 Naomi Hartopp
Naomi Hartopp Dawn H W. Lau
Dawn H W. Lau Sandra M. Martin-Guerrero
Sandra M. Martin-Guerrero Andrea Markovinovic
Andrea Markovinovic Gábor M. Mórotz
Gábor M. Mórotz Jenny Greig
Jenny Greig Elizabeth B. Glennon
Elizabeth B. Glennon Claire Troakes
Claire Troakes Patricia Gomez-Suaga*
Patricia Gomez-Suaga* Wendy Noble*
Wendy Noble* Christopher C.J. Miller*
Christopher C.J. Miller*- Department of Basic and Clinical Neuroscience. Institute of Psychiatry, Psychology and Neuroscience, King’s College London, London, United Kingdom
Signaling between the endoplasmic reticulum (ER) and mitochondria regulates many neuronal functions that are perturbed in amyotrophic lateral sclerosis (ALS) and perturbation to ER-mitochondria signaling is seen in cell and transgenic models of ALS. However, there is currently little evidence that ER-mitochondria signaling is altered in human ALS. ER-mitochondria signaling is mediated by interactions between the integral ER protein VAPB and the outer mitochondrial membrane protein PTPIP51 which act to recruit and “tether” regions of ER to the mitochondrial surface. The VAPB-PTPI51 tethers are now known to regulate a number of ER-mitochondria signaling functions. These include delivery of Ca2+ from ER stores to mitochondria, mitochondrial ATP production, autophagy and synaptic activity. Here we investigate the VAPB-PTPIP51 tethers in post-mortem control and ALS spinal cords. We show that VAPB protein levels are reduced in ALS. Proximity ligation assays were then used to quantify the VAPB-PTPIP51 interaction in spinal cord motor neurons in control and ALS cases. These studies revealed that the VAPB-PTPIP51 tethers are disrupted in ALS. Thus, we identify a new pathogenic event in post-mortem ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease and involves progressive loss of motor neurons resulting in muscle wasting and ultimately paralysis. ALS is now known to be clinically, pathologically and genetically linked to frontotemporal dementia (FTD). FTD is the second most common cause of presenile dementia after Alzheimer’s disease (Ling et al., 2013; Robberecht and Philips, 2013). Thus, many FTD patients display clinical ALS features and likewise many ALS patients develop clinical symptoms of FTD (Ringholz et al., 2005; Wheaton et al., 2007). Both diseases can display similar pathological phenotypes and notably, the accumulation of abnormal aggregates of TAR DNA-binding protein 43 (TDP43) in affected neurons (Arai et al., 2006; Neumann et al., 2006). Finally, both diseases have strong genetic components and mutations in the same genes can cause dominant familial inherited forms of ALS and FTD. Mutant genes causing both ALS and FTD include TARDBP encoding TDP43, FUS encoding fused in sarcoma and C9orf72; the disease causing mutations in C9orf72 involve expansion of an intronic hexanucleotide repeat which is translated into neurotoxic dipeptide repeat proteins (DPRs) (Ling et al., 2013; Robberecht and Philips, 2013; Abramzon et al., 2020).
There are no cures or effective disease modifying treatments for ALS. Developing new therapies can include correcting damaged molecular, cellular and physiological processes but this is complicated as a large number of changes are seen in ALS. Thus, damage to mitochondria, the endoplasmic reticulum (ER), Ca2+ signaling, lipid metabolism, axonal transport, autophagy and inflammatory responses are all features of ALS (Paillusson et al., 2016; Lau et al., 2018; Dafinca et al., 2021; Markovinovic et al., 2022). The biological conundrum is how so many apparently disparate physiological processes are perturbed collectively. The therapeutic challenge is selecting which of these different perturbed processes to prioritize for drug discovery.
Recently, attention has focussed on signaling between the ER and mitochondria and this is because ER-mitochondria signaling regulates many of the functions that are damaged in ALS (Paillusson et al., 2016; Csordas et al., 2018; Lau et al., 2018; Dafinca et al., 2021; Markovinovic et al., 2022). ER-mitochondria signaling involves close contacts between the two organelles (up to approximately 30 nm distances) and the regions of ER in contact with mitochondria are termed mitochondria associated ER membranes (MAM) (Paillusson et al., 2016; Csordas et al., 2018; Lau et al., 2018; Dafinca et al., 2021; Markovinovic et al., 2022). The mechanisms by which ER membranes are recruited to the mitochondrial surface are not fully understood but it is widely accepted that the process involves “tethering proteins” which act to scaffold the two organelles in close proximity. One well characterised tether involves an interaction between the integral ER protein, vesicle-associated membrane protein-associated protein B (VAPB) and the outer mitochondrial membrane protein, protein tyrosine phosphatase interacting protein-51 (PTPIP51) (also known as regulator of microtubule dynamics-3 and family with sequence similarity 82 member A2) (De Vos et al., 2012; Stoica et al., 2014). The VAPB-PTPIP51 tethers are known to control a number of ER-mitochondria regulated functions including inositol 1,4,5-trisphosphate (IP3) receptor delivery of Ca2+ from ER stores to mitochondria, mitochondrial ATP production, autophagy, phospholipid synthesis and synaptic activity (De Vos et al., 2012; Stoica et al., 2014; Galmes et al., 2016; Stoica et al., 2016; Gomez-Suaga et al., 2017; Paillusson et al., 2017; Gomez-Suaga et al., 2019; Puri et al., 2019; Yeo et al., 2021; Gomez-Suaga et al., 2022). Loss of synaptic activity is a key feature of ALS and other neurodegenerative diseases (Herms and Dorostkar, 2016; Spires-Jones et al., 2017).
Such findings have prompted investigations into the VAPB-PTPIP51 tethers in ALS and this has revealed them to be disrupted in cell and transgenic mouse models involving mutant TDP43, FUS, and C9orf72 (Stoica et al., 2014; Stoica et al., 2016; Gomez-Suaga et al., 2022). However, as yet there is no evidence that the VAPB-PTPIP51 interaction is altered in human ALS patients. This is an important omission. Firstly, because transgenic mouse and cell models of ALS do not always fully recapitulate human disease; for example some C9orf72 transgenic mouse models display hippocampal rather than motor neuron loss (Jiang et al., 2016). More importantly, if correcting disrupted ER-mitochondria signaling and VAPB-PTPIP51 tethering is to be a valid drug target for ALS, it is essential we know whether these features are actually damaged in human disease. Here we address this issue by examining the ER-mitochondria tethering proteins VAPB and PTPIP51 in post-mortem ALS and control tissues.
Materials and methods
Antibodies
The following primary antibodies were used in this study: Rabbit and rat antibodies to VAPB and PTPIP51 were as described (De Vos et al., 2012). Rabbit anti-PTPIP51 antibody (Anti-RMDN3, HPA009975) and rabbit anti-IP3 receptor type-3 (HPA003915) were from Atlas Antibodies. Rabbit anti-voltage-dependent anion channel-1 (VDAC1) (ab14734) was from Abcam. Mouse anti-neuron specific enolase (NSE) (BBS/NC/VI-H14 -M0873) was from Dako.
Human tissues
Post-mortem human spinal cord samples from control and clinically and pathologically confirmed cases of ALS were obtained from the London Neurodegenerative Diseases Brain Bank, King’s College London. All tissue collection and processing were carried out under the regulations and licensing of the Human Tissue Authority, and in accordance with the Human Tissue Act, 2004. The ALS cases analysed all contained TDP43 positive inclusions.
SDS-PAGE and immunoblotting
Frozen human lumbar spinal cord tissues were prepared for SDS-PAGE as described previously (Lau et al., 2020). Protein concentrations were determined using a bicinchoninic acid protein concentration assay kit (Pierce) according to the manufacturer’s instructions and samples stored at −80°C until required. Samples were separated by SDS-PAGE using Novex 4–12% Tris-glycine gels (Invitrogen) and transferred to Protran nitrocellulose membranes (0.45 μm pore; G.E. Healthcare) using an Invitrogen X-Cell blot II transfer system. After transfer, membranes were blocked in Tris–HCl-buffered saline (TBS, pH 7.3), 0.1% (v/v) Tween-20 containing either 5% (w/v) BSA (for probing for and IP3 receptor type-3) or 5% (w/v) non-fat dried milk powder (for probing all other proteins). Membranes were incubated with primary antibodies in blocking buffer overnight at 4°C, washed and incubated with secondary antibodies and then processed for chemiluminescent detection using a 1:1 dilution of ECL blotting reagents 1 and 2 (GE Healthcare). Chemiluminescent signals were detected with a Bio-Rad ChemiDoc imaging system and analysed using ImageJ; protein signals were normalised to NSE signals from the same sample. Atlas PTPIP51 antibody was used for immunoblots. All primary antibodies were used at 1:2000 apart from anti-NSE which was used at 1:10000 concentration.
Proximity ligations assays (PLAs) and microscopy
VAPB-PTPIP51 PLAs were performed on 7 μm sections of paraffin wax embedded formalin fixed post-mortem human lumbar spinal cord tissues using rabbit VAPB and rat PTPIP51 antibodies essentially as described previously for studies of post-mortem Alzheimer’s disease tissues, and using Duolink In Situ Detection Brightfield kits (Sigma) (Lau et al., 2020). Donkey anti-rabbit in situ PLA probes were purchased directly; donkey anti-rat PLA probes were prepared using Duolink In Situ Probemaker kits (Sigma). Primary antibodies were used at 1:200 concentration. Following PLAs, sections were counterstained with haematoxylin, dehydrated in graded alcohols and xylene, and mounted using DPX mounting reagent. Experimental controls to demonstrate specificity of the PLAs involved omission of VAPB, PTPIP51 or both VAPB and PTPIP51 antibodies.
Sections were imaged using an Olympus VS.120 slide scanner using an Olympus 40x UPlanSApo NA 0.95 lens and driven by Olympus L100 VS-ASW software. Motor neurons were identified by morphology. They are located in the anterior horn of the lumbar spinal cord and are the largest cells in the spinal cord so are easily identified. Images were analysed as previously described using Visiopharm 2018.4 Image Analyses software with an analyse package protocol created with Author Module (Lau et al., 2020). Briefly, the perimeter of each motor neuron was marked which enabled the relative area and the number of PLA dots within each cell to be calculated by the software.
Statistical analyses
Statistical analysis was performed using Excel (Microsoft Corporation) and Prism software (version 9; GraphPad Software Inc.). Statistical significance was determined as described in the figure legends. Correlation analyses were performed as previously described (Lau et al., 2020). Briefly, VAPB-PTPIP51 PLA dot numbers per case were correlated with age and post-mortem delay by generating correlation coefficients and significance was established using parametric, two-tailed Pearson tests.
Results
VAPB levels are reduced in post-mortem ALS spinal cord
Firstly, we investigated the levels of key ER-mitochondria signaling proteins in post-mortem control and ALS spinal cord tissues. We studied VAPB and PTPIP51 since they function to tether ER domains with mitochondria so as to permit signaling, and IP3 receptor and VDAC1 since they represent the major channel for delivery of Ca2+ from ER stores to mitochondria; ER-mitochondria Ca2+ exchange controls several functions perturbed in ALS such as mitochondrial ATP production, autophagy and synaptic activity (De Vos et al., 2012; Stoica et al., 2014; Stoica et al., 2016; Gomez-Suaga et al., 2017; Paillusson et al., 2017; Csordas et al., 2018; Gomez-Suaga et al., 2019; Puri et al., 2019; Gomez-Suaga et al., 2022; Markovinovic et al., 2022). There are 3 isoforms of IP3 receptor (type−1, −2 and −3) which all function to transport Ca2+ to mitochondria (Bartok et al., 2019). These isoforms show different expression patterns in the nervous system. IP3 receptor type-1 is highly expressed in neurons in the cortex, hippocampus and cerebellum, IP3 receptor type-2 is primarily expressed in glia, and IP3 receptor type-3 is the major isoform in brain stem and spinal cord, including motor neurons, but is largely absent in cortex and hippocampus (De Smedt et al., 1994; Sharp et al., 1999; Watanabe et al., 2016). We therefore studied IP3 receptor type-3 levels.
To quantify the levels of these proteins, we performed immunoblots of post-mortem control and ALS spinal cord tissues. Details of these human cases are shown in Table 1 and involve tissues from 16 control and 15 ALS patients. The levels of each protein were normalised to the levels of NSE as described by others in similar studies of human post-mortem neurodegenerative disease tissues (Tiwari et al., 2015; Kurbatskaya et al., 2016; Lau et al., 2016; Tiwari et al., 2016; Morotz et al., 2019a; Morotz et al., 2019b; Lau et al., 2020). Compared to controls, VAPB levels were significantly reduced in the ALS tissues but there were no changes in the levels of PTPIP51, IP3 receptor type-3 or VDAC1 (Figure 1).
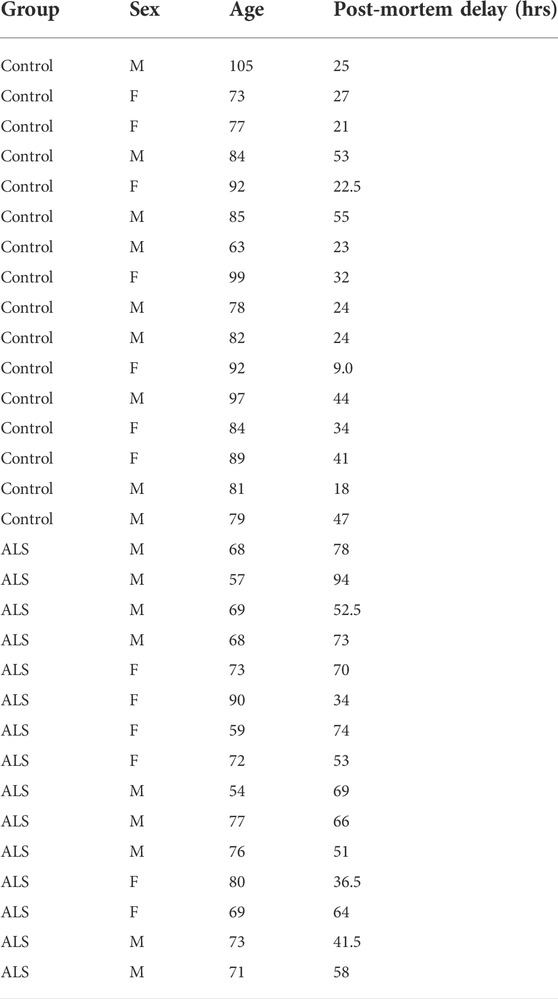
TABLE 1. Data for human post-mortem samples.
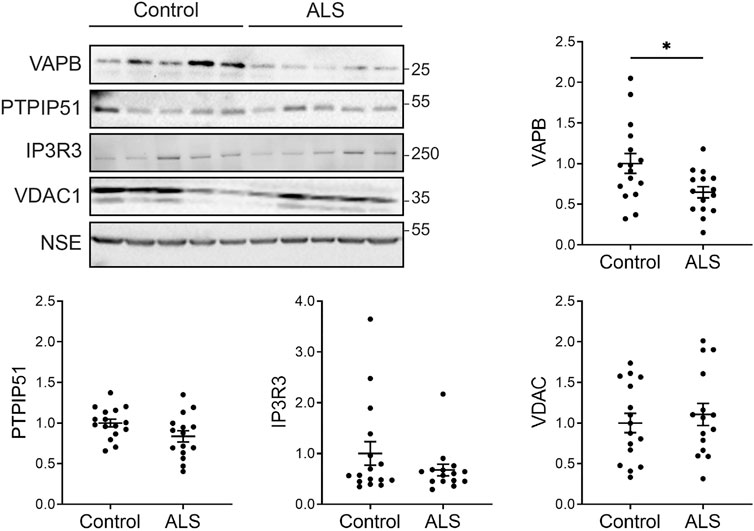
FIGURE 1. Expression of VAPB, PTPIP51, IP3 receptor type-3 and VDAC1 proteins in post-mortem control and ALS spinal cords. Representative immunoblots are shown. Graphs show quantification of protein levels in the different samples following normalisation to NSE levels in the same sample. N = 16 control and 15 ALS cases. Data were analysed by unpaired t-test. Error bars are standard error of means (s.e.m.); *p < 0.05.
The VAPB-PTPIP51 interaction is disrupted in spinal cord motor neurons in post-mortem ALS
To determine whether the VAPB-PTPIP51 interaction is disrupted in human ALS, we used in situ PLAs to quantify their binding in spinal cord motor neurons in the control and ALS tissues. The distances detected by PLAs are up to about 30 nm which makes these assays suitable for quantifying ER-mitochondria contacts (Soderberg et al., 2006; Paillusson et al., 2016). PLAs including ones for VAPB and PTPIP51 have already been used to quantify ER-mitochondria contacts and signaling in models of ALS, Parkinson’s disease and Alzheimer’s disease (De Vos et al., 2012; Hedskog et al., 2013; Bernard-Marissal et al., 2015; Stoica et al., 2016; Paillusson et al., 2017; Gomez-Suaga et al., 2019; Gomez-Suaga et al., 2022). Most recently, such studies have been extended to analyses of the VAPB-PTPIP51 interaction in human post-mortem Alzheimer’s disease brains (Lau et al., 2020).
Firstly, we demonstrated the specificity of the PLAs in control experiments where primary VAPB and/or PTPIP51 antibodies were omitted. Such omission produced none or only very few signals whereas inclusion of both antibodies generated significant positivity (Figure 2). These findings are in agreement with several previous studies including studies of human post-mortem Alzheimer’s disease brains (De Vos et al., 2012; Stoica et al., 2016; Lau et al., 2020).
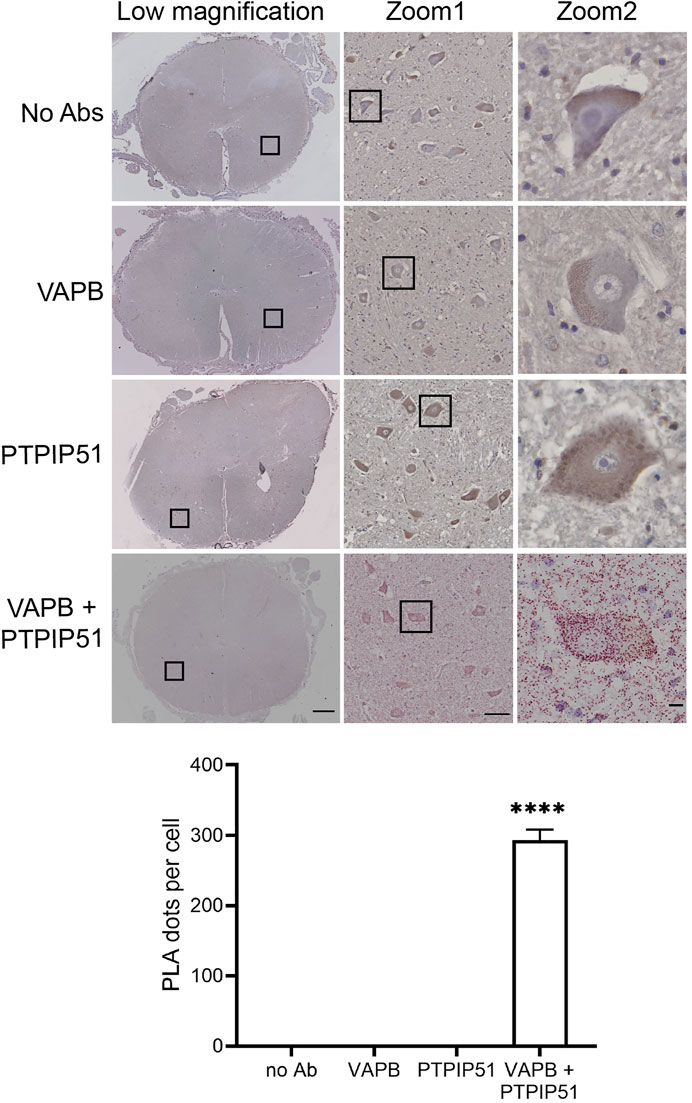
FIGURE 2. Control experiments demonstrating the specificity of VAPB-PTPIP51 PLAs on human post-mortem spinal cord tissues. Controls involved omission of VAPB, PTPIP51, or both VAPB and PTPIP51 primary antibodies (no primary Ab). The graph shows the number of PLA dots per spinal cord motor neuron in the different experiments. Data were analysed by ANOVA and Tukey post hoc test. N = 20–106 per condition, error bars are s. e.m.; ****p < 0.0001. Scale bars: 1,000 μm (Low magnification), 100 μm (Zoom 1) and 10 μm (Zoom 2).
We then quantified the VAPB-PTPIP51 PLA dots in the motor neurons of the 16 control and 15 ALS spinal cords. The number of PLA positive dots per cell were normalised to the area of each cell so as to correct for any changes in neuron size in the ALS cases. Thus, any difference in PLA signal detected in the ALS motor neurons cannot be the consequence of changes in cell size. These studies revealed that compared to controls, VAPB-PTPIP51 PLA signal numbers/cell were significantly reduced in ALS motor neurons (Figure 3A). We also performed correlation analyses to determine whether the number of PLA dots correlated with post-mortem delay or age in the cases studied. These analyses revealed that there were no significant correlations between the number of PLA dots and either of these parameters (post-mortem delay, r = −0.102, p = 0.58; age r = −0.112, p = 0.55).
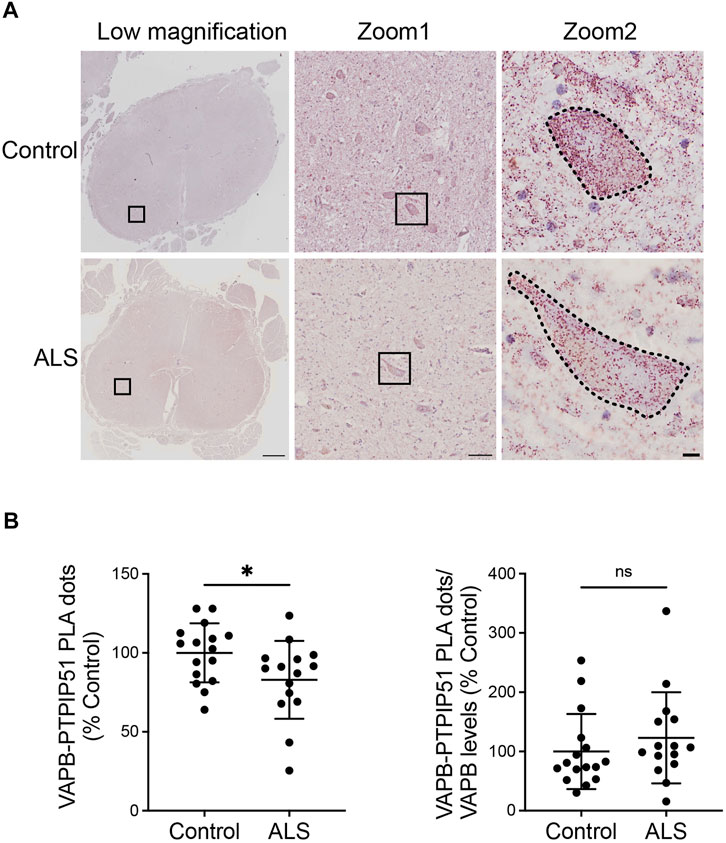
FIGURE 3. The VAPB-PTPIP51 interaction is reduced in ALS spinal cord motor neurons. (A) Representative images of VAPB-PTPIP51 PLAs in control and ALS tissues. Low magnification and two zoom images are shown for each sample; motor neurons are outlined in the highest zoom images. The graph shows the mean number of VAPB-PTPIP51 PLA dots per motor neuron for each case. VAPB-PTPIP51 PLA numbers were normalised to the area of each cell so as to correct for any changes in neuron size in the ALS cases as described in Materials and Methods. (B) Graph showing mean numbers of VAPB-PTPIP51 PLA dots following normalisation to VAPB protein levels. N = 16 control and 15 ALS cases. Data were analysed by unpaired t-test; Error bars are s. e.m., *p < 0.05, ns not significant. Scale bars; 1,000 μm (Low magnification), 100 μm (Zoom 1) and 10 μm (Zoom 2).
Finally, since we detected a significant reduction in VAPB protein levels in the ALS cases, we analysed the impact of this reduction on the VAPB-PTPIP51 interaction by normalising the VAPB-PTPIP51 PLA dot numbers to VAPB protein levels. The significant reduction in VAPB-PTPIP51 PLA dots in the ALS cases was lost following this normalisation (Figure 3B). This suggest that loss of VAPB may contribute to the reduced VAPB-PTPIP51 interaction in ALS.
Discussion
A number of studies have now shown that ER-mitochondria signaling is perturbed in ALS (Lautenschlager et al., 2013; Stoica et al., 2014; Bernard-Marissal et al., 2015; Dafinca et al., 2016; Gregianin et al., 2016; Stoica et al., 2016; Watanabe et al., 2016; Tadic et al., 2017; Dafinca et al., 2020; Dafinca et al., 2021; Gomez-Suaga et al., 2022). However, these studies largely focused on experimental models and to date, there is little evidence that ER-mitochondria contacts and signaling are altered in human ALS patient tissues. Here, we utilised PLA technology to study the VAPB-PTPIP51 interaction in post-mortem ALS spinal cord motor neurons.
Our analysis involved 16 control and 15 ALS cases and so represents a highly powered study. Compared to controls, we detected a significant decrease in VAPB-PTPIP51 PLA signals in the ALS motor neurons. Studies of induced pluripotent stem cell neurons derived from patients carrying pathogenic TDP43 and C9orf72 mutations also support a role for perturbation to ER-mitochondria signaling in ALS and this includes disruption to the VAPB-PTPIP51 interaction (Dafinca et al., 2016; Dafinca et al., 2020; Dafinca et al., 2021; Gomez-Suaga et al., 2022). Our findings thus complement and extend these prior studies.
The mechanisms that underlie the disruption to the VAPB-PTPIP51 tethers in ALS are not clear. Clearly the expression levels of VAPB and PTPIP51 affect their interaction and this in turn has been shown to influence ER-mitochondria contacts and linked functions. Thus, siRNA loss of VAPB and/or PTPIP51 reduce ER-mitochondria contacts, IP3 receptor mediated delivery of Ca2+ to mitochondria, and downstream functions of this Ca2+ delivery (De Vos et al., 2012; Stoica et al., 2014; Gomez-Suaga et al., 2017; Gomez-Suaga et al., 2019). Our finding that VAPB levels are reduced in ALS spinal cord suggests that this loss may contribute to the decrease in the VAPB-PTPIP51 interaction we detect in ALS. Interestingly, others have also reported decreased levels of VAPB in ALS post-mortem tissues (Anagnostou et al., 2010). Indeed, we found that following normalisation of VAPB-PTPIP51 PLA dot numbers to VAPB protein levels, the reduction in the VAPB-PTPIP51 interaction we detected in the ALS cases was lost. This supports the notion that loss of VAPB contributes to the reduced VAPB-PTPIP51 interaction in ALS. Interestingly, there is evidence that VAPB may have other functions aside from ER-mitochondria tethering and that it may act to tether regions of ER with other organelles (Kors et al., 2022). Loss of VAPB may contribute to ALS via mechanisms other than ER-mitochondria tethering.
An alternative possibility is that the ALS linked perturbation of the VAPB-PTPIP51 tethers is linked to activation of glycogen synthase kinase-3β (GSK3β). GSK3β is a negative regulator of the VAPB-PTPIP51 interaction and its activation has been linked to disruption of ER-mitochondria tethering and signaling by ALS mutant TDP43, FUS, and C9orf72 in experimental models (Stoica et al., 2014; Stoica et al., 2016; Gomez-Suaga et al., 2022). However, studying GSK3β activity in post-mortem human tissues is difficult and similar analyses of GSK3β in post-mortem Alzheimer’s disease brains have generated highly conflicting data with some reporting increased and some decreased activity in Alzheimer’s disease (Ferrer et al., 2002; Swatton et al., 2004; Leroy et al., 2007). Such studies have led to the conclusion that it is technically difficult, if not impossible, to measure GSK3β enzymatic activity in post-mortem neurodegenerative disease tissues (Hooper et al., 2008). Analyses of ALS tissues with very short post-mortem times may assist such analyses in future studies.
Whatever the precise mechanisms, our findings demonstrate, for the first time, that the VAPB-PTPIP51 ER-mitochondria tethers are perturbed in human ALS post-mortem motor neurons. As such, they provide clinical support for prior experimental studies which highlighted the role of damaged ER-mitochondria signaling in ALS.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by London Neurodegenerative Diseases Brain Bank, King’s College London. . The patients/participants provided their written informed consent to participate in this study.
Author contributions
CM, WN, and NH planned the study. NH and DL performed most experiments. SM-G, AM, PG-S, and GM analysed data and performed statistical analyses, JG performed additional experiments, EG assisted experiments and CT provided input and expertise for post-mortem tissues. CM, NH, WN, SM-G, AM, PG-S, and GM wrote the manuscript. All authors edited the manuscript.
Funding
This work was supported by grants from the United Kingdom Medical Research Council (MR/R022666/1), Alzheimer’s Research United Kingdom, the Alzheimer’s Society, a Motor Neurone Disease Association Fellowship to PG-S and a King’s College Guy’s and St Thomas’s studentship to NH.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abramzon, Y. A., Fratta, P., Traynor, B. J., and Chia, R. (2020). The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci. 14, 42. doi:10.3389/fnins.2020.00042
Anagnostou, G., Akbar, M. T., Paul, P., Angelinetta, C., Steiner, T. J., and de Belleroche, J. (2010). Vesicle associated membrane protein B (VAPB) is decreased in ALS spinal cord. Neurobiol. Aging 31, 969–985. doi:10.1016/j.neurobiolaging.2008.07.005
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi:10.1016/j.bbrc.2006.10.093
Bartok, A., Weaver, D., Golenar, T., Nichtova, Z., Katona, M., Bansaghi, S., et al. (2019). IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 10 (1), 3726. doi:10.1038/s41467-019-11646-3
Bernard-Marissal, N., Medard, J. J., Azzedine, H., and Chrast, R. (2015). Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138, 875–890. doi:10.1093/brain/awv008
Csordas, G., Weaver, D., and Hajnoczky, G. (2018). Endoplasmic reticulum-mitochondrial contactology: Structure and signaling functions. Trends Cell. Biol. 28, 523–540. doi:10.1016/j.tcb.2018.02.009
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A., et al. (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell. Rep. 14 (5), 892–908. doi:10.1016/j.stemcr.2020.03.023
Dafinca, R., Barbagallo, P., and Talbot, K. (2021). The role of mitochondrial dysfunction and ER stress in TDP-43 and C9ORF72 ALS. Front. Cell. Neurosci. 15, 653688. doi:10.3389/fncel.2021.653688
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34, 2063–2078. doi:10.1002/stem.2388
De Smedt, H., Missiaen, L., Parys, J. B., Bootman, M. D., Mertens, L., Van Den Bosch, L., et al. (1994). Determination of relative amounts of inositol trisphosphate receptor mRNA isoforms by ratio polymerase chain reaction. J. Biol. Chem. 269 (34), 21691–21698. doi:10.1016/s0021-9258(17)31861-6
De Vos, K. J., Morotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi:10.1093/hmg/ddr559
Ferrer, I., Barrachina, M., and Puig, B. (2002). Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer's disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 104, 583–591. doi:10.1007/s00401-002-0587-8
Galmes, R., Houcine, A., van Vliet, A. R., Agostinis, P., Jackson, C. L., and Giordano, F. (2016). ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 17, 800–810. doi:10.15252/embr.201541108
Gomez-Suaga, P., Mórotz, G. M., Markovinovic, A., Martín-Guerrero, S. M., Preza, E., Arias, N., et al. (2022). Disruption of ER-mitochondria tethering and signalling in C9orf72-associated amyotrophic lateral sclerosis and frontotemporal dementia. Aging Cell. 21 (2), e13549. doi:10.1111/acel.13549
Gomez-Suaga, P., Paillusson, S., Stoica, R., Noble, W., Hanger, D. P., and Miller, C. C. (2017). The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 27, 371–385. doi:10.1016/j.cub.2016.12.038
Gomez-Suaga, P., Perez-Nievas, B. G., Glennon, E. B., Lau, D. H. W., Paillusson, S., Morotz, G. M., et al. (2019). The VAPB-PTPIP51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathol. Commun. 7, 35. doi:10.1186/s40478-019-0688-4
Gregianin, E., Pallafacchina, G., Zanin, S., Crippa, V., Rusmini, P., Poletti, A., et al. (2016). Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum. Mol. Genet. 25, 3741–3753. doi:10.1093/hmg/ddw220
Hedskog, L., Pinho, C. M., Filadi, R., Ronnback, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer's disease and related models. Proc. Natl. Acad. Sci. U. S. A. 110, 7916–7921. doi:10.1073/pnas.1300677110
Herms, J., and Dorostkar, M. M. (2016). Dendritic spine pathology in neurodegenerative diseases. Annu. Rev. Pathol. 11, 221–250. doi:10.1146/annurev-pathol-012615-044216
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer's disease. J. Neurochem. 104, 1433–1439. doi:10.1111/j.1471-4159.2007.05194.x
Jiang, J., Zhu, Q., Gendron, T. F., Saberi, S., McAlonis-Downes, M., Seelman, A., et al. (2016). Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550. doi:10.1016/j.neuron.2016.04.006
Kors, S., Costello, J. L., and Schrader, M. (2022). VAP proteins - from organelle tethers to pathogenic host interactors and their role in neuronal disease. Front. Cell. Dev. Biol. 10, 895856. doi:10.3389/fcell.2022.895856
Kurbatskaya, K., Phillips, E. C., Croft, C. L., Dentoni, G., Hughes, M. M., Wade, M. A., et al. (2016). Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer's disease brain. Acta Neuropathol. Commun. 4, 34. doi:10.1186/s40478-016-0299-2
Lau, D. H., Hogseth, M., Phillips, E. C., O'Neill, M. J., Pooler, A. M., Noble, W., et al. (2016). Critical residues involved in tau binding to fyn: Implications for tau phosphorylation in alzheimer's disease. Acta Neuropathol. Commun. 4, 49. doi:10.1186/s40478-016-0317-4
Lau, D. H. W., Hartopp, N., Welsh, N. J., Mueller, S., Glennon, E. B., Morotz, G. M., et al. (2018). Disruption of ER-mitochondria signalling in fronto-temporal dementia and related amyotrophic lateral sclerosis. Cell. Death Dis. 9, 327. doi:10.1038/s41419-017-0022-7
Lau, D. H. W., Paillusson, S., Hartopp, N., Rupawala, H., Mórotz, G. M., Gomez-Suaga, P., et al. (2020). Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer's disease brain. Neurobiol. Dis. 143, 105020. doi:10.1016/j.nbd.2020.105020
Lautenschlager, J., Prell, T., Ruhmer, J., Weidemann, L., Witte, O. W., and Grosskreutz, J. (2013). Overexpression of human mutated G93A SOD1 changes dynamics of the ER mitochondria calcium cycle specifically in mouse embryonic motor neurons. Exp. Neurol. 247, 91–100. doi:10.1016/j.expneurol.2013.03.027
Leroy, K., Yilmaz, Z., and Brion, J. P. (2007). Increased level of active GSK-3beta in Alzheimer's disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 33, 43–55. doi:10.1111/j.1365-2990.2006.00795.x
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi:10.1016/j.neuron.2013.07.033
Markovinovic, A., Greig, J., Martín-Guerrero, S. M., Salam, S., and Paillusson, S. (2022). Endoplasmic reticulum-mitochondria signaling in neurons and neurodegenerative diseases. J. Cell. Sci. 135 (3), jcs248534. doi:10.1242/jcs.248534
Morotz, G. M., Glennon, E. B., Gomez-Suaga, P., Lau, D. H. W., Robinson, E. D., Sedlak, E., et al. (2019a). LMTK2 binds to kinesin light chains to mediate anterograde axonal transport of cdk5/p35 and LMTK2 levels are reduced in Alzheimer's disease brains. Acta Neuropathol. Commun. 7, 73. doi:10.1186/s40478-019-0715-5
Morotz, G. M., Glennon, E. B., Greig, J., Lau, D. H. W., Bhembre, N., Mattedi, F., et al. (2019b). Kinesin light chain-1 serine-460 phosphorylation is altered in Alzheimer's disease and regulates axonal transport and processing of the amyloid precursor protein. Acta Neuropathol. Commun. 7 (1), 200. doi:10.1186/s40478-019-0857-5
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi:10.1126/science.1134108
Paillusson, S., Gomez-Suaga, P., Stoica, R., Little, D., Gissen, P., Devine, M. J., et al. (2017). α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 134, 129–149. doi:10.1007/s00401-017-1704-z
Paillusson, S., Stoica, R., Gomez-Suaga, P., Lau, D. H., Mueller, S., Miller, T., et al. (2016). There's something wrong with my MAM; the ER-Mitochondria axis and neurodegenerative diseases. Trends Neurosci. 39, 146–157. doi:10.1016/j.tins.2016.01.008
Puri, R., Cheng, X. T., Lin, M. Y., Huang, N., and Sheng, Z. H. (2019). Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat. Commun. 10 (1), 3645. doi:10.1038/s41467-019-11636-5
Ringholz, G. M., Appel, S. H., Bradshaw, M., Cooke, N. A., Mosnik, D. M., and Schulz, P. E. (2005). Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65, 586–590. doi:10.1212/01.wnl.0000172911.39167.b6
Robberecht, W., and Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 14, 248–264. doi:10.1038/nrn3430
Sharp, A. H., Nucifora, F. C., Blondel, O., Sheppard, C. A., Zhang, C., Snyder, S. H., et al. (1999). Differential cellular expression of isoforms of inositol 1, 4, 5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 406, 207–220. doi:10.1002/(sici)1096-9861(19990405)406:2<207::aid-cne6>3.0.co;2-7
Soderberg, O., Gullberg, M., Jarvius, M., Ridderstrale, K., Leuchowius, K. J., Jarvius, J., et al. (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000. doi:10.1038/nmeth947
Spires-Jones, T. L., Attems, J., and Thal, D. R. (2017). Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 134, 187–205. doi:10.1007/s00401-017-1709-7
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5, 3996. doi:10.1038/ncomms4996
Stoica, R., Paillusson, S., Gomez-Suaga, P., Mitchell, J. C., Lau, D. H., Gray, E. H., et al. (2016). ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 17, 1326–1342. doi:10.15252/embr.201541726
Swatton, J. E., Sellers, L. A., Faull, R. L., Holland, A., Iritani, S., and Bahn, S. (2004). Increased MAP kinase activity in Alzheimer's and Down syndrome but not in schizophrenia human brain. Eur. J. Neurosci. 19, 2711–2719. doi:10.1111/j.0953-816X.2004.03365.x
Tadic, V., Malci, A., Goldhammer, N., Stubendorff, B., Sengupta, S., Prell, T., et al. (2017). Sigma 1 receptor activation modifies intracellular calcium exchange in the G93AhSOD1 ALS model. Neuroscience 359, 105–118. doi:10.1016/j.neuroscience.2017.07.012
Tiwari, S. S., d'Orange, M., Troakes, C., Shurovi, B. N., Engmann, O., Noble, W., et al. (2015). Evidence that the presynaptic vesicle protein CSPalpha is a key player in synaptic degeneration and protection in Alzheimer's disease. Mol. Brain 8, 6. doi:10.1186/s13041-015-0096-z
Tiwari, S. S., Mizuno, K., Ghosh, A., Aziz, W., Troakes, C., Daoud, J., et al. (2016). Alzheimer-related decrease in CYFIP2 links amyloid production to tau hyperphosphorylation and memory loss. Brain 139, 2751–2765. doi:10.1093/brain/aww205
Watanabe, S., Ilieva, H., Tamada, H., Nomura, H., Komine, O., Endo, F., et al. (2016). Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 8, 1421–1437. doi:10.15252/emmm.201606403
Wheaton, M. W., Salamone, A. R., Mosnik, D. M., McDonald, R. O., Appel, S. H., Schmolck, H. I., et al. (2007). Cognitive impairment in familial ALS. Neurology 69, 1411–1417. doi:10.1212/01.wnl.0000277422.11236.2c
Keywords: VAPB, PTPIP51, endoplasmic reticulum, mitochondria, amyotrophic lateral sclerosis
Citation: Hartopp N, Lau DHW, Martin-Guerrero SM, Markovinovic A, Mórotz GM, Greig J, Glennon EB, Troakes C, Gomez-Suaga P, Noble W and Miller CCJ (2022) Disruption of the VAPB-PTPIP51 ER-mitochondria tethering proteins in post-mortem human amyotrophic lateral sclerosis. Front. Cell Dev. Biol. 10:950767. doi: 10.3389/fcell.2022.950767
Received: 23 May 2022; Accepted: 18 July 2022;
Published: 16 August 2022.
Edited by:
Riccardo Filadi, National Research Council (CNR), ItalyReviewed by:
Xiangming Wang, Institute of Biophysics (CAS), ChinaGiorgia Pallafacchina, University of Padua, Italy
Copyright © 2022 Hartopp, Lau, Martin-Guerrero, Markovinovic, Mórotz, Greig, Glennon, Troakes, Gomez-Suaga, Noble and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patricia Gomez-Suaga, cGF0cmljaWEuZ29tZXotc3VhZ2FAa2NsLmFjLnVr; Wendy Noble, d2VuZHkubm9ibGVAa2NsLmFjLnVr; Christopher C.J. Miller, Y2hyaXMubWlsbGVyQGtjbC5hYy51aw==