 Wei Su
Wei Su Stan W. van Wijk
Stan W. van Wijk Bianca J. J. M. Brundel
Bianca J. J. M. Brundel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 09 September 2022
Sec. Cellular Biochemistry
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.986718
This article is part of the Research TopicProteostasis in Cardiac Health and DiseaseView all 5 articles
Desmin (DES) is a classical type III intermediate filament protein encoded by the DES gene. Desmin is abundantly expressed in cardiac, skeletal, and smooth muscle cells. In these cells, desmin interconnects several protein-protein complexes that cover cell-cell contact, intracellular organelles such as mitochondria and the nucleus, and the cytoskeletal network. The extra- and intracellular localization of the desmin network reveals its crucial role in maintaining the structural and mechanical integrity of cells. In the heart, desmin is present in specific structures of the cardiac conduction system including the sinoatrial node, atrioventricular node, and His-Purkinje system. Genetic variations and loss of desmin drive a variety of conditions, so-called desminopathies, which include desmin-related cardiomyopathy, conduction system-related atrial and ventricular arrhythmias, and sudden cardiac death. The severe cardiac disease outcomes emphasize the clinical need to understand the molecular and cellular role of desmin driving desminopathies. As the role of desmin in cardiomyopathies has been discussed thoroughly, the current review is focused on the role of desmin impairment as a trigger for cardiac arrhythmias. Here, the molecular and cellular mechanisms of desmin to underlie a healthy cardiac conduction system and how impaired desmin triggers cardiac arrhythmias, including atrial fibrillation, are discussed. Furthermore, an overview of available (genetic) desmin model systems for experimental cardiac arrhythmia studies is provided. Finally, potential implications for future clinical treatments of cardiac arrhythmias directed at desmin are highlighted.
For a healthy function of the heart, the crucial importance of intermediate filament (IF) proteins to maintain balanced communication within and between neighbouring cardiomyocytes has been recognized (Hnia et al., 2015; Henning and Brundel, 2017; Brodehl et al., 2018). Desmin is a key IF subunit expressed in specialized cardiac cell subpopulations related to the cardiac conduction system, including the sinoatrial node (Mavroidis et al., 2020), atrioventricular node (Benvenuti et al., 2012), and His-Purkinje system (Yuri et al., 2007). As such, desmin controls the structural and mechanical integrity of the contractile apparatus and is involved in the conduction of electrical signals within the heart (Tsikitis et al., 2018).
The important function of desmin in the cardiac conduction system is related to the distinctive property of desmin to form networks that connect and anchor various cell structures and organelles including desmosomes, costameres, Z-bands, the cytoskeleton, mitochondria, and nuclei (Henning and Brundel, 2017). In addition, desmin binds to various proteins within cardiomyocytes, modulating a variety of (Henning and Brundel, 2017) signaling pathways to maintain a healthy cardiomyocyte function. As a consequence, variations in the desmin (DES) gene have been reported in a number of cardiac diseases including atrial and ventricular arrhythmias, as well as hypertrophic-, restrictive-, dilated-, and non-compaction cardiomyopathy (Protonotarios et al., 2021). Although various papers describe the role of DES variants underlying cardiomyopathies, limited information is available on the molecular origin of DES variants, loss of expression, and the onset of cardiac arrhythmias, including atrial fibrillation (AF). As cardiomyocytes within the cardiac conduction system are particularly prone to age-related desmin dysfunction and consequently structural and functional impairment, desmin dysfunction increases the likelihood of arrhythmias and the requirement for pacemaker implantation in the growing aging population (Goldfarb et al., 2004). This review will provide up-to-date insight into the molecular and cellular mechanisms of desmin to underlie a healthy cardiac conduction system and how impaired desmin triggers cardiac arrhythmias, including AF. Furthermore, an overview of available genetic desmin model systems for experimental cardiac arrhythmia is provided. Finally, potential implications for future clinical treatments of cardiac arrhythmias directed at desmin are discussed.
The distribution pattern of high desmin levels in the heart elucidates its role in the cardiac conduction system (Yamamoto et al., 2011). Immunohistochemical staining of human embryonic hearts described the spatial expression of desmin at different developmental stages. According to Carnegie stages, desmin is first expressed in the myocardial wall of the atrioventricular canal and the upper region of the primary ring at stage 11. During stage 12 through stage 13 of cardiac development, desmin is expressed in the primordium of the sinus node and right venous valve. When septation of the heart is almost complete, at late embryo stage 20, desmin is widely expressed in the primordium of the atrioventricular node, the atrioventricular bundle, the bundle branches, and at the entire ventricular trabeculations (Liu et al., 2020). The distribution of desmin throughout the conduction system and the key role of the desmin network in the maintenance of cardiomyocyte structure and mechanical function provides a direct morphological basis for investigating the mechanism of arrhythmogenesis caused by desmin impairment. This is supported by previous studies, which showed that genetic variations of DES and loss of desmin expression trigger cardiac arrhythmia (Liu et al., 2020).
Desmin is abundantly expressed in cardiomyocytes and represents one of the type III intermediate filaments. The important function of desmin in the cardiac conduction system is related to the distinctive property of desmin to form intra- and intercellular networks by connecting and anchoring various cytoskeletal structures and organelles, including desmosomes, mitochondria, nuclei, costameres, and Z-bands (Figure 1) (Herrmann and Aebi, 2004; Brodehl et al., 2018).
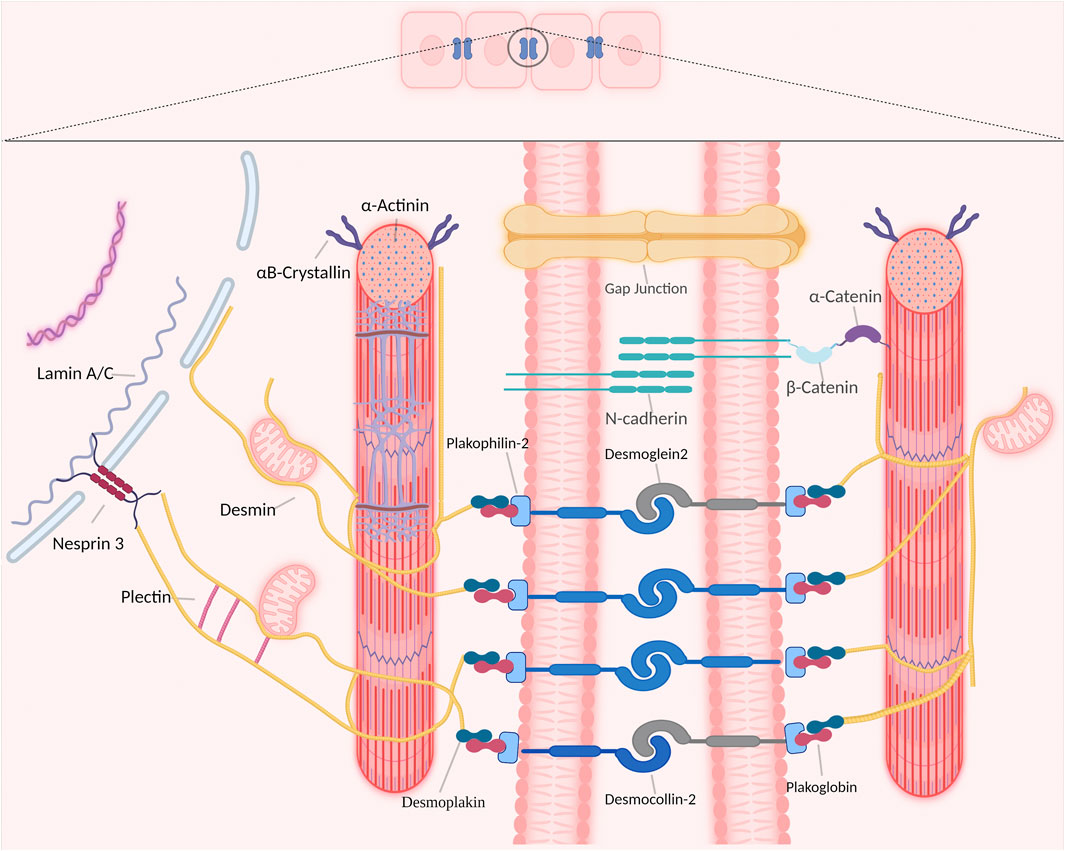
FIGURE 1. Overview of desmin network in and between cardiomyocytes. Desmin interacts with different organelles (including nucleus and mitochondria) and connects the entire contractile apparatus to different membranous compartments (such as sarcolemma, costameres and desmosomes of the intercalated disks) to form a continuous cytoskeletal intermediate filaments network.
In cardiomyocytes, the desmin network plays an important role in striated myocardium development and maintenance by integrating and coordinating most cellular components necessary for proper mechanochemical signaling, organelle cross-talk, energy production, and trafficking processes required for proper tissue homeostasis (Capetanaki et al., 2015). Desmin interacts directly with various proteins within cardiomyocytes, as such desmin modulates a diversity of signaling pathways to maintain a healthy cardiomyocyte function. Desmin interacts directly with other members of the intermediate filaments family, costameres, cytolinkers bridging organelles and cytoskeleton, and the LINC-complex protein nesprin-3 that is present in the nuclear membrane (Figure 1) (Hol and Capetanaki, 2017). In addition, desmin binds indirectly to posttranslational modifications and signaling pathways that are important for proper skeletal or cardiac muscle functions (Figure 1).
Although it has been thoroughly described that desmin interacts with mitochondria, the nature of this interaction is not fully understood. Binding can be indirect through desmin-associated proteins, such as plectin. Moreover, the direct binding of desmin with the lipid phosphatase myotubularin has been shown to regulate mitochondrial dynamics, morphology, and function (Hnia et al., 2011). However, recent data shows that mitochondria can directly interact with desmin in vitro (Dayal et al., 2020). Mitochondrial function and structure abnormalities seem to be the earliest detected defects in desmin knockout cardiomyocytes. These defects include morphological aberrations in the form of mitochondrial swelling, increased mitochondrial size, disrupted cristae structure, loss in respiration, abnormal activation of mitochondrial permeability transition pore (mPTP), and dissipation of the mitochondrial membrane potential (Diokmetzidou et al., 2016). These alterations are known to play a role in cardiac arrhythmias, therefore a possible association between desmin disruption and the onset of cardiac arrhythmias has been suggested (Rutledge and Dudley, 2013; Pool et al., 2021).
Recently, the various binding partners of desmin have been elucidated (Hnia et al., 2015). Based on published data (Hnia et al., 2015; Hol and Capetanaki, 2017) and interaction databases BioGRID4.4, the interactome of desmin binding partners has been depicted (Figure 2). Here, we introduce three categories of desmin protein partners in detail: intermediate filaments, intercalated discs, and αB-crystallin.
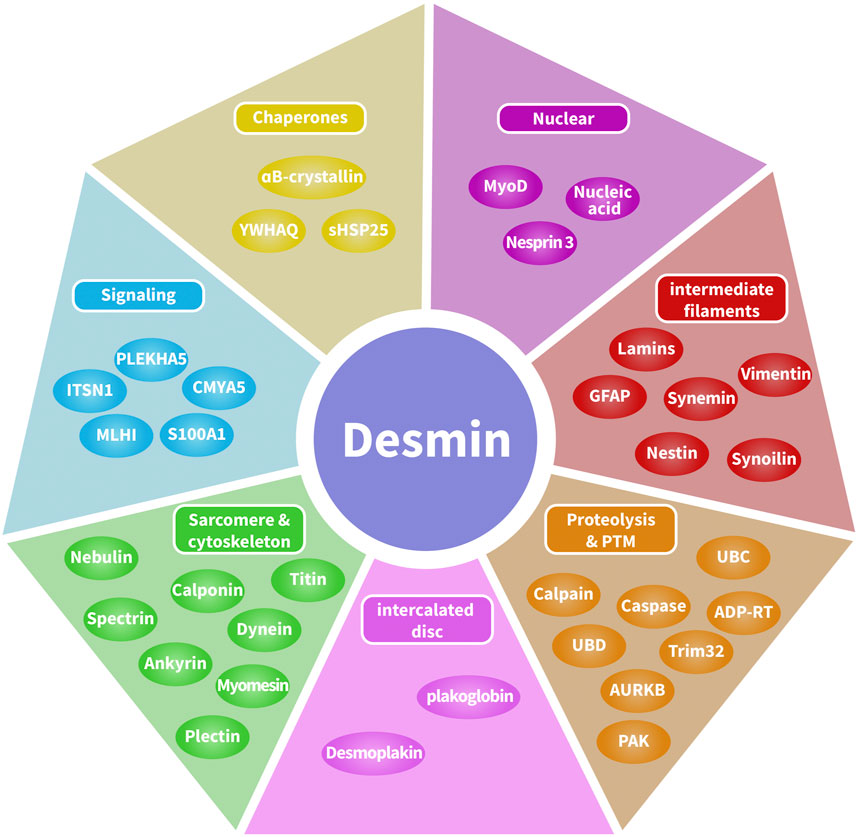
FIGURE 2. Desmin interactome map. Desmin-associated proteins include chaperones, DNA-related proteins, intermediate filaments, proteolysis and post-translational modifications, cytolinkers bridging sarcomeres and the cytoskeleton, and cellular signaling. Desmin-associated proteins are localized in multiple compartments, including costameres, contractile apparatus, intercalated discs, lysosomes, mitochondria, and the nucleus.
In muscle cells, desmin interacts with type III intermediate filament protein vimentin (Shahraz et al., 2017) and syncoilin (Poon et al., 2002), type IV intermediate filament protein synemin (Granger and Lazarides, 1980) and nestin (Cízková et al., 2009), and type V intermediate filament protein lamin (Cartaud et al., 1995). Lamins consist of two types, type A and type B, according to their structural similarities and isoelectric points. Lamin type A consists of lamin A and lamin C, both of them transcribed from LMNA and turned into lamin A and lamin C by alternative splicing. Lamin A/C is located in the nuclear interior. Lamin B is transcribed from LMNB, and along with heterochromatin, is anchored to the inner surface of the nuclear membrane by the lamin B receptor. Lamin B has been suggested to be a direct binding partner of desmin (Georgatos and Blobel, 1987). Interestingly, a desmin depletion leads to infolding of the nuclear envelope, loss of the nuclear integrity, increased amount of DNA damage, and diminished contractile function (Heffler et al., 2020). Similarly, the anchoring of desmin to the nucleus is lost in a Lamin A/C (Lmna−/−) knockout mice model mice, which causes disorganization of the desmin (Nikolova et al., 2004). Furthermore, a known pathogenic lamin A/C variations can lead to desmin dysfunction as was shown in a cardiomyopathy LmnaH222P/H222P mouse model. Disturbance in the nuclear lamina introduced due to the Lmna variation leads to mislocalization of desmin at the Z-disk and intercalated disk, and desmin aggregate formation. Interestingly, overexpression of the small heat shock protein αB-crystallin or reduction of endogenous desmin, improved LmnaH222P/H222P cardiac function and pathology (Galata et al., 2018). Syncoilin is found at the neuromuscular junction, sarcolemma, and Z-lines, which are involved in the anchoring of the IF network at the sarcolemma and the neuromuscular junction. The dysfunction of syncoilin may result in the disruption of the IF network (Poon et al., 2002). Vimentin is expressed in mesenchymal cells, vimentin together with desmin and nestin play a significant role in the construction and restoration of skeletal myofibers (Sjöberg et al., 1994).
Desmin interacts with the desmosomes of the intercalated discs through desmoplakin (Capetanaki et al., 2015). Intercalated discs consist of three types of cellular junctions: adherent junctions, desmosomes, and gap junctions. Intercalated discs are structures that connect adjacent cardiomyocytes, which are crucial for cell-cell mechanical and electrical connections, and as such are fundamental for cardiac function. In desmin knockout mice, changes in the morphology of intercalated discs were observed (Thornell et al., 1997). By immunohistological analysis of cardiac tissue heterozygous for the DES p.A120D mutation, Brodehl et al. (2013) found that desmin localization is completely lost at the intercalated discs. An impaired desmin network may slow electrical conduction, enhance conduction heterogeneity, and predispose patients to develop reentrant arrhythmias (Capetanaki et al., 2015).
Transient transfection of H36CE cells with the small heat shock protein αB-crystallin (encoded by CRYAB) indicate that αB-crystallin and desmin form a functional complex (Elliott et al., 2013). The association of desmin with the αB-crystallin, and the fact that both the CRYAB p.R120G variant and DES variants lead to cardiac arrhythmias in mice (Jiao et al., 2014), and CRYAB variant p.D109G and DES variants lead to restrictive cardiomyopathy (Brodehl et al., 2017), suggest a potential compensatory interplay between the two in cardioprotection. Both proteins co-localize at the mitochondria-SR contact sites (Diokmetzidou et al., 2016), and disruption of the mitochonrida-SR contact sites has been suggested to underlie AF (Henning and Brundel, 2017; Wiersma et al., 2017; Li et al., 2019). Overexpression of αB-crystallin in desmin-deficient mice hearts ameliorates all mitochondrial defects and improves cardiac function significantly (Diokmetzidou et al., 2016).
Because desmin is located in different human tissues, the clinical phenotypes associated with DES variants are diverse. So far, over 60 different pathogenic DES variants have been described (Hnia et al., 2015). Most patients with DES variants develop combined skeletal and cardiac myopathy.
A limited amount of variants in desmin genes have been identified that affect exclusively cardiac function (for details, see Table 1). A meta-analysis including 40 different DES variants revealed that 50% of the carriers developed cardiomyopathy and around 60% cardiac conduction disease (CCD) or arrhythmias, with the atrioventricular block (AVB) as an important hallmark (van Spaendonck-Zwarts et al., 2011). DES variants with a pathogenic single nucleotide polymorphism located in coil 2B or near the carboxyl terminus of the gene, usually result into missense mutations that associate with conduction diseases and arrhythmias (Figure 3; Table 1). Moreover, several studies showed that single nucleotide polymorphisms (SNPs) around DES c.375G leads to a splice variant which excludes exon 3 (p.D214-E245del) (Ojrzyńska et al., 2017; Fan et al., 2019; Brodehl et al., 2021; Chen et al., 2021). The carboxyl tail of desmin has previously been suggested to play an important role in filament-filament interactions, and variations lead to filament flexibility and stiffening of the filament. Furthermore, Bär et al. (2010) suggest that variations in the carboxyl tail lead to impaired cellular mechanosensing and intracellular mechanotransduction (Bär et al., 2007; Bär et al., 2010). The rod part of desmin is made up of four coil domains and consists of a heptad-repeat arrangement which enables desmin to form stable parallel, two-stranded α-helical coiled-coil dimers. Notably, the end of the coil 2B region of desmin contains the “IF-consensus” motif: ‘“TYRKLLEGEESRI” (amino acid 404–416) which is important in filament assembly, stability, and contains multiple pathogenic variants (Figure 3; Table 1) (Bär et al., 2004; Herrmann and Aebi, 2004). So, gene variants located in coil 2B and/or carboxyl terminus of desmin are associated with clinical arrhythmias. As these locations are involved in filament stability and interactions, loss in filament network may trigger DES variant-induced cardiac arrhythmias. In addition, a previous study showed that DES p.N116S, a variant located in the conserved IF ‘LNDR’-motif which is located in the coil 1A region, leads to disturbed desmin filament formation and fuels aggresome formation (Figure 3).
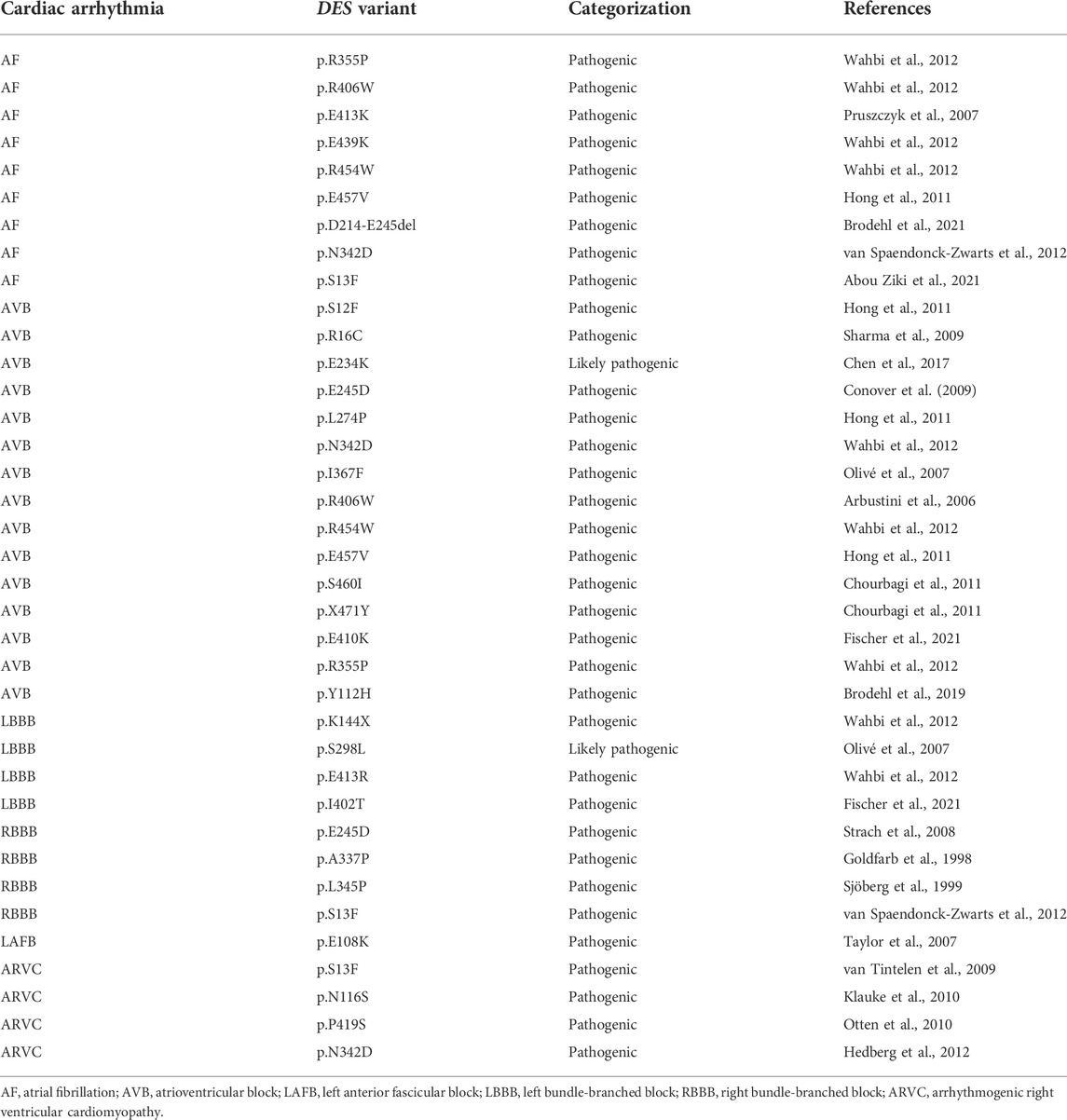
TABLE 1. Overview of different types of cardiac conduction disease and arrhythmias related to DES variant carriers.
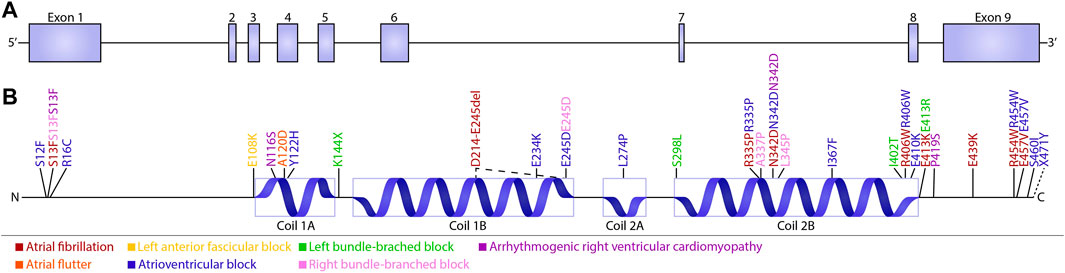
FIGURE 3. Schematic overview of cardiac conduction defects and arrhythmias associated with DES variants. (A) Schematic overview of the DES gene consisting of nine exons. (B) Schematic domain organization of desmin and the localization of the known conduction defects and arrhythmias associated with DES variants. Variants are subdivided into five groups depending on their related phenotype: Atrial fibrillation (brown), Left anterior fascicular block (yellow), Left bundle-branched block (green), Arrhythmogenic right ventricular cardiomyopathy (purple), Atrial flutter (orange), atrioventricular block (blue), and Right bundle-branched block (pink).
By utilizing DES variant model systems, the most obvious pathological hallmark for cardiac dysfuntion is an abnormal cytoplasmic configuration of the desmin network and desmin aggregation formation. Desmin aggregation has been observed in subsarcolemmal, intermyofibrillar, and perinuclear regions (Carlsson et al., 2002). In mice overexpressing desmin with a seven amino acid deletion, toxic aggregates of desmin were observed. As desmin normally interacts with other cytoskeletal proteins and organelles, loss in this network will disrupt the continuity and overall organization of cell structure from the sarcolemma to the nuclear envelope (Wang et al., 2001). One patient with a homozygous missense mutation in the DES gene (p.Y122H) was diagnosed with restrictive cardiomyopathy and AVB. By generating an induced pluripotent stem cell (iPSC) model expressing p.Y122H in combination with functional analysis, Brodehl et al. (2019) showed that variant p.Y122H caused severe filament assembly defect and desmin aggregation, which may drive AVB.
Regarding cardiac conduction diseases, AVB may be associated with anatomical interruption of the atrioventricular conduction system. This pathological phenotype has been reported in the cardiac conduction system of one patient with p.A337P variation in the DES gene (Figure 4). This is an autopsy case from a Japanese man who showed His bundle calcification and left and right bundle branches with sporadic calcium deposits (Yuri et al., 2007). Conversely, another autopsy case from Brazil showed no calcification of the atrioventricular junction and His bundle, there was extensive fibrosis of the terminal portions of the branching bundle and the beginning of the left and right bundles at the top of the ventricular septum (Benvenuti et al., 2012). Further studies are needed to elucidate the role of desmin disruption, calcification and onset of AVB.
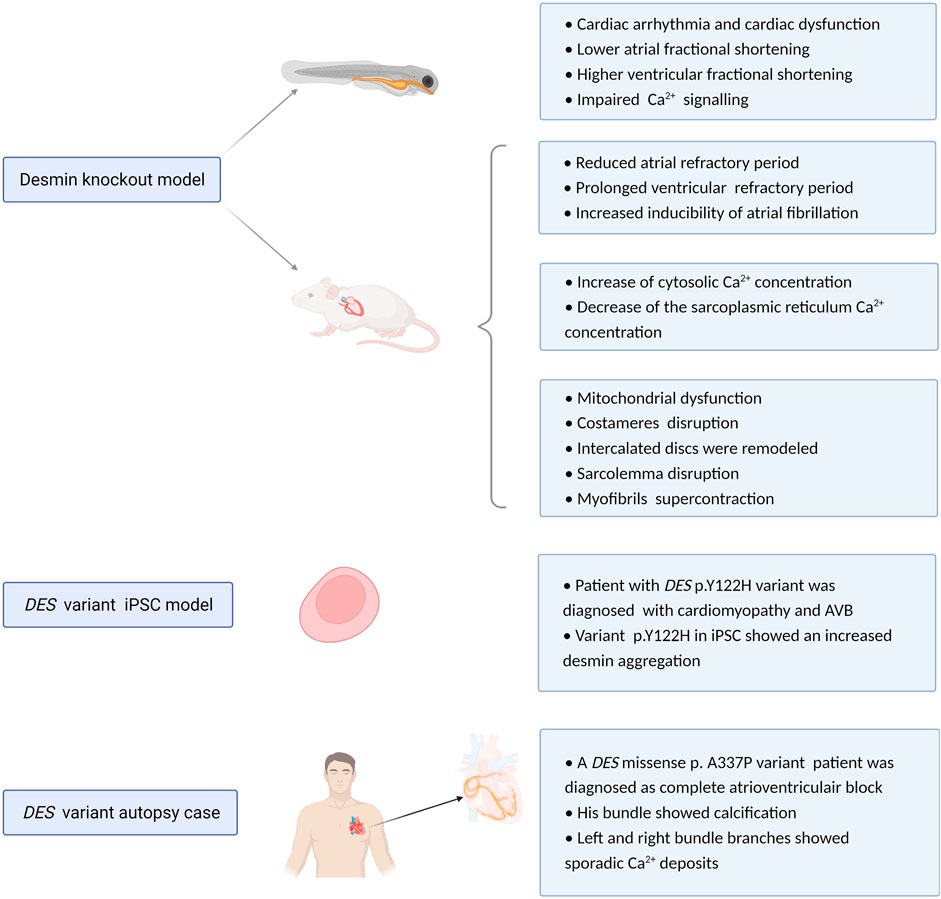
FIGURE 4. Overview potential pathophysiological mechanisms related to DES variants or loss. Experimental desmin knockout and variant model systems reveal a role for desmin impairment in development of cardiac conduction defects and arrhythmias. Proposed mechanism of action is via altered refractory periods, cytosolic Ca2+ concentrations, loss in desmin network, protein aggregation, and calcification.
Previous studies utilizing DES knockout mouse models showed that these mice develop and reproduce normally and display no obvious anatomical defects (Li et al., 1997). However, ultrastructural studies of heart tissue samples from DES knockout mice reveal damage including swollen, disintegrated and abnormal distribution of mitochondria, all features that are indicative of mitochondrial dysfunction (Agbulut et al., 2001). Furthermore, costameres were found disrupted, especially at Z-domains (O'Neill et al., 2002). Five days post-partum, cardiomyofibers degenerate and 10 days post-partum, this was complemented with an accumulation of macrophages, fibrosis, and calcification of the inter-ventricular septum and the free wall of the right ventricle (Sprinkart et al., 2012). Also, at the ultrastructural level, intercalated discs were remodeled, sarcolemma disruption and myofibrils showed super contraction phenotype (Thornell et al., 1997).
In electrocardiography studies, desmin deficient mice present a significantly reduced atrial but prolonged ventricular refractory period, indicating increased inducibility of atrial arrhythmias but diminished susceptibility to ventricular arrhythmias (Schrickel et al., 2010). Moreover, knockdown of desmin in cardiomyocytes results in an abnormal distribution of Ca2+. Here, a marked increase in cytosolic Ca2+ concentration and a decrease in the sarcoplasmic reticulum Ca2+ concentration was found. As cytosolic Ca2+ overload is a trigger for AF (Brundel et al., 2002a; Brundel et al., 2002b) and Ca2+ is a critical element in the electrical excitation of cardiomyocytes, abnormal Ca2+ homeostasis may represent a mechanism by which desmin loss participates in cardiac arrhythmia (Chen et al., 2020).
Compared with mouse models, utilization of zebrafish as a model systems is easier to analyze the effect of desmin knock out on desmin aggregation in the heart. Ramspacher et al. (2015) generated two zebrafish models to compare a desmin loss of function model with desmin aggregate formation model. They found that both models led to cardiac arrhythmia and cardiac dysfunction. These defects are related to abnormal Ca2+ flux due to the disruption of excitation-contraction coupling machinery and abnormal subcellular localization of ryanodine receptor (Figure 4).
Desmin aggregation in cardiomyocytes is the most significant histopathological hallmark of desmin cardiomyopathies. Moreover, desmin aggregation leads to cardiomyopathy phenotypes (Ramspacher et al., 2015). One of the critical steps for a future therapeutic approach to desmin cardiomyopathies is to characterize representative animal models (e.g., mice, zebrafish) that phenocopy desmin aggregation in patients. Small heat shock proteins, including HSP27, αA-crystallin, αB-crystallin, and HSP22 prevent protein accumulation and aggregation formation (Garrido et al., 2012). One previous study showed that the non-toxic HSP inducer geranylgeranylacetone (GGA), a nontoxic antiulcer drug and inducer of small HSPs can inhibit desmin-related cardiomyopathy progression (Sanbe et al., 2009). This study indicated that GGA can induce expression of HSPB8 and HSPB1 inhibit protein aggregation. GGA led to a reduction in heart size and inhibition of interstitial fibrosis, and recovery of cardiac function as well as improved survival (Sanbe et al., 2009). Additionally, manipulating cell signaling pathways (i.e., PAK1, Rac1, PKC, or NSC23766), activating autophagy (mTOR inhibitor PP242), and using antioxidants (α-tocopherols or trolox) efficiently reduces up to 75% of aggregation of desmin variants in muscle cells (Cabet et al., 2015).
Targeting DES gene regulation could be a kind of effective treatment that leads to a decreased expression of the mutant DES allele. Such as RNA-targeted therapeutics, and methods based on DNA genome editing using CRISPR-Cas9 (Abdelnour et al., 2021). Nicorandil, a vasodilatory drug, was shown to prevent ventricular tachyarrhythmias induction by normalizing Cx43 expression in this desmin-related cardiomyopathy mouse model (Matsushita et al., 2014).
Although there are no clear and effective treatment methods and drugs for impaired desmin-induced cardiac arrhythmia, some complications can be prevented. For patients with early onset cardiac arrhythmia, early whole genome sequencing, 24-hour holter monitoring, and treatment of cardiac arrhythmias and conduction defects are important, since early diagnosis allows to make treatment plans at an early stage, reducing the incidence of serious heart failure. At the same time implantation of a pacemaker can be lifesaving. Modulating mitochondrial function such as nicotinamide mononucleotide, mitochondria-targeted peptides, gene, and stem-cell therapy or muscle-specific gene transfer approaches are active areas of research that promise effective treatments in the future (Goldfarb et al., 2004; Clemen et al., 2013).
The desmin network plays a significant role in maintaining the structural and mechanical integrity of cardiomyoctyes. Moreover, desmin is involved in cardiomyocyte function by modulating cellular signaling and calcium homeostasis. As desmin is highly expressed in structures of the cardiac conduction system, the majority of pathogenic DES variants cause cardiac arrhythmia and cardiac conduction defects. On the molecular level, impaired desmin causes severe filament assembly defects, desmin aggregation, and abnormal distribution of Ca2+, that collectively may drive cardiac arrhythmias and cardiac conduction defects. Finally, further studies should elucidate the exact molecular mechanism how desmin affects specific cardiac conduction system structures. This knowledge will aid in the identification of druggable targets which may fuel development of effective mechanism-based therapies to treat cardiac conduction defects and arrhythmias.
Conceptualization, WS and BB; writing—original draft preparation, WS, SW, and BB; writing—review and editing, WS, SW, and BB.; visualization WS, SW, and BB.; supervision, BB.; funding acquisition, BB. All authors have read and agreed to the published version of the manuscript.
This research was funded by the Dutch Heart Foundation (DnAFix project, 2020B003, 2020), Atrial Fibrillation Innovation Platform (www.AFIPonline.org) and NWO (CIRCULAR project, NWA.1389.20.157).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdelnour, S. A., Xie, L., Hassanin, A. A., Zuo, E., and Lu, Y. (2021). The potential of CRISPR/Cas9 gene editing as a treatment strategy for inherited diseases. Front. Cell. Dev. Biol. 9, 699597. doi:10.3389/fcell.2021.699597
Abou Ziki, M. D., Bhat, N., Neogi, A., Driscoll, T. P., Ugwu, N., Liu, Y., et al. (2021). Epistatic interaction of PDE4DIP and DES mutations in familial atrial fibrillation with slow conduction. Hum. Mutat. 42 (10), 1279–1293. doi:10.1002/humu.24265
Agbulut, O., Li, Z., Périé, S., Ludosky, M. A., Paulin, D., Cartaud, J., et al. (2001). Lack of desmin results in abortive muscle regeneration and modifications in synaptic structure. Cell. Motil. Cytoskelet. 49 (2), 51–66. doi:10.1002/cm.1020
Arbustini, E., Pasotti, M., Pilotto, A., Pellegrini, C., Grasso, M., Previtali, S., et al. (2006). Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur. J. Heart Fail. 8 (5), 477–483. doi:10.1016/j.ejheart.2005.11.003
Bär, H., Goudeau, B., Wälde, S., Casteras-Simon, M., Mücke, N., Shatunov, A., et al. (2007). Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum. Mutat. 28 (4), 374–386. doi:10.1002/humu.20459
Bär, H., Schopferer, M., Sharma, S., Hochstein, B., Mücke, N., Herrmann, H., et al. (2010). Mutations in desmin's carboxy-terminal "tail" domain severely modify filament and network mechanics. J. Mol. Biol. 397 (5), 1188–1198. doi:10.1016/j.jmb.2010.02.024
Bär, H., Strelkov, S. V., Sjöberg, G., Aebi, U., and Herrmann, H. (2004). The biology of desmin filaments: How do mutations affect their structure, assembly, and organisation? J. Struct. Biol. 148 (2), 137–152. doi:10.1016/j.jsb.2004.04.003
Benvenuti, L. A., Aiello, V. D., Falcão, B. A., and Lage, S. G. (2012). Atrioventricular block pathology in cardiomyopathy by desmin deposition. Arq. Bras. Cardiol. 98 (1), e3–6. doi:10.1590/s0066-782x2012000100017
Brodehl, A., Dieding, M., Klauke, B., Dec, E., Madaan, S., Huang, T., et al. (2013). The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ. Cardiovasc. Genet. 6 (6), 615–623. doi:10.1161/circgenetics.113.000103
Brodehl, A., Gaertner-Rommel, A., Klauke, B., Grewe, S. A., Schirmer, I., Peterschröder, A., et al. (2017). The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 38 (8), 947–952. doi:10.1002/humu.23248
Brodehl, A., Gaertner-Rommel, A., and Milting, H. (2018). Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 10 (4), 983–1006. doi:10.1007/s12551-018-0429-0
Brodehl, A., Hain, C., Flottmann, F., Ratnavadivel, S., Gaertner, A., Klauke, B., et al. (2021). The desmin mutation DES-c.735G>C causes severe restrictive cardiomyopathy by inducing in-frame skipping of exon-3. Biomedicines 9 (10), 1400. doi:10.3390/biomedicines9101400
Brodehl, A., Pour Hakimi, S. A., Stanasiuk, C., Ratnavadivel, S., Hendig, D., Gaertner, A., et al. (2019). Restrictive cardiomyopathy is caused by a novel homozygous desmin (DES) mutation p.Y122H leading to a severe filament assembly defect. Genes (Basel) 10 (11), E918. doi:10.3390/genes10110918
Brundel, B. J., Ausma, J., van Gelder, I. C., Van der Want, J. J., van Gilst, W. H., Crijns, H. J., et al. (2002a). Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc. Res. 54 (2), 380–389. doi:10.1016/s0008-6363(02)00289-4
Brundel, B. J., Henning, R. H., Kampinga, H. H., Van Gelder, I. C., and Crijns, H. J. (2002b). Molecular mechanisms of remodeling in human atrial fibrillation. Cardiovasc. Res. 54 (2), 315–324. doi:10.1016/s0008-6363(02)00222-5
Cabet, E., Batonnet-Pichon, S., Delort, F., Gausserès, B., Vicart, P., and Lilienbaum, A. (2015). Antioxidant treatment and induction of autophagy cooperate to reduce desmin aggregation in a cellular model of desminopathy. PLoS One 10 (9), e0137009. doi:10.1371/journal.pone.0137009
Capetanaki, Y., Papathanasiou, S., Diokmetzidou, A., Vatsellas, G., and Tsikitis, M. (2015). Desmin related disease: A matter of cell survival failure. Curr. Opin. Cell. Biol. 32, 113–120. doi:10.1016/j.ceb.2015.01.004
Carlsson, L., Fischer, C., Sjöberg, G., Robson, R. M., Sejersen, T., and Thornell, L. E. (2002). Cytoskeletal derangements in hereditary myopathy with a desmin L345P mutation. Acta Neuropathol. 104 (5), 493–504. doi:10.1007/s00401-002-0583-z
Cartaud, A., Jasmin, B. J., Changeux, J. P., and Cartaud, J. (1995). Direct involvement of a lamin-B-related (54 kDa) protein in the association of intermediate filaments with the postsynaptic membrane of the Torpedo marmorata electrocyte. J. Cell. Sci. 108, 153–160. doi:10.1242/jcs.108.1.153
Chen, L., Wang, L., Li, X., Wang, C., Hong, M., Li, Y., et al. (2020). The role of desmin alterations in mechanical electrical feedback in heart failure. Life Sci. 241, 117119. doi:10.1016/j.lfs.2019.117119
Chen, R., Qiu, Z., Wang, J., Yao, Y., Huang, K., and Zhu, F. (2021). DES mutation associated with cardiac hypertrophy and alternating bundle branch block. Hear. Case Rep. 7 (1), 16–20. doi:10.1016/j.hrcr.2020.10.003
Chen, Y., Barajas-Martinez, H., Zhu, D., Wang, X., Chen, C., Zhuang, R., et al. (2017). Novel trigenic CACNA1C/DES/MYPN mutations in a family of hypertrophic cardiomyopathy with early repolarization and short QT syndrome. J. Transl. Med. 15 (1), 78. doi:10.1186/s12967-017-1180-1
Chourbagi, O., Bruston, F., Carinci, M., Xue, Z., Vicart, P., Paulin, D., et al. (2011). Desmin mutations in the terminal consensus motif prevent synemin-desmin heteropolymer filament assembly. Exp. Cell. Res. 317 (6), 886–897. doi:10.1016/j.yexcr.2011.01.013
Cízková, D., Soukup, T., and Mokrý, J. (2009). Expression of nestin, desmin and vimentin in intact and regenerating muscle spindles of rat hind limb skeletal muscles. Histochem. Cell. Biol. 131 (2), 197–206. doi:10.1007/s00418-008-0523-7
Clemen, C. S., Herrmann, H., Strelkov, S. V., and Schröder, R. (2013). Desminopathies: Pathology and mechanisms. Acta Neuropathol. 125 (1), 47–75. doi:10.1007/s00401-012-1057-6
Conover, G. M., Henderson, S. N., and Gregorio, C. C. (2009). A myopathy-linked desmin mutation perturbs striated muscle actin filament architecture. Mol. Biol. Cell. 20 (3), 834–845. doi:10.1091/mbc.e08-07-0753
Dayal, A. A., Medvedeva, N. V., Nekrasova, T. M., Duhalin, S. D., Surin, A. K., and Minin, A. A. (2020). Desmin interacts directly with mitochondria. Int. J. Mol. Sci. 21 (21), 8122. doi:10.3390/ijms21218122
Diokmetzidou, A., Soumaka, E., Kloukina, I., Tsikitis, M., Makridakis, M., Varela, A., et al. (2016). Desmin and αB-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. J. Cell. Sci. 129 (20), 3705–3720. doi:10.1242/jcs.192203
Elliott, J. L., Der Perng, M., Prescott, A. R., Jansen, K. A., Koenderink, G. H., and Quinlan, R. A. (2013). The specificity of the interaction between αB-crystallin and desmin filaments and its impact on filament aggregation and cell viability. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368 (1617), 20120375. doi:10.1098/rstb.2012.0375
Fan, P., Lu, C. X., Dong, X. Q., Zhu, D., Yang, K. Q., Liu, K. Q., et al. (2019). A novel phenotype with splicing mutation identified in a Chinese family with desminopathy. Chin. Med. J. 132 (2), 127–134. doi:10.1097/cm9.0000000000000001
Fischer, B., Dittmann, S., Brodehl, A., Unger, A., Stallmeyer, B., Paul, M., et al. (2021). Functional characterization of novel alpha-helical rod domain desmin (DES) pathogenic variants associated with dilated cardiomyopathy, atrioventricular block and a risk for sudden cardiac death. Int. J. Cardiol. 329, 167–174. doi:10.1016/j.ijcard.2020.12.050
Galata, Z., Kloukina, I., Kostavasili, I., Varela, A., Davos, C. H., Makridakis, M., et al. (2018). Amelioration of desmin network defects by αB-crystallin overexpression confers cardioprotection in a mouse model of dilated cardiomyopathy caused by LMNA gene mutation. J. Mol. Cell. Cardiol. 125, 73–86. doi:10.1016/j.yjmcc.2018.10.017
Garrido, C., Paul, C., Seigneuric, R., and Kampinga, H. H. (2012). The small heat shock proteins family: The long forgotten chaperones. Int. J. Biochem. Cell. Biol. 44 (10), 1588–1592. doi:10.1016/j.biocel.2012.02.022
Georgatos, S. D., and Blobel, G. (1987). Lamin B constitutes an intermediate filament attachment site at the nuclear envelope. J. Cell. Biol. 105 (1), 117–125. doi:10.1083/jcb.105.1.117
Goldfarb, L. G., Park, K. Y., Cervenáková, L., Gorokhova, S., Lee, H. S., Vasconcelos, O., et al. (1998). Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. 19 (4), 402–403. doi:10.1038/1300
Goldfarb, L. G., Vicart, P., Goebel, H. H., and Dalakas, M. C. (2004). Desmin myopathy. Brain 127 (4), 723–734. doi:10.1093/brain/awh033
Granger, B. L., and Lazarides, E. (1980). Synemin: A new high molecular weight protein associated with desmin and vimentin filaments in muscle. Cell. 22 (3), 727–738. doi:10.1016/0092-8674(80)90549-8
Hedberg, C., Melberg, A., Kuhl, A., Jenne, D., and Oldfors, A. (2012). Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur. J. Hum. Genet. 20 (9), 984–985. doi:10.1038/ejhg.2012.39
Heffler, J., Shah, P. P., Robison, P., Phyo, S., Veliz, K., Uchida, K., et al. (2020). A balance between intermediate filaments and microtubules maintains nuclear architecture in the cardiomyocyte. Circ. Res. 126 (3), e10–e26. doi:10.1161/circresaha.119.315582
Henning, R. H., and Brundel, B. (2017). Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 14 (11), 637–653. doi:10.1038/nrcardio.2017.89
Herrmann, H., and Aebi, U. (2004). Intermediate filaments: Molecular structure, assembly mechanism, and integration into functionally distinct intracellular scaffolds. Annu. Rev. Biochem. 73, 749–789. doi:10.1146/annurev.biochem.73.011303.073823
Hnia, K., Ramspacher, C., Vermot, J., and Laporte, J. (2015). Desmin in muscle and associated diseases: Beyond the structural function. Cell. Tissue Res. 360 (3), 591–608. doi:10.1007/s00441-014-2016-4
Hnia, K., Tronchère, H., Tomczak, K. K., Amoasii, L., Schultz, P., Beggs, A. H., et al. (2011). Myotubularin controls desmin intermediate filament architecture and mitochondrial dynamics in human and mouse skeletal muscle. J. Clin. Investig. 121 (1), 70–85. doi:10.1172/jci44021
Hol, E. M., and Capetanaki, Y. (2017). Type III intermediate filaments desmin, glial fibrillary acidic protein (GFAP), vimentin, and peripherin. Cold Spring Harb. Perspect. Biol. 9 (12), a021642. doi:10.1101/cshperspect.a021642
Hong, D., Wang, Z., Zhang, W., Xi, J., Lu, J., Luan, X., et al. (2011). A series of Chinese patients with desminopathy associated with six novel and one reported mutations in the desmin gene. Neuropathol. Appl. Neurobiol. 37 (3), 257–270. doi:10.1111/j.1365-2990.2010.01112.x
Jiao, Q., Sanbe, A., Zhang, X., Liu, J. P., and Minamisawa, S. (2014). αB-Crystallin R120G variant causes cardiac arrhythmias and alterations in the expression of Ca(2+) -handling proteins and endoplasmic reticulum stress in mice. Clin. Exp. Pharmacol. Physiol. 41 (8), 589–599. doi:10.1111/1440-1681.12253
Klauke, B., Kossmann, S., Gaertner, A., Brand, K., Stork, I., Brodehl, A., et al. (2010). De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 19 (23), 4595–4607. doi:10.1093/hmg/ddq387
Li, J., Zhang, D., Brundel, B., and Wiersma, M. (2019). Imbalance of ER and mitochondria interactions: Prelude to cardiac ageing and disease? Cells 8 (12), E1617. doi:10.3390/cells8121617
Li, Z., Mericskay, M., Agbulut, O., Butler-Browne, G., Carlsson, L., Thornell, L. E., et al. (1997). Desmin is essential for the tensile strength and integrity of myofibrils but not for myogenic commitment, differentiation, and fusion of skeletal muscle. J. Cell. Biol. 139 (1), 129–144. doi:10.1083/jcb.139.1.129
Liu, H. X., Jing, Y. X., Wang, J. J., Yang, Y. P., Wang, Y. X., Li, H. R., et al. (2020). Expression patterns of intermediate filament proteins desmin and lamin A in the developing conduction system of early human embryonic hearts. J. Anat. 236 (3), 540–548. doi:10.1111/joa.13108
Matsushita, N., Hirose, M., Sanbe, A., Kondo, Y., Irie, Y., and Taira, E. (2014). Nicorandil improves electrical remodelling, leading to the prevention of electrically induced ventricular tachyarrhythmia in a mouse model of desmin-related cardiomyopathy. Clin. Exp. Pharmacol. Physiol. 41 (1), 89–97. doi:10.1111/1440-1681.12185
Mavroidis, M., Athanasiadis, N. C., Rigas, P., Kostavasili, I., Kloukina, I., Te Rijdt, W. P., et al. (2020). Desmin is essential for the structure and function of the sinoatrial node: Implications for increased arrhythmogenesis. Am. J. Physiol. Heart Circ. Physiol. 319 (3), H557-H570–h570. doi:10.1152/ajpheart.00594.2019
Nikolova, V., Leimena, C., McMahon, A. C., Tan, J. C., Chandar, S., Jogia, D., et al. (2004). Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Investig. 113 (3), 357–369. doi:10.1172/jci19448
O'Neill, A., Williams, M. W., Resneck, W. G., Milner, D. J., Capetanaki, Y., and Bloch, R. J. (2002). Sarcolemmal organization in skeletal muscle lacking desmin: Evidence for cytokeratins associated with the membrane skeleton at costameres. Mol. Biol. Cell. 13 (7), 2347–2359. doi:10.1091/mbc.01-12-0576
Ojrzyńska, N., Bilińska, Z. T., Franaszczyk, M., Płoski, R., and Grzybowski, J. (2017). Restrictive cardiomyopathy due to novel desmin gene mutation. Kardiol. Pol. 75 (7), 723. doi:10.5603/kp.2017.0129
Olivé, M., Armstrong, J., Miralles, F., Pou, A., Fardeau, M., Gonzalez, L., et al. (2007). Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul. Disord. 17 (6), 443–450. doi:10.1016/j.nmd.2007.02.009
Otten, E., Asimaki, A., Maass, A., van Langen, I. M., van der Wal, A., de Jonge, N., et al. (2010). Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart rhythm. 7 (8), 1058–1064. doi:10.1016/j.hrthm.2010.04.023
Pool, L., Wijdeveld, L. F. J. M., de Groot, N. M. S., and Brundel, B. J. J. M. (2021). The role of mitochondrial dysfunction in atrial fibrillation: Translation to druggable target and biomarker discovery. Int. J. Mol. Sci. 22 (16), 8463. doi:10.3390/ijms22168463
Poon, E., Howman, E. V., Newey, S. E., and Davies, K. E. (2002). Association of syncoilin and desmin: Linking intermediate filament proteins to the dystrophin-associated protein complex. J. Biol. Chem. 277 (5), 3433–3439. doi:10.1074/jbc.M105273200
Protonotarios, A., Brodehl, A., Asimaki, A., Jager, J., Quinn, E., Stanasiuk, C., et al. (2021). The novel desmin variant p.Leu115Ile is associated with a unique form of biventricular arrhythmogenic cardiomyopathy. Can. J. Cardiol. 37 (6), 857–866. doi:10.1016/j.cjca.2020.11.017
Pruszczyk, P., Kostera-Pruszczyk, A., Shatunov, A., Goudeau, B., Dramiñska, A., Takeda, K., et al. (2007). Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int. J. Cardiol. 117 (2), 244–253. doi:10.1016/j.ijcard.2006.05.019
Ramspacher, C., Steed, E., Boselli, F., Ferreira, R., Faggianelli, N., Roth, S., et al. (2015). Developmental alterations in heart biomechanics and skeletal muscle function in desmin mutants suggest an early pathological root for desminopathies. Cell. Rep. 11 (10), 1564–1576. doi:10.1016/j.celrep.2015.05.010
Rutledge, C., and Dudley, S. (2013). Mitochondria and arrhythmias. Expert Rev. cardiovasc. Ther. 11 (7), 799–801. doi:10.1586/14779072.2013.811969
Sanbe, A., Daicho, T., Mizutani, R., Endo, T., Miyauchi, N., Yamauchi, J., et al. (2009). Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS One 4 (4), e5351. doi:10.1371/journal.pone.0005351
Schrickel, J. W., Stöckigt, F., Krzyzak, W., Paulin, D., Li, Z., Lübkemeier, I., et al. (2010). Cardiac conduction disturbances and differential effects on atrial and ventricular electrophysiological properties in desmin deficient mice. J. Interv. Card. Electrophysiol. 28 (2), 71–80. doi:10.1007/s10840-010-9482-8
Shahraz, S., Pittas, A. G., Saadati, M., Thomas, C. P., Lundquist, C. M., and Kent, D. M. (2017). Change in testing, awareness of hemoglobin A1c result, and glycemic control in US adults, 2007-2014. Jama 318 (18), 1825–1827. doi:10.1001/jama.2017.11927
Sharma, S., Mücke, N., Katus, H. A., Herrmann, H., and Bär, H. (2009). Disease mutations in the "head" domain of the extra-sarcomeric protein desmin distinctly alter its assembly and network-forming properties. J. Mol. Med. 87 (12), 1207–1219. doi:10.1007/s00109-009-0521-9
Sjöberg, G., Jiang, W. Q., Ringertz, N. R., Lendahl, U., and Sejersen, T. (1994). Colocalization of nestin and vimentin/desmin in skeletal muscle cells demonstrated by three-dimensional fluorescence digital imaging microscopy. Exp. Cell. Res. 214 (2), 447–458. doi:10.1006/excr.1994.1281
Sjöberg, G., Saavedra-Matiz, C. A., Rosen, D. R., Wijsman, E. M., Borg, K., Horowitz, S. H., et al. (1999). A missense mutation in the desmin rod domain is associated with autosomal dominant distal myopathy, and exerts a dominant negative effect on filament formation. Hum. Mol. Genet. 8 (12), 2191–2198. doi:10.1093/hmg/8.12.2191
Sprinkart, A. M., Block, W., Träber, F., Meyer, R., Paulin, D., Clemen, C. S., et al. (2012). Characterization of the failing murine heart in a desmin knock-out model using a clinical 3 T MRI scanner. Int. J. Cardiovasc. Imaging 28 (7), 1699–1705. doi:10.1007/s10554-011-9990-3
Strach, K., Sommer, T., Grohé, C., Meyer, C., Fischer, D., Walter, M. C., et al. (2008). Clinical, genetic, and cardiac magnetic resonance imaging findings in primary desminopathies. Neuromuscul. Disord. 18 (6), 475–482. doi:10.1016/j.nmd.2008.03.012
Taylor, M. R., Slavov, D., Ku, L., Di Lenarda, A., Sinagra, G., Carniel, E., et al. (2007). Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 115 (10), 1244–1251. doi:10.1161/circulationaha.106.646778
Thornell, L., Carlsson, L., Li, Z., Mericskay, M., and Paulin, D. (1997). Null mutation in the desmin gene gives rise to a cardiomyopathy. J. Mol. Cell. Cardiol. 29 (8), 2107–2124. doi:10.1006/jmcc.1997.0446
Tsikitis, M., Galata, Z., Mavroidis, M., Psarras, S., and Capetanaki, Y. (2018). Intermediate filaments in cardiomyopathy. Biophys. Rev. 10 (4), 1007–1031. doi:10.1007/s12551-018-0443-2
van Spaendonck-Zwarts, K. Y., van der Kooi, A. J., van den Berg, M. P., Ippel, E. F., Boven, L. G., Yee, W. C., et al. (2012). Recurrent and founder mutations in The Netherlands: The cardiac phenotype of DES founder mutations p.S13F and p.N342D. Neth. Heart J. 20 (5), 219–228. doi:10.1007/s12471-011-0233-y
van Spaendonck-Zwarts, K. Y., van Hessem, L., Jongbloed, J. D., de Walle, H. E., Capetanaki, Y., van der Kooi, A. J., et al. (2011). Desmin-related myopathy. Clin. Genet. 80 (4), 354–366. doi:10.1111/j.1399-0004.2010.01512.x
van Tintelen, J. P., Van Gelder, I. C., Asimaki, A., Suurmeijer, A. J., Wiesfeld, A. C., Jongbloed, J. D., et al. (2009). Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart rhythm. 6 (11), 1574–1583. doi:10.1016/j.hrthm.2009.07.041
Wahbi, K., Béhin, A., Charron, P., Dunand, M., Richard, P., Meune, C., et al. (2012). High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: A 10-year longitudinal study. Neuromuscul. Disord. 22 (3), 211–218. doi:10.1016/j.nmd.2011.10.019
Wang, X., Osinska, H., Dorn, G. W., Nieman, M., Lorenz, J. N., Gerdes, A. M., et al. (2001). Mouse model of desmin-related cardiomyopathy. Circulation 103 (19), 2402–2407. doi:10.1161/01.cir.103.19.2402
Wiersma, M., Meijering, R. A. M., Qi, X. Y., Zhang, D., Liu, T., Hoogstra-Berends, F., et al. (2017). Endoplasmic reticulum stress is associated with autophagy and cardiomyocyte remodeling in experimental and human atrial fibrillation. J. Am. Heart Assoc. 6 (10), e006458. doi:10.1161/jaha.117.006458
Yamamoto, M., Abe, S., Rodríguez-Vázquez, J. F., Fujimiya, M., Murakami, G., and Ide, Y. (2011). Immunohistochemical distribution of desmin in the human fetal heart. J. Anat. 219 (2), 253–258. doi:10.1111/j.1469-7580.2011.01382.x
Keywords: desmin, cardiac arrhtyhmias, cardiac conduction system, desmosomes, DES gene variants
Citation: Su W, van Wijk SW and Brundel BJJM (2022) Desmin variants: Trigger for cardiac arrhythmias?. Front. Cell Dev. Biol. 10:986718. doi: 10.3389/fcell.2022.986718
Received: 05 July 2022; Accepted: 22 August 2022;
Published: 09 September 2022.
Edited by:
Ashraf Yusuf Rangrez, University of Kiel, GermanyReviewed by:
Andreas Brodehl, Heart and Diabetes Center North Rhine-Westphalia, GermanyCopyright © 2022 Su, van Wijk and Brundel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bianca J. J. M. Brundel, Yi5icnVuZGVsQGFtc3RlcmRhbXVtYy5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.