
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 29 August 2022
Sec. Cellular Biochemistry
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.966950
This article is part of the Research TopicNew Perspectives on Osteoclasts in Health and DiseaseView all 14 articles
 Maria Mangini1†
Maria Mangini1† Rosa D’Angelo1
Rosa D’Angelo1 Caterina Vinciguerra2Christine Payré3
Caterina Vinciguerra2Christine Payré3 Gérard Lambeau3
Gérard Lambeau3 Barbara Balestrieri4
Barbara Balestrieri4 Julia F. Charles5
Julia F. Charles5 Stefania Mariggiò1*†
Stefania Mariggiò1*†Increasing evidence points to the involvement of group IIA secreted phospholipase A2 (sPLA2-IIA) in pathologies characterized by abnormal osteoclast bone-resorption activity. Here, the role of this moonlighting protein has been deepened in the osteoclastogenesis process driven by the RANKL cytokine in RAW264.7 macrophages and bone-marrow derived precursor cells from BALB/cJ mice. Inhibitors with distinct selectivity toward sPLA2-IIA activities and recombinant sPLA2-IIA (wild-type or catalytically inactive forms, full-length or partial protein sequences) were instrumental to dissect out sPLA2-IIA function, in conjunction with reduction of sPLA2-IIA expression using small-interfering-RNAs and precursor cells from Pla2g2a knock-out mice. The reported data indicate sPLA2-IIA participation in murine osteoclast maturation, control of syncytium formation and resorbing activity, by mechanisms that may be both catalytically dependent and independent. Of note, these studies provide a more complete understanding of the still enigmatic osteoclast multinucleation process, a crucial step for bone-resorbing activity, uncovering the role of sPLA2-IIA interaction with a still unidentified receptor to regulate osteoclast fusion through p38 SAPK activation. This could pave the way for the design of specific inhibitors of sPLA2-IIA binding to interacting partners implicated in osteoclast syncytium formation.
Among the lipolytic enzymes, phospholipases A2 (PLA2) are acyl esterases, originally defined by their positional specificity to cleave the sn-2 ester bond of glycerophospholipids, to release fatty acids and lysophospholipids (Dennis, 1994). Currently, more than 50 mammalian proteins are classified within the PLA2 family, based on the homology with prototypical enzymes, even if they do not necessarily exert a PLA2 activity (Murakami et al., 2020). Based on their structural relationships, PLA2s are classified into secreted (Lambeau and Gelb, 2008), cytosolic (Kita et al., 2019), calcium-independent PLA2s (Ramanadham et al., 2015), and within other groups that include platelet-activating factor acetylhydrolase (Kono and Arai, 2019), lysosomal PLA2 (Shayman and Tesmer, 2019), PLA/acyltransferase (Hussain et al., 2017), α/β hydrolase (Thomas et al., 2014), and glycosylphosphatidylinositol-specific PLA2 subfamilies (Murakami et al., 2020).
In mammals, PLA2s participate in almost all biological processes, regulating pathophysiological functions related to membrane homeostasis, energy production and generation of body surface barriers, among others (Murakami, 2019). The peculiar tissue and cellular distribution of the different PLA2s (Murakami et al., 2017), in conjunction with their substrate specificities toward the fatty acyl and/or headgroup moieties of the membrane phospholipids (Astudillo et al., 2019), and the availability of functional cofactors or regulatory proteins (Murakami et al., 2020), ensures a variety of distinct functions among these enzymes (Murakami, 2019). The recent advent of lipidomics, combined with studies of gene-manipulated mice or human diseases caused by mutations of PLA2 enzymes, supported the definition of in-vivo functions of individual PLA2s (Murakami et al., 2010; Murakami et al., 2015; Yamamoto et al., 2017; Kita et al., 2019; Murakami et al., 2019; Murakami et al., 2020; Sun et al., 2021).
The subfamily of secreted (s)PLA2s comprises 12 mammalian proteins, namely IB, IIA, IIC (present in mice and rats, but pseudogene in humans; Tischfield et al., 1996), IID, IIE, IIF, III, V, X, XIIA, XIIB (catalytically inactive), and otoconin-95 (catalytically inactive structural protein in the inner ear; Lambeau and Gelb, 2008). Secreted PLA2s are typically characterized by a low molecular mass (13–18 kDa), a conserved three-dimensional helical structure with high disulfide bond content, and a calcium-dependent enzymatic activity through the conserved His-Asp catalytic dyad (Lambeau and Gelb, 2008; Murakami et al., 2010; Murakami et al., 2020). The most studied member, sPLA2-IIA, has a demonstrated primary function in host defense against invading pathogens (Doré and Boilard, 2019; Scott et al., 2021; Miki et al., 2022). As a secreted lipolytic enzyme, sPLA2-IIA shows bactericidal activity through hydrolysis of Gram-positive membranes (Weinrauch et al., 1998), and anti-parasitic function through hydrolysis of oxidized phospholipids in the plasma lipoproteins of malaria patients, thus promoting Plasmodium falciparum killing effect (Dacheux et al., 2019; Dacheux et al., 2021). Secreted PLA2-IIA is also referred as an inflammatory enzyme and amplifier of innate immune responses (Doré and Boilard, 2019; Doré et al., 2022). Indeed, by its hydrolysis of specific phospholipid substrates present in extracellular vesicles released by immune cells (Fourcade et al., 1995; Papadopoulos et al., 2020), or in extracellular mitochondria released by damaged organs or tissues, sPLA2-IIA generates lysophospholipids and non-esterified fatty acids, among which are eicosanoid precursors (Murakami et al., 2020; Kudo et al., 2022).
Secreted PLA2-IIA acts prevalently on extracellular lipids, and its membrane binding preference (phosphatidylglycerol > phosphatidylserine > phosphatidylcholine) suggests that it is almost inactive toward the intact plasma membrane of mammalian cells (Snitko et al., 1997; Koduri et al., 1998; Singer et al., 2002; Doré and Boilard, 2019). Loss of membrane asymmetry, due to glycerophospholipid scrambling, has been demonstrated during apoptosis (Atsumi et al., 1997), blood platelet activation (Suzuki et al., 2010), viral infection (Arganaraz et al., 2020; Snider et al., 2021), and the osteoclastogenesis of bone marrow-derived cells (Kang et al., 2020). This process can render the plasma membranes of mammalian cells susceptible to sPLA2-IIA attack (Olson et al., 2010).
The sPLA2-IIA cellular functions account for the enzymatic hydrolysis of glycerophospholipids, but can also occur in a catalysis-independent manner (Murakami et al., 2017; Doré and Boilard, 2019; Scott et al., 2021; Ivanusec et al., 2022). Several cellular and soluble receptors for sPLA2-IIA have been characterized until now, starting from the cell surface M-type receptor for secreted phospholipases (Cupillard et al., 1999; Lambeau and Lazdunski, 1999), and these may be involved in the activation of downstream cellular signaling pathways with hydrolytic activity-independent mechanisms (Saegusa et al., 2008; Ivanusec et al., 2022). On the other hand, the sPLA2-IIA protein is highly basic and can easily associate to the cell surface via heparin sulfated proteoglycans (Koduri et al., 1998; Murakami et al., 1999; Boilard et al., 2003; Boilard et al., 2006), and its interaction with the cellular membranes has been proposed to be sufficient to alter the lipid bilayer structure with consequent pathophysiological functions (Cirino et al., 1994; Lomonte et al., 1995).
Several studies have related sPLA2-IIA to bone metabolism, mainly under pathological conditions. Abnormal osteoclast activity is the primary cause of the pathophysiology of Paget’s disease of bone, for which pharmacological inhibition of osteoclast cell-to-cell fusion remains the best therapeutic strategy to mitigate bone loss and related fractures associated with this disorder (Coulthard et al., 2011; Ferraz-de-Souza and Correa, 2013). De Brum-Fernandes and collaborators showed that sPLA2-IIA is highly expressed in bones with high turnover, and in particular in Pagetic bone samples, and that sPLA2-IIA inhibition abrogates osteoclast differentiation and function (Allard-Chamard et al., 2014b). Other groups found that sPLA2-IIA inhibition prevents bone loss after ovariectomy in rats (Gregory et al., 2006), as well as bone erosion in rheumatoid arthritis (Bradley et al., 2005; Thwin et al., 2009). However, the precise role of sPLA2-IIA in osteoclast formation and activity remained to be determined, as well as the role of its metabolites.
Here we provide evidence of a multimodal regulation of the osteoclastogenesis and of mature osteoclast activity by sPLA2-IIA, using RAW264.7 murine macrophages as osteoclast precursors, or primary precursor cells from bone marrow of wild-type (wt) and Pla2g2a-knockout (ko) BALB/cJ mice. We also collected insights about the catalytically independent participation of sPLA2-IIA in the selective regulation of osteoclast cell-to-cell fusion, through the activation of p38 SAPK-dependent pathways.
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with high glucose and GlutaMAX, Eagle’s alpha-modified medium (α-MEM) supplemented with GlutaMAX, fetal bovine serum (cod. 10270), Hank’s balanced salt solution supplemented without (HBSS−) or with calcium and magnesium (HBSS++), and phosphate-buffered saline (PBS) were from Gibco (Life Technologies Italia, Monza, Italy). Ammonium chloride (NH4Cl), β-glycerophosphate, bovine serum albumin, Hoechst, l-glutamine, penicillin–streptomycin, pyrogallol, potassium bicarbonate (KHCO3), saponin, silver nitrate, sodium azide, sodium deoxycholate, sodium dodecyl sulphate, sodium fluoride, sodium orthovanadate, Triton X-100, Tween-20, Mowiol 4–88, bromoenol lactone (BEL, cod. B1552), cPLA2α inhibitor (cPLA2α–Inh., cod. 525143), SB203580 (cod. 559389), sPLA2-IIA Inhibitor I (Inhib-I, cod. 525145), KH064 (sPLA2 inhibitor, cod. S3319; or sPLA2-IIA inhibitor II, cod. 525146-M), LY311727 (cod. L6795), 2,4′-dibromoacetophenone (BPB, p-bromophenacyl bromide, cod. D38308), trifluoroacetic acid (TFA, cod. 91707) were from Sigma-Aldrich (St. Louis, MO, United States). Methyl arachidonyl fluorophosphonate (MAFP, cod. Ab141763) was from Abcam (Cambridge, UK). The purified recombinant mouse (mGIIA) and human sPLA2-IIA (hGIIA), the human sPLA2-IIA H48Q and mouse group V sPLA2 (mGV) (Edwards et al., 2002; Ghomashchi et al., 2017), as well as the control and anti‒mouse sPLA2-IIA IgG fractions were produced as described (Nevalainen et al., 2005; Eerola et al., 2006). All sPLA2 preparations had a purity higher than 99%, and were routinely checked for LPS contamination (Limulus amebocyte test, MP Biomedicals, Illkirch, France; or other sensitive assays), and discarded if the LPS concentration was above the detection limit of the assay (0.125 EU/mL). The synthetic cyclic peptides: FLSYK, WDIYR, LLKYK, with an amide bond between N- and C-terminus, were synthesized with a purity higher than 98.8% from Caslo ApS (Lyngby, Denmark). All other reagents were obtained at the highest purities available from Merck Life Science (Milano, Italy).
All the experiments with animals were performed at the Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States. The wild-type BALB/cJ mice were purchased from the Jackson Laboratory (Bar Harbor, ME, United States). The Pla2g2a-ko mice with a BALB/cJ genetic background were generated as previously described (Balestrieri et al., 2006), and backcrossed for 11 generations to a BALB/cJ background (Boilard et al., 2010). All the animal procedures were approved by the Institutional Animal Care and Use Committee for Brigham and Women’s Hospital, and conformed to relevant guidelines and laws.
The RAW264.7 murine monocytes/macrophages (cod. TIB-71) were from American Type Culture Collection (ATCC, Manassas, VA, United States), and cultured in DMEM with 10% heat-inactivated (30 min at 55°C) fetal bovine serum, 2 mM l-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin. RAW264.7 cells were used between passages 10 and 30.
For transient interference, RAW264.7 cells were plated at 300,000 cells/well in 12-well plate in 1 mL growth medium without antibiotics. After 24 and 48 h, cells were double transfected with 125 pmol/well of small-interfering (si)RNAs against sPLA2-IIA (si–sPLA2-IIA) (murine group II sPLA2 siRNA, sc-43818, from Santa Cruz Biotecnology, San Diego, CA, United States), or with non-targeting siRNAs (si–NT) (siGENOME non-targeting siRNA pool #2, cat. No. D-001206–14 from Dharmacon, Chicago, IL, United States), using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, United States), according to the manufacturer instructions. One day after the second interference, cells were detached (with 600 μM EDTA in PBS), and plated for the in-vitro osteoclastogenesis assays (as reported below), or for RNA extraction to determine the interference efficiency at 72 h of siRNA treatment.
RAW264.7 cells were plated in differentiation medium (α-MEM without nucleosides supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin) at a density of 1,250 cells/well in 96-well plates; at 5,000 cells/well on coverslips in 24-well plates for morphological analysis; or at 10,000 cells/well in 12-well plates for RNA extraction. Twenty-four hours later, and after every 48 h, the medium was replaced, and the cells were treated without or with 15–30 ng/mL ‘Receptor activator of nuclear factor kappa-Β ligand’ (RANKL) (cod. 310–01 from Peprotech, London, UK), in the absence or presence of different agents diluted in the differentiation medium. The RANKL concentration used was adjusted according to batch-to-batch variability of the cytokine. The range of RANKL concentration specified in several figure/table legends implies that all the different treatments shown were analyzed under the same differentiation conditions of control samples. When differentiation was complete, cells were harvested in lysis buffer for RNA extraction or processed for the different assays (as reported below).
The ex-vivo osteoclastogenesis assays were carried out at the Brigham and Women’s Hospital, according to published protocols (Charles et al., 2012; O'Brien et al., 2016). After euthanasia of mice, the femurs and tibias were removed and washed in 70% ethanol, and then with HBSS−. The bone ends were excised, and the bone marrow flushed out. The extracted bone marrow cells were collected by centrifugation. Subsequently, the red blood cells were lysed by 2-min treatment with lysis solution (0.15 M NH4Cl, 1 mM KHCO3, in water), and the reaction was stopped by addition of fetal bovine serum and subsequent centrifugation. Total bone marrow cells (BMC) were plated at a density of 50,000 cells/well in 96-well plates to be differentiated for 4 days in differentiation medium for primary cells (α-MEM with nucleosides supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin), containing 20 ng/mL M-CSF and 5 ng/mL RANKL, and then fixed for TRAP staining. In parallel, BMC were treated with 40 ng/mL M-CSF for 4 days in α-MEM with nucleosides supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin, to expand the osteoclast precursor cells (OPC). After expansion, adherent OPC were detached using the Gibco cell dissociation buffer, and were plated at 10,000 cells/well in 96-well plates for TRAP staining and degradation assays (on Osteo Assay Surface), or at 40,000 cells/well in 24-well plates for immunofluorescence analysis, or at 60,000 cells/well in 12-well plates for RNA extraction. These cells were treated with 20 ng/mL M-CSF without or with 2.5 ng/mL RANKL in differentiation medium for primary cells, and the medium was replenished every other day. Differentiation was usually complete after 3–6 days. At the end, the cells were fixed with 4% (w/v) paraformaldehyde for TRAP staining and immunofluorescence analysis, or harvested in lysis buffer for RNA extraction (as reported below).
Total RNA was extracted using RNeasy isolation kits, cDNAs were obtained using QuantiTect Reverse Transcription kits, and real-time qPCR was performed with QuantiTect SYBR Green PCR kits, all according to the manufacturer instructions (all the kits were from Qiagen, Hilden, Germany). The primers used for real-time qPCR (LightCycler 480 Instrument II; Roche, Indianapolis, IN, United States) are listed in Supplementary Table S1. β2-Microglobulin was used as housekeeping gene. The real-time qPCR program consisted of an initial step of 15 min at 95°C, and then 45 cycles, as follows: 94°C for 15 s, annealing temperature (see Supplementary Table S1) of each primer for 30 s, and 72°C for 30 s. The osteoclastogenesis marker mRNA levels, in cells subjected to pharmacological treatments, were quantified by real-time qPCR as fold of control levels, i.e., mRNA levels in cells treated with RANKL alone.
The sPLA2 enzymatic activity was determined using a commercially available colorimetric kit, sPLA2 Assay kit from the Cayman Chemical Company (cod. 765001, Ann Arbor, MI, United States). Briefly, cells were scraped in assay buffer (25 mM Tris-HCl pH 7.5, 10 mM CaCl2, 100 mM KCl, 0.3 mM Triton X-100) and lysed by sonication. The protein concentration was determined using the Bradford assay, and 250 μg total cell lysate was used to determine the catalytic activity, following the kit manufacturer instructions.
After treatment with or without RANKL, cells were rinsed with HBSS++ and fixed in 4% (w/v) paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, United States) for 10 min at room temperature, and then washed three times with HBSS++. Fixed cells were incubated for 1 h with blocking solution (50 mM NH4Cl, 0.5% bovine serum albumin, 0.1% saponin, 0.02% sodium azide, in PBS), with 33 nM Alexa546-labeled phalloidin (Invitrogen) for filamentous actin visualization, and 2 μg/mL Hoechst for nucleus staining. Finally, the cells were washed three times with PBS plus 0.02% Tween-20, and the coverslips were mounted with Mowiol 4–88 and examined by confocal microscopy (LSM 510; Zeiss, Oberkochen, Germany). For the evaluation of osteoclast syncytium formation, the nuclei of multinucleated cells were counted in a blinded manner using a 63× objective, moving across the coverslip in the vertical and horizontal directions. No evident differences in cell numbers, due to cell toxicity or changes in proliferation rates, were observed for the differentiated cells treated in the absence or presence of the different treatments. As Inhib-I was purchased as trifluoroacetate (TFA) salt, the absence of cell toxicity induced by TFA was verified treating RAW264.7 cells with 50 µM TFA for up to 120 h (with 3 additions and medium changes every 48 h) in differentiation medium (in absence of RANKL), and analyzing annexin-V staining (Annexin V-FITC kit, from Milteny Biotech GmbH, Germany), and propidium iodide incorporation by FACS analysis.
For TRAP staining, fixed cells were incubated with staining solution (1.5% sodium tartrate, 0.1% naphthol AS-MX phosphate, 1% N,N-dimethylformamide, 0.6% Fast Red LB Violet Salt in 0.1 M acetate buffer) for 15 min at 37°C. The reaction was stopped by washing cells three times with HBSS++. Osteoclasts were quantified by counting TRAP-positive cells with three or more nuclei.
To measure resorptive function, osteoclasts were grown on Osteo Assay Surface plates from Corning (New York, United States) for 7 days. When differentiation was complete, the cells were removed (using 10% bleach) and the wells were stained with 5% silver nitrate for 1 h. The staining was stopped by addition of 1% pyrogallol. Images were taken with a Nikon TMS-F inverted microscope (Nikon Instruments Inc., Melville, NY, United States) at a final magnification of ×5 (BMC and OCP), or with EVOS XL Core microscope (Thermo Fisher Scientific, Waltham, MA United States) at a final magnification of ×2 (RAW264.7 derived osteoclasts), and the total resorbed areas were quantified using ImageJ (NIH).
Cell lysates were obtained by scraping cells into phospho-lysis buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulphate, 40 mM β-glycerophosphate, 50 mM sodium fluoride, 200 µM sodium orthovanadate, plus phosphatase and protease inhibitors from Roche). Following gentle homogenization by 20 passages through a 26-gauge needle, the lysates were centrifuged at 10,000× g for 5 min at 4°C, and the supernatants were collected.
Protein lysates were subjected to SDS-PAGE, and after electrophoresis, the proteins were transferred to a nitrocellulose membrane (PerkinElmer Life Science, Boston, MA, United States). For immunoblotting, the membranes were blocked with 5% non-fat dry milk in TBS (10 mM Tris-HCl, pH 7.4, 150 mM NaCl) plus 0.1% Tween-20 (T-TBS) for 30 min at room temperature, and incubated with primary antibodies (as listed below) in T-TBS plus 3% bovine serum albumin for 2 h at room temperature, or overnight at 4°C. The membranes were washed twice in T-TBS for 7 min, and then incubated with secondary antibodies conjugated to horseradish peroxidase (1:5,000) (Calbiochem, San Diego, CA, United States) in T-TBS with 5% non-fat milk for 30 min at room temperature. The membranes were then washed twice with T-TBS, and once with TBS for 5 min, and the signals were detected by ECL (Amersham Pharmacia, Piscataway, NJ, United States). The rabbit anti-phospho AKT (Ser473; D9E, cod. 4060), anti phospho-p44/42 MAPK (Thr202/Tyr204; Erk1/2, cod. 9101), anti-phospho p38 MAPK (Thr180/Tyr182; 3D7, cod. 9215), anti-p38 MAPK (cod. 9212), anti-phospho MAPKAPK-2 (T334; 27B7, cod. 3007), anti-MAPKAPK-2 (D1E11, cod. 12155), all used at dilution 1:1,000 were from Cell Signaling Technology (Danvers, MA, United States). The rabbit anti-AKT (B-1, cod. sc-5298, diluted 1:1,000), and anti-ERK1 (K-23, cod. sc-94, diluted 1:5,000) were from Santa Cruz Biotechnology.
Statistical analysis was performed with the GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, United States). Comparisons between groups were performed using Student’s t-test and Analysis of Variance (ANOVA) with 95% confidence interval. p values < 0.05 were considered as statistically significant.
The osteoclastogenesis process was assessed with the RAW264.7 monocyte/macrophage cell line (Collin-Osdoby and Osdoby, 2012; Mosca et al., 2021) and with primary osteoclast precursors from bone marrow of BALB/cJ mice (Charles et al., 2012; O'Brien et al., 2016). ‘Receptor activator of nuclear factor kappa-Β ligand’ (RANKL) alone or in the presence of ‘macrophage colony-stimulating factor’ (M-CSF) was used to induce osteoclast differentiation of RAW264.7 cells or bone marrow-derived precursors, respectively.
Transcriptional analysis of most PLA2 family members, as assessed by real-time qPCR, indicated that the mRNA levels of sPLA2-IIA, -IIC, -IIE, and -V increased upon RANKL-induced differentiation of RAW264.7 cells, while that of sPLA2-IB, -III and of intracellular cPLA2α (PLA2-IVA) and iPLA2β (PLA2-VIA) decreased (Table 1). Other sPLA2s (-XIIA, -XIIB) and PLA2-XVI showed almost unchanged mRNA levels, while sPLA2-IID, -IIF, -X were not detectable (Table 1). These modulations were comparable to those observed within the osteoclastogenesis of bone marrow-derived M-CSF–expanded osteoclast precursors (OCP) from wt mice, with few exceptions: sPLA2-IIC was not detectable in the primary system, sPLA2-III was not modulated, and the iPLA2β levels increased during the osteoclastogenesis of wt OCP (Table 1; data from Pla2g2a-ko mice are commented in paragraph 3.3).
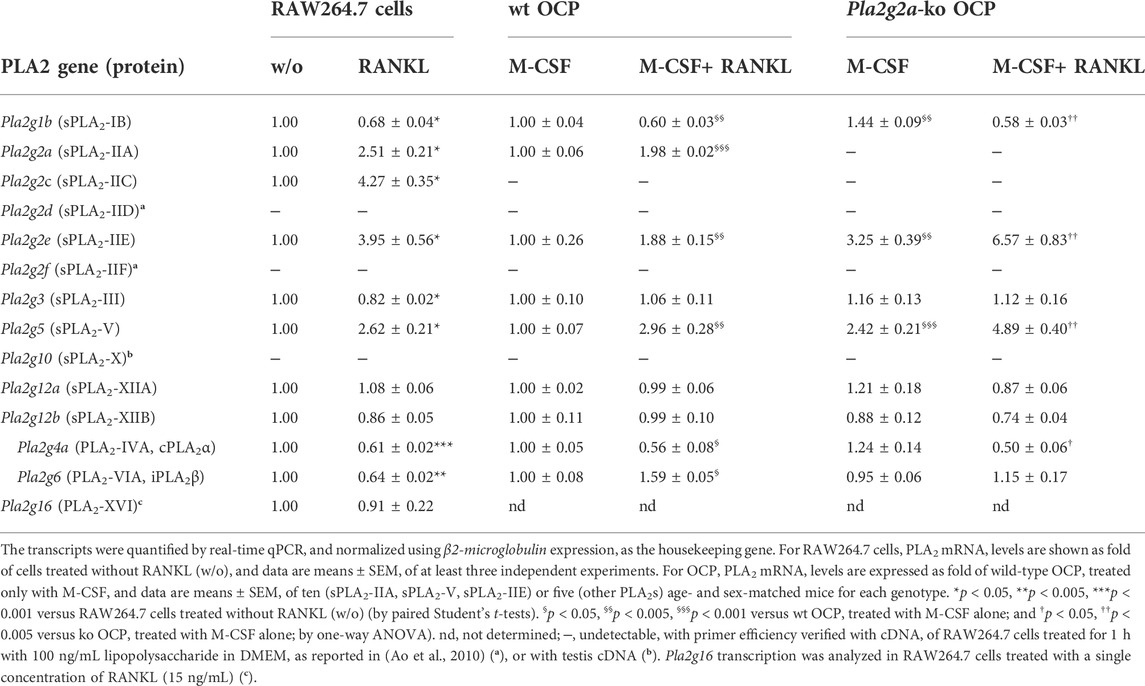
TABLE 1. Modulation of PLA2 mRNA levels during murine osteoclastogenesis. RAW264.7 cells were treated without (w/o) or with 15–30 ng/mL RANKL for 72 h, while M-CSF–expanded OCP from wild-type (wt) and Pla2g2a-ko mice were treated with 20 ng/mL M-CSF alone (M-CSF) or with 2.5 ng/mL RANKL (M-CSF+RANKL) for 3–6 days, before RNA extraction.
Phospholipase A2 activity was assayed in cell extracts from undifferentiated or RANKL-treated RAW264.7 macrophages with a commercially available kit (see Section 2 for details), based on the use of the 1,2-dithio analog of diheptanoyl phosphatidylcholine, which serves as a substrate for most PLA2s (Hendrickson et al., 1983; Reynolds et al., 1992). A 6-fold increase in PLA2 activity was measured in osteoclasts compared to undifferentiated cells (Table 2). This activity was not blocked by inhibitors of intracellular PLA2s, like the cPLA2α specific inhibitor (cPLA2α–Inh.; San Pietro et al., 2009; Seno et al., 2000); or the iPLA2β irreversible inhibitor (BEL; Ackermann et al., 1995; Zizza et al., 2012). Instead, various inhibitors of sPLA2s were efficient. Indeed, Inhib-I (Church et al., 2001; Zizza et al., 2012) reduced by 40%, while KH064 (Hansford et al., 2003; Kim et al., 2020) and the potent and covalent inhibitor of sPLA2-IIA and other sPLA2s (BPB; Kim et al., 2020; Marchi-Salvador et al., 2009) almost abolished the measured activity of RANKL lysates (Table 2). Moreover, a significant inhibition was seen with 10 μM LY311727, a quite specific inhibitor of sPLA2-IIA versus other mouse and human sPLA2s (Singer et al., 2002; Kim et al., 2020), which showed an apparent IC50 of 6.8 μM, and by a specific anti-mouse sPLA2-IIA IgG fraction (Eerola et al., 2006) (Table 2, Supplementary Figure S1).

TABLE 2. Secreted PLA2 activity measured in cell lysates. RAW264.7 cells were treated without (w/o) or with 15 ng/mL RANKL for 72 h, in the absence or presence of 10 nM BPB. Phospholipase A2 activity was analyzed in cell lysates (250 μg) in absence (‒) or presence of the indicated PLA2 inhibitors.
The monitored catalytic activity, together with the expression profile in the above settings, was suggestive of a sPLA2 role in RANKL-induced differentiation of murine precursor cells, reasonably that of sPLA2-IIA.
In addition to sPLA2s (Bradley et al., 2005; Gregory et al., 2006; Allard-Chamard et al., 2014b), cytosolic PLA2s were reported to participate in the osteoclastogenesis process (Bonventre, 2004; Kim et al., 2010; Allard-Chamard et al., 2014a). Therefore, inhibitors targeting the three major types of PLA2s were used to unveil their role (if any) in the osteoclastogenesis process of RAW264.7 macrophages. Real-time qPCR analysis of the osteoclastogenesis markers showed that the sPLA2 inhibitor (Inhib-I) was efficient to reduce the RANKL-induced transcription of all the analyzed markers (Nfatc1, Cathepsin-k, Mmp-9, Trap and Ctr) by at least 50%, and was the only treatment able to significantly impair the osteoclast syncytium formation (Supplementary Figures S2, S3). Specific cPLA2α inhibition, by cPLA2α–Inh., reduced by 40% only Nfatc1 mRNA levels, while iPLA2β blockage with BEL reduced Nfatc1 and Trap mRNA levels by 35% and 45%, respectively, in RANKL-differentiated cells (Supplementary Figure S2). Instead, the active site directed, irreversible inhibitor of both cPLA2α and iPLA2β (MAFP; Balsinde and Dennis, 1996; Lio et al., 1996), was not able to inhibit the RANKL-induced transcription of any marker nor the osteoclast fusion (Supplementary Figures S2, S3).
Although long-term treatment of cells with pharmacological inhibitors has to be taken with extreme caution, these data supported the hypothesis of sPLA2 involvement, seemingly sPLA2-IIA, in the regulation of the osteoclastogenesis of RAW264.7 macrophages. Instead, intracellular PLA2s did not seem to play a relevant role, as their inhibitors mainly reduced RANKL-induced transcription of Nfatc1, while the other marker mRNA levels and the osteoclast-syncytium formation were almost comparable to that of cells treated with RANKL alone. The absence of any effect by the less specific inhibitor MAFP could be consequent to the blockade of both positive and negative pathways regulated by distinct PLA2 isoforms.
The involvement of sPLA2-IIA was deepened using cells with reduced or abolished sPLA2-IIA expression. RAW264.7 macrophages were transfected with specific small interfering (si)RNAs that significantly decreased by 50% the sPLA2-IIA mRNA expression (Figure 1A). This treatment did not affect the mRNA levels of intracellular PLA2s, sPLA2-IIC and -IIE, but slightly reduced that of sPLA2-V (Supplementary Table S2). The RANKL-induced differentiation of siRNA transfected cells showed a significantly reduced transcription, by about 40%, of the main osteoclastogenesis markers (Nfatc1, Cathepsin-k, Mmp-9, Trap and Ctr), upon sPLA2-IIA interference (Figures 1B–F). The osteoclast-syncytium formation was also impaired by sPLA2-IIA interference, with lower numbers of osteoclasts with more than 10 nuclei, compared to cells interfered with the no-targeting siRNAs (Figures 1G,H).
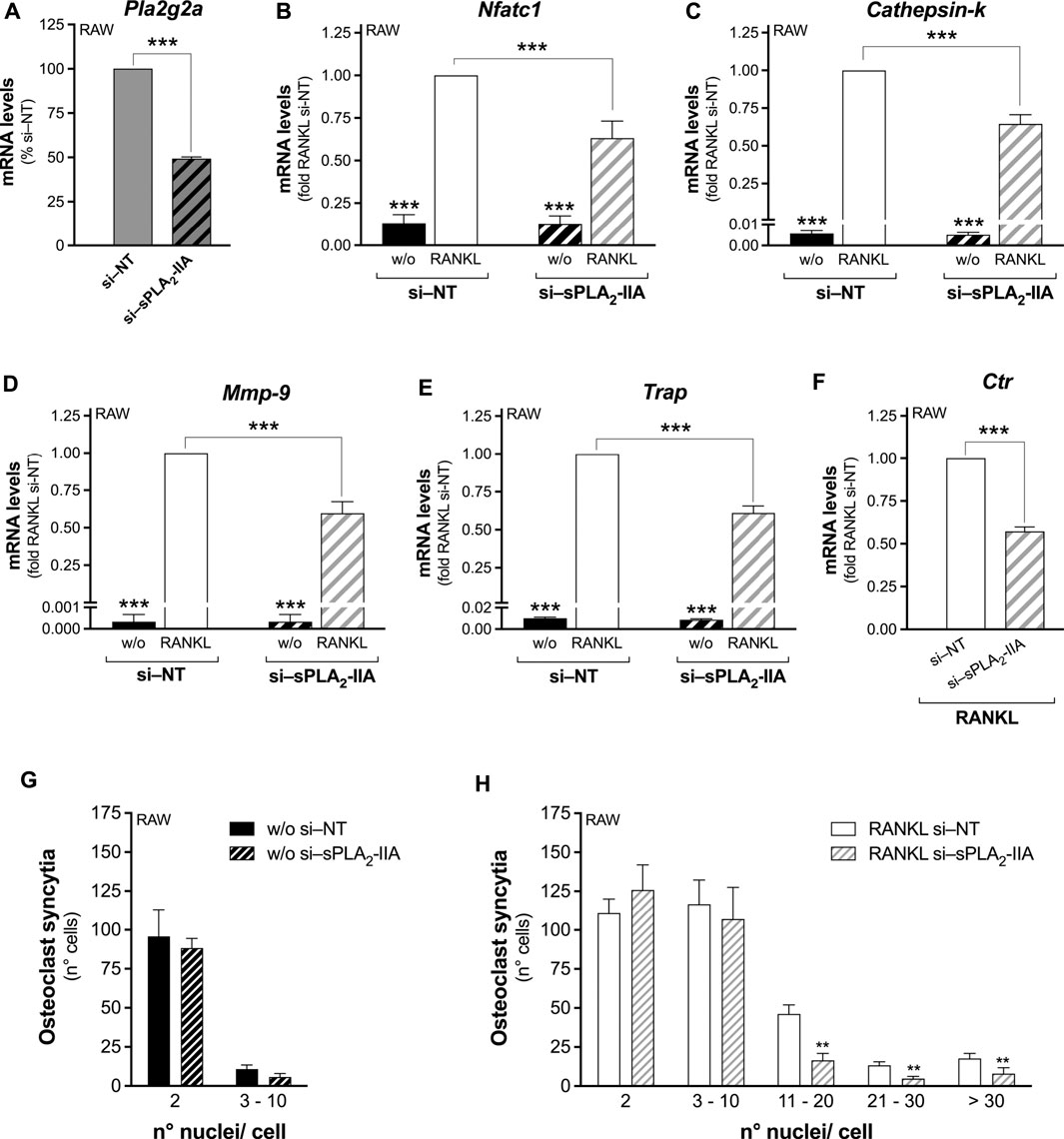
FIGURE 1. Secreted PLA2-IIA interference in RAW264.7 precursor cells inhibits RANKL-induced osteoclast differentiation. RAW264.7 cells were interfered for 72 h with no-targeting (si–NT) or sPLA2-IIA specific (si–sPLA2-IIA) siRNAs, and subsequently treated in the absence (w/o) or presence of 15–30 ng/mL RANKL for the following 48 h. (A) Pla2g2a mRNA levels of 72-h interfered RAW264.7 cells were quantified by real-time qPCR, and normalized using β2-microglobulin expression, as the housekeeping gene. Data are means ± SEM from six independent experiments. ***p < 0.005, paired Student’s t-test. (B–F) After 48 h of RANKL treatment, the differentiation markers were quantified by real-time qPCR, and normalized using β2-microglobulin expression, as the housekeeping gene. The mRNA levels of Ctr were undetectable in undifferentiated cells. Data are expressed as fold of RANKL-differentiated si–NT cells (RANKL si–NT), and are shown as means ± SEM of three independent experiments. ***p < 0.005 versus RANKL si–NT (one-way ANOVA or paired Student’s t-tests). (G,H) Osteoclast syncytium formation was determined as number of nuclei/cell, by fluorescence microscopy. Data are means ± SEM of three independent experiments performed in duplicates. **p < 0.01 versus correspondent RANKL si–NT (paired Student’s t-tests).
Furthermore, ex-vivo osteoclast differentiation was carried out with primary osteoclast precursors obtained from bone marrow of wt and Pla2g2a-ko BALB/cJ mice (Balestrieri et al., 2006; Boilard et al., 2010). The PLA2 expression profile in M-CSF–expanded OCP from Pla2g2a-ko mice was similar to the wt counterpart, with the exception of Pla2g1b, Pla2g2e and Pla2g5 that were expressed at higher levels, and the obvious lack of Pla2g2a (Table 1). Osteoclasts obtained from total bone marrow cells (BMC) or M-CSF–expanded OCP of Pla2g2a-ko mice, were less numerous (evaluated as TRAP-positive cells), and with lower degrading activity, compared to differentiated precursors from wt mice (Figure 2). In accordance with the RANKL-induced differentiation of sPLA2-IIA–interfered RAW264.7 macrophages, the differentiation of OCP from Pla2g2a-ko mice showed highly reduced marker transcription, and inhibition of osteoclast fusion, with lower numbers of osteoclasts containing more than 10 nuclei, compared to wt osteoclasts (Supplementary Figure S4).
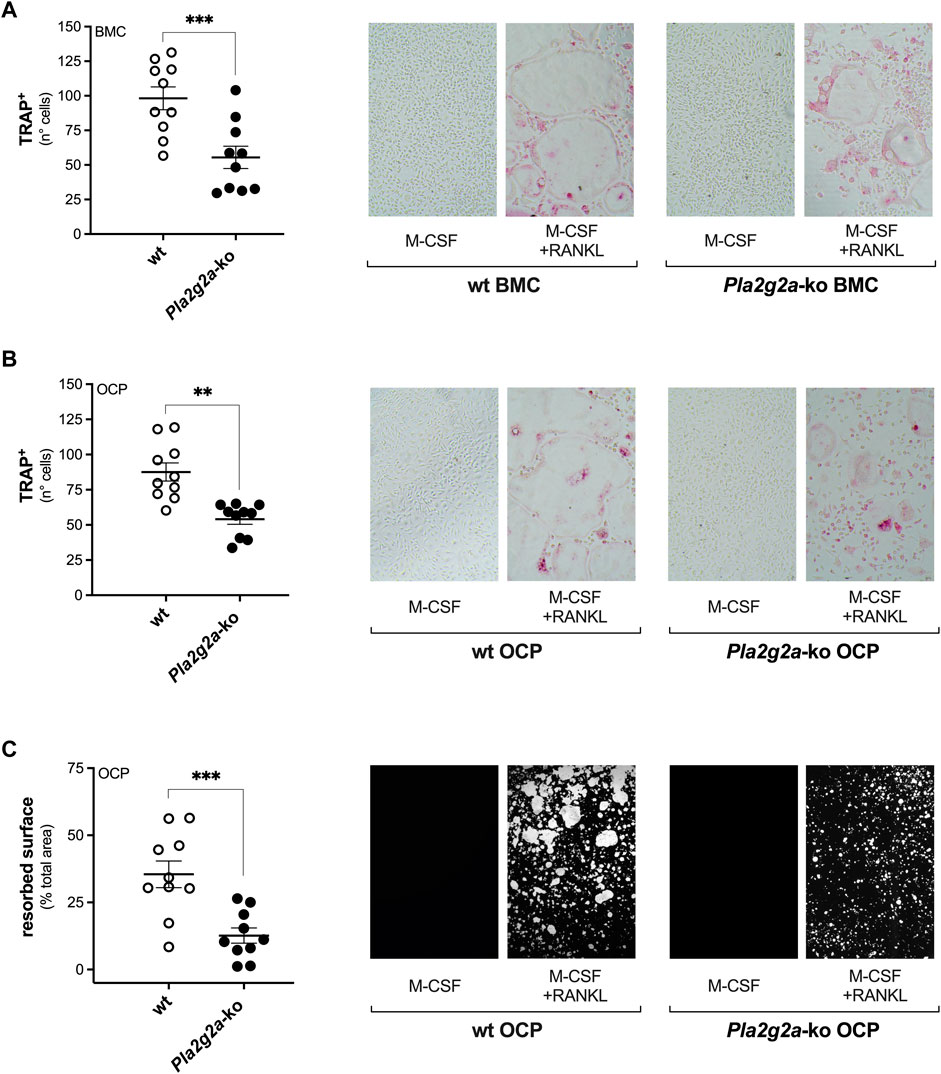
FIGURE 2. Osteoclast precursor cells derived from bone marrows of Pla2g2a-ko mice show reduced ex-vivo osteoclast differentiation and activity. (A) Freshly isolated total bone marrow cells (BMC) were differentiated with 20 ng/mL M-CSF and 5 ng/mL RANKL for 4 days, then fixed and stained for TRAP. (B,C) BMC were expanded with 40 ng/mL M-CSF for 4 days, and the obtained osteoclast precursor cells (OCP) were differentiated with 20 ng/mL M-CSF and 2.5 ng/mL RANKL for further 3–6 days. Then, OCP were fixed and stained for TRAP (B), or their degrading activity was evaluated on Osteo Assay Surface plates (C). Data are means ± SEM of ten age- and sex-matched mice for each genotype, analyzed in triplicates (A,B), or duplicates (C). On the right of each graph are representative images of TRAP staining (A,B), and of Osteo Assay Surface plates analyzed for the degrading activity (C), acquired with Nikon TMS-F inverted microscope, at a final magnification of ×5. **p < 0.01; ***p < 0.005 versus wt (unpaired Student’s t-tests).
Altogether, both pharmacological and molecular tools indicated the prevalent role of sPLA2-IIA in RANKL-induced differentiation of RAW264.7 macrophages. The impaired maturation, fusion and activity of osteoclasts derived from Pla2g2a-ko bone-marrow precursors paralleled the data obtained with the sPLA2-IIA–interfered RAW264.7 cell line, despite the increased transcription of Pla2g2e and Pla2g5 in the Pla2g2a-ko OCP.
The crystal structures of sPLA2-IIA in complex with KH064 and LY311727 showed that these competitive inhibitors bind to the active site of sPLA2-IIA and also perturbate the binding surface at the entrance of the cavity (Lee et al., 2013). Therefore, these small molecules are able to block not only the sPLA2-IIA enzymatic activity, but also its possible interaction with binding partners. This was shown to be the case for methyl-indoxam, another active site sPLA2 inhibitor, which perturbates the interaction of various mouse sPLA2s to PLA2R1, shifting the affinity for this latter by up to 300-fold (Boilard et al., 2006).
Differentiation assays carried out in the presence of the small molecule KH064 showed that it acted similarly to the peptide Inhib-I, impairing RANKL-induced osteoclastogenesis. Indeed, both inhibitors significantly reduced the RANKL-stimulated transcription of all five osteoclastogenesis markers monitored, either with RAW264.7-macrophage precursors (Figures 3A–E), and with wt OCP (Supplementary Figures S5A–E). In addition, both inhibitors impaired osteoclast cell-to-cell fusion, as indicated by the decreased number of osteoclasts with more than 10 nuclei among RANKL-differentiated RAW264.7 cells (Figure 3F), and with more than 3 nuclei with wt OPC (Supplementary Figures S5F,G). As further confirmation, a significantly reduced formation of TRAP-positive cells from wt OCP was obtained in the presence of both KH064 and Inhib-I (Supplementary Figure S6). Strikingly, all these inhibitory effects were not seen differentiating Pla2g2a-ko OCP (Supplementary Figures S5, S6).
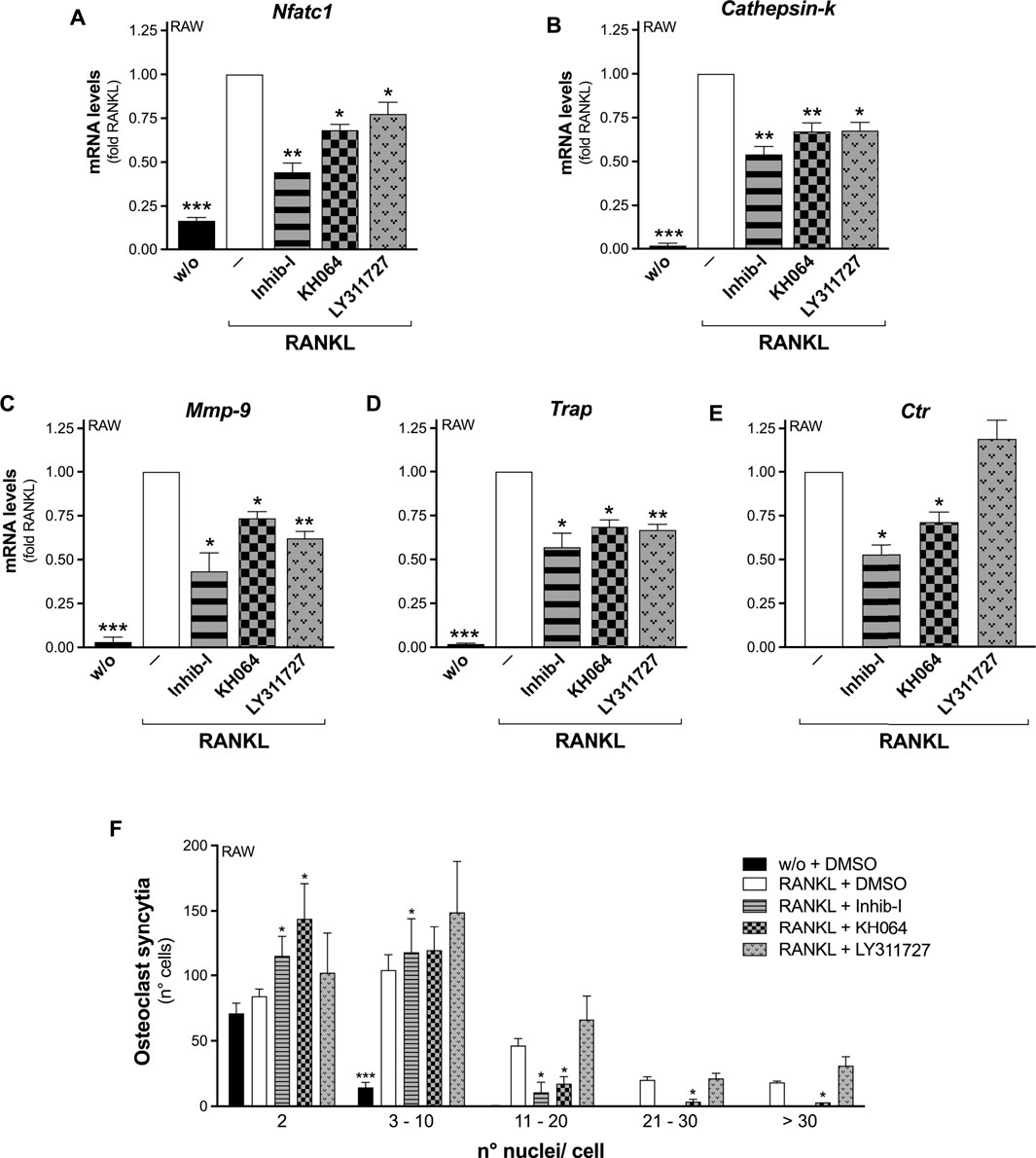
FIGURE 3. Bifunctional inhibitors of sPLA2-IIA impair RANKL-induced osteoclastogenesis of RAW264.7 macrophages. (A–E) RAW264.7 cells were treated without (w/o) or with 30 ng/mL RANKL for 72 h, in presence of the indicated sPLA2-IIA inhibitors (20 μM Inhib-I, 40 μM KH064, 30 μM LY311727) or with DMSO as carrier (‒). The differentiation markers were quantified by real-time qPCR, and normalized using β2-microglobulin expression, as the housekeeping gene. Data are expressed as fold of RANKL, and are means ± SEM of three independent experiments. (F) RAW264.7 cells were treated without (w/o) or with 15–30 ng/mL RANKL for 72–96 h, in presence of the above indicated sPLA2-IIA inhibitors or with DMSO as carrier. Osteoclast syncytium formation was determined as number of nuclei/cell, by fluorescence microscopy. Data are means ± SE of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 versus correspondent RANKL (paired Student’s t-tests).
The LY311727 inhibitor was also capable to inhibit the osteoclastogenesis of RAW264.7 cells, but less potently, as it reduced RANKL-triggered Nfatc1, Cathepsin-k, Mmp-9, and Trap transcription, but not that of Ctr, nor the osteoclast-syncytium formation (Figure 3).
The results obtained with two structurally different inhibitors of sPLA2-IIA, namely Inhib-I and KH064, further support the involvement of this protein in the different steps of the murine osteoclastogenesis process, from the transcriptional remodeling of key genes to the osteoclast fusion. The absence of any effect with Pla2g2a-ko OPC confirmed the specificity of these inhibitors toward sPLA2-IIA, at least under our assay conditions.
The results shown so far provide the rationale to analyze in more detail the mechanism by which sPLA2-IIA regulates the osteoclastogenesis process. To this end, we took advantage of purified recombinant sPLA2-IIA of murine and human origin, including a H48Q catalytic site mutant which has less than 5% of wt activity (Edwards et al., 2002). The active site and catalytic mechanism of these two sPLA2-IIA orthologs are conserved and both enzymes share the same interfacial kinetic properties and substrate specificities (Cupillard et al., 1999; Valentin et al., 1999; Valentin et al., 2000; Singer et al., 2002). However, they have a rather low identity at the protein level (69%), especially on the protein surface, with a number of different amino acids that likely explain their different binding properties to proteins including PLA2R1 and Factor Xa (Cupillard et al., 1999; Mounier et al., 2000). Experimental evidence indicated that the 70–74 region of human sPLA2-IIA is relevant for sPLA2-IIA binding to membranes or putative signaling partners (Tseng et al., 1996). In addition, peptides derived from this sPLA2-IIA primary sequence were reported to competitively antagonize the sPLA2-IIA binding activity, and consequent sPLA2-IIA cellular functions, in a species-specific mode (Church et al., 2001).
The addition of recombinant mouse sPLA2-IIA (mGIIA) significantly reinforced the RANKL-induced transcription of all the differentiation markers analyzed, by more than 1.5-fold, during the differentiation of RAW264.7 macrophages (Figure 4). Even if with lower potency, the human enzyme (hGIIA) also increased the transcription of Nfatc1, Cathepsin-k, Trap and Ctr. Conversely, the catalytically inactive mutant H48Q (hGIIA H48Q) was without any effect (Figure 4). In parallel, the mouse sPLA2-V (mGV) was found to stimulate only the transcription of Cathepsin-k and Trap, but at a higher concentration of 100 nM (Figure 4). The Mmp-9 transcription was stimulated only by mGIIA, and not by the human enzymes (active and mutated), nor by mGV (Figure 4). Finally, the RANKL-induced osteoclast syncytium formation was modulated only by mGIIA, with a significantly increased number of osteoclasts with more than 10 nuclei (Figure 5).
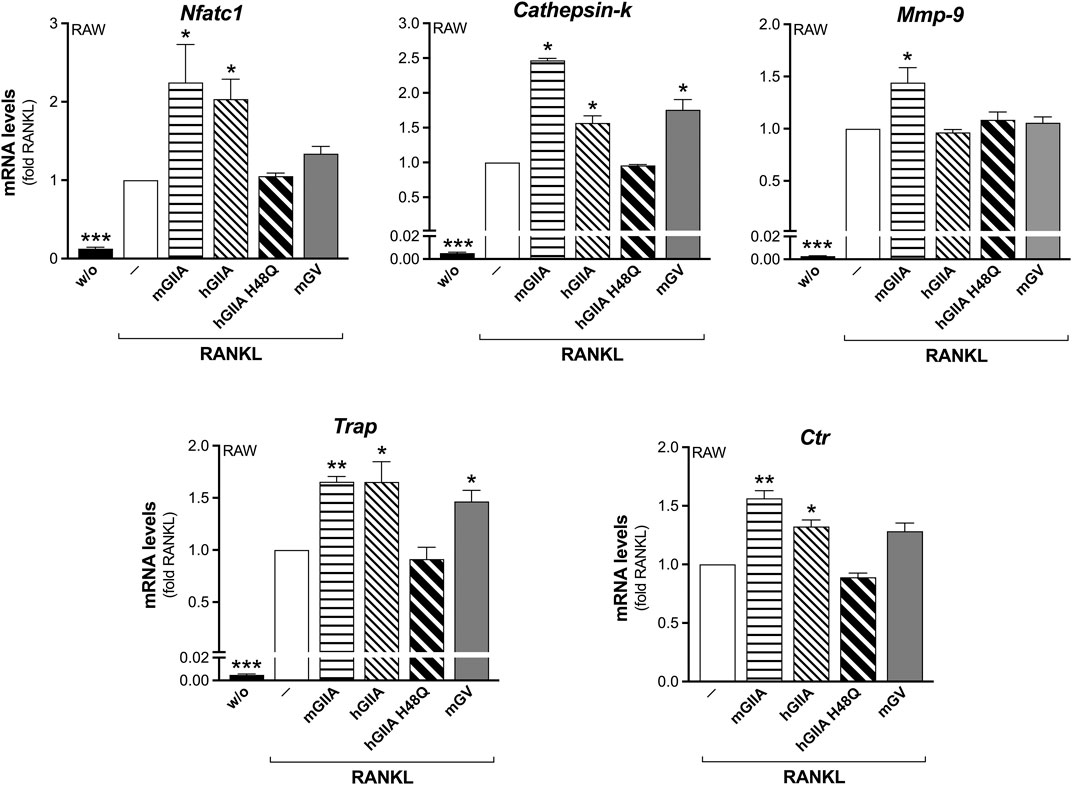
FIGURE 4. Murine recombinant sPLA2-IIA stimulates RANKL-induced osteoclastogenesis of RAW264.7 macrophages. RAW264.7 cells were treated without (w/o) or with 15 ng/mL RANKL for 72 h, in presence of the indicated recombinant proteins: 30 nM murine sPLA2-IIA (mGIIA), 30 nM human sPLA2-IIA (hGIIA), 30 nM human sPLA2-IIA H48Q (hGIIA H48Q), 100 nM murine sPLA2-V (mGV); or with PBS (‒) as carrier. The differentiation markers were quantified by real-time qPCR, and normalized using β2-microglobulin expression, as the housekeeping gene. Data are expressed as fold of RANKL, and are means ± SEM of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 versus RANKL (paired Student’s t-tests).
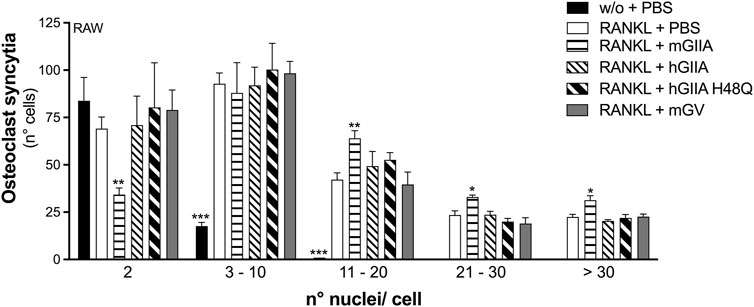
FIGURE 5. Murine recombinant sPLA2-IIA stimulates RANKL-induced osteoclast fusion of RAW264.7 macrophages. RAW264.7 cells were treated without (w/o) or with 15 ng/mL RANKL for 72 h, in presence of the indicated recombinant proteins: 30 nM murine sPLA2-IIA (mGIIA), 30 nM human sPLA2-IIA (hGIIA), 30 nM human sPLA2-IIA H48Q (hGIIA H48Q), 100 nM murine sPLA2-V (mGV); or with PBS as carrier. Osteoclast syncytium formation was determined as number of nuclei/cell, by fluorescence microscopy. Data are means ± SE of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 versus correspondent RANKL (paired Student’s t-tests).
Three cyclic pentapeptides were synthesized based on the FLSYK sequence of human sPLA2-IIA (70FLSYK74; Church et al., 2001; Tseng et al., 1996), and the corresponding one in C. durissus terrificus venom sPLA2-IIA (70WDIYR74; Church et al., 2001; Tseng et al., 1996), and in mouse sPLA2-IIA (84LLKYK89) (Figure 6A). The LLKYK murine peptide, but not that from other species, was able to reduce the RANKL-induced Mmp-9 transcription and cell-to-cell fusion, leaving unaffected the Nfatc1 and Ctr transcription (Figures 6B–E). As the fusion step is crucial for the osteoclast activity, the effect of these peptides on the degrading activity was analyzed. The murine cyclic pentapeptide, LLKYK, was able to reduce by more than 50% the resorbing activity of mature osteoclasts on hydroxyapatite-coated plates, while the human FLSYK was ineffective (Figure 7).
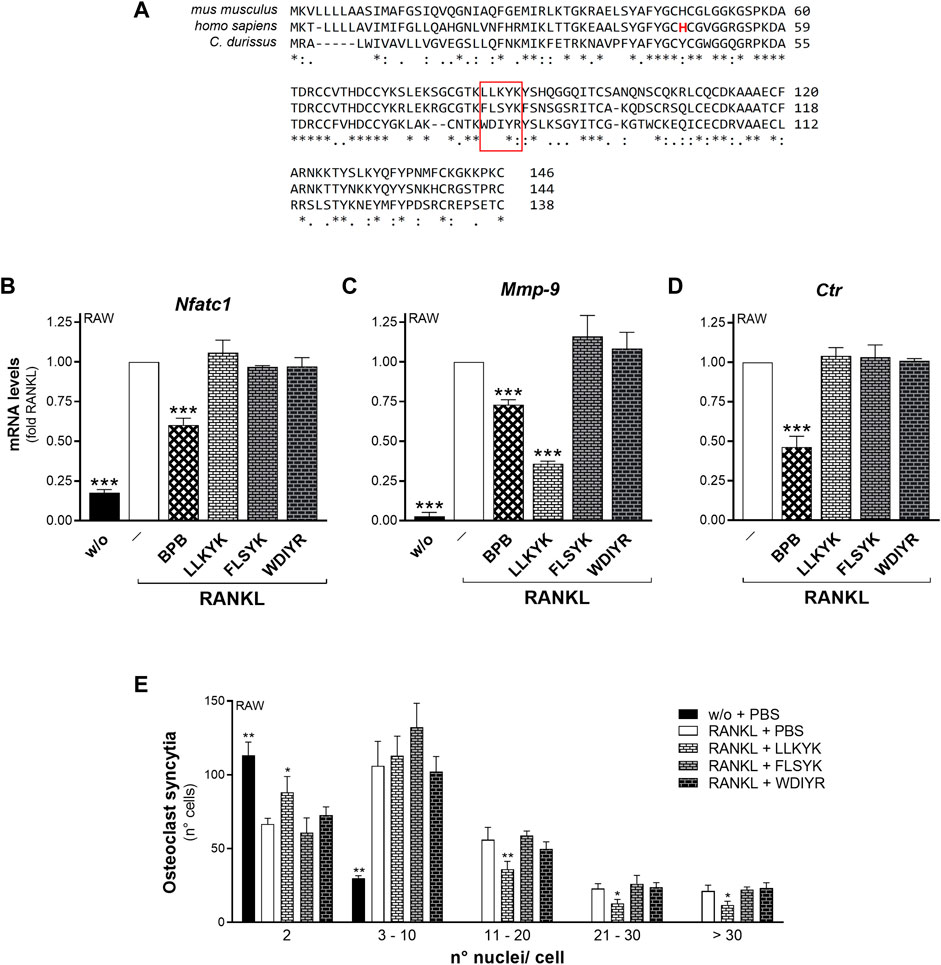
FIGURE 6. The LLKYK cyclic peptide inhibits RANKL-induced Mmp-9 transcription and osteoclast fusion of RAW264.7 macrophages. (A) Alignment of sPLA2-IIA sequences from mus musculus (UniProtKB - P31482), homo sapiens (UniProtKB - P14555), and C. durissus (UniProtKB - P24027). Consensus symbols indicate residue conservation, as calculated with the Clustal Omega program. The pentapeptide sequences are boxed in red, and the catalytic histidine residue in the human sPLA2-IIA sequence is shown in red-letter code. (B–D) RAW264.7 cells were treated without (w/o) or with 15–30 ng/mL RANKL for 72 h, in presence of the carriers (‒), or 10 nM BPB, or 250 μM of the indicated peptides (LLKYK from the murine sPLA2-IIA sequence, FLSYK from that of human sPLA2-IIA, WDIYR from that of C. durissus). The differentiation markers were quantified by real-time qPCR analysis, and normalized using β2-microglobulin expression, as the housekeeping gene. Data are expressed as fold of RANKL, and are means ± SEM of at least three independent experiments. (E) RAW264.7 cells were treated without (w/o) or with of 15 ng/mL RANKL for 72 h, in presence of the indicated peptides (250 μM), or with PBS as carrier. Osteoclast syncytium formation was determined as number of nuclei/cell, by fluorescence microscopy. Data are means ± SE of six independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 versus correspondent RANKL (paired Student’s t-tests).
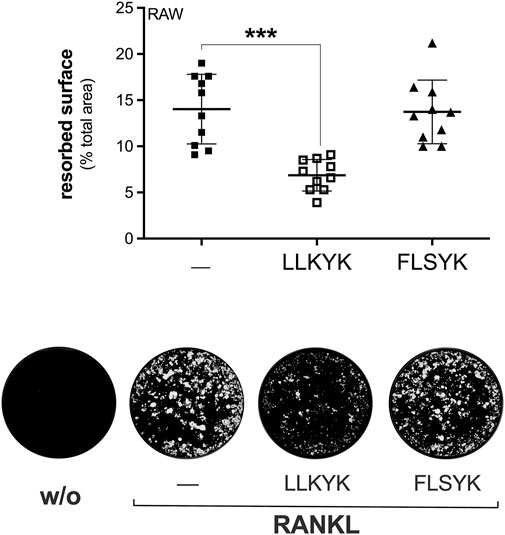
FIGURE 7. The LLKYK cyclic peptide inhibits RANKL-induced osteoclast resorbing activity of RAW264.7 macrophages. RAW264.7 cells plated on Osteo Assay Surface plates were treated without (w/o) or with 15 ng/mL RANKL for 7 days, in presence of PBS (‒) or 250 μM of the indicated cyclic pentapeptides (LLKYK from murine sPLA2-IIA sequence, and FLSYK from that of human sPLA2-IIA). Below are shown representative images of the degrading activity, acquired with the EVOS XL Core microscope at a final magnification of ×2. Data are means ± SEM of three independent experiments performed in triplicates. ***p < 0.005, paired Student’s t-test.
In addition to the above experiments with the H48Q mutant, the role of mouse sPLA2-IIA enzymatic activity in osteoclastogenesis was confirmed by the use of BPB. This alkylating agent covalently binds to the histidine residue of the active site, but preserves the sPLA2-IIA surface properties and sPLA2-IIA binding properties to cells (Lee et al., 2013). Figure 6 shows that BPB treatment of RAW264.7 macrophages led to a clear inhibition of transcription for various RANKL-induced markers of the osteoclastogenesis, with reduction of Nfatc1, Mmp-9 and Ctr levels by 40%, 30%, and 60%, respectively. In addition, Cathepsin-k and Trap transcription were significantly reduced by about 50% in RANKL-treated cells, and the osteoclast fusion process was not inhibited at all (Supplementary Figure S7).
Altogether, these results were indicative of the sole requirement of sPLA2-IIA catalytic activity for RANKL-induced marker transcription, with the exception of Mmp-9. Instead, sPLA2-IIA regulation of the osteoclast fusion occurred independently of its enzymatic activity, and was specific for mouse recombinant sPLA2-IIA.
The syncytium formation constitutes a crucial step for the osteoclast bone resorbing activity (Soe, 2020), and the unveiled catalysis-independent regulation of this process by sPLA2-IIA pushed toward the definition of the signaling pathways involved. Evaluation of RANKL downstream effectors in RAW264.7 macrophages, by Western blot analysis, displayed a significant activation of p38 SAPK and of one of its downstream substrates (MAPKAPK-2), while the phosphorylation levels of ERK1/2 MAPKs and AKT did not significantly differed from that of undifferentiated cells (Figure 8). Cell treatment with the different sPLA2-IIA inhibitors revealed that Inhib-I and KH064 counteracted the RANKL-induced phosphorylation of p38 SAPK and of MAPKAPK-2, while BPB and LY311727 were without effect (Figure 8).
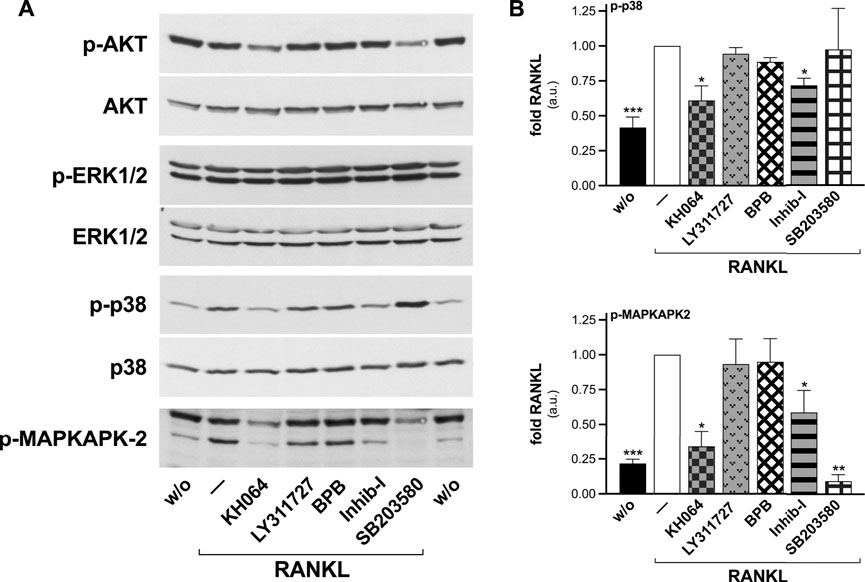
FIGURE 8. KH064 and Inhib-I counteract the RANKL-induced activation of p38 SAPK in differentiated RAW264.7 macrophages. RAW264.7 cells were treated without (w/o) or with 15–30 ng/mL RANKL for 72–96 h, in presence of the indicated sPLA2-IIA inhibitors (40 μM KH064, 30 μM LY311727, 10 nM BPB, 20 μM Inhib-I, 25 μM SB203580), or with DMSO as carrier (‒). (A) Western blotting of phosphorylated (p-AKT, p-ERK1/2, p-p38, p-MAPKAPK-2), and total AKT, ERK1/2, and p38 are shown, from an experiment representative of at least three independents. (B) Densitometric analysis by arbitrary units (a.u.) of p38 (top) and 50-kDa MAPKAPK-2 (bottom) phosphorylation levels, normalized for the correspondent protein levels. Data are expressed as fold of cells treated with RANKL, and are means ± SE of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 versus RANKL (paired Student’s t-tests).
The osteoclastogenesis of RAW264.7 macrophages in the presence of p38 inhibition by SB203580, showed a foreseen blunted RANKL-induced phosphorylation of MAPKAPK-2 (Figure 8), but also a selectively impaired osteoclast fusion (Figure 9). Consistently, ex-vivo differentiation of M-CSF–expanded OPC, both wt and Pla2g2a-ko, upon SB203580 treatment produced significantly less TRAP-positive cells, maintaining unaffected the rate of the osteoclastogenesis marker transcription (Supplementary Figure S8).
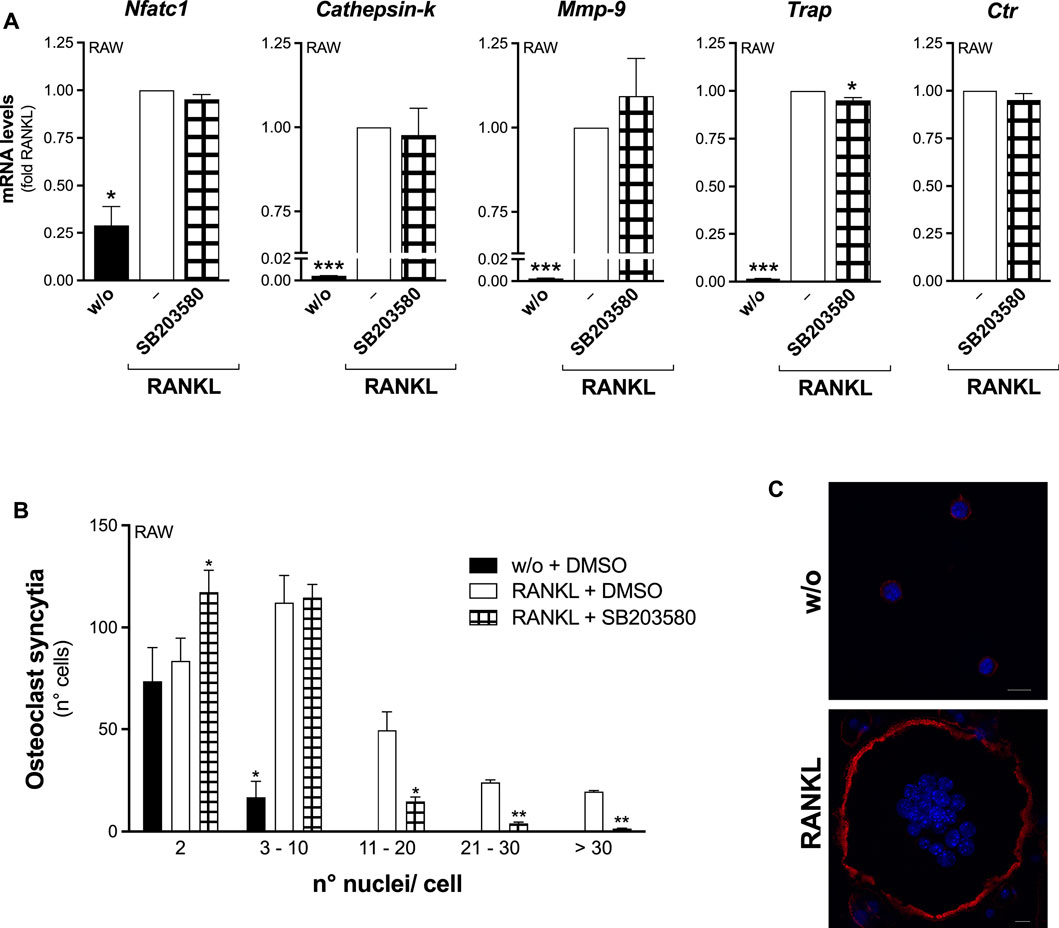
FIGURE 9. The p38 SAPK inhibitor selectively reduces RANKL-induced osteoclast fusion of RAW264.7 macrophages. (A) RAW264.7 cells were treated without (w/o) or with 15–30 ng/mL RANKL for 72 h, in presence of 25 μM SB203580 or with DMSO as carrier (‒). The differentiation markers were quantified by real-time qPCR, and normalized using β2-microglobulin expression, as the housekeeping gene. Data are expressed as fold of RANKL, and are means ± SEM of three independent experiments. (B) Osteoclast syncytium formation was determined as number of nuclei/cell, by fluorescence microscopy. Data are means ± SE of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 versus correspondent RANKL (paired Student’s t-tests). (C) Representative confocal images of RAW264.7 cells treated without (w/o) or with RANKL for 72 h, fixed and stained for confocal imaging. Phalloidin staining allows visualization of filamentous actin (red), while Hoechst staining reveals cell nuclei (blue, see Section 2 for details). Scale bars, 10 µm.
These results suggested that p38 SAPK, a kinase part of the RANKL-triggered signaling (Park et al., 2017), mediated the osteoclast fusion but not the osteoclastogenesis marker transcription. In addition, p38 SAPK seemed to be downstream of the sPLA2-IIA catalysis-independent modulation of the osteoclastogenesis, selectively required for a fully functional osteoclast multinucleation.
The findings here summarized prove the sPLA2-IIA multimodal regulation of RANKL-induced differentiation of murine osteoclast precursor cells. Whereas the osteoclastogenesis marker transcription requires sPLA2 hydrolytic activity, sPLA2-IIA participation in the osteoclast fusion and degrading activity occurs in a catalysis-independent manner and leads to the increased activation of p38 SAPK, presumably through the interaction with a still unidentified receptor.
This experimental evidence arises from in-vitro osteoclastogenesis assays performed with the well-validated and simplified model, based on the use of RAW264.7 macrophages as osteoclast precursor cells (Cuetara et al., 2006). The results obtained with this Abelson leukemia virus-transformed cell line, established from a BALB/c mouse strain (BAB/14; Raschke et al., 1978), were validated in ex-vivo osteoclastogenesis assays with primary precursor cells from BALB/cJ mice. Within these model systems, the transcriptional remodeling induced by RANKL results in increased sPLA2-IIA, -IIE, and -V mRNA levels, suggesting their involvement in the differentiation process or in mature osteoclast activity. These sPLA2s are all located in the distal part of the mouse chromosome 4 and lie within the so-called sPLA2 gene cluster (Valentin et al., 2000). The RANKL-triggered transcription of sPLA2-IIA could be consequent to an increased NF-κB activity, as this transcription factor is downstream the RANKL-activated signaling (Park et al., 2017), and has been reported to mediate the sPLA2-IIA transcription under inflammatory conditions. Indeed, cytokines such as TNF-α and IL-1β that are upregulated during inflammatory processes were shown to promote sPLA2-IIA expression in different cellular lineages, through the involvement of the transcription factor NF-κB (Vadas et al., 1991; Couturier et al., 1999; Beck et al., 2003). Though unable to detect sPLA2-IIA protein expression, with RAW264.7-cell lysates we succeeded in monitoring a PLA2 enzymatic activity that increased upon RANKL-induced differentiation, and that was impaired by inhibitors and a blocking antibody specific for sPLA2-IIA. This observation, together with the reported evidence of sPLA2-IIA involvement in pathologies of the bone system, led us to investigate the role of this enzyme in the osteoclastogenesis process.
The unique genetic background of inbred BALB/cJ mice allowed the production of a Pla2g2a knock out from 129Sv mice (Balestrieri et al., 2006; Boilard et al., 2010). Indeed, a number of mouse strains generally used for research purposes (C57BL/6, 129Sv, A/J, C58/J, P/J, and B10. RIII) are naturally knock out for Pla2g2a, due to a frameshift mutation in the gene (Kennedy et al., 1995; MacPhee et al., 1995). Here, both Pla2g2a-ko primary cells and sPLA2-IIA–interfered RAW264.7 macrophages were instrumental to unveil sPLA2-IIA role in both osteoclast maturation and fusion. In addition, the use of inhibitors selective for the different sPLA2-IIA activities dissected the underlying mechanisms of action.
The first open question that remains to be answered is how sPLA2-IIA could regulate osteoclast maturation via its enzymatic activity in a cell-autonomous context. The preferential cellular substrates for this enzyme are prevalently distributed in the inner (cytosolic) leaflet of normal plasma membranes (as phosphatidylserine, phosphatidylethanolamine or phosphatidylglycerol), and are thus not accessible in resting cells, also because the required conditions for sPLA2-IIA hydrolysis are not met in the cytosol (Bezzine et al., 2002; Singer et al., 2002). Of note, phosphatidylserine externalization has been reported to occur in pre-osteoclasts obtained from mouse bone marrow-derived cells, and was shown to be crucial for their M-CSF/RANKL-induced multinucleation (Kang et al., 2020). Interestingly, we observed an increased transcription of TMEM16F, a lipid scramblase and major contributor to the process of phosphatidylserine exposure (Pedemonte and Galietta, 2014), during the first 24 h of RANKL-induced differentiation of RAW264.7 macrophages (data not shown). A hallmark of TMEM16 scramblases is their lack of specificity, as phosphatidylinositol transbilayer movement across proteoliposomes reconstituted with purified fungal Nectria haematococca TMEM16 has been reported (Wang et al., 2018). Among the metabolites mediating sPLA2-IIA cellular effects, the lysophosphatidylinositol could be a reasonable candidate, as several studies have already highlighted its involvement in the osteoclastogenesis process (Whyte et al., 2009; Mosca et al., 2021). In particular, we have recently showed that lysophosphatidylinositol stimulates the osteoclast maturation of RAW264.7 macrophages, with increased transcription of Nfatc1, Cathepsin-k, Trap, and Ctr, without any effect on the osteoclast syncytium formation, through GPR55-mediated signal transduction (Mosca et al., 2021). This is consistent with the requirement of sPLA2-IIA catalytic activity exclusively for the regulation of the osteoclastogenesis marker transcription in our assay conditions. Indeed, both murine and human recombinant sPLA2-IIA increased the RANKL-induced transcription of Nfatc1, Cathepsin-k, Trap, and Ctr, but not the catalytically inactive sPLA2-IIA H48Q mutant. In addition, treatment of RAW264.7 macrophages with the alkylating agent BPB, which halts the sPLA2-IIA enzymatic activity without changing the overall conformation of the protein surface (Lee et al., 2013), induced a significant reduction of the osteoclastogenesis marker transcription, without affecting the osteoclast fusion. Although this inhibitor is a general alkylating agent and its administration to intact cells cannot guarantee for a sPLA2-IIA specific targeting (Scott et al., 2021), we verified that under BPB treatment the sPLA2-IIA enzymatic activity is significantly impaired in RANKL-differentiated RAW264.7 macrophages (Table 2). However, off-target effects cannot be completely ruled out.
Substantial progress has been made recently in the identification of the molecular machinery involved in osteoclast fusion, but the role of the lipid components in osteoclast syncytium formation is still under investigation. In different cell-to-cell fusion models, changes in membrane lipid composition have been shown to have relevant roles through the involvement of phospholipase activity (Chernomordik and Kozlov, 2008). For instance, in sperm fertilization of oocytes, changes in the acyl-chain composition of the main membrane glycerophospholipids may depend on another secreted phospholipase A2: sPLA2-X (Escoffier et al., 2010). Inhibition of osteoclast functions by sPLA2-IIA blockade has been reported by different groups, both in vitro and in animal models. In Pagetic bone samples, sPLA2-IIA was shown to be expressed at high levels and to stimulate the osteoclast differentiation and function by a mechanism that may be independent of its enzymatic activity (Allard-Chamard et al., 2014b). Consistently, under our assay conditions, sPLA2-IIA regulation of osteoclast cell-to-cell fusion seems to occur independently of its enzymatic activity. Indeed, not only two specific sPLA2-IIA inhibitors (Inhib-I and KH064), but mostly the cyclic pentapetide (LLKYK) derived from the murine sPLA2-IIA primary sequence, were able to impair the RANKL-induced syncytium formation and the osteoclast functional activity. Of note, the LLKYK peptide was not able to affect the osteoclastogenesis marker transcription, which seems to depend exclusively on sPLA2-IIA catalytic activity, with the exception of Mmp-9 transcription that is regulated by sPLA2-IIA through multiple mechanisms. Differently from KH064, LY311727 did not affect the osteoclast fusion event. An explanation for the different behavior of these two sPLA2-IIA inhibitors in regulating the osteoclast fusion could reside in their peculiar ability to perturbate sPLA2-IIA interaction with a putative binding partner (Boilard et al., 2006), or their different cell-membrane permeability (Mounier et al., 2004; Jemel et al., 2011). Indeed, the sPLA2-IIA exhibits compartmentalization, not only for the enzymatic activity primarily occurring in the extracellular environment with a requirement of millimolar calcium concentrations. Even if the binding with a PLA2 receptor on the cell surface can occur, several other sPLA2-IIA interactions occur upon its internalization (Scott et al., 2021).
Studies over the last decade have revealed that dysregulated lipid metabolism is one of the fundamental metabolic alterations that enable cancer cell survival and sustain rapid tumor growth and cell proliferation (Kroemer and Pouyssegur, 2008). The expression of several sPLA2s is altered in various cancer cells, and in the neighboring stromal and immune cells at primary or metastatic tumor sites (Cummings, 2007; Murakami et al., 2011; Brglez et al., 2014; Sukocheva et al., 2019; Scott et al., 2021). Once secreted, sPLA2s can have both autocrine and paracrine roles, to induce metabolic and signaling changes in a tumor (Kamphorst et al., 2013). The expression of sPLA2-IIA is increased in the serum and tumors of patients with prostate, esophageal and lung cancers, and it is associated with poorer patient survival. Thus, the elevated levels of sPLA2-IIA in the plasma of these cancer patients has led to the suggestion of a biomarker role for sPLA2-IIA (Kupert et al., 2011). However, it remains unclear whether increased serum sPLA2-IIA levels are cancer-specific or whether they reflect inflammatory reactions during malignancy (Kupert et al., 2011). Of note, bone metastases are a common complication of epithelial cancers, of which breast, prostate and lung carcinomas are the most common (Kan et al., 2016). Serum levels of sPLA2-IIA can also rise from 100-fold to 1,000-fold during acute inflammatory disorders, such as sepsis, acute pancreatitis, peritonitis and chronic rheumatoid arthritis (Triggiani et al., 2005). Intriguingly, inflammatory pathologies such as rheumatoid arthritis and spondyloarthritides have also been associated with localized bone resorption and generalized osteoporosis (Szentpetery et al., 2017).
Many of the observed effects of sPLA2-IIA on biological systems, particularly in pathological settings characterized by persistent aberrant expression of this protein, may be attributed to direct or indirect perturbation of intracellular cell signaling pathways mediated by protein-protein interactions (Scott et al., 2021). Among the potential candidates, the M-type receptor PLA2R1 is able to bind murine sPLA2-IIA at low nanomolar concentrations, even if with an affinity 6–10-fold lower compared to sPLA2-IB (Cupillard et al., 1999; Rouault et al., 2007). In the lung, the proposed role for PLA2R1 is the clearance of PLA2-IB from the bronchoalveolar fluid by endocytosis and lysosomal degradation in airway smooth muscle cells (Tamaru et al., 2013). Otherwise, PLA2-IB binding to PLA2R1 may promote downstream signaling pathways, with activation of stress kinases and key enzymes part of the phospholipid and sphingolipid metabolism, able to generate pro-inflammatory bioactive lipid derivatives (Mandal et al., 2001). Furthermore, the highly basic nature of sPLA2-IIA allows its interaction with the negatively-charged heparan sulfates found at the cell surface (Murakami et al., 1999; Boilard et al., 2003), and with components of the coagulation cascade such as Factors Va and Xa (Mounier et al., 2000). The sPLA2-IIA interfacial binding site has been reported to be relevant for vimentin binding at the surface of apoptotic human T cells (Boilard et al., 2003). Instead, the arginine residues 74 and 100 in the human sPLA2-IIA sequence are crucial for the interaction with integrins αVβ3 and α4β1 that leads to integrin activation and monocytic cell proliferation (Takada and Fujita, 2017). Intriguingly, several of these sPLA2-IIA binding partners are not only main components of the osteoclast adhesion zone, but play a crucial role in the activation of osteoclasts to resorb the bone, as it is the case for vimentin and integrins (αVβ3, αVβ5, and α2β1, among others) (Marchisio et al., 1984; Karanth et al., 2021).
More work will be necessary to dissect out each of the many regulatory pathways responsible for sPLA2-IIA actions during the osteoclast fusion and to identify the putative sPLA2-IIA receptor involved. These studies could pave the way for therapeutic application of sPLA2-IIA inhibitors. An ideal blocker would be required to selectively inhibit sPLA2-IIA binding to the specific receptor implicated in the osteoclast fusion. This will probably guarantee a tissue and cellular specific action. Several PLA2 inhibitors, including sPLA2 blockers, have been evaluated in clinical trials for the treatment of osteoarthritis, rheumatoid arthritis, atherosclerosis, sepsis and atopic dermatitis, among others, but none has reached the market (Nikolaou et al., 2019). The main limitation seems to be a lack of selectivity among the different human sPLA2s upon systemic administration, also as a consequence of a lack of a comprehensive knowledge on cellular functions connected with each sPLA2 subtype. Despite the fact that the rational design of selective inhibitors for the enzymatic activity of the different PLA2s is a challenging issue, due to the highly conserved catalytic domain within this family of enzymes (Lambeau and Gelb, 2008; Oslund et al., 2008; Burke and Dennis, 2009; Cao et al., 2013), selective targeting of sPLA2 ligand function has higher potentialities. This expectation could be fulfilled by monoclonal antibodies specifically raised against each enzyme (Pothlichet et al., 2020). Such sPLA2-interaction inhibitors could be proposed as alternatives to the bisphosphonates (Center et al., 2020; D'Oronzo et al., 2021), the actual treatment of choice for several osteoclast disorders, since bisphosphonates are contraindicated in older patients with kidney impairment or cardiac disease (Dos Santos Ferreira et al., 2017; Rogers et al., 2020; de Roij van Zuijdewijn et al., 2021).
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee for Brigham and Women’s Hospital.
MM, RD’A, and CV performed experiments and contributed to data analysis. BB, JC designed experiments with the primary cell cultures, performed the data analysis, interpreted the data and contributed to writing of the manuscript. GL and CP produced the recombinant sPLA2-IIA proteins and antibodies; GL interpreted the data and contributed to writing of the manuscript. SM designed and performed experiments, analyzed and interpreted the data, conducted the project, and wrote the manuscript. All authors reviewed the manuscript.
This work was supported by the P.O.R. FESR Campania SATIN and PRIN project 2012CK5RPF_05 (to SM); and by the Agence Nationale de la Recherche (ANR-20-CE14-0024-01) and the Fondation de la Recherche Médicale (DEQ20180339193) (to GL). MM was supported by the Young Researcher Mobility Program of Fondazione Berlucchi and by the University of Campania “Luigi Vanvitelli” for her visit of the JC and BB laboratories at the Brigham and Women’s Hospital-Harvard Medical School (Boston, MA, United States).
We thank all colleagues who kindly provided reagents.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.966950/full#supplementary-material
Ackermann, E. J., Conde-Frieboes, K., and Dennis, E. A. (1995). Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J. Biol. Chem. 270 (1), 445–450. doi:10.1074/jbc.270.1.445
Allard-Chamard, H., Dufort, P., Haroun, S., and de Brum-Fernandes, A. J. (2014a). Cytosolic phospholipase A2 and eicosanoids modulate life, death and function of human osteoclasts in vitro. Prostagl. Leukot. Essent. Fat. Acids 90 (4), 117–123. doi:10.1016/j.plefa.2013.12.009
Allard-Chamard, H., Haroun, S., and de Brum-Fernandes, A. J. (2014b). Secreted phospholipase A2 type II is present in Paget's disease of bone and modulates osteoclastogenesis, apoptosis and bone resorption of human osteoclasts independently of its catalytic activity in vitro. Prostagl. Leukot. Essent. Fat. Acids 90 (2-3), 39–45. doi:10.1016/j.plefa.2013.12.007
Ao, C., Huo, Y., Qi, L., Xiong, Z., Xue, L., and Qi, Y. (2010). Pioglitazone suppresses the lipopolysaccharide-induced production of inflammatory factors in mouse macrophages by inactivating NF-kappaB. Cell Biol. Int. 34 (7), 723–730. doi:10.1042/CBI20090005
Arganaraz, G. A., Palmeira, J. D. F., and Arganaraz, E. R. (2020). Phosphatidylserine inside out: A possible underlying mechanism in the inflammation and coagulation abnormalities of COVID-19. Cell Commun. Signal. 18 (1), 190. doi:10.1186/s12964-020-00687-7
Astudillo, A. M., Balboa, M. A., and Balsinde, J. (2019). Selectivity of phospholipid hydrolysis by phospholipase A2 enzymes in activated cells leading to polyunsaturated fatty acid mobilization. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1864 (6), 772–783. doi:10.1016/j.bbalip.2018.07.002
Atsumi, G., Murakami, M., Tajima, M., Shimbara, S., Hara, N., and Kudo, I. (1997). The perturbed membrane of cells undergoing apoptosis is susceptible to type II secretory phospholipase A2 to liberate arachidonic acid. Biochim. Biophys. Acta 1349 (1), 43–54. doi:10.1016/s0005-2760(97)00082-9
Balestrieri, B., Hsu, V. W., Gilbert, H., Leslie, C. C., Han, W. K., Bonventre, J. V., et al. (2006). Group V secretory phospholipase A2 translocates to the phagosome after zymosan stimulation of mouse peritoneal macrophages and regulates phagocytosis. J. Biol. Chem. 281 (10), 6691–6698. doi:10.1074/jbc.M508314200
Balsinde, J., and Dennis, E. A. (1996). Distinct roles in signal transduction for each of the phospholipase A2 enzymes present in P388D1 macrophages. J. Biol. Chem. 271 (12), 6758–6765. doi:10.1074/jbc.271.12.6758
Beck, S., Lambeau, G., Scholz-Pedretti, K., Gelb, M. H., Janssen, M. J., Edwards, S. H., et al. (2003). Potentiation of tumor necrosis factor α-induced secreted phospholipase A2 (sPLA2)-IIA expression in mesangial cells by an autocrine loop involving sPLA2 and peroxisome proliferator-activated receptor α activation. J. Biol. Chem. 278 (32), 29799–29812. doi:10.1074/jbc.M211763200
Berrie, C. P., Iurisci, C., Piccolo, E., Bagnati, R., and Corda, D. (2007). Analysis of phosphoinositides and their aqueous metabolites. Methods Enzymol. 434, 187–232. doi:10.1016/S0076-6879(07)34011-1
Bezzine, S., Bollinger, J. G., Singer, A. G., Veatch, S. L., Keller, S. L., and Gelb, M. H. (2002). On the binding preference of human groups IIA and X phospholipases A2 for membranes with anionic phospholipids. J. Biol. Chem. 277 (50), 48523–48534. doi:10.1074/jbc.M203137200
Boilard, E., Bourgoin, S. G., Bernatchez, C., Poubelle, P. E., and Surette, M. E. (2003). Interaction of low molecular weight group IIA phospholipase A2 with apoptotic human T cells: Role of heparan sulfate proteoglycans. FASEB J. 17 (9), 1068–1080. doi:10.1096/fj.02-0938com
Boilard, E., Lai, Y., Larabee, K., Balestrieri, B., Ghomashchi, F., Fujioka, D., et al. (2010). A novel anti-inflammatory role for secretory phospholipase A2 in immune complex-mediated arthritis. EMBO Mol. Med. 2 (5), 172–187. doi:10.1002/emmm.201000072
Boilard, E., Rouault, M., Surrel, F., Le Calvez, C., Bezzine, S., Singer, A., et al. (2006). Secreted phospholipase A2 inhibitors are also potent blockers of binding to the M-type receptor. Biochemistry 45 (44), 13203–13218. doi:10.1021/bi061376d
Bonventre, J. (2004). Cytosolic phospholipase A2α reigns supreme in arthritis and bone resorption. Trends Immunol. 25 (3), 116–119. doi:10.1016/j.it.2004.01.006
Bradley, J. D., Dmitrienko, A. A., Kivitz, A. J., Gluck, O. S., Weaver, A. L., Wiesenhutter, C., et al. (2005). A randomized, double-blinded, placebo-controlled clinical trial of LY333013, a selective inhibitor of group II secretory phospholipase A2, in the treatment of rheumatoid arthritis. J. Rheumatol. 32 (3), 417–423.
Brglez, V., Lambeau, G., and Petan, T. (2014). Secreted phospholipases A2 in cancer: Diverse mechanisms of action. Biochimie 107 Pt, 114–123. doi:10.1016/j.biochi.2014.09.023
Burke, J. E., and Dennis, E. A. (2009). Phospholipase A2 biochemistry. Cardiovasc. Drugs Ther. 23 (1), 49–59. doi:10.1007/s10557-008-6132-9
Cao, J., Burke, J. E., and Dennis, E. A. (2013). Using hydrogen/deuterium exchange mass spectrometry to define the specific interactions of the phospholipase A2 superfamily with lipid substrates, inhibitors, and membranes. J. Biol. Chem. 288 (3), 1806–1813. doi:10.1074/jbc.R112.421909
Center, J. R., Lyles, K. W., and Bliuc, D. (2020). Bisphosphonates and lifespan. Bone 141, 115566. doi:10.1016/j.bone.2020.115566
Charles, J. F., Hsu, L. Y., Niemi, E. C., Weiss, A., Aliprantis, A. O., and Nakamura, M. C. (2012). Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J. Clin. Invest. 122 (12), 4592–4605. doi:10.1172/JCI60920
Chernomordik, L. V., and Kozlov, M. M. (2008). Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 15 (7), 675–683. doi:10.1038/nsmb.1455
Church, W. B., Inglis, A. S., Tseng, A., Duell, R., Lei, P. W., Bryant, K. J., et al. (2001). A novel approach to the design of inhibitors of human secreted phospholipase A2 based on native peptide inhibition. J. Biol. Chem. 276 (35), 33156–33164. doi:10.1074/jbc.M101272200
Cirino, G., Cicala, C., and Sorrentino, L. (1994). Human recombinant platelet phospholipase A2 exacerbates poly-L-arginine induced rat paw edema. Inflammation 18 (1), 59–66. doi:10.1007/BF01534598
Collin-Osdoby, P., and Osdoby, P. (2012). RANKL-mediated osteoclast formation from murine RAW 264.7 cells. Methods Mol. Biol. 816, 187–202. doi:10.1007/978-1-61779-415-5_13
Coulthard, L. G., Costello, J., Robinson, B., Shiels, I. A., Taylor, S. M., and Woodruff, T. M. (2011). Comparative efficacy of a secretory phospholipase A2 inhibitor with conventional anti-inflammatory agents in a rat model of antigen-induced arthritis. Arthritis Res. Ther. 13 (2), R42. doi:10.1186/ar3278
Couturier, C., Brouillet, A., Couriaud, C., Koumanov, K., Bereziat, G., and Andreani, M. (1999). Interleukin 1β induces type II-secreted phospholipase A2 gene in vascular smooth muscle cells by a nuclear factor κB and peroxisome proliferator-activated receptor-mediated process. J. Biol. Chem. 274 (33), 23085–23093. doi:10.1074/jbc.274.33.23085
Cuetara, B. L., Crotti, T. N., O'Donoghue, A. J., and McHugh, K. P. (2006). Cloning and characterization of osteoclast precursors from the RAW264.7 cell line. Vitro Cell. Dev. Biol. Anim. 42 (7), 182–188. doi:10.1290/0510075.1
Cummings, B. S. (2007). Phospholipase A2 as targets for anti-cancer drugs. Biochem. Pharmacol. 74 (7), 949–959. doi:10.1016/j.bcp.2007.04.021
Cupillard, L., Mulherkar, R., Gomez, N., Kadam, S., Valentin, E., Lazdunski, M., et al. (1999). Both group IB and group IIA secreted phospholipases A2 are natural ligands of the mouse 180-kDa M-type receptor. J. Biol. Chem. 274 (11), 7043–7051. doi:10.1074/jbc.274.11.7043
D'Oronzo, S., Wood, S., and Brown, J. E. (2021). The use of bisphosphonates to treat skeletal complications in solid tumours. Bone 147, 115907. doi:10.1016/j.bone.2021.115907
Dacheux, M., Chaouch, S., Joy, A., Labat, A., Payre, C., Petit-Paitel, A., et al. (2021). Role of human group IIA secreted phospholipase A2 in malaria pathophysiology: Insights from a transgenic mouse model. Biochimie 189, 120–136. doi:10.1016/j.biochi.2021.06.009
Dacheux, M., Sinou, V., Payre, C., Jeammet, L., Parzy, D., Grellier, P., et al. (2019). Antimalarial activity of human group IIA secreted phospholipase A2 in relation to enzymatic hydrolysis of oxidized lipoproteins. Infect. Immun. 87 (11), e00556-19. doi:10.1128/IAI.00556-19
de Roij van Zuijdewijn, C., van Dorp, W., Florquin, S., Roelofs, J., and Verburgh, K. (2021). Bisphosphonate nephropathy: A case series and review of the literature. Br. J. Clin. Pharmacol. 87 (9), 3485–3491. doi:10.1111/bcp.14780
Dennis, E. A. (1994). Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem. 269 (18), 13057–13060. doi:10.1016/s0021-9258(17)36794-7
Doré, E., and Boilard, E. (2019). Roles of secreted phospholipase A2 group IIA in inflammation and host defense. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1864 (6), 789–802. doi:10.1016/j.bbalip.2018.08.017
Doré, E., Joly-Beauparlant, C., Morozumi, S., Mathieu, A., Levesque, T., Allaeys, I., et al. (2022). The interaction of secreted phospholipase A2-IIA with the microbiota alters its lipidome and promotes inflammation. JCI Insight 7 (2), e152638. doi:10.1172/jci.insight.152638
Dos Santos Ferreira, D., Jesus de Oliveira Pinto, B. L., Kumar, V., Cardoso, V. N., Fernandes, S. O., Souza, C. M., et al. (2017). Evaluation of antitumor activity and cardiac toxicity of a bone-targeted pH-sensitive liposomal formulation in a bone metastasis tumor model in mice. Nanomedicine 13 (5), 1693–1701. doi:10.1016/j.nano.2017.03.005
Edwards, S. H., Thompson, D., Baker, S. F., Wood, S. P., and Wilton, D. C. (2002). The crystal structure of the H48Q active site mutant of human group IIA secreted phospholipase A2 at 1.5 A resolution provides an insight into the catalytic mechanism. Biochemistry 41 (52), 15468–15476. doi:10.1021/bi020485z
Eerola, L. I., Surrel, F., Nevalainen, T. J., Gelb, M. H., Lambeau, G., and Laine, V. J. (2006). Analysis of expression of secreted phospholipases A2 in mouse tissues at protein and mRNA levels. Biochim. Biophys. Acta 1761 (7), 745–756. doi:10.1016/j.bbalip.2006.04.002
Escoffier, J., Jemel, I., Tanemoto, A., Taketomi, Y., Payre, C., Coatrieux, C., et al. (2010). Group X phospholipase A2 is released during sperm acrosome reaction and controls fertility outcome in mice. J. Clin. Invest. 120 (5), 1415–1428. doi:10.1172/JCI40494
Ferraz-de-Souza, B., and Correa, P. H. (2013). Diagnosis and treatment of paget's disease of bone: A mini-review. Arq. Bras. Endocrinol. Metabol. 57 (8), 577–582. doi:10.1590/s0004-27302013000800001
Fourcade, O., Simon, M. F., Viode, C., Rugani, N., Leballe, F., Ragab, A., et al. (1995). Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 80 (6), 919–927. doi:10.1016/0092-8674(95)90295-3
Ghomashchi, F., Brglez, V., Payre, C., Jeammet, L., Bezzine, S., Gelb, M. H., et al. (2017). Preparation of the full set of recombinant mouse- and human-secreted phospholipases A2. Methods Enzymol. 583, 35–69. doi:10.1016/bs.mie.2016.10.034
Gregory, L. S., Kelly, W. L., Reid, R. C., Fairlie, D. P., and Forwood, M. R. (2006). Inhibitors of cyclo-oxygenase-2 and secretory phospholipase A2 preserve bone architecture following ovariectomy in adult rats. Bone 39 (1), 134–142. doi:10.1016/j.bone.2005.12.017
Hansford, K. A., Reid, R. C., Clark, C. I., Tyndall, J. D., Whitehouse, M. W., Guthrie, T., et al. (2003). D-Tyrosine as a chiral precusor to potent inhibitors of human nonpancreatic secretory phospholipase A2 (IIa) with antiinflammatory activity. Chembiochem 4 (2-3), 181–185. doi:10.1002/cbic.200390029
Hendrickson, H. S., Hendrickson, E. K., and Dybvig, R. H. (1983). Chiral synthesis of a dithiolester analog of phosphatidylcholine as a substrate for the assay of phospholipase A2. J. Lipid Res. 24 (11), 1532–1537. doi:10.1016/s0022-2275(20)37877-9
Hussain, Z., Uyama, T., Tsuboi, K., and Ueda, N. (2017). Mammalian enzymes responsible for the biosynthesis of N-acylethanolamines. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1862 (12), 1546–1561. doi:10.1016/j.bbalip.2017.08.006
Ivanusec, A., Sribar, J., and Krizaj, I. (2022). Secreted phospholipases A2 - not just enzymes: Revisited. Int. J. Biol. Sci. 18 (2), 873–888. doi:10.7150/ijbs.68093
Jemel, I., Ii, H., Oslund, R. C., Payre, C., Dabert-Gay, A. S., Douguet, D., et al. (2011). Group X secreted phospholipase A2 proenzyme is matured by a furin-like proprotein convertase and releases arachidonic acid inside of human HEK293 cells. J. Biol. Chem. 286 (42), 36509–36521. doi:10.1074/jbc.M111.268540
Kamphorst, J. J., Cross, J. R., Fan, J., de Stanchina, E., Mathew, R., White, E. P., et al. (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. U. S. A. 110 (22), 8882–8887. doi:10.1073/pnas.1307237110
Kan, C., Vargas, G., Pape, F. L., and Clezardin, P. (2016). Cancer cell colonisation in the bone microenvironment. Int. J. Mol. Sci. 17 (10), E1674. doi:10.3390/ijms17101674
Kang, J. H., Ko, H. M., Han, G. D., Lee, S. Y., Moon, J. S., Kim, M. S., et al. (2020). Dual role of phosphatidylserine and its receptors in osteoclastogenesis. Cell Death Dis. 11 (7), 497. doi:10.1038/s41419-020-2712-9
Karanth, D. S., Martin, M. L., and Holliday, L. S. (2021). Plasma membrane receptors involved in the binding and response of osteoclasts to noncellular components of the bone. Int. J. Mol. Sci. 22 (18), 10097. doi:10.3390/ijms221810097
Kennedy, B. P., Payette, P., Mudgett, J., Vadas, P., Pruzanski, W., Kwan, M., et al. (1995). A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J. Biol. Chem. 270 (38), 22378–22385. doi:10.1074/jbc.270.38.22378
Kim, R. R., Chen, Z., Mann, T. J., Bastard, K., K, F. S., and Church, W. B. (2020). Structural and functional aspects of targeting the secreted human group IIA phospholipase A2. Molecules 25 (19), E4459. doi:10.3390/molecules25194459
Kim, Y. S., Min, K. S., Lee, H. D., Oh, H. W., and Kim, E. C. (2010). Effect of cytosolic phospholipase A2 on proinflammatory cytokine-induced bone resorptive genes including receptor activator of nuclear factor kappa B ligand in human dental pulp cells. J. Endod. 36 (4), 636–641. doi:10.1016/j.joen.2009.12.009
Kita, Y., Shindou, H., and Shimizu, T. (2019). Cytosolic phospholipase A2 and lysophospholipid acyltransferases. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1864 (6), 838–845. doi:10.1016/j.bbalip.2018.08.006
Koduri, R. S., Baker, S. F., Snitko, Y., Han, S. K., Cho, W., Wilton, D. C., et al. (1998). Action of human group IIa secreted phospholipase A2 on cell membranes. Vesicle but not heparinoid binding determines rate of fatty acid release by exogenously added enzyme. J. Biol. Chem. 273 (48), 32142–32153. doi:10.1074/jbc.273.48.32142
Kono, N., and Arai, H. (2019). Platelet-activating factor acetylhydrolases: An overview and update. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1864 (6), 922–931. doi:10.1016/j.bbalip.2018.07.006
Kroemer, G., and Pouyssegur, J. (2008). Tumor cell metabolism: cancer's achilles' heel. Cancer Cell 13 (6), 472–482. doi:10.1016/j.ccr.2008.05.005
Kudo, K., Miki, Y., Carreras, J., Nakayama, S., Nakamoto, Y., Ito, M., et al. (2022). Secreted phospholipase A2 modifies extracellular vesicles and accelerates B cell lymphoma. Cell Metab. 34 (4), 615–633.e8. e618. doi:10.1016/j.cmet.2022.02.011
Kupert, E., Anderson, M., Liu, Y., Succop, P., Levin, L., Wang, J., et al. (2011). Plasma secretory phospholipase A2-IIa as a potential biomarker for lung cancer in patients with solitary pulmonary nodules. BMC Cancer 11, 513. doi:10.1186/1471-2407-11-513
Lambeau, G., and Gelb, M. H. (2008). Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 77, 495–520. doi:10.1146/annurev.biochem.76.062405.154007
Lambeau, G., and Lazdunski, M. (1999). Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol. Sci. 20 (4), 162–170. doi:10.1016/s0165-6147(99)01300-0
Lee, L. K., Bryant, K. J., Bouveret, R., Lei, P. W., Duff, A. P., Harrop, S. J., et al. (2013). Selective inhibition of human group IIA-secreted phospholipase A2 (hGIIA) signaling reveals arachidonic acid metabolism is associated with colocalization of hGIIA to vimentin in rheumatoid synoviocytes. J. Biol. Chem. 288 (21), 15269–15279. doi:10.1074/jbc.M112.397893
Lio, Y. C., Reynolds, L. J., Balsinde, J., and Dennis, E. A. (1996). Irreversible inhibition of Ca2+-independent phospholipase A2 by methyl arachidonyl fluorophosphonate. Biochim. Biophys. Acta 1302 (1), 55–60. doi:10.1016/0005-2760(96)00002-1
Lomonte, B., Tarkowski, A., and Hanson, L. A. (1995). Phospholipase A2 and inflammation. Mol. Med. Today 1 (1), 9. doi:10.1016/1357-4310(95)80011-5
MacPhee, M., Chepenik, K. P., Liddell, R. A., Nelson, K. K., Siracusa, L. D., and Buchberg, A. M. (1995). The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell 81 (6), 957–966. doi:10.1016/0092-8674(95)90015-2
Mandal, A. K., Zhang, Z., Chou, J. Y., and Mukherjee, A. B. (2001). Pancreatic phospholipase A2 via its receptor regulates expression of key enzymes of phospholipid and sphingolipid metabolism. FASEB J. 15 (10), 1834–1836. doi:10.1096/fj.00-0831fje
Marchi-Salvador, D. P., Fernandes, C. A., Silveira, L. B., Soares, A. M., and Fontes, M. R. (2009). Crystal structure of a phospholipase A2 homolog complexed with p-bromophenacyl bromide reveals important structural changes associated with the inhibition of myotoxic activity. Biochim. Biophys. Acta 1794 (11), 1583–1590. doi:10.1016/j.bbapap.2009.07.005
Marchisio, P. C., Cirillo, D., Naldini, L., Primavera, M. V., Teti, A., and Zambonin-Zallone, A. (1984). Cell-substratum interaction of cultured avian osteoclasts is mediated by specific adhesion structures. J. Cell Biol. 99 (5), 1696–1705. doi:10.1083/jcb.99.5.1696
Miki, Y., Taketomi, Y., Kidoguchi, Y., Yamamoto, K., Muramatsu, K., Nishito, Y., et al. (2022). Group IIA secreted phospholipase A2 controls skin carcinogenesis and psoriasis by shaping the gut microbiota. JCI Insight 7 (2), e152611. doi:10.1172/jci.insight.152611
Mosca, M. G., Mangini, M., Cioffi, S., Barba, P., and Mariggiò, S. (2021). Peptide targeting of lysophosphatidylinositol-sensing GPR55 for osteoclastogenesis tuning. Cell Commun. Signal. 19 (1), 48. doi:10.1186/s12964-021-00727-w
Mounier, C. M., Ghomashchi, F., Lindsay, M. R., James, S., Singer, A. G., Parton, R. G., et al. (2004). Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A2 occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A2-α. J. Biol. Chem. 279 (24), 25024–25038. doi:10.1074/jbc.M313019200
Mounier, C. M., Luchetta, P., Lecut, C., Koduri, R. S., Faure, G., Lambeau, G., et al. (2000). Basic residues of human group IIA phospholipase A2 are important for binding to factor Xa and prothrombinase inhibition comparison with other mammalian secreted phospholipases A2. Eur. J. Biochem. 267 (16), 4960–4969. doi:10.1046/j.1432-1327.2000.01523.x
Murakami, M., Kambe, T., Shimbara, S., Yamamoto, S., Kuwata, H., and Kudo, I. (1999). Functional association of type IIA secretory phospholipase A2 with the glycosylphosphatidylinositol-anchored heparan sulfate proteoglycan in the cyclooxygenase-2-mediated delayed prostanoid-biosynthetic pathway. J. Biol. Chem. 274 (42), 29927–29936. doi:10.1074/jbc.274.42.29927
Murakami, M., Miki, Y., Sato, H., Murase, R., Taketomi, Y., and Yamamoto, K. (2019). Group IID, IIE, IIF and III secreted phospholipase A2s. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1864 (6), 803–818. doi:10.1016/j.bbalip.2018.08.014
Murakami, M., Nakatani, Y., Atsumi, G. I., Inoue, K., and Kudo, I. (2017). Regulatory functions of phospholipase A2. Crit. Rev. Immunol. 37 (2-6), 127–195. doi:10.1615/CritRevImmunol.v37.i2-6.20
Murakami, M. (2019). Novel functions of phospholipase A2s: Overview. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1864 (6), 763–765. doi:10.1016/j.bbalip.2019.02.005
Murakami, M., Sato, H., Miki, Y., Yamamoto, K., and Taketomi, Y. (2015). A new era of secreted phospholipase A2. J. Lipid Res. 56 (7), 1248–1261. doi:10.1194/jlr.R058123
Murakami, M., Sato, H., and Taketomi, Y. (2020). Updating phospholipase A2 biology. Biomolecules 10 (10), E1457. doi:10.3390/biom10101457
Murakami, M., Taketomi, Y., Girard, C., Yamamoto, K., and Lambeau, G. (2010). Emerging roles of secreted phospholipase A2 enzymes: Lessons from transgenic and knockout mice. Biochimie 92 (6), 561–582. doi:10.1016/j.biochi.2010.03.015
Murakami, M., Taketomi, Y., Sato, H., and Yamamoto, K. (2011). Secreted phospholipase A2 revisited. J. Biochem. 150 (3), 233–255. doi:10.1093/jb/mvr088
Nevalainen, T. J., Eerola, L. I., Rintala, E., Laine, V. J., Lambeau, G., and Gelb, M. H. (2005). Time-resolved fluoroimmunoassays of the complete set of secreted phospholipases A2 in human serum. Biochim. Biophys. Acta 1733 (2-3), 210–223. doi:10.1016/j.bbalip.2004.12.012
Nikolaou, A., Kokotou, M. G., Vasilakaki, S., and Kokotos, G. (2019). Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1864 (6), 941–956. doi:10.1016/j.bbalip.2018.08.009
O'Brien, W., Fissel, B. M., Maeda, Y., Yan, J., Ge, X., Gravallese, E. M., et al. (2016). RANK-independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol. 68 (12), 2889–2900. doi:10.1002/art.39837
Olson, E. D., Nelson, J., Griffith, K., Nguyen, T., Streeter, M., Wilson-Ashworth, H. A., et al. (2010). Kinetic evaluation of cell membrane hydrolysis during apoptosis by human isoforms of secretory phospholipase A2. J. Biol. Chem. 285 (14), 10993–11002. doi:10.1074/jbc.M109.070797
Oslund, R. C., Cermak, N., and Gelb, M. H. (2008). Highly specific and broadly potent inhibitors of mammalian secreted phospholipases A2. J. Med. Chem. 51 (15), 4708–4714. doi:10.1021/jm800422v
Papadopoulos, S., Kazepidou, E., Antonelou, M. H., Leondaritis, G., Tsapinou, A., Koulouras, V. P., et al. (2020). Secretory phospholipase A2-IIA protein and mRNA pools in extracellular vesicles of bronchoalveolar lavage fluid from patients with early acute respiratory distress syndrome: A new perception in the dissemination of inflammation? Pharm. (Basel) 13 (11), E415. doi:10.3390/ph13110415
Park, J. H., Lee, N. K., and Lee, S. Y. (2017). Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol. Cells 40 (10), 706–713. doi:10.14348/molcells.2017.0225
Pedemonte, N., and Galietta, L. J. (2014). Structure and function of TMEM16 proteins (anoctamins). Physiol. Rev. 94 (2), 419–459. doi:10.1152/physrev.00039.2011
Pothlichet, J., Rose, T., Bugault, F., Jeammet, L., Meola, A., Haouz, A., et al. (2020). PLA2G1B is involved in CD4 anergy and CD4 lymphopenia in HIV-infected patients. J. Clin. Invest. 130 (6), 2872–2887. doi:10.1172/JCI131842
Ramanadham, S., Ali, T., Ashley, J. W., Bone, R. N., Hancock, W. D., and Lei, X. (2015). Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 56 (9), 1643–1668. doi:10.1194/jlr.R058701
Raschke, W. C., Baird, S., Ralph, P., and Nakoinz, I. (1978). Functional macrophage cell lines transformed by Abelson leukemia virus. Cell 15 (1), 261–267. doi:10.1016/0092-8674(78)90101-0
Reynolds, L. J., Hughes, L. L., and Dennis, E. A. (1992). Analysis of human synovial fluid phospholipase A2 on short chain phosphatidylcholine-mixed micelles: Development of a spectrophotometric assay suitable for a microtiterplate reader. Anal. Biochem. 204 (1), 190–197. doi:10.1016/0003-2697(92)90160-9
Rogers, M. J., Monkkonen, J., and Munoz, M. A. (2020). Molecular mechanisms of action of bisphosphonates and new insights into their effects outside the skeleton. Bone 139, 115493. doi:10.1016/j.bone.2020.115493
Rouault, M., Le Calvez, C., Boilard, E., Surrel, F., Singer, A., Ghomashchi, F., et al. (2007). Recombinant production and properties of binding of the full set of mouse secreted phospholipases A2 to the mouse M-type receptor. Biochemistry 46 (6), 1647–1662. doi:10.1021/bi062119b
Saegusa, J., Akakura, N., Wu, C. Y., Hoogland, C., Ma, Z., Lam, K. S., et al. (2008). Pro-inflammatory secretory phospholipase A2 type IIA binds to integrins αvβ3 and α4β1 and induces proliferation of monocytic cells in an integrin-dependent manner. J. Biol. Chem. 283 (38), 26107–26115. doi:10.1074/jbc.M804835200
San Pietro, E., Capestrano, M., Polishchuk, E. V., DiPentima, A., Trucco, A., Zizza, P., et al. (2009). Group IV phospholipase A2α controls the formation of inter-cisternal continuities involved in intra-Golgi transport. PLoS Biol. 7 (9), e1000194. doi:10.1371/journal.pbio.1000194
Scott, K. F., Mann, T. J., Fatima, S., Sajinovic, M., Razdan, A., Kim, R. R., et al. (2021). Human group IIA phospholipase A2–Three decades on from its discovery. Molecules 26 (23), 7267. doi:10.3390/molecules26237267
Seno, K., Okuno, T., Nishi, K., Murakami, Y., Watanabe, F., Matsuura, T., et al. (2000). Pyrrolidine inhibitors of human cytosolic phospholipase A2. J. Med. Chem. 43 (6), 1041–1044. doi:10.1021/jm9905155
Shayman, J. A., and Tesmer, J. J. G. (2019). Lysosomal phospholipase A2. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 1864 (6), 932–940. doi:10.1016/j.bbalip.2018.07.012
Singer, A. G., Ghomashchi, F., Le Calvez, C., Bollinger, J., Bezzine, S., Rouault, M., et al. (2002). Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J. Biol. Chem. 277 (50), 48535–48549. doi:10.1074/jbc.M205855200
Snider, J. M., You, J. K., Wang, X., Snider, A. J., Hallmark, B., Zec, M. M., et al. (2021). Group IIA secreted phospholipase A2 is associated with the pathobiology leading to COVID-19 mortality. J. Clin. Invest. 131 (19), e149236. doi:10.1172/JCI149236
Snitko, Y., Koduri, R. S., Han, S. K., Othman, R., Baker, S. F., Molini, B. J., et al. (1997). Mapping the interfacial binding surface of human secretory group IIa phospholipase A2. Biochemistry 36 (47), 14325–14333. doi:10.1021/bi971200z
Soe, K. (2020). Osteoclast fusion: Physiological regulation of multinucleation through heterogeneity-potential implications for drug sensitivity. Int. J. Mol. Sci. 21 (20), E7717. doi:10.3390/ijms21207717
Sukocheva, O., Menschikowski, M., Hagelgans, A., Yarla, N. S., Siegert, G., Reddanna, P., et al. (2019). Current insights into functions of phospholipase A2 receptor in normal and cancer cells: More questions than answers. Semin. Cancer Biol. 56, 116–127. doi:10.1016/j.semcancer.2017.11.002
Sun, G. Y., Geng, X., Teng, T., Yang, B., Appenteng, M. K., Greenlief, C. M., et al. (2021). Dynamic role of phospholipases A2 in health and diseases in the central nervous system. Cells 10 (11), 2963. doi:10.3390/cells10112963
Suzuki, J., Umeda, M., Sims, P. J., and Nagata, S. (2010). Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468 (7325), 834–838. doi:10.1038/nature09583
Szentpetery, A., Horvath, A., Gulyas, K., Petho, Z., Bhattoa, H. P., Szanto, S., et al. (2017). Effects of targeted therapies on the bone in arthritides. Autoimmun. Rev. 16 (3), 313–320. doi:10.1016/j.autrev.2017.01.014
Takada, Y., and Fujita, M. (2017). Secreted phospholipase A2 type IIA (sPLA2-IIA) activates integrins in an allosteric manner. Adv. Exp. Med. Biol. 925, 103–115. doi:10.1007/5584_2016_95
Tamaru, S., Mishina, H., Watanabe, Y., Watanabe, K., Fujioka, D., Takahashi, S., et al. (2013). Deficiency of phospholipase A2 receptor exacerbates ovalbumin-induced lung inflammation. J. Immunol. 191 (3), 1021–1028. doi:10.4049/jimmunol.1300738
Thomas, G., Brown, A. L., and Brown, J. M. (2014). In vivo metabolite profiling as a means to identify uncharacterized lipase function: Recent success stories within the alpha beta hydrolase domain (ABHD) enzyme family. Biochim. Biophys. Acta 1841 (8), 1097–1101. doi:10.1016/j.bbalip.2014.01.004
Thwin, M. M., Douni, E., Arjunan, P., Kollias, G., Kumar, P. V., and Gopalakrishnakone, P. (2009). Suppressive effect of secretory phospholipase A2 inhibitory peptide on interleukin-1β-induced matrix metalloproteinase production in rheumatoid synovial fibroblasts, and its antiarthritic activity in hTNFtg mice. Arthritis Res. Ther. 11 (5), R138. doi:10.1186/ar2810
Tischfield, J. A., Xia, Y. R., Shih, D. M., Klisak, I., Chen, J., Engle, S. J., et al. (1996). Low-molecular-weight, calcium-dependent phospholipase A2 genes are linked and map to homologous chromosome regions in mouse and human. Genomics 32 (3), 328–333. doi:10.1006/geno.1996.0126
Triggiani, M., Granata, F., Giannattasio, G., and Marone, G. (2005). Secretory phospholipases A2 in inflammatory and allergic diseases: Not just enzymes. J. Allergy Clin. Immunol. 116 (5), 1000–1006. doi:10.1016/j.jaci.2005.08.011
Tseng, A., Inglis, A. S., and Scott, K. F. (1996). Native peptide inhibition. Specific inhibition of type II phospholipases A2 by synthetic peptides derived from the primary sequence. J. Biol. Chem. 271 (39), 23992–23998. doi:10.1074/jbc.271.39.23992
Vadas, P., Pruzanski, W., Stefanski, E., Ellies, L. G., Aubin, J. E., Sos, A., et al. (1991). Extracellular phospholipase A2 secretion is a common effector pathway of interleukin-1 and tumour necrosis factor action. Immunol. Lett. 28 (3), 187–193. doi:10.1016/0165-2478(91)90002-r
Valentin, E., Ghomashchi, F., Gelb, M. H., Lazdunski, M., and Lambeau, G. (1999). On the diversity of secreted phospholipases A2. Cloning, tissue distribution, and functional expression of two novel mouse group II enzymes. J. Biol. Chem. 274 (44), 31195–31202. doi:10.1074/jbc.274.44.31195
Valentin, E., Singer, A. G., Ghomashchi, F., Lazdunski, M., Gelb, M. H., and Lambeau, G. (2000). Cloning and recombinant expression of human group IIF-secreted phospholipase A2. Biochem. Biophys. Res. Commun. 279 (1), 223–228. doi:10.1006/bbrc.2000.3908
Wang, L., Iwasaki, Y., Andra, K. K., Pandey, K., Menon, A. K., and Butikofer, P. (2018). Scrambling of natural and fluorescently tagged phosphatidylinositol by reconstituted G protein-coupled receptor and TMEM16 scramblases. J. Biol. Chem. 293 (47), 18318–18327. doi:10.1074/jbc.RA118.004213
Weinrauch, Y., Abad, C., Liang, N. S., Lowry, S. F., and Weiss, J. (1998). Mobilization of potent plasma bactericidal activity during systemic bacterial challenge. Role of group IIA phospholipase A2. J. Clin. Invest. 102 (3), 633–638. doi:10.1172/JCI3121
Whyte, L. S., Ryberg, E., Sims, N. A., Ridge, S. A., Mackie, K., Greasley, P. J., et al. (2009). The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc. Natl. Acad. Sci. U. S. A. 106 (38), 16511–16516. doi:10.1073/pnas.0902743106
Yamamoto, K., Miki, Y., Sato, H., Murase, R., Taketomi, Y., and Murakami, M. (2017). Secreted phospholipase A2 specificity on natural membrane phospholipids. Methods Enzymol. 583, 101–117. doi:10.1016/bs.mie.2016.09.007
Keywords: group IIA secreted phospholipase A2 (sPLA2-IIA), p38 SAPK, osteoclastogenesis, osteoclast syncytia, LLKYK cyclic peptide
Citation: Mangini M, D’Angelo R, Vinciguerra C, Payré C, Lambeau G, Balestrieri B, Charles JF and Mariggiò S (2022) Multimodal regulation of the osteoclastogenesis process by secreted group IIA phospholipase A2. Front. Cell Dev. Biol. 10:966950. doi: 10.3389/fcell.2022.966950
Received: 11 June 2022; Accepted: 25 July 2022;
Published: 29 August 2022.
Edited by:
Jesús Balsinde, Instituto de Biología y Genética Molecular (CSIC), SpainReviewed by:
Kieran F. Scott, Western Sydney University, Penrith, AustraliaCopyright © 2022 Mangini, D’Angelo, Vinciguerra, Payré, Lambeau, Balestrieri, Charles and Mariggiò. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefania Mariggiò, c3RlZmFuaWEubWFyaWdnaW9AaWJiYy5jbnIuaXQ=
†Present address: Maria Mangini, Institute of Endocrinology and Experimental Oncology “G. Salvatore”- IEOS, Second Unit, National Research Council, Naples, Italy Stefania Mariggiò, Institute of Biochemistry and Cell Biology, National Research Council, Naples, Italy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.