 Umer Farooq1,2*
Umer Farooq1,2* Dimple Notani1*
Dimple Notani1*- 1Genetics and Development, National Centre for Biological Sciences, Tata Institute for Fundamental Research, Bangalore, India
- 2The University of Trans-Disciplinary Health Sciences and Technology, Bangalore, India
9p21 locus is one of the most reproducible regions in genome-wide association studies (GWAS). The region harbors CDKN2A/B genes that code for p16INK4a, p15INK4b, and p14ARF proteins, and it also harbors a long gene desert adjacent to these genes. The polymorphisms that are associated with several diseases and cancers are present in these genes and the gene desert region. These proteins are critical cell cycle regulators whose transcriptional dysregulation is strongly linked with cellular regeneration, stemness, aging, and cancers. Given the importance of this locus, intense scientific efforts on understanding the regulation of these genes via promoter-driven mechanisms and recently, via the distal regulatory mechanism have provided major insights. In this review, we describe these mechanisms and propose the ways by which this locus can be targeted in pathologies and aging.
Introduction
The INK4/ARF locus functions are attributed to three distinct but related proteins, namely, p14ARF, p16INK4a, and p15INK4b. These proteins are coded by two genes; CDKN2A and CDKN2B. p14ARF and p16INK4a are transcribed from the CDKN2A gene, whereas p15INK4b is transcribed from the CDKN2B gene (Figure 1). The initial exons of p14ARF (exon1β) and p16INK4a (exon1α) are different, but the second and third exons are identical. While the mRNA sequences of p14ARF and p16INK4a are relatively similar, the resultant proteins do not share any sequence similarity due to the alternative reading frames; thus, these proteins are not isoforms. On the other hand, p15INK4b and p16INK4a have a high degree of amino acid similarity (about 80%) and are thought to have emerged from a gene duplication event (Lopez et al., 2017). Additionally, there is a CDKN2BAS gene that transcribes a non-coding RNA known as ANRIL. Because ANRIL is transcribed in the antisense direction relative to CDKN2B, the gene is termed CDKN2BAS. Together, these proteins regulate the cell cycle progression and are known to operate as a barrier to the reprogramming of somatic cells. Inactivation of this locus due to homozygous deletions or epigenetic alterations such as transcriptional silencing by DNA methylation or polycomb-mediated suppression is a frequent event that occurs in a wide spectrum of cancers. Furthermore, single nucleotide polymorphisms (SNPs) in this locus are associated with several aging-related disorders, including coronary artery disease (CAD), type 2 diabetes, and atherosclerosis. The majority of the SNPs in this locus are located within the genes and the ∼0.3 Mb long adjacent gene desert region, but the mechanisms of their action are largely unknown. Thus, the identification of molecular pathways that regulate this locus in different diseases is of great therapeutic relevance.
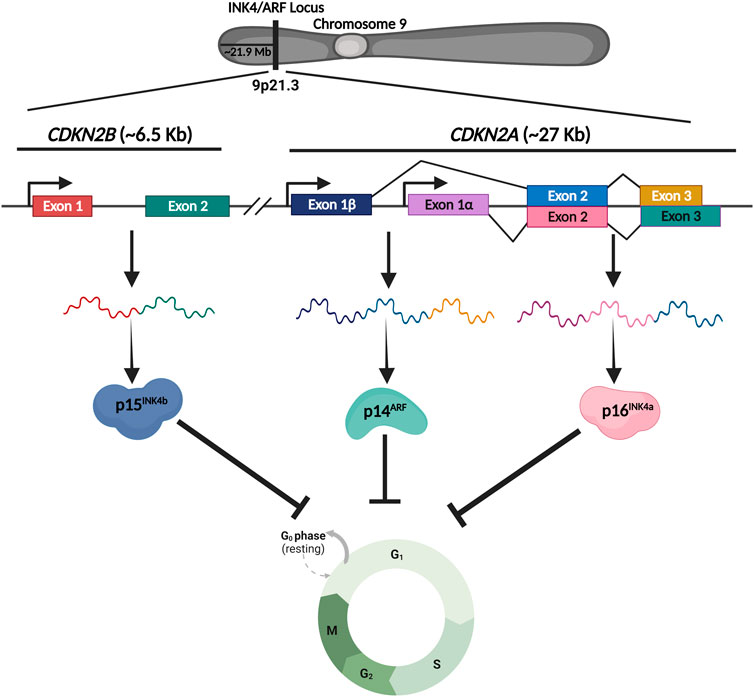
FIGURE 1. Schematic representing the genomic structure of INK4/ARF locus. INK4/ARF locus harbors two genes, CDKN2A and CDKN2B, that code for three critical cell cycle regulators. CDKN2A gene produces two proteins, p161NK4a and p14ARF, whereas the CDKN2B gene produces p151NK4b. Together these genes regulate the cell cycle under various conditions.
Cell cycle regulation by INK4/ARF proteins
Several stress signals including oncogene overexpression, DNA damage, oxidative stress, etc., induce the expression of INK4/ARF genes (Romagosa et al., 2011). Once activated, these genes trigger a cascade of signaling events that effectively bring the cell cycle to a halt (Ivanchuk et al., 2001). Mechanistically, p53 (a well-studied tumor suppressor that blocks the cell cycle at the G1 phase) is a downstream effector of the p14ARF pathway (Sherr 2001). The interaction of MDM2 with p53 alters the stability and cellular localization of p53 (Kubbutat, Jones, and Vousden 1997). MDM2 acts as an E3 ubiquitin ligase and mediates the proteasomal degradation of p53 by ubiquitinating its C-terminal domain (Wade, Wang, and Wahl 2010). Multiple domains of p53 interact with MDM2, including the DNA binding domain (DBD), the transactivation domain (TAD), and the carboxy-terminal domain (CTD). MDM2, on the other hand, interacts with p53 via its N-terminal hydrophobic domain (HD) and acid domain (AD) (Chi et al., 2005; Yu et al., 2006; Poyurovsky et al., 2010). When expressed, p14ARF interacts with the acid domain of MDM2, preventing it from interacting with p53. This interaction alters the conformation of MDM2 that exposes its Nucleolar localization signal (NoLS) present in the RING domain (RD), leading to sequestration of the MDM2-p14ARF complex in the nucleolus (Weber et al., 1999; Maggi et al., 2014). Sequestration of MDM2 in the nucleolus prevents MDM2-mediated export of p53 to the cytoplasm, hence preventing its degradation (Maggi et al., 2014). These events lead to p53 translocation into the nucleus thereby, activating genes that cause the cell cycle to arrest at the G1 phase (Weber et al., 1999) (Figure 2A).
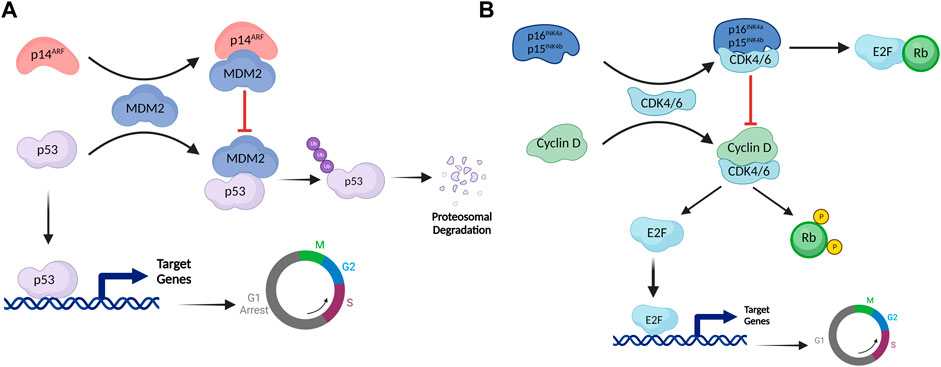
FIGURE 2. The INK4/ARF cell cycle regulatory network. (A) The p53-MDM2 pathway is controlled by the upstream effector protein p14ARF. By establishing a complex with MDM2, p14ARF permits p53 to activate its transcriptional targets. MDM2 ubiquitinates p53, which mediates proteasomal degradation in normal conditions. However, when MDM2 interacts with p14ARF, NoLS of MDM2 is exposed, resulting in MDM2 sequestration in the nucleolus. MDM2 sequestration prevents degradation of p53, allowing it to activate its transcriptional targets and arrest the cell cycle in the G1 phase. (B) The retinoblastoma pathway is regulated by p16INK4a and p15INK4b. E2F is a transcription factor that activates genes involved in the transition from G1 to M phase. Rb inhibits this function of E2F by establishing a complex with it. Under normal conditions, the cyclin D-CDK4/6 complex phosphorylates Rb. Phosphorylated Rb doesn't engage with E2F, as a result, E2F binds to target genes to activate them. Once expressed, p16INK4a/p15INK4b inhibits cyclin D-CDK4/6 complex formation, keeping Rb hypophosphorylated. Hypophosphorylated Rb forms a complex with E2F, inhibiting its transcriptional activity.
p16INK4a and p15INK4b, on the other hand, regulate the retinoblastoma (Rb) pathway. These proteins activate Rb, a tumor suppressor protein that blocks the cell cycle at the G1 phase (Kim and Sharpless 2006). CDK4/6 typically forms an active complex with cyclin D that binds to and phosphorylates Rb (Cobrinik 2005). Rb loses its ability to interact with the E2F transcription factor in the phosphorylated state (Dimova and Dyson 2005). E2F activates genes involved in the cell cycle transition from G1 to S (Giacinti and Giordano 2006). When stress signals activate p16INK4a/p15INK4b, these proteins bind to CDK4/CDK6, causing an allosteric shift in the latter proteins, preventing them from forming the active complex with cyclin D, thereby maintaining Rb in a hypophosphorylated state (Hannon and Beach 1994; Russo et al., 1998). Hypophosphorylated Rb binds with the transactivation domain of E2; this complex subsequently recruits HDAC1 and SUV39H1 to the E2F target genes, thereby inhibiting them and preventing the G1 to S phase transition (Giacinti and Giordano 2006) (Figure 2B). These proteins being high in cellular senescence, permanently inhibit cell division. However, HPV-positive cancer cells express significant levels of p16INK4a, p14ARF, and p15INK4b without undergoing cell cycle arrest, attributed to two HPV-encoded oncoproteins, E6 and E7 (Kanao et al., 2004). These proteins inhibit the downstream effectors of p14ARF and p16INK4a genes, thereby preventing cell cycle arrest. E7 interacts with Rb, leading to its inactivation, whereas E6 induces the degradation of p53 protein (Munger et al., 1992).
Implication of INK4/ARF locus in aging, cancer, and regeneration
INK4/ARF locus in senescence/aging
Senescence is an innate cellular response in which normally proliferating cells cease to divide permanently in response to specific intrinsic and extrinsic stimuli. Senescent cells exhibit morphological and physiological changes, the formation of senescence-associated heterochromatin foci (SAHF), and the release of senescence-associated secretory phenotype (SASP), etc., (van Deursen 2014). This irreversible cell cycle halt is thought to be the first line of defense against cancer by preventing the division of abnormal cells (Prieto and Baker 2019). Senescence, on the other hand, plays a significant role in aging-related pathologies, as it impairs tissue repair and regeneration (McHugh and Gil 2018). Several recent studies have expanded our understanding of the role of senescence in other complex biological processes such as development, and tissue repair, among others (Herranz and Gil 2018). p16INK4a is the fundamental driver and a well-established biomarker of senescence (Krishnamurthy et al., 2004; Rayess, Wang, and Srivatsan 2012). Studies have demonstrated that ectopic expression of oncogenes like Ras and Raf, increases p16INK4a expression, triggering premature senescence in various cell types (Lin et al., 1998; Zhu et al., 1998). For example, fibroblasts, epithelial cells, and T lymphocytes, express higher p16INK4a when they approach replicative senescence (Lin et al., 1998; Zhu et al., 1998; Mirzayans et al., 2012). In summary, the INK4/ARF locus regulates oncogene-induced and replicative senescence in several cell types (Mirzayans et al., 2012).
INK4/ARF locus in cancer
Cancer cells proliferate abnormally and do not respond to signals that regulate cell growth and division. Most frequently, cancer cells contain mutations in genes that regulate the cell cycle; once altered, these genes lose their ability to control the cell cycle (Papp and Plath 2011). As mentioned previously, INK4/ARF genes are cell cycle regulators that arrest the cell cycle at various stages in response to stress signals such as DNA damage. These tumor suppressor genes must be silenced for cancer to progress. Thus, INK4/ARF locus harbors homozygous deletions in several malignancies, silencing the expression of all three cell cycle regulator genes (Sherr 2012). Similarly, loss of p16INK4a expression through specific point mutations has been reported in several cancers (Forbes et al., 2006). The suppression of this locus by DNA hypermethylation at the promoters or through histone modifications mediated by the PRC2 complex is also prevalent in cancers. In animal studies, mice lacking either INK4a or ARF gene are more susceptible to certain tumors than mice lacking the INK4b gene. On the other hand, overexpression of the INK4/ARF genes results in a threefold reduction in tumor incidence in mice (Matheu et al., 2004).
INK4/ARF locus in cellular reprogramming
Cellular plasticity facilitates the reprogramming of somatic cells to a more pluripotent state. This reprogramming process considerably alters the epigenetic and chromatin landscapes of the cells (Papp and Plath 2011). A few critical transcription factors, like Oct4, Sox2, Klf4, Nanog, and others, can transform a somatic cell into a pluripotent cell (Papp and Plath 2011). However, the primary limitation of reprogramming is its significantly lower efficiency (approx. 1%). In the fast-dividing embryonic stem cells and induced pluripotent stem cells (iPSCs), the INK4/ARF locus is repressed. This locus, however, is activated during the reprogramming process as a result of highly mitogenic cell culture conditions (Sharpless 2005). As a result of the activation of this locus in somatic cells, reprogramming efficiency decreases significantly. Conversely, mouse embryonic fibroblasts (MEFs) lacking the INK4/ARF locus reprogram more efficiently with 15-fold higher efficiency (Li et al., 2009). While silencing INK4a or ARF alone improves reprogramming efficiency, double silencing results in increased efficiency, as seen in INK4/ARF null cell lines (Li et al., 2009). Not only is the efficiency increased, but the rate at which iPSC colonies develop is also increased in INK4/ARF defective cells. Interestingly, ARF is the primary regulator of cell reprogramming in murine cells, but INK4a is the dominant regulator in humans (Li et al., 2009).
Transcriptional regulation of INK4/ARF locus
Repression of INK4/ARF locus via PRC complexes
Polycomb group (PcG) proteins are epigenetic modifiers that play a crucial role in transcriptional repression and therefore regulate cell proliferation, differentiation, embryonic development, cellular memory, and other vital cellular functions (Wang et al., 2015). PcGs form two major protein complexes, the Polycomb repressive complex 1 (PRC1) and the Polycomb repressive complex 2 (PRC2). PRC2 exerts inhibition by adding trimethyl marks to lysine 27 of histone 3 (H3K27me3). The trimethyl mark serves as a docking site for PRC1, which recognizes this mark and monoubiquitinates Histone 2A at lysine 119 (H2AK119ub) (Chittock et al., 2017). The H2AK119ub further enhances H3K27me3 deposition by PRC2 and subsequent recruitment of PRC1 (Chittock et al., 2017). Both PRC1 and PRC2 are multimeric protein complexes with several core subunits and a few auxiliary subunits (Kerppola 2009). The PRC1 core consists of RING1A/B, PCGF2/4, CBX2/4/6/7/8, PHC1/2/3 subunits, while PRC2 contains Suz12, Ezh2, RbAp46/48, and Eed as core subunits (Chittock et al., 2017). Additionally, various auxiliary subunits aid or improve the activity of these complexes (Chittock et al., 2017). Ezh2, a SET domain-containing protein, is the enzymatic component of the PRC2 complex responsible for the trimethylation of H3K27. At the INK4/ARF locus, PcG inhibits the promoters by trimethylating H3K27 to increase cell proliferation (Bracken et al., 2007). PcGs have been demonstrated to repress all three INK4a, ARF, and INK4b genes in some instances, but only INK4a and INK4b in others (Bruggeman et al., 2005; Bracken et al., 2007; Kheradmand Kia et al., 2009). Ectopic expression of PcG subunits such as Bmi1, Ezh2, CBX7, and CBX8 has been shown to downregulate INK4a and INK4b expression to bypass senescence (Jacobs et al., 1999; Gil et al., 2004; Dietrich et al., 2007). In contrast, depletion of the PcG subunits activates this locus, resulting in cell growth inhibition and senescence (Bracken et al., 2007; Dietrich et al., 2007).
Several transcription factors facilitate PcG binding to INK4/ARF promoters; for example, Zfp277, a zinc finger protein, interacts with the Bm1 subunit of PRC1 to recruit PRC1 to these promoters in MEFs (Negishi et al., 2010). Zfp277 depletion causes the PRC1 complex to displace from the promoters, activating INK4a/ARF gene and early senescence (Negishi et al., 2010). Similarly, Homeobox proteins such as HLX1 and HOXA9 play an essential role in suppressing INK4a. These proteins cooperate with PRC2 and HDACs at the INK4a promoter to mediate the repression (Martin et al., 2013). Haematopoietically expressed homeobox gene (Hhex) is vital in maintaining acute myeloid leukemia (AML), as its deletion causes upregulation of INK4a and ARF. Further, Hhex, like HLX1 and HOXA9, facilitates PRC2 binding to the promoters by interacting with the Suz12 subunit, thereby repressing the genes (Shields et al., 2016). In neonatal human diploid fibroblasts (HDFs), PRC2 binding to the INK4a promoter and the upstream region of the INK4b promoter induces a long-range interaction (repressive chromatin loop) between these promoters (Kheradmand Kia et al., 2009). Similar long-range interaction between the INK4a and INK4b promoters has been observed in hematopoietic progenitor cells and malignant rhabdoid tumors (MRTs) (Kheradmand Kia et al., 2009). In mature HDFs, however, the chromatin architecture of these genes is noticeably different where the looping between INK4a and INK4b is lost. Under such alterations, transcriptional activation and senescence induction occurs due to the concomitant loss of Ezh2 binding on promoters (Kheradmand Kia et al., 2009).
JMJD3-mediated transcriptional activation of INK4/ARF locus
Jumonji domain-containing D3 protein (JMJD3) is a lysine-specific histone demethylase. Its role in development, cancer progression, infectious diseases, immune disorders, and other conditions has been extensively studied (Xiang et al., 2007; Zhang X et al., 2019). JMJD3 belongs to the Jumonji (JmjC) domain-containing protein family, and this domain enzymatically catalyzes the removal of trimethyl marks from Histone 3 at lysine 27 (H3K27me3). Ubiquitously transcribed TPR protein on the X chromosome (UTX) is another demethylase that also demethylates H3K27me3 (Agger et al., 2007). While UTX is ubiquitously expressed, JMJD3 is induced in response to certain signaling events such as stress, etc (Swigut and Wysocka 2007). Due to its antagonistic role relative to PcG proteins, JMJD3 is a positive regulator of the INK4/ARF during the onset of cellular senescence (Agger et al., 2009; Barradas et al., 2009). Many cellular signals have been implicated in the induction of JMJD3 expression and subsequent activation of the INK4/ARF genes. For example, oncogene-mediated upregulation of JMJD3 causes activation of INK4/ARF genes in various cell types like fetal lung fibroblasts (IMR90), MEFs, etc., which results in INK4a-mediated growth arrest in these cells (Agger et al., 2009). By activating the INK4/ARF locus, JMJD3 prevents Schwann cells from proliferating uncontrollably in response to tumorigenic signals or following injury (Gomez-Sanchez et al., 2013). Under these conditions, JMJD3 binds to and demethylates the INK4/ARF promoters, activating these genes and initiating senescence. These cells lose the cell cycle control and continue to proliferate, resulting in neurofibromas when this pathway is disturbed (Gomez-Sanchez et al., 2013). As mentioned in previous section, INK4/ARF locus functions as a barrier to MEFs and keratinocyte reprogramming, and its silencing enhances reprogramming efficiency (Li et al., 2009). JMJD3 increases p16INK4a and p14ARF expression, limiting MEF reprogramming (Zhao et al., 2013). Therefore, JMJD3 silencing inhibits INK4/ARF-mediated cellular senescence, improving reprogramming efficiency. Moreover, double knockdown of JMJD3 and INK4a or ARF further enhances the reprogramming efficiency (Zhao et al., 2013).
KDM2B-mediated transcriptional repression of INK4/ARF locus
KDM2B is an epigenetic modifier that preferentially demethylates trimethylated lysine 4 (H3K4me3) and dimethylated lysine 36 of histone H3 (H3K36me2) (Frescas et al., 2007). It regulates numerous biological processes, including cellular senescence, differentiation, and stem cell self-renewal (He et al., 2008; Liang et al., 2012; He et al., 2013). Furthermore, it is highly expressed in various cancers and plays a crucial role in cancer progression, especially in leukemia (Yan et al., 2018). KDM2B associates with the promoters of the INK4/ARF genes and demethylates histones H3K36me2 and H3K4me3. Demethylation results in a decrease in PolII binding and an increase in H3K27me3. KDM2B suppresses this locus by epigenetic modifications of histones and also by preventing the downregulation of Ezh2 (Tzatsos et al., 2009). Consequently, KDM2B protects MEFs from replicative and oncogenic senescence, and its knockdown decreases proliferation and induces senescence. Another study showed that KDM2B functions as a proto-oncogene and inhibits senescence by negatively regulating INK4b. Similarly, KDM2B achieves repression of INK4b by removing the active H3K36me2 mark near the promoter and the gene body, whereas its knockdown causes increased expression of INK4b (He et al., 2008).
DNA methylation-mediated transcriptional repression of INK4/ARF locus
In addition to the aforementioned mechanisms, DNA methylation is another epigenetic mechanism to silence INK4/ARF genes. DNA methylation is catalyzed by DNA methyltransferases (Dnmts), which transfer the methyl group from S-adenosyl methionine (SAM) to carbon 5 of cytosine to generate 5-methylcytosine (5mC) (Lyko 2018). CpG islands in the promoters of tumor suppressor genes undergo abnormal hypermethylation in cancers (Robertson and Jones 1998). Notably, INK4a was one of the first genes discovered to be silenced in human cancers as a result of DNA methylation (Esteller et al., 2001). Numerous malignancies have been linked to aberrant CpG island methylation in the promoter region of the INK4/ARF genes. CpG islands are present near the promoter of ARF and exon1α of INK4a (Robertson and Jones 1998). Aberrant methylation of the ARF promoter is more prevalent than INK4a (Dominguez et al., 2003). Numerous types of cancer, including colon cancer, Merkel cell carcinoma, breast cancer, bladder tumors, and oligodendrogliomas, harbor abnormal DNA methylation of these genes (Watanabe et al., 2001; Tsujimoto et al., 2002; Lee et al., 2006; Lassacher et al., 2008).
Chromatin remodelling of INK4/ARF locus via SWI/SNF complex
SWI/SNF is a multi-subunit ATP-dependent complex. This complex is largely involved in chromatin remodelling, which facilitates gene transcription by allowing transcription factors to access their DNA binding sites (Wilson and Roberts 2011). Abnormal expression and mutations in the SWI/SNF components can cause cancer (Klochendler-Yeivin, Muchardt, and Yaniv 2002; Orlando et al., 2019). Malignant rhabdoid tumors (MRTs) exhibit the loss of the hSNF5 gene, which encodes one of the subunits of the SWI/SNF complex (Biegel et al., 1999; Sevenet et al., 1999; Roberts and Orkin 2004). On the other hand, ectopic expression of hSNF5 inhibits cell growth and induces cellular senescence (Oruetxebarria et al., 2004). It was found that hSNF5 exerts these effects via the p16INK4a/Rb pathway, as the re-expression of hSNF5 in MRT cells activated INK4b and INK4a, but not ARF (Chai et al., 2005; Kia et al., 2008). hSNF5 activates INK4a in these cells by recruiting the SWI/SNF complex to its promoter. As a result of its recruitment, the PRC1 and PRC2 complexes are displaced from the promoter (Kia et al., 2008).
Transcription factors involved in the regulation of INK4/ARF locus
Many transcription factors have been implicated in the regulation of the INK4/ARF locus. The majority of these transcription factors have been shown to act directly on the promoters of these genes. A few of them have been found to operate via upstream enhancer elements. While several transcription factors are required for the activation of the INK4/ARF genes, some have been demonstrated to inhibit their transcription (Figure 3). Due to the limited scope of this review, we have described only a few transcription factors involved in the activation of this locus.
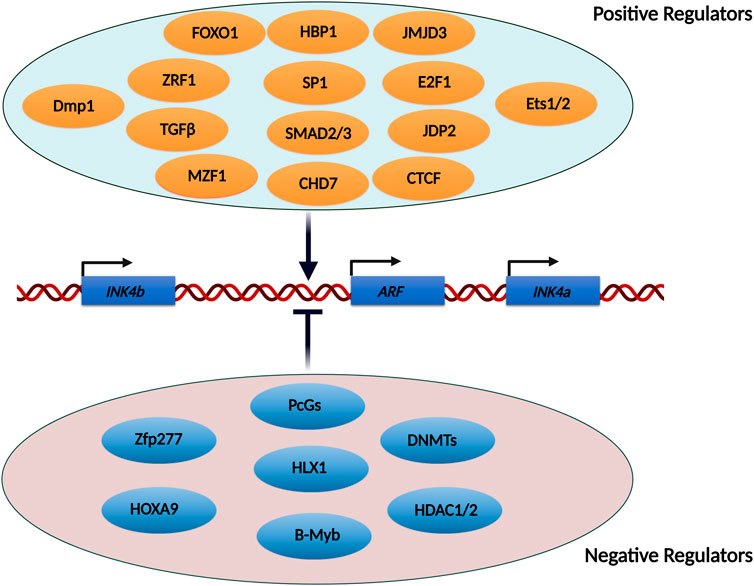
FIGURE 3. List of transcription factors and epigenetic modifiers known to regulate the INK4/ARF locus. The INK4/ARF locus is regulated by a number of transcription factors and epigenetic modifiers. Some of the factors stimulate transcription from this locus, whereas others repress it. Some of these factors act directly on the promoters, facilitating the binding of RNA polymerase, while others activate enhancers located upstream of the genes.
FOXO1. FOXO1 is a tumor suppressor protein that inhibits Myc-induced lymphomagenesis in mice by activating the ARF gene. FOXO1 directly regulates ARF expression by binding to a motif located in the intron between exon1β and exon1α (Bouchard et al., 2007).
ZRF1. Zuotin-related factor 1 (ZRF1), a ubiquitin recognition domain-containing transcription factor, promotes the expression of PRC1-repressed genes during differentiation by competing for H2AK119Ub with PRC1 (Richly et al., 2010). ZRF1 expression is enhanced in MEFs and is recruited to the INK4/ARF promoters following hRas overexpression. Ectopic expression of ZRF1 activates INK4a and INK4b in IMR90, but not ZRF1delUBD, showing that ZRF1 binding to H2AK119Ub is necessary for its recruitment during senescence. (Ribeiro et al., 2013).
Dmp1. The deletion of Dmp1, a well-characterized tumor suppressor, accelerates tumor growth in mice. It acts as a link between Ras/Raf overexpression and INK4/ARF gene activation. Dmp1 expression is promoted by Ras overexpression, and it enhances ARF transcription by directly binding to Dmp1/ETS motif present in its promoter (Sreeramaneni et al., 2005).
JDP2. Jun dimerization protein 2 (JDP2) is a transcription factor that binds to JDP2 response regions and prevents histone acetylation and methylation (Huang et al., 2011). JDP2 is required for normal cell differentiation and proliferation, as MEFs lacking JDP2 do not undergo replicative senescence. Its overexpression inhibits MEF proliferation by increasing the expression of INK4a and ARF (Nakade et al., 2009).
CTCF. In U2OS cells, CTCF binds to a DNA sequence near the ANRIL promoter, and its silencing results in down-regulation of all three INK4/ARF genes. CTCF binding is lost when its DNA motif is methylated, resulting in the downregulation of these genes (Rodriguez et al., 2010).
CHD7. Chromodomain helicase DNA binding protein 7 (CHD7) is an ATP-dependent chromatin remodeler that plays a critical role in Ras-mediated senescence. It is essential for the activation of INK4a following Ras overexpression (Su et al., 2018). Transcription factors like c-Jun and Ets1 promote Myeloid zinc finger 1 (MZF1) expression during Ras-induced senescence, which in turn recruits CHD7 to the promoter of INK4a for its upregulation (Wu et al., 2022).
HBP1. HMG box-containing protein 1 (HBP1) transcription factor is a downstream effector protein in the Ras signaling pathway. INK4a promoter contains a putative binding motif for this transcription factor between positions −426 and −433. Its binding to this motif triggers cellular senescence (Li et al., 2010). HBP1 promotes acetylation of the INK4a promoter by assisting in the recruitment of histone acetyltransferase p300 and CREB-binding protein (CBP) (Wang et al., 2012). Furthermore, ectopic expression of HBP1 induces premature cellular senescence in normal fibroblasts via INK4a, while its knockdown delays senescence and senescence-associated phenotypes (Wang et al., 2012).
SP1. INK4a promoter has numerous GC-rich regions that are required for its induction upon senescence onset (Wu et al., 2007). SP1, a transcription factor, with a strong affinity for GC-rich motifs binds to these regions to enhance INK4a expression. In human fibroblasts, ectopic expression of SP1 upregulates the INK4a (Wu et al., 2007). Furthermore, SP1, like HBP1 physically interacts with p300/CBP to promote INK4a expression. (Wang et al., 2008).
Transcriptional regulation of INK4/ARF locus by distal regulatory elements
As stated earlier, the gene desert region upstream of the CDKN2A/B genes contains several SNPs that are strongly associated with the risk of CAD and type 2 diabetes in humans. In mice, deletion of this CAD (70 kb) interval resulted in a substantial decrease in the cardiac expression of CDKN2A/B genes, significantly increased mortality upon high cholesterol diet and ARF-dependent developmental abnormalities (Visel et al., 2010). Primary cells isolated from such mice showed increased proliferation compared to wild-type cells and exhibited no signs of senescence over subsequent passages (Visel et al., 2010). Furthermore, allele-specific expression analysis in heterozygous mice carrying a CAD interval deletion on one chromosome revealed that the cdkn2b gene was preferentially expressed from the allele with a wild-type CAD interval, but the expression of the allele bearing the CAD deletion was dramatically reduced in the heart and other organs, implying that CAD interval may regulate these genes through a distant-acting cis-regulatory mechanism (Visel et al., 2010). Further work indicated that the mice lacking the CAD interval developed primary vitreous hyperplasia at the E13.5 developmental stage. It is well established that TGFβ regulates ARF expression in developing eyes and MEFs (Freeman-Anderson et al., 2009). It leads to ARF induction in MEFs and HeLa cells (Zheng et al., 2010).
In pursuit of understanding how this interval regulates the expression of INK4/ARF genes and to biologically underpin the genetic variations in the interval seen in several diseases, the interval was tested for the presence of distal regulatory elements known as enhancers. Towards this, a landmark study established the presence of several enhancers in the gene desert region of this locus (Harismendy et al., 2011). A relationship between the CAD-associated genetic variations (rs10811656 and rs10757278) in one of the enhancer elements (ECAD9) where STAT1 binds upon IFNγ stimulation was established. STAT1 binding on the homozygous CAD risk allele was reduced in lymphoblastoid cells (LCL) therefore, the knockdown of STAT1 in LCLs that were homozygous for the non-risk CAD allele upregulated CDKN2BAS suggesting a repressive role of STAT1 on CDKN2BAS expression. However, HUVEC cells exhibited an activatory role of STAT1 on the expression of CDKN2BAS suggesting, the effects of CAD risk allele on INK4/ARF genes could be cell-type specific (Harismendy et al., 2011). Notably, the CAD risk interval contains a cis-acting enhancer that collaborates with TGFβ to promote ARF expression during development (Zheng et al., 2013), and mice lacking the CAD interval don’t show such induction of ARF, implying that TGFβ works on ARF via the enhancers in CAD interval. Furthermore, TGFβ induces three H3K27ac peaks at the 110 kb distance from the CDKN2A promoter in HeLa cells and the deletion of a 20 kb area spanning all three peaks significantly lowers ARF and INK4b expression (Liu et al., 2019). These findings imply that TGFβ affects the transcription of these genes by activating the enhancers upstream of the genes (Liu et al., 2019). Macrophages derived from mice with an atherosclerosis susceptibility locus express significantly lower levels of INK4a and ARF (p19 in mice), but not INK4b (Kuo et al., 2011). Furthermore, individuals with the risk allele rs10757278, which has been related to an increased risk of atherosclerosis, have lower expression of all three INK4/ARF genes and even ANRIL in peripheral blood T-cells (Liu et al., 2009). Another study discovered a cis-regulatory region adjacent to the ARF promoter that represses INK4a gene expression. This element loops with the promoter of INK4a to repress its transcription. Perturbation of the element stimulated the transcription of the INK4a gene. (Zhang Y et al., 2019). All of these studies show a connection between disease-associated SNPs in the gene desert interval and INK4/ARF gene expression. Taken together, the risk alleles for CAD and atherosclerosis are primarily associated with lower expression of the INK4/ARF genes and these effects are cell-type specific.
Recently, we showed that the gene desert region upstream of the INK4/ARF genes contains 21 potential enhancer elements in the HeLa cells. Among these enhancers, 15 enhancers exhibited marks of active enhancers such as H3K27ac, PolII, and eRNA transcription. Out of these, only five active enhancers interacted with the CDKN2A/B gene promoters. However, disruption of any of these interacting enhancers but not non-interacting enhancer impacted the expression of INK4a, ARF, and INK4b at similar levels (Farooq et al., 2021). Interestingly, the interacting and non-interacting enhancers were indistinguishable at the levels of enhancer marks such as levels of H3K27ac, p300, and eRNA expression. This indicates that the bio-chemical marking of enhancers alone fails to predict enhancer activity (Farooq et al., 2021) (Figure 4). However, how SNPs in these enhancers regulate the locus in various diseases requires more efforts focused on functional studies to molecularly underpin the genetic variation and associated diseases in this locus. The resultant mechanistic understanding will pave the way for future therapeutic interventions.
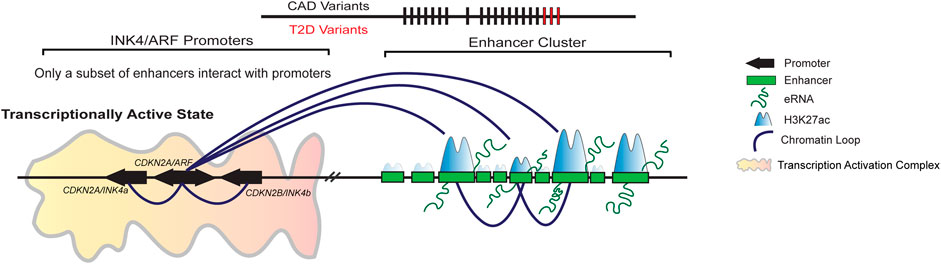
FIGURE 4. A subset of enhancers in the enhancer cluster upstream regulates INK4/ARF locus. Gene desert upstream of INK4/ARF genes contains 21 enhancers in HeLa cells. Only 15 of these enhancers are active, displaying both H3K27ac and H3K4me1 marks. Out of 15 active enhancers, only a subset of enhancers interacts with the promoters of INK4/ARF genes. The promoter interacting enhancers are critical for the regulation of these genes. The deletion of a single enhancer causes down regulation of gene transcription and EZH2 loading on the promoters. Furthermore, the deletion of interacting enhancers has an effect on the other enhancers in the cluster, indicating that the enhancers are interdependent.
LncRNAs as transcriptional regulators of the INK4/ARF locus
Long noncoding RNAs (lncRNAs) are a subclass of RNAs that are longer than 200 nucleotides and do not code for any protein product. They play critical roles in gene regulation, chromatin organization, translational regulation, etc. Several lncRNAs have been reported to influence the INK4/ARF locus expression (Puvvula 2019). Most of them are repressive and act by recruiting the PcG complexes onto the promoters of these genes. Recently, certain lncRNAs have been described to activate this locus (Figure 5).
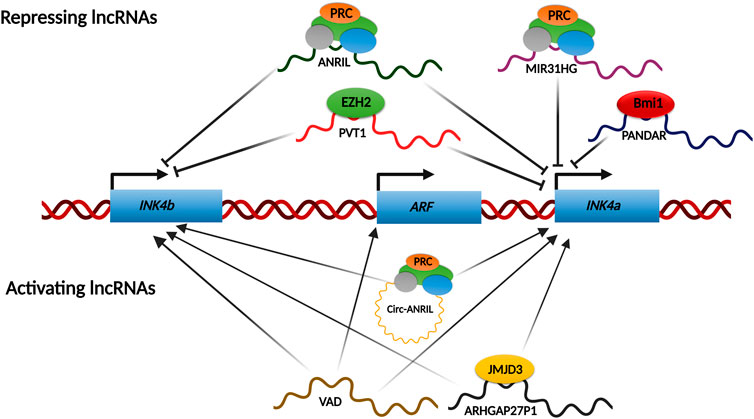
FIGURE 5. lncRNAs network regulating INK4/ARF locus. Various lncRNAs regulate the INK4a/ARF locus under certain conditions. Most of the known regulatory lncRNAs shut down the transcription from this locus by recruiting PcGs onto the promoters. However, some lncRNAs have been shown to activate this locus transcriptionally by either recruiting chromatin modifiers such as JMJD3 onto the promoters or by removing the repressive complexes like PcGs from the promoters.
LncRNAs-mediated repression of INK4/ARF locus
INK4/ARF locus contains a lncRNA, ANRIL, which is transcribed antisense to the genes. ANRIL is ∼3.8 kb and has over 20 different splice variants. These splice variants of ANRIL play a differential protective role depending on the presence or absence of CAD risk interval (Lo Sardo et al., 2018). ANRIL is required for INK4/ARF silencing in growing cells, as its expression in these cells is inversely correlated to gene expression (Yap et al., 2010; Kotake et al., 2011). This repression is a result of PRC2 loading on the INK4a promoter by nascently transcribing ANRIL RNA (Yap et al., 2010). Another study demonstrated similar recruitment of PRC2 to the INK4b promoter (Kotake et al., 2011). Therefore, ANRIL expression decreases as senescence progresses for the activation of this locus. Through RNA binding experiments, ANRIL was shown to interact with the CBX7 component of PRC1 to enhance INK4/ARF gene silencing (Yap et al., 2010). Subsequently, ANRIL binding to SUZ12, a component of the PRC2 complex, was shown to enhance the silencing of INK4b, but not INK4a (Kotake et al., 2011). In contrast to these observations, ANRIL expression is positively linked with INK4a/ARF expression, in cervical cancers. In a recent study, we report that the PRC2 complex can bind to the INK4/ARF promoters independent of ANRIL levels in cervical cancer cell lines (Farooq et al., 2021). Another lncRNA, MIR31HG, which is transcribed from the short arm of chr9 itself, has been shown to recruit PRC complex on the INK4a promoter (Montes et al., 2015). Interestingly, during OIS (Oncogene induced senescence), MIR31HG localizes solely to the cytoplasm. This leads to the loss of the PRC complex from the INK4a promoter, resulting in transcriptional activation of INK4a (Montes et al., 2015). PANDAR (promoter of CDKN1A antisense DNA damage-activated RNA) is elevated in breast cancer tissues and cell lines. PANDAR interacts with Bmi1, a PRC1 subunit, inhibiting INK4a transcription by loading Bmi1 to its promoter. PANDAR silencing reduces cell proliferation and colony formation in MCF7 cells and causes G1/S arrest in a p16INK4a-dependent manner (Sang et al., 2016). PVT1 is critical for gastric cancer progression. It accomplishes this in part by forming a complex with EZH2 and directing it to the promoters of INK4b and INK4a, suppressing their expression in gastric cancer (Kong et al., 2015). ANROC, a recently discovered lncRNA, is found downstream of the INK4a gene, and its silencing results in overexpression of all three genes, indicating that ANROC is a repressive RNA (Kotake and Tsuruda 2020).
LncRNAs-mediated activation of INK4/ARF locus
LncRNA ARHGAP27P1 is downregulated in gastric cancer cells, and when overexpressed, it inhibits gastric cancer cell proliferation, migration, and other functions in a p16INK4a and p15INK4b- dependent manner. This lncRNA regulates INK4/ARF expression by interacting with and directing the histone demethylase JMJD3 to the promoters for removal of the repressing H3K27me3 mark (Zhang G et al., 2019). AUF1 is an RNA-binding lncRNA that has been found to enhance the degradation of various RNAs. P14AS was identified using RNA capture sequencing as a novel RNA with its promoter located on the antisense strand of the fragment near CDKN2A exon1β. P14AS binds to AUF1, preventing ANRIL/INK4a RNA from interacting with AUF1. This competitive interaction between P14AS and AUF1 promotes ARF, INK4b, and INK4a gene expression (Ma et al., 2020). During OIS, VAD (Vlinc RNA Antisense to DDAH1) is highly upregulated and required to maintain senescence characteristics. VAD functions in trans on the INK4/ARF locus, and its depletion causes downregulation of ARF, INK4b, and INK4a. VAD promotes the expression of these genes by removing H2A.Z from their promoters. H2A.Z deposition represses these genes by promoting the recruitment of the PRC complex to the promoters (Lazorthes et al., 2015). Several circular ANRIL isoforms have been identified that activate the INK4/ARF genes rather than inhibiting them. They switch from repressors to activators of these genes during RAF1-mediated senescence. These circular isoforms engage with Polycomb subunits and displace EZH2 from the INK4b and INK4a promoters, stimulating transcription of these genes. As a result of the PRC2 dislocation, H3K27me3 levels at these promoters drop (Muniz et al., 2021). Similarly, TUBA4b is downregulated in CRC tissues and cells, and its overexpression inhibits CRC cell proliferation by upregulating INK4a and INK4b (Zhou, Sun, and Zhou 2020). The precise mechanisms by which TUBA4b long noncoding RNA activates these genes are unknown.
Discussion
Since the products of the INK4/ARF genes are implicated in a wide range of cancers and age-related diseases, they hold immense promise for treating or mitigating the consequences of these diseases. Regulation of the INK4/ARF locus is multi-layered, with a plethora of factors involved. As a result, the greatest challenge in harnessing this locus for therapeutic purposes is identifying the critical regulatory elements that can be targeted in a particular disease. Targeting transcription factors or epigenetic modifiers involved in its regulation has very broad effects, affecting not just this locus but others as well. We recently uncovered a few enhancers in the upstream enhancer cluster that regulate these genes. These DNA regulatory elements can be altered to provide a more precise and targeted effect. Since this enhancer cluster contains multiple enhancers, these enhancers may act in a tissue type-specific manner. Thus, determining which enhancers regulate these genes under various physiological conditions is critical for the therapeutic use of enhancers or enhancer products like eRNAs. Additionally, SNPs in upstream enhancer regions have been associated with several diseases. These SNPs can facilitate the identification of regulatory enhancers in various cell types. Thus, a functional genomics approach is required to decipher how these SNPs result in changes in gene regulation.
Author contributions
UF and DN have prepared the manuscript.
Acknowledgments
We thank members of DN lab for discussion and suggestions. We thank Sudha Swaminathan and Salsabeel Khan for critical comments on review. DN acknowledges funding support from, India-Alliance DBT-Wellcome (IA: IA/1/14/2/501539), SERB (CRG/2019/005714). Department of Atomic Energy, Government of India, under project no.12-R&D-TFR-5.04-0800 and intramural funds from NCBS-TIFR. DN also acknowledges support from EMBO global investigator program. UF is supported by the fellowship from CSIR, India.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agger, K., Cloos, P. A., Christensen, J., Pasini, D., Rose, S., Rappsilber, J., et al. (2007). UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734. doi:10.1038/nature06145
Agger, K., Cloos, P. A., Rudkjaer, L., Williams, K., Andersen, G., Christensen, J., et al. (2009). The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176. doi:10.1101/gad.510809
Barradas, M., Anderton, E., Acosta, J. C., Li, S., Banito, A., Rodriguez-Niedenfuhr, M., et al. (2009). Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 23, 1177–1182. doi:10.1101/gad.511109
Biegel, J. A., Zhou, J. Y., Rorke, L. B., Stenstrom, C., Wainwright, L. M., and Fogelgren, B. (1999). Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 59, 74–79.
Bouchard, C., Lee, S., Paulus-Hock, V., Loddenkemper, C., Eilers, M., and Schmitt, C. A. (2007). FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev. 21, 2775–2787. doi:10.1101/gad.453107
Bracken, A. P., Kleine-Kohlbrecher, D., Dietrich, N., Pasini, D., Gargiulo, G., Beekman, C., et al. (2007). The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 21, 525–530. doi:10.1101/gad.415507
Bruggeman, S. W., Valk-Lingbeek, M. E., van der Stoop, P. P., Jacobs, J. J., Kieboom, K., Tanger, E., et al. (2005). Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 19, 1438–1443. doi:10.1101/gad.1299305
Chai, J., Charboneau, A. L., Betz, B. L., and Weissman, B. E. (2005). Loss of the hSNF5 gene concomitantly inactivates p21CIP/WAF1 and p16INK4a activity associated with replicative senescence in A204 rhabdoid tumor cells. Cancer Res. 65, 10192–10198. doi:10.1158/0008-5472.CAN-05-1896
Chi, S. W., Lee, S. H., Kim, D. H., Ahn, M. J., Kim, J. S., Woo, J. Y., et al. (2005). 'Structural details on mdm2-p53 interaction. J. Biol. Chem. 280, 38795–38802. doi:10.1074/jbc.M508578200
Chittock, E. C., Latwiel, S., Miller, T. C., and Muller, C. W. (2017). Molecular architecture of polycomb repressive complexes. Biochem. Soc. Trans. 45, 193–205. doi:10.1042/BST20160173
Cobrinik, D. (2005). Pocket proteins and cell cycle control. Oncogene 24, 2796–2809. doi:10.1038/sj.onc.1208619
Dietrich, N., Bracken, A. P., Trinh, E., Schjerling, C. K., Koseki, H., Rappsilber, J., et al. (2007). Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. EMBO J. 26, 1637–1648. doi:10.1038/sj.emboj.7601632
Dimova, D. K., and Dyson, N. J. (2005). The E2F transcriptional network: Old acquaintances with new faces. Oncogene 24, 2810–2826. doi:10.1038/sj.onc.1208612
Dominguez, G., Silva, J., Garcia, J. M., Silva, J. M., Rodriguez, R., Munoz, C., et al. (2003). Prevalence of aberrant methylation of p14ARF over p16INK4a in some human primary tumors. Mutat. Res. 530, 9–17. doi:10.1016/s0027-5107(03)00133-7
Esteller, M., Corn, P. G., Baylin, S. B., and Herman, J. G. (2001). A gene hypermethylation profile of human cancer. Cancer Res. 61, 3225–3229.
Farooq, U., Saravanan, B., Islam, Z., Walavalkar, K., Singh, A. K., Jayani, R. S., et al. (2021). An interdependent network of functional enhancers regulates transcription and EZH2 loading at the INK4a/ARF locus. Cell Rep. 34, 108898. doi:10.1016/j.celrep.2021.108898
Forbes, S., Clements, J., Dawson, E., Bamford, S., Webb, T., Dogan, A., et al. (2006). COSMIC 2005. Br. J. Cancer 94, 318–322. doi:10.1038/sj.bjc.6602928
Freeman-Anderson, N. E., Zheng, Y., McCalla-Martin, A. C., Treanor, L. M., Zhao, Y. D., Garfin, P. M., et al. (2009). 'Expression of the Arf tumor suppressor gene is controlled by Tgfbeta2 during development. Development 136, 2081–2089. doi:10.1242/dev.033548
Frescas, D., Guardavaccaro, D., Bassermann, F., Koyama-Nasu, R., and Pagano, M. (2007). JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature 450, 309–313. doi:10.1038/nature06255
Giacinti, C., and Giordano, A. (2006). RB and cell cycle progression. Oncogene 25, 5220–5227. doi:10.1038/sj.onc.1209615
Gil, J., Bernard, D., Martinez, D., and Beach, D. (2004). Polycomb CBX7 has a unifying role in cellular lifespan. Nat. Cell Biol. 6, 67–72. doi:10.1038/ncb1077
Gomez-Sanchez, J. A., Gomis-Coloma, C., Morenilla-Palao, C., Peiro, G., Serra, E., Serrano, M., et al. (2013). Epigenetic induction of the Ink4a/Arf locus prevents Schwann cell overproliferation during nerve regeneration and after tumorigenic challenge. Brain. 136, 2262–2278. doi:10.1093/brain/awt130
Hannon, G. J., and Beach, D. (1994). p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 371, 257–261. doi:10.1038/371257a0
Harismendy, O., Notani, D., Song, X., Rahim, N. G., Tanasa, B., Heintzman, N., et al. (2011). 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature 470, 264–268. doi:10.1038/nature09753
He, J., Kallin, E. M., Tsukada, Y., and Zhang, Y. (2008). The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat. Struct. Mol. Biol. 15, 1169–1175. doi:10.1038/nsmb.1499
He, J., Shen, L., Wan, M., Taranova, O., Wu, H., and Zhang, Y. (2013). Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 15, 373–384. doi:10.1038/ncb2702
Herranz, N., and Gil, J. (2018). Mechanisms and functions of cellular senescence. J. Clin. Invest. 128, 1238–1246. doi:10.1172/JCI95148
Huang, Y. C., Hasegawa, H., Wang, S. W., Ku, C. C., Lin, Y. C., Chiou, S. S., et al. (2011). Jun dimerization protein 2 controls senescence and differentiation via regulating histone modification. J. Biomed. Biotechnol., 569034. doi:10.1155/2011/569034
Ivanchuk, S. M., Mondal, S., Dirks, P. B., and Rutka, J. T. (2001). The INK4A/ARF locus: Role in cell cycle control and apoptosis and implications for glioma growth. J. Neurooncol. 51, 219–229. doi:10.1023/a:1010632309113
Jacobs, J. J., Kieboom, K., Marino, S., DePinho, R. A., and van Lohuizen, M. (1999). 'The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164–168. doi:10.1038/16476
Kanao, H., Enomoto, T., Ueda, Y., Fujita, M., Nakashima, R., Ueno, Y., et al. (2004). Correlation between p14(ARF)/p16(INK4A) expression and HPV infection in uterine cervical cancer. Cancer Lett. 213, 31–37. doi:10.1016/j.canlet.2004.03.030
Kerppola, T. K. (2009). Polycomb group complexes--many combinations, many functions. Trends Cell Biol. 19, 692–704. doi:10.1016/j.tcb.2009.10.001
Kheradmand Kia, S., Solaimani Kartalaei, P., Farahbakhshian, E., Pourfarzad, F., von Lindern, M., and Verrijzer, C. P. (2009). EZH2-dependent chromatin looping controls INK4a and INK4b, but not ARF, during human progenitor cell differentiation and cellular senescence. Epigenetics Chromatin 2, 16. doi:10.1186/1756-8935-2-16
Kia, S. K., Gorski, M. M., Giannakopoulos, S., and Verrijzer, C. P. (2008). SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 28, 3457–3464. doi:10.1128/MCB.02019-07
Kim, W. Y., and Sharpless, N. E. (2006). The regulation of INK4/ARF in cancer and aging. Cell 127, 265–275. doi:10.1016/j.cell.2006.10.003
Klochendler-Yeivin, A., Muchardt, C., and Yaniv, M. (2002). SWI/SNF chromatin remodeling and cancer. Curr. Opin. Genet. Dev. 12, 73–79. doi:10.1016/s0959-437x(01)00267-2
Kong, R., Zhang, E. B., Yin, D. D., You, L. H., Xu, T. P., Chen, W. M., et al. (2015). Long noncoding RNA PVT1 indicates a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically regulating p15 and p16. Mol. Cancer 14, 82. doi:10.1186/s12943-015-0355-8
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M., et al. (2011). Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30, 1956–1962. doi:10.1038/onc.2010.568
Kotake, Y., and Tsuruda, T. (2020). Long noncoding RNA ANROC on the INK4 locus functions to suppress cell proliferation. Cancer Genomics Proteomics 17, 425–430. doi:10.21873/cgp.20201
Krishnamurthy, J., Torrice, C., Ramsey, M. R., Kovalev, G. I., Al-Regaiey, K., Su, L., et al. (2004). Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 114, 1299–1307. doi:10.1172/JCI22475
Kubbutat, M. H., Jones, S. N., and Vousden, K. H. (1997). Regulation of p53 stability by Mdm2. Nature 387, 299–303. doi:10.1038/387299a0
Kuo, C. L., Murphy, A. J., Sayers, S., Li, R., Yvan-Charvet, L., Davis, J. Z., et al. (2011). Cdkn2a is an atherosclerosis modifier locus that regulates monocyte/macrophage proliferation. Arterioscler. Thromb. Vasc. Biol. 31, 2483–2492. doi:10.1161/ATVBAHA.111.234492
Lassacher, A., Heitzer, E., Kerl, H., and Wolf, P. (2008). p14ARF hypermethylation is common but INK4a-ARF locus or p53 mutations are rare in Merkel cell carcinoma. J. Invest. Dermatol. 128, 1788–1796. doi:10.1038/sj.jid.5701256
Lazorthes, S., C., Vallot, S., Briois, M., Aguirrebengoa, J.Y., Thuret, G., St Laurent, C., Rougeulle, P., Kapranov, C., Mann, D., Trouche, D., and E., Nicolas. 2015. A vlincRNA participates in senescence maintenance by relieving H2AZ-mediated repression at the INK4 locus. Nat Commun, 6: 5971.
Lee, M., Sup Han, W., Kyoung Kim, O., Hee Sung, S., Sun Cho, M., Lee, S. N., et al. (2006). Prognostic value of p16INK4a and p14ARF gene hypermethylation in human colon cancer. Pathol. Res. Pract. 202, 415–424. doi:10.1016/j.prp.2005.11.011
Li, H., Collado, M., Villasante, A., Strati, K., Ortega, S., Canamero, M., et al. (2009). The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460, 1136–1139. doi:10.1038/nature08290
Li, H., Wang, W., Liu, X., Paulson, K. E., Yee, A. S., and Zhang, X. (2010). Transcriptional factor HBP1 targets P16(INK4A), upregulating its expression and consequently is involved in Ras-induced premature senescence. Oncogene 29, 5083–5094. doi:10.1038/onc.2010.252
Liang, G., He, J., and Zhang, Y. (2012). Kdm2b promotes induced pluripotent stem cell generation by facilitating gene activation early in reprogramming. Nat. Cell Biol. 14, 457–466. doi:10.1038/ncb2483
Lin, A. W., Barradas, M., Stone, J. C., van Aelst, L., Serrano, M., and Lowe, S. W. (1998). Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 12, 3008–3019. doi:10.1101/gad.12.19.3008
Liu, Yan, Sanoff, Hanna K., Cho, Hyunsoon, Burd, Christin E., Torrice, Chad, Mohlke, Karen L., et al. (2009). INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One 4, e5027. doi:10.1371/journal.pone.0005027
Liu, Y. T., Xu, L., Bennett, L., Hooks, J. C., Liu, J., Zhou, Q., et al. (2019). Identification of de novo enhancers activated by TGFβ to drive expression of CDKN2A and B in HeLa cells. Mol. Cancer Res. 17, 1854–1866. doi:10.1158/1541-7786.MCR-19-0289
Lo Sardo, V., Chubukov, P., Ferguson, W., Kumar, A., Teng, E. L., Duran, M., et al. (2018). Unveiling the role of the most impactful cardiovascular risk locus through haplotype editing. Cell 175, 1796–1810.e20. doi:10.1016/j.cell.2018.11.014
Lopez, F., Sampedro, T., Llorente, J. L., Hermsen, M., and Alvarez-Marcos, C. (2017). Alterations of p14 (ARF) , p15 (INK4b) , and p16 (INK4a) genes in primary laryngeal squamous cell carcinoma. Pathol. Oncol. Res. 23, 63–71. doi:10.1007/s12253-016-0083-4
Lyko, F. (2018). The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 19, 81–92. doi:10.1038/nrg.2017.80
Ma, W., Qiao, J., Zhou, J., Gu, L., and Deng, D. (2020). Characterization of novel LncRNA P14AS as a protector of ANRIL through AUF1 binding in human cells. Mol. Cancer 19, 42. doi:10.1186/s12943-020-01150-4
Maggi, L. B., Winkeler, C. L., Miceli, A. P., Apicelli, A. J., Brady, S. N., Kuchenreuther, M. J., et al. (2014). ARF tumor suppression in the nucleolus. Biochim. Biophys. Acta, 831–839. doi:10.1016/j.bbadis.2014.01.016
Martin, N., Popov, N., Aguilo, F., O'Loghlen, A., Raguz, S., Snijders, A. P., et al. (2013). Interplay between Homeobox proteins and Polycomb repressive complexes in p16INK⁴a regulation. EMBO J. 32, 982–995. doi:10.1038/emboj.2013.37
Matheu, A., Pantoja, C., Efeyan, A., Criado, L. M., Martin-Caballero, J., Flores, J. M., et al. (2004). Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev. 18, 2736–2746. doi:10.1101/gad.310304
McHugh, D., and Gil, J. (2018). Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 217, 65–77. doi:10.1083/jcb.201708092
Mirzayans, R., Andrais, B., Hansen, G., and Murray, D. (2012). Role of p16(INK4A) in replicative senescence and DNA damage-induced premature senescence in p53-deficient human cells. Biochem. Res. Int., 951574. doi:10.1155/2012/951574
Montes, M., Nielsen, M. M., Maglieri, G., Jacobsen, A., Hojfeldt, J., Agrawal-Singh, S., et al. (2015). The lncRNA MIR31HG regulates p16(INK4A) expression to modulate senescence. Nat. Commun. 6, 6967. doi:10.1038/ncomms7967
Munger, K., Scheffner, M., Huibregtse, J. M., and Howley, P. M. (1992). Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv. 12, 197–217.
Muniz, L., Lazorthes, S., Delmas, M., Ouvrard, J., Aguirrebengoa, M., Trouche, D., et al. (2021). Circular ANRIL isoforms switch from repressors to activators of p15/CDKN2B expression during RAF1 oncogene-induced senescence. RNA Biol. 18, 404–420. doi:10.1080/15476286.2020.1812910
Nakade, K., Pan, J., Yamasaki, T., Murata, T., Wasylyk, B., and Yokoyama, K. K. (2009). JDP2 (Jun Dimerization Protein 2)-deficient mouse embryonic fibroblasts are resistant to replicative senescence. J. Biol. Chem. 284, 10808–10817. doi:10.1074/jbc.M808333200
Negishi, M., Saraya, A., Mochizuki, S., Helin, K., Koseki, H., and Iwama, A. (2010). A novel zinc finger protein Zfp277 mediates transcriptional repression of the Ink4a/arf locus through polycomb repressive complex 1. PLoS One 5, e12373. doi:10.1371/journal.pone.0012373
Orlando, K. A., Nguyen, V., Raab, J. R., Walhart, T., and Weissman, B. E. (2019). Remodeling the cancer epigenome: Mutations in the SWI/SNF complex offer new therapeutic opportunities. Expert Rev. Anticancer Ther. 19, 375–391. doi:10.1080/14737140.2019.1605905
Oruetxebarria, I., Venturini, F., Kekarainen, T., Houweling, A., Zuijderduijn, L. M., Mohd-Sarip, A., et al. (2004). P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J. Biol. Chem. 279, 3807–3816. doi:10.1074/jbc.M309333200
Papp, B., and Plath, K. (2011). Reprogramming to pluripotency: Stepwise resetting of the epigenetic landscape. Cell Res. 21, 486–501. doi:10.1038/cr.2011.28
Poyurovsky, M. V., Katz, C., Laptenko, O., Beckerman, R., Lokshin, M., Ahn, J., et al. (2010). The C terminus of p53 binds the N-terminal domain of MDM2. Nat. Struct. Mol. Biol. 17, 982–989. doi:10.1038/nsmb.1872
Prieto, L. I., and Baker, D. J. (2019). Cellular senescence and the immune system in cancer. Gerontology 65, 505–512. doi:10.1159/000500683
Puvvula, P. K. (2019). LncRNAs regulatory networks in cellular senescence. Int. J. Mol. Sci. 20, E2615. doi:10.3390/ijms20112615
Rayess, H., Wang, M. B., and Srivatsan, E. S. (2012). Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 130, 1715–1725. doi:10.1002/ijc.27316
Ribeiro, J. D., Morey, L., Mas, A., Gutierrez, A., Luis, N. M., Mejetta, S., et al. (2013). ZRF1 controls oncogene-induced senescence through the INK4-ARF locus. Oncogene 32, 2161–2168. doi:10.1038/onc.2012.241
Richly, H., Rocha-Viegas, L., Ribeiro, J. D., Demajo, S., Gundem, G., Lopez-Bigas, N., et al. (2010). Transcriptional activation of polycomb-repressed genes by ZRF1. Nature 468, 1124–1128. doi:10.1038/nature09574
Roberts, C. W., and Orkin, S. H. (2004). The SWI/SNF complex--chromatin and cancer. Nat. Rev. Cancer 4, 133–142. doi:10.1038/nrc1273
Robertson, K. D., and Jones, P. A. (1998). The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol. Cell. Biol. 18, 6457–6473. doi:10.1128/MCB.18.11.6457
Rodriguez, C., Borgel, J., Court, F., Cathala, G., Forne, T., and Piette, J. (2010). CTCF is a DNA methylation-sensitive positive regulator of the INK/ARF locus. Biochem. Biophys. Res. Commun. 392, 129–134. doi:10.1016/j.bbrc.2009.12.159
Romagosa, C., Simonetti, S., Lopez-Vicente, L., Mazo, A., Lleonart, M. E., Castellvi, J., et al. (2011). p16(Ink4a) overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 30, 2087–2097. doi:10.1038/onc.2010.614
Russo, A. A., Tong, L., Lee, J. O., Jeffrey, P. D., and Pavletich, N. P. (1998). Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395, 237–243. doi:10.1038/26155
Sang, Y., Tang, J., Li, S., Li, L., Tang, X., Cheng, C., et al. (2016). LncRNA PANDAR regulates the G1/S transition of breast cancer cells by suppressing p16(INK4A) expression. Sci. Rep. 6, 22366. doi:10.1038/srep22366
Sevenet, N., Sheridan, E., Amram, D., Schneider, P., Handgretinger, R., and Delattre, O. (1999). Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am. J. Hum. Genet. 65, 1342–1348. doi:10.1086/302639
Sharpless, N. E. (2005). INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. 576, 22–38. doi:10.1016/j.mrfmmm.2004.08.021
Sherr, C. J. (2012). Ink4-Arf locus in cancer and aging. Wiley Interdiscip. Rev. Dev. Biol. 1, 731–741. doi:10.1002/wdev.40
Sherr, C. J. (2001). The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2, 731–737. doi:10.1038/35096061
Shields, B. J., Jackson, J. T., Metcalf, D., Shi, W., Huang, Q., Garnham, A. L., et al. (2016). 'Acute myeloid leukemia requires Hhex to enable PRC2-mediated epigenetic repression of Cdkn2a. Genes Dev. 30, 78–91. doi:10.1101/gad.268425.115
Sreeramaneni, R., Chaudhry, A., McMahon, M., Sherr, C. J., and Inoue, K. (2005). 'Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol. Cell. Biol. 25, 220–232. doi:10.1128/MCB.25.1.220-232.2005
Su, W., Hong, L., Xu, X., Huang, S., Herpai, D., Li, L., et al. (2018). miR-30 disrupts senescence and promotes cancer by targeting both p16(INK4A) and DNA damage pathways. Oncogene 37, 5618–5632. doi:10.1038/s41388-018-0358-1
Swigut, T., and Wysocka, J. (2007). H3K27 demethylases, at long last. Cell 131, 29–32. doi:10.1016/j.cell.2007.09.026
Tsujimoto, H., Hagiwara, A., Sugihara, H., Hattori, T., and Yamagishi, H. (2002). Promoter methylations of p16INK4a and p14ARF genes in early and advanced gastric cancer. Correlations of the modes of their occurrence with histologic type. Pathol. Res. Pract. 198, 785–794. doi:10.1078/0344-0338-00337
Tzatsos, A., Pfau, R., Kampranis, S. C., and Tsichlis, P. N. (2009). Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proc. Natl. Acad. Sci. U. S. A. 106, 2641–2646. doi:10.1073/pnas.0813139106
van Deursen, J. M. (2014). The role of senescent cells in ageing. Nature 509, 439–446. doi:10.1038/nature13193
Visel, A., Zhu, Y., May, D., Afzal, V., Gong, E., Attanasio, C., et al. (2010). Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 464, 409–412. doi:10.1038/nature08801
Wade, M., Wang, Y. V., and Wahl, G. M. (2010). The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 20, 299–309. doi:10.1016/j.tcb.2010.01.009
Wang, W., Pan, K., Chen, Y., Huang, C., and Zhang, X. (2012). The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4A expression. Nucleic Acids Res. 40, 981–995. doi:10.1093/nar/gkr818
Wang, W., Qin, J. J., Voruganti, S., Nag, S., Zhou, J., and Zhang, R. (2015). Polycomb group (PcG) proteins and human cancers: Multifaceted functions and therapeutic implications. Med. Res. Rev. 35, 1220–1267. doi:10.1002/med.21358
Wang, X., Pan, L., Feng, Y., Wang, Y., Han, Q., Han, L., et al. (2008). P300 plays a role in p16(INK4a) expression and cell cycle arrest. Oncogene 27, 1894–1904. doi:10.1038/sj.onc.1210821
Watanabe, T., Nakamura, M., Yonekawa, Y., Kleihues, P., and Ohgaki, H. (2001). Promoter hypermethylation and homozygous deletion of the p14ARF and p16INK4a genes in oligodendrogliomas. Acta Neuropathol. 101, 185–189. doi:10.1007/s004010000343
Weber, J. D., Taylor, L. J., Roussel, M. F., Sherr, C. J., and Bar-Sagi, D. (1999). Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1, 20–26. doi:10.1038/8991
Wilson, B. G., and Roberts, C. W. (2011). SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492. doi:10.1038/nrc3068
Wu, D., Tan, H., Su, W., Cheng, D., Wang, G., Wang, J., et al. (2022). MZF1 mediates oncogene-induced senescence by promoting the transcription of p16(INK4A). Oncogene 41, 414–426. doi:10.1038/s41388-021-02110-y
Wu, J., Xue, L., Weng, M., Sun, Y., Zhang, Z., Wang, W., et al. (2007). Sp1 is essential for p16 expression in human diploid fibroblasts during senescence. PLoS One 2, e164. doi:10.1371/journal.pone.0000164
Xiang, Y., Zhu, Z., Han, G., Lin, H., Xu, L., and Chen, C. D. (2007). JMJD3 is a histone H3K27 demethylase. Cell Res. 17, 850–857. doi:10.1038/cr.2007.83
Yan, M., Yang, X., Wang, H., and Shao, Q. (2018). The critical role of histone lysine demethylase KDM2B in cancer. Am. J. Transl. Res. 10, 2222–2233.
Yap, K. L., Li, S., Munoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., et al. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674. doi:10.1016/j.molcel.2010.03.021
Yu, G. W., Rudiger, S., Veprintsev, D., Freund, S., Fernandez-Fernandez, M. R., and Fersht, A. R. (2006). The central region of HDM2 provides a second binding site for p53. Proc. Natl. Acad. Sci. U. S. A. 103, 1227–1232. doi:10.1073/pnas.0510343103
Zhang G, G., Xu, Y., Zou, C., Tang, Y., Lu, J., Gong, Z., et al. (2019). Long noncoding RNA ARHGAP27P1 inhibits gastric cancer cell proliferation and cell cycle progression through epigenetically regulating p15 and p16. Aging (Albany NY) 11, 9090–9110. doi:10.18632/aging.102377
Zhang, X., Liu, L., Yuan, X., Wei, Y., and Wei, X. (2019). JMJD3 in the regulation of human diseases. Protein Cell 10, 864–882. doi:10.1007/s13238-019-0653-9
Zhang, Y., Hyle, J., Wright, S., Shao, Y., Zhao, X., Zhang, H., et al. (2019). A cis-element within the ARF locus mediates repression of p16 (INK4A) expression via long-range chromatin interactions. Proc Natl Acad Sci U S A 116, 26644-26652. doi:10.1073/pnas.1909720116
Zhao, W., Li, Q., Ayers, S., Gu, Y., Shi, Z., Zhu, Q., et al. (2013). Jmjd3 inhibits reprogramming by upregulating expression of INK4a/Arf and targeting PHF20 for ubiquitination. Cell 152, 1037–1050. doi:10.1016/j.cell.2013.02.006
Zheng, Y., Devitt, C., Liu, J., Mei, J., and Skapek, S. X. (2013). A distant, cis-acting enhancer drives induction of Arf by Tgfβ in the developing eye. Dev. Biol. 380, 49–57. doi:10.1016/j.ydbio.2013.05.003
Zheng, Y., Zhao, Y. D., Gibbons, M., Abramova, T., Chu, P. Y., Ash, J. D., et al. (2010). Tgfbeta signaling directly induces Arf promoter remodeling by a mechanism involving Smads 2/3 and p38 MAPK. J. Biol. Chem. 285, 35654–35664. doi:10.1074/jbc.M110.128959
Zhou, Y. G., Sun, F., and Zhou, Y. F. (2020). Low expression of lncRNA TUBA4B promotes proliferation and inhibits apoptosis of colorectal cancer cells via regulating P15 and P16 expressions. Eur. Rev. Med. Pharmacol. Sci. 24, 3023–3029. doi:10.26355/eurrev_202003_20666
Keywords: INK4/ARF, enhancer, 9p21, gene desert, p15INK4b, p16INK4a, ANRIL, CDKN2BAS
Citation: Farooq U and Notani D (2022) Transcriptional regulation of INK4/ARF locus by cis and trans mechanisms. Front. Cell Dev. Biol. 10:948351. doi: 10.3389/fcell.2022.948351
Received: 19 May 2022; Accepted: 09 August 2022;
Published: 09 September 2022.
Edited by:
Chandrima Das, Saha Institute of Nuclear Physics (SINP), IndiaReviewed by:
Salvatore Saccone, University of Catania, ItalyManuel Serrano, Institute for Research in Biomedicine, Spain
Copyright © 2022 Farooq and Notani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Umer Farooq, dW1lcmZAbmNicy5yZXMuaW4=; Dimple Notani, ZG5vdGFuaUBuY2JzLnJlcy5pbg==