 Grant C. Walters
Grant C. Walters Yuriy M. Usachev
Yuriy M. Usachev
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 24 January 2023
Sec. Molecular and Cellular Pathology
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1094356
This article is part of the Research TopicMitochondria at the Nexus of Metabolism, Aging, and DiseaseView all 5 articles
Mitochondria are essential for proper cellular function through their critical roles in ATP synthesis, reactive oxygen species production, calcium (Ca2+) buffering, and apoptotic signaling. In neurons, Ca2+ buffering is particularly important as it helps to shape Ca2+ signals and to regulate numerous Ca2+-dependent functions including neuronal excitability, synaptic transmission, gene expression, and neuronal toxicity. Over the past decade, identification of the mitochondrial Ca2+ uniporter (MCU) and other molecular components of mitochondrial Ca2+ transport has provided insight into the roles that mitochondrial Ca2+ regulation plays in neuronal function in health and disease. In this review, we discuss the many roles of mitochondrial Ca2+ uptake and release mechanisms in normal neuronal function and highlight new insights into the Ca2+-dependent mechanisms that drive mitochondrial dysfunction in neurologic diseases including epilepsy, Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. We also consider how targeting Ca2+ uptake and release mechanisms could facilitate the development of novel therapeutic strategies for neurological diseases.
Mitochondria are colloquially referred to as the powerhouse of the cell because of their critical role in the production of ATP through oxidative phosphorylation (OXPHOS) (Brookes et al., 2004; Nunnari and Suomalainen, 2012). The production of ATP through OXPHOS is especially important within the brain, which accounts for nearly 20% of the body’s resting metabolism (Attwell and Laughlin, 2001; Howarth et al., 2012). Due to the high energy demand of neurons, they are very susceptible to mtDNA mutations that disrupt the electron transport chain; these mutations can result in a variety of neurological dysfunctions including seizures, blindness, and intellectual disabilities (DiMauro and Schon, 2003). In addition to producing ATP, reactive oxygen species (ROS) are produced as a byproduct of OXPHOS.
Mitochondrial ROS production is associated with damage to lipids, proteins, and nucleic acids, and excessive ROS are implicated in neurodegeneration (Nunnari and Suomalainen, 2012; Beckhauser et al., 2016). Super oxide free radicals (O2−) produced through OXPHOS are converted to H2O2 by enzymes such as superoxide dismutase and subsequently broken down into harmless byproducts (Beckhauser et al., 2016; Angelova and Abramov, 2018). When ROS are not broken down, the resulting oxidative stress can lead to activation of apoptotic signaling cascades and thereby to cell death (Fleury et al., 2002; Ueda et al., 2002). However, ROS also play an important physiological role, regulating signaling pathways that are critical for a wide variety of cellular functions. In neurons, ROS signaling is critical for axon development (Beckhauser et al., 2016), it modulates expression of AMPA and NMDA glutamate receptors (Esteras et al., 2022), and regulates various forms of synaptic plasticity including long-term potentiation (LTP) (Klann, 1998; Beckhauser et al., 2016). Despite the importance of ROS for normal neuronal function, proper regulation of their production is critical, because overproduction and subsequent oxidative stress are implicated in multiple pathologies. These include Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, Epilepsy, and schizophrenia (Bezprozvanny, 2009; Liochev, 2013; Puttachary et al., 2015; Beckhauser et al., 2016; Angelova and Abramov, 2018; Dagda, 2018).
In addition to ATP synthesis and ROS generation, mitochondria also play a critical role in Ca2+ signaling. Mitochondria efficiently buffer Ca2+ entering the cell during neuronal excitation and then slowly release accumulated Ca2+ back to the cytosol; this limits the amplitude and prolongs the duration of cytosolic Ca2+ responses. Consequently, mitochondrial have a tremendous ability to control and modulate many Ca2+-dependent functions in neurons, including neuronal excitability, neurotransmitter release, synaptic plasticity, gene expression, and cell survival (Mattson et al., 2008; Pivovarova and Andrews, 2010; Brini et al., 2014; Devine and Kittler, 2018; Jung et al., 2020) (Figure 1). Ca2+ uptake into the mitochondrial matrix regulates ATP synthesis by activating multiple components of the OXPHOS pathway, including ATP synthase, α-ketoglutarate dehydrogenase, isocitrate dehydrogenase, and pyruvate dehydrogenase (Denton et al., 1972; Brookes et al., 2004; Denton, 2009; Tarasov et al., 2012). ATP production depends on the concentration of Ca2+ within the mitochondrial matrix, with submicromolar mitochondrial Ca2+ concentrations stimulating ATP synthesis, and mitochondrial Ca2+ concentrations above 10 μM suppressing ATP production (Fink et al., 2017). While the Ca2+ dependent production of ATP is conserved across most cells within the body, neurons have unique functions that are dependent upon Ca2+ signaling as discussed below.
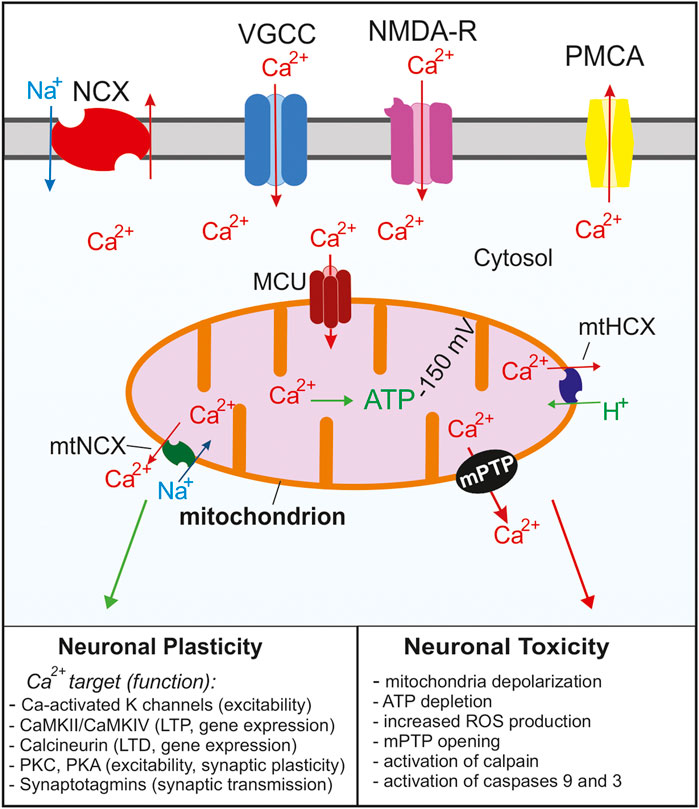
FIGURE 1. Summary of the Ca2+ signaling mechanisms in neurons that regulate cytosolic and mitochondrial Ca2+ concentrations. Ca2+ enters the cell through voltage gated Ca2+ channels (VGCC) following depolarization or through glutamate NMDA receptors. As the cytosolic Ca2+ concentration increases, the MCU complex opens and Ca2+ enters the mitochondria. Ca2+ is rapidly removed from the cytosol via Na+/Ca2+ exchangers (NCX) and plasma membrane Ca2+ ATPases (PMCA). As the cytosolic Ca2+ concentration decreases Ca2+ is slowly released from the mitochondria through Na+/Ca2+ (mtNCX) exchangers and H+/Ca2+ exchangers (mtHCX). Proper Ca2+ buffering is critical for normal neuronal functions and neuronal plasticity. Impaired buffering and Ca2+ overload leads to the opening of the mPTP and activation of apoptotic pathways and is implicated in many neurological diseases. For simplicity, the ER and other Ca2+ organelles are omitted from the model.
In neurons at rest, total calcium concentration in the matrix is very low, usually <100 nM (Pivovarova et al., 1999; Stanika et al., 2012). During electrical or synaptic stimulation, total mitochondrial calcium concentration can increase to millimolar levels within seconds (Pivovarova et al., 1999; Pivovarova et al., 2002). In the case of modest to strong stimulation, the rate of mitochondrial Ca2+ uptake in intact neurons can be as high as 200 μM/s relative to the mitochondrial volume (Pivovarova et al., 1999; Colegrove et al., 2000a). Ca2+ transport from the cytosol into the mitochondrial matrix requires crossing the outer and inner mitochondrial membranes (OMM and IMM, respectively). The OMM is permeable to molecules smaller than 5 kDa, including Ca2+. This permeability is ascribed to voltage-dependent anion channels (VDACs) also known as mitochondrial porins that are highly expressed in the OMM (Spat et al., 2008; Shoshan-Barmatz et al., 2010; Rizzuto et al., 2012). It is generally assumed that Ca2+ concentration in the intermembrane space is the same as in the cytosol. Ca2+ transport across the IMM is mediated by a designated transporter, mitochondrial Ca2+ uniporter, and is driven by a steep electrical potential across the IMM (ΔΨmt ∼ −150 mV) (Friel, 2000; Thayer et al., 2002; Nicholls, 2005). Recent electrophysiological and molecular studies strongly suggest that the mitochondrial Ca2+ uniporter functions as a Ca2+-selective ion channel, which is consistent with the very high rate of Ca2+ uptake by mitochondria (Kirichok et al., 2004; De Stefani et al., 2011; Fieni et al., 2012; Chaudhuri et al., 2013). The mitochondrial Ca2+ uniporter channel is gated by Ca2+, and the rate of mitochondrial Ca2+ uptake depends on the cytosolic Ca2+ concentration [Ca2+]i as a power function of ∼2–2.5 (Gunter and Pfeiffer, 1990; Colegrove et al., 2000a; Nicholls, 2012; Shutov et al., 2013). Earlier studies in isolated mitochondria suggested that the EC50 of the [Ca2+]i for uniporter activation is ∼10–20 μM (Gunter and Pfeiffer, 1990). Such a requirement implied that mitochondria are involved in Ca2+ transport only in a setting of intense stimulation or pathological [Ca2+]i elevations caused by toxic conditions. However, most recent studies using intact neurons indicate that mitochondrial Ca2+ uptake can be induced by [Ca2+]i elevations as low as 200–300 nM, and that mitochondrial Ca2+ transport contributes to many aspects of Ca2+ signaling in neurons, both under physiological and pathological conditions (Figure 1) (Werth and Thayer, 1994; Stout et al., 1998; David, 1999; Pivovarova et al., 1999; Colegrove et al., 2000a; Pivovarova et al., 2004; Medvedeva et al., 2008; Kim and Usachev, 2009; Chouhan et al., 2010; Shutov et al., 2013).
Within the matrix, free Ca2+ is rapidly buffered by its incorporation into Ca-phosphate complexes, primarily in the forms of Ca3(PO4)2 and CaHPO4 (Chalmers and Nicholls, 2003; Kristian et al., 2007). Heavy Ca2+ buffering in the matrix helps to explain why mitochondrial Ca2+ concentration typically does not exceed 1–5 μM in neurons even after strong stimulation (David, 1999; Billups and Forsythe, 2002; Chalmers and Nicholls, 2003; David et al., 2003). The ratio of bound:free Ca is a dynamic value that increases as the Ca load of mitochondria increases (Chalmers and Nicholls, 2003). In neurons at rest or with a low mitochondrial Ca load, the ratio is estimated to be ∼4,000:1, and as mitochondrial Ca approaches toxic levels the ratio increases to as much as 150,000:1 (Babcock et al., 1997; Chalmers and Nicholls, 2003; Pivovarova et al., 2004; Stanika et al., 2012).
Ca2+ accumulated in mitochondria can be released via at least three mechanisms, Na+/Ca2+ exchanger, H+/Ca2+ exchanger, and the mitochondrial permeability transition pore (mPTP; Figure 1) (Bernardi, 1999; Thayer et al., 2002; Nicholls, 2005; Pivovarova and Andrews, 2010; Jung et al., 2020). It is generally agreed that under physiological conditions the mitochondrial Na+/Ca2+ exchanger (mtNCX) is the main pathway mediating Ca2+ efflux from mitochondria in neurons (Friel, 2000; Thayer et al., 2002; Nicholls, 2005; Jung et al., 2020; Rysted et al., 2021; Yang et al., 2021). Because the rate of Ca2+ efflux from mitochondria is significantly slower than that of Ca2+ uptake, Ca2+ rapidly accumulates in mitochondria following its entry into the cell during stimulation. Using an approach combining pharmacological tools and mathematical modeling, Friel and colleagues estimated that Ca2+ is removed from neuronal mitochondria at 35–75 nM/s, a rate ∼1000-fold slower than the rate of mitochondrial Ca2+ uptake (Colegrove et al., 2000a; b). However, EPMA-based examination of sympathetic neurons showed that total matrix Ca2+ recovered from depolarization-induced peak value of 22 mM–0.9 mM within 15 min, which yields a much faster rate value of ∼23 μM/s (Pivovarova et al., 1999). Removal of 1 Ca2+ by mtNCX is coupled to mitochondrial uptake of 3 Na+ (Bernardi, 1999; Palty and Sekler, 2012). Thus, mtNCX is electrogenic, and its activity is predicted to be reduced by mitochondrial depolarization.
While uptake of Ca2+ ions into the mitochondria has been studied for many decades, the molecular mechanisms by which Ca2+ enters and exits the mitochondria have been discovered only over the past 10 years and are discussed below (Drago et al., 2011; Kamer and Mootha, 2015; De Stefani et al., 2016; Garbincius and Elrod, 2022).
In 2004, Kirichok and Clapham demonstrated that mitochondrial Ca2+ uniporter functions as a Ca2+-selective ion channel (Kirichok et al., 2004). In 2011, two groups identified MCU (mitochondrial Ca2+ uniporter) as a pore-forming subunit of the greater uniporter complex (Baughman et al., 2011; De Stefani et al., 2011), and two years later, a MCU paralog and negative regulator of the complex, MCUb, was identified (Raffaello et al., 2013). MCU heteromerizes with MCUb to form a tetrameric pore structure within the inner mitochondrial membrane (Raffaello et al., 2013; Baradaran et al., 2018; Wang et al., 2019) (Figure 2). Several additional components essential for the function of the MCU complex were recently discovered. These include: mitochondria Ca2+ uptake isoforms 1, 2, and 3 (MICU1, MICU2, and MICU3), which are auxiliary subunits that regulate Ca2+-dependent gating of the channel; the mitochondrial Ca2+ uniporter regulator 1 (MCUR1), and an essential MCU regulator (EMRE), the protein critical for the assembly of MCU complex (Mallilankaraman et al., 2012a; Mallilankaraman et al., 2012b; Csordás et al., 2013; Sancak et al., 2013; Tomar et al., 2016; Patron et al., 2019; Garg et al., 2021). The functions of these subunits are discussed in greater detail later.
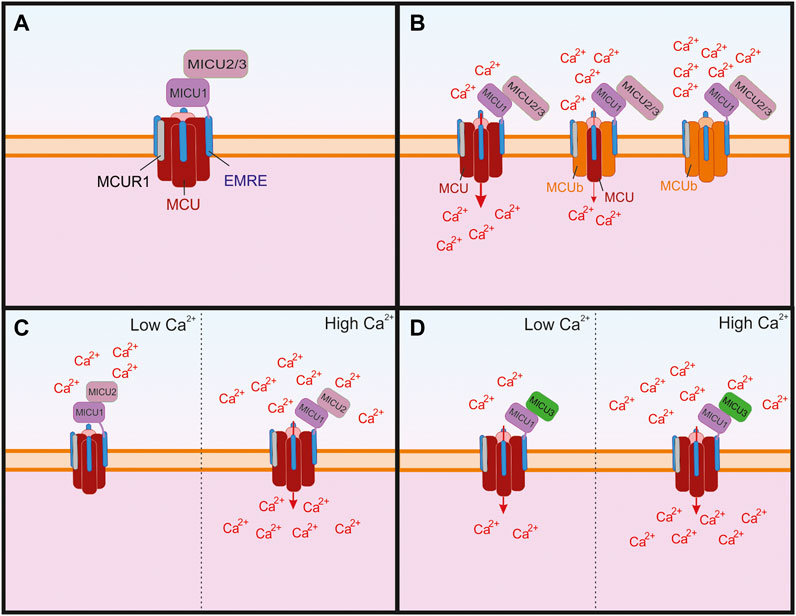
FIGURE 2. The structure of the greater mitochondrial Ca2+ uniporter (MCU) complex and its subunits that can regulate its permeability. (A) The assembled complex containing a tetramer of pore forming MCU and four subunits of the essential MCU regulator (EMRE), which are critical for the proper assembly of the transmembrane MCU to the mitochondrial Ca2+ uptake isoforms 1, 2, and 3 (MICU 1–3). The MCU complex also contains the MCU regulator 1 (MCUR1), which is thought to bind to EMRE. (B) The MCU paralog MCUb has been shown to replace subunits of pore forming MCU, which leads to reduced Ca2+ through the channel and as more MCUb subunits replace MCU the Ca2+ conductivity is reduced. (C) MICU1 and MICU two are critical for the Ca2+ sensing function of the channel and MICU2 has been shown to regulate Ca2+ sensitivity. When Ca2+ is low, MICU1 blocks the pore, but in the presence of high Ca2+ the conformational changes in MICU1 and MICU2 result in opening of the channel and allow Ca2+ to enter the mitochondria. (D) In neurons, MICU3 is more abundant and has been shown to replace MICU2 in dimers with MICU1. This replacement leads to increeased sensitivity to cytosolic Ca2+ of the MCU complex and results in MCU channel opening to lower concentrations of Ca2+ than compared to MICU2.
In addition to determining tertiary structure of the MCU pore complex, recent studies explored the ion selectivity regions that are conserved across multiple species, with the aim of identifying the selectivity filters within MCU and MCUb. These studies revealed that the cation selectivity of the MCU subunits is determined by the critical residues Asp240 (D) and Glu243 (E) within the carboxylate rings at the mouth of the channel, which form a so-called DXXE motif that is critical for Ca2+ selectivity (Oxenoid et al., 2016; Cao et al., 2017). Notably, whereas the substitution of Asp240 to Glu results in a partially functional channel, the substitution of Glu243 to Asp completely inhibits Ca2+ uptake (Oxenoid et al., 2016). Previous work also found that mutation of Asp240 or Glu243 to alanine (A) completely inhibited Ca2+ uptake through MCU (Baughman et al., 2011).
Defining the roles of MCU in the central and peripheral nervous systems in vivo remains a critical task in the field of mitochondrial biology. Although many in vitro studies (e.g., in cultured neurons or brain slices) suggested that mitochondrial Ca2+ transport is critical to neuronal function, studies in MCU knockout (KO) mice revealed that MCU deletion had surprisingly limited effects in vivo (Pan et al., 2013). The development of viable MCU KO mice was a surprise due to the importance of MCU for Ca2+ passage into the mitochondria. MCU KO mice were only viable on a CD1 background and KO offspring were not born at Mendelian ratios, but C57BL/6 mice were not viable due to embryonic lethality between E11.5-E13.5 (Pan et al., 2013; Murphy et al., 2014; Murphy and Steenbergen, 2021). The exact cause of embryonic lethality is not known, but it is hypothesized to be due to myocardial development issues (Murphy et al., 2014). Mitochondria from MCU KO mice did not uptake Ca2+, but it was not protective from cardiac ischemic reperfusion injuries (Pan et al., 2013; Murphy and Steenbergen, 2021). However, deletion of MCU from adult mice using a tamoxifen inducible model results in protection from cardiac ischemic reperfusion injuries (Kwong et al., 2015; Luongo et al., 2015). Mitochondria from global MCU KO mice have been shown to have MCU independent Ca2+ uptake, which could explain its therapeutic limitations in the ischemic reperfusion model (Hamilton et al., 2018). MCU independent Ca2+ uptake is blocked by the MCU inhibitor Ru360, and therefore it is hypothesized that the other pore forming component MCUb may be capable of compensating for the loss of MCU (Hamilton et al., 2018). MCU independent uptake could also possibly occur through mtNCX functioning in reverse mode and Letm1 (Palty et al., 2010; Jiang et al., 2013). Interestingly, MCU KO mice did not show any deficit in motor function or cognitive performance (Szibor et al., 2020), suggesting that MCU is not critical for baseline neuronal function, but plays a more important role during states of high neuronal activity, stress, neuronal injury, or in disease states. Indeed, a recent study using a mouse high-fat diet model of peripheral diabetic neuropathy showed that the deletion of MCU from peripheral neurons prevented axonal degeneration. Specifically, the deletion of MCU prevented the accumulation of Ca2+ in the mitochondria, subsequent Ca2+ induced mitochondrial disruption, and eventual loss of nociceptive fibers (George et al., 2021). Inducible neuron-specific MCU KO also protected neurons from hypoxic-ischemic injury (Nichols et al., 2018), in contrast to findings in mice with global deletion of MCU (Pan et al., 2013). The exact mechanisms by which global and inducible MCU KO differ are not known, but compensatory mechanisms, which were discussed above, may occur over time and lead to the differences observed in these studies. In another study, deletion of MCU from mice carrying the Cdh23ahl allele, which causes hearing loss, led to earlier onset of high frequency hearing loss and subsequent significant hearing loss when compared to controls. By 3 months of age, there was also a loss of close to 18% of outer hair cells, but the development of the cochlea was normal (Manikandan et al., 2021). Collectively, these studies suggest that MCU either promote or reduce neuronal survival, with the outcome depending on the developmental state, cellular context, and disease conditions.
The pore forming subunit of MCU is surrounded by multiple proteins that make up the greater MCU complex (Figure 2). One of these is MCUb, another pore-forming subunit sharing many structural similarities with MCU (Raffaello et al., 2013). Like MCU, it contains two transmembrane domains. Its protein sequence shares 50% similarity with that of MCU, and it can form hetero-oligomers with MCU. A substitution within the DXXE motif of MCUb is hypothesized to affect its Ca2+ permeability. Therefore, MCUb has been proposed to function as a dominant negative regulator of the MCU complex (Raffaello et al., 2013). A recent study revealed a potential new structural role for MCUb in the greater MCU complex (Lambert et al., 2019). Specifically, it showed that the presence of MCUb disrupts the tertiary structure of the complex by competitively displacing MCU from the greater complex and reducing the association of MCU with regulatory proteins MICU1/2. This coincided with reduced function of the complex and disrupted Ca2+ uptake into the mitochondria (Lambert et al., 2019; Garbincius and Elrod, 2022). A recent in vivo study provided additional support for MCUb as a negative regulator of Ca2+ uptake into mitochondria (Huo et al., 2020). In this study, the deletion of MCUb in mice led to increased susceptibility to damage from ischemic-reperfusion injury in the heart, whereas overexpression of MCUb conferred some protection from cardiac remodeling and damage from the infarct (Huo et al., 2020). Studies also revealed that MCUb expression increases following cardiac injury. This led to the hypothesis that the upregulation of MCUb has evolved to prevent mitochondrial Ca2+ overload in response to ischemic damage (Lambert et al., 2019; Huo et al., 2020). In contrast to these findings, MCUb is upregulated in a type 2 diabetic mouse heart, and the introduction of a dominant-negative MCUb transgene into the type 2 diabetes mouse model reversed the cardiac defects, as well as the metabolic aspect of the phenotype suggesting detrimental effects of MCUb expression in this model (Cividini et al., 2021). These results are somewhat contrary to the suggestions in previous MCUb studies that the overexpression of MCUb is cytoprotective. It could be that the overexpression of MCUb is initially protective, but that chronic overexpression results in deficits in energy production leading to negative outcomes.
MCUb is also found in brain mitochondria (Hamilton et al., 2018), although its role in neurons remains unknown. Given the protective function of MCUb in the heart (Lambert et al., 2019; Huo et al., 2020) and the detrimental role of mitochondrial Ca2+ overload in neurons (Mattson et al., 2008; Briston et al., 2019; Jung et al., 2020; Modesti et al., 2021), it is logical to predict that MCUb plays neuroprotective roles in stroke and neurodegenerative diseases. Future work will determine if this is the case.
One of the regulatory subunits of the MCU complex was described before MCU was discovered and is encoded by the gene CBARA1, which was renamed to mitochondrial Ca2+ uptake 1 (MICU1). It is ubiquitously expressed throughout the body including the brain (Plovanich et al., 2013; Patron et al., 2019). MICU1 interacts with MCU via EMRE and has two EF hand domains that are critical for its Ca2+ sensing function (Perocchi et al., 2010; Baughman et al., 2011). Knockdown of MICU1 leads to inhibition of mitochondrial Ca2+ uptake in response to stimulation, yet deletion of MICU1 results in chronically elevated Ca2+ concentrations within the mitochondrial matrix (Mallilankaraman et al., 2012b; Csordás et al., 2013). The physiological importance of MICU1 has been demonstrated in both humans and mice. Indeed, loss-of-function mutations in MICU1 are associated with learning and motor deficits in humans (Logan et al., 2014). Similarly, neuron-specific deletion of MICU1 in mice leads to abnormal motor and cognitive phenotypes, which are likely caused by neuronal degeneration in the spinal cord and the brain (Liu et al., 2016; Singh et al., 2022). Mechanistically, the loss or deficit in MICU1 function results in chronic elevation of mitochondrial Ca2+ concentration and impairment of mitochondrial Ca2+ signaling, which leads to altered neuronal excitability, increased susceptibility to excitotoxic stress and neuronal death dependent on mitochondrial permeability transition pore (Logan et al., 2014; Singh et al., 2022). Thus, MICU1 represents a critical component of mitochondrial Ca2+ signaling in neurons.
MICU1 has two paralogs, MICU2 and MICU3 (Plovanich et al., 2013). MICU2 is ubiquitously expressed throughout the body, whereas MICU3 is found primarily in the nervous system (Plovanich et al., 2013; Patron et al., 2019; Ashrafi et al., 2020). Both proteins interact with MICU1 to control Ca2+-dependent gating of the MCU channel complex (Kamer and Mootha, 2015; De Stefani et al., 2016; Garbincius and Elrod, 2022) (Figure 2).
MICU1 and MICU2 exist as a heterodimer and have opposing regulatory effects on MCU. When the cytosolic Ca2+ concentration is low, MICU2 reduces MCU activity, but when the Ca2+ concentration is higher MICU1 increases MCU activity (Patron et al., 2014). Notably, the presence of a disulfide bond between MICU1 and MICU2 is critical for its biochemical behavior, enabling them to physically adjust their positions in response to changes in Ca2+ levels; when the disulfide bond is removed mitochondrial Ca2+ uptake increases (Petrungaro et al., 2015). A recent structural analysis of the MCU complex showed that at low Ca2+ concentrations MICU1 interacts closely with MCU to seal the pore, and that MICU2 is bound to MICU1 but does not directly interact with MCU (Fan et al., 2020). The same study showed that at high levels of Ca2+ (2 mM) the MCU complex forms a dimer with a second MCU complex and in this arrangement the MICU2 subunits interact directly. This also results in MICU1 moving away from the pore of the MCU complex, allowing for Ca2+ uptake (Fan et al., 2020). In addition to playing a critical role in the function of the MCU complex, MICU1 is critical for directing the assembly of the MCU complex in the presence of Ca2+ (Gottschalk et al., 2019). Interestingly, MICU1 is also critical for the induction of cold-induced cell death. In a cold-induced stress model, the deletion of MICU1 prevents hyperpolarization of the mitochondria, which in turn reduces lipid peroxidation and prevents subsequent ferroptosis (Nakamura et al., 2021).
In contrast to MICU1 and MICU2, MICU3 received little attention until recently, when it was found to modulate mitochondria specifically in brain tissue (Patron et al., 2019). Interestingly, the authors found that it interacted with MICU1 and formed disulfide bonds, which were similar to those between MICU1 and MICU2. However, unlike the case with MICU2, the expression of MICU3 was shown to lead to enhanced uptake of Ca2+ into the mitochondria during intensive neuronal activity, which is explained by a higher Ca2+ affinity of MICU3 than that of MICU2 (Figure 2). These findings led to the proposal that MICU3 functions similarly to MICU2 by forming heterodimers with MICU1 to control gating of the MCU complex, and that the MICU2/MICU3 ratio determines the Ca2+ sensitivity of the MCU complex (Patron et al., 2019; Garbincius and Elrod, 2022). Recent experiments showed that MICU3 is responsible for a higher sensitivity of mitochondrial Ca2+ uptake to cytosolic Ca2+ in neurons as compared to non-neuronal cells such e.g., HEK293 cells that likely lack MICU3 and rely on the MICU1-MICU2 complex to gate the MCU channel (Ashrafi et al., 2020). Notably, this MICU3-dependent sensitization of mitochondrial Ca2+ uptake plays a critical role as the synapses by boosting Ca2+-dependent ATP synthesis in presynaptic terminals (Ashrafi et al., 2020). Additional knowledge of the role of MICU3 within the MCU complex will be important for understanding the mechanism by which it influences neuronal activity, but the available data show that it is critical for normal neuronal activity.
Another important component of the uniporter complex is EMRE, which was named for its role as an essential MCU regulator (Sancak et al., 2013; Tsai et al., 2016). This protein is critical for normal uniporter function. It is a 10 kD protein with only one transmembrane domain. When EMRE is deleted the MCU pore forms, but it is not functional and it fails to interact with MICU1 and MICU2 (Sancak et al., 2013; Tsai et al., 2016). EMRE is encoded only in metazoan genomes with no homologs in other organisms (Sancak et al., 2013; Tsai et al., 2016; Liu et al., 2017). The MCU/EMRE complex assembles at a 1:1 stoichiometry, and it does so on the membrane facing side of the MCU tetramer within the transmembrane domains (Wang et al., 2019). This stoichiometry was further supported by recent identification of the assembled MCU complex with EMRE, MICU1, and MICU2 all present. Interestingly, the structure of the EMRE containing MCU complex is similar regardless of whether or not MICU proteins are bound (Fan et al., 2020). This shows that proper MCU/EMRE assembly is not dependent upon MICUs. Recently, EMRE KO mice were characterized and found to be viable and to have normal metabolism, however, the birth rates were low. The EMRE KO mice had reduced mitochondrial Ca2+ uptake and were resistant to Ca2+ driven mitochondrial dysfunction, but they were not protected from ischemic reperfusion injury (Liu et al., 2020). These mice will be a useful model for further determining the role of Ca2+ uptake via EMRE/MCU complex formation in other diseases in which mitochondrial Ca2+ overload occurs.
Under physiological conditions, mitochondrial Na+/Ca2+ (mtNCX) and H+/Ca2+ (mtHCX) exchangers mediate Ca2+ extrusion from mitochondria (De Stefani et al., 2016). In excitable cells, such as neurons and muscles, mtNCX is the main contributor to this transport (Crompton et al., 1978; Thayer et al., 2002; Medvedeva et al., 2008; Jadiya et al., 2019; Rysted et al., 2021; Yang et al., 2021; Hagenston et al., 2022). It was discovered that when Ca2+ uptake was blocked, Ca2+ extrusion from the mitochondria occurs in a Na+ dependent manner (Nicholls, 1978). Interestingly, the release of Ca2+ by mitochondria was not inhibited by the inhibitor of Ca2+ uptake ruthenium red, suggesting that a reversal of the uptake mechanism is not responsible for the Ca2+ release (Rossi et al., 1973; Nicholls, 1978). An early study, performed in isolated mitochondria, identified the exchanger responsible for Ca2+ extrusion as 110 kD in size (Li et al., 1992).
In 2010, Sekler and colleagues proposed that a so-called Na+/Ca2+/Li+ exchanger (NCLX), a product of gene Slc8b1, represents an essential component of the mitochondrial Na+/Ca2+ exchanger (Palty et al., 2010). Historically, NCLX was first described as the sixth member of the family of Na+/Ca2+/K+ exchangers, and it was named NCKX6 (Cai and Lytton, 2004b). It was found to express as two splice isoforms, with the short isoform being localized to the plasma membrane and the full-length isoform being localized to the ER (Cai and Lytton, 2004b). At about the same time it was shown that NCKX6 was able to exchange Li+ for Ca2+ across the plasma membrane as effectively as it could exchange Na+ for Ca2+, and the protein was renamed to NCLX (Palty et al., 2004; Palty et al., 2006). Later, the same group showed that in addition to the plasma membrane and ER, NCLX was also enriched in the mitochondrial fraction (Palty et al., 2010). It was also found to partially localize to the mitochondria using gold-particle transmission electron microscopy, although some particles were located outside of the mitochondria as well. In addition to the localization data, these functional assays revealed that mitochondrial Ca2+ efflux increased when NCLX was overexpressed and that it decreased when NCLX was silenced using an shRNA approach (Palty et al., 2010). All this led to the proposal that NCLX is an essential contributor to the mitochondrial Na+/Ca2+ exchange (Palty et al., 2010).
NCLX is broadly expressed throughout the body including the brain and spinal cord (Palty et al., 2010; Jadiya et al., 2019; Patron et al., 2019). The in vivo role of NCLX was first demonstrated in the cardiomyocytes of adult mice (Luongo et al., 2017). Cardiomyocyte-specific deletion of NCLX resulted in severe lethality (87%) within two weeks, due to heart failure; whereas overexpression of NCLX resulted in protection from ischemic heart damage (Luongo et al., 2017). The same group also provided evidence for the role of NCLX in aging brain (Jadiya et al., 2019). They showed that in the 3xTg-AD mouse model of Alzheimer’s disease, the neuron-restricted deletion of NCLX increased tau and amyloid pathology and increased the rate of cognitive decline in mice. Restoration of NCLX expression reduced the acceleration of Alzheimer’s pathology (Jadiya et al., 2019). Similarly, disruption of NCLX expression was shown to sensitize hippocampal neurons to excitotoxic stress (Hagenston et al., 2022). A recent study has reported that a human recessive missense mutation in the gene encoding NCLX, SLC8B1, impairs its function and is associated with severe intellectual disability (Stavsky et al., 2021). The same study also demonstrated that deletion of NCLX in mice impaired synaptic transmission and long-term synaptic plasticity (Stavsky et al., 2021). Collectively, these studies highlight a key role of NCLX in regulating synaptic function and neuronal survival.
Despite the appreciation of NCLX as a mediator of Ca2+ extrusion from mitochondria, several studies showed that NCLX is also localized on the plasma membrane, endoplasmic reticulum (ER), and possibly other organelles (Haworth and Hunter, 1979; Bernardi, 1999; Cai and Lytton, 2004a; Palty et al., 2004; Palty et al., 2006; Han et al., 2015; Rysted et al., 2021). There is also inconsistency between the ability of NCLX to exchange Ca2+ for Li+ nearly as effectively as for Na+ (Palty et al., 2004; Roy et al., 2017) and the observations that Li+-driven mitochondrial Ca2+ efflux is only 10–25% of that driven by Na+ (Crompton et al., 1977; Tokunaga et al., 2000; Rysted et al., 2021). In addition, recent studies showed that NCLX knockdown or knockout had little effect on mitochondrial Ca2+ extrusion in airways smooth muscle and B lymphocytes, respectively (Emrich et al., 2022; Johnson et al., 2022). These findings raise several important questions for future research regarding the mechanisms of mitochondrial Ca2+ extrusion and the roles of NCLX, such as: 1) What are the mechanisms of cellular trafficking of NCLX? 2) What are the functions of NCLX on the plasma membrane, ER and possibly other organelles? 3) Are there additional yet to be identified molecules that mediate Na+/Ca2+ exchange across the mitochondrial membranes?
Although Ca2+ uptake into the mitochondria is critical for normal cellular function, the entry of excessive amount of Ca2+ into the mitochondria can result in Ca2+ overload and lead to activation of the mitochondrial permeability transition pore (mPTP); this phenomenon has been associated with many different disease states, and thus the mPTP is considered a promising target for therapeutic intervention (Duchen et al., 1993; Irwin et al., 2003; Du et al., 2008; Martin et al., 2009; Martin et al., 2014). Ca2+ dependent activation of the mPTP, in isolated mitochondria, results in the formation of a large non-selective ion channel that is permeable to solutes up to approximately 1,500 Da in size, which allows for the passage of internalized Ca2+ and many proapoptotic proteins out of the mitochondria (Haworth and Hunter, 1979; Bernardi, 1999; Pivovarova and Andrews, 2010; Jung et al., 2020). Prolonged opening of the mPTP results in the loss of ΔΨmt, which in turn prevents the production of ATP within the mitochondria and leads to cell death (Bernardi, 1999; Briston et al., 2019). Transient, low-conductance opening or “flickering” of the mPTP has been shown to occur in response to increased [Ca2+]mt and helps to reduce the accumulation of Ca2+ into the mitochondria (Bernardi and Petronilli, 1996; Ichas et al., 1997; Bernardi, 1999; Loupatatzis et al., 2002; Rasola and Bernardi, 2011; Garbincius and Elrod, 2022). In further support of the Ca2+ dependent activation of mPTP, preventing Ca2+ uptake into the mitochondria by deleting MCU reduces mPTP activation in response to high levels of Ca2+ (Hamilton et al., 2021). Despite the presence of the mPTP being known for many years, the identity of the proteins that make up the mPTP remains highly debated.
Early hypotheses proposed that the mPTP could form as the result of contact between inner and outer mitochondrial proteins, specifically between voltage-dependent anion-selective channels (VDAC) on the outer membrane and adenine nucleotide translocators (ANT) on the inner mitochondrial membrane (Zamzami and Kroemer, 2001). Although, deletion of each of these proteins failed to inhibit formation of the mPTP (Giorgio et al., 2018). More recently, compelling evidence has pointed towards a role for the F0F1 ATP synthase in forming the mPTP (Giorgio et al., 2013; Alavian et al., 2014; Giorgio et al., 2018; Mnatsakanyan et al., 2019; Neginskaya et al., 2019; Carrer et al., 2021). Using blue native gel electrophoresis and immunoprecipitation, Giorgio and colleagues found that the essential mPTP regulator cyclophilin D interacts with F0F1 ATP synthase (Giorgio et al., 2009). Subsequent immunoprecipitation experiments identified that the oligomycin sensitivity conferring protein (OSCP) subunit of ATP synthase specifically interacts with cyclophilin D through the same binding site as the ATP synthase inhibitor benzodiazepine 423 (Stelzer et al., 2010; Giorgio et al., 2013). Following these findings, experiments suggested that the c-subunit of F0 was able to form a voltage-sensitive channel that also appeared to be sensitive to Ca2+ exposure (Alavian et al., 2014), and had conductance consistent with activated mPTP (Mnatsakanyan et al., 2022). When c-subunit expression was reduced using an shRNA, mPTP activation was reduced and the survival of HeLa cells increased (Alavian et al., 2014). The deletion of the c-subunit from HeLa cells prevented mPTP activation in response to ionomycin as efficiently as the pre-application of cyclosporin A and overexpression of the c-subunit increased the rate of mPTP transition (Bonora et al., 2013). In HAP1 cells, knocking out the c-subunit of F0 was also shown to significantly alter, but not eliminate, mPTP activity. In this context, channels formed and remained sensitive to the mPTP inhibitor cyclosporine A and to the ANT inhibitor, bongkrekate (Neginskaya et al., 2019). Deletion of specific subunits of the ATPase impaired the formation of the mPTP. Specifically, the deletion of the g subunit, which also reduced subunit e expression, impaired mPTP channel opening in both mitoplasts and in HeLa cells (Carrer et al., 2021). However, some studies suggested that the mPTP is not ATP synthase, by demonstrating mPTP activation even after the deletion of ATP synthase subunits (He et al., 2017a; He et al., 2017b; Carroll et al., 2019). In support of these findings, the deletion of the b and OSCP subunits in HAP1 cells did not interfere with functional mPTP currents that were not blocked by cyclosporin A (Carrer et al., 2021). It is possible that in the absence of ATPase subunits, the ANT could form a functional mPTP, consistent with the finding that addition of bongkrekate impaired mPTP opening in these cells (Neginskaya et al., 2019; Carrer et al., 2021). However, a recent study using intact HAP1 cells suggests that both ANT and ATPase are required for mPTP activation, but that mitochondrial depolarization occurs through low-conductance, cyclosporin A sensitive permeability transition (Neginskaya et al., 2022). These findings suggest that while mPTP activation requires both ANT and ATPase, the low-conductance or “flickering” activity may occur through either of these molecules or through different mechanisms altogether.
Collectively, the available data on the mPTP represent compelling evidence that the F0F1 ATPase plays a critical role in its formation, but also suggest that certain subunits of the ATPase can be compensated for by other mechanisms. The combination of the results for and against the notion that F0F1 ATPase is the mPTP suggest that cell-type specific differences exist and that the mPTP can form via several mechanisms (He et al., 2017a; He et al., 2017b; Carrer et al., 2021). There are also other proposed Ca2+ sensitive, pore-forming components of mPTP including the mitochondrial phosphate carrier (PiC) (Leung et al., 2008) and spastic paraplegia 7 (SPG7) (Shanmughapriya et al., 2015). However other studies suggest these proteins may have a regulatory role as opposed to a role in forming the pore (Briston et al., 2019; Hurst et al., 2019; Boyenle et al., 2022). The key cytotoxic role of mPTP in many diseases, makes this molecular complex an attractive therapeutic target for neuroprotection, and more generally, cytoprotection. However, the existence of compensatory mechanisms helps explain why targeting this system for the development of therapeutics is so challenging. Even if additional potential inhibitors are identified, they may have significant side effects given the importance of the ATPase for normal cellular function.
Ca2+ signaling plays an especially important role in neurons because it regulates many aspects of neurotransmitter release and synaptic function (Neher and Sakaba, 2008) (Figure 1). For example, Ca2+ triggers synchronous transmitter release via the low-affinity presynaptic Ca2+ sensors synaptotagmin 1, 2, and 9 (Xu et al., 2007; Bacaj et al., 2013). In addition, the subsequent endocytotic retrieval of synaptic vesicles is regulated by Ca2+- and calcineurin-dependent dephosphorylation of dynamin-1 and other endocytotic proteins (Cousin and Robinson, 2001; Clayton and Cousin, 2009). Presynaptic Ca2+ also regulates short-term synaptic plasticity by controlling the size of the readily releasable pool (RRP) of synaptic vesicles.
Presynaptic terminals are enriched for mitochondria, an observation that reflects high energy demand at the site of transmitter release (De Camilli et al., 2001; Hollenbeck, 2005; Wimmer et al., 2006). Ca2+ buffering by mitochondria limits the amplitude of the activity-driven elevation of presynaptic [Ca2+]i and regulates synaptic strength at many different synapses, including central synapses, nerve-muscular junction, and the first sensory synapse (Tang and Zucker, 1997; David, 1999; David and Barrett, 2000; Billups and Forsythe, 2002; David and Barrett, 2003; Lee et al., 2007; Shutov et al., 2013; Kwon et al., 2016). Indeed, inhibition of mitochondrial Ca2+ uptake resulted in a larger presynaptic [Ca2+]i elevation, an increase in the frequency of asynchronous release, and accelerated synaptic depression at the neuro-muscular junction (David, 1999; David and Barrett, 2000; 2003). Similar effects were described at the central synapses (Billups and Forsythe, 2002; Lee et al., 2007). In the case of GABAergic synaptic transmission between retinal amacrine cells, mitochondrial depolarization led to a reduction in evoked transmitter release that was attributed to an increase in Ca2+-dependent inactivation of presynaptic voltage-gated Ca2+ channels in the absence of Ca2+ buffering by mitochondria (Medler and Gleason, 2002).
Mitochondrial Ca2+ transport also contributes to post-tetanic potentiation (PTP) at neuro-muscular and central synapses (Tang and Zucker, 1997; Zhong et al., 2001; Storozhuk et al., 2005; García-Chacón et al., 2006; Lee et al., 2007). PTP is a form of short-term synaptic plasticity that is induced by high-frequency (tetanic) stimulation of the presynaptic terminal, and is characterized by an enhancement of synaptic strength that lasts for many minutes (Zucker and Regehr, 2002). This phenomenon relies on a prolonged presynaptic [Ca2+]i elevation (residual [Ca2+]i or [Ca2+]i plateau) following the initial [Ca2+]i rise to the micromolar levels in response to intense stimulation (Zucker and Regehr, 2002; Neher and Sakaba, 2008). In many instances prolonged Ca2+ release from presynaptic mitochondria accounts for the residual [Ca2+]i (Tang and Zucker, 1997; Zhong et al., 2001; García-Chacón et al., 2006; Lee et al., 2007; Medvedeva et al., 2008). For example, high-frequency presynaptic stimulation produced a prolonged residual presynaptic [Ca2+]i elevation and PTP in nerve muscular junctions of crayfish and mouse, as well at mossy fiber synapses of the rat hippocampus that were blocked by inhibitors of mitochondrial Ca2+ transport (Tang and Zucker, 1997; Zhong et al., 2001; García-Chacón et al., 2006; Lee et al., 2007). A similar mechanism was shown to contribute to short-term facilitation at the calyx of Held synapse (Yang et al., 2021). Mitochondria-dependent PTP was also reported for GABAergic synaptic transmission between neocortical neurons, although presynaptic [Ca2+]i was not measured in this study (Storozhuk et al., 2005). It is most likely that Ca2+ release from presynaptic mitochondria is mediated by the mitochondrial Na+/Ca2+ exchanger (García-Chacón et al., 2006; Lee et al., 2007; Medvedeva et al., 2008). However alternative mechanisms may also be involved at some synapses (Zhong et al., 2001).
Findings from recent studies in Drosophila motor terminals suggest that mitochondrial Ca2+ uptake enhances energy metabolism by presynaptic mitochondria (Chouhan et al., 2012; Ivannikov and Macleod, 2013). These observations are consistent with the ability of mitochondrial Ca2+ to stimulate Ca2+-dependent dehydrogenases, pyruvate dehydrogenase, isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, thereby increasing the input of critical substrates into the oxidative phosphorylation reaction and ATP synthesis (Denton, 2009; Glancy and Balaban, 2012). Given that [Ca2+]mt rarely exceeds 5 μM, it is likely that pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, each of which has Ca2+ Kd of ∼0.8 μM (for comparison, isocitrate dehydrogenase has a Ca2+ Kd of∼40 μM) (McCormack et al., 1990), are the primary targets of matrix Ca2+. Such a mechanism may be an important coordinator of ATP supply and demand at the synapse. Indeed, increased synaptic activity is associated with larger amounts of Ca2+ entering the presynaptic terminal, which in turn increases Ca2+ flux into the mitochondria and stimulates ATP-synthesis. Thus, mitochondrial Ca2+ transport regulates synaptic transmission through at least two general mechanisms, with one contributing to the shaping of presynaptic [Ca2+]i signals, and the other impacting the ATP synthesis at the synapse.
Recent work started exploring the roles of specific molecular components of the mitochondrial Ca2+ transport in synaptic transmission. Inhibiting MCU in the Drosophila mushroom body neurons caused impairment of synaptic function and olfactory memory (Drago and Davis, 2016). In rat hippocampal neurons, MCU and MICU3 were found to contribute to presynaptic ATP synthesis during synaptic activity (Ashrafi et al., 2020). The same study also found that MCU silencing slowed the rate of endocytosis of synaptic vesicles (Ashrafi et al., 2020). However, an opposite effect of MCU silencing on the rate of synaptic vesicle endocytosis in hippocampal neurons was also reported (Marland et al., 2016). Thus, despite recent progress many questions remain about the role of MCU and other components of the MCU complex at synapses, including their specific roles in regulating synaptic transmission and synaptic plasticity at various excitatory and inhibitory synapses.
One of the essential roles of Ca2+ as a second messenger is to regulate neuronal excitability (Figure 1). This is achieved through the gating of three types of ion channels: Ca2+-activated K+, Ca2+-activated Cl− and Ca2+-activated cation channels (Partridge and Swandulla, 1988; Marty, 1989; Scott et al., 1995; Sah, 1996; Hartzell et al., 2005). The small- and large-conductance Ca2+-activated K+ channels, SK and BK, respectively, are widely expressed in neurons and play important roles in regulating their excitability (Sah, 1996; Wei et al., 2005). Ca2+-activated cation and Cl− channels require [Ca2+]i elevations in the 0.1–1 μM range to open, and the former channels contribute to neuronal depolarization, whereas the latter can either depolarize or hyperpolarize the membrane depending on the transmembrane Cl− gradient and membrane potential (Partridge and Swandulla, 1988; Scott et al., 1995; Hartzell et al., 2005). Ca2+ also modulates many other channels and receptors including voltage-gated Ca2+ channels, TRP channels, and NMDA glutamate receptors (McBain and Mayer, 1994; Catterall, 2000; Caterina and Julius, 2001; Budde et al., 2002; Clapham, 2003; Chuang et al., 2004; Catterall et al., 2013).
Mitochondria buffering and the release of Ca2+ have complex effects on Ca2+-activated and -modulated conductances and neuronal excitability. For example, blocking mitochondrial Ca2+ uptake by applying protonophores or inhibitors of electron transport resulted in hyperpolarization mediated by Ca2+-activated K+ channels in hippocampal and myenteric neurons (Nowicky and Duchen, 1998; Vanden Berghe et al., 2002). Similarly, intracellular application of the MCU selective inhibitor Ru360 facilitated slow afterhyperpolarization (AHP) in CA1 hippocampal pyramidal neurons (Groten and MacVicar, 2022). Several reports also showed that blocking mitochondrial Ca2+ uptake reduced voltage-gated Ca2+ currents, likely due to an increase in Ca2+-dependent inactivation of the Ca2+ channels (Hernandez-Guijo et al., 2001; Medler and Gleason, 2002; Shutov et al., 2013). Thus, under normal conditions, mitochondrial Ca2+ uptake limits Ca2+-dependent activation of BK and SK channels, and minimizes Ca2+-dependent inactivation of voltage-gated Ca2+ channels. The latter effect is especially critical for the synapse, where Ca2+ flux through these channels trigger transmitter release.
A recent study has demonstrated a specific role of MCU in regulating so-called Ca2+-release activated Ca2+ (CRAC) channels and Ca2+ signaling (Yoast et al., 2021). CRAC channels are activated in response to agonist stimulation and inositol-1,4,5-trisphosphate-induced Ca2+ release from ER stores and play important roles in Ca2+ signaling in a broad variety of non-excitable and excitable cells (Prakriya and Lewis, 2015; Emrich et al., 2021). In neurons, CRAC channels can control both excitability and synaptic transmission (Gemes et al., 2011; Ureshino et al., 2019; Hori et al., 2020; Maneshi et al., 2020). In their work, Yoast et al., found that MCU deletion led to increased Ca2+-dependent inactivation of CRAC channels and reduced Ca2+ currents through the channels. Yet, the net result was an overall amplification of cytosolic Ca2+ signaling caused by elimination of mitochondrial Ca2+ buffering in the absence of MCU (Yoast et al., 2021; Walters and Usachev, 2022). Although this effect was documented in non-excitable cell lines, given that CRAC channels and MCU are present in the nervous tissues, it is predicted that a similar mechanism could contribute to the regulation of neuronal excitability (Walters and Usachev, 2022).
Epilepsy is one of the most common neurologic conditions worldwide, with close to 70 million people currently diagnosed and approximately 1 in 26 people expected to develop epilepsy in their lifetime (Hesdorffer et al., 2011; Friedman et al., 2018). For many of these patients, available pharmaceutical interventions reduce the prevalence of seizures. Unfortunately, 30% of patients have epilepsy that is poorly controlled by current pharmaceuticals, a condition referred to as refractory epilepsy (Kwan and Brodie, 2000). Patients with refractory epilepsy are at a much greater risk of experiencing sudden unexpected death in epilepsy (SUDEP, 1/150) than those with non-refractory epilepsy (1/1,000) (Thurman et al., 2014; Friedman et al., 2018). The increased risk of mortality associated with refractory epilepsy justifies further research of mechanisms underlying epilepsy; such knowledge will be required to identify novel therapeutic targets for epilepsy.
The changes in neuronal networks that result in epilepsy can have multiple causes. For example, lesions that occur following strokes or traumatic brain injuries result in structural changes that increase the risk of epilepsy. Genetic mutations are another common cause for epilepsy. In many cases, however, epilepsy is of unknown etiology, i.e., no clear underlying cause can be identified (Berg et al., 2010). Multiple forms of epilepsy have been associated with genetic mutations that lead to metabolic and mitochondrial dysfunction; mutations in both mitochondrial and nuclear DNA have been implicated in these cases (Bindoff and Engelsen, 2012; Zsurka and Kunz, 2015; Pearson-Smith and Patel, 2017). One such example is myoclonic epilepsy and ragged-red fibers (MERRF) syndrome, for which myoclonus is a defining feature that presents along with other neurologic defects. Studies of cultured cells into which the patient mitochondrial DNA mutation had been introduced showed that this resulted in decreased ATP-dependent protease activity and increased ROS production (Wu et al., 2010). MERRF mutations also resulted in disrupted mitochondrial Ca2+ handling but did not alter cytosolic Ca2+ regulation. The authors observed reduced ATP production in these cells following stimulation with histamine. They hypothesized that the reduction in ATP production resulted from a reduction in Ca2+ entry into the mitochondria, and that this led to reduced activation of Ca2+-dependent enzymes (Brini et al., 1999). Other examples of genetic diseases that lead to epilepsy by interfering with metabolic function include Alpers-Huttenlocher syndrome (AHS) and Leigh syndrome; in each, mutations lead to reduced ATP production and refractory epilepsy (Bindoff and Engelsen, 2012; Zsurka and Kunz, 2015; Pearson-Smith and Patel, 2017). Although, disrupted energy production results in many different forms of epilepsy, a loss-of-function mutation in MICU1 in humans instead results in myopathy, movement disorders, and learning disabilities (Logan et al., 2014). The propensity for mitochondrial diseases to result in epilepsy identify the mitochondria as targets for new therapies for epilepsy.
Epilepsy occurs when brain networks transition into a state of hyperexcitability, a process known as epileptogenesis (Scharfman, 2007). Hyperexcitability of neuronal networks is a key change that occurs in individuals who develop epilepsy. It can be caused by changes in gene expression, posttranslational modification, or synaptic circuits (Kinjo et al., 2018). These processes are frequently Ca2+ dependent. For example, dendritic arborization and synaptogenesis are dependent upon changes in gene expression following increases in intracellular Ca2+ levels (Lohmann and Wong, 2005; Redmond and Ghosh, 2005). Mitochondrial Ca2+ regulation has been shown to be important for neurotransmission and altered mitochondrial function can disrupt synaptogenesis (MacAskill et al., 2010; Sun et al., 2013). Proper Ca2+ regulation by mitochondria is essential for normal neuronal processes including synaptogenesis, excitability, neurotransmission, and ATP synthesis (Billups and Forsythe, 2002; Thayer et al., 2002; Mattson et al., 2008; Usachev, 2015). During seizures, excess Ca2+ can build up within the cytosol and eventually lead to mitochondrial Ca2+ overload, which is known to lead to cell death (Figure 3).
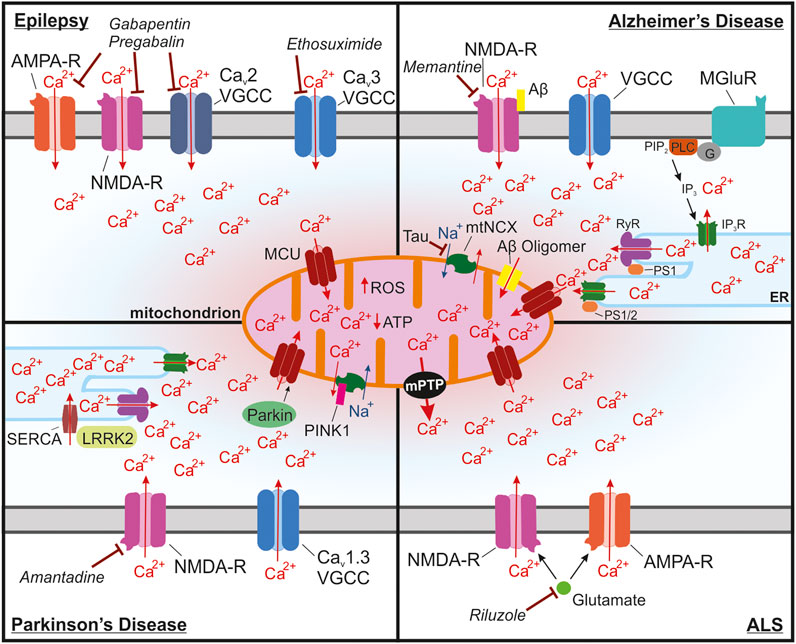
FIGURE 3. Mitochondrial Ca2+ overload leading to activation of the mPTP is a common pathway in many neurological diseases. (A) In epilepsy, seizures lead to buildup of cytosolic and mitochondrial Ca2+ through NMDA receptors, Ca2+ permeable AMPA receptors and Cav2 and Cav3 voltage-gated Ca2+ channels (VGCCs) in neurons that are in a state of hyperexcitability. Long seizures or repeated seizures can lead to Ca2+ overload in the mitochondria and trigger activation of the mPTP, ultimately leading to neuronal toxicity. Commonly prescribed antiepileptic drugs target these channels including: ethosuximide, which is a Cav3 VGCC inhibitor, as well as gabapentin and pregabalin, which act by binding to the with α2δ-1 subunit of voltage-gated Ca2+ channels, which could disrupt plasma membrane expression and function of Cav2 VGCCs, NMDA receptors, and Ca2+ permeable AMPA receptors. (B) In Alzheimer’s disease, Ca2+ overload in the mitochondria occurs through multiple mechanisms. Ca2+ entry into the cytosol can be enhanced by binding of Aβ to NMDA receptors. Internal release of Ca2+ from the ER through activation of metabotropic glutamate receptors (mGluR) also increases the accumulation of Ca2+ into the mitochondria. Enhanced Ca2+ extrusion from the ER has also been shown to occur through interactions between presenilin 1/2 (PS1/2) and inositol trisphosphate receptors (IP3R) and ryanodine receptors (RyR). This interaction seems to occur at mitochondria associated membranes (MAMs), which due to the proximity to the mitochondria, drives Ca2+ uptake into the mitochondria. Aβ oligomers also can form a pore in the mitochondria that can lead to increases in Ca2+ in the mitochondria. Ca2+ extrusion through the mitochondrial Na+/Ca2+ exchanger (mtNCX) has been shown to be impaired by tau accumulation leading to excessive Ca2+ accumulation in the mitochondria. Memantine, one of the FDA approved drugs for AD, inhibits NMDA receptors, which could block excessive Ca2+ uptake and to reduce glutamate induced excitotoxicity. (C) In Parkinson’s disease (PD), substantia nigra dopaminergic neurons are at risk of Ca2+ overload due to the presence of voltage-gated Cav1.3 channels, which are critical for the pacemaking activity of these neurons. One common form of autosomal recessive familial PD occurs through mutations in PARK2, the gene for Parkin. Parkin is responsible for regulating mitochondrial Ca2+ uptake isoform 1 (MICU1) degradation and may lead to disrupted MCU assembly altering mitochondrial Ca2+ uptake. Another form of autosomal recessive PD is caused by mutations in PINK1, which can impair mtNCX function and increase Ca2+ accumulation into the mitochondria. In astrocytes, ER Ca2+ release at MAMs may be enhanced in PD due to mutations in leucine-rich repeat kinase 2 (LRRK2), which enhance Ca2+ uptake into the ER through activation of sarcoplasmic reticulum Ca2+ -ATPase (SERCA). The NMDA receptor antagonist amantadine is used to prevent dyskinesia in PD patients. (D) In amyotrophic lateral sclerosis (ALS), motor neurons of the spinal cord and brain begin to degenerate. These neurons have been shown to have increased Ca2+ uptake through glutamate NMDA and Ca2+ permeable AMPA receptors, which enhances their susceptibility to mitochondrial Ca2+ overload. Excitotoxic glutamate, which signals through NMDA and GluA2-lacking Ca2+ permeable AMPA receptors, is the target of Riluzole, one of the few FDA approved drugs for ALS.
Indeed, hippocampal sclerosis and neuronal loss in the hippocampus and other limbic and mesial cortical structures is the very common pathological finding in patients with temporal lobe epilepsy, the most prevalent form of epilepsy in humans (Levesque and Avoli, 2013; Tai et al., 2018). In fact, frequent generalized convulsive seizures are a strong predictor of cognitive decline as well as progressive development of behavioral and psychiatric abnormalities in patients with epilepsy, all of which is linked to neuronal damage in the limbic and mesial cortical regions (Kovac et al., 2017; Tai et al., 2018; Farrell et al., 2019). Cell death is particularly apparent following status epilepticus, sustained generalized convulsive seizures lasting 5 min or more (Walker, 2018). These prolonged seizures result in Ca2+ overload in neurons and astrocytes (Tian et al., 2005; Fellin et al., 2006; Ding et al., 2007). Neurotoxic build-up of glutamate released from firing neurons and astrocytes is thought to be the major trigger of neuronal damage (i.e., excitotoxicity) caused by prolonged seizures, as well as in other pathological conditions (e.g., stroke, and neurodegenerative diseases) (Nicholls and Ward, 2000; Mattson et al., 2008; Moskowitz et al., 2010; Kovac et al., 2017). Glutamate-induced Ca2+ entry, largely via NMDA as well as Ca2+-permeable AMPA glutamate receptors, mitochondrial Ca2+ overload and Ca2+ deregulation initiate neurotoxic cascades through many mechanisms, including rupture of mitochondrial membranes, mitochondrial depolarization, ROS generation and opening of mitochondrial permeability transition pore (mPTP) and release of cytochrome c and other proapoptotic factors (Figure 3) (Nicholls and Ward, 2000; Newmeyer and Ferguson-Miller, 2003; Orrenius et al., 2003; Munoz-Pinedo et al., 2006; Mattson et al., 2008; Moskowitz et al., 2010; Guo and Ma, 2021). Accordingly, selective antagonists of glutamate NMDA or Ca2+-permeable AMPA receptors reduced seizure-induced neuronal toxicity in animal models of status epilepticus and temporal lobe epilepsy (Ding et al., 2007; Kovac et al., 2017; Walker, 2018; Konen et al., 2020).
Antiepileptic drugs such as gabapentin and pregabalin were also shown to reduce hippocampal neuronal loss in the rodent models of temporal lobe epilepsy and to improve mitochondrial function (Huang et al., 2013; Rossi et al., 2013; Finsterer and Scorza, 2017). Although originally designed as GABAmimetics, it is now well accepted that the anticonvulsant action of these gabapentinoids is mediated by mechanisms distinct from GABAergic activity. Instead, it has been proposed that these drugs interact with α2δ-1 subunit of voltage-gated Ca2+ channels, which in turn interferes with α2δ-1-dependent insertion and retention of α1 subunits of the channels (Dolphin, 2012; Sills and Rogawski, 2020) (Figure 3). An additional mechanism likely involves interaction of α2δ-1 subunit with glutamate NMDA receptor (Sills and Rogawski, 2020), and disrupting this interaction by gabapentin was shown to inhibit neuronal toxicity caused by ischemic stroke (Luo et al., 2018). The α2δ-1 subunit can also interact with the GluA1 and GluA2 subunits of glutamate AMPA receptors, which promotes expression of GluA2-lacking Ca2+-permeable AMPA receptors, the process that can be disrupted by gabapentin (Li et al., 2021). Thus, it is plausible that the described neuroprotective effects of gabapentin and pregabalin are mediated by their inhibitory action on NMDA and Ca2+-permeable GluA2-lacking AMPA receptors (Figure 3).
The mitochondrial Ca2+ overload due to mitochondrial Ca2+ uptake is a major trigger of excitotoxicity and neuronal death downstream of Ca2+ entry into neurons via glutamate NMDA receptors and other Ca2+ channels (Nicholls and Ward, 2000; Mattson et al., 2008; Kovac et al., 2017). Consequently, it is predicted that inhibiting mitochondrial Ca2+ uptake is neuroprotective. To this end, a recent study reported that systemically administered MCU inhibitor Ru360 reduced hippocampal neuronal death in a rat model of pilocarpine-induced status epilepticus (Wang et al., 2015). The findings of this work however, should be interpreted with caution because Ru360 poorly penetrates through the plasma membrane, which is required for inhibiting MCU (Hajnoczky et al., 2006; Woods and Wilson, 2020; Marta et al., 2021). In addition, Ru360 is very unstable for in vivo work, and can affect various ion channels of the plasma membrane when applied extracellularly (Hajnoczky et al., 2006; Woods and Wilson, 2020; Marta et al., 2021). Thus, additional studies are needed to test the specific roles of the mitochondrial Ca2+ uptake and release mechanisms transport in neurotoxicity associated with seizures and epilepsy.
In summary, mitochondrial Ca2+ cycling can affect neural activity and susceptibility to seizures and epilepsy via several mechanisms, including regulation of neuronal excitability, synaptic transmission, bioenergetics, and excitotoxicity (Figures 1, 3). A better understanding of neuronal Ca2+ regulation may prove essential for further elucidation of the mechanisms that underlie epilepsy, and thus for identifying potential therapies.
Alzheimer’s disease (AD) is a debilitating neurodegenerative disorder and the most common form of dementia in the elderly. It is characterized by progressive impairment of memory and cognitive functions and may lead to a completely vegetative state and early death (Querfurth and LaFerla, 2010; Scheltens et al., 2016; Corriveau et al., 2017). AD currently affects approximately 5.8 million Americans who are 65 years of age and older and current estimates predict that this number will increase to 13.8 million by 2050 (Alzheimer's and Dementia, 2020a).
AD pathology is characterized by an accumulation of β-amyloid (Aβ) and the formation of neurofibrillary tangles from accumulated tau protein (Hyman et al., 2012). According to the amyloid hypothesis of AD, progressive accumulation and deposition of Aβ42 plaques leads to a cascade of pathological changes in the brain that ultimately lead to the synaptic and neuronal loss and progressing dementia (Selkoe and Hardy, 2016; Golde et al., 2018). Such changes include inflammation, the activation of microglia and astrocytes, an accumulation of hyperphosphorylated tau and neurofibrillary tangles, altered ionic homeostasis, and oxidative injury (Selkoe and Hardy, 2016; Golde et al., 2018). Neuronal Ca2+ deregulation is thought to play critical roles during various phases of AD and can manifest in many forms, including chronic elevation of the cytosolic and mitochondrial Ca2+ concentrations, enhanced Ca2+ release from intracellular Ca2+ stores, spontaneous Ca2+ spikes, and enhanced activity of glutamate NMDA receptors (Bezprozvanny and Mattson, 2008; Mattson et al., 2008; Querfurth and LaFerla, 2010; Workgroup and Khachaturian, 2017) (Figure 3). Although various mechanisms contribute to Ca2+ deregulation in AD, most of them ultimately lead to a common outcome–mitochondrial Ca2+ overload. This in turn, is thought to trigger neuronal demise through a range of neurotoxic mechanisms, including mPTP opening and release of proapoptotic factors, mitochondrial depolarization and ROS generation (Bezprozvanny and Mattson, 2008; Mattson et al., 2008; Querfurth and LaFerla, 2010).
Mitochondrial dysfunction in AD may be driven by multiple factors, but in this review we focus on the role of Ca2+ dysregulation in the progression of AD. Ca2+ dysregulation by mitochondria in the context of AD pathology was proposed to be a consequence of the aggregation of Aβ within the mitochondria by forming functional channels that have a high affinity for Ca2+ ions (Figure 3) (Arispe et al., 1993; Jang et al., 2007; Hansson Petersen et al., 2008; Rosales-Corral et al., 2012). Direct channel formation into the mitochondria has received limited attention since, but Aβ induced Ca2+ release from ER has been proposed to contribute to mitochondrial Ca2+ overload (Jensen et al., 2013). In both Xenopus oocytes and cultured SH-SY5Y cells, ER Ca2+ release was triggered by soluble Aβ via generation of inositol triphosphate (IP3) (Demuro and Parker, 2013; Jensen et al., 2013). Interestingly, in SH-SY5Y cells an increase in ER Ca2+ was also observed in the absence of IP3 receptors, suggesting that a more direct effect of Aβ on the ER accounts for the increased cytotoxicity of Aβ oligomers (Jensen et al., 2013). ER Ca2+ regulation is also disrupted by mutations in presenilin 1 (PS1) and presenilin 2 (PS2). Mutated PS1 and PS2 modulate IP3R activity and increase Ca2+ release (Cheung et al., 2008; Modesti et al., 2021). PS1 mutants have also been shown to interact with ryanodine receptors and increase their activity (Rybalchenko et al., 2008; Chakroborty et al., 2009; Modesti et al., 2021), which combined with increased release from IP3-sensitive Ca2+ stores might contribute to elevated cytosolic Ca2+, excessive mitochondrial Ca2+ load, and Ca2+ dysregulation associated with AD (Figure 3). The store-operated Ca2+ entry into the ER is also disrupted in “aging” neurons and is further deregulated by the presence of Aβ oligomers, which drives increases in cytosolic Ca2+ concentration (Calvo-Rodriguez et al., 2019). Injections of Aβ oligomers into the rat hippocampus leads to increased expression of type 2 ryanodine receptors at mitochondria associated membranes, which leads to increased Ca2+ release from the ER in response to Ca2+ signals and drives increased cytosolic Ca2+ concentrations (More et al., 2018). In addition to its potential direct actions on internal Ca2+ stores, Aβ is thought to modulate NMDA receptor activity and has been shown to contribute to Ca2+-induced cytotoxicity (Popugaeva et al., 2018). Glutamate induced excitotoxicity through NMDA receptors is also the target of one of the few FDA approved therapies for AD, memantine (Sonkusare et al., 2005). However, memantine’s effectiveness has been limited to symptomatic improvements in cognition of patients with moderate-to-severe AD (Areosa et al., 2005; Li et al., 2019).
It has been proposed that many forms of Ca2+ dysregulation in AD and other neurodegenerative disorders ultimately converge on mitochondrial Ca2+ overload, a crucial event that triggers mPTP opening, release of proapoptotic factors, ROS generation, and cell death (Bezprozvanny and Mattson, 2008; Mattson et al., 2008; Plotegher et al., 2021). Consequently, inhibiting MCU is predicted to be neuroprotective. Recent studies support this idea. Indeed, MCU was shown to be upregulated in the brain of 3xTg-AD (APP/PS1/tau model of AD) mice, whereas knocking down MCU led to reduced synaptic loss, increased mitochondrial mass and improved behavioral performance in memory tasks in 3xTg-AD mice (Cai et al., 2022). Another group recently reported that application of soluble Aβ peptides onto neurons increased mitochondrial Ca2+ concentration and promoted neuronal death, which could be reversed by extracellularly applied MCU inhibitor Ru360 (Calvo-Rodriguez et al., 2020). Complicating the interpretation of these findings is the observations that Ru360 poorly penetrates through the plasma membrane, which is required for inhibiting MCU, and can also affect various ion channels on the plasma membrane (Hajnoczky et al., 2006; Woods and Wilson, 2020; Marta et al., 2021). Thus, additional studies using more suitable pharmacological tools and genetic approaches will be needed to thoroughly test the role of MCU and other components of the MCU complex in AD models.
Along with Ca2+ uptake machinery, mitochondrial Ca2+ extrusion mechanisms are also potentially critical in AD progression. Recently, tau has been shown to impair Ca2+ extrusion out of the mitochondria through mtNCX and resulted in faster mitochondrial depolarization in response to repeated stimulations (Britti et al., 2020; Esteras and Abramov, 2020). NCLX, which has been proposed to be molecular determinant of the mtNCX transport, has reduced expression in the frontal cortex of patients with AD and overexpression of NCLX in animal models of AD slows disease progression (Jadiya et al., 2019; Garbincius and Elrod, 2022). While Ca2+ cycling within the cytosol, ER, and mitochondria are key factors in AD progression (Figure 3), further investigation is required to identify how the relationship between these cellular machineries is driving Ca2+ overload, mitochondrial dysfunction, and apoptotic signaling.
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that is defined by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) (Braak et al., 2003; Bezprozvanny, 2009; Surmeier et al., 2017; Fu et al., 2018). Before the substantia nigra deteriorates, neurons in multiple brainstem nuclei (including the dorsal motor nucleus, the raphe nucleus, and the locus coeruleus) begin to die (Braak et al., 2003; Fu et al., 2018). Another common hallmark of PD is Lewy body pathology, which spreads to some common brain regions as the disease progresses (Surmeier et al., 2017). Despite the variability of Lewy pathology, one common trend occurs, which is the neuronal loss within the SNpc (Surmeier, 2007). Dopaminergic neurons of the SNpc are uniquely susceptible to perturbations that lead to the development of PD, when compared to dopaminergic neurons of the ventral tegmental area (VTA), which has been shown due to the dependence of SNpc neurons on Cav1.3 for their rhythmicity (Chan et al., 2007; Surmeier, 2007; Chan et al., 2009; Fu et al., 2018). This unique dependence on Cav1.3 likely leads to greater accumulation of Ca2+ into the mitochondria and subsequent Ca2+ overload when combined with other challenges such as genetic mutations or environmental toxins (Figure 3).
Many genetic causes of PD point to mitochondrial Ca2+-overload as a key target in the treatment of PD (Verma et al., 2018; Jung et al., 2020; Lim et al., 2021a; Modesti et al., 2021; Soman and Dagda, 2021). PINK1, which is encoded by a gene whose mutation causes autosomal recessive PD, is a mitochondrially targeted protein that was shown to regulate Ca2+ efflux from the mitochondria (Gandhi et al., 2009). Genetic knockdown or knockout of PINK1 resulted in impairment of Ca2+ extrusion from the mitochondria in neurons, coupled with increases in ROS production (Gandhi et al., 2009) (Figure 3). Mitochondria purified from PINK1 knockout mice had reduced Ca2+ loading capacities and enhanced mPTP activation (Akundi et al., 2011). PINK1 KO midbrain neurons are susceptible to excitotoxic dopamine treatment, but were shown to be protected by NCLX overexpression in vitro (Kostic et al., 2015). Deletion of MCU in zebrafish prevented loss of DA neurons in the PINK1 and MPTP models of PD, supporting the potential of targeting MCU for the prevention of PD progression (Soman et al., 2017; Soman et al., 2019). Further support of the role of MCU and mitochondrial Ca2+ regulation in PD comes from recent work reporting that MICU1 is regulated by the E3 ubiquitin ligase Parkin, whose mutation leads to another form of familial PD (Matteucci et al., 2018). Overexpression of Parkin reduces MICU1 expression and leads to reduced MICU2 expression due to its dependence on forming dimers with MICU1. Given the critical role of Parkin in regulating MICU1 expression, its mutations could lead to disrupted assembly of the MCU complex and abnormal handling of mitochondrial Ca2+ (Matteucci et al., 2018) (Figure 3).
The most common familial form of PD is caused by mutations in leucine-rich repeat kinase 2 (LRRK2) (Lee et al., 2019; Kovacs et al., 2021). Experiments in astrocytes identified that mutant LRRK2 modulates sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) function and reduces ER Ca2+ uptake (Lee et al., 2019). The reduced uptake of Ca2+ into the ER increases ER stress and leads to mitochondrial Ca2+ overload (Figure 3) (Lee et al., 2019; Kovacs et al., 2021; Modesti et al., 2021). Human fibroblasts from patients with LRRK2 mutations show upregulation of MCU and MICU1, and mitochondria have increased Ca2+ uptake (Verma et al., 2017; Modesti et al., 2021). In SH-SY5Y cells with mutant LRRK2, targeting MCU with inhibitors or genetic knockdown reduced Ca2+ uptake and improved neurite growth and stability (Verma et al., 2017). These studies suggest that targeting mitochondrial Ca2+ uptake or release may be beneficial in delaying the onset or slowing the progression of PD in humans.
The above-described experiments show the importance of Ca2+ regulation and the impact that its dysfunction can have in the progression of PD (Figure 3). Although targeting mechanisms of Ca2+ regulation to inhibit PD progression holds promise, additional work is needed to determine how effective this would be in preventing the PD progression. The Cav1.3 inhibitor isradipine has shown to prevent the onset and progression of PD in rodent models of PD (Chan et al., 2009), but has failed to delay clinical progression and onset in clinical trials (2020b; Venuto et al., 2021). Studies have suggested that certain L-type Ca2+-channel inhibitors used in patients with hypertension to some extent reduce the risk of PD, but currently no FDA approved drugs available to selectively target Cav1.3 in humans (Chan et al., 2009). Another way of preventing Ca2+ overload and excitotoxicity is by reducing NMDA receptor activity. The drug amantadine is a NMDA receptor antagonist that is approved for the treatment of PD induced dyskinesia (Schwab et al., 1969; Olanow et al., 2009). While the clinical use of amantadine is to help reducing symptoms of PD, it could aid in delaying disease progression due to its potential effect on reducing excitotoxic Ca2+ signaling through NMDA receptors (Olanow et al., 2009). It is also possible that targeting the MCU complex would be useful in the prevention of PD progression, the approach that showed efficacy in zebrafish models of PD (Soman et al., 2017; Soman et al., 2019). Drug targets of MCU and their efficacy in PD is also discussed in recent reviews (Dey et al., 2020; Woods and Wilson, 2020). However, further testing of these hypotheses in mouse models of PD and humans will require more selective inhibitors of Ca2+ regulating proteins.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder that results in progressive loss of motor neurons throughout the brain and spinal cord, leading to death typically within 2.5–3 years after its initial diagnosis (Rosen et al., 1993; Bezprozvanny, 2009; Fu et al., 2018). Although most cases of ALS occur sporadically, approximately 5–10% cases of ALS are familial, with mutations in the Cu/Zn superoxide dismutase (SOD1) gene accounting for approximately 20% of familial ALS cases (Rosen et al., 1993; Cook and Petrucelli, 2019). These mutations lead to toxic accumulation of SOD1 on the mitochondria leading to cell death (Van Den Bosch et al., 2006; Bezprozvanny, 2009). Notably, in mice and rats mutations in SOD1 lead to motor neuron disease similar to familial ALS (Gurney et al., 1994; Howland et al., 2002). Distinct populations of motor neurons are vulnerable in ALS. The neurons most affected are fast-fatigable motor neurons; the most resistant are slow motor neurons, and distinct brainstem motor nuclei populations also remain mostly unaffected (Fu et al., 2018). These findings suggest that, as is the case for PD, a unique neuronal physiology may lead to vulnerability to Ca2+ overload in ALS. Spinal motor neurons express lower levels of the Ca2+ binding proteins calbindin D28K and parvalbumin (Shaw and Eggett, 2000; Appel et al., 2001). Consistent with this notion, the motor neurons of the brainstem nuclei that are spared from ALS degeneration have higher levels of Ca2+ binding proteins (Van Den Bosch et al., 2006; Bezprozvanny, 2009). Further support for the importance of Ca2+ binding proteins in motor neurons is that the overexpression of rat parvalbumin in SOD1 mutant mice both delayed the onset of symptoms and prolonged survival of a mouse model of familial ALS (Beers et al., 2001). Increasing the expression of calbindin D28K, with glia derived neurotrophic factors, protected motor neurons of spinal cord slices from glutamate induced toxicity (Spruill and Kuncl, 2015).
In addition to the differences in the expression of Ca2+-binding proteins, motor neurons have been shown to contain glutamate AMPA receptors that are more permeable to Ca2+, thereby adding to their susceptibility to excitotoxicity (Van Den Bosch et al., 2006). This was found to be due to a reduction in the number of GluA2 receptor subunits present within the motor neurons (Shaw and Eggett, 2000; Van Den Bosch et al., 2006). The level of GluA2 subunit expression has been shown to alter lifespan in SOD1 ALS mice. Crossing GluA2 knockout mice with SOD1 mice shortens their lifespan and overexpressing GluA2 increases their lifespan (Van Den Bosch et al., 2006). Along with decreases in the expression of the GluA2 subunits, iPSC derived motor neurons containing the familial C9orf72 mutation showed increased expression of GluA1 subunits, further contributing to a reduced GluA2/GluA1 ratio and increased Ca2+ permeability of AMPA receptors (Selvaraj et al., 2018). A recent assessment of post-mortem tissue from ALS patients found that AMPA receptor subunits were disrupted in both familial and sporadic ALS cases (Gregory et al., 2020). Familial C9orf72 mutations and sporadic cases showed increased GluA1 expression, whereas SOD1 mutation cases showed reduced GluA2 expression (Gregory et al., 2020). While there appear to be differences in the AMPA receptor subunits expressed in ALS patients, the common denominator is a reduced GluA2/GluA1 ratio that leads to enhanced Ca2+ permeability of the receptors (Figure 3).
The excessive Ca2+ influx associated with ALS combined with lower expression of Ca2+-binding proteins in motor neurons, leave mitochondria in these neurons at high risk of Ca2+ overload (Figure 3). Mitochondria Ca2+ buffering is dysfunctional in SOD1 mutant mice (Damiano et al., 2006). In mitochondria of the brain and spinal cord, dysfunction in the form of decreased Ca2+ loading capacity occurs much earlier than the onset of symptoms and significant motor neuron loss (Damiano et al., 2006). This was supported by a subsequent study that found that mitochondria of motor neurons of pre-symptomatic SOD1 mutant mice became more depolarized from Ca2+ entry in response to repeated stimulations (Nguyen et al., 2009). Using motor neurons derived from induced pluripotent stem cells of patients with two different ALS causing mutations (C9orf72 and TARDBP), it was determined that the neurons had enhanced Ca2+ release following stimulation and both mutations also led to upregulation of Ca2+ permeable AMPA and NMDA receptors (Dafinca et al., 2020). Interestingly, both mutations resulted in higher levels of MICU1 compared to MICU2, with C9orf72 mutations reducing MCU and MICU2 expression, whereas TARDBP increasing MICU1 expression, altogether resulting in reduced uptake of Ca2+ into the mitochondria (Dafinca et al., 2020). In hypoglossal motor neurons, mitochondrial Ca2+ uptake is impaired later in disease progression, and cytosolic Ca2+ clearance is enhanced relative to that in neurons resistant to ALS pathology (Fuchs et al., 2013). An additional analysis showed that in late stage SOD1 mutant mouse hypoglossal motor neurons, not only are MCU and MICU1 upregulated, but so are Letm1, UCP2, and NCX1, one of the plasma membrane Na+/Ca2+ exchangers (Mühling et al., 2014). As the disease progresses motor neurons attempt to compensate for increased cytosolic Ca2+ concentrations by upregulating mechanisms to improve mitochondrial buffering, but also extrusion across the plasma membrane. However, excessive mitochondrial Ca2+ load can lead to activation of the mPTP. Notably, in SOD1 mutant mice the mPTP-promoting protein cyclophilin D is upregulated, the mitochondria are swollen, and mPTP can be activated. Deleting a single copy of cyclophilin D gene in SOD1 mutant mice led to delayed disease onset and prolonged lifespan when compared SOD1 mutant mice alone (Martin et al., 2009). These latter findings suggest that inhibiting mitochondrial Ca2+ uptake and mPTP activation may be beneficial in preventing the onset and progression of ALS.
The challenge with ALS is that many potential treatments have failed in clinical trials. For example, the FDA approved drug Riluzole has led to only a modest increase in lifespan (about 3 months) (Bezprozvanny, 2009; Smith et al., 2019). The mechanism of action of this drug is not completely clear, but it appears to inhibit glutamate NMDA receptors, an effect that can reduce the overall influx of Ca2+ into the cell (Bezprozvanny, 2009; Smith et al., 2019). Many other approaches targeting mitochondrial function and ROS production have failed in clinical trials (Smith et al., 2019). The importance of Ca2+ overload in ALS has been extensively discussed, and while studies have shown changes in MCU/MICU1/MICU2 expression in ALS (Fuchs et al., 2013; Mühling et al., 2014; Dafinca et al., 2020), studies targeting mitochondrial Ca2+ uptake and release mechanisms have been limited (Tadić et al., 2019). One of the only studies targeting MCU in SOD1 mutant mouse neurons showed that MCU expression is higher in cultured SOD1 mutant motor neurons vs. control motor neurons, but MCU is downregulated in adulthood (Tadić et al., 2019). The application of KN-62, a selective inhibitor of the Ca2+/calmodulin-dependent protein kinase II (CaMKII), protected against kainic acid-induced excitotoxity in vitro, and this appeared to involve a reduction in MCU expression (Tadić et al., 2019). Although this study holds promise for targeting the MCU complex in ALS, further studies are needed to determine its potential as a therapeutic target.
Mitochondrial Ca2+ buffering is essential for normal neuronal function and dysfunctional Ca2+ buffering and subsequent Ca2+ overload is common to many neurological diseases. Thanks to the recent identification of the components of the MCU complex, targeting the mitochondrial Ca2+ uptake and release machinery has become possible and has helped to refine the roles of these processes in neuronal function, as well as enable investigation of the effectiveness of targeting these proteins in disease prevention. Despite progress, many major questions remain unanswered in defining the roles of specific components of mitochondrial Ca2+ uptake and release systems in neurodegenerative disorders, epilepsy, stroke and other neurological conditions. Recent advancements in genetic targeting of all the major components of mitochondrial Ca2+ transport have provided important tools for addressing these questions. Thus, an important task for future research would be to systematically examine the impact of deletion or overexpression of MCU, MCUb, MICU3 and other components of mitochondrial Ca2+ transport in animal models of AD, PD, ALS, epilepsy, and other neurological conditions.
Even though this review focused primarily on neurons, it is important to point out that glial cells, such as astrocytes and microglia, also play crucial roles in epilepsy and neurodegenerative diseases as highlighted in many excellent reviews on this topic (Salter and Stevens, 2017; Patel et al., 2019; Santello et al., 2019; Escartin et al., 2021). Equally important is that Ca2+ signaling and mitochondria Ca2+ transport play crucial roles in these cells in the context of many neurological diseases (Shigetomi et al., 2019; Lim et al., 2021b; Heuser and Enger, 2021; Sobolczyk and Boczek, 2022). Thus, another task for future studies would be to define cell-type specific roles of MCU and other molecular components of mitochondrial Ca2+ transport in neurological diseases by genetically targeting these molecules in neurons, glia, and immune cells.
Another important line of future research would be development of potent and selective pharmacological tools that target MCU. Although a commonly used MCU inhibitor Ru360 is both potent and selective in in vitro assays, its poor plasma membrane permeability and low stability in aqueous solutions limit its applicability for in vivo research (Hajnoczky et al., 2006; Woods and Wilson, 2020; Marta et al., 2021). A recently developed analog of Ru360, Ru265, shows improved potency and much more readily penetrates through plasma membrane (Woods and Wilson, 2020). The latter property is mediated by a ubiquitously expressed organic cation transporter 3 (OCT3) (Woods et al., 2020). Notably, administration of Ru265 provided a neuroprotective effect in vivo in mice subjected to hypoxic/ischemic brain injury (Novorolsky et al., 2020). Currently, only a few of the available pharmacological inhibitors of MCU are translatable to human medicine. One FDA-approved drug has been shown to inhibit MCU is mitoxantrone (Arduino et al., 2017), but it is also an inhibitor of topoisomerase II and chemotherapeutic with high cytotoxicity (Woods and Wilson, 2020). Mitoxantrone is approved as a treatment for relapsing-remitting multiple sclerosis but may carry some risk for cardiotoxicity as well as risk for the development of cancers (Scott and Figgitt, 2004; Buttmann et al., 2016; Chartier et al., 2018). Interestingly, mitoxantrone improved neurological symptoms and delayed the progression of multiple sclerosis in patients while also being reasonably well tolerated (Scott and Figgitt, 2004). Given its ability to inhibit MCU, it could be an interesting candidate to test in ALS due to its similarities with multiple sclerosis. Another recent screening of FDA-approved compounds has identified two drugs that modulate MCU activity: amorolfine, which promotes mitochondrial uptake of Ca2+, and benzethonium, which inhibits this process (De Mario et al., 2021). Since mitochondrial Ca2+ overload is a common neurotoxic mechanism implicated AD, PD, ALS, and epilepsy (Figure 3), it will be critical for future experiments to test the efficacy of mitoxantrone, amorolfine, and benzethonium in animal models of these neurological diseases. The major bottleneck in investigating the efficacy of targeting Ca2+ uptake in human disease is that only a few drugs and small-molecule inhibitors of the uptake and release mechanisms are currently available. Thus, there is a critical need to screen for such agents, and to develop small-molecule inhibitors that could potentially be effective in treating neurologic diseases in which Ca2+ overload is a key problem.
GW and YU wrote and edited the manuscript.
This work was supported by National Institutes of Health grants NS113189 and NS125884 to YU. GW was supported by a predoctoral fellowship through NIH T32 Grant GM067795.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Akundi, R. S., Huang, Z., Eason, J., Pandya, J. D., Zhi, L., Cass, W. A., et al. (2011). Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One 6 (1), e16038. doi:10.1371/journal.pone.0016038
Alavian, K. N., Beutner, G., Lazrove, E., Sacchetti, S., Park, H.-A., Licznerski, P., et al. (2014). An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. 111(29), 10580–10585. doi:10.1073/pnas.1401591111
Alzheimer's and Dementia (2020a). Alzheimer's disease facts and figures. Alzheimer's Dementia 16 (3), 391–460. doi:10.1002/alz.12068
Angelova, P. R., and Abramov, A. Y. (2018). Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 592 (5), 692–702. doi:10.1002/1873-3468.12964
Appel, S. H., Beers, D., Siklos, L., Engelhardt, J. I., and Mosier, D. R. (2001). Calcium: The darth vader of ALS. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2 (1), 47–54. doi:10.1080/146608201300079418
Arduino, D. M., Wettmarshausen, J., Vais, H., Navas-Navarro, P., Cheng, Y., Leimpek, A., et al. (2017). Systematic identification of MCU modulators by orthogonal interspecies chemical screening. Mol. Cell 67 (4), 711–723. e717. doi:10.1016/j.molcel.2017.07.019
Areosa, S. A., Sherriff, F., and McShane, R. (2005). Memantine for dementia. Cochrane database Syst. Rev. 2005, CD003154. doi:10.1002/14651858.CD003154.pub4
Arispe, N., Pollard, H. B., and Rojas, E. (1993). Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc. Natl. Acad. Sci. 90 (22), 10573–10577. doi:10.1073/pnas.90.22.10573
Ashrafi, G., de Juan-Sanz, J., Farrell, R. J., and Ryan, T. A. (2020). Molecular tuning of the axonal mitochondrial Ca2+ uniporter ensures metabolic flexibility of neurotransmission. Neuron 105 (4), 678–687. e675. doi:10.1016/j.neuron.2019.11.020
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metabolism 21 (10), 1133–1145. doi:10.1097/00004647-200110000-00001
Babcock, D. F., Herrington, J., Goodwin, P. C., Park, Y. B., and Hille, B. (1997). Mitochondrial participation in the intracellular Ca2+ network. J. Cell Biol. 136 (4), 833–844. doi:10.1083/jcb.136.4.833
Bacaj, T., Wu, D., Yang, X., Morishita, W., Zhou, P., Xu, W., et al. (2013). Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80 (4), 947–959. doi:10.1016/j.neuron.2013.10.026
Baradaran, R., Wang, C., Siliciano, A. F., and Long, S. B. (2018). Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature 559 (7715), 580–584. doi:10.1038/s41586-018-0331-8
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476 (7360), 341–345. doi:10.1038/nature10234
Beckhauser, T. F., Francis-Oliveira, J., and De Pasquale, R. (2016). Reactive oxygen species: Physiological and physiopathological effects on synaptic plasticity. J. Exp. Neurosci. 10 (1), 23–48. doi:10.4137/JEN.S39887
Beers, D. R., Ho, B.-K., Siklós, L., Alexianu, M. E., Mosier, D. R., Mohamed, A. H., et al. (2001). Parvalbumin overexpression alters immune-mediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J. Neurochem. 79 (3), 499–509. doi:10.1046/j.1471-4159.2001.00582.x
Berg, A. T., Berkovic, S. F., Brodie, M. J., Buchhalter, J., Cross, J. H., Van Emde Boas, W., et al. (2010). Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 51 (4), 676–685. doi:10.1111/j.1528-1167.2010.02522.x
Bernardi, P. (1999). Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 79 (4), 1127–1155. doi:10.1152/physrev.1999.79.4.1127
Bernardi, P., and Petronilli, V. (1996). The permeability transition pore as a mitochondrial calcium release channel: A critical appraisal. J. Bioenergetics Biomembr. 28 (2), 131–138. doi:10.1007/BF02110643
Bezprozvanny, I. (2009). Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 15 (3), 89–100. doi:10.1016/j.molmed.2009.01.001
Bezprozvanny, I., and Mattson, M. P. (2008). Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 31 (9), 454–463. doi:10.1016/j.tins.2008.06.005
Billups, B., and Forsythe, I. D. (2002). Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J. Neurosci. 22 (14), 5840–5847. doi:10.1523/JNEUROSCI.22-14-05840.2002
Bindoff, L. A., and Engelsen, B. A. (2012). Mitochondrial diseases and epilepsy. Epilepsia 53 (4), 92–97. doi:10.1111/j.1528-1167.2012.03618.x
Bonora, M., Bononi, A., De Marchi, E., Giorgi, C., Lebiedzinska, M., Marchi, S., et al. (2013). Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12 (4), 674–683. doi:10.4161/cc.23599
Boyenle, I. D., Oyedele, A. K., Ogunlana, A. T., Adeyemo, A. F., Oyelere, F. S., Akinola, O. B., et al. (2022). Targeting the mitochondrial permeability transition pore for drug discovery: Challenges and opportunities. Mitochondrion 63, 57–71. doi:10.1016/j.mito.2022.01.006
Braak, H., Tredici, K. D., Rüb, U., de Vos, R. A. I., Jansen Steur, E. N. H., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24 (2), 197–211. doi:10.1016/S0197-4580(02)00065-9
Brini, M., Calì, T., Ottolini, D., and Carafoli, E. (2014). Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 71 (15), 2787–2814. doi:10.1007/s00018-013-1550-7
Brini, M., Pinton, P., King, M. P., Davidson, M., Schon, E. A., and Rizzuto, R. (1999). A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat. Med. 5 (8), 951–954. doi:10.1038/11396
Briston, T., Selwood, D. L., Szabadkai, G., and Duchen, M. R. (2019). Mitochondrial permeability transition: A molecular lesion with multiple drug targets. Trends Pharmacol. Sci. 40 (1), 50–70. doi:10.1016/j.tips.2018.11.004
Britti, E., Ros, J., Esteras, N., and Abramov, A. Y. (2020). Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 86, 102150. doi:10.1016/j.ceca.2019.102150
Brookes, P. S., Yoon, Y., Robotham, J. L., Anders, M. W., and Sheu, S.-S. (2004). Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiology - Cell Physiology 287 (4), C817–C833. doi:10.1152/ajpcell.00139.2004
Budde, T., Meuth, S., and Pape, H. C. (2002). Calcium-dependent inactivation of neuronal calcium channels. Nat. Rev. Neurosci. 3 (11), 873–883. doi:10.1038/nrn959
Buttmann, M., Seuffert, L., Mäder, U., and Toyka, K. V. (2016). Malignancies after mitoxantrone for multiple sclerosis: A retrospective cohort study. A Retrosp. cohort study 86 (23), 2203–2207. doi:10.1212/wnl.0000000000002745
Cai, H., Qiao, J., Chen, S., Yang, J., Hölscher, C., Wang, Z., et al. (2022). MCU knockdown in hippocampal neurons improves memory performance of an Alzheimer's disease mouse model. Acta Biochimica Biophysica Sinica 54 (1672-9145), 1528–1539. doi:10.3724/abbs.2022138
Cai, X., and Lytton, J. (2004b). Molecular cloning of a sixth member of the K+-dependent Na+/Ca2+ exchanger gene family, NCKX6. J. Biol. Chem. 279 (7), 5867–5876. doi:10.1074/jbc.M310908200
Cai, X., and Lytton, J. (2004a). The cation/Ca2+ exchanger superfamily: Phylogenetic analysis and structural implications. Mol. Biol. Evol. 21 (9), 1692–1703. doi:10.1093/molbev/msh177
Calvo-Rodriguez, M., Hernando-Perez, E., Nuñez, L., and Villalobos, C. (2019). Amyloid β oligomers increase ER-mitochondria Ca2+ cross talk in young hippocampal neurons and exacerbate aging-induced intracellular Ca2+ remodeling. Front. Cell. Neurosci. 13, 22. doi:10.3389/fncel.2019.00022
Calvo-Rodriguez, M., Hou, S. S., Snyder, A. C., Kharitonova, E. K., Russ, A. N., Das, S., et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer's disease. Nat. Commun. 11 (1), 2146. doi:10.1038/s41467-020-16074-2
Cao, C., Wang, S., Cui, T., Su, X.-C., and Chou, J. J. (2017). Ion and inhibitor binding of the double-ring ion selectivity filter of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. 114 (14), E2846–E2851. doi:10.1073/pnas.1620316114
Carrer, A., Tommasin, L., Šileikytė, J., Ciscato, F., Filadi, R., Urbani, A., et al. (2021). Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F-ATP synthase. Nat. Commun. 12 (1), 4835. doi:10.1038/s41467-021-25161-x
Carroll, J., He, J., Ding, S., Fearnley, I. M., and Walker, J. E. (2019). Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc. Natl. Acad. Sci. 116(26), 12816–12821. doi:10.1073/pnas.1904005116
Caterina, M. J., and Julius, D. (2001). The vanilloid receptor: A molecular gateway to the pain pathway. Annu. Rev. Neurosci. 24, 487–517. doi:10.1146/annurev.neuro.24.1.487
Catterall, W. A., Leal, K., and Nanou, E. (2013). Calcium channels and short-term synaptic plasticity. J. Biol. Chem. 288 (15), 10742–10749. doi:10.1074/jbc.R112.411645
Catterall, W. A. (2000). Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555. doi:10.1146/annurev.cellbio.16.1.521
Chakroborty, S., Goussakov, I., Miller, M. B., and Stutzmann, G. E. (2009). Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci. 29 (30), 9458–9470. doi:10.1523/jneurosci.2047-09.2009
Chalmers, S., and Nicholls, D. G. (2003). The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 278 (21), 19062–19070. doi:10.1074/jbc.M212661200
Chan, C. S., Gertler, T. S., and Surmeier, D. J. (2009). Calcium homeostasis, selective vulnerability and Parkinson's disease. Trends Neurosci. 32 (5), 249–256. doi:10.1016/j.tins.2009.01.006
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447 (7148), 1081–1086. doi:10.1038/nature05865
Chartier, N., Epstein, J., Soudant, M., Dahan, C., Michaud, M., Pittion-Vouyovitch, S., et al. (2018). Clinical follow-up of 411 patients with relapsing and progressive multiple sclerosis 10 years after discontinuing mitoxantrone treatment: A real-life cohort study. Eur. J. Neurology 25 (12), 1439–1445. doi:10.1111/ene.13748
Chaudhuri, D., Sancak, Y., Mootha, V. K., and Clapham, D. E. (2013). MCU encodes the pore conducting mitochondrial calcium currents. eLife 2, e00704. doi:10.7554/eLife.00704
Cheung, K.-H., Shineman, D., Müller, M., Cárdenas, C., Mei, L., Yang, J., et al. (2008). Mechanism of Ca2+ disruption in alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58 (6), 871–883. doi:10.1016/j.neuron.2008.04.015
Chouhan, A. K., Ivannikov, M. V., Lu, Z., Sugimori, M., Llinas, R. R., and Macleod, G. T. (2012). Cytosolic calcium coordinates mitochondrial energy metabolism with presynaptic activity. J. Neurosci. 32 (4), 1233–1243. doi:10.1523/JNEUROSCI.1301-11.2012
Chouhan, A. K., Zhang, J., Zinsmaier, K. E., and Macleod, G. T. (2010). Presynaptic mitochondria in functionally different motor neurons exhibit similar affinities for Ca2+ but exert little influence as Ca2+ buffers at nerve firing rates in situ. J. Neurosci. 30 (5), 1869–1881. doi:10.1523/JNEUROSCI.4701-09.2010
Chuang, H. H., Neuhausser, W. M., and Julius, D. (2004). The super-cooling agent icilin reveals a mechanism of coincidence detection by a temperature-sensitive TRP channel. Neuron 43 (6), 859–869. doi:10.1016/j.neuron.2004.08.038
Cividini, F., Scott, B. T., Suarez, J., Casteel, D. E., Heinz, S., Dai, A., et al. (2021). Ncor2/PPARα-Dependent upregulation of MCUb in the type 2 diabetic heart impacts cardiac metabolic flexibility and function. Diabetes 70 (3), 665–679. doi:10.2337/db20-0779
Clapham, D. E. (2003). TRP channels as cellular sensors. Nature 426 (6966), 517–524. doi:10.1038/nature02196
Clayton, E. L., and Cousin, M. A. (2009). Quantitative monitoring of activity-dependent bulk endocytosis of synaptic vesicle membrane by fluorescent dextran imaging. J. Neurosci. Methods 185 (1), 76–81. doi:10.1016/j.jneumeth.2009.09.016
Colegrove, S. L., Albrecht, M. A., and Friel, D. D. (2000a). Dissection of mitochondrial Ca2+ uptake and release fluxes in situ after depolarization-evoked [Ca2+]i elevations in sympathetic neurons. J. Gen. Physiol. 115 (3), 351–370. doi:10.1085/jgp.115.3.351
Colegrove, S. L., Albrecht, M. A., and Friel, D. D. (2000b). Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neurons - reconstruction of the recovery after depolarization-evoked [Ca2+](i) elevations. J. General Physiology 115 (3), 371–388. doi:10.1085/jgp.115.3.371
Cook, C., and Petrucelli, L. (2019). Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron 101 (6), 1057–1069. doi:10.1016/j.neuron.2019.02.032
Corriveau, R. A., Koroshetz, W. J., Gladman, J. T., Jeon, S., Babcock, D., Bennett, D. A., et al. (2017). Alzheimer's disease–related dementias summit 2016: National research priorities. Neurology 89 (23), 2381–2391. doi:10.1212/wnl.0000000000004717
Cousin, M. A., and Robinson, P. J. (2001). The dephosphins: Dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci. 24 (11), 659–665. doi:10.1016/S0166-2236(00)01930-5
Crompton, M., Künzi, M., and Carafoli, E. (1977). The calcium-induced and sodium-induced effluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier. Eur. J. Biochem. 79 (2), 549–558. doi:10.1111/j.1432-1033.1977.tb11839.x
Crompton, M., Moser, R., Lüdi, H., and Carafoli, E. (1978). The interrelations between the transport of sodium and calcium in mitochondria of various mammalian tissues. Eur. J. Biochem. 82 (1), 25–31. doi:10.1111/j.1432-1033.1978.tb11993.x
Csordás, G., Golenár, T., Seifert, E. L., Kamer, K. J., Sancak, Y., Perocchi, F., et al. (2013). MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca²⁺ uniporter. Cell Metab. 17 (6), 976–987. doi:10.1016/j.cmet.2013.04.020
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A., et al. (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Rep. 14 (5), 892–908. doi:10.1016/j.stemcr.2020.03.023
Dagda, R. K. (2018). Role of mitochondrial dysfunction in degenerative brain diseases, an overview. Brain Sci. 8 (10), 178. doi:10.3390/brainsci8100178
Damiano, M., Starkov, A. A., Petri, S., Kipiani, K., Kiaei, M., Mattiazzi, M., et al. (2006). Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 96 (5), 1349–1361. doi:10.1111/j.1471-4159.2006.03619.x
David, G., and Barrett, E. F. (2003). Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J. Physiol. 548 (2), 425–438. doi:10.1113/jphysiol.2002.035196
David, G., and Barrett, E. F. (2000). Stimulation-evoked increases in cytosolic [Ca2+] in mouse motor nerve terminals are limited by mitochondrial uptake and are temperature-dependent. J. Neurosci. 20 (19), 7290–7296. doi:10.1523/JNEUROSCI.20-19-07290.2000
David, G. (1999). Mitochondrial clearance of cytosolic Ca2+ in stimulated lizard motor nerve terminals proceeds without progressive elevation of mitochondrial matrix [Ca2+]. J. Neurosci. 19 (17), 7495–7506. doi:10.1523/JNEUROSCI.19-17-07495.1999
David, G., Talbot, J., and Barrett, E. F. (2003). Quantitative estimate of mitochondrial [Ca2+] in stimulated motor nerve terminals. Cell Calcium 33 (3), 197–206. doi:10.1016/s0143-4160(02)00229-4
De Camilli, P., Haucke, V., Takey, K., and Mugnaini, E. (2001). “The structure of synapses,” in Synapses. Editors M. W. Cowan, T. C. Sudhof, and C. F. Stevens (Baltimore London: The Johns Hopkins University Press), 89–133.
De Mario, A., Tosatto, A., Hill, J. M., Kriston-Vizi, J., Ketteler, R., Vecellio Reane, D., et al. (2021). Identification and functional validation of FDA-approved positive and negative modulators of the mitochondrial calcium uniporter. Cell Rep. 35 (12), 109275. doi:10.1016/j.celrep.2021.109275
De Stefani, D., Raffaello, A., Teardo, E., Szabo, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476 (7360), 336–340. doi:10.1038/nature10230
De Stefani, D., Rizzuto, R., and Pozzan, T. (2016). Enjoy the trip: Calcium in mitochondria back and forth. Annu. Rev. Biochem. 85, 161–192. doi:10.1146/annurev-biochem-060614-034216
Demuro, A., and Parker, I. (2013). Cytotoxicity of intracellular Aβ42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 33 (9), 3824–3833. doi:10.1523/jneurosci.4367-12.2013
Denton, R. M., Randle, P. J., and Martin, B. R. (1972). Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem. J. 128 (1), 161–163. doi:10.1042/bj1280161
Denton, R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787 (11), 1309–1316. doi:10.1016/j.bbabio.2009.01.005
Devine, M. J., and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19 (2), 63–80. doi:10.1038/nrn.2017.170
Dey, K., Bazala, M. A., and Kuznicki, J. (2020). Targeting mitochondrial calcium pathways as a potential treatment against Parkinson’s disease. Cell Calcium 89, 102216. doi:10.1016/j.ceca.2020.102216
DiMauro, S., and Schon, E. A. (2003). Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 348 (26), 2656–2668. doi:10.1056/NEJMra022567
Ding, S., Fellin, T., Zhu, Y., Lee, S.-Y., Auberson, Y. P., Meaney, D. F., et al. (2007). Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J. Neurosci. 27 (40), 10674–10684. doi:10.1523/jneurosci.2001-07.2007
Dolphin, A. C. (2012). Calcium channel auxiliary α2δ and β subunits: Trafficking and one step beyond. Nat. Rev. Neurosci. 13 (8), 542–555. doi:10.1038/nrn3311
Drago, I., and Davis, R. L. (2016). Inhibiting the mitochondrial calcium uniporter during development impairs memory in adult Drosophila. Cell Rep. 16 (10), 2763–2776. doi:10.1016/j.celrep.2016.08.017
Drago, I., Pizzo, P., and Pozzan, T. (2011). After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 30 (20), 4119–4125. doi:10.1038/emboj.2011.337
Du, H., Guo, L., Fang, F., Chen, D., A Sosunov, A., M McKhann, G., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 14 (10), 1097–1105. doi:10.1038/nm.1868
Duchen, M. R., McGuinness, O., Brown, L. A., and Crompton, M. (1993). On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc. Res. 27 (10), 1790–1794. doi:10.1093/cvr/27.10.1790
Emrich, S. M., Yoast, R. E., Fike, A. J., Bricker, K. N., Xin, P., Zhang, X., et al. (2022). The mitochondrial sodium/calcium exchanger NCLX (Slc8b1) in B lymphocytes. Cell Calcium 108, 102667. doi:10.1016/j.ceca.2022.102667
Emrich, S. M., Yoast, R. E., and Trebak, M. (2021). Physiological functions of CRAC channels. Annu. Rev. Physiol. 84, 355–379. doi:10.1146/annurev-physiol-052521-013426
Escartin, C., Galea, E., Lakatos, A., O'Callaghan, J. P., Petzold, G. C., Serrano-Pozo, A., et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24 (3), 312–325. doi:10.1038/s41593-020-00783-4
Esteras, N., and Abramov, A. Y. (2020). Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells 9 (9), 2135. doi:10.3390/cells9092135
Esteras, N., Kopach, O., Maiolino, M., Lariccia, V., Amoroso, S., Qamar, S., et al. (2022). Mitochondrial ROS control neuronal excitability and cell fate in frontotemporal dementia. Alzheimer's Dementia 18 (2), 318–338. doi:10.1002/alz.12394
Fan, M., Zhang, J., Tsai, C.-W., Orlando, B. J., Rodriguez, M., Xu, Y., et al. (2020). Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 582 (7810), 129–133. doi:10.1038/s41586-020-2309-6
Farrell, J. S., Nguyen, Q. A., and Soltesz, I. (2019). Resolving the micro-macro disconnect to address core features of seizure networks. Neuron 101 (6), 1016–1028. doi:10.1016/j.neuron.2019.01.043
Fellin, T., Gomez-Gonzalo, M., Gobbo, S., Carmignoto, G., and Haydon, P. G. (2006). Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J. Neurosci. 26 (36), 9312–9322. doi:10.1523/jneurosci.2836-06.2006
Fieni, F., Lee, S. B., Jan, Y. N., and Kirichok, Y. (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 3, 1317. doi:10.1038/ncomms2325
Fink, B. D., Bai, F., Yu, L., and Sivitz, W. I. (2017). Regulation of ATP production: Dependence on calcium concentration and respiratory state. Am. J. Physiology-Cell Physiology 313 (2), C146–C153. doi:10.1152/ajpcell.00086.2017
Finsterer, J., and Scorza, F. A. (2017). Effects of antiepileptic drugs on mitochondrial functions, morphology, kinetics, biogenesis, and survival. Epilepsy Res. 136, 5–11. doi:10.1016/j.eplepsyres.2017.07.003
Fleury, C., Mignotte, B., and Vayssière, J.-L. (2002). Mitochondrial reactive oxygen species in cell death signaling. Biochimie 84 (2), 131–141. doi:10.1016/S0300-9084(02)01369-X
Friedman, D., Kannan, K., Faustin, A., Shroff, S., Thomas, C., Heguy, A., et al. (2018). Cardiac arrhythmia and neuroexcitability gene variants in resected brain tissue from patients with sudden unexpected death in epilepsy (SUDEP). npj Genomic Med. 3 (1), 9. doi:10.1038/s41525-018-0048-5
Friel, D. D. (2000). Mitochondria as regulators of stimulus-evoked calcium signals in neurons. Cell Calcium 28 (5-6), 307–316. doi:10.1054/ceca.2000.0172
Fu, H., Hardy, J., and Duff, K. E. (2018). Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 21 (10), 1350–1358. doi:10.1038/s41593-018-0221-2
Fuchs, A., Kutterer, S., Mühling, T., Duda, J., Schütz, B., Liss, B., et al. (2013). Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Physiology 591 (10), 2723–2745. doi:10.1113/jphysiol.2012.247981
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. (2009). PINK1-Associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33 (5), 627–638. doi:10.1016/j.molcel.2009.02.013
Garbincius, J. F., and Elrod, J. W. (2022). Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 102 (2), 893–992. doi:10.1152/physrev.00041.2020
García-Chacón, L. E., Nguyen, K. T., David, G., and Barrett, E. F. (2006). Extrusion of Ca(2+) from mouse motor terminal mitochondria via a Na(+)–Ca(2+) exchanger increases post-tetanic evoked release. J. Physiology 574 (3), 663–675. doi:10.1113/jphysiol.2006.110841
Garg, V., Suzuki, J., Paranjpe, I., Unsulangi, T., Boyman, L., Milescu, L. S., et al. (2021). The mechanism of MICU-dependent gating of the mitochondrial Ca2+uniporter. eLife 10, e69312. doi:10.7554/eLife.69312
Gemes, G., Bangaru, M. L., Wu, H. E., Tang, Q., Weihrauch, D., Koopmeiners, A. S., et al. (2011). Store-operated Ca2+ entry in sensory neurons: Functional role and the effect of painful nerve injury. J. Neurosci. 31 (10), 3536–3549. doi:10.1523/JNEUROSCI.5053-10.2011
George, D. S., Hackelberg, S., Jayaraj, N. D., Ren, D., Edassery, S. L., Rathwell, C., et al. (2021). Mitochondrial calcium uniporter deletion prevents painful diabetic neuropathy by restoring mitochondrial morphology and dynamics. PAIN 163, 560–578. doi:10.1097/j.pain.0000000000002391
Giorgio, V., Bisetto, E., Soriano, M. E., Dabbeni-Sala, F., Basso, E., Petronilli, V., et al. (2009). Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 284 (49), 33982–33988. doi:10.1074/jbc.M109.020115
Giorgio, V., Guo, L., Bassot, C., Petronilli, V., and Bernardi, P. (2018). Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 70, 56–63. doi:10.1016/j.ceca.2017.05.004
Giorgio, V., von Stockum, S., Antoniel, M., Fabbro, A., Fogolari, F., Forte, M., et al. (2013). Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. 110 (15), 5887–5892. doi:10.1073/pnas.1217823110
Glancy, B., and Balaban, R. S. (2012). Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51 (14), 2959–2973. doi:10.1021/bi2018909
Golde, T. E., DeKosky, S. T., and Galasko, D. (2018). Alzheimer's disease: The right drug, the right time. Science 362(6420), 1250–1251. doi:10.1126/science.aau0437
Gottschalk, B., Klec, C., Leitinger, G., Bernhart, E., Rost, R., Bischof, H., et al. (2019). MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun. 10 (1), 3732. doi:10.1038/s41467-019-11692-x
Gregory, J. M., Livesey, M. R., McDade, K., Selvaraj, B. T., Barton, S. K., Chandran, S., et al. (2020). Dysregulation of AMPA receptor subunit expression in sporadic ALS post-mortem brain. J. Pathology 250 (1), 67–78. doi:10.1002/path.5351
Groten, C. J., and MacVicar, B. A. (2022). Mitochondrial Ca2+ uptake by the MCU facilitates pyramidal neuron excitability and metabolism during action potential firing. Commun. Biol. 5 (1), 900. doi:10.1038/s42003-022-03848-1
Gunter, T. E., and Pfeiffer, D. R. (1990). Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 258, C755–C786. doi:10.1152/ajpcell.1990.258.5.C755
Guo, C., and Ma, Y. Y. (2021). Calcium permeable-AMPA receptors and excitotoxicity in neurological disorders. Front. Neural Circuits 15, 711564. doi:10.3389/fncir.2021.711564
Gurney, M. E., Pu, H., Chiu, A. Y., Canto, M. C. D., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264(5166), 1772–1775. doi:10.1126/science.8209258
Hagenston, A. M., Yan, J., Bas-Orth, C., Tan, Y., Sekler, I., and Bading, H. (2022). Disrupted expression of mitochondrial NCLX sensitizes neuroglial networks to excitotoxic stimuli and renders synaptic activity toxic. J. Biol. Chem. 298 (2), 101508. doi:10.1016/j.jbc.2021.101508
Hajnoczky, G., Csordas, G., Das, S., Garcia-Perez, C., Saotome, M., Sinha Roy, S., et al. (2006). Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40 (5-6), 553–560. doi:10.1016/j.ceca.2006.08.016
Hamilton, J., Brustovetsky, T., and Brustovetsky, N. (2021). The effect of mitochondrial calcium uniporter and cyclophilin D knockout on resistance of brain mitochondria to Ca2+-induced damage. J. Biol. Chem. 296, 100669. doi:10.1016/j.jbc.2021.100669
Hamilton, J., Brustovetsky, T., Rysted, J. E., Lin, Z., Usachev, Y. M., and Brustovetsky, N. (2018). Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J. Biol. Chem. 293 (40), 15652–15663. doi:10.1074/jbc.RA118.002926
Han, Y.-E., Ryu, S.-Y., Park, S.-H., Lee, K.-H., Lee, S.-H., and Ho, W.-K. (2015). Ca2+ clearance by plasmalemmal NCLX, Li+-permeable Na+/Ca2+ exchanger, is required for the sustained exocytosis in rat insulinoma INS-1 cells. Pflügers Archiv - Eur. J. Physiology 467 (12), 2461–2472. doi:10.1007/s00424-015-1715-3
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., et al. (2008). The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. 105 (35), 13145–13150. doi:10.1073/pnas.0806192105
Hartzell, C., Putzier, I., and Arreola, J. (2005). Calcium-activated chloride channels. Annu. Rev. Physiology 67, 719–758. doi:10.1146/annurev.physiol.67.032003.154341
Haworth, R. A., and Hunter, D. R. (1979). The Ca2+-induced membrane transition in mitochondria: II. Nature of the Ca2+ trigger site. Archives Biochem. Biophysics 195 (2), 460–467. doi:10.1016/0003-9861(79)90372-2
He, J., Carroll, J., Ding, S., Fearnley, I. M., and Walker, J. E. (2017a). Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc. Natl. Acad. Sci. 114(34), 9086–9091. doi:10.1073/pnas.1711201114
He, J., Ford, H. C., Carroll, J., Ding, S., Fearnley, I. M., and Walker, J. E. (2017b). Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. 114(13), 3409–3414. doi:10.1073/pnas.1702357114
Hernandez-Guijo, J. M., Maneu-Flores, V. E., Ruiz-Nuno, A., Villarroya, M., Garcia, A. G., and Gandia, L. (2001). Calcium-dependent inhibition of L, N, and P/Q Ca2+ channels in chromaffin cells: Role of mitochondria. J. Neurosci. 21 (8), 2553–2560. doi:10.1523/JNEUROSCI.21-08-02553.2001
Hesdorffer, D. C., Logroscino, G., Benn, E. K. T., Katri, N., Cascino, G., and Hauser, W. A. (2011). Estimating risk for developing epilepsy: A population-based study in rochester, Minnesota. A population-based study Rochester, Minn. 76 (1), 23–27. doi:10.1212/WNL.0b013e318204a36a
Heuser, K., and Enger, R. (2021). Astrocytic Ca(2+) signaling in epilepsy. Front. Cell Neurosci. 15, 695380. doi:10.3389/fncel.2021.695380
Hollenbeck, P. J. (2005). Mitochondria and neurotransmission: Evacuating the synapse. Neuron 47 (3), 331–333. doi:10.1016/j.neuron.2005.07.017
Hori, K., Tsujikawa, S., Novakovic, M. M., Yamashita, M., and Prakriya, M. (2020). Regulation of chemoconvulsant-induced seizures by store-operated Orai1 channels. J. Physiol. 598 (23), 5391–5409. doi:10.1113/JP280119
Howarth, C., Gleeson, P., and Attwell, D. (2012). Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow Metabolism 32 (7), 1222–1232. doi:10.1038/jcbfm.2012.35
Howland, D. S., Liu, J., She, Y., Goad, B., Maragakis, N. J., Kim, B., et al. (2002). Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. 99(3), 1604–1609. doi:10.1073/pnas.032539299
Huang, C. W., Lai, M. C., Cheng, J. T., Tsai, J. J., Huang, C. C., and Wu, S. N. (2013). Pregabalin attenuates excitotoxicity in diabetes. PLoS One 8 (6), e65154. doi:10.1371/journal.pone.0065154
Huo, J., Lu, S., Kwong, J. Q., Bround, M. J., Grimes, K. M., Sargent, M. A., et al. (2020). MCUb induction protects the heart from post-ischemic remodeling. Circulation Res. 0, 379. doi:10.1161/CIRCRESAHA.119.316369
Hurst, S., Baggett, A., Csordas, G., and Sheu, S.-S. (2019). SPG7 targets the m-AAA protease complex to process MCU for uniporter assembly, Ca2+ influx, and regulation of mitochondrial permeability transition pore opening. J. Biol. Chem. 294 (28), 10807–10818. doi:10.1074/jbc.RA118.006443
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer's Dementia 8 (1), 1–13. doi:10.1016/j.jalz.2011.10.007
Ichas, F., Jouaville, L. S., and Mazat, J.-P. (1997). Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89 (7), 1145–1153. doi:10.1016/S0092-8674(00)80301-3
Irwin, W. A., Bergamin, N., Sabatelli, P., Reggiani, C., Megighian, A., Merlini, L., et al. (2003). Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 35 (4), 367–371. doi:10.1038/ng1270
Ivannikov, M. V., and Macleod, G. T. (2013). Mitochondrial free Ca²⁺ levels and their effects on energy metabolism in Drosophila motor nerve terminals. Biophysical J. 104 (11), 2353–2361. doi:10.1016/j.bpj.2013.03.064
Jadiya, P., Kolmetzky, D. W., Tomar, D., Di Meco, A., Lombardi, A. A., Lambert, J. P., et al. (2019). Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 10 (1), 3885. doi:10.1038/s41467-019-11813-6
Jang, H., Zheng, J., and Nussinov, R. (2007). Models of β-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophysical J. 93 (6), 1938–1949. doi:10.1529/biophysj.107.110148
Jensen, L., Bultynck, G., Luyten, T., Amijee, H., Bootman, M., and Roderick, H. (2013). Alzheimer’s disease-associated peptide Aβ42 mobilizes ER Ca 2+ via InsP3R-dependent andindependent mechanisms. Front. Mol. Neurosci. 6, 36. doi:10.3389/fnmol.2013.00036
Jiang, D., Zhao, L., Clish, C. B., and Clapham, D. E. (2013). Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf–Hirschhorn syndrome. Proc. Natl. Acad. Sci. 110 (24), E2249–E2254. doi:10.1073/pnas.1308558110
Johnson, M. T., Benson, J. C., Pathak, T., Xin, P., McKernan, A. S., Emrich, S. M., et al. (2022). The airway smooth muscle sodium/calcium exchanger NCLX is critical for airway remodeling and hyperresponsiveness in asthma. J. Biol. Chem. 298 (8), 102259. doi:10.1016/j.jbc.2022.102259
Jung, H., Kim, S. Y., Canbakis Cecen, F. S., Cho, Y., and Kwon, S. K. (2020). Dysfunction of mitochondrial Ca(2+) regulatory machineries in brain aging and neurodegenerative diseases. Front. Cell Dev. Biol. 8, 599792. doi:10.3389/fcell.2020.599792
Kamer, K. J., and Mootha, V. K. (2015). The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 16, 545–553. doi:10.1038/nrm4039
Kim, M. S., and Usachev, Y. M. (2009). Mitochondrial Ca2+ cycling facilitates activation of the transcription factor NFAT in sensory neurons. J. Neurosci. 29 (39), 12101–12114. doi:10.1523/JNEUROSCI.3384-09.2009
Kinjo, E. R., Rodríguez, P. X. R., dos Santos, B. A., Higa, G. S. V., Ferraz, M. S. A., Schmeltzer, C., et al. (2018). New insights on temporal lobe epilepsy based on plasticity-related network changes and high-order statistics. Mol. Neurobiol. 55 (5), 3990–3998. doi:10.1007/s12035-017-0623-2
Kirichok, Y., Krapivinsky, G., and Clapham, D. E. (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427 (6972), 360–364. doi:10.1038/nature02246
Klann, E. (1998). Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J. Neurophysiology 80 (1), 452–457. doi:10.1152/jn.1998.80.1.452
Konen, L. M., Wright, A. L., Royle, G. A., Morris, G. P., Lau, B. K., Seow, P. W., et al. (2020). A new mouse line with reduced GluA2 Q/R site RNA editing exhibits loss of dendritic spines, hippocampal CA1-neuron loss, learning and memory impairments and NMDA receptor-independent seizure vulnerability. Mol. Brain 13 (1), 27. doi:10.1186/s13041-020-0545-1
Kostic, M., Ludtmann, M. H. R., Bading, H., Hershfinkel, M., Steer, E., Chu, C. T., et al. (2015). PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep. 13 (2), 376–386. doi:10.1016/j.celrep.2015.08.079
Kovac, S., Dinkova Kostova, A., Herrmann, A., Melzer, N., Meuth, S., and Gorji, A. (2017). Metabolic and homeostatic changes in seizures and acquired epilepsy—mitochondria, calcium dynamics and reactive oxygen species. Int. J. Mol. Sci. 18 (9), 1935. doi:10.3390/ijms18091935
Kovacs, G., Reimer, L., and Jensen, P. H. (2021). Endoplasmic reticulum-based calcium dysfunctions in synucleinopathies. Front. Neurology 12, 742625. doi:10.3389/fneur.2021.742625
Kristian, T., Pivovarova, N. B., Fiskum, G., and Andrews, S. B. (2007). Calcium-induced precipitate formation in brain mitochondria: Composition, calcium capacity, and retention. J. Neurochem. 102 (4), 1346–1356. doi:10.1111/j.1471-4159.2007.04626.x
Kwan, P., and Brodie, M. J. (2000). Early identification of refractory epilepsy. N. Engl. J. Med. 342 (5), 314–319. doi:10.1056/nejm200002033420503
Kwon, S. K., Sando, R., Lewis, T. L., Hirabayashi, Y., Maximov, A., and Polleux, F. (2016). LKB1 regulates mitochondria-dependent presynaptic calcium clearance and neurotransmitter release properties at excitatory synapses along cortical axons. PLoS Biol. 14 (7), e1002516. doi:10.1371/journal.pbio.1002516
Kwong, J. Q., Lu, X., Correll, R. N., Schwanekamp, J. A., Vagnozzi, R. J., Sargent, M. A., et al. (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 12 (1), 15–22. doi:10.1016/j.celrep.2015.06.002
Lambert, J. P., Luongo, T. S., Tomar, D., Jadiya, P., Gao, E., Zhang, X., et al. (2019). MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation 140 (21), 1720–1733. doi:10.1161/CIRCULATIONAHA.118.037968
Lee, D., Lee, K. H., Ho, W. K., and Lee, S. H. (2007). Target cell-specific involvement of presynaptic mitochondria in post-tetanic potentiation at hippocampal mossy fiber synapses. J. Neurosci. 27 (50), 13603–13613. doi:10.1523/JNEUROSCI.3985-07.2007
Lee, J. H., Han, J.-h., Kim, H., Park, S. M., Joe, E.-h., and Jou, I. (2019). Parkinson’s disease-associated LRRK2-G2019S mutant acts through regulation of SERCA activity to control ER stress in astrocytes. Acta Neuropathol. Commun. 7 (1), 68. doi:10.1186/s40478-019-0716-4
Leung, A. W. C., Varanyuwatana, P., and Halestrap, A. P. (2008). The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 283 (39), 26312–26323. doi:10.1074/jbc.M805235200
Levesque, M., and Avoli, M. (2013). The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav Rev. 37, 2887–2899. doi:10.1016/j.neubiorev.2013.10.011
Li, D.-D., Zhang, Y.-H., Zhang, W., and Zhao, P. (2019). Meta-analysis of randomized controlled trials on the efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease. Front. Neurosci. 13, 472. doi:10.3389/fnins.2019.00472
Li, L., Chen, S. R., Zhou, M. H., Wang, L., Li, D. P., Chen, H., et al. (2021). α2δ-1 switches the phenotype of synaptic AMPA receptors by physically disrupting heteromeric subunit assembly. Cell Rep. 36 (3), 109396. doi:10.1016/j.celrep.2021.109396
Li, W., Shariat-Madar, Z., Powers, M., Sun, X., Lane, R. D., and Garlid, K. D. (1992). Reconstitution, identification, purification, and immunological characterization of the 110-kDa Na+/Ca2+ antiporter from beef heart mitochondria. J. Biol. Chem. 267 (25), 17983–17989. doi:10.1016/s0021-9258(19)37140-6
Lim, D., Dematteis, G., Tapella, L., Genazzani, A. A., Cali, T., Brini, M., et al. (2021a). Ca(2+) handling at the mitochondria-ER contact sites in neurodegeneration. Cell Calcium 98, 102453. doi:10.1016/j.ceca.2021.102453
Lim, D., Semyanov, A., Genazzani, A., and Verkhratsky, A. (2021b). Calcium signaling in neuroglia. Int. Rev. Cell Mol. Biol. 362, 1–53. doi:10.1016/bs.ircmb.2021.01.003
Liochev, S. I. (2013). Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 60, 1–4. doi:10.1016/j.freeradbiomed.2013.02.011
Liu, J. C., Liu, J., Holmstrom, K. M., Menazza, S., Parks, R. J., Fergusson, M. M., et al. (2016). MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 16 (6), 1561–1573. doi:10.1016/j.celrep.2016.07.011
Liu, J. C., Parks, R. J., Liu, J., Stares, J., Rovira, I. I., Murphy, E., et al. (2017). “The in vivo Biology of the mitochondrial calcium uniporter,” in Mitochondrial dynamics in cardiovascular medicine. Editor G. Santulli (Cham: Springer International Publishing), 49–63.
Liu, J. C., Syder, N. C., Ghorashi, N. S., Willingham, T. B., Parks, R. J., Sun, J., et al. (2020). EMRE is essential for mitochondrial calcium uniporter activity in a mouse model. JCI Insight 5 (4), e134063. doi:10.1172/jci.insight.134063
Logan, C. V., Szabadkai, G., Sharpe, J. A., Parry, D. A., Torelli, S., Childs, A.-M., et al. (2014). Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 46 (2), 188–193. doi:10.1038/ng.2851
Lohmann, C., and Wong, R. O. L. (2005). Regulation of dendritic growth and plasticity by local and global calcium dynamics. Cell Calcium 37 (5), 403–409. doi:10.1016/j.ceca.2005.01.008
Loupatatzis, C., Seitz, G., Schönfeld, P., Lang, F., and Siemen, D. (2002). Single-channel currents of the permeability transition pore from the inner mitochondrial membrane of rat liver and of a human hepatoma cell line. Cell Physiol. Biochem. 12 (5-6), 269–278. doi:10.1159/000067897
Luo, Y., Ma, H., Zhou, J. J., Li, L., Chen, S. R., Zhang, J., et al. (2018). Focal cerebral ischemia and reperfusion induce brain injury through α2δ-1-bound NMDA receptors. Stroke 49 (10), 2464–2472. doi:10.1161/STROKEAHA.118.022330
Luongo, T. S., Lambert, J. P., Gross, P., Nwokedi, M., Lombardi, A. A., Shanmughapriya, S., et al. (2017). The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 545 (7652), 93–97. doi:10.1038/nature22082
Luongo, T. S., Lambert, J. P., Yuan, A., Zhang, X., Gross, P., Song, J., et al. (2015). The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 12 (1), 23–34. doi:10.1016/j.celrep.2015.06.017
MacAskill, A. F., Atkin, T. A., and Kittler, J. T. (2010). Mitochondrial trafficking and the provision of energy and calcium buffering at excitatory synapses. Eur. J. Neurosci. 32(2), 231–240. doi:10.1111/j.1460-9568.2010.07345.x
Mallilankaraman, K., Cárdenas, C., Doonan, P. J., Chandramoorthy, H. C., Irrinki, K. M., Golenár, T., et al. (2012a). MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 14, 1336. doi:10.1038/ncb2622
Mallilankaraman, K., Doonan, P., Cárdenas, C., Chandramoorthy, H. C., Müller, M., Miller, R., et al. (2012b). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 151 (3), 630–644. doi:10.1016/j.cell.2012.10.011
Maneshi, M. M., Toth, A. B., Ishii, T., Hori, K., Tsujikawa, S., Shum, A. K., et al. (2020). Orai1 channels are essential for amplification of glutamate-evoked Ca(2+) signals in dendritic spines to regulate working and associative memory. Cell Rep. 33 (9), 108464. doi:10.1016/j.celrep.2020.108464
Manikandan, M., Walker, S., Deshmukh, A. R., Perea, E., Wang, D., Alagramam, K. N., et al. (2021). Mitochondrial calcium uniporter is essential for hearing and hair cell preservation in congenic FVB/NJ mice. Sci. Rep. 11 (1), 9660. doi:10.1038/s41598-021-88841-0
Marland, J. R., Hasel, P., Bonnycastle, K., and Cousin, M. A. (2016). Mitochondrial calcium uptake modulates synaptic vesicle endocytosis in central nerve terminals. J. Biol. Chem. 291 (5), 2080–2086. doi:10.1074/jbc.M115.686956
Marta, K., Hasan, P., Rodriguez-Prados, M., Paillard, M., and Hajnoczky, G. (2021). Pharmacological inhibition of the mitochondrial Ca(2+) uniporter: Relevance for pathophysiology and human therapy. J. Mol. Cell Cardiol. 151, 135–144. doi:10.1016/j.yjmcc.2020.09.014
Martin, L. J., Gertz, B., Pan, Y., Price, A. C., Molkentin, J. D., and Chang, Q. (2009). The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 218 (2), 333–346. doi:10.1016/j.expneurol.2009.02.015
Martin, L. J., Semenkow, S., Hanaford, A., and Wong, M. (2014). Mitochondrial permeability transition pore regulates Parkinson's disease development in mutant α-synuclein transgenic mice. Neurobiol. Aging 35 (5), 1132–1152. doi:10.1016/j.neurobiolaging.2013.11.008
Marty, A. (1989). The physiological role of calcium-dependent channels. Trends Neurosci. 12 (11), 420–424. doi:10.1016/0166-2236(89)90090-8
Matteucci, A., Patron, M., Vecellio Reane, D., Gastaldello, S., Amoroso, S., Rizzuto, R., et al. (2018). Parkin-dependent regulation of the MCU complex component MICU1. Sci. Rep. 8 (1), 14199. doi:10.1038/s41598-018-32551-7
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60 (5), 748–766. doi:10.1016/j.neuron.2008.10.010
McBain, C. J., and Mayer, M. L. (1994). N-methyl-D-aspartic acid receptor structure and function. Physiol. Rev. 74 (3), 723–760. doi:10.1152/physrev.1994.74.3.723
McCormack, J. G., Halestrap, A. P., and Denton, R. M. (1990). Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391–425. doi:10.1152/physrev.1990.70.2.391
Medler, K., and Gleason, E. L. (2002). Mitochondrial Ca2+ buffering regulates synaptic transmission between retinal amacrine cells. J. Neurophysiology 87 (3), 1426–1439. doi:10.1152/jn.00627.2001
Medvedeva, Y. V., Kim, M. S., and Usachev, Y. M. (2008). Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J. Neurosci. 28 (20), 5295–5311. doi:10.1523/JNEUROSCI.4810-07.2008
Mnatsakanyan, N., Llaguno, M. C., Yang, Y., Yan, Y., Weber, J., Sigworth, F. J., et al. (2019). A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat. Commun. 10 (1), 5823. doi:10.1038/s41467-019-13766-2
Mnatsakanyan, N., Park, H.-A., Wu, J., He, X., Llaguno, M. C., Latta, M., et al. (2022). Mitochondrial ATP synthase c-subunit leak channel triggers cell death upon loss of its F1 subcomplex. Cell Death Differ. 29, 1874–1887. doi:10.1038/s41418-022-00972-7
Modesti, L., Danese, A., Angela Maria Vitto, V., Ramaccini, D., Aguiari, G., Gafà, R., et al. (2021). Mitochondrial Ca2+ signaling in health, disease and therapy. Cells 10 (6), 1317. doi:10.3390/cells10061317
More, J., Galusso, N., Veloso, P., Montecinos, L., Finkelstein, J. P., Sanchez, G., et al. (2018). N-acetylcysteine prevents the spatial memory deficits and the redox-dependent RyR2 decrease displayed by an Alzheimer’s disease rat model. Front. Aging Neurosci. 10, 399. doi:10.3389/fnagi.2018.00399
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: Mechanisms in search of treatments. Neuron 67 (2), 181–198. doi:10.1016/j.neuron.2010.07.002
Mühling, T., Duda, J., Weishaupt, J. H., Ludolph, A. C., and Liss, B. (2014). Elevated mRNA-levels of distinct mitochondrial and plasma membrane Ca2+ transporters in individual hypoglossal motor neurons of endstage SOD1 transgenic mice. Front. Cell. Neurosci. 8, 353. doi:10.3389/fncel.2014.00353
Munoz-Pinedo, C., Guio-Carrion, A., Goldstein, J. C., Fitzgerald, P., Newmeyer, D. D., and Green, D. R. (2006). Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc. Natl. Acad. Sci. U. S. A. 103 (31), 11573–11578. doi:10.1073/pnas.0603007103
Murphy, E., Pan, X., Nguyen, T., Liu, J., Holmström, K. M., and Finkel, T. (2014). Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophysical Res. Commun. 449 (4), 384–385. doi:10.1016/j.bbrc.2014.04.144
Murphy, E., and Steenbergen, C. (2021). Regulation of mitochondrial Ca2+ uptake. Annu. Rev. Physiology 83 (1), 107–126. doi:10.1146/annurev-physiol-031920-092419
Nakamura, T., Ogawa, M., Kojima, K., Takayanagi, S., Ishihara, S., Hattori, K., et al. (2021). The mitochondrial Ca2+ uptake regulator, MICU1, is involved in cold stress-induced ferroptosis. EMBO Rep. 22 (5), e51532. doi:10.15252/embr.202051532
Neginskaya, M. A., Morris, S. E., and Pavlov, E. V. (2022). Both ANT and ATPase are essential for mitochondrial permeability transition but not depolarization. iScience 25 (11), 105447. doi:10.1016/j.isci.2022.105447
Neginskaya, M. A., Solesio, M. E., Berezhnaya, E. V., Amodeo, G. F., Mnatsakanyan, N., Jonas, E. A., et al. (2019). ATP synthase C-Subunit-Deficient mitochondria have a small cyclosporine A-sensitive channel, but lack the permeability transition pore. Cell Rep. 26 (1), 11–17. e12. doi:10.1016/j.celrep.2018.12.033
Neher, E., and Sakaba, T. (2008). Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59 (6), 861–872. doi:10.1016/j.neuron.2008.08.019
Newmeyer, D. D., and Ferguson-Miller, S. (2003). Mitochondria: Releasing power for life and unleashing the machineries of death. Cell 112 (4), 481–490. doi:10.1016/s0092-8674(03)00116-8
Nguyen, K. T., García-Chacón, L. E., Barrett, J. N., Barrett, E. F., and David, G. (2009). The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc. Natl. Acad. Sci. U. S. A. 106 (6), 2007–2011. doi:10.1073/pnas.0810934106
Nicholls, D. G. (1978). Calcium transport and porton electrochemical potential gradient in mitochondria from Guinea-pig cerebral cortex and rat heart. Biochem. J. 170 (3), 511–522. doi:10.1042/bj1700511
Nicholls, D. G. (2005). Mitochondria and calcium signaling. Cell Calcium 38 (3-4), 311–317. doi:10.1016/j.ceca.2005.06.011
Nicholls, D. G. (2012). “Mitochondria, sodium, and calcium in neuronal dysfunction,” in Mitochondrial dysfunction in neurodegenerative disorders. Editors A. K. Reeve, K. J. Krishnan, M. R. Duchen, and D. M. Turnbull (London: Springer), 113–125.
Nicholls, D. G., and Ward, M. W. (2000). Mitochondrial membrane potential and neuronal glutamate excitotoxicity: Mortality and millivolts. Trends Neurosci. 23 (4), 166–174. doi:10.1016/s0166-2236(99)01534-9
Nichols, M., Pavlov, E. V., and Robertson, G. S. (2018). Tamoxifen-induced knockdown of the mitochondrial calcium uniporter in Thy1-expressing neurons protects mice from hypoxic/ischemic brain injury. Cell Death Dis. 9 (6), 606. doi:10.1038/s41419-018-0607-9
Novorolsky, R. J., Nichols, M., Kim, J. S., Pavlov, E. V., J, J. W., Wilson, J. J., et al. (2020). The cell-permeable mitochondrial calcium uniporter inhibitor Ru265 preserves cortical neuron respiration after lethal oxygen glucose deprivation and reduces hypoxic/ischemic brain injury. J. Cereb. Blood Flow. Metab. 40 (6), 1172–1181. doi:10.1177/0271678x20908523
Nowicky, A. V., and Duchen, M. R. (1998). Changes in [Ca2+]i and membrane currents during impaired mitochondrial metabolism in dissociated rat hippocampal neurons. J. physiology 507 (1), 131–145. doi:10.1111/j.1469-7793.1998.131bu.x
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: In sickness and in health. Cell 148 (6), 1145–1159. doi:10.1016/j.cell.2012.02.035
Olanow, C. W., Stern, M. B., and Sethi, K. (2009). The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology 72, S1–S136. doi:10.1212/WNL.0b013e3181a1d44c
Orrenius, S., Zhivotovsky, B., and Nicotera, P. (2003). Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4 (7), 552–565. doi:10.1038/nrm1150
Oxenoid, K., Dong, Y., Cao, C., Cui, T., Sancak, Y., Markhard, A. L., et al. (2016). Architecture of the mitochondrial calcium uniporter. Nature 533, 269–273. doi:10.1038/nature17656
Palty, R., Hershfinkel, M., Yagev, O., Saar, D., Barkalifa, R., Khananshvili, D., et al. (2006). Single α-domain constructs of the Na+/Ca2+ exchanger, NCLX, oligomerize to form a functional exchanger. Biochemistry 45 (39), 11856–11866. doi:10.1021/bi060633b
Palty, R., Ohana, E., Hershfinkel, M., Volokita, M., Elgazar, V., Beharier, O., et al. (2004). Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 279 (24), 25234–25240. doi:10.1074/jbc.M401229200
Palty, R., and Sekler, I. (2012). The mitochondrial Na(+)/Ca(2+) exchanger. Cell Calcium 52 (1), 9–15. doi:10.1016/j.ceca.2012.02.010
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., Parnis, J., et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. 107 (1), 436–441. doi:10.1073/pnas.0908099107
Pan, X., Liu, J., Nguyen, T., Liu, C., Sun, J., Teng, Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464. doi:10.1038/ncb2868
Parkinson Study Group STEADY-PD III Investigators (2020). Isradipine versus placebo in early Parkinson disease. Ann. Intern. Med. 172 (9), 591–598. doi:10.7326/m19-2534./m.32227247
Partridge, L. D., and Swandulla, D. (1988). Calcium-activated non-specific cation channels. Trends Neurosci. 11 (2), 69–72. doi:10.1016/0166-2236(88)90167-1
Patel, D. C., Tewari, B. P., Chaunsali, L., and Sontheimer, H. (2019). Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 20 (5), 282–297. doi:10.1038/s41583-019-0126-4
Patron, M., Checchetto, V., Raffaello, A., Teardo, E., Vecellio Reane, D., Mantoan, M., et al. (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53 (5), 726–737. doi:10.1016/j.molcel.2014.01.013
Patron, M., Granatiero, V., Espino, J., Rizzuto, R., and De Stefani, D. (2019). MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 26 (1), 179–195. doi:10.1038/s41418-018-0113-8
Pearson-Smith, J. N., and Patel, M. (2017). Metabolic dysfunction and oxidative stress in epilepsy. Int. J. Mol. Sci. 18 (11), 2365. doi:10.3390/ijms18112365
Perocchi, F., Gohil, V. M., Girgis, H. S., Bao, X. R., McCombs, J. E., Palmer, A. E., et al. (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467 (7313), 291–296. doi:10.1038/nature09358
Petrungaro, C., Zimmermann, K. M., Küttner, V., Fischer, M., Dengjel, J., Bogeski, I., et al. (2015). The Ca(2+)-dependent release of the mia40-induced MICU1-MICU2 dimer from MCU regulates mitochondrial Ca(2+) uptake. Cell Metab. 22 (4), 721–733. doi:10.1016/j.cmet.2015.08.019
Pivovarova, N. B., and Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277 (18), 3622–3636. doi:10.1111/j.1742-4658.2010.07754.x
Pivovarova, N. B., Hongpaisan, J., Andrews, S. B., and Friel, D. D. (1999). Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: Spatial and temporal characteristics. J. Neurosci. 19 (15), 6372–6384. doi:10.1523/JNEUROSCI.19-15-06372.1999
Pivovarova, N. B., Nguyen, H. V., Winters, C. A., Brantner, C. A., Smith, C. L., and Andrews, S. B. (2004). Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J. Neurosci. 24 (24), 5611–5622. doi:10.1523/JNEUROSCI.0531-04.2004
Pivovarova, N. B., Pozzo-Miller, L. D., Hongpaisan, J., and Andrews, S. B. (2002). Correlated calcium uptake and release by mitochondria and endoplasmic reticulum of CA3 hippocampal dendrites after afferent synaptic stimulation. J. Neurosci. 22 (24), 10653–10661. doi:10.1523/JNEUROSCI.22-24-10653.2002
Plotegher, N., Filadi, R., Pizzo, P., and Duchen, M. R. (2021). Excitotoxicity revisited: Mitochondria on the verge of a nervous breakdown. Trends Neurosci. 44 (5), 342–351. doi:10.1016/j.tins.2021.01.001
Plovanich, M., Bogorad, R. L., Sancak, Y., Kamer, K. J., Strittmatter, L., Li, A. A., et al. (2013). MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 8 (2), e55785. doi:10.1371/journal.pone.0055785
Popugaeva, E., Pchitskaya, E., and Bezprozvanny, I. (2018). Dysregulation of intracellular calcium signaling in alzheimer's disease. Antioxidants Redox Signal. 29 (12), 1176–1188. doi:10.1089/ars.2018.7506
Prakriya, M., and Lewis, R. S. (2015). Store-operated calcium channels. Physiol. Rev. 95 (4), 1383–1436. doi:10.1152/physrev.00020.2014
Puttachary, S., Sharma, S., Stark, S., and Thippeswamy, T. (2015). Seizure-induced oxidative stress in temporal lobe epilepsy. Biomed. Res. Int. 2015, 745613. doi:10.1155/2015/745613
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer's disease. N. Engl. J. Med. 362 (4), 329–344. doi:10.1056/NEJMra0909142
Raffaello, A., De Stefani, D., Sabbadin, D., Teardo, E., Merli, G., Picard, A., et al. (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32 (17), 2362–2376. doi:10.1038/emboj.2013.157
Rasola, A., and Bernardi, P. (2011). Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50 (3), 222–233. doi:10.1016/j.ceca.2011.04.007
Redmond, L., and Ghosh, A. (2005). Regulation of dendritic development by calcium signaling. Cell Calcium 37 (5), 411–416. doi:10.1016/j.ceca.2005.01.009
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13 (9), 566–578. doi:10.1038/nrm3412
Rosales-Corral, S., Acuna-Castroviejo, D., Tan, D. X., López-Armas, G., Cruz-Ramos, J., Munoz, R., et al. (2012). Accumulation of exogenous amyloid-beta peptide in hippocampal mitochondria causes their dysfunction: A protective role for melatonin. Oxidative Med. Cell. Longev. 2012, 843649. doi:10.1155/2012/843649
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362 (6415), 59–62. doi:10.1038/362059a0
Rossi, A. R., Angelo, M. F., Villarreal, A., Lukin, J., and Ramos, A. J. (2013). Gabapentin administration reduces reactive gliosis and neurodegeneration after pilocarpine-induced status epilepticus. PLoS One 8 (11), e78516. doi:10.1371/journal.pone.0078516
Rossi, C. S., Vasington, F. D., and Carafoli, E. (1973). The effect of ruthenium red on the uptake and release of Ca 2+ by mitochondria. Biochem. Biophys. Res. Commun. 50 (3), 846–852. doi:10.1016/0006-291x(73)91322-3
Roy, S., Dey, K., Hershfinkel, M., Ohana, E., and Sekler, I. (2017). Identification of residues that control Li+ versus Na+ dependent Ca2+ exchange at the transport site of the mitochondrial NCLX. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1864 (6), 997–1008. doi:10.1016/j.bbamcr.2017.01.011
Rybalchenko, V., Hwang, S.-Y., Rybalchenko, N., and Koulen, P. (2008). The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 40 (1), 84–97. doi:10.1016/j.biocel.2007.06.023
Rysted, J. E., Lin, Z., Walters, G. C., Rauckhorst, A. J., Noterman, M., Liu, G., et al. (2021). Distinct properties of Ca2+ efflux from brain, heart and liver mitochondria: The effects of Na+, Li+ and the mitochondrial Na+/Ca2+ exchange inhibitor CGP37157. Cell Calcium 96, 102382. doi:10.1016/j.ceca.2021.102382
Sah, P. (1996). Ca2+-activated K+ currents in neurones: Types, physiological roles and modulation. Trends Neurosci. 19 (4), 150–154. doi:10.1016/s0166-2236(96)80026-9
Salter, M. W., and Stevens, B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23 (9), 1018–1027. doi:10.1038/nm.4397
Sancak, Y., Markhard, A. L., Kitami, T., Kovács-Bogdán, E., Kamer, K. J., Udeshi, N. D., et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342 (6164), 1379–1382. doi:10.1126/science.1242993
Santello, M., Toni, N., and Volterra, A. (2019). Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 22 (2), 154–166. doi:10.1038/s41593-018-0325-8
Scharfman, H. E. (2007). The neurobiology of epilepsy. Curr. neurology Neurosci. Rep. 7 (4), 348–354. doi:10.1007/s11910-007-0053-z
Scheltens, P., Blennow, K., Breteler, M. M. B., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer's disease. Lancet 388 (10043), 505–517. doi:10.1016/S0140-6736(15)01124-1
Schwab, R. S., England, A. C., Poskanzer, D. C., and Young, R. R. (1969). Amantadine in the treatment of Parkinson's disease. JAMA 208 (7), 1168–1170. doi:10.1001/jama.1969.03160070046011
Scott, L. J., and Figgitt, D. P. (2004). Mitoxantrone: A review of its use in multiple sclerosis. CNS Drugs 18 (6), 379–396. doi:10.2165/00023210-200418060-00010
Scott, R. H., Sutton, K. G., Griffin, A., Stapleton, S. R., and Currie, K. P. (1995). Aspects of calcium-activated chloride currents: A neuronal perspective. Pharmacol. Ther. 66 (3), 535–565. doi:10.1016/0163-7258(95)00018-c
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8 (6), 595–608. doi:10.15252/emmm.201606210
Selvaraj, B. T., Livesey, M. R., Zhao, C., Gregory, J. M., James, O. T., Cleary, E. M., et al. (2018). C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 9 (1), 347. doi:10.1038/s41467-017-02729-0
Shanmughapriya, S., Rajan, S., Hoffman, N. E., Higgins, A. M., Tomar, D., Nemani, N., et al. (2015). SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol. Cell 60 (1), 47–62. doi:10.1016/j.molcel.2015.08.009
Shaw, P. J., and Eggett, C. J. (2000). Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J. Neurol. 247, I17–I27. doi:10.1007/bf03161151
Shigetomi, E., Saito, K., Sano, F., and Koizumi, S. (2019). Aberrant calcium signals in reactive astrocytes: A key process in neurological disorders. Int. J. Mol. Sci. 20 (4), 996. doi:10.3390/ijms20040996
Shoshan-Barmatz, V., De Pinto, V., Zweckstetter, M., Raviv, Z., Keinan, N., and Arbel, N. (2010). VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. aspects Med. 31 (3), 227–285. doi:10.1016/j.mam.2010.03.002
Shutov, L. P., Kim, M. S., Houlihan, P. R., Medvedeva, Y. V., and Usachev, Y. M. (2013). Mitochondria and plasma membrane Ca2+-ATPase control presynaptic Ca2+ clearance in capsaicin-sensitive rat sensory neurons. J. Physiol. 591 (10), 2443–2462. doi:10.1113/jphysiol.2012.249219
Sills, G. J., and Rogawski, M. A. (2020). Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 168, 107966. doi:10.1016/j.neuropharm.2020.107966
Singh, R., Bartok, A., Paillard, M., Tyburski, A., Elliott, M., and Hajnoczky, G. (2022). Uncontrolled mitochondrial calcium uptake underlies the pathogenesis of neurodegeneration in MICU1-deficient mice and patients. Sci. Adv. 8 (11), eabj4716. doi:10.1126/sciadv.abj4716
Smith, E. F., Shaw, P. J., and De Vos, K. J. (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710, 132933. doi:10.1016/j.neulet.2017.06.052
Sobolczyk, M., and Boczek, T. (2022). Astrocytic calcium and cAMP in neurodegenerative diseases. Front. Cell Neurosci. 16, 889939. doi:10.3389/fncel.2022.889939
Soman, S. K., Bazała, M., Keatinge, M., Bandmann, O., and Kuznicki, J. (2019). Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson's disease. Biol. Open 8 (10), bio044347. doi:10.1242/bio.044347
Soman, S. K., and Dagda, R. K. (2021). Role of cleaved PINK1 in neuronal development, synaptogenesis, and plasticity: Implications for Parkinson's disease. Front. Neurosci. 15, 769331. doi:10.3389/fnins.2021.769331
Soman, S., Keatinge, M., Moein, M., Da Costa, M., Mortiboys, H., Skupin, A., et al. (2017). Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1−/− zebrafish. Eur. J. Neurosci. 45 (4), 528–535. doi:10.1111/ejn.13473
Sonkusare, S. K., Kaul, C. L., and Ramarao, P. (2005). Dementia of Alzheimer’s disease and other neurodegenerative disorders—Memantine, a new hope. Pharmacol. Res. 51 (1), 1–17. doi:10.1016/j.phrs.2004.05.005
Spat, A., Szanda, G., Csordas, G., and Hajnoczky, G. (2008). High- and low-calcium-dependent mechanisms of mitochondrial calcium signalling. Cell Calcium 44 (1), 51–63. doi:10.1016/j.ceca.2007.11.015
Spruill, M. M., and Kuncl, R. W. (2015). Calbindin-D28K is increased in the ventral horn of spinal cord by neuroprotective factors for motor neurons. J. Neurosci. Res. 93 (8), 1184–1191. doi:10.1002/jnr.23562
Stanika, R. I., Villanueva, I., Kazanina, G., Andrews, S. B., and Pivovarova, N. B. (2012). Comparative impact of voltage-gated calcium channels and NMDA receptors on mitochondria-mediated neuronal injury. J. Neurosci. 32 (19), 6642–6650. doi:10.1523/JNEUROSCI.6008-11.2012
Stavsky, A., Stoler, O., Kostic, M., Katoshevsky, T., Assali, E. A., Savic, I., et al. (2021). Aberrant activity of mitochondrial NCLX is linked to impaired synaptic transmission and is associated with mental retardation. Commun. Biol. 4 (1), 666. doi:10.1038/s42003-021-02114-0
Stelzer, A. C., Frazee, R. W., Van Huis, C., Cleary, J., Opipari, A. W., Glick, G. D., et al. (2010). NMR studies of an immunomodulatory benzodiazepine binding to its molecular target on the mitochondrial F(1)F(0)-ATPase. Biopolymers 93 (1), 85–92. doi:10.1002/bip.21306
Storozhuk, M. V., Ivanova, S. Y., Balaban, P. M., and Kostyuk, P. G. (2005). Possible role of mitochondria in posttetanic potentiation of GABAergic synaptic transmission in rat neocortical cell cultures. Synapse 58 (1), 45–52. doi:10.1002/syn.20186
Stout, A. K., Raphael, H. M., Kanterewicz, B. I., Klann, E., and Reynolds, I. J. (1998). Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1 (5), 366–373. doi:10.1038/1577
Sun, T., Qiao, H., Pan, P.-Y., Chen, Y., and Sheng, Z.-H. (2013). Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 4 (3), 413–419. doi:10.1016/j.celrep.2013.06.040
Surmeier, D. J. (2007). Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurology 6 (10), 933–938. doi:10.1016/S1474-4422(07)70246-6
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18 (2), 101–113. doi:10.1038/nrn.2016.178
Szibor, M., Gizatullina, Z., Gainutdinov, T., Endres, T., Debska-Vielhaber, G., Kunz, M., et al. (2020). Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply. J. Biol. Chem. 295 (14), 4383–4397. doi:10.1074/jbc.RA119.011902
Tadić, V., Adam, A., Goldhammer, N., Lautenschlaeger, J., Oberstadt, M., Malci, A., et al. (2019). Investigation of mitochondrial calcium uniporter role in embryonic and adult motor neurons from G93AhSOD1 mice. Neurobiol. Aging 75, 209–222. doi:10.1016/j.neurobiolaging.2018.11.019
Tai, X. Y., Bernhardt, B., Thom, M., Thompson, P., Baxendale, S., Koepp, M., et al. (2018). Review: Neurodegenerative processes in temporal lobe epilepsy with hippocampal sclerosis: Clinical, pathological and neuroimaging evidence. Neuropathol. Appl. Neurobiol. 44 (1), 70–90. doi:10.1111/nan.12458
Tang, Y. G., and Zucker, R. S. (1997). Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron 18 (3), 483–491. doi:10.1016/s0896-6273(00)81248-9
Tarasov, A. I., Griffiths, E. J., and Rutter, G. A. (2012). Regulation of ATP production by mitochondrial Ca(2+). Cell calcium 52 (1), 28–35. doi:10.1016/j.ceca.2012.03.003
Thayer, S. A., Usachev, Y. M., and Pottorf, W. J. (2002). Modulating Ca2+ clearance from neurons. Front. Biosci. 7, D1255–D1279. doi:10.2741/A838
Thurman, D. J., Hesdorffer, D. C., and French, J. A. (2014). Sudden unexpected death in epilepsy: Assessing the public health burden. Epilepsia 55(10), 1479–1485. doi:10.1111/epi.12666
Tian, G.-F., Azmi, H., Takano, T., Xu, Q., Peng, W., Lin, J., et al. (2005). An astrocytic basis of epilepsy. Nat. Med. 11 (9), 973–981. doi:10.1038/nm1277
Tokunaga, H., Hollenberg, N. K., and Graves, S. W. (2000). Sodium-dependent calcium release from vascular smooth muscle mitochondria. Hypertens. Res. 23 (1), 39–45. doi:10.1291/hypres.23.39
Tomar, D., Dong, Z., Shanmughapriya, S., Koch, D. A., Thomas, T., Hoffman, N. E., et al. (2016). MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 15 (8), 1673–1685. doi:10.1016/j.celrep.2016.04.050
Tsai, M. F., Phillips, C. B., Ranaghan, M., Tsai, C. W., Wu, Y., Willliams, C., et al. (2016). Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 5, e15545. doi:10.7554/eLife.15545
Ueda, S., Masutani, H., Nakamura, H., Tanaka, T., Ueno, M., and Yodoi, J. (2002). Redox control of cell death. Antioxidants Redox Signal. 4 (3), 405–414. doi:10.1089/15230860260196209
Ureshino, R. P., Erustes, A. G., Bassani, T. B., Wachilewski, P., Guarache, G. C., Nascimento, A. C., et al. (2019). The interplay between Ca(2+) signaling pathways and neurodegeneration. Int. J. Mol. Sci. 20 (23), 6004. doi:10.3390/ijms20236004
Usachev, Y. M. (2015). “Mitochondrial Ca2+ transport in the control of neuronal functions: Molecular and cellular mechanisms,” in The functions, disease-related dysfunctions, and therapeutic targeting of neuronal mitochondria. Editors V. K. G. J. M. Hardwick, and E. A. Jonas (New York: Wiley), 103–129.
Van Den Bosch, L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1762 (11), 1068–1082. doi:10.1016/j.bbadis.2006.05.002
Vanden Berghe, P., Kenyon, J. L., and Smith, T. K. (2002). Mitochondrial Ca2+ uptake regulates the excitability of myenteric neurons. J. Neurosci. 22 (16), 6962–6971. doi:10.1523/JNEUROSCI.22-16-06962.2002
Venuto, C. S., Yang, L., Javidnia, M., Oakes, D., James Surmeier, D., and Simuni, T. (2021). Isradipine plasma pharmacokinetics and exposure–response in early Parkinson’s disease. Ann. Clin. Transl. Neurology 8 (3), 603–612. doi:10.1002/acn3.51300
Verma, M., Callio, J., Otero, P. A., Sekler, I., Wills, Z. P., and Chu, C. T. (2017). Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-Associated LRRK2 mutants. J. Neurosci. 37 (46), 11151–11165. doi:10.1523/jneurosci.3791-16.2017
Verma, M., Wills, Z., and Chu, C. T. (2018). Excitatory dendritic mitochondrial calcium toxicity: Implications for Parkinson's and other neurodegenerative diseases. Front. Neurosci. 12, 523. doi:10.3389/fnins.2018.00523
Walker, M. C. (2018). Pathophysiology of status epilepticus. Neurosci. Lett. 667, 84–91. doi:10.1016/j.neulet.2016.12.044
Walters, G. C., and Usachev, Y. M. (2022). MCU (mitochondrial Ca(2+) uniporter) makes the calcium go round. J. Biol. Chem. 298 (2), 101604. doi:10.1016/j.jbc.2022.101604
Wang, C., Xie, N., Wang, Y., Li, Y., Ge, X., and Wang, M. (2015). Role of the mitochondrial calcium uniporter in rat hippocampal neuronal death after pilocarpine-induced status epilepticus. Neurochem. Res. 40 (8), 1739–1746. doi:10.1007/s11064-015-1657-3
Wang, Y., Nguyen, N. X., She, J., Zeng, W., Yang, Y., Bai, X.-c., et al. (2019). Structural mechanism of EMRE-dependent gating of the human mitochondrial calcium uniporter. Cell 177 (5), 1252–1261. e1213. doi:10.1016/j.cell.2019.03.050
Wei, A. D., Gutman, G. A., Aldrich, R., Chandy, K. G., Grissmer, S., and Wulff, H. (2005). International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol. Rev. 57 (4), 463–472. doi:10.1124/pr.57.4.9
Werth, J. L., and Thayer, S. A. (1994). Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. official J. Soc. Neurosci. 14 (1), 348–356. doi:10.1523/JNEUROSCI.14-01-00348.1994
Wimmer, V. C., Horstmann, H., Groh, A., and Kuner, T. (2006). Donut-like topology of synaptic vesicles with a central cluster of mitochondria wrapped into membrane protrusions: A novel structure-function module of the adult calyx of Held. J. Neurosci. 26 (1), 109–116. doi:10.1523/JNEUROSCI.3268-05.2006
Woods, J. J., Lovett, J., Lai, B., Harris, H. H., and Wilson, J. J. (2020). Redox stability controls the cellular uptake and activity of ruthenium-based inhibitors of the mitochondrial calcium uniporter (MCU). Angew. Chem. Int. Ed. 59 (16), 6482–6491. doi:10.1002/anie.202000247
Woods, J. J., and Wilson, J. J. (2020). Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Curr. Opin. Chem. Biol. 55, 9–18. doi:10.1016/j.cbpa.2019.11.006
Workgroup, A. s. A. C. H., and Khachaturian, Z. S. (2017). Calcium hypothesis of alzheimer's disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer's Dementia 13 (2), 178–182. e117. doi:10.1016/j.jalz.2016.12.006
Wu, S. B., Ma, Y. S., Wu, Y. T., Chen, Y. C., and Wei, Y. H. (2010). Mitochondrial DNA mutation-elicited oxidative stress, oxidative damage, and altered gene expression in cultured cells of patients with MERRF syndrome. Mol. Neurobiol. 41 (2-3), 256–266. doi:10.1007/s12035-010-8123-7
Xu, J., Mashimo, T., and Südhof, T. C. (2007). Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54 (4), 567–581. doi:10.1016/j.neuron.2007.05.004
Yang, C. H., Lee, K. H., Ho, W. K., and Lee, S. H. (2021). Inter-spike mitochondrial Ca(2+) release enhances high frequency synaptic transmission. J. Physiol. 599 (5), 1567–1594. doi:10.1113/JP280351
Yoast, R. E., Emrich, S. M., Zhang, X., Xin, P., Arige, V., Pathak, T., et al. (2021). The Mitochondrial Ca(2+) uniporter is a central regulator of interorganellar Ca(2+) transfer and NFAT activation. J. Biol. Chem. 297 (4), 101174. doi:10.1016/j.jbc.2021.101174
Zamzami, N., and Kroemer, G. (2001). The mitochondrion in apoptosis: How pandora's box opens. Nat. Rev. Mol. Cell Biol. 2 (1), 67–71. doi:10.1038/35048073
Zhong, N., Beaumont, V., and Zucker, R. S. (2001). Roles for mitochondrial and reverse mode Na+/Ca2+ exchange and the plasmalemma Ca2+ ATPase in post-tetanic potentiation at crayfish neuromuscular junctions. J. Neurosci. 21 (24), 9598–9607. doi:10.1523/JNEUROSCI.21-24-09598.2001
Zsurka, G., and Kunz, W. S. (2015). Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurology 14 (9), 956–966. doi:10.1016/S1474-4422(15)00148-9
Keywords: mitochondria, calcium, MCU, neurodegeneration, neuronal calcium homeostasis
Citation: Walters GC and Usachev YM (2023) Mitochondrial calcium cycling in neuronal function and neurodegeneration. Front. Cell Dev. Biol. 11:1094356. doi: 10.3389/fcell.2023.1094356
Received: 10 November 2022; Accepted: 12 January 2023;
Published: 24 January 2023.
Edited by:
Karthik Babu Mallilankaraman, National University of Singapore, SingaporeReviewed by:
Cecilia Hidalgo, University of Chile, ChileCopyright © 2023 Walters and Usachev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grant C. Walters, Z3JhbnQtd2FsdGVyc0B1aW93YS5lZHU=; Yuriy M. Usachev, eXVyaXktdXNhY2hldkB1aW93YS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.