 Maria Livia Sassano
Maria Livia Sassano Blanca Felipe-Abrio1,2
Blanca Felipe-Abrio1,2 Patrizia Agostinis
Patrizia Agostinis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 16 August 2022
Sec. Cell Death and Survival
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.988014
This article is part of the Research TopicThe Proceedings of Mitochondria Apoptosis and Cancer (MAC 2021) Virtual SymposiumView all 6 articles
Membrane contact sites (MCS) between organelles of eukaryotic cells provide structural integrity and promote organelle homeostasis by facilitating intracellular signaling, exchange of ions, metabolites and lipids and membrane dynamics. Cataloguing MCS revolutionized our understanding of the structural organization of a eukaryotic cell, but the functional role of MSCs and their role in complex diseases, such as cancer, are only gradually emerging. In particular, the endoplasmic reticulum (ER)-mitochondria contacts (EMCS) are key effectors of non-vesicular lipid trafficking, thereby regulating the lipid composition of cellular membranes and organelles, their physiological functions and lipid-mediated signaling pathways both in physiological and diseased conditions. In this short review, we discuss key aspects of the functional complexity of EMCS in mammalian cells, with particular emphasis on their role as central hubs for lipid transport between these organelles and how perturbations of these pathways may favor key traits of cancer cells.
Membrane contact sites (MCS) operate as molecular bridges empowering the flow of communication within membrane-bound organelles with specialized cellular functions. Perturbations of MCS are emerging traits of a broad spectrum of diseases, including lysosomal storage diseases, neurodegeneration and cancer (Gómez-Suaga et al., 2018; Peretti et al., 2019; Ballabio and Bonifacino, 2020; Gil-Hernández et al., 2020; Vrijsen et al., 2022). Despite this recognition, the full biological relevance of MCS is only beginning to emerge in its complexity. Possibly all organelles can form heterotypic membrane contact sites in eukaryotic cells. Since the ER is the largest membrane-bound organelle of the eukaryotic cells, it operates as key communication center by making contact with all vital organelles, such as mitochondria, Golgi, endosomes, lysosomes, peroxisomes and the plasma membrane (Wu et al., 2018; Vance, 2020). However, MCS that do not involve the ER, such as lipid droplets (LDs)-peroxisomes, mitochondria-peroxisomes and mitochondria-LDs contacts have also been described recently (Scorrano et al., 2019). Because of their essential role in many aspects of cellular homeostasis, including, but not limited to-energy production, proteostasis and Ca2+ signaling- ER-mitochondria contacts (ERMES in yeast or EMCS in mammalian cells) have been characterized to a greater extent compared to other inter-organelle appositions.
EMCS are highly dynamic contact regions (usually in the range of 10–80 nm) between the smooth ER and the mitochondrial outer membrane, which are tethered by proteins, without complete fusion of the membranes of both organelles (Scorrano et al., 2019). EMCS regulate numerous biological functions including lipid transfer, Ca2+ homeostasis, ROS signaling, autophagy, and mitochondrial dynamics (for a comprehensive discussion about EMCS, we refer to these recent reviews; (Giamogante et al., 2020; Prinz et al., 2020; Wilson and Metzakopian, 2021). The recognition that critical functions of EMCS are dysregulated in pathological conditions such as metabolic diseases, neurodegeneration and cancer, has sparked an increasing interest in these subdomains, which have become a hot topic in biomedical research (Sassano et al., 2017; Rieusset, 2018; Xu et al., 2020; Wilson and Metzakopian, 2021). In cancer cells, perturbations of the signaling functions (i.e., dysfunction) of EMCS by oncogenes reprogram both cancer cell-autonomous (e.g., glucose metabolism, Ca2+ fluxes, mitochondria dynamics, redox signaling, cell death) and non-autonomous (inflammation, innate immunity) processes favoring tumor progression (Sassano et al., 2017; Morciano et al., 2018). However, how cancer cell-associated changes of EMCS affect lipid trafficking and reprogramming of lipid metabolism, which is an emerging trait of aggressive cancer, remains poorly understood.
Here we briefly discuss the role of EMCS in Ca2+ signaling and lipid transport and highlight how key signaling pathways regulating EMCS functions are harnessed by cancer cells to support the plasticity of their metabolic traits.
EMCS play a crucial role in shaping cellular Ca2+ fluxes by establishing an intimate interaction between the ER and the mitochondria (Figure 1). The ER is the main Ca2+ storage of the cell from which mitochondria take up Ca2+, reaching concentration values above those of the bulk cytosol, which in resting conditions are in the range of 100 nM (Rizzuto et al., 1993). The fact that Ca2+ enters the mitochondrial matrix via the Ca2+ low-affinity mitochondrial Ca2+ uniporter (MCU), raised the question of how mitochondria could display such high Ca2+ concentration ([Ca2+]mit) after ER Ca2+ release (Rizzuto et al., 1992; Hajnóczky et al., 1995). The discrepancy was solved once in 1998 EMCS were discovered as microdomains of high [Ca2+], which is 10-fold higher than the cytosolic ranges (Rizzuto et al., 1998; Csordás et al., 2010). Two main ER-resident proteins are responsible for regulating ER Ca2+ homeostasis. The sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump, which actively pumps Ca2+ from the cytosol into the ER lumen to maintain steady-state [Ca2+]ER within the 400–1,000 μM range, and the inositol 1,4,5-trisphosphate receptors (IP3R) (Camello et al., 2002). Both transmembrane proteins have been found enriched at the EMCS. In particular, the SERCA2b and the IP3R3 isoforms are preferentially involved in the regulation of Ca2+ fluxes at EMCS (Mendes et al., 2005; Lynes et al., 2012). The IP3R channels are responsible for transferring Ca2+ from the ER lumen into the mitochondria, by interacting with the mitochondrial voltage-dependent anion channel 1 (VDAC1), located in the mitochondrial outer membrane (OMM), and the ER chaperone glucose-related regulated protein 75 (Grp75) bridging their interaction (Szabadkai et al., 2006).
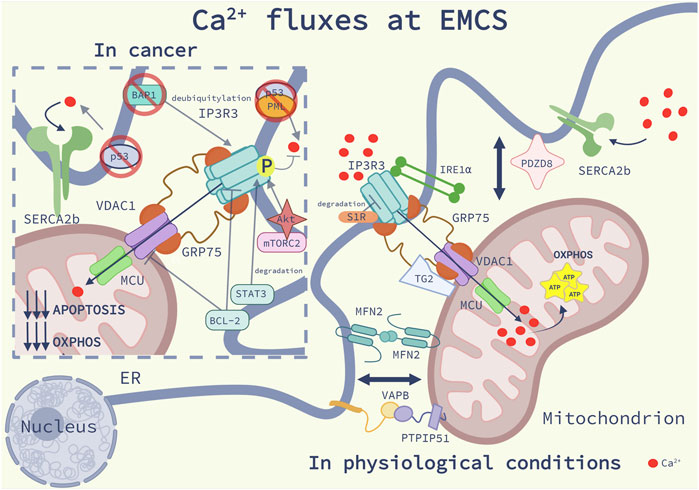
FIGURE 1. Ca2+ fluxes at EMCS: Schematic representation of the main modulators of Ca2+ homeostasis at ER-mitochondria contact sites (EMCS) in physiological conditions (on the right) and in cancer (on the left), as discussed in the main text. On the right side of the image, are represented the main Ca2+ regulatory systems at EMCS: the inositol 1,4,5-trisphosphate receptors 3 (IP3R3)- glucose-related regulated protein 75 (Grp75)-voltage-dependent anion channel 1 (VDAC1) signaling complex, delivering Ca2+ to the mitochondria, the Ca2+ uniporter (MCU), allowing Ca2+entry into the mitochondrial matrix, and the sarco/endoplasmic reticulum Ca2+ ATPase 2b (SERCA2b), which pumps Ca2+ from the cytosol to the ER. Maintenance of mitochondrial Ca2+ homeostasis through the integrity of the EMCS, promotes mitochondrial bioenergetics. Sigma-1Receptor (S1R) binds to IP3R3 and decreases its degradation; the ER stress sensor Inositol-requiring enzyme 1 (IRE1α) interacts and brings IP3R3 at EMCS. Transglutaminase type 2 (TG2) binds to Grp75 and promotes mitochondrial Ca2+ uptake. The complex formed by the protein tyrosine phosphatase interacting protein 51 (PTPIP51) and the vesicle-associated membrane protein-associated protein B (VAPB), PDZ domain-containing protein 8 (PDZD8) and Mitofusin 2 (MFN2) homodimers favor Ca2+ fluxes by regulating the EMCS proximity. On the left side of the image we describe Ca2+ dysregulation in cancer. The tumor suppressor p53 at EMCS binds to SERCA pump preventing its ROS-mediated inactivation. P53 interacts with promyelocytic leukemia (PML) protein, promoting the dephosphorylation and inactivation of the proto-oncogene serine/threonine kinase Akt, which together with the Rapamycin complex 2 (mTORC2) phosphorylate and inhibit IP3R3. The proto-oncogene B-cell lymphoma 2 (Bcl-2) interacts and inhibits VDAC1 and IP3R3 suppressing Ca2+ fluxes. IP3R3 stability is regulated by the oncogenic signal transducer and activator of transcription 3 (STAT3) which promotes its degradation and the tumor suppressor deubiquitylase BRCA1-associated protein 1 (BAP1), which prevents its ubiquitylation. The mechanisms described here contribute to a reduction of mitochondrial Ca2+ import, and consequently to a lower oxidative phosphorylation (OXPHOS) and reduced apoptosis sensitivity.
This molecular machinery shapes Ca2+ fluxes between the ER and mitochondria thereby sustaining cellular bioenergetics and mitochondria metabolism (reviewed in (Silva-Pavez et al., 2021), in physiological conditions. Indeed, the transfer of Ca2+ from the ER to the mitochondria sustains the activity of Ca2+-sensitive mitochondrial dehydrogenases, including pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (IDH) and α-ketoglutarate dehydrogenase (αKGDH) (Hajnóczky et al., 1995). Additionally ER-mitochondria Ca2+ transfer regulates vital cellular processes such as autophagy, ER stress and apoptosis (Rowland and Voeltz, 2012; Fan and Simmen, 2019). Given the importance of this Ca2+ transferring axis in cellular physiology, EMCS Ca2+ dynamics are finely tuned by functional or structural proteins which directly or indirectly modulate Ca2+ fluxes (Lee and Min, 2018). Several proteins can affect either the stability, activity or the subcellular localization of the main players of the EMCS Ca2+ regulatory system. For example, a notable function of the EMCS-associated ER-chaperone Sigma-1Receptor (S1R), is to attenuate IP3R3 degradation (Hayashi and Su, 2007). More recent studies have underscored that other proteins with enzymatic activities involved in other signaling pathways regulate Ca2+ fluxes by localizing at the EMCS. Transglutaminase Type 2 (TG2), by interacting with Grp75, functions as a scaffold for IP3R-Grp75-VDAC1-mediated mitochondrial Ca2+transfer (D’Eletto et al., 2018). The ER stress sensor inositol-requiring enzyme 1 (IRE1α), which is one of the main mediators of the unfolded protein response (UPR) (Hetz, 2012) scaffolds the IP3R3 at the EMCS and promotes its ER-mitochondrial Ca2+ transfer thereby sustaining mitochondria metabolism under resting conditions (Carreras-Sureda et al., 2019).
As other functions exerted through the EMCS, ER-mitochondria Ca2+ homeostasis is also regulated by the distance between these organelles. Disruption of the ER-mitochondria architecture perturbs mitochondrial Ca2+ uptake, upon ER Ca2+ depletion (Rizzuto et al., 1998; Csordás et al., 2006). In mammalian cells, the ER-mitochondria proximity is modulated by many structural proteins, which guarantee, or disrupt when lost, EMCS integrity and consequently proper Ca2+ signaling (we refer to these recent reviews for a broader overview on tethering proteins (Csordás et al., 2018; Lee and Min, 2018; Scorrano et al., 2019; Prinz et al., 2020). The dynamin-like GTPase MFN2, which resides both at the ER and the OMM and is able to form homo-oligomers, is a key regulator of EMCS proximity and Ca2+ fluxes between the ER and mitochondria (de Brito and Scorrano, 2008; Filadi et al., 2015; Naon et al., 2016). A well-established tethering complex is formed by the mitochondrial outer membrane protein tyrosine phosphatase interacting protein 51 (PTPIP51) and the integral ER vesicle-associated membrane protein-associated protein B (VAPB) (De Vos et al., 2012). Under conditions causing release of Ca2+ from the ER, loss of either VAPB or PTPIP51 is sufficient to blunt the uptake of Ca2+ by mitochondria (De Vos et al., 2012). More recently, the PDZ domain-containing protein 8 (PDZD8), a Synaptotagmin-like Mitochondrial lipid-binding Proteins (SMP) domain-containing ER transmembrane protein, was identified as ER-mitochondria tethering protein required to regulate Ca2+ dynamics in neurons (Hirabayashi et al., 2017). Interestingly, PDZD8 can also bring the ER and mitochondria in contact with the late endosomes, by interacting with the late-endosomal GTPase Rab7 and the ER transmembrane protein Protrudin (Elbaz-Alon et al., 2020).
While these and other studies (Prinz et al., 2020) support the emerging view that molecular entities of distinct membrane contact sites can be dynamically shared and favor the formation of tripartite organelle contacts, in the following section e focus on discussing how cancer cells reprogram Ca2+ signaling at EMCS. The cellular implications of Ca2+ in physiological functions have been broadly explained in these studies (Marchi et al., 2017, 2018; Vecellio Reane et al., 2020).
Remodeling of the functions, molecular compositions and dynamics of inter-organellar communication, allow cancer cell to rapidly respond to the fluctuating intrinsic metabolic cues and the tumor microenvironmental stress. Over the past years an increasing number of studies indicated that dynamic re-organization of EMCS supports several hallmarks of cancer cells, including, but not limited to, metabolic rewiring, resistance to cell death, migration and invasiveness and responses to inflammatory signals. The full complexity of the role played by MCS in cancer has been discussed in recent reviews (Doghman-Bouguerra and Lalli, 2019; Simoes et al., 2020; Silva-Pavez et al., 2021). Here we briefly discuss some emerging paradigms from the recent literature highlighting the relevance of Ca2+ transfer between the ER and mitochondria contacts as critical determinant of cancer cell fate decisions (Figure 1).
Different key tumor suppressors such as p53, the phosphatase tensin homolog (PTEN), promyelocytic leukemia (PML) protein, proto-oncogenes such as the serine/threonine kinase Akt kinase, breast/ovarian cancer susceptibility gene 1 (BRCA1), and various members of the B-cell lymphoma 2 (Bcl-2) family including B-cell lymphoma 2 (Bcl-2), B-cell lymphoma-extra large (Bcl-XL) and BCL-2 ovarian killer (BOK), have been found to localize to ECMS (Thomenius and Distelhorst, 2003; Monaco et al., 2015; Lalier et al., 2021). A major function of their crowded presence at the ER-mitochondria interface is the spatiotemporal modulation of EMCS-resident molecular and signaling complexes, most importantly those formed by the Ca2+ releasing IP3R channels and the SERCA pump.
Congruently, several studies have revealed that cancer cells are highly dependent on ER-mitochondrial Ca2+ transfer for the maintenance of energy balance, the ability to switch between oxidative phosphorylation (OXPHOS), aerobic glycolysis or other metabolic pathways, and for the supply of metabolic intermediates for the biosynthesis of lipid, proteins and nucleic acid required to proliferate and adapt to the metabolically stressed environment (reviewed in (Dejos et al., 2020; Silva-Pavez et al., 2021).
However, the interplay between mitochondrial Ca2+ and cellular metabolic pathways is complex and likely shaped by the cancer cell’s specific metabolic requirements and adaptations to nutrients availability. While several cancer subtypes rely primarily on aerobic glycolysis (e.g. Warburg effect), they do not completely shut down mitochondria oxidative phosphorylation. Interestingly, in cells with impaired OXPHOS, mitochondria Ca2+ flux through EMCS supports reductive carboxylation and cell survival, by sustaining the activity of the Ca2+ sensitive enzyme αKGDH and NADH (Cardenas et al., 2020). On the other hand, the overexpression of mitochondrial calcium uptake 1 (MICU1), a key component of the mitochondrial Ca2+ transport system and a negative regulator of MCU, in ovarian cancers drives aerobic glycolysis and is associated to poor survival (Chakraborty et al., 2017). Silencing MICU1 increases mitochondrial Ca2+transfer, oxygen consumption by the stimulation of pyruvate dehydrogenase (PDH), which by converting pyruvate to acetyl CoA catalyzes the rate-limiting step of the metabolic fate between glycolysis versus OXPHOS, and results in the inhibition of ovarian cancer growth in vivo (Chakraborty et al., 2017). These studies exemplify the complex interplay between mitochondrial Ca2+ and metabolic rewiring in cancer through the regulation of Ca2+ sensitive matrix dehydrogenases and their metabolites.
Recent studies have revealed a cancer cell dependency on IP3R-Bcl-2 interaction for their survival. EMCS-localized Bcl-2 suppresses ER-mitochondria Ca2+ fluxes and mitochondrial apoptosis, by interacting through different domains with VDAC1 or the IP3R3 (the main EMCS-enriched IP3R isoform) via specific aa within their BH4 domain (Monaco et al., 2012; Lalier et al., 2021; Rosa et al., 2021). Disrupting the interaction of Bcl-2 and IP3R3 through a BH4-domain targeting peptide, BIRD2, induces Bax/Bak dependent apoptosis in diffuse large B-cell lymphoma (DLBCL) and chronic lymphocytic leukemia (CLL) cells, through mitochondria Ca2+overload driving mitochondrial transition pore opening (mPTP) (Kerkhofs et al., 2021).
The mechanistic target of Rapamycin complex 2 (mTORC2)-Akt axis controls the phosphorylation-mediated inhibition of IP3R, which by preventing the transfer of Ca2+ from the ER to the mitochondria attenuates mitochondrial apoptosis, while it favors cancer cell’s aerobic glycolysis (Warburg effect) by phosphorylating hexokinase 2 (HK2), recently found as an essential component of EMCS in cancer cells (Betz et al., 2013; Ciscato et al., 2020). This oncogenic mechanism is counterbalanced by a transcriptional independent function of p53. A fraction of p53 localizes at the ER-mitochondria contact sites and establishes a functional interaction with PML (Missiroli et al., 2016). Notably, EMCS-localized PML recruits protein phosphatase 2 A (PP2A) to the complex with the IP3R3 and Akt. PP2A-mediated dephosphorylation of Akt rescues Ca2+ flux from ER to mitochondria and Ca2+-dependent apoptosis (Giorgi et al., 2010). Interestingly, disruption of the p53-PML interaction at the ER-mitochondria appositions, by hampering the constitutive ER-Ca2+−release, compromises mitochondrial respiration and ATP production, resulting in the stimulation of autophagy through the activation of AMPK (Missiroli et al., 2016). Hence the loss of p53 and consequent removal of PML from these ER membrane subdomains provides a mean to promote tumor growth, by increasing resistance to apoptotic stimuli and increasing adaptation to metabolic stress and anticancer therapy-mediated cellular damage, by stimulating autophagy (Missiroli et al., 2016). Furthermore, EMCS-associated p53 binds to and prevents ROS-inactivation of SERCA. This mechanism maintains ER-Ca2+ levels and favors pro-apoptotic Ca2+ transfer, which primes cancer cells for mitochondrial apoptosis following oxidative stress or chemotherapy (Giorgi et al., 2015). In contrast, the binding of the thioredoxin-related transmembrane protein (TMX1) to the SERCA2b pump (the housekeeping SERCA isoform at EMCS), results in a lower ER-Ca2+ load and low-level constitutive IP3-mediated Ca2+ release, which rewires metabolism toward aerobic glycolysis and favors tumorigenesis (Lynes et al., 2012).
Beyond phosphorylation and redox-dependent mechanisms, the stability of IP3R at the EMCS is controlled by its ubiquitylation status. Recently, the tumor suppressor and deubiquitylase BRCA1-associated protein 1 (BAP1), has been shown to have extranuclear functions in the cytoplasm by localizing at the ER. BAP1 operates as an IP3R3 deubiquitylating enzyme and supports pro-apoptotic Ca2+ signaling under conditions of cellular stress, a mechanism that contributes to the powerful ability of BAP1 in thwarting oncogenesis (Bononi et al., 2017). In particular, BAP1 prevents IP3R3 ubiquitylation and its subsequent proteasomal degradation (Bononi et al., 2017) by the -box protein L2 (FBXL2), a subunit of the SCF (SKP1-cullin-F-box) ubiquitin-protein ligase complex, which outcompetes PTEN for IP3R3 binding (Kuchay et al., 2017). IP3R3 stabilization in breast cancer cells is further regulated by an ER-localized constitutively active form of the oncogenic transcription factor signal transducer and activator of transcription 3 (STAT3). EMCS-associated STAT3 interacts with IP3R3 and decreases Ca2+ transfer to mitochondria by inducing its degradation (Avalle et al., 2019), thereby protecting cancer cells from cell death induced by oxidative damaging agents.
Interestingly, a selective peptide capable to delocalize the mitochondrial outer membrane bound glycolytic enzyme HK2 from EMCS, results in IP3R3-mediated mitochondrial Ca2+overload, killing of patient derived chronic lymphocytic leukemia B cells and reduced cancer growth in mice, without affecting healthy tissues (Ciscato et al., 2020). Together these studies support the notion that targeting molecular mechanisms and mediators of the aberrant ER-mitochondria Ca2+ transfer in cancer cells, may represent an effective actionable anti-cancer strategy.
EMCS form a molecular platform for the recruitment of the autophagy machinery (Hamasaki et al., 2013), a fundamental pro-survival process that is often heightened in cancer cells for recent reviews see (Li et al., 2020; Miller and Thorburn, 2021). Hence, altering the expression of EMCS-associated tethers, tumor suppressors or oncogenes, may contribute to control the amplitude of autophagy in cancer cells.
For example, PML was shown to repress pro-tumorigenic autophagy as part of its tumor suppressor activities (Missiroli et al., 2016). In line with this, in response to the loss of PML tumor development is supported by cancer cell-intrinsic autophagy, as a mechanism promoting cell survival during stress conditions (Missiroli et al., 2016). Blockade of the constitutive ER-to-mitochondrial Ca2+ transfer lowers OXPHOS, ATP production and results in AMPK activation, which induces autophagy in both normal and cancer cells. However, whereas autophagy promotes the survival of untransformed cells, it may be insufficient to maintain cancer cell viability (Cárdenas et al., 2016). Hence, the functional link between autophagy and dynamics of ER-mitochondria contacts/mitochondrial Ca2+ transfer remains complex and it is likely subjected to differential regulation by the network of oncogenes and tumor suppressors formed at the EMCS, the type of cancer cells and their dependency on autophagy for survival and growth.
Growing evidence linking ER-mitochondria appositions and cancer are constantly emerging as more EMCS-resident or associated proteins with growth promoting activity are found to be dysregulated in different cancer types. However, not all these studies have provided conclusive experimental evidence that these proteins favor tumor growth by their specific role as tethers at the ER-mitochondria contact sites.
In conclusion, while future investigations are needed to untangle the role of the growing list of EMCS-associated proteins in cancer cells, the available data suggests that targeting the ER-mitochondria interface may provide novel therapeutic strategies to halt tumor growth and improve therapeutic outcomes.
The existence of an internal system to shuttle lipids between organelles was at first considered after the observation that the unique organelle lipid composition differed from that of the main site of bulk lipid production, the ER (Balla et al., 2019). Lipid homeostasis was known to be maintained by vesicular trafficking between organelles but the simple fact that mitochondria were not integrated into these classical routes, fostered the studies of an alternative pathway (Holthuis and Menon, 2014). In 1990 EMCS were isolated and first identified as the site of phospholipid synthesis and non-vesicular transfer between the ER and mitochondria (Vance, 1990). The “crude mitochondria contaminated by ER-derived membranes” fraction was found enriched in lipid synthesis enzymes (Vance, 1990), thus supporting the thought that EMCS served to regulate membranes lipid production and composition in response to cellular demands (Petrungaro and Kornmann, 2019).
A notable function of EMCS is to harbor the synthesis of phosphatidylserine (PS), catalyzed by EMCS-associated phosphatidylserine (PS) synthase 1 and 2 (PSS1; PSS2) (Stone and Vance, 2000). PSS1 converts phosphatidylcholine (PC) into PS while PSS2 synthetizes PS from phosphatidylethanolamine (PE). EMCS synthesis of PS from PSS1 and PSS2 is crucial for mitochondrial PE enrichment. Mitochondria do not directly import PE synthetized in the ER (Vance and Tasseva, 2013; 2013b), but they rather rely on the PS transfer occurring at EMCS, which is then rapidly converted in the mitochondria into PE by PS decarboxylase enzyme (PSD) (Petrungaro and Kornmann, 2019). The active role of the ER-mitochondria platform in determining the abundance of PE in the mitochondria was further confirmed by the massive buildup of PE at EMCS upon inhibition of PSD (Ardail et al., 1991).
Controlling the conversion of PS to mitochondrial PE is essential not only to ensure a local gradient that favors PS transport but also to maintain mitochondrial fitness (Horvath and Daum, 2013). Altering mitochondrial PE levels strongly impairs mitochondrial dynamics, morphology and respiration (Verkleij et al., 1984; Steenbergen et al., 2005; Tasseva et al., 2013; Mårtensson et al., 2017; Zhao and Wang, 2020). Furthermore, PE is a key modulator of autophagy and it is conjugated to LC3 (Atg8 in yeast) through a ubiquitin-like system (Ichimura et al., 2000), suggesting that mitochondrial PE formed through the agency of EMCS may play a role in autophagy-mediated membrane dynamics (Thomas et al., 2018). Mitochondria-derived PE can be transferred back to the ER/EMCS membranes where it is converted into PC by PE-N-methyltransferase (Cui et al., 1993). Curiously, although PC is the most abundant mitochondrial PL (around 40–50%), mitochondria are not able to synthesize it, thus depending entirely on PC import from ER membranes, the site of PC bulk production (Horvath and Daum, 2013). Altering the efficiency of PC transport into the mitochondria impairs cristae formation and destabilizes mitochondrial respiration (Horibata and Sugimoto, 2010; Horibata et al., 2017).
EMCS regulates the homeostasis of another crucial mitochondrial lipid, the anionic phospholipid cardiolipin (CL), which in healthy cells is found in the matrix-facing inner leaflet of the inner mitochondria membrane. Although mitochondria own enzymes to synthesize the precursor of CL, phosphatidic acid (PA), most of the PA converted into CL comes from the ER via EMCS (Osman et al., 2011; Potting et al., 2013). CL is involved in the regulation of several physiological processes that occur in the mitochondria. In particular, CL is together with PE a master regulator of mitochondrial respiration (Belikova et al., 2006; Raemy and Martinou, 2014; Hsu et al., 2015). PE and CL bind to and modulate the stability and activity of ETC complexes and respiratory supercomplexes (RSCs) (Acín-Pérez et al., 2008; Böttinger et al., 2012; Horvath and Daum, 2013; Acoba et al., 2020). Specifically, PE and CL bind respectively to complexes I-IV and complexes I-V (Lange et al., 2001; Sun et al., 2005; Sharpley et al., 2006; Schwall et al., 2012; Tasseva et al., 2013; Calzada et al., 2019), regulating mitochondrial bioenergetics and membrane potential (Basu Ball et al., 2018). Consistent with these observations, CL or PE deficient mitochondria exhibit reduced energetic coupling and because of their role in reducing the activity of cytochrome c oxidase (complex IV), diminished mitochondrial membrane potential (Jiang et al., 2000; Böttinger et al., 2012; He et al., 2013; Tasseva et al., 2013; Vance, 2020). The individual components of the mitochondrial respiratory chain assemble into RSCs to stabilize the single components of the ETC and minimize the production of reactive oxygen species. Importantly, while CL by providing a flexible rather than rigid interface between subunits is crucial for the assembly of the RSCs (Fyfe et al., 2001), PE destabilizes it, thus playing an opposite role in their functionalities (Böttinger et al., 2012). In contrast to depletion of CL, disturbance of PE level in mitochondria does not destabilize RSCs but rather favors the assembly of “megacomplexes”, larger RSCs. On the other hand, another study reports that PE does not affect the stability of RSCs, suggesting that PE is required for the activities of ETC complexes while CL regulates both the activity and formation of RSCs (Baker et al., 2016). However, disturbances of mitochondrial levels of PE or CL result in aberrant cristae shape and length (Tasseva et al., 2013; Rampelt et al., 2018; Kondadi et al., 2020).
Beyond maintaining mitochondria respiration, CL plays a pivotal role in initiating apoptosis and the clearance of mitochondria through the process of mitophagy. Under conditions of mitochondrial stress, externalization of CL to the outer membrane serves as an “eat me” signal to target unhealthy mitochondria to the autophagosome, through a direct interaction between CL and LEC3 (Atg8) (Chu et al., 2013). Moreover, in response to cell death stimuli, outer membrane bound CL can serve as a platform for caspase 8 recruitment to the mitochondria. This leads to the caspase-8 mediated cleavage of the BH3 interacting domain death agonist (BID) into its truncated pro-death fragment tBID (Gonzalvez et al., 2008). tBID, together with CL-mediated BAX/BAK oligomerization, fosters mitochondrial cristae remodeling, permeabilization of the OMM and cytochrome c (CYTC) release into the cytosol, triggering apoptosis (Raemy and Martinou, 2014). Moreover, upon ROS production, the CL oxygenase activity of CL-bound CYTC (Kagan et al., 2005) drives the peroxidation of the CL polyunsaturated fatty acid (PUFA) chains, which causes the permeabilization of the OMM and the release of pro-death factors from the mitochondria (Kagan et al., 2005). A close link between EMCS integrity, CL oxidation and apoptosis was suggested in previous studies. In a paradigm of ROS-mediated cell death, weakening the ER-mitochondria contacts by the removal of the Ser/Thr kinase RNA-dependent protein kinase (PKR)-like ER kinase (PERK), another key member of UPR with unconventional tethering function at the EMCS (van Vliet et al., 2018), impaired early CL oxidation and pro-apoptotic cytosolic CYTC release (Verfaillie et al., 2012; van Vliet et al., 2018). Hence, EMCS integrity could serve as an early checkpoint in cell death by promoting CL peroxidation.
EMCS also act as a hotspot for other lipid biosynthetic pathways, which are involved in key cellular processes. The enrichment of cholesteryl esters (CE) and triacylglycerols (TG) synthesizing enzymes, such as acyl-CoA cholesterol acyltransferase-1 (ACAT1) and diacylglycerol acyltransferase 2 (DGAT2) respectively, to EMCS suggest their functional implication in LDs biogenesis (Rusiñol et al., 1994; Stone and Vance, 2000; Stone et al., 2009). Lipid droplets, which bud from the ER membranes, serve as storage sites for neutral lipids and as a supply of fatty acids to mitochondria, to maintain fatty acid oxidation and the tricarboxylic acid (TCA) cycle (Renne and Hariri, 2021). Recently, the existence of a close proximity between LDs, ER and mitochondria, led to the suggestion that EMCS-associated proteins may coordinate the formation of LDs (Benador et al., 2018; Thiam and Ikonen, 2021). In support of this hypothesis, the OMM protein Mitoguardin 2 (MIGA2), which tethers the EMCS by forming a complex with VAPA/B, has been shown to promote the synthesis of TG from non-lipid precursors by linking mitochondria to the ER and LDs (Freyre et al., 2019) and facilitating efficient lipid storage in LDs.
Non-vesicular trafficking of PLs at EMCS (Balla et al., 2019) requires lipid transfer proteins (LTPs), which serve as hydrophilic shuttles to transport PLs between membranes through their hydrophobic cavities (Wong et al., 2019; Prinz et al., 2020). A characteristic feature of LTPs recruited at MCS is the FFAT domain, a short motif made of two phenylalanines in an acidic tract that binds the ER-resident VAP proteins (Murphy and Levine, 2016). Although it is important to distinguish proteins that are structurally in charge of EMCS maintenance from proteins that promote PL transfer functions, it is becoming evident that tethers and LTPs are functionally linked. Several LTPs bind to both the interacting membranes, therefore stabilizing as well the contact. On the other hand, tethers that directly anchor two membranes often display a second motif or domain, which binds lipids and/or lipid binding proteins thus directly or indirectly contributing to the lipid dynamics at EMCS (Scorrano et al., 2019; Wong et al., 2019). The VAPB-PTPIP51 complex has been recently shown to regulate PL transfer at EMCS, in particular the transport of PA (Yeo et al., 2021). Other FFAT-motif proteins are the ER-anchored oxysterol-binding protein-related protein (ORP) 5 and 8, which transfer PS at the ER-PM contact sites (Chung et al., 2015). It has been recently found that ORP5/8 requires the interaction of PTPIP51 and their lipid binding domain [SBP-related lipid-binding (ORD)] to localize at EMCS, suggesting a possible role of this complex in PS transfer at EMCS. Notably, the co-expression of PTPIP51 and ORP5 or 8 reinforces the ER-mitochondria association, therefore functioning as a stabilizing tether (Galmes et al., 2016).
MIGA2 harbors a non-conventional FFAT motif which is required for its interaction with VAPA/B and for the transport of lipid at the EMCS (Freyre et al., 2019). Mitofusin 2 (MFN2) is a master regulator of the contact site proximity (de Brito and Scorrano, 2008; Filadi et al., 2015). Recent evidence shows that MFN2 directly binds and transfers PS across the ER-mitochondria interface (Hernández-Alvarez et al., 2019). Interestingly, PERK is an interactor partner of MFN2 (Muñoz et al., 2013). While the PERK-MFN2 regulatory role under stress conditions has been described, whether both proteins interact to maintain the homeostatic functions of the EMCS remains unclear. Interestingly, PERK is endowed with a lipid kinase activity responsible for diacylglycerol (DAG) phosphorylation and PA production (Bobrovnikova-Marjon et al., 2012). Moreover, PERK interacts with VAPB, although the biological function of their interaction at the EMCS has not been explored yet (Sassano et al., 2021). Together these findings raise the intriguing possibility that PERK might contribute to the regulation of lipid transport at EMCS, a hypothesis that needs experimental confirmation.
Collectively, these observations indicate that a complex regulatory network composed of lipid carrying shuttles, LTPs and multiple tethering complexes, dynamically interact to govern lipid homeostasis, mitochondrial metabolism and cell fate decisions (Figure 2).
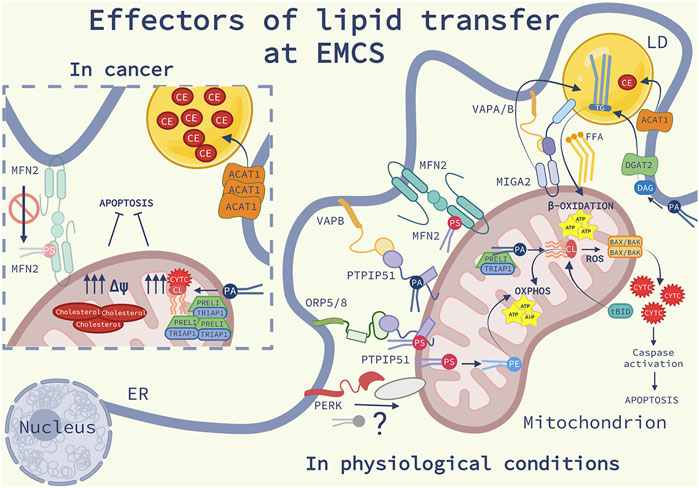
FIGURE 2. Effectors of lipid transfer at EMCS: Schematic representation of the lipid homeostasis at ER-mitochondria contact sites (EMCS) and lipid droplets (LD), in physiological conditions (on the right) and in cancer (on the left). On the right side of the image, are represented the main lipid transfer proteins (LTP) discussed in the review (see the main text for further explanations); Mitofusin 2 (MFN2) homodimers, bind and transfer phosphatidylserine (PS), the oxysterol-binding protein-related protein (ORP) 5 and 8-protein- tyrosine phosphatase interacting protein 51 (PTPIP51) complex, the vesicle-associated membrane protein-associated protein B (VAPB)- PTPIP51 complex, responsible of the transfer of phosphatidic acid (PA). The Ser/Thr kinase RNA-dependent protein kinase (PKR)-like ER kinase (PERK) is shown to interact with an unknown mitochondrial protein, possibly facilitating phospholipid (PL) transport. In the inner mitochondrial membrane (IMM) PS is converted into PE and PA and transferred into the mitochondrial intermembrane space (IMS) by TP53-regulated inhibitor of apoptosis gene 1 TRIAP1-PRELI complex. PA is converted into cardiolipin (CL), an anionic PL which promotes mitochondrial oxidative phosphorylation (OXPHOS). In response to cell death stimuli, CL interacts with truncated BH3 interacting-domain death agonist (tBID) promoting BAX/BAK oligomerization, release of cytochrome c (CYTC), triggering caspase activation and apoptosis. Mitoguardin 2 (MIGA2)-VAPA/B complex tethers the EMCS to LD and facilitates the production of triglycerides (TG). TG is synthesized into the ER from PA via the diacylglycerol acyltransferase 2 (DGAT2) enriched at EMCS. The enzyme acyl-CoA cholesterol acyltransferase-1 (ACAT1) localized at EMCS synthetizes cholesteryl esters (CE). TG and CE are stored in LD. TG hydrolysis provides free fatty acids (FFA) as fuel for β-oxidation occurring in the mitochondria. On the left panel, some molecular mechanisms involving lipid signaling at EMCS, which are altered in cancer cells, are illustrated: ACAT1 expression is increased, consequently leading to the accumulation of elevated levels of CE into LD; the complex TRIPA1-PRELI is upregulated, PA is converted into CL, impairing the release of CYTC and the sensitivity to apoptosis; cholesterol is highly accumulated into the IMM, affecting the mitochondrial potential (Δψ) and favoring resistance to apoptosis. Deficiency of MFN2 compromises the transferring of PS at EMCS causing ER stress, inflammation, fibrosis and cancer.
Given the crucial role of lipids in mitochondrial metabolism and signaling, it comes as no surprise that cancer cells often present abnormalities in the lipid content by altering their synthesis, storage or transport. In general cancer cells, due to their greater energy demands for survival and growth, increase the de novo fatty acid synthesis, necessary for the formation of new structural membranes and as a source of energy (Medes et al., 1953; Simoes et al., 2020).
Since EMCS as discussed above, represent the domains for lipid synthesis and play a pivotal role in cell fate decisions (Balla et al., 2019), they are an ideal platform for cancer lipid remodeling (Figure 2). Cancer cells often present alterations in the levels of inner membrane CL and cholesterol, which may favor resistance to apoptosis or metabolic reprogramming (Kiebish et al., 2008, Kiebish et al., 2009; Monteiro et al., 2013). For example, the tumor suppressor p53 regulates the transport of PA into the mitochondria via EMCS (Potting et al., 2013). p53-mediated expression of TP53-regulated inhibitor of apoptosis gene 1 (TRIAP1) and the PRELI complex, was found to promote PA transfer into the mitochondrial intermembrane space (IMS). Disruption of the TRIAP1-PRELI complex reduces levels of PA transfer from the ER into the mitochondrial inner membrane and CL production, which facilitates the release of CYTC and apoptosis (Potting et al., 2013), a mechanism that possibly explains why TRIAP1 is upregulated in multiple myeloma (Park and Nakamura, 2005; Felix et al., 2009).
Additionally, although mitochondria present a low content of cholesterol compared to other organelles, the transfer of cholesterol relies on the ER-mitochondria interconnection to reach the inner mitochondrial membrane (IMM). High cholesterol content affects the IMM permeability in cancer cells, therefore compromising their sensitivity to apoptosis (Sassano et al., 2017; Peretti et al., 2019). In basal state, membrane-bound free cholesterol is converted by the EMCS-localized ACAT1 enzyme into CE and stored in LDs (Puglielli et al., 2001). Breast cancer cells show high levels of ACAT1 and show elevated levels of CE in LDs (Koizume and Miyagi, 2016). Additionally, ACAT1-mediated accumulation of CE in cancer is often associated with proliferation, metastasis and bad prognosis (Koizume and Miyagi, 2016; Li et al., 2016; Stopsack et al., 2017), suggesting that LDs formation is associated with cancer proliferation and progression. Moreover, besides regulating LDs synthesis, in acute myeloid leukemia (AML), cancer cells control autophagy-mediated degradation of LDs through the regulation of the EMCS integrity. In line with this, EMCS have been recently proposed to serve as an autophagic platform supplying free fatty acids (FFAs) from LDs to fuel OXPHOS pathway, necessary for AML cell survival and proliferation (Singh et al., 2009; Bosc et al., 2020).
Cancer cells also alter the expression of various LTPs or EMCS-associated proteins with a role in facilitating lipid transfer between the ER and mitochondria. As mentioned above, PS can be transferred at the EMCS by the ORP5/8 complex. Some studies have correlated the increased expression levels of ORPs to cancer development (Du et al., 2018). In particular, ORP5 expression has been associated with increased cancer cell invasion and metastasis, possibly because of its PS transferring function at EMCS (Koga et al., 2008; Nagano et al., 2015). However, further studies are needed to validate the role of ORP5 in tumor progression. Additionally, other LTPs have been linked to cancer development, such as VAPB and PTPIP51, whose high expression levels are associated with tumor growth in breast cancer (Peretti et al., 2019). Intriguingly, a recent study disclosed MFN2 as a key factor in the development of non-alcoholic fatty liver disease (NAFLD) and liver cancer, due to its role in transferring PS at the EMCS. Hepatic MFN2 deficiency reduces the transfer of PS at EMCS which leads to a decrease in PS synthesis and ER stress, consequently causing inflammation, fibrosis and liver cancer (Hernández-Alvarez et al., 2019).
Together, these studies illustrate the intricate interplay between mitochondrial phospholipid dynamics, relying on the ER-mitochondria communication and mitochondrial homeostasis as a vital node of cancer metabolic adaptation. Nevertheless, further mechanistic analysis is needed to understand how dysfunctional EMCS enable aberrant lipid signaling that supports cancer growth and whether normalizing the EMCS-lipid crosstalk may offer a strategy to halt cancer progression.
Although in the last decade the complex architecture of contact sites has become more and more clear and new family components have been characterized, several outstanding questions still remain to be solved. In general, given the increasing number of newly-identified structural elements part of EMCS, it became important to better understand their mutual regulation and selectivity. Why would organelles display a wide variety of protein complexes regulating the same biological function at EMCS? Are certain protein complexes prioritized and taking over others depending on the cellular stimulus? Do different types of contact sites display different specialized domains? Recent evidence seems to indicate that the majority of proteins targeted to membrane contact sites display a broad range of functions, selectively executed on the bases of their localization, dynamic protein-protein interactions and cellular necessities.
In particular, while studies on Ca2+ signaling at EMCS have been quite conclusive and the molecular identities of the main ER-mitochondria Ca2+ players have been extensively characterized, molecular components regulating the lipid landscape of the EMCS are largely elusive. How are certain lipids preferentially more prone to vesicular pathways rather than the endomembrane system? Is the assigned route PL-dependent or based on the metabolic state of the cell and cellular stress? Is the close proximity between membranes sufficient for an efficient lipid remodeling at EMCS or are LTPs the “conditio sine qua non”? Answering these biological questions urge a deeper understanding of the lipid dynamics at MCS, requiring complex techniques not fully available nowadays but certainly in rapid progress.
Most importantly, although persuasive evidence indicates that oncogenes or tumor suppressors utilize the EMCS platform to interact with the major protein regulators of Ca2+ signaling to promote their survival, the molecular pathways that allow cancer cells to accurately regulate lipid reshuffling at EMCS and their implication in the cancer context are still unclear. Is the remodeling of EMCS proximity also a way for cancer cells to affect lipid cancer metabolism?
Ultimately, a better characterization of Ca2+ and lipid signaling and identification of new players at EMCS will facilitate the identification of new targets/signaling pathways hijacked by cancer cells at EMCS, which may help design new therapeutic approaches against cancer.
MLS wrote the manuscript with the support of BF-A, PA contributed to the writing, edited and corrected the manuscript.
PA is supported by the C1 KU Leuven Consortium InterAction C14/21/095, grants by the Flemish Research Foundation (FWO-Vlaanderen; G0A3320N, G094922N), the EOS MetaNiche consortium N° 40007532 and the iBOF/21/053 ATLANTIS consortium.
We apologize to the authors whose research contributed significantly to the development of the field but could not be cited due to space limitations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acín-Pérez, R., Fernández-Silva, P., Peleato, M. L., Pérez-Martos, A., and Enriquez, J. A. (2008). Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539. doi:10.1016/j.molcel.2008.10.021
Acoba, M. G., Senoo, N., and Claypool, S. M. (2020). Phospholipid ebb and flow makes mitochondria go. J. Cell Biol. 219, e202003131. doi:10.1083/jcb.202003131
Ardail, D., Lerme, F., and Louisot, P. (1991). Involvement of contact sites in phosphatidylserine import into liver mitochondria. J. Biol. Chem. 266, 7978–7981. doi:10.1016/s0021-9258(18)92926-1
Avalle, L., Camporeale, A., Morciano, G., Caroccia, N., Ghetti, E., Orecchia, V., et al. (2019). STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca2+ fluxes and apoptotic responses. Cell Death Differ. 26, 932–942. doi:10.1038/s41418-018-0171-y
Baker, C. D., Basu Ball, W., Pryce, E. N., and Gohil, V. M. (2016). Specific requirements of nonbilayer phospholipids in mitochondrial respiratory chain function and formation. Mol. Biol. Cell 27, 2161–2171. doi:10.1091/mbc.E15-12-0865
Balla, T., Kim, Y. J., Alvarez-Prats, A., and Pemberton, J. (2019). Lipid dynamics at contact sites between the endoplasmic reticulum and other organelles. Annu. Rev. Cell Dev. Biol. 35, 85–109. doi:10.1146/annurev-cellbio-100818-125251
Ballabio, A., and Bonifacino, J. S. (2020). Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118. doi:10.1038/s41580-019-0185-4
Basu Ball, W., Neff, J. K., and Gohil, V. M. (2018). The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett. 592, 1273–1290. doi:10.1002/1873-3468.12887
Belikova, N. A., Vladimirov, Y. A., Osipov, A. N., Kapralov, A. A., Tyurin, V. A., Potapovich, M. V., et al. (2006). Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry 45, 4998–5009. doi:10.1021/bi0525573
Benador, I. Y., Veliova, M., Mahdaviani, K., Petcherski, A., Wikstrom, J. D., Assali, E. A., et al. (2018). Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 27, 869–885.e6. e6. doi:10.1016/j.cmet.2018.03.003
Betz, C., Stracka, D., Prescianotto-Baschong, C., Frieden, M., Demaurex, N., and Hall, M. N. (2013). Feature article: mTOR complex 2-akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. U. S. A. 110, 12526–12534. doi:10.1073/pnas.1302455110
Bobrovnikova-Marjon, E., Pytel, D., Riese, M. J., Vaites, L. P., Singh, N., Koretzky, G. A., et al. (2012). PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate Akt activation, and promote adipocyte differentiation. Mol. Cell. Biol. 32, 2268–2278. doi:10.1128/MCB.00063-12
Bononi, A., Giorgi, C., Patergnani, S., Larson, D., Verbruggen, K., Tanji, M., et al. (2017). BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 546, 549–553. doi:10.1038/nature22798
Bosc, C., Broin, N., Fanjul, M., Saland, E., Farge, T., Courdy, C., et al. (2020). Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun. 11, 4056. doi:10.1038/s41467-020-17882-2
Böttinger, L., Horvath, S. E., Kleinschroth, T., Hunte, C., Daum, G., Pfanner, N., et al. (2012). Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J. Mol. Biol. 423, 677–686. doi:10.1016/j.jmb.2012.09.001
Calzada, E., Avery, E., Sam, P. N., Modak, A., Wang, C., McCaffery, J. M., et al. (2019). Phosphatidylethanolamine made in the inner mitochondrial membrane is essential for yeast cytochrome bc1 complex function. Nat. Commun. 10, 1432. doi:10.1038/s41467-019-09425-1
Camello, C., Lomax, R., Petersen, O. H., and Tepikin, A. V. (2002). Calcium leak from intracellular stores--the enigma of calcium signalling. Cell Calcium 32, 355–361. doi:10.1016/s0143416002001926
Cardenas, C., Lovy, A., Silva-Pavez, E., Urra, F., Mizzoni, C., Ahumada-Castro, U., et al. (2020). Cancer cells with defective oxidative phosphorylation require endoplasmic reticulum-to-mitochondria Ca2+ transfer for survival. Sci. Signal. 13, eaay1212. doi:10.1126/scisignal.aay1212
Cárdenas, C., Müller, M., McNeal, A., Lovy, A., Jaňa, F., Bustos, G., et al. (2016). Selective vulnerability of cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep. 14, 2313–2324. doi:10.1016/j.celrep.2016.02.030
Carreras-Sureda, A., Jaña, F., Urra, H., Durand, S., Mortenson, D. E., Sagredo, A., et al. (2019). Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 21, 755–767. doi:10.1038/s41556-019-0329-y
Chakraborty, P. K., Mustafi, S. B., Xiong, X., Dwivedi, S. K. D., Nesin, V., Saha, S., et al. (2017). MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 8, 14634. doi:10.1038/ncomms14634
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi:10.1038/ncb2837
Chung, J., Torta, F., Masai, K., Lucast, L., Czapla, H., Tanner, L. B., et al. (2015). INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 349, 428–432. doi:10.1126/science.aab1370
Ciscato, F., Filadi, R., Masgras, I., Pizzi, M., Marin, O., Damiano, N., et al. (2020). Hexokinase 2 displacement from mitochondria-associated membranes prompts Ca2+ -dependent death of cancer cells. EMBO Rep. 21, e49117. doi:10.15252/embr.201949117
Csordás, G., Renken, C., Várnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi:10.1083/jcb.200604016
Csordás, G., Várnai, P., Golenár, T., Roy, S., Purkins, G., Schneider, T. G., et al. (2010). Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121–132. doi:10.1016/j.molcel.2010.06.029
Csordás, G., Weaver, D., and Hajnóczky, G. (2018). Endoplasmic reticulum-mitochondrial contactology: Structure and signaling functions. Trends Cell Biol. 28, 523–540. doi:10.1016/j.tcb.2018.02.009
Cui, Z., Vance, J. E., Chen, M. H., Voelker, D. R., and Vance, D. E. (1993). Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. a specific biochemical and cytological marker for a unique membrane fraction in rat liver. J. Biol. Chem. 268, 16655–16663. doi:10.1016/s0021-9258(19)85468-6
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi:10.1038/nature07534
De Vos, K. J., Mórotz, G. M., Stoica, R., Tudor, E. L., Lau, K.-F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi:10.1093/hmg/ddr559
Dejos, C., Gkika, D., and Cantelmo, A. R. (2020). The two-way relationship between calcium and metabolism in cancer. Front. Cell Dev. Biol. 8, 573747. doi:10.3389/fcell.2020.573747
D’Eletto, M., Rossin, F., Occhigrossi, L., Farrace, M. G., Faccenda, D., Desai, R., et al. (2018). Transglutaminase type 2 regulates ER-mitochondria contact sites by interacting with GRP75. Cell Rep. 25, 3573–3581.e4. e4. doi:10.1016/j.celrep.2018.11.094
Doghman-Bouguerra, M., and Lalli, E. (2019). ER-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta. Mol. Cell Res. 1866, 650–662. doi:10.1016/j.bbamcr.2019.01.009
Du, X., Turner, N., and Yang, H. (2018). The role of oxysterol-binding protein and its related proteins in cancer. Semin. Cell Dev. Biol. 81, 149–153. doi:10.1016/j.semcdb.2017.07.017
Elbaz-Alon, Y., Guo, Y., Segev, N., Harel, M., Quinnell, D. E., Geiger, T., et al. (2020). PDZD8 interacts with Protrudin and Rab7 at ER-late endosome membrane contact sites associated with mitochondria. Nat. Commun. 11, 3645. doi:10.1038/s41467-020-17451-7
Fan, Y., and Simmen, T. (2019). Mechanistic connections between endoplasmic reticulum (ER) redox control and mitochondrial metabolism. Cells 8.
Felix, R. S., Colleoni, G. W. B., Caballero, O. L., Yamamoto, M., Almeida, M. S. S., Andrade, V. C. C., et al. (2009). SAGE analysis highlights the importance of p53csv, ddx5, mapkapk2 and ranbp2 to multiple myeloma tumorigenesis. Cancer Lett. 278, 41–48. doi:10.1016/j.canlet.2008.12.022
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U. S. A. 112, E2174–E2181. doi:10.1073/pnas.1504880112
Freyre, C. A. C., Rauher, P. C., Ejsing, C. S., and Klemm, R. W. (2019). MIGA2 links mitochondria, the ER, and lipid droplets and promotes de novo lipogenesis in adipocytes. Mol. Cell 76, 811–825.e14. e14. doi:10.1016/j.molcel.2019.09.011
Fyfe, P. K., McAuley, K. E., Roszak, A. W., Isaacs, N. W., Cogdell, R. J., and Jones, M. R. (2001). Probing the interface between membrane proteins and membrane lipids by X-ray crystallography. Trends biochem. Sci. 26, 106–112. doi:10.1016/s0968-0004(00)01746-1
Galmes, R., Houcine, A., van Vliet, A. R., Agostinis, P., Jackson, C. L., and Giordano, F. (2016). ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 17, 800–810. doi:10.15252/embr.201541108
Giamogante, F., Barazzuol, L., Brini, M., and Calì, T. (2020). ER-mitochondria contact sites reporters: strengths and weaknesses of the available approaches. Int. J. Mol. Sci. 21, E8157. doi:10.3390/ijms21218157
Gil-Hernández, A., Arroyo-Campuzano, M., Simoni-Nieves, A., Zazueta, C., Gomez-Quiroz, L. E., and Silva-Palacios, A. (2020). Relevance of membrane contact sites in cancer progression. Front. Cell Dev. Biol. 8, 622215. doi:10.3389/fcell.2020.622215
Giorgi, C., Bonora, M., Sorrentino, G., Missiroli, S., Poletti, F., Suski, J. M., et al. (2015). p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 112, 1779–1784. doi:10.1073/pnas.1410723112
Giorgi, C., Ito, K., Lin, H.-K., Santangelo, C., Wieckowski, M. R., Lebiedzinska, M., et al. (2010). PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330, 1247–1251. doi:10.1126/science.1189157
Gómez-Suaga, P., Bravo-San Pedro, J. M., González-Polo, R. A., Fuentes, J. M., and Niso-Santano, M. (2018). ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis. 9, 337. doi:10.1038/s41419-017-0079-3
Gonzalvez, F., Schug, Z. T., Houtkooper, R. H., MacKenzie, E. D., Brooks, D. G., Wanders, R. J. A., et al. (2008). Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 183, 681–696. doi:10.1083/jcb.200803129
Hajnóczky, G., Robb-Gaspers, L. D., Seitz, M. B., and Thomas, A. P. (1995). Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415–424. doi:10.1016/0092-8674(95)90430-1
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. doi:10.1038/nature11910
Hayashi, T., and Su, T.-P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610. doi:10.1016/j.cell.2007.08.036
He, Q., Wang, M., Harris, N., and Han, X. (2013). Tafazzin knockdown interrupts cell cycle progression in cultured neonatal ventricular fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 305, H1332–H1343. doi:10.1152/ajpheart.00084.2013
Hernández-Alvarez, M. I., Sebastián, D., Vives, S., Ivanova, S., Bartoccioni, P., Kakimoto, P., et al. (2019). Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease. Cell 177, 881–895.e17. e17. doi:10.1016/j.cell.2019.04.010
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi:10.1038/nrm3270
Hirabayashi, Y., Kwon, S.-K., Paek, H., Pernice, W. M., Paul, M. A., Lee, J., et al. (2017). ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 358, 623–630. doi:10.1126/science.aan6009
Holthuis, J. C. M., and Menon, A. K. (2014). Lipid landscapes and pipelines in membrane homeostasis. Nature 510, 48–57. doi:10.1038/nature13474
Horibata, Y., Ando, H., Satou, M., Shimizu, H., Mitsuhashi, S., Shimizu, Y., et al. (2017). Identification of the N-terminal transmembrane domain of StarD7 and its importance for mitochondrial outer membrane localization and phosphatidylcholine transfer. Sci. Rep. 7, 8793. doi:10.1038/s41598-017-09205-1
Horibata, Y., and Sugimoto, H. (2010). StarD7 mediates the intracellular trafficking of phosphatidylcholine to mitochondria. J. Biol. Chem. 285, 7358–7365. doi:10.1074/jbc.M109.056960
Horvath, S. E., and Daum, G. (2013). Lipids of mitochondria. Prog. Lipid Res. 52, 590–614. doi:10.1016/j.plipres.2013.07.002
Hsu, P., Liu, X., Zhang, J., Wang, H.-G., Ye, J.-M., and Shi, Y. (2015). Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 11, 643–652. doi:10.1080/15548627.2015.1023984
Ichimura, Y., Kirisako, T., Takao, T., Satomi, Y., Shimonishi, Y., Ishihara, N., et al. (2000). A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492. doi:10.1038/35044114
Jiang, F., Ryan, M. T., Schlame, M., Zhao, M., Gu, Z., Klingenberg, M., et al. (2000). Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394. doi:10.1074/jbc.M909868199
Kagan, V. E., Tyurin, V. A., Jiang, J., Tyurina, Y. Y., Ritov, V. B., Amoscato, A. A., et al. (2005). Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 1, 223–232. doi:10.1038/nchembio727
Kerkhofs, M., La Rovere, R., Welkenhuysen, K., Janssens, A., Vandenberghe, P., Madesh, M., et al. (2021). BIRD-2, a BH4-domain-targeting peptide of Bcl-2, provokes Bax/Bak-independent cell death in B-cell cancers through mitochondrial Ca2+-dependent mPTP opening. Cell Calcium 94, 102333. doi:10.1016/j.ceca.2020.102333
Kiebish, M. A., Han, X., Cheng, H., Chuang, J. H., and Seyfried, T. N. (2008). Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: lipidomic evidence supporting the warburg theory of cancer. J. Lipid Res. 49, 2545–2556. doi:10.1194/jlr.M800319-JLR200
Kiebish, M. A., Han, X., Cheng, H., and Seyfried, T. N. (2009). In vitro growth environment produces lipidomic and electron transport chain abnormalities in mitochondria from non-tumorigenic astrocytes and brain tumours. ASN Neuro 1, e00011. doi:10.1042/AN20090011
Koga, Y., Ishikawa, S., Nakamura, T., and Masuda, T. (2008). Oxysterol binding protein‐related protein‐5 is related to invasion and poor prognosis in pancreatic cancer. Cancer 99, 2387. doi:10.1111/j.1349-7006.2008.00987.x
Koizume, S., and Miyagi, Y. (2016). Lipid droplets: a key cellular organelle associated with cancer cell survival under Normoxia and Hypoxia. Int. J. Mol. Sci. 17, E1430. doi:10.3390/ijms17091430
Kondadi, A. K., Anand, R., and Reichert, A. S. (2020). Cristae membrane dynamics - a paradigm change. Trends Cell Biol. 30, 923–936. doi:10.1016/j.tcb.2020.08.008
Kuchay, S., Giorgi, C., Simoneschi, D., Pagan, J., Missiroli, S., Saraf, A., et al. (2017). PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 546, 554–558. doi:10.1038/nature22965
Lalier, L., Mignard, V., Joalland, M.-P., Lanoé, D., Cartron, P.-F., Manon, S., et al. (2021). TOM20-mediated transfer of Bcl2 from ER to MAM and mitochondria upon induction of apoptosis. Cell Death Dis. 12, 182. doi:10.1038/s41419-021-03471-8
Lange, C., Nett, J. H., Trumpower, B. L., and Hunte, C. (2001). Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J. 20, 6591–6600. doi:10.1093/emboj/20.23.6591
Lee, S., and Min, K.-T. (2018). The interface between ER and mitochondria: molecular compositions and functions. Mol. Cells 41, 1000–1007. doi:10.14348/molcells.2018.0438
Li, J., Gu, D., Lee, S. S. Y., Song, B., Bandyopadhyay, S., Chen, S., et al. (2016). Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 35, 6378–6388. doi:10.1038/onc.2016.168
Li, X., He, S., and Ma, B. (2020). Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19, 12. doi:10.1186/s12943-020-1138-4
Lynes, E. M., Bui, M., Yap, M. C., Benson, M. D., Schneider, B., Ellgaard, L., et al. (2012). Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 31, 457–470. doi:10.1038/emboj.2011.384
Marchi, S., Bittremieux, M., Missiroli, S., and Morganti, C. (2017). Endoplasmic reticulum-mitochondria communication through Ca2+ signaling: The importance of mitochondria-associated membranes (MAMs). Organelle Contact 997, 49–67. doi:10.1007/978-981-10-4567-7_4
Marchi, S., Patergnani, S., Missiroli, S., Morciano, G., Rimessi, A., Wieckowski, M. R., et al. (2018). Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72. doi:10.1016/j.ceca.2017.05.003
Mårtensson, C. U., Doan, K. N., and Becker, T. (2017). Effects of lipids on mitochondrial functions. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids, 102–113. doi:10.1016/j.bbalip.2016.06.015
Medes, G., Thomas, A., and Weinhouse, S. (1953). Metabolism of neoplastic tissue.' ' IV. a study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 13, 27–29.
Mendes, C. C. P., Gomes, D. A., Thompson, M., Souto, N. C., Goes, T. S., Goes, A. M., et al. (2005). The type III inositol 1, 4, 5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 280, 40892–40900. doi:10.1074/jbc.M506623200
Miller, D. R., and Thorburn, A. (2021). Autophagy and organelle homeostasis in cancer. Dev. Cell 56, 906–918. doi:10.1016/j.devcel.2021.02.010
Missiroli, S., Bonora, M., Patergnani, S., Poletti, F., Perrone, M., Gafà, R., et al. (2016). PML at mitochondria-associated membranes is critical for the repression of autophagy and cancer development. Cell Rep. 16, 2415–2427. doi:10.1016/j.celrep.2016.07.082
Monaco, G., Decrock, E., Akl, H., Ponsaerts, R., Vervliet, T., Luyten, T., et al. (2012). Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 19, 295–309. doi:10.1038/cdd.2011.97
Monaco, G., Decrock, E., Arbel, N., van Vliet, A. R., La Rovere, R. M., De Smedt, H., et al. (2015). The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J. Biol. Chem. 290, 9150–9161. doi:10.1074/jbc.M114.622514
Monteiro, J. P., Oliveira, P. J., and Jurado, A. S. (2013). Mitochondrial membrane lipid remodeling in pathophysiology: a new target for diet and therapeutic interventions. Prog. Lipid Res. 52, 513–528. doi:10.1016/j.plipres.2013.06.002
Morciano, G., Marchi, S., Morganti, C., Sbano, L., Bittremieux, M., Kerkhofs, M., et al. (2018). Role of mitochondria-associated ER membranes in calcium regulation in cancer-specific settings. Neoplasia 20, 510–523. doi:10.1016/j.neo.2018.03.005
Muñoz, J. P., Ivanova, S., Sánchez-Wandelmer, J., Martínez-Cristóbal, P., Noguera, E., Sancho, A., et al. (2013). Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361. doi:10.1038/emboj.2013.168
Murphy, S. E., and Levine, T. P. (2016). VAP, a versatile access point for the endoplasmic reticulum: review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta 1861, 952–961. doi:10.1016/j.bbalip.2016.02.009
Nagano, K., Imai, S., Zhao, X., Yamashita, T., Yoshioka, Y., Abe, Y., et al. (2015). Identification and evaluation of metastasis-related proteins, oxysterol binding protein-like 5 and calumenin, in lung tumors. Int. J. Oncol. 47, 195–203. doi:10.3892/ijo.2015.3000
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. U. S. A. 113, 11249–11254. doi:10.1073/pnas.1606786113
Osman, C., Voelker, D. R., and Langer, T. (2011). Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 192, 7–16. doi:10.1083/jcb.201006159
Park, W.-R., and Nakamura, Y. (2005). p53CSV, a novel p53-inducible gene involved in the p53-dependent cell-survival pathway. Cancer Res. 65, 1197–1206. doi:10.1158/0008-5472.CAN-04-3339
Peretti, D., Kim, S., Tufi, R., and Lev, S. (2019). Lipid transfer proteins and membrane contact sites in human cancer. Front. Cell Dev. Biol. 7, 371. doi:10.3389/fcell.2019.00371
Petrungaro, C., and Kornmann, B. (2019). Lipid exchange at ER-mitochondria contact sites: a puzzle falling into place with quite a few pieces missing. Curr. Opin. Cell Biol. 57, 71–76. doi:10.1016/j.ceb.2018.11.005
Potting, C., Tatsuta, T., König, T., Haag, M., Wai, T., Aaltonen, M. J., et al. (2013). TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 18, 287–295. doi:10.1016/j.cmet.2013.07.008
Prinz, W. A., Toulmay, A., and Balla, T. (2020). The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 21, 7–24. doi:10.1038/s41580-019-0180-9
Puglielli, L., Konopka, G., and Pack-Chung, E. (2001). Acyl-coenzyme a: Cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nat. Cell. 3, 905. doi:10.1038/ncb1001-905
Raemy, E., and Martinou, J.-C. (2014). Involvement of cardiolipin in tBID-induced activation of BAX during apoptosis. Chem. Phys. Lipids 179, 70–74. doi:10.1016/j.chemphyslip.2013.12.002
Rampelt, H., Wollweber, F., Gerke, C., de Boer, R., van der Klei, I. J., Bohnert, M., et al. (2018). Assembly of the mitochondrial cristae organizer Mic10 is regulated by mic26-mic27 antagonism and cardiolipin. J. Mol. Biol. 430, 1883–1890. doi:10.1016/j.jmb.2018.04.037
Renne, M. F., and Hariri, H. (2021). Lipid droplet-organelle contact sites as hubs for fatty acid metabolism, trafficking, and metabolic channeling. Front. Cell Dev. Biol. 9, 726261. doi:10.3389/fcell.2021.726261
Rieusset, J. (2018). The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death Dis. 9, 388. doi:10.1038/s41419-018-0416-1
Rizzuto, R., Brini, M., Murgia, M., and Pozzan, T. (1993). Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262, 744–747. doi:10.1126/science.8235595
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. doi:10.1126/science.280.5370.1763
Rizzuto, R., Simpson, A. W., Brini, M., and Pozzan, T. (1992). Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358, 325–327. doi:10.1038/358325a0
Rosa, N., Ivanova, H., Wagner, L. E., Kale, J., La Rovere, R., Welkenhuyzen, K., et al. (2021). Bcl-xL acts as an inhibitor of IP3R channels, thereby antagonizing Ca2+-driven apoptosis. Cell Death Differ. 29, 788–805. doi:10.1038/s41418-021-00894-w
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625. doi:10.1038/nrm3440
Rusiñol, A. E., Cui, Z., Chen, M. H., and Vance, J. E. (1994). A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 269, 27494–27502. doi:10.1016/s0021-9258(18)47012-3
Sassano, M. L., Derua, R., Waelkens, E., Agostinis, P., and van Vliet, A. R. (2021). Interactome analysis of the ER stress sensor perk uncovers key components of ER-mitochondria contact sites and Ca2+ signalling. Contact 4, 251525642110523. doi:10.1177/25152564211052392
Sassano, M. L., van Vliet, A. R., and Agostinis, P. (2017). Mitochondria-associated membranes as networking platforms and regulators of cancer cell fate. Front. Oncol. 7, 174. doi:10.3389/fonc.2017.00174
Schwall, C. T., Greenwood, V. L., and Alder, N. N. (2012). The stability and activity of respiratory complex II is cardiolipin-dependent. Biochim. Biophys. Acta 1817, 1588–1596. doi:10.1016/j.bbabio.2012.04.015
Scorrano, L., De Matteis, M. A., Emr, S., Giordano, F., Hajnóczky, G., Kornmann, B., et al. (2019). Coming together to define membrane contact sites. Nat. Commun. 10, 1287. doi:10.1038/s41467-019-09253-3
Sharpley, M. S., Shannon, R. J., Draghi, F., and Hirst, J. (2006). Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248. doi:10.1021/bi051809x
Silva-Pavez, E., Ahumada-Castro, U., Lovy, A., and Cárdenas, J. C. (2021). Ca2+ transfer to mitochondria: a spark of life in unexpected conditions. Mol. Cell. Oncol. 8, 1839341. doi:10.1080/23723556.2020.1839341
Simoes, I. C. M., Morciano, G., Lebiedzinska-Arciszewska, M., Aguiari, G., Pinton, P., Potes, Y., et al. (2020). The mystery of mitochondria-ER contact sites in physiology and pathology: A cancer perspective. Biochim. Biophys. Acta. Mol. Basis Dis. 1866, 165834. doi:10.1016/j.bbadis.2020.165834
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. doi:10.1038/nature07976
Steenbergen, R., Nanowski, T. S., Beigneux, A., Kulinski, A., Young, S. G., and Vance, J. E. (2005). Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 280, 40032–40040. doi:10.1074/jbc.M506510200
Stone, S. J., Levin, M. C., Zhou, P., Han, J., Walther, T. C., and Farese, R. V. (2009). The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem. 284, 5352–5361. doi:10.1074/jbc.M805768200
Stone, S. J., and Vance, J. E. (2000). Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 275, 34534–34540. doi:10.1074/jbc.M002865200
Stopsack, K. H., Gerke, T. A., Andrén, O., Andersson, S.-O., Giovannucci, E. L., Mucci, L. A., et al. (2017). Cholesterol uptake and regulation in high-grade and lethal prostate cancers. Carcinogenesis 38, 806–811. doi:10.1093/carcin/bgx058
Sun, F., Huo, X., Zhai, Y., Wang, A., Xu, J., Su, D., et al. (2005). Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121, 1043–1057. doi:10.1016/j.cell.2005.05.025
Szabadkai, G., Bianchi, K., Várnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. doi:10.1083/jcb.200608073
Tasseva, G., Bai, H. D., Davidescu, M., Haromy, A., Michelakis, E., and Vance, J. E. (2013). Phosphatidylethanolamine deficiency in mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 288, 4158–4173. doi:10.1074/jbc.M112.434183
Thiam, A. R., and Ikonen, E. (2021). Lipid droplet nucleation. Trends Cell Biol. 31, 108–118. doi:10.1016/j.tcb.2020.11.006
Thomas, H. E., Zhang, Y., Stefely, J. A., Veiga, S. R., Thomas, G., Kozma, S. C., et al. (2018). Mitochondrial complex I activity is required for maximal autophagy. Cell Rep. 24, 2404–2417.e8. e8. doi:10.1016/j.celrep.2018.07.101
Thomenius, M. J., and Distelhorst, C. W. (2003). Bcl-2 on the endoplasmic reticulum: protecting the mitochondria from a distance. J. Cell Sci. 116, 4493–4499. doi:10.1242/jcs.00829
van Vliet, A. R., Sassano, M. L., and Agostinis, P. (2018). The unfolded protein response and membrane contact sites: tethering as a matter of life and death? Contact 1, 251525641877051. doi:10.1177/2515256418770512
Vance, J. E. (2020). Inter-organelle membrane contact sites: implications for lipid metabolism. Biol. Direct 15, 24. doi:10.1186/s13062-020-00279-y
Vance, J. E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256. doi:10.1016/s0021-9258(19)39106-9
Vance, J. E., and Tasseva, G. (2013). Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta 1831, 543–554. doi:10.1016/j.bbalip.2012.08.016
Vecellio Reane, D., Rizzuto, R., and Raffaello, A. (2020). The ER-mitochondria tether at the hub of Ca2+ signaling. Curr. Opin. Physiol. 17, 261–268. doi:10.1016/j.cophys.2020.08.013
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J. P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 19, 1880–1891. doi:10.1038/cdd.2012.74
Verkleij, A. J., Leunissen-Bijvelt, J., de Kruijff, B., Hope, M., and Cullis, P. R. (1984). Non-bilayer structures in membrane fusion. Ciba Found. Symp. 103, 45–59. doi:10.1002/9780470720844.ch4
Vrijsen, S., Vrancx, C., Del Vecchio, M., Swinnen, J. V., Agostinis, P., Winderickx, J., et al. (2022). Inter-organellar communication in Parkinson’s and alzheimer's disease: looking beyond endoplasmic reticulum-mitochondria contact sites. Front. Neurosci. 16, 900338. doi:10.3389/fnins.2022.900338
Wilson, E. L., and Metzakopian, E. (2021). ER-Mitochondria contact sites in neurodegeneration: genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ. 28, 1804–1821. doi:10.1038/s41418-020-00705-8
Wong, L. H., Gatta, A. T., and Levine, T. P. (2019). Lipid transfer proteins: the lipid commute via shuttles, bridges and tubes. Nat. Rev. Mol. Cell Biol. 20, 85–101. doi:10.1038/s41580-018-0071-5
Wu, H., Carvalho, P., and Voeltz, G. K. (2018). Here, there, and everywhere: the importance of ER membrane contact sites. Science 361, eaan5835. doi:10.1126/science.aan5835
Xu, L., Wang, X., and Tong, C. (2020). Endoplasmic reticulum-mitochondria contact sites and neurodegeneration. Front. Cell Dev. Biol. 8, 428. doi:10.3389/fcell.2020.00428
Yeo, H. K., Park, T. H., Kim, H. Y., Jang, H., Lee, J., Hwang, G.-S., et al. (2021). Phospholipid transfer function of PTPIP51 at mitochondria-associated ER membranes. EMBO Rep. 22, e51323. doi:10.15252/embr.202051323
Keywords: ER-mitochondria contact sites, lipids, lipid transfer protein, Ca2+ signaling, cancer
Citation: Sassano ML, Felipe-Abrio B and Agostinis P (2022) ER-mitochondria contact sites; a multifaceted factory for Ca2+ signaling and lipid transport . Front. Cell Dev. Biol. 10:988014. doi: 10.3389/fcell.2022.988014
Received: 06 July 2022; Accepted: 22 July 2022;
Published: 16 August 2022.
Edited by:
Jiri Neuzil, Griffith University, AustraliaReviewed by:
Nataliia Naumova, Université Paris-Sud, FranceCopyright © 2022 Sassano, Felipe-Abrio and Agostinis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrizia Agostinis, cGF0cml6aWEuYWdvc3RpbmlzQGt1bGV1dmVuLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.