 Emma J. Doyle1
Emma J. Doyle1 Lluis Morey
Lluis Morey Eric Conway
Eric Conway- 1School of Biomolecular and Biomedical Science, Conway Institute, University College Dublin, Dublin, Ireland
- 2Sylvester Comprehensive Cancer Centre, Miami, FL, United States
- 3Department of Human Genetics, Biomedical Research Building, University of Miami Miller School of Medicine, Miami, FL, United States
- 4Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland
Chromatin is spatially and temporally regulated through a series of orchestrated processes resulting in the formation of 3D chromatin structures such as topologically associating domains (TADs), loops and Polycomb Bodies. These structures are closely linked to transcriptional regulation, with loss of control of these processes a frequent feature of cancer and developmental syndromes. One such oncogenic disruption of the 3D genome is through recurrent dysregulation of Polycomb Group Complex (PcG) functions either through genetic mutations, amplification or deletion of genes that encode for PcG proteins. PcG complexes are evolutionarily conserved epigenetic complexes. They are key for early development and are essential transcriptional repressors. PcG complexes include PRC1, PRC2 and PR-DUB which are responsible for the control of the histone modifications H2AK119ub1 and H3K27me3. The spatial distribution of the complexes within the nuclear environment, and their associated modifications have profound effects on the regulation of gene transcription and the 3D genome. Nevertheless, how PcG complexes regulate 3D chromatin organization is still poorly understood. Here we glean insights into the role of PcG complexes in 3D genome regulation and compaction, how these processes go awry during tumorigenesis and the therapeutic implications that result from our insights into these mechanisms.
Introduction
Cell fate regulation is a critical developmental process, governed carefully by transcriptional control. This results in the ability of a given cell to activate and repress specific gene sets, culminating in a great diversity of cell types and transcriptional phenotypes. While it has long been appreciated that the 3D structure of chromatin is critical to the determination of cell fate and maintenance of cellular identity, technological advances of the last decade have facilitated a renaissance in the molecular dissection of genome architecture (Jerkovic and Cavalli, 2021). Moreover, there is a burgeoning understanding of the protein complexes and molecular mechanisms responsible for regulating chromatin architecture. These layers of topological genome regulation require precise temporal and spatial coordination to control gene expression.
Foremost among those protein complexes that control genome architecture and transcription are the Polycomb group complexes (PcG). These are a family of highly conserved histone modifying multi-subunit complexes that are essential for development (Lagger et al., 2002; Schuettengruber et al., 2017). Moreover, PcG dysfunction has been implicated as a driving force in several human diseases and syndromes (Conway et al., 2015; Deevy and Bracken, 2019; Tamburri et al., 2022).
The ever-expanding repertoire of genomics techniques to study the regulation of genome architecture has allowed a deep molecular understanding of the modes of PcG-mediated compaction (Jerkovic and Cavalli, 2021). In this review, we will summarize the contribution of the PcG complexes and their respective catalytic activities to the control of genome topology and how this is perturbed in cancer. We speculate on the future direction of research into the role of PcG in chromatin structure and the therapeutic opportunities that a mechanistic understanding of PcG-dysregulated cancers can provide.
Catalytic activities of Polycomb group complexes
The PcG machinery can be divided into three major complexes: Polycomb Repressive Complex 1 and 2 (PRC1 and PRC2) and Polycomb Repressive Deubiquitinase complex (PR-DUB). Traditionally, PcG complexes are associated with maintenance of gene repression, mainly via histone-modifying activities. PRC2 catalyses methylation at lysine 27 of histone H3 (H3K27me1/2/3) through its catalytic core of EZH1/2, SUZ12, EED, and RBBP4/7 (Cao et al., 2002; Ferrari et al., 2014). PRC1 deposits a ubiquitin group on lysine 119 of histone H2A (H2AK119ub1) via the E3-ligases RING1A/B which heterodimerize with one of the six PcG RING finger domain (PCGF1–6) paralogs (Wang et al., 2004a; de Napoles et al., 2004; Gao et al., 2012). In mammals, PRC1 can be further subdivided into two main complexes, namely canonical or variant PRC1 (cPRC1 and vPRC1), though all PRC1 complexes contain either RING1A or RING1B (Gao et al., 2012). H2AK119ub1 is erased through the activity of deubiquitinating enzymes such as BAP1, USP16 and MYSM1 (Joo et al., 2007; Zhu et al., 2007; Scheuermann et al., 2010; Yang et al., 2014; Rong et al., 2022). BAP1 forms part of the PR-DUB complex, which consists of a catalytic core of BAP1 and one of three ASXL paralogs (ASXL1-3). PR-DUB has a particularly prominent role in oncogenesis (Carbone et al., 2013).
These PcG complexes are responsible for two distinct forms of chromatin structural features (Figures 1A,B); either proximal compaction or distal looping. The proximal form, ‘PcG compacted domains’ can perhaps be better considered as compaction of the linear 2D genome (Figure 1A), through careful ordering of neighboring nucleosomes in regions enriched by PcG modifications. This coordinated process, accompanied by accumulation of the linker histone H1, promotes local compaction. These proximally compacted sites correlate well with ‘B compartments’ which are inactive regions that tend to be in close proximity to other inactive regions (Jerkovic and Cavalli, 2021; Willcockson et al., 2021; Yusufova et al., 2021). Such domains are interspersed by ‘A compartments’ that are enriched in active genes or broad domains enriched in the H3K36me2 modification that is associated with more permissive or open chromatin (Figure 1A).
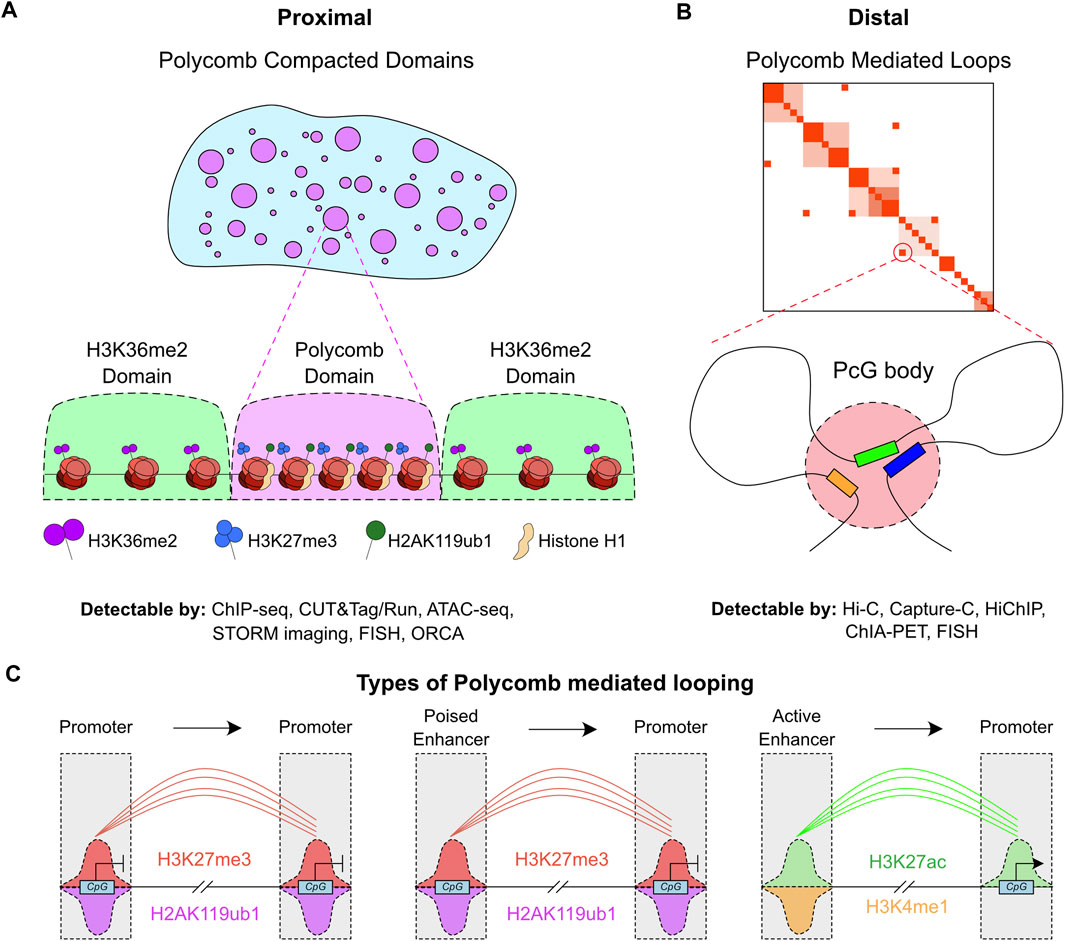
FIGURE 1. Divergent mechanisms of chromatin structure regulation by PcG. (A) Illustration of nuclear (blue) clusters containing Polycomb domains (purple). These proximal domains are occupied by H3K27me3, H2AK119ub1 and Histone H1, while boundary domains are enriched in H3K36me2 (green). Techniques that can be used to measure Polycomb domains and their associated compaction and histone modifications are listed underneath the graphic. (B) Illustration of distal Polycomb mediated loops, with example chromatin capture contact map, which involve multiple regions that are distal in 3D-space contacting each other in stable Polycomb bodies. Techniques that can be used to measure Polycomb mediated loops are listed underneath the graphic. (C) Cartoon of the different genomic features that undergo Polycomb mediated looping, and the histone modifications associated with these features.
The distal form of ‘PcG mediated loops’, more commonly termed PcG bodies, involves mid/long-range loops from one PcG bound region to another. These can be observed at distances of up to 100 MB (Figure 1B) (Schoenfelder et al., 2015; Kraft et al., 2022; Ngan et al., 2020). This type of compaction is perhaps better studied than the proximal form, but its precise contribution to transcriptional regulation is still an open question. A number of forms of these PcG-mediated loops have been reported, with the general mechanism reporting loops between PcG bound promoters (Figure 1C) (Ngan et al., 2020). However, studies to date have been limited to certain cell-types which may prevent a complete understanding of the dynamic role of PcG-mediated loops in transcription and development. Similar loops have been discovered between ‘poised’ or PcG marked enhancers with transcriptionally repressed genes (Figure 1C) (Rada-Iglesias et al., 2011; Pachano et al., 2021).
There is huge diversity in the proteomic composition of the PcG complexes in mammals (Gao et al., 2012; Morey et al., 2012; Hauri et al., 2016; Gahan et al., 2020; Kang et al., 2022) with specific activities and biological functions. For instance, we and others have shown that cPRC1 and vPRC1 complexes have distinct biological functions (Morey et al., 2013; Rose et al., 2016; Fursova et al., 2019; Scelfo et al., 2019). Notably, their specific gene targets, recruitment mechanisms, and enzymatic activities are context dependent, and they exert specialized functions during the early stages of development. These differential activities are generally driven by the specific combinatorial assembly of PcG accessory proteins in each sub-complex. The molecular function of many PcG subunits have been well defined, with three general means through which a subunit can contribute to the function of a PcG complex: chromatin recruitment, catalytic regulation, and compaction/structural functions (Figures 2A–C). Here we will focus on the catalytic and compaction roles of PcG subunits as they are the direct contributors to PcG-mediated 3D genome architecture. The mechanisms of PcG recruitment to chromatin have been reviewed in depth elsewhere (Glancy et al., 2021; Tamburri et al., 2022).
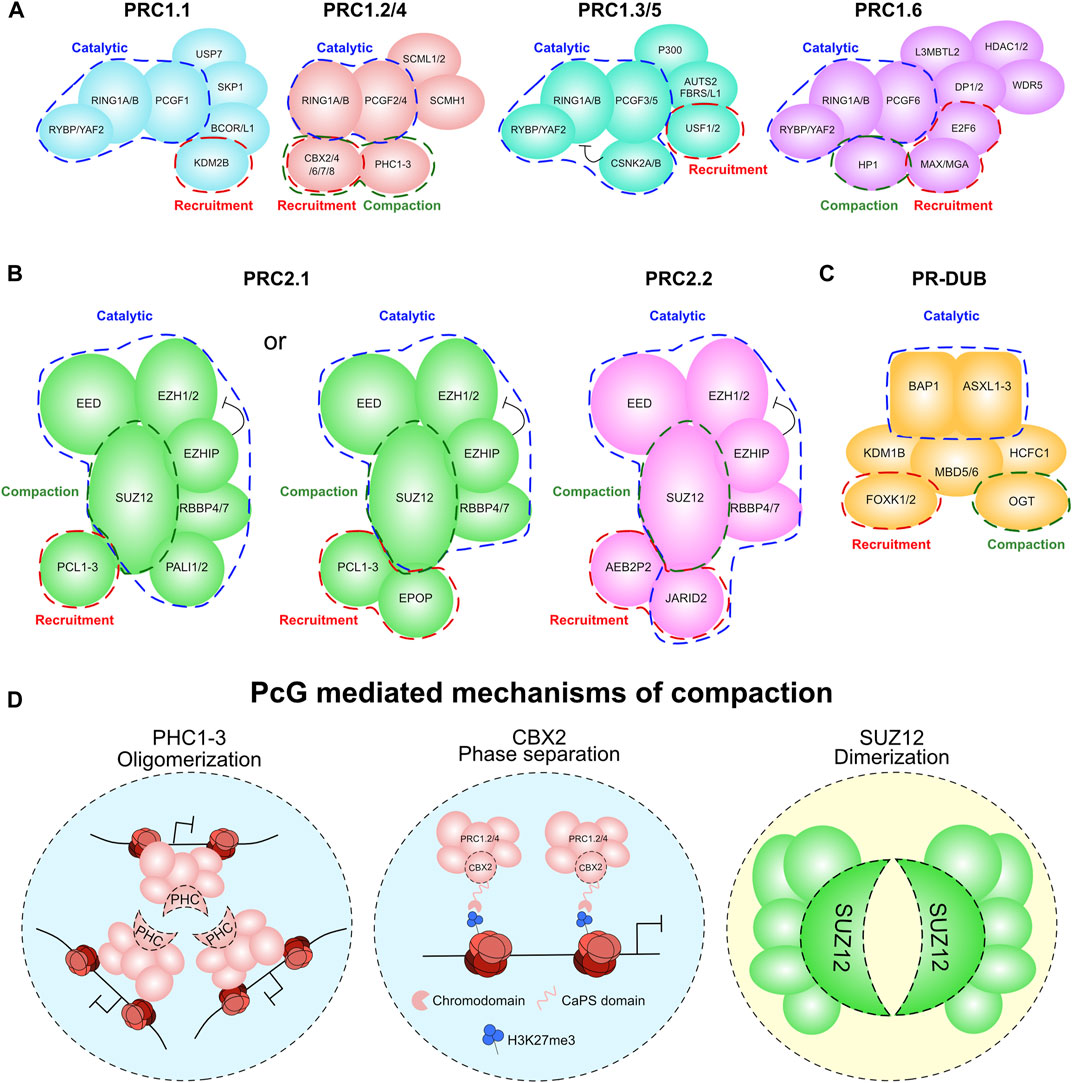
FIGURE 2. PcG complexes and their contribution to compaction. (A) The variable compositions of the PRC1 complex. Compaction related subunits are highlighted in green. Catalytic related subunits are highlighted in blue. Recruitment related subunits are highlighted in red. (B) The variable compositions of the PRC2 complex. Compaction related subunits are highlighted in green. Catalytic related subunits are highlighted in blue. Recruitment related subunits are highlighted in red. (C) The composition of the PR-DUB complex. Compaction related subunits are highlighted in green. Catalytic related subunits are highlighted in blue. Recruitment related subunits are highlighted in red. (D) Model of the compaction mechanism for PcG proteins PHC1-3, CBX2 and SUZ12.
While the catalytic products of these complexes are clearly correlated with transcriptional repression of nearby genes, the precise contribution of each histone modification in maintaining or initiating repression is still incompletely understood (Pengelly et al., 2013; Pengelly et al., 2015; Blackledge et al., 2020; Tamburri et al., 2020; Dobrinic et al., 2021). Catalytic inactivation of the PRC1 complex in embryonic stem cells (ESC) rapidly depletes H2AK119ub1 levels in the cell, resulting in concurrent upregulation of PRC1 target genes (Blackledge et al., 2020; Tamburri et al., 2020; Dobrinic et al., 2021). This highlights a clear direct contribution to transcriptional repression. However, this process is apparently independent of PRC2 complex activity as H3K27me3 levels are largely maintained. In fact, knockout or deletion of PRC2 fails to induce transcriptional upregulation in ESC (Tamburri et al., 2020; Dobrinic et al., 2021), despite clear importance in transcriptional repression in adult stem cells, differentiation and development (Pengelly et al., 2013; Chiacchiera et al., 2016; Sankar et al., 2022). Together this suggests that the role of H2AK119ub1 is at least partially independent of PRC2. While there are many reported sensors of H2AK119ub1, it is as yet unclear which, if any, of these contribute to the role of this modification in repression (Tamburri et al., 2022).
Dynamic functions of PcG
The PcG complex cores act as the scaffold upon which auxiliary subunits bind to confer their distinct functions, including proteins that are involved in recruitment such as CBXs for cPRC1, KDM2B or MAX/MGA for vPRC1, PCL1-3 or JARID2 for PRC2 and FOXK1/2 for PR-DUB (Figures 2A–C) (Farcas et al., 2012; Endoh et al., 2017; Healy et al., 2019; Kolovos et al., 2020). Importantly, while these targeting proteins are required for maintaining stable tethering of complexes to target sites such as promoters or enhancers, emerging evidence shows that the function of PcG complexes is not limited to those long-term residences. H2AK119ub1, H3K27me2 and H3K27me3, although specifically enriched at these tethered sites, are also broadly distributed throughout the genome (Ferrari et al., 2014; Lee et al., 2015; Fursova et al., 2019). This is in stark contrast to the occupancy of the PcG complexes from ChIP studies, suggesting a low/temporary residency of these complexes at chromatin. Indeed, live-cell imaging studies have allowed the dissection of the dynamics of PRC1 and PRC2 complexes (Youmans et al., 2018; Huseyin and Klose, 2021). Remarkably, only 10%–20% of PRC1 complexes and ∼20% of PRC2 complexes are bound to chromatin at any given time in ESC. This supports a ‘hit and run’ model in which the PcG complexes are diffusing throughout the nucleus generating sparse ‘blanket’ levels of their modifications, and when they reach a site for which they have a higher affinity (such as CpG islands) they can become stably tethered, but the majority of PcG complexes are in a catalytically active state of flux.
While the precise function of these intergenic, diffuse histone modifications is yet to be fully understood, work to date suggests that they can contribute to the maintenance of proximal PcG compacted domains. Whether this is through a direct role of the modifications, or their established antagonisms with other epigenetic modifications such as H3K36me2, DNA methylation or RNA molecules remains to be fully elucidated.
Intra-complex rheostats of catalytic potency in PcG
The biochemical diversity of PcG provides these complexes with tools to allosterically modulate the activities of their catalytic cores. These rheostats within the PcG complexes fine tune histone modifying activity to maintain an equilibrium of H2AK119ub1 and H3K27me3 at specific regions in the genome. PRC2 is a prime example of this, with a positive regulator of allosteric activity within each of its two subcomplexes, PALI1/2 within PRC2.1 and JARID2 within PRC2.2 (Figure 2B) (Sanulli et al., 2015; Conway et al., 2018; Zhang et al., 2021a). Each of these are methylated themselves by EZH2 (at residue K1241 for PALI1 and K116 for JARID2), these modifications are recognised by the aromatic cage of EED which promotes allosteric activity of the PRC2 complex. Genetic approaches aimed at perturbing the allosteric activity of PRC2 complexes will shed light into how PRC2 modulates its own enzymatic activity and will determine the phenotypic implications of these intra-complex rheostats in controlling cell fate determination and cellular homeostasis.
PRC2 can also associate with an inhibitory subunit, EZHIP (Figure 2B). EZHIP is mainly expressed in gonads, where it inhibits PRC2 activity through a domain mimicking the H3K27M oncohistone (Jain et al., 2019; Piunti et al., 2019; Ragazzini et al., 2019). This mechanism blocks PRC2 activity in trans and prevents its spreading from nucleation sites at CpG islands (Jain et al., 2020b). It is not yet known whether other mammalian proteins contain the same domain of EZHIP responsible for PRC2 inhibition.
PRC1 shares similar positive and negative regulators of catalytic activity. RYBP and YAF2 subunits, which can be a part of any of the vPRC1 complexes, promote catalytic activity, though the structural basis for this is not yet known (Figure 2A) (Morey et al., 2013; Rose et al., 2016; Zhao et al., 2020). This possibly utilises the H2AK119ub1 binding affinity for the paralogous RYBP/YAF2 subunits in order to facilitate spreading of H2AK119ub1 from existing modified sites (Kalb et al., 2014; Cooper et al., 2016). Moreover, chromatin compaction dependent on the linker histone H1 plays a critical role in the stable propagation of H2AK119ub1 by RYBP/YAF2–PRC1 (Zhao et al., 2020). The negative regulators of PRC1 exist specifically within the PCGF3 and PCGF5-containing PRC1.3/5 complexes (Figure 2A). These are the two most catalytically active forms of the complex in vitro and contribute to the bulk of H2AK119ub1 in ESC (Taherbhoy et al., 2015; Fursova et al., 2019; Conway et al., 2021). However, the PRC1.3/5 complexes contain the CSNK2A and CSNK2B kinases which can phosphorylate RING1B at S168 resulting in diminished catalytic function in vitro (Gao et al., 2014). It is unknown whether this inhibitory phosphorylation occurs in a physiological setting, indeed its relevance to PRC1 biology also remains to be fully elucidated.
Contribution of Polycomb group complexes to compaction
Each of the three PcG complexes can serve structural roles that facilitate chromatin compaction and structural maintenance such as through PcG bodies. PcG bodies are foci in the nuclei containing H2AK119ub1 and H3K27me3 (Isono et al., 2013). Imaging and high-throughput chromosome conformation capture experiments revealed that PcG bodies contain PcG bound loci that interact in 3D space, even though they can be separated by very large distances across the genome (Boyle et al., 2020). The key player in PcG body formation is PRC1 (Francis et al., 2004). This is supported by PRC1 knockout studies showing loss of local and distal compaction/looping, and decompacted nuclear architecture in its absence (Schoenfelder et al., 2015; Boyle et al., 2020; Rhodes et al., 2020).
More specifically, cPRC1 containing either PCGF2/4, CBX (CBX2/4/6/7/8) and the Polyhomeotic (PHC1-3) subunits (Gao et al., 2012; Morey et al., 2012) are recruited to chromatin through the affinity of the chromodomain within each CBX subunit for the PRC2-mediated modification H3K27me3 (Fischle et al., 2003; Wang L. et al., 2004). The role of cPRC1 in distal regulation of compaction and looping is generally considered to be through their PHC subunits. These are essential for normal embryonic development, with a dose-dependent function in maintaining homeotic gene repression (Isono et al., 2005). PHC1-3 contribute to cPRC1 mediated repression through hetero-oligomerisation via their Sterile Alpha Motif (SAM) domain (Figure 2D). This results in compaction of cPRC1 marked regions into distinct PcG body speckles (Isono et al., 2013; Boettiger et al., 2016; Kundu et al., 2017). This PHC1-3 mediated aggregation controls looping of PcG-bound loci to each other, most strikingly at HOX genes which are particularly prominent PcG target loci. Most significantly, mutation of the PHC2 SAM domain prevents cPRC1 aggregation and induces homeotic transformations in developing mice, showing that this role of PHC1-3 is critical to PRC1 function (Isono et al., 2013).
The CBX subunits of cPRC1 are primarily involved in cPRC1 recruitment to chromatin. However, the CBX2 subunit has unique properties in proximal chromatin compaction that seemingly cause phase separation of PcG target loci (Figure 2D) (Plys et al., 2019; Tatavosian et al., 2019). In Drosophila, this function is controlled by the Psc (PCGF ortholog) subunit, but in mammals is unique to CBX2 (Grau et al., 2011). This function of CBX2 is conferred by an intrinsically disordered Compaction and Phase Separation (CaPS) domain. This domain is enriched in basic residues that are essential for the capacity of CBX2 to phase separate (Plys et al., 2019; Tatavosian et al., 2019). Intriguingly, this domain is also critical for appropriate repression of PcG target genes during embryogenesis, with mutations in these basic residues causing homeotic transformations (Lau et al., 2017). This CaPS domain also confers an impaired ability to remove PcG, suggesting that loci in these condensates have altered dynamics of transcriptional regulation (Jaensch et al., 2021). This is consistent with the observed immobility of CBX2 on chromatin relative to the other CBX proteins, and indeed the rest of cPRC1 in live cell imaging studies (Zhen et al., 2014). While the function of this ability of CBX2 to form condensates is still incompletely understood, it does afford CBX2-PRC1 the capacity to condense DNA and chromatin into discrete particles (Tatavosian et al., 2019).
Therefore, the PHC1-3 and CBX2 subunits confer distinct modes through which cPRC1 can regulate chromatin compaction. Despite this, the precise contribution of cPRC1 to PcG mediated repression remains a matter of debate, relative to its more catalytically active variant (vPRC1) forms containing PCGF1/3/5/6 (Taherbhoy et al., 2015; Fursova et al., 2019). In fact, acute perturbation of cPRC1 via PCGF2 and PCGF4 double knockout in ESC fails to induce major gene expression changes (Fursova et al., 2019). The same is clear in adult stem cells such as skin stem cells, where double loss of PCGF2 and PCGF4 does not recapitulate the RING1A/B knockout phenotype (Cohen et al., 2018). Notwithstanding these mild phenotypes in acute systems, constitutive mutants of cPRC1 result in homeotic transformations and defects in differentiation (Akasaka et al., 1996; Jacobs et al., 1999; Akasaka et al., 2001; Morey et al., 2015). Together this may suggest that cPRC1 is more important during long-term cell fate transitions and is dispensable for transcriptional repression in static model systems. Therefore, short-term PcG repression is maintained by factors other than compaction but establishing de novo PcG repressive domains may require cPRC1 mediated compaction.
PR-DUB contributes to PcG-mediated repression through the catalytic activity of BAP1 by removing H2AK119ub1 (Scheuermann et al., 2010; Sahtoe et al., 2016). However, it can also contribute to the function of cPRC1 complexes in chromatin looping. Drosophila lacking the glycosyltransferase enzyme Ogt exhibit failure to repress PcG targets akin to Ph mutants (the Drosophila PHC paralog) (Gambetta and Muller, 2014). Biochemical analysis attributed this phenotype to a requirement for glycosylation of a serine/threonine rich domain on Ph to prevent non-productive aggregation of Ph or PHC proteins (Gambetta and Muller, 2014). Therefore, OGT contributes to PcG repression through prevention of aberrant PHC hetero-oligomerisation, maintaining PcG compaction in a precisely regulated manner (Figure 2D). In mammals, OGT is a subunit of the PR-DUB complex (Hauri et al., 2016; Kloet et al., 2016). However, while Ogt is clearly important for PcG-mediated repression, it remains to be seen whether this is a PR-DUB related function, and what physiological function the glycosylation of PHC has on PcG body formation in mammals.
The role of PRC2 in directly controlling compaction is less well defined than PRC1. Although, emerging biochemical data suggests two distinct modes through which PRC2 can contribute to chromatin compaction. These are through i) SUZ12-mediated dimerisation and ii) direct nucleosomal compaction by EZH1 (Figure 2D). While it has been known for some time that PRC2 is capable of dimerisation (Davidovich et al., 2014), the structural basis for this has recently been elucidated. SUZ12 can undergo ‘domain switching’ between its C2 domain and the WDB1 domain on another molecule of SUZ12 resulting in dimerisation of two core PRC2 complexes (Chen et al., 2020; Grau et al., 2021). Moreover, loss of this dimerisation capacity leads to defects in H3K27me3 deposition at PcG target genes and compaction of nucleosomal arrays.
EZH1 can promote compaction of nucleosomal arrays independently of catalytic activity and interestingly, its compaction function is stronger than that of its more catalytically active paralog EZH2 (Margueron et al., 2008; Lee et al., 2018; Grau et al., 2021). Structural studies have revealed that this function of EZH1 can be attributed to a divergent basic patch in the MSL2 loop of EZH1. This domain promotes nucleosome binding and indeed compaction in vitro. This divergent role of EZH1 in compaction is strikingly similar to the divergent evolution of PRC1 subcomplexes with cPRC1 having weaker enzymatic potency than vPRC1 but instead maintaining functions in chromatin compaction (Blackledge et al., 2014; Gahan et al., 2020), much like the differences in the compaction and catalytic activities of EZH1 and EZH2 (Margueron et al., 2008; Lee et al., 2018; Lavarone et al., 2019; Grau et al., 2021).
The biological contributions of the OGT, SUZ12 and EZH1-mediated compaction mechanisms to PcG biology has yet to be fully uncovered as the studies to date have generally been in vitro. Discrete mutagenesis experiments will allow the dissection of their contributions to PcG-mediated compaction and transcriptional repression.
Polycomb body disruption in cancer
An ever-expanding concept in cancer genetics over the last decade is the recurrent disruption of transcriptional and epigenetic machinery (Vogelstein et al., 2013). The components of the PcG complexes are a strong example of this, with mutations in their subunits driving many cancers in a context-dependent fashion (Conway et al., 2015). The effect of these mutations on PcG-mediated 3D-structure has been under explored to date. This is particularly urgent as fully elucidating these mechanisms may facilitate the discovery of epigenetic targeted therapies to restore chromatin architecture to a healthy state.
PcG bodies that control distal looping of PcG bound elements are primarily regulated through the cPRC1 complex. Disruption of these avenues of looping or aggregation can occur through a myriad of mutations affecting the PcG recruitment pathway. A simplified version of this pathway is that: i) vPRC1 is recruited to chromatin through DNA binding at CpG islands or transcription factor binding sites (Farcas et al., 2012; Wu et al., 2013; Endoh et al., 2017; Scelfo et al., 2019). ii) These complexes catalyse H2AK119ub1 which promotes PRC2 recruitment either directly through PRC2.2 sampling H2AK119ub1 or PRC2.1 affinity for CpG islands (Cooper et al., 2016; Choi et al., 2017; Healy et al., 2019; Hojfeldt et al., 2019; Blackledge et al., 2020; Tamburri et al., 2020). iii) These PRC2 subcomplexes in turn catalyse H3K27me3, thus prompting recruitment of CBX-containing cPRC1 and causing aggregation of these sites into PcG bodies (Figure 3A) (Fischle et al., 2003; Min et al., 2003; Blackledge and Klose, 2021).
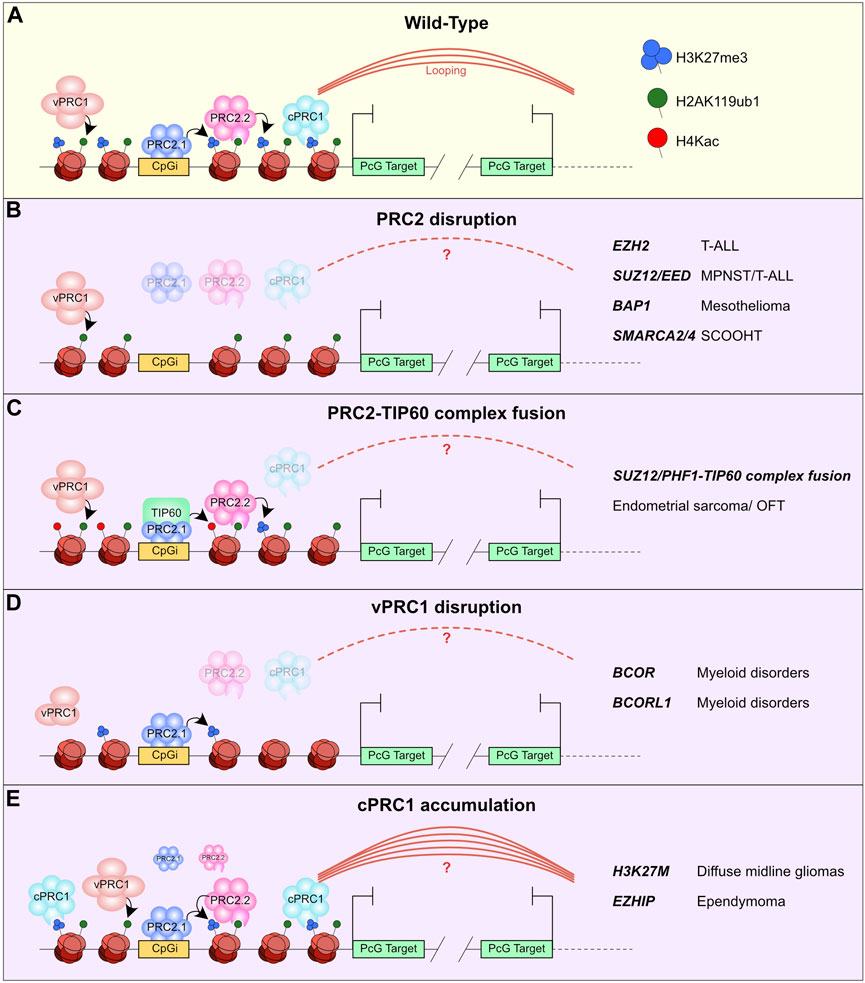
FIGURE 3. Malformation of PcG bodies in cancer. (A) Schematic of recruitment model for PRC1 and PRC2 complexes. i) vPRC1 is recruited by transcription factors or CpGi readers and catalyses H2AK119ub1. ii) PRC2.2 reads H2AK119ub1 and PRC2.1 is recruited to CpGi, these complexes catalyse H3K27me3. iii) cPRC1 reads H3K27me3 and promotes Polycomb mediated looping to other cPRC1 marked regions. (B–E) Illustration of how different mutations of PcG proteins in diverse cancer types affects the PcG recruitment pathway and in turn cPRC1 mediated looping. This includes (B) PRC2 disruption, (C) PRC2-TIP60 complex fusion, (D) vPRC1 disruption, (E) and cPRC1 accumulation. The genes mutated and the cancer they are associated with are listed on the right.
Loss-of-function (LOF) mutations of EZH2 occur in several cancers including T-cell Acute Lymphoblastic Leukemia (T-ALL) and Acute Myeloid Leukemia (AML) (Ntziachristos et al., 2012; Zhang et al., 2012; Dohner et al., 2017; Liu et al., 2017). The SUZ12 and EED genes also feature LOF mutations in T-ALL and are particularly frequent in Malignant Peripheral Nerve Sheath tumours (MPNST) (Lee et al., 2014; Zhang et al., 2014). These PRC2 LOF mutant cancers have reduced H3K27me3 (Ntziachristos et al., 2012; Lee et al., 2014; Wassef et al., 2019) and therefore may be deficient in cPRC1 mediated looping, as loss of H3K27me3 can prevent cPRC1 recruitment and aggregation (Figure 3B).
A similar disruption of PcG distal looping may occur through other chromatin modifier-mutated cancers such as BAP1 in mesothelioma, uveal melanoma, and renal clear cell carcinomas (Figure 3B) (Testa et al., 2011; Carbone et al., 2013). Recent studies have shown that PR-DUB and the catalytic function of BAP1 are required for maintenance of PRC2 target gene occupancy and PcG-mediated repression in ESC and cancer cells (Scheuermann et al., 2010; Abdel-Wahab et al., 2012; Conway et al., 2021; Fursova et al., 2021). Diffuse accumulation of H2AK119ub1 in the absence of BAP1 causes titration of PRC2 and cPRC1 from their target sites (Conway et al., 2021). Although BAP1 loss causes widespread compaction, the specific effect of BAP1 loss on PcG bodies remains to be investigated. Supporting the idea that BAP1 mutant cancers actually confers loss of PcG repression rather than the previously assumed hyperactivation of PRC1 and PRC2 activities, is the lack of response of BAP1 mutant mesothelioma patients to the EZH2 inhibitor Tazemetostat (Zauderer et al., 2022).
The PcG complex genes SUZ12, PHF1, EZHIP and BCOR undergo chromosomal translocations resulting in fusion proteins in endometrial stromal sarcoma and ossifying fibromyoxoid tumours (OFT) (Hrzenjak, 2016; Sudarshan et al., 2022). Intriguingly, their fusion protein partners can vary but are consistently part of the TIP60 (NuA4) complex family. The most common of these fusions being JAZF1-SUZ12 fusions, but these PcG genes have also been reported to be translocated with other TIP60 complex subunits (Sudarshan et al., 2022). TIP60 is a histone acetyltransferase for histone H4 and is primarily found at sites of active transcription. These fusions have a profound effect on the histone modification landscape at PcG target genes with a reduction in H3K27me3 at target genes and gain in H4Kac (Sudarshan et al., 2022; Tavares et al., 2022). These alterations are likely caused by eviction of accessory subunits from the PcG complexes such as loss of EPOP, PALI1 and JARID2 (Chen et al., 2018; Piunti et al., 2019; Tavares et al., 2022). This biochemical disruption of the PRC2 complex disturbs PcG complex redistribution and gene repression during ESC differentiation. The reductions in H3K27me3 at PcG targets make it likely that PcG body formation is also disrupted with potential gains in active enhancer-promoter looping mediated by increased acetylation (Figure 3C), such as the observed gains in intra-TAD acetylation at the HOXD locus (Sudarshan et al., 2022).
Disruption of PcG bodies is not limited to cancers with mutations in PcG family members. In fact, contrary to canonical models of PcG-BAF complex antagonism (Kadoch et al., 2017), loss of SMARCA2/4 causes displacement of cPRC1 and PRC2 complexes from their targets and decompaction of PcG target loci including HOX gene clusters (Weber et al., 2021). This is intriguing as mutation of SMARCA2 and SMARCA4 occurs in several cancer types such as small cell carcinoma of the ovary hyper-calcaemic Type (SCCOHT) (Pan et al., 2019). This new data suggests that BAF mutant cancers may also reduce PcG target occupancy and therefore impair its looping activity (Figure 3B). The mechanism for this has not been fully elucidated but is reminiscent of the displacement of cPRC1 and PRC2 in the BAP1 mutant context, which causes an imbalance in H3K27me3 and H2AK119ub1 that titrate the complexes away from their target sites.
Several components involved in catalysing/erasing/reading H2AK119ub1 are recurrently mutated in myeloid disorders such as AML, including ASXL1, DNMT3A, BCOR and BCORL1 (Dohner et al., 2017). BCOR and BCORL1 are critical components of the PCGF1-containing vPRC1 complexes that catalyse promoter proximal H2AK119ub1 (Farcas et al., 2012; Wu et al., 2013). The BCOR and BCORL1 nonsense and frameshift mutations occurring in AML patients result in C-terminally truncated proteins lacking the PUFD domain. This causes an uncoupling of KDM2B and BCOR/L1 from the rest of the complex, thus impairing vPRC1 recruitment and its catalytic activity at target loci (Schaefer et al., 2022). The subsequent reductions in PRC2 binding are in keeping with the established recruitment model and suggest that cPRC1 driven looping may be indirectly impaired by these mutations due to reductions in H3K27me3 (Figure 3D).
There are also several oncogenic events that can cause gain- or change-of-function to PcG complex activities. These include the H3K27M oncohistone mutation that occurs in diffuse midline gliomas, or expression of the gonad specific PRC2 subunit EZHIP in ependymoma (Sturm et al., 2012; Wu et al., 2012; Lewis et al., 2013; Pajtler et al., 2018). These events share a mechanistic impact on PcG function as they can broadly inhibit the catalytic activity of the PRC2 complex (Lewis PW 2013; Mohammad et al., 2017; Jain et al., 2019; Ragazzini et al., 2019), therefore they were initially thought to act as LOF mutations. Precise epigenomic mapping of PcG activities in these contexts has instead revealed that H3K27M mutation or EZHIP expression in fact allows maintenance of H3K27me3 at discrete PcG target loci, including tumour suppressor genes, throughout the genome, but there is a subsequent failure to spread H3K27me3 beyond these nucleating sites (Mohammad et al., 2017; Jain et al., 2020b; Brien et al., 2021). The consequence of this is increased PRC2 and cPRC1 binding at target loci (Brien et al., 2021). Therefore, a likely scenario is that these alterations have a hyper-compacting influence on PcG body formation, driven by high levels of cPRC1, preventing activation of differentiation stimuli under PcG repression (Figure 3E).
Taken together there are a myriad of mechanisms through which PcG-mediated looping can be directly or indirectly disrupted in cancer. Whether these compaction changes are themselves involved in oncogenesis or driving aberrant gene expression changes remains largely unexplored. However, exciting new technologies and more cost-efficient methodologies (Jerkovic and Cavalli, 2021) makes investigating these mechanisms a high priority for the future of both PcG and cancer fields.
Polycomb at distal regulatory elements to support gene transcription
While PcG complexes have been classically associated with gene repression, especially in pluripotent cells, recent evidence suggests that formation of PcG domains is also required for bringing enhancers within close proximity of gene promoters that are rapidly induced by differentiation stimuli (Figure 1C). This proposes a model in which PcG complexes are also required for rapid induction of gene transcription (Cruz-Molina et al., 2017; Ngan et al., 2020; Pachano et al., 2021). Similarly, an elegant study by Koseki and colleagues showed that RING1B is required for both repression and activation of Meis2 during early midbrain mouse development by regulating the promoter and enhancer interaction in a spatial and temporal manner (Kondo et al., 2014). Moreover, during neuronal differentiation, RING1B associates with active enhancers and enhancer-promoter interactions (Loubiere et al., 2020) and can associate with AUTS2 and p300 to drive neuronal differentiation by the transcription factor nuclear respiratory factor 1 (NRF1) (Figure 4A) (Gao et al., 2014; Liu et al., 2021).
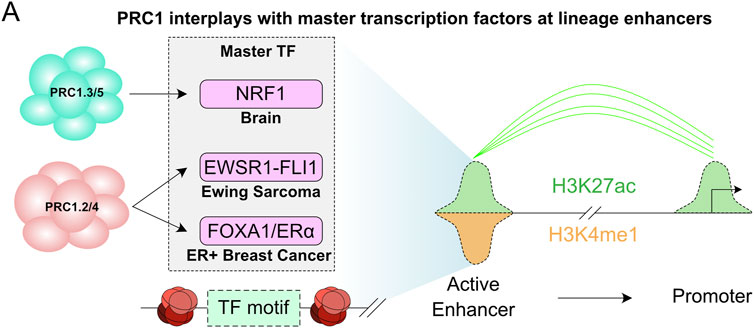
FIGURE 4. Polycomb at distal regulatory elements to support gene transcription. (A) Illustration of the interplay between PRC1 and master transcription factors showing their requirement for enhancer activity in different disease and tissue contexts.
Association of PRC1 with gene transcription has also been linked to adult stem cell differentiation. Work on epidermal stem cells suggested that PRC1 complexes promote the expression of skin regulatory genes. Although PRC1 still exerts its repression function in epidermal stem cells, a large cluster of PRC1 occupied sites contain histone modifications associated with active enhancers and promoters (H3K27ac, H3K4me3 and H3K4me1) (Cohen et al., 2018). Therefore, PRC1 may be required to promote transcription of key skin regulatory genes but it might also be associated with active enhancers and 3D chromatin organization to maintain cellular homeostasis. Similarly, RING1B is required for gene activation in resting B cells, and analysis of RING1B binding shows occupancy of RING1B at promoters and distal sites, suggesting a role of RING1B in regulating enhancer-promoter interactions to promote gene transcription (Frangini et al., 2013).
During tumorigenesis it has been shown that PRC1 complexes are recruited to both transcriptionally repressed and active genes, as well as at active enhancers. Interestingly, PRC1 is co-recruited with oncogenic transcription factors (TFs) to active enhancers in estrogen receptor positive (ER+), triple-negative breast cancer (TNBC), leukemia, hepatocellular carcinoma cell lines and Ewing sarcomas (Chan et al., 2018; Sánchez-Molina et al., 2020). This suggests that PRC1 is a master epigenetic regulator that associates with master TFs to drive tumorigenesis. For instance, in Ewing sarcomas expressing the oncogenic transcription factor EWSR1-FLI1, RING1B colocalizes with EWSR1-FLI1 at active enhancers to regulate its recruitment to chromatin and promote transcription (Figure 4A) (Sánchez-Molina et al., 2020). Similarly, in breast cancer cells RING1B is not the main H2AK119ub1 E3-ligase, and RING1A seems to be more enzymatically active (Chan et al., 2018). In ER+ breast cancer, the steroid hormone estrogen recruits PRC1 to ERα target genes. RING1B depletion demonstrated that it is required for full ERα recruitment, estrogen-mediated transcription, R-loop formation, and enhancer-promoter interactions upon estrogen stimulation (Figure 4A) (Zhang et al., 2020; Zhang et al., 2021b). Although the exact PRC1-mediated mechanism that promotes gene transcription in breast cancer cells is not fully understood, similarly to PRC2 (Long et al., 2020), RNA is required for PRC1 stabilization at chromatin upon estrogen induction (Zhang et al., 2021b). Whether other steroid hormones, such androgens and progesterone, regulate PRC1 recruitment to activate oncogenic pathways in other cancer types is not known.
Polycomb domain boundaries in stem cells and cancer
While H3K27me3 and H2AK119ub1 are particularly enriched at repressed promoters (in proximity to sites of stable PcG binding and CpG islands) a large proportion of these modifications are distributed at low “blanket”-like levels throughout the intergenic genome (Bracken et al., 2006; Ferrari et al., 2014; Lee et al., 2015; Fursova et al., 2019). These diffuse levels of H3K27me3 and H2AK119ub1 maintain “PcG domains” and often co-occur with high levels of H3K27me2, another catalytic product of PRC2 (Ferrari et al., 2014). It is thought that these diffuse modifications are catalysed by dynamic untethered PRC1 or PRC2 that are in a constant state of flux in the nucleus (Youmans et al., 2018; Huseyin and Klose, 2021). Another hypothesis is that these modifications can be generated shortly following DNA replication as PRC1 and PRC2 complexes have been observed in close proximity to the replication fork machinery (Hansen et al., 2008; Alabert et al., 2014; Piunti et al., 2014). Functional effectors for these diffuse or ‘blanket’ modifications are not well understood, with reader proteins such as CBX7 and JARID2 only stably binding to the highly enriched promoter proximal sites (Healy et al., 2019; Scelfo et al., 2019). Nevertheless, these PcG domains are precisely regulated through antagonistic interplay between PcG modifications and another epigenetic modification located at intergenic chromatin, H3K36me2. While the contribution of this careful balance of distal modifications to transcription is unknown, it has high relevance to chromatin structure and cancer as many of the proteins regulating this equilibrium of modifications are recurrently disrupted in cancer.
H3K36me2 is a modification deposited by the NSD1/2/3 histone methyltransferases (Deevy and Bracken, 2019). Structural studies have suggested a model in which NSD protein contacts with nucleosomes are sterically blocked by the presence of H2AK119ub1 (Figure 5A) (Li et al., 2021). Supporting this, H2AK119ub1 modified nucleosomes serve as a poor enzymatic substrate for NSD proteins compared to unmodified nucleosomes (Yuan et al., 2013; Li et al., 2021). A reciprocal antagonism is true for H3K36me2-modified nucleosomes which are an inefficient substrate for PRC2 complex activity (Yuan et al., 2011; Jain et al., 2020a). The presence of H3K36me2 on the histone tail reduces access of the SET domain of EZH2 to the H3K27 residue, as unmodified H3K36 is required to orient the H3 N-terminal tail correctly for optimal PRC2 activity (Finogenova et al., 2020). This reduction in efficiency of PRC2 in the presence of H3K36me2 explains the low correlation between H3K27me3 and H3K36me2 in the genome (Figure 5B), although H3K27me2 and H3K36me2 do coexist at intergenic sites (Streubel et al., 2018). This may be due to the rapid enzyme kinetics required for PRC2 catalysis of H3K27 di-methylation compared to long kinetics required for tri-methylation (Sneeringer et al., 2010; McCabe et al., 2012a). Accordingly, experiments that disrupt either PcG or NSD activities result in a switch in the balance of histone modification domains. For instance, in the absence of NSD1, H3K27me3 becomes predominant (Streubel et al., 2018), while in BAP1 null cells, broad accumulation of H2AK119ub1 causes reductions in H3K36me2 (Conway et al., 2021).
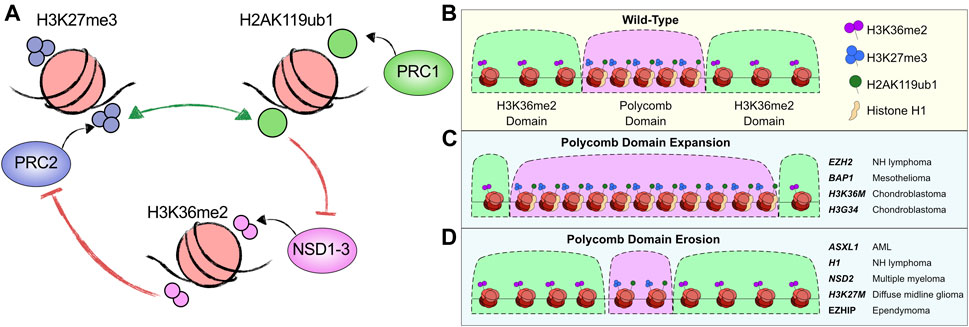
FIGURE 5. Dysregulated PcG domain formation in cancer. (A) Depiction of the protagonistic and antagonistic relationships between the histone modifications H3K27me3, H2AK119ub1 and H3K36me2 and the enzymatic complexes that catalyse them. (B) Schematic of Polycomb domains (B compartments) enriched in blanket levels of H3K27me3, H2AK119ub1 and occupied by Histone H1. These domains are boundaried by ‘A’ compartment related H3K36me2 enriched domains. (C) Illustration of how different mutations in PcG and other epigenetic machinery causes Polycomb domain expansion. The genes mutated and the cancer they are associated with are listed on the right. (D) Illustration of how different mutations in PcG and other epigenetic machinery causes Polycomb domain erosion. The genes mutated and the cancer they are associated with are listed on the right.
A further layer of regulation to the PcG and H3K36me2 domains comes in the form of histone H1. The linker histone H1 binds to nucleosomes at the dyad and maintains ordered structure of the surrounding chromatin. This in turn has been shown to promote catalytic function of PRC1 as it facilitates spreading of PcG modifications across nucleosomes (Zhao et al., 2020). Accordingly, histone H1 loss causes invasion of PcG domains by H3K36me2 (Willcockson et al., 2021; Yusufova et al., 2021). The structural basis for this promotion of PRC1 catalytic activity has not yet been evaluated. However, histone H1 can stabilise the H2A C-terminal tail and promote its contact with DNA near the chromatosome dyad (Arimura et al., 2021; Zhou et al., 2021), which is in close proximity to the H2AK119 residue, perhaps implicating a direct role in promoting catalytic activity through structural manipulation of histone tails.
Invasive Polycomb domains
The balance of PcG domains has been directly implicated in oncogenesis of follicular lymphoma patients bearing EZH2 gain-of-function mutations (e.g., Y641/A677) (Morin et al., 2010; McCabe et al., 2012a). These mutations alter the substrate preference for EZH2 from unmodified H3K27 to H3K27me1/2 facilitating an enzymatic progression to H3K27me2 by the wild-type EZH2 allele followed by rapid catalysis of H3K27me3 by the mutant EZH2 (McCabe et al., 2012a). These mutations were one of the earliest discovered PRC2 related dysregulations in cancer, and they feature global hyper-H3K27me3 (McCabe et al., 2012b). Importantly, the gains in H3K27me3 are not localised to promoter or CpG island regions but instead are primarily found in the ‘blanket’ intergenic regions (Souroullas et al., 2016; Donaldson-Collier et al., 2019). This coincides with increases in local compaction within TADs, inducing downregulation of germinal centre B-cell differentiation pathway genes such as PRDM1 (Béguelin et al., 2016; Donaldson-Collier et al., 2019). Therefore, an altered PcG domain pathway causes a differentiation blockade through hyper-compaction of broad regions of chromatin (Figure 5C). Patients with these mutations are already benefiting from EZH2 inhibitors with early clinical results indicating promising response rates (Italiano et al., 2018).
Somatic missense mutations in genes encoding for canonical and variant histone H3 have been recently identified in a myriad of cancers (Sturm et al., 2012; Nacev et al., 2019). Mutations are often found in residues located in the histone N-terminal tail which plays fundamental roles in development, cancer, and gene regulation. Common mutations in genes encoding canonical histone H3 (H3.1) and variant histone H3 (H3.3, encoded by H3F3A and H3F3B) include lysine 36 to methionine (H3K36M) found in chondroblastomas, lysine 27 to methionine (H3K27M) found in pediatric gliomas, and glycine 34 to tryptophan/leucine (H3G34W/L) mutations found in giant cell tumors of the bone (Lewis et al., 2013; Lu et al., 2016; Jain et al., 2020a). Expression of H3K36M or H3G34W/L inhibits NSD proteins leading to global reduction of H3K36 methylation concomitantly with an accumulation of H3K27me3 at intergenic regions (Lu et al., 2016; Jain et al., 2020a). Notably, these oncohistones induce a similar phenotype to EZH2 GOF (gain-of-function) mutations through removal or reduction in H3K36 methylation levels. These reductions in H3K36me2 cause widespread accumulation of PcG domains via diffuse spreading of H3K27me3 in turn promoting hyper-repression of distal regulatory elements (Figure 5C) (Jain et al., 2020a).
PcG domains are similarly altered upon BAP1 LOF (loss-of-function) mutations and EZH2 GOF mutations (Conway et al., 2021; Fursova et al., 2021). BAP1 null or catalytic mutant cells exhibit broad increases in H2AK119ub1 and H3K27me3, due to the displacement of PcG complexes from promoters. This in turn enhances either their dynamism, their catalytic activity or perhaps both. Domains featuring gains in H3K27me3 in BAP1 null contexts exhibit concurrent losses of H3K36me2 (Conway et al., 2021). Indeed, this coincides with global gains in compaction and nucleosomal reorganisation into packed clusters, and an increase in the levels of histone H1 on chromatin. Together with H3K36M, H3G34W/L and EZH2 mutant cancers, these data highlight a parallel mechanism for PcG domain expansion/invasion and blockade of differentiation pathways in distinct cancer types (Figure 5C).
Polycomb domain erosion
The PR-DUB subunit ASXL1 is frequently mutated in myeloid malignancies such as AML (Dohner et al., 2017). Most ASXL1 mutations are nonsense or frameshift mutations that result in a truncated protein lacking a C-terminal PHD domain (Tamburri et al., 2022). Unlike the LOF BAP1 mutations observed in cancer, these truncating ASXL1 mutations result in hyper-deubiquitination activity for PR-DUB complexes (Balasubramani et al., 2015). The mechanisms proposed for this increase in catalytic activity have been quite divergent with some reports suggesting that truncated ASXL1 either stabilises BAP1 protein levels (Bai et al., 2021; Wang et al., 2021) or loses interaction with the PR-DUB co-factors FOXK1/2 (Xia et al., 2021). FOXK1/2 are required for stably targeting PR-DUB to chromatin, but do not contribute to the catalytic activity of the complex (Kolovos et al., 2020). Together this may suggest that the ASXL1-truncated PR-DUB could have increased rates of dynamic activity at diffuse “blanket sites” or PcG domains. The effect of these mutations has yet to be fully established and whether hyper-DUB activity is restricted to PRC1 bound sites or broad PcG domains remains to be elucidated. If the latter, then it is possible that AML patients harbouring these ASXL1 truncations have restricted PcG domains with low levels of H2AK119ub1 and H3K27me3, potentially causing decompaction of chromatin into a naïve stem-like cell state (Figure 5D).
The H3K27M mutations in diffuse midline glioma (DMG) and EZHIP overexpression in ependymoma, while featuring increased levels of H3K27me3 at tumor suppressor genes such as CDKN2A, have broadly reduced levels of H3K27me3 and H3K27me2. This is caused by the inability to spread these modifications from PcG nucleation sites at promoters (Jain et al., 2020b). Concurrently, hyper-acetylation of H3K27 has been observed broadly in the absence of diffuse PcG activity (Krug et al., 2019; Brien et al., 2021). This results in the aberrant transcription of repetitive elements (Krug et al., 2019) and likely leads to a decompacted and stem-like chromatin state which is permissive to uncontrolled proliferation. Similarly, linker histone H1 encoding genes H1-2, H1-3 and H1-4 are recurrently mutated in follicular lymphoma. These mutations result in the degradation of PcG domains and invasion of these regions by H3K36me2 (Figure 5D) (Willcockson et al., 2021; Yusufova et al., 2021). This coincides with a switch from B to A compartments at these loci supporting their decompaction in the absence of PcG activity and histone H1 (Yusufova et al., 2021).
The NSD2 gene exhibits recurrent GOF mutations in acute lymphocytic leukemia (Oyer et al., 2014; Swaroop et al., 2019). This NSD2 E1099K mutation disrupts an autoinhibitory loop allowing enhanced catalytic activity (Sato et al., 2021). NSD2 is also recurrently overexpressed in multiple myeloma through a t(4; 14) translocation that places the NSD2 gene under the control of the IgH immunoglobulin promoter (Lhoumaud et al., 2019). Both events promote hyper-activity of NSD2, increasing H3K36me2 domains (Lhoumaud et al., 2019; Narang et al., 2022). This causes the erosion of PcG domains through inhibition of H3K27me3 and further coincides with large-scale changes in genome architecture (Figure 5D). This includes a general shift from B compartments (inactive) to A compartments (active), coinciding with those regions that gain H3K36me2 and lose H3K27me3 (Lhoumaud et al., 2019; Narang et al., 2022). These sites tend to become more transcriptionally active, and there is also an increase in enhancer activity. A further mechanism through which the balance of H3K36me2 and PcG domains may be affected is through DNMT3A mutations that occur in AML (Dohner et al., 2017). DNMT3A can read both H2AK119ub1 or H3K36me2 through its UIM and PWWP domains respectively (Weinberg et al., 2019; Weinberg et al., 2021; Gu et al., 2022). However, the myeloid mutations occur in its cytosine methyltransferase domain so it is not yet clear what effects these mutations can have on PcG domains. It seems likely that they cause a spreading of PcG domains due to the previously identified antagonism between DNA methylation and PcG.
Taken together, a careful balance of PcG and H3K36me2 domains appears to be important for the regulation of chromatin structure, plasticity, and differentiation potential. This is best highlighted by the fact that PRC2 and PR-DUB have both gain- and loss-of-function mutations in different cancer contexts (Morin et al., 2010; Ntziachristos et al., 2012; Balasubramani et al., 2015; Conway et al., 2021). Indeed, efficacy of EZH2 inhibitors in malignant rhabdoid tumours is dependent on the activity of NSD1, reinforcing the importance of this balance of Polycomb and H3K36me2 domains across different cancers (Drosos et al., 2022). The precise contribution of broad histone modification domains, largely devoid of epigenetic complex binding, remain unknown. Despite this, the many avenues through which these domains are recurrently disrupted in cancer highlights the importance of further investigation in this area in order to understand the oncogenic mechanisms and tailor appropriate epigenetic therapies to patients harbouring these mutations.
Drugging the epigenome to restore PcG structural regulation
As epigenetic machineries are often deregulated or mutated in cancer (Chan and Morey, 2019), efforts in medicinal chemistry have led to the development of epidrugs to combat these dysregulated functions. Given the importance of the epigenome in promoting and maintaining human diseases, it is curious that to date only nine epidrugs are FDA-approved. These include inhibitors of EZH2, IDH, HDACs, and DNA hypomethylating agents. Nevertheless, during the last decade, dozens of small molecules that inhibit the enzymatic activity of epigenetic machineries have been developed. Notably, several epidrugs are undergoing clinical trials for treating solid and hematological malignancies. In this chapter, we will discuss recent progress on the development of small molecules that target specific PRC1 and PRC2 subunits (Table 1).
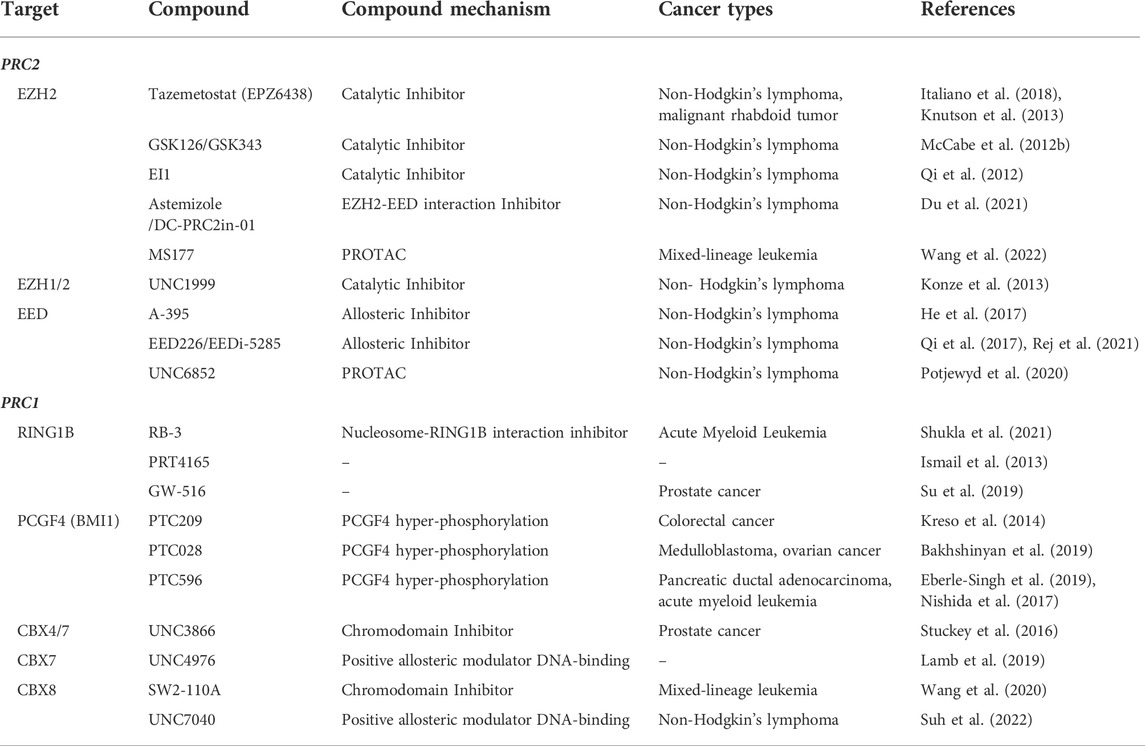
TABLE 1. PRC1 and PRC2 complex targeting compounds. Table detailing the published small molecule compounds that target PRC1 or PRC2 complex members and the mechanism of their targeting activity.
Targeting PRC2 complexes
In homeostasis and non-malignant conditions, PRC2 suppresses expression of tumor suppressor genes and maintains cellular identity. EZH2 mutations and overexpression of core PRC2 subunits have been found in multiple solid cancers, including prostate and breast cancer, as well as in hematological malignancies (Comet et al., 2016; Chan and Morey, 2019). Therefore, PRC2 complexes and subunits are considered promising targets in cancer.
Notably, PRC2 can be targeted on multiples fronts and strategies should be tailored for each cancer type. For instance, initial studies were focused on targeting the enzymatic activity of EZH2. This led to the discovery of highly potent and selective compounds, including tazemetostat (McCabe et al., 2012b; Qi et al., 2012; Knutson et al., 2013; Konze et al., 2013; Italiano et al., 2018), which has been recently approved by the FDA. Alternatively, given the key role of EED in propagating H3K27me3 and maintaining PRC2 complex integrity, multiple small molecules targeting EED have been developed. EED directly interacts with EZH2, therefore Orkin and colleagues developed peptides that selectively disrupt the EED/EZH2 interaction, thereby reducing EZH2 protein levels as well as H3K27me3 in MLL-AF9-mediated leukemia (Kim et al., 2013). Also, small-molecule compounds that interfere with the EED/EZH2 interaction have been developed (e.g., astemizole and DC-PRC2in-01) and tested in PRC2-dependent follicular lymphomas (Du et al., 2021). Alternatively, recognition of H3K27me3 by EED suggested the possibility of developing allosteric inhibitors. Indeed, EED226 and A-395 bind to the H3K27me3 binding pocket of EED to allosterically inhibit PRC2 activity by inducing a conformational change of EED. Notably, both inhibitors reduced the growth of EZH2 GOF mutant follicular lymphoma (He et al., 2017; Qi et al., 2017). Most importantly, these compounds are still effective in cancer cells that have acquired resistance to EZH2 inhibitors. Recently, analogs of EED226 (e.g., EEDi-5285) have been further developed and optimized with an enhanced binding affinity for EED and the ability to inhibit growth in mouse xenograft models of human follicular lymphoma (He et al., 2017; Rej et al., 2021).
Small molecules targeting EZH2, EED/EZH2 or EED/H3K27me3, although effective do not achieve complete tumor regression. Although an obvious consideration would be that a combination of PRC2 inhibitors with either other epidrugs or standard of care therapies would increase their efficacy, another possibility is that EZH2 has acquired non-canonical functions outside the context of PRC2. Indeed, it has been shown that in prostate cancer, breast cancer and in leukemia, EZH2 is also recruited to oncogenes to promote gene activation independently of its enzymatic activity (Lee et al., 2011; Xu et al., 2012; Chan and Morey, 2019; Wang et al., 2022). Therefore, compounds targeting PRC2 enzymatic activity or H3K27me3 propagation are unable to block aberrant EZH2 functions. This problem could be circumvented by the development of proteolysis targeting chimeras (PROTACs). PROTACs are bifunctional molecules containing a target protein ligand and an E3 ligase ligand. They induce selective degradation of their targeted proteins via the ubiquitin-proteasome system. Jin and Wang laboratories recently reported that the PROTAC MS177 achieved effective depletion of EZH2 in the context of both its canonical and non-canonical activities in MLL leukemias (Wang et al., 2022). Notably, they showed that tumor burden was more significantly reduced when mice were treated with MS177 instead of with UNC6852, a PROTAC that selectively targets EED (Potjewyd et al., 2020) and therefore the PRC2 activity but not the PRC2-independent activity of EZH2. Finally, given the specific roles of PRC2.1 and PRC2.2 complexes in development, cancer, and gene regulation, PROTACS targeting JARID2, and PCL proteins will certainly be developed in the near future.
Targeting PRC1 complexes
Although the majority of genes encoding for PRC1 subunits are not mutated in cancer, they are often deregulated (Chan et al., 2018; Chan and Morey, 2019). As mentioned above, dozens of PRC1 complexes can be assembled and simultaneously present in a given cell type. Thus, it is of great interest to develop small molecules and PROTACS that would either specifically target a discrete PRC1 complex or at least discriminate between cPRC1 and vPRC1 complexes, particularly in the context of cancers with high levels of H2AK119ub1, like BAP1 mutant tumors. To date, small molecules targeting RING1B, PCGF4 and CBX proteins have been reported. Thus, compounds specifically targeting vPRC1 complexes have not yet been developed. We envision that PROTACs targeting RYBP, PCGF1, PCGF3, PCGF5 and PCGF6 will be reported in the coming years.
RING1B inhibitors: During the last decade, great efforts have been directed towards targeting RING1B, the core component of PRC1. To date, three small molecules targeting RING1B have been reported. Cierpicki and colleagues developed RB-3, the only available compound that directly interacts with RING1B (Shukla et al., 2021). RB-3 treatment leads to a reduction of H2AK119ub1 in AML cells. Notably, xenograft experiments showed that RB-3 was effective in reducing leukemias. Whether RB-3 is also effective in reducing tumor burden in cancers addicted to RING1B, such as breast and pancreatic cancers (Chen et al., 2014; Chan et al., 2018; Zhang Z. et al., 2021), remains to be investigated. PRT4165 and GW-516 were proposed to inhibit the E3-ligase activity of RING1B in vitro, although their mechanism of action is not known (Ismail et al., 2013; Su et al., 2019). While these compounds directly or indirectly regulate H2AK119ub1, high concentrations are required to inhibit RING1B’s activity, and therefore it is expected that more powerful analogs will be developed.
BMI1 (PCGF4) inhibitors: Several compounds that reduce BMI1 protein levels have been described. High-throughput screenings designed to identify compounds that reduced BMI1 transcript levels identified PTC209 (Kreso et al., 2014). Although PTC209 does not bind to BMI1, it reduces both BMI1 and H2AK119ub1 levels and inhibits the self-renewal of colorectal cancer-initiating cells, resulting in the abrogation of colorectal tumors. PTC028, an analog of PTC209, showed reduction in both primary medulloblastoma tumors and their metastatic sites, without neurotoxicity and led to increased survival in mice (Bakhshinyan et al., 2019). The growth of ovarian cancer cells, but not normal cells, and the tumor burden of an orthotopic ovarian cancer model are also reduced by PTC028 treatment. Mechanistically, BMI1 modulators inhibit the anaphase promoting complex APC/C leading to persistent cyclin-dependent kinase (CDK)1/2 activity and BMI1 hyper-phosphorylation (Dey et al., 2018). PTC596 also reduced BMI1 levels and H2AK119ub1 in pancreatic ductal adenocarcinoma (PDA) and AML (Nishida et al., 2017; Eberle-Singh et al., 2019), but recent studies challenged the specificity of this compound. Crystal structure studies revealed that PTC596 interacts with tubulin and inhibits microtubule polymerization (Jernigan et al., 2021). Moreover, elegant genetic studies using Bmi1 null PDA cells, demonstrated that PTC596 induced mitotic arrest and had an antiproliferative effect independently of BMI1 (Eberle-Singh et al., 2019). Nevertheless, PTC596 is currently in clinical trials. A phase study in patients with advanced cancer (NCT02404480) has been completed. PTC596 was tolerable with gastrointestinal side effects. Currently, three phase 1b trials are ongoing in patients with high grade glioma (NCT03605550), advanced leiomyosarcoma (NCT03761095) and ovarian cancer (NCT03206645).
CBX ligand inhibitors: The ability of the Polycomb CBXs’ chromodomain to bind to H3K27me3 and the specific roles of these CBX proteins in development and disease, inspired medicinal chemists to develop compounds that disrupt this interaction. Work from the Frye lab and others have provided a catalog of compounds that target CBX4, CBX7 or CBX8. UNC3866 is a potent antagonist of CBX4 and CBX7, although it possesses affinity for other Polycomb CBX proteins and the CDY family of chromodomains (Stuckey et al., 2016). ChIP-seq studies in ESC showed that UNC3866 has a minor effect on cPRC1 stability on chromatin, yet its analog UNC4976 significantly affects cPRC1 recruitment to chromatin without affecting PRC2 activity (Lamb et al., 2019). UNC4976 is more specific towards CBX7 than UNC3866 and has a unique mechanism of action as a positive allosteric modulator of DNA and the ncRNA ANRIL binding to CBX7 (Lamb et al., 2019). This mechanism is not fully understood. To date, two specific CBX8 inhibitors have been discovered. SW2-110A has a 5-fold specificity for CBX8 compared to other Polycomb CBX proteins and reduces the binding of CBX8 to chromatin. It also inhibits cellular proliferation of leukemia cells expressing the MLL-AF9 translocation and alleviates the expression of HOXA9, an MLL-AF9 and CBX8 target (Wang et al., 2020). More recently, Bell and colleagues reported UNC7040, a specific CBX8 compound that blocks CBX8 and RING1B recruitment to PRC2 targets while, similarly to the UNC4976, enhancing CBX8 affinity to DNA. Nonetheless, UNC7040 results in cPRC1 displacement from H3K27me3-enriched genes resulting in reduced cell proliferation of cancer cells (Suh et al., 2022).
Discussion
The importance of chromatin regulator dysfunction in cancer has become increasingly evident over the past decade, with PcG proteins being a prime example of this. Understanding the mechanisms through which the healthy and oncogenic PcG proteins control the epigenome is already yielding clinically important results (Italiano et al., 2018). However, many questions remain unanswered about their contribution to chromatin structure and function. Foremost of which is whether PHC1-3 or CBX2 mediated compaction directly contribute to repression, as acute cPRC1 knockout has little effect on transcription (Fursova et al., 2019). Importantly, cPRC1 is developmentally essential (Akasaka et al., 1996; Akasaka et al., 2001; Isono et al., 2005; Lau et al., 2017), so perhaps dynamic/differentiation model systems are required for fully elucidating its role in transcription. The contribution of phase separation to these developmental processes is still not fully appreciated as separation-of-function mutations of phase separation phenomena from other biochemical roles has proven difficult in the chromatin field. In fact, emerging data suggests phase separation does not contribute to transcriptional control in certain contexts (Trojanowski et al., 2022).
Whether the compaction functions of SUZ12, EZH1 and OGT are maintained, and physiologically contribute to gene silencing in vivo is also a burning question. Biochemical models supporting their compaction activities have largely been in vitro (Gambetta and Muller, 2014; Chen et al., 2020; Grau et al., 2021). Functional studies on PRC2 and PR-DUB compaction mutants may allow careful dissection of their contribution to chromatin compaction and gene repression in a biological system.
The contribution of histone modifications to transcriptional repression and chromatin structural organisation is becoming increasingly clear with improved genome editing and PROTAC methods for gene/protein perturbation (Dobrinic et al., 2021; Sankar et al., 2022). However, it is still unclear how promoter enriched H2AK119ub1 promotes repression in a PRC2 independent fashion (Tamburri et al., 2020; Dobrinic et al., 2021). These questions will be key to understanding how defects in PcG body formation in different cancer contexts contribute to pathogenesis and how we can leverage this to identify targeted therapeutics for patients through the multitude of available epigenetic targeted inhibitors.
It is clear that in development and oncogenic models that an imbalance in PcG and H3K36me2 domains can affect chromatin topology and, in turn, differentiation trajectories. Nonetheless, many questions remain over the functional output of these ‘blanket’ domains of histone modification. How do diffuse levels of H3K27me3 and H2AK119ub1 result in compaction (Donaldson-Collier et al., 2019; Conway et al., 2021) in the absence of accumulation of their readers CBX and JARID2/AEBP2 (Fischle et al., 2003; Kalb et al., 2014; Cooper et al., 2016; Kasinath et al., 2021)? Is this process regulated through some of the other reported readers for H2AK119ub1 such as SS18-SSX, DNMT3A or indeed the inhibitory sensing of H2AK119ub1 by NSD1/2/3 enzymes (McBride et al., 2020; Li et al., 2021; Gu et al., 2022)?
A curious commonality between the genes that regulate PcG domains is that they are often recurrently mutated in neurodevelopmental syndromes. Weavers (EZH2), Cohen-Gibson (EED), SOTOS (NSD1), Bohring-Opitz (ASXL1), Bainbridge-Roper (ASXL3) and Tatton-Brown-Raman (DNMT3A) syndromes all feature syndrome-defining mutations in chromatin regulators that control the PcG-H3K36me2 domain axis (Deevy and Bracken, 2019; Tamburri et al., 2022). This feature further highlights the importance of in-depth molecular characterisation of how these domains are regulated in order to provide clinically relevant information. The next-generation of genomics and molecular biology technologies such as PROTAC, micro-C and single cell -omics will facilitate a deeper molecular understanding of these pathways in order to maximise clinical benefit (Kaya-Okur et al., 2019; Wimalasena et al., 2020; Jerkovic and Cavalli, 2021).
Author contributions
EC conceived the topic of the review. EC and LM wrote the draft of the manuscript and prepared figures, with inputs from ED. All authors contributed to manuscript revision, read, edited and approved the submitted version.
Funding
The work of the Conway laboratory was supported by an SFI/IRC Pathway Program grant (21/PATH-S/9384). The work of the Morey laboratory is supported by SCCC funds, the Florida Health Bankhead-Coley Cancer Research Program (20B15), the V Foundation (DEC 2020–009), the Lampert Breast Cancer Research Fund, and R01GM141349 from the National Institute of General Medical Sciences. Research in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA240139. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
We would like to thank all members of the Conway laboratory and Aisling Y. Coughlan for helpful discussion and critical reading.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Jacobs, J. J., Kieboom, K., Marino, S., Depinho, R. A., and Van Lohuizen, M. (1999). The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164–168. doi:10.1038/16476
Abdel-Wahab, O., Adli, M., Lafave, L. M., Gao, J., Hricik, T., Shih, A. H., et al. (2012). ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22, 180–193. doi:10.1016/j.ccr.2012.06.032
Akasaka, T., Kanno, M., Balling, R., Mieza, M. A., Taniguchi, M., and Koseki, H. (1996). A role for mel-18, a Polycomb group-related vertebrate gene, during theanteroposterior specification of the axial skeleton. Development 122, 1513–1522. doi:10.1242/dev.122.5.1513
Akasaka, T., Van Lohuizen, M., Van Der Lugt, N., Mizutani-Koseki, Y., Kanno, M., Taniguchi, M., et al. (2001). Mice doubly deficient for the Polycomb Group genes Mel18 and Bmi1 reveal synergy and requirement for maintenance but not initiation of Hox gene expression. Development 128, 1587–1597. doi:10.1242/dev.128.9.1587
Alabert, C., Bukowski-Wills, J.-C., Lee, S.-B., Kustatscher, G., Nakamura, K., Alves, F. D. L., et al. (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 16, 281–293. doi:10.1038/ncb2918
Arimura, Y., Shih, R. M., Froom, R., and Funabiki, H. 2021. Structural features of nucleosomes in interphase and metaphase chromosomes. Mol. Cell 81, 4377–4397 e12.
Bai, J., Chen, Z., Chen, C., Zhang, M., Zhang, Y., Song, J., et al. (2021). Reducing hyperactivated BAP1 attenuates mutant ASXL1-driven myeloid malignancies in human haematopoietic cells. Cancer Lett. 519, 78–90. doi:10.1016/j.canlet.2021.06.019
Bakhshinyan, D., Venugopal, C., Adile, A. A., Garg, N., Manoranjan, B., Hallett, R., et al. (2019). BMI1 is a therapeutic target in recurrent medulloblastoma. Oncogene 38, 1702–1716. doi:10.1038/s41388-018-0549-9
Balasubramani, A., Larjo, A., Bassein, J. A., Chang, X., Hastie, R. B., Togher, S. M., et al. (2015). Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat. Commun. 6, 7307. doi:10.1038/ncomms8307
Béguelin, W., Teater, M., Gearhart, M. D., Calvo Fernández, M. T., Goldstein, R. L., Cárdenas, M. G., et al. (2016). EZH2 and BCL6 cooperate to assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell 30, 197–213. doi:10.1016/j.ccell.2016.07.006
Blackledge, N. P., Farcas, A. M., Kondo, T., King, H. W., Mcgouran, J. F., Hanssen, L. L., et al. (2014). Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157, 1445–1459. doi:10.1016/j.cell.2014.05.004
Blackledge, N. P., Fursova, N. A., Kelley, J. R., Huseyin, M. K., Feldmann, A., and Klose, R. J. (2020). PRC1 catalytic activity is central to polycomb system function. Mol. Cell 77, 857–874. doi:10.1016/j.molcel.2019.12.001
Blackledge, N. P., and Klose, R. J. (2021). The molecular principles of gene regulation by Polycomb repressive complexes. Nat. Rev. Mol. Cell Biol. 22, 815–833. doi:10.1038/s41580-021-00398-y
Boettiger, A. N., Bintu, B., Moffitt, J. R., Wang, S., Beliveau, B. J., Fudenberg, G., et al. (2016). Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422. doi:10.1038/nature16496
Boyle, S., Flyamer, I. M., Williamson, I., Sengupta, D., Bickmore, W. A., and Illingworth, R. S. (2020). A central role for canonical PRC1 in shaping the 3D nuclear landscape. Genes Dev. 34, 931–949. doi:10.1101/gad.336487.120
Bracken, A. P., Dietrich, N., Pasini, D., Hansen, K. H., and Helin, K. (2006). Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 20, 1123–1136. doi:10.1101/gad.381706
Brien, G. L., Bressan, R. B., Monger, C., Gannon, D., Lagan, E., Doherty, A. M., et al. (2021). Simultaneous disruption of PRC2 and enhancer function underlies histone H3.3-K27M oncogenic activity in human hindbrain neural stem cells. Nat. Genet. 53, 1221–1232. doi:10.1038/s41588-021-00897-w
Comet, I., Riising, E. M., Leblanc, B., and Helin, K. (2016). Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 16, 803–810. doi:10.1038/nrc.2016.83
Cao, R., Wang, L., Wang, H., Xia, L., Erdjument-Bromage, H., Tempst, P., et al. (2002). Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043. doi:10.1126/science.1076997
Carbone, M., Yang, H., Pass, H. I., Krausz, T., Testa, J. R., and Gaudino, G. (2013). BAP1 and cancer. Nat. Rev. Cancer 13, 153–159. doi:10.1038/nrc3459
Chan, H. L., Beckedorff, F., Zhang, Y., Garcia-Huidobro, J., Jiang, H., Colaprico, A., et al. (2018). Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun. 9, 3377. doi:10.1038/s41467-018-05728-x
Chan, H. L., and Morey, L. (2019). Emerging roles for polycomb-group proteins in stem cells and cancer. Trends biochem. Sci. 44, 688–700. doi:10.1016/j.tibs.2019.04.005
Chen, J., Xu, H., Zou, X., Wang, J., Zhu, Y., Chen, H., et al. (2014). Snail recruits Ring1B to mediate transcriptional repression and cell migration in pancreatic cancer cells. Cancer Res. 74, 4353–4363. doi:10.1158/0008-5472.CAN-14-0181
Chen, S., Jiao, L., Liu, X., Yang, X., and Liu, X. (2020). A dimeric structural scaffold for PRC2-PCL targeting to CpG island chromatin. Mol. Cell 77, 1265–1278. doi:10.1016/j.molcel.2019.12.019
Chen, S., Jiao, L., Shubbar, M., Yang, X., and Liu, X. (2018). Unique structural platforms of Suz12 dictate distinct classes of PRC2 for chromatin binding. Mol. Cell 69, 840–852. doi:10.1016/j.molcel.2018.01.039
Chiacchiera, F., Rossi, A., Jammula, S., Zanotti, M., and Pasini, D. (2016). PRC2 preserves intestinal progenitors and restricts secretory lineage commitment. Embo J. 35, 2301–2314. doi:10.15252/embj.201694550
Choi, J., Bachmann, A. L., Tauscher, K., Benda, C., Fierz, B., and Muller, J. (2017). DNA binding by PHF1 prolongs PRC2 residence time on chromatin and thereby promotes H3K27 methylation. Nat. Struct. Mol. Biol. 24, 1039–1047. doi:10.1038/nsmb.3488
Cohen, I., Zhao, D., Bar, C., Valdes, V. J., Dauber-Decker, K. L., Nguyen, M. B., et al. (2018). PRC1 fine-tunes gene repression and activation to safeguard skin development and stem cell specification. Cell Stem Cell 22, 726–739. doi:10.1016/j.stem.2018.04.005
Conway, E., Healy, E., and Bracken, A. P. (2015). PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr. Opin. Cell Biol. 37, 42–48. doi:10.1016/j.ceb.2015.10.003
Conway, E., Jerman, E., Healy, E., Ito, S., Holoch, D., Oliviero, G., et al. (2018). A family of vertebrate-specific polycombs encoded by the LCOR/LCORL genes balance PRC2 subtype Activities. Mol. Cell 70, 408–421. doi:10.1016/j.molcel.2018.03.005
Conway, E., Rossi, F., Fernandez-Perez, D., Ponzo, E., Ferrari, K. J., Zanotti, M., et al. (2021). BAP1 enhances Polycomb repression by counteracting widespread H2AK119ub1 deposition and chromatin condensation. Mol. Cell 81, 3526–3541. doi:10.1016/j.molcel.2021.06.020
Cooper, S., Grijzenhout, A., Underwood, E., Ancelin, K., Zhang, T., Nesterova, T. B., et al. (2016). Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 7, 13661. doi:10.1038/ncomms13661
Cruz-Molina, S., Respuela, P., Tebartz, C., Kolovos, P., Nikolic, M., Fueyo, R., et al. (2017). PRC2 facilitates the regulatory topology required for poised enhancer function during pluripotent stem cell differentiation. Cell Stem Cell 20, 689–705 e9. doi:10.1016/j.stem.2017.02.004
Davidovich, C., Goodrich, K. J., Gooding, A. R., and Cech, T. R. (2014). A dimeric state for PRC2. Nucleic Acids Res. 42, 9236–9248. doi:10.1093/nar/gku540
De Napoles, M., Mermoud, J. E., Wakao, R., Tang, Y. A., Endoh, M., Appanah, R., et al. (2004). Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 7, 663–676. doi:10.1016/j.devcel.2004.10.005
Deevy, O., and Bracken, A. P. (2019). PRC2 functions in development and congenital disorders. Development 146, dev181354. doi:10.1242/dev.181354
Dey, A., Xiong, X., Crim, A., Dwivedi, S. K. D., Mustafi, S. B., Mukherjee, P., et al. (2018). Evaluating the mechanism and therapeutic potential of PTC-028, a novel inhibitor of BMI-1 function in ovarian cancer. Mol. Cancer Ther. 17, 39–49. doi:10.1158/1535-7163.MCT-17-0574
Dobrinic, P., Szczurek, A. T., and Klose, R. J. (2021). PRC1 drives Polycomb-mediated gene repression by controlling transcription initiation and burst frequency. Nat. Struct. Mol. Biol. 28, 811–824. doi:10.1038/s41594-021-00661-y
Dohner, H., Estey, E., Grimwade, D., Amadori, S., Appelbaum, F. R., Buchner, T., et al. (2017). Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447. doi:10.1182/blood-2016-08-733196
Donaldson-Collier, M. C., Sungalee, S., Zufferey, M., Tavernari, D., Katanayeva, N., Battistello, E., et al. (2019). EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 51, 517–528. doi:10.1038/s41588-018-0338-y
Drosos, Y., Myers, J. A., Xu, B., Mathias, K. M., Beane, E. C., Radko-Juettner, S., et al. (2022). NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition. Mol. Cell 82, 2472–2489. doi:10.1016/j.molcel.2022.04.015
Du, D., Xu, D., Zhu, L., Stazi, G., Zwergel, C., Liu, Y., et al. (2021). Structure-guided development of small-molecule PRC2 inhibitors targeting EZH2-EED interaction. J. Med. Chem. 64, 8194–8207. doi:10.1021/acs.jmedchem.0c02261
Eberle-Singh, J. A., Sagalovskiy, I., Maurer, H. C., Sastra, S. A., Palermo, C. F., Decker, A. R., et al. (2019). Effective delivery of a microtubule polymerization inhibitor synergizes with standard regimens in models of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 25, 5548–5560. doi:10.1158/1078-0432.CCR-18-3281
Endoh, M., Endo, T. A., Shinga, J., Hayashi, K., Farcas, A., Ma, K. W., et al. (2017). PCGF6-PRC1 suppresses premature differentiation of mouse embryonic stem cells by regulating germ cell-related genes. Elife 6, e21064. doi:10.7554/eLife.21064
Farcas, A. M., Blackledge, N. P., Sudbery, I., Long, H. K., Mcgouran, J. F., Rose, N. R., et al. (2012). KDM2B links the polycomb repressive complex 1 (PRC1) to recognition of CpG islands. Elife 1, e00205. doi:10.7554/eLife.00205
Ferrari, K. J., Scelfo, A., Jammula, S., Cuomo, A., Barozzi, I., Stutzer, A., et al. (2014). Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 53, 49–62. doi:10.1016/j.molcel.2013.10.030
Finogenova, K., Bonnet, J., Poepsel, S., Schafer, I. B., Finkl, K., Schmid, K., et al. (2020). Structural basis for PRC2 decoding of active histone methylation marks H3K36me2/3. Elife 9, e61964. doi:10.7554/eLife.61964
Fischle, W., Wang, Y., Jacobs, S. A., Kim, Y., Allis, C. D., and Khorasanizadeh, S. (2003). Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 17, 1870–1881. doi:10.1101/gad.1110503
Francis, N. J., Kingston, R. E., and Woodcock, C. L. (2004). Chromatin compaction by a polycomb group protein complex. Science 306, 1574–1577. doi:10.1126/science.1100576
Frangini, A., Sjoberg, M., Roman-Trufero, M., Dharmalingam, G., Haberle, V., Bartke, T., et al. (2013). The aurora B kinase and the polycomb protein ring1B combine to regulate active promoters in quiescent lymphocytes. Mol. Cell 51, 647–661. doi:10.1016/j.molcel.2013.08.022
Fursova, N. A., Blackledge, N. P., Nakayama, M., Ito, S., Koseki, Y., Farcas, A. M., et al. (2019). Synergy between variant PRC1 complexes defines polycomb-mediated gene repression. Mol. Cell 74, 1020–1036. doi:10.1016/j.molcel.2019.03.024
Fursova, N. A., Turberfield, A. H., Blackledge, N. P., Findlater, E. L., Lastuvkova, A., Huseyin, M. K., et al. (2021). BAP1 constrains pervasive H2AK119ub1 to control the transcriptional potential of the genome. Genes Dev. 35, 749–770. doi:10.1101/gad.347005.120
Gahan, J. M., Rentzsch, F., and Schnitzler, C. E. (2020). The genetic basis for PRC1 complex diversity emerged early in animal evolution. Proc. Natl. Acad. Sci. U. S. A. 117, 22880–22889. doi:10.1073/pnas.2005136117
Gambetta, M. C., and Muller, J. (2014). O-GlcNAcylation prevents aggregation of the Polycomb group repressor polyhomeotic. Dev. Cell 31, 629–639. doi:10.1016/j.devcel.2014.10.020
Gao, Z., Lee, P., Stafford, J. M., Von Schimmelmann, M., Schaefer, A., and Reinberg, D. (2014). An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354. doi:10.1038/nature13921
Gao, Z., Zhang, J., Bonasio, R., Strino, F., Sawai, A., Parisi, F., et al. (2012). PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 45, 344–356. doi:10.1016/j.molcel.2012.01.002
Glancy, E., Ciferri, C., and Bracken, A. P. (2021). Structural basis for PRC2 engagement with chromatin. Curr. Opin. Struct. Biol. 67, 135–144. doi:10.1016/j.sbi.2020.10.017
Grau, D. J., Chapman, B. A., Garlick, J. D., Borowsky, M., Francis, N. J., and Kingston, R. E. (2011). Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev. 25, 2210–2221. doi:10.1101/gad.17288211
Grau, D., Zhang, Y., Lee, C. H., Valencia-Sanchez, M., Zhang, J., Wang, M., et al. (2021). Structures of monomeric and dimeric PRC2:EZH1 reveal flexible modules involved in chromatin compaction. Nat. Commun. 12, 714. doi:10.1038/s41467-020-20775-z
Gu, T., Hao, D., Woo, J., Huang, T. W., Guo, L., Lin, X., et al. (2022). The disordered N-terminal domain of DNMT3A recognizes H2AK119ub and is required for postnatal development. Nat. Genet. 54, 625–636. doi:10.1038/s41588-022-01063-6
Hrzenjak, A. (2016). JAZF1/SUZ12 gene fusion in endometrial stromal sarcomas. Orphanet J. Rare Dis. 11, 15. doi:10.1186/s13023-016-0400-8
Hansen, K. H., Bracken, A. P., Pasini, D., Dietrich, N., Gehani, S. S., Monrad, A., et al. (2008). A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 10, 1291–1300. doi:10.1038/ncb1787
Hauri, S., Comoglio, F., Seimiya, M., Gerstung, M., Glatter, T., Hansen, K., et al. (2016). A high-density map for navigating the human polycomb complexome. Cell Rep. 17, 583–595. doi:10.1016/j.celrep.2016.08.096
He, Y., Selvaraju, S., Curtin, M. L., Jakob, C. G., Zhu, H., Comess, K. M., et al. (2017). The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 13, 389–395. doi:10.1038/nchembio.2306
Healy, E., Mucha, M., Glancy, E., Fitzpatrick, D. J., Conway, E., Neikes, H. K., et al. (2019). PRC2.1 and PRC2.2 synergize to coordinate H3K27 trimethylation. Mol. Cell 76, 437–452. doi:10.1016/j.molcel.2019.08.012
Hojfeldt, J. W., Hedehus, L., Laugesen, A., Tatar, T., Wiehle, L., and Helin, K. (2019). Non-core subunits of the PRC2 complex are collectively required for its target-site specificity. Mol. Cell 76, 423–436. doi:10.1016/j.molcel.2019.07.031
Huseyin, M. K., and Klose, R. J. (2021). Live-cell single particle tracking of PRC1 reveals a highly dynamic system with low target site occupancy. Nat. Commun. 12, 887. doi:10.1038/s41467-021-21130-6
Ismail, I. H., Mcdonald, D., Strickfaden, H., Xu, Z., and Hendzel, M. J. (2013). A small molecule inhibitor of polycomb repressive complex 1 inhibits ubiquitin signaling at DNA double-strand breaks. J. Biol. Chem. 288, 26944–26954. doi:10.1074/jbc.M113.461699
Isono, K., Endo, T. A., Ku, M., Yamada, D., Suzuki, R., Sharif, J., et al. (2013). SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell 26, 565–577. doi:10.1016/j.devcel.2013.08.016
Isono, K., Fujimura, Y., Shinga, J., Yamaki, M., J, O. W., Takihara, Y., et al. (2005). Mammalian polyhomeotic homologues Phc2 and Phc1 act in synergy to mediate polycomb repression of Hox genes. Mol. Cell. Biol. 25, 6694–6706. doi:10.1128/MCB.25.15.6694-6706.2005
Italiano, A., Soria, J.-C., Toulmonde, M., Michot, J.-M., Lucchesi, C., Varga, A., et al. (2018). Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet. Oncol. 19, 649–659. doi:10.1016/S1470-2045(18)30145-1
Jaensch, E. S., Zhu, J., Cochrane, J. C., Marr, S. K., Oei, T. A., Damle, M., et al. (2021). A Polycomb domain found in committed cells impairs differentiation when introduced into PRC1 in pluripotent cells. Mol. Cell 81, 4677–4691. doi:10.1016/j.molcel.2021.09.018
Jain, S. U., Do, T. J., Lund, P. J., Rashoff, A. Q., Diehl, K. L., Cieslik, M., et al. (2019). PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat. Commun. 10, 2146. doi:10.1038/s41467-019-09981-6
Jain, S. U., Khazaei, S., Marchione, D. M., Lundgren, S. M., Wang, X., Weinberg, D. N., et al. (2020a). Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. U. S. A. 117, 27354–27364. doi:10.1073/pnas.2006076117
Jain, S. U., Rashoff, A. Q., Krabbenhoft, S. D., Hoelper, D., Do, T. J., Gibson, T. J., et al. (2020b). H3 K27M and EZHIP impede H3K27-methylation spreading by inhibiting allosterically stimulated PRC2. Mol. Cell 80, 726–735. doi:10.1016/j.molcel.2020.09.028
Jerkovic, I., and Cavalli, G. (2021). Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 22, 511–528. doi:10.1038/s41580-021-00362-w
Jernigan, F., Branstrom, A., Baird, J. D., Cao, L., Dali, M., Furia, B., et al. (2021). Preclinical and early clinical development of PTC596, a novel small-molecule tubulin-binding agent. Mol. Cancer Ther. 20, 1846–1857. doi:10.1158/1535-7163.MCT-20-0774
Joo, H. Y., Zhai, L., Yang, C., Nie, S., Erdjument-Bromage, H., Tempst, P., et al. (2007). Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature 449, 1068–1072. doi:10.1038/nature06256
Kadoch, C., Williams, R. T., Calarco, J. P., Miller, E. L., Weber, C. M., Braun, S. M., et al. (2017). Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 49, 213–222. doi:10.1038/ng.3734
Kalb, R., Latwiel, S., Baymaz, H. I., Jansen, P. W., Muller, C. W., Vermeulen, M., et al. (2014). Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 21, 569–571. doi:10.1038/nsmb.2833
Kang, H., Cabrera, J. R., Zee, B. M., Kang, H. A., Jobe, J. M., Hegarty, M. B., et al. 2022. Variant Polycomb complexes in Drosophila consistent with ancient functional diversity. bioRxiv. doi:10.1101/2022.04.29.490092
Kasinath, V., Beck, C., Sauer, P., Poepsel, S., Kosmatka, J., Faini, M., et al. (2021). JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science 371, eabc3393. doi:10.1126/science.abc3393
Kaya-Okur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., et al. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930. doi:10.1038/s41467-019-09982-5
Kim, W., Bird, G. H., Neff, T., Guo, G., Kerenyi, M. A., Walensky, L. D., et al. (2013). Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol. 9, 643–650. doi:10.1038/nchembio.1331
Kloet, S. L., Makowski, M. M., Baymaz, H. I., Van Voorthuijsen, L., Karemaker, I. D., Santanach, A., et al. (2016). The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol. 23, 682–690. doi:10.1038/nsmb.3248
Knutson, S. K., Warholic, N. M., Wigle, T. J., Klaus, C. R., Allain, C. J., Raimondi, A., et al. (2013). Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. U. S. A. 110, 7922–7927. doi:10.1073/pnas.1303800110
Kolovos, P., Nishimura, K., Sankar, A., Sidoli, S., Cloos, P. A., Helin, K., et al. (2020). PR-DUB maintains the expression of critical genes through FOXK1/2- and ASXL1/2/3-dependent recruitment to chromatin and H2AK119ub1 deubiquitination. Genome Res. 30, 1119–1130. doi:10.1101/gr.261016.120
Kondo, T., Isono, K., Kondo, K., Endo, T. A. K. A. H. O. A., Itohara, S., Vidal, M., et al. (2014). Polycomb potentiates Meis2 activation in midbrain by mediating interaction of the promoter with a tissue-specific enhancer. Dev. Cell 28, 94–101. doi:10.1016/j.devcel.2013.11.021
Konze, K. D., Ma, A., Li, F., Barsyte-Lovejoy, D., Parton, T., Macnevin, C. J., et al. (2013). An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 8, 1324–1334. doi:10.1021/cb400133j
Kraft, K., Yost, K. E., Murphy, S., Magg, A., Long, Y., Corces, M. R., et al. 2022. Polycomb-mediated genome architecture enables long-range spreading of H3K27 methylation. Proc. Natl. Acad. Sci. USA 119, e2201883119.
Kreso, A., Van Galen, P., Pedley, N. M., Lima-Fernandes, E., Frelin, C., Davis, T., et al. (2014). Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 20, 29–36. doi:10.1038/nm.3418
Krug, B., De Jay, N., Harutyunyan, A. S., Deshmukh, S., Marchione, D. M., Guilhamon, P., et al. (2019). Pervasive H3K27 acetylation leads to ERV expression and a therapeutic vulnerability in H3K27M gliomas. Cancer Cell 35, 782–797. doi:10.1016/j.ccell.2019.04.004
Kundu, S., Ji, F., Sunwoo, H., Jain, G., Lee, J. T., Sadreyev, R. I., et al. (2017). Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol. Cell 65, 432–446. doi:10.1016/j.molcel.2017.01.009
Lagger, G., O'Carroll, D., Rembold, M., Khier, H., Tischler, J., Weitzer, G., et al. (2002). Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. Embo J. 21, 2672–2681. doi:10.1093/emboj/21.11.2672
Lamb, K. N., Bsteh, D., Dishman, S. N., Moussa, H. F., Fan, H., Stuckey, J. I., et al. (2019). Discovery and characterization of a cellular potent positive allosteric modulator of the polycomb repressive complex 1 chromodomain, CBX7. Cell Chem. Biol. 26, 1365–1379. doi:10.1016/j.chembiol.2019.07.013
Lau, M. S., Schwartz, M. G., Kundu, S., Savol, A. J., Wang, P. I., Marr, S. K., et al. (2017). Mutation of a nucleosome compaction region disrupts Polycomb-mediated axial patterning. Science 355, 1081–1084. doi:10.1126/science.aah5403
Lavarone, E., Barbieri, C. M., and Pasini, D. (2019). Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat. Commun. 10, 1679. doi:10.1038/s41467-019-09624-w
Lee, C. H., Holder, M., Grau, D., Saldana-Meyer, R., Yu, J. R., Ganai, R. A., et al. (2018). Distinct stimulatory mechanisms regulate the catalytic activity of polycomb repressive complex 2. Mol. Cell 70, 435–448. doi:10.1016/j.molcel.2018.03.019
Lee, H. G., Kahn, T. G., Simcox, A., Schwartz, Y. B., and Pirrotta, V. (2015). Genome-wide activities of Polycomb complexes control pervasive transcription. Genome Res. 25, 1170–1181. doi:10.1101/gr.188920.114
Lee, S. T., Li, Z., Wu, Z., Aau, M., Guan, P., Karuturi, R. K., et al. (2011). Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol. Cell 43, 798–810. doi:10.1016/j.molcel.2011.08.011
Lee, W., Teckie, S., Wiesner, T., Ran, L., Prieto Granada, C. N., Lin, M., et al. (2014). PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 46, 1227–1232. doi:10.1038/ng.3095
Lewis Pw, E. A., Muller, M. M., Koletsky, M. S., Cordero, F., Lin, S., Banaszynski, L. A., et al. (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. doi:10.1126/science.1232245
Lewis, P. W., Muller, M. M., Koletsky, M. S., Cordero, F., Lin, S., Banaszynski, L. A., et al. (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. doi:10.1126/science.1232245
Lhoumaud, P., Badri, S., Rodriguez-Hernaez, J., Sakellaropoulos, T., Sethia, G., Kloetgen, A., et al. (2019). NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat. Commun. 10, 4843. doi:10.1038/s41467-019-12811-4
Li, W., Tian, W., Yuan, G., Deng, P., Sengupta, D., Cheng, Z., et al. (2021). Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature 590, 498–503. doi:10.1038/s41586-020-03069-8
Liu, S., Aldinger, K. A., Cheng, C. V., Kiyama, T., Dave, M., Mcnamara, H. K., et al. (2021). NRF1 association with AUTS2-Polycomb mediates specific gene activation in the brain. Mol. Cell 81, 4663–4676. doi:10.1016/j.molcel.2021.09.020
Liu, Y., Easton, J., Shao, Y., Maciaszek, J., Wang, Z., Wilkinson, M. R., et al. (2017). The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 49, 1211–1218. doi:10.1038/ng.3909
Long, Y., Hwang, T., Gooding, A. R., Goodrich, K. J., Rinn, J. L., and Cech, T. R. (2020). RNA is essential for PRC2 chromatin occupancy and function in human pluripotent stem cells. Nat. Genet. 52, 931–938. doi:10.1038/s41588-020-0662-x
Loubiere, V., Papadopoulos, G. L., Szabo, Q., Martinez, A. M., and Cavalli, G. (2020). Widespread activation of developmental gene expression characterized by PRC1-dependent chromatin looping. Sci. Adv. 6, eaax4001. doi:10.1126/sciadv.aax4001
Lu, C., Jain, S. U., Hoelper, D., Bechet, D., Molden, R. C., Ran, L., et al. (2016). Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849. doi:10.1126/science.aac7272
Margueron, R., Li, G., Sarma, K., Blais, A., Zavadil, J., Woodcock, C. L., et al. (2008). Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 32, 503–518. doi:10.1016/j.molcel.2008.11.004
Mcbride, M. J., Mashtalir, N., Winter, E. B., Dao, H. T., Filipovski, M., D'Avino, A. R., et al. (2020). The nucleosome acidic patch and H2A ubiquitination underlie mSWI/SNF recruitment in synovial sarcoma. Nat. Struct. Mol. Biol. 27, 836–845. doi:10.1038/s41594-020-0466-9
Mccabe, M. T., Graves, A. P., Ganji, G., Diaz, E., Halsey, W. S., Jiang, Y., et al. (2012a). Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. U. S. A. 109, 2989–2994. doi:10.1073/pnas.1116418109
Mccabe, M. T., Ott, H. M., Ganji, G., Korenchuk, S., Thompson, C., Van Aller, G. S., et al. (2012b). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. doi:10.1038/nature11606
Min, J., Zhang, Y., and Xu, R. M. (2003). Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 17, 1823–1828. doi:10.1101/gad.269603
Mohammad, F., Weissmann, S., Leblanc, B., Pandey, D. P., Hojfeldt, J. W., Comet, I., et al. (2017). EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 23, 483–492. doi:10.1038/nm.4293
Morey, L., Aloia, L., Cozzuto, L., Benitah, S. A., and Di Croce, L. (2013). RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells. Cell Rep. 3, 60–69. doi:10.1016/j.celrep.2012.11.026
Morey, L., Pascual, G., Cozzuto, L., Roma, G., Wutz, A., Benitah, S. A., et al. (2012). Nonoverlapping functions of the Polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell 10, 47–62. doi:10.1016/j.stem.2011.12.006
Morey, L., Santanach, A., Blanco, E., Aloia, L., Nora, E. P., Bruneau, B. G., et al. (2015). Polycomb regulates mesoderm cell fate-specification in embryonic stem cells through activation and repression mechanisms. Cell Stem Cell 17, 300–315. doi:10.1016/j.stem.2015.08.009
Morin, R. D., Johnson, N. A., Severson, T. M., Mungall, A. J., An, J., Goya, R., et al. (2010). Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42, 181–185. doi:10.1038/ng.518
Nacev, B. A., Feng, L., Bagert, J. D., Lemiesz, A. E., Gao, J., Soshnev, A. A., et al. (2019). The expanding landscape of 'oncohistone' mutations in human cancers. Nature 567, 473–478. doi:10.1038/s41586-019-1038-1
Narang, S., Evensen, N. A., Saliba, J., Pierro, J., Loh, M. L., Brown, P. A., et al. (2022). NSD2 E1099K drives relapse in pediatric acute lymphoblastic leukemia by disrupting 3D chromatin organization. bioRxiv.
Ngan, C. Y., Wong, C. H., Tjong, H., Wang, W., Goldfeder, R. L., Choi, C., et al. (2020). Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat. Genet. 52, 264–272. doi:10.1038/s41588-020-0581-x
Nishida, Y., Maeda, A., Kim, M. J., Cao, L., Kubota, Y., Ishizawa, J., et al. (2017). The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 7, e527. doi:10.1038/bcj.2017.8
Ntziachristos, P., Tsirigos, A., Van Vlierberghe, P., Nedjic, J., Trimarchi, T., Flaherty, M. S., et al. (2012). Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 18, 298–301. doi:10.1038/nm.2651
Oyer, J. A., Huang, X., Zheng, Y., Shim, J., Ezponda, T., Carpenter, Z., et al. (2014). Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 28, 198–201. doi:10.1038/leu.2013.204
Pachano, T., Sanchez-Gaya, V., Ealo, T., Mariner-Fauli, M., Bleckwehl, T., Asenjo, H. G., et al. (2021). Orphan CpG islands amplify poised enhancer regulatory activity and determine target gene responsiveness. Nat. Genet. 53, 1036–1049. doi:10.1038/s41588-021-00888-x
Pajtler, K. W., Wen, J., Sill, M., Lin, T., Orisme, W., Tang, B., et al. (2018). Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 136, 211–226. doi:10.1007/s00401-018-1877-0
Pan, J., Mckenzie, Z. M., D'Avino, A. R., Mashtalir, N., Lareau, C. A., St Pierre, R., et al. (2019). The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity-independent genomic targeting. Nat. Genet. 51, 618–626. doi:10.1038/s41588-019-0363-5
Pengelly, A. R., Copur, O., Jackle, H., Herzig, A., and Muller, J. (2013). A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 339, 698–699. doi:10.1126/science.1231382
Pengelly, A. R., Kalb, R., Finkl, K., and Muller, J. (2015). Transcriptional repression by PRC1 in the absence of H2A monoubiquitylation. Genes Dev. 29, 1487–1492. doi:10.1101/gad.265439.115
Piunti, A., Rossi, A., Cerutti, A., Albert, M., Jammula, S., Scelfo, A., et al. (2014). Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat. Commun. 5, 3649. doi:10.1038/ncomms4649
Piunti, A., Smith, E. R., Morgan, M. A. J., Ugarenko, M., Khaltyan, N., Helmin, K. A., et al. (2019). Catacomb: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci. Adv. 5, eaax2887. doi:10.1126/sciadv.aax2887
Plys, A. J., Davis, C. P., Kim, J., Rizki, G., Keenen, M. M., Marr, S. K., et al. (2019). Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 33, 799–813. doi:10.1101/gad.326488.119
Potjewyd, F., Turner, A. W., Beri, J., Rectenwald, J. M., Norris-Drouin, J. L., Cholensky, S. H., et al. (2020). Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem. Biol. 27, 47–56. doi:10.1016/j.chembiol.2019.11.006
Qi, W., Chan, H., Teng, L., Li, L., Chuai, S., Zhang, R., et al. (2012). Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. U. S. A. 109, 21360–21365. doi:10.1073/pnas.1210371110
Qi, W., Zhao, K., Gu, J., Huang, Y., Wang, Y., Zhang, H., et al. (2017). An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 13, 381–388. doi:10.1038/nchembio.2304
Rada-Iglesias, A., Bajpai, R., Swigut, T., Brugmann, S. A., Flynn, R. A., and Wysocka, J. (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. doi:10.1038/nature09692
Ragazzini, R., Perez-Palacios, R., Baymaz, I. H., Diop, S., Ancelin, K., Zielinski, D., et al. (2019). EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat. Commun. 10, 3858. doi:10.1038/s41467-019-11800-x
Rej, R. K., Wang, C., Lu, J., Wang, M., Petrunak, E., Zawacki, K. P., et al. (2021). Discovery of EEDi-5273 as an exceptionally potent and orally efficacious EED inhibitor capable of achieving complete and persistent tumor regression. J. Med. Chem. 64, 14540–14556. doi:10.1021/acs.jmedchem.1c01059
Rhodes, J. D. P., Feldmann, A., Hernandez-Rodriguez, B., Diaz, N., Brown, J. M., Fursova, N. A., et al. (2020). Cohesin disrupts polycomb-dependent chromosome interactions in embryonic stem cells. Cell Rep. 30, 820–835 e10. doi:10.1016/j.celrep.2019.12.057
Rong, Y., Zhu, Y. Z., Yu, J. L., Wu, Y. W., Ji, S. Y., Zhou, Y., et al. (2022). USP16-mediated histone H2A lysine-119 deubiquitination during oocyte maturation is a prerequisite for zygotic genome activation. Nucleic Acids Res. 50, 5599–5616. doi:10.1093/nar/gkac468
Rose, N. R., King, H. W., Blackledge, N. P., Fursova, N. A., Ember, K. J., Fischer, R., et al. (2016). RYBP stimulates PRC1 to shape chromatin-based communication between Polycomb repressive complexes. Elife 5, e18591. doi:10.7554/eLife.18591
Sahtoe, D. D., Van Dijk, W. J., Ekkebus, R., Ovaa, H., and Sixma, T. K. (2016). BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat. Commun. 7, 10292. doi:10.1038/ncomms10292
Sánchez-Molina, S., Figuerola-Bou, E., Blanco, E., Sánchez-Jiménez, M., Táboas, P., Gómez, S., et al. (2020). RING1B recruits EWSR1-FLI1 and cooperates in the remodeling of chromatin necessary for Ewing sarcoma tumorigenesis. Sci. Adv. 6, eaba3058. doi:10.1126/sciadv.aba3058
Sankar, A., Mohammad, F., Sundaramurthy, A. K., Wang, H., Lerdrup, M., Tatar, T., et al. (2022). Histone editing elucidates the functional roles of H3K27 methylation and acetylation in mammals. Nat. Genet. 54, 754–760. doi:10.1038/s41588-022-01091-2
Sanulli, S., Justin, N., Teissandier, A., Ancelin, K., Portoso, M., Caron, M., et al. (2015). Jarid2 methylation via the PRC2 complex regulates H3K27me3 deposition during cell differentiation. Mol. Cell 57, 769–783. doi:10.1016/j.molcel.2014.12.020
Sato, K., Kumar, A., Hamada, K., Okada, C., Oguni, A., Machiyama, A., et al. (2021). Structural basis of the regulation of the normal and oncogenic methylation of nucleosomal histone H3 Lys36 by NSD2. Nat. Commun. 12, 6605. doi:10.1038/s41467-021-26913-5
Scelfo, A., Fernandez-Perez, D., Tamburri, S., Zanotti, M., Lavarone, E., Soldi, M., et al. (2019). Functional landscape of PCGF proteins reveals both RING1A/B-Dependent-and RING1A/B-Independent-Specific activities. Mol. Cell 74, 1037–1052. doi:10.1016/j.molcel.2019.04.002
Schaefer, E. J., Wang, H. C., Karp, H. Q., Meyer, C. A., Cejas, P., Gearhart, M. D., et al. (2022). BCOR and BCORL1 mutations drive epigenetic reprogramming and oncogenic signaling by unlinking PRC1.1 from target genes. Blood Cancer Discov. 3, 116–135. doi:10.1158/2643-3230.BCD-21-0115
Scheuermann, J. C., De Ayala Alonso, A. G., Oktaba, K., Ly-Hartig, N., Mcginty, R. K., Fraterman, S., et al. (2010). Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465, 243–247. doi:10.1038/nature08966
Schoenfelder, S., Sugar, R., Dimond, A., Javierre, B. M., Armstrong, H., Mifsud, B., et al. (2015). Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 47, 1179–1186. doi:10.1038/ng.3393
Schuettengruber, B., Bourbon, H. M., Di Croce, L., and Cavalli, G. (2017). Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell 171, 34–57.
Shukla, S., Ying, W., Gray, F., Yao, Y., Simes, M. L., Zhao, Q., et al. (2021). Small-molecule inhibitors targeting Polycomb repressive complex 1 RING domain. Nat. Chem. Biol. 17, 784–793. doi:10.1038/s41589-021-00815-5
Sneeringer, C. J., Scott, M. P., Kuntz, K. W., Knutson, S. K., Pollock, R. M., Richon, V. M., et al. (2010). Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. U. S. A. 107, 20980–20985. doi:10.1073/pnas.1012525107
Souroullas, G. P., Jeck, W. R., Parker, J. S., Simon, J. M., Liu, J. Y., Paulk, J., et al. (2016). An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 22, 632–640. doi:10.1038/nm.4092
Streubel, G., Watson, A., Jammula, S. G., Scelfo, A., Fitzpatrick, D. J., Oliviero, G., et al. (2018). The H3K36me2 methyltransferase Nsd1 demarcates PRC2-mediated H3K27me2 and H3K27me3 domains in embryonic stem cells. Mol. Cell 70, 371–379. doi:10.1016/j.molcel.2018.02.027
Stuckey, J. I., Dickson, B. M., Cheng, N., Liu, Y., Norris, J. L., Cholensky, S. H., et al. (2016). A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat. Chem. Biol. 12, 180–187. doi:10.1038/nchembio.2007
Sturm, D., Witt, H., Hovestadt, V., Khuong-Quang, D. A., Jones, D. T., Konermann, C., et al. (2012). Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437. doi:10.1016/j.ccr.2012.08.024
Su, W., Han, H. H., Wang, Y., Zhang, B., Zhou, B., Cheng, Y., et al. (2019). The polycomb repressor complex 1 drives double-negative prostate cancer metastasis by coordinating stemness and immune suppression. Cancer Cell 36, 139–155 e10. doi:10.1016/j.ccell.2019.06.009
Sudarshan, D., Avvakumov, N., Lalonde, M.-E., Alerasool, N., Joly-Beauparlant, C., Jacquet, K., et al. (2022). Recurrent chromosomal translocations in sarcomas create a megacomplex that mislocalizes NuA4/TIP60 to Polycomb target loci. Genes Dev. 36, 664–983.
Suh, J. L., Bsteh, D., Hart, B., Si, Y., Weaver, T. M., Pribitzer, C., et al. (2022). Reprogramming CBX8-PRC1 function with a positive allosteric modulator. Cell Chem. Biol. 29, 555–571. doi:10.1016/j.chembiol.2021.10.003
Swaroop, A., Oyer, J. A., Will, C. M., Huang, X., Yu, W., Troche, C., et al. (2019). An activating mutation of the NSD2 histone methyltransferase drives oncogenic reprogramming in acute lymphocytic leukemia. Oncogene 38, 671–686. doi:10.1038/s41388-018-0474-y
Taherbhoy, A. M., Huang, O. W., and Cochran, A. G. (2015). BMI1-RING1B is an autoinhibited RING E3 ubiquitin ligase. Nat. Commun. 6, 7621. doi:10.1038/ncomms8621
Tamburri, S., Conway, E., and Pasini, D. (2022). Polycomb-dependent histone H2A ubiquitination links developmental disorders with cancer. Trends Genet. 38, 333–352. doi:10.1016/j.tig.2021.07.011
Tamburri, S., Lavarone, E., Fernandez-Perez, D., Conway, E., Zanotti, M., Manganaro, D., et al. (2020). Histone H2AK119 mono-ubiquitination is essential for polycomb-mediated transcriptional repression. Mol. Cell 77, 840–856 e5. doi:10.1016/j.molcel.2019.11.021
Tatavosian, R., Kent, S., Brown, K., Yao, T., Duc, H. N., Huynh, T. N., et al. (2019). Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem. 294, 1451–1463. doi:10.1074/jbc.RA118.006620
Tavares, M., Khandelwal, G., Muter, J., Viiri, K., Beltran, M., Brosens, J. J., et al. (2022). JAZF1-SUZ12 dysregulates PRC2 function and gene expression during cell differentiation. Cell Rep. 39, 110889. doi:10.1016/j.celrep.2022.110889
Testa, J. R., Cheung, M., Pei, J., Below, J. E., Tan, Y., Sementino, E., et al. (2011). Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 43, 1022–1025. doi:10.1038/ng.912
Trojanowski, J., Frank, L., Rademacher, A., Mucke, N., Grigaitis, P., and Rippe, K. (2022). Transcription activation is enhanced by multivalent interactions independent of phase separation. Mol. Cell 82, 1878–1893 e10. doi:10.1016/j.molcel.2022.04.017
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A., and Kinzler, K. W. (2013). Cancer genome landscapes. Science 339, 1546–1558. doi:10.1126/science.1235122
Wang, H., Wang, L., Erdjument-Bromage, H., Vidal, M., Tempst, P., Jones, R. S., et al. (2004a). Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878. doi:10.1038/nature02985
Wang, J., Yu, X., Gong, W., Liu, X., Park, K. S., Ma, A., et al. (2022). EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat. Cell Biol. 24, 384–399. doi:10.1038/s41556-022-00850-x
Wang, L., Birch, N. W., Zhao, Z., Nestler, C. M., Kazmer, A., Shilati, A., et al. (2021). Epigenetic targeted therapy of stabilized BAP1 in ASXL1 gain-of-function mutated leukemia. Nat. Cancer 2, 515–526. doi:10.1038/s43018-021-00199-4
Wang, L., Brown, J. L., Cao, R., Zhang, Y., Kassis, J. A., and Jones, R. S. (2004b). Hierarchical recruitment of polycomb group silencing complexes. Mol. Cell 14, 637–646. doi:10.1016/j.molcel.2004.05.009
Wang, S., Denton, K. E., Hobbs, K. F., Weaver, T., Mcfarlane, J. M. B., Connelly, K. E., et al. (2020). Optimization of ligands using focused DNA-encoded libraries to develop a selective, cell-permeable CBX8 chromodomain inhibitor. ACS Chem. Biol. 15, 112–131. doi:10.1021/acschembio.9b00654
Wassef, M., Luscan, A., Aflaki, S., Zielinski, D., Jansen, P., Baymaz, H. I., et al. (2019). EZH1/2 function mostly within canonical PRC2 and exhibit proliferation-dependent redundancy that shapes mutational signatures in cancer. Proc. Natl. Acad. Sci. U. S. A. 116, 6075–6080. doi:10.1073/pnas.1814634116
Weber, C. M., Hafner, A., Kirkland, J. G., Braun, S. M. G., Stanton, B. Z., Boettiger, A. N., et al. (2021). mSWI/SNF promotes Polycomb repression both directly and through genome-wide redistribution. Nat. Struct. Mol. Biol. 28, 501–511. doi:10.1038/s41594-021-00604-7
Weinberg, D. N., Papillon-Cavanagh, S., Chen, H., Yue, Y., Chen, X., Rajagopalan, K. N., et al. (2019). The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286. doi:10.1038/s41586-019-1534-3
Weinberg, D. N., Rosenbaum, P., Chen, X., Barrows, D., Horth, C., Marunde, M. R., et al. (2021). Two competing mechanisms of DNMT3A recruitment regulate the dynamics of de novo DNA methylation at PRC1-targeted CpG islands. Nat. Genet. 53, 794–800. doi:10.1038/s41588-021-00856-5
Willcockson, M. A., Healton, S. E., Weiss, C. N., Bartholdy, B. A., Botbol, Y., Mishra, L. N., et al. (2021). H1 histones control the epigenetic landscape by local chromatin compaction. Nature 589, 293–298. doi:10.1038/s41586-020-3032-z
Wimalasena, V. K., Wang, T., Sigua, L. H., Durbin, A. D., and Qi, J. (2020). Using chemical epigenetics to target cancer. Mol. Cell 78, 1086–1095. doi:10.1016/j.molcel.2020.04.023
Wu, G., Broniscer, A., Mceachron, T. A., Lu, C., Paugh, B. S., Becksfort, J., et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253. doi:10.1038/ng.1102
Wu, X., Johansen, J. V., and Helin, K. (2013). Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol. Cell 49, 1134–1146. doi:10.1016/j.molcel.2013.01.016
Xia, Y. K., Zeng, Y. R., Zhang, M. L., Liu, P., Liu, F., Zhang, H., et al. (2021). Tumor-derived neomorphic mutations in ASXL1 impairs the BAP1-ASXL1-FOXK1/K2 transcription network. Protein Cell 12, 557–577. doi:10.1007/s13238-020-00754-2
Xu, K., Wu, Z. J., Groner, A. C., He, H. H., Cai, C., Lis, R. T., et al. (2012). EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science 338, 1465–1469. doi:10.1126/science.1227604
Yang, W., Lee, Y. H., Jones, A. E., Woolnough, J. L., Zhou, D., Dai, Q., et al. (2014). The histone H2A deubiquitinase Usp16 regulates embryonic stem cell gene expression and lineage commitment. Nat. Commun. 5, 3818. doi:10.1038/ncomms4818
Youmans, D. T., Schmidt, J. C., and Cech, T. R. (2018). Live-cell imaging reveals the dynamics of PRC2 and recruitment to chromatin by SUZ12-associated subunits. Genes Dev. 32, 794–805. doi:10.1101/gad.311936.118
Yuan, G., Ma, B., Yuan, W., Zhang, Z., Chen, P., Ding, X., et al. (2013). Histone H2A ubiquitination inhibits the enzymatic activity of H3 lysine 36 methyltransferases. J. Biol. Chem. 288, 30832–30842. doi:10.1074/jbc.M113.475996
Yuan, W., Xu, M., Huang, C., Liu, N., Chen, S., and Zhu, B. (2011). H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 286, 7983–7989. doi:10.1074/jbc.M110.194027
Yusufova, N., Kloetgen, A., Teater, M., Osunsade, A., Camarillo, J. M., Chin, C. R., et al. (2021). Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 589, 299–305. doi:10.1038/s41586-020-3017-y
Zauderer, M. G., Szlosarek, P. W., Le Moulec, S., Popat, S., Taylor, P., Planchard, D., et al. (2022). EZH2 inhibitor tazemetostat in patients with relapsed or refractory, BAP1-inactivated malignant pleural mesothelioma: A multicentre, open-label, phase 2 study. Lancet. Oncol. 23, 758–767. doi:10.1016/S1470-2045(22)00277-7
Zhang, J., Ding, L., Holmfeldt, L., Wu, G., Heatley, S. L., Payne-Turner, D., et al. (2012). The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163.
Zhang, M., Wang, Y., Jones, S., Sausen, M., Mcmahon, K., Sharma, R., et al. (2014). Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 46, 1170–1172. doi:10.1038/ng.3116
Zhang, Q., Agius, S. C., Flanigan, S. F., Uckelmann, M., Levina, V., Owen, B. M., et al. (2021a). PALI1 facilitates DNA and nucleosome binding by PRC2 and triggers an allosteric activation of catalysis. Nat. Commun. 12, 4592. doi:10.1038/s41467-021-24866-3
Zhang, Y., Chan, H. L., Garcia-Martinez, L., Karl, D. L., Weich, N., Slingerland, J. M., et al. (2020). CRL4DCAF1/VprBP E3 ubiquitin ligase controls ribosome biogenesis, cell proliferation, and development. Sci. Adv. 6, eabd6078. doi:10.1126/sciadv.abd6078
Zhang, Y., Liu, T., Yuan, F., Garcia-Martinez, L., Lee, K. D., Stransky, S., et al. (2021b). The Polycomb protein RING1B enables estrogen-mediated gene expression by promoting enhancer-promoter interaction and R-loop formation. Nucleic Acids Res. 49, 9768–9782. doi:10.1093/nar/gkab723
Zhang, Z., Luo, L., Xing, C., Chen, Y., Xu, P., Li, M., et al. (2021c). RNF2 ablation reprograms the tumor-immune microenvironment and stimulates durable NK and CD4(+) T-cell-dependent antitumor immunity. Nat. Cancer 2, 1018–1038. doi:10.1038/s43018-021-00263-z
Zhao, J., Wang, M., Chang, L., Yu, J., Song, A., Liu, C., et al. (2020). RYBP/YAF2-PRC1 complexes and histone H1-dependent chromatin compaction mediate propagation of H2AK119ub1 during cell division. Nat. Cell Biol. 22, 439–452. doi:10.1038/s41556-020-0484-1
Zhen, C. Y., Duc, H. N., Kokotovic, M., Phiel, C. J., and Ren, X. (2014). Cbx2 stably associates with mitotic chromosomes via a PRC2- or PRC1-independent mechanism and is needed for recruiting PRC1 complex to mitotic chromosomes. Mol. Biol. Cell 25, 3726–3739. doi:10.1091/mbc.E14-06-1109
Zhou, B. R., Feng, H., Kale, S., Fox, T., Khant, H., De Val, N., et al. (2021). Distinct structures and dynamics of chromatosomes with different human linker histone isoforms. Mol. Cell 81, 166–182 e6. doi:10.1016/j.molcel.2020.10.038
Keywords: Polycomb, H2AK119ub1, H3K27me3, chromatin architecture, cancer, compaction, transcription
Citation: Doyle EJ, Morey L and Conway E (2022) Know when to fold ‘em: Polycomb complexes in oncogenic 3D genome regulation. Front. Cell Dev. Biol. 10:986319. doi: 10.3389/fcell.2022.986319
Received: 05 July 2022; Accepted: 04 August 2022;
Published: 29 August 2022.
Edited by:
Prasad Pethe, Symbiosis International University, IndiaReviewed by:
Cristina Pina, Brunel University London, United KingdomHengbin Wang, Virginia Commonwealth University, United States
Copyright © 2022 Doyle, Morey and Conway. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lluis Morey, bG1vcmV5QG1lZC5taWFtaS5lZHU=; Eric Conway, ZXJpYy5jb253YXlAdWNkLmll