
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 22 August 2022
Sec. Cell Death and Survival
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.948097
This article is part of the Research TopicThe Proceedings of Mitochondria Apoptosis and Cancer (MAC 2021) Virtual SymposiumView all 6 articles
 Hong Toan Lai1
Hong Toan Lai1 Nataliia Naumova1
Nataliia Naumova1 Antonin Marchais2,3
Antonin Marchais2,3 Nathalie Gaspar2,3Birgit Geoerger2,3
Nathalie Gaspar2,3Birgit Geoerger2,3 Catherine Brenner1*
Catherine Brenner1*Osteosarcoma (OS) is a pediatric malignant bone tumor that predominantly affects adolescent and young adults. It has high risk for relapse and over the last four decades no improvement of prognosis was achieved. It is therefore crucial to identify new drug candidates for OS treatment to combat drug resistance, limit relapse, and stop metastatic spread. Two acquired hallmarks of cancer cells, mitochondria-related regulated cell death (RCD) and metabolism are intimately connected. Both have been shown to be dysregulated in OS, making them attractive targets for novel treatment. Promising OS treatment strategies focus on promoting RCD by targeting key molecular actors in metabolic reprogramming. The exact interplay in OS, however, has not been systematically analyzed. We therefore review these aspects by synthesizing current knowledge in apoptosis, ferroptosis, necroptosis, pyroptosis, and autophagy in OS. Additionally, we outline an overview of mitochondrial function and metabolic profiles in different preclinical OS models. Finally, we discuss the mechanism of action of two novel molecule combinations currently investigated in active clinical trials: metformin and the combination of ADI-PEG20, Docetaxel and Gemcitabine.
Osteosarcoma (OS) is considered a rare disease and there has been no improvement in survival rate over the last 40 years. OS is the most frequent primary malignant bone tumor worldwide and predominantly affects children and young adults (Gill and Gorlick, 2021). For 80% of patients with OS, primary tumors arise in the metaphysis of long bones at extremities (distal femur, proximal tibia, proximal femur, and proximal humerus), which are the location of rapid bone growth (Isakoff et al., 2015) (Bielack et al., 2002). The disease demonstrates a high degree of malignancy and has a tendency to metastasize at early stages (Bielack et al., 2002). OS has two peaks of incidence: one between the ages of 5–24 years and the second peak from 60 to 85 years (Mirabello et al., 2009) (Whelan et al., 2012). Recent epidemiologic studies show that OS accounts 3%–5% of all pediatric cancers and 20%–40% of all bone tumors with an incidence rate of 2–4,2 cases/million per year (Rojas et al., 2021). Exemplary for other international trials, as part of the French OS2006/sarcoma-09 study, 376 patients underwent surgery after receiving intensive preoperative chemotherapy. 5-year survival rate outcomes were 1) 86% for localized disease and good histological response is; 2) 68% for localized disease and poor histological response; 3) 68% for metastatic disease and good histological response and 4) 24% for metastatic disease and poor histological response is (Gaspar et al., 2018) (Marchandet et al., 2021). Despite groundbreaking advances in other fields of pediatric oncology, therapeutic approaches for OS continue to demonstrate insufficient adequacy (Casali et al., 2018) (Mirabello et al., 2009). OS is considered as one of the most complex cancers and shows a high propensity to develop metastasis, mostly to the lung. This is one of the reasons for limited progress in treatment of this disease (Sheng et al., 2021).
To date, the main pillars of therapeutic management of OS are made of neoadjuvant chemotherapy with five major drugs: cisplatin (CIS), doxorubicin (DOX) and methotrexate (MTX), ifosfamide (IFOS) and etoposide (ETOP) (Gaspar et al., 2018), followed by surgical resection of the primary tumor along with the additional adjuvant chemotherapy after the surgery (Isakoff et al., 2015) (Edmonson et al., 1984) (Link et al., 1991) (Piperno-Neumann et al., 2016). Some patients acquire chemoresistance that impacts the survival, together with the development of metastasis (Whelan and Davis, 2018). Therefore, further studies are needed to understand OS tumorigenesis and progression, as well as for the development of OS-focused therapeutic and metastasis preventative approaches.
Across different cancer types, many basic and translational studies focused on the role of mitochondria in cancer cells, because these organelles are a central node in both, cellular metabolism, and cell fate decisions (Strasser and Vaux, 2020) (Carneiro and El-Deiry, 2020). Indeed, mitochondria are crucial for the maintenance of cellular homeostasis, energy production, regulating Ca2+ homeostasis and signaling, redox homeostasis, and cell survival or death decisions (Galluzzi et al., 2012) (Vasan et al., 2020). It is well established that mitochondria are the powerhouse of adenosine triphosphate (ATP) production via oxidative phosphorylation (OXPHOS) and the source of many biosynthetic intermediates (Mitchell, 1961). They function in a dynamic interconnected network with continuous communication to other cellular compartments such as the endoplasmic reticulum and the nucleus (Lai et al., 2022) (Galluzzi et al., 2012) (Green and Evan, 2002). Its functions are well described beyond the cellular level influencing whole organism physiology by regulating interactions between cells and tissues (Nunnari and Suomalainen, 2012).
Cancer metabolism is a flexible network of anabolic and catabolic reactions that allow cancer cells to respond to the cellular demands for rapid proliferation and adapt to harsh conditions of survival (Hanahan, 2022). Metabolic reprogramming influence many tumor stages, from transformation initiation to cancer progression (Faubert et al., 2020). To fulfill the energy requirements for cell growth and survival, including for the biosynthesis of proteins, lipids, and nucleotides, cancer cells must rewire their metabolism. The most famous phenomenon is the so called “Warburg effect,” classically used to describe the preference of use of cancer cells for glycolysis and lactate production under aerobic conditions (Warburg et al., 1927) (Liberti and Locasale, 2016) (Vander Heiden et al., 2009) (Schmidt et al., 2021). Besides enhanced glycolysis, there are other common metabolic adaptations, including prominent glutaminolysis to fuel the citric acid cycle and lipid biosynthesis (Cluntun et al., 2017). Increased pentose phosphate pathway activity aims to provide NADPH and pentose phosphate for nucleotide synthesis (Phan et al., 2014). Other metabolic pathways shown to contribute to tumorigenesis include fatty acid β-oxidation, folate-dependent serine and glycine metabolism, and the methionine cycle (Koundouros and Poulogiannis, 2020), (Rosenzweig et al., 2018). In line with these findings, metabolic reprogramming in cancer constitutes a decisive Achilles’ heel that attracts scientists and pharmacists to develop new classes of metabolic inhibitors to specifically target metabolic reprogramming. Understanding how metabolic reprogramming is acquired at different stages of tumor development and understanding its essentiality opens novel therapeutic opportunities by inducing cancer cell death in a personalized cancer concept (Stuani et al., 2019).
In this review, we provide a thorough overview of the current knowledge on the mitochondria-related RCD machinery in OS, focusing on the molecular mechanisms of apoptotic and non-apoptotic RCD. More specifically, we address apoptosis, ferroptosis, necroptosis, pyroptosis, and autophagy. Second, we will focus on the role of mitochondria and metabolic dysregulations in OS supported by fundamental findings by metabolomic and lipidomic analysis. We will finish by discussing on-going clinical trials of promising metabolism-related therapeutic targets showing the ability to induce cell death in OS.
The Nomenclature Committee on Cell Death states that cell death pathways can be categorized into two distinct forms, accidental cell death and RCD. RCD includes both apoptotic and non-apoptotic cell death (ferroptosis, pyroptosis, necroptosis, etc.) (Galluzzi et al., 2018). Apoptosis is well-known to be the main form of RCD (Kerr et al., 1972). There are two principal apoptotic signaling pathways upon the activation of caspase-3 and caspase-8: cell death receptor (extrinsic) pathway and mitochondrial apoptotic (intrinsic) pathway (Carneiro and El-Deiry, 2020). It has been established that mitochondria are central initiators of the apoptotic intrinsic pathways, but they may also participate in other forms of RCD such as necroptosis, and ferroptosis (Nikoletopoulou et al., 2013).
The intrinsic apoptosis pathway is triggered by various injurious stimuli such as oxidative stress, hypoxia, radiation, etc. (Elmore, 2007) (Danial and Korsmeyer, 2004). This type of programmed cell death requires the disruption of the outer mitochondrial membrane (OMM), known as mitochondrial outer membrane permeabilization (MOMP), allowing the release of pro-apoptotic proteins from the intermembrane space to the cytosol. However, since it is unclear how these apoptotic initiators cross the OMM and are thus released into the cytosol, several hypotheses have been proposed. These include opening of the PTP in response to over-production of ROS or Ca2+ overload (Biasutto et al., 2016), a large channel formed by Bax and/or Bak oligomers (Antignani and Youle, 2006) (Lovell et al., 2008), a channel formed by hetero-oligomers of VDAC1 and Bax (Banerjee and Ghosh, 2004) (Shimizu and Tsujimoto, 2000), or VDAC1 oligomers (Keinan et al., 2010) (Shoshan-Barmatz et al., 2013) (Keinan et al., 2013) (Weisthal et al., 2014) (Zalk et al., 2005). Despite the unclear mechanism, MOMP results in the release of cytochrome c (CytC) that, therefore, binds to and activates apoptotic protease activating factor 1 (Apaf-1) in cytosol. The heptameric quaternary structure of Apaf-1, thus, recruits cytoplasmic inactive pro-caspase-9 to form the apoptosome. Apoptosome is considered as the platform for the activation of pro-caspase-9, and other effectors such as caspase-3, -6, and-7 (Jiang and Wang, 2000, p. 1) (Bratton et al., 2001) (Chipuk et al., 2006). Moreover, MOMP induces the release of other apoptogenic factors that interact with other cellular organelles where they induce different pro-apoptotic functions. These apoptogenic factors include second mitochondria-derived activator of caspase (Smac)/direct inhibitor of apoptosis-binding protein with low pI (DIABLO), apoptosis-inducing factor (AIF), endonuclease G, and high-temperature requirement protein A2 (HtrA2/Omi) (Liu et al., 1996) (Verhagen et al., 2000) (Li et al., 2001) (Hegde et al., 2002). Importantly, once MOMP is induced and the transmembrane inner potential (ΔΨm) disrupted, a bioenergetic catastrophe occurs, manifesting by cessation of mitochondrial bioenergetic activities, ATP production arrest, ATP consumption, ROS production and pH changes (Kroemer et al., 2007).
The intrinsic apoptosis pathway is positively regulated by several pro-apoptotic members and negatively by anti-apoptotic proteins. Interestingly, for the past 30 years, a large set of proteins including both pro- and anti-apoptotic have been identified and categorized. These include proteins of the B-cell lymphoma 2 (Bcl-2) family with three subfamilies: anti-apoptotic, pore-forming proteins, and Bcl-2 homology 3 (BH3)-only proteins, whereby the classification is depending on the presence of the Bcl-2 homology domains. Anti-apoptotic members including, e.g., Bcl-2, B-cell lymphoma-extra-large (Bcl-XL), B-cell lymphoma-w (Bcl-W), and myeloid cell leukemia 1 (Mcl-1) prevent the formation and activation of the apoptosome (Nijhawan et al., 2003) (Kharbanda et al., 1997) (Yang et al., 1997). Pore-forming proteins, in contrast, possess pro-apoptotic activities and include Bcl-2-associated X protein (Bax), Bcl-2 antagonist/killer (Bak) or Bcl-2 related ovarian killer (Box). The work by forming pores in the MOM, what is required for MOMP formation, as discussed below (Bleicken et al., 2014) (Borner and Andrews, 2014). The third group of proteins, namely BH3-only proteins, also has pro-apoptotic activities. Examples include Bcl-2-interacting mediator of cell death (BIM), BH3-interacting domain death agonist (BID), and p53-upregulated modulator of apoptosis (Puma). This family contains only one Bcl-2 homology domain that either activates pore-forming proteins and/or inhibits anti-apoptotic proteins to strictly regulate intrinsic apoptosis (Singh et al., 2019) (Zha et al., 1996, p. 2) (Figure 1).

FIGURE 1. Molecular mechanisms of apoptosis.
Considered as an important apoptosis signal transduction pathway in cancer cells, extrinsic apoptosis pathway can be triggered by diverse DNA-damaging agents such as DOX, CIS, and ETOP (Müller et al., 1998) (Friesen et al., 1996). This type of apoptosis requires the binding of extracellular death ligands to death receptors. To date, there are six mammalian death receptors: tumor necrosis factor receptor 1 (TNFR1), Fas receptor, death receptor 3 (DR3), DR4 [tumor-necrosis-factor related apoptosis inducing ligand receptor (TRAILR1)], DR5 (TRAILR2), and DR6 (Nagata, 1997) (Nagata and Golstein, 1995) (Schneider et al., 1997) (Wiley et al., 1995) (Feinstein et al., 1995). This type of RCD requires generally three main steps: 1) binding of death ligands on death receptors; 2) formation of death-inducing signaling complex (DISC) by recruiting adaptor proteins including Fas-associated death domain-containing protein (FADD) and the initiator caspases. Cells can either promote the non-cytotoxic signaling-mediated cell survival via mitogen-activated protein kinases (MAPK) and/or nuclear factor-kappa B (NF-kB) surviving pathways (complex I) or cytotoxic signaling-mediated apoptosis (complex II) (Hsu et al., 1995) (Carneiro and El-Deiry, 2020). When NF-kB surviving pathway of complex I is activated, the cytotoxic complex II will be inhibited by caspase-8 inhibitor cellular FLICE-inhibitory protein (c-FLIPL) (Irmler et al., 1997). Complex II will only take place when the surviving pathway is insufficiently activated. Upon activating the cytotoxic signaling-mediated apoptosis axis, the assembly of the DISC complex stimulates the dimerization of two pro-caspase-8 molecules, results in self-cleavage and thus releases the active form of caspase-8 (Ozören and El-Deiry, 2002). Active caspase-8 then interact with the mitochondrial membrane and cleaves BID to become truncated BID (t-BID) by exposing its BH3-domain. This activates MOMP-mediated caspases to induce cell death (Schug et al., 2011) (Li et al., 1998) (Figure 1).
1) Extrinsic apoptosis: Binding of death ligands to their death receptors, induces the assembly of DISC which consists of adaptor proteins and pro-caspases (pro-caspase-8 and pro-caspase-10). Assembly of the DISC complex results in the activation of caspase-8 and caspase-10 and thus, caspase-3 and caspase-7. The crosstalk of extrinsic apoptosis and mitochondrial intrinsic apoptosis is mediated by the activation of caspase-8 and the cleavage of BID to tBID. tBID activates the MOMP-mediated caspases to induce cell death.
2) Intrinsic apoptosis can be activated by various intracellular stimuli that lead to the activation of pro-apoptotic proteins (including pore-forming proteins and BH3-only proteins). It induces the release of apoptogenic factors such as cytochrome c, AIF and SMAC/Diablo, followed by activation of the apoptosome including caspase-9, caspase-3, caspase-7 into the cytosol.
In OS, the dysregulated expressions of Bcl-2 family members have been observed and linked to patient prognosis, indicating that apoptosis could be a biomarker as well as an interesting therapeutic target. This was confirmed in a study of 49 high-grade OS patients, in which immunohistochemistry revealed that metastatic patients express higher levels of anti-apoptotic Bcl-2 in the primary tumor than non-metastatic patients. Moreover, low Bcl-2 expressing patients had a better prognosis than the high Bcl-2 expressing patients (TRIEB et al., 2013). Another study conducted by Wang et al. (2010) showed the protein and mRNA overexpression of anti-apoptotic Bcl-XL in high metastatic osteosarcoma cell line (M8), in contrary to low metastatic osteosarcoma cell lines (Saos-2, MG63 and U2OS). Furthermore, high-expressing Bcl-XL mRNA tissues from 72 OS patients without prior chemotherapy are correlated with lower prognosis. Interestingly, not only Bcl-XL but other anti-apoptotic members (Bcl-2 and Mcl-1) also showed significantly higher protein expression in OS patient tissue samples, compared to non-tumor tissues samples. By knocking-down Bcl-XL, OS cell lines showed a significant reduction of cell proliferation and higher apoptotic rate, confirmed by the increase of caspase-3. In vitro, targeting apoptosis in OS has been shown to enhance chemosensitivity. Results supporting this observation include that genetically (siRNA) or pharmacologically (ABT-737, WEHI-539) inhibiting anti-apoptotic members such as Bcl-2 and Bcl-XL, in various OS cell line (MG-63, Saos-2, M8, U2OS, etc.) induced sensitivity to CIS and DOX treatment. In contrast, overexpression of pore-forming proteins such as Bax, induced chemosensitivity of the Saos2 OS cell line (Baranski et al., 2015) (Masuelli et al., 2020) (Eliseev et al., 2008) (Wang et al., 2010) (Baranski et al., 2015). Recently, drug combinations between Bcl-XL-inhibitor (A-1331852), Mcl-1 inhibitor (S63845) and/or Bcl-2 inhibitor (ABT-199/Venetoclax) were tested on a panel of pediatric cancer cell lines, including U2O2, MG-63 cell lines. Surprisingly, only dual inhibition of Bcl-XL and Mcl-1 displayed a significant higher anti-tumoral effects in all tested pediatric cell lines and in vivo embryonic chicken model, rather than the combination with Bcl-2 inhibitor (Kehr et al., 2020). Despite the efficacy of those inhibitors in preclinical models, Venetoclax and dual inhibition of A-1331852/S63845 have been separately reported to cause acute liver toxicity in lung squamous cell carcinoma in vivo model and in clinical trial for hematological malignancies (NCT02265731), since these two proteins control the hepatic integrity in a gene-dose dependent fashion (Hikita et al., 2009) (Salem et al., 2019) (Weeden et al., 2018). In recent years, many new development of Mcl-1 inhibitors have been designed and entered clinical trial for different types of cancer as mono or combined treatment with Venetoclax (AstraZeneca, 2022) (AbbVie, 2021) (Institut de Recherches Internationales Servier, 2022). However, no clinical trial was launched, to our knowledge, to target those inhibitors in OS. Therefore, further studies are required to successfully target anti-apoptotic members to treat OS.
Non-apoptotic RCD processes are crucially involved in the induction of cancer development and progression, including OS (Tait et al., 2014). Although about 11 different RCD modalities have been described, mainly three have been associated with OS progression: ferroptosis, necroptosis, pyroptosis, and autophagy. Therefore, these RCD pathways can be considered as promising targets for OS treatment (Zhao et al., 2021) (Li et al., 2016). We will therefore limit this section to these three processes, excluding immunogenic cell death, among others, that are beyond the scope of this review.
Ferroptosis is an important RCD process involved in various biological and pathophysiological processes (Dixon et al., 2012). Ferroptosis is driven by erastin-induced iron-dependent lipid peroxidation. Unlike apoptosis, ferroptosis is characterized by the rupture of the plasma membrane to expose inner components to the extracellular environment (Dolma et al., 2003) (Yang and Stockwell, 2008) (Yagoda et al., 2007).
The most important features of ferroptosis are the dys-homeostasis of lipid hydroperoxides and ferrous ion (Fe2+), which are induced by excessive reactive oxygen species (ROS), inducing oxidative stress (Galadari et al., 2017). As a guardian of redox balance, the glutathione (GSH) metabolic pathway participates in the regulation of ferroptosis (Kennedy et al., 2020). Glutathione peroxidase 4 (GPX4) is responsible for reducing the conversion from lipid peroxides into lipid alcohols in the presence of GSH and therefore acts as an important negative regulator of ferroptosis (Yang et al., 2014, p. 4). In this metabolic pathway, cystine is imported into the cytosol via the antiporter system (SLC7A11/SLC3A2), which is then converted into cysteine to produce GSH (Stockwell et al., 2017). Serving as a cofactor of GPX4, GSH is required for protecting the cells against oxidative stress and ferroptosis (Toyokuni et al., 2017). Depletion of either GPX4, SLC7A11, or GSH levels could lead to ROS overproduction and the accumulation of lipid peroxides (Friedmann Angeli et al., 2014) (Lachaier et al., 2014) (Eling et al., 2015). In the contrary increasing the abundance of intracellular cysteine, and GPX4 activity could lead to ferroptosis resistance. Beside the increased levels of Fe2+ and ROS, the increase of lipid oxidation products such as lipid hydroperoxides and subsequent reactive aldehydes [e.g., malondialdehyde (MDA) and 4-hydroxynonenal (4HNE)] are commonly used to evaluate ferroptosis (Shimada and Stockwell, 2016) (Lei et al., 2019) (Tang et al., 2021) (Figure 2).
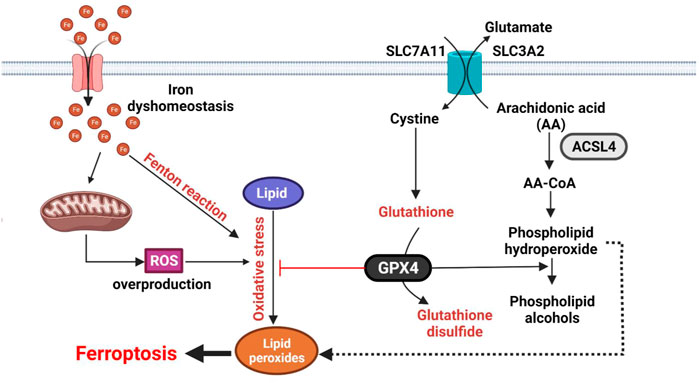
FIGURE 2. Molecular mechanisms of ferroptosis. Ferroptosis is characterized by oxidation of lipids upon inhibition of SLC7A11/SLC3A2 (Xc- system) transport complex or GPX4 activity. The reaction involved ROS and Fe2+. Ferroptosis is initiated by inhibition of the SLC7A11/SLC3A2 (Xc- system) transport complex or GPX4 activity. ROS and Fe2+ directly catalyze lipid peroxides to form damaging free radicals via the Fenton reaction. The SLC7A11/SLC3A2 antiporter is responsible for GSL biosynthesis by importing cystine into cytosol. GPX4 inhibits ferroptosis by transforming lipid peroxides into lipid alcohols.
Recent studies confirmed that ferroptosis can be induced by some natural anti-tumoral compounds [e.g., Bavachin and Phenethyl isothiocyanate (PEITC)] in in vitro and in vivo in OS models (Luo et al., 2021) (Lv et al., 2020). It was demonstrated that Bavachin treatment in OS cellular models (HOS and MG-63) significantly increased the intracellular ferrous ion, ROS, and MDA levels. Therefore, the ferroptosis regulators were found to be modulated in treated OS cell lines. In these experiments, it was also shown that Bavachin can inhibit SLC7A11, GPX4, phosphorylated signal transducer and activator of transcription 3 (p-STAT3). To explain the underlying molecular mechanisms, it was proposed that Bavachin can inhibit p-STAT3, subsequently stimulate tumor suppressor p53 and negatively regulate cysteine antiporter system (SLC7A11/SLC3A2) (Luo et al., 2021). Similar observations were found in PEITC-treated K7M2 OS cell line, showing the redox imbalance and the dysregulated Fe2+ metabolism (Lv et al., 2020). Interestingly, PEITC treatment also induces autophagy, which was confirmed by upregulation of autophagy markers such as Beclin1, conversion of LC3I to LC3II and p62. Hence, autophagy regulators such as mTOR and p-STAT3 were examined, demonstrating the inhibition of phosphorylated form of mTOR and STAT3. Given the important anti-tumoral effect of PEITC on OS cell lines, different doses of PEITC (from 30 to 90 mg/kg) were administrated in syngeneic orthotopic OS mouse model. Interestingly, tumor regression was significantly observed with 30 and 60 mg/kg doses, compared to the control group (Lv et al., 2020). All these results demonstrated that ferroptosis inducers could be proposed as a promising OS treatment strategy (Liang et al., 2019). However, ferroptosis inducers could be used only after detailed consideration and precise molecular screening, considering that ferroptosis-focused strategy may induce further complications in cancer patients. Indeed, it is worth noting that tumor-suppressor gene TP53 can promote the accumulation of lipid peroxidation products, therefore promoting ferroptosis, and is broadly mutated in OS cell lines (Jiang et al., 2015).
Ferroptosis is closely related to autophagy and various interconnections between other forms of RCD have been demonstrated (Hou et al., 2016) (Gao et al., 2016). It is currently believed that the autophagy pathway can contribute to development of drug resistance, while the ferroptosis pathway can reverse this resistance (Zhou et al., 2019). To our knowledge, induction of ferroptosis in the treatment of OS are limited to chemotherapeutics, while alternative options, such as radiotherapy and immunotherapy, known to efficiently induce ferroptosis in other solid tumors, have not yet been reported to our knowledge (Zhao et al., 2021). Further studies are needed to evaluate the translational potential of inducing ferroptosis as a treatment strategy for OS.
Necroptosis has previously been considered as the passive unprogrammed cell death, induced by external factors (toxins, various infections, physical traumatic influence, etc.) (Trump et al., 1997) (Jouan-Lanhouet et al., 2014). Based on current knowledge, however, it represents a form of RCD pathway and can be regulated similar to apoptosis (Christofferson and Yuan, 2010) (Robinson et al., 2019). Necroptosis and apoptosis share numerous molecular elements in the signaling transduction process, and could be modulated by the same effectors (Gupta et al., 2018) (Lin et al., 1999). Necroptosis is typically characterized by specific morphological characteristics such as swelling of the subcellular structures, permeabilization of the plasma membrane, mitochondrial depolarization, inflammation in the surrounding tissues (Khoury et al., 2020) (Figure 3). Understanding necroptosis signaling pathways could therefore contribute to the treatment of different pathologies, including cancers such as OS.
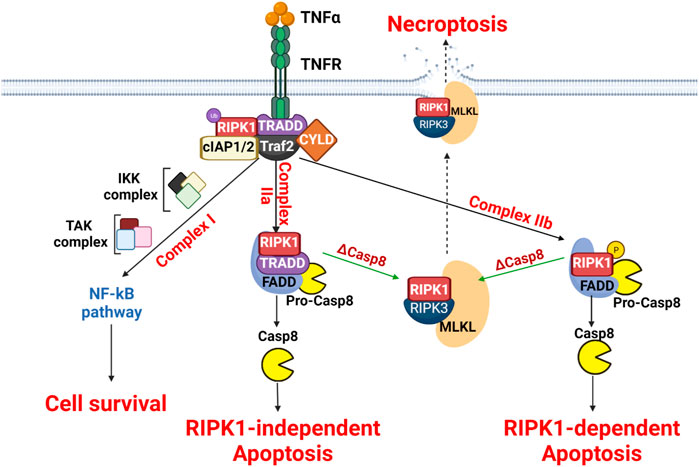
FIGURE 3. Molecular mechanisms of necroptosis. The scheme presents the three signaling pathways downstream of TNFR involving complex I, complex IIa and complex IIb formation. These complexes influence differentially cell fate. In the absence of caspase 8, necroptosis is then induced.
Necroptosis is initiated by the interaction between a wide range of stimuli such as tumor necrosis factor α (TNFα), interferon γ (IFγ) or lipopolysaccharide (LPS) and its receptors (Chen et al., 2019). The most studied prototype is the TNFα -/receptor TNFR pathway that has been thoroughly explored on the molecular level to induce necroptosis (Fulda, 2013). The binding of TNFα to its receptor TNFR results in the formation of the membrane signaling necroptotic complex I, containing tumor the necrosis factor receptor type 1-associated death domain protein (TRADD), FAS-associated death domain protein (FADD), RIPK1, cellular inhibitor of apoptosis protein 1/2 (cIAP1/2), and TNF receptor-associated factor 2 (TRAF2/5) (Han et al., 2011) (Vandenabeele et al., 2010). The stabilization of the complex is ensured by ubiquitination of RIPK1, that is mediated by cIAP1/2 and TRAF2/5. The ubiquitinated RIPK1 recruits IκB kinase (IKK) and TGFβ-activated kinase (TAK) complexes, leading to the activation of nuclear factor kappa B (NF-κB) pathway which promotes cell survival (Newton et al., 2014). In consequence, the formation of necroptotic complex I is considered an important checkpoint between cell survival and necroptosis (Oberst et al., 2011). To activate necroptosis, there are two crucial conditions 1) RIPK3 mandatory expression and 2) the inhibition of caspase-8 (Holler et al., 2000) (Vercammen et al., 1998). Following the inactivation of caspase-8, RIPK1 will be deubiquitinated by lysine 63 deubiquitinase (CYLD), thus forming the necroptotic complex II (O’Donnell et al., 2011). Two different forms of necroptotic complex II are formed, (IIa and IIb), depending on the composition; complex IIa contains TRADD, RIPK1, FADD, and caspase-8 while complex IIb only contains RIPK1, FADD, and caspase-8 (Amin et al., 2018). For the complex IIa, the presence of caspase-8 induces RIPK1-independent apoptosis while for the complex IIb, without TRADD, phosphorylated RIPK1 activates caspase-8, resulting in RIPK1-dependent apoptosis (Micheau and Tschopp, 2003). Interesting, only when caspase-8 is inhibited the necrosome forms with the presence of RIPK1, RIPK3, and MLKL, thus promoting necroptosis-associated pore formation (Pasparakis and Vandenabeele, 2015).
Upon TNFα binding to TNFR, complex I is formed by recruiting adaptor proteins (RIPK1, Traf2, cIAP1/2, and CYLD) using TRADD as a platform. The ubiquitination of RIPK1 results in the recruitment of the IKK complex and/or TAK complex, promoting the NF-κB survival pathway. The complex II activation requires the deubiquitylation of RIPK1, thus forming the complex IIa (containing RIPK1, TRADD, and FADD) and pro-caspase-8 or the complex IIb (containing RIPK1, FADD, and pro-caspase-8). Both complex IIa and IIb can induce apoptosis. Therefore, activation of necroptosis is obtained only when caspase-8 is inhibited or depleted by recruiting RIPK1, RIPK3, and MLKL to form the necrosome. MLKL-mediated necroptosome causes the disruption of plasma membrane and cell lysis.
Necroptosis can be considered as a mitochondrial-mediated RCD, because the receptor-interacting protein kinase-1/-2/pseudokinase mixed lineage kinase domain-like (RIPK1/3/MLKL) necrosome complex induces mitochondrial dysfunction and thereby necroptosis (Wang et al., 2012) (Marshall and Baines, 2014) (Tian et al., 2020). The RIPK1/3/MLKL necrosome complex depends on mitochondrial functioning and can enhance oxidative respiration, ROS generation, translocation of Bax/Bak to mitochondria, and various metabolic enzymes in the mitochondrial matrix (Van Herreweghe et al., 2010) (Gan et al., 2019) (Tischner et al., 2012). Targeting necrosome complex could offer effective therapeutic strategies for OS treatment, according to the existing in vitro and in vivo studies (Sun et al., 2015) (Fu et al., 2013). For example, Shikonin, a natural compound, extracted from Chinese medicinal herbs, was demonstrated to induce necroptosis in both primary and metastatic OS by upregulating RIPK1 and RIPK2 (Fu et al., 2013).
However, necroptosis involvement in OS progression is still ill-defined and open new opportunities for researchers to deeply investigate.
Pyroptosis is a regulated cell death type, which was discovered relatively recently and was characterized as morphologically different from apoptosis, ferroptosis and necrosis (Ding et al., 2016) (Shi et al., 2017). Pyroptosis is considered as the inflammatory type of regulated cell death that requires membrane pore-forming proteins and is associated with the morphological changes of cells, including swelling, with cell lysis and release of various proinflammatory factors (Xia et al., 2019). It is believed that pyroptosis could generate a tumor-promoting milieu due to the circulation of numerous inflammatory factors. In parallel, pyroptosis could prevent tumor growth by inducing a regulated cancer cell death (Xia et al., 2019) (Wu et al., 2021).
Pyroptosis depends on the formation of plasma membrane pores by the pore-forming proteins, in particular, gasdermin protein family (Galluzzi et al., 2018). Gasdermin D (GSDMD) as a key member of the gasdermin protein family is a primary substrate for the caspase family and after cleavage by activated caspases, the N-terminal fragment of GSDMD (N-GSDMD) could be oligomerized, leading to the pores formation and then, pyroptosis initiation (Lu et al., 2021). It is shown that genes linked to the pyroptosis are associated with the proliferation and invasion of tumor cells in different cancer types, strongly indicating the cancer prognosis (Lin et al., 2020) (Ye et al., 2021) (Tang et al., 2020) (Wang et al., 2018). It is obvious that pyroptosis-associated genes are involved in the osteosarcoma progression, however, the precise molecular mechanisms are still needed to be clarified.
Canonical inflammasome-induced pyroptosis can be induced by various pathogenic signals and is associated with the formation of inflammasome complex. Several inflammasomes (NLRP1, NLRP3, NLRC4, AIM2, and pyrin domain) were identified to interact with caspase activation and recruitment domain (PYD/CARD) (Shi et al., 2017) (Wu et al., 2021). Inflammasomes interact with the adaptor protein apoptosis associated speck like proteins (ASC) and pro-caspase-1 (He et al., 2016). Activation of pro-caspase-1 is needed for the cleavage of GSDMD and N-GSDMD production (Shi et al., 2015) (Van Opdenbosch and Lamkanfi, 2019). In addition, active Caspase-1 could cleave and activate pro-IL-1β and pro-IL-18 allowing their release via the membrane pores (Lacey et al., 2018) (Figure 4).
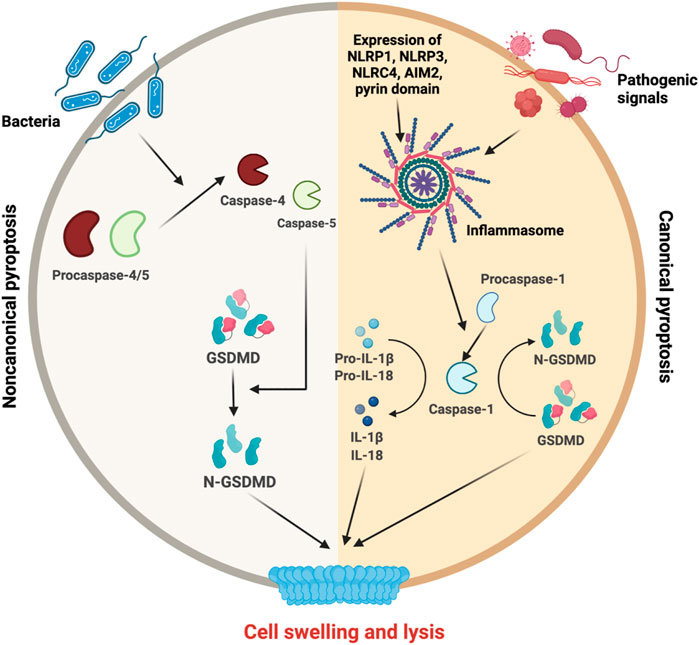
FIGURE 4. Molecular mechanisms of pyroptosis. Upon the initiation of noncanonical pyroptosis or canonical pyroptosis, different inflammatory actors are triggered. On the left side, pro-caspase 4/5 are cleaved for their maturation and participate in noncanocical pathway to cleave GSDMD into its active form N-GSDMD. On the right side, canonical pathway requires inflammasome formation to activate the caspase-1. The caspase-1 is necessary for GSDMD cleavage and IL-1β and IL-18 maturation, release and final steps of cell swelling and lysis.
In contrast, noncanonical inflammasome-induced pyroptosis could be triggered by lipopolysaccharide (LPS) and bacteria and is independent of the inflammasome pathway. It is associated with the activation of caspase-4/5 via their oligomerization. Finally, GSDMD is cleaved in N-GSDMD leading to cell swelling and lysis (Yang et al., 2015) (Shi et al., 2014) (Fang et al., 2020).
Recently, even is the number of patients investigated is limited, differential expression analysis of the microarray data from the TARGET and GTEx databases identified 46 differentially expressed pyroptosis-related genes in osteosarcoma (Zhang et al., 2021). This evaluation of the association between the pyroptosis-related signature scores and clinical characteristics with immune microenvironment in osteosarcoma indicates that the pyroptosis-associated prognostic signature could be crucial for the osteosarcoma diagnosis and prognosis (Zhang et al., 2021).
To date, the role of mitochondria in OS-induced pyroptosis is still unknown. As it has been shown that the Ragulator-Rag-mTORC1 pathway and mitochondrial ROS are required for pyroptosis in macrophages, it could be important in the future to examine more deeply how mitochondria could affect this cell death modality in OS (Evavold et al., 2021).
Autophagy is a catabolic pathway that is activated by numerous factors including cellular stress such as nutrient deprivation, hypoxia or ROS overproduction (Levine and Kroemer, 2008). This process is characterized by the degradation of cytoplasmic components and is regulated in defined sequential steps: 1) induction of autophagy by cellular stress; 2) phagophore formation for damaged organelles; 3) autophagosome formation by elongating and closing the phagophore; 4) autolysosome formation by fusing the autophagosome with the lysosome to degrade the damaged organelles (Glick et al., 2010) (Figure 5). For over 30 years, a large set of autophagy-related genes (Atg) has been discovered and categorized into five groups according to their functions: 1) Atg1/UNC-51-like kinase (ULK-1) complex to initiate autophagy, 2) class III phosphatidylinositol three kinase (PI3K)/Beclin-1 complex to form the autophagosome, 3) the Atg12/Atg5/Atg6 conjugation system for promoting the formation of autophagy precursors by elongating the vesicle, 4) the Atg8 (LC3)/phosphatidylethanolamine (PE) conjugation system to form a tight membrane-associated form, and 5) Atg9 for the autophagosomal membrane expansion (Nakatogawa et al., 2009) (Matoba et al., 2020). In the absence of cell stress, the mammalian target of rapamycin (mTOR) signaling pathway negatively inhibits ULK1, thus preventing autophagy. Upon cellular stress, mTOR is inhibited, releasing ULK1 triggering autophagic activity (Díaz-Troya et al., 2008) (Figure 5). In cancer cells, the overactivation of oncogenic mTOR signaling pathway is frequently observed and mTOR inhibitors have been actively developed in recent years (Hua et al., 2019). In OS, the activation of mTOR pathways is associated with tumor growth and proliferation, metastasis, inhibition of apoptosis and suppression of autophagy, hence becoming an attractive therapeutic target (Kuijjer et al., 2014) (Houghton et al., 2012) (Gordon et al., 2008) (Mu et al., 2013) (Wang et al., 2014) (Song et al., 2015). While mTOR inhibitor has a limited efficacy in cancer cells, the combination of mTOR inhibitors and other drugs demonstrated synergistic effects. Different combinations were explored in the clinical trials. For example, Cixutumumab [humanized IgG1-monoclonal-antibody-targeting insulin-like growth factor-1 (IGF-1R)] combined with Temsirolimus (mTOR inhibitor) showed no robust differences in median progression-free survival end point for both IGF-1R positive patients and IGF-1R negative patients (Schwartz et al., 2013). Another clinical study for unresectable high-grade OS after standard treatment with Sorafenib (multikinase inhibitor) and Everolimus (mTOR inhibitor) also failed to demonstrate 50% of 6-month progression free survival endpoint as previously designed in the study (Grignani et al., 2015). Although several pre-clinical data suggested that mTOR inhibitors showed anti-tumoral effects in OS models, more clinical trials are nonetheless needed to confirm this attractive target.
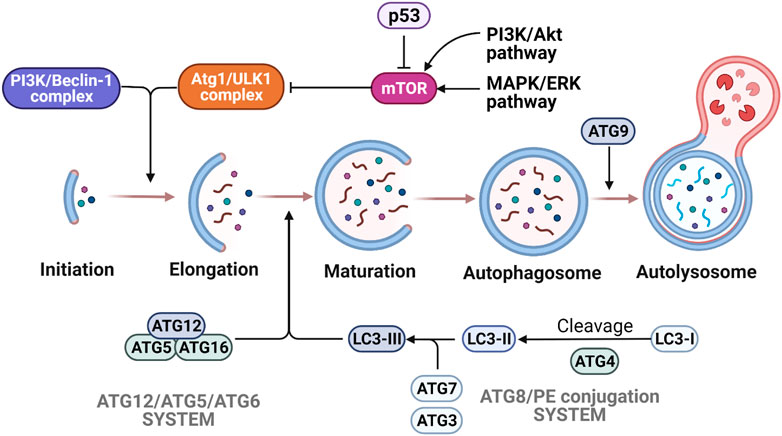
FIGURE 5. Molecular mechanisms of autophagy. The various steps of autophagy process are depicted following mTORC1 regulation by p53, AMPK and MAPK/ERK signaling. Autophagy is activated by the initiation complex ULK1/Atg1 and PIK3/Beclin-1 complex, which enables the autophagosome formation. The elongation and maturation depend on two ubiquitin-like conjugation systems (Atg12/Atg5/Atg6 system and Atg8/PE system). Many signaling pathways are involved in the regulation of autophagy. Due to the activation of p53 signaling or AMPK, activity of the mTORC1 complex is decreased and triggers autophagy activation. On the other hand, the activation of PI3K/Akt and MAPK/Erk pathways induce the increase of mTORC1 activity that results in the inhibition of autophagy.
According to many studies, autophagy is known to play a dual role as a promoter of cell survival and chemosensitivity. In normal conditions, autophagy prevents organelle dysfunction and maintains normal cellular biosynthesis. Chemo-resistant OS cells exhibit a high autophagy rate, especially CIS-resistant cell lines (Jiang et al., 2017) (Mukherjee et al., 2017). Through autophagy induction, they also observed the stimulation of many resistance-mediated factors such as high mobility group box 1 protein (HMGB1), Heat Shock Protein 90 Alpha Family Class A Member 1 (HSP90AA1), and Glial cell-derived neurotrophic factor family receptor alpha 1 protein (GFRA1). Moreover, knock-down of Beclin-1, an essential actor for autophagosome formation displays reduced cell invasion and metastasis rates. These results were confirmed in vivo in OS mouse xenograft models resulting in reduced tumor growth. Beclin-1 knocked-down cell lines show significantly increased sensitivity to classic anticancer agents used to treat OS, such as DOX, CIS, and MTX as proven in HOS, MG63, and U2OS cell lines (Zhang et al., 2015). Similarly, Atg14 knock-down Saos2 cell lines rendered more sensitive to CIS (Zhao et al., 2014), suggesting that the high autophagic rate can strongly contribute to OS chemoresistance. Thus, many autophagy inhibitors are recently under investigation in combination with conventional drugs such as CIS and DOX to sensitize OS cell death (Sui et al., 2013).
While the protective role of autophagy is well known to provide many advantages for OS development, the role of autophagy to induce cell death is still under debate. Increasing evidence suggests that autophagy also has anti-tumoral roles under certain conditions. Through mechanisms, yet to be confirmed, autophagy also is able to promote apoptosis (Thorburn, 2008). Many drugs stimulate both autophagy and apoptosis but pharmacological inhibition of autophagy results in a decrease of apoptotic markers, such as caspase-3 and poly (ADP-ribose) polymerase (PARP) cleavage, showing the effect of autophagy-induced apoptosis (Wang et al., 2013) (Li et al., 2015). Recently, by analyzing 394 tumor specimens from 260 OS patients in various stages of OS development, Livingston et al. (2018), suggested that positivity for puncta of the autophagy marker LC3B + can be used as prognostic markers for better survival after neoadjuvant chemotherapy. They therefore highlighted the beneficial role of autophagy in this context.
In many cancers, the interconnections between metabolism and cell death are mainly mediated by mitochondrial dysfunction 1) pro-apoptotic MOMP, 2) ROS- or Ca2+-dependent mitochondrial permeability transition pore (mPTP), 3) ATP production by OXPHOS and glycolysis, 4) synthesis of certain biosynthetic intermediates (Green et al., 2014), opening the perspective to identify new factors/checkpoints connecting these processes. Unfortunately, knowledge is limited in OS in comparison to other cancer types and deserves more research efforts. Thus, the search for connection between cell death and metabolism might be stimulated by the bioinformatic analysis of genomic and genetic pediatric OS patients’ data (Berlanga et al., 2022).
Glucose and glutamine are important fuel sources for cancer cells (DeNicola and Cantley, 2015). Whereas this is almost exclusive in cell culture, also in vivo they remain important contributors across a wide variety of cancer types (Hui et al., 2017). In metabolic stress conditions, especially nutrient starvation, cells undergo cell cycle arrest and MOMP-driven apoptosis. For example, glucose deprivation first reduces ATP production rate and increases the AMP/ATP ratio, inducing the activation of AMP-activated protein kinase (AMPK) to arrest the cell cycle (Kim et al., 2010). In parallel, AMPK activation also causes the phosphorylation of p53 to induce p53-dependent apoptosis (Okoshi et al., 2008). Together with AMPK, p53 increases levels of pro-apoptotic BH3-only proteins (BIM and Puma) and pore-forming protein (Bax) to sensitize cells to mitochondria-mediated apoptosis (Zhao et al., 2008) (Chi et al., 2000). Similarly, glutamine deprivation induces both extrinsic and intrinsic apoptotic pathways but these events are cell type-specific (Fuchs and Bode, 2006). This can stimulate the activation of different caspases either -2, -3, -8, and/or -9, cleaved-PARP expression, translocating pro-apoptotic protein Bax or releasing apoptogenic factor CytC, depending on the cell line (Paquette et al., 2005) (Fuchs et al., 2004).
The mitochondrial permeability transition (mPT) is a pathophysiological state of the mitochondrial inner membrane (MIM). It is related to specific permeability conditions, including Ca2+ overload, oxidative stress, increased phosphate concentrations, decreased ATP availability or various stress conditions (Zoratti and Szabò, 1995) (Baumgartner et al., 2009) (Seidlmayer et al., 2015) (Crompton, 1999). The mPT is mediated by the multi-protein pore between MIM and MOM called the mPTP. Opening of mPTP results in loss of ΔΨm and ATP synthesis interruption, being directly regulated by the concentration of mitochondrial Ca2+ (mtCa2+) levels (Marzo et al., 1998) (Bernardi et al., 2015) (Duchen, 2000). The functional dualism of mtCa2+ is an important factor of mPTP induction: physiological levels of mtCa2+ could activate transient opening of mPTP, while mtCa2+ overload leads to significant pathological changes, resulting in sustained mPTP opening. This subsequently induces mitochondrial dysfunction and cell death (Morciano et al., 2021). Of note, modulation of mtCa2+ caused by the dysfunctions of Ca2+ transporters or oncogenic signaling pathways can regulate the opening of mPTP activity. Moreover, mPTP opening is strictly regulated by series of regulators, including voltage-dependent anion channel 1 (VDAC1) on the MOM, adenine nucleotide translocator (ANT) on the MIM, Hexokinase II (HKII), glycogen synthase kinase 3 β (GSK3β), cyclophilin D (CypD) and also members of apoptotic Bcl-2 family (Baines et al., 2005) (Kroemer et al., 2007) (Bonora et al., 2022).
In solid tumors, including OS, cancer cells avoid the opening of the mPTP, however, the precise molecular mechanisms have not been identified. One possible mechanism is related to the Warburg effect with an increase in aerobic glycolytic ATP production (Warburg et al., 1927). The oxygen independent continuous ATP production through glycolysis avoids ATP depletion and at the same time promotes lactate accumulation. This secondary product of the Warburg effect, lowers the extracellular pH levels, thus inhibiting mPTP activation and contributing to evade cell death (Bonora and Pinton, 2014). Hypoxia-inducible factor 1-alpha (HIF1α) is another potential mechanism to explain the mPTP-mediated cell death avoidance. HIF1α acts as a transcription factor that stimulates the expression of glycolysis-related genes, including HKII and therefore promoting the Warburg effect (Gogvadze et al., 2008). Recent studies reported that HIF1α-mediated HKII over-expression, results in mPTP inhibition (Gwak et al., 2005). Therefore, HKII becomes an attractive target for cancer therapy. HKII can interact with other regulators of the mPTP, namely VDAC1, to facilitate VDAC interaction with the ANT to form a contact site between MOM and MIM facilitating thereby the exchange of cytosolic ADP for mitochondrial ATP. Subsequently, ATP from mitochondria bind to active sites on HKII, resulting in activation of HKII for glycolysis (Azoulay-Zohar et al., 2004) (Beutner et al., 1996). Moreover, VDAC1 is an important effector due to its role in transporting metabolites, Ca2+, anions, cations, etc.; across the MOM and is also known for its interaction with anti-apoptotic proteins (Bcl-2 or Bcl-XL) (Shoshan-Barmatz et al., 2017). Targeting HKII and/or VDAC1 by chemical agents, genetic manipulation, microRNA or peptide-based therapies have been demonstrated induce significant inhibition of glycolysis, loss of ΔΨm, and induce cell death (Galluzzi et al., 2008) (Li et al., 2017, p. 2) (Sun and Zhang, 2017, p. 2) (DeWaal et al., 2018).
Immunohistochemical analysis of 30 OS patient derived specimens showed the overexpression of HKII in 83.3% OS specimen, compared with normal bone tissues. This overexpression has been also found in a panel of OS cell lines (MG63, HOS, Saos2, and U2OS). Therefore, knock-down of siRNA in MG63 and U2OS led to decrease Warburg effect and remarkably increases apoptotic rates. However, 2-Deoxy-D-Glucose (2-DG)—a glycolysis inhibitor through HKII did not display any significant changes in cell viability. The authors suggested that the current inhibitors such as 2-DG or 3-bromopyruvate are not efficient due to their low specificity. Subsequently, targeting the interaction of HKII and VDAC could be beneficial to reduce Warburg effect observed in OS (Zhuo et al., 2015). Beside those chemical inhibitors, the research of miRNA–endogenous RNA has been elucidated in many human cancers and showed the potential clinical application, including OS. Recently, two miRNAs, namely miR-615 and miR-185 were discovered to target HKII and reduce OS cell viability and metastasis. MiR615 was found to be significantly downregulated in 92 OS patient tissues and correlated with poor clinical outcomes. Mimicking miR-615 in HOS cell line reduce cell proliferation and metastatic rates by directly targeting HKII while inhibiting miR-615 showed the opposite effects. Therefore, epithelial-mesenchymal transition (EMT) and PI3K/Akt pathway were found to be dysregulated in miR-615 mimicking HOS cell line (Sun et al., 2020, p. 2). In other study, total of 30 OS tissues were compared with adjacent normal tissues, showing the downregulation of miR-185 in OS tissues. This observation was also confirmed in OS cell lines (MG-36, U2OSS, HOS, Saos2), compared with human osteoplastic cell line (NHOst). MiR-185 is also able to directly target HKII and reduce glycolytic profile and cell viability of OS cell lines (C. Liu et al., 2019a, p. 2). Taken together, these two miRNAs could become novel therapeutic candidates for OS treatment in the future. However, there are still many challenges of miRNA application in the future to overcome, such as, technical development and unclear understanding of biological characteristics of miRNAs (Cui et al., 2019). Taken together, understanding the interplay between metabolism and cell death could be useful to identify critical cell fate checkpoint and help the development of anticancer metabolic inhibitors.
Cancer cell lines have been instrumental models to discover tumor proliferation mechanisms, characterize functional profiles, and as models for drug discovery over sever decades (Masters, 2002). Unfortunately, the number of established OS cell lines is relatively low, compared to other cancer types (Mohseny et al., 2011). Therefore, the knowledge of heterogeneity in metabolic adaptation of OS is limited.
Recently, the metabolic comparison between the noncancerous osteoblast cell line (hFOB), parental OS cell lines (Saos2 and HOS), and its metastatic subtypes (LM7 and 143B), has helped deepen our understanding (Table 1). By examining the morphology of mitochondria, hFOB, Saos2, and HOS cell lines display elongated mitochondria. Bioenergetic values such as OCR and ECAR were not significantly different between hFOB, Saos2, and HOS with a partial decrease of ΔΨm and impaired mitochondrial integrity, thus hinting towards involvement of mitochondrial dysfunction in OS oncogenesis. Furthermore, apoptotic parameters of Saos2 and HOS were examined, showing significantly decreased cytosolic CytC protein expression in the HOS and Saos2 cell lines, compared with hFOB, following the increase of anti-apoptotic Bcl-2 protein expression while caspase-3 remained unchanged (Giang et al., 2013).
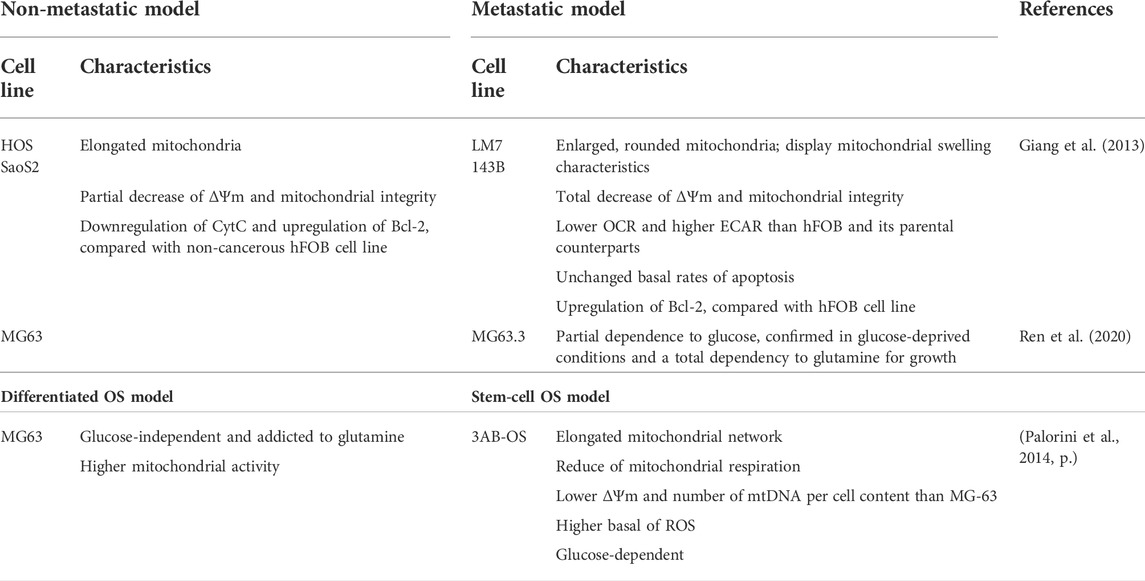
TABLE 1. Characteristics of dysregulated energy metabolism and substrate energy use in OS models.
In contrast, metastatic LM7 and 143B cell line models contain many enlarged rounded mitochondria, showing mitochondrial swelling characteristics. These results were also confirmed in mouse 143B xenografts and diagnosed human OS biopsy specimens without any previous treatments. Therefore, metastatic LM7 and 143B cell lines showed significantly lower OCR and higher ECAR, compared to their non-cancerous and parental counterparts, indicating the presence of Warburg effect in metastatic OS cell lines. Furthermore, the ΔΨm and the mitochondrial membrane integrity were totally reduced, indicating mitochondrial depolarization, compared with these counterparts (Giang et al., 2013). As a consequence of these changes, the opening of mPTP and the release of apoptogenic factor—CytC were expected (Li et al., 1997) (Kalkavan and Green, 2018). Surprisingly, however, the protein expression of cytosolic CytC, caspase-3 was not significantly changed in 143B and LM7 cell lines. This was in line with the unchanged basal rates of apoptosis. Bcl-2 overexpression was also observed in LM7 and 143B, indicating the inhibition of mPTP opening mechanism and CytC release to induce apoptosis. Interestingly, by using an mPTP inhibitor named sanglifehrin A (SfA), the Warburg effect was significantly reversed in LM7 and 143B cell lines, accompanied by the increase of OCR and decrease of ECAR. As expected, the ΔΨm and integrity were consequently increased. These results indicate an interplay between mPTP, Warburg effect and mitochondrial dysfunction in OS (Giang et al., 2013).
Moreover, it has recently been shown that the cell energetic profile measured as the OXPHOS/glycolysis ratio might reflect the stemness of OS cells. Della Sala et al. (2022), compared the differentiated MG63 cell line with its OS subclone, the mesenchymal stem cell line 3AB-OS. This comparison showed that the stem cell like 3AB-OS cells display a more glycolytic profile. Interestingly, the reduced mitochondrial respiration of 3AB-OS cell line also correlates with a lower ΔΨm and mtDNA copy number, whereas MG-63 shows an elongated, and thus more functional mitochondrial network. The 3AB-OS cell line also produces higher basal levels of total ROS than MG63. Finally, by suppressing nutrient availability, two distinct energetic dependencies were observed, revealing that the 3AB-OS cell line is highly dependent on glycolysis while MG63 cells are addicted to glutamine (Palorini et al., 2014). These observations are similar to normal stem cells that often rely mainly on glycolysis and might be linked to the self-renewal capacity of 3AB-OS (Ito and Suda, 2014). To activate differentiation, stem cells need to reprogram metabolism towards OXPHOS. In line with this finding, high basal levels of ROS production in 3AB-OS enhance the glycolytic profile and inhibit the OXPHOS activity. With this mechanism they produce sufficient ATP for energy-consuming processes while maintaining stemness characteristics (Faubert et al., 2020) (Zhang et al., 2012) (Intlekofer and Finley, 2019). Contrary to 3AB-OS stem cells, differentiated MG63 cells are glucose-independent, but display glutamine addiction and higher mitochondrial activity (Palorini et al., 2014). Taken together, understanding of how OS stem cell metabolically reprogram could contribute to discover the new therapeutics directly targeting this chemoresistant sub-population in OS.
Amino acid and fatty acid metabolisms have also been described to be changed during OS oncogenesis. This includes 3-phosphoglycerate dehydrogenase (PHGDH) that has recently been identified as a potential target for OS treatment (Rathore et al., 2021). PHGDH is responsible for the conversion of the glycolytic intermediate 3-phosphoglycerate into 3-phosphohydroxypyruvate, an intermediate of de novo serine biosynthesis. Upregulation of PHGDH has been observed in many cancers, including melanoma, breast cancer, leukemia and colorectal cancer (Locasale et al., 2011) (Fan et al., 2015) (Polet et al., 2016) (Jia et al., 2016) (Zhao et al., 2020). This upregulation is thought to be linked to increased demands for folate and methionine cycle-related processes such as DNA synthesis, biomass production and epigenetic modifications (Rathore et al., 2020). Targeting the folate cycle is a mainstay of treatment in several cancers, including OS, with MTX being one of the oldest and still most widely used chemotherapeutics (Ghodke-Puranik et al., 2015). Given the role of MTX in inhibiting dihydrofolate reductase (DHFR), a component of the folate cycle, targeting PHGDH could support the use of MTX either to produce synergistic activity or to prevent current resistance issues in OS treatment, given that the molecules are non-cytotoxic. By analyzing 260 OS patient samples using both immunohistochemistry and RNA-sequencing analysis, Rathore et al. (2021) observed that patients expressing high PHGDH levels have worse outcomes and disease-free survival than low-PHGDH expressing patients. Moreover, a panel of OS cell lines was detected with three to ten times higher PHGDH protein expression, compared with precursor mesenchymal stem cell (hMSC). Those include MG63, HOS/MNNG, Saos2, U2OS, and NOS1. Interestingly, pharmacologically or shRNA targeting PHGDH showed decreased proliferation rates in OS cell lines, but no effects were shown in breast cancer cell lines with low PHGDH expression, thus confirming a potential therapeutic interest. Metabolic evaluation of PHGDH-inhibited OS cell lines showed a decrease of mitochondrial OCR and an increase of the glycolytic rate. Metabolomics analysis of those cells displayed a decrease of metabolites involved in the TCA cycle, suggesting that there might be a decreased glucose consumption to enter the TCA cycle for mitochondrial OXPHOS. In line with the glutamine addiction that we discussed above, the accumulation of glutamine and glutamate suggested that PHGDH-inhibited OS cell lines decrease glutamine consumption as a oxidative fuel source. To determine the alternative energy sources in PHGDH inhibitor (NCT-503)-treated OS cell lines, intracellular [U-13C] tracing analysis revealed the metabolic switch from glutamine utilization to fatty acid oxidation as a principal source of non-glucose-derived acetyl-coenzyme A (Acetyl-CoA), confirmed by the accumulation of unsaturated fatty acid by lipidomic analysis. Most strikingly, this was not sufficiently incorporated in the TCA cycle. The lipidomic analysis demonstrated that the increase of unsaturated fatty acids inside PHGDH-inhibited OS cell line was likely from dysfunctional mitochondria, rather than fatty acid biosynthesis. The exact mechanism, however, needs to be shown. In addition, inhibition of PHGDH also resulted in an increase of branched-chain amino acid (BCAA) metabolite levels (Rathore et al., 2021). Given the cell capacity to sense lipid, BCAA, methionine cycle, and nucleotide level changes, likely via GATOR complexes, mTORC1 and autophagic actor Atg4 were found to be activated to prevent mitochondria dysfunction and toxicity induced by pharmacological PHGDH inhibitor NCT-503 (Gu et al., 2017) (Kim et al., 2015). To counteract these compensatory effects, the mTORC inhibitor (rapamycin) was combined with NCT-503. In practice, however, this drug combination showed no difference in OS cell death, compared to monotherapy. Taking into account the metabolic compensation, observed with the accumulation of unsaturated fatty acid, while treated with the PHGDH inhibitor; perhexiline, the inhibitor of β-oxidation, and carnitine palmitoyltransferases 1/2 (CPT1/2) was combined with NCT-503 (Rathore et al., 2021). CPT1 and CPT2 are mitochondrial enzymes, responsible for the transport of acyl-carnitines into IMS to participate in fatty acid β-oxidation (Bonnefont et al., 2004). It was shown, that mono-treatment by perhexiline did not induce significant cell death while the combination of perhexiline with NCT-503 or of other PHGDH inhibitor, namely PKIMDL, caused significant cell death in OS cell line and also in U2OS xenograft models (Rathore et al., 2021). Despite the promising effects of PHGDH inhibitor in pre-clinical models and the discovery of new PHGDH inhibitors during this past decade, none of these inhibitors was demonstrated in clinical trials due to their high concentrations (µM range) and lack of sensitivity, thus leading to high toxicity. Therefore, the next challenging step of PHGDH inhibitor development is to identify new target action sites of that subsequently lower the concentration of those compounds and reduce the toxicity. In line with this concept, these results highlight the notion of metabolism flexibility in OS, thus constituting a potential Achilles Heel that can be leveraged to target metabolic vulnerabilities at different OS development stages. Despite a still very preliminary stage, this concept currently raises many therapeutic opportunities to precisely target metabolism (Wolpaw and Dang, 2018).
Recently, three human OS cell lines with the same genetic background, but at three different stages of OS development were compared by using metabolomic, proteomic, and lipidomic analysis, namely the benign HOS cell line, the malignant N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) treated-HOS (MNNG/HOS) line and the metastatic HOS-derived 143B line (Table 2). Malignant MNNG/HOS and metastatic 143B cell lines exhibited the increase of glycolytic metabolites and the upregulation of glycolysis-involved enzymes than benign HOS cell lines with a non-significant difference between MNNG/HOS and 143B cell lines. Concerning the TCA cycle, MNNG/HOS displayed a higher level of TCA metabolites and TCA cycle-related enzymes than metastatic 143B cells. 143B cells also showed higher levels of both when compared to the benign HOS cells (MNNG/HOS >143B >HOS). The same classification was found in amino acid intermediate metabolites and enzymes responsible for amino acid metabolism. In the MNNG/HOS cell line, a significant 50% increase in lipid species was observed, in comparison to metastatic 143B cells. A pulse isotope approach was designed to trace the incorporation of 13C-glucose and 13C-glutamine. After feeding three cell lines with 13C-glucose, metastatic 143B cells showed significantly higher incorporation in glycolytic flux than their benign and malignant counterparts (Fritsche-Guenther et al., 2020). High levels of 13C-glutamine incorporation into the TCA cycle was also found in 143B, with predominant labeling of citric acid, suggesting a reductive carboxylation flux in the TCA cycle fuel Acetyl-CoA production (Martínez-Reyes and Chandel, 2020). Under glucose or glutamine deprivation, no significant change in cell viability was found in 143B, confirming the flexibility in using either glucose or glutamine for proliferation. In contrast the viability of HOS and MNNG/HOS was found to be reduced in glutamine-deprived conditions (Fritsche-Guenther et al., 2020).
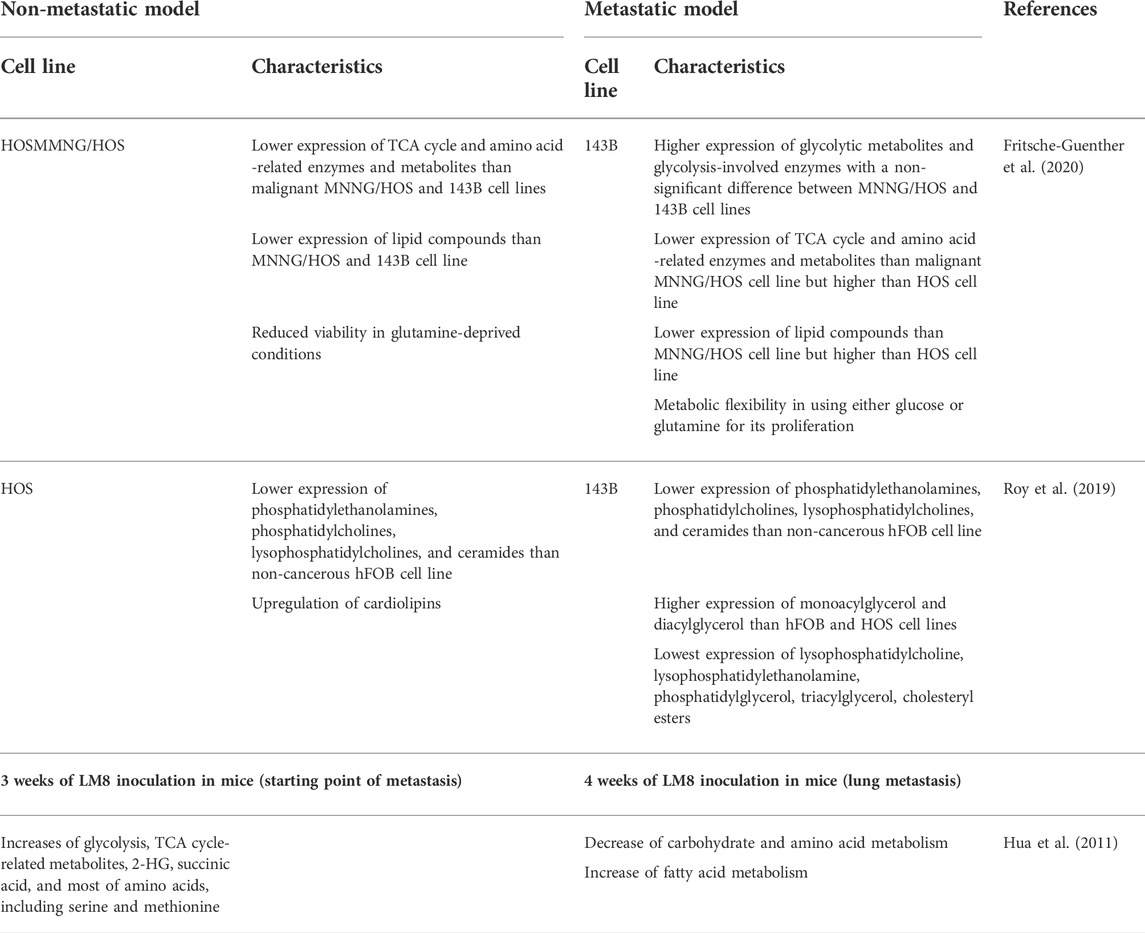
TABLE 2. Metabolic reprogramming profiling by omics- analysis in OS.
Further research on nutrient dependencies showed that the OS MG63-derived metastatic MG3.3 cell line shows a partial dependency on glucose and a total dependency to glutamine for growth. Upon treatment with a glutamine synthase (GLS) inhibitor, only a partial reduction of cell growth was observed, suggesting the capacity of these cells to switch the primary nutrient source. Increased, growth inhibition was observed in vitro as well as in an orthotopic xenograft model, by combining the GLS inhibitor with metformin, a mitochondrial Complex I (CI) inhibitor (Ren et al., 2020). The mechanisms of action of metformin in OS treatment will be discussed below in more detail.
In addition to the analysis of tumor tissue, Hua et al. (2011) performed a metabonomic study of serum in mice carrying subcutaneous tumors of murine LM8 OS cell lines. In this model all mice developed lung metastasis after 4 weeks of LM8 inoculation, their serum metabolic profiles revealed dynamic changes associated with the development of OS metastasis. In the third week, considered as the starting point of metastasis, many glycolysis and TCA cycle-related metabolites were up-regulated in serum which is consistent with the Warburg effect previously observed in the comparison of Saos2, HOS cell lines and its metastatic subtypes LM7, 143B (Hua et al., 2011) (Giang et al., 2013). Among dysregulated metabolites, the level of 2-hydroxyglutaric acid (2-HG) and succinic acid were significantly upregulated in the third week. Moreover, most of the amino acids were upregulated, including serine and methionine which are correlated with studies of Rathore et al. (2021), thus constructing a specific metabolic signature of OS. At week 4 when the lung metastasis is fully developed, metabolic reprogramming was observed with the decrease of carbohydrate and amino acid metabolism whereas fatty acid metabolism was increased, indicating a role of lipid metabolism in lung metastasis in this model (Hua et al., 2011).
2-HG is considered as an oncometabolite and strongly contributes to tumorigenesis in many cancers (Parsons et al., 2008) (Mardis et al., 2009) (Shelar et al., 2018). Mutated forms of isocitrate dehydrogenase 1/2 (IDHmt1/2) that catalyze the conversion from α-ketoglutarate to the D enantiomer of 2-HG (D-2-HG) were famously identified as potential targets with the development of the first-in-class metabolic inhibitors (Parsons et al., 2008) (Dang et al., 2009). In 2017 and 2018, these metabolic inhibitors, namely Enasidenib and Ivosidenib, have been approved by the US Food and Drug Administration (Kim, 2017) (Dhillon, 2018). Currently, Ivosidenib is explored in the Phase II clinical trial in advanced solid pediatric tumors, for pediatric patient containing IDH1 genetic alterations, including OS (NCT04195555) (National Cancer Institute (NCI), 2022) and Enasidenib in IDH2-mutated malignancies in the European AcSé-ESMART trial (NCT02813135) (Gustave Roussy, Cancer Campus, Grand Paris, 2021) (Gatz et al., 2019). Despite many different studies reporting that no mutations of neither IDH1 nor IDH2 were detected in OS patient samples, Liu et al. (2013) nonetheless demonstrated that IDH2-R172S was still detected in 25% of OS patients by using their anti-mutated monoclonal antibody MsMab-1 (Amary et al., 2011). Taking into account that for acute myeloid leukemia, serum 2-HG levels have been discovered as a diagnostic and prognostic tool to predict IDHmt1/2 and also clinical outcome, the detection of IDHmt1/2 needs to be re-examined by different methods in OS (DiNardo et al., 2013). Of note, only D-2-HG is increased in IDHmt1/2, the metabolic difference that Hua and colleagues reported, could be the total amount of D-2-HG and L-2-HG. Therefore, the ratio of these two enantiomers or the separated quantification of D-2-HG becomes critical for specific clinical test development to avoid false-positive results in the future (Struys, 2013).
A detailed lipidomic analysis of the non-cancerous hFOB, non-metastatic HOS, and metastatic 143B was performed by Roy et al. (2019). Many chief lipid classes were found to be differentially regulated. Briefly, phosphatidylethanolamines, phosphatidylcholines, lysophosphatidylcholines, and ceramides displayed the highest expression, while monoacylglycerol and diacylglycerol displayed the lowest expression in hFOB, compared with HOS and 143B cell lines. When evaluating the functional consequences, they could show that inhibiting phospholipase C (PLC), whicht catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol and IP3, reduces 143B cell viability and migration. Moreover, several interesting observations were obtained that could be further investigated in the future: 1) cardiolipins are upregulated in the HOS cell line, while there was no significant difference between hFOB and 143B, and 2) lysophosphatidylcholine, lysophosphatidylethanolamine, phosphatidylglycerol, triacylglycerol, cholesteryl esters display the lowest poolsize in 143B cells. Upon more detailed examination of cholesterol species levels (cholesterol, free cholesterol, and cholesteryl ester), no significant changes between cell lines were observed (Roy et al., 2019). The only significant change was the decrease of esterified cholesterol in 143B, compared with hFOB and HOS. The authors hypothesized that this decrease was due to the lack of adipocytes in their cellular model and the result could not reflect the in vivo condition. In conclusion, further investigation of lipidomic profiling into in vivo primary and metastatic OS models could confirm their observations and enable targeting lipid synthesis in OS treatment (Roy et al., 2019).
In this review, we have searched in ClinicalTrials.gov with “Osteosarcoma” for condition or disease, “Metabolism” for other terms and selected “Recruiting” and “Active, not recruiting” for recruitment status. Thus, five studies were found to be relevant with these terms. Among these five studies, there are only Metformin and combination of ADI-PEG20 with Gemcitabine and Docetaxel that showed the complete matched criteria. As consequence, we finally decided to decipher Metformin and ADI-PEG20 because of their active clinical status to summarize the real-time advances in using metabolic inhibitors in OS treatment.
Metformin has originally been developed as a biguanide antidiabetic agent to treat type II diabetes mellitus (Rena et al., 2017). In recent years, metformin has been evaluated in a wide variety of cancer models for its effect on cell viability and growth. Despite the very high concentration of metformin was reported to have effect on tumor growth in vitro, it is still one of the most prescribed drugs worldwide for this indication. Metformin is transported into the cell via organic cation transporter 3 (OCT-3) and OCT-1 (Jonker and Schinkel, 2004). The first known target of Metformin is complex I of mitochondrial electron transport chain (mtETC), exhibiting the antitumor effects through the decrease of respiration chain activity and oxygen consumption rate (El-Mir et al., 2000). Moreover, it has been shown that Metformin treatment influences multiple signaling pathways involved in cellular proliferation and survival such as AMPK signaling pathway and mTORC1 pathway. By inhibiting mitochondrial complex I, metformin treatment increases the ADP: ATP and AMP:ATP ratios, leading to the activation of AMPK signaling pathway. The AMPK pathway activation results in suppression of hepatic gluconeogenesis, inhibition of lipogenesis by reducing the conversion of acetyl-CoA to malonyl-coA (Musi et al., 2002) (Foretz et al., 2014) (Whitburn et al., 2017). According to the literature, the anti-cancer effects of metformin were also described by the APMK-induced inhibition of mTOR activity, that, subsequently, inhibits the protein synthesis (Howell et al., 2017). Accumulated evidences suggested that metformin could target the HK-VDAC1 interaction by allosterically inhibiting HK enzymatic activity, leading to detach HK from mitochondrial VDAC1 and promotes apoptotic cell death (Marini et al., 2013) (Abu-Hamad et al., 2008) (Shoshan-Barmatz et al., 2021).
Several clinical studies in different cancer types have been using metformin as mono-treatment, or combination with other molecules to improve therapeutic response (J. Li et al., 2020b). NCT01101438 is an active phase III clinical trial that showed the improvement of patient’ progression-free survival while treated with Metformin for more than 3,000 breast cancer patients (Canadian Cancer Trials Group, 2022). Several on-going clinical trials are promisingly conducted by combinating Metformin with targeted therapy such as BRAF tyrosine-kinase inhibitors (TKI) for melanoma (NCT01638676) (NCT02143050) (Chesney, 2021a) (Chesney, 2021b). Temsirolimus (mTOR inhibitor) for advanced cancers patients that are refractory to standard therapies (NCT01529593) (M.D. Anderson Cancer Center, 2022a), Sapanisertib (mTOR1/2 inhibitors) for metastatic, recurrent and refractory cancers (NCT03017833) (M.D. Anderson Cancer Center, 2022b). Combining Metformin with standard chemotherapy has also shown the synergetic effects and been conducted in several on-going clinical trials for breast cancer (NCT03238495) (Khan, 2021) and prostate cancer (NCT02640534) (Swiss Group for Clinical Cancer Research, 2022). Of note, Metformin has no significant side effects; however, the overdose of Metformin can cause the symptoms of lactic acidosis that likely occur in patients with pre-existing medical conditions and those older than 80 years old (Nasri and Rafieian-Kopaei, 2014). In the population-based study of type II diabetes patients, nearly 25% of patients have contraindications to Metformin treatment, but rarely results in discontinuation of therapy (Emslie-Smith et al., 2001, p.). However, the benefit-risk balance of metformin treatment in cancer therapy still requires to be further investigated.
In OS cell lines, metformin exhibited a dose- and time-dependent inhibition of cell cycle, tumor proliferation, and invasion in vitro and in xenograft models, as shown by three different research groups (Ko et al., 2016) (B. Li et al., 2020a) (Li et al., 2018). In these studies, high concentration (mM range, far higher than doses used in patients) of metformin could trigger cell cycle arrest by G2/M accumulation, which was confirmed by the upregulation of cell cycle-related protein Cyclin D1. Metformin-induced apoptosis was also found as indicated by loss of ΔΨm, shrinkage of the cells, fragmentation of nuclei, and the cleavage of caspase-3 and PARP. Interestingly, metformin-treated OS cell lines also manifested higher levels of autophagy-related actors such as LC3B-II, p62 and Beclin-1. To determine the crosstalk between apoptosis and autophagy they co-administered an autophagy inhibitor with metformin. This further improved the anti-tumor effect of metformin on OS tumor growth, indicating an anti-apoptotic effect of autophagy in OS treatment. Similarly, in other model systems, it has been proven that mitochondrial ROS play a crucial role in the activation of apoptosis and autophagy via the ROS/JNK/c-Jun pathway. In analogy, mitochondrial ROS production was increased in metformin-treated OS cell lines (B. Li et al., 2020a). According to these results, metformin activates JNK/c-Jun pathway-mediated autophagy and apoptosis by stimulating the phosphorylation of JNK and c-Jun (Redza-Dutordoir and Averill-Bates, 2016) (Wang et al., 2016).
Metastasis development is strictly linked to the EMT mechanisms and the loss of cellular adhesion that facilitates cell migration. On immunohistochemistry, this is characterized by decreased expression of epithelial markers (E-cadherin and ZO-1) and increased expression of mesenchymal markers (N-cadherin and vimentin) (Greenburg and Hay, 1982) (Stoker and Perryman, 1985). Beyond the known role of metformin on mitochondrial CI, metformin-treated OS cell lines showed an increase of epithelial marker E-cadherin and a decrease of the mesenchymal marker vimentin, suggesting that metformin could negatively regulate the EMT mechanism (Ren et al., 2020) (Li et al., 2018). A proposition of explanation of the mechanism of action of high doses of metformin would be that phosphatase and tensin (PTEN) tumor suppressor activity is upregulated and thus, phosphorylated Akt (p-Akt) were found to be decreased (Li et al., 2018). It is known that PTEN/PI3K/Akt pathway hyperactivation is crucial in many cancers, including OS and that the genomic deletion of PTEN gene is frequently observed in OS cell lines (Freeman et al., 2008) (Zheng et al., 2020) (Nielsen-Preiss et al., 2003). In further detail, immunohistochemical stainings of PTEN of nearly 100 OS patients were analyzed and divided into two groups: 1) chemotherapy naive biopsies (BXs) and 2) tumor resections after neoadjuvant chemotherapy (RXs). BXs significantly displayed a positive staining for two thirds of samples whereas RXs after receiving chemotherapy, showed the decrease of PTEN marker. Moreover, patients with low PTEN expression are associated with poorer prognosis in both BXs and RXs (Robl et al., 2015). As a well-defined pathway, a product of PI3K - phosphatidylinositol-3-phosphates (PIP-3) level is maintained low by PTEN activity. Consequently, the loss of PTEN increases PIP-3 level, and the accumulation of PIP-3 recruits and hyper-activates several proteins containing pleckstrin homology (PH) domain, including Akt (Di Cristofano and Pandolfi, 2000). Indeed, increased proliferation, migration and invasive rates were found in PTEN-knocked-down Metformin-treated OS cell lines, compared with only Metformin-treated OS cell lines, thus indicating that the anti-tumor effect of metformin was suppressed by prior PTEN knock-down using siRNA (Li et al., 2018).
Moreover, metformin can sensitize OS stem cells to CIS by inhibiting the pyruvate kinase isoenzyme M2 (PKM2) which is consistent with many different studies in other cancers (Shang et al., 2017, p. 2) (Chen et al., 2015) (M. Liu et al., 2019b, p. 2). Distinct mechanistic investigations were conducted in different cancer models. In gastric cancer, metformin decreases PKM2 protein expression via HIF1α inhibition, while in renal cancer cell, metformin promotes the activation of AMPK under glucose deprivation, thus resulting in PKM2 recruitement to form AMPK/PKM2/β-Catenin complex that subsequently stimulates the transcription of c-Myc and CCND1 genes (Chen et al., 2015) (M. Liu et al., 2019b, p. 2). By sorting OS cell lines (HOS, Saos2 and MG-63) with CD133 markers, Shang et al. (2017) were able to separate CD133-positive OS stem cells and CD133-negative OS non-stem cells. In this study, it was observed that CD133-negative OS non-stem cells responded better to CIS than CD133-positive OS stem cells. In OS stem cells, the expression of PKM2 is significantly higher than in OS non-stem cells. To confirm the role of PKM2 in CIS resistance, PKM2-knocked-down OS stem cells showed significantly higher sensitivity to CIS treatment. In line with PKM2-involved CIS resistance, metformin treatment appears to sensitize OS stem cells to CIS by inhibiting PKM2 in both in vitro and in vivo models. Regarding metabolic impacts, metformin was able to lower glucose flux, as indicated by reduced glucose uptake, lactate production, and ATP production. This might partially explain the effect of metformin on tumor proliferation. Combining metformin and CIS resulted in higher expression of cleaved caspase-9 and cleaved caspase-3, effectors of apoptosis (Shang et al., 2017, p. 2) (Figure 6).
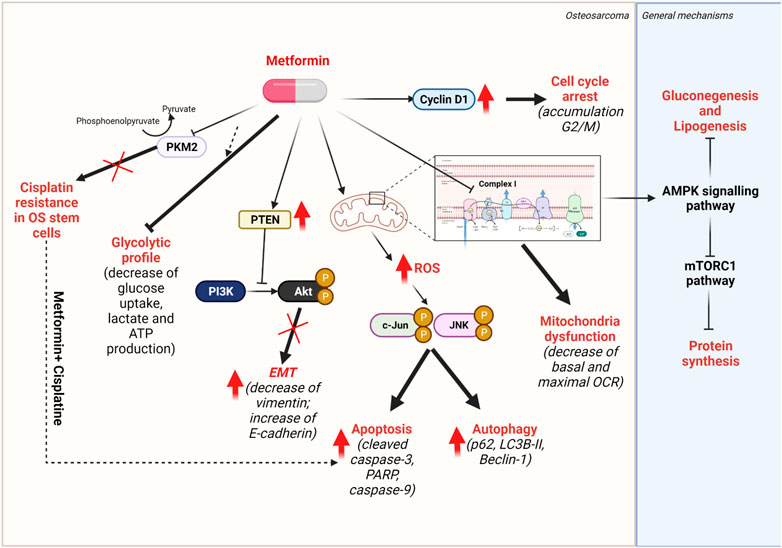
FIGURE 6. Mechanisms of action of metformin. Metformin targets mitochondrial complex I of respiratory chain, thus induces the activation of AMPK signalling pathway, resulting in the inhibition of glucogenesis and lipogenesis. The overactivation of AMPK pathway also inhibits the mTORC1 pathways, thus, protein synthesis.
Based on different preclinical studies of metformin, a phase-II single-arm clinical trial of metformin as 3 years maintenance treatment for OS patients is currently recruiting (NCT04758000). The study is divided into two groups, the first group of poor responders for post neoadjuvant chemotherapy and the second group of patients (including OS patients and Ewing sarcoma patients) in complete remission after the first relapse to clinically evaluate the event-free survival and toxicity of metformin (Longhi, 2021).
Metformin resensitizes CIS-resistant OS stem cells to CIS and promotes apoptosis by increasing levels of cleaved caspase-3 and caspase-9; it induces reduced glycolytic flux as indicated by decreased glucose uptake, lactate, and ATP production; it prevents the EMT-mediated metastasis by the loss of mesenchymal marker vimentin and the gain of epithelial marker E-cadherin via the increase of PTEN, resulting in inhibiting cell survival PI3K/Akt pathway. Metformin targets the complex I of mtETC and alters mitochondrial activity. Thus, an over-production of ROS was found to promote both apoptosis and protective autophagy via the JNK/c-Jun pathway. Cell cycle arrest, characterized by the cell accumulation in the G2/M phase, was found in metformin-treated OS cell lines by elevating Cyclin D1 protein expression.
Due to the deficiency of cellular arginine synthesis, many cancer cells depend on the uptake of exogenous arginine when citrulline or ornitine uptake is not possible (Cheng et al., 2018). Whereas the full urea cycle incorporates carbamoyl phosphate that stems from the assimilation of ammonia with bicarbonate; also, partial cycles exist starting with citrulline, taken up from circulation. Citrulline then enters the urea cycle and generates arginine via two reactions, catalyzed by argininosuccinate (ASS) for the conversion from citrulline + aspartate to argininosuccinate and argininosuccinase (ASL) for the conversion from argininosuccinate to arginine + fumarate (Figure 7).
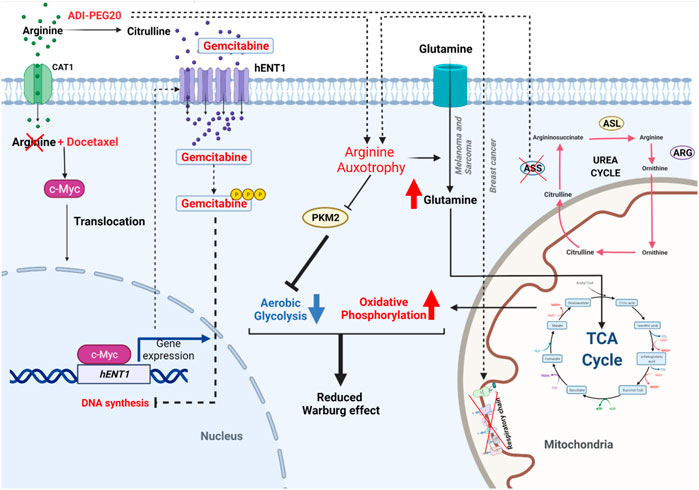
FIGURE 7. Mechanisms of action of ADI-PEG20 combined with Docetaxel and Gemcitabine in Ongoing Clinical for ASS1-deficient cancer (adapted from Prudner et al., 2019). Shown on the left are the synergic molecular mechanism of ADI-PEG20, Docetaxel, and Gemcitabine in ASS1-deficient OS. Together with Docetaxel, the arginine starvation induced by ADI-PEG20 induces c-Myc translocation. c-Myc acts as a transcription factor that stimulates the expression of the hENT1 gene which is important for Gemcitabine uptake to inhibit DNA synthesis in OS. Shown on the right are the metabolic impacts in arginine auxotrophic conditions, where the loss of arginine inhibits the activity of glycolysis-mediated enzyme PKM2, resulting in decrease of aerobic glycolysis. Moreover, in ASS1-deficient breast cancer, ADI-PEG20 treatment inhibits the mitochondrial activities, resulting in the inhibition of mitochondrial respiratory complexes. While in melanoma and sarcoma, arginine starvation causes the increase of extracellular glutamine uptake with higher expression of glutaminolysis-related actors, resulting in the metabolic reprogramming for the consumption of glutamine as a fuel source via TCA cycle, and OXPHOS. This metabolic profile represents the reduced Warburg effect.
Focusing on the rate-limiting enzyme ASS, the isoform ASS1 is commonly lost in high-grade sarcoma, its role being crucial for arginine production. Therefore, the loss of ASS1 is considered a metabolic vulnerability because, growth of ASS1-deficient cancer cells under arginine depletion is strongly impaired (Bean et al., 2016). To investigate this treatment strategy, starvation of exogenous arginine was induced by using arginine deiminase (ADI-PEG20). This enzyme degrades extracellular arginine to citrulline, and results in loss of exogenous arginine uptake via CAT1 transporter (Closs et al., 2004). ADI-PEG20 has been investigated in many different cancer types (Kobayashi et al., 2010) (Huang et al., 2013) (Bowles et al., 2008).
In ASS1- deficient breast cancer, arginine starvation by ADI-PEG 20 displayed the dysregulated mitochondrial bioenergetics by decreasing 50% of total ATP levels and inhibiting oxygen consumption rate in MDA-MB-231 cells. Transcriptomic analysis comparison between treated and un-treated MDA-MB-231 cell lines showed several mitochondrial candidates such as members of complex I, complex II, complex III, complex IV and complex V of mtETC, thus confirming the effect of ADI-PEG20 on mitochondrial respiratory function. Moreover, ADI-PEG20 treatment results in ROS over-production, decrease of mitochondrial membrane potential, mitochondrial fragmentation in ASS1-deficient MDA-MB-231 cells. Furthermore, in this breast cancer model, Qiu et al. (2014) combined ADI-PEG20 with autophagy inducer (rapamycin) or autophagic flux inhibitor (chloroquine) to observe the requirement of autophagy in ADI-PEG-induced cell death. Interestingly, chloroquine, not rapamycin was able to decrease ADI-PEG20-induced cell death. As consequence, they proposed that prolonged ADI-PEG20 treatment induces cytotoxic autophagy-dependent cell death. In contrast, the biological differences between melanoma and sarcoma versus breast cancer were observed in other study conducted by Kremer et al. (2017) in leiomyosarcoma and melanoma cell lines. Upon ADI-PEG20 treatment, the glucose uptake was reduced with the lower expression of PKM2, whereas the OXPHOS activity was increased that may explain. In line with this study, intracellular [U-13C] tracing analysis revealed that intermediate metabolite levels of pentose phosphate pathway (6-phosphogluconolactone), citrate and lactate were significantly decreased, indicating the inhibition of the Warburg effect and cellular glucose dependence in ASS1-deficient cell lines. Moreover, arginine starvation also caused the increase of extracellular glutamine uptake with higher expression of glutaminolysis-related actors such as GLS and glutamate dehydrogenase. This process determines the metabolic switch from aerobic glycolysis to glutamine metabolism to fuel OXPHOS activity through the TCA cycle in ADI-PEG20 treated cell lines. This metabolic vulnerability was then explored using the combination of ADI-PEG20 and GLS inhibitor bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), resulting in better reduction in a broad range of ASS1-deficient cell lines, including OS cell line.
However, the only concern to be taken into account is that ADI-PEG20 can stimulate the re-expression of ASS1 gene due to metabolic adaptation, resulting in the resistance mechanism of ASS1-deficient cancer cells (Shen et al., 2003) (Feun and Savaraj, 2006). The study conducted by Prudner et al. (2019) showed the promising combination of arginine deiminase (ADI-PEG20), Gemcitabine, and Docetaxel on a panel of ASS1-low to -null expressing cancers, both in vitro and in vivo. This study cohort included not only OS, but a larger group of sarcomas including fibrosarcoma, uterine spindle cell sarcoma, leiomyosarcoma, melanoma, Ewing sarcoma and rhabdomyosarcoma. Of note, Gemcitabine is well known as a nucleoside analog that incorporates into the DNA and induces cell deaths, used for many cancer treatments (de Sousa Cavalcante and Monteiro, 2014) (Toschi et al., 2005). Docetaxel is a microtubule-binding agent, causing cell-cycle arrest and apoptosis (Montero et al., 2005). The combination of Gemcitabine and Docetaxel is used as a second-line therapy for advanced soft-tissue sarcoma (Maki et al., 2007, p. 002) (Seddon et al., 2017). Gemcitabine resistance is associated to the dysregulation of the Gemcitabine metabolism-related proteins, including the decrease of Human Equilibrative Nucleoside Transporter 1 (hENT1)—the main plasma membrane transporter of Gemcitabine; decrease of deoxycytidine kinase (dCK)—a kinase responsible for the phosphorylation of Gemcitabine and increase of ribonucleotide reductase subunit M1 and 2 (RRM1/2) that can convert active Gemcitabine to its inactive form (Jia and Xie, 2015). Interestingly, in ASS1-deficient pancreatic cell lines, ADI-PEG20 can inhibit RRM2 while this effect was not observed in other ASS1-expressing cell lines. Of note, RRM2 gene is negatively regulated by transcription factor E2F-1. Therefore, ADI-PEG20 increases E2F-1 and inhibits RRM2 transcription. Thus, the combination of ADI-PEG20 and Gemcitabine exhibited an increased growth reduction, leading to tumor regression both in vitro and in vivo (Daylami et al., 2014). Other studies showed that the arginine starvation by ADI-PEG20 combined with the cMyc stabilization by Docetaxel increases expression of hENT1. The combination of these three drugs promotes the entry of Gemcitabine in cancer cells and results in higher cell death rate (Figure 7). Taking into account the re-expression of ASS1 as a resistant mechanism, the research group showed that the long-term treatment with ADI-PEG20 remains effective (Prudner et al., 2019).
Based on these findings, a phase II clinical trial is now active, named “ADI-PEG 20” for combination with Docetaxel and Gemcitabine for the treatment of soft tissue sarcoma as the first cohort and OS, Ewing’s Sarcoma, Small Cell Lung Cancer as the second cohort for progression/relapse disease after failure of standard treatment (NCT03449901) (Washington University School of Medicine, 2021) (Figure 7). Of note, in the previous clinical evaluation of ADI-PEG20 in advanced hepatocellular carcinoma, the most frequent adverse event is fatigue and there is no statistical difference in term of adverse event between ADI-PEG20 treated patients versus placebo cohort (Abou-Alfa et al., 2018, p. 0).
Metabolic reprogramming is considered a central hallmark in cancer. It allows not only utilization of various energy sources depending on their availability in the tumor microenvironment, but most importantly fuel biomass synthesis. In line with the increased attention to this field across disciplines, we reflect in this summary on the current knowledge of cell death and metabolic reprogramming at different OS stages.
In this review, we discuss how mitochondria-related RCD and metabolism directly impact the proliferation of benign, malignant, and metastatic OS in vitro and in vivo models. We summarize the evidence that supports the interconnectivity of different mitochondria-related RCD pathways, with a focus on fundamental notions of apoptosis, ferroptosis, necroptosis, and autophagy and their involvement in cell fate, progression, and metastasis. The development of RCD inducers has recently emphasized the importance of a balance between apoptosis and autophagy, which is crucial for cell fate and drug efficacy. Autophagy also plays an essential role in the induction of ferroptosis and necroptosis by regulating metabolic pathways and ROS generation. Furthermore, necroptosis and extrinsic apoptosis share many common actors within the signal transduction pathways. Therefore, the crosstalk between apoptosis, ferroptosis, necroptosis, pyroptosis and autophagy creates an orchestra of RCD, emphasizing many new targets that could become useful suggestions for the development of OS therapeutic strategies in the future.
Many RCD and metabolism inducers or inhibitors are actively under investigation and have recently opened new areas for potential mono- or combinatorial OS therapies. However, at this stage undoubtedly further studies are needed to build fundamental knowledge of mitochondria-related RCDs and metabolic adaptation for novel OS treatments. Because of the high incidence of OS metastasis, relapse, and drug resistance, novel pharmacological approaches are urgently needed in this patient category. Currently, metformin and the combination of ADI-PEG20, Docetaxel, and Gemcitabine are in active clinical studies for OS treatment. This will thus provide a first benchmark of translational relevance of these findings and hopefully open the door to further novel treatment strategies for OS.
The question of how OS cells can precisely modulate cell death machineries and metabolism remains to be fully elucidated. Notably, comparing OS mitochondrial metabolism with that of other cancer types remains a future challenge and could be instrumental to develop novel OS therapeutic approaches in the near future.
The conceptualization of this work was carried out by HL, NN, and CB. HL and NN wrote the original draft of the manuscript. HL and NN participated in interpretation of the literature. Proofreading and editing by NN, AM, NG, BG, and CB. All authors revised the draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
CB and HL’s research was funded by Société française de lutte contre les cancers et les leucémies de l’enfant et de l’adolescent (SFCE), grant number ECS 20 and Institut National du Cancer (INCa), grant number INCA_ 16344. NN was funded by Pause Program and CNRS.
We thank Prisca Josephine Sanusi and Daniela Martins Dias Pedroso for their feedback and useful suggestions. Figures were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AbbVie, (2021). A first in human study of the MCL-1 inhibitor, ABBV-467 (clinical trial registration No. NCT04178902). clinicaltrials.gov.
Abou-Alfa, G. K., Qin, S., Ryoo, B.-Y., Lu, S.-N., Yen, C.-J., Feng, Y.-H., et al. (2018). Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann. Oncol. 29, 1402–1408. doi:10.1093/annonc/mdy101
Abu-Hamad, S., Zaid, H., Israelson, A., Nahon, E., and Shoshan-Barmatz, V. (2008). Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 283, 13482–13490. doi:10.1074/jbc.M708216200
Amary, M. F., Bacsi, K., Maggiani, F., Damato, S., Halai, D., Berisha, F., et al. (2011). IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 224, 334–343. doi:10.1002/path.2913
Amin, P., Florez, M., Najafov, A., Pan, H., Geng, J., Ofengeim, D., et al. (2018). Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. U. S. A. 115, E5944–E5953. doi:10.1073/pnas.1806973115
Antignani, A., and Youle, R. J. (2006). How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol. 18, 685–689. doi:10.1016/j.ceb.2006.10.004
AstraZeneca, (2022). A phase 1/1b/2a, 3-part, open-label, multicentre study to assess the safety, tolerability, pharmacokinetics and preliminary antitumor activity of ascending doses of AZD5991 monotherapy and in combination with Venetoclax in subjects with relapsed or refractory haematologic malignancies (clinical trial registration No. NCT03218683). clinicaltrials.gov.
Azoulay-Zohar, H., Israelson, A., Abu-Hamad, S., and Shoshan-Barmatz, V. (2004). In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 377, 347–355. doi:10.1042/BJ20031465
Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., et al. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662. doi:10.1038/nature03434
Banerjee, J., and Ghosh, S. (2004). Bax increases the pore size of rat brain mitochondrial voltage-dependent anion channel in the presence of tBid. Biochem. Biophys. Res. Commun. 323, 310–314. doi:10.1016/j.bbrc.2004.08.094
Baranski, Z., de Jong, Y., Ilkova, T., Peterse, E. F. P., Cleton-Jansen, A.-M., van de Water, B., et al. (2015). Pharmacological inhibition of Bcl-xL sensitizes osteosarcoma to doxorubicin. Oncotarget 6, 36113–36125. doi:10.18632/oncotarget.5333
Baumgartner, H. K., Gerasimenko, J. V., Thorne, C., Ferdek, P., Pozzan, T., Tepikin, A. V., et al. (2009). Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 284, 20796–20803. doi:10.1074/jbc.M109.025353
Bean, G. R., Kremer, J. C., Prudner, B. C., Schenone, A. D., Yao, J.-C., Schultze, M. B., et al. (2016). A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death Dis. 7, e2406. doi:10.1038/cddis.2016.232
Berlanga, P., Pierron, G., Lacroix, L., Chicard, M., Adam de Beaumais, T., Marchais, A., et al. (2022). The European MAPPYACTS trial: Precision medicine program in pediatric and adolescent patients with recurrent malignancies. Cancer Discov. 12, 1266–1281. doi:10.1158/2159-8290.CD-21-1136
Bernardi, P., Rasola, A., Forte, M., and Lippe, G. (2015). The mitochondrial permeability transition pore: Channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 95, 1111–1155. doi:10.1152/physrev.00001.2015
Beutner, G., Ruck, A., Riede, B., Welte, W., and Brdiczka, D. (1996). Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 396, 189–195. doi:10.1016/0014-5793(96)01092-7
Biasutto, L., Azzolini, M., Szabò, I., and Zoratti, M. (2016). The mitochondrial permeability transition pore in AD 2016: An update. Biochim. Biophys. Acta 1863, 2515–2530. doi:10.1016/j.bbamcr.2016.02.012
Bielack, S. S., Kempf-Bielack, B., Delling, G., Exner, G. U., Flege, S., Helmke, K., et al. (2002). Prognostic factors in high-grade osteosarcoma of the extremities or trunk: An analysis of 1, 702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J. Clin. Oncol. 20, 776–790. doi:10.1200/JCO.2002.20.3.776
Bleicken, S., Jeschke, G., Stegmueller, C., Salvador-Gallego, R., García-Sáez, A. J., Bordignon, E., et al. (2014). Structural model of active Bax at the membrane. Mol. Cell 56, 496–505. doi:10.1016/j.molcel.2014.09.022
Bonnefont, J.-P., Djouadi, F., Prip-Buus, C., Gobin, S., Munnich, A., Bastin, J., et al. (2004). Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Asp. Med. 25, 495–520. doi:10.1016/j.mam.2004.06.004
Bonora, M., Giorgi, C., and Pinton, P. (2022). Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 23, 266–285. doi:10.1038/s41580-021-00433-y
Bonora, M., and Pinton, P. (2014). The mitochondrial permeability transition pore and cancer: Molecular mechanisms involved in cell death. Front. Oncol. 4, 302. doi:10.3389/fonc.2014.00302
Borner, C., and Andrews, D. W. (2014). The apoptotic pore on mitochondria: Are we breaking through or still stuck? Cell Death Differ. 21, 187–191. doi:10.1038/cdd.2013.169
Bowles, T. L., Kim, R., Galante, J., Parsons, C. M., Virudachalam, S., Kung, H.-J., et al. (2008). Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. Int. J. Cancer 123, 1950–1955. doi:10.1002/ijc.23723
Bratton, S. B., Walker, G., Srinivasula, S. M., Sun, X. M., Butterworth, M., Alnemri, E. S., et al. (2001). Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 20, 998–1009. doi:10.1093/emboj/20.5.998
Canadian Cancer Trials Group (2022). A phase III randomized trial of metformin versus placebo on recurrence and survival in early stage breast cancer (clinical trial registration No. NCT01101438). clinicaltrials.gov.
Carneiro, B. A., and El-Deiry, W. S. (2020). Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17, 395–417. doi:10.1038/s41571-020-0341-y
Casali, P. G., Bielack, S., Abecassis, N., Aro, H. T., Bauer, S., Biagini, R., et al. (2018). ESMO guidelines committee, PaedCan and ERN EURACANBone sarcomas: ESMO-PaedCan-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 29, iv79–iv95. doi:10.1093/annonc/mdy310
Chen, G., Feng, W., Zhang, S., Bian, K., Yang, Y., Fang, C., et al. (2015). Metformin inhibits gastric cancer via the inhibition of HIF1α/PKM2 signaling. Am. J. Cancer Res. 5, 1423–1434.
Chen, J., Kos, R., Garssen, J., and Redegeld, F. (2019). Molecular insights into the mechanism of necroptosis: The necrosome as a potential therapeutic target. Cells 8, E1486. doi:10.3390/cells8121486
Cheng, C.-T., Qi, Y., Wang, Y.-C., Chi, K. K., Chung, Y., Ouyang, C., et al. (2018). Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun. Biol. 1, 178. doi:10.1038/s42003-018-0178-4
Chesney, J. (2021b). A phase I/II trial of dabrafenib, trametinib and metformin administered to unresectable stage IIIC and stage IV BRAF V600E + melanoma patients (clinical trial registration No. NCT02143050). clinicaltrials.gov.
Chesney, J. (2021a). V600E+ melanoma patients (clinical trial registration No. NCT01638676). clinicaltrials.gov.A phase I/II trial of vemurafenib and metformin to unresectable stage IIIC and stage IV BRAF
Chi, M. M.-Y., Pingsterhaus, J., Carayannopoulos, M., and Moley, K. H. (2020). Decreased glucose transporter expression triggers BAX-dependent apoptosis in the murine blastocyst. J. Biol. Chem. 275, 40252–40257. doi:10.1074/jbc.M005508200
Chipuk, J. E., Bouchier-Hayes, L., and Green, D. R. (2006). Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 13, 1396–1402. doi:10.1038/sj.cdd.4401963
Christofferson, D. E., and Yuan, J. (2010). Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 22, 263–268. doi:10.1016/j.ceb.2009.12.003
Closs, E. I., Simon, A., Vékony, N., and Rotmann, A. (2004). Plasma membrane transporters for arginine. J. Nutr. 134, 2752S–2759S. doi:10.1093/jn/134.10.2752S
Cluntun, A. A., Lukey, M. J., Cerione, R. A., and Locasale, J. W. (2017). Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer 3, 169–180. doi:10.1016/j.trecan.2017.01.005
Crompton, M. (1999). The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341 (Pt 2), 233–249. doi:10.1042/bj3410233
Cui, M., Wang, H., Yao, X., Zhang, D., Xie, Y., Cui, R., et al. (2019). Circulating MicroRNAs in cancer: Potential and challenge. Front. Genet. 10, 626. doi:10.3389/fgene.2019.00626
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. doi:10.1038/nature08617
Danial, N. N., and Korsmeyer, S. J. (2004). Cell death: Critical control points. Cell 116, 205–219. doi:10.1016/s0092-8674(04)00046-7
Daylami, R., Muilenburg, D. J., Virudachalam, S., and Bold, R. J. (2014). Pegylated arginine deiminase synergistically increases the cytotoxicity of gemcitabine in human pancreatic cancer. J. Exp. Clin. Cancer Res. 33, 102. doi:10.1186/s13046-014-0102-9
de Sousa Cavalcante, L., and Monteiro, G. (2014). Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 741, 8–16. doi:10.1016/j.ejphar.2014.07.041
Della Sala, G., Pacelli, C., Agriesti, F., Laurenzana, I., Tucci, F., Tamma, M., et al. (2022). Unveiling metabolic vulnerability and plasticity of human osteosarcoma stem and differentiated cells to improve cancer therapy. Biomedicines 10, 28. doi:10.3390/biomedicines10010028
DeNicola, G. M., and Cantley, L. C. (2015). Cancer’s fuel choice: New flavors for a picky eater. Mol. Cell 60, 514–523. doi:10.1016/j.molcel.2015.10.018
DeWaal, D., Nogueira, V., Terry, A. R., Patra, K. C., Jeon, S.-M., Guzman, G., et al. (2018). Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 9, 446. doi:10.1038/s41467-017-02733-4
Dhillon, S. (2018). Ivosidenib: First global approval. Drugs 78, 1509–1516. doi:10.1007/s40265-018-0978-3
Di Cristofano, A., Pandolfi, P. P., and Di CristofAno, A. (2000). The multiple roles of PTEN in tumor suppression. Cell 100, 387–390. doi:10.1016/s0092-8674(00)80674-1
Díaz-Troya, S., Pérez-Pérez, M. E., Florencio, F. J., and Crespo, J. L. (2008). The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 4, 851–865. doi:10.4161/auto.6555
DiNardo, C. D., Propert, K. J., Loren, A. W., Paietta, E., Sun, Z., Levine, R. L., et al. (2013). Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood 121, 4917–4924. doi:10.1182/blood-2013-03-493197
Ding, J., Wang, K., Liu, W., She, Y., Sun, Q., Shi, J., et al. (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. doi:10.1038/nature18590
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. doi:10.1016/s1535-6108(03)00050-3
Duchen, M. R. (2000). Mitochondria and Ca2+in cell physiology and pathophysiology. Cell Calcium 28, 339–348. doi:10.1054/ceca.2000.0170
Edmonson, J. H., Green, S. J., Ivins, J. C., Gilchrist, G. S., Creagan, E. T., Pritchard, D. J., et al. (1984). A controlled pilot study of high-dose methotrexate as postsurgical adjuvant treatment for primary osteosarcoma. J. Clin. Oncol. 2, 152–156. doi:10.1200/JCO.1984.2.3.152
El-Mir, M. Y., Nogueira, V., Fontaine, E., Avéret, N., Rigoulet, M., Leverve, X., et al. (2000). Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 275, 223–228. doi:10.1074/jbc.275.1.223
Eling, N., Reuter, L., Hazin, J., Hamacher-Brady, A., and Brady, N. R. (2015). Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2, 517–532. doi:10.18632/oncoscience.160
Eliseev, R., Dong, Y.-F., Sampson, E., Zuscik, M., Schwarz, E., O’Keefe, R., et al. (2008). Runx2-mediated activation of the Bax gene increases osteosarcoma cell sensitivity to apoptosis. Oncogene 27, 3605–3614. doi:10.1038/sj.onc.1211020
Elmore, S. (2007). Apoptosis: A review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi:10.1080/01926230701320337
Emslie-Smith, A. M., Boyle, D. I., Evans, J. M., Sullivan, F., and Morris, A. D. (2001). DARTS/MEMO CollaborationContraindications to metformin therapy in patients with Type 2 diabetes-a population-based study of adherence to prescribing guidelines. Diabet. Med. 18, 483–488. doi:10.1046/j.1464-5491.2001.00509.x
Evavold, C. L., Hafner-Bratkovič, I., Devant, P., D’Andrea, J. M., Ngwa, E. M., Boršić, E., et al. (2021). Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 184, 4495–4511.e19. e19. doi:10.1016/j.cell.2021.06.028
Fan, J., Teng, X., Liu, L., Mattaini, K. R., Looper, R. E., Vander Heiden, M. G., et al. (2015). Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem. Biol. 10, 510–516. doi:10.1021/cb500683c
Fang, Y., Tian, S., Pan, Y., Li, W., Wang, Q., Tang, Y., et al. (2020). Pyroptosis: A new frontier in cancer. Biomedicine & Pharmacotherapy 121.
Faubert, B., Solmonson, A., and DeBerardinis, R. J. (2020). Metabolic reprogramming and cancer progression. Science 368, eaaw5473. doi:10.1126/science.aaw5473
Feinstein, E., Kimchi, A., Wallach, D., Boldin, M., and Varfolomeev, E. (1995). The death domain: A module shared by proteins with diverse cellular functions. Trends biochem. Sci. 20, 342–344. doi:10.1016/s0968-0004(00)89070-2
Feun, L., and Savaraj, N. (2006). Pegylated arginine deiminase: A novel anticancer enzyme agent. Expert Opin. Investig. Drugs 15, 815–822. doi:10.1517/13543784.15.7.815
Foretz, M., Guigas, B., Bertrand, L., Pollak, M., and Viollet, B. (2014). Metformin: From mechanisms of action to therapies. Cell Metab. 20, 953–966. doi:10.1016/j.cmet.2014.09.018
Freeman, S. S., Allen, S. W., Ganti, R., Wu, J., Ma, J., Su, X., et al. (2008). Copy number gains in EGFR and copy number losses in PTEN are common events in osteosarcoma tumors. Cancer 113, 1453–1461. doi:10.1002/cncr.23782
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi:10.1038/ncb3064
Friesen, C., Herr, I., Krammer, P. H., and Debatin, K.-M. (1996). Involvement of the CD95 (APO–1/Fas) receptor/ligand system in drug–induced apoptosis in leukemia cells. Nat. Med. 2, 574–577. doi:10.1038/nm0596-574
Fritsche-Guenther, R., Gloaguen, Y., Kirchner, M., Mertins, P., Tunn, P.-U., Kirwan, J. A., et al. (2020). Progression-dependent altered metabolism in osteosarcoma resulting in different nutrient source dependencies. Cancers 12, 1371. doi:10.3390/cancers12061371
Fu, Z., Deng, B., Liao, Y., Shan, L., Yin, F., Wang, Z., et al. (2013). The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 13, 580. doi:10.1186/1471-2407-13-580
Fuchs, B. C., and Bode, B. P. (2006). Stressing out over survival: Glutamine as an apoptotic modulator. J. Surg. Res. 131, 26–40. doi:10.1016/j.jss.2005.07.013
Fuchs, B. C., Perez, J. C., Suetterlin, J. E., Chaudhry, S. B., and Bode, B. P. (2004). Inducible antisense RNA targeting amino acid transporter ATB0/ASCT2 elicits apoptosis in human hepatoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G467–G478. doi:10.1152/ajpgi.00344.2003
Fulda, S. (2013). The mechanism of necroptosis in normal and cancer cells. Cancer Biol. Ther. 14, 999–1004. doi:10.4161/cbt.26428
Galadari, S., Rahman, A., Pallichankandy, S., and Thayyullathil, F. (2017). Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 104, 144–164. doi:10.1016/j.freeradbiomed.2017.01.004
Galluzzi, L., Kepp, O., Tajeddine, N., and Kroemer, G. (2008). Disruption of the hexokinase-VDAC complex for tumor therapy. Oncogene 27, 4633–4635. doi:10.1038/onc.2008.114
Galluzzi, L., Kepp, O., Trojel-Hansen, C., and Kroemer, G. (2012). Mitochondrial control of cellular life, stress, and death. Circ. Res. 111, 1198–1207. doi:10.1161/CIRCRESAHA.112.268946
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541. doi:10.1038/s41418-017-0012-4
Gan, I., Jiang, J., Lian, D., Huang, X., Fuhrmann, B., Liu, W., et al. (2019). Mitochondrial permeability regulates cardiac endothelial cell necroptosis and cardiac allograft rejection. Am. J. Transpl. 19, 686–698. doi:10.1111/ajt.15112
Gao, M., Monian, P., Pan, Q., Zhang, W., Xiang, J., Jiang, X., et al. (2016). Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032. doi:10.1038/cr.2016.95
Gaspar, N., Occean, B.-V., Pacquement, H., Bompas, E., Bouvier, C., Brisse, H. J., et al. (2018). SFCE (Société Française des Cancers de l’Enfant et l’adolescent), GSF-GETO (Groupe Sarcome Français), UNICANCER sarcoma groupResults of methotrexate-etoposide-ifosfamide based regimen (M-EI) in osteosarcoma patients included in the French OS2006/sarcome-09 study. Eur. J. Cancer 88, 57–66. doi:10.1016/j.ejca.2017.09.036
Gatz, S. A., Rubino, J., Rossoni, C., Andre, N., Aerts, I., Thebaud, E., et al. (2019). AcSé-ESMART: European proof of concept therapeutic stratification trial of molecular anomalies in relapsed or refractory tumors in children and adolescents–arm D: Olaparib and irinotecan. JCO 37, 10047. doi:10.1200/JCO.2019.37.15_suppl.10047
Ghodke-Puranik, Y., Puranik, A. S., Shintre, P., Joshi, K., Patwardhan, B., Lamba, J., et al. (2015). Folate metabolic pathway single nucleotide polymorphisms: A predictive pharmacogenetic marker of methotrexate response in Indian (asian) patients with rheumatoid arthritis. Pharmacogenomics 16, 2019–2034. doi:10.2217/pgs.15.145
Giang, A.-H., Raymond, T., Brookes, P., de Mesy Bentley, K., Schwarz, E., O’Keefe, R., et al. (2013). Mitochondrial dysfunction and permeability transition in osteosarcoma cells showing the Warburg effect. J. Biol. Chem. 288, 33303–33311. doi:10.1074/jbc.M113.507129
Gill, J., and Gorlick, R. (2021). Advancing therapy for osteosarcoma. Nat. Rev. Clin. Oncol. 18, 609–624. doi:10.1038/s41571-021-00519-8
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: Cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi:10.1002/path.2697
Gogvadze, V., Orrenius, S., and Zhivotovsky, B. (2008). Mitochondria in cancer cells: What is so special about them? Trends Cell Biol. 18, 165–173. doi:10.1016/j.tcb.2008.01.006
Gordon, I. K., Ye, F., and Kent, M. S. (2008). Evaluation of the mammalian target of rapamycin pathway and the effect of rapamycin on target expression and cellular proliferation in osteosarcoma cells from dogs. Am. J. Vet. Res. 69, 1079–1084. doi:10.2460/ajvr.69.8.1079
Green, D. R., and Evan, G. I. (2002). A matter of life and death. Cancer Cell 1, 19–30. doi:10.1016/s1535-6108(02)00024-7
Green, D. R., Galluzzi, L., and Kroemer, G. (2014). Cell biology. Metabolic control of cell death. Science 345, 1250256. doi:10.1126/science.1250256
Greenburg, G., and Hay, E. D. (1982). Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 95, 333–339. doi:10.1083/jcb.95.1.333
Grignani, G., Palmerini, E., Ferraresi, V., D’Ambrosio, L., Bertulli, R., Asaftei, S. D., et al. (2015). Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet. Oncol. 16, 98–107. doi:10.1016/S1470-2045(14)71136-2
Gu, X., Orozco, J. M., Saxton, R. A., Condon, K. J., Liu, G. Y., Krawczyk, P. A., et al. (2017). SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358, 813–818. doi:10.1126/science.aao3265
Gupta, K., Phan, N., Wang, Q., and Liu, B. (2018). Necroptosis in cardiovascular disease - a new therapeutic target. J. Mol. Cell. Cardiol. 118, 26–35. doi:10.1016/j.yjmcc.2018.03.003
Gustave Roussy, Cancer Campus, Grand Paris (2021). European proof-of-concept therapeutic stratification trial of molecular anomalies in relapsed or refractory tumors (clinical trial registration No. NCT02813135). Villejuif: clinicaltrials.gov.
Gwak, G.-Y., Yoon, J.-H., Kim, K. M., Lee, H.-S., Chung, J. W., Gores, G. J., et al. (2005). Hypoxia stimulates proliferation of human hepatoma cells through the induction of hexokinase II expression. J. Hepatol. 42, 358–364. doi:10.1016/j.jhep.2004.11.020
Han, J., Zhong, C.-Q., and Zhang, D.-W. (2011). Programmed necrosis: Backup to and competitor with apoptosis in the immune system. Nat. Immunol. 12, 1143–1149. doi:10.1038/ni.2159
Hanahan, D. (2022). Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46. doi:10.1158/2159-8290.CD-21-1059
He, Y., Hara, H., and Núñez, G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends biochem. Sci. 41, 1012–1021. doi:10.1016/j.tibs.2016.09.002
Hegde, R., Srinivasula, S. M., Zhang, Z., Wassell, R., Mukattash, R., Cilenti, L., et al. (2002). Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 277, 432–438. doi:10.1074/jbc.M109721200
Hikita, H., Takehara, T., Shimizu, S., Kodama, T., Li, W., Miyagi, T., et al. (2009). Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology 50, 1217–1226. doi:10.1002/hep.23126
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495. doi:10.1038/82732
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi:10.1080/15548627.2016.1187366
Houghton, P. J., Gorlick, R., Kolb, E. A., Lock, R., Carol, H., Morton, C. L., et al. (2012). Initial testing (stage 1) of the mTOR kinase inhibitor AZD8055 by the pediatric preclinical testing program. Pediatr. Blood Cancer 58, 191–199. doi:10.1002/pbc.22935
Howell, J. J., Hellberg, K., Turner, M., Talbott, G., Kolar, M. J., Ross, D. S., et al. (2017). Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 25, 463–471. doi:10.1016/j.cmet.2016.12.009
Hsu, H., Xiong, J., and Goeddel, D. V. (1995). The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 81, 495–504. doi:10.1016/0092-8674(95)90070-5
Hua, H., Kong, Q., Zhang, H., Wang, J., Luo, T., Jiang, Y., et al. (2019). Targeting mTOR for cancer therapy. J. Hematol. Oncol. 12, 71. doi:10.1186/s13045-019-0754-1
Hua, Y., Qiu, Y., Zhao, A., Wang, X., Chen, T., Zhang, Z., et al. (2011). Dynamic metabolic transformation in tumor invasion and metastasis in mice with LM-8 osteosarcoma cell transplantation. J. Proteome Res. 10, 3513–3521. doi:10.1021/pr200147g
Huang, H.-Y., Wu, W.-R., Wang, Y.-H., Wang, J.-W., Fang, F.-M., Tsai, J.-W., et al. (2013). ASS1 as a novel tumor suppressor gene in myxofibrosarcomas: Aberrant loss via epigenetic DNA methylation confers aggressive phenotypes, negative prognostic impact, and therapeutic relevance. Clin. Cancer Res. 19, 2861–2872. doi:10.1158/1078-0432.CCR-12-2641
Hui, S., Ghergurovich, J. M., Morscher, R. J., Jang, C., Teng, X., Lu, W., et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. doi:10.1038/nature24057
Institut de Recherches Internationales Servier (2022). An international phase ib multicentre study to characterize the safety and tolerability of intravenously administered S64315, a selective mcl-1 inhibitor, in combination with orally administered Venetoclax, a selective bcl-2 inhibitor in patients with acute myeloid leukaemia (AML). (Clinical trial registration No. NCT03672695). clinicaltrials.gov.
Intlekofer, A. M., and Finley, L. W. S. (2019). Metabolic signatures of cancer cells and stem cells. Nat. Metab. 1, 177–188. doi:10.1038/s42255-019-0032-0
Irmler, M., Thome, M., Hahne, M., Schneider, P., Hofmann, K., Steiner, V., et al. (1997). Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195. doi:10.1038/40657
Isakoff, M. S., Bielack, S. S., Meltzer, P., and Gorlick, R. (2015). Osteosarcoma: Current treatment and a collaborative pathway to success. J. Clin. Oncol. 33, 3029–3035. doi:10.1200/JCO.2014.59.4895
Ito, K., and Suda, T. (2014). Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 15, 243–256. doi:10.1038/nrm3772
Jia, X.-Q., Zhang, S., Zhu, H.-J., Wang, W., Zhu, J.-H., Wang, X.-D., et al. (2016). Increased expression of PHGDH and prognostic significance in colorectal cancer. Transl. Oncol. 9, 191–196. doi:10.1016/j.tranon.2016.03.006
Jia, Y., and Xie, J. (2015). Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes Dis. 2, 299–306. doi:10.1016/j.gendis.2015.07.003
Jiang, K., Zhang, C., Yu, B., Chen, B., Liu, Z., Hou, C., et al. (2017). Autophagic degradation of FOXO3a represses the expression of PUMA to block cell apoptosis in cisplatin-resistant osteosarcoma cells. Am. J. Cancer Res. 7, 1407–1422.
Jiang, L., Kon, N., Li, T., Wang, S.-J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. doi:10.1038/nature14344
Jiang, X., and Wang, X. (2000). Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to apaf-1. J. Biol. Chem. 275, 31199–31203. doi:10.1074/jbc.C000405200
Jonker, J. W., and Schinkel, A. H. (2004). Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3). J. Pharmacol. Exp. Ther. 308, 2–9. doi:10.1124/jpet.103.053298
Jouan-Lanhouet, S., Riquet, F., Duprez, L., Vanden Berghe, T., Takahashi, N., Vandenabeele, P., et al. (2014). Necroptosis, in vivo detection in experimental disease models. Semin. Cell Dev. Biol. 35, 2–13. doi:10.1016/j.semcdb.2014.08.010
Kalkavan, H., and Green, D. R. (2018). MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 25, 46–55. doi:10.1038/cdd.2017.179
Kehr, S., Haydn, T., Bierbrauer, A., Irmer, B., Vogler, M., Fulda, S., et al. (2020). Targeting BCL-2 proteins in pediatric cancer: Dual inhibition of BCL-XL and MCL-1 leads to rapid induction of intrinsic apoptosis. Cancer Lett. 482, 19–32. doi:10.1016/j.canlet.2020.02.041
Keinan, N., Pahima, H., Ben-Hail, D., and Shoshan-Barmatz, V. (2013). The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim. Biophys. Acta 1833, 1745–1754. doi:10.1016/j.bbamcr.2013.03.017
Keinan, N., Tyomkin, D., and Shoshan-Barmatz, V. (2010). Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 30, 5698–5709. doi:10.1128/MCB.00165-10
Kennedy, L., Sandhu, J. K., Harper, M.-E., and Cuperlovic-Culf, M. (2020). Role of glutathione in cancer: From mechanisms to therapies. Biomolecules 10, E1429. doi:10.3390/biom10101429
Kerr, J. F., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. doi:10.1038/bjc.1972.33
Khan, Q. (2021). Randomized trial of neo-adjuvant chemotherapy with or without metformin for HER2 positive operable breast cancer (clinical trial registration No. NCT03238495). clinicaltrials.gov.
Kharbanda, S., Pandey, P., Schofield, L., Israels, S., Roncinske, R., Yoshida, K., et al. (1997). Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc. Natl. Acad. Sci. U. S. A. 94, 6939–6942. doi:10.1073/pnas.94.13.6939
Khoury, M. K., Gupta, K., Franco, S. R., and Liu, B. (2020). Necroptosis in the pathophysiology of disease. Am. J. Pathol. 190, 272–285. doi:10.1016/j.ajpath.2019.10.012
Kim, E. S. (2017). Enasidenib: First global approval. Drugs 77, 1705–1711. doi:10.1007/s40265-017-0813-2
Kim, J. S., Ro, S.-H., Kim, M., Park, H.-W., Semple, I. A., Park, H., et al. (2015). Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci. Rep. 5, 9502. doi:10.1038/srep09502
Kim, M.-J., Park, I.-J., Yun, H., Kang, I., Choe, W., Kim, S.-S., et al. (2010). AMP-Activated protein kinase antagonizes pro-apoptotic extracellular signal-regulated kinase activation by inducing dual-specificity protein phosphatases in response to glucose deprivation in HCT116 carcinoma. J. Biol. Chem. 285, 14617–14627. doi:10.1074/jbc.M109.085456
Ko, Y., Choi, A., Lee, M., and Lee, J. A. (2016). Metformin displays in vitro and in vivo antitumor effect against osteosarcoma. Korean J. Pediatr. 59, 374–380. doi:10.3345/kjp.2016.59.9.374
Kobayashi, E., Masuda, M., Nakayama, R., Ichikawa, H., Satow, R., Shitashige, M., et al. (2010). Reduced argininosuccinate synthetase is a predictive biomarker for the development of pulmonary metastasis in patients with osteosarcoma. Mol. Cancer Ther. 9, 535–544. doi:10.1158/1535-7163.MCT-09-0774
Koundouros, N., and Poulogiannis, G. (2020). Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22. doi:10.1038/s41416-019-0650-z
Kremer, J. C., Prudner, B. C., Lange, S. E. S., Bean, G. R., Schultze, M. B., Brashears, C. B., et al. (2017). Arginine deprivation inhibits the Warburg effect and upregulates glutamine anaplerosis and serine biosynthesis in ASS1-deficient cancers. Cell Rep. 18, 991–1004. doi:10.1016/j.celrep.2016.12.077
Kroemer, G., Galluzzi, L., and Brenner, C. (2007). Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163. doi:10.1152/physrev.00013.2006
Kuijjer, M. L., van den Akker, B. E. W. M., Hilhorst, R., Mommersteeg, M., Buddingh, E. P., Serra, M., et al. (2014). Kinome and mRNA expression profiling of high-grade osteosarcoma cell lines implies Akt signaling as possible target for therapy. BMC Med. Genomics 7, 4. doi:10.1186/1755-8794-7-4
Lacey, C. A., Mitchell, W. J., Dadelahi, A. S., and Skyberg, J. A. (2018). Caspase-1 and caspase-11 mediate pyroptosis, inflammation, and control of Brucella joint infection. Infect. Immun. 86, e00361–18. doi:10.1128/IAI.00361-18
Lachaier, E., Louandre, C., Godin, C., Saidak, Z., Baert, M., Diouf, M., et al. (2014). Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 34, 6417–6422.
Lai, H.-T., Canoy, R. J., Campanella, M., Vassetzky, Y., and Brenner, C. (2022). Ca2+ transportome and the interorganelle communication in hepatocellular carcinoma. Cells 11, 815. doi:10.3390/cells11050815
Lei, P., Bai, T., and Sun, Y. (2019). Mechanisms of ferroptosis and relations with regulated cell death: A review. Front. Physiol. 10, 139. doi:10.3389/fphys.2019.00139
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi:10.1016/j.cell.2007.12.018
Li, B., Zhou, P., Xu, K., Chen, T., Jiao, J., Wei, H., et al. (2020a). Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int. J. Biol. Sci. 16, 74–84. doi:10.7150/ijbs.33787
Li, H.-Y., Zhang, J., Sun, L.-L., Li, B.-H., Gao, H.-L., Xie, T., et al. (2015). Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: An in vitro and in vivo study. Cell Death Dis. 6, e1604. doi:10.1038/cddis.2014.543
Li, H., Zhu, H., Xu, C. J., and Yuan, J. (1998). Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501. doi:10.1016/s0092-8674(00)81590-1
Li, J., Eu, J. Q., Kong, L. R., Wang, L., Lim, Y. C., Goh, B. C., et al. (2020b). Targeting metabolism in cancer cells and the tumour microenvironment for cancer therapy. Molecules 25, E4831. doi:10.3390/molecules25204831
Li, J., Yang, Z., Li, Y., Xia, J., Li, D., Li, H., et al. (2016). Cell apoptosis, autophagy and necroptosis in osteosarcoma treatment. Oncotarget 7, 44763–44778. doi:10.18632/oncotarget.8206
Li, L. Y., Luo, X., and Wang, X. (2001). Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412, 95–99. doi:10.1038/35083620
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S., et al. (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. doi:10.1016/s0092-8674(00)80434-1
Li, W., Zheng, M., Wu, S., Gao, S., Yang, M., Li, Z., et al. (2017). Benserazide, a dopadecarboxylase inhibitor, suppresses tumor growth by targeting hexokinase 2. J. Exp. Clin. Cancer Res. 36, 58. doi:10.1186/s13046-017-0530-4
Li, Z., Wang, L., Luo, N., Zhao, Y., Li, J., Chen, Q., et al. (2018). Metformin inhibits the proliferation and metastasis of osteosarcoma cells by suppressing the phosphorylation of Akt. Oncol. Lett. 15, 7948–7954. doi:10.3892/ol.2018.8297
Liang, C., Zhang, X., Yang, M., and Dong, X. (2019). Recent progress in ferroptosis inducers for cancer therapy. Adv. Mat. 31, 1904197. doi:10.1002/adma.201904197
Liberti, M. V., and Locasale, J. W. (2016). The Warburg effect: How does it benefit cancer cells? Trends biochem. Sci. 41, 211–218. doi:10.1016/j.tibs.2015.12.001
Lin, R., Wei, H., Wang, S., Huang, Z., Chen, H., Zhang, S., et al. (2020). Gasdermin D expression and clinicopathologic outcome in primary osteosarcoma patients. Int. J. Clin. Exp. Pathol. 13, 3149–3157.
Lin, Y., Devin, A., Rodriguez, Y., and Liu, Z. G. (1999). Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526. doi:10.1101/gad.13.19.2514
Link, M. P., Goorin, A. M., Horowitz, M., Meyer, W. H., Belasco, J., Baker, A., et al. (1991). Adjuvant chemotherapy of high-grade osteosarcoma of the extremity. Updated results of the Multi-Institutional Osteosarcoma Study. Clin. Orthop. Relat. Res. 270, 8–14. doi:10.1097/00003086-199109000-00003
Liu, C., Cai, L., and Li, H. (2019a). miR-185 regulates the growth of osteosarcoma cells via targeting Hexokinase 2. Mol. Med. Rep. 20, 2774–2782. doi:10.3892/mmr.2019.10534
Liu, M., Zhang, Z., Wang, H., Chen, X., and Jin, C. (2019b). Activation of AMPK by metformin promotes renal cancer cell proliferation under glucose deprivation through its interaction with PKM2. Int. J. Biol. Sci. 15, 617–627. doi:10.7150/ijbs.29689
Liu, X., Kato, Y., Kaneko, M. K., Sugawara, M., Ogasawara, S., Tsujimoto, Y., et al. (2013). Isocitrate dehydrogenase 2 mutation is a frequent event in osteosarcoma detected by a multi-specific monoclonal antibody MsMab-1. Cancer Med. 2, 803–814. doi:10.1002/cam4.149
Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and Wang, X. (1996). Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 86, 147–157. doi:10.1016/s0092-8674(00)80085-9
Livingston, J. A., Wang, W.-L., Tsai, J.-W., Lazar, A. J., Leung, C. H., Lin, H., et al. (2018). Analysis of HSP27 and the autophagy marker LC3B+ puncta following preoperative chemotherapy identifies high-risk osteosarcoma patients. Mol. Cancer Ther. 17, 1315–1323. doi:10.1158/1535-7163.MCT-17-0901
Locasale, J. W., Grassian, A. R., Melman, T., Lyssiotis, C. A., Mattaini, K. R., Bass, A. J., et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874. doi:10.1038/ng.890
Longhi, M. D, A. (2021). Metformin as maintenance therapy in patients with bone sarcoma and high risk of relapse (clinical trial registration No. NCT04758000). clinicaltrials.gov.
Lovell, J. F., Billen, L. P., Bindner, S., Shamas-Din, A., Fradin, C., Leber, B., et al. (2008). Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084. doi:10.1016/j.cell.2008.11.010
Lu, X., Guo, T., and Zhang, X. (2021). Pyroptosis in cancer: Friend or foe? Cancers (Basel) 13, 3620. doi:10.3390/cancers13143620
Luo, Y., Gao, X., Zou, L., Lei, M., Feng, J., Hu, Z., et al. (2021). Bavachin induces ferroptosis through the STAT3/P53/slc7a11 Axis in osteosarcoma cells. Oxid. Med. Cell. Longev. 2021, 1783485. doi:10.1155/2021/1783485
Lv, H.-H., Zhen, C.-X., Liu, J.-Y., and Shang, P. (2020). PEITC triggers multiple forms of cell death by GSH-iron-ROS regulation in K7M2 murine osteosarcoma cells. Acta Pharmacol. Sin. 41, 1119–1132. doi:10.1038/s41401-020-0376-8
Maki, R. G., Wathen, J. K., Patel, S. R., Priebat, D. A., Okuno, S. H., Samuels, B., et al. (2007). Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: Results of sarcoma alliance for research through collaboration study 002 [corrected]. J. Clin. Oncol. 25, 2755–2763. doi:10.1200/JCO.2006.10.4117
Marchandet, L., Lallier, M., Charrier, C., Baud’huin, M., Ory, B., Lamoureux, F., et al. (2021). Mechanisms of resistance to conventional therapies for osteosarcoma. Cancers 13, 683. doi:10.3390/cancers13040683
Mardis, E. R., Ding, L., Dooling, D. J., Larson, D. E., McLellan, M. D., Chen, K., et al. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066. doi:10.1056/NEJMoa0903840
Marini, C., Salani, B., Massollo, M., Amaro, A., Esposito, A. I., Orengo, A. M., et al. (2013). Direct inhibition of hexokinase activity by metformin at least partially impairs glucose metabolism and tumor growth in experimental breast cancer. Cell Cycle 12, 3490–3499. doi:10.4161/cc.26461
Marshall, K. D., and Baines, C. P. (2014). Necroptosis: Is there a role for mitochondria? Front. Physiol. 5, 323. doi:10.3389/fphys.2014.00323
Martínez-Reyes, I., and Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102. doi:10.1038/s41467-019-13668-3
Marzo, I., Brenner, C., Zamzami, N., Susin, S. A., Beutner, G., Brdiczka, D., et al. (1998). The permeability transition pore complex: A target for apoptosis regulation by caspases and bcl-2–related proteins. J. Exp. Med. 187, 1261–1271. doi:10.1084/jem.187.8.1261
Masters, J. R. (2002). HeLa cells 50 years on: The good, the bad and the ugly. Nat. Rev. Cancer 2, 315–319. doi:10.1038/nrc775
Masuelli, L., Benvenuto, M., Izzi, V., Zago, E., Mattera, R., Cerbelli, B., et al. (2020). In vivo and in vitro inhibition of osteosarcoma growth by the pan Bcl-2 inhibitor AT-101. Invest. New Drugs 38, 675–689. doi:10.1007/s10637-019-00827-y
Matoba, K., Kotani, T., Tsutsumi, A., Tsuji, T., Mori, T., Noshiro, D., et al. (2020). Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 27, 1185–1193. doi:10.1038/s41594-020-00518-w
M.D. Anderson Cancer Center (2022b). Phase I study of TAK-228 (MLN0128) in combination with metformin in patients with advanced cancers (clinical trial registration No. NCT03017833). clinicaltrials.gov.
M.D. Anderson Cancer Center (2022a). Phase I study of Temsirolimus in combination with metformin in patients with advanced cancers (clinical trial registration No. NCT01529593). clinicaltrials.gov.
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190. doi:10.1016/s0092-8674(03)00521-x
Mirabello, L., Troisi, R. J., and Savage, S. A. (2009). Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the surveillance, epidemiology, and end results program. Cancer 115, 1531–1543. doi:10.1002/cncr.24121
Mitchell, P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148. doi:10.1038/191144a0
Mohseny, A. B., Machado, I., Cai, Y., Schaefer, K.-L., Serra, M., Hogendoorn, P. C. W., et al. (2011). Functional characterization of osteosarcoma cell lines provides representative models to study the human disease. Lab. Invest. 91, 1195–1205. doi:10.1038/labinvest.2011.72
Montero, A., Fossella, F., Hortobagyi, G., and Valero, V. (2005). Docetaxel for treatment of solid tumours: A systematic review of clinical data. Lancet. Oncol. 6, 229–239. doi:10.1016/S1470-2045(05)70094-2
Morciano, G., Naumova, N., Koprowski, P., Valente, S., Sardão, V. A., Potes, Y., et al. (2021). The mitochondrial permeability transition pore: An evolving concept critical for cell life and death. Biol. Rev. Camb. Philos. Soc. 96, 2489–2521. doi:10.1111/brv.12764
Mu, X., Isaac, C., Schott, T., Huard, J., and Weiss, K. (20132013). Rapamycin inhibits ALDH activity, resistance to oxidative stress, and metastatic potential in murine osteosarcoma cells. Sarcoma, 480713. doi:10.1155/2013/480713
Mukherjee, S., Dash, S., Lohitesh, K., and Chowdhury, R. (2017). The dynamic role of autophagy and MAPK signaling in determining cell fate under cisplatin stress in osteosarcoma cells. PLoS One 12, e0179203. doi:10.1371/journal.pone.0179203
Müller, M., Wilder, S., Bannasch, D., Israeli, D., Lehlbach, K., Li-Weber, M., et al. (1998). p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 188, 2033–2045. doi:10.1084/jem.188.11.2033
Musi, N., Hirshman, M. F., Nygren, J., Svanfeldt, M., Bavenholm, P., Rooyackers, O., et al. (2002). Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51, 2074–2081. doi:10.2337/diabetes.51.7.2074
Nagata, S., and Golstein, P. (1995). The Fas death factor. Science 267, 1449–1456. doi:10.1126/science.7533326
Nakatogawa, H., Suzuki, K., Kamada, Y., and Ohsumi, Y. (2009). Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 10, 458–467. doi:10.1038/nrm2708
Nasri, H., and Rafieian-Kopaei, M. (2014). Metformin: Current knowledge. J. Res. Med. Sci. 19, 658–664.
National Cancer Institute (NCI) (2022). NCI-COG pediatric MATCH (molecular analysis for therapy choice) - phase 2 subprotocol of AG-120 (Ivosidenib) in patients with tumors harboring IDH1 mutations (clinical trial registration No. NCT04195555). clinicaltrials.gov.
Newton, K., Dugger, D. L., Wickliffe, K. E., Kapoor, N., de Almagro, M. C., Vucic, D., et al. (2014). Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360. doi:10.1126/science.1249361
Nielsen-Preiss, S. M., Silva, S. R., and Gillette, J. M. (2003). Role of PTEN and Akt in the regulation of growth and apoptosis in human osteoblastic cells. J. Cell. Biochem. 90, 964–975. doi:10.1002/jcb.10709
Nijhawan, D., Fang, M., Traer, E., Zhong, Q., Gao, W., Du, F., et al. (2003). Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 17, 1475–1486. doi:10.1101/gad.1093903
Nikoletopoulou, V., Markaki, M., Palikaras, K., and Tavernarakis, N. (2013). Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 1833, 3448–3459. doi:10.1016/j.bbamcr.2013.06.001
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: In sickness and in health. Cell 148, 1145–1159. doi:10.1016/j.cell.2012.02.035
Oberst, A., Dillon, C. P., Weinlich, R., McCormick, L. L., Fitzgerald, P., Pop, C., et al. (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. doi:10.1038/nature09852
O’Donnell, M. A., Perez-Jimenez, E., Oberst, A., Ng, A., Massoumi, R., Xavier, R., et al. (2011). Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 13, 1437–1442. doi:10.1038/ncb2362
Okoshi, R., Ozaki, T., Yamamoto, H., Ando, K., Koida, N., Ono, S., et al. (2008). Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J. Biol. Chem. 283, 3979–3987. doi:10.1074/jbc.M705232200
Ozören, N., and El-Deiry, W. S. (2002). Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia 4, 551–557. doi:10.1038/sj.neo.7900270
Palorini, R., Votta, G., Balestrieri, C., Monestiroli, A., Olivieri, S., Vento, R., et al. (2014). Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J. Cell. Biochem. 115, 368–379. doi:10.1002/jcb.24671
Paquette, J. C., Guérin, P. J., and Gauthier, E. R. (2005). Rapid induction of the intrinsic apoptotic pathway by L-glutamine starvation. J. Cell. Physiol. 202, 912–921. doi:10.1002/jcp.20194
Parsons, D. W., Jones, S., Zhang, X., Lin, J. C.-H., Leary, R. J., Angenendt, P., et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812. doi:10.1126/science.1164382
Pasparakis, M., and Vandenabeele, P. (2015). Necroptosis and its role in inflammation. Nature 517, 311–320. doi:10.1038/nature14191
Phan, L. M., Yeung, S.-C. J., and Lee, M.-H. (2014). Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 11, 1–19. doi:10.7497/j.issn.2095-3941.2014.01.001
Piperno-Neumann, S., Le Deley, M.-C., Rédini, F., Pacquement, H., Marec-Bérard, P., Petit, P., et al. 2016. Zoledronate in combination with chemotherapy and surgery to treat osteosarcoma (OS2006): A randomised, multicentre, open-label, phase 3 trial. Lancet. Oncol. 17, 1070–1080. doi:10.1016/S1470-2045(16)30096-1
Polet, F., Corbet, C., Pinto, A., Rubio, L. I., Martherus, R., Bol, V., et al. (2016). Reducing the serine availability complements the inhibition of the glutamine metabolism to block leukemia cell growth. Oncotarget 7, 1765–1776. doi:10.18632/oncotarget.6426
Prudner, B. C., Rathore, R., Robinson, A. M., Godec, A., Chang, S. F., Hawkins, W. G., et al. (2019). Arginine starvation and docetaxel induce c-myc-driven hENT1 surface expression to overcome gemcitabine resistance in ASS1-negative tumors. Clin. Cancer Res. 25, 5122–5134. doi:10.1158/1078-0432.CCR-19-0206
Qiu, F., Chen, Y.-R., Liu, X., Chu, C.-Y., Shen, L.-J., Xu, J., et al. (2014). Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer cells. Sci. Signal. 7, ra31. doi:10.1126/scisignal.2004761
Rathore, R., Caldwell, K. E., Schutt, C., Brashears, C. B., Prudner, B. C., Ehrhardt, W. R., et al. (2021). Metabolic compensation activates pro-survival mTORC1 signaling upon 3-phosphoglycerate dehydrogenase inhibition in osteosarcoma. Cell Rep. 34, 108678. doi:10.1016/j.celrep.2020.108678
Rathore, R., Schutt, C. R., and Van Tine, B. A. (2020). PHGDH as a mechanism for resistance in metabolically-driven cancers. Cancer Drug resist. 3, 762–774. doi:10.20517/cdr.2020.46
Redza-Dutordoir, M., and Averill-Bates, D. A. (2016). Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 1863, 2977–2992. doi:10.1016/j.bbamcr.2016.09.012
Ren, L., Ruiz-Rodado, V., Dowdy, T., Huang, S., Issaq, S. H., Beck, J., et al. (2020). Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 8, 4. doi:10.1186/s40170-020-0209-8
Rena, G., Hardie, D. G., and Pearson, E. R. (2017). The mechanisms of action of metformin. Diabetologia 60, 1577–1585. doi:10.1007/s00125-017-4342-z
Robinson, N., Ganesan, R., Hegedűs, C., Kovács, K., Kufer, T. A., Virág, L., et al. (2019). Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 26, 101239. doi:10.1016/j.redox.2019.101239
Robl, B., Pauli, C., Botter, S. M., Bode-Lesniewska, B., and Fuchs, B. (2015). Prognostic value of tumor suppressors in osteosarcoma before and after neoadjuvant chemotherapy. BMC Cancer 15, 379. doi:10.1186/s12885-015-1397-4
Rojas, G. A., Hubbard, A. K., Diessner, B. J., Ribeiro, K. B., and Spector, L. G. (2021). International trends in incidence of osteosarcoma (1988-2012). Int. J. Cancer 149, 1044–1053. doi:10.1002/ijc.33673
Rosenzweig, A., Blenis, J., and Gomes, A. P. (2018). Beyond the Warburg effect: How do cancer cells regulate one-carbon metabolism? Front. Cell Dev. Biol. 6, 90. doi:10.3389/fcell.2018.00090
Roy, J., Dibaeinia, P., Fan, T. M., Sinha, S., and Das, A. (2019). Global analysis of osteosarcoma lipidomes reveal altered lipid profiles in metastatic versus nonmetastatic cells. J. Lipid Res. 60, 375–387. doi:10.1194/jlr.M088559
Salem, A. H., Dave, N., Marbury, T., Hu, B., Miles, D., Agarwal, S. K., et al. (2019). Pharmacokinetics of the BCL-2 inhibitor Venetoclax in subjects with hepatic impairment. Clin. Pharmacokinet. 58, 1091–1100. doi:10.1007/s40262-019-00746-4
Schmidt, C. A., Fisher-Wellman, K. H., and Neufer, P. D. (2021). From OCR and ECAR to energy: Perspectives on the design and interpretation of bioenergetics studies. J. Biol. Chem. 297, 101140. doi:10.1016/j.jbc.2021.101140
Schneider, P., Bodmer, J. L., Holler, N., Mattmann, C., Scuderi, P., Terskikh, A., et al. (1997). Characterization of Fas (Apo-1, cd95)-fas ligand interaction. J. Biol. Chem. 272, 18827–18833. doi:10.1074/jbc.272.30.18827
Schug, Z. T., Gonzalvez, F., Houtkooper, R. H., Vaz, F. M., and Gottlieb, E. (2011). BID is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 18, 538–548. doi:10.1038/cdd.2010.135
Schwartz, G. K., Tap, W. D., Qin, L.-X., Livingston, M. B., Undevia, S. D., Chmielowski, B., et al. (2013). Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: A multicentre, open-label, phase 2 trial. Lancet. Oncol. 14, 371–382. doi:10.1016/S1470-2045(13)70049-4
Seddon, B., Strauss, S. J., Whelan, J., Leahy, M., Woll, P. J., Cowie, F., et al. (2017). Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): A randomised controlled phase 3 trial. Lancet. Oncol. 18, 1397–1410. doi:10.1016/S1470-2045(17)30622-8
Seidlmayer, L. K., Juettner, V. V., Kettlewell, S., Pavlov, E. V., Blatter, L. A., Dedkova, E. N., et al. (2015). Distinct mPTP activation mechanisms in ischaemia-reperfusion: Contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 106, 237–248. doi:10.1093/cvr/cvv097
Shang, D., Wu, J., Guo, L., Xu, Y., Liu, L., Lu, J., et al. (2017). Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int. J. Oncol. 50, 1848–1856. doi:10.3892/ijo.2017.3950
Shelar, S., Shim, E.-H., Brinkley, G. J., Kundu, A., Carobbio, F., Poston, T., et al. (2018). Biochemical and epigenetic insights into L-2-hydroxyglutarate, a potential therapeutic target in renal cancer. Clin. Cancer Res. 24, 6433–6446. doi:10.1158/1078-0432.CCR-18-1727
Shen, L.-J., Lin, W.-C., Beloussow, K., and Shen, W.-C. (2003). Resistance to the anti-proliferative activity of recombinant arginine deiminase in cell culture correlates with the endogenous enzyme, argininosuccinate synthetase. Cancer Lett. 191, 165–170. doi:10.1016/s030-43835(02)00693-6
Sheng, G., Gao, Y., Yang, Y., and Wu, H. (2021). Osteosarcoma and metastasis. Front. Oncol. 11, 780264. doi:10.3389/fonc.2021.780264
Shi, J., Gao, W., and Shao, F. (2017). Pyroptosis: Gasdermin-Mediated programmed necrotic cell death. Trends biochem. Sci. 42, 245–254. doi:10.1016/j.tibs.2016.10.004
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi:10.1038/nature15514
Shi, J., Zhao, Y., Wang, Y., Gao, W., Ding, J., Li, P., et al. (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. doi:10.1038/nature13683
Shimada, K., and Stockwell, B. R. (2016). tRNA synthase suppression activates de novo cysteine synthesis to compensate for cystine and glutathione deprivation during ferroptosis. Mol. Cell. Oncol. 3, e1091059. doi:10.1080/23723556.2015.1091059
Shimizu, S., and Tsujimoto, Y. (2000). Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc. Natl. Acad. Sci. U. S. A. 97, 577–582. doi:10.1073/pnas.97.2.577
Shoshan-Barmatz, V., Anand, U., Nahon-Crystal, E., Di Carlo, M., and Shteinfer-Kuzmine, A. (2021). Adverse effects of metformin from diabetes to COVID-19, cancer, neurodegenerative diseases, and aging: Is VDAC1 a common target? Front. Physiol. 12, 730048. doi:10.3389/fphys.2021.730048
Shoshan-Barmatz, V., Krelin, Y., Shteinfer-Kuzmine, A., and Arif, T. (2017). Voltage-dependent anion channel 1 as an emerging drug target for novel anti-cancer therapeutics. Front. Oncol. 7, 154. doi:10.3389/fonc.2017.00154
Shoshan-Barmatz, V., Mizrachi, D., and Keinan, N. (2013). Oligomerization of the mitochondrial protein VDAC1: From structure to function and cancer therapy. Prog. Mol. Biol. Transl. Sci. 117, 303–334. doi:10.1016/B978-0-12-386931-9.00011-8
Singh, R., Letai, A., and Sarosiek, K. (2019). Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 20, 175–193. doi:10.1038/s41580-018-0089-8
Song, R., Tian, K., Wang, W., and Wang, L. (2015). P53 suppresses cell proliferation, metastasis, and angiogenesis of osteosarcoma through inhibition of the PI3K/AKT/mTOR pathway. Int. J. Surg. 20, 80–87. doi:10.1016/j.ijsu.2015.04.050
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi:10.1016/j.cell.2017.09.021
Stoker, M., and Perryman, M. (1985). An epithelial scatter factor released by embryo fibroblasts. J. Cell Sci. 77, 209–223. doi:10.1242/jcs.77.1.209
Strasser, A., and Vaux, D. L. (2020). Cell death in the origin and treatment of cancer. Mol. Cell 78, 1045–1054. doi:10.1016/j.molcel.2020.05.014
Struys, E. A. (2013). 2-Hydroxyglutarate is not a metabolite; D-2-hydroxyglutarate and L-2-hydroxyglutarate are. Proc. Natl. Acad. Sci. U. S. A. 110, E4939. doi:10.1073/pnas.1318777110
Stuani, L., Sabatier, M., and Sarry, J.-E. (2019). Exploiting metabolic vulnerabilities for personalized therapy in acute myeloid leukemia. BMC Biol. 17, 57. doi:10.1186/s12915-019-0670-4
Sui, X., Chen, R., Wang, Z., Huang, Z., Kong, N., Zhang, M., et al. (2013). Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 4, e838. doi:10.1038/cddis.2013.350
Sun, L., Wang, P., Zhang, Z., Zhang, K., Xu, Z., Li, S., et al. (2020). MicroRNA-615 functions as a tumor suppressor in osteosarcoma through the suppression of HK2. Oncol. Lett. 20, 226. doi:10.3892/ol.2020.12089
Sun, M., Zhou, C., Zeng, H., Puebla-Osorio, N., Damiani, E., Chen, J., et al. (2015). Hiporfin-mediated photodynamic therapy in preclinical treatment of osteosarcoma. Photochem. Photobiol. 91, 533–544. doi:10.1111/php.12424
Sun, X., and Zhang, L. (2017). MicroRNA-143 suppresses oral squamous cell carcinoma cell growth, invasion and glucose metabolism through targeting hexokinase 2. Biosci. Rep. 37, BSR20160404. doi:10.1042/BSR20160404
Swiss Group for Clinical Cancer Research (2022). Investigation of metformin in patients with castration resistant prostate cancer in combination with enzalutamide vs. Enzalutamide alone (improve trial): A randomized, open label, phase II trial (clinical trial registration No. NCT02640534). clinicaltrials.gov.
Tait, S. W. G., Ichim, G., and Green, D. R. (2014). Die another way-non-apoptotic mechanisms of cell death. J. Cell Sci. 127, 2135–2144. doi:10.1242/jcs.093575
Tang, D., Chen, X., Kang, R., and Kroemer, G. (2021). Ferroptosis: Molecular mechanisms and health implications. Cell Res. 31, 107–125. doi:10.1038/s41422-020-00441-1
Tang, Z., Ji, L., Han, M., Xie, J., Zhong, F., Zhang, X., et al. (2020). Pyroptosis is involved in the inhibitory effect of FL118 on growth and metastasis in colorectal cancer. Life Sci. 257, 118065. doi:10.1016/j.lfs.2020.118065
Thorburn, A. (2008). Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis 13, 1–9. doi:10.1007/s10495-007-0154-9
Tian, Q., Qin, B., Gu, Y., Zhou, L., Chen, S., Zhang, S., et al. (2020). ROS-mediated necroptosis is involved in iron overload-induced osteoblastic cell death. Oxid. Med. Cell. Longev. 2020, 1295382. doi:10.1155/2020/1295382
Tischner, D., Manzl, C., Soratroi, C., Villunger, A., and Krumschnabel, G. (2012). Necrosis-like death can engage multiple pro-apoptotic Bcl-2 protein family members. Apoptosis 17, 1197–1209. doi:10.1007/s10495-012-0756-8
Toschi, L., Finocchiaro, G., Bartolini, S., Gioia, V., and Cappuzzo, F. (2005). Role of gemcitabine in cancer therapy. Future Oncol. 1, 7–17. doi:10.1517/14796694.1.1.7
Toyokuni, S., Ito, F., Yamashita, K., Okazaki, Y., and Akatsuka, S. (2017). Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic. Biol. Med. 108, 610–626. doi:10.1016/j.freeradbiomed.2017.04.024
Trieb, K., Sulzbacher, I., and Kubista, B. (2013). Bcl-2 correlates with localization but not outcome in human osteosarcoma. Oncol. Lett. 6, 559–561. doi:10.3892/ol.2013.1395
Trump, B. F., Berezesky, I. K., Chang, S. H., and Phelps, P. C. (1997). The pathways of cell death: Oncosis, apoptosis, and necrosis. Toxicol. Pathol. 25, 82–88. doi:10.1177/019262339702500116
Van Herreweghe, F., Festjens, N., Declercq, W., and Vandenabeele, P. (2010). Tumor necrosis factor-mediated cell death: To break or to burst, that’s the question. Cell. Mol. Life Sci. 67, 1567–1579. doi:10.1007/s00018-010-0283-0
Van Opdenbosch, N., and Lamkanfi, M. (2019). Caspases in cell death, inflammation, and disease. Immunity 50, 1352–1364. doi:10.1016/j.immuni.2019.05.020
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi:10.1038/nrm2970
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324, 1029–1033. doi:10.1126/science.1160809
Vasan, K., Werner, M., and Chandel, N. S. (2020). Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 32, 341–352. doi:10.1016/j.cmet.2020.06.019
Vercammen, D., Beyaert, R., Denecker, G., Goossens, V., Van Loo, G., Declercq, W., et al. (1998). Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187, 1477–1485. doi:10.1084/jem.187.9.1477
Verhagen, A. M., Ekert, P. G., Pakusch, M., Silke, J., Connolly, L. M., Reid, G. E., et al. (2000). Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102, 43–53. doi:10.1016/s0092-8674(00)00009-x
Wang, H., Zhang, T., Sun, W., Wang, Z., Zuo, D., Zhou, Z., et al. (2016). Erianin induces G2/M-phase arrest, apoptosis, and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 7, e2247. doi:10.1038/cddis.2016.138
Wang, W. J., Chen, D., Jiang, M. Z., Xu, B., Li, X. W., Chu, Y., et al. (2018). Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle-related proteins. J. Dig. Dis. 19, 74–83. doi:10.1111/1751-2980.12576
Wang, X., Lai, P., Zhang, Z., Huang, M., Wang, L., Yin, M., et al. (2014). Targeted inhibition of mTORC2 prevents osteosarcoma cell migration and promotes apoptosis. Oncol. Rep. 32, 382–388. doi:10.3892/or.2014.3182
Wang, Y., Ji, Y., Hu, Z., Jiang, H., Zhu, F., Yuan, H., et al. (2013). Riccardin D induces cell death by activation of apoptosis and autophagy in osteosarcoma cells. Toxicol. Vitro 27, 1928–1936. doi:10.1016/j.tiv.2013.06.001
Wang, Z.-X., Yang, J.-S., Pan, X., Wang, J.-R., Li, J., Yin, Y.-M., et al. (2010). Functional and biological analysis of Bcl-xL expression in human osteosarcoma. Bone 47, 445–454. doi:10.1016/j.bone.2010.05.027
Wang, Z., Jiang, H., Chen, S., Du, F., and Wang, X. (2012). The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243. doi:10.1016/j.cell.2011.11.030
Warburg, O., Wind, F., and Negelein, E. (1927). The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530. doi:10.1085/jgp.8.6.519
Washington University School of Medicine (2021). A phase II trial of ADI-PEG 20 in combination with gemcitabine and Docetaxel for the treatment of soft tissue sarcoma osteosarcoma, ewing’s sarcoma, and Small cell lung cancer (clinical trial registration No. NCT03449901). clinicaltrials.gov.
Weeden, C. E., Ah-Cann, C., Holik, A. Z., Pasquet, J., Garnier, J.-M., Merino, D., et al. (2018). Dual inhibition of BCL-XL and MCL-1 is required to induce tumour regression in lung squamous cell carcinomas sensitive to FGFR inhibition. Oncogene 37, 4475–4488. doi:10.1038/s41388-018-0268-2
Weisthal, S., Keinan, N., Ben-Hail, D., Arif, T., and Shoshan-Barmatz, V. (2014). Ca(2+)-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta 1843, 2270–2281. doi:10.1016/j.bbamcr.2014.03.021
Whelan, J., McTiernan, A., Cooper, N., Wong, Y. K., Francis, M., Vernon, S., et al. (2012). Incidence and survival of malignant bone sarcomas in England 1979-2007. Int. J. Cancer 131, E508–E517. doi:10.1002/ijc.26426
Whelan, J. S., and Davis, L. E. (2018). Osteosarcoma, chondrosarcoma, and chordoma. J. Clin. Oncol. 36, 188–193. doi:10.1200/JCO.2017.75.1743
Whitburn, J., Edwards, C. M., and Sooriakumaran, P. (2017). Metformin and prostate cancer: A new role for an old drug. Curr. Urol. Rep. 18, 46. doi:10.1007/s11934-017-0693-8
Wiley, S. R., Schooley, K., Smolak, P. J., Din, W. S., Huang, C. P., Nicholl, J. K., et al. (1995). Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3, 673–682. doi:10.1016/1074-7613(95)90057-8
Wolpaw, A. J., and Dang, C. V. (2018). Exploiting metabolic vulnerabilities of cancer with precision and accuracy. Trends Cell Biol. 28, 201–212. doi:10.1016/j.tcb.2017.11.006
Wu, D., Wei, C., Li, Y., Yang, X., and Zhou, S. (2021). Pyroptosis, a new breakthrough in cancer treatment. Front. Oncol. 11, 698811. doi:10.3389/fonc.2021.698811
Xia, X., Wang, X., Cheng, Z., Qin, W., Lei, L., Jiang, J., et al. (2019). The role of pyroptosis in cancer: Pro-cancer or pro-"host. Cell Death Dis. 10, 650. doi:10.1038/s41419-019-1883-8
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. doi:10.1038/nature05859
Yang, D., He, Y., Muñoz-Planillo, R., Liu, Q., and Núñez, G. (2015). Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic Shock. Immunity 43, 923–932. doi:10.1016/j.immuni.2015.10.009
Yang, J., Liu, X., Bhalla, K., Kim, C. N., Ibrado, A. M., Cai, J., et al. (1997). Prevention of apoptosis by bcl-2: Release of cytochrome c from mitochondria blocked. Science 275, 1129–1132. doi:10.1126/science.275.5303.1129
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi:10.1016/j.cell.2013.12.010
Yang, W. S., and Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245. doi:10.1016/j.chembiol.2008.02.010
Ye, Y., Dai, Q., and Qi, H. (2021). A novel defined pyroptosis-related gene signature for predicting the prognosis of ovarian cancer. Cell Death Discov. 7, 71. doi:10.1038/s41420-021-00451-x
Zalk, R., Israelson, A., Garty, E. S., Azoulay-Zohar, H., and Shoshan-Barmatz, V. (2005). Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem. J. 386, 73–83. doi:10.1042/BJ20041356
Zha, H., Aimé-Sempé, C., Sato, T., and Reed, J. C. (1996). Proapoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2. J. Biol. Chem. 271, 7440–7444. doi:10.1074/jbc.271.13.7440
Zhang, J., Nuebel, E., Daley, G. Q., Koehler, C. M., and Teitell, M. A. (2012). Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 11, 589–595. doi:10.1016/j.stem.2012.10.005
Zhang, W., Li, Q., Song, C., and Lao, L. (2015). Knockdown of autophagy-related protein 6, Beclin-1, decreases cell growth, invasion, and metastasis and has a positive effect on chemotherapy-induced cytotoxicity in osteosarcoma cells. Tumour Biol. 36, 2531–2539. doi:10.1007/s13277-014-2868-y
Zhang, Y., He, R., Lei, X., Mao, L., Jiang, P., Ni, C., et al. (2021). A novel pyroptosis-related signature for predicting prognosis and indicating immune microenvironment features in osteosarcoma. Front. Genet. 12, 780780. doi:10.3389/fgene.2021.780780
Zhao, J., Zhao, Y., Ma, X., Zhang, B., and Feng, H. (2021). Targeting ferroptosis in osteosarcoma. J. Bone Oncol. 30, 100380. doi:10.1016/j.jbo.2021.100380
Zhao, X., Fu, J., Du, J., and Xu, W. (2020). The role of D-3-phosphoglycerate dehydrogenase in cancer. Int. J. Biol. Sci. 16, 1495–1506. doi:10.7150/ijbs.41051
Zhao, Y., Coloff, J. L., Ferguson, E. C., Jacobs, S. R., Cui, K., Rathmell, J. C., et al. (2008). Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J. Biol. Chem. 283, 36344–36353. doi:10.1074/jbc.M803580200
Zhao, Z., Tao, L., Shen, C., Liu, B., Yang, Z., Tao, H., et al. (2014). Silencing of Barkor/ATG14 sensitizes osteosarcoma cells to cisplatin-induced apoptosis. Int. J. Mol. Med. 33, 271–276. doi:10.3892/ijmm.2013.1578
Zheng, C., Tang, F., Min, L., Hornicek, F., Duan, Z., Tu, C., et al. (2020). PTEN in osteosarcoma: Recent advances and the therapeutic potential. Biochim. Biophys. Acta. Rev. Cancer 1874, 188405. doi:10.1016/j.bbcan.2020.188405
Zhou, Y., Shen, Y., Chen, C., Sui, X., Yang, J., Wang, L., et al. (2019). The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 16, 630–646. doi:10.20892/j.issn.2095-3941.2019.0158
Zhuo, B., Li, Y., Li, Z., Qin, H., Sun, Q., Zhang, F., et al. (2015). PI3K/Akt signaling mediated Hexokinase-2 expression inhibits cell apoptosis and promotes tumor growth in pediatric osteosarcoma. Biochem. Biophys. Res. Commun. 464, 401–406. doi:10.1016/j.bbrc.2015.06.092
Keywords: osteosarcoma, mitochondria, regulated cell death, metabolic reprogramming, Metformin, ADI-PEG20
Citation: Lai HT, Naumova N, Marchais A, Gaspar N, Geoerger B and Brenner C (2022) Insight into the interplay between mitochondria-regulated cell death and energetic metabolism in osteosarcoma. Front. Cell Dev. Biol. 10:948097. doi: 10.3389/fcell.2022.948097
Received: 19 May 2022; Accepted: 04 July 2022;
Published: 22 August 2022.
Edited by:
Ildikò Szabò, University of Padua, ItalyReviewed by:
Varda Shoshan-Barmatz, Ben-Gurion University of the Negev, IsraelCopyright © 2022 Lai, Naumova, Marchais, Gaspar, Geoerger and Brenner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine Brenner, Y2F0aGVyaW5lLmJyZW5uZXJAdW5pdmVyc2l0ZS1wYXJpcy1zYWNsYXkuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.