 Chunmei Xiu1†
Chunmei Xiu1† Jianquan Chen
Jianquan Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 02 September 2022
Sec. Molecular and Cellular Pathology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.997838
This article is part of the Research Topic Editors' Showcase: Insights into Molecular and Cellular Pathology View all 9 articles
Hedgehog (Hh) signaling plays multiple critical roles in regulating chondrocyte proliferation and differentiation during epiphyseal cartilage development. However, it is still unclear whether Hh signaling in chondrocytes is required for growth plate maintenance during juvenile growth, and whether sustained activation of Hh signaling in chondrocytes promotes limb elongation. In this study, we first utilized Hh reporter mice to reveal that Hh signaling was activated in resting and columnar chondrocytes in growth plates of juvenile and adult mice. Next, we genetically modulated Hh signaling by conditionally deleting Smo or Sufu in all or a subpopulation of growth plate chondrocytes, and found that ablation of either Smo or Sufu in chondrocytes of juvenile mice caused premature closure of growth plates and shorter limbs, whereas Osx-Cre-mediated deletion of either of these two genes in prehypertrophic chondrocytes did not lead to obvious growth plate defects, indicating that Hh signaling mainly functions in resting and/or columnar chondrocytes to maintain growth plates at the juvenile stage. At the cellular level, we found that chondrocyte-specific ablation of Smo or Sufu accelerated or suppressed chondrocyte hypertrophy, respectively, whereas both decreased chondrocyte proliferation and survival. Thus, our study provided the first genetic evidence to establish the essential cell-autonomous roles for tightly-regulated Hh signaling in epiphyseal growth plate maintenance and limb elongation during juvenile growth.
Longitudinal skeletal growth in children is primarily orchestrated by activity of growth plates, the specialized epiphyseal cartilages sandwiched between primary spongiosa and secondary ossification center (Hallett et al., 2019; Chagin and Newton, 2020; Matsushita et al., 2020; Koyama et al., 2021). Similar to other epiphyseal cartilages, the juvenile growth plates are mainly composed of three histologically distinct layers of chondrocytes, including the top resting zone containing the progenitor/stem cells, the middle proliferating zone with flat column-forming chondrocytes, and the bottom hypertrophic zone containing terminally differentiated chondrocytes that could either undergo apoptosis or convert into osteoblasts directly or via marrow-associated skeletal stem and progenitor cells (Yang et al., 2014; Aghajanian and Mohan, 2018; Hallett et al., 2019; Long et al., 2022). However, unlike other epiphyseal cartilages, formation of juvenile growth plates is accompanied by maturation of the secondary ossification center (SOC), which could influence growth plate growth and maintenance. Indeed, bony epiphyses in the SOC were recently shown to function as a niche that generated and maintained self-renewing growth plate skeletal stem cells (gpSSCs) within the resting zone (Mizuhashi et al., 2018; Newton et al., 2019; Chagin and Newton, 2020; Kurenkova et al., 2020). Accordingly, chondrocytes in juvenile growth plates are produced from asymmetric division of gpSSCs, rather than consumption of chondroprogenitors observed in fetal/neonatal mice (Mizuhashi et al., 2018; Newton et al., 2019; Chagin and Newton, 2020). Moreover, this unique SOC niche appears to alter behaviors of growth plate chondrocytes, since growth plate chondrocytes exhibit differential responses to modulators of Hedgehog signaling before and after onset of SOC formation (Mizuhashi et al., 2018; Newton et al., 2019; Kurenkova et al., 2020). Despite these differences, our current understanding of the mechanism regulating chondrocyte proliferation and differentiation is mainly derived from genetic studies of cartilage development at fetal and neonate stages before the onset of SOC formation (Kurenkova et al., 2020; Ohba, 2020). Whether and how these mechanisms are involved in growth plate maintenance during juvenile growth remain to be specifically tested.
Indian hedgehog (Ihh) signaling plays multiple critical roles in the regulation of chondrocyte proliferation and differentiation during epiphyseal cartilage development at both embryonic and neonatal stages (St-Jacques et al., 1999; Long et al., 2001; Razzaque et al., 2005; Maeda et al., 2007; Amano et al., 2015; Ohba, 2020). Interestingly, Ihh continues to be expressed in prehypertrophic chondrocytes of the growth plates and some osteoblast-like cells at the chondro-osseous junction in juvenile mice (Maeda et al., 2007; Zhang et al., 2022). In addition, Sonic hedgehog (Shh) molecules are expressed by several types of cells residing in the SOC (Newton et al., 2019). These two Hh ligands might activate Hh signaling in the growth plate chondrocytes of juvenile mice. In agreement with this prediction, activity of Hh signaling was indeed detected in the growth plate chondrocytes in juvenile and adult mice (Shi et al., 2017; Haraguchi et al., 2018; Newton et al., 2019; Zhang et al., 2022), implying a direct requirement of Hh signaling in chondrocytes. Consistently, pharmacological inhibition of Hh signaling during or after SOC formation led to premature closure of long bone growth plates (Kimura et al., 2008), confirming the functional significance of Hh signaling in epiphyseal growth plate maintenance during juvenile growth. However, different treatment regimens appeared to exert differential cellular effects on chondrocytes or their progenitors. Treatment of mice with Smo antagonists HhAntag for 5 days from postnatal (P) 10–14 days (P10-14) or LDE225 (sonidegib) for 2 days (P22-P23) both led to impaired columnar chondrocyte proliferation and accelerated chondrocyte hypertrophy (Kimura et al., 2008; Koyama et al., 2021). In contrast, 6 administrations of Smo antagonist vismodegib at P31-P34 did not affect columnar chondrocyte proliferation and hypertrophy (Newton et al., 2019). Similarly, mice subjected to 2 doses of LDE225 (sonidegib) exhibited a decrease in the number of reserve progenitors (Koyama et al., 2021), whereas pharmacological inhibition or activation of Hh signaling (via 6 administrations of vismodegib or Smo agonist SAG at P31-P34, respectively) did not alter the number of CD73+ epiphyseal stem cells within the resting zone, but instead affect their cycling (Newton et al., 2019). Thus, while the above pharmacological studies have established the essential role of Hh signaling in juvenile growth plates, the underlying cellular mechanisms are still unclear. Furthermore, since chondrocyte-specific ablation of Smo, the gene encoding the G protein-coupled receptor Smoothened (Smo) that is essential for cells responding to Hh signals, caused early lethality and severe defects in endochondral bone development (Long et al., 2001), while genetic deletion of Smo in hypertrophic chondrocytes did not result in notable growth plate defects in juvenile mice (Wang et al., 2022). Therefore, it remains to be determined whether the effects of Hh molecules on growth plates is mediated by the direct activation of Hh signaling in chondrocytes.
Hh signaling is tightly regulated by several key regulators, one of which is suppressor of fused (Sufu) (Wu et al., 2017; Ohba, 2020). Sufu inhibits production and transcriptional activity of full-length Gli transcriptional activators, while facilitating formation of truncated Gli transcriptional repressors (Jiwani et al., 2020). Thus, through reducing Gli activator to repressor ratio, Sufu restrains excessive Hh signaling, whereas inactivation of Sufu leads to constitutively activated Hh signaling. Intriguingly, Sufu could exert either inhibitory or stimulatory effect on cell proliferation and differentiation in different tissues, suggesting that cellular effects of Sufu-mediated restraint of Hh signaling is highly context-dependent (Hsu et al., 2011; Li et al., 2017; Noguchi et al., 2019; Yung et al., 2019; Feng et al., 2020; Jiwani et al., 2020). During embryonic epiphyseal cartilage development, Sufu promotes chondrocyte proliferation, while inhibiting chondrocyte hypertrophy (Hsu et al., 2011). However, its role in growth plate chondrocytes during juvenile growth is unclear.
In this study, we explored the role of chondrocyte-specific Hh signaling in growth plate maintenance during juvenile growth. By utilizing Hh reporter mice, we revealed that Hh signaling is activated in resting and columnar chondrocytes in the epiphyseal growth plates of juvenile and adult mice. By genetically modulating Hh signaling in all or a subpopulation of growth plate chondrocytes, we demonstrated that Hh signaling mainly functions in immature chondrocytes to regulate chondrocyte turnover and hypertrophy during juvenile life in mice, and that Sufu-mediated restraint of Hh activity is critical for maintaining juvenile growth plates.
Sufu conditional knockout mice (Sufuf/f) (Li et al., 2015) were generated and kindly provided by Dr. Zunyi Zhang (Hangzhou Normal University, China). Gli1-LacZ mice, Ptc1-LacZ, Smo conditional knockout mice (Smof/f), Agc1-CreERT2, and Osx-Cre mice were described previously (Goodrich et al., 1997; Long et al., 2001; Bai et al., 2002; Rodda and McMahon, 2006; Henry et al., 2009). All animal used in this study were maintained in a standard specific pathogen-free (SPF) barrier facility at Laboratory Animal Center of Soochow University. All animal experiments were performed following the protocol approved by the Ethics Committee of Soochow University.
Tamoxifen (T5648, Sigma) was dissolved in corn oil (C8267, Sigma) at a concentration of 10 mg/ml and stored in dark place. To specifically ablate Smo or Sufu in growth plate chondrocytes of juvenile mice, 2-week-old Agc1-CreERT2; Smofl/fl mice (SmoAgc1), Agc1-CreERT2; Sufufl/fl (SufuAgc1) mice or their respective littermate controls were injected intraperitoneally with 100 mg/kg body weight of tamoxifen once daily for 5 consecutive days.
Hindlimbs were dissected from mice and fixed with 10% neutral buffered formalin (Sangon Biotech, China) for 48 h. After being thoroughly washed with PBS (pH7.4), samples were subjected to μCT scanning as previously described (Sun et al., 2022). Measurement of tibia or femur length was performed on coronal images using DataViewer software.
X-gal/LacZ staining was performed to monitor β-galactosidase (β-gal, a product of the LacZ gene) activity as previously described (Han and Feng, 2014). Briefly, tibiae from mice carrying Gli1-LacZ or Ptc1-LacZ allele were fixed with 1% PFA at 4°C overnight and decalcified with 0.1 M EDTA solution at 37°C for 3 d with gentle agitation. Subsequently, samples were processed for O.C.T. embedding and cryosectioned at 6 μm thickness. Sections were washed three times with LacZ washing buffer (PBS containing 0.02% Nonidet P-40, 0.01% sodium deoxycholate, and 2 mM MgCl2), followed by incubation with LacZ staining buffer (LacZ washing buffer supplemented with 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, and 0.5 mg/ml X-gal) at 37°C overnight. Afterwards, sections were counterstained with nuclear fast red solution, dehydrated in 75 and 100% ethanol, cleared in xylenes, and then mounted with neutral resin.
Paraffin or frozen sections of long bones were prepared as we previously described (Zhang et al., 2022). Hematoxylin and eosin (H&E) staining was performed following the standard protocol. For Safranin O/Fast green staining, sections were first stained with hematoxylin for 2 min, followed by differentiation in 1% acid alcohol. Subsequently, the samples were stained with 0.02% Fast Green (F7252, Sigma) for 2 min, rinsed with 1% acetic acid for 30 s, and then stained with 1% Safranin O (S8884, Sigma) for 30 min. Next, slides were dehydrated in graded ethanol series, cleared in xylene, applied with one drop of neutral balsam mounting medium, and finally covered with a coverslip. Images were acquired using a Zeiss Axio Imager Z2 upright microscope (Carl Zeiss Microscopy).
To detect the apoptotic cells, TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining was performed on paraffin sections of long bones using a commercial kit (In Situ Cell Death Detection Kit, TMR Red; Roche, Mannheim, Germany) according to the manufacturer’ protocol. Briefly, tibial sections were de-paraffinized, rehydrated, and then permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate at room temperature for 8 min. Afterwards, slides were incubated with TUNEL reaction mixture (9:1 mixture of label solution and enzyme solution provided by the kit) at 37°C for 1 h. After washing twice with phosphate buffer solution (PBS), samples were counter-stained with DAPI (C1005, Beyotime Biotechnology, Shanghai, China). Finally, slides were mounted with anti-fade mounting medium and analyzed under a fluorescence microscope.
For BrdU (5-bromo-2′-deoxyuridine) labelling, mice were intraperitoneally injected with 10 μl per gram body weight of BrdU labeling reagent (#000103, Invitrogen) 6 h before being sacrificed. The hindlimbs were then harvested and processed for paraffin sections. To detect BrdU-labeled proliferating cells, tibial sections were dewaxed, rehydrated, and then denatured with 2 N HCl, prior to antigen retrieval with 0.125% trypsin in PBS at 37°C for 10 min. After blocking with 10% goat serum for 1 h, sections were incubated with rat anti-BrdU antibody (ab6326, Abcam, 1:100) at 4°C overnight, followed by incubation with Alexa Fluor 647-conjugated donkey anti-rat (ab150155, Abcam,1:200) or Alexa Fluor 488-conjugated goat anti-rat secondary antibody (ab150157, Abcam, 1:200). Finally, nuclei were counterstained with DAPI (C1005, Beyotime Biotechnology, Shanghai, China) to stain the cell nuclei, and subsequently mounted with anti-fade mounting medium.
To assess ALP activity, ALP staining was performed on frozen sections of tibias. Briefly, tibial sections were washed three times with PBS, and then incubated with 0.1 M Tris-HCl solution (pH8.5) containing 0.1 mg/ml naphthol AS-MX phosphate, 0.5% N, N-dimethylformamide, 2 mM MgCl2, and 0.6 mg/ml of fast blue BB salt for 20–60 min. Next, sections were dehydrated in graded ethanol series, cleared in xylene, applied with one drop of neutral balsam mounting medium, and finally covered with a coverslip. Images were acquired using a Zeiss Axio Imager Z2 upright microscope (Carl Zeiss Microscopy).
IF was performed on frozen sections. To unmask antigen epitopes for Collagen II, Collagen X, and Mmp13 antibodies, section samples were pretreated with 2 mg/ml hyaluronidase (Sigma, H3506) at 58°C for 2 h. After antigen retrieval, sections were incubated with 10% goat serum in PBS to block nonspecific binding, followed by incubation with primary antibodies in a humidified chamber at 4°C overnight. On the next day, Cy3-conjugated goat anti-rabbit secondary antibody (GB21303, Servicebio, 1:400) were applied to detect primary antibodies, and then DAPI was used to stain cell nuclei.
IHC was carried out on paraffin sections following the standard protocol. Briefly, paraffin sections were deparaffinized, rehydrated, and then subjected to antigen retrieval as described above. Subsequently, 3% hydrogen peroxide in methanol was used to quench endogenous peroxidase activity, prior to blocking nonspecific antigens with 10% goat serum. Slides were then incubated with properly diluted primary antibodies in a humidified chamber at 4°C overnight, followed by incubation with HRP-conjugated goat anti-rabbit secondary antibody (D110058-0025, BBI Life Science, 1:400). For visualizing antibody binding, sections were covered with 3,3′-diaminobenzidine (DAB) freshly-prepared from a DAB substrate kit (ZLI-9018, ZSGB-BIO, Beijing, China) until brown color developed. The stained sections were counterstained with hematoxylin solution, dehydrated in ethanol, cleared in xylene, and finally sealed with a coverslip.
Primary antibody against Collagen II was purchased from Fitzgerald (70R, 1:200). Primary antibodies against Collagen X (ab58632, 1:750) and Mmp13 (ab39012, 1:200) were both obtained from Abcam. Images were acquired using a Zeiss Axio Imager Z2 upright microscope (Carl Zeiss Microscopy).
Data plotting and statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA). Data were plotted as dot-plots showing individual data points together with mean ± standard deviation. Each data dot represents the value from one mouse. Comparison of means between two groups and among multiple groups were assessed by Student’s t-test and by one-way ANOVA with Tukey’s post-hoc test, respectively. In both cases, a p value <0.05 was considered to be statistically significant, and degrees of statistical significance were further designated by different numbers of asterisk (*) in the figures: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
We previously showed that Hh signaling was activated in the chondrocytes of epiphyseal growth plates from 2- and 12-month-old mice (Zhang et al., 2022). To further characterize Hh-responsive cells in the epiphyseal growth plates during postnatal skeletal growth, we first examined the expression of Gli1, a transcriptional target of Hh signaling activity in juvenile and adult mice. We utilized Gli1-LacZ knock-in reporter mice, in which the bacterial LacZ gene is controlled by the endogenous Gli1 locus, and therefore its activity can be used as a readout of Gli1 expression. LacZ staining of tibial sections from two-week-old Gli1-LacZ reporter mouse revealed strong Gli1-LacZ activity in nearly all chondrocytes in the columnar proliferative zone (PZ) (Figure 1A). Similarly, the vast majority of chondrocytes within the resting zone (RZ), a region that contains growth plate skeletal stem cells, were also LacZ-positive, although the LacZ signal in these cells tended to be slightly weaker than columnar chondrocytes at this stage ((Figure 1A). In contrast, the hypertrophic zone (HZ) exhibited significantly less LacZ-positive cells as well as weaker LacZ signal in these positive cells (Figure 1A). The similar LacZ expression patterns were also observed in the tibial growth plates of 1-, 2-, and 4-month-old Gli1-LacZ mice (Figures 1B–D). As the epiphyseal growth plates matured from 2 weeks to 4 months of age, the numbers of LacZ-positive chondrocytes in both resting and columnar zones tended to decrease gradually (Figures 1A–D). To confirm the distribution of Hh-responsive cells in the epiphyseal growth plates, we then monitored the expression of Ptc1, another target gene of Hh signaling, using Ptc1-LacZ knock-in reporter mice. Similar to the results from Gli1-LacZ, strong LacZ activity was detected in most of columnar chondrocytes and some resting chondrocytes, whereas almost no LacZ expression was observed in hypertrophic zones, in the tibial growth plates from 1-and 4-month-old Ptc1-LacZ mice (Figures 1E,F). Collectively, these results demonstrated that the columnar and resting chondrocytes are major Hh-responsive cells in the epiphyseal growth plates of juvenile and adult mice.
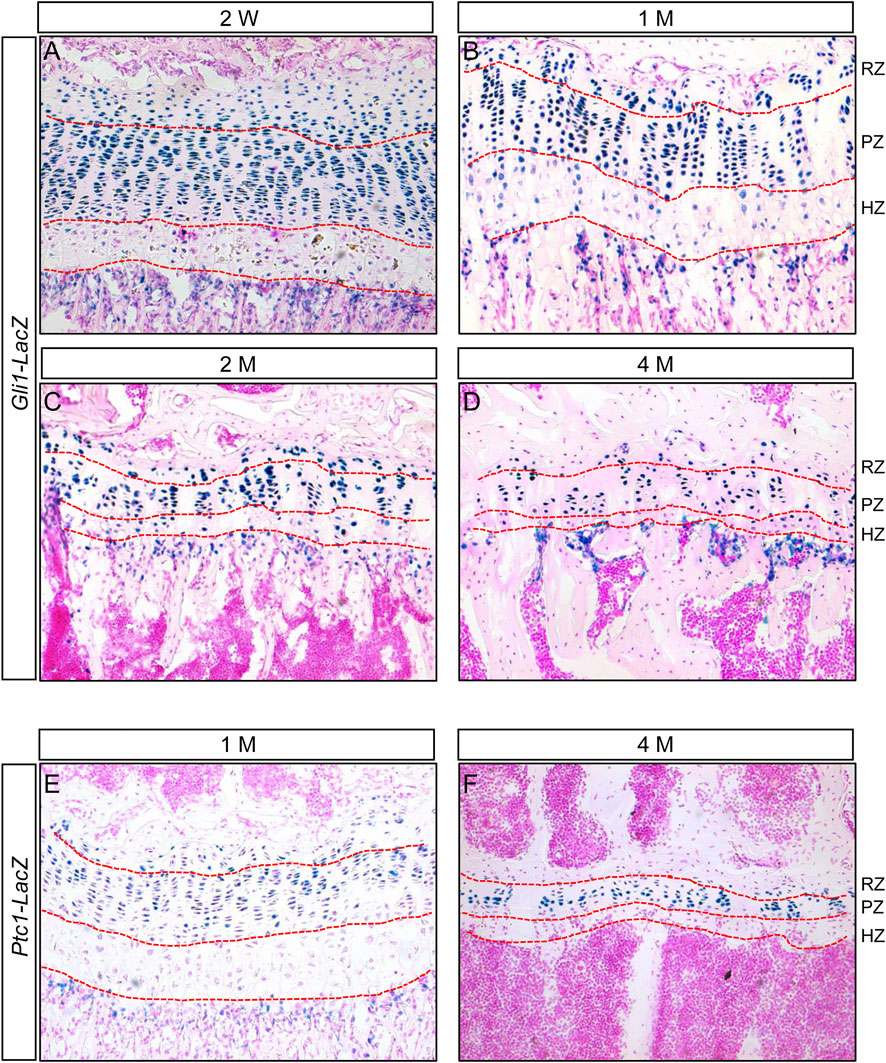
FIGURE 1. Hh signaling is active in columnar and resting chondrocytes, but downregulated in hypertrophic chondrocytes, in the epiphyseal growth plates of juvenile and adult mice (A–D) LacZ staining of longitudinal tibial sections from 2-week-old (A), 1-month-old (B), 2-month-old (C), and 4-month-old (D) Gli1-LacZ reporter mice (E,F) LacZ staining of longitudinal tibial sections from 1-month-old (E) and 4-month-old (F) Ptc1-LacZ reporter mice. RZ, resting zone; PZ, proliferative zone, HZ, hypertrophic zone.
Since Hh signaling is activated in growth plate chondrocytes of juvenile and adult mice, we next investigated whether Hh-responsiveness in these cells was required for maintenance of epiphyseal growth plates after onset of SOC formation. Smoothened (Smo) is a G protein-coupled receptor essential for cells to respond to Hh signals. We therefore blocked Hh responsiveness in chondrocytes of juvenile mice by genetically removing Smo gene with tamoxifen-inducible Agc1-CreERT2 allele. By using the R26-tdTomato reporter mice, we first confirmed that Agc1-CreERT2 can efficiently target growth plate chondrocytes (Supplementary Figure S1), when induced in mice at 2 weeks of ages, a stage when the SOC has largely formed. We then used tamoxifen to induce Cre activity in 2-week-old Agc1-CreERT2; Smofl/fl (hereafter SmoAgc1) mice and examined the effects of Smo deletion on epiphyseal growth plates. μCT analyses of tamoxifen-induced SmoAgc1 mice identified obvious defects in the growth plates of the proximal tibiae and distal femurs. At P30 (12 days after tamoxifen injections), the growth plates were clearly visible in radiographical images of tibiae and femurs from both SmoAgc1 and control mice (Figures 2A,B). However, their sizes appeared to be significantly reduced in mutant mice, when compared to control mice (Figures 2A,B). By P120, the growth plates from mutant mice exhibited a variable degree of defects. In the most severely-affected mutants, the growth plates of tibiae and femurs were both prematurely fused (Figures 2C,D). By contrast, the growth plates from control mice were still recognizable at this stage (Figure 2C), although became narrower compared to those at P30. To confirm these findings, we next performed histological analyses of long bones from SmoAgc1 and control mice at P30 and P120 (12 and 102 days from the last dose of tamoxifen, respectively). Safranin O staining revealed that Smo deletion progressively impaired morphology and organization of growth plate chondrocytes in both tibiae and femurs (Figures 2E–J). Proliferating chondrocytes in control mice were arranged in longitudinal columns at P30, whereas this columnar organization of chondrocytes was notably disrupted at P30. Moreover, the composition of hypertrophic layer of chondrocytes were significantly reduced at this stage (Supplementary Figure S2). By P120, the integrity of growth plates in mutant mice was severely impaired (Figures 2K–N). In particular, the growth plates in distal femurs from mutant mice were all prematurely fused. Some growth plates from proximal tibiae were also similarly affected. Collectively, these results demonstrated that Hh signaling is cell-autonomously required for maintenance of the epiphyseal growth plates in juvenile mice.
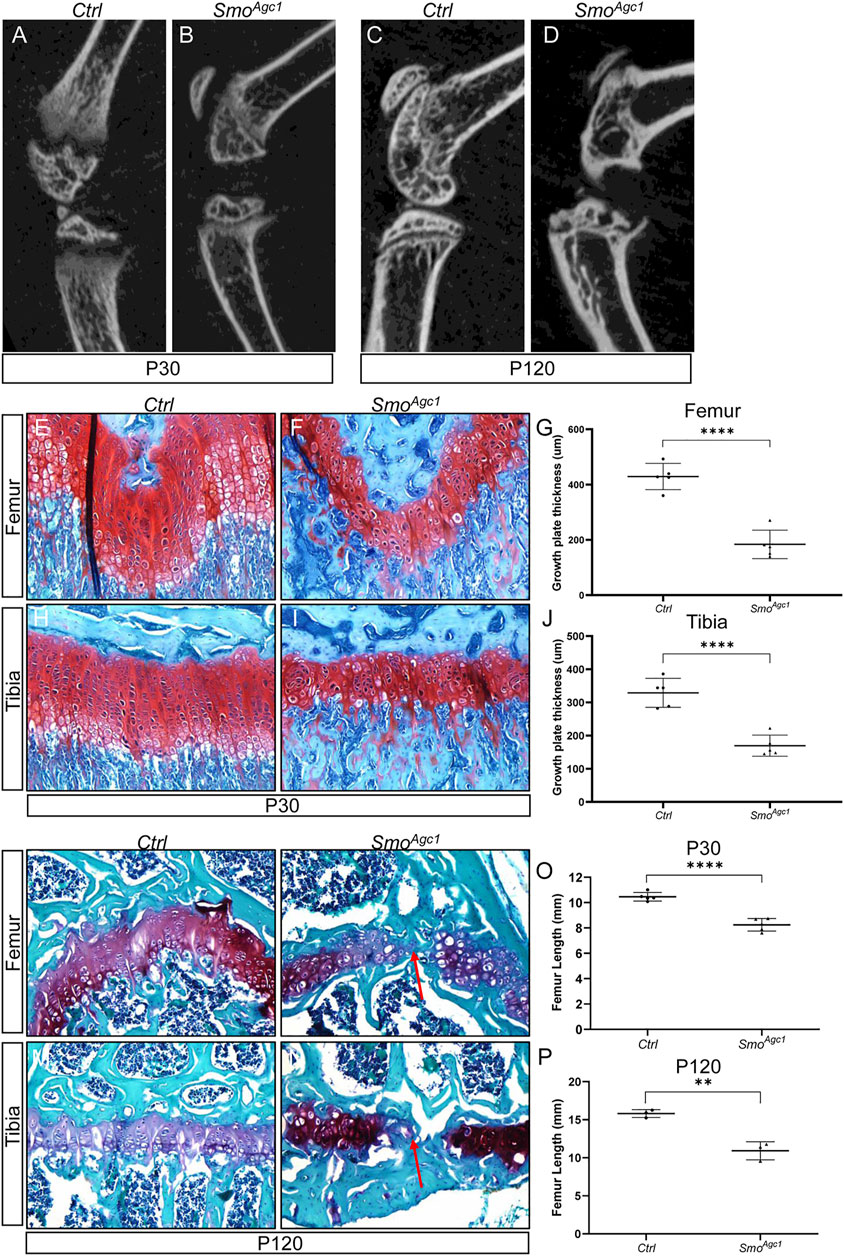
FIGURE 2. Chondrocyte-specific deletion of Smo in juvenile mice led to defects in epiphyseal growth plate and limb elongation (A–D) Representative uCT slices of the knee joint region in Agc1-CreERT2; Smof/f (SmoAgc1) (A,C) and control mice (Ctrl) (B,D) treated with tamoxifen at P14-P18 and harvested at P30 (A,B) or P120 (C,D) (E,F) Safranin O staining of distal femoral growth plates from P30 Ctrl and SmoAgc1 mice (G) Quantification of average thickness of distal femoral growth plates (H,I) Safranin O staining of proximal tibial growth plates from P30 Ctrl and SmoAgc1 mice (J). Quantification of average thickness of proximal tibial growth plates (K–N) Safranin O staining of growth plates in distal femurs (K,L) or proximal tibiae (M,N) from P120 Ctrl and SmoAgc1 mice. Red arrows indicated the fusion of the growth plates in the mutant distal femurs and proximal tibiae (O,P). Measurement of average lengths of femurs from Ctrl and SmoAgc1 mice at P30 (O) and P120 (P). All quantitative data were shown as individual data points from each mouse together with mean ± standard deviation. p values were obtained by Student’s t-test and degrees of statistical significance were designated by different numbers of asterisk (*) in the figures: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
To determine whether the changes in growth plate caused by Smo deletion had detrimental consequences on overall limb growth and elongation, we performed μCT-based analyses of femur length of SmoAgc1 and control mice at different ages. At P30, femurs of mutant mice were shortened by 21%, when compared to those of control littermates (Figure 2O). By 120, this reduction in femur length was further enlarged to 30% (Figure 2P). Together, our results indicated that disruption of Hh signaling in growth plate chondrocytes of juvenile mice not only caused growth plate defects, but also impaired limb elongation.
Chondrocytes proliferation and survival are important contributors of growth plate maintenance in juvenile mice. To assess how deletion of the Smo in juvenile chondrocytes caused the growth plate defects, we first examined status of chondrocyte proliferation in Smo mutant mice at 12 days after tamoxifen administration. BrdU staining of tibial sections revealed abundant BrdU-positive chondrocytes in the growth plates of control mice (Figure 3A). By contrast, only a few proliferating cells were detected in the mutant growth plates at this stage (Figure 3B). Histomorphometric analysis confirmed that the number of proliferative chondrocytes, when normalized to areas of growth plates, was dramatically decreased in mutant growth plate when compared with controls (Figure 3C). Since a morphological recognizable columnar zone was largely absent in the growth plates of P30 mutants (Figure 2), it was infeasible to calculate the percentage of BrdU-positive columnar chondrocytes at this stage. Therefore, we examined chondrocyte proliferation at an earlier time when the mutant growth plates still contained the morphologically distinct columnar zones. The results showed that the percentage of BrdU-positive chondrocytes in the columnar region were significantly reduced in P21 SmoAgc1 mice when compared to their littermate controls (Figures 3D–F). Collectively, these results demonstrated that Hh signaling is cell-autonomously required for chondrocyte proliferation.
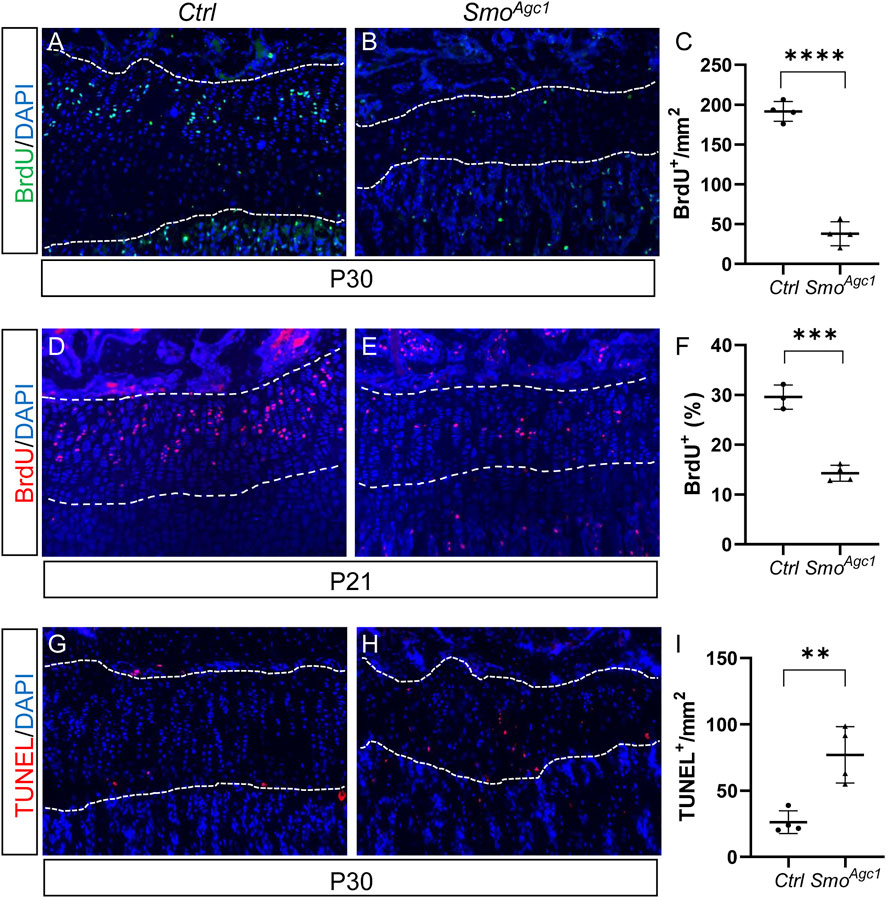
FIGURE 3. Chondrocyte-specific deletion of Smo in juvenile mice led to reduced chondrocyte proliferation and increased chondrocyte apoptosis (A,B) BrdU staining of proximal tibial growth plates from P30 Ctrl (A) and SmoAgc1 mice (B). BrdU-labeled cells were shown in green and nuclei were stained in blue (C) Quantification of BrdU-positive cells normalized to areas of proximal tibial growth plates in P30 Ctrl and SmoAgc1 mice (D,E) BrdU staining of proximal tibial growth plates from P21 Ctrl (D) and SmoAgc1 mice (E). BrdU-labeled cells were shown in red and nuclei were stained in blue (F) Quantification of the percentage of BrdU-positive cells in columnar zones of proximal tibial growth plates from P21 Ctrl and SmoAgc1 mice (G,H) TUNEL assay of proximal tibial growth plates from P30 Ctrl (G) and SmoAgc1 mice (H). Apoptotic cells were shown in red and nuclei were counterstained in blue (I) Quantification of TUNEL-positive cells normalized to areas of proximal tibial growth plates in P30 Ctrl and SmoAgc1 mice. All mice were treated with 5 daily doses of tamoxifen starting at P14. All quantitative data were shown as individual data points from each mouse together with mean ± standard deviation. p values were obtained by Student’s t-test and degrees of statistical significance were designated by different numbers of asterisk (*) in the figures: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Next, we evaluated whether increased chondrocyte apoptosis contributed to growth plate abnormality. To this end, we performed TUNEL assays on tibial sections from SmoAgc1 and control mice at 12 d after tamoxifen administration. In line with the fact that apoptosis normally occurs in terminally differentiated hypertrophic chondrocytes, TUNEL-positive cells were detected at the chondro-osseous junction but were largely absent in other regions of the growth plate in control mice (Figure 3G). By contrast, apoptotic cells were observed not only at the chondro-osseous junction, but also in the middle of growth plates in mutants (Figure 3H). Quantitative analysis confirmed an increase in number of TUNEL-positive cells in the growth plates of mutant mice, when compared to control mice (Figure 3I). Thus, the growth chondrocytes in juvenile mice, unlike their counterparts at embryonic stages, requires direct Hh input to maintain their survival.
Postnatal growth plate is maintained not only by chondrocyte proliferation and survival, but also by chondrocyte differentiation. We therefore investigated whether chondrocyte differentiation was altered by Smo deletion. Histological analysis of the proximal tibial growth plates of SmoAgc1 mice revealed that almost no chondrocytes with enlarged morphology were detected at P30 (Figures 4A,B). Immunofluorescent staining showed that nearly all chondrocytes in the mutant growth plates expressed collagen type Ⅱ (Col II) and collagen type X (Col X) proteins (Figures 4C–F), the latter of which is normally restricted to the lower hypertrophic zone of the growth plates. Moreover, ALP, a marker normally expressed by all stages of hypertrophic chondrocytes, was ectopically expressed in chondrocytes throughout the growth plates in mutant mice (Figures 4G,H). In contrast, expression of Mmp13, a marker for terminal differentiated hypertrophic chondrocytes, was similar between controls and mutants (Figures 4I,J). Together, these results demonstrated that postnatal deletion of Smo from chondrocytes accelerated earlier steps of hypertrophic differentiation, but did not affect terminal stages of chondrocyte hypertrophy.
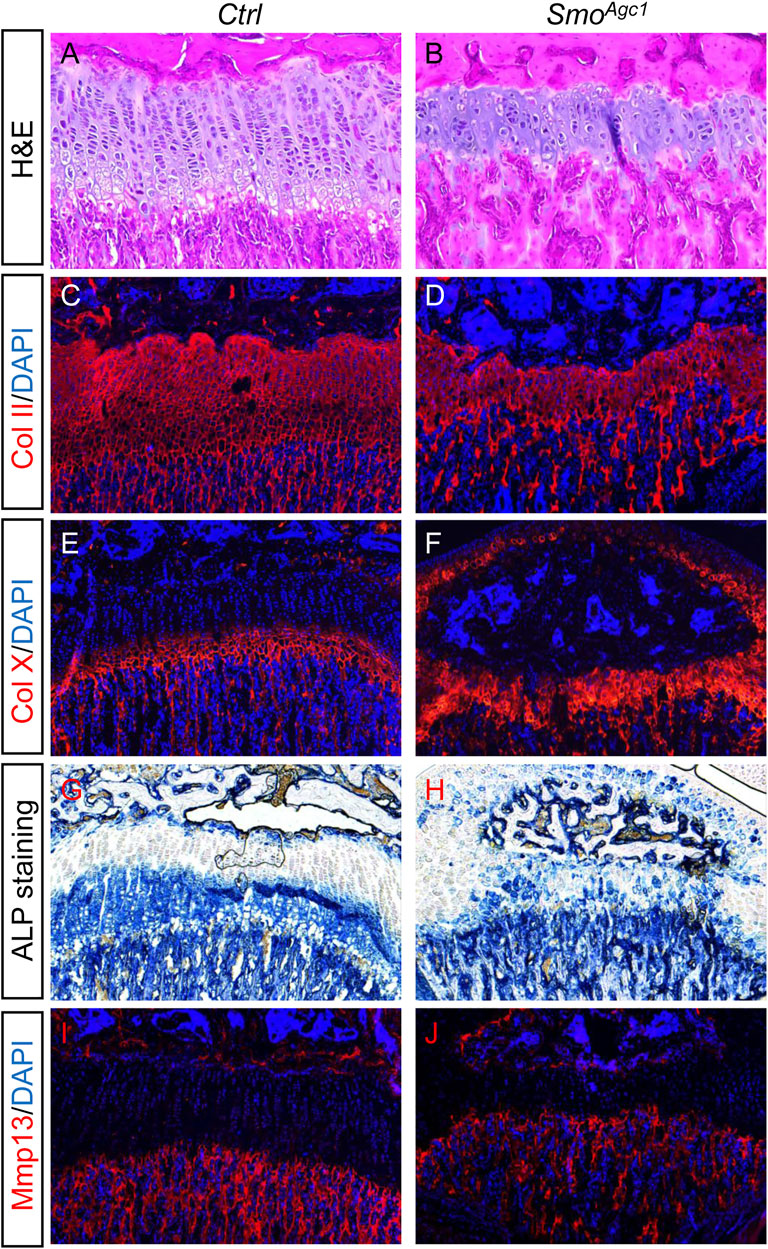
FIGURE 4. Chondrocyte-specific deletion of Smo in juvenile mice led to premature hypertrophic differentiation (A,B) H&E staining of the proximal tibial growth plates from P30 Ctrl (A) and SmoAgc1 mice (B) (C–F) Immunofluorescent staining of collagen type Ⅱ (Col II) (C,D) and collagen type X (Col X) (E,F) on frozen sections of the proximal tibial growth plates from P30 Ctrl (C,E) and SmoAgc1 (D,F) mice (G,H) ALP staining of the proximal tibial growth plates from P30 Ctrl (G) and SmoAgc1 mice (H) (I,J) Immunofluorescent staining of Mmp13 on frozen sections of the proximal tibial growth plates from P30 Ctrl (I) and SmoAgc1 (J) mice. All mice were treated with 5 daily doses of tamoxifen starting at P14.
Hh signaling is normally tightly regulated, whereas either inactivation or hyperactivation could lead to defects in tissue development and homeostasis. Therefore, we next tested whether restraint of Hh activity level was critical for epiphyseal growth plate maintenance and limb elongation in juvenile mice. To this end, we conditionally deleted Sufu, a negative regulator of Hh signaling, to forcedly activate Hh signaling in growth plate chondrocytes of juvenile mice. We administered 5 daily doses of tamoxifen to Agc1-CreERT2; Sufufl/fl (hereafter SufuAgc1) mice starting at P14 and performed radiographic and histological analyses of epiphyseal growth plates at P30 and P120. μCT imaging of tibiae and femurs revealed that the growth plates were abnormally formed in SufuAgc1 mice (Figures 5A–D). At P30, the growth plates of tibia and femurs in mutant mice, as shown by the translucent area between primary and secondary ossification centers in the μCT images, were obviously expanded compared with control mice (Figures 5A,B). However, by P120, the expansion of growth plate was no longer observed in radiographical images of mutant mice that instead exhibited the complete or partial closure of growth plates (Figures 5C,D). To confirm these observations, we next performed histological analyses. Safranin O staining showed that Sufu-deficient mice had enlarged growth plates of both tibiae and femurs, compared to wild-type littermates at P30 (Figures 5E–J). In addition, the morphology and organization of the growth plates in mutant mice were strikingly different from controls. The control growth plates were composed of three morphologically distinct zones of chondrocytes: the round chondrocytes at the top resting zone, the flat column-forming chondrocytes in the middle zone, and the hypertrophic chondrocytes at the bottom zone (Figures 5E,H). By contrast, this orderly structure was lost in the mutant growth plate, which was instead occupied by a poorly-organized mass of chondrocytes with heterogenous morphology (Figures 5F,I). These changes appeared to negatively affect growth plates in the long term. At P120, the growth plates from mutant mice were prematurely closed, as evidenced by a significant loss of Safranin O-stained cartilages in the mutant growth plates (Figures 5K–N; Supplementary Figure S3). As a control, morphologically intact growth plates remained evident in both tibiae and femurs from tamoxifen-injected control mice (Figures 5K–N; Supplementary Figure S3). Thus, chondrocyte-specific deletion of Sufu in juvenile mice caused a transient expansion of growth plate followed by its premature fusion.
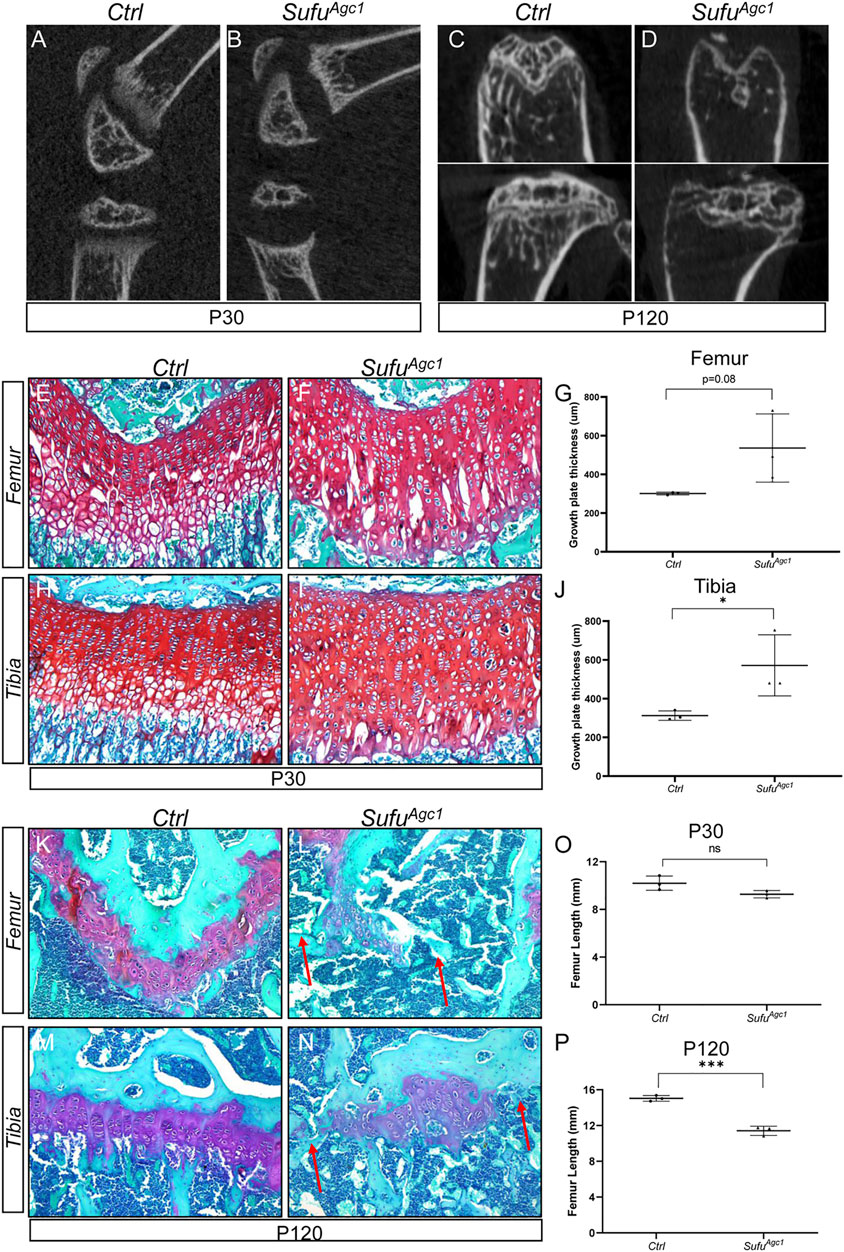
FIGURE 5. Chondrocyte-specific deletion of Sufu in juvenile mice caused a transient expansion of growth plates followed by their premature fusion (A–D) Representative uCT slices of growth plates in Agc1-CreERT2; Sufuf/f (SufuAgc1) (A,C) and control mice (Ctrl) (B,D) at P30 (A,B) or P120 (C,D) (E,F) Safranin O staining of distal femoral growth plates from P30 Ctrl and SufuAgc1 mice (G) Quantification of average thickness of distal femoral growth plates from P30 Ctrl and SufuAgc1 mice (H,I) Safranin O staining of proximal tibial growth plates from P30 Ctrl and SufuAgc1 mice (J) Quantification of average thickness of proximal tibial growth plates from P30 Ctrl and SufuAgc1 mice (K–N) Safranin O staining of growth plates in distal femurs (K,L) or proximal tibiae (M,N) from P120 Ctrl and SufuAgc1 mice. Red arrows indicated the fusion of the growth plates in the mutant distal femurs and proximal tibiae (O,P). Measurement of average lengths of femurs from Ctrl and SufuAgc1 mice at P30 (O) and P120 (P). All quantitative data were shown as individual data points from each mouse together with mean ± standard deviation. p values were obtained by Student’s t-test and degrees of statistical significance were designated by different numbers of asterisk (*) in the figures: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
To evaluate the consequences of growth plates defects in Sufu-deficient mice on their overall limb growth and elongation, we also utilized μCT images to measure length of femurs from SufuAgc1 and control mice at P30 and P120. The results showed that femur length of P30 SufuAgc1 mice was slightly reduced when compared with those of control littermates, although this difference did not reach the statistical significance (Figure 5O). Moreover, femurs from mutant mice became significantly shorter than those of control mice by P120 (Figure 5P), indicating that loss of Sufu impaired limb elongation. Together, these results indicated that Sufu-mediated restraint of Hh activity level is critical for epiphyseal growth plate maintenance and limb elongation in juvenile mice.
To assess the effect of Sufu deletion on chondrocyte proliferation, we performed BrdU assays at 12 d after tamoxifen injection. As expected, BrdU-positive proliferating chondrocytes were mainly localized in the columnar zone of the control growth plates (Figure 6A). By contrast, these cells were scattered across the entire growth plates from mutant mice (Figure 6B). Surprisingly, despite the remarkably increased numbers of total chondrocytes, the number of proliferative chondrocytes were significantly decreased in mutant growth plate compared to the controls (Figure 6C). Consequently, the rate of chondrocyte differentiation, as calculated by the percentage of proliferative chondrocytes out of total immature chondrocytes, was significantly reduced in Sufu-deficient mice (Figure 6D), indicating that Sufu-mediated restraint of Hh signaling in chondrocytes is critical for their proliferation. Next, we performed TUNEL assays on tibial sections from P30 SmoAgc1 and control mice to evaluate the impact of Sufu deletion on chondrocyte apoptosis. As expected, apoptotic cells were mainly localized at the border between the growth plate and primary spongiosa in control mice (Figure 6E). In contrast, TUNEL-positive cells were distributed throughout the enlarged growth plates in mutants (Figure 6F). Quantitative analysis further revealed that the number of TUNEL-positive chondrocytes was strikingly induced by Sufu deletion (Figure 6G), indicating that activity of Hh signaling in chondrocytes needs to be tightly restrained by Sufu for their survival. Collectively, these results demonstrated that Sufu-mediated restraint of Hh signaling in chondrocytes plays an essential role in promoting chondrocyte proliferation and suppressing chondrocyte apoptosis in juvenile mice.
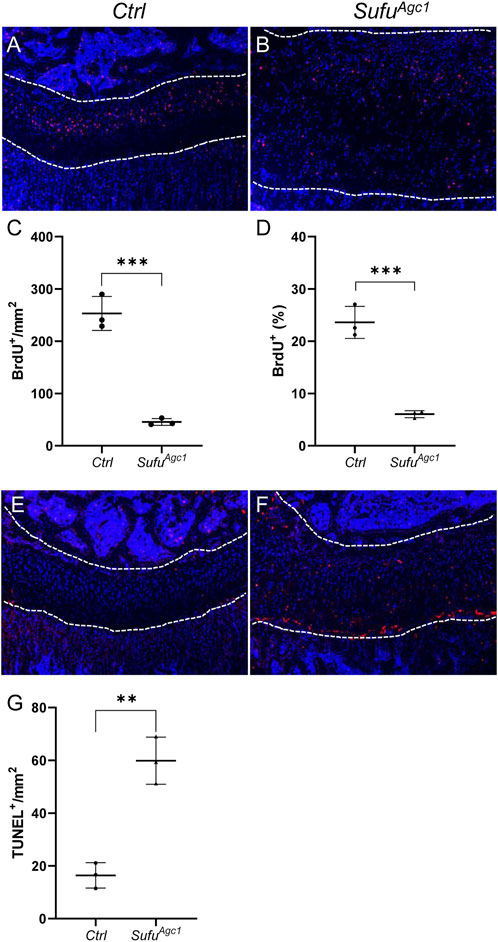
FIGURE 6. Chondrocyte-specific deletion of Sufu in juvenile mice impaired chondrocyte proliferation and promoted cell apoptosis (A,B) BrdU staining of proximal tibial growth plates from P30 Ctrl (A) and SufuAgc1mice (B). BrdU-labeled cells were shown in red and nuclei were stained in blue (C,D) Quantification of the number (C) and percentage (D) of BrdU-positive cells in resting and columnar zones of proximal tibial growth plates from P30 Ctrl and SufuAgc1 mice (E,F) TUNEL assays of proximal tibial growth plates from P30 Ctrl (E) and SufuAgc1 mice (F). Apoptotic cells were shown in red and nuclei were counterstained in blue (G) Quantification of TUNEL-positive cells normalized to areas of proximal tibial growth plates in P30 Ctrl and SufuAgc1 mice. All mice were treated with 5 daily doses of tamoxifen starting at P14. All quantitative data were shown as individual data points from each mouse together with mean ± standard deviation. p values were obtained by Student’s t-test and degrees of statistical significance were designated by different numbers of asterisk (*) in the figures: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Next, we evaluated the status of chondrocyte differentiation in Sufu-deficient mice. H&E staining of tibial growth plates revealed that although the mutant growth plates have significantly more cells than controls, they did not appear to form morphologically distinct hypertrophic chondrocytes (Figures 7A,B), suggesting that chondrocyte-specific deletion of Sufu could suppress formation of hypertrophic chondrocytes. Furthermore, consistent with less BrdU-positive proliferating cells, no obvious increase in the number of flat column-forming cells was observed in the mutant growth plates (Figures 7A,B). Histomorphometrical analyses of tibial growth plates revealed that Sufu ablation led to increased composition of resting layers of chondrocytes, but reduced composition of hypertrophic zones without significantly altering the portion of columnar chondrocytes (Supplementary Figure S4). To confirm these observations at the molecular levels, we performed immunostaining of longitudinal sections from tibias harvested at P30 to examine the expression of chondrocyte differentiation markers. Growth plates from control mice exhibited high level of ColII protein in proliferative zones, but significantly lower level in hypertrophic zones (Figure 7C). In comparison, the mutant growth plates exhibited strong and uniform expression of Col II in the entire growth plate (Figure 7D). On the other hand, both IHC and IF staining showed that the number of cells expressing ColX was remarkably reduced in mutant growth plates when compared to controls (Figures 7E–H). Collectively, these morphological and molecular analyses demonstrated that postnatal deletion of Sufu from chondrocytes inhibited chondrocyte hypertrophy. However, unlikely ColⅡ and ColX, Mmp13 was similarly detected in the last row of the hypertrophic zone of growth plates from both controls and mutants (Figures 7I,J), indicating that terminal differentiation of chondrocytes was not altered by Sufu deletion. Thus, the expansion of the growth plate observed in Sufu-deficient mice is due to accumulation of immature chondrocytes partly attributable to impaired chondrocyte hypertrophy.
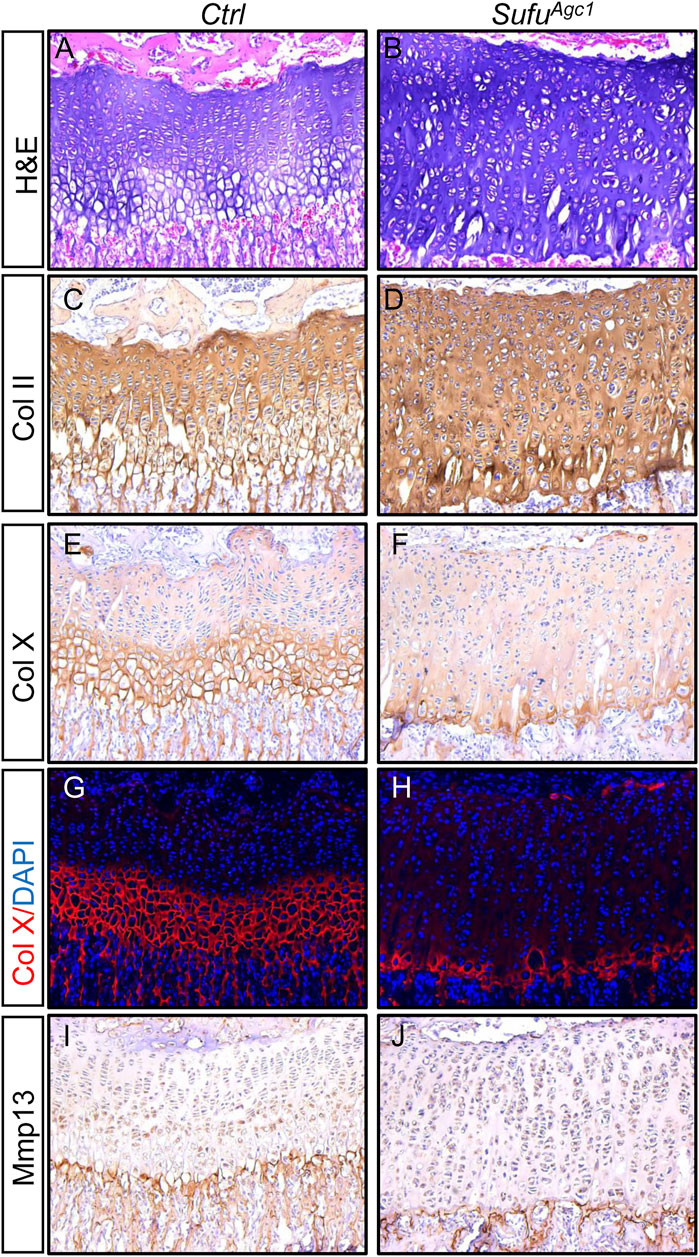
FIGURE 7. Chondrocyte-specific deletion of Sufu in juvenile mice inhibited hypertrophic differentiation (A,B) H&E staining of the proximal tibial growth plates from P30 Ctrl (A) and SufuAgc1 mice (B) (C–F) Immunohistochemical staining of collagen type Ⅱ (Col II) (C,D) and collagen type X (Col X) (E,F) on frozen sections of the proximal tibial growth plates from P30 Ctrl (C,E) and SufuAgc1 (D,F) mice (G,H) Immunofluorescent staining of Col X on frozen sections of the proximal tibial growth plates from P30 Ctrl (G) and SufuAgc1 (H) mice (I,J) Immunohistochemical staining of Mmp13 on frozen sections of the proximal tibial growth plates from P30 Ctrl (I) and SufuAgc1 (J) mice. All mice were treated with 5 daily doses of tamoxifen starting at P14 and analyzed at indicate time.
Thus far, we have demonstrated that undisturbed Hh signaling in chondrocytes is essential for growth plate maintenance in juvenile mice. In contrast, a recent study showed that modulating Hh signaling by conditionally ablating either Smo or Ptch1 in Col10a1-expressing hypertrophic chondrocytes did not impair growth plate cartilage (Wang et al., 2022), suggesting that Hh signaling might mainly function in immature chondrocytes. To further explore this possibility in our hand, we utilized Osx-Cre to ablate Smo or Sufu in prehypertrophic/hypertrophic chondrocytes, since we have previously showed that Osx-Cre can efficiently mediate recombination in these cells in postnatal growing mice (Zhang et al., 2022). In line with the finding by Wang et al., histological analyses of tibiae from 4-month-old Osx-Cre; Smof/f (SmoOsx) and their littermate controls revealed that Osx-Cre-mediated ablation of Smo did not overtly affect tibial growth plates (Figures 8A–D). Similarly, the enlarged or prematurely fused growth plates was not observed in tibial growth plates from 3-month-old Osx-Cre; Sufuf/f (SufuOsx) mice (Figures 8E–H). Together, our results demonstrated that Hh signaling mainly functions in immature chondrocytes, not but in hypertrophic chondrocytes, to maintain growth plates at the juvenile stage.
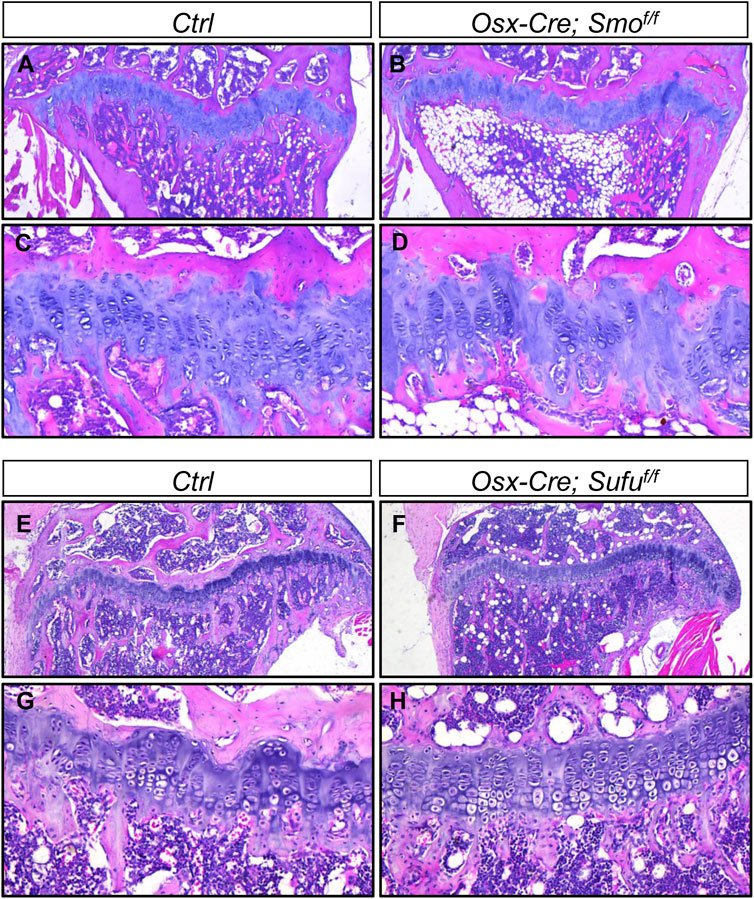
FIGURE 8. Osx-Cre-mediated ablation of either Smo or Sufu in hypertrophic chondrocytes did not overtly affect growth plates (A–D) H&E staining of the proximal tibial growth plates from P120 Ctrl (A,C) and Osx-Cre; Smof/f mice (B,D) shown at low magnification (A,B) and higher magnification (C,D) (E–H) H&E staining of the proximal tibial growth plates from P90 Ctrl (E,G) and Osx-Cre;Sufuf/f mice (F,H) shown at low magnification (E,F) and higher magnification (G,H).
Genetic studies have established the cell-autonomous and cell-non-cell-autonomous roles of Hh signaling in regulating epiphyseal cartilage development (Ohba, 2020). However, a chondrocyte-specific requirement of Hh signaling for growth plate maintenance during juvenile growth was yet to be established. In the present study, we utilized genetic approaches to specifically inactivate or activate Hh signaling in growth plate chondrocytes of juvenile mice, and made two major findings:1) Hh signaling functions in immature chondrocytes to promote chondrocyte proliferation and survival, while inhibiting chondrocyte hypertrophy during juvenile life in mice; 2) Sufu-mediated restraint of Hh activity is critical for maintaining juvenile growth plates. This study provided the first genetic evidence to establish the essential cell-autonomous role of fine-tuned Hh signaling in regulating multiple aspects of growth plate chondrocytes in juvenile mice.
Previous studies have shown that the tibial growth plates were essentially closed at 8 days after 2 doses of LDE225 (administered daily from P22 to P23) (Koyama et al., 2021). By contrast, we found that Agc1-CreERT2-mediated ablation of Smo in growth plates of 2-week-old mice only led to partial fusion of tibial growth plates by P120. The cause why transient inhibition of Hh signaling caused more deleterious effect on tibial growth plates than permanent ablation of Smo in chondrocytes is still unclear. One possibility is that Agc1-CreERT2, when induced in mice at 2 weeks of age, was not efficient to ablate Smo in growth plate chondrocytes. As a result, Smo inhibitors suppressed Hh signaling activity in chondrocytes to a greater degree than Smo ablation, therefore leading to more severer growth plate defects. Alternatively, this difference may reflect the important role of Hh signaling in regulating chondrocyte production from chondroprogenitors outside the growth plate. In line with this possibility, Axin2-positive perichondrial cells in the Ranvier’s groove (RG) were shown to contribute to the growth plates during juvenile growth (Usami et al., 2019). Importantly, Hh signaling, as indicated by LacZ signal in Gli1-LacZ reporter mice, was also activated in the RG (data not shown). Thus, it is possible that treatment with Smo inhibitors, but not chondrocyte-specific ablation of Smo, suppressed Hh signaling in the RG, and therefore undermined their ability to contribute to growth plates. Further studies are warranted to explore these possibilities.
Previous pharmacological studies reported inconsistent results about the role of Hh signaling in regulating chondrocyte hypertrophy during the juvenile period of growth. Premature chondrocyte hypertrophy was detected in mice treated with Smo antagonists HhAntag for 5 days (P10-14) or LDE225 (sonidegib) for 2 days (P22-P23), but not in mice treated with 6 doses of Smo antagonist vismodegib (Kimura et al., 2008; Newton et al., 2019; Koyama et al., 2021). In this study, we showed that chondrocyte-specific deletion of Smo in 2-week-old mice led to premature hypertrophic differentiation at P30. Therefore, our results strongly supported an essential role of Hh signaling in inhibiting chondrocyte hypertrophy during juvenile growth. This inhibitory function of Hh signaling in juvenile mice is consistent with its roles during cartilage development where Hh signaling restricts chondrocyte hypertrophy via an indirect mechanism involving PTHrP expression in periarticular cells (Long et al., 2001). Interestingly, PTHrP is also expressed in the resting chondrocytes in juvenile mice, and conditional ablation of PTHrP receptor in postnatal chondrocytes results in accelerated chondrocyte hypertrophy (Hirai et al., 2011). Thus, it is likely that Hh signaling suppresses hypertrophic differentiation of chondrocytes by promoting PTHrP expression in the resting chondrocytes. In addition to this inhibitory role, Ihh signaling can directly promote chondrocyte hypertrophy independently of PTHrP during embryonic skeletal development (Mak et al., 2008). It will be interesting to determine whether such a mechanism also functions in the postnatal cartilage.
Our LacZ staining results showed that hypertrophic chondrocytes of Gli1-and Ptc1-LacZ reporter mice had very low levels of LacZ expression, suggesting a low level of Hh signaling activity in these cells. However, despite ectopic expression of markers of hypertrophic chondrocytes in resting/proliferating chondrocytes, SmoAgc1 mice developed a clear loss of hypertrophic zones. The similar results were also observed in mice treated with LDE225 (sonidegib) for 2 days (P22-P23) (Koyama et al., 2021). These seemingly conflicting results could be explained by the fact that Hh signaling promotes chondrocyte proliferation and survival, but restricts their maturation. Thus, while inactivation of Hh signaling accelerates hypertrophic differentiation of proliferating chondrocytes, it also causes a rapid depletion of growth plate chondrocytes due to reduced proliferation and increased apoptosis. As a result, the growth plates in SmoAgc1 mice cannot continuously generate sufficient hypertrophic chondrocytes, and subsequently the hypertrophic zones in these mice are eroded rapidly by the chondroclasts.
Our data showed that ablation of either Smo or Sufu in chondrocytes led to impaired chondrocyte proliferation and survival, which could partially explain the premature fusion of growth plates in these mice. Similar phenomenon was also observed in pharmacological studies, in which both Smo agonist (SAG) and inhibitor (LDE225) treatments significantly decreased number of columns in the proliferating zone (Mizuhashi et al., 2018). Currently, the reason why inactivation and over-activation of Hh signaling exerted the same detrimental effects on chondrocytes is still unclear. Previous studies have shown that chondrocyte proliferation can be positively and negatively regulated by IGF1 and FGF18/FGFR3 signaling pathways, whereas BMP signaling is critical for both chondrocyte proliferation and survival (Hallett et al., 2019). Interestingly, Hh signaling has been shown to interact with the above signaling pathways in certain contexts (Hallett et al., 2019). However, it is unclear whether the similar interactions occur in the context of chondrocyte proliferation or survival in juvenile mice. Similarly, Hh signaling can directly promote chondrocyte proliferation during growth plate development by increasing expression of cyclin D1, a regulator of cell cycle progression (Hallett et al., 2019). However, whether this mechanism works in juvenile growth plates remains to be tested. Despite these uncertainties, our results clearly indicated that Hh signaling needs to be tightly regulated for its proper function in chondrocytes.
One limitation of our study is that we did not evaluate the effects of Hh manipulation on growth plate skeletal stem cells (gpSSC). Previous studies have suggested that Shh in the bony epiphyses may function as a niche to maintain SSC within the resting zone (Newton et al., 2019). However, the genetic evidence is still needed to support such a role of Hh signaling. Another limitation of our study is that we did not thoroughly investigate the downstream mechanism that mediates the effects of Hh signaling on chondrocytes. However, we did generate compound knockout mice with genetic deletion of both Smo and Sufu in chondrocytes, and found that these mice exhibited the same growth plate phenotypes as Sufu knockout mice (data not shown). Given that Sufu mainly function as a modulator of Gli activator and repressor, these genetic results suggested that Hh-Smo signaling likely regulates growth plate chondrocytes through Gli proteins. Clearly, the relative contribution of different Gli proteins remains to be determined. In summary, while further investigation is surely needed to address the above questions, the present study clearly demonstrated that the fine-tuned Hh signaling activity in immature chondrocytes is essential for epiphyseal growth plate maintenance and limb elongation in juvenile mice.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Ethics Committee of Soochow University.
JC and LZ conceived and designed the experiments. CX, LZ, NL, LM, and TG performed the experiments and analyzed the data. JC wrote the original draft. All authors reviewed, edited and approved the final manuscript.
This study was supported in part by the grant from the National Natural Science Foundation of China (81974344), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.997838/full#supplementary-material
Aghajanian, P., and Mohan, S. (2018). The art of building bone: Emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 6, 19. doi:10.1038/s41413-018-0021-z
Amano, K., Densmore, M. J., and Lanske, B. (2015). Conditional deletion of Indian hedgehog in limb mesenchyme results in complete loss of growth plate formation but allows mature osteoblast differentiation. J. Bone Min. Res. 30, 2262–2272. doi:10.1002/jbmr.2582
Bai, C. B., Auerbach, W., Lee, J. S., Stephen, D., and Joyner, A. L. (2002). Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753–4761. doi:10.1242/dev.129.20.4753
Chagin, A. S., and Newton, P. T. (2020). Postnatal skeletal growth is driven by the epiphyseal stem cell niche: Potential implications to pediatrics. Pediatr. Res. 87, 986–990. doi:10.1038/s41390-019-0722-z
Feng, H., Xing, W., Han, Y., Sun, J., Kong, M., Gao, B., et al. (2020). Tendon-derived cathepsin K-expressing progenitor cells activate Hedgehog signaling to drive heterotopic ossification. J. Clin. Invest. 130, 6354–6365. doi:10.1172/JCI132518
Goodrich, L. V., Milenkovic, L., Higgins, K. M., and Scott, M. P. (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113. doi:10.1126/science.277.5329.1109
Hallett, S. A., Ono, W., and Ono, N. (2019). growth plate chondrocytes: Skeletal development, growth and beyond. Int. J. Mol. Sci. 20, E6009. doi:10.3390/ijms20236009
Han, X. L., and Feng, J. Q. (2014). Beta-galactosidase staining in the skeleton. Methods Mol. Biol. 1130, 185–191. doi:10.1007/978-1-62703-989-5_13
Haraguchi, R., Kitazawa, R., Imai, Y., and Kitazawa, S. (2018). Growth plate-derived hedgehog-signal-responsive cells provide skeletal tissue components in growing bone. Histochem. Cell Biol. 149, 365–373. doi:10.1007/s00418-018-1641-5
Henry, S. P., Jang, C. W., Deng, J. M., Zhang, Z., Behringer, R. R., and de Crombrugghe, B. (2009). Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis 47, 805–814. doi:10.1002/dvg.20564
Hirai, T., Chagin, A. S., Kobayashi, T., Mackem, S., and Kronenberg, H. M. (2011). Parathyroid hormone/parathyroid hormone-related protein receptor signaling is required for maintenance of the growth plate in postnatal life. Proc. Natl. Acad. Sci. U. S. A. 108, 191–196. doi:10.1073/pnas.1005011108
Hsu, S. H., Zhang, X., Yu, C., Li, Z. J., Wunder, J. S., Hui, C. C., et al. (2011). Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory function of Sufu. Development 138, 3791–3801. doi:10.1242/dev.069492
Jiwani, T., Kim, J. J., and Rosenblum, N. D. (2020). Suppressor of fused controls cerebellum granule cell proliferation by suppressing Fgf8 and spatially regulating Gli proteins. Development 147, dev170274. doi:10.1242/dev.170274
Kimura, H., Ng, J. M., and Curran, T. (2008). Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 13, 249–260. doi:10.1016/j.ccr.2008.01.027
Koyama, E., Mundy, C., Saunders, C., Chung, J., Catheline, S. E., Rux, D., et al. (2021). Premature growth plate closure caused by a hedgehog cancer drug is preventable by Co-administration of a retinoid antagonist in mice. J. Bone Min. Res. 36, 1387–1402. doi:10.1002/jbmr.4291
Kurenkova, A. D., Medvedeva, E. V., Newton, P. T., and Chagin, A. S. (2020). Niches for skeletal stem cells of mesenchymal origin. Front. Cell Dev. Biol. 8, 592. doi:10.3389/fcell.2020.00592
Li, J., Wang, Q., Cui, Y., Yang, X., Li, Y., Zhang, X., et al. (2015). Suppressor of fused is required for determining digit number and identity via gli3/fgfs/gremlin. PLoS One 10, e0128006. doi:10.1371/journal.pone.0128006
Li, J., Cui, Y., Xu, J., Wang, Q., Yang, X., Li, Y., et al. (2017). Suppressor of Fused restraint of Hedgehog activity level is critical for osteogenic proliferation and differentiation during calvarial bone development. J. Biol. Chem. 292, 15814–15825. doi:10.1074/jbc.M117.777532
Long, F., Zhang, X. M., Karp, S., Yang, Y., and McMahon, A. P. (2001). Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128, 5099–5108. doi:10.1242/dev.128.24.5099
Long, J. T., Leinroth, A., Liao, Y., Ren, Y., Mirando, A. J., Nguyen, T., et al. (2022). Hypertrophic chondrocytes serve as a reservoir for marrow-associated skeletal stem and progenitor cells, osteoblasts, and adipocytes during skeletal development. Elife 11, e76932. doi:10.7554/eLife.76932
Maeda, Y., Nakamura, E., Nguyen, M. T., Suva, L. J., Swain, F. L., Razzaque, M. S., et al. (2007). Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc. Natl. Acad. Sci. U. S. A. 104, 6382–6387. doi:10.1073/pnas.0608449104
Mak, K. K., Kronenberg, H. M., Chuang, P. T., Mackem, S., and Yang, Y. (2008). Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 135, 1947–1956. doi:10.1242/dev.018044
Matsushita, Y., Ono, W., and Ono, N. (2020). Growth plate skeletal stem cells and their transition from cartilage to bone. Bone 136, 115359. doi:10.1016/j.bone.2020.115359
Mizuhashi, K., Ono, W., Matsushita, Y., Sakagami, N., Takahashi, A., Saunders, T. L., et al. (2018). Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 563, 254–258. doi:10.1038/s41586-018-0662-5
Newton, P. T., Li, L., Zhou, B., Schweingruber, C., Hovorakova, M., Xie, M., et al. (2019). A radical switch in clonality reveals a stem cell niche in the epiphyseal growth plate. Nature 567, 234–238. doi:10.1038/s41586-019-0989-6
Noguchi, H., Castillo, J. G., Nakashima, K., and Pleasure, S. J. (2019). Suppressor of fused controls perinatal expansion and quiescence of future dentate adult neural stem cells. Elife 8, e42918. doi:10.7554/eLife.42918
Ohba, S. (2020). Hedgehog signaling in skeletal development: Roles of Indian hedgehog and the mode of its action. Int. J. Mol. Sci. 21, E6665. doi:10.3390/ijms21186665
Razzaque, M. S., Soegiarto, D. W., Chang, D., Long, F., and Lanske, B. (2005). Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J. Pathol. 207, 453–461. doi:10.1002/path.1870
Rodda, S. J., and McMahon, A. P. (2006). Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133, 3231–3244. doi:10.1242/dev.02480
Shi, Y., He, G., Lee, W. C., McKenzie, J. A., Silva, M. J., and Long, F. (2017). Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat. Commun. 8, 2043. doi:10.1038/s41467-017-02171-2
St-Jacques, B., Hammerschmidt, M., and McMahon, A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086. doi:10.1101/gad.13.16.2072
Sun, S., Xiu, C., Chai, L., Chen, X., Zhang, L., Liu, Q., et al. (2022). HDAC inhibitor quisinostat prevents estrogen deficiency-induced bone loss by suppressing bone resorption and promoting bone formation in mice. Eur. J. Pharmacol. 927, 175073. doi:10.1016/j.ejphar.2022.175073
Usami, Y., Gunawardena, A. T., Francois, N. B., Otsuru, S., Takano, H., Hirose, K., et al. (2019). Possible contribution of wnt-responsive chondroprogenitors to the postnatal murine growth plate. J. Bone Min. Res. 34, 964–974. doi:10.1002/jbmr.3658
Wang, H., Zheng, C., Lu, W., He, T., Fan, J., Wang, C., et al. (2022). Hedgehog signaling orchestrates cartilage-to-bone transition independently of Smoothened. Matrix Biol. 110, 76–90. doi:10.1016/j.matbio.2022.04.006
Wu, F., Zhang, Y., Sun, B., McMahon, A. P., and Wang, Y. (2017). Hedgehog signaling: From basic Biology to cancer therapy. Cell Chem. Biol. 24, 252–280. doi:10.1016/j.chembiol.2017.02.010
Yang, L., Tsang, K. Y., Tang, H. C., Chan, D., and Cheah, K. S. (2014). Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. U. S. A. 111, 12097–12102. doi:10.1073/pnas.1302703111
Yung, T., Poon, F., Liang, M., Coquenlorge, S., McGaugh, E. C., Hui, C. C., et al. (2019). Sufu- and Spop-mediated downregulation of Hedgehog signaling promotes beta cell differentiation through organ-specific niche signals. Nat. Commun. 10, 4647. doi:10.1038/s41467-019-12624-5
Keywords: hedgehog signaling, epiphyseal growth plate, skeletal stem cell, suppressor of fused, chondrocyte proliferation and hypertrophy
Citation: Xiu C, Gong T, Luo N, Ma L, Zhang L and Chen J (2022) Suppressor of fused-restrained Hedgehog signaling in chondrocytes is critical for epiphyseal growth plate maintenance and limb elongation in juvenile mice. Front. Cell Dev. Biol. 10:997838. doi: 10.3389/fcell.2022.997838
Received: 19 July 2022; Accepted: 12 August 2022;
Published: 02 September 2022.
Edited by:
Liu Yang, Fourth Military Medical University, ChinaReviewed by:
Jinbo Li, Hebei Medical University, ChinaCopyright © 2022 Xiu, Gong, Luo, Ma, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianquan Chen, Y2hlbmpxQHp1Y2MuZWR1LmNu; Lei Zhang, MjAyMTQwNTAwMDFAc3R1LnN1ZGEuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.