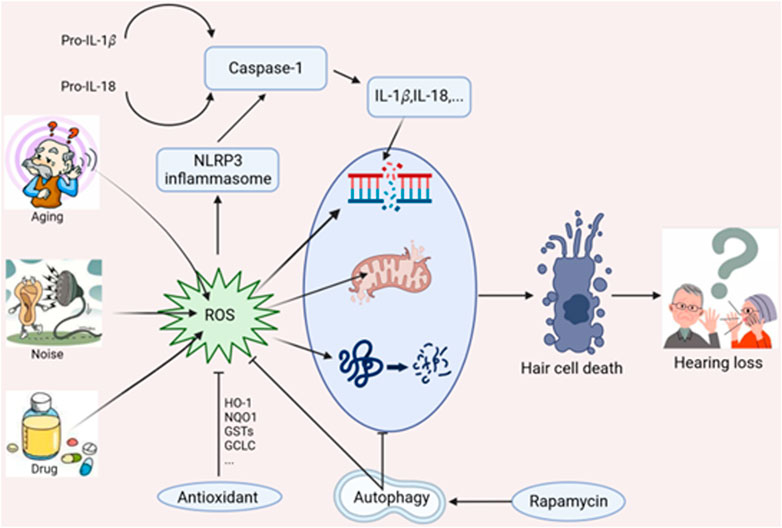
FIGURE 1. Interplay of oxidative stress, autophagy and NLRP3 inflammasome in hearing loss.
2 Mitochondria and reactive oxygen species
2.1 ROS generation
ROS, such as superoxide anions, hydrogen peroxide, and hydroxyl radicals, are reactive molecules containing oxygen and are mainly generated in the mitochondria (Han and Someya, 2013; Shrestha et al., 2021). Various stress conditions could markedly induce mitochondrial ROS production, increase metabolic rates, hypoxia, and membrane damage. The mitochondrial electron transport chains generate the electrochemical gradient to drive oxidative phosphorylation for ATP synthesis. The electron leakage in the respiratory chain results in the production of superoxide (Chance et al., 1979; Turrens, 2003; Brand, 2010). Seven precise sites have been identified for mitochondrial ROS production, complex I (NADH), complex III (cytochrome c oxidoreductase), glycerol 3-phosphate dehydrogenase, NADH-Q oxidoreductase, pyruvate dehydrogenase, and 2-oxoglutarate dehydrogenase. Of these sites, complex I-III are well shown to display the maximum capacities to produce ROS (Turrens et al., 1985; Muller et al., 2004; Pryde and Hirst, 2011). Antioxidant enzymes (catalase, SOD, and glutathione peroxidase) exert the cytoprotective effect by scavenging ROS. Oxidative stress represents an imbalance between ROS production and the antioxidant defense system. Overproduction of ROS can destroy biological membranes, attack DNA, cause gene mutations and protein denaturation, and ultimately cause various human diseases, neurodegenerative diseases, carcinogenesis, and aging-related diseases (Fujimoto and Yamasoba, 2019).
2.2 Mitochondrial ROS in hearing loss
Mitochondria ROS overproduction plays a key role in cochlear damage and hearing impairment (Pryde and Hirst, 2011). ROS-induced oxidative stress cause direct damage in cochlear hair cells, SGNs, and stria vascularis. ROS generation also leads to inflammation and pro-inflammatory cytokines secretion (Wong and Ryan, 2015). There are approximately 16,000 hair cells, and 35,000 transmissive SGNs in the human cochlea (Ehret and Frankenreiter, 1977; Schettino and Lauer, 2013). The cochlear hair cell and SGN refer to the foundation of sensory, and they do not regenerate after death. The most common histological sign of SNHL is cochlear sensory cell loss and damage (Schuknecht et al., 1973).
Oxidative stress induced by ROS can cause NIHL (Choi and Choi, 2015). ROS production is immediately detected in the cochlear tissue after exposure to high-intensity sound, and could be detected for several days (Ohlemiller et al., 1999). ROS-induced lipid peroxidation (malondialdehyde and 4-hydroxynonenal) can lead to apoptosis and vasoactive lipid peroxidation, and reduce cochlear blood flow (Yamashita et al., 2004; Fetoni et al., 2019). Noise-related ischemia and reperfusion further potentiate ROS generation (Wong and Ryan, 2015). In addition, noise trauma could induce elevate mitochondrial calcium levels and metabolic demand in hair cells, subsequently increasing ROS production (Henderson et al., 2006; Peng and Jou, 2010). Moreover, the antioxidants have been shown to attenuate NIHL when given either before or after noise exposure (Le et al., 2017). Ototoxic drugs are general associated with ototoxicity in clinical application. Hair cells are mechanoreceptors uniquely containing mechanotransducer (MET) channels on stereo ciliary bundles (Beurg et al., 2009; Wagner and Shin, 2019). Aminoglycoside transports into the hair cell via the MET channel, results in cell demise (Guthrie, 2008). Cisplatin-induced hearing loss is mainly caused by inflammation. Inflammation leads to the overproduction of NADPH oxidases subunits, impaired antioxidant defense systems. The subsequent ROS accumulation results in cell death in multiple manners, apoptosis, autophagy, pyroptosis, and necroptosis (Sheth et al., 2017; Gentilin et al., 2019; Nan et al., 2019). Cisplatin induced chronic changes affect outer hair cells, stria vascularis, and SGNs (Meech et al., 1998; Alam et al., 2000; Bowers et al., 2002). The free radical theory of aging believes that ROS attacking life macromolecules and causing tissue and cell damage is the fundamental cause of body aging (Beckman and Ames, 1998). Aging stress causes mitochondrial DNA damage, ROS overproduction, antioxidant function decreasing, and subsequent cochlear senescence (Fujimoto and Yamasoba, 2014). Mouse models of age-related hearing loss (ARHL) are known to harbor excessive ROS levels mitochondrial DNA mutations and. Increased ROS levels leads to lower mitochondrial membrane potential, affecting hair cell survival and hearing loss (Han and Someya, 2013; Yamasoba et al., 2013). SOD1-null mice display premature ARHL due to hair cell loss (McFadden et al., 1999).
In summary, noise trauma, ototoxic drugs or aging could first induce elevate mitochondrial calcium levels and metabolic demand, or lead to ischemia and reperfusion, or causes mitochondrial DNA damage. Then ROS is overproduced and leads to lower mitochondrial membrane potential, lipid peroxidation, pro-inflammatory cytokines secretion. Finally, senescence and cell death occur in the cochlea.
2.3 Antioxidants strategy in hearing loss
Antioxidants have the function of scavenging ROS and can be used to treat oxidative stress-related hearing loss. Mitochondrial-targeted antioxidants are expected to prevent or treat mitochondria-related disorder (Fujimoto and Yamasoba, 2019). Currently, the novel effective antioxidants, MitoQ, and SkQR1 exhibit protective effects against hearing loss in mouse auditory cell lines and animal models. MitoQ is a ubiquinone derivative that covalently binds to lipophilic triphenylphosphine (TPP) ions through aliphatic carbon chains and targets mitochondria (Kelso et al., 2001). The efficacy of mitochondria target antioxidants is more relying on their ability to cross the phospholipid bilayer and mitochondrial ROS elimination. The positive charge and hydrophilicity of TPP cation enable MitoQ accumulate to several hundred-fold in negatively charged mitochondria (Murphy and Smith, 2007). SkQ1 and SkQR1 are designed from MitoQ and display greater permeability in membrane transportation than MitoQ (Antonenko et al., 2008). The two antioxidants could also inhibit mitochondrial ROS formation. Treatment with MitoQ or SkQR1 protects against gentamicin-induced ototoxicity in animal models (Jankauskas et al., 2012; Ojano-Dirain et al., 2014). However, the efficacy of MitoQ in human studies is limited (Snow et al., 2010), and more clinical trials are required to evaluate its therapeutic effects on hearing loss patients. In addition, another antioxidant astaxanthin exerts powerful activities for ROS scavenging due to its unique membrane function and ability to permeate the blood-brain barrier (Nan et al., 2019). Collectively, these novel antioxidants may be a feasible method to alleviate and prevent ROS-related hearing loss.
3 Antioxidative role of mitochondria autophagy on hearing loss
3.1 Mitochondria autophagy mechanism
Autophagy is the process through which cells degraded cytoplasmic contents in the lysosome. Despite autophagy is once considered a non-selective process that mediated the bulk degradation of cytoplasmic components, current studies have demonstrated it can specifically target damaged organelles, such as mitochondria, ruptured lysosomes, peroxisomes, ER, lipid droplets (Levine and Kroemer, 2019). More than 40 autophagy-related (ATG) genes have been reported in yeast, of which ATG11 and ATG101 are considered core genes. These autophagic factors are recruited at the initiation, elongation, and closure of autophagosome. Mostly, the cellular cargo either containing an LC3-interacting region (LIR) or labeled with a ubiquitin tag could be recruited to adaptor proteins, which serves as a bridge between substances and LC3 (or GABARAP) motif and conjugate to the autophagosome membrane. Moreover, specific proteins bind to tripartite motif (TRIM) members for alternative autophagic degradation (Kimura et al., 2017). During the past decade, emerging studies have reported the involvement of autophagy in human diseases, particularly in neurodegenerative disorders, autoimmune diseases, and cancers (Mizushima and Levine, 2020).
The selective elimination of abnormal mitochondria via the autophagy pathway is termed mitophagy. Mitochondria degradation is triggered under the conditions of basal mitochondrial quality control, dysfunction, and developmental processes (Twig et al., 2008; Sato and Sato, 2011). The most well-studied mitophagy pathways are termed ubiquitin- and receptor-mediated mitophagy. The first pathway is mainly involved in PINK/Parkin, while receptor mitophagy includes NIX/BNIP3L, BNIP3, and FUNDC1. PINK1, a serine/threonine-protein kinase, is mainly associated with oxidative stress, autophagy, and apoptosis in cell (Trempe et al., 2013). Physiological PINK1 is transported into mitochondria via the mitochondrial target signal (MTS) and the membrane potential (ΔΨ m) (Yamano and Youle, 2013). The whole length of PINK1 is cleaved by matrix processing peptidase and PINK-associated rhomboid-like protease (PARL), releasing into the cytoplasm and degraded through ubiquitin-proteasome system (Deas et al., 2011). PINK1 transportation to the inner mitochondrial membrane is impaired in damaged or depolarized mitochondrial, causing the PINK1 accumulation on the outer mitochondrial to form dimers. Phosphorylation of PINK1 dimers recruits Parkin (an E3 ubiquitin ligase) through direct interaction, further activating Parkin and initiating mitophagy (Sarraf et al., 2013; Gladkova et al., 2018). Damaged mitochondria are enclosed into phagosome, and delivered to lysosome for depredating, as well as the PINK1 and Parkin protein degradation (Li et al., 2022a).
Bcl-2 and adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L/NIX) are homologous members of the Bcl-2 family, which are initially reported as pro-apoptotic proteins (Imazu et al., 1999). Both of the proteins are expressed on the mitochondria outer membrane, and contain classical LIR domains as essential components for mitophagy initiation. NIX/BNIP3L is required for excess mitochondria removal during reticulocyte maturation (Schweers et al., 2007). Phosphorylation of BNIP3 and NIX at serine residue sites near the LIR motif could stabilize their interactions with LC3, and promote mitophagy (Zhu et al., 2013; Rogov et al., 2017).
FUNDC1 is another well-known mitophagy receptor localized to the outer membrane of mitochondria. Similar to BNIP3 and BNIP3L, FUNDC1 also contains an LIR motif for interacting with the LC3 region (Liu et al., 2012). Under normal condition, FUNDC1 is phosphorylated at Try18 and Ser 13 by Src kinase and CK2, respectively. Hypoxia stress causes FUNDC1 dephosphorylation via inhibiting Src kinase and CK2 activity (Liu et al., 2012). Dephosphorylated FUNDC1 has a significantly higher affinity to LC3. Moreover, phosphoglycerate mutase family member 5 (PGAM5) also response for FUNDC1 dephosphorylation under hypoxia or mitochondrial uncoupling (Chen et al., 2014).
3.2 Mitochondria ROS and autophagy
The complex interplay between mitochondria oxidative stress and autophagy has been extensively reported. Excessive ROS level triggers general autophagy over mitophagy (Frank et al., 2012), while moderate ROS level triggers mitophagy through specific signaling activation. In turn, the redox signaling with mitophagy possess a cytoprotective protective effect to promote cell survival (Zhang et al., 2021). Dynein-related protein 1 (DRP-1) is the key factor in controlling mitochondria division and mitophagy initiation. Inhibiting DRP-1-dependent mitophagy can cause damaged mitochondria accumulation and ATP metabolic dysfunction, leading to cochlear hair cell senescence (Lin et al., 2019).
Increased ROS levels can regulate mitophagy via several pathways, NF-κB, mTOR, p38-MAPK, SIRT, etc., For example, ROS (H2O2) accumulation results in NF-κB inhibitor releasing in H2O2 oxidation manner, which activate NF-κB signaling (Sies et al., 2017). NF-κB promotes mitophagy through upregulating p62 expression, and attenuates NLRP3 inflammasome-mediated mitochondrial damage. p38-MAPK belong to the MAPK family and is induced by stress stimuli, such as inflammatory cytokines and oxidative stress. p38-MAPK affect the Parkin-mediated mitophagy in Parkin/PINK1-dependent pathway (Xiao et al., 2017). Sirtuins are a family of NAD-dependent deacetylases, which is response to activated NAD + function. SIRT1 affects Parkin translocation to mitochondrial inner membrane, and is slightly related to NAD +/NADH ratio alterations (Di Sante et al., 2015). SIRT1 could deacetylate FOXO1/3 and enhances mitophagy via activating the PINK1-Parkin axis (Zhao et al., 2021a). Inhibition of miR-34a/SIRT1 signaling enhance mitophagy and attenuate ROS-related hair cell death in the hearing loss context, implicating complicated interplay between ROS and mitophagy (Xiong et al., 2019).
3.3 Autophagy impairment contributes to ototoxic drugs, noise exposure, and aging-related hearing loss
Autophagy is an essential process participating in normal cochlear development and normal function of inner ear cells. Several autophagic genes (e.g., ATG4, ATG5) are expressed in the mouse cochlea from the embryonic phage until the adult phage (Aburto et al., 2012). ATG4-null mice show the common pathological features of the inner ear (Mariño et al., 2010). Deletion of Atg5 results in HCs degeneration and severe congenital hearing loss in mice due to the accumulation of undesired autophagic substrates (Fujimoto et al., 2017).
Dysfunction autophagy is linked to ototoxic hearing loss. Autophagic protein formation in the early stage of cisplatin treatment exerts cytoprotective activity, and induce HEI-OC1 cell death in the late stage of cisplatin treatment (Youn et al., 2015; Li et al., 2018). Neomycin treatment could induce HC death via mitophagy suppression (He et al., 2017; Zhang et al., 2022). Systemically aminoglycoside administration preferentially induces cochlear hair cell death and results in irreversible hearing loss. Gentamicin triggers RIPOR2 translocation from cochlear stereocilia to the pericuticular area in murine hair cells; RIPOR2 interacts with the autophagic protein GABARAP to activate autophagy, resulting in hair cells death (Li et al., 2022c). Downregulated RIPOR2 or GABARAP could prevent hair cell death and alleviate hearing loss in mice, suggesting autophagy components may be therapeutic targets to prevent ototoxicity (Li et al., 2022b).
Presbycusis is a common sensory disorder associated with aging. Autophagy deficiency occurs both in the aging model of HEI-OC1 cells and cochlear explant cultures (He et al., 2020). Furthermore, SGN from premature deaf mice model also display autophagic decreasing and accumulated lipofuscin (Menardo et al., 2012). Upregulation of autophagy promotes aging HC survival and slows the degeneration of auditory cells (Xiong et al., 2019). Rapamycin, an mTOR inhibitor, enhances SGNs autophagy via inhibiting the mTOR pathway, resulting in ARHL amelioration (Liu et al., 2022). Moreover, mitophagy refers to a specific autophagy process for damaged mitochondria degradation and cellular homeostasis maintaining. Mitophagy proteins (PINK1/Parkin, and BNIP3) is downregulated in the mouse auditory cortex and inferior colliculus region with aging (Youn et al., 2020); additionally, colocalization of autophagosome and lysosome is also decreased in the auditory system of aged mouse, indicating mitophagy impairment in the central auditory system.
ROS-induced oxidative damage is a major element of NIHL. The interplay between autophagy and ROS generation in NIHL has been detected. TTS-noise induced low-level oxidative stress activates autophagy that exerts a protective effect on outer hair cell survival, while excessive oxidative stress overwhelms the beneficial potential of autophagy, leading to outer hair cell death (Yuan et al., 2015). This point is supported by the results: noise-induced oxidative marker elevations is noise-dose-dependent in outer hair cells; whereas, autophagy marker is sharply increased after TTS, but slightly elevated in PTS and unaltered in sPTS noise (Yuan et al., 2015). Moreover, several antioxidant proteins or autophagic activators display the capacity to alleviate NIHL. SESN2 (sestrin 2) is an endogenous antioxidant protein. SESN2 interact with Unc-51-like protein kinase 1 (ULK1) to promote Beclin1 phosphorylation, Parkin mitochondrial translocation, and further facilitate mitophagy (Kumar and Shaha, 2018), indicating it might be as a therapeutic target against noise-induced cochlear injury. Pejvakin is a peroxisome-associated protein from the gasdermin family and exhibit a protective effect against harmful oxidative stress. Pejvakin-mediated selective autophagic degradation (pexophagy) could protect auditory hair cells against noise-induced damage via modulating the recruitment of autophagosome-associated protein MAP1LC3B (LC3B) (Defourny et al., 2019). Calcineurin inhibitor FK506 is also reported as an autophagic activator. It can activate autophagy via binding to ATPase catalytic subunit in neuronal cells, and alleviate neurodegenerative diseases (Kim et al., 2017). Treatment with FK506 (tacrolimus) could also reduce noise-induced hair cell damage via activating autophagy, and alleviated NIHL in adult CBA/J mice (He et al., 2021).
4 NLRP3 inflammasome in hearing loss
Inflammasomes are likely responsible for elevated ROS production in immune cells. The cochlea was once thought to have immune privileges. However, the immune privileged status changed when it was discovered that lymphocytes, such as macrophages can infiltrate into the endolymphatic sac of guinea pigs (Watson et al., 2017).
4.1 NLRP3 inflammasome activation
NLRP3 inflammasome is the most studied inflammasome comprised of NLRP3, an apoptosis associated speck-like protein containing a CARD (ASC) and procaspase (Agostini et al., 2004). NLRP3 contains an N-terminal pyrin domain (PYD), nucleotide-binding oligomerization (NACHT) domain, and C-terminal leucine-rich repeat (LRR) domain. Under the healthy condition, NLRP3 displays auto-repressed via the internal interaction of the NACHT and LRRs domain. PAMPs from microorganisms or DAMPs from endogenous lead to the removal of auto-repressed (Bryant and Fitzgerald, 2009). Exposure of the PYD domain mediates ASC and pro-caspase one recruitment, triggering the caspase-1 activation, and pro-inflammatory cytokines maturation (such as IL-1β and IL-18). NLRP3 is a general sensor of cellular stress that could be activated by ROS proximity to the inflammasome. NLRP3 inflammasome activation has been tightly regulated, and participates in several physiological processes, including immune system response and host defenses. The activated mechanisms included three panels, ionic flux, lysosomal damage, and ROS-mediated mitochondrial dysfunction (Tschopp and Schroder, 2010).
4.2 NLRP3 inflammasome in hearing loss
Mitochondria ROS has been confirmed as the crucial mechanism of ototoxic drugs. However, it is undesirable for the clinical application of antioxidants to alleviate the cisplatin-induced hearing loss. Cisplatin can trigger the assembly of NLRP3 inflammasome, induce marginal cell pyroptosis and cochlear damage (Yu et al., 2022). The pathological morphology changes and NLRP3 expression could be suppressed by inhibiting the upstream signal TXNIP. In addition, the NLRP3 inflammasome activation triggered by ROS was also reported in the ARHL and NIHL. The mechanisms are mainly involved in downstream inflammatory cytokines secretion (Shi et al., 2017; Zhuang et al., 2018; Sai et al., 2022).
Gain-of-function mutation in NLRP3 causes a spectrum of autoinflammatory diseases termed cryopyrin-associated periodic syndromes (CAPS) (Yu and Leslie, 2011). Abnormal NLRP3 inflammasome activation and excessive IL-1β secretion is the major reason of CAPS, and has been demonstrated in three clinical subtypes: neonatal-onset multisystem inflammatory disease (NOMID), Muckle-Wells syndrome (MWS), and familial cold autoinflammatory syndrome (FCAS). These phenotypes share general features, recurrent fever, rash, headache, etc., (Moltrasio et al., 2022). Hearing loss is one of the most common symptoms of NOMID and MWS, but is rare in FCAS. Notably, postcontrast MRI examination observed pathologic cochlear enhancement in most NOMID and MWS patients (Ahmadi et al., 2011), indicating the blood-labyrinth barrier is permeable by inflammation. Genetic studies of CAPSs have reported more than 80 NLRP3 variants, the majority of which are missense mutations located in exon 3, encoding the conserved NACHT domain (Conforti Andreoni et al., 2011). Whereas, minority NLRP3 mutations in other regions (e.g., LRR domain) are related to syndromic and non-syndromic hearing loss, such as deafness autosomal dominant 34 (DFN34) and keratitis fugax hereditaria (KFH) diseases. DFNA34 caused by the missense substitution p.Arg918Gln (c.2753G > A) in exon 7 (encoding the LRR domain) has been reported in two unrelated families (Nakanishi et al., 2017). Subjects in one family exhibited hearing loss accompanied by autoinflammatory features, while another family patients displayed hearing loss segregated without any other organ symptoms. Pathologic cochlea enhancement is identified in these subjects, implicating the cochlear inflammation in hearing loss. The hearing loss in several members is alleviated or completely resolved after treatment by IL-1β inhibitor anakinra (Nakanishi et al., 2017). A further study demonstrated the NLRP3 inflammasome were also activated in inner ear macrophages, and cochlea-infiltrated macrophages contribute to NLRP3-related hearing loss in the murine model (Nakanishi et al., 2017; Nakanishi et al., 2020).
4.3 Interplay between autophagy, NLRP3 inflammasome, and ROS generation
ROS induces NLRP3 inflammasome and results in cell damage. The mitophagy/autophagy system may eliminate mitochondrial ROS, thereby inhibiting the of NLRP3 inflammasome activation (Zhou et al., 2011). Autophagy can protect cells from inflammatory damage by inhibiting the activation of the inflammasome and pro-inflammatory signaling pathways (He et al., 2017). However, under starvation conditions of yeast, autophagy can promote inflammation via ATG-5 dependent non-classical manner (Dupont et al., 2011; Cao et al., 2019). In turn, NLRP3 inflammasome exerts an inhibitory effect on autophagy by cleaving the downstream signal molecule TRIF (Lai et al., 2018). Moreover, NLRP3 inflammasome activation mediate IL-1β releasing, which represents the main neurotoxicity in neuronal diseases, including brain stroke, Parkinson, and Alzheimer’s Disease (Wang et al., 2017; Han et al., 2019; Zhao et al., 2021b).
5 Another mechanisms of hearing loss
ERG (ether-a-go-go-related gene) channels are the members of the voltage-dependent potassium channel family, which have three subtypes, as ERG1 (Kv 11.1), ERG2 (Kv 11.2), and ERG3 (Kv11.3). The results of Ramazan Bal et al. show that the ERG channels appear to contribute to setting action potential (AP) frequency, threshold for AP induction, and, possibly, resting membrane potentials in this cells, which plays an important role in the formation of hearing (Yildirim and Bal, 2018).
Acid-sensing ion channels (ASICs) are voltage-independent and proton-gated channels. Bao-Ming Wu and Tian-Dong Leng demonstrate that oxidative stress increases ASIC1a expression/activation through the JNK signaling pathway, which may provide insight into the pathogenesis of neurological disorders that involve both ROS and activation of ASIC1a (Cakir et al., 2019; Wu et al., 2021).
Oxidative stress-induced Ca2+ permeable transient receptor potential melastatin 2 (TRPM2) channels are expressed at high levels in the brain, which seems to link neuronal excitability to cellular metabolism and participate in the pathogenesis of neurodegenerative disorders, such as SNHL (Bal et al., 2020). Activation of TRPM2 by reactive oxygen/nitrogen species (ROS/RNS) occurs following the production of ADPR, an intracellular activator. TRPM2 activation has been associated with cell death in the presence of increased oxidative and nitrosative stress, such as during oxygen–glucose deprivation. In the cochlear nucleus (CN), among these redox-sensitive TRP channels, TRPM2 is a potential candidate sensor of oxidative stress.
All in all, these studies have shown that oxidative stress-induced ion channels, including TRPM2 cation channels (Bal et al., 2020), ASICs (Cakir et al., 2019), ATP-sensitive potassium channels (Bal et al., 2018) and ERG channels (Yildirim and Bal, 2018), play an important role in hearing loss. These channels seem to transduce the increase in levels of reactive oxygen species exceeding physiological limits into specific cellular responses, such as triggering the apoptosis pathway, and ultimately leading to hearing loss.
6 Conclusion
In summary, ROS-mediated mitochondrial dysfunction combined with NLRP3 inflammasome activation contribute to progression of neurodegenerative diseases, including SNHL. ROS-induced oxidative stress and NLRP3-activated pro-inflammatory cytokines can damage the cochlea structure of auditory hair cells and SGNs. Novel efficient antioxidants can remove ROS and protect auditory cells. In addition, autophagy not only eliminate damaged proteins and organelle but also reduce ROS formation, and alleviate hearing loss. In most conditions, autophagy can ameliorate inflammatory diseases via inhibiting the NLRP3 inflammasome activation. Whereas, the pro-inflammatory effect of autophagy should be also noted in some cases. There remain some details to be solved. For example, the potential regulatory mechanism of autophagy in NLRP3 inflammasome remain unclear in SNHL progression. Moreover, intervention time in inflammatory reactions in the early or advanced stages of hearing loss should also be investigated. These conclusions provide therapeutic targets for inner ear diseases, such as targeting mitochondrial ROS, neutralizing pro-inflammatory cytokines, and appropriately increasing autophagy levels.
Author contributions
PL and SL wrote the draft, and ZL revised the manuscript and figure. YZ and FY contributed substantially to discussions of its content. LW, HL, and YW undertook review and editing of the manuscript before submission. HL and XW helped with the revision and figure preparation. All the authors read, amended, polished, and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the China Postdoctoral Science Foundation (NO.2022M712892) and the Joint project of Henan Province Medical Science and Technology Project (LHGJ20220358).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ATG, autophagy-related gene; ARHL, age-related hearing loss; DAMP, damage-associated molecular pattern; FCAS, familial cold autoinflammatory syndrome; LIR, LC3-interacting region; MET, mechanotransducer; MRI, magnetic resonance diffusion tension imaging; MWS, Muckle-Wells syndrome; NAD, nicotinamide adenine dinucleotide; NIHL, noise-induced hearing loss; LC3B, microtubule-associated protein one light chain 3β; PAMP, Pathogen associated molecular pattern; ROS, reactive oxygen species; SNHL, sensorineural hearing loss; SODs, superoxide dismutase; SGNs, spiral ganglion neurons; NOMID, neonatal-onset multisystem inflammatory disease; ULK1, unc-51-like protein kinase 1; 3MA, 3-methyladenine.
References
Aburto, M. R., SáNCHEZ-CalderóN, H., Hurlé, J. M., Varela-Nieto, I., and MagariñOS, M. (2012). Early otic development depends on autophagy for apoptotic cell clearance and neural differentiation. Cell Death Dis. 3, e394. doi:10.1038/cddis.2012.132
Agostini, L., Martinon, F., Burns, K., Mcdermott, M. F., Hawkins, P. N., and Tschopp, J. (2004). NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325. doi:10.1016/s1074-7613(04)00046-9
Ahmadi, N., Brewer, C. C., Zalewski, C., King, K. A., Butman, J. A., Plass, N., et al. (2011). Cryopyrin-associated periodic syndromes: Otolaryngologic and audiologic manifestations. Otolaryngol. Head. Neck Surg. 145, 295–302. doi:10.1177/0194599811402296
Alam, S. A., Ikeda, K., Oshima, T., Suzuki, M., Kawase, T., Kikuchi, T., et al. (2000). Cisplatin-induced apoptotic cell death in Mongolian gerbil cochlea. Hear Res. 141, 28–38. doi:10.1016/s0378-5955(99)00211-7
Antonenko, Y. N., Roginsky, V. A., Pashkovskaya, A. A., Rokitskaya, T. I., Kotova, E. A., Zaspa, A. A., et al. (2008). Protective effects of mitochondria-targeted antioxidant SkQ in aqueous and lipid membrane environments. J. Membr. Biol. 222, 141–149. doi:10.1007/s00232-008-9108-6
Bal, R., Ozturk, G., Etem, E. O., Eraslan, E., and Ozaydin, S. (2020). Modulation of the excitability of stellate neurons in the ventral cochlear nucleus of mice by TRPM2 channels. Eur. J. Pharmacol. 882, 173163. doi:10.1016/j.ejphar.2020.173163
Bal, R., Ozturk, G., Etem, E. O., Him, A., Cengiz, N., Kuloglu, T., et al. (2018). Modulation of excitability of stellate neurons in the ventral cochlear nucleus of mice by ATP-sensitive potassium channels. J. Membr. Biol. 251, 163–178. doi:10.1007/s00232-017-0011-x
Beckman, K. B., and Ames, B. N. (1998). The free radical theory of aging matures. Physiol. Rev. 78, 547–581. doi:10.1152/physrev.1998.78.2.547
Beurg, M., Fettiplace, R., Nam, J. H., and Ricci, A. J. (2009). Localization of inner hair cell mechanotransducer channels using high-speed calcium imaging. Nat. Neurosci. 12, 553–558. doi:10.1038/nn.2295
Bowers, W. J., Chen, X., Guo, H., Frisina, D. R., Federoff, H. J., and Frisina, R. D. (2002). Neurotrophin-3 transduction attenuates cisplatin spiral ganglion neuron ototoxicity in the cochlea. Mol. Ther. 6, 12–18. doi:10.1006/mthe.2002.0627
Brand, M. D. (2010). The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472. doi:10.1016/j.exger.2010.01.003
Bryant, C., and Fitzgerald, K. A. (2009). Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 19, 455–464. doi:10.1016/j.tcb.2009.06.002
Cakir, Z., Yildirim, C., Buran, I., Önalan, E. E., and Bal, R. (2019). Acid-sensing ion channels (ASICs) influence excitability of stellate neurons in the mouse cochlear nucleus. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 205, 769–781. doi:10.1007/s00359-019-01365-x
Cao, Z., Wang, Y., Long, Z., and He, G. (2019). Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim. Biophys. Sin. 51, 1087–1095. doi:10.1093/abbs/gmz098
Chance, B., Sies, H., and Boveris, A. (1979). Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 59, 527–605. doi:10.1152/physrev.1979.59.3.527
Chen, G., Han, Z., Feng, D., Chen, Y., Chen, L., Wu, H., et al. (2014). A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 54, 362–377. doi:10.1016/j.molcel.2014.02.034
Choi, S. H., and Choi, C. H. (2015). Noise-induced neural degeneration and therapeutic effect of antioxidant drugs. J. Audiol. Otol. 19, 111–119. doi:10.7874/jao.2015.19.3.111
Conforti Andreoni, C., Ricciardi-Castagnoli, P., and Mortellaro, A. (2011). The inflammasomes in health and disease: From genetics to molecular mechanisms of autoinflammation and beyond. Cell Mol. Immunol. 8, 135–145. doi:10.1038/cmi.2010.81
Cunningham, L. L., and Tucci, D. L. (2017). Hearing loss in adults. N. Engl. J. Med. 377, 2465–2473. doi:10.1056/NEJMra1616601
Deas, E., Plun-Favreau, H., Gandhi, S., Desmond, H., Kjaer, S., Loh, S. H., et al. (2011). PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 20, 867–879. doi:10.1093/hmg/ddq526
Defourny, J., Aghaie, A., Perfettini, I., Avan, P., Delmaghani, S., and Petit, C. (2019). Pejvakin-mediated pexophagy protects auditory hair cells against noise-induced damage. Proc. Natl. Acad. Sci. U. S. A. 116, 8010–8017. doi:10.1073/pnas.1821844116
DI Sante, G., Pestell, T. G., Casimiro, M. C., Bisetto, S., Powell, M. J., Lisanti, M. P., et al. (2015). Loss of Sirt1 promotes prostatic intraepithelial neoplasia, reduces mitophagy, and delays PARK2 translocation to mitochondria. Am. J. Pathol. 185, 266–279. doi:10.1016/j.ajpath.2014.09.014
Dupont, N., Jiang, S., Pilli, M., Ornatowski, W., Bhattacharya, D., and Deretic, V. (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. Embo J. 30, 4701–4711. doi:10.1038/emboj.2011.398
Ehret, G., and Frankenreiter, M. (1977). Quantitative analysis of cochlear structures in the house mouse in relation to mechanisms of acoustical information processing. J. Comp. physiology 122, 65–85. doi:10.1007/bf00611249
Fetoni, A. R., Paciello, F., Rolesi, R., Paludetti, G., and Troiani, D. (2019). Targeting dysregulation of redox homeostasis in noise-induced hearing loss: Oxidative stress and ROS signaling. Free Radic. Biol. Med. 135, 46–59. doi:10.1016/j.freeradbiomed.2019.02.022
Frank, M., Duvezin-Caubet, S., Koob, S., Occhipinti, A., Jagasia, R., Petcherski, A., et al. (2012). Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 12, 16.
Fujimoto, C., Iwasaki, S., Urata, S., Morishita, H., Sakamaki, Y., Fujioka, M., et al. (2017). Autophagy is essential for hearing in mice. Cell Death Dis. 8, e2780. doi:10.1038/cddis.2017.194
Fujimoto, C., and Yamasoba, T. (2019). Mitochondria-targeted antioxidants for treatment of hearing loss: A systematic review. Antioxidants 8, 109. doi:10.3390/antiox8040109
Fujimoto, C., and Yamasoba, T. (2014). Oxidative stresses and mitochondrial dysfunction in age-related hearing loss. Oxid. Med. Cell Longev. 582849, 3.
Gentilin, E., Simoni, E., Candito, M., Cazzador, D., and Astolfi, L. (2019). Cisplatin-induced ototoxicity: Updates on molecular targets. Trends Mol. Med. 25, 1123–1132. doi:10.1016/j.molmed.2019.08.002
Gladkova, C., Maslen, S. L., Skehel, J. M., and Komander, D. (2018). Mechanism of parkin activation by PINK1. Nature 559, 410–414. doi:10.1038/s41586-018-0224-x
Guthrie, O. (2008). Aminoglycoside induced ototoxicity. Toxicology 249, 91–96. doi:10.1016/j.tox.2008.04.015
Han, C., and Someya, S. (2013). Mouse models of age-related mitochondrial neurosensory hearing loss. Mol. Cell Neurosci. 55, 95–100. doi:10.1016/j.mcn.2012.07.004
Han, X., Sun, S., Sun, Y., Song, Q., Zhu, J., Song, N., et al. (2019). Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: Implications for Parkinson disease. Autophagy 15, 1860–1881. doi:10.1080/15548627.2019.1596481
He, Z., Guo, L., Shu, Y., Fang, Q., Zhou, H., Liu, Y., et al. (2017). Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy 13, 1884–1904. doi:10.1080/15548627.2017.1359449
He, Z. H., Pan, S., Zheng, H. W., Fang, Q. J., Hill, K., and Sha, S. H. (2021). Treatment with calcineurin inhibitor FK506 attenuates noise-induced hearing loss. Front. Cell Dev. Biol. 9, 648461. doi:10.3389/fcell.2021.648461
He, Z. H., Zou, S. Y., Li, M., Liao, F. L., Wu, X., Sun, H. Y., et al. (2020). The nuclear transcription factor FoxG1 affects the sensitivity of mimetic aging hair cells to inflammation by regulating autophagy pathways. Redox Biol. 28, 101364. doi:10.1016/j.redox.2019.101364
Henderson, D., Bielefeld, E. C., Harris, K. C., and Hu, B. H. (2006). The role of oxidative stress in noise-induced hearing loss. Ear Hear 27, 1–19. doi:10.1097/01.aud.0000191942.36672.f3
Imazu, T., Shimizu, S., Tagami, S., Matsushima, M., Nakamura, Y., Miki, T., et al. (1999). Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene 18, 4523–4529. doi:10.1038/sj.onc.1202722
Jankauskas, S. S., Plotnikov, E. Y., Morosanova, M. A., Pevzner, I. B., Zorova, L. D., Skulachev, V. P., et al. (2012). Mitochondria-targeted antioxidant SkQR1 ameliorates gentamycin-induced renal failure and hearing loss. Biochemistry 77, 666–670. doi:10.1134/S0006297912060144
Keithley, E. M. (2020). Pathology and mechanisms of cochlear aging. J. Neurosci. Res. 98, 1674–1684. doi:10.1002/jnr.24439
Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K., Ledgerwood, E. C., et al. (2001). Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596. doi:10.1074/jbc.M009093200
Kim, D., Hwang, H. Y., Kim, J. Y., Lee, J. Y., Yoo, J. S., Marko-Varga, G., et al. (2017). FK506, an immunosuppressive drug, induces autophagy by binding to the V-ATPase catalytic subunit A in neuronal cells. J. Proteome Res. 16, 55–64. doi:10.1021/acs.jproteome.6b00638
Kimura, T., Jia, J., Kumar, S., Choi, S. W., Gu, Y., Mudd, M., et al. (2017). Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. Embo J. 36, 42–60. doi:10.15252/embj.201695081
Kumar, A., and Shaha, C. (2018). SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Sci. Rep. 8, 615–19102. doi:10.1038/s41598-017-19102-2
Lai, M., Yao, H., Shah, S. Z. A., Wu, W., Wang, D., Zhao, Y., et al. (2018). The NLRP3-caspase 1 inflammasome negatively regulates autophagy via TLR4-TRIF in prion peptide-infected microglia. Front. aging Neurosci. 10, 116. doi:10.3389/fnagi.2018.00116
LE, T. N., Straatman, L. V., Lea, J., and Westerberg, B. (2017). Current insights in noise-induced hearing loss: A literature review of the underlying mechanism, pathophysiology, asymmetry, and management options. J. Otolaryngol. Head. Neck Surg. 46, 41–0219. doi:10.1186/s40463-017-0219-x
Levine, B., and Kroemer, G. (2019). Biological functions of autophagy genes: A disease perspective. Cell 176, 11–42. doi:10.1016/j.cell.2018.09.048
Li, A., Gao, M., Liu, B., Qin, Y., Chen, L., Liu, H., et al. (2022a). Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 13, 444–04906. doi:10.1038/s41419-022-04906-6
Li, H., Song, Y., He, Z., Chen, X., Wu, X., Li, X., et al. (2018). Meclofenamic acid reduces reactive oxygen species accumulation and apoptosis, inhibits excessive autophagy, and protects hair cell-like HEI-OC1 cells from cisplatin-induced damage. Front. Cell Neurosci. 12, 139. doi:10.3389/fncel.2018.00139
Li, J., Liu, C., MüLLER, U., and Zhao, B. (2022b). Autophagy proteins are essential for aminoglycoside-induced hearing loss. Autophagy 2, 1–2. doi:10.1080/15548627.2022.2127525
Li, J., Liu, C., MüLLER, U., and Zhao, B. (2022c). RIPOR2-mediated autophagy dysfunction is critical for aminoglycoside-induced hearing loss. Dev. Cell 57, 2204–2220.e6. doi:10.1016/j.devcel.2022.08.011
Lin, H., Xiong, H., Su, Z., Pang, J., Lai, L., Zhang, H., et al. (2019). Inhibition of DRP-1-dependent mitophagy promotes cochlea hair cell senescence and exacerbates age-related hearing loss. Front. Cell Neurosci. 13, 550. doi:10.3389/fncel.2019.00550
Liu, H., Li, F., Li, X., Wu, Q., and Dai, C. (2022). Rapamycin ameliorates age-related hearing loss in C57BL/6J mice by enhancing autophagy in the SGNs. Neurosci. Lett. 772, 136493. doi:10.1016/j.neulet.2022.136493
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi:10.1038/ncb2422
MariñO, G., FernáNDEZ, A. F., Cabrera, S., Lundberg, Y. W., Cabanillas, R., RodríGUEZ, F., et al. (2010). Autophagy is essential for mouse sense of balance. J. Clin. Invest 120, 2331–2344. doi:10.1172/JCI42601
Martinon, F. (2010). Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 40, 616–619. doi:10.1002/eji.200940168
Mcfadden, S. L., Ding, D., Reaume, A. G., Flood, D. G., and Salvi, R. J. (1999). Age-related cochlear hair cell loss is enhanced in mice lacking copper/zinc superoxide dismutase. Neurobiol. Aging 20, 1–8. doi:10.1016/s0197-4580(99)00018-4
Meech, R. P., Campbell, K. C., Hughes, L. P., and Rybak, L. P. (1998). A semiquantitative analysis of the effects of cisplatin on the rat stria vascularis. Hear Res. 124, 44–59. doi:10.1016/s0378-5955(98)00116-6
Menardo, J., Tang, Y., Ladrech, S., Lenoir, M., Casas, F., Michel, C., et al. (2012). Oxidative stress, inflammation, and autophagic stress as the key mechanisms of premature age-related hearing loss in SAMP8 mouse Cochlea. Antioxid. Redox Signal 16, 263–274. doi:10.1089/ars.2011.4037
Mizushima, N., and Levine, B. (2020). Autophagy in human diseases. N. Engl. J. Med. 383, 1564–1576. doi:10.1056/NEJMra2022774
Moltrasio, C., Romagnuolo, M., and Marzano, A. V. (2022). NLRP3 inflammasome and NLRP3-related autoinflammatory diseases: From cryopyrin function to targeted therapies. Front. Immunol. 13, 1007705. doi:10.3389/fimmu.2022.1007705
Muller, F. L., Liu, Y., and VAN Remmen, H. (2004). Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 279, 49064–49073. doi:10.1074/jbc.M407715200
Murphy, M. P., and Smith, R. A. (2007). Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 47, 629–656. doi:10.1146/annurev.pharmtox.47.120505.105110
Nakanishi, H., Kawashima, Y., Kurima, K., Chae, J. J., Ross, A. M., Pinto-Patarroyo, G., et al. (2017). NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc. Natl. Acad. Sci. U. S. A. 114, E7766–E7775. doi:10.1073/pnas.1702946114
Nakanishi, H., Prakash, P., Ito, T., Kim, H. J., Brewer, C. C., Harrow, D., et al. (2020). Genetic hearing loss associated with autoinflammation. Front. Neurol. 11, 141. doi:10.3389/fneur.2020.00141
Nan, B., Gu, X., and Huang, X. (2019). The role of the reactive oxygen species scavenger agent, astaxanthin, in the protection of cisplatin-treated patients against hearing loss. Drug Des. Devel Ther. 13, 4291–4303. doi:10.2147/DDDT.S212313
Ohlemiller, K. K., Wright, J. S., and Dugan, L. L. (1999). Early elevation of cochlear reactive oxygen species following noise exposure. Audiol. Neurootol 4, 229–236. doi:10.1159/000013846
Ojano-Dirain, C. P., Antonelli, P. J., and LE Prell, C. G. (2014). Mitochondria-targeted antioxidant MitoQ reduces gentamicin-induced ototoxicity. Otol. Neurotol. 35, 533–539. doi:10.1097/MAO.0000000000000192
Peng, T. I., and Jou, M. J. (2010). Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 1201, 183–188. doi:10.1111/j.1749-6632.2010.05634.x
Pryde, K. R., and Hirst, J. (2011). Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 286, 18056–18065. doi:10.1074/jbc.M110.186841
Rogov, V. V., Suzuki, H., Marinković, M., Lang, V., Kato, R., Kawasaki, M., et al. (2017). Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 7, 1131–01258. doi:10.1038/s41598-017-01258-6
Rousset, F., Nacher-Soler, G., Coelho, M., Ilmjarv, S., Kokje, V. B. C., Marteyn, A., et al. (2020). Redox activation of excitatory pathways in auditory neurons as mechanism of age-related hearing loss. Redox Biol. 30, 101434. doi:10.1016/j.redox.2020.101434
Sai, N., Yang, Y. Y., Ma, L., Liu, D., Jiang, Q. Q., Guo, W. W., et al. (2022). Involvement of NLRP3-inflammasome pathway in noise-induced hearing loss. Neural Regen. Res. 17, 2750–2754. doi:10.4103/1673-5374.339499
Sarraf, S. A., Raman, M., Guarani-Pereira, V., Sowa, M. E., Huttlin, E. L., Gygi, S. P., et al. (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. doi:10.1038/nature12043
Sato, M., and Sato, K. (2011). Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 334, 1141–1144. doi:10.1126/science.1210333
Schettino, A. E., and Lauer, A. M. (2013). The efficiency of design-based stereology in estimating spiral ganglion populations in mice. Hear Res. 304, 153–158. doi:10.1016/j.heares.2013.07.007
Schuknecht, H. F., Kimura, R. S., and Naufal, P. M. (1973). The pathology of sudden deafness. Acta Otolaryngol. 76, 75–97. doi:10.3109/00016487309121486
Schweers, R. L., Zhang, J., Randall, M. S., Loyd, M. R., Li, W., Dorsey, F. C., et al. (2007). NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. U. S. A. 104, 19500–19505. doi:10.1073/pnas.0708818104
Sheffield, A. M., and Smith, R. J. H. (2019). The epidemiology of deafness. Cold Spring Harb. Perspect. Med. 9, a033258. doi:10.1101/cshperspect.a033258
Sheth, S., Mukherjea, D., Rybak, L. P., and Ramkumar, V. (2017). Mechanisms of cisplatin-induced ototoxicity and otoprotection. Front. Cell Neurosci. 11, 338. doi:10.3389/fncel.2017.00338
Shi, X., Qiu, S., Zhuang, W., Yuan, N., Wang, C., Zhang, S., et al. (2017). NLRP3-inflammasomes are triggered by age-related hearing loss in the inner ear of mice. Am. J. Transl. Res. 9, 5611–5618.
Shrestha, R., Johnson, E., and Byrne, F. L. (2021). Exploring the therapeutic potential of mitochondrial uncouplers in cancer. Mol. Metab. 51, 101222. doi:10.1016/j.molmet.2021.101222
Sies, H., Berndt, C., and Jones, D. P. (2017). Oxidative stress. Annu. Rev. Biochem. 86, 715–748. doi:10.1146/annurev-biochem-061516-045037
Snow, B. J., Rolfe, F. L., Lockhart, M. M., Frampton, C. M., O'Sullivan, J. D., Fung, V., et al. (2010). A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov. Disord. 25, 1670–1674. doi:10.1002/mds.23148
Sorbara, M. T., and Girardin, S. E. (2011). Mitochondrial ROS fuel the inflammasome. Cell Res. 21, 558–560. doi:10.1038/cr.2011.20
Turrens, J. F., Alexandre, A., and Lehninger, A. L. (1985). Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 237, 408–414. doi:10.1016/0003-9861(85)90293-0
Trempe, J. F., Sauvé, V., Grenier, K., Seirafi, M., Tang, M. Y., MéNADE, M., et al. (2013). Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455. doi:10.1126/science.1237908
Tschopp, J., and Schroder, K. (2010). NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10, 210–215. doi:10.1038/nri2725
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344. doi:10.1113/jphysiol.2003.049478
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 27, 433–446. doi:10.1038/sj.emboj.7601963
Vernon, P. J., and Tang, D. (2013). Eat-me: Autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid. Redox Signal 18, 677–691. doi:10.1089/ars.2012.4810
Wagner, E. L., and Shin, J. B. (2019). Mechanisms of hair cell damage and repair. Trends Neurosci. 42, 414–424. doi:10.1016/j.tins.2019.03.006
Wang, C. W., and Klionsky, D. J. (2003). The molecular mechanism of autophagy. Mol. Med. 9, 65–76. doi:10.1007/bf03402040
Wang, D., Zhang, J., Jiang, W., Cao, Z., Zhao, F., Cai, T., et al. (2017). The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy 13, 914–927. doi:10.1080/15548627.2017.1293766
Watson, N., Ding, B., Zhu, X., and Frisina, R. D. (2017). Chronic inflammation - inflammaging - in the ageing cochlea: A novel target for future presbycusis therapy. Ageing Res. Rev. 40, 142–148. doi:10.1016/j.arr.2017.10.002
Wong, A. C., and Ryan, A. F. (2015). Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front. Aging Neurosci. 7, 58. doi:10.3389/fnagi.2015.00058
Wu, B. M., Bargaineer, J., Zhang, L., Yang, T., Xiong, Z. G., and Leng, T. D. (2021). Upregulation of acid sensing ion channel 1a (ASIC1a) by hydrogen peroxide through the JNK pathway. Acta Pharmacol. Sin. 42, 1248–1255. doi:10.1038/s41401-020-00559-3
Xiao, B., Deng, X., Lim, G. G. Y., Xie, S., Zhou, Z. D., Lim, K.-L., et al. (2017). Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis. 8, e3097. doi:10.1038/cddis.2017.463
Xiong, H., Chen, S., Lai, L., Yang, H., Xu, Y., Pang, J., et al. (2019). Modulation of miR-34a/SIRT1 signaling protects cochlear hair cells against oxidative stress and delays age-related hearing loss through coordinated regulation of mitophagy and mitochondrial biogenesis. Neurobiol. Aging 79, 30–42. doi:10.1016/j.neurobiolaging.2019.03.013
Yamano, K., and Youle, R. J. (2013). PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769. doi:10.4161/auto.24633
Yamashita, D., Jiang, H. Y., Schacht, J., and Miller, J. M. (2004). Delayed production of free radicals following noise exposure. Brain Res. 3, 201–209. doi:10.1016/j.brainres.2004.05.104
Yamasoba, T., Lin, F. R., Someya, S., Kashio, A., Sakamoto, T., and Kondo, K. (2013). Current concepts in age-related hearing loss: Epidemiology and mechanistic pathways. Hear Res. 303, 30–38. doi:10.1016/j.heares.2013.01.021
Ye, B., Fan, C., Shen, Y., Wang, Q., Hu, H., and Xiang, M. (2019). The antioxidative role of autophagy in hearing loss. Front. Neurosci. 12, 1010. doi:10.3389/fnins.2018.01010
Yildirim, C., and Bal, R. (2018). ERG channels regulate excitability in stellate and bushy cells of mice ventral cochlear nucleus. J. Membr. Biol. 251, 711–722. doi:10.1007/s00232-018-0048-5
Youn, C. K., Jun, Y., Jo, E. R., and Cho, S. I. (2020). Age-related hearing loss in C57bl/6J mice is associated with mitophagy impairment in the central auditory system. Int. J. Mol. Sci. 21, 7202. doi:10.3390/ijms21197202
Youn, C. K., Kim, J., Park, J. H., Do, N. Y., and Cho, S. I. (2015). Role of autophagy in cisplatin-induced ototoxicity. Int. J. Pediatr. Otorhinolaryngol. 79, 1814–1819. doi:10.1016/j.ijporl.2015.08.012
Yu, J. R., and Leslie, K. S. (2011). Cryopyrin-associated periodic syndrome: An update on diagnosis and treatment response. Curr. Allergy Asthma Rep. 11, 12–20. doi:10.1007/s11882-010-0160-9
Yu, W., Zong, S., Zhou, P., Wei, J., Wang, E., Ming, R., et al. (2022). Cochlear marginal cell pyroptosis is induced by cisplatin via NLRP3 inflammasome activation. Front. Immunol. 13, 823439. doi:10.3389/fimmu.2022.823439
Yuan, H., Wang, X., Hill, K., Chen, J., Lemasters, J., Yang, S. M., et al. (2015). Autophagy attenuates noise-induced hearing loss by reducing oxidative stress. Antioxid. Redox Signal 22, 1308–1324. doi:10.1089/ars.2014.6004
Zhang, C., Nie, P., Zhou, C., Hu, Y., Duan, S., Gu, M., et al. (2021). Oxidative stress-induced mitophagy is suppressed by the miR-106b-93-25 cluster in a protective manner. Cell Death Dis. 12, 209–03484. doi:10.1038/s41419-021-03484-3
Zhang, Y., Fang, Q., Wang, H., Qi, J., Sun, S., Liao, M., et al. (2022). Increased mitophagy protects cochlear hair cells from aminoglycoside-induced damage. Autophagy 26, 75–91. doi:10.1080/15548627.2022.2062872
Zhao, N., Xia, J., and Xu, B. (2021a). Physical exercise may exert its therapeutic influence on Alzheimer's disease through the reversal of mitochondrial dysfunction via SIRT1-FOXO1/3-PINK1-Parkin-mediated mitophagy. J. Sport Health Sci. 10, 1–3. doi:10.1016/j.jshs.2020.08.009
Zhao, S., Li, X., Wang, J., and Wang, H. (2021b). The role of the effects of autophagy on NLRP3 inflammasome in inflammatory nervous system diseases. Front. Cell Dev. Biol. 9, 657478. doi:10.3389/fcell.2021.657478
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. doi:10.1038/nature09663
Zhu, Y., Massen, S., Terenzio, M., Lang, V., Chen-Lindner, S., Eils, R., et al. (2013). Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 288, 1099–1113. doi:10.1074/jbc.M112.399345
Keywords: oxidative stress, autophagy, NLRP3 inflammasome, hearing loss, mitochondrial dysfunction
Citation: Li P, Li S, Wang L, Li H, Wang Y, Liu H, Wang X, Zhu X, Liu Z, Ye F and Zhang Y (2023) Mitochondrial dysfunction in hearing loss: Oxidative stress, autophagy and NLRP3 inflammasome. Front. Cell Dev. Biol. 11:1119773. doi: 10.3389/fcell.2023.1119773
Received: 13 December 2022; Accepted: 07 February 2023;
Published: 20 February 2023.
Edited by:
Bilal Çiğ, Ahi Evran University, TürkiyeReviewed by:
Jiann-Ruey Hong, National Cheng Kung University, TaiwanAhmi Öz, Süleyman Demirel University, Türkiye
Copyright © 2023 Li, Li, Wang, Li, Wang, Liu, Wang, Zhu, Liu, Ye and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhangsuo Liu, emhhbmdzdW9saXVAenp1LmVkdS5jbg==; Fanglei Ye, eWVmYW5nbGVpMDAwQHNpbmEuY29t; Yuan Zhang, emhhbmd5dWFuYnN5c0AxNjMuY29t
†These authors have contributed equally to this work