 Zulong Chen
Zulong Chen Jessica K. Tyler
Jessica K. Tyler- Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, New York City, NY, United States
DNA double-strand breaks (DSBs), the most deleterious DNA lesions, are primarily repaired by two pathways, namely homologous recombination (HR) and non-homologous end joining (NHEJ), the choice of which is largely dependent on cell cycle phase and the local chromatin landscape. Recent studies have revealed that post-translational modifications on histones play pivotal roles in regulating DSB repair pathways including repair pathway choice. In this review, we present our current understanding of how these DSB repair pathways are employed in various chromatin landscapes to safeguard genomic integrity. We place an emphasis on the impact of different histone post-translational modifications, characteristic of euchromatin or heterochromatin regions, on DSB repair pathway choice. We discuss the potential roles of damage-induced chromatin modifications in the maintenance of genome and epigenome integrity. Finally, we discuss how RNA transcripts from the vicinity of DSBs at actively transcribed regions also regulate DSB repair pathway choice.
Introduction
Genome integrity is repeatedly challenged by a variety of endogenous and environmental stresses including, but not limited to, replication stress, reactive oxygen stress (ROS), ionizing radiation (IR), ultraviolet (UV) light, and various chemicals, all of which can induce DNA lesions (Tubbs and Nussenzweig, 2017). Among the various lesions, DNA double-strand breaks (DSB) are the most deleterious and can cause loss of chromosomal arms and cell death if left unrepaired and can lead to chromosomal translocations and deletions if inaccurately repaired (Elbakry and Lobrich, 2021). Defects in DSB repair lead to a variety of human diseases such as developmental defects, immunodeficiency, premature aging, neurodegenerative disorders, and cancer (Jackson and Bartek, 2009). To safeguard genome integrity, cells have evolved several DSB repair pathways, among which homologous recombination (HR) and non-homologous end joining (NHEJ) are the most predominant mechanisms (Chapman et al., 2012b; Hustedt and Durocher, 2016; Scully et al., 2019).
HR is the most faithful DSB repair mechanism and requires the presence of homologous sequences, usually the sister chromatid, as a template for DNA synthesis to accurately repair the DSB. Therefore, HR is restricted to the S and G2 phases of the cell cycle (Escribano-Diaz et al., 2013; Chen et al., 2018). Extensive 5′–3′ resection of the broken DNA ends is a critical step for HR repair, which is initiated by the Mre11-Rad50-Nbs1 (MRN) complex and C-terminal binding protein interacting protein (CtIP), followed by extensive resection by EXO1 or DNA2-BLM (Symington, 2014). The 3′ single-stranded DNA (ssDNA) overhangs are bound and protected by replication protein A (RPA), which is subsequently removed and replaced with the ssDNA protein RAD51 to form the ssDNA/RAD51 nucleoprotein filament. The ssDNA/RAD51 nucleofilament performs strand invasion of the homologous DNA sequence, which leads to templated DNA synthesis from the 3′ ends of the ssDNA, enabling the subsequent completion of HR (Chapman et al., 2012b).
In contrast, NHEJ demands minimal or no homology to join the DNA ends and occurs throughout the cell cycle in an error-prone manner (Escribano-Diaz et al., 2013; Hustedt and Durocher, 2016). The broken DNA ends are rapidly recognized and bound by the heterodimer of Ku70/Ku80 and form the DNA-PK complex upon binding of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) which activates a DNA damage response (DDR) signaling pathway (Blackford and Jackson, 2017). During NHEJ, the two broken DNA ends are directly joined by DNA Ligase 4 (Lig 4), through the X-ray repair cross-complementing protein 4 (XRCC4) and XRCC4-like factor (XLF) complex, with no or minimal end processing by Artemis (Lieber, 2010; Panier and Boulton, 2014).
The eukaryotic genome is packaged into chromatin within the nucleus. The fundamental unit of chromatin, the nucleosome, is composed of two copies of each core histone (H3, H4, H2A, and H2B) wrapped by 147 base pairs of DNA (Kouzarides, 2007). The unstructured core histone tails are subject to numerous different post-translational modifications (PTMs), among which phosphorylation, methylation, acetylation, and ubiquitylation have been most extensively studied (Tan et al., 2011; Rothbart and Strahl, 2014). These histone PTMs, as well as DNA methylation, help to partition the genome into distinct domains such as euchromatin and heterochromatin (which can be facultative or constitutive). Euchromatin is an open chromatin state, is associated with active transcription, and is enriched in H3K4me2/3, H3K36me3 and histone hyperacetylation (Bannister and Kouzarides, 2011; Zhang et al., 2015; Talbert and Henikoff, 2021). In contrast, heterochromatin is more highly compacted and less accessible to the transcription machinery and is enriched in repressive histone PTMs and thus transcriptionally inactive. Facultative heterochromatin is formed at regions that contain genes which are developmentally regulated and are enriched in repressive histone PTMs such as H3K9me2/3 and H3K27me2/3 (Trojer and Reinberg, 2007). Facultative heterochromatin is dispersed throughout the genome and dictates gene silencing, for example in X-chromosome inactivation, autosomal imprinted genomic loci and HOX gene clusters (Feil and Berger, 2007; Trojer and Reinberg, 2007). Constitutive heterochromatin is composed of highly repetitive sequences and is usually at gene poor regions such as centromeres, peri-centromeres and telomeres, and is characterized by repressive histone PTMs such as H3K9me3 which recruits Heterochromatin Protein 1 (HP1) (Janssen et al., 2018). Centromeric heterochromatin uniquely includes the histone H3 variant, CENP-A that epigenetically defines the location of centromeres, and is interspersed with the active histone PTMs H3K4me1/2 and H3K36me2/3, but is depleted of H3K9me3 (Bloom, 2014), which correlates with non-coding transcriptional activity at centromeric regions (Arunkumar and Melters, 2020).
The dynamics of histone PTMs on chromatin is precisely controlled by proteins that write and erase these modifications (Hyun et al., 2017). Enzymes that add and remove the chemical modifications on histones are termed “writers” and “erasers,” respectively. Proteins that recognize one specific or combination of histone PTMs are called “readers.” In response to various developmental and environmental cues, histone PTMs are actively incorporated or removed, which leads to alterations in the chromatin and gene expression. A growing body of evidence has shown that histone PTMs play crucial roles in the DNA damage response and repair (Table 1) (Chapman et al., 2012b; Dabin et al., 2016).
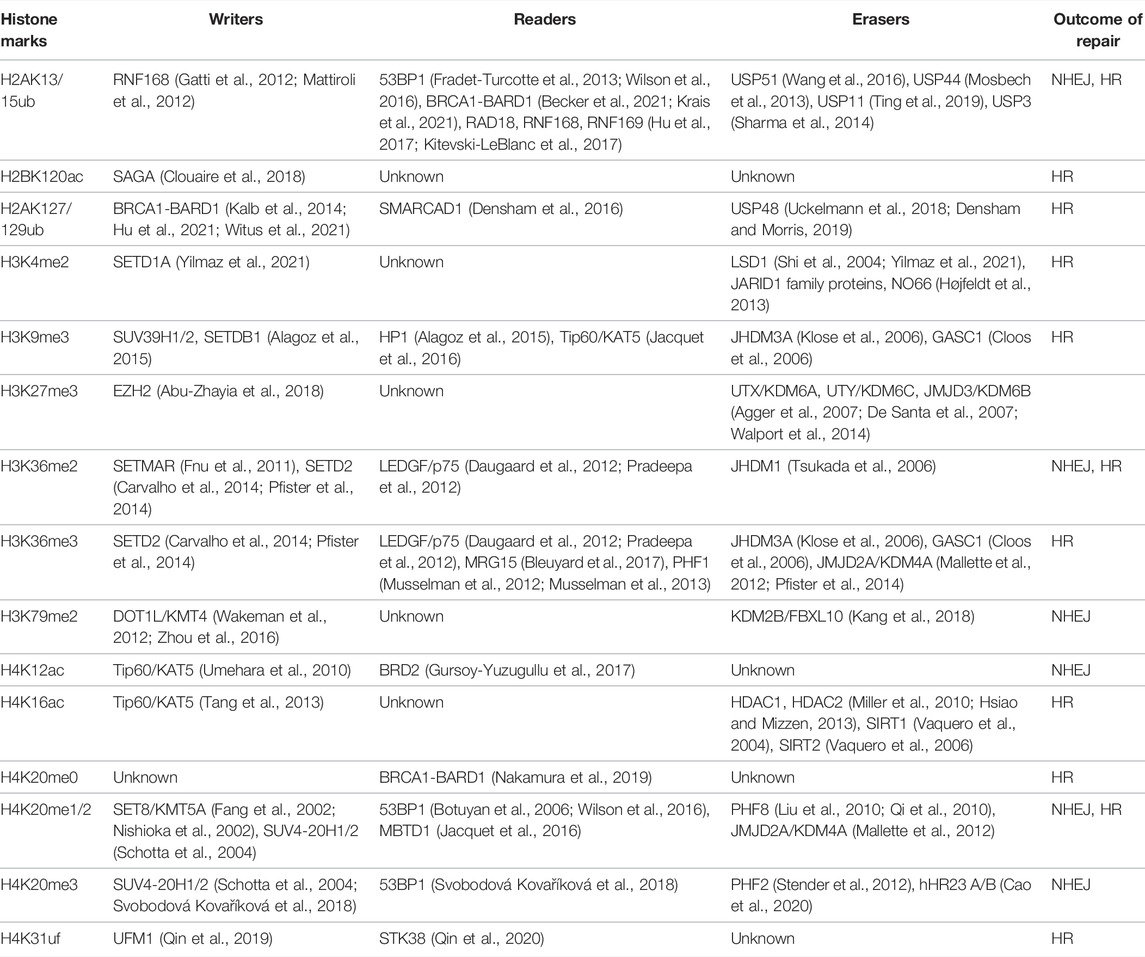
TABLE 1. Histone Marks that are involved in DSB repair choice in mammalian cells.
In a broad sense, the chromatin landscape comprises not only DNA and histone proteins but also nascent RNA transcripts that interact with the chromatin, as well as the modifications on these nucleic acids and proteins (Black and Whetstine, 2011). The spatiotemporal dynamics of these factors accurately control DNA replication, gene transcription and genome stability. DSBs occur in the context of different chromatin landscapes, which in turn activate DDR signaling pathways to remodel chromatin structure and modulate nucleosome organization such as histone variant exchange and histone post-translational modification, to facilitate DSB repair (Price and D’Andrea, 2013). Specifically, after DSB induction in mammalian cells, the chromatin structure is rapidly and transiently compacted to repress transcription, followed by the relaxation of the chromatin structure to enable access of the repair machinery, the mechanisms of which will not be covered here but have been extensively reviewed recently (Densham and Morris, 2019). Instead, here we will focus on how different chromatin structures and pre-existing and DSB-induced histone modifications orchestrate whether HR or NHEJ is used to repair a DSB, termed DSB repair pathway choice.
Histone Modifications Orchestrate BRCA1-BARD1-and 53BP1-Mediated Double-Strand Break Repair Pathway Choice
Upon DSB induction, a cascade of histone and protein PTMs are induced which are critical for recruitment of DNA repair proteins. Initially, the PIKK family kinase ATM is recruited to the chromatin and activated through its interaction with NBS1 in the MRE11-RAD50-NBS1 (MRN) complex (Blackford and Jackson, 2017; Britton et al., 2013), followed by ATM phosphorylating Serine 139 on the histone variant H2A.X (γH2A.X), MDC1 and other proteins. The phosphorylated MDC1 in turn recruits an E3 ligase, RNF8, to promote K63-linked polyubiquitylation of chromatin proteins (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007; Mattiroli and Penengo, 2021). Meanwhile, RNF8, and UBC13, an E2 ubiquitin-conjugating enzyme, mediate K63-linked polyubiquitylation on the linker histone H1, which sequentially recruits another E3 ligase RNF168 to specifically mono-ubiquitylate H2A at lysines 13 and 15 (H2AK13ub and H2AK15ub) (Mattiroli et al., 2012; Thorslund et al., 2015; Uckelmann and Sixma, 2017). Mono-ubiquitylation on H2AK13/15 in turn contributes to the recruitment of the pro-NHEJ factor p53-binding protein 1 (53BP1) (Fradet-Turcotte et al., 2013) and the pro-HR factor, the heterodimer of Breast cancer type 1 susceptibility protein (BRCA1) and BRCA1-associated RING domain protein 1 (BARD1), to DSB sites (as discussed below) (Figure 1). The mono-ubiquitylated H2AK13/15 is then further extended by RNF8 to form K63-linked ubiquitin chains to recruit downstream factors to fulfill DNA damage repair (Doil et al., 2009; Stewart et al., 2009; Mattiroli et al., 2012; Mattiroli and Penengo, 2021). In addition to 53BP1 and BARD1, H2AK15ub is also recognized by other histone readers such as RAD18, RNF169, and RNF168 itself (Hu et al., 2017; Kitevski-LeBlanc et al., 2017). Both RAD18 and RNF169 are involved in DSB repair and promote HR. Intriguingly, RNF169 and RAD18 have a much higher affinity for H2AK13/15ub, as compared to 53BP1, suggesting that RNF169 and RAD18 may be able to shift the balance between the choice of HR vs. NHEJ repair (Mattiroli and Penengo, 2021). Interestingly, DSB induced ubiquitylation or K63-linked polyubiquitylation on H2A leads to ATM-dependent transcriptional silencing. H2A deubiquitylation by the deubiquitylase USP16 is required for rapid restoration of transcription around DSB sites (Shanbhag et al., 2010). Concomitantly, DSBs within chromatin are rapidly recognized by the Ku70/80 heterodimer, which recruits and activates DNA-PKcs to promote NHEJ (Blackford and Jackson, 2017).
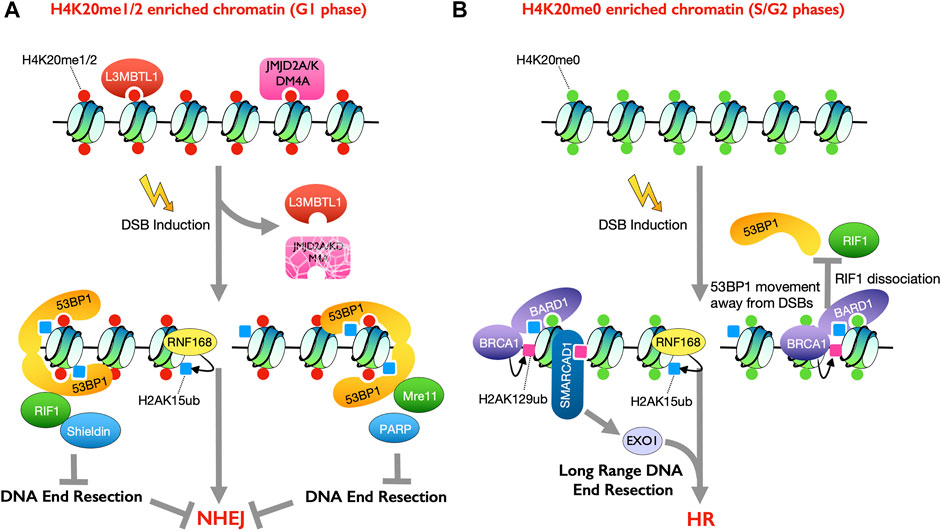
FIGURE 1. Histone modifications involved in dictating the cell cycle phase specific DSB repair pathway choice. (A) Histone mark H4K20me1/2 is enriched in G1 phase of the cell cycle. 53BP1 binds to nucleosomes containing both H4K20me1/2 and H2AK15ub, mediated by RNF168 upon DSB induction, to promote DSB repair by NHEJ (B) Unmethylated H4K20me0 is enriched in S/G2 phases of the cell cycle. BRCA1-BARD1 heterodimer recognizes H4K20me0 and H2AK15ub to facilitate HR repair (right). BRCA1 mediated H2AK129ub recruits SMARCAD1 to promote DNA end resection and thus HR (left).
H2AK15ub and H4K20me1/2 Promote 53BP1 Recruitment to Double-Strand Breaks
Whether or not DNA end resection occurs is the critical decision step in DSB repair pathway choice, as extensive DNA end resection necessitates repair by HR and blocks NHEJ (Chapman et al., 2012b). Meanwhile limited or no DNA end resection prevents HR and drives NHEJ. 53BP1 can be rapidly recruited to the vicinity of DSBs upon DSB induction, where it blocks DNA end resection (Schultz et al., 2000; Anderson et al., 2001; Rappold et al., 2001). The role of 53BP1 in blocking end resection was uncovered from a series of studies showing loss of 53BP1 rescues homologous recombination in Brca1-deficient mice and cells (Cao et al., 2009; Bouwman et al., 2010; Bunting et al., 2010). RIF1 was found, by several independent groups, to also prevent end resection at DSBs and promotes 53BP1-dependent NHEJ (Chapman et al., 2013; Di Virgilio et al., 2013; Escribano-Diaz et al., 2013; Zimmermann et al., 2013). More recently, multiple research groups identified the Shieldin complex as acting downstream of RIF1 to block DNA end resection (Dev et al., 2018; Findlay et al., 2018; Gao et al., 2018; Ghezraoui et al., 2018; Gupta et al., 2018; Mirman et al., 2018; Noordermeer et al., 2018). Once recruited to DSBs, 53BP1/RIF1/Shieldin function together to play a critical role in promoting NHEJ and blocking HR.
The recruitment of 53BP1 to DSBs, and thus NHEJ promotion, is dependent on specific histone PTMs. 53BP1 has several histone modification recognition domains (Chapman et al., 2012b). The tandem Tudor domain (TTD) in 53BP1 recognizes the mono- or di-methylated states of H4 on lysine 20 (H4K20me1/2) (Botuyan et al., 2006). Point mutation in the TTD domain of 53BP1 completely abolished its accumulation at DSBs (Huyen et al., 2004). As such 53BP1 accumulates at DSB sites marked with H4K20me2 (Pellegrino et al., 2017). However, H4K20me2 is a highly abundant histone mark that is present on the vast majority of nucleosomes in late G2 and G1 phases (Pesavento et al., 2008). Meanwhile, 53BP1 is recruited to the DNA in the vicinity of the DSBs, which are visualized in the cell as DNA repair foci upon DSB induction. As such, the DSB specific recruitment of 53BP1 must be regulated by factors in addition to H4K20me2. Indeed, structural studies showed that L3MBTL1, the human homolog of the Drosophila melanogaster tumor-suppressor protein l (3)mbt, and JMJD2A/KDM4A, the human histone demethylase, bound to methylated H4K20 and masked the histone interface in undamaged chromatin to prevent 53BP1 from binding to the chromatin (Huang et al., 2006; Min et al., 2007; Lee et al., 2008; Acs et al., 2011; Mallette et al., 2012). However, in response to DNA damage, both L3MBTL1 and JMJD2A/KDM4A are evicted, in a manner dependent on activation of the RNF8-RNF168 pathway upon DSB induction, which exposes H4K20me2 to enable 53BP1 accumulation at DSBs (Acs et al., 2011; Mallette et al., 2012) (Figure 1A).
Recognition of H4K20me2 is necessary, but not sufficient, for 53BP1 to be recruited to DSB sites (Zgheib et al., 2009). Accumulation of the 53BP1 orthologue in fission yeast Crb2 at DSBs relies on dual recognition of two histone marks, H4K20me2 and γH2A.X, via its tandem Tudor domains and C-terminal BRCT domains, respectively (Du et al., 2006; Sanders et al., 2004; Stucki et al., 2005). Damage-induced phosphorylation on the histone variant H2A.X is dispensable for 53BP1 recruitment in metazoan cells but instead the accumulation of 53BP1 on the damaged chromatin requires RNF8-RNF168-mediated histone ubiquitylation in metazoans, as mentioned above (Celeste et al., 2003; Doil et al., 2009; Stewart et al., 2009). Indeed, 53BP1 directly binds to the DSB-induced and RNF168-mediated ubiquitylation at lysine 15 on H2A (H2AK15ub) through its ubiquitylation-dependent recruitment (UDR) motif (Fradet-Turcotte et al., 2013; Wilson et al., 2016). Point mutations in the UDR motif abolish the binding of 53BP1 to H2AK15ub and attenuate 53BP1 recruitment (Fradet-Turcotte et al., 2013), demonstrating the importance of this interaction. Ultimately, the 53BP1 protein recognizes mononucleosomes containing both H4K20me2 and damage induced H2AK15ub marks, which ensures the specific recruitment of 53BP1 to damaged DNA sites in G1 phase cells (Figure 1A) (Panier and Boulton, 2014; Wilson et al., 2016). Cell cycle phase-dependent changes in H4K20 methylation enhance the recruitment of 53BP1 in G1 to promote NHEJ and reduce the recruitment of 53BP1 during S/G2 phases to promote HR. Using nascent chromatin capture (NCC), newly synthesized H4 histone was found to be exclusively unmethylated at lysine 20 (H4K20me0), which is a signature of post-replicative chromatin (Alabert et al., 2014; Alabert et al., 2015; Saredi et al., 2016). This unmethylated H4K20me0 exists from S phase (when most newly synthesized H4 is incorporated into the chromatin) until late G2/M phase, at which time the SET domain-containing protein 8 (SET8) methyltransferase catalyzes mono-methylation of H4K20 (H4K20me1) and subsequently the suppressor of variegation 4–20 homologue ½ (SUV4-20H1/2) converts the mono-methylation to di- and tri-methylation (H4K20me2/3). In this manner, most of the H4 lysine 20 is methylated by G1 phase (Beck et al., 2012; Jørgensen et al., 2013; Saredi et al., 2016), which promotes 53BP1 recruitment to block HR and promote NHEJ of DSBs in G1 phase cells (Figure 1A). This is key, because G1 phase cells lack sister chromatids and if HR was allowed to occur, it would result in chromosomal deletions or translocations, depending on whether the homology was on the same or a different chromosome.
H2AK15ub and H4K20me0 Recruit BRCA1-BARD1 to Facilitate Double-Strand Break Repair by Homologous Recombination
The BRCA1-BARD1 heterodimer antagonizes 53BP1 accumulation at DSB sites in the S/G2 cell cycle phases (Chapman et al., 2012a; Pellegrino et al., 2017), in multiple different ways, to promote HR repair. The ankyrin repeat domain (ARD) in TONSL, a protein that forms a heterodimer with MMS22L to maintain genomic stability during replication, was identified as a reader of the unmethylated state of lysine 20 on H4 (H4K20me0) (Saredi et al., 2016). Recognition of H4K20me0 is required for the accumulation of TONSL-MMS22L at damaged replication forks and DNA lesions, which promotes RAD51 loading (Saredi et al., 2016). The ARD domains in TONSL and BARD1 are highly conserved, such that the ARD domain of BARD1 can also specifically bind to H4K20me0 (Fox et al., 2008; Saredi et al., 2016; Nakamura et al., 2019). Indeed, the presence of H4K20me0 is required for BRCA1-BARD1 recruitment to chromatin. Mutation of the predicted H4K20me0 binding motif on the ARD domain abolished BARD1 binding to nucleosomes and failed to antagonize 53BP1 accumulation at DSB sites in BARD1 deficient cells (Nakamura et al., 2019). Consistently, depletion of SET8, which in principle eliminates all methylation from H4K20, increased H4K20me0 levels and enriched BRCA1-BARD1 binding to chromatin in G2 and G1 phases to attenuate NHEJ repair (Nakamura et al., 2019).
Given that half of H4K20 is unmethylated on post-replicative chromatin in late S and G2 phases, how do cells ensure that the BRCA1-BARD1 complex is recruited specifically to damaged DNA to promote HR? A tandem BRCT-domain-associated ubiquitin-dependent recruitment motif (BUDR) in BARD1 was identified to recruit BRCA1 to DSB sites through its binding to H2AK15ub (Becker et al., 2021; Krais et al., 2021). Notably, as discussed above, this is one of the histone PTMs that recruits 53BP1, suggesting that 53BP1 and BRCA1-BARD1 may physically compete for binding to H2AK15ub (Figure 1). In vitro pull-down assays showed the interaction between GST–BARD1 (ARD–BRCT) and recombinant nucleosome variants were strongly stimulated by H2AK15ub but inhibited by methylation on H4K20 (Becker et al., 2021). On the contrary, both modifications on the histones were required for the interaction between GST–53BP1(TTD–UDR) and recombinant nucleosomes (Becker et al., 2021). Indeed, the cooperation of the ARD and BUDR domains in BARD1 to bind H4K20me0 and H2AK15ub, respectively, is required for high affinity recognition of DSB lesions by the BRCA1-BARD1 complex and for its activity in promoting HR repair on post-replicative chromatin (Becker et al., 2021; Dai et al., 2021).
BRCA1-Mediated H2AK129ub Antagonizes 53BP1 Recruitment to Double-Strand Breaks
The BRCA1-mediated H2AK129 ubiquitylation promotes end resection via its ability to recruit SMARCAD1, an ATP dependent nucleosome remodeler that facilitates Exo1-mediated extensive DNA end resection (Chen et al., 2012; Costelloe et al., 2012; Densham et al., 2016; Eapen et al., 2012; Kalb et al., 2014; Adkins et al., 2017). Indeed, the balance between RNF168 mediated H2AK15 ubiquitylation and Brca1-mediated H2AK129 ubiquitylation is thought to determine the pathway choice between NHEJ and HR, where H2AK15ub recruits 53BP1 to promote NHEJ and H2AK129ub recruits SMARCAD1 to promote DNA end resection. On the other hand, the deubiquitylation enzyme (DUB) USP48 specifically removes the BRCA1-mediated H2Aub modifications to limit SMARCAD1 interaction to prevent over resection to limit the use of the mutagenic single-strand annealing repair pathway (Densham and Morris, 2019; Uckelmann et al., 2018) (Figure 1B). Whether USP48 is regulated in a cell cycle specific manner is unclear. Also, the DUB USP51 was shown in vitro to directly bind to H2A-H2B and deubiquitylate H2AK13/15ub to regulate DSB repair (Wang et al., 2016). Overexpression of USP51 suppressed IR-induced foci formation of both 53BP1 and BRCA1 (Wang et al., 2016). However, it seems that USP51 does not play a role in determining the choice between DSB repair by NHEJ or HR as it removes the PTMs on H2A that are required for both the recruitment of 53BP1 and BRCA1. Another mechanism to limit excessive resection is via incorporation of H2AZ at sites of damage (Xu Y. et al., 2012). H2AZ has a shorter C-terminal tail than H2A and lacks the C-terminal lysines K125/K127/K129 present on H2A, and thus H2AZ may thus be refractory to BRCA1-mediated modification and thus SMARCAD1-mediated nucleosome remodeling.
BRCA1 not only colocalizes with γH2A.X at DSB sites but also catalyzes ubiquitin conjugation to H2A.X at lysine 127, which may promote eviction of 53BP1 from γH2A.X containing chromatin (Densham et al., 2016; Hu et al., 2021; Witus et al., 2021). However, it remains unclear to what extent the ubiquitylation on H2AX by BRCA1-BARD1 contributes to DNA repair pathway choice. The ubiquitin binding protein Rap80 forms a complex with BRCA1, and this also facilitates BRCA1-BARD1 accumulation at DNA damage sites (Kim et al., 2007; Sobhian et al., 2007; Wang et al., 2007). Furthermore, the interaction between BARD1 and the nucleosome core particle inhibits K63 polyubiquitination at H2AK13ub and H2AK15ub (Hu et al., 2021). K63-linked polyubiquitylation is specifically recognized by Rap80, which recruits the Abraxas complex (ARISC) to limit end resection (Wang et al., 2007; Coleman and Greenberg, 2011; Hu et al., 2011). Consequently, because of the BARD1-mediated inhibition of K63-linked polyubiquitylation, recruitment of ARISC is compromised and HR is enhanced. As such, BRCA1 and 53BP1 antagonize each other in multiple different ways to regulate DSB repair pathway choice to promote NHEJ in G1 phase and HR in S/G2 phases.
BRCA1’s Influence on Spaciotemporal Dynamics of 53BP1 and RIF1 Through the Cell Cycle
Histone PTMs mediate the recruitment of NHEJ- and HR- promoting factors to the chromatin flanking DSBs, which leads to changes on the spatial level of 53BP1 and BRCA1-BARD1 at DNA repair foci, as seen by microscopy. 53BP1 exists in dense foci around DSBs in G1 phase cells, whereas 53BP1 appears more dispersed from the DNA repair foci center in S-phase cells. Meanwhile, BRCA1, CtIP, and RPA are found at the center of DNA repair foci in S-phase cells (Chapman et al., 2012a; Kakarougkas et al., 2013). Upon experimental loss of BRCA1, 53BP1 relocalizes to the center of the S phase repair foci, resembling a G1 phase focus. As such, BRCA1 is responsible for the spatial positioning of 53BP1 away from DSBs in S-phase cells, and this depends on its E3 ubiquitin ligase activity (Densham et al., 2016) (Figure 1B). Recently, more details of the spaciotemporal dynamics of key DSB repair mediators during end resection at single-ended double-strand breaks (seDSB) at collapsed replication forks have been revealed by single molecule super-resolution microscopy (Whelan and Rothenberg, 2021). This study showed that 53BP1 was recruited to seDSB foci during S phase (even though seDSBs are usually repaired by HR) along with HR machinery factors such as CtIP, MRE11, BRCA1, EXO1, and DNA2. However, in contrast to the HR proteins that were retained at the damage sites for several hours, 53BP1 was rapidly removed from the seDSB foci after recruitment (Whelan and Rothenberg, 2021).
The influence of BRCA1 ligase activity on positioning 53BP1 away from DSBs in S phase cells is not the only way that BRCA1 negatively influences end protection factors. BRCA1 can also counteract RIF1 recruitment in S phase under conditions where no impact on 53BP1 is apparent (Chapman et al., 2013; Escribano-Diaz et al., 2013; Feng et al., 2013; Zimmermann et al., 2013). Specifically, BRCA1 promotes recruitment of the protein phosphatase 4°C (PP4C) to dephosphorylate 53BP1 and release RIF1 (Feng et al., 2015; Isono et al., 2017). Furthermore, BRCA1 promotes the recruitment of the protein Ub-like with PHD and RING finger domains 1 (UHRF1), which mediates K63-linked polyubiquitylation of RIF1 that results in its dissociation from 53BP1, facilitating DNA end resection and HR (Zhang et al., 2016) (Figure 1B).
Histone Post-Translational Modifications Associated With Transcriptional Activity Promote Homologous Recombination in Euchromatin
Mounting evidence suggests that histone PTMs that promote transcription also facilitate HR repair (Figure 2A). This is clearly the case with di- and tri-methylated lysine 36 on H3 (H3K36me2/3). The PWWP domain-containing protein lens epithelium–derived growth factor (LEDGF), the p75 splice variant of protein coding gene Psip1, is constitutively associated with chromatin via its preferential binding to H3K36me2/3 (Ge et al., 1998; Daugaard et al., 2012; Pradeepa et al., 2012). Depletion of LEDGF impairs CtIP and RAD51 recruitment to actively transcribed regions on chromatin and thus inhibits HR, which suggests that H3K36me2/3 plays a direct role in DSB repair pathway choice by promoting HR (Daugaard et al., 2012; Aymard et al., 2014). Consistently, SETD2, the main histone methyltransferase for H3K36me3, is required for HR through facilitating CtIP recruitment and end resection (Carvalho et al., 2014; Pfister et al., 2014). Further evidence for H3K36me2/3 in promoting HR comes from the fact that depletion of SETD2 impairs RAD51 recruitment and HR at DSBs within euchromatin (Aymard et al., 2014). Furthermore, overexpression of JMJD2A/KDM4A, an H3K36me2/3 demethylase, diminished HR efficiency (Pfister et al., 2014). However, neither SETD2 recruitment nor changes in H3K36me3 levels were observed at DSB sites, suggesting the pre-existing H3K36me3 at active genes promotes HR repair (Carvalho et al., 2014; Pfister et al., 2014). Conversely, DSB induction by AsiSI, an endonuclease that targets an 8-bp recognition sequence, at sites that were repaired by NHEJ, was accompanied by a significant increase in H3K36me3 and H4K20me1 (Clouaire et al., 2018), which might be due to the different approaches used to induce DSBs in these studies. Moreover, the partner and localizer of BRCA2 (PALB2) is a physical link between BRCA1 and BRCA2, thus playing a key role in DSB repair by HR (Zhang et al., 2009). Beyond being a major binding partner to BRCA2, PALB2 is associated with transcriptionally active chromatin through its interaction with MRG15 that recognizes H3K36me3 (Bleuyard et al., 2017). Another H3K36me3 reader, The PHD finger protein 1 (PHF1), is recruited to DSB sites in a manner dependent on the Ku70/Ku80 heterodimer. The recognition of H3K36me3 by PHF1 inhibits trimethylation of H3K27, which is a marker of transcriptionally silent chromatin, in vitro and in vivo (Musselman et al., 2012). Finally, binding of the PHF1 Tudor domain to H3K36me3 enhances nucleosome accessibility (Musselman et al., 2013), which may help to keep chromatin in an open state to promote HR. Therefore, H3K36me2/3, either pre-existing or induced by DSBs, plays a critical role in promoting HR repair in euchromatin through multiple different mechanisms.
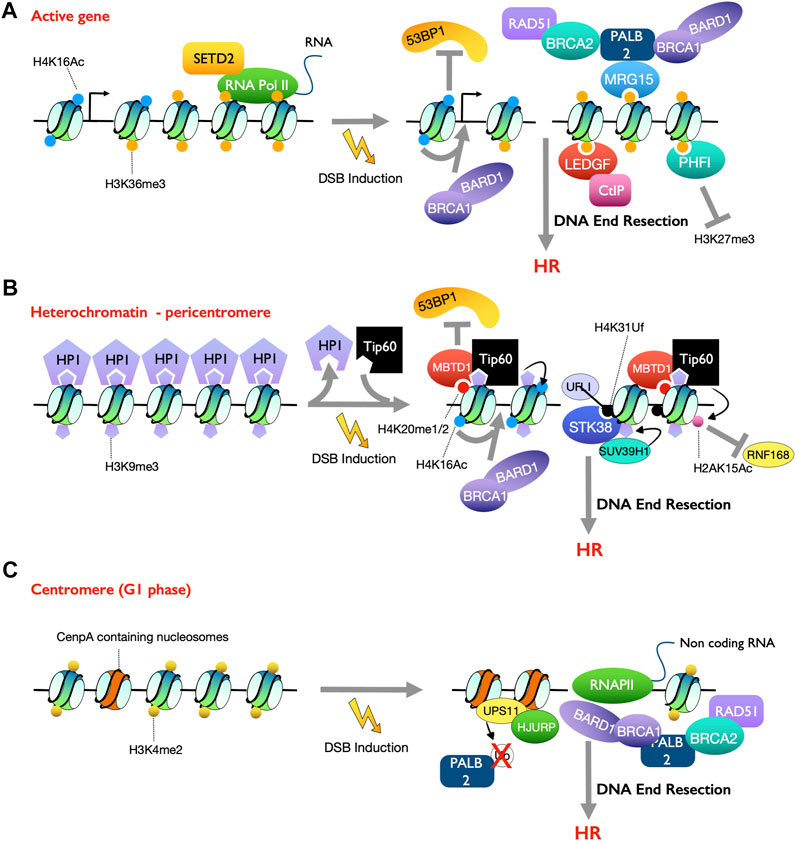
FIGURE 2. Summary of different chromatin environments that promote DSB repair by HR. (A) In euchromatin, transcriptionally active histone mark H3K36me3 and H4K16ac promote DSB repair by HR (B) At peri-centromeric heterochromatin, histone repressive mark H3K9me3 interacts with Tip60 to acetylate H4K16 to attenuate 53BP1 recruitment. MBTD1, a subunit of Tip60/NuA4 complex, competes with 53BP1 form H4K20me1/2 to promote HR repair. Upon DSB induction, UFL1 mediates ufmylation on H4K31, which recruit histone lysine methyltransferase SUV39H1 to increase H3K9me3 and thereby promote HR (C) At centromeres in G1 phase, H3K4me2 promotes non-coding RNA transcription and RNA-DNA hybrids formation, which recruits HR factors to DSBs. The centromere specific histone variant CENP-A promotes the recruitment of Rad51 for HR repair. The deubiquitylase USP11 stabilizes CENP-A chaperone HJURP and PALB2 to recruit RAD51 and BRCA1, respectively, for HR repair.
In other studies, H3K36me2 was found to promote NHEJ repair at DSBs. The H3K36me2 level was markedly increased, mediated by the DNA repair protein Metnase/SETMAR containing the SET histone methylase domain, at DSB sites induced by the endonuclease I-SceI (Fnu et al., 2011). Conversely, H3K36me2 modification was reduced at DSBs induced by AsiSI (Clouaire et al., 2018). The reason for the differences in results depending on which endonuclease was induced is unclear. In fission yeast, the SETD2 homolog Set2, which mediates all three forms of H3K36 methylation, limits end resection and promotes DSB repair by NHEJ (Jha and Strahl, 2014; Pai et al., 2014). Therefore, the choice of repair pathway by NHEJ and HR appears to be carefully controlled by the methylation status of H3K36 and clearly more work remains to be done to fully understand all the nuances of H3K36 methylation in DSB repair pathway choice.
Another PTM that is linked to transcriptionally active chromatin, H4K16ac, plays an important role in tipping the balance from 53BP1 binding towards BRCA1-BARD1 binding at DSBs. Damage induced acetylation on H4K16, mediated by Tip60/KAT5, counteracts 53BP1 binding to H4K20me2 due to the disruption of the interaction between 53BP1 and H4K16 (Hsiao and Mizzen, 2013; Tang et al., 2013) (Figure 2A). Consistently, HDAC1 and HDAC2 (which deacetylate H4K16ac) rapidly accumulate at DSBs and their inactivation, either by inhibition or siRNA knockdown, reduces 53BP1 foci formation on damaged chromatin (Miller et al., 2010; Hsiao and Mizzen, 2013; Tang et al., 2013). Conversely, less H4K16ac, due to deficiency in Tip60 acetyltransferase activity, reduces BRCA1 occupancy at DSBs (Tang et al., 2013). Thus, H4K16ac promotes BRCA-BARD1 recruitment at DSBs and promotes repair by HR. Intriguingly, active transcription also promotes HR repair at DSBs that occur at regions with highly repetitive sequences such as the rDNA locus and centromeres via recruiting the HR machinery irrespective of the cell cycle phase (van Sluis and McStay, 2017; Yilmaz et al., 2021) (Figure 2).
ROLES FOR DAMAGE‐INDUCED HISTONE POST-TRANSLATIONAL MODIFICATIONS IN DOUBLE‐STRAND BREAK REPAIR PATHWAY CHOICE
Histone Post-Translational Modifications Influencing Other Histone Post-Translational Modifications in Double-Strand Break Repair Pathway Choice
It is of great interest to interrogate if DSBs can induce de novo PTMs on histones or removal of pre-existing histone PTMs. If so, how do these PTM changes regulate DSB repair and control repair pathway choice? H3K36me2, H3K9me3 and H4K20me3 are induced at DSBs (Fnu et al., 2011; Svobodová Kovaříková et al., 2018; Qin et al., 2020). Recently, a comprehensive mapping of histone modifications, using ChIP-seq, at DSBs induced by AsiSI was performed to gain a more complete picture of histone PTM changes during DSB repair. RAD51 ChIP-seq peaks and XRCC4 ChIP-seq peaks were defined as HR-prone sites and NHEJ-prone sites, respectively. Twenty histone PTMs and histones were interrogated in the study, among which 6 were significantly decreased (H3K79me2, H3, H3K36me2, H4K12ac, H2AZ, and H2BK120ub) and 5 were significantly increased (H4S1P, H4K20me1, macroH2A, H2BK120ac, and ubiquitin) surrounding the DSB sites (Clouaire et al., 2018). Interestingly, the switch from H2BK120 ubiquitylation (H2BK120ub) to H2BK120 acetylation (H2BK120ac) occurred at both types of DSB sites, irrespective of their being HR-prone or NHEJ-prone (Clouaire et al., 2018). The reduced H3K79me2 around DSBs may be related to the reduced H2BK120Ub because H2BK120ub stimulates DOT1L (the H3K79 methyltransferase) catalytic activity, leading to efficient methylation on H3K79 (McGinty et al., 2008; Zhou et al., 2016). Suppression of DOT1L leads to decreased recruitment of 53BP1 to DSBs (Huyen et al., 2004). Consistently, H3K79me2 is required for IR-induced 53BP1 foci formation when H4K20me2 levels are low during G1/G2 phases (Wakeman et al., 2012). Thus, the DSB induced transition of ubiquitylation to acetylation on H2BK120 and subsequent decreases in H3K79me2 may promote repair by HR if the DSB occurred at the same sites in the future. However, future studies will be required to determine to what extent these PTM changes contribute to DSB repair pathway choice.
Intriguingly, decreases in H3K36 dimethylation and H4K12/K16 acetylation after DSB induction were significant at HR-prone DSBs, whereas a significant increase in H3K36me3 and H4K20me1 was detected at NHEJ-prone sites (Clouaire et al., 2018). This is consistent with the fact that H3K36me2 facilitates NHEJ but is inconsistent with H4K16ac promoting HR, discussed above. The reduced level of H4K12ac at HR-prone sites could be related to the following mechanism by which H4K12ac indirectly recruits 53BP1: H4K12 is acetylated by Tip60 at DSBs, which recruits the bromodomain protein BRD2 to spread the acetylation state through the interaction with a second bromodomain protein ZMYND8 on the flanking chromatin (Umehara et al., 2010; Gursoy-Yuzugullu et al., 2017). BRD2 limits binding of the L3MBTL1 repressor to expose H4K20me1/2 and thus promotes 53BP1 recruitment (Gursoy-Yuzugullu et al., 2017). In line with this report, ZMYND8 was shown to be recruited to damaged chromatin by Tip60 mediated acetylation on H4 in transcriptionally active chromatin (Gong et al., 2015). However, ZMYND8 has also been reported to promote DSB repair by HR at breaks induced by AsiSI (Gong et al., 2015; Gong et al., 2017), which suggests that there is more to learn before we understand the intricate dynamics between NHEJ and HR repair. The increase in H3K36me3 following DSB induction at the NHEJ-prone sites is consistent with the reports of H3K363 promoting NHEJ (although as discussed above, some reports propose that H3K36me3 promotes HR), while induction of H4K20me1 around NHEJ-prone DSBs is consistent with elevated 53BP1 recruitment and NHEJ.
The Role of Histone Variant macroH2A1 in Double-Strand Break Repair Pathway Choice
The histone H2A variant macroH2A1 has two alternative splicing isoforms, macroH2A1.1 and macroH2A1.2, with distinct biological functions (Kim et al., 2018). MacroH2A1.1 was found to accumulate at DSBs dependent on PARP1 (Timinszky et al., 2009; Xu C. et al., 2012). In agreement, incorporation of macroH2A1 into chromatin was significantly increased at AsiSI induced DSBs (Clouaire et al., 2018). Interestingly, either overexpression or depletion of macroH2A1.1 leads to impaired NHEJ repair due to a reduction in the recruitment of the Ku70/Ku80 heterodimer or 53BP1, respectively (Timinszky et al., 2009; Xu C. et al., 2012). MacroH2A1.1 was shown to cooperate with PARP-1 to stimulate H2BK120 acetylation, which is consistent with the ubiquitylation to acetylation transition on H2BK120 after DSB repair (Chen et al., 2014; Clouaire et al., 2018). Recently, macroH2A1.1 was shown to promote DSB repair by micro homology mediated end joining (MMEJ), also termed alternative end joining, in mice (Sebastian et al., 2020). Likewise, the macroH2A1.2 variant was also recruited to DSBs, mediated by ATM, and recruits BRCA1 to facilitate HR repair (Khurana et al., 2014). Taken together, both macroH2A1 variants play important roles in regulating DSB repair pathway choice. In this study (Clouaire et al., 2018), whether macroH2A1, macroH2A1.1 or macroH2A1.2, or both were increased at the vicinity of DSB sites is unclear since a macroH2A1 antibody was used to conduct the chromatin immunoprecipitation assay. In the future, it is worth differentiating the changes of levels of macroH2A1.1 and macroH2A1.2 at DSBs, as it appears likely to control the balance of DSB repair pathway choice given they promote NHEJ and HR, respectively.
Double-Strand Break Induced Novel Histone Post-Translational Modifications Regulate Repair
Novel histone PTMs have also been discovered at DSB sites that regulate repair. UFM1 specific ligase 1 (UFL1), an ufmylation E3 ligase, was identified to be recruited to DSB sites by the MRN complex (Qin et al., 2019). The recruitment of UFL1 and ufmylation on MRE11 is important for ATM activation, while serine 462 on UFL1 is phosphorylated by ATM to enhance its E3 ligase activity (Qin et al., 2019; Wang et al., 2019). UFL1 also monoufmylates histone H4 at lysine 31 (H4K31uf), which is recognized by the serine/threonine kinase 38 (STK38) to recruit SUV39H1, through the HP1/KAP-1 complex, leading to an increase in the heterochromatin mark H3K9me3 (Qin et al., 2020) (Figure 2B). Moreover, the zinc-finger domain containing proteins, ZMYM2 and ZMYM3, were identified as antagonizers of 53BP1 recruitment to facilitate HR factor recruitment at DSBs (Lee et al., 2022). Recruitment of ZMYM2 to DSB sites requires the SUMO E3 ligase PIAS4 and SUMO binding activity of ZMYM2 (Lee et al., 2022), suggesting that either sumoylated HR proteins or histones around the DSBs may mediate DSB repair pathway choice. Indeed, H2A.X is sumoylated by PIAS4 at DSBs (Chen et al., 2013). Also, sumoylation was identified, in vivo and in vitro, on histone H4 but it is unknown if it is stimulated by DNA damage and how it regulates repair (Shiio and Eisenman, 2003). It is likely that other new types of histone PMTs will be found that regulate DSB repair pathway choice in the future.
Double-Strand Break Repair Pathway Choice in Heterochromatin Is Coordinated by Chromatin Decompaction and Histone PTMs
H3K9me3 Promotes Homologous Recombination Repair While H3K27me3 Promotes Non-Homologous End Joining Within Heterochromatin
H3K9me3 promotes HR repair and reduces NHEJ, seemingly by a combination of mechanisms. H3K9me3 is enriched at heterochromatin regions, but also increases at DNA damage sites to form transient repressive chromatin in euchromatin (Ayrapetov et al., 2014; Tsouroula et al., 2016). Several H3K9me3 writer proteins, SUV39H1/2, and SETDB1, and reader proteins, HP1, and TIP60, were shown to promote HR repair (Alagoz et al., 2015; Jacquet et al., 2016). Furthermore, the interaction between H3K9me3 and Tip60 at DSB sites, together with phosphorylation of Tip60, activates the acetylase activity of Tip60, which in turn acetylates histone H4K16 to attenuate 53BP1 binding to chromatin (Sun et al., 2005; Sun et al., 2009) (as discussed above). MBTD1, a stable subunit of the TIP60/NuA4 complex that acetylates H4K16, competes with 53BP1 for binding to H4K20me1/2. Finally, TIP60 acetylates H2AK15 and blocks its ubiquitylation by RNF168, which leads to blocking 53BP1 binding to the chromatin and promoting HR (Tang et al., 2013; Jacquet et al., 2016) (Figure 2B). In this manner, upon DSB induction, H3K9me3 promotes H4K16ac and H2AK15ac which reduce 53BP1 binding to limit NHEJ in constitutive heterochromatin.
In facultative heterochromatin, the repressive histone mark H3K27me3 is increased after DSB induction (Abu-Zhayia et al., 2018). This occurs by the DSB induced recruitment of the methyltransferase enhancer of zeste 2 (EZH2) by chromodomain Y-like (CDYL1) to catalyze methylation on H3K27 (Abu-Zhayia et al., 2018). Conversely, DSBs induced by CRISPR-Cas9 guided cutting in H3K27me3 enriched chromatin undergo MMEJ while inhibition of EZH2 leads to NHEJ repair (Sallmyr and Tomkinson, 2018; Schep et al., 2021). Moreover, another heterochromatin enriched histone PTM H4K20me3 is induced by irradiation in a manner dependent on SUV39H1/H2, which may promote 53BP1 recruitment (Svobodová Kovaříková et al., 2018).
There also exist mechanisms to limit HR within heterochromatin, including mechanisms to move the DSBs to the edge of the heterochromatin domain. Heterochromatin is enriched for highly repetitive sequences, where HR repair may lead to mutagenic recombination (Janssen et al., 2018). Drosophila lysine demethylase 4a (dKDM4a) is recruited to DSBs in pericentromeric heterochromatin, but not in euchromatin, and promotes the demethylation of heterochromatic histone marks, H3K9me3 and H3K56me3, to limit HR in heterochromatin domains (Janssen et al., 2019). This may promote the chromatin decompaction that is required for efficient DSB repair at heterochromatic regions. In Drosophila, DSBs in heterochromatin are relocated in the nucleus, driven by F-actin and myosins, to the periphery of the heterochromatin domains and then move to the nuclear pore for repair (Chiolo et al., 2011; Ryu et al., 2015; Caridi et al., 2018). In mammalian cells, relocation of heterochromatic DSBs to the periphery of heterochromatin was also observed, indicating that this movement of heterochromatic DSBs is conserved (Jakob et al., 2011; Tsouroula et al., 2016). The purpose for this relocation of DSBs within repeated sequence to the periphery of heterochromatin enables RAD51 association, because RAD51 is recruited only after relocation of the DSB out of the heterochromatin domain to promote HR repair. The mechanism whereby RAD51 is prevented from binding to DSBs within repeated sequences while they are located within the heterochromatin domain is mediated by the Smc5/6 complex, presumably to prevent inaccurate recombination which will occur if the broken DNA repeat is repaired within the repeat-rich environment of heterochromatin domains (Chiolo et al., 2011).
H3K4me2 and CENP-A Facilitate Double-Strand Break Repair by Homologous Recombination at Centromeres in G1 Phase Cells
Intriguingly, DSB repair at centromeres and peri-centromeres, though both defined as constitutive heterochromatin, is strikingly different. At pericentromeric heterochromatin, DSBs induced by CRISPR-Cas9 in G1 are not relocated out of the heterochromatin domain and recruit NHEJ proteins, while in G2 they relocate to the periphery of heterochromatin for HR repair (Tsouroula et al., 2016). However, DSBs at centromeric regions recruit both NHEJ and HR factors throughout the cell cycle (Tsouroula et al., 2016). It is possible that these differences are related to the fact that peri-centromeres are enriched for the repressive PTM H3K9me3 and its HP1 reader, while centromeres include CENP-A and active chromatin PTMs such H3K4me2, H3K36 methylation and H3 acetylation, but are depleted of H3K9me3 (Chan and Wong, 2012; Bloom, 2014).
HR repair requires the presence of sister chromatids and is usually suppressed in the G1 phase of the cell cycle. Interestingly, the HR machinery was recently reported to also be recruited to DSBs at centromeres in G1 phase even in the absence of sister chromatids (Yilmaz et al., 2021). The active histone mark H3K4me2 at centromeres promotes non-coding RNA transcription. This was shown by the depletion of the histone methyltransferase SETD1A or tethering of the H3K4me2 demethylase LSD1 to centromeres leading to a substantial decrease in transcription of centromeric non-coding RNAs (Yilmaz et al., 2021). Interestingly, DSB breaks at centromeres, but not peri-centromeres, in G1 phase increased centromeric transcription and concomitantly the H3K4me2 level, which in turn facilitated RNA-DNA hybrid formation (Aguilera and Gómez-González, 2017; Yilmaz et al., 2021). The RNA-DNA hybrids are required for the recruitment of HR components including BRCA1, RPA, and RAD51 to promote end resection and HR repair in G1. In addition to promoting transcription at centromeres, H3K4me2 was also shown to be required for HJURP (the histone H3 variant CENP-A chaperone)-mediated CENP-A assembly onto an epigenetically engineered human kinetochore (Bergmann et al., 2011). Depletion of the centromeric histone H3 variant CENP-A, its chaperone HJURP or the cofactor MIS18 leads to a significant decrease in recruitment of RAD51 at DSBs in centromeres (Yilmaz et al., 2021). This suggests that CENPA is important for HR, via RAD51 recruitment, to DSBs within the centromere in G1 cells. Indeed, dCas9-mediated tethering of CENP-A or HJURP at pericentromeric DSBs increased the recruitment of RAD51 in G1, suggesting that CENP-A and HJURP are sufficient to recruit RAD51 (Yilmaz et al., 2021). Ubiquitylation on PALB2 in G1 phase suppresses its interaction with BRCA1 and thus RAD51 recruitment, which is counteracted by the deubiquitylase USP11 (Orthwein et al., 2015). USP11 is rapidly turned over in G1, especially upon DSB induction (Orthwein et al., 2015). Interestingly, USP11 was found to be specifically recruited to centromeric, but not peri-centromeric, DSBs in G1 and interacts with both CENP-A and HJURP to promote RAD51 recruitment (Yilmaz et al., 2021). Furthermore, USP11 deubiquitylates HJURP which is normally ubiquitylated to facilitate CENP-A incorporation at centromeric chromatin (Yilmaz et al., 2021), which thereby reinforces HR repair at centromeres in G1. Indeed, depletion of USP11 leads to impairment in RAD51 recruitment and HR repair at centromeres in G1 (Yilmaz et al., 2021). Mechanistically, the presence of H3K4me2 and HJURP-CENP-A at centromeres promotes HR in G1 phase (Yilmaz et al., 2021). In summary, there are complex mechanisms to regulate DSB pathway choice in centromeric and peri-centromeric heterochromatin (Figures 2B,C).
Double-Strand Break Induced RNA Transcripts Play Pivotal Roles in Double-Strand Break Repair Pathway Choice
Non-Coding RNAs: diRNAs/DDRNAs and dilncRNAs in Double-Strand Break Repair Pathway Choice
A growing body of evidence shows that local DSB induced non-coding transcripts play important roles in HR (Durut and Mittelsten Scheid, 2019; Ohsawa et al., 2013). Small RNAs derived from the vicinity of DSBs, named DSB-induced RNAs (diRNAs) or DICER- and DROSHA-dependent small RNAs (DDRNAs), have been identified in both plants and vertebrates (Francia et al., 2012; Wei et al., 2012). DiRNAs promote HR repair in a manner dependent on the effector protein Argonaute 2 (Ago2) that is required for RAD51 recruitment (Gao et al., 2014). The production of diRNAs/DDRNAs indicates activation of non-coding transcription surrounding DSBs. Indeed, RNA polymerase II (RNAPII) is recruited to DSB sites, via its binding to the MRN complex, and generates damage-induced long non-coding RNAs (dilncRNAs). Most of the dilncRNAs are transcribed in the direction away from the DSB while less are transcribed towards the DSB to generate, which act not only as precursors to DDRNAs but also scaffolds for DDRNAs recruitment to the dilncRNA-from molecules (Michelini et al., 2017) (Figure 3). On the other hand, dilncRNAs and DDRNAs facilitate DDR focus formation and interact with 53BP1 (Michelini et al., 2017), suggesting a role of DDRNAs in promoting NHEJ repair (Figure 3).
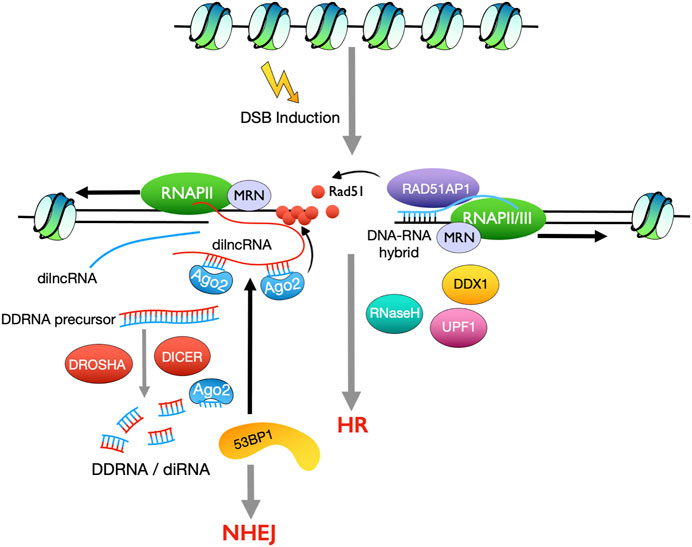
FIGURE 3. Summary of how non-coding RNAs induced by DSBs, promote DSB repair by NHEJ and HR. Small non-coding RNAs (diRNAs/DDRNAs) and long non-coding RNAs (dilncRNAs) generated from the vicinity of DSB sites promote 53BP1 recruitment and thus NHEJ repair (left). RNA-DNA hybrids, produced by RNAPII or RNAPIII around DSBs, promote DSB repair by HR.
RNA-DNA Hybrids Promote Double-Strand Break Repair by Homologous Recombination
In fission yeast, RNAPII was shown to be rapidly recruited to a site-specific DSB induced by the endonuclease I-PpoI (Ohle et al., 2016). Remarkably, a robust increase of RNAPII around the DSB was detected, but augmentation of RNA transcripts was not observed by RT-qPCR at this region, which suggests the nascent transcripts immediately form RNA-DNA hybrids with the DNA template and are stabilized in these structures. Overexpression of RNaseH, rnh1, impairs DSB repair efficiency, indicating that the RNA-DNA hybrids act as a functional intermediate during HR repair (Ohle et al., 2016). Intriguingly, depletion of both RNaseH proteins, rnh1 and rnh201, leads to the accumulation of RNA-DNA hybrids and inhibits HR-mediated DSB repair through impairing RPA recruitment to the ssDNA strand (Ohle et al., 2016). Therefore, the RNA-DNA hybrid intermediates must be tightly regulated, spatially and temporally, during HR repair. Furthermore, formation of RNA-DNA hybrids was observed at DSB sites in mammalian cells (Li et al., 2016). Consistently, depletion of the RNA-unwinding protein DEAD box 1 (DDX1) elevates the RNA-DNA hybrid levels and decreases RAD51 focus formation at DSBs, resulting in impaired DSB repair by HR (Li et al., 2016) (Figure 3).
It remains unclear how these RNA-DNA hybrids are formed and regulate DSB repair mediated by HR. Recently, RNA polymerase III (RNAPIII) was characterized as an essential factor in HR repair (Liu et al., 2021). RNAPIII is recruited to DSBs by the MRN complex and is required for the synthesis of the RNA strand, which protects the 3’ ssDNA overhang from degradation through the formation of RNA-DNA hybrids (Liu et al., 2021). Consistently, inhibition of RNA-DNA hybrid formation, by either knockdown of RNAPIII subunits or chemical inhibitors, significantly impairs end resection and focus formation of RPA and RAD51, which leads to defects in HR repair and thus loss of genetic material at DSBs (Liu et al., 2021). Interestingly, the formation of RNAPIII RNA-DNA hybrids requires the nuclease activity of CtIP and MRN and is restricted to S and G2/M phases (Liu et al., 2021), which is consistent with their roles in promoting HR repair. In this study, the nascent RNA strand produced by RNAPIII is proposed to be degraded for subsequent formation of ssDNA/RAD51 nucleoprotein filaments (Liu et al., 2021), whereas a role of RPA in this model needs further interrogation. Moreover, an in vitro system, in which mRNA synthesis occurs in the absence of promoters, shows that dilncRNAs are produced by RNAPII, but not RNAPIII, to form RNA-DNA hybrids at DSB sites (Sharma et al., 2021). Interestingly, DNA end melting mediated by MRN is required for dilncRNA synthesis, while the nucleolytic activity of MRN is dispensable for RNAPII transcription at DSBs (Sharma et al., 2021). At DSB sites induced by AsiSI, DROSHA promotes the production of RNA-DNA hybrids, probably by RNAPII, to facilitate DSB repair by HR or NHEJ (Castel et al., 2014; Lu et al., 2018). Paradoxically, no diRNAs/DDRNAs were detected using equal sequencing coverage and depth as seen in previous studies at DSB sites (Francia et al., 2012; Wei et al., 2012; Lu et al., 2018), which might be due to the different DSB induction systems that were utilized or the transient nature of these RNA species.
During HR repair, a ssDNA/Rad51 nucleoprotein filament invades the donor dsDNA homolog and forms a heteroduplex DNA joint called the D-loop (Petukhova et al., 2000; Van Komen et al., 2000). When the transcription machinery collides with the machinery mediating other physiological processes, such as DNA replication or DNA repair, the nascent RNA transcript forms hybrids with the template DNA and displaces the non-template ssDNA in the double helix, which generates a three-stranded structure called an R-loop (Rinaldi et al., 2020). Unscheduled R-loop formation is taken as a threat to genome stability, which has been thoroughly reviewed recently (Rinaldi et al., 2020). Some light has been shed on the mechanisms of how transcription enhances DNA end resection and thus HR repair via formation of D-loop and R-loop hybrids (DR-loops) (Ngo et al., 2021; Ouyang et al., 2021). In the study by Ouyang et al. (2021) the constitutive promoter of sceGFP in the direct-repeat GFP (DR-GFP) reporter was replaced with a tetracycline-inducible (Tet-On) promoter (Ouyang et al., 2021). When sceGFP was actively transcribed, HR efficiency was markedly enhanced although HR products were still detectable when transcription was inactive (Ouyang et al., 2021). In agreement, with Cas9-mediated DSB induction and transcriptional activation of a neuronal-specific gene, ASCL1, HR products were dramatically increased when transcription was active. Further, RNA transcripts were shown to stimulate HR by annealing with DNA mediated by the RAD51-associated protein RAD51AP1 (Ouyang et al., 2021). RAD51AP1 accumulates at DSB sites and promotes R-loop formation, which forms DR-loops together with D-loops generated by RAD51 strand invasion, to facilitate HR (Ouyang et al., 2021). At sub-telomeric DSB sites, the formation of R-loops and RNA-DNA hybrids, driven by an RNA/DNA helicase UPF1, stimulated end resection and thus HR (Ngo et al., 2021). R-loops and RNA-DNA hybrids are clearly generated via different mechanisms because the generation of R-loops is independent of DNA end resection but is induced at RNA-DNA hybrids that forms on ssDNA generated by resection (Ngo et al., 2021). In centromeres, RNA-DNA hybrids were also found to promote HR repair at DSBs in G1 phase (Yilmaz et al., 2021). These studies raise many interesting questions, for example how is the resolution of RNA-DNA hybrids regulated after DNA end resection? How are RPA and RNA-DNA hybrids coordinated to protect the 3’ ssDNAs and facilitate RAD51 deposition during HR repair? Do the RNA-DNA hybrids play a role in inhibiting NHEJ?
Conclusion and Perspectives
In the last two decades, our knowledge of how epigenetic modifications regulate DSB repair, and DSB repair pathway choice has grown exponentially. Our knowledge of the interplay between the pro-NHEJ factor 53BP1 and pro-HR heterodimer BRCA1-BARD1 has increased extensively. The recruitment of 53BP1-RIF1-Shieldin to DSBs plays a critical role in promoting NHEJ repair. We now have an improved appreciation of how pre-existing and DSB-induced histone PTMs play pivotal roles in regulating DSB repair pathway choice in different phases of the cell cycle: Upon DSB induction, RNF168 is recruited to DSB sites to monoubiquitylate H2AK15, which is a prerequisite for both 53BP1 and BRCA1-BARD1 recruitment. In G1 phase, the pre-existing H4K20me1/2 on chromatin is recognized by 53BP1 to promote DSB repair by NHEJ. However, H4K20 is unmethylated on the post-replicative chromatin in S/G2 phase, which enhances the recruitment of BRCA1-BARD1 to facilitate HR repair. We have learned that in euchromatin and heterochromatin regions histone PTMs that promote transcriptional activation such as H3K36me and H3K4me2 and those that promote transcriptional repression such as H3K9me3, both promote DSB repair by HR through interactions with distinct factors. Intriguingly, we now appreciate that DSB-induced RNAs are involved in DSB repair regulation. The nascent RNA transcripts can also form R-loops with DNA ends at the breaks to promote HR repair.
It remains largely unknown how the intricate chromatin modification network, namely the chromatin landscape, is orchestrated to regulate DSB repair pathway choices. More studies are required to address whether other histone modifications, such as crotonylation and glycosylation, also contribute to DSB repair pathway choice. In addition, it is tempting to speculate whether DSB-induced small RNAs, diRNAs/DDRNAs, and dilincRNAs can stimulate heritable epigenetic changes on DNA molecules, such as DNA methylation, in the chromatin landscape surrounding the DSB sites, which may act as an “epigenetic memory” to prime the repaired region for more efficient repair if DSBs occur at the same sites. Indeed, DSBs do initiate the transient recruitment of the DNA methyltransferases DNMT1 and DNMT3B, in a SIRT1 dependent manner, and subsequent methylation at promoter regions (O'Hagan et al., 2008).
Why do the histone PTMs involved in DSB repair pathway choice, regardless of whether they occur in euchromatin or heterochromatin regions, tend to promote HR repair? Does this indicate that NHEJ repair is the default mechanism of DSB repair throughout the cell cycle and specific histone modifications are required to overcome the NHEJ default to promote HR? Indeed, NHEJ was found, by live imaging and fluorescent labeling of components in HR and NHEJ repair pathways, to be the dominant repair pathway not only in G1 but also in G2 phase where both HR and NHEJ repair pathways are functional (Karanam et al., 2012). Why then do cells choose a relatively more error-prone pathway as the main DSB repair mechanism? Compared to HR repair, NHEJ requires less processing at broken DNA ends such that it is more rapid and efficient, which helps to protect our genome from deleterious DSB-induced translocations or chromosome loss. Moreover, only ∼2% of our genome is protein-coding and the rest comprises mostly non-coding regions or regulatory elements such as introns, promoters, and enhancers. Even if nucleotide sequence changes are introduced at these non-coding regions by NHEJ, it is less likely to lead to mutation in genes or more detrimental consequences. On the other hand, it is interesting to envision why HR repair over NHEJ is used at centromeres in G1 phase without the presence of sister chromatids. Utilization of HR repair at repetitive sequences is thought to be detrimental to genome stability as it usually leads to repeat contraction and expansion (Khristich and Mirkin, 2020). However, Yilmaz et al. (2021) proposed activation of HR at centromeres in G1 counteracts the engagement of alternative mutagenic repair pathways, resulting in the prevention of chromosomal abnormalities (Yilmaz et al., 2021).
Interestingly, the highest occurrence of HR is at the peak of active replication with the longest half-lives of DSBs in mid S phase (Karanam et al., 2012), where sister chromatids are widely accessible due to the transient absence of histones on DNA. The proportion and rates of active HR vary widely at the DSBs among individual cells even at the same cell cycle phase (Karanam et al., 2012), which suggests that the choice between HR and NHEJ repair pathways could possibly be channeled by the unique chromatin landscapes surrounding the DSB sites in individual cells. Clearly the situation is very complex and hopefully future studies will provide a better understanding of the intriguing regulation of DSB repair processes.
Author Contributions
ZC wrote the first draft of the review and conceived the figures. JT edited the review and made the final figures.
Funding
This work was supported by JT from NIH R35 GM139816 and CA9564.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abu-Zhayia, E. R., Awwad, S. W., Ben-Oz, B. M., Khoury-Haddad, H., and Ayoub, N. (2018). CDYL1 Fosters Double-Strand Break-Induced Transcription Silencing and Promotes Homology-Directed Repair. J. Mol. Cell Biol. 10, 341–357. doi:10.1093/jmcb/mjx050
Acs, K., Luijsterburg, M. S., Ackermann, L., Salomons, F. A., Hoppe, T., and Dantuma, N. P. (2011). The AAA-ATPase VCP/p97 Promotes 53BP1 Recruitment by Removing L3MBTL1 from DNA Double-Strand Breaks. Nat. Struct. Mol. Biol. 18, 1345–1350. doi:10.1038/nsmb.2188
Adkins, N. L., Swygert, S. G., Kaur, P., Niu, H., Grigoryev, S. A., Sung, P., et al. (2017). Nucleosome-like, Single-Stranded DNA (ssDNA)-Histone Octamer Complexes and the Implication for DNA Double Strand Break Repair. J. Biol. Chem. 292, 5271–5281. doi:10.1074/jbc.M117.776369
Agger, K., Cloos, P. A. C., Christensen, J., Pasini, D., Rose, S., Rappsilber, J., et al. (2007). UTX and JMJD3 Are Histone H3K27 Demethylases Involved in HOX Gene Regulation and Development. Nature 449, 731–734. doi:10.1038/nature06145
Aguilera, A., and Gómez-González, B. (2017). DNA-RNA Hybrids: the Risks of DNA Breakage during Transcription. Nat. Struct. Mol. Biol. 24, 439–443. doi:10.1038/nsmb.3395
Alabert, C., Barth, T. K., Reverón-Gómez, N., Sidoli, S., Schmidt, A., Jensen, O. N., et al. (2015). Two Distinct Modes for Propagation of Histone PTMs across the Cell Cycle. Genes Dev. 29, 585–590. doi:10.1101/gad.256354.114
Alabert, C., Bukowski-Wills, J.-C., Lee, S.-B., Kustatscher, G., Nakamura, K., de Lima Alves, F., et al. (2014). Nascent Chromatin Capture Proteomics Determines Chromatin Dynamics during DNA Replication and Identifies Unknown Fork Components. Nat. Cell Biol. 16, 281–291. doi:10.1038/ncb2918
Alagoz, M., Katsuki, Y., Ogiwara, H., Ogi, T., Shibata, A., Kakarougkas, A., et al. (2015). SETDB1, HP1 and SUV39 Promote Repositioning of 53BP1 to Extend Resection during Homologous Recombination in G2 Cells. Nucleic Acids Res. 43, 7931–7944. doi:10.1093/nar/gkv722
Anderson, L., Henderson, C., and Adachi, Y. (2001). Phosphorylation and Rapid Relocalization of 53BP1 to Nuclear Foci upon DNA Damage. Mol. Cell Biol. 21, 1719–1729. doi:10.1128/MCB.21.5.1719-1729.2001
Arunkumar, G., and Melters, D. P. (2020). Centromeric Transcription: A Conserved Swiss-Army Knife. Genes 11, 911. doi:10.3390/genes11080911
Aymard, F., Bugler, B., Schmidt, C. K., Guillou, E., Caron, P., Briois, S., et al. (2014). Transcriptionally Active Chromatin Recruits Homologous Recombination at DNA Double-Strand Breaks. Nat. Struct. Mol. Biol. 21, 366–374. doi:10.1038/nsmb.2796
Ayrapetov, M. K., Gursoy-Yuzugullu, O., Xu, C., Xu, Y., and Price, B. D. (2014). DNA Double-Strand Breaks Promote Methylation of Histone H3 on Lysine 9 and Transient Formation of Repressive Chromatin. Proc. Natl. Acad. Sci. U.S.A. 111, 9169–9174. doi:10.1073/pnas.1403565111
Bannister, A. J., and Kouzarides, T. (2011). Regulation of Chromatin by Histone Modifications. Cell Res. 21, 381–395. doi:10.1038/cr.2011.22
Beck, D. B., Oda, H., Shen, S. S., and Reinberg, D. (2012). PR-Set7 and H4K20me1: at the Crossroads of Genome Integrity, Cell Cycle, Chromosome Condensation, and Transcription. Genes Dev. 26, 325–337. doi:10.1101/gad.177444.111
Becker, J. R., Clifford, G., Bonnet, C., Groth, A., Wilson, M. D., and Chapman, J. R. (2021). BARD1 Reads H2A Lysine 15 Ubiquitination to Direct Homologous Recombination. Nature 596, 433–437. doi:10.1038/s41586-021-03776-w
Bergmann, J. H., Rodríguez, M. G., Martins, N. M. C., Kimura, H., Kelly, D. A., Masumoto, H., et al. (2011). Epigenetic Engineering Shows H3K4me2 Is Required for HJURP Targeting and CENP-A Assembly on a Synthetic Human Kinetochore. EMBO J. 30, 328–340. doi:10.1038/emboj.2010.329
Blackford, A. N., and Jackson, S. P. (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 66, 801–817. doi:10.1016/j.molcel.2017.05.015
Bleuyard, J.-Y., Fournier, M., Nakato, R., Couturier, A. M., Katou, Y., Ralf, C., et al. (2017). MRG15-mediated Tethering of PALB2 to Unperturbed Chromatin Protects Active Genes from Genotoxic Stress. Proc. Natl. Acad. Sci. U.S.A. 114, 7671–7676. doi:10.1073/pnas.1620208114
Bloom, K. S. (2014). Centromeric Heterochromatin: the Primordial Segregation Machine. Annu. Rev. Genet. 48, 457–484. doi:10.1146/annurev-genet-120213-092033
Botuyan, M. V., Lee, J., Ward, I. M., Kim, J.-E., Thompson, J. R., Chen, J., et al. (2006). Structural Basis for the Methylation State-specific Recognition of Histone H4-K20 by 53BP1 and Crb2 in DNA Repair. Cell 127, 1361–1373. doi:10.1016/j.cell.2006.10.043
Bouwman, P., Aly, A., Escandell, J. M., Pieterse, M., Bartkova, J., van der Gulden, H., et al. (2010). 53BP1 Loss Rescues BRCA1 Deficiency and Is Associated with Triple-Negative and BRCA-Mutated Breast Cancers. Nat. Struct. Mol. Biol. 17, 688–695. doi:10.1038/nsmb.1831
Britton, S., Coates, J., and Jackson, S. P. (2013). A New Method for High-Resolution Imaging of Ku Foci to Decipher Mechanisms of DNA Double-Strand Break Repair. J. Cell Biol. 202, 579–595. doi:10.1083/jcb.201303073
Bunting, S. F., Callén, E., Wong, N., Chen, H.-T., Polato, F., Gunn, A., et al. (2010). 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 141, 243–254. doi:10.1016/j.cell.2010.03.012
Cao, L., Xu, X., Bunting, S. F., Liu, J., Wang, R.-H., Cao, L. L., et al. (2009). A Selective Requirement for 53BP1 in the Biological Response to Genomic Instability Induced by Brca1 Deficiency. Mol. Cell 35, 534–541. doi:10.1016/j.molcel.2009.06.037
Cao, X., Chen, Y., Wu, B., Wang, X., Xue, H., Yu, L., et al. (2020). Histone H4K20 Demethylation by Two hHR23 Proteins. Cell Rep. 30, 4152–4164. e4156. doi:10.1016/j.celrep.2020.03.001
Caridi, C. P., D’Agostino, C., Ryu, T., Zapotoczny, G., Delabaere, L., Li, X., et al. (2018). Nuclear F-Actin and Myosins Drive Relocalization of Heterochromatic Breaks. Nature 559, 54–60. doi:10.1038/s41586-018-0242-8
Carvalho, S., Vítor, A. C., Sridhara, S. C., Martins, F. B., Raposo, A. C., Desterro, J. M., et al. (2014). SETD2 Is Required for DNA Double-Strand Break Repair and Activation of the P53-Mediated Checkpoint. Elife 3, e02482. doi:10.7554/eLife.02482
Castel, S. E., Ren, J., Bhattacharjee, S., Chang, A.-Y., Sánchez, M., Valbuena, A., et al. (2014). Dicer Promotes Transcription Termination at Sites of Replication Stress to Maintain Genome Stability. Cell 159, 572–583. doi:10.1016/j.cell.2014.09.031
Celeste, A., Fernandez-Capetillo, O., Kruhlak, M. J., Pilch, D. R., Staudt, D. W., Lee, A., et al. (2003). Histone H2AX Phosphorylation Is Dispensable for the Initial Recognition of DNA Breaks. Nat. Cell Biol. 5, 675–679. doi:10.1038/ncb1004
Chan, F. L., and Wong, L. H. (2012). Transcription in the Maintenance of Centromere Chromatin Identity. Nucleic Acids Res. 40, 11178–11188. doi:10.1093/nar/gks921
Chapman, J. R., Barral, P., Vannier, J. B., Borel, V., Steger, M., Tomas-Loba, A., et al. (2013). RIF1 Is Essential for 53BP1-dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol. Cell 49, 858–871. doi:10.1016/j.molcel.2013.01.002
Chapman, J. R., Sossick, A. J., Boulton, S. J., and Jackson, S. P. (2012a). BRCA1-associated Exclusion of 53BP1 from DNA Damage Sites Underlies Temporal Control of DNA Repair. J. Cell Sci. 125, 3529–3534. doi:10.1242/jcs.105353
Chapman, J. R., Taylor, M. R. G., and Boulton, S. J. (2012b). Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 47, 497–510. doi:10.1016/j.molcel.2012.07.029
Chen, C.-C., Feng, W., Lim, P. X., Kass, E. M., and Jasin, M. (2018). Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2, 313–336. doi:10.1146/annurev-cancerbio-030617-050502
Chen, H., Ruiz, P. D., Novikov, L., Casill, A. D., Park, J. W., and Gamble, M. J. (2014). MacroH2A1.1 and PARP-1 Cooperate to Regulate Transcription by Promoting CBP-Mediated H2B Acetylation. Nat. Struct. Mol. Biol. 21, 981–989. doi:10.1038/nsmb.2903
Chen, W. T., Alpert, A., Leiter, C., Gong, F., Jackson, S. P., and Miller, K. M. (2013). Systematic Identification of Functional Residues in Mammalian Histone H2AX. Mol. Cell Biol. 33, 111–126. doi:10.1128/MCB.01024-12
Chen, X., Cui, D., Papusha, A., Zhang, X., Chu, C.-D., Tang, J., et al. (2012). The Fun30 Nucleosome Remodeller Promotes Resection of DNA Double-Strand Break Ends. Nature 489, 576–580. doi:10.1038/nature11355
Chiolo, I., Minoda, A., Colmenares, S. U., Polyzos, A., Costes, S. V., and Karpen, G. H. (2011). Double-strand Breaks in Heterochromatin Move outside of a Dynamic HP1a Domain to Complete Recombinational Repair. Cell 144, 732–744. doi:10.1016/j.cell.2011.02.012
Cloos, P. A. C., Christensen, J., Agger, K., Maiolica, A., Rappsilber, J., Antal, T., et al. (2006). The Putative Oncogene GASC1 Demethylates Tri- and Dimethylated Lysine 9 on Histone H3. Nature 442, 307–311. doi:10.1038/nature04837
Clouaire, T., Rocher, V., Lashgari, A., Arnould, C., Aguirrebengoa, M., Biernacka, A., et al. (2018). Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 72, 250–262. e256. doi:10.1016/j.molcel.2018.08.020
Coleman, K. A., and Greenberg, R. A. (2011). The BRCA1-RAP80 Complex Regulates DNA Repair Mechanism Utilization by Restricting End Resection. J. Biol. Chem. 286, 13669–13680. doi:10.1074/jbc.M110.213728
Costelloe, T., Louge, R., Tomimatsu, N., Mukherjee, B., Martini, E., Khadaroo, B., et al. (2012). The Yeast Fun30 and Human SMARCAD1 Chromatin Remodellers Promote DNA End Resection. Nature 489, 581–584. doi:10.1038/nature11353
Dabin, J., Fortuny, A., and Polo, S. E. (2016). Epigenome Maintenance in Response to DNA Damage. Mol. Cell 62, 712–727. doi:10.1016/j.molcel.2016.04.006
Dai, L., Dai, Y., Han, J., Huang, Y., Wang, L., Huang, J., et al. (2021). Structural Insight into BRCA1-BARD1 Complex Recruitment to Damaged Chromatin. Mol. Cell 81, 2765–2777. e2766. doi:10.1016/j.molcel.2021.05.010
Daugaard, M., Baude, A., Fugger, K., Povlsen, L. K., Beck, H., Sørensen, C. S., et al. (2012). LEDGF (P75) Promotes DNA-End Resection and Homologous Recombination. Nat. Struct. Mol. Biol. 19, 803–810. doi:10.1038/nsmb.2314
De Santa, F., Totaro, M. G., Prosperini, E., Notarbartolo, S., Testa, G., and Natoli, G. (2007). The Histone H3 Lysine-27 Demethylase Jmjd3 Links Inflammation to Inhibition of Polycomb-Mediated Gene Silencing. Cell 130, 1083–1094. doi:10.1016/j.cell.2007.08.019
Densham, R. M., Garvin, A. J., Stone, H. R., Strachan, J., Baldock, R. A., Daza-Martin, M., et al. (2016). Human BRCA1-BARD1 Ubiquitin Ligase Activity Counteracts Chromatin Barriers to DNA Resection. Nat. Struct. Mol. Biol. 23, 647–655. doi:10.1038/nsmb.3236
Densham, R. M., and Morris, J. R. (2019). Moving Mountains-The BRCA1 Promotion of DNA Resection. Front. Mol. Biosci. 6, 79. doi:10.3389/fmolb.2019.00079
Densham, R. M., and Morris, J. R. (2017). The BRCA1 Ubiquitin Ligase Function Sets a New Trend for Remodelling in DNA Repair. Nucleus 8, 116–125. doi:10.1080/19491034.2016.1267092
Dev, H., Chiang, T.-W. W., Lescale, C., de Krijger, I., Martin, A. G., Pilger, D., et al. (2018). Shieldin Complex Promotes DNA End-Joining and Counters Homologous Recombination in BRCA1-Null Cells. Nat. Cell Biol. 20, 954–965. doi:10.1038/s41556-018-0140-1
Di Virgilio, M., Callen, E., Yamane, A., Zhang, W., Jankovic, M., Gitlin, A. D., et al. (2013). Rif1 Prevents Resection of DNA Breaks and Promotes Immunoglobulin Class Switching. Science 339, 711–715. doi:10.1126/science.1230624
Doil, C., Mailand, N., Bekker-Jensen, S., Menard, P., Larsen, D. H., Pepperkok, R., et al. (2009). RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 136, 435–446. doi:10.1016/j.cell.2008.12.041
Du, L.-L., Nakamura, T. M., and Russell, P. (2006). Histone Modification-dependent and -independent Pathways for Recruitment of Checkpoint Protein Crb2 to Double-Strand Breaks. Genes Dev. 20, 1583–1596. doi:10.1101/gad.1422606
Durut, N., and Mittelsten Scheid, O. (2019). The Role of Noncoding RNAs in Double-Strand Break Repair. Front. Plant Sci. 10, 1155. doi:10.3389/fpls.2019.01155
Eapen, V. V., Sugawara, N., Tsabar, M., Wu, W.-H., and Haber, J. E. (2012). The Saccharomyces cerevisiae Chromatin Remodeler Fun30 Regulates DNA End Resection and Checkpoint Deactivation. Mol. Cell Biol. 32, 4727–4740. doi:10.1128/MCB.00566-12
Elbakry, A., and Löbrich, M. (2021). Homologous Recombination Subpathways: A Tangle to Resolve. Front. Genet. 12, 723847. doi:10.3389/fgene.2021.723847
Escribano-Díaz, C., Orthwein, A., Fradet-Turcotte, A., Xing, M., Young, J. T. F., Tkáč, J., et al. (2013). A Cell Cycle-dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 49, 872–883. doi:10.1016/j.molcel.2013.01.001
Fang, J., Feng, Q., Ketel, C. S., Wang, H., Cao, R., Xia, L., et al. (2002). Purification and Functional Characterization of SET8, a Nucleosomal Histone H4-Lysine 20-specific Methyltransferase. Curr. Biol. 12, 1086–1099. doi:10.1016/s0960-9822(02)00924-7
Feil, R., and Berger, F. (2007). Convergent Evolution of Genomic Imprinting in Plants and Mammals. Trends Genet. 23, 192–199. doi:10.1016/j.tig.2007.02.004
Feng, L., Fong, K.-W., Wang, J., Wang, W., and Chen, J. (2013). RIF1 Counteracts BRCA1-Mediated End Resection during DNA Repair. J. Biol. Chem. 288, 11135–11143. doi:10.1074/jbc.M113.457440
Feng, L., Li, N., Li, Y., Wang, J., Gao, M., Wang, W., et al. (2015). Cell Cycle-dependent Inhibition of 53BP1 Signaling by BRCA1. Cell Discov. 1, 15019. doi:10.1038/celldisc.2015.19
Findlay, S., Heath, J., Luo, V. M., Malina, A., Morin, T., Coulombe, Y., et al. (2018). SHLD 2/FAM 35A Co‐operates with REV 7 to Coordinate DNA Double‐strand Break Repair Pathway Choice. EMBO J. 37, e100158. doi:10.15252/embj.2018100158
Fnu, S., Williamson, E. A., De Haro, L. P., Brenneman, M., Wray, J., Shaheen, M., et al. (2011). Methylation of Histone H3 Lysine 36 Enhances DNA Repair by Nonhomologous End-Joining. Proc. Natl. Acad. Sci. U.S.A. 108, 540–545. doi:10.1073/pnas.1013571108
Fox, D., Le Trong, I., Rajagopal, P., Brzovic, P. S., Stenkamp, R. E., and Klevit, R. E. (2008). Crystal Structure of the BARD1 Ankyrin Repeat Domain and its Functional Consequences. J. Biol. Chem. 283, 21179–21186. doi:10.1074/jbc.M802333200
Fradet-Turcotte, A., Canny, M. D., Escribano-Díaz, C., Orthwein, A., Leung, C. C. Y., Huang, H., et al. (2013). 53BP1 Is a Reader of the DNA-Damage-Induced H2A Lys 15 Ubiquitin Mark. Nature 499, 50–54. doi:10.1038/nature12318
Francia, S., Michelini, F., Saxena, A., Tang, D., de Hoon, M., Anelli, V., et al. (2012). Site-specific DICER and DROSHA RNA Products Control the DNA-Damage Response. Nature 488, 231–235. doi:10.1038/nature11179
Gao, M., Wei, W., Li, M.-M., Wu, Y.-S., Ba, Z., Jin, K.-X., et al. (2014). Ago2 Facilitates Rad51 Recruitment and DNA Double-Strand Break Repair by Homologous Recombination. Cell Res. 24, 532–541. doi:10.1038/cr.2014.36
Gao, S., Feng, S., Ning, S., Liu, J., Zhao, H., Xu, Y., et al. (2018). An OB-fold Complex Controls the Repair Pathways for DNA Double-Strand Breaks. Nat. Commun. 9, 3925. doi:10.1038/s41467-018-06407-7
Gatti, M., Pinato, S., Maspero, E., Soffientini, P., Polo, S., and Penengo, L. (2012). A Novel Ubiquitin Mark at the N-Terminal Tail of Histone H2As Targeted by RNF168 Ubiquitin Ligase. Cell Cycle 11, 2538–2544. doi:10.4161/cc.20919
Ge, H., Si, Y., and Roeder, R. G. (1998). Isolation of cDNAs Encoding Novel Transcription Coactivators P52 and P75 Reveals an Alternate Regulatory Mechanism of Transcriptional Activation. EMBO J. 17, 6723–6729. doi:10.1093/emboj/17.22.6723
Ghezraoui, H., Oliveira, C., Becker, J. R., Bilham, K., Moralli, D., Anzilotti, C., et al. (2018). 53BP1 Cooperation with the REV7-Shieldin Complex Underpins DNA Structure-specific NHEJ. Nature 560, 122–127. doi:10.1038/s41586-018-0362-1
Gong, F., Chiu, L.-Y., Cox, B., Aymard, F., Clouaire, T., Leung, J. W., et al. (2015). Screen Identifies Bromodomain Protein ZMYND8 in Chromatin Recognition of Transcription-Associated DNA Damage that Promotes Homologous Recombination. Genes Dev. 29, 197–211. doi:10.1101/gad.252189.114
Gong, F., Clouaire, T., Aguirrebengoa, M., Legube, G., and Miller, K. M. (2017). Histone Demethylase KDM5A Regulates the ZMYND8-NuRD Chromatin Remodeler to Promote DNA Repair. J. Cell Biol. 216, 1959–1974. doi:10.1083/jcb.201611135
Gupta, R., Somyajit, K., Narita, T., Maskey, E., Stanlie, A., Kremer, M., et al. (2018). DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 173, 972–988. e923. doi:10.1016/j.cell.2018.03.050
Gursoy-Yuzugullu, O., Carman, C., and Price, B. D. (2017). Spatially Restricted Loading of BRD2 at DNA Double-Strand Breaks Protects H4 Acetylation Domains and Promotes DNA Repair. Sci. Rep. 7, 12921. doi:10.1038/s41598-017-13036-5
Højfeldt, J. W., Agger, K., and Helin, K. (2013). Histone Lysine Demethylases as Targets for Anticancer Therapy. Nat. Rev. Drug Discov. 12, 917–930. doi:10.1038/nrd4154
Hsiao, K.-Y., and Mizzen, C. A. (2013). Histone H4 Deacetylation Facilitates 53BP1 DNA Damage Signaling and Double-Strand Break Repair. J. Mol. cell Biol. 5, 157–165. doi:10.1093/jmcb/mjs066
Hu, Q., Botuyan, M. V., Cui, G., Zhao, D., and Mer, G. (2017). Mechanisms of Ubiquitin-Nucleosome Recognition and Regulation of 53BP1 Chromatin Recruitment by RNF168/169 and RAD18. Mol. Cell 66, 473–487. e479. doi:10.1016/j.molcel.2017.04.009
Hu, Q., Botuyan, M. V., Zhao, D., Cui, G., Mer, E., and Mer, G. (2021). Mechanisms of BRCA1-BARD1 Nucleosome Recognition and Ubiquitylation. Nature 596, 438–443. doi:10.1038/s41586-021-03716-8
Hu, Y., Scully, R., Sobhian, B., Xie, A., Shestakova, E., and Livingston, D. M. (2011). RAP80-directed Tuning of BRCA1 Homologous Recombination Function at Ionizing Radiation-Induced Nuclear Foci. Genes Dev. 25, 685–700. doi:10.1101/gad.2011011
Huang, Y., Fang, J., Bedford, M. T., Zhang, Y., and Xu, R.-M. (2006). Recognition of Histone H3 Lysine-4 Methylation by the Double Tudor Domain of JMJD2A. Science 312, 748–751. doi:10.1126/science.1125162
Huen, M. S. Y., Grant, R., Manke, I., Minn, K., Yu, X., Yaffe, M. B., et al. (2007). RNF8 Transduces the DNA-Damage Signal via Histone Ubiquitylation and Checkpoint Protein Assembly. Cell 131, 901–914. doi:10.1016/j.cell.2007.09.041
Hustedt, N., and Durocher, D. (2016). The Control of DNA Repair by the Cell Cycle. Nat. Cell Biol. 19, 1–9. doi:10.1038/ncb3452
Huyen, Y., Zgheib, O., DiTullio Jr, R. A., Gorgoulis, V. G., Zacharatos, P., Petty, T. J., et al. (2004). Methylated Lysine 79 of Histone H3 Targets 53BP1 to DNA Double-Strand Breaks. Nature 432, 406–411. doi:10.1038/nature03114
Hyun, K., Jeon, J., Park, K., and Kim, J. (2017). Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 49, e324. doi:10.1038/emm.2017.11
Isono, M., Niimi, A., Oike, T., Hagiwara, Y., Sato, H., Sekine, R., et al. (2017). BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell Rep. 18, 520–532. doi:10.1016/j.celrep.2016.12.042
Jackson, S. P., and Bartek, J. (2009). The DNA-Damage Response in Human Biology and Disease. Nature 461, 1071–1078. doi:10.1038/nature08467
Jacquet, K., Fradet-Turcotte, A., Avvakumov, N., Lambert, J.-P., Roques, C., Pandita, R. K., et al. (2016). The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol. Cell 62, 409–421. doi:10.1016/j.molcel.2016.03.031
Jakob, B., Splinter, J., Conrad, S., Voss, K. O., Zink, D., Durante, M., et al. (2011). DNA Double-Strand Breaks in Heterochromatin Elicit Fast Repair Protein Recruitment, Histone H2AX Phosphorylation and Relocation to Euchromatin. Nucleic Acids Res. 39, 6489–6499. doi:10.1093/nar/gkr230
Janssen, A., Colmenares, S. U., and Karpen, G. H. (2018). Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 34, 265–288. doi:10.1146/annurev-cellbio-100617-062653
Janssen, A., Colmenares, S. U., Lee, T., and Karpen, G. H. (2019). Timely Double-Strand Break Repair and Pathway Choice in Pericentromeric Heterochromatin Depend on the Histone Demethylase dKDM4A. Genes Dev. 33, 103–115. doi:10.1101/gad.317537.118
Jha, D. K., and Strahl, B. D. (2014). An RNA Polymerase II-Coupled Function for Histone H3K36 Methylation in Checkpoint Activation and DSB Repair. Nat. Commun. 5, 3965. doi:10.1038/ncomms4965
Jorgensen, S., Schotta, G., and Sorensen, C. S. (2013). Histone H4 Lysine 20 Methylation: Key Player in Epigenetic Regulation of Genomic Integrity. Nucleic Acids Res. 41, 2797–2806. doi:10.1093/nar/gkt012
Kakarougkas, A., Ismail, A., Klement, K., Goodarzi, A. A., Conrad, S., Freire, R., et al. (2013). Opposing Roles for 53BP1 during Homologous Recombination. Nucleic Acids Res. 41, 9719–9731. doi:10.1093/nar/gkt729
Kalb, R., Mallery, D. L., Larkin, C., Huang, J. T. J., and Hiom, K. (2014). BRCA1 Is a Histone-h2a-specific Ubiquitin Ligase. Cell Rep. 8, 999–1005. doi:10.1016/j.celrep.2014.07.025
Kang, J. Y., Kim, J. Y., Kim, K. B., Park, J. W., Cho, H., Hahm, J. Y., et al. (2018). KDM2B Is a Histone H3K79 Demethylase and Induces Transcriptional Repression via Sirtuin‐1‐mediated Chromatin Silencing. FASEB J. 32, 5737–5750. doi:10.1096/fj.201800242R
Karanam, K., Kafri, R., Loewer, A., and Lahav, G. (2012). Quantitative Live Cell Imaging Reveals a Gradual Shift between DNA Repair Mechanisms and a Maximal Use of HR in Mid S Phase. Mol. Cell 47, 320–329. doi:10.1016/j.molcel.2012.05.052
Khristich, A. N., and Mirkin, S. M. (2020). On the Wrong DNA Track: Molecular Mechanisms of Repeat-Mediated Genome Instability. J. Biol. Chem. 295, 4134–4170. doi:10.1074/jbc.REV119.007678
Khurana, S., Kruhlak, M. J., Kim, J., Tran, A. D., Liu, J., Nyswaner, K., et al. (2014). A Macrohistone Variant Links Dynamic Chromatin Compaction to BRCA1-dependent Genome Maintenance. Cell Rep. 8, 1049–1062. doi:10.1016/j.celrep.2014.07.024
Kim, H., Chen, J., and Yu, X. (2007). Ubiquitin-binding Protein RAP80 Mediates BRCA1-dependent DNA Damage Response. Science 316, 1202–1205. doi:10.1126/science.1139621
Kim, J., Oberdoerffer, P., and Khurana, S. (2018). The Histone Variant macroH2A1 Is a Splicing-Modulated Caretaker of Genome Integrity and Tumor Growth. Mol. Cell. Oncol. 5, e1441629. doi:10.1080/23723556.2018.1441629
Kitevski-LeBlanc, J., Fradet-Turcotte, A., Kukic, P., Wilson, M. D., Portella, G., Yuwen, T., et al. (2017). The RNF168 Paralog RNF169 Defines a New Class of Ubiquitylated Histone Reader Involved in the Response to DNA Damage. Elife 6, e23872. doi:10.7554/eLife.23872
Klose, R. J., Yamane, K., Bae, Y., Zhang, D., Erdjument-Bromage, H., Tempst, P., et al. (2006). The Transcriptional Repressor JHDM3A Demethylates Trimethyl Histone H3 Lysine 9 and Lysine 36. Nature 442, 312–316. doi:10.1038/nature04853
Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., Sweeney, F. D., et al. (2007). Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase. Science 318, 1637–1640. doi:10.1126/science.1150034
Kouzarides, T. (2007). Chromatin Modifications and Their Function. Cell 128, 693–705. doi:10.1016/j.cell.2007.02.005
Krais, J. J., Wang, Y., Patel, P., Basu, J., Bernhardy, A. J., and Johnson, N. (2021). RNF168-mediated Localization of BARD1 Recruits the BRCA1-PALB2 Complex to DNA Damage. Nat. Commun. 12, 5016. doi:10.1038/s41467-021-25346-4
Lee, D., Apelt, K., Lee, S. O., Chan, H. R., Luijsterburg, M. S., Leung, J. W. C., et al. (2022). ZMYM2 Restricts 53BP1 at DNA Double-Strand Breaks to Favor BRCA1 Loading and Homologous Recombination. Nucleic. Acids Res. doi:10.1093/nar/gkac160
Lee, J., Thompson, J. R., Botuyan, M. V., and Mer, G. (2008). Distinct Binding Modes Specify the Recognition of Methylated Histones H3K4 and H4K20 by JMJD2A-Tudor. Nat. Struct. Mol. Biol. 15, 109–111. doi:10.1038/nsmb1326
Li, L., Germain, D. R., Poon, H.-Y., Hildebrandt, M. R., Monckton, E. A., McDonald, D., et al. (2016). DEAD Box 1 Facilitates Removal of RNA and Homologous Recombination at DNA Double-Strand Breaks. Mol. Cell Biol. 36, 2794–2810. doi:10.1128/MCB.00415-16
Lieber, M. R. (2010). The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 79, 181–211. doi:10.1146/annurev.biochem.052308.093131
Liu, S., Hua, Y., Wang, J., Li, L., Yuan, J., Zhang, B., et al. (2021). RNA Polymerase III Is Required for the Repair of DNA Double-Strand Breaks by Homologous Recombination. Cell 184, 1314–1329. e1310. doi:10.1016/j.cell.2021.01.048
Liu, W., Tanasa, B., Tyurina, O. V., Zhou, T. Y., Gassmann, R., Liu, W. T., et al. (2010). PHF8 Mediates Histone H4 Lysine 20 Demethylation Events Involved in Cell Cycle Progression. Nature 466, 508–512. doi:10.1038/nature09272
Lu, W.-T., Hawley, B. R., Skalka, G. L., Baldock, R. A., Smith, E. M., Bader, A. S., et al. (2018). Drosha Drives the Formation of DNA:RNA Hybrids Around DNA Break Sites to Facilitate DNA Repair. Nat. Commun. 9, 532. doi:10.1038/s41467-018-02893-x
Mailand, N., Bekker-Jensen, S., Faustrup, H., Melander, F., Bartek, J., Lukas, C., et al. (2007). RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 131, 887–900. doi:10.1016/j.cell.2007.09.040
Mallette, F. A., Mattiroli, F., Cui, G., Young, L. C., Hendzel, M. J., Mer, G., et al. (2012). RNF8- and RNF168-dependent Degradation of KDM4A/JMJD2A Triggers 53BP1 Recruitment to DNA Damage Sites. EMBO J. 31, 1865–1878. doi:10.1038/emboj.2012.47
Mattiroli, F., and Penengo, L. (2021). Histone Ubiquitination: An Integrative Signaling Platform in Genome Stability. Trends Genet. 37, 566–581. doi:10.1016/j.tig.2020.12.005
Mattiroli, F., Vissers, J. H. A., van Dijk, W. J., Ikpa, P., Citterio, E., Vermeulen, W., et al. (2012). RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 150, 1182–1195. doi:10.1016/j.cell.2012.08.005
McGinty, R. K., Kim, J., Chatterjee, C., Roeder, R. G., and Muir, T. W. (2008). Chemically Ubiquitylated Histone H2B Stimulates hDot1L-Mediated Intranucleosomal Methylation. Nature 453, 812–816. doi:10.1038/nature06906
Michelini, F., Pitchiaya, S., Vitelli, V., Sharma, S., Gioia, U., Pessina, F., et al. (2017). Damage-induced lncRNAs Control the DNA Damage Response through Interaction with DDRNAs at Individual Double-Strand Breaks. Nat. Cell Biol. 19, 1400–1411. doi:10.1038/ncb3643
Miller, K. M., Tjeertes, J. V., Coates, J., Legube, G., Polo, S. E., Britton, S., et al. (2010). Human HDAC1 and HDAC2 Function in the DNA-Damage Response to Promote DNA Nonhomologous End-Joining. Nat. Struct. Mol. Biol. 17, 1144–1151. doi:10.1038/nsmb.1899
Min, J., Allali-Hassani, A., Nady, N., Qi, C., Ouyang, H., Liu, Y., et al. (2007). L3MBTL1 Recognition of Mono- and Dimethylated Histones. Nat. Struct. Mol. Biol. 14, 1229–1230. doi:10.1038/nsmb1340
Mirman, Z., Lottersberger, F., Takai, H., Kibe, T., Gong, Y., Takai, K., et al. (2018). 53BP1-RIF1-shieldin Counteracts DSB Resection through CST- and Polα-dependent Fill-In. Nature 560, 112–116. doi:10.1038/s41586-018-0324-7
Mosbech, A., Lukas, C., Bekker-Jensen, S., and Mailand, N. (2013). The Deubiquitylating Enzyme USP44 Counteracts the DNA Double-Strand Break Response Mediated by the RNF8 and RNF168 Ubiquitin Ligases. J. Biol. Chem. 288, 16579–16587. doi:10.1074/jbc.M113.459917
Musselman, C. A., Avvakumov, N., Watanabe, R., Abraham, C. G., Lalonde, M.-E., Hong, Z., et al. (2012). Molecular Basis for H3K36me3 Recognition by the Tudor Domain of PHF1. Nat. Struct. Mol. Biol. 19, 1266–1272. doi:10.1038/nsmb.2435
Musselman, C. A., Gibson, M. D., Hartwick, E. W., North, J. A., Gatchalian, J., Poirier, M. G., et al. (2013). Binding of PHF1 Tudor to H3K36me3 Enhances Nucleosome Accessibility. Nat. Commun. 4, 2969. doi:10.1038/ncomms3969
Nakamura, K., Saredi, G., Becker, J. R., Foster, B. M., Nguyen, N. V., Beyer, T. E., et al. (2019). H4K20me0 Recognition by BRCA1-BARD1 Directs Homologous Recombination to Sister Chromatids. Nat. Cell Biol. 21, 311–318. doi:10.1038/s41556-019-0282-9
Ngo, G. H. P., Grimstead, J. W., and Baird, D. M. (2021). UPF1 Promotes the Formation of R Loops to Stimulate DNA Double-Strand Break Repair. Nat. Commun. 12, 3849. doi:10.1038/s41467-021-24201-w
Nishioka, K., Rice, J. C., Sarma, K., Erdjument-Bromage, H., Werner, J., Wang, Y., et al. (2002). PR-Set7 Is a Nucleosome-specific Methyltransferase that Modifies Lysine 20 of Histone H4 and Is Associated with Silent Chromatin. Mol. Cell 9, 1201–1213. doi:10.1016/s1097-2765(02)00548-8
Noordermeer, S. M., Adam, S., Setiaputra, D., Barazas, M., Pettitt, S. J., Ling, A. K., et al. (2018). The Shieldin Complex Mediates 53BP1-dependent DNA Repair. Nature 560, 117–121. doi:10.1038/s41586-018-0340-7
O'Hagan, H. M., Mohammad, H. P., and Baylin, S. B. (2008). Double Strand Breaks Can Initiate Gene Silencing and SIRT1-dependent Onset of DNA Methylation in an Exogenous Promoter CpG Island. PLoS Genet. 4, e1000155. doi:10.1371/journal.pgen.1000155
Ohle, C., Tesorero, R., Schermann, G., Dobrev, N., Sinning, I., and Fischer, T. (2016). Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell 167, 1001–1013. e1007. doi:10.1016/j.cell.2016.10.001
Ohsawa, R., Seol, J.-H., and Tyler, J. K. (2013). At the Intersection of Non-coding Transcription, DNA Repair, Chromatin Structure, and Cellular Senescence. Front. Genet. 4, 136. doi:10.3389/fgene.2013.00136
Orthwein, A., Noordermeer, S. M., Wilson, M. D., Landry, S., Enchev, R. I., Sherker, A., et al. (2015). A Mechanism for the Suppression of Homologous Recombination in G1 Cells. Nature 528, 422–426. doi:10.1038/nature16142
Ouyang, J., Yadav, T., Zhang, J.-M., Yang, H., Rheinbay, E., Guo, H., et al. (2021). RNA Transcripts Stimulate Homologous Recombination by Forming DR-Loops. Nature 594, 283–288. doi:10.1038/s41586-021-03538-8
Pai, C.-C., Deegan, R. S., Subramanian, L., Gal, C., Sarkar, S., Blaikley, E. J., et al. (2014). A Histone H3K36 Chromatin Switch Coordinates DNA Double-Strand Break Repair Pathway Choice. Nat. Commun. 5, 4091. doi:10.1038/ncomms5091
Panier, S., and Boulton, S. J. (2014). Double-strand Break Repair: 53BP1 Comes into Focus. Nat. Rev. Mol. Cell Biol. 15, 7–18. doi:10.1038/nrm3719
Pellegrino, S., Michelena, J., Teloni, F., Imhof, R., and Altmeyer, M. (2017). Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative Chromatin. Cell Rep. 19, 1819–1831. doi:10.1016/j.celrep.2017.05.016
Pesavento, J. J., Yang, H., Kelleher, N. L., and Mizzen, C. A. (2008). Certain and Progressive Methylation of Histone H4 at Lysine 20 during the Cell Cycle. Mol. Cell Biol. 28, 468–486. doi:10.1128/MCB.01517-07
Petukhova, G., Sung, P., and Klein, H. (2000). Promotion of Rad51-dependent D-Loop Formation by Yeast Recombination Factor Rdh54/Tid1. Genes Dev. 14, 2206–2215. doi:10.1101/gad.826100
Pfister, S. X., Ahrabi, S., Zalmas, L.-P., Sarkar, S., Aymard, F., Bachrati, C. Z., et al. (2014). SETD2-dependent Histone H3K36 Trimethylation Is Required for Homologous Recombination Repair and Genome Stability. Cell Rep. 7, 2006–2018. doi:10.1016/j.celrep.2014.05.026
Pradeepa, M. M., Sutherland, H. G., Ule, J., Grimes, G. R., and Bickmore, W. A. (2012). Psip1/Ledgf P52 Binds Methylated Histone H3K36 and Splicing Factors and Contributes to the Regulation of Alternative Splicing. PLoS Genet. 8, e1002717. doi:10.1371/journal.pgen.1002717
Price, B. D., and D’Andrea, A. D. (2013). Chromatin Remodeling at DNA Double-Strand Breaks. Cell 152, 1344–1354. doi:10.1016/j.cell.2013.02.011
Qi, H. H., Sarkissian, M., Hu, G.-Q., Wang, Z., Bhattacharjee, A., Gordon, D. B., et al. (2010). Histone H4K20/H3K9 Demethylase PHF8 Regulates Zebrafish Brain and Craniofacial Development. Nature 466, 503–507. doi:10.1038/nature09261
Qin, B., Yu, J., Nowsheen, S., Wang, M., Tu, X., Liu, T., et al. (2019). UFL1 Promotes Histone H4 Ufmylation and ATM Activation. Nat. Commun. 10, 1242. doi:10.1038/s41467-019-09175-0
Qin, B., Yu, J., Nowsheen, S., Zhao, F., Wang, L., and Lou, Z. (2020). STK38 Promotes ATM Activation by Acting as a Reader of Histone H4 Ufmylation. Sci. Adv. 6, eaax8214. doi:10.1126/sciadv.aax8214
Rappold, I., Iwabuchi, K., Date, T., and Chen, J. (2001). Tumor Suppressor P53 Binding Protein 1 (53BP1) Is Involved in DNA Damage-Signaling Pathways. J. Cell Biol. 153, 613–620. doi:10.1083/jcb.153.3.613
Rinaldi, C., Pizzul, P., Longhese, M. P., and Bonetti, D. (2020). Sensing R-Loop-Associated DNA Damage to Safeguard Genome Stability. Front. Cell Dev. Biol. 8, 618157. doi:10.3389/fcell.2020.618157
Rothbart, S. B., and Strahl, B. D. (2014). Interpreting the Language of Histone and DNA Modifications. Biochimica Biophysica Acta (BBA) - Gene Regul. Mech. 1839, 627–643. doi:10.1016/j.bbagrm.2014.03.001
Ryu, T., Spatola, B., Delabaere, L., Bowlin, K., Hopp, H., Kunitake, R., et al. (2015). Heterochromatic Breaks Move to the Nuclear Periphery to Continue Recombinational Repair. Nat. Cell Biol. 17, 1401–1411. doi:10.1038/ncb3258
Sallmyr, A., and Tomkinson, A. E. (2018). Repair of DNA Double-Strand Breaks by Mammalian Alternative End-Joining Pathways. J. Biol. Chem. 293, 10536–10546. doi:10.1074/jbc.TM117.000375
Sanders, S. L., Portoso, M., Mata, J., Bähler, J., Allshire, R. C., and Kouzarides, T. (2004). Methylation of Histone H4 Lysine 20 Controls Recruitment of Crb2 to Sites of DNA Damage. Cell 119, 603–614. doi:10.1016/j.cell.2004.11.009
Saredi, G., Huang, H., Hammond, C. M., Alabert, C., Bekker-Jensen, S., Forne, I., et al. (2016). H4K20me0 Marks Post-replicative Chromatin and Recruits the TONSL-Mms22l DNA Repair Complex. Nature 534, 714–718. doi:10.1038/nature18312
Schep, R., Brinkman, E. K., Leemans, C., Vergara, X., van der Weide, R. H., Morris, B., et al. (2021). Impact of Chromatin Context on Cas9-Induced DNA Double-Strand Break Repair Pathway Balance. Mol. Cell 81, 2216–e10. e2210. doi:10.1016/j.molcel.2021.03.032
Schotta, G., Lachner, M., Sarma, K., Ebert, A., Sengupta, R., Reuter, G., et al. (2004). A Silencing Pathway to Induce H3-K9 and H4-K20 Trimethylation at Constitutive Heterochromatin. Genes Dev. 18, 1251–1262. doi:10.1101/gad.300704
Schultz, L. B., Chehab, N. H., Malikzay, A., and Halazonetis, T. D. (2000). p53 Binding Protein 1 (53BP1) Is an Early Participant in the Cellular Response to DNA Double-Strand Breaks. J. Cell Biol. 151, 1381–1390. doi:10.1083/jcb.151.7.1381
Scully, R., Panday, A., Elango, R., and Willis, N. A. (2019). DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 20, 698–714. doi:10.1038/s41580-019-0152-0
Sebastian, R., Hosogane, E. K., Sun, E. G., Tran, A. D., Reinhold, W. C., Burkett, S., et al. (2020). Epigenetic Regulation of DNA Repair Pathway Choice by MacroH2A1 Splice Variants Ensures Genome Stability. Mol. Cell 79, 836–845. e837. doi:10.1016/j.molcel.2020.06.028
Shanbhag, N. M., Rafalska-Metcalf, I. U., Balane-Bolivar, C., Janicki, S. M., and Greenberg, R. A. (2010). ATM-dependent Chromatin Changes Silence Transcription in Cis to DNA Double-Strand Breaks. Cell 141, 970–981. doi:10.1016/j.cell.2010.04.038
Sharma, N., Zhu, Q., Wani, G., He, J., Wang, Q.-E., and Wani, A. A. (2014). USP3 Counteracts RNF168 via Deubiquitinating H2A and γH2AX at Lysine 13 and 15. Cell Cycle 13, 106–114. doi:10.4161/cc.26814
Sharma, S., Anand, R., Zhang, X., Francia, S., Michelini, F., Galbiati, A., et al. (2021). MRE11-RAD50-NBS1 Complex Is Sufficient to Promote Transcription by RNA Polymerase II at Double-Strand Breaks by Melting DNA Ends. Cell Rep. 34, 108565. doi:10.1016/j.celrep.2020.108565
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 119, 941–953. doi:10.1016/j.cell.2004.12.012
Shiio, Y., and Eisenman, R. N. (2003). Histone Sumoylation is Associated With Transcriptional Repression. Proc. Natl. Acad. Sci. U S A 100, 13225–13230. doi:10.1073/pnas.1735528100
Sobhian, B., Shao, G., Lilli, D. R., Culhane, A. C., Moreau, L. A., Xia, B., et al. (2007). RAP80 Targets BRCA1 to Specific Ubiquitin Structures at DNA Damage Sites. Science 316, 1198–1202. doi:10.1126/science.1139516
Stender, J. D., Pascual, G., Liu, W., Kaikkonen, M. U., Do, K., Spann, N. J., et al. (2012). Control of Proinflammatory Gene Programs by Regulated Trimethylation and Demethylation of Histone H4K20. Mol. Cell 48, 28–38. doi:10.1016/j.molcel.2012.07.020
Stewart, G. S., Panier, S., Townsend, K., Al-Hakim, A. K., Kolas, N. K., Miller, E. S., et al. (2009). The RIDDLE Syndrome Protein Mediates a Ubiquitin-dependent Signaling Cascade at Sites of DNA Damage. Cell 136, 420–434. doi:10.1016/j.cell.2008.12.042
Stucki, M., Clapperton, J. A., Mohammad, D., Yaffe, M. B., Smerdon, S. J., and Jackson, S. P. (2005). MDC1 Directly Binds Phosphorylated Histone H2AX to Regulate Cellular Responses to DNA Double-Strand Breaks. Cell 123, 1213–1226. doi:10.1016/j.cell.2005.09.038
Sun, Y., Jiang, X., Chen, S., Fernandes, N., and Price, B. D. (2005). A Role for the Tip60 Histone Acetyltransferase in the Acetylation and Activation of ATM. Proc. Natl. Acad. Sci. U.S.A. 102, 13182–13187. doi:10.1073/pnas.0504211102
Sun, Y., Jiang, X., Xu, Y., Ayrapetov, M. K., Moreau, L. A., Whetstine, J. R., et al. (2009). Histone H3 Methylation Links DNA Damage Detection to Activation of the Tumour Suppressor Tip60. Nat. Cell Biol. 11, 1376–1382. doi:10.1038/ncb1982
Svobodová Kovaříková, A., Legartová, S., Krejčí, J., and Bártová, E. (2018). H3K9me3 and H4K20me3 Represent the Epigenetic Landscape for 53BP1 Binding to DNA Lesions. Aging 10, 2585–2605. doi:10.18632/aging.101572
Symington, L. S. (2014). End Resection at Double-Strand Breaks: Mechanism and Regulation. Cold Spring Harb. Perspect. Biol. 6, a016436. doi:10.1101/cshperspect.a016436
Talbert, P. B., and Henikoff, S. (2021). The Yin and Yang of Histone Marks in Transcription. Annu. Rev. Genom. Hum. Genet. 22, 147–170. doi:10.1146/annurev-genom-120220-085159
Tan, M., Luo, H., Lee, S., Jin, F., Yang, J. S., Montellier, E., et al. (2011). Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 146, 1016–1028. doi:10.1016/j.cell.2011.08.008
Tang, J., Cho, N. W., Cui, G., Manion, E. M., Shanbhag, N. M., Botuyan, M. V., et al. (2013). Acetylation Limits 53BP1 Association with Damaged Chromatin to Promote Homologous Recombination. Nat. Struct. Mol. Biol. 20, 317–325. doi:10.1038/nsmb.2499
Thorslund, T., Ripplinger, A., Hoffmann, S., Wild, T., Uckelmann, M., Villumsen, B., et al. (2015). Histone H1 Couples Initiation and Amplification of Ubiquitin Signalling after DNA Damage. Nature 527, 389–393. doi:10.1038/nature15401
Timinszky, G., Till, S., Hassa, P. O., Hothorn, M., Kustatscher, G., Nijmeijer, B., et al. (2009). A Macrodomain-Containing Histone Rearranges Chromatin upon Sensing PARP1 Activation. Nat. Struct. Mol. Biol. 16, 923–929. doi:10.1038/nsmb.1664
Ting, X., Xia, L., Yang, J., He, L., Si, W., Shang, Y., et al. (2019). USP11 Acts as a Histone Deubiquitinase Functioning in Chromatin Reorganization during DNA Repair. Nucleic Acids Res. 47, 9721–9740. doi:10.1093/nar/gkz726
Trojer, P., and Reinberg, D. (2007). Facultative Heterochromatin: Is There a Distinctive Molecular Signature? Mol. Cell 28, 1–13. doi:10.1016/j.molcel.2007.09.011
Tsouroula, K., Furst, A., Rogier, M., Heyer, V., Maglott-Roth, A., Ferrand, A., et al. (2016). Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 63, 293–305. doi:10.1016/j.molcel.2016.06.002
Tsukada, Y.-i., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P., et al. (2006). Histone Demethylation by a Family of JmjC Domain-Containing Proteins. Nature 439, 811–816. doi:10.1038/nature04433
Tubbs, A., and Nussenzweig, A. (2017). Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 168, 644–656. doi:10.1016/j.cell.2017.01.002
Uckelmann, M., Densham, R. M., Baas, R., Winterwerp, H. H. K., Fish, A., Sixma, T. K., et al. (2018). USP48 Restrains Resection by Site-specific Cleavage of the BRCA1 Ubiquitin Mark from H2A. Nat. Commun. 9, 229. doi:10.1038/s41467-017-02653-3
Uckelmann, M., and Sixma, T. K. (2017). Histone Ubiquitination in the DNA Damage Response. DNA Repair 56, 92–101. doi:10.1016/j.dnarep.2017.06.011
Umehara, T., Nakamura, Y., Wakamori, M., Ozato, K., Yokoyama, S., and Padmanabhan, B. (2010). Structural Implications for K5/K12-Di-Acetylated Histone H4 Recognition by the Second Bromodomain of BRD2. FEBS Lett. 584, 3901–3908. doi:10.1016/j.febslet.2010.08.013
Van Komen, S., Petukhova, G., Sigurdsson, S., Stratton, S., and Sung, P. (2000). Superhelicity-driven Homologous DNA Pairing by Yeast Recombination Factors Rad51 and Rad54. Mol. Cell 6, 563–572. doi:10.1016/s1097-2765(00)00055-1
van Sluis, M., and McStay, B. (2017). Nucleolar Reorganization in Response to rDNA Damage. Curr. Opin. Cell Biol. 46, 81–86. doi:10.1016/j.ceb.2017.03.004
Vaquero, A., Scher, M. B., Lee, D. H., Sutton, A., Cheng, H.-L., Alt, F. W., et al. (2006). SirT2 Is a Histone Deacetylase with Preference for Histone H4 Lys 16 during Mitosis. Genes Dev. 20, 1256–1261. doi:10.1101/gad.1412706
Vaquero, A., Scher, M., Lee, D., Erdjument-Bromage, H., Tempst, P., and Reinberg, D. (2004). Human SirT1 Interacts with Histone H1 and Promotes Formation of Facultative Heterochromatin. Mol. Cell 16, 93–105. doi:10.1016/j.molcel.2004.08.031
Wakeman, T. P., Wang, Q., Feng, J., and Wang, X.-F. (2012). Bat3 Facilitates H3K79 Dimethylation by DOT1L and Promotes DNA Damage-Induced 53BP1 Foci at G1/G2 Cell-Cycle Phases. EMBO J. 31, 2169–2181. doi:10.1038/emboj.2012.50
Walport, L. J., Hopkinson, R. J., Vollmar, M., Madden, S. K., Gileadi, C., Oppermann, U., et al. (2014). Human UTY(KDM6C) Is a Male-specific Nϵ-Methyl Lysyl Demethylase. J. Biol. Chem. 289, 18302–18313. doi:10.1074/jbc.M114.555052
Wang, B., Matsuoka, S., Ballif, B. A., Zhang, D., Smogorzewska, A., Gygi, S. P., et al. (2007). Abraxas and RAP80 Form a BRCA1 Protein Complex Required for the DNA Damage Response. Science 316, 1194–1198. doi:10.1126/science.1139476
Wang, Z., Gong, Y., Peng, B., Shi, R., Fan, D., Zhao, H., et al. (2019). MRE11 UFMylation Promotes ATM Activation. Nucleic. Acids Res. 47, 4124–4135. doi:10.1093/nar/gkz110
Wang, Z., Zhang, H., Liu, J., Cheruiyot, A., Lee, J.-H., Ordog, T., et al. (2016). USP51 Deubiquitylates H2AK13,15ub and Regulates DNA Damage Response. Genes Dev. 30, 946–959. doi:10.1101/gad.271841.115
Wei, W., Ba, Z., Gao, M., Wu, Y., Ma, Y., Amiard, S., et al. (2012). A Role for Small RNAs in DNA Double-Strand Break Repair. Cell 149, 101–112. doi:10.1016/j.cell.2012.03.002
Whelan, D. R., and Rothenberg, E. (2021). Super-resolution Mapping of Cellular Double-Strand Break Resection Complexes during Homologous Recombination. Proc. Natl. Acad. Sci. U.S.A. 118, e2021963118. doi:10.1073/pnas.2021963118
Wilson, M. D., Benlekbir, S., Fradet-Turcotte, A., Sherker, A., Julien, J.-P., McEwan, A., et al. (2016). The Structural Basis of Modified Nucleosome Recognition by 53BP1. Nature 536, 100–103. doi:10.1038/nature18951
Witus, S. R., Burrell, A. L., Farrell, D. P., Kang, J., Wang, M., Hansen, J. M., et al. (2021). BRCA1/BARD1 Site-specific Ubiquitylation of Nucleosomal H2A Is Directed by BARD1. Nat. Struct. Mol. Biol. 28, 268–277. doi:10.1038/s41594-020-00556-4
Xu, C., Xu, Y., Gursoy-Yuzugullu, O., and Price, B. D. (2012a). The Histone Variant macroH2A1.1 Is Recruited to DSBs through a Mechanism Involving PARP1. FEBS Lett. 586, 3920–3925. doi:10.1016/j.febslet.2012.09.030
Xu, Y., Ayrapetov, M. K., Xu, C., Gursoy-Yuzugullu, O., Hu, Y., and Price, B. D. (2012b). Histone H2A.Z Controls a Critical Chromatin Remodeling Step Required for DNA Double-Strand Break Repair. Mol. Cell 48, 723–733. doi:10.1016/j.molcel.2012.09.026
Yilmaz, D., Furst, A., Meaburn, K., Lezaja, A., Wen, Y., Altmeyer, M., et al. (2021). Activation of Homologous Recombination in G1 Preserves Centromeric Integrity. Nature 600, 748–753. doi:10.1038/s41586-021-04200-z
Zgheib, O., Pataky, K., Brugger, J., and Halazonetis, T. D. (2009). An Oligomerized 53BP1 Tudor Domain Suffices for Recognition of DNA Double-Strand Breaks. Mol. Cell Biol. 29, 1050–1058. doi:10.1128/MCB.01011-08
Zhang, F., Ma, J., Wu, J., Ye, L., Cai, H., Xia, B., et al. (2009). PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr. Biol. 19, 524–529. doi:10.1016/j.cub.2009.02.018
Zhang, H., Liu, H., Chen, Y., Yang, X., Wang, P., Liu, T., et al. (2016). A Cell Cycle-dependent BRCA1-UHRF1 Cascade Regulates DNA Double-Strand Break Repair Pathway Choice. Nat. Commun. 7, 10201. doi:10.1038/ncomms10201
Zhang, T., Cooper, S., and Brockdorff, N. (2015). The Interplay of Histone Modifications - Writers that Read. EMBO Rep. 16, 1467–1481. doi:10.15252/embr.201540945
Zhou, L., Holt, M. T., Ohashi, N., Zhao, A., Müller, M. M., Wang, B., et al. (2016). Evidence that Ubiquitylated H2B Corrals hDot1L on the Nucleosomal Surface to Induce H3K79 Methylation. Nat. Commun. 7, 10589. doi:10.1038/ncomms10589
Keywords: repair pathway choice, euchromatin, heterochromatin, histone modifications, transcription, RNA-DNA hybrids, DNA doube-strand breaks
Citation: Chen Z and Tyler JK (2022) The Chromatin Landscape Channels DNA Double-Strand Breaks to Distinct Repair Pathways. Front. Cell Dev. Biol. 10:909696. doi: 10.3389/fcell.2022.909696
Received: 31 March 2022; Accepted: 17 May 2022;
Published: 08 June 2022.
Edited by:
Lovisa Lundholm, Stockholm University, SwedenReviewed by:
Alessandro A. Sartori, University of Zurich, SwitzerlandAmelie Fradet-Turcotte, Laval University, Canada
Copyright © 2022 Chen and Tyler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica K. Tyler, amV0MjAyMUBtZWQuY29ybmVsbC5lZHU=