
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 25 October 2022
Sec. Cell Death and Survival
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.986835
This article is part of the Research Topic Mitophagy in Health and Disease, volume II View all 5 articles
 Alfonso Schiavi1,2†
Alfonso Schiavi1,2† Alessandra Runci1,2†
Alessandra Runci1,2† Teresa Maiorino3‡Francesco Davide Naso3
Teresa Maiorino3‡Francesco Davide Naso3 Marta Barenys1‡
Marta Barenys1‡ Ellen Fritsche1
Ellen Fritsche1 Flavie Strappazzon3,4
Flavie Strappazzon3,4 Natascia Ventura1,2*
Natascia Ventura1,2*Severe oxygen and iron deficiencies have evolutionarily conserved detrimental effects, leading to pathologies in mammals and developmental arrest as well as neuromuscular degeneration in the nematode Caenorhabditis elegans. Yet, similar to the beneficial effects of mild hypoxia, non-toxic levels of iron depletion, achieved with the iron chelator bipyridine or through frataxin silencing, extend C. elegans lifespan through hypoxia-like induction of mitophagy. While the positive health outcomes of hypoxia preconditioning are evident, its practical application is rather challenging. Here, we thus test the potential beneficial effects of non-toxic, preconditioning interventions acting on iron instead of oxygen availability. We find that limiting iron availability through the iron competing agent cobalt chloride has evolutionarily conserved dose-dependent beneficial effects: while high doses of cobalt chloride have toxic effects in mammalian cells, iPS-derived neurospheres, and in C. elegans, sub-lethal doses protect against hypoxia- or cobalt chloride-induced death in mammalian cells and extend lifespan and delay age-associated neuromuscular alterations in C. elegans. The beneficial effects of cobalt chloride are accompanied by the activation of protective mitochondrial stress response pathways.
Severe hypoxia has evolutionarily conserved and widely described deleterious consequences, ranging from ischemic stroke and myocardial infarction in mammals to developmental arrest and neuromuscular degeneration in the nematode Caenorhabditis elegans (C. elegans) (Rodriguez et al., 2013; Lee et al., 2019). In normoxic conditions, HIF1α (hypoxia-inducible-factor) is hydroxylated by enzymes that require O2, Fe++, and 2-oxoglutarate: at proline by PHD1-3 (prolyl-4-hydroxylase domain-containing enzymes) or at asparagine by FIH (factor inhibiting HIF). This leads to the subsequent ubiquitination and degradation of HIF1α via the proteasome. Low oxygen and iron concentrations diminish PHD and FIH hydroxylase activities and prevent HIF1α degradation. This favors HIF1α translocation into the nucleus and binding to co-activators, which ultimately induces its transcriptional activity resulting in the modulation of a variety of downstream genes containing hypoxia response elements (HRE) (Wenger, 2002; Powell-Coffman, 2010). Accordingly, iron chelators such as deferoxamine (DFO) or bipyridine (BP), and iron-competing metals such as cobalt chloride (CoCl2) or nickel chloride (NiCl2) have been shown to work as hypoxia mimetics by deactivating the hydroxylases (Goldberg et al., 1988). Furthermore, similar to severe hypoxia, severe iron depletion is also involved in the pathogenesis of different human disorders and it leads to developmental arrest in C. elegans (Schiavi et al., 2015; Schiavi et al., 2020). Iron is an essential factor for several enzymes and cellular reactions, and therefore, its homeostasis must be finely tuned. Not only iron deprivation but also its excessive accumulation has evolutionarily conserved detrimental effects. Iron overload concurs to the pathogenesis of age-associated neuronal pathologies such as Parkinson’s or Alzheimer’s diseases, and it is a typical hallmark of the aging process, thereby shortening lifespan in C. elegans (Lee et al., 2006; Schiavi et al., 2015).
Friedreich’s ataxia (FRDA) is the most frequently inherited recessive ataxia, and it is ascribed to severe deficiency of frataxin, a nuclear-encoded mitochondrial protein involved in iron–sulfur cluster (ISC) protein biogenesis and iron homeostasis. Accumulation of iron within mitochondria, with consequent alteration of ISC formation and induction of a cytosolic iron-starvation response, has been recognized as a consequence of frataxin deficiency involved in disease pathogenesis (Anzovino et al., 2014). In line with the disease state, and similar to severe oxygen and iron deprivation, severe or complete frataxin deficiency also displays evolutionarily conserved detrimental effects, leading to lethality in mice and to developmental arrest in C. elegans (Puccio et al., 2001; Ventura et al., 2005). Nonetheless, non-toxic levels of iron depletion, achieved with the iron chelator BP or through frataxin (frh-1) silencing, extend C. elegans lifespan through hypoxia-like induction of mitophagy (Schiavi et al., 2015). Moreover, exposure to non-lethal levels of hypoxia (or hypoxia preconditioning, HP) has been shown to prevent the detrimental effects of severe hypoxia-induced neuronal degeneration in an evolutionarily conserved manner (Dasgupta et al., 2007; Liu et al., 2021). While the potential beneficial health effects of HP are evident, its actual exploitation is hampered by the low feasibility of its practical application. In this study, we thus asked whether non-toxic, preconditioning interventions acting on iron instead of oxygen availability may provide beneficial effects too. We found that similar to HP, limiting iron availability through CoCl2 treatment has evolutionarily conserved beneficial effects, protecting against severe hypoxia- and age-induced neuromuscular alterations. We thus provide evidence that limiting iron availability represents a potential feasible strategy to delay aging and associated neuromuscular pathologies possibly via autophagy/mitophagy induction.
Iron is a vital cofactor for numerous proteins and enzymatic reactions and its intracellular concentration has to be finely tuned for appropriate organism homeostasis (Schiavi et al., 2020). Accordingly, both excess iron accumulation and depletion have toxic effects across species. Interestingly, we have previously shown that contrary to its severe deficiency, sub-lethal iron depletion through the iron chelator BP or through frh-1 silencing extends C. elegans lifespan (Schiavi et al., 2015). Limiting iron availability acts as a hypoxia mimetic, and exploiting hypoxia mimetics to prevent or protect against hypoxia- or age-induced neuromuscular pathologies may provide different advantages over hypoxia preconditioning. We thus sought to investigate the beneficial effects of additional modulators of iron availability. Consistent with the detrimental role of severe iron depletion especially during development, we found that the iron chelators DFO and BP as well as the iron-competing metal CoCl2 significantly reduce neuronal differentiation (Figures 1A–D) and viability (Figures 1E–H) in mouse neural progenitor cell (NPC)-derived neurospheres, a very sensitive and powerful system for developmental neurotoxicity studies (Fritsche et al., 2018).
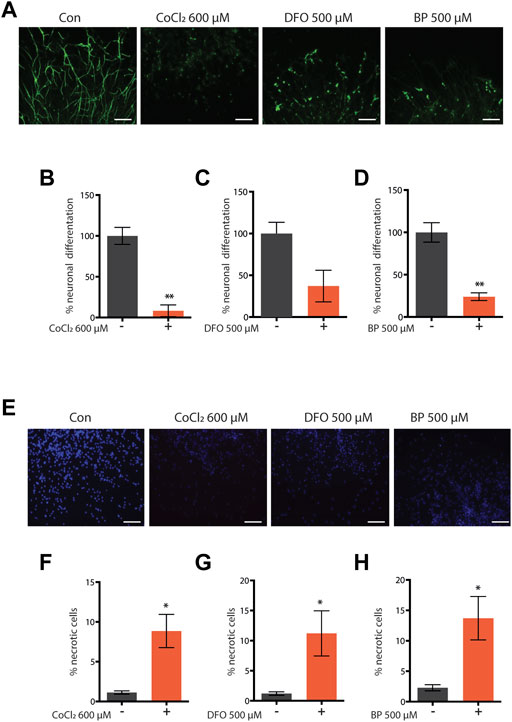
FIGURE 1. (A–H) Representative pictures and quantification of neuronal differentiation (A–D) and necrotic cell death (E–H) of mouse embryo iPS-derived neurospheres left untreated or treated with cobalt chloride 600 µM (CoCl—600 μM) (A–F) for 48 h, deferoxamine 500 µM (DFO 500 μM) (C,G) for 72 h, or 2,2′-dipyridyl 500 µM (BP 500 μM) (D,F) for 72 h during neuronal precursor cells (NPC) differentiation. Bar graph represents means ± SEM from n = 3 independent replicas where N = 5 randomly chosen fields were scored. Unpaired t-test, *p value < 0.05 and **p value < 0.01 versus untreated. Above the bar graphs are shown the representative fluorescence pictures of the corresponding conditions. Scale bar are 100 µm. Green fluorescence pictures show neurons stained with beta-III-tubulin (A), and blue fluorescence pictures show nuclei stained with Hoechst 33342 (E).
Similar to complete frh-1 deficiency (Ventura et al., 2005) or high doses of BP (Schiavi et al., 2015), a high concentration of CoCl2 also arrested development in C. elegans (Figure 2A). Most notably, we found that non-toxic doses of CoCl2 extend lifespan and healthspan (motility period) in a stage- and dose-dependent manner (Figures 2B,C; Table 1). This is in line with what has been previously observed with BP treatment or depletion of different mitochondrial electron transport chain (ETC) regulatory proteins including frh-1 (Dillin et al., 2002; Rea et al., 2007; Schiavi et al., 2015). Pro-longevity doses of CoCl2 also significantly reduced the animals’ size (Supplementary Figure S1), increased their resistance to heat shock (Figure 2D), and extended their fertility period without modifying the overall brood size (Figure 2E). These are all parameters also associated with mitochondrial-stress extension of lifespan.
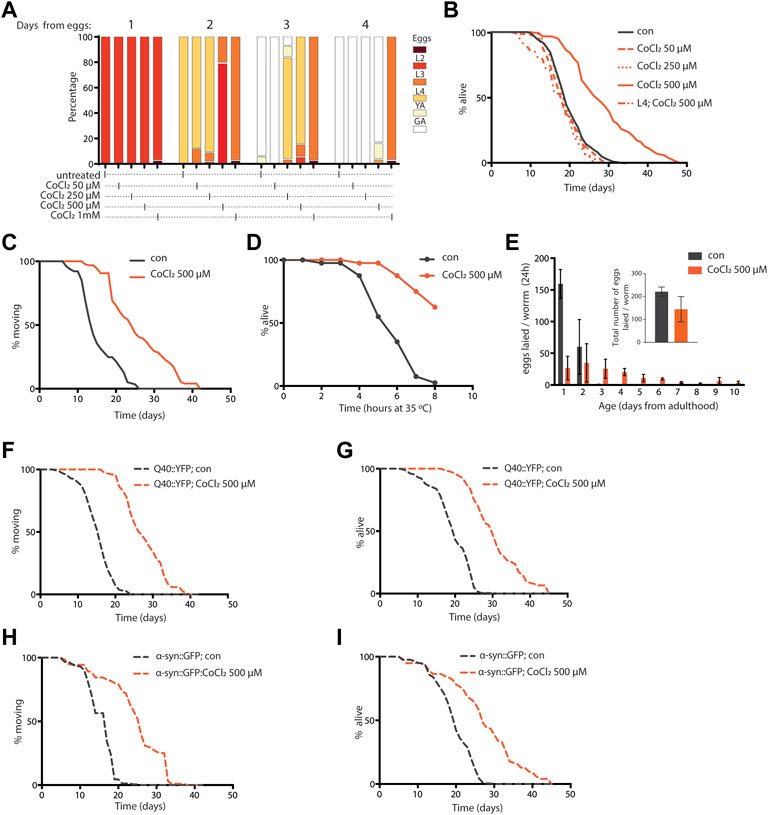
FIGURE 2. (A) Developmental rate of wild-type animals—from eggs through four different larval stages, until young (YA), and finally gravid (GA) adults—either left untreated or treated from eggs with 50 µM, 250 µM, 500 µM, and 1 mM cobalt chloride (CoCl2 50 µM, CoCl2 250 µM, CoCl2 500 µM, and CoCl2 1 mM), and followed for 4 days after hatching at 20°C. (B) Kaplan–Meier survival curves at 20°C of wild-type worms, left untreated (con) or treated from eggs with 50 µM, 250 µM, and 500 µM CoCl2 and starting from L4 larval stage with 500 µM CoCl2. Comparison between curves was performed using the log‐rank test (Table 1). (C,D) Animals’ locomotion activity (C) and survival against heat shock (D) of wild-type strain left untreated (con) or treated with 500 µM cobalt chloride (CoCl2 500 µM). Comparison between curves was performed using the log-rank test. (E) Fertility and brood size over time of wild-type worms treated as in (C). (F–I) Animal locomotion activity (F,H) and survival against heat shock (G,I) of unc-54p::Q40::YFP (F,G) or unc-54p::a-syn::YFP (H,I) strain left untreated (con) or treated with 500 µM cobalt chloride (CoCl2 500 µM). Comparison between curves was performed using the log-rank test.
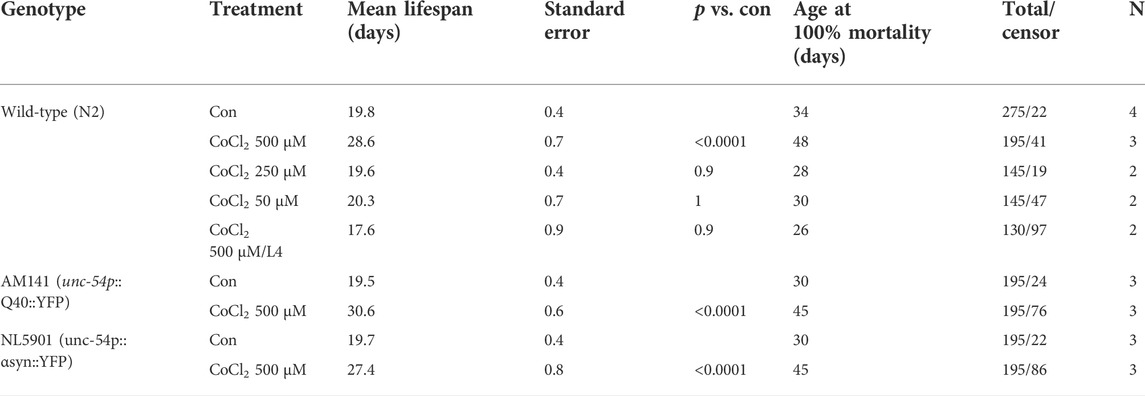
TABLE 1. Summary of lifespan analysis.
Excess iron is a typical hallmark of the aging process which concurs to the pathogenesis of age-associated neurodegenerative diseases, and progressive accumulation and toxicity of aggregation-prone proteins in these diseases are worsened by excess iron levels (Lee and Andersen, 2006; Schiavi et al., 2020). We thus wondered whether CoCl2 could promote beneficial effects in C. elegans strains expressing human aggregation-prone proteins, poly-Q40 and α-synuclein (under muscle-specific promote, unc-54), and widely exploited as model systems to respectively study Huntington’s and Parkinson’s diseases (Alexander et al., 2014; Caldwell et al., 2020; Liang et al., 2020). Notably, CoCl2 also significantly extended lifespan and locomotion ability in the Huntington’s and Parkinson’s disease models (Figures 2F–I). Moreover, we observed an age-dependent accumulation of poly-Q40 and α-synuclein aggregates, which was significantly reduced by pro-longevity CoCl2 treatment (Supplementary Figure S1). Thus, in C. elegans, CoCl2 extends health- and lifespan in a wild-type background as well as in age-associated disease models.
Given the striking similarity between animals’ phenotypes upon CoCl2 treatment and pro-longevity mitochondrial stress (Rea et al., 2007; Ventura et al., 2009), we then wondered whether the beneficial effects of CoCl2 preconditioning in C. elegans could be ascribed to the induction of mitochondrial stress response signaling. Interestingly, like frh-1 silencing (Ventura et al., 2009; Schiavi et al., 2015), pro-longevity doses of CoCl2 in C. elegans significantly increased the expression of genes normally induced by mitochondrial stress such as gst-4 (glutathione-S-transferase), hsp-6 (mitochondrial unfolded protein response), and nhr-57 (hypoxia–inducible gene) and reduced the expression of the mitophagy marker dct-1/BNIP3 (Figures 3A–D). CoCl2 also strongly induced the nuclear translocation of the autophagy regulator transcription factor hlh-30/TFEB, but only marginally affected the number of lgg-1/LC3 and of sqst-1/p62 foci, the other two central autophagy regulatory genes (Figures 3E–G). These changes suggest CoCl2 treatment induces mitochondrial stress that activates the autophagic machinery.
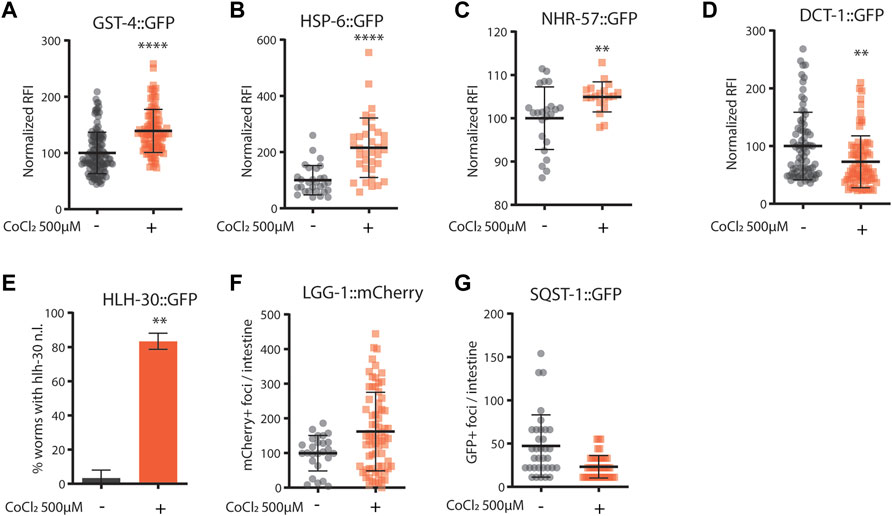
FIGURE 3. (A–G) Quantification of fluorescence reported strains in adult worms (4 days old) left untreated (−) or treated from eggs with CoCl2 500 µM (+). Relative fluorescence intensity normalized to animals’ size was quantified in GST-4::GFP (A), HSP-6::GFP (B), NHR-57::GFP (C), and DCT-1::GFP (D). Percentage of worms with HLH-30::GFP nuclear localization (E). Fluorescence foci number was quantified in LGG-1: mCherry (F) and SQST-1::GFP (G). Bar graph represents means ± SD (n = 3, N = 10–15); lines in the scatter plots represent means ± SD (n = 3, N = 10–15). Comparison between con and CoCl2 was performed using the unpaired t-test. **p value < 0.01 and **** p value < 0.0001, versus con.
In mammals, Bnip3/dct-1-regulated mitochondrial autophagy is induced in response to hypoxia in a HIF-1-dependent manner (Zhang et al., 2008; Bellot et al., 2009), and in C. elegans, frh-1 silencing extends lifespan via mitophagy activation and in a hif-1-dependent manner (Schiavi et al., 2015). Here, we find that the C. elegans HIF-1 homolog, hif-1, is required for the survival of CoCl2-treated animals. Indeed, while C. elegans wild-type animals treated with pro-longevity doses of CoCl2 reached the fertile stage slower than untreated animals (Figure 2A), hif-1 mutants completely arrested their development upon CoCl2 treatment (Supplementary Figure S2). Thus, CoCl2 activates a mitochondrial stress response likely in a hif-1-dependent manner.
To further investigate the potential protective effects of CoCl2 preconditioning, we then turned to mammalian cells. As expected, treatments with either high doses of CoCl2 (1 mM for 48 h) or severe hypoxia (<0, 1% O2 for 48 h) induced apoptosis in HeLa cells (Figures 4A,C). Notably, milder doses of CoCl2 provided protection against apoptosis either when applied as a pretreatment (250 uM for 24 h, followed by a recovery period of 4 h) before exposure to higher doses of CoCl2 (Figures 4A,B) or when co-applied (250 uM for 48 h) during severe hypoxia treatment (Figures 4C,D).
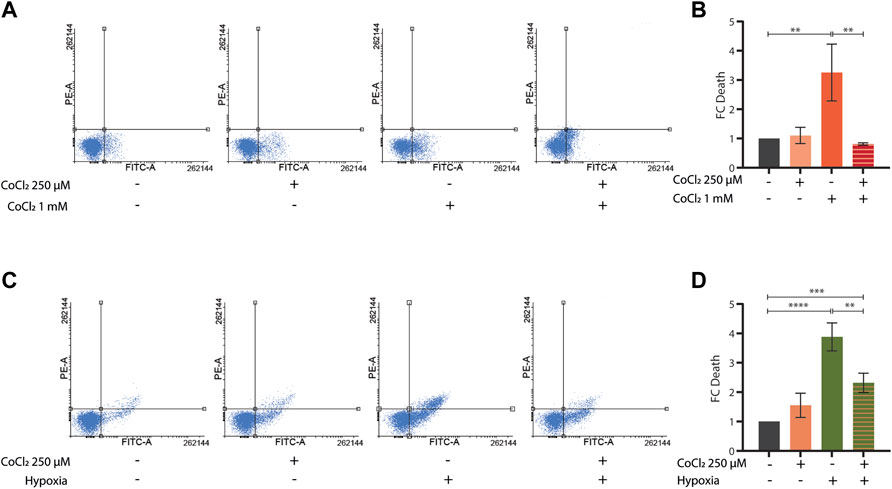
FIGURE 4. (A–D) Representative FACS plot (A,C) and quantification (B,D) of cell death assays performed in HeLa cells left untreated or treated with CoCl2 1 mM for 48 h (A,B) or severe hypoxia (<1% O2 for 48 h) (C,D) alone or in combination with either a CoCl2 250 µM pre-treatment for 24 h followed by 4 h recovery (A,B) or a CoCl2 250 µM co-treatment for 48 h, respectively. Bar graph represents means ± SD (n = 3); two-way ANOVA (Tukey’s multiple comparison test). **p value < 0.01 and **** p value < 0.0001.
Induction of autophagy and/or mitophagy is an attractive common determinant underlying the protection against aging and hypoxia induced by CoCl2 preconditioning across species. Indeed, these fundamental cellular turnover processes are required to protect cells against hypoxia-induced cell death (Zhang et al., 2008) or against proteotoxicity (Florez-McClure et al., 2007; Guerrero-Gomez et al., 2019) and to extend C. elegans lifespan upon other iron depleting interventions such as frh-1 RNAi or the iron chelator BP (Schiavi et al., 2013; Schiavi et al., 2015). Thus, to assess the effect of CoCl2 preconditioning on autophagy activity, we analyzed the LC3II status [a well-known autophagosome marker (Klionsky et al., 2016)] by Western blot in mammalian cells, at early time points (before apoptotic cell death was observed), upon mild doses of CoCl2 either alone or in combination with chloroquine [CQ, an inhibitor of the fusion between autophagosomes and lysosomes used to ascertain the integrity of the autophagic flux (Klionsky et al., 2016)]. Mild doses of CoCl2 significantly increased LC3II protein levels, which was further increased in the presence of CQ, thus clearly supporting the induction of intact autophagy flux by CoCl2 preconditioning (Figure 5A). We then wondered whether mild doses of CoCl2 also modulate the expression of p62/SQSTM1 protein, a targeted receptor for autophagic degradation of ubiquitinated substrates, which is often used as a reporter of autophagy activity (Klionsky et al., 2016). Surprisingly, LC3II accumulation upon CoCl2 preconditioning was paralleled by a chloroquine-insensitive induction of p62 (Figure 5B). This indicates that the autophagic flux induced by CoCl2 preconditioning is not sufficient to decrease the high levels of expression of p62 likely ascribed to a strong increase in its transcriptional expression and/or stability. Consistent with this possibility, exposure of preconditioned cells to higher doses of CoCl2 (1 mM) significantly increased LC3 turnover at early time points and decreased p62 expression, indicating a strong induction of autophagy compared to CoCl2 preconditioning alone (Figures 5C,D). The expressions of COXII and COXIV, two classical markers to reveal mitochondrial turnover (Klionsky et al., 2021), were instead inconsistently modulated (Figures 5E,F). To ascertain the effect of CoCl2 preconditioning in prompting autophagy, we then compared the effect of 1mM CoCl2 in HeLa cells pre-exposed or not to milder doses of CoCl2. Our data confirmed that the preconditioning favors LC3II conversion compared to cells treated only with 1 mM CoCl2 as well as a stronger decrease of p62 (Supplementary Figures S3A,B). Moreover, we noticed that 1 mM CoCl2 alone stimulates the production of COXII and COXIV after 16 h of treatment (Supplementary Figures S3C,D). Notably, at this early time point used to analyze autophagy, we could already observe morphological changes hinting CoCl2 preconditioning-induced protection in cells treated with 1 mM of CoCl2 compared to cells undergoing only the severe treatment (Supplementary Figure S4). Thus, according to a hormetic paradigm, CoCl2 preconditioning promotes healthspan likely via its hypoxia mimetic activity (compete with iron in HIF-1 activation), and autophagy induction may represent its underlying mechanism, which is in common with other treatments which limit iron availability (i.e., frh-1 RNAi, BP).
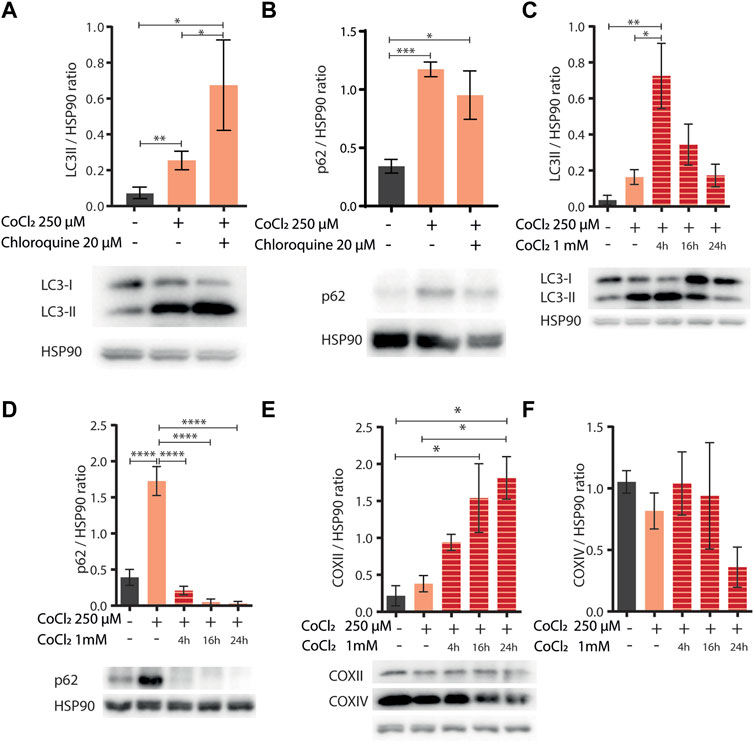
FIGURE 5. (A,B) Western blot analysis of LC3II (A) and p62 (B) proteins extracted from HeLa cells left untreated (−) or treated (+) with 250 µM of CoCl2 alone or in combination with 20 µM of chloroquine. Proteins were normalized using HSP90; below the bar graph are shown the representatives Western blot scan. Bar graphs represent means ± SD (N = 3), two-way ANOVA (Tukey’s multiple comparison test). *p value < 0.05 and **p value < 0.01. (C–F) Western blot analysis of LC3II (C), p62 (D), and COXII (E) and COXIV (F) proteins extracted from HeLa cells left untreated (−) or treated (+) with 250 µM of CoCl2 alone or in combination with 1 mM of CoCl2 at different exposure time 4, 16, and 24 h. Proteins were normalized using HSP90, and below the bar graph (C–E) are shown the representatives Western blot scan. Bar graph represents means ± SD (N = 3), two-way ANOVA (Tukey’s multiple comparison test). *p value < 0.05, **p value < 0.01, ***p value < 0.001, and ****p value < 0.0001.
Overall, our work provides evidence that CoCl2 preconditioning displayed evolutionarily conserved beneficial effects: it protects C. elegans against age- and proteotoxicity-induced neuromuscular damage and functional decline as well as mammalian cells against hypoxia-induced death. While we showed in the past that reducing the expression of the mitochondrial protein FRH-1 (via frh-1 silencing) extends lifespan through an iron starvation response and in a hif-1-dependent manner, whether CoCl2 promotes health span via hif-1-activation (through its iron competing effect) and/or also by inducing mitochondrial stress remains to be evaluated. Nonetheless, the ultimate effect of reducing iron availability is an improvement in health span, and this has important repercussions in light of the emerging role of hypoxia preconditioning in preventing neuromuscular deficits. Indeed, while the potential therapeutic benefits of hypoxia preconditioning are evident and have been suggested (Li et al., 2017; Caricati-Neto et al., 2019), fine-tuning oxygen levels to trigger beneficial effects without surpassing the critical threshold is not trivial and has low practical applications. Thus, the identification of more finely tunable interventions mimicking hypoxia, such as iron chelators already used in the clinic or undergoing clinical trials (Wijesinghe et al., 2021; Yang et al., 2021; Reddy et al., 2022) or modulators of downstream molecular players mediating their beneficial effects, may suggest more practicable strategies to interfere with the detrimental features associated with aging and severe hypoxia. Different phenotypic features are concurrently associated with the pro-longevity effect induced by mild doses of CoCl2 such as reduced animals’ size and egg-lay rate, prolonged fertility period, and increased resistance to stress (Ventura et al., 2005; Rea et al., 2007) and delayed neuromuscular decline during aging (Maglioni et al., 2014), which strikingly resemble those induced by mild mitochondrial stress. Notably, we showed that limiting iron availability with CoCl2 displays beneficial effects only when applied at non-toxic doses. This threshold effect has been previously demonstrated for HP, mitochondria hormesis (e.g., with frh-1 silencing), and the iron chelator BP, which only display beneficial properties at mild doses (Rea et al., 2007; Schiavi et al., 2015; Merry and Ristow, 2016; Li et al., 2017; Caricati-Neto et al., 2019). While HP has known beneficial effects against severe hypoxia-induced neuromuscular damage across species (Dasgupta et al., 2007), we show here that mild treatment with CoCl2 may also similarly provide protection against severe hypoxia, aging, or accumulation of proteotoxic proteins. It is worth noting that the beneficial effect of the different iron depleting agents is dose-dependent and similar to mitochondrial stress (Dillin et al., 2002; Rea et al., 2007), it is elicited only when the agents are applied at sub-lethal doses starting during animal development. This clearly indicates that mitochondria preconditioning-associated metabolic remodeling must occur early in life to promote protection against aging and age-associated features (Maglioni et al., 2019).
In search of a common molecular mechanism mediating the beneficial effects of the different iron modulating interventions, we initially considered hif-1-regulated autophagy or mitophagy. Indeed, autophagy protects against apoptosis (Marino et al., 2014) and against the toxicity of aggregation-prone proteins during aging (Florez-McClure et al., 2007; Guerrero-Gomez et al., 2019). Autophagy is also induced by hypoxia and by frataxin silencing across species and it is required to mediate the pro-longevity effect of frh-1-depletion in C. elegans (Schiavi et al., 2013; Schiavi et al., 2015). Our results in C. elegans clearly suggest that CoCl2 acts in a hif-1-dependent manner. Moreover, the combined data in worms and cells support the induction of bulk autophagy as a likely downstream mediator of the beneficial effect of CoCl2 preconditioning, but whether this is activated in a hif-1-dependent manner, via mitochondrial stress responses or other mechanisms, remains to be systematically investigated. Very interestingly, our data suggest that low doses of CoCl2 “prepare” the cells for better autophagy activation by subsequent higher doses of hypoxia-like stress [which would otherwise have deleterious effects on the cells (Pourpirali et al., 2015)]. This is a classical hormetic scenario (Cypser and Johnson, 2002; Gems and Partridge, 2008), and it will be important to establish whether and how CoCl2 preconditioning is also protective in other pathological or stressful contexts.
Paradoxically, our data point to a strong accumulation of p62 as a potential underlying mechanism of the CoCl2 preconditioning effect. While an increase p62 may indicate a block of autophagy flux, which in a more severe hypoxic scenario (higher doses of CoCl2) was linked to induction of apoptosis (Pourpirali et al., 2015), in our preconditioning paradigm, it could instead favor autophagy activation by additional stress. The overexpression of p62 by CoCl2 preconditioning could be ascribed on the one hand to its increased stability via different post-translational modifications, which might prevent its association with the E3 ligase Keap1 (Kelch-like ECH-associated protein 1) followed by poly-ubiquitination and degradation in the proteasome (Pan et al., 2016; Sun et al., 2016; Xu et al., 2019; Feng et al., 2021). On the other hand, overexpression of p62 could be induced by transcriptional activation for instance by the redox transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2). Since Nrf2 is also a substrate for Keap1, the binding of p62 to Keap1 results in a positive feedback loop leading to Nrf2 activation (Jain et al., 2010; Komatsu et al., 2010). Notably, we showed that CoCl2 in C. elegans increases the expression of gst-4 and of dct-1, two genes induced by mitochondrial stress in an Nrf2/skn-1-dependent manner (Ventura and Rea, 2007; Palikaras et al., 2015). While our combined findings in nematode and cells point to prompted activation of autophagy by CoCl2 preconditioning followed by more severe treatment, they do not unambiguously support the induction of mitophagy. These results may be ascribed by the masking effects of cell-specific or cell-non-autonomous processes but also to context-specific roles of p62. Indeed, p62 is not always required to mediate mitophagy (Narendra et al., 2010) and it may also modulate other forms of cell death such as ferroptosis [e.g., via Nrf2 activation (Sun et al., 2020; Chen et al., 2022; Tang et al., 2022)].
Interestingly, while frh-1 silencing induces both autophagy and mitophagy, the Nrf2/skn-1 pathway and extends lifespan in a hif-1-dependent manner, the iron chelator BP only induces mitochondrial but not bulk autophagy, induces the Nrf2/skn-1 pathway, and extends lifespan in a hif-1-independent manner (Schiavi et al., 2013; Schiavi et al., 2015). Moreover, while CoCl2 modulates gene expression in a hif-1-dependent manner across species (Triantafyllou et al., 2006; Padmanabha et al., 2015), it can also induce hypoxia-regulated genes, such as nhr-57, in a hif-1-independent manner (Padmanabha et al., 2015). Thus, while reducing iron availability can mimic the beneficial effects of HP, the common underlying molecular mechanism of the different hypoxia-mimetics may in fact be through hif-1-independent but Nrf2-related pathways.
Further studies will be required to actually clarify whether autophagy, mitophagy, or other forms of iron-regulated processes such as ferritinophagy or ferroptosis are involved in the protective effects of CoCl2 or other interventions reducing iron availability. In conclusion, we identified CoCl2 as a new agent protecting against hypoxia- and age-induced neuromuscular degeneration, possibly via hif-1- and/or Nrf2-regulated pathways.
The following strains were used in this study: N2 wild type, AM141: rmIs133 [unc-54p::Q40::YFP], NL5901: pkIs2386 [unc-54p::alpha-synuclein::YFP + unc-119(+)], CL2166: dvIs19[pAF15(gst-4p::GFP::NLS)], SJ4100: zcIs13[hsp-6p::GFP], ZG120: ials07[nhr-57p::GFP, unc-119(+)], JIN1679: jinEx10 [hlh-30p::hlh-30::GFP + rol-6(su1006)], vkEx1093: [nhx-2p::mCherry::lgg-1], HZ589: bpIs151[sqst-1p::SQST-1::GFP; unc-76(+)]IV; him-5(e1490)V, IR1431: N2;Ex001[dct-1p::DCT-1::GFP], ZG31: hif-1(ia4).
All strains were maintained and kept synchronized by egg lay at 20°C on nematode growth media (NGM) agar supplemented with Escherichia coli OP50 unless otherwise described.
Cobalt chloride (CoCl2 C8661, Sigma-Aldrich) was dissolved at 0.1 M stock solution in ddH2O, sterilized using a 0.22-μM filter, and supplemented to the NGM after autoclaving at the concentration of interest.
2,2′ dipyridyl (BP, 4153 Carl Roth) was dissolved in ddH2O and added to NGM to the indicated concentrations.
Survival analyses were carried out with the standard procedure in the field as routinely carried out in our laboratory (Torgovnick et al., 2018). Briefly, a synchronized population of 60–80 worms was used to start the lifespan analysis. Animals were transferred on fresh plates every day during the fertile phase and at the end of the fertile period, worms were transferred every other day. Animals not able to move upon prodding and with no pharyngeal pumping were scored as dead animals and were scored as not moving when no sinusoid locomotory activity was observed upon prodding. Survival analysis was performed in OASIS 2 using the Kaplan–Meier estimator. Statistical differences were evaluated using the log-rank test between the pooled population of worms, and p values were adjusted for multiple comparisons by the Bonferroni method.
Movement was assessed in the same populations used for the lifespan assay. Worms able to crawl spontaneously or after manual stimulus were considered as moving, while dead worms or animals without crawling behavior were considered as not moving. Statistical analysis was performed as described for lifespan.
The fecundity of animals was estimated by counting the eggs laid in a period of 4 h; three worms on four different plates were used in each condition and repeated in three independent trials.
Twenty wild-type animals for each condition were maintained from eggs at 20°C on plates with or without cobalt chloride 500 μM. On the first day of adulthood, they were moved to 35°C and the survival rate was counted every hour for 8 h.
The nematodes were placed in a 15 µl S-Basal plus levamisole 10 mM drop on a microscope glass slide, covered with a cover slide, and immediately imaged. Pictures were acquired with an Imager 2 Zeiss fluorescence microscope, with magnification 25-fold. The images were analyzed with the software ImageJ (http://imagej.nih.gov/ij/). The bright field images were used to generate the mask to select each single worm pictured, and consequently to measure the fluorescence in the correspondent fluorescent filter.
mCherry-positive foci from the Pnhx-2::mCherry::LGG-1 transgenic strain were counted in the intestine of young adult worms. To quantify p62::GFP puncta, the number of GFP foci was quantified in the anterior pharyngeal bulb of the Psqst-1:SQST-1::GFP transgenic strain. Worms were mounted on agarose pads, pictures were acquired using 100-fold magnification on a Zeiss Axio Imager 2 microscope, and the foci were analyzed with Fiji (Schindelin et al., 2012).
The number of worms showing HLH-30::GFP nuclear localization was visually counted using a Zeiss Axio Imager 2 (100-fold magnification). Worms were anesthetized using 10 mM levamisole in S-Basal and acquired within 5 min of mounting on glass slides. The percentage of worms with HLH-30 nuclear localization was calculated for each condition and plotted as mean ± SEM.
Poly-Q40 formations were acquired using a 25-fold magnification on a Zeiss Axio Imager 2 microscope. Worms were acquired at 4,7, and 10 days after hatching, and left untreated or treated from eggs with CoCl2 500 µM. The number of poly-Q40 aggregates was counted using the “analyze particles…” tools in ImageJ after thresholding and converting the acquired images in a binary form. The number of poly-Q40 was counted per worm by manually selecting the single worms. Worms were anesthetized using 10 mM levamisole in S-Basal and acquired within 5 min.
The human cervix carcinoma (HeLa) cell line was used in this study. The cells were incubated at 37°C with 5% CO2 and maintained in DMEM with 1.0 g/L glucose (PAN-Biotech GmbH, Germany) as a standard medium, supplemented with fetal bovine serum (FBS, Sigma-Aldrich), 10% and Penicillin/Streptomycin/Amphotericin B Mix (PAN-Biotech GmbH, Germany), 1%.
For cobalt chloride (CoCl2 15862, Sigma-Aldrich) treatment, it was directly added to the plates at the desired concentration. To generate the hypoxia status, the seeded cells were placed into a modular incubator chamber (Billups-Rothenberg). A gas mixture (95% N2 and 5% CO2) was flushed (15 l/min) into the chamber for 10 min and then incubated for the indicated time. After the different treatments, the cells were immediately collected for cell death detection.
Autophagosome–lysosome fusion was blocked with chloroquine (CQ) at 20 μM for 1 h (Sigma-Aldrich, United States). Images of HeLa cells after CoCl2 treatments were acquired using the ZOE Fluorescent Cell Imager (Bio-Rad, United States).
HeLa cells, untreated or treated with cobalt chloride and/or hypoxia, were washed with PBS and collected by centrifugation after trypsinization. The cell pellet was suspended in binding buffer (Annexin V-FITC kit; BioVision, Milpitas, CA, United States), followed by the addition of 5 μl Annexin V-labeling solution (BioVision) and 5 μl propidium iodide (BioVision). Cells were then incubated for 5 min at room temperature and analyzed via fluorescence-activated cell sorting (FACS; BD FACSCanto II).
HeLa cells were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris with pH 7.4, 1% Triton, 0.5% Nonidet P40, 10% glycerol, and 2.5% sodium deoxycholate) plus protease and phosphatase inhibitors (Roche Diagnostic, Germany, 11836153001). Protein concentrations were determined with the Bio-Rad Protein Assay Dye kit (Bio-Rad, United States, 5000006). Cell extracts were resolved by SDS-PAGE and transferred onto PVDF membranes (Millipore, Germany, Immobilon-P IPVH00010). Blocking was performed at room temperature in TBS 1X 0.1% Tween 5% low-fat milk for 1 h. Membranes were incubated with primary antibodies overnight at +4°C, followed by incubation with horseradish peroxidase-conjugate secondary antibodies (Bio-Rad) and revealed with western chemoluminescent HRP substrate (Millipore, Immobilon WBKLS0500). Chemoluminescent signals were acquired with the iBright CL1000 Imaging System (Thermo Fisher, United States). Quantitative analysis was performed using ImageJ software. Primary antibodies used were rabbit anti-p62 (MBL International Corporation, United States, PM045), mouse anti-COXII (Abcam, United Kingdom, ab110258), mouse anti-COXIV (Abcam, United Kingdom, ab33985), rabbit anti-LC3 A/B (Cell Signaling, United States, D3U4C, 12741S), rabbit anti-HSP90 (Cell Signalling; United States, E289, 4875), and mouse anti-vinculin (Santa Cruz Biotechnology, United States, 7F9, sc-73614).
Cryopreserved mouse neural progenitor cells were cultured at 37°C and 5% CO2 as a suspension culture in a proliferation medium consisting of Dulbecco’s modified Eagle medium (DMEM) supplemented with B27 (Invitrogen GmbH, Karlsruhe, Germany). When spheres reached 0.5 mm in diameter, they were chopped up to passage 3 with a McIlwain tissue chopper. Differentiation was initiated by growth factor withdrawal in a differentiation medium supplemented with N2 (insulin, transferrin, sodium selenite, putrescine, and progesterone; Invitrogen), plated onto poly-d-lysine/laminin–coated chamber slides (BD Bioscience, Erembodegem, Belgium). Cobalt chloride, deferoxamine, and 2,2′-dipyridyl were added at the desired concentration directly into the differentiation medium. The rate of neuronal differentiation and cell death were detected after 48 or 72 h of drug treatment.
Afterward, cells were fixed with 4% PFA (Sigma-Aldrich, Germany) for 30 min at 37°C and washed with PBS. Cells were stained with the primary antibody mouse-anti-βIII-tubulin for 1 h at 37°C. After washing with PBS, cells were incubated with the secondary antibody antimouse-Alexa-448 (Invitrogen, United States) for 30 min at 37°C. Nuclei were stained with Hoechst 33258 (Sigma-Aldrich, Germany). Samples were analyzed using a fluorescent microscope (Carl Zeiss, Germany) and AxioVision Rel.4.8 software (Carl Zeiss, Germany).
For the quantification of neuronal differentiation, the number of βIII-tubulin + cells in a given area was manually counted and divided by the total number of nuclei of the same area, which were counted automatically with ImageJ, and the result was expressed in percentage. Afterward, the values obtained for all exposed groups were expressed in percentage of the value of the control of the corresponding experiment. Cell death (necrosis) quantification was performed by counting cells stained with propidium iodide normalized to the total number of cells stained with Hoechst.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
NV conceived and supervised the study. AS, AR, MB, FS, and NV designed the experiments. AS, AR, TM, FDN performed the experiments. AS, AR, FDN, TM, FS, and NV analyzed the data. AS and NV wrote the paper. FS and EF edited the manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Laura Nimtz and Farina Bendt for technical help with the iPS-derived mouse neurospheres. This work was possible thanks to financial support to NV from the German Research Foundation (DFG grants VE366/3-1; VE366/6-1) and the Federal Ministry of Education and Research (JPI-HDHL, Grant no. 01EA1602). FS is supported by fondazione AIRC (Grant IG MFAG-2020 24467). We also thank the Caenorhabditis Genetics Center (funded by the National Institutes of Health Office of Research Infrastructure Programs: P40OD010440) as well as the National Bioresource Project (NBRP) for C. elegans strains.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.986835/full#supplementary-material
Supplementary Figure S1 | (A,B) Representative pictures (A) and quantification (B) of the poly-Q40 reporter strain AM141: rmIs133 [unc-54p::Q40::YFP] in 4-, 7-, and 10-days-old adult worms left untreated (−) or treated from eggs with CoCl2 500 µM (+). The number of poly-Q40 aggregates was counted using the “analyze particles…” tools in ImageJ after thresholding and converting the acquired images (right side of panel A) into a binary form (left side of panel A). (B) Bar graph represents means ± SD (n = 3, N=10–15). Comparison between untreated and CoCl2 at the same time point was performed using the unpaired t-test. *p value < 0.05 and **** p value < 0.0001. Scale bar = 100 µm.
Supplementary Figure S2 | Developmental rate of wild-type (WT) and hif-1 KO animals from eggs through four different larval stages, until young (YA) and gravid (GA) adults, either left untreated (−) or treated from eggs with 500 µM CoCl2 (+) and followed for 5 days after hatching at 20°C.
Supplementary Figure S3 | (A–D) Western blot analysis of LC3II (A), p62 (B), COXII (C), and COXIV (D) proteins extracted from HeLa cells left untreated (−) or treated (+) with 250 µM alone or in combination with 1mM of CoCl2 at different time points (4, 16, and 24 hours). Proteins were normalized using vinculin. Representative Western blots are shown. Bar graph represents means ± SD (N = 3). Comparison between groups was performed using the unpaired t-test. *p value < 0.05.
Supplementary Figure S4 | Representative pictures of HeLa cells left untreated (−) or treated (+) with 250 µM alone or in combination with 1 mM of CoCl2 (+) at different time points (4, 16, and 24 hours). White squares represent the magnified crop (2x) shown on the right. Scale bar = 100 µm.
Alexander, A. G., Marfil, V., and Li, C. (2014). Use of Caenorhabditis elegans as a model to study Alzheimer's disease and other neurodegenerative diseases. Front. Genet. 5, 279. doi:10.3389/fgene.2014.00279
Anzovino, A., Huang, M. L. H., and Richardson, D. R. (2014). Fixing frataxin: 'ironing out' the metabolic defect in friedreich's ataxia. Br. J. Pharmacol. 171 (8), 2174–2190. doi:10.1111/bph.12470
Bellot, G., Garcia-Medina, R., Gounon, P., Chiche, J., Roux, D., Pouyssegur, J., et al. (2009). Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29 (10), 2570–2581. doi:10.1128/MCB.00166-09
Caldwell, K. A., Willicott, C. W., and Caldwell, G. A. (2020). Modeling neurodegeneration in Caenorhabditis elegans. Dis. Model. Mech. 13 (10), dmm046110. doi:10.1242/dmm.046110
Caricati-Neto, A., Errante, P. R., and Menezes-Rodrigues, F. S. (2019). Recent advances in pharmacological and non-pharmacological strategies of cardioprotection. Int. J. Mol. Sci. 20 (16), E4002. doi:10.3390/ijms20164002
Chen, J., Zhang, J., Chen, T., Bao, S., Li, J., Wei, H., et al. (2022). Xiaojianzhong decoction attenuates gastric mucosal injury by activating the p62/Keap1/Nrf2 signaling pathway to inhibit ferroptosis. Biomed. Pharmacother. 155, 113631. doi:10.1016/j.biopha.2022.113631
Cypser, J. R., and Johnson, T. E. (2002). Multiple stressors in Caenorhabditis elegans induce stress hormesis and extended longevity. J. Gerontol. A Biol. Sci. Med. Sci. 57 (3), B109–B114. doi:10.1093/gerona/57.3.b109
Dasgupta, N., Patel, A. M., Scott, B. A., and Crowder, C. M. (2007). Hypoxic preconditioning requires the apoptosis protein CED-4 in C. elegans. Curr. Biol. 17 (22), 1954–1959. doi:10.1016/j.cub.2007.10.017
Dillin, A., Hsu, A. L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., et al. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298 (5602), 2398–2401. doi:10.1126/science.1077780
Feng, L., Chen, M., Li, Y., Li, M., Hu, S., Zhou, B., et al. (2021). Sirt1 deacetylates and stabilizes p62 to promote hepato-carcinogenesis. Cell. Death Dis. 12 (4), 405. doi:10.1038/s41419-021-03666-z
Florez-McClure, M. L., Hohsfield, L. A., Fonte, G., Bealor, M. T., and Link, C. D. (2007). Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy 3 (6), 569–580. doi:10.4161/auto.4776
Fritsche, E., Barenys, M., Klose, J., Masjosthusmann, S., Nimtz, L., Schmuck, M., et al. (2018). Current availability of stem cell-based in vitro methods for developmental neurotoxicity (DNT) testing. Toxicol. Sci. 165 (1), 21–30. doi:10.1093/toxsci/kfy178
Gems, D., and Partridge, L. (2008). Stress-response hormesis and aging: "that which does not kill us makes us stronger. Cell. Metab. 7 (3), 200–203. doi:10.1016/j.cmet.2008.01.001
Goldberg, M. A., Dunning, S. P., and Bunn, H. F. (1988). Regulation of the erythropoietin gene: Evidence that the oxygen sensor is a heme protein. Science 242 (4884), 1412–1415. doi:10.1126/science.2849206
Guerrero-Gomez, D., Mora-Lorca, J. A., Saenz-Narciso, B., Naranjo-Galindo, F. J., Munoz-Lobato, F., Parrado-Fernandez, C., et al. (2019). Loss of glutathione redox homeostasis impairs proteostasis by inhibiting autophagy-dependent protein degradation. Cell. Death Differ. 26 (9), 1545–1565. doi:10.1038/s41418-018-0270-9
Jain, A., Lamark, T., Sjottem, E., Larsen, K. B., Awuh, J. A., Overvatn, A., et al. (2010). p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 285 (29), 22576–22591. doi:10.1074/jbc.M110.118976
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12 (1), 1–222. doi:10.1080/15548627.2015.1100356
Klionsky, D. J., Abdel-Aziz, A K, Abdelfatah, S, Abdellatif, M, Abdoli, A, Abel, S, et al. (2021). Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy 17 (1), 1–382.doi:10.1080/15548627.2020.1797280
Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell. Biol. 12 (3), 213–223. doi:10.1038/ncb2021
Lee, D. W., Andersen, J. K., and Kaur, D. (2006). Iron dysregulation and neurodegeneration: The molecular connection. Mol. Interv. 6 (2), 89–97. doi:10.1124/mi.6.2.6
Lee, D. W., and Andersen, J. K. (2006). Role of HIF-1 in iron regulation: Potential therapeutic strategy for neurodegenerative disorders. Curr. Mol. Med. 6 (8), 883–893. doi:10.2174/156652406779010849
Lee, J. W., Ko, J., Ju, C., and Eltzschig, H. K. (2019). Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 51 (6), 1–13. doi:10.1038/s12276-019-0235-1
Li, S., Hafeez, A., Noorulla, F., Geng, X., Shao, G., Ren, C., et al. (2017). Preconditioning in neuroprotection: From hypoxia to ischemia. Prog. Neurobiol. 157, 79–91. doi:10.1016/j.pneurobio.2017.01.001
Liang, J. J. H., McKinnon, I. A., and Rankin, C. H. (2020). The contribution of C. elegans neurogenetics to understanding neurodegenerative diseases. J. Neurogenet. 34 (3-4), 527–548. doi:10.1080/01677063.2020.1803302
Liu, J., Gu, Y., Guo, M., and Ji, X. (2021). Neuroprotective effects and mechanisms of ischemic/hypoxic preconditioning on neurological diseases. CNS Neurosci. Ther. 27 (8), 869–882. doi:10.1111/cns.13642
Maglioni, S., Mello, D. F., Schiavi, A., Meyer, J. N., and Ventura, N. (2019). Mitochondrial bioenergetic changes during development as an indicator of C. elegans health-span. Aging (Albany NY) 11 (16), 6535–6554. doi:10.18632/aging.102208
Maglioni, S., Schiavi, A., Runci, A., Shaik, A., and Ventura, N. (2014). Mitochondrial stress extends lifespan in C. elegans through neuronal hormesis. Exp. Gerontol. 56, 89–98. doi:10.1016/j.exger.2014.03.026
Marino, G., Niso-Santano, M., Baehrecke, E. H., and Kroemer, G. (2014). Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 15 (2), 81–94. doi:10.1038/nrm3735
Merry, T. L., and Ristow, M. (2016). Mitohormesis in exercise training. Free Radic. Biol. Med. 98, 123–130. doi:10.1016/j.freeradbiomed.2015.11.032
Narendra, D., Kane, L. A., Hauser, D. N., Fearnley, I. M., and Youle, R. J. (2010). p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6 (8), 1090–1106. doi:10.4161/auto.6.8.13426
Padmanabha, D., Padilla, P. A., You, Y. J., and Baker, K. D. (2015). A HIF-independent mediator of transcriptional responses to oxygen deprivation in Caenorhabditis elegans. Genetics 199 (3), 739–748. doi:10.1534/genetics.114.173989
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi:10.1038/nature14300
Pan, J. A., Sun, Y., Jiang, Y. P., Bott, A. J., Jaber, N., Dou, Z., et al. (2016). TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol. Cell. 61 (5), 720–733. doi:10.1016/j.molcel.2016.02.007
Pourpirali, S., Valacca, C., Merlo, P., Rizza, S., D'Amico, S., and Cecconi, F. (2015). Prolonged pseudohypoxia targets Ambra1 mRNA to P-bodies for translational repression. PLoS One 10 (6), e0129750. doi:10.1371/journal.pone.0129750
Powell-Coffman, J. A. (2010). Hypoxia signaling and resistance in C. elegans. Trends Endocrinol. Metab. 21 (7), 435–440. doi:10.1016/j.tem.2010.02.006
Puccio, H., Simon, D., CosseeM., , Criqui-FiliPe, P., TizianoF., , Melki, J., et al. (2001). Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 27 (2), 181–186. doi:10.1038/84818
Rea, S. L., Ventura, N., and Johnson, T. E. (2007). Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 5 (10), e259. doi:10.1371/journal.pbio.0050259
Reddy, P. S., Locke, M., and Badawy, S. M. (2022). A systematic review of adherence to iron chelation therapy among children and adolescents with thalassemia. Ann. Med. 54 (1), 326–342. doi:10.1080/07853890.2022.2028894
Rodriguez, M., Snoek, L. B., De Bono, M., and Kammenga, J. E. (2013). Worms under stress: C. elegans stress response and its relevance to complex human disease and aging. Trends Genet. 29 (6), 367–374. doi:10.1016/j.tig.2013.01.010
Schiavi, A., Maglioni, S., Palikaras, K., Shaik, A., Strappazzon, F., Brinkmann, V., et al. (2015). Iron-starvation-induced mitophagy mediates lifespan extension upon mitochondrial stress in C. elegans. Curr. Biol. 25 (14), 1810–1822. doi:10.1016/j.cub.2015.05.059
Schiavi, A., Strappazzon, F., and Ventura, N. (2020). Mitophagy and iron: Two actors sharing the stage in age-associated neuronal pathologies. Mech. Ageing Dev. 188, 111252. doi:10.1016/j.mad.2020.111252
Schiavi, A., Torgovnick, A., Kell, A., Megalou, E., Castelein, N., Guccini, I., et al. (2013). Autophagy induction extends lifespan and reduces lipid content in response to frataxin silencing in C. elegans. Exp. Gerontol. 48 (2), 191–201. doi:10.1016/j.exger.2012.12.002
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9 (7), 676–682. doi:10.1038/nmeth.2019
Sun, X., Ou, Z., Chen, R., Niu, X., Chen, D., Kang, R., et al. (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63 (1), 173–184. doi:10.1002/hep.28251
Sun, Y., He, L., Wang, T., Hua, W., Qin, H., Wang, J., et al. (2020). Activation of p62-keap1-nrf2 pathway protects 6-hydroxydopamine-induced ferroptosis in dopaminergic cells. Mol. Neurobiol. 57 (11), 4628–4641. doi:10.1007/s12035-020-02049-3
Tang, K., Chen, Q., Liu, Y., Wang, L., and Lu, W. (2022). Combination of metformin and sorafenib induces ferroptosis of hepatocellular carcinoma through p62-keap1-nrf2 pathway. J. Cancer 13 (11), 3234–3243. doi:10.7150/jca.76618
Torgovnick, A., Schiavi, A., Shaik, A., Kassahun, H., Maglioni, S., Rea, S. L., et al. (2018). BRCA1 and BARD1 mediate apoptotic resistance but not longevity upon mitochondrial stress in Caenorhabditis elegans. EMBO Rep. 19 (12), e45856. doi:10.15252/embr.201845856
Triantafyllou, A., Liakos, P., Tsakalof, A., Georgatsou, E., Simos, G., and Bonanou, S. (2006). Cobalt induces hypoxia-inducible factor-1alpha (HIF-1alpha) in HeLa cells by an iron-independent, but ROS-PI-3K- and MAPK-dependent mechanism. Free Radic. Res. 40 (8), 847–856. doi:10.1080/10715760600730810
Ventura, N., Rea, S., Henderson, S. T., Condo, I., Johnson, T. E., and Testi, R. (2005). Reduced expression of frataxin extends the lifespan of Caenorhabditis elegans. Aging Cell. 4 (2), 109–112. doi:10.1111/j.1474-9726.2005.00149.x
Ventura, N., and Rea, S. L. (2007). Caenorhabditis elegans mitochondrial mutants as an investigative tool to study human neurodegenerative diseases associated with mitochondrial dysfunction. Biotechnol. J. 2 (5), 584–595. doi:10.1002/biot.200600248
Ventura, N., Rea, S. L., Schiavi, A., Torgovnick, A., Testi, R., and Johnson, T. E. (2009). p53/CEP-1 increases or decreases lifespan, depending on level of mitochondrial bioenergetic stress. Aging Cell. 8 (4), 380–393. doi:10.1111/j.1474-9726.2009.00482.x
Wenger, R. H. (2002). Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 16 (10), 1151–1162. doi:10.1096/fj.01-0944rev
Wijesinghe, T. P., Dharmasivam, M., Dai, C. C., and Richardson, D. R. (2021). Innovative therapies for neuroblastoma: The surprisingly potent role of iron chelation in up-regulating metastasis and tumor suppressors and down-regulating the key oncogene, N-myc. Pharmacol. Res. 173, 105889. doi:10.1016/j.phrs.2021.105889
Xu, D., Li, X., Shao, F., Lv, G., Lv, H., Lee, J. H., et al. (2019). The protein kinase activity of fructokinase A specifies the antioxidant responses of tumor cells by phosphorylating p62. Sci. Adv. 5 (4), eaav4570. doi:10.1126/sciadv.aav4570
Keywords: aging, C. elegans, cobalt chloride, iron, healthspan, hormesis, neurodegeneration
Citation: Schiavi A, Runci A, Maiorino T, Naso FD, Barenys M, Fritsche E, Strappazzon F and Ventura N (2022) Cobalt chloride has beneficial effects across species through a hormetic mechanism. Front. Cell Dev. Biol. 10:986835. doi: 10.3389/fcell.2022.986835
Received: 05 July 2022; Accepted: 05 October 2022;
Published: 25 October 2022.
Edited by:
Konstantinos Palikaras, University of Athens, GreeceReviewed by:
Nikolaos Charmpilas, University of Cologne, GermanyCopyright © 2022 Schiavi, Runci, Maiorino, Naso, Barenys, Fritsche, Strappazzon and Ventura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natascia Ventura, bmF0YXNjaWEudmVudHVyYUB1bmktZHVlc3NlbGRvcmYuZGU=
‡Present Address: Marta Barenys, Department of Pharmacology, Toxicology and Therapeutic Chemistry, University of Barcelona, Barcelona, SpainTeresa Maiorino, Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università degli Studi di Napoli Federico II, Napoli, Italy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.