 Non-Nuoc Tran
Non-Nuoc Tran Byung-Hoon Lee
Byung-Hoon Lee
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 09 September 2022
Sec. Cellular Biochemistry
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.931968
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease in which motor neurons in spinal cord and motor cortex are progressively lost. About 15% cases of ALS also develop the frontotemporal dementia (FTD), in which the frontotemporal lobar degeneration (FTLD) occurs in the frontal and temporal lobes of the brain. Among the pathologic commonalities in ALS and FTD is ubiquitin-positive cytoplasmic aggregation of TDP-43 that may reflect both its loss-of-function and gain-of-toxicity from proteostasis impairment. Deep understanding of how protein quality control mechanisms regulate TDP-43 proteinopathies still remains elusive. Recently, a growing body of evidence indicates that ubiquitinating and deubiquitinating pathways are critically engaged in the fate decision of aberrant or pathological TDP-43 proteins. E3 ubiquitin ligases coupled with deubiquitinating enzymes may influence the TDP-43-associated proteotoxicity through diverse events, such as protein stability, translocation, and stress granule or inclusion formation. In this article, we recapitulate our current understanding of how ubiquitinating and deubiquitinating mechanisms can modulate TDP-43 protein quality and its pathogenic nature, thus shedding light on developing targeted therapies for ALS and FTD by harnessing protein degradation machinery.
Amyotrophic lateral sclerosis (ALS) is among the most prevalent and fatal neurodegenerative diseases in which motor neurons in spinal cord and motor cortex are progressively lost. Patients diagnosed with ALS suffer from the gradual respiratory dysfunction, having the mean survival time of 3–5 years due to no effective therapeutic treatment (Mejzini et al., 2019). Approximately 90–95% of patients show sporadic ALS, while only less than 10% belong to familial cases (Ling et al., 2013). Frontotemporal lobar degeneration (FTLD), the second-highest incidence rates of early-onset dementia after Alzheimer’s disease, is characterized by the cumulative neuronal loss in the frontal and temporal lobes of the brain, manifesting behavioral abnormalities, personality changes, and gradual language inability (Van Mossevelde et al., 2018; Ferrari et al., 2019). It was reported that about 15% cases of FTLD show ALS symptoms, meanwhile as much as 15% of ALS patients also develop classic features of FTLD such as cognitive impairment (Ringholz et al., 2005; Wheaton et al., 2007). Such shared modality can be explained by the fact that there is a distinctively broad but also overlapping spectrum of ALS and FTLD in terms of clinical aspects, neuropathological mechanisms, and genetic mutations (Figure 1A) (Ling et al., 2013; Ferrari et al., 2019). In fact, TAR DNA-binding protein-43 (TDP-43) was found to account for the connecting pathology of more than 90% of ALS and about 50% of FTD (i.e., FTLD-TDP) cases (Ling et al., 2013). Strikingly, ubiquitinated and hyper-phosphorylated TDP-43 was identified as a primary constituent of the mislocalized and insoluble cytoplasmic inclusions in the patient brain samples (Arai et al., 2006; Neumann et al., 2006). This observation may support the idea that the proteolytic control of TDP-43 is intimately connected to the pathomechanisms in ALS and FTD (Gao et al., 2018). However, it still remains as questions of how the TDP-43 pathogenesis upends this quality controlling protein clearance system. In this review, we briefly summarize the recent progress of ubiquitination and deubiquitination mechanisms that can modulate TDP-43 protein stability, localization and stress response, which may provide the mechanistic insights into developing proteolysis-based treatment for TDP-43 proteinopathies. Detailed physiology and pathology of TDP-43 have been extensively discussed recently (Prasad et al., 2019; Keating et al., 2022).
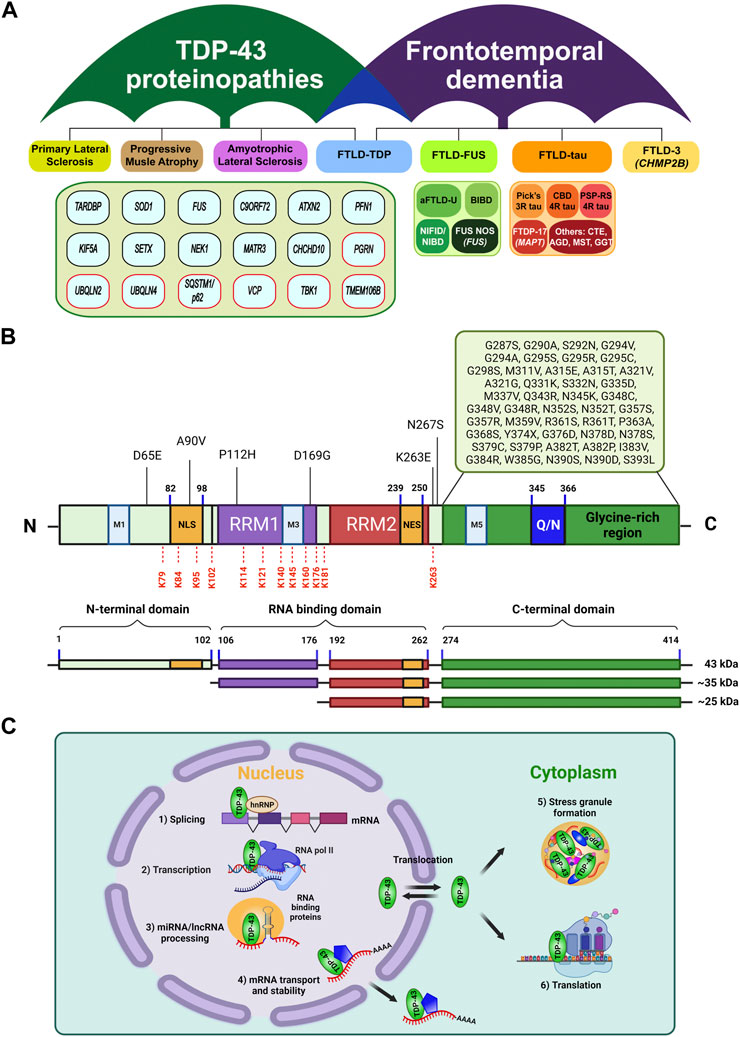
FIGURE 1. Classification of TDP-43 proteinopathies, and its structural features and general functions. (A) Umbrella diagram of TDP-43 proteinopathies and frontotemporal lobar degeneration (FTLD) that are classified by neuropathy types and causal genetic factors or pathological signature factors (modified from Prasad et al., 2019 and Hofmann et al., 2019). Genes in red boxes highlight the UPS or autophagy components. Note that the genetic factors under TDP-43 proteinopathies were primarily identified from ALS and FTLD-TDP. (B) Structural organization of TDP-43. TDP-43 is composed of an N-terminal domain (NTD) with a nuclear localization signal (NLS), two RNA-recognition motifs (RRM1, RRM2) with a nuclear export signal (NES) in RRM2, and a glycine-rich C-terminal domain (CTD) containing a glutamine/asparagine-rich region (Q/N). Approximately 50 missense mutations were located in the glycine-rich region of CTD (Buratti, 2015; Harrison and Shorter, 2017). Under pathological conditions, the full length TDP-43 can undergo truncation into the C-terminal fragments of 35 kDa or 25 kDa via caspase or calpain (Zhang et al., 2009; Xiao et al., 2015; Berning and Walker, 2019). In addition, a number of ubiquitination sites in TDP-43 have been reported, including K79, K84, K95, K102, K114, K121, K140, K145, K160, K176, K181, and K263, which are marked in red dash lines (Kim et al., 2011; Dammer et al., 2012; Kametani et al., 2016; Hans et al., 2018). (C) TDP-43 is mainly localized in nucleus where it performs multiple functions relating to RNA metabolic pathways, including 1) mRNA splicing, 2) transcription, 3) miRNA biogenesis, and lncRNA processing. Also, it shuttles between nucleus and cytoplasm, where it participates in 4) & 6) mRNA transport, stability, protein translation, and 5) stress granule assembly.
A 414-amino acid TDP-43 protein contains an N-terminal domain with a nuclear localization signal (NLS), two RNA recognition motifs (RRM1 and RRM2) with a nuclear export signal (NES), mitochondria localization signals (M1-M5), and a glycine-rich C-terminal domain where multiple ALS-associated mutations were reported (Figure 1B) (Guerrero et al., 2016; Prasad et al., 2019). This feature defines the predominant localization of TDP-43 in the nucleus where it regulates RNA metabolism, including transcription, splicing, translation as well as stability (Pinarbasi et al., 2018; Weskamp and Barmada, 2018; Bjork et al., 2022), while being also capable of translocation to the cytoplasm to mediate mRNA transport (Chu et al., 2019) or stress granule formation (Khalfallah et al., 2018) (Figure 1C). Indeed, deletion or A90V mutation in the NLS promotes the insoluble cytoplasmic TDP-43 mislocalization and aggregation (Winton et al., 2008a; Winton et al., 2008b; Barmada et al., 2010). Two RRM domains and possibly their dimerization are necessary for the proper binding of TDP-43 to DNA/RNA molecules with high specificity towards TG/UG-rich sequences. Several studies indicated that mutations in these regions impair TDP-43’s RNA binding and splicing activities (Lukavsky et al., 2013; Kuo et al., 2014; Furukawa et al., 2016), confirming its critical role in RNA metabolism. Notably, most of ubiquitination sites on TDP-43 so far have been identified within or near these RRM domains (Figure 1B) (Kim et al., 2011; Dammer et al., 2012; Kametani et al., 2016; Hans et al., 2018). The C-terminal domain (CTD) represents a highly disordered and low-complexity domain (LCD) which consists of a glycine-rich region separated by a glutamine and asparagine (Q/N)-enriched segment (Figure 1B). The CTD has been of great interest due to its intrinsic aggregation-prone property like the prion-like domain (King et al., 2012) which apparently contributes to TDP-43-induced pathological inclusions and neurotoxicity in ALS or FTLD-TDP (Afroz et al., 2017; Santamaria et al., 2017). Furthermore, most of ALS-causing mutations, cytotoxic truncated forms, and phosphorylation sites have been associated with the CTD of TDP-43 (Figure 1B) (Hasegawa et al., 2008; Pesiridis et al., 2009; Zhang et al., 2009; Xiao et al., 2015; Berning and Walker, 2019). The N-terminal domain (NTD) also exhibits its own oligomerization or aggregation propensity (Tsoi et al., 2017), and intriguingly, the part of NTD adopts a novel ubiquitin-like fold that can bind to ssDNA (Qin et al., 2014). It should be further investigated whether this structural element can also provide extra avidity for forming aggregates or association with ubiquitin binding proteins under pathological conditions. Overall, these structural features endow multi-dimensional regulation of TDP-43 protein, in which ubiquitin signaling may also serve as a critical surveillance system for its proper functioning through proteostasis networks.
Failure of TDP-43 regulation culminates in the fatal outcome of neuronal defect obviously because its pleiotropic nature may represent the sum from any of proteostasis, RNA homeostasis, liquid condensate homeostasis (as in stress granule), and organelle homeostasis (as in mitochondria) (Keating et al., 2022). Under physiological conditions, TDP-43 predominantly exerts nuclear functions in RNA-related processes but also shuttles between nucleus and cytoplasm to participate in stress granule formation and mRNA translation (Figure 1C). By contrast, under pathological mutations or stressors, nuclear depletion of TDP-43 seems to precede, and then entails its cytoplasmic mislocalization, aggregation, and inclusion formation (Tomé et al., 2020). Also, the pathological hallmark of TDP-43 proteinopathies highlights the aberrant deposition of ubiquitinated and hyper-phosphorylated TDP-43 in the cytoplasmic inclusions, which indicates that the proteolytic control of TDP-43 must have been severely compromised (Klaips et al., 2018). Therefore, for TDP-43-induced neurotoxicity, TDP-43 loss-of-function likely occurs due to its nuclear depletion accompanying cytoplasmic accumulation, which in turn progressively drives the pathogenic aggregation and deposition of insoluble TDP-43 inclusions (Lee et al., 2012). Probably, such dramatic changes of the molecular signature inevitably drive the progression of TDP-43 pathogenesis by altering or disturbing its bona fide protein (and also RNA) interaction networks. Above certain threshold that can be held by protein quality control mechanisms–such as ubiquitin-proteasome system (UPS), heat shock response, or autophagy-lysosomal degradation pathway, TDP-43 proteopathies can become runaway or even aggravated by faulty proteostasis pathways. Moreover, aberrant accumulation of pathological TDP-43 species sequesters the critical UPS components, directly impairs the proteasome activity, induces the accumulation of insoluble polyubiquitinated proteins, and also depletes free ubiquitin pool, thereby leading to perturbed protein and ubiquitin homeostasis (Cicardi et al., 2018; Lee et al., 2020b; Farrawell et al., 2020; Riemenschneider et al., 2022). In consistent with this view, dysregulation of TDP-43 in ALS and FTLD-TDP disrupts multiple physiological events and manifests a complex set of pathomechanisms–for example, 1) the loss-of-TDP-43 function impairs a range of its nuclear functions in RNA metabolism such as alternative or exon splicing, RNA biogenesis or stability, and polyadenylation (Yang et al., 2014; Mitra et al., 2019; Ni et al., 2021); 2) the gain-of-toxicity of TDP-43 may further exacerbate the pathogenesis by sequestering other important biomolecules and organelles through undesirable interactions (Igaz et al., 2011; Zuo et al., 2021). Accumulation of cytoplasmic TDP-43 also represses the global protein synthesis in neuroblastoma models and FTD brain samples (Russo et al., 2017; Charif et al., 2020). In addition, a growing list of evidences indicate that TDP-43-induced neuroinflammation and innate immune responses are critically associated with the TDP-43 pathogenesis via NF-κβ/p65, cGAS-STING, NLRP3 inflammasome and PTP1B pathways (Zhao et al., 2015; Lee et al., 2020a; Dutta et al., 2020; Yu et al., 2020; Bright et al., 2021).
So far, over 50 missense mutations in TARDBP gene (encoding TDP-43) have been associated with ALS and FTLD-TDP (Buratti, 2015; Harrison and Shorter, 2017), most of which being clustered within the aggregation-prone CTD region (Figure 1B). Remarkably, although TARDBP mutations account for only 5–10% of familial ALS and the remaining over 90% are attributable to other genes such as C9ORF72, SOD1, FUS, and UBQLN2 (Kim et al., 2020), the majority of ALS cases (about 97%) also exhibit TDP-43-induced pathology (Ling et al., 2013). TDP-43 mutations were proposed to alter the protein stability or increase the propensity of mislocalization or aggregation. The reported half-lives of TDP-43 wild-type and mutants have been somewhat inconsistent among the literatures: while some studies observed the more prolonged turnover rate of TDP-43 mutants (Ling et al., 2010; Watanabe et al., 2013; Austin et al., 2014), others reported the faster degradation of the mutants and the C-terminal truncated forms (Araki et al., 2014; Scotter et al., 2014). Disease-associated TDP-43 mutants (e.g., G376D, G335D, G343R, A315T, and M337V) were also reported to increase the cytoplasmic TDP-43 mislocalization and stress granule or inclusion formation, which may become deleterious to neuronal cells (Jiang et al., 2016; Mitsuzawa et al., 2018; Ding et al., 2021).
Ubiquitination is a cascade of ATP-dependent conjugation reaction that attaches the ubiquitin tag on the target substrate by the sequential cooperation of E1 (ubiquitin activating enzyme), E2 (ubiquitin conjugating enzyme), and E3 (ubiquitin ligase) enzymes (Hershko and Ciechanover, 1998). By generating diverse configurations and linkage types of chains, the ubiquitination pathways are critically involved in both proteolytic (as in UPS and autophagy-lysosomal degradation) and non-proteolytic processes (as in signal transduction and membrane trafficking) (Yau and Rape, 2016; Pohl and Dikic, 2019). Strikingly, ubiquitin-positive inclusion represents one of the pathological hallmarks of many types of neurodegenerative diseases (Schmidt et al., 2021), suggesting that the protein quality control mechanisms must have been dysfunctional in general. Likewise, ubiquitinated TDP-43 has been also enriched in the ALS and FTD brain inclusions (Arai et al., 2006; Neumann et al., 2006).
Indeed, the maintenance of TDP-43 proteostasis seems to be critical for its proper functioning because either upregulation or downregulation of TDP-43 can drive the neurotoxicity in various model systems (Ash et al., 2010; Li et al., 2010; Miguel et al., 2011; Zhang et al., 2012; Ling et al., 2013). TDP-43 level is also tightly regulated through autoregulation for which TDP-43 protein represses its own mRNA translation by binding to the 3’ untranslated region (Figure 2) (Ayala et al., 2011). The coordinated operation of UPS, chaperones, and autophagy likely influences the balanced turnover of TDP-43 protein. Thus, it is not surprising that a number of ALS- and FTLD-TDP-associated mutations have been identified in the UPS or autophagic components, such as UBQLN, VCP, p62, and OPTN (Figure 1A) (Medinas et al., 2017; Keating et al., 2022). It was suggested that the soluble TDP-43 (either wild-type or the pathologic forms) may undergo efficient degradation by the UPS, but once the cytotoxic oligomers or aggregates being formed, they are preferentially cleared by autophagy-lysosomal degradation pathway (Scotter et al., 2014).
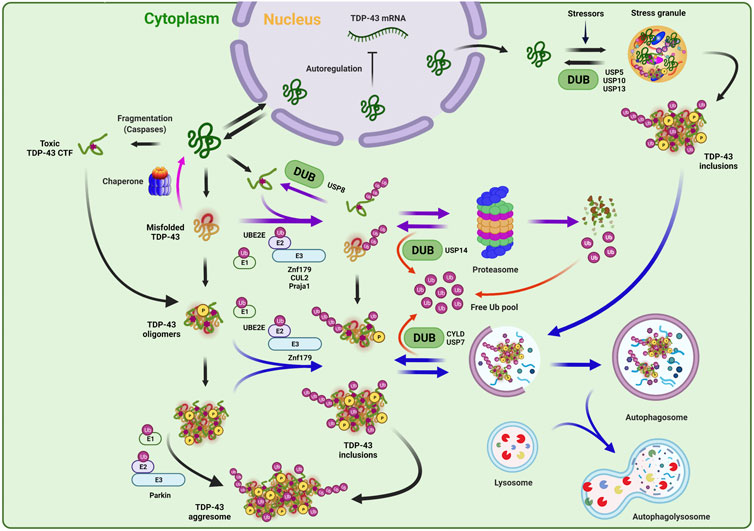
FIGURE 2. Ubiquitination and deubiquitination mechanisms in TDP-43 regulation. TDP-43 protein level can be tight regulated by multi-dimensional proteostasis surveillance systems. In the normal conditions, the TDP-43 amount in the cell is maintained via a negative feedback loop mechanism in which TDP-43 autoregulates and suppresses its own mRNA expression. Under the pathological conditions, TDP-43 translocates from nucleus to cytoplasm to be accumulated, and can be turned into various pathological species, such as misfolded, oligomeric, or truncated toxic C-terminal TDP-43 fragment. Under the prolonged stress conditions, TDP-43 also can be recruited into aberrant stress granules, forming toxic aggregates or insoluble inclusions. TDP-43 can be refolded by chaperones or recognized and eliminated by ubiquitin-proteasome system and autophagy pathway. E2 (UBE2E) and E3 (Parkin, Znf179, CUL2, and Praja 1) enzymes, which have been reported to regulate the quality control or fate decision of TDP-43, are indicated. Note that depending on the ubiquitination events by given enzymes, the ubiquitin conjugates are doomed to be degraded by proteasome or autophagy, or rather to form the pathological aggregates or inclusions. This ubiquitination process can be reversed by deubiquitinating enzymes (DUBs) (USP8, USP14, USP7, and CYLD), thereby regulating TDP-43 stability, editing ubiquitin architecture, and maintaining free ubiquitin pool. DUBs that are involved in stress granule dynamics (USP5, USP10, and USP13) were also indicated.
UBE2E3 was identified to be an E2 ubiquitin-conjugating enzyme that is involved in the ubiquitination process of TDP-43. This E2 enzyme physically interacts with TDP-43 and promotes its ubiquitination and insolubility (Figure 2) (Hans et al., 2014). A few of E3 ubiquitin ligase enzymes have been also reported to participate in the regulation of TDP-43 proteinopathies (Figure 2). Hebron et al. have reported that the E3 ligase Parkin and TDP-43 form a multi-protein complex with HDAC6, and this interaction facilitates the cytoplasmic accumulation of TDP-43 and also mediates its K48- and K63-linked polyubiquitination. Interestingly, this Parkin-induced TDP-43 ubiquitination only promotes its cytoplasmic mislocalization and inclusion formation without any indication of the proteolytic clearance (Hebron et al., 2013). In 2016, Uchida et al. revealed that VHL/CUL2 E3 ligase ubiquitinates and enhances the degradation of C-terminal TDP-43 fragments of 35 and 25 kDa by favorably recognizing misfolded form of TDP-43 at E246 site in RRM2 domain. Unexpectedly, excess VHL instead stabilizes TDP-43 and enhances its inclusion formation, suggesting the importance of balanced proteolytic control by VHL/CUL2 complex in ALS (Uchida et al., 2016). Another RING E3 ubiquitin ligase, Znf179 was identified to interact with and polyubiquitinate TDP-43 in vitro and in vivo. The Znf179-mediated ubiquitination enhances TDP-43 protein degradation and also antagonizes its cytoplasmic mislocalization and insoluble aggregation. Conversely, Znf179-knockout in mouse brain accumulates insoluble and cytoplasmic TDP-43 inclusions in neuronal tissues (Lee et al., 2018). Recently, one study also reported that the Praja 1 E3 ligase exerts remarkably suppressive effects on phosphorylation and aggregation of pathological cytoplasmic TDP-43 C-terminal fragment (CTF) both in vitro and in vivo, although it was not determined whether Praja 1 is able to ubiquitinate TDP-43 (Watabe et al., 2020). Furthermore, Praja 1 was also found to interact with E2 ubiquitin-conjugating enzyme UBE2E3, implying that the pair of Praja1/UBE2E3 may induce the ubiquitination of TDP-43 against the pathogenic process, contrary to the outcome from only UBE2E3-mediated ubiquitination (Hans et al., 2014; Watabe et al., 2020). Overall, aberrant ubiquitination and defective degradation of TDP-43 may underly the pathological mechanisms in TDP-43 proteopathies, in which reduced proteasomal and autophagic activities along with augmented proteolysis demand by TDP-43 accumulation render the vicious cycle into progressively aggravating the pathogenesis (Figure 2).
Deubiquitinating enzymes (DUBs) can exclusively reverse the ubiquitination process and thus are capable of stabilizing the target proteins. Besides, DUBs also play essential roles in ubiquitin recycling and ubiquitin chain editing, which may lead to altered subcellular localization of substrates and various responses to cellular signaling (Komander et al., 2009; Clague et al., 2019). Although DUBs may represent potential drug targets for modulating TDP-43 protein turnover, only a few have been implicated in TDP-43 regulation or stress granule formation including USP5, USP7, USP8, USP10, USP13, USP14, and CYLD (Figure 2) (Lee et al., 2010; Hans et al., 2014; Xie et al., 2018; Dobson-Stone et al., 2020; Zhang et al., 2020).
USP14, one of the major DUBs on the proteasome, can deubiquitinate and stabilize the protein substrates that are ubiquitinated at multiple sites (Lee et al., 2010; Lee et al., 2016). Given the remarkable nature of USP14–i.e., highly activated only when bound to the proteasome and potent suppression of substrate degradation, small-molecule USP14 inhibitors (e.g., IU1 series) have been also developed as drug-like molecules (Lee et al., 2010; Boselli et al., 2017). In fact, overexpression of USP14 in MEF cells substantially stabilized the protein level of TDP-43, whereas USP14 inhibitor treatment remarkably enhanced the turnover of neurotoxic substrates including TDP-43 (Lee et al., 2010; Boselli et al., 2017). USP8/UBPY was identified to modulate TDP-43-induced neurotoxicity (Hans et al., 2014). This DUB was previously known to regulate endocytosis via interactions with ESCRT-associated protein components such as EGFRs or STAM (Clague and Urbé, 2006). By employing yeast two-hybrid screening and co-immunoprecipitation, Hans et al. revealed that UBPY is a novel interacting partner of pathogenic TDP-43. Deficiency of UBPY noticeably promotes TDP-43 ubiquitination but the resulting conjugates may serve as unfavorable proteasomal substrates or just overburden the proteasome. Consequently, UBPY-silenced TDP-43 ubiquitination accelerates insoluble TDP-43 accumulation and aggregation as well as neurotoxicity in vivo fly model. Thus, in this case, UBPY may serve as a protective factor against TDP-43-induced neurotoxicity (Hans et al., 2014). DUBs may be also involved in autophagy-lysosome pathway for TDP-43 regulation. Recently, Dobson-Stone et al. reported that a missense mutation in CYLD (CYLDM719V), which was genetically identified from ALS and FTLD disease locus, exhibits significantly elevated K63-linkage specific deubiquitinase activity and caused the impairment in autophagosome-lysosome fusion pathway. This CYLDM719V mutant also increased cytoplasmic mislocalization of TDP-43 and reduced the axonal length (Dobson-Stone et al., 2020). Similarly, USP7 was identified as a negative regulator of autophagy via deubiquitination of NEDD4L, an E3 ubiquitin ligase, and inactivation of TGFβ-SMAD pathway. Pharmacological inhibition or genetic suppression of USP7 significantly reduced the levels of misfolded SOD1 and TDP-43 (wild-type and Q331K), leading to attenuation of mutant SOD1 or TDP-43-induced neurotoxicity in fruit flies (Zhang et al., 2020).
Aberrant stress granule dynamics represent another pathological feature of TDP-43 proteinopathy (Hallegger et al., 2021). Notably, a handful of DUBs have been involved in stress granule formation or liquid-liquid phase separation (LLPS). USP10 was found to be localized in stress granule and also interacts with TDP-43 and G3BP1, a well-known stress granule marker (Soncini et al., 2001; Freibaum et al., 2010). USP10 can suppress the formation of aberrant cytoplasmic TDP-43/TDP-35 aggregates by enhancing the clearance of stress granules in neuronal cells (Takahashi et al., 2022). Two other DUBs, USP5 and USP13, can be also recruited to heat-induced stress granules and regulate their assembly or disassembly (Figure 2) (Xie et al., 2018). Due to the apparent difference in their mode of activities, USP5 regulates the stress granule dynamics by preferentially cleaving unanchored ubiquitin chains, whereas USP13 does so through deubiquitination of protein-conjugated ubiquitin chains (Xie et al., 2018). However, it should be further examined whether USP5 or USP13 could also functionally regulate TDP-43-positive stress granules in ALS and FTD models.
While ubiquitin-enriched cytoplasmic inclusion of TDP-43 is considered to be the proteopathic signature of ALS and FTD, the pathophysiological meaning of the ubiquitination has been a long-standing question. Dramatic alteration of TDP-43 localization, stability, and post-translational modifications under the pathological conditions may disturb its genuine functional interaction networks by losing nuclear functions or gaining cytoplasmic toxicity. On top of TDP-43’s cytoplasmic mislocalization and aggregation-prone property, the initial pathogenic cue may trigger ‘butterfly effect’ and eventually the TDP-43 proteinopathy would prevail. Reduced proteasomal or autophagic activity and free ubiquitin pool depletion will badly influence the quality control of TDP-43 protein, or rather aberrant or undesirable ubiquitination of TDP-43 from proteostasis failure may only further exacerbate the disease phenotype. In fact, ubiquitination-rich pathogenic inclusions or liquid condensates, if the modification is not cleared properly, may act as avid and irreversible absorbent chambers to sequester many important proteolysis components such as ubiquitin binding proteins and proteasome. In this sense, pharmacological modulation of the pathogenic TDP-43-specific ubiquitination or the protein turnover itself may provide promising therapeutic opportunities to cope with TDP-43 proteinopathies. Remodeling or editing the ubiquitin chains or fine control of TDP-43 half-life may favor its proteolysis flux or antagonize the disease progression. It should be also noted that the total depletion of TDP-43 is not a viable option because of its essentiality. Indeed, pharmacological activation of the UPS or lysosomal pathways have been shown to bear the promise for TDP-43 clearance. For example, as noted above, a small-molecule inhibitor targeting proteasome-bound USP14 was reported to accelerate TDP-43 turnover by enhancing the proteasome activity (Lee et al., 2010). Similarly, forskolin-activated cAMP-PKA pathway increased proteasome function and remarkably reduced the levels of TDP-43 WT and its pathological mutants (Lokireddy et al., 2015). In addition, rapamycin, which works as an autophagic activator, strongly decreased the pathogenic TDP-43 species and attenuated TDP-43-induced neurotoxicity in ALS and FTD models (Cheng et al., 2015; Lattante et al., 2015). Of note, recently emerging targeted protein degradation or proteolysis targeting chimera (PROTAC) approaches have been showing great success in induced proteolysis of the undruggable targets including neurotoxic proteins (Moon and Lee, 2018). In fact, TDP-43 PROTACs were also recently reported, and await further improvement and validation (Gao et al., 2019; Hyun and Shin, 2021). In any cases, deep understanding of ubiquitinating and deubiquitinating mechanisms in TDP-43 pathology should provide the fundamental basis to develop the proteolysis-controlling therapeutic strategies for TDP-43-associated neurodegenerative diseases.
B-HL planned and directed the overall process of manuscript preparation. N-NT and B-HL wrote the manuscript.
This work was supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) and Korea Dementia Research Center (KDRC), funded by the Ministry of Health & Welfare and Ministry of Science and ICT, Korea (HU21C0027), the National Research Foundation of Korea (NRF) grant (2022R1A4A2000703), and DGIST R&D Programs (22-CoE-BT-04 and 22-HRHR+-04) of the Ministry of Science and ICT, Korea.
The authors apologize to researchers whose citations are not included due to space limit. We thank other lab members for nice co-operation. The Figures were in part created with BioRender.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Afroz, T., Hock, E.-M., Ernst, P., Foglieni, C., Jambeau, M., Gilhespy, L. A., et al. (2017). Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 8 (1), 45–15. doi:10.1038/s41467-017-00062-0
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351 (3), 602–611. doi:10.1016/j.bbrc.2006.10.093
Araki, W., Minegishi, S., Motoki, K., Kume, H., Hohjoh, H., Araki, Y. M., et al. (2014). Disease-associated mutations of TDP-43 promote turnover of the protein through the proteasomal pathway. Mol. Neurobiol. 50 (3), 1049–1058. doi:10.1007/s12035-014-8644-6
Ash, P. E., Zhang, Y.-J., Roberts, C. M., Saldi, T., Hutter, H., Buratti, E., et al. (2010). Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 19 (16), 3206–3218. doi:10.1093/hmg/ddq230
Austin, J. A., Wright, G. S., Watanabe, S., Grossmann, J. G., Antonyuk, S. V., Yamanaka, K., et al. (2014). Disease causing mutants of TDP-43 nucleic acid binding domains are resistant to aggregation and have increased stability and half-life. Proc. Natl. Acad. Sci. U. S. A. 111 (11), 4309–4314. doi:10.1073/pnas.1317317111
Ayala, Y. M., De Conti, L., Avendaño-Vázquez, S. E., Dhir, A., Romano, M., D'ambrogio, A., et al. (2011). TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30 (2), 277–288. doi:10.1038/emboj.2010.310
Barmada, S. J., Skibinski, G., Korb, E., Rao, E. J., Wu, J. Y., and Finkbeiner, S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 30 (2), 639–649. doi:10.1523/JNEUROSCI.4988-09.2010
Berning, B. A., and Walker, A. K. (2019). The pathobiology of TDP-43 C-terminal fragments in ALS and FTLD. Front. Neurosci. 13, 335. doi:10.3389/fnins.2019.00335
Bjork, R. T., Mortimore, N. P., Loganathan, S., and Zarnescu, D. (2022). Dysregulation of translation in TDP-43 proteinopathies: deficits in the RNA supply chain and local protein production. Front. Neurosci. 254, 840357. doi:10.3389/fnins.2022.840357
Boselli, M., Lee, B.-H., Robert, J., Prado, M. A., Min, S.-W., Cheng, C., et al. (2017). An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem. 292 (47), 19209–19225. doi:10.1074/jbc.M117.815126
Bright, F., Chan, G., van Hummel, A., Ittner, L. M., and Ke, Y. D. (2021). TDP-43 and inflammation: implications for amyotrophic lateral sclerosis and frontotemporal dementia. Int. J. Mol. Sci. 22 (15), 7781. doi:10.3390/ijms22157781
Buratti, E. (2015). Functional significance of TDP-43 mutations in disease. Adv. Genet. 91, 1–53. doi:10.1016/bs.adgen.2015.07.001
Charif, S. E., Luchelli, L., Vila, A., Blaustein, M., and Igaz, L. M. (2020). Cytoplasmic expression of the ALS/FTD-related protein TDP-43 decreases global translation both in vitro and in vivo. Front. Cell. Neurosci. 14, 594561. doi:10.3389/fncel.2020.594561
Cheng, C.-W., Lin, M.-J., and Shen, C.-K. J. (2015). Rapamycin alleviates pathogenesis of a new Drosophila model of ALS-TDP. J. Neurogenet. 29 (2-3), 59–68. doi:10.3109/01677063.2015.1077832
Chu, J.-F., Majumder, P., Chatterjee, B., Huang, S.-L., and Shen, C.-K. J. (2019). TDP-43 regulates coupled dendritic mRNA transport-translation processes in co-operation with FMRP and Staufen1. Cell Rep. 29 (10), 3118–3133. doi:10.1016/j.celrep.2019.10.061
Cicardi, M. E., Cristofani, R., Rusmini, P., Meroni, M., Ferrari, V., Vezzoli, G., et al. (2018). Tdp-25 routing to autophagy and proteasome ameliorates its aggregation in amyotrophic lateral sclerosis target cells. Sci. Rep. 8 (1), 12390–12416. doi:10.1038/s41598-018-29658-2
Clague, M. J., and Urbé, S. (2006). Endocytosis: the DUB version. Trends Cell Biol. 16 (11), 551–559. doi:10.1016/j.tcb.2006.09.002
Clague, M. J., Urbé, S., and Komander, D. (2019). Breaking the chains: deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 20 (6), 338–352. doi:10.1038/s41580-019-0099-1
Dammer, E. B., Fallini, C., Gozal, Y. M., Duong, D. M., Rossoll, W., Xu, P., et al. (2012). Coaggregation of RNA-binding proteins in a model of TDP-43 proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination. PloS one 7 (6), e38658. doi:10.1371/journal.pone.0038658
Ding, Q., Chaplin, J., Morris, M. J., Hilliard, M. A., Wolvetang, E., Ng, D. C., et al. (2021). TDP-43 mutation affects stress granule dynamics in differentiated NSC-34 motoneuron-like cells. Front. Cell Dev. Biol. 9, 611601. doi:10.3389/fcell.2021.611601
Dobson-Stone, C., Hallupp, M., Shahheydari, H., Ragagnin, A. M., Chatterton, Z., Carew-Jones, F., et al. (2020). CYLD is a causative gene for frontotemporal dementia–amyotrophic lateral sclerosis. Brain 143 (3), 783–799. doi:10.1093/brain/awaa039
Dutta, K., Thammisetty, S. S., Boutej, H., Bareil, C., and Julien, J.-P. (2020). Mitigation of ALS pathology by neuron-specific inhibition of nuclear factor kappa B signaling. J. Neurosci. 40 (26), 5137–5154. doi:10.1523/JNEUROSCI.0536-20.2020
Farrawell, N. E., McAlary, L., Lum, J. S., Chisholm, C. G., Warraich, S. T., Blair, I. P., et al. (2020). Ubiquitin homeostasis is disrupted in TDP-43 and FUS cell models of ALS. Iscience 23 (11), 101700. doi:10.1016/j.isci.2020.101700
Ferrari, R., Manzoni, C., and Hardy, J. (2019). Genetics and molecular mechanisms of frontotemporal lobar degeneration: an update and future avenues. Neurobiol. Aging 78, 98–110. doi:10.1016/j.neurobiolaging.2019.02.006
Freibaum, B. D., Chitta, R. K., High, A. A., and Taylor, J. P. (2010). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9 (2), 1104–1120. doi:10.1021/pr901076y
Furukawa, Y., Suzuki, Y., Fukuoka, M., Nagasawa, K., Nakagome, K., Shimizu, H., et al. (2016). A molecular mechanism realizing sequence-specific recognition of nucleic acids by TDP-43. Sci. Rep. 6 (1), 20576–20612. doi:10.1038/srep20576
Gao, J., Wang, L., Huntley, M. L., Perry, G., and Wang, X. (2018). Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 146 (1), 7–20. doi:10.1111/jnc.14327
Gao, N., Huang, Y.-P., Chu, T.-T., Li, Q.-Q., Zhou, B., Chen, Y.-X., et al. (2019). TDP-43 specific reduction induced by Di-hydrophobic tags conjugated peptides. Bioorg. Chem. 84, 254–259. doi:10.1016/j.bioorg.2018.11.042
Guerrero, E. N., Wang, H., Mitra, J., Hegde, P. M., Stowell, S. E., Liachko, N. F., et al. (2016). TDP-43/FUS in motor neuron disease: complexity and challenges. Prog. Neurobiol. 145, 78–97. doi:10.1016/j.pneurobio.2016.09.004
Hallegger, M., Chakrabarti, A. M., Lee, F. C., Lee, B. L., Amalietti, A. G., Odeh, H. M., et al. (2021). TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cell 184 (18), 4680–4696. doi:10.1016/j.cell.2021.07.018
Hans, F., Eckert, M., von Zweydorf, F., Gloeckner, C. J., and Kahle, P. J. (2018). Identification and characterization of ubiquitinylation sites in TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 293 (41), 16083–16099. doi:10.1074/jbc.RA118.003440
Hans, F., Fiesel, F. C., Strong, J. C., Jäckel, S., Rasse, T. M., Geisler, S., et al. (2014). UBE2E ubiquitin-conjugating enzymes and ubiquitin isopeptidase Y regulate TDP-43 protein ubiquitination. J. Biol. Chem. 289 (27), 19164–19179. doi:10.1074/jbc.M114.561704
Harrison, A. F., and Shorter, J. (2017). RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 474 (8), 1417–1438. doi:10.1042/BCJ20160499
Hasegawa, M., Arai, T., Nonaka, T., Kametani, F., Yoshida, M., Hashizume, Y., et al. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64 (1), 60–70. doi:10.1002/ana.21425
Hebron, M. L., Lonskaya, I., Sharpe, K., Weerasinghe, P. P., Algarzae, N. K., Shekoyan, A. R., et al. (2013). Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem. 288 (6), 4103–4115. doi:10.1074/jbc.M112.419945
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67 (1), 425–479. doi:10.1146/annurev.biochem.67.1.425
Hofmann, J. W., Seeley, W. W., and Huang, E. J. (2019). RNA binding proteins and the pathogenesis of frontotemporal lobar degeneration. Annual review of pathology 14, 469
Hyun, S., and Shin, D. (2021). Chemical-mediated targeted protein degradation in neurodegenerative diseases. Life 11 (7), 607. doi:10.3390/life11070607
Igaz, L. M., Kwong, L. K., Lee, E. B., Chen-Plotkin, A., Swanson, E., Unger, T., et al. (2011). Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest. 121 (2), 726–738. doi:10.1172/JCI44867
Jiang, L.-L., Zhao, J., Yin, X.-F., He, W.-T., Yang, H., Che, M.-X., et al. (2016). Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci. Rep. 6 (1), 23928–24011. doi:10.1038/srep23928
Kametani, F., Obi, T., Shishido, T., Akatsu, H., Murayama, S., Saito, Y., et al. (2016). Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci. Rep. 6 (1), 23281–23315. doi:10.1038/srep23281
Keating, S. S., San Gil, R., Swanson, M. E., Scotter, E. L., and Walker, A. K. (2022). TDP-43 pathology: from noxious assembly to therapeutic removal. Prog. Neurobiol. 211, 102229. doi:10.1016/j.pneurobio.2022.102229
Khalfallah, Y., Kuta, R., Grasmuck, C., Prat, A., Durham, H. D., and Vande Velde, C. (2018). TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep. 8 (1), 7551–7613. doi:10.1038/s41598-018-25767-0
Kim, G., Gautier, O., Tassoni-Tsuchida, E., Ma, X. R., and Gitler, A. D. (2020). ALS genetics: gains, losses, and implications for future therapies. Neuron 108 (5), 822–842. doi:10.1016/j.neuron.2020.08.022
Kim, W., Bennett, E. J., Huttlin, E. L., Guo, A., Li, J., Possemato, A., et al. (2011). Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44 (2), 325–340. doi:10.1016/j.molcel.2011.08.025
King, O. D., Gitler, A. D., and Shorter, J. (2012). The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80. doi:10.1016/j.brainres.2012.01.016
Klaips, C. L., Jayaraj, G. G., and Hartl, F. U. (2018). Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 217 (1), 51–63. doi:10.1083/jcb.201709072
Komander, D., Clague, M. J., and Urbé, S. (2009). Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10 (8), 550–563. doi:10.1038/nrm2731
Kuo, P.-H., Chiang, C.-H., Wang, Y.-T., Doudeva, L. G., and Yuan, H. S. (2014). The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG-and TG-rich nucleic acids. Nucleic Acids Res. 42 (7), 4712–4722. doi:10.1093/nar/gkt1407
Lattante, S., de Calbiac, H., Le Ber, I., Brice, A., Ciura, S., and Kabashi, E. (2015). Sqstm1 knock-down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum. Mol. Genet. 24 (6), 1682–1690. doi:10.1093/hmg/ddu580
Lee, B.-H., Lee, M. J., Park, S., Oh, D.-C., Elsasser, S., Chen, P.-C., et al. (2010). Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467 (7312), 179–184. doi:10.1038/nature09299
Lee, B.-H., Lu, Y., Prado, M. A., Shi, Y., Tian, G., Sun, S., et al. (2016). USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 532 (7599), 398–401. doi:10.1038/nature17433
Lee, E. B., Lee, V. M.-Y., and Trojanowski, J. Q. (2012). Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13 (1), 38–50. doi:10.1038/nrn3121
Lee, S., Kim, S., Kang, H.-Y., Lim, H. R., Kwon, Y., Jo, M., et al. (2020a). The overexpression of TDP-43 in astrocytes causes neurodegeneration via a PTP1B-mediated inflammatory response. J. Neuroinflammation 17 (1), 299–322. doi:10.1186/s12974-020-01963-6
Lee, S., Kwon, Y., Kim, S., Jo, M., Jeon, Y.-M., Cheon, M., et al. (2020b). The role of HDAC6 in TDP-43-induced neurotoxicity and UPS impairment. Front. Cell Dev. Biol. 8, 581942. doi:10.3389/fcell.2020.581942
Lee, Y. C., Huang, W. C., Lin, J. H., Kao, T. J., Lin, H. C., Lee, K. H., et al. (2018). Znf179 E3 ligase-mediated TDP-43 polyubiquitination is involved in TDP-43- ubiquitinated inclusions (UBI) (+)-related neurodegenerative pathology. J. Biomed. Sci. 25 (1), 76. doi:10.1186/s12929-018-0479-4
Li, Y., Ray, P., Rao, E. J., Shi, C., Guo, W., Chen, X., et al. (2010). A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U. S. A. 107 (7), 3169–3174. doi:10.1073/pnas.0913602107
Ling, S.-C., Albuquerque, C. P., Han, J. S., Lagier-Tourenne, C., Tokunaga, S., Zhou, H., et al. (2010). ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. U. S. A. 107 (30), 13318–13323. doi:10.1073/pnas.1008227107
Ling, S.-C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79 (3), 416–438. doi:10.1016/j.neuron.2013.07.033
Lokireddy, S., Kukushkin, N. V., and Goldberg, A. L. (2015). cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. U. S. A. 112 (52), E7176–E7185. doi:10.1073/pnas.1522332112
Lukavsky, P. J., Daujotyte, D., Tollervey, J. R., Ule, J., Stuani, C., Buratti, E., et al. (2013). Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20 (12), 1443–1449. doi:10.1038/nsmb.2698
Medinas, D. B., Valenzuela, V., and Hetz, C. (2017). Proteostasis disturbance in amyotrophic lateral sclerosis. Hum. Mol. Genet. 26 (R2), R91–R104. doi:10.1093/hmg/ddx274
Mejzini, R., Flynn, L. L., Pitout, I. L., Fletcher, S., Wilton, S. D., and Akkari, P. A. (2019). ALS genetics, mechanisms, and therapeutics: where are we now? Front. Neurosci. 13, 1310. doi:10.3389/fnins.2019.01310
Miguel, L., Frébourg, T., Campion, D., and Lecourtois, M. (2011). Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol. Dis. 41 (2), 398–406. doi:10.1016/j.nbd.2010.10.007
Mitra, J., Guerrero, E. N., Hegde, P. M., Liachko, N. F., Wang, H., Vasquez, V., et al. (2019). Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. U. S. A. 116 (10), 4696–4705. doi:10.1073/pnas.1818415116
Mitsuzawa, S., Akiyama, T., Nishiyama, A., Suzuki, N., Kato, M., Warita, H., et al. (2018). TARDBP p. G376D mutation, found in rapid progressive familial ALS, induces mislocalization of TDP-43. Eneurologicalsci 11, 20–22. doi:10.1016/j.ensci.2018.04.001
Moon, S., and Lee, B.-H. (2018). Chemically induced cellular proteolysis: an emerging therapeutic strategy for undruggable targets. Mol. Cells 41 (11), 933–942. doi:10.14348/molcells.2018.0372
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314 (5796), 130–133. doi:10.1126/science.1134108
Ni, J., Ren, Y., Su, T., Zhou, J., Fu, C., Lu, Y., et al. (2021). Loss of TDP-43 function underlies hippocampal and cortical synaptic deficits in TDP-43 proteinopathies. Mol. Psychiatry, 1–15. doi:10.1038/s41380-021-01346-0
Pesiridis, G. S., Lee, V. M. Y., and Trojanowski, J. Q. (2009). Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum. Mol. Genet. 18 (R2), R156–R162. doi:10.1093/hmg/ddp303
Pinarbasi, E. S., Cağatay, T., Fung, H. Y. J., Li, Y. C., Chook, Y. M., and Thomas, P. J. (2018). Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci. Rep. 8 (1), 7083–7116. doi:10.1038/s41598-018-25008-4
Pohl, C., and Dikic, I. (2019). Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366 (6467), 818–822. doi:10.1126/science.aax3769
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12, 25. doi:10.3389/fnmol.2019.00025
Qin, H., Lim, L.-Z., Wei, Y., and Song, J. (2014). TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl. Acad. Sci. U. S. A. 111 (52), 18619–18624. doi:10.1073/pnas.1413994112
Riemenschneider, H., Guo, Q., Bader, J., Frottin, F., Farny, D., Kleinberger, G., et al. (2022). Gel-like inclusions of C-terminal fragments of TDP-43 sequester stalled proteasomes in neurons. EMBO Rep. 23, e53890. doi:10.15252/embr.202153890
Ringholz, G., Appel, S. H., Bradshaw, M., Cooke, N., Mosnik, D., and Schulz, P. (2005). Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65 (4), 586–590. doi:10.1212/01.wnl.0000172911.39167.b6
Russo, A., Scardigli, R., La Regina, F., Murray, M. E., Romano, N., Dickson, D. W., et al. (2017). Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum. Mol. Genet. 26 (8), 1407–1418. doi:10.1093/hmg/ddx035
Santamaria, N., Alhothali, M., Alfonso, M. H., Breydo, L., and Uversky, V. N. (2017). Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 74 (7), 1297–1318. doi:10.1007/s00018-016-2416-6
Schmidt, M. F., Gan, Z. Y., Komander, D., and Dewson, G. (2021). Ubiquitin signalling in neurodegeneration: mechanisms and therapeutic opportunities. Cell Death Differ. 28 (2), 570–590. doi:10.1038/s41418-020-00706-7
Scotter, E. L., Vance, C., Nishimura, A. L., Lee, Y.-B., Chen, H.-J., Urwin, H., et al. (2014). Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 127 (6), 1263–1278. doi:10.1242/jcs.140087
Soncini, C., Berdo, I., and Draetta, G. (2001). Ras–GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene 20 (29), 3869–3879. doi:10.1038/sj.onc.1204553
Takahashi, M., Kitaura, H., Kakita, A., Kakihana, T., Katsuragi, Y., Onodera, O., et al. (2022). USP10 inhibits aberrant cytoplasmic aggregation of TDP-43 by promoting stress granule clearance. Mol. Cell. Biol. 42, 3. doi:10.1128/mcb.00393-21
Tomé, S. O., Vandenberghe, R., Ospitalieri, S., Van Schoor, E., Tousseyn, T., Otto, M., et al. (2020). Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: relationship with clinical phenotypes. Acta Neuropathol. Commun. 8 (1), 61–22. doi:10.1186/s40478-020-00934-5
Tsoi, P. S., Choi, K. J., Leonard, P. G., Sizovs, A., Moosa, M. M., MacKenzie, K. R., et al. (2017). The N-terminal domain of ALS-linked TDP-43 assembles without misfolding. Angew. Chem. Int. Ed. Engl. 129 (41), 12590–12593. doi:10.1002/anie.201706769
Uchida, T., Tamaki, Y., Ayaki, T., Shodai, A., Kaji, S., Morimura, T., et al. (2016). CUL2-mediated clearance of misfolded TDP-43 is paradoxically affected by VHL in oligodendrocytes in ALS. Sci. Rep. 6 (1), 19118–19119. doi:10.1038/srep19118
Van Mossevelde, S., Engelborghs, S., van der Zee, J., and Van Broeckhoven, C. (2018). Genotype–phenotype links in frontotemporal lobar degeneration. Nat. Rev. Neurol. 14 (6), 363–378. doi:10.1038/s41582-018-0009-8
Watabe, K., Kato, Y., Sakuma, M., Murata, M., Niida-Kawaguchi, M., Takemura, T., et al. (2020). Praja1 RING-finger E3 ubiquitin ligase suppresses neuronal cytoplasmic TDP-43 aggregate formation. Neuropathology 40 (6), 570–586. doi:10.1111/neup.12694
Watanabe, S., Kaneko, K., and Yamanaka, K. (2013). Accelerated disease onset with stabilized familial amyotrophic lateral sclerosis (ALS)-linked mutant TDP-43 proteins. J. Biol. Chem. 288 (5), 3641–3654. doi:10.1074/jbc.M112.433615
Weskamp, K., and Barmada, S. J. (2018). TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Res. 1693, 67–74. doi:10.1016/j.brainres.2018.01.015
Wheaton, M., Salamone, A., Mosnik, D., McDonald, R., Appel, S. H., Schmolck, H., et al. (2007). Cognitive impairment in familial ALS. Neurology 69 (14), 1411–1417. doi:10.1212/01.wnl.0000277422.11236.2c
Winton, M. J., Igaz, L. M., Wong, M. M., Kwong, L. K., Trojanowski, J. Q., and Lee, V. M.-Y. (2008a). Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283 (19), 13302–13309. doi:10.1074/jbc.M800342200
Winton, M. J., Van Deerlin, V. M., Kwong, L. K., Yuan, W., Wood, E. M., Yu, C.-E., et al. (2008b). A90V TDP-43 variant results in the aberrant localization of TDP-43 in vitro. FEBS Lett. 582 (15), 2252–2256. doi:10.1016/j.febslet.2008.05.024
Xiao, S., Sanelli, T., Chiang, H., Sun, Y., Chakrabartty, A., Keith, J., et al. (2015). Low molecular weight species of TDP-43 generated by abnormal splicing form inclusions in amyotrophic lateral sclerosis and result in motor neuron death. Acta Neuropathol. 130 (1), 49–61. doi:10.1007/s00401-015-1412-5
Xie, X., Matsumoto, S., Endo, A., Fukushima, T., Kawahara, H., Saeki, Y., et al. (2018). Deubiquitylases USP5 and USP13 are recruited to and regulate heat-induced stress granules through their deubiquitylating activities. J. Cell Sci. 131 (8), jcs210856. doi:10.1242/jcs.210856
Yang, C., Wang, H., Qiao, T., Yang, B., Aliaga, L., Qiu, L., et al. (2014). Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 111 (12), E1121–E1129. doi:10.1073/pnas.1322641111
Yau, R., and Rape, M. (2016). The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18 (6), 579–586. doi:10.1038/ncb3358
Yu, C.-H., Davidson, S., Harapas, C. R., Hilton, J. B., Mlodzianoski, M. J., Laohamonthonkul, P., et al. (2020). TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183 (3), 636–649. doi:10.1016/j.cell.2020.09.020
Zhang, T., Hwang, H.-Y., Hao, H., Talbot, C., and Wang, J. (2012). Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. J. Biol. Chem. 287 (11), 8371–8382. doi:10.1074/jbc.M111.311977
Zhang, T., Periz, G., Lu, Y.-N., and Wang, J. (2020). USP7 regulates ALS-associated proteotoxicity and quality control through the NEDD4L–SMAD pathway. Proc. Natl. Acad. Sci. U. S. A. 117 (45), 28114–28125. doi:10.1073/pnas.2014349117
Zhang, Y.-J., Xu, Y.-F., Cook, C., Gendron, T. F., Roettges, P., Link, C. D., et al. (2009). Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U. S. A. 106 (18), 7607–7612. doi:10.1073/pnas.0900688106
Zhao, W., Beers, D. R., Bell, S., Wang, J., Wen, S., Baloh, R. H., et al. (2015). TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp. Neurol. 273, 24–35. doi:10.1016/j.expneurol.2015.07.019
Keywords: ubiquitinating enzyme, deubiquitinating enzyme, ubiquitin-proteasome system, TDP-43, protein quality control, proteinopathy, frontotemporal lobar degeneration, amyotrophic lateral sclerosis
Citation: Tran N-N and Lee B-H (2022) Functional implication of ubiquitinating and deubiquitinating mechanisms in TDP-43 proteinopathies. Front. Cell Dev. Biol. 10:931968. doi: 10.3389/fcell.2022.931968
Received: 29 April 2022; Accepted: 23 August 2022;
Published: 09 September 2022.
Edited by:
Key-Hwan Lim, Korea Brain Research Institute, South KoreaReviewed by:
Hyung-Jun Kim, Korea Brain Research Institute, South KoreaCopyright © 2022 Tran and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Byung-Hoon Lee, Ynl1bmctaG9vbl9sZWVAZGdpc3QuYWMua3I=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.