 Patrick Ernst
Patrick Ernst Philip A. Bidwell
Philip A. Bidwell Michaela Dora2
Michaela Dora2 Forum Kamdar
Forum Kamdar
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 18 January 2023
Sec. Stem Cell Research
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.986107
This article is part of the Research Topic Mechanistic Understanding and Approaches to Promote Human Heart Regeneration View all 6 articles
Human induced pluripotent stem cell cardiomyocytes (hiPSC-CMs) are based on ground-breaking technology that has significantly impacted cardiovascular research. They provide a renewable source of human cardiomyocytes for a variety of applications including in vitro disease modeling and drug toxicity testing. Cardiac calcium regulation plays a critical role in the cardiomyocyte and is often dysregulated in cardiovascular disease. Due to the limited availability of human cardiac tissue, calcium handling and its regulation have most commonly been studied in the context of animal models. hiPSC-CMs can provide unique insights into human physiology and pathophysiology, although a remaining limitation is the relative immaturity of these cells compared to adult cardiomyocytes Therefore, this field is rapidly developing techniques to improve the maturity of hiPSC-CMs, further establishing their place in cardiovascular research. This review briefly covers the basics of cardiomyocyte calcium cycling and hiPSC technology, and will provide a detailed description of our current understanding of calcium in hiPSC-CMs.
Since Ringer’s initial discovery of the critical contribution of extracellular calcium (Ca2+) to cardiac contractility in 1833, extensive research using fluorescent calcium indicators has delineated the ever-expanding role of Ca2+ in muscle, and the heart in particular, beyond the second messenger controlling contraction to a regulator of critical processes such as energetics and cell survival in healthy and diseased hearts. (Ringer, 1883). e. Aberrant Ca2+ homeostasis is a nearly universal characteristic of both acquired and genetic forms of human heart failure, an observation reinforced by an extensive array of experimental models of disease. Dysregulation of Ca2+ handling not only depresses the mechanical function of the heartbut also triggers numerous signaling pathways that contributes to the progression of heart failure and cardiovascular disease.
Our understanding of cardiac Ca2+ handling has been built upon extensive experimentation on various large and small animal models, given the exceedingly limited availability of human cardiac tissue. Early efforts emphasized larger animal models (e.g., porcine and canine) that most closely mimic human physiology on a cellular and organ level (Bers, 2008; Eisner et al., 2017). In recent decades, murine models have been at the forefront of preclinical cardiovascular research, with genetic modification (knockout, overexpression, introduction of mutations) has proven invaluable in delineating the role of individual proteins and their function within complex signaling pathways and networks under physiological and pathophysiological conditions. A major caveat of this research is the significant difference in the size and mechanics of the mouse and human hearts.
The limitations of small animal models and heterologous expression systems codify the need to faithfully recapitulate the genetic, epigenetic, and molecular framework of the human system, especially when considering the validity of cardiovascular models. Recent developments in human induced pluripotent stem cell (hiPSC) technology and somatic reprogramming of hiPSCs, using the Yamanaka factors (Oct3/4, Sox2, Klf4, and c-Myc) (Takahashi and Yamanaka, 2006), offer a partial solution. The subsequent capacity to differentiate into cardiomyocytes (hiPSC-CMs) has provided a new avenue for investigation of cardiac cell physiology (Yang et al., 2008). A distinct advantage over previous models is the ability to generate patient-derived hiPSC lines that display the exact genetic architecture of an individual, enabling study of nuances of phenotype variability. Patient-derived hiPSCs have become a primary tool in precision medicine, including drug toxicity screening. While hiPSC-CMs technology overcomes the need to translate research between species to assess the human condition, hiPSC-CMs are also relatively immature, not fully reflecting adult cardiomyocytes, and have limited ability to assess organ and systemic effects of cardiac function and disease (Karakikes et al., 2015a; Kamdar et al., 2015; Karbassi et al., 2020). However, the field is continually making advances in differentiation strategies to drive enhanced maturity of these cells. Above all, further research on cardiac Ca2+ handling will require a multifaceted approach, utilizing several model systems, of which hiPSC-CMs will be a major component.
This review briefly details the major mechanisms of cardiac Ca2+ handling and provides an overview of the basics and current state of hiPSC-CMs technology, where numerous other reviews provide in-depth detail. This review focuses on the current understanding of Ca2+-handling proteins expression and function in hiPSC-CMs, and how this relates to existing knowledge from other model systems. Because this is a nascent and rapidly changing field with little standardization, much of this information is limited and scattered, so direct apples-to-apples comparisons are often difficult.
Cardiomyocyte Ca2+ cycling directly mediates cardiac muscle contraction and relaxation. Briefly, sarcolemmal depolarization triggers a Ca2+-induced Ca2+ release through opening and release of Ca2+ through the Cav1.2 subunit of the L-type Ca2+ channel (LTCC), which activates the release of Ca2+ stores from the sarcoplasmic reticulum (SR) through opening of the ryanodine receptor 2 (RyR2) (Bers, 2001; Setterberg et al., 2021). Elevated cytosolic Ca2+ then activates myofibril contraction. Ca2+ is then removed from the cytosol through several mechanisms to allow muscle relaxation. The predominant transport occurs through the cardiac sarcoplasmic reticulum calcium ATPase (SERCA2a, which hydrolyzes ATP to pump Ca2+ from the cytosol into the SR (Figure 1), which is also necessary for subsequent Ca2+ release events through RyR2 (Bers, 2001; Haghighi et al., 2014). Ca2+ can also be transported across the plasma membrane by the Na/Ca2+ exchanger (NCX) and the plasma membrane Ca2+ ATPase (PMCA) (Bers, 2001; Shattock et al., 2015), and into the mitochondria by the mitochondrial Ca2+ uniporter (MCU) (Garbincius et al., 2020; Boyman et al., 2021) (Figure 1). In human cardiomyocytes, approximately 70% of Ca2+ is removed by SERCA2a, compared to approximately 90% in rodent cells (Bers, 2001), highlighting a fundamental difference in Ca2+ cycling that must be accounted for in these extensively used rodent experimental models.
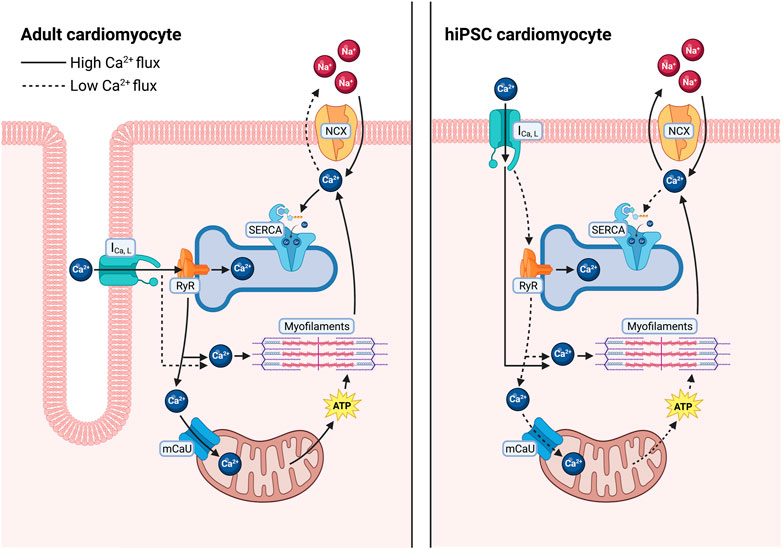
FIGURE 1. Differences in calcium regulation between adult human cardiomyocytes and in hiPSC-derived cardiomyocytes. In adult human cardiomyocytes (left), T-tubules result in close proximity between L-type Ca2+ channels and RyR2 allowing for a larger release of Ca2+ from the SR, quickly diffusing into the cytosol and going to the myofilaments to facilitate contraction as well as into closely tethered mitochondria to stimulate ATP production. Ca2+ is then primarily sequestered back into the SR via SERCA. In hiPSC-CMs (right), increased distance between L-type calcium channels and RyR2 results in less Ca2+ release from the SR, requiring the cells to rely more heavily on L-type Ca2+ influx to facilitate contraction. After relaxation a larger fraction of the cytosolic Ca2+ is removed via the Na+/Ca2+ exchanger.
Ca2+ cycling is physiologically regulated by the adrenergic system for the fight-or-flight response, through increases in Ca2+ release and uptake capabilities. SERCA is inhibited by the 52-aa transmembrane protein phospholamban (PLB), which lowers the binding affinity of Ca2+ (Haghighi et al., 2014). Physiologic activation of adrenergic G-protein-coupled receptors elicits downstream activation of protein kinases, protein kinase A (PKA), and calmodulin kinase II (CaMKII), thus phosphorylating PLB at residues Ser16 and/or Thr17 and alleviating inhibition on SERCA (Haghighi et al., 2014). The resulting increased SERCA activity shortens the duration of Ca2+ transient for faster relaxation, enabling adequate chamber filling during accelerated beating as a compensatory mechanism. Enhanced SERCA2a activity also elevates SR Ca2+ content for greater Ca2+ release, yielding faster and stronger myofibril contractions. This elevated Ca2+ release is also enhanced by RyR phosphorylation, leading to greater pore opening (Bers, 2014).
In addition to PLB, several other binding partners modulate SERCA2a activity and cardiomyocyte function. HS-1 associated protein X-1 (HAX-1) binds PLB, enhancing its inhibition of SERCA2a, and it (Zhao et al., 2009) is responsible for approximately half of the basal physiological inhibition of SERCA2a by PLB (Bidwell et al., 2018). A recently discovered integral membrane protein, dwarf open reading frame (DWORF), homologous to PLB, probably binds in a similar manner but activates SERCA activity, thereby increasing cardiomyocyte contractility (Nelson et al., 2016; Li A. et al., 2021). SERCA activity and regulation has been predominantly studied in the context of ventricular function, with Ca2+ transport less understood in the atrium, although several key differences have been observed. SERCA expression and Ca2+ pumping ability are greater in the atrium compared to the ventricle. In the atrium of the cardiomyocyte, PLB levels are lower, but an additional protein regulator, sarcolipin (SLN) is also expressed (Shaikh et al., 2016). SLN is a 31 amino acid protein, which similarly reduces the Ca2+ affinity of SERCA, but can also decrease the maximum velocity of enzymatic turnover (Bal and Periasamy, 2020).
Cardiac pathology is commonly associated with the disruption of this finely tuned Ca2+ transport apparatus. In chronic heart failure, a state characterized by pathological chronic sympathetic activation, there is reduction in SERCA2a expression and activity, as well as an increase in the inhibitory function of PLB. These alterations in the heart failure state lead to increased intracellular cardiac Ca2+ and reduced SR Ca2+ stores, and consequently reduced systolic calcium concentrations (Kranias and Hajjar, 2012; Denniss et al., 2020). Subsequently, these changes stemming from the reduction in SERCA2a activity and expression result in abnormal excitation-contraction coupling, activation of stress signaling pathways, and reduced cardiac contractility (Lipskaia et al., 2007; Kawase and Hajjar, 2008; Kranias and Hajjar, 2012; Denniss et al., 2020). RyR phosphorylation is chronically increased in disease, which is associated with arrhythmogenesis due to increased Ca2+ leak (Bers, 2014). Thus, addressing changes in calcium regulation, specifically SERCA2a activation, has been a therapeutic interest in heart failure.
In addition, mitochondria play an important role in cardiac function and cardiac Ca2+ regulation. In cardiomyocytes, the mitochondria are in close proximity to the Ca2+ store of the SR. (Frederick and Shaw, 2007). The high Ca2+ concentration microenvironment results in the influx of Ca2+ into mitochondria through the MCU. This mitochondrial Ca2 influx is crucial as the mitochondrial Ca2+ influx stimulates respiration. Furthermore, inhibiting MCU activity in cardiac mitochondria has been associated with a resulting increase in cytosolic Ca2+ transient amplitudes (Drago et al., 2012; Boyman et al., 2014). In addition to the MCU, mitochondrial calcium influx is possible through the rapid mode of Ca2+ update (RaM). Although RaM allows for more rapid calcium influx, it is inactivated at calcium concentrations well below those seen in active cardiomyocytes and can take well over a minute to recover from inactivation. As a result, the likelihood of its involvement in beat-by-beat cardiac calcium regulation is slim (Buntinas et al., 2001). Mitochondrial calcium efflux occurs primarily through the mitochondrial Na+/Ca2+-exchanger (mNCX), and although Na+-independent means of mitochondrial calcium efflux are more prominent in other tissues (e.g., kidneys and liver), their activity has been found to be relatively low in the heart (Carafoli et al., 1974; Crompton et al., 1977).
hiPSC differentiation to cardiomyocytes draws upon insights from human embryonic development. hiPSC-CM differentiation relies upon generation of cardiac mesoderm and, ultimately, cardiomyocytes, through recapitulating the stage-specific release of signaling factors. To date, three main methodological approaches have shown success in generating hiPSC-derived cardiomyocytes in the laboratory: Embryoid body formation, inductive co-culture, and adherent monolayer culture. Here, we will review monolayer culture, which is the predominant method for differentiation.
In-depth characterization of signaling components regulating cardiac development, including fibroblast growth factors (FGF), Wnt, Nodal/Activin A and bone morphogenic proteins (BMPs), served as the foundation for directed differentiation (Yang et al., 2008). It was then possible to reenact the sequence of events leading to CM specification in vitro through the fine-tuned application of growth factors and small molecules that possess the capacity to modulate cardiac signaling. To date, Activin A, BMP2/4, and Wnt regulators (IWP-2, CHIR99021) have been identified as the most essential auxiliary components for optimal differentiation of hiPSC-CMs (Burridge et al., 2011; Lian et al., 2012).
The increased utilization of hiPSC-CMs has also renewed the interest in understanding cardiac development. During heart development, cells undergo specification, morphogenesis, and lastly maturation to form an adult heart. Maturation is defined by a developmental program that leads to the conversion of cardiomyocyte gene expression, function, cell structure, and metabolism from an immature fetal to mature adult cardiomyocyte (Karbassi et al., 2020) (Figure 2). While hiPSC-CMs have revolutionized the study of cardiovascular physiology and disease modeling, the well-acknowledged limitation of hiPSC-CMs is their relative immaturity compared to adult cardiomyocytes. hiPSC-CMs most closely resemble immature fetal cardiomyocytes which are characterized by less sarcomeric organization, spontaneous beating, reduced contractility, and dependence on glycolytic metabolism (Guo and Pu, 2020). With regards to calcium handling, hiPSC-CMs have express lower levels of calcium handling genes and proteins, increased time to peak, slower calcium signal decay, less well-developed calcium handling, absence of T-tubules, and lack of ultrastructural development and relationship between the SR and mitochondria, resulting in reduced coordination of cell-wide calcium (Zhang and Morad, 2016). In contrast, mature cardiomyocytes have higher expression of SERCA2a, RyR2, and other calcium handling genes and proteins, mature calcium regulation, T-tubule formation, organized SR, and low automaticity (Hwang et al., 2015; Zhang and Morad, 2016; Guo and Pu, 2020).
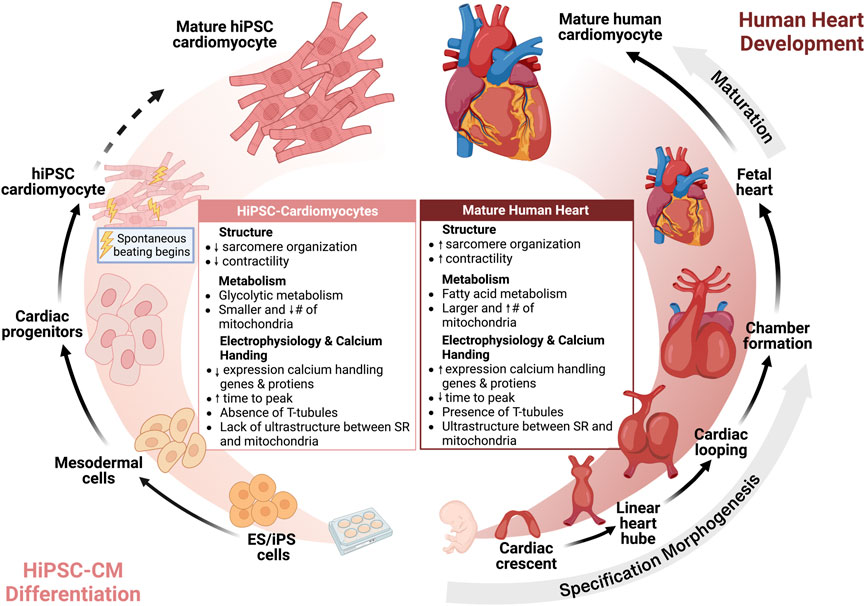
FIGURE 2. Human induced pluripotent stem cell cardiomyocytes are immature but reflect human heart development. Insights from cardiac morphogenesis has facilitated differentiation of hiPSC to hiPSC-CMs. Currently, hiPSC-CMs are immature compared to adult human cardiomyocytes, but further strategies for maturation may advance the maturity of hiPSC-CMs towards an adult phenotype.
Thus, the cardiovascular stem cell field is highly invested in harnessing our understanding of cardiovascular development to drive maturity enhanced maturity, and importantly more robust Ca2+ handling in hiPSC-CMs. To this end, Ca2+ transient parameters, assessed by real-time detection of fluorescent Ca2+ dyes and indicators, are often used as positive markers for this improved cellular physiology.
The initial approach to maturation of hiPSC-CMs was to increase the duration of time from differentiation to mimic temporal maturation of the human cardiomyocyte (Snir et al., 2003; Kamakura et al., 2013; Lundy et al., 2013). Characterization of hiPSC-CM differentiated and maintained for 1 and up to 4 months has shown an increase in maturation including sarcomeric organization, cell size and shape, and improved calcium amplitude and faster decay (Lundy et al., 2013; Pioner et al., 2019) (Figure 3). While hiPSC-CMs have been cultured for up to 1 year and have demonstrated an in sarcomeric organization, some aspects of maturation such as development of T-tubules have not be achieved with long term culture (Snir et al., 2003; Kamakura et al., 2013; Lundy et al., 2013). Some challenges of these early studies were the utilization of a single hiPSC line, variable differentiation protocols, and limited quantitative calcium imaging.
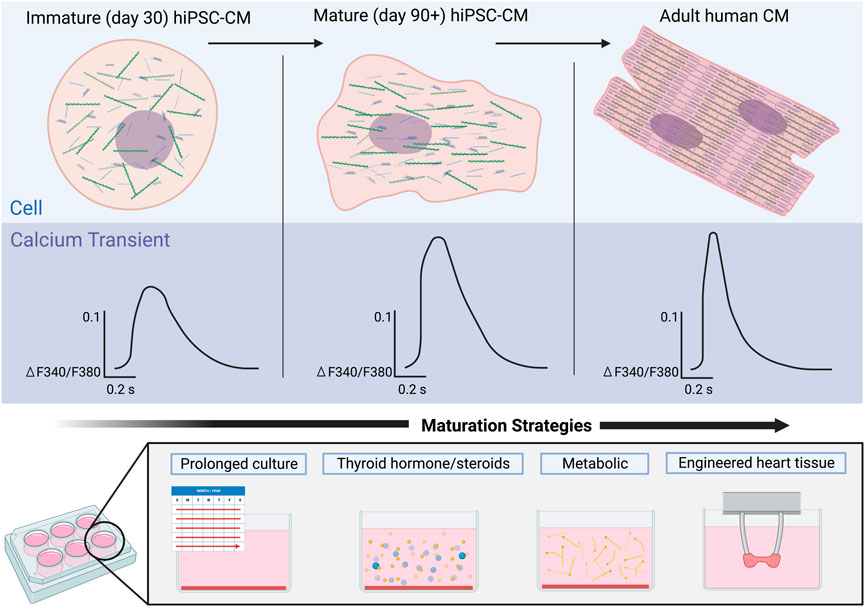
FIGURE 3. Maturation of calcium signaling in hiPSC-CMs over time in comparison to adult human cardiomyocytes. Early hiPSC-CMs (left) lack the structural organization of adult CMs (right), an immaturity reflected in their respective Ca2+ transients. Ca2+ transients of immature hiPSC-CMs exhibit a smaller amplitude as well as much slower rise times and recovery rates. Maturation of the hiPSC-CMs (middle) results in increased structural organization of the cells and Ca2+ transients with larger amplitudes and faster rise times and recovery rates. Strategies to mature Ca2+ handling in hiPSC-CMs will be critical to achieve more adult human like Ca2+ transients.
While there is maturation of hiPSC-CMs with long-term culture, the long-term culture is time-consuming and impractical for routine downstream experiments, thus to address these limitations the field has harnessed other aspects of cardiovascular development to drive the maturity of hiPSC-CMs in a shorter duration. Here we review maturation strategies that have been employed to mature Ca2+ handling including metabolic, hormonal, electrical, and engineered heart tissues (Table 1).
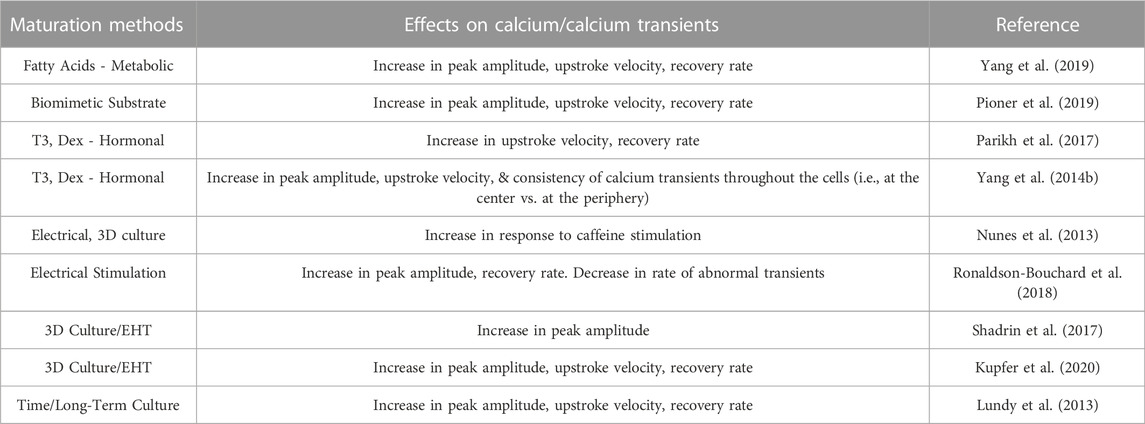
TABLE 1. Impact of maturation strategies on hiPSC calcium cycling.
A common strategy to drive maturity is through metabolic regulation, with supplementation of culture media or manipulation of metabolic signaling pathways. As adult cardiomyocytes rely predominantly on long-chain fatty acids to meet metabolic demand, their addition to culture media was theorized to yield a more mature energetic environment and enable improved Ca2+ handling. Indeed, the addition of palmitate, oleate and lineolate increases Ca2+ transient amplitudes and kinetics, which is consistent with the observation that inhibition of HIF-1α, and subsequent increase in fatty acid oxidation, enhance Ca2+ transient parameters and rates of cellular contraction and relaxation (Gentillon et al., 2019). However, the use of high glucose media also increases Ca2+ transient amplitudes and shortened decays (Balistreri et al., 2017), which may suggest that standard hiPSC-CM culture media may need to be adapted to provide the appropriate energetic fuel to maximize calcium handling maturity.
Thyroid and glucocorticoid hormones are essential for normal in vivo cardiac development (Kruger et al., 2008; Lee et al., 2010; Rog-Zielinska et al., 2014; Rog-Zielinska et al., 2015). Tri-iodo-L-thyronine (T3) is the active form of thyroid hormone and is a strong transcriptional regulator of cardiac specific genes, including several involved in Ca2+ handling (SERCA2a, PLB, and NCX) (Chattergoon et al., 2012; Rog-Zielinska et al., 2013; Li et al., 2014), and supplementation has been found to enhance cardiomyocyte maturity (Yang et al., 2014b; Birket et al., 2015; Rog-Zielinska et al., 2015). Specifically, Yang et al. (2014b) showed that T3 treatment for 1 week, 3 weeks post-differentiation, resulted in greater contractile force, along with faster contraction and relaxation rates (Yang et al., 2014b). While T3-treated cells had comparable Ca2+ transient amplitudes to control cells, they showed much faster upstroke velocity and decay rates, with transients much more closely resembling those of adult cardiomyocytes. T3-treated cells also showed large increases in a-MHC and SERCA2a expression, and a significant increase in cell area and sarcomere length, as well as a decrease in cell circularity. T3 treatment also resulted in improvements in mitochondrial function, with oxygen consumption rates consistently higher than control cells in all instances.
In addition to thyroid hormones, glucocorticoids similarly regulate critical genetic programming necessary for development in vivo (Rog-Zielinska et al., 2013; Rog-Zielinska et al., 2014; Rog-Zielinska et al., 2015). In an experimental setting, treatment with T3 together with the synthetic glucocorticoid dexamethasone (Dex) further improves the functional maturity of hiPSC-CMs beyond T3 treatment alone (Yang et al., 2014b) (Parikh et al., 2017). Treatment with T3+Dex treatment resulted in a significant increase in T-tubule density in hiPSC-CMs, and improved intracellular Ca2+ handling, with Ca2+ transients having larger amplitudes and faster rise times. The Ca2+ propagation of treated cells was also much more consistent across cells, with similar Ca2+ transients in the center of the cell compared to in the periphery, a characteristic that is distinctly lacking in control cells. This improved coordination is consistent with improved ultrastructural development of cell-wide SR and mitochondrial networks. Ca2+ imaging, before and after blocking the release of SR Ca2+ with thapsigargin and ryanodine, showed that the release of SR Ca2+ contributes to a much larger amount of the cytosolic Ca2+ transient in the hiPSC-CMs treated with T3+Dex, again indicating a more developed SR structure. However, the T-tubules were not comparable in abundance or organization to adult cardiomyocytes.
The heart is a dynamic tissue that undergoes continuous electrical depolarization and mechanical strain on a beat-to-beat basis, which has a tremendous impact on cellular structure and gene regulation during development and progression to an adult phenotype. These factors are then likely essential to fully drive hiPSC-CM maturity (Yang et al., 2014a). While standard differentiation protocols do produce cell-to-cell contacts (monolayers and EBs) that facilitate electrical conduction, they are significantly slower compared to the adult heart. hiPSC-CMs are also effectively unloaded, experiencing minimal cell stretch and physiological strain-induced genetic regulation (Lasher et al., 2012; Ruan et al., 2015; Veerman et al., 2015). Thus, several research groups have explored the application of mechanical stress and electrical stimulation, mimicking a more in vivo cellular environment of an adult cardiomyocyte. Such conditions do, in fact, improve Ca2+ transient kinetics (Kroll et al., 2017), and in some cases directly related to an increase in SERCA2a and RyR2 expression (Ruan et al., 2016). Spontaneous beating, a hallmark of immaturity, is thought to be related to the low expression of potassium inward rectifier channels, which stabilize the membrane potential (Barbuti et al., 2016). Therefore, increasing the expression of the potassium inward rectifier channel in hiPSC-CMs resulted in more mature hiPSC-CMs that had a loss of spontaneous beating, stable resting membrane potential and increased Ca2+ transient amplitudes (Vaidyanathan et al., 2016).
Biophysical stimulation through electrical pacing has been used to enhance the maturation of hESC-CMs and hiPSC-CM (Chan et al., 2013; Nunes et al., 2013; Kroll et al., 2017). Cells that received electrical stimulation developed a higher percentage of rod-like cells, sarcomeric alignment, more mature electrophysiology parameters, and enhanced Ca2+ transients (Chan et al., 2013; Nunes et al., 2013; Kroll et al., 2017).
In another study, to assess the effects of electrical stimulation on hiPSC-CM maturity (Ronaldson-Bouchard et al., 2018), cells were divided into three treatment groups: control (no stimulation), constant (2 Hz for 3 weeks) and intensity training (frequency increasing from 2 Hz to 6 Hz by 0.33 Hz per day, followed by a final week at 2 Hz). After constant stimulation, cells showed greater contractile force across a wide range of extracellular Ca2+ concentrations compared to controls, which increased further in the intensity training group. This difference in contractile force was much more pronounced in cells that underwent electrical stimulation immediately post-differentiation than in CMs that were cultured for 4 weeks prior to electrical stimulation. Ca2+ transients of cells in the intensity training group were much more consistent than in control cells, with larger peak amplitudes and shorter durations. Both the constant and intensity training stimulation resulted in hiPSC-CMs exhibiting a positive force-frequency relationship (FFR), while control cells continued to display a negative FFR (Ronaldson-Bouchard et al., 2018).
Engineered heart tissues and cardiac organoids contain hiPSC-CM and scaffold or extracellular matrix, and possibly other cells in the heart that better recapitulate aspects of the human heart (Weinberger et al., 2017). Zhang D. et al.( 2013) demonstrated that SERCA2A and CASQ2 genes increased significantly in 3D fibrin-based cardiac patch cultures of human embryonic stem cells compared to 2D human embryonic stem cell derived cardiomyocytes (Zhang D. et al., 2013). Cardiomyocytes are thought to mature in the presence of multiple cell types in cardiac organoids. Therefore, Richards et al. developed scaffold-free hiPSC organoids and included human ventricular cardiac fibroblasts and human umbilical vein endothelial cells and demonstrated that these organoids have functional calcium handling in response to verapamil, a calcium channel blocker, ryanodine, and isoproterenol (Richards et al., 2017). Similarly, cardiac patches that were generated by seeding a mixture of cells (86% hiPSC-CMs, the remaining 14% made up of smooth muscle cells and fibroblasts) in hydrogel squares (Shadrin et al., 2017). Following 3 weeks in culture, the cardiac patches consisted of densely packed multilayered hiPSC-CMs that exhibited organized striations and gap junctions surrounded by smooth muscle cells and fibroblasts. During those 3 weeks, hiPSC-CMs consistently showed increased levels of expression of genes indicating structural maturity (TNNI3, MYL2, MYOM2, MYOM3) and metabolic maturity (COX6A2, CKMT2, CKM), as well as E-C coupling (CASQ2, S100A1, PLN). Ca2+ imaging showed an increase in Ca2+ transient amplitudes as the 3D culture time also increased. Upon implantation in the rat epicardia, these cardiac patches displayed a conduction velocity comparable to that of the rat heart tissue pre-implantation and did not interrupt electrical propagation across the heart (Shadrin et al., 2017).
Kupfer et al.(2020) utilized bioprinting with a specialized bio-ink and undifferentiated hiPSC with in situ cardiac differentiation to generate a chambered human muscle pump. They demonstrated that calcium transients were similar at 2 weeks and 6 weeks and had an expected response to isoproterenol (Kupfer et al., 2020). Further advances in cardiac tissue engineering and comparison to 2D hiPSC-CM will be important for head to head comparisons of cardiac calcium handling maturation.
The wide variations in differentiation and maturation protocols yield a hiPSC-CM quality that differs significantly between studies. However, cells generally have sufficient Ca2+ cycling machinery to study Ca2+ transient defects in numerous inherited conditions (Kamdar et al., 2015) such as Brugada syndrome (Liang et al., 2016), catecholaminergic polymorphic ventricular tachycardia (Fatima et al., 2011), Friedreich ataxia (Crombie et al., 2017), and Duchenne muscular dystrophy (DMD) cardiomyopathy (Kamdar et al., 2020), in addition to several others detailed later in this review (Table 2). CRISPR/Cas9 has been used to correct DMD-causing dystrophin mutations in hiPSC lines, restore Ca2+ transient parameters, and reduced arrhythmic events (Kyrychenko et al., 2017). Limb-girdle muscular dystrophy arrhythmias can be recapitulated in patient-derived hiPSC-CM (30–60 days) associated with reduced Cav1.2 expression and conductance (El-Battrawy et al., 2018). Pathological phenotypes with no known molecular mechanisms are also excellent candidates for study by hiPSC-CM, including stress cardiomyopathy, which were observed to have enhanced β-adrenergic responsiveness and toxicity. Furthermore, hiPSC-CMs, despite their immature phenotype, have been used to recapitulate ischemia/reperfusion injury (Wei et al., 2017; Hakli et al., 2021). Although hiPSC-CMs are a revolutionary tool for studying cardiovascular diseases, the goal remains to mature hiPSC-CM towards an adult phenotype to enhance our knowledge of human cardiac physiology and pathophysiology.
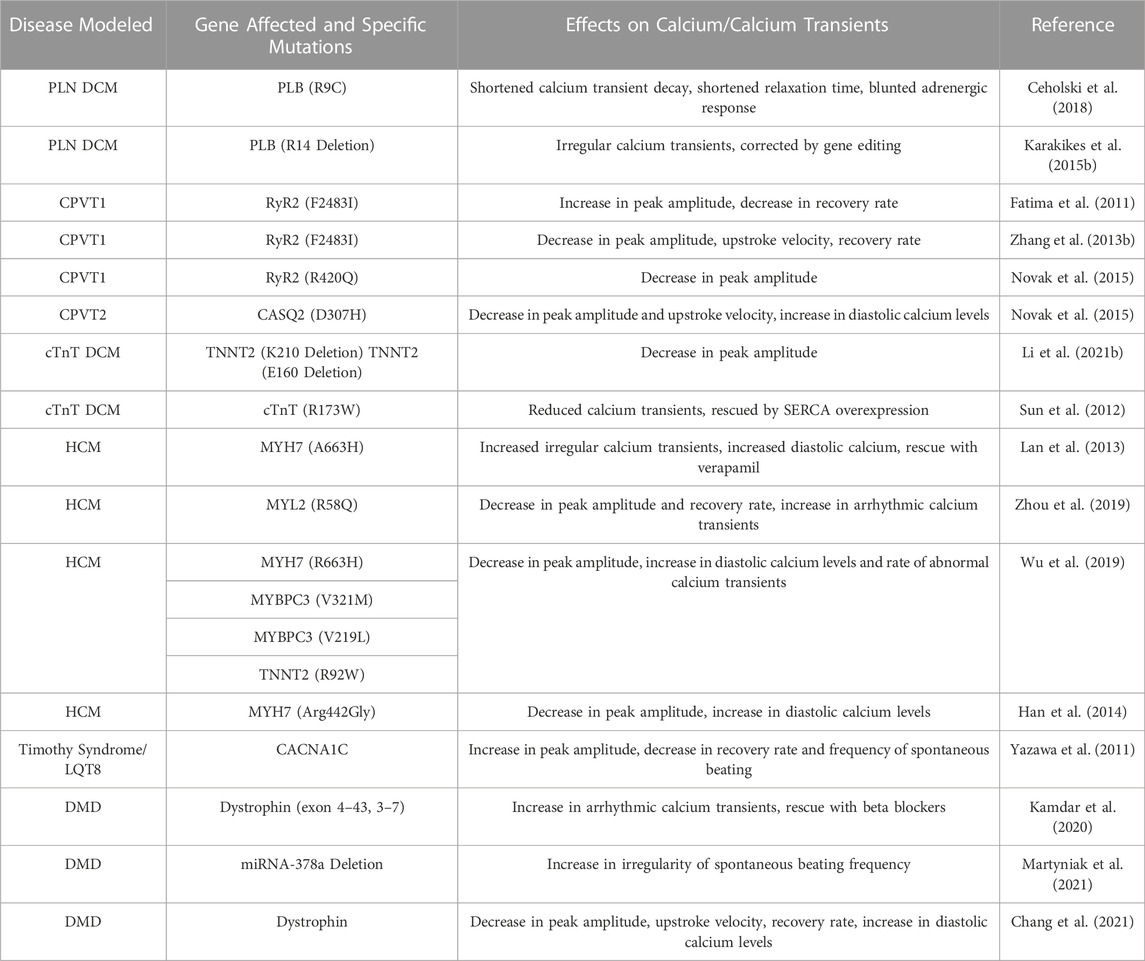
TABLE 2. Summary of hiPSC calcium changes in cardiovascular disease modeling.
To assess the utility of hiPSC-CM for cardiovascular disease modeling, it is critical to assess and understand the differences in calcium regulation between hiPSC-CMs and the adult human heart. Overall, hiPSC-CMs express the major components involved in Ca2+ handling including SERCA2a, PLB, RYR2, and calsequestrin, though hiPSC-CM Ca2+ transients are less mature than adult cardiomyocytes (Germanguz et al., 2011; Itzhaki et al., 2011; Lee et al., 2011; Rao et al., 2013). In this section, we review the differences in calcium regulation between hiPSC-CM and adult cardiomyocytes by calcium transport proteins and relevant disease models.
SERCA2a and PLB are expressed in hiPSC-CMs, though at lower levels than in hESC-CMs and adult cardiomyocytes (Itzhaki et al., 2011; Lee et al., 2011). In contrast to adult cardiomyocytes, hiPSC-CMs are dependent on both sarcolemmal Ca2+ transport and intracellular Ca2+ store release, which is comparable to fetal and post-natal cardiomyocyte and attributed to a less developed SR (Lee et al., 2011). While SERCA2a is expressed in hiPSC-CM, it was critical to assess whether SERCA2a was functional in hiPSC-CMs. Thapsigargin, a SERCA2a inhibitor, was applied to hiPSC-CMs to determine the contribution of SERCA2a to the Ca2 transient. 1–10 µM thapsigargin decreased the Ca2 transient amplitude in a dose-dependent manner, and 20 µM thapsigargin ablated the Ca2 transient (Itzhaki et al., 2011). Moreover, to determine if thapsigargin inhibition resulted from a decrease in the SR Ca2 load due to SERCA2a uptake inhibition, caffeine was applied to hiPSC-CM in the presence of thapsigargin. In hiPSC-CMs treated with thapsigargin, there was a minimal effect of caffeine, which demonstrates the inability of the SR to reload due to thapsigargin inhibiting SERCA (Itzhaki et al., 2011).
Furthermore, increased Ca2+ transient amplitude, shortened decay, and increased SR Ca2+ load (all attributable to increased SERCA activity) have been used as markers to assess improved differentiation maturity (Lee et al., 2011; Ng et al., 2011; Streckfuss-Bomeke et al., 2013) even when SERCA expression is not directly assessed. A two-fold or more change can be observed in these parameters, indicating a wide range in developmental variation of SERCA activity.
Beyond modification of pharmacological protocols and increased differentiation time, driving structural development through mechanical approaches greatly improves SR Ca2+ handling. Culturing cells on a micropatterned surface to generate cellular alignment increases Ca2+ transient amplitude and decay indicative of increased SERCA activity (Khan et al., 2015). Similar Ca2+ transient effects are observed in engineered tissue (Gao et al., 2017), suggesting that improved cellular organization is critical for the development of Ca2+ transport, likely through improved SR membrane structure and content.
PLB is a critical downstream target of adrenergic stimulation, where PKA and CaMKII can phosphorylate PLB, thereby tapping an activity reserve capacity of SERCA. Consistent with this, low PLB expression in immature cells contributes to a blunted ionotropic response to β-agonist (Chen et al., 2015), and in some early studies, a complete lack of PLB is observed (Dolnikov et al., 2006). In numerous other studies where PLB is not directly quantified, a faster Ca2+ transient decay is observed in response to isoproterenol, probably associated with increased phosphorylation of PLB (Cao et al., 2012; Lian et al., 2012; Correia et al., 2014; Hayakawa et al., 2014; Song et al., 2015). However, frequently in these studies, relative maturity is difficult to discern, as days post-differentiation are not indicated. PLB expression has been observed to increase over time in cultured cells (Lian et al., 2012; Shadrin et al., 2017; Cai et al., 2019) and through incorporation into engineered tissue (Cai et al., 2019; Murphy et al., 2019), likely contributing to enhanced adrenergic responsiveness.
Although SERCA2a and PLB are not major loci for disease causing mutations, a few mutations have been identified with PLB, with the disease phenotype extending to mutant hiPSC cell lines. In engineered hiPSC cardiac tissue, the PLB R9C mutation causes a shortened Ca2+ transient decay in cells and shortened relaxation time along with a blunted adrenergic response (Ceholski et al., 2018; Stroik et al., 2020). Patient-derived hiPSC-CMs from a carrier of PLN R14 deletion mutation exhibit irregular Ca2+ transients associated with higher beating rate and occurrences of Ca2+ waves (Karakikes et al., 2015b).
A key molecular dysfunction identified in human dilated cardiomyopathy and arrhythmogenesis involves the transport of calcium needed for the relaxation of cardiomyocytes in diastole, which is usually associated with the impaired activity of SERCA2a (Hasenfuss et al., 1994; Schwinger et al., 1995; Zarain-Herzberg et al., 1996; Davia et al., 1997; Tweedie et al., 2000; Haghighi et al., 2006). Structure-function studies of human SERCA2a overexpressed in human embryonic kidney cells indicate that large-scale movements of SERCA2a′s domains couple ATP hydrolysis to Ca transport (Moller et al., 2010). In cardiomyopathy, deficient Ca2+ removal from the cytoplasm is associated with decreased SERCA2a activity and an increased ratio of PLB to SERCA2a (MacLennan and Kranias, 2003). SERCA2a gene therapy in preclinical models and initial cardiomyopathy was successful in improving heart failure and arrhythmias, definitely showing that SERCA2a activation is a promising approach to treating heart failure. However, larger clinical trials of SERCA2a gene therapy failed, primarily due to the difficulty of delivering the large SERCA2a gene to patients on a large scale (Zsebo et al., 2014; Greenberg et al., 2016).
Fluorescent spectroscopic probes have been developed to understand the structural dynamics and function of SERCA2a and PLB (Schaaf et al., 2017a; Schaaf et al., 2017b; Lo et al., 2017; Schaaf et al., 2018). Fluorescent SERCA2a biosensors have been used in HEK293 cells to create a living cell biosensor. Time-resolved fluorescence resonance energy transfer (FRET) allows for detection of structural changes and activity in SERCA2a in living cells (Robia et al., 2007; Muretta et al., 2010; Gruber et al., 2012; Hou et al., 2012; Cornea et al., 2013; Pallikkuth et al., 2013; Gruber et al., 2014b; Schaaf et al., 2017a; Schaaf et al., 2018). SERCA2a activity is determined by the conformational state of SERCA2a, which can be detected by measuring the intramolecular distance between the acceptor and donor fluorescent proteins domains located on the cytoplasmic aspect (Cornea et al., 2013; Gruber et al., 2014a; Schaaf et al., 2017a; Schaaf et al., 2018). Prospective pharmacologic agents can be added to determine the impact of these agents on SERCA2a activity. These FRET biosensors have been limited to non-cardiac HEK293 cells, which has limited the utility of therapeutic discovery and understanding disease states, as the key structural and functional cardiac proteins needed for cardiac excitation and coupling are not expressed. The application of this technology in hiPSC-CM is currently being implemented and can allow functional and physiological measurements of calcium handling in disease models, high-throughput drug screening to identify allosteric SERCA2a activators, and drug toxicity assessment.
RyR2 has been observed to increase over time and thus, has been used as a marker of hiPSC-CM quality for cells (Cao et al., 2012) and engineered tissue (Gao et al., 2017), indicating maturation towards adult like Ca2+ handling. Caffeine releases the SR calcium stores that are gated by RyR, and in hiPSC-CMs caffeine-induced Ca2+ store release was similar to those seen in adult small animal cardiomyocytes (Hwang et al., 2015). Application of ryanodine, a RyR2 inhibitor, reduced the amplitude, maximal upstroke, and decay velocity of hiPSC-CM Ca2+ transientsCa2+ release in hiPSC-CMs (Lee et al., 2011; Ng et al., 2011; Zhang and Morad, 2016). These data suggest that hiPSC-CMs have functional SR Ca2+ stores, but may have immature mechanisms of release.
RyR2 is a well established locus for arrhythmogenic mutations, through perturbation of Ca2+ gating and aberrant Ca2+ release. Gain-of-function pathogenic RyR2 mutantations appear particularly well suited to being modeled in hiPSC-CM, commonly recapitulating basic patient phenotypes. Catecholaminergic polymorphic ventricular tachycardia type 1 (CPVT1) is known to arise from mutations to RyR2. CPVT1 is characterized by abnormal intracellular Ca2+ handling and in conjunction with increased catecholamine levels result in DADs and ventricular arrhythmias. It is unknown whether the arrhythmic activity in CPVT1 is due to SR Ca2+ leak following RyR2 phosphorylation or increased Ca2+ sensitivity leading to diastolic Ca2+ release. Numerous groups have utilized hiPSC-CM to study specific RyR2 mutations causing CPVT1, from cells directly collected from patients, or through gene editing to introduce known CPVT1 mutations.
hiPSC-CMs derived from patients with the F2483I variant in RyR2 exhibited diminished Ca2+ release upon exposure to caffeine, indicating smaller stores of Ca2+, as well as a similarly diminished INCX (Fatima et al., 2011; Zhang X. H. et al., 2013). However, the cells seemed to have compensated for their depleted Ca2+ stores by increasing the fractional release of Ca2+ resulting from calcium induced calcium release during spontaneous beating. This was accompanied by a significantly longer duration of Ca2+ transients, consistent with abnormalities in SR Ca2+ release in cells with RyR2 mutations that underlie CPVT1.The F24831 variant hiPSC-CMs also showed longer and farther-reaching Ca2+ sparks capable of activating nearby release sites with Ca2+ overload. F2483I variant gene edited hiPSC-CMs also exhibited longer and wandering Ca2+ sparks, elevated diastolic Ca2+ leak, and smaller SR Ca2+ stores, similar to that of F2483I patient-derived cells (Wei et al., 2018).
To understand how CPVT1 mutations in different domains of RYR2 inpact Ca2+ handling, hiPSC were gene edited to have RYR2 mutations in the N-terminal (R420Q), C-terminal (Q4201R), and central-domain (F42831). hiPSC-CMs from all three RYR2 variant lines had wandering sparks with abberrant focal Ca2+ release. The magnitude of Ca2+ leak was larger and SR content was larger in the F2483I and Q4201R variants than the R420Q mutant hiPSC-CMs. The increase in spark frequency and large leaks in the F2783I could be due to its position near the FK506-binding protein, that results in dissociation of the FK506-binding protein from RyR2 to result in larger leaks. The location of the mutation impacts the calcium induced calcium release gain, SR Ca2+ content, leakiness, and resposne to arrhythmic medications (Zhang et al., 2021). The S406L variant resulted in hiPSC-CMs that exhibited similar Ca2+ activity to control cells under resting conditions. However, isoproterenol stimulation of these mutant hiPSC-CMs caused elevated diastolic Ca2+ levels and Ca2+ spark frequency, resulting in an increase in DADs (Jung et al., 2012).
Investigation of other hiPSC-CMs with a variety of pathogenic RYR2 mutations have alson demonstrated an increase in DADs, arrhythmias, SR Ca2+ leak, and increased RyR2 phosporylation (Itzhaki et al., 2012; Di Pasquale et al., 2013; Paavola et al., 2016; Preininger et al., 2016; Acimovic et al., 2018; Mehta et al., 2018; Wei et al., 2018; Polonen et al., 2020; Word et al., 2021). Thus, evaluating different RYR2 mutations can lead to mechanistic molecular understanding of disease progression, and provide a platform for precision medicine.
Sarcolemmal Ca2+ transport represents an important aspect of Ca2+ regulation and excitation-coupling, including for hiPSC-CMs. Application of nifedipine, a L-type calcium channel blocker (LTCC) o hiPSC-CMs led to the elimination of the whole cell Ca2+ transient, which suggests that Ca2+ entry via the LTCC is important for generation of the whole cell transient in hiPSC-CMs (Itzhaki et al., 2011)
Long QT Syndrome 8 (LQT8), or Timothy Syndrome, is caused by a missense mutation in CACNA1C, which encodes the Cav1.2 component of the L-type Ca2+ channe and can lead to fatal-ventricular arrhythmias. Insights into the pathophysiology has been limtied due to the LQTS mouse having different electrical properties, however LQTS8 has been modeled using hiPSC-CMs (Huikuri et al., 2001).
Patient-derived LQT8 hiPSC-CMs spontaneously beat at half the frequency of wild-type hiPSC-CMs. They also exhibited delays in L-type Ca2+ current inactivation and alterations to intracellular Ca2+ handling as evidenced by Ca2+ transients with larger and more inconsistent amplitudes as well as longer durations. LQTS hiPSC-CM, also showed a prolonged action potential and DADs. The LQTS8 electrical and Ca2+ handling abnormalities could be ameliorated by roscovitine, a small molecule, that increases voltage-dependent inactivation of CaV1.2. (Yazawa et al., 2011).
LTCC current response to Mg increases over time, reflecting maturity similar to adult cardiomyocytes (Nguemo et al., 2014). As would be expected with adult cells, LTCC current inhibtion is arrythmogenic in hiPSC-CM (Wu et al., 2017). Pharmacological induction of ER stress also down-regulates Cav1.2, contributing to arrhythmogenesis via the unfolded protein response (Liu et al., 2018).
As outlined in prior sections, Ca2+ cycling has a critical role in regulation of excitation-contraction coupling in the cardiomyocyte (Bers, 2008; Eisner et al., 2017), and a major component of calcium regulation and homeostasis is Ca2+ buffering both in the SR and cytoplasm (Smith and Eisner, 2019). In this section, the role of the SR Ca2+ buffer calsequestrin (Csq2) and cytoplasmic Ca2+ buffer, namely cardiac troponin C, will be discussed including the impact of dysregulated Ca2+ buffering and insights from hiPSC-CM disease modeling.
CASQ2 encodes cardiac calsequestrin (Csq2), a high-capacity, low-affinity Ca2+ binding protein, that is the major cardiac SR Ca2+ buffer (Ikemoto et al., 1972; Yano and Zarain-Herzberg, 1994). The ability of Csq2 to buffer SR Ca2+ facilitates the repeated cardiomyocyte contraction necessary to support cardiac output. Thus, alterations in SR Ca2+ buffering via Csq2 have a significant impact on intracellular Ca2+, as CASQ2 mutations result in catacholaminergic polymorphic ventricular tachycardia 2 (CPVT2) (Ng et al., 2020). In both CPVT1 and CPVT2 there is abnormal Ca2+ from the SR, but in contrast in CPVT2 the mechanism is secondary to a loss of function mutation in CASQ2. CPVT2 clinically results in DADs and catecholamine triggered ventricular tachycardia, however the current guideline directed therapy, β-blockers, can fail to prevent fatal arrhythmias, thus there is a need to identify novel therapies (Priori et al., 2013).
Casq2 expression has been identified in hiPSC-CM and increase with time in cultured cells and through incorporation into engineered tissue, likely contributing to improved Ca2+ transient kinetics (Itzhaki et al., 2011; Gao et al., 2017; Cai et al., 2019). Patient-derived CPVT2 hiPSC-CMs with a CASQ2 D307H mutation, exhibited a decrease in the Ca2+ transient amplitude, an increase in diastolic Ca2+ levels, although they also showed a faster rise in Ca2+ transient. In control cells, caffeine exposure resulted in a sharp increase in diastolic Ca2+ levels, which returned to a steadystate within approximately 20 s. However, in the CPVT2-hiPSC-CMs, diastolic Ca2+ levels showed a much larger increase and remained elevated for much longer, only returning to a steady-state after 50–60 s (Novak et al., 2015). Similar results are also observed with other CPVT2 CSQ2 mutations: the CASQ2-G112+5X mutation exhibits DADs in patient-derived hiPSC and can be corrected by non-mutant Csq2 overexpression (Lodola et al., 2016) and the D307H mutation in CSQ in patient-derived hiPSC causes EADs (Maizels et al., 2017).
Sarcomeric proteins, including cardiac troponin C and β-myosin heavy chain, play a critical role in cardiomyocyte contractility. Contraction is dependent on the interaction of the thin actin and the thick myosin filament, which is initiated by electrical activation of the cardiomyocyte and the resultant increase in intracellular Ca2+ (Huxley, 1957). During activation the majority of intracellular Ca2+ is bound to calcium buffers, including SERCA2a and troponin C (Smith and Eisner, 2019). Increasing buffering results in changes in a decreased Ca2+transient amplitude and the rate constant of Ca2+ decay (Diaz et al., 2001). Thus, while sarcomeric proteins do not participate directly in Ca2+ transport, they play a crucial role in cardiomyocyte Ca2+ homeostasis. β-adrenergic stimulation also results in sarcomeric protein phosphorylation via protein kinase and in combination with SERCA2a activation results in increased intracellular Ca2+ to promote inotropic, chronotropic, and lusitropic responses in the myocardium (Bers, 2002). The critical nature of sarcomeric proteins is revealed when patients have mutations in sarcomeric proteins that can lead to genetic dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy. Although patients with DCM and HCM have different phenotypes, a commonality of the phenotypes includes contractile defects, ventricular arrhythmias, and sudden cardiac death, all of which have aberrant Ca2+ cycling involvement (Kamisago et al., 2000).
DCM results in reduced contractile function leading to left ventricular dilatation, ventricular arrhythmias, and systolic heart failure. Genetic DCM accounts for over a third of non-ischemic cardiomyopathies (Hershberger et al., 2013). Genetic DCM is most commonly due to gene encoding sarcomeric proteins, including titin (TTN), myosin heavy chain (MYH7), and cardiac troponin T mutations (CNNT). (McNally and Mestroni, 2017).
Several groups have modeled genetic and acquired DCM using hiPSC-CM to uncover molecular insights into the pathophysiology (Siu et al., 2012; Sun et al., 2012; Hinson et al., 2015; Wu et al., 2015; Yang et al., 2018; McDermott-Roe et al., 2019; Li B. et al., 2021). Consistent with the clinical manifestations, hiPSC-CMs with DCM mutations have been identified to have abnormal myocardial structure and dysregulated Ca2+ signaling. Cardiac troponin T, encoded by TNNT2, is a common DCM mutation that has altered Ca2+ handling and response to adrenergic signaling (McNally and Mestroni, 2017). Patient-derived hiPSC-CMs with a TNNT2 R173W mutation were used to recapitulate the DCM phenotype and decreased contractility, reduced Ca2+ transient parameters consistent with reduced SR Ca2+ load, and abnormal α-actinin distribution, which could be corrected with the β-blocker metoprolol (Sun et al., 2012). Subsequently, the role of β-adrenergic signaling was further evaluated in the TNNT2 R173W hiPSC-CMs, and a blunted β-adrenergic pathway was identified in these DCM hiPSC-CMs. Furthermore, they discovered that this mutation could promote nuclear localization of TNNT2 and modify the epigenetic regulation of key β-adrenergic signaling genes (Wu et al., 2015). Cardiac troponin T mutations can result in both DCM and hypertrophic cardiomyopathy (HCM). Li et al.(2014) sought to compare two TNNT2 mutations, K210, which causes DCM, and E160, which results in HCM through the generation of TALEN-mediated hESC lines with both heterozygous or homozygous mutations. Distinct phenotypes were identified between mutations. hiPSC-CMs with the heterozygous TNNT2 K210 mutation exhibited many physical hallmarks of DCM such as fewer myofibrils, disrupted sarcomere organization, and punctate cardiac troponin T distribution. Furthermore, these hiPSC-CMshad abnormalCa2+ transients, with a more irregular frequency and shorter durations. In the homozygous TNNT2 the structural flaws were much more pronounced in cells with the homozygousK210 mutation, and their Ca2+ transients had much smaller amplitudes. In contrast, hiPSC-CMs with the heterozygous E160 mutation had denser myofibrils, thicker Z-lines, and increased contractility consistent with HCM. Although all mutant hiPSC-CMs demonstrated irregular beating corresponding to arrhythmias, the TNNT2 E160 hiPSC-CMs had a lower frequency and amplitude with slower rise times and longer recovery (Li B. et al., 2021).
In contrast to DCM, HCM is characterized by severe hypertrophy of the left ventricle leading to left ventricular outflow obstruction, heart failure, ventricular arrhythmias, and sudden cardiac death (Maron et al., 2022a; Maron et al., 2022b; Maron et al., 2022c). hiPSC-CMs have also been utilized to model a variety of HCM mutations. Patient-derived hiPSC-CM carrying a MYH7 R663H mutation demonstrated cell enlargement, arrhythmias exacerbated by isoproterenol, and increased intracellular Ca2+ (Lan et al., 2013). Since calcium handling abnormalities appeared to precede the hypertrophic phenotype, it was hypothesized that calcium handling may be central to the phenotype. To test this hypothesis, HCM hiPSC-derived CMs were treated with calcium channel blockers verapamil, nifedipine, and diltiazem; and the results demonstrated prevention of myocyte hypertrophy, arrhythmias, and calcium handling abnormalities. This study demonstrated that aberrant calcium handling is a central mechanism in HCM (Lan et al., 2013).
In another study, hiPSC-CMs were generated from four HCM patient-derived hiPSC lines that including a variety of sarcomeric mutations including:MYH7 R663H, MYBPC3 V321M, MYBPC3 V219L, and TNNT2 R92W (Wu et al., 2019). All HCM hiPSC-CMs recapitulated diastolic dysfunction and arrhythmic phenotype, including diastolic Ca2+ overload, increased myofilament Ca2+ sensitivity, andshorter Ca2+ transient peaks while being paced. The HCM phenotype could further be exacerbated through β-adrenergic stimulation and partially rescued with the calcium channel blocker diltiazem, and nearly fully rescued with verapamil and the sodium channel inhibitor ranolazine (Wu et al., 2019).
Others have also modeled a variety of HCM mutations using hiPSC-CMs and have also recapitulated the HCM phenotype, including aberrant calcium regulation and arrhythmias. (Toepfer et al., 2019; Zhou et al., 2019; Toepfer et al., 2020; Hsieh et al., 2022). Further investigation of the pathophysiology of HCM using hiPSC-CM will be important to better understand the genotype-phenotype relationship, the impact of novel myosin inhibitors on calcium regulation, and identification of novel disease-specific therapies.
Cytoskeletal proteins play a critical role in cardiac maintenance of mechanical integrity, mechanotransduction, sarcomere stabilization, but also modulation of signaling pathways in the myocardium, and mutations in cytoskeletal genes can result in cardiomyopathy (Kostin et al., 2000). Duchenne muscular dystrophy (DMD) is characterized by the absence of the full-length membrane associated cytoskeletal protein, dystrophin, leading to severe muscle weakness, cardiomyopathy, and premature death (Kamdar and Garry, 2016). Ventricular myocytes from muscular dystrophy mice show an increase in stretch-related intracellular calcium overload and dysregulation in calcium handling (Yasuda et al., 2005; Williams and Allen, 2007). The mechanism of Ca2+ dysregulation in the absence of dystrophin may be multifactorial due to increased intracellular sarcolemmal tears and alterations in the function of key Ca2+ cycling proteins; however the precise mechanism is not yet known (Law et al., 2020). However, DMD hiPSC-CMs have been increasingly used to assess Ca2+ dysregulation in DMD cardiomyopathy. Patient-derived DMD hiPSC-CMs (Kamdar et al., 2020) were used to model DMD cardiomyopathy. The control hiPSC-CMs exhibited regular Ca2+ transients with consistent amplitudes at baseline. Treatment with isopropterenol increased both the frequency and the time to peak of Ca2+ transients, while subsequent addition of propranolol led to a sharp decrease in the frequency of Ca2+ transients. At baseline, DMD-hiPSC-CMs exhibited highly irregular Ca2+ transients, displaying high variability in cycle length, as well as high amount of arrhythmic activity, including double beats and DADs. Treatment with isoproterenol similarly led to an increase in Ca2+ transient frequency, along with a large increase in arrhythmic activity. Subsequent treatment with propranolol significantly decreased Ca2+ transient frequency and reduced arrhythmic activity to levels below those at baseline. Moreover, propranolol treatment prior to isoproterenol maintained arrhythmic activity similar to that of baseline. DMD null isogenic control lines (iDMD-hiPSC-CMs) exhibited similar arrhythmic Ca2+ activity and responses to isoproterenol and propranolol as seen in the DMD-hiPSC-CMs derived from reprogrammed patient cells (Kamdar et al., 2020). Similarly, Chang et al.(2021) identified that hiPSC-CMs from multiple DMD patient lines exhibited irregular calcium activity, including elevated diastolic calcium levels, decreased transient amplitude, and longer recovery rates. Following a period of moderate mechanical stress intended to mimic fibrotic stiffening of the heart, DMD-hiPSC-CMs notably exhibited weaker contractile force compared to control hiPSC-CMs given the same stress, however, no such discrepancy was observed in cells subjected to a milder stimulus. Moderately stressed DMD-hiPSC-CMs also showed decreases in contraction and relaxation velocities, as well as overall cell size (Chang et al., 2021). Additionally, the microRNA-378a has been found to be altered in the serum of DMD patients, and deletion in hiPSC-CM depresses Ca2+ transients (Martyniak et al., 2021). Further investigation of calcium dysregulation in DMD cardiomyopathy using DMD hiPSC-CMs, other model systems, and patient samples will help to elucidate the major mechanisms and potential treatments.
In conclusion, our understanding of the physiological and pathological regulation of cardiac calcium has been largely based on the utilization of isolated animal model cardiac tissue or cardiomyocytes. However, the discovery of hiPSC has been revolutionary in science, and the subsequent differentiation to cardiomyocytes in the last decade has provided a new platform of renewable human cardiac cells to deepen our understanding of the mechanisms of cardiovascular physiology and disease and the screening for therapeutics. In particular, inherited cardiovascular disease, including genetic arrhythmias and cardiomyopathies, have become more accessible to research with hiPSC-CM. While the molecular mechanisms of cardiovascular diseases are different, aberrant calcium signaling is common to the majority of them. Here, we have reviewed the important differences in calcium regulation between adult human cardiomyocytes, especially in relation to cardiovascular disease modeling.
Compared to adult human cardiomyocytes, hiPSC-CMs are less mature. While hiPSC-CMs are a promising translational tool for understanding cardiovascular disease, the maturity of hiPSC-CMs must be taken into account, especially their ability to regulate calcium, to model human cardiovascular disease. Although hiPSC-CM are relatively immature, they may be useful in understanding early stage and developmental changes in calcium regulation, and hiPSC-CMs have many qualitative calcium handling properties similar to those of human cardiomyocytes. The majority of our understanding of calcium regulation in the heart stems from studies done on animal cardiomyocytes or diseased human hearts, and this should also be taken into consideration when evaluating hiPSC-CM calcium regulation.
These differences in calcium handling, especially related to maturity, are important for the field overall to understand the limitations of current hiPSC-CM technology and subsequently develop more mature hiPSC-CM that better recapitulate the human myocardium that is needed for disease modeling, drug toxicity screening, and clinical therapeutic applications.
hiPSC-CMs are a unique tool, but in order for hiPSC-CM to be utilized to understand mechanisms of calcium handling at baseline the maturity of hiPSC-CM including the ability to differentiate mature chamber specific CM is needed. Additionally, establishing expected parameters for calcium regulation at different stages of differentiation can better characterize and standardize hiPSC-CMs to evaluate the impact of maturation strategies and ensure appropriate disease modeling.
There are still many gaps in the knowledge regarding calcium regulation in hiPSC-CMs. A number of questions remain especially in regards to the structure and function of various calcium handling components and the coupling of components needed for excitation contraction coupling in hiPSC-CM.
The combination of hiPSC-CMs with biosensors, such as fluorescence resonance energy transfer (FRET) biosensors for SERCA2a which have typically been utilized in non-cardiac cells, can inform the field about the structure and function of these calcium handling components at baseline and in disease states. Additionally, hiPSC-CMs that express FRET biosensors for SERCA2a can be used for high-throughput drug screening to identify novel therapeutics that could be used to increase SERCA2a activity, which is reduced in heart failure states (Schaaf et al., 2020). These hiPSC-CM containing SERCA2a FRET biosensors could also be used to test drug toxicity of new therapies on SERCA2A function, which may help better understand impact of new drugs on calcium regulation and excitation coupling in the heart.
Similarly, other biosensors can be utilized in hiPSC-CMs to understand the contribution of mitochondrial calcium in the context of cytosolic calcium at baseline and in disease states, which is not currently well established (Ernst et al., 2021).
Gene editing hiPSC-CMs with CRISPR/Cas9 and newer precise editing methods can be utilized for both basic investigation and translational development, by assessing the impact of mutations in various calcium handling genes and their regulators, systematically assessing novel therapies, and for precision medicine to determine if specific mutations result in a phenotype. Genetic engineering strategies have great potential to help elucidate mechanisms of early cardiac calcium regulation and is an exciting area for the field.
PE and PB contributed equally to writing of the first draft of the manuscript, PE and MD contributed to revisions and generation of figures, DT provided guidance. FK conceived the idea, substantially revised and critically edited the manuscript.
This work was supported by a Doris Duke Charitable Foundation Clinical Scientist Development Award 2020103A (FK) and NIH T32 HL144472 (PE).
These figures were adapted and created with Biorender. Figure 2 is adapted from “Cardiomyocyte Differentiation Markers” & “From iPSCs to cardiomyocytes”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acimovic, I., Refaat, M. M., Moreau, A., Salykin, A., Reiken, S., Sleiman, Y., et al. (2018). Post-translational modifications and diastolic calcium leak associated to the novel RyR2-d3638a mutation lead to CPVT in patient-specific hiPSC-derived cardiomyocytes. J. Clin. Med. 7, 423. doi:10.3390/jcm7110423
Bal, N. C., and Periasamy, M. (2020). Uncoupling of sarcoendoplasmic reticulum calcium ATPase pump activity by sarcolipin as the basis for muscle non-shivering thermogenesis. Philos. Trans. R. Soc. Lond B Biol. Sci. 375, 20190135. doi:10.1098/rstb.2019.0135
Balistreri, M., Davis, J. A., Campbell, K. F., Da Rocha, A. M., Treadwell, M. C., and Herron, T. J. (2017). Effect of glucose on 3D cardiac microtissues derived from human induced pluripotent stem cells. Pediatr. Cardiol. 38, 1575–1582. doi:10.1007/s00246-017-1698-2
Barbuti, A., Benzoni, P., Campostrini, G., and Dell'era, P. (2016). Human derived cardiomyocytes: A decade of knowledge after the discovery of induced pluripotent stem cells. Dev. Dyn. 245, 1145–1158. doi:10.1002/dvdy.24455
Bers, D. M. (2008). Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49. doi:10.1146/annurev.physiol.70.113006.100455
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi:10.1038/415198a
Bers, D. M. (2014). Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127. doi:10.1146/annurev-physiol-020911-153308
Bers, D. M. (2001). Excitation-contraction coupling and cardiac contractile force. Dordrecht ; Boston: Kluwer Academic Publishers.
Bidwell, P. A., Haghighi, K., and Kranias, E. G. (2018). The antiapoptotic protein HAX-1 mediates half of phospholamban's inhibitory activity on calcium cycling and contractility in the heart. J. Biol. Chem. 293, 359–367. doi:10.1074/jbc.RA117.000128
Birket, M. J., Ribeiro, M. C., Kosmidis, G., Ward, D., Leitoguinho, A. R., Van De Pol, V., et al. (2015). Contractile defect caused by mutation in MYBPC3 revealed under conditions optimized for human PSC-cardiomyocyte function. Cell. Rep. 13, 733–745. doi:10.1016/j.celrep.2015.09.025
Boyman, L., Chikando, A. C., Williams, G. S., Khairallah, R. J., Kettlewell, S., Ward, C. W., et al. (2014). Calcium movement in cardiac mitochondria. Biophys. J. 107, 1289–1301. doi:10.1016/j.bpj.2014.07.045
Boyman, L., Greiser, M., and Lederer, W. J. (2021). Calcium influx through the mitochondrial calcium uniporter holocomplex, MCUcx. J. Mol. Cell. Cardiol. 151, 145–154. doi:10.1016/j.yjmcc.2020.10.015
Buntinas, L., Gunter, K. K., Sparagna, G. C., and Gunter, T. E. (2001). The rapid mode of calcium uptake into heart mitochondria (RaM): Comparison to RaM in liver mitochondria. Biochim. Biophys. Acta 1504, 248–261. doi:10.1016/s0005-2728(00)00254-1
Burridge, P. W., Thompson, S., Millrod, M. A., Weinberg, S., Yuan, X., Peters, A., et al. (2011). A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS One 6, e18293. doi:10.1371/journal.pone.0018293
Cai, W., Zhang, J., De Lange, W. J., Gregorich, Z. R., Karp, H., Farrell, E. T., et al. (2019). An unbiased proteomics method to assess the maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 125, 936–953. doi:10.1161/CIRCRESAHA.119.315305
Cao, N., Liu, Z., Chen, Z., Wang, J., Chen, T., Zhao, X., et al. (2012). Ascorbic acid enhances the cardiac differentiation of induced pluripotent stem cells through promoting the proliferation of cardiac progenitor cells. Cell. Res. 22, 219–236. doi:10.1038/cr.2011.195
Carafoli, E., Tiozzo, R., Lugli, G., Crovetti, F., and Kratzing, C. (1974). The release of calcium from heart mitochondria by sodium. J. Mol. Cell. Cardiol. 6, 361–371. doi:10.1016/0022-2828(74)90077-7
Ceholski, D. K., Turnbull, I. C., Kong, C. W., Koplev, S., Mayourian, J., Gorski, P. A., et al. (2018). Functional and transcriptomic insights into pathogenesis of R9C phospholamban mutation using human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 119, 147–154. doi:10.1016/j.yjmcc.2018.05.007
Chan, Y. C., Ting, S., Lee, Y. K., Ng, K. M., Zhang, J., Chen, Z., et al. (2013). Electrical stimulation promotes maturation of cardiomyocytes derived from human embryonic stem cells. J. Cardiovasc Transl. Res. 6, 989–999. doi:10.1007/s12265-013-9510-z
Chang, A. C. Y., Pardon, G., Chang, A. C. H., Wu, H., Ong, S. G., Eguchi, A., et al. (2021). Increased tissue stiffness triggers contractile dysfunction and telomere shortening in dystrophic cardiomyocytes. Stem Cell. Rep. 16, 2169–2181. doi:10.1016/j.stemcr.2021.04.018
Chattergoon, N. N., Giraud, G. D., Louey, S., Stork, P., Fowden, A. L., and Thornburg, K. L. (2012). Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J. 26, 397–408. doi:10.1096/fj.10-179895
Chen, G., Li, S., Karakikes, I., Ren, L., Chow, M. Z., Chopra, A., et al. (2015). Phospholamban as a crucial determinant of the inotropic response of human pluripotent stem cell-derived ventricular cardiomyocytes and engineered 3-dimensional tissue constructs. Circ. Arrhythm. Electrophysiol. 8, 193–202. doi:10.1161/CIRCEP.114.002049
Cornea, R. L., Gruber, S. J., Lockamy, E. L., Muretta, J. M., Jin, D., Chen, J., et al. (2013). High-throughput FRET assay yields allosteric SERCA activators. J. Biomol. Screen 18, 97–107. doi:10.1177/1087057112456878
Correia, C., Serra, M., Espinha, N., Sousa, M., Brito, C., Burkert, K., et al. (2014). Combining hypoxia and bioreactor hydrodynamics boosts induced pluripotent stem cell differentiation towards cardiomyocytes. Stem Cell. Rev. Rep. 10, 786–801. doi:10.1007/s12015-014-9533-0
Crombie, D. E., Curl, C. L., Raaijmakers, A. J., Sivakumaran, P., Kulkarni, T., Wong, R. C., et al. (2017). Friedreich's ataxia induced pluripotent stem cell-derived cardiomyocytes display electrophysiological abnormalities and calcium handling deficiency. Aging (Albany NY) 9, 1440–1452. doi:10.18632/aging.101247
Crompton, M., Kunzi, M., and Carafoli, E. (1977). The calcium-induced and sodium-induced effluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier. Eur. J. Biochem. 79, 549–558. doi:10.1111/j.1432-1033.1977.tb11839.x
Davia, K., Davies, C. H., and Harding, S. E. (1997). Effects of inhibition of sarcoplasmic reticulum calcium uptake on contraction in myocytes isolated from failing human ventricle. Cardiovasc Res. 33, 88–97. doi:10.1016/s0008-6363(96)00187-3
Denniss, A. L., Dashwood, A. M., Molenaar, P., and Beard, N. A. (2020). Sarcoplasmic reticulum calcium mishandling: Central tenet in heart failure? Biophys. Rev. 12, 865–878. doi:10.1007/s12551-020-00736-y
Di Pasquale, E., Lodola, F., Miragoli, M., Denegri, M., Avelino-Cruz, J. E., Buonocore, M., et al. (2013). CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell. Death Dis. 4, e843. doi:10.1038/cddis.2013.369
Diaz, M. E., Trafford, A. W., and Eisner, D. A. (2001). The role of intracellular Ca buffers in determining the shape of the systolic Ca transient in cardiac ventricular myocytes. Pflugers Arch. 442, 96–100. doi:10.1007/s004240000509
Dolnikov, K., Shilkrut, M., Zeevi-Levin, N., Gerecht-Nir, S., Amit, M., Danon, A., et al. (2006). Functional properties of human embryonic stem cell-derived cardiomyocytes: Intracellular Ca2+ handling and the role of sarcoplasmic reticulum in the contraction. Stem Cells 24, 236–245. doi:10.1634/stemcells.2005-0036
Drago, I., De Stefani, D., Rizzuto, R., and Pozzan, T. (2012). Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 109, 12986–12991. doi:10.1073/pnas.1210718109
Eisner, D. A., Caldwell, J. L., Kistamas, K., and Trafford, A. W. (2017). Calcium and excitation-contraction coupling in the heart. Circ. Res. 121, 181–195. doi:10.1161/CIRCRESAHA.117.310230
El-Battrawy, I., Zhao, Z., Lan, H., Li, X., Yucel, G., Lang, S., et al. (2018). Ion Channel dysfunctions in dilated cardiomyopathy in limb-girdle muscular dystrophy. Circ. Genom Precis. Med. 11, e001893. doi:10.1161/CIRCGEN.117.001893
Ernst, P., Chen, K., Tang, Y., Kim, S., Guan, J., He, J., et al. (2021). Investigation into the difference in mitochondrial-cytosolic calcium coupling between adult cardiomyocyte and hiPSC-CM using a novel multifunctional genetic probe. Pflugers Arch. 473, 447–459. doi:10.1007/s00424-021-02524-3
Fatima, A., Xu, G., Shao, K., Papadopoulos, S., Lehmann, M., Arnaiz-Cot, J. J., et al. (2011). In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell. Physiol. Biochem. 28, 579–592. doi:10.1159/000335753
Frederick, R. L., and Shaw, J. M. (2007). Moving mitochondria: Establishing distribution of an essential organelle. Traffic 8, 1668–1675. doi:10.1111/j.1600-0854.2007.00644.x
Gao, L., Kupfer, M. E., Jung, J. P., Yang, L., Zhang, P., Da Sie, Y., et al. (2017). Myocardial tissue engineering with cells derived from human-induced pluripotent stem cells and a native-like, high-resolution, 3-dimensionally printed scaffold. Circ. Res. 120, 1318–1325. doi:10.1161/CIRCRESAHA.116.310277
Garbincius, J. F., Luongo, T. S., and Elrod, J. W. (2020). The debate continues - what is the role of MCU and mitochondrial calcium uptake in the heart? J. Mol. Cell. Cardiol. 143, 163–174. doi:10.1016/j.yjmcc.2020.04.029
Gentillon, C., Li, D., Duan, M., Yu, W. M., Preininger, M. K., Jha, R., et al. (2019). Targeting HIF-1α in combination with PPARα activation and postnatal factors promotes the metabolic maturation of human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 132, 120–135. doi:10.1016/j.yjmcc.2019.05.003
Germanguz, I., Sedan, O., Zeevi-Levin, N., Shtrichman, R., Barak, E., Ziskind, A., et al. (2011). Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells. J. Cell. Mol. Med. 15, 38–51. doi:10.1111/j.1582-4934.2009.00996.x
Greenberg, B., Butler, J., Felker, G. M., Ponikowski, P., Voors, A. A., Desai, A. S., et al. (2016). Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 387, 1178–1186. doi:10.1016/S0140-6736(16)00082-9
Gruber, S. J., Cornea, R. L., Li, J., Peterson, K. C., Schaaf, T. M., Gillispie, G. D., et al. (2014a). Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J. Biomol. Screen. 19, 215–222. doi:10.1177/1087057113510740
Gruber, S. J., Cornea, R. L., Li, J., Peterson, K. C., Schaaf, T. M., Gillispie, G. D., et al. (2014b). Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J. Biomol. Screen 19, 215–222. doi:10.1177/1087057113510740
Gruber, S. J., Haydon, S., and Thomas, D. D. (2012). Phospholamban mutants compete with wild type for SERCA binding in living cells. Biochem. Biophys. Res. Commun. 420, 236–240. doi:10.1016/j.bbrc.2012.02.125
Guo, Y., and Pu, W. T. (2020). Cardiomyocyte maturation: New phase in development. Circ. Res. 126, 1086–1106. doi:10.1161/CIRCRESAHA.119.315862
Haghighi, K., Bidwell, P., and Kranias, E. G. (2014). Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J. Mol. Cell. Cardiol. 77, 160–167. doi:10.1016/j.yjmcc.2014.10.005
Haghighi, K., Kolokathis, F., Gramolini, A. O., Waggoner, J. R., Pater, L., Lynch, R. A., et al. (2006). A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 103, 1388–1393. doi:10.1073/pnas.0510519103
Hakli, M., Kreutzer, J., Maki, A. J., Valimaki, H., Lappi, H., Huhtala, H., et al. (2021). Human induced pluripotent stem cell-based platform for modeling cardiac ischemia. Sci. Rep. 11, 4153. doi:10.1038/s41598-021-83740-w
Han, L., Li, Y., Tchao, J., Kaplan, A. D., Lin, B., Li, Y., et al. (2014). Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc Res. 104, 258–269. doi:10.1093/cvr/cvu205
Hasenfuss, G., Reinecke, H., Studer, R., Meyer, M., Pieske, B., Holtz, J., et al. (1994). Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ. Res. 75, 434–442. doi:10.1161/01.res.75.3.434
Hayakawa, T., Kunihiro, T., Ando, T., Kobayashi, S., Matsui, E., Yada, H., et al. (2014). Image-based evaluation of contraction-relaxation kinetics of human-induced pluripotent stem cell-derived cardiomyocytes: Correlation and complementarity with extracellular electrophysiology. J. Mol. Cell. Cardiol. 77, 178–191. doi:10.1016/j.yjmcc.2014.09.010
Hershberger, R. E., Hedges, D. J., and Morales, A. (2013). Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547. doi:10.1038/nrcardio.2013.105
Hinson, J. T., Chopra, A., Nafissi, N., Polacheck, W. J., Benson, C. C., Swist, S., et al. (2015). HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. doi:10.1126/science.aaa5458
Hou, Z., Hu, Z., Blackwell, D. J., Miller, T. D., Thomas, D. D., and Robia, S. L. (2012). 2-Color calcium pump reveals closure of the cytoplasmic headpiece with calcium binding. PLoS One 7, e40369. doi:10.1371/journal.pone.0040369
Hsieh, J., Becklin, K. L., Givens, S., Komosa, E. R., Llorens, J. E. A., Kamdar, F., et al. (2022). Myosin heavy chain converter domain mutations drive early-stage changes in extracellular matrix dynamics in hypertrophic cardiomyopathy. Front. Cell. Dev. Biol. 10, 894635. doi:10.3389/fcell.2022.894635
Huikuri, H. V., Castellanos, A., and Myerburg, R. J. (2001). Sudden death due to cardiac arrhythmias. N. Engl. J. Med. 345, 1473–1482. doi:10.1056/NEJMra000650
Huxley, A. F. (1957). Muscle structure and theories of contraction. Prog. Biophys. Biophys. Chem. 7, 255–318. doi:10.1016/s0096-4174(18)30128-8
Hwang, H. S., Kryshtal, D. O., Feaster, T. K., Sanchez-Freire, V., Zhang, J., Kamp, T. J., et al. (2015). Comparable calcium handling of human iPSC-derived cardiomyocytes generated by multiple laboratories. J. Mol. Cell. Cardiol. 85, 79–88. doi:10.1016/j.yjmcc.2015.05.003
Ikemoto, N., Bhatnagar, G. M., Nagy, B., and Gergely, J. (1972). Interaction of divalent cations with the 55, 000-dalton protein component of the sarcoplasmic reticulum. J. Biol. Chem. 247, 7835–7837. doi:10.1016/s0021-9258(19)44598-5
Itzhaki, I., Maizels, L., Huber, I., Gepstein, A., Arbel, G., Caspi, O., et al. (2012). Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J. Am. Coll. Cardiol. 60, 990–1000. doi:10.1016/j.jacc.2012.02.066
Itzhaki, I., Rapoport, S., Huber, I., Mizrahi, I., Zwi-Dantsis, L., Arbel, G., et al. (2011). Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS One 6, e18037. doi:10.1371/journal.pone.0018037
Jung, C. B., Moretti, A., Mederos Y Schnitzler, M., Iop, L., Storch, U., Bellin, M., et al. (2012). Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol. Med. 4, 180–191. doi:10.1002/emmm.201100194
Kamakura, T., Makiyama, T., Sasaki, K., Yoshida, Y., Wuriyanghai, Y., Chen, J., et al. (2013). Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ. J. 77, 1307–1314. doi:10.1253/circj.cj-12-0987
Kamdar, F., Das, S., Gong, W., Klaassen Kamdar, A., Meyers, T. A., Shah, P., et al. (2020). Stem cell-derived cardiomyocytes and beta-adrenergic receptor blockade in Duchenne muscular dystrophy cardiomyopathy. J. Am. Coll. Cardiol. 75, 1159–1174. doi:10.1016/j.jacc.2019.12.066
Kamdar, F., and Garry, D. J. (2016). Dystrophin-deficient cardiomyopathy. J. Am. Coll. Cardiol. 67, 2533–2546. doi:10.1016/j.jacc.2016.02.081
Kamdar, F., Klaassen Kamdar, A., Koyano-Nakagawa, N., Garry, M. G., and Garry, D. J. (2015). Cardiomyopathy in a dish: Using human inducible pluripotent stem cells to model inherited cardiomyopathies. J. Card. Fail 21, 761–770. doi:10.1016/j.cardfail.2015.04.010
Kamisago, M., Sharma, S. D., Depalma, S. R., Solomon, S., Sharma, P., Mcdonough, B., et al. (2000). Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 343, 1688–1696. doi:10.1056/NEJM200012073432304
Karakikes, I., Ameen, M., Termglinchan, V., and Wu, J. C. (2015a). Human induced pluripotent stem cell-derived cardiomyocytes: Insights into molecular, cellular, and functional phenotypes. Circ. Res. 117, 80–88. doi:10.1161/CIRCRESAHA.117.305365
Karakikes, I., Stillitano, F., Nonnenmacher, M., Tzimas, C., Sanoudou, D., Termglinchan, V., et al. (2015b). Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 6, 6955. doi:10.1038/ncomms7955
Karbassi, E., Fenix, A., Marchiano, S., Muraoka, N., Nakamura, K., Yang, X., et al. (2020). Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 17, 341–359. doi:10.1038/s41569-019-0331-x
Kawase, Y., and Hajjar, R. J. (2008). The cardiac sarcoplasmic/endoplasmic reticulum calcium ATPase: A potent target for cardiovascular diseases. Nat. Clin. Pract. Cardiovasc Med. 5, 554–565. doi:10.1038/ncpcardio1301
Khan, M., Xu, Y., Hua, S., Johnson, J., Belevych, A., Janssen, P. M., et al. (2015). Evaluation of changes in morphology and function of human induced pluripotent stem cell derived cardiomyocytes (HiPSC-CMs) cultured on an aligned-nanofiber cardiac patch. PLoS One 10, e0126338. doi:10.1371/journal.pone.0126338
Kostin, S., Hein, S., Arnon, E., Scholz, D., and Schaper, J. (2000). The cytoskeleton and related proteins in the human failing heart. Heart Fail Rev. 5, 271–280. doi:10.1023/A:1009813621103
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. doi:10.1161/CIRCRESAHA.111.259754
Kroll, K., Chabria, M., Wang, K., Hausermann, F., Schuler, F., and Polonchuk, L. (2017). Electro-mechanical conditioning of human iPSC-derived cardiomyocytes for translational research. Prog. Biophys. Mol. Biol. 130, 212–222. doi:10.1016/j.pbiomolbio.2017.07.003
Kruger, M., Sachse, C., Zimmermann, W. H., Eschenhagen, T., Klede, S., and Linke, W. A. (2008). Thyroid hormone regulates developmental titin isoform transitions via the phosphatidylinositol-3-kinase/AKT pathway. Circ. Res. 102, 439–447. doi:10.1161/CIRCRESAHA.107.162719
Kupfer, M. E., Lin, W. H., Ravikumar, V., Qiu, K., Wang, L., Gao, L., et al. (2020). In situ expansion, differentiation, and electromechanical coupling of human cardiac muscle in a 3D bioprinted, chambered organoid. Circ. Res. 127, 207–224. doi:10.1161/CIRCRESAHA.119.316155
Kyrychenko, V., Kyrychenko, S., Tiburcy, M., Shelton, J. M., Long, C., Schneider, J. W., et al. (2017). Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2, e95918. doi:10.1172/jci.insight.95918
Lan, F., Lee, A. S., Liang, P., Sanchez-Freire, V., Nguyen, P. K., Wang, L., et al. (2013). Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell. Stem Cell. 12, 101–113. doi:10.1016/j.stem.2012.10.010
Lasher, R. A., Pahnke, A. Q., Johnson, J. M., Sachse, F. B., and Hitchcock, R. W. (2012). Electrical stimulation directs engineered cardiac tissue to an age-matched native phenotype. J. Tissue Eng. 3, 2041731412455354. doi:10.1177/2041731412455354
Law, M. L., Cohen, H., Martin, A. A., Angulski, A. B. B., and Metzger, J. M. (2020). Dysregulation of calcium handling in Duchenne muscular dystrophy-associated dilated cardiomyopathy: Mechanisms and experimental therapeutic strategies. J. Clin. Med. 9, 520. doi:10.3390/jcm9020520
Lee, Y. K., Ng, K. M., Chan, Y. C., Lai, W. H., Au, K. W., Ho, C. Y., et al. (2010). Triiodothyronine promotes cardiac differentiation and maturation of embryonic stem cells via the classical genomic pathway. Mol. Endocrinol. 24, 1728–1736. doi:10.1210/me.2010-0032
Lee, Y. K., Ng, K. M., Lai, W. H., Chan, Y. C., Lau, Y. M., Lian, Q., et al. (2011). Calcium homeostasis in human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell. Rev. Rep. 7, 976–986. doi:10.1007/s12015-011-9273-3
Li, A., Yuen, S. L., Stroik, D. R., Kleinboehl, E., Cornea, R. L., and Thomas, D. D. (2021a). The transmembrane peptide DWORF activates SERCA2a via dual mechanisms. J. Biol. Chem. 296, 100412. doi:10.1016/j.jbc.2021.100412
Li, B., Zhan, Y., Liang, Q., Xu, C., Zhou, X., Cai, H., et al. (2021b). Isogenic human pluripotent stem cell disease models reveal ABRA deficiency underlies cTnT mutation-induced familial dilated cardiomyopathy. Protein Cell. 13, 65–71. doi:10.1007/s13238-021-00843-w
Li, M., Iismaa, S. E., Naqvi, N., Nicks, A., Husain, A., and Graham, R. M. (2014). Thyroid hormone action in postnatal heart development. Stem Cell. Res. 13, 582–591. doi:10.1016/j.scr.2014.07.001
Lian, X., Hsiao, C., Wilson, G., Zhu, K., Hazeltine, L. B., Azarin, S. M., et al. (2012). Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. U. S. A. 109, E1848–E1857. doi:10.1073/pnas.1200250109
Liang, P., Sallam, K., Wu, H., Li, Y., Itzhaki, I., Garg, P., et al. (2016). Patient-specific and genome-edited induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of Brugada syndrome. J. Am. Coll. Cardiol. 68, 2086–2096. doi:10.1016/j.jacc.2016.07.779
Lipskaia, L., Ly, H., Kawase, Y., Hajjar, R. J., and Lompre, A. M. (2007). Treatment of heart failure by calcium cycling gene therapy. Future Cardiol. 3, 413–423. doi:10.2217/14796678.3.4.413
Liu, M., Shi, G., Zhou, A., Rupert, C. E., Coulombe, K. L. K., and Dudley, S. C. (2018). Activation of the unfolded protein response downregulates cardiac ion channels in human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 117, 62–71. doi:10.1016/j.yjmcc.2018.02.011
Lo, C. H., Vunnam, N., Lewis, A. K., Chiu, T. L., Brummel, B. E., Schaaf, T. M., et al. (2017). An innovative high-throughput screening approach for discovery of small molecules that inhibit TNF receptors. SLAS Discov. 22, 950–961. doi:10.1177/2472555217706478
Lodola, F., Morone, D., Denegri, M., Bongianino, R., Nakahama, H., Rutigliano, L., et al. (2016). Adeno-associated virus-mediated CASQ2 delivery rescues phenotypic alterations in a patient-specific model of recessive catecholaminergic polymorphic ventricular tachycardia. Cell. Death Dis. 7, e2393. doi:10.1038/cddis.2016.304
Lundy, S. D., Zhu, W. Z., Regnier, M., and Laflamme, M. A. (2013). Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 22, 1991–2002. doi:10.1089/scd.2012.0490
Maclennan, D. H., and Kranias, E. G. (2003). Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell. Biol. 4, 566–577. doi:10.1038/nrm1151
Maizels, L., Huber, I., Arbel, G., Tijsen, A. J., Gepstein, A., Khoury, A., et al. (2017). Patient-specific drug screening using a human induced pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia type 2. Circ. Arrhythm. Electrophysiol. 10, e004725. doi:10.1161/CIRCEP.116.004725
Maron, B. J., Desai, M. Y., Nishimura, R. A., Spirito, P., Rakowski, H., Towbin, J. A., et al. (2022a). Management of hypertrophic cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 79, 390–414. doi:10.1016/j.jacc.2021.11.021
Maron, B. J., Desai, M. Y., Nishimura, R. A., Spirito, P., Rakowski, H., Towbin, J. A., et al. (2022b). Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 79, 372–389. doi:10.1016/j.jacc.2021.12.002
Maron, B. J., Rowin, E. J., and Maron, M. S. (2022c). Hypertrophic cardiomyopathy: New concepts and therapies. Annu. Rev. Med. 73, 363–375. doi:10.1146/annurev-med-042220-021539
Martyniak, A., Andrysiak, K., Motais, B., Coste, S., Podkalicka, P., Ferdek, P., et al. (2021). Generation of microRNA-378a-deficient hiPSC as a novel tool to study its role in human cardiomyocytes. J. Mol. Cell. Cardiol. 160, 128–141. doi:10.1016/j.yjmcc.2021.07.007
Mcdermott-Roe, C., Lv, W., Maximova, T., Wada, S., Bukowy, J., Marquez, M., et al. (2019). Investigation of a dilated cardiomyopathy-associated variant in BAG3 using genome-edited iPSC-derived cardiomyocytes. JCI Insight 4, e128799. doi:10.1172/jci.insight.128799
Mcnally, E. M., and Mestroni, L. (2017). Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 121, 731–748. doi:10.1161/CIRCRESAHA.116.309396
Mehta, A., Ramachandra, C. J. A., Singh, P., Chitre, A., Lua, C. H., Mura, M., et al. (2018). Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur. Heart J. 39, 1446–1455. doi:10.1093/eurheartj/ehx394
Moller, J. V., Olesen, C., Winther, A. M., and Nissen, P. (2010). The sarcoplasmic Ca2+-ATPase: Design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 43, 501–566. doi:10.1017/S003358351000017X
Muretta, J. M., Kyrychenko, A., Ladokhin, A. S., Kast, D. J., Gillispie, G. D., and Thomas, D. D. (2010). High-performance time-resolved fluorescence by direct waveform recording. Rev. Sci. Instrum. 81, 103101. doi:10.1063/1.3480647
Murphy, J. F., Mayourian, J., Stillitano, F., Munawar, S., Broughton, K. M., Agullo-Pascual, E., et al. (2019). Adult human cardiac stem cell supplementation effectively increases contractile function and maturation in human engineered cardiac tissues. Stem Cell. Res. Ther. 10, 373. doi:10.1186/s13287-019-1486-4
Nelson, B. R., Makarewich, C. A., Anderson, D. M., Winders, B. R., Troupes, C. D., Wu, F., et al. (2016). A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351, 271–275. doi:10.1126/science.aad4076
Ng, K. M., Lee, Y. K., Lai, W. H., Chan, Y. C., Fung, M. L., Tse, H. F., et al. (2011). Exogenous expression of human apoA-I enhances cardiac differentiation of pluripotent stem cells. PLoS One 6, e19787. doi:10.1371/journal.pone.0019787
Ng, K., Titus, E. W., Lieve, K. V., Roston, T. M., Mazzanti, A., Deiter, F. H., et al. (2020). An international multicenter evaluation of inheritance patterns, arrhythmic risks, and underlying mechanisms of CASQ2-catecholaminergic polymorphic ventricular tachycardia. Circulation 142, 932–947. doi:10.1161/CIRCULATIONAHA.120.045723
Nguemo, F., Semmler, J., Reppel, M., and Hescheler, J. (2014). Modulation of L-type calcium current by intracellular magnesium in differentiating cardiomyocytes derived from induced pluripotent stem cells. Stem Cells Dev. 23, 1316–1327. doi:10.1089/scd.2013.0549
Novak, A., Barad, L., Lorber, A., Gherghiceanu, M., Reiter, I., Eisen, B., et al. (2015). Functional abnormalities in iPSC-derived cardiomyocytes generated from CPVT1 and CPVT2 patients carrying ryanodine or calsequestrin mutations. J. Cell. Mol. Med. 19, 2006–2018. doi:10.1111/jcmm.12581
Nunes, S. S., Miklas, J. W., Liu, J., Aschar-Sobbi, R., Xiao, Y., Zhang, B., et al. (2013). Biowire: A platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat. Methods 10, 781–787. doi:10.1038/nmeth.2524
Paavola, J., Vaananen, H., Larsson, K., Penttinen, K., Toivonen, L., Kontula, K., et al. (2016). Slowed depolarization and irregular repolarization in catecholaminergic polymorphic ventricular tachycardia: A study from cellular Ca2+ transients and action potentials to clinical monophasic action potentials and electrocardiography. Europace 18, 1599–1607. doi:10.1093/europace/euv380
Pallikkuth, S., Blackwell, D. J., Hu, Z., Hou, Z., Zieman, D. T., Svensson, B., et al. (2013). Phosphorylated phospholamban stabilizes a compact conformation of the cardiac calcium-ATPase. Biophys. J. 105, 1812–1821. doi:10.1016/j.bpj.2013.08.045
Parikh, S. S., Blackwell, D. J., Gomez-Hurtado, N., Frisk, M., Wang, L., Kim, K., et al. (2017). Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ. Res. 121, 1323–1330. doi:10.1161/CIRCRESAHA.117.311920
Pioner, J. M., Santini, L., Palandri, C., Martella, D., Lupi, F., Langione, M., et al. (2019). Optical investigation of action potential and calcium handling maturation of hiPSC-cardiomyocytes on biomimetic substrates. Int. J. Mol. Sci. 20, 3799. doi:10.3390/ijms20153799
Polonen, R. P., Swan, H., and Aalto-Setala, K. (2020). Mutation-specific differences in arrhythmias and drug responses in CPVT patients: Simultaneous patch clamp and video imaging of iPSC derived cardiomyocytes. Mol. Biol. Rep. 47, 1067–1077. doi:10.1007/s11033-019-05201-y
Preininger, M. K., Jha, R., Maxwell, J. T., Wu, Q., Singh, M., Wang, B., et al. (2016). A human pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia recapitulates patient-specific drug responses. Dis. Model. Mech. 9, 927–939. doi:10.1242/dmm.026823
Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C., et al. (2013). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in may 2013 and by ACCF, AHA, PACES, and AEPC in june 2013. Heart rhythm. 10, 1932–1963. doi:10.1016/j.hrthm.2013.05.014
Rao, C., Prodromakis, T., Kolker, L., Chaudhry, U. A., Trantidou, T., Sridhar, A., et al. (2013). The effect of microgrooved culture substrates on calcium cycling of cardiac myocytes derived from human induced pluripotent stem cells. Biomaterials 34, 2399–2411. doi:10.1016/j.biomaterials.2012.11.055
Richards, D. J., Coyle, R. C., Tan, Y., Jia, J., Wong, K., Toomer, K., et al. (2017). Inspiration from heart development: Biomimetic development of functional human cardiac organoids. Biomaterials 142, 112–123. doi:10.1016/j.biomaterials.2017.07.021
Ringer, S. (1883). A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J. Physiol. 4, 29–42. doi:10.1113/jphysiol.1883.sp000120
Robia, S. L., Campbell, K. S., Kelly, E. M., Hou, Z., Winters, D. L., and Thomas, D. D. (2007). Forster transfer recovery reveals that phospholamban exchanges slowly from pentamers but rapidly from the SERCA regulatory complex. Circ. Res. 101, 1123–1129. doi:10.1161/CIRCRESAHA.107.159947
Rog-Zielinska, E. A., Craig, M. A., Manning, J. R., Richardson, R. V., Gowans, G. J., Dunbar, D. R., et al. (2015). Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: A role for PGC-1α. Cell. Death Differ. 22, 1106–1116. doi:10.1038/cdd.2014.181
Rog-Zielinska, E. A., Richardson, R. V., Denvir, M. A., and Chapman, K. E. (2014). Glucocorticoids and foetal heart maturation; implications for prematurity and foetal programming. J. Mol. Endocrinol. 52, R125–R135. doi:10.1530/JME-13-0204
Rog-Zielinska, E. A., Thomson, A., Kenyon, C. J., Brownstein, D. G., Moran, C. M., Szumska, D., et al. (2013). Glucocorticoid receptor is required for foetal heart maturation. Hum. Mol. Genet. 22, 3269–3282. doi:10.1093/hmg/ddt182
Ronaldson-Bouchard, K., Ma, S. P., Yeager, K., Chen, T., Song, L., Sirabella, D., et al. (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243. doi:10.1038/s41586-018-0016-3
Ruan, J. L., Tulloch, N. L., Razumova, M. V., Saiget, M., Muskheli, V., Pabon, L., et al. (2016). Mechanical stress conditioning and electrical stimulation promote contractility and force maturation of induced pluripotent stem cell-derived human cardiac tissue. Circulation 134, 1557–1567. doi:10.1161/CIRCULATIONAHA.114.014998
Ruan, J. L., Tulloch, N. L., Saiget, M., Paige, S. L., Razumova, M. V., Regnier, M., et al. (2015). Mechanical stress promotes maturation of human myocardium from pluripotent stem cell-derived progenitors. Stem Cells 33, 2148–2157. doi:10.1002/stem.2036
Schaaf, T. M., Kleinboehl, E., Yuen, S. L., Roelike, L. N., Svensson, B., Thompson, A. R., et al. (2020). Live-cell cardiac-specific high-throughput screening platform for drug-like molecules that enhance Ca(2+) transport. Cells 9.
Schaaf, T. M., Li, A., Grant, B. D., Peterson, K., Yuen, S., Bawaskar, P., et al. (2018). Red-shifted FRET biosensors for high-throughput fluorescence lifetime screening. Biosens. (Basel) 8, 99. doi:10.3390/bios8040099
Schaaf, T. M., Peterson, K. C., Grant, B. D., Bawaskar, P., Yuen, S., Li, J., et al. (2017a). High-throughput spectral and lifetime-based FRET screening in living cells to identify small-molecule effectors of SERCA. SLAS Discov. 22, 262–273. doi:10.1177/1087057116680151
Schaaf, T. M., Peterson, K. C., Grant, B. D., Thomas, D. D., and Gillispie, G. D. (2017b). Spectral unmixing plate reader: High-throughput, high-precision FRET assays in living cells. SLAS Discov. 22, 250–261. doi:10.1177/1087057116679637
Schwinger, R. H., Bohm, M., Schmidt, U., Karczewski, P., Bavendiek, U., Flesch, M., et al. (1995). Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca(2+)-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation 92, 3220–3228. doi:10.1161/01.cir.92.11.3220
Setterberg, I. E., Le, C., Frisk, M., Li, J., and Louch, W. E. (2021). Corrigendum: The physiology and pathophysiology of T-tubules in the heart. Front. Physiol. 12, 790227. doi:10.3389/fphys.2021.790227
Shadrin, I. Y., Allen, B. W., Qian, Y., Jackman, C. P., Carlson, A. L., Juhas, M. E., et al. (2017). Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat. Commun. 8, 1825. doi:10.1038/s41467-017-01946-x
Shaikh, S. A., Sahoo, S. K., and Periasamy, M. (2016). Phospholamban and sarcolipin: Are they functionally redundant or distinct regulators of the sarco(Endo)Plasmic reticulum calcium ATPase? J. Mol. Cell. Cardiol. 91, 81–91. doi:10.1016/j.yjmcc.2015.12.030
Shattock, M. J., Ottolia, M., Bers, D. M., Blaustein, M. P., Boguslavskyi, A., Bossuyt, J., et al. (2015). Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 593, 1361–1382. doi:10.1113/jphysiol.2014.282319
Siu, C. W., Lee, Y. K., Ho, J. C., Lai, W. H., Chan, Y. C., Ng, K. M., et al. (2012). Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging (Albany NY) 4, 803–822. doi:10.18632/aging.100503
Smith, G. L., and Eisner, D. A. (2019). Calcium buffering in the heart in Health and disease. Circulation 139, 2358–2371. doi:10.1161/CIRCULATIONAHA.118.039329
Snir, M., Kehat, I., Gepstein, A., Coleman, R., Itskovitz-Eldor, J., Livne, E., et al. (2003). Assessment of the ultrastructural and proliferative properties of human embryonic stem cell-derived cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 285, H2355–H2363. doi:10.1152/ajpheart.00020.2003
Song, L., Awari, D. W., Han, E. Y., Uche-Anya, E., Park, S. H., Yabe, Y. A., et al. (2015). Dual optical recordings for action potentials and calcium handling in induced pluripotent stem cell models of cardiac arrhythmias using genetically encoded fluorescent indicators. Stem Cells Transl. Med. 4, 468–475. doi:10.5966/sctm.2014-0245
Streckfuss-Bomeke, K., Wolf, F., Azizian, A., Stauske, M., Tiburcy, M., Wagner, S., et al. (2013). Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur. Heart J. 34, 2618–2629. doi:10.1093/eurheartj/ehs203
Stroik, D. R., Ceholski, D. K., Bidwell, P. A., Mleczko, J., Thanel, P. F., Kamdar, F., et al. (2020). Viral expression of a SERCA2a-activating PLB mutant improves calcium cycling and synchronicity in dilated cardiomyopathic hiPSC-CMs. J. Mol. Cell. Cardiol. 138, 59–65. doi:10.1016/j.yjmcc.2019.11.147
Sun, N., Yazawa, M., Liu, J., Han, L., Sanchez-Freire, V., Abilez, O. J., et al. (2012). Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 4, 130ra47. doi:10.1126/scitranslmed.3003552
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126, 663–676. doi:10.1016/j.cell.2006.07.024
Toepfer, C. N., Garfinkel, A. C., Venturini, G., Wakimoto, H., Repetti, G., Alamo, L., et al. (2020). Myosin sequestration regulates sarcomere function, cardiomyocyte energetics, and metabolism, informing the pathogenesis of hypertrophic cardiomyopathy. Circulation 141, 828–842. doi:10.1161/CIRCULATIONAHA.119.042339
Toepfer, C. N., Sharma, A., Cicconet, M., Garfinkel, A. C., Mucke, M., Neyazi, M., et al. (2019). SarcTrack. Circ. Res. 124, 1172–1183. doi:10.1161/CIRCRESAHA.118.314505
Tweedie, D., Harding, S. E., and Macleod, K. T. (2000). Sarcoplasmic reticulum Ca content, sarcolemmal Ca influx and the Genesis of arrhythmias in isolated Guinea-pig cardiomyocytes. J. Mol. Cell. Cardiol. 32, 261–272. doi:10.1006/jmcc.1999.1070
Vaidyanathan, R., Markandeya, Y. S., Kamp, T. J., Makielski, J. C., January, C. T., and Eckhardt, L. L. (2016). Ik1-enhanced human-induced pluripotent stem cell-derived cardiomyocytes: An improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am. J. Physiol. Heart Circ. Physiol. 310, H1611–H1621. doi:10.1152/ajpheart.00481.2015
Veerman, C. C., Kosmidis, G., Mummery, C. L., Casini, S., Verkerk, A. O., and Bellin, M. (2015). Immaturity of human stem-cell-derived cardiomyocytes in culture: Fatal flaw or soluble problem? Stem Cells Dev. 24, 1035–1052. doi:10.1089/scd.2014.0533
Wei, H., Zhang, X. H., Clift, C., Yamaguchi, N., and Morad, M. (2018). CRISPR/Cas9 Gene editing of RyR2 in human stem cell-derived cardiomyocytes provides a novel approach in investigating dysfunctional Ca(2+) signaling. Cell. Calcium 73, 104–111. doi:10.1016/j.ceca.2018.04.009
Wei, W., Liu, Y., Zhang, Q., Wang, Y., Zhang, X., and Zhang, H. (2017). Danshen-enhanced cardioprotective effect of cardioplegia on ischemia reperfusion injury in a human-induced pluripotent stem cell-derived cardiomyocytes model. Artif. Organs 41, 452–460. doi:10.1111/aor.12801
Weinberger, F., Mannhardt, I., and Eschenhagen, T. (2017). Engineering cardiac muscle tissue: A maturating field of research. Circ. Res. 120, 1487–1500. doi:10.1161/CIRCRESAHA.117.310738
Williams, I. A., and Allen, D. G. (2007). Intracellular calcium handling in ventricular myocytes from mdx mice. Am. J. Physiol. Heart Circ. Physiol. 292, H846–H855. doi:10.1152/ajpheart.00688.2006
Word, T. A., Quick, A. P., Miyake, C. Y., Shak, M. K., Pan, X., Kim, J. J., et al. (2021). Efficacy of RyR2 inhibitor EL20 in induced pluripotent stem cell-derived cardiomyocytes from a patient with catecholaminergic polymorphic ventricular tachycardia. J. Cell. Mol. Med. 25, 6115–6124. doi:10.1111/jcmm.16521
Wu, H., Lee, J., Vincent, L. G., Wang, Q., Gu, M., Lan, F., et al. (2015). Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised beta-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell. Stem Cell. 17, 89–100. doi:10.1016/j.stem.2015.04.020
Wu, H., Yang, H., Rhee, J. W., Zhang, J. Z., Lam, C. K., Sallam, K., et al. (2019). Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur. Heart J. 40, 3685–3695. doi:10.1093/eurheartj/ehz326
Wu, J., Wang, X., Chung, Y. Y., Koh, C. H., Liu, Z., Guo, H., et al. (2017). L-type calcium channel inhibition contributes to the proarrhythmic effects of aconitine in human cardiomyocytes. PLoS One 12, e0168435. doi:10.1371/journal.pone.0168435
Yang, K. C., Breitbart, A., De Lange, W. J., Hofsteen, P., Futakuchi-Tsuchida, A., Xu, J., et al. (2018). Novel adult-onset systolic cardiomyopathy due to MYH7 E848G mutation in patient-derived induced pluripotent stem cells. JACC Basic Transl. Sci. 3, 728–740. doi:10.1016/j.jacbts.2018.08.008
Yang, L., Soonpaa, M. H., Adler, E. D., Roepke, T. K., Kattman, S. J., Kennedy, M., et al. (2008). Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 453, 524–528. doi:10.1038/nature06894
Yang, X., Pabon, L., and Murry, C. E. (2014a). Engineering adolescence: Maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 114, 511–523. doi:10.1161/CIRCRESAHA.114.300558
Yang, X., Rodriguez, M. L., Leonard, A., Sun, L., Fischer, K. A., Wang, Y., et al. (2019). Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell. Rep. 13, 657–668. doi:10.1016/j.stemcr.2019.08.013
Yang, X., Rodriguez, M., Pabon, L., Fischer, K. A., Reinecke, H., Regnier, M., et al. (2014b). Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell. Cardiol. 72, 296–304. doi:10.1016/j.yjmcc.2014.04.005
Yano, K., and Zarain-Herzberg, A. (1994). Sarcoplasmic reticulum calsequestrins: Structural and functional properties. Mol. Cell. Biochem. 135, 61–70. doi:10.1007/BF00925961
Yasuda, S., Townsend, D., Michele, D. E., Favre, E. G., Day, S. M., and Metzger, J. M. (2005). Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 436, 1025–1029. doi:10.1038/nature03844
Yazawa, M., Hsueh, B., Jia, X., Pasca, A. M., Bernstein, J. A., Hallmayer, J., et al. (2011). Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471, 230–234. doi:10.1038/nature09855
Zarain-Herzberg, A., Afzal, N., Elimban, V., and Dhalla, N. S. (1996). Decreased expression of cardiac sarcoplasmic reticulum Ca(2+)-pump ATPase in congestive heart failure due to myocardial infarction. Mol. Cell. Biochem. 163-164, 285–290. doi:10.1007/BF00408669
Zhang, D., Shadrin, I. Y., Lam, J., Xian, H. Q., Snodgrass, H. R., and Bursac, N. (2013a). Tissue-engineered cardiac patch for advanced functional maturation of human ESC-derived cardiomyocytes. Biomaterials 34, 5813–5820. doi:10.1016/j.biomaterials.2013.04.026
Zhang, X. H., Haviland, S., Wei, H., Saric, T., Fatima, A., Hescheler, J., et al. (2013b). Ca2+ signaling in human induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted subjects. Cell. Calcium 54, 57–70. doi:10.1016/j.ceca.2013.04.004
Zhang, X. H., and Morad, M. (2016). Calcium signaling in human stem cell-derived cardiomyocytes: Evidence from normal subjects and CPVT afflicted patients. Cell. Calcium 59, 98–107. doi:10.1016/j.ceca.2015.12.002
Zhang, X. H., Wei, H., Xia, Y., and Morad, M. (2021). Calcium signaling consequences of RyR2 mutations associated with CPVT1 introduced via CRISPR/Cas9 gene editing in human-induced pluripotent stem cell-derived cardiomyocytes: Comparison of RyR2-R420Q, F2483I, and Q4201R. Heart rhythm. 18, 250–260. doi:10.1016/j.hrthm.2020.09.007
Zhao, W., Waggoner, J. R., Zhang, Z. G., Lam, C. K., Han, P., Qian, J., et al. (2009). The anti-apoptotic protein HAX-1 is a regulator of cardiac function. Proc. Natl. Acad. Sci. U. S. A. 106, 20776–20781. doi:10.1073/pnas.0906998106
Zhou, W., Bos, J. M., Ye, D., Tester, D. J., Hrstka, S., Maleszewski, J. J., et al. (2019). Induced pluripotent stem cell-derived cardiomyocytes from a patient with MYL2-r58q-mediated apical hypertrophic cardiomyopathy show hypertrophy, myofibrillar disarray, and calcium perturbations. J. Cardiovasc Transl. Res. 12, 394–403. doi:10.1007/s12265-019-09873-6
Keywords: calcium, stem cells, cardiomyocytes, disease modeling, maturation, human induced pluripotent stem cells, genetic cardiovascular diseases
Citation: Ernst P, Bidwell PA, Dora M, Thomas DD and Kamdar F (2023) Cardiac calcium regulation in human induced pluripotent stem cell cardiomyocytes: Implications for disease modeling and maturation. Front. Cell Dev. Biol. 10:986107. doi: 10.3389/fcell.2022.986107
Received: 04 July 2022; Accepted: 23 December 2022;
Published: 18 January 2023.
Edited by:
Amaresh Ranjan, Midwestern University, United StatesReviewed by:
Lei Ye, University of Alabama at Birmingham, United StatesCopyright © 2023 Ernst, Bidwell, Dora, Thomas and Kamdar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Forum Kamdar, a2FtZDAwMDFAdW1uLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.