
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cell Dev. Biol., 17 June 2022
Sec. Epigenomics and Epigenetics
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.909097
This article is part of the Research TopicDeregulations in Chromatin Topology, Transcription and EpigeneticsView all 5 articles
 Eugenia Bezzecchi
1,2
†
Eugenia Bezzecchi
1,2
†
 Giulia Pagani
1
†
Giulia Pagani
1
†
 Barbara Forte
3
Barbara Forte
3 Stefano Percio3
Stefano Percio3
 Nadia Zaffaroni
3
Nadia Zaffaroni
3
 Diletta Dolfini
1
Diletta Dolfini
1
 Paolo Gandellini
1*
Paolo Gandellini
1*Aside serving as host gene for miR-205, MIR205HG transcribes for a chromatin-associated long noncoding RNA (lncRNA) able to restrain the differentiation of prostate basal cells, thus being reannotated as LEADR (Long Epithelial Alu-interacting Differentiation-related RNA). We previously showed the presence of Alu sequences in the promoters of genes modulated upon MIR205HG/LEADR manipulation. Notably, an Alu element also spans the first and second exons of MIR205HG/LEADR, suggesting its possible involvement in target selection/binding. Here, we performed ChIRP-seq to map MIR205HG/LEADR chromatin occupancy at genome-wide level in prostate basal cells. Our results confirmed preferential binding to regions proximal to gene transcription start site (TSS). Moreover, enrichment of triplex-forming sequences was found upstream of MIR205HG/LEADR-bound genes, peaking at −1,500/−500 bp from TSS. Triplexes formed with one or two putative DNA binding sites within MIR205HG/LEADR sequence, located just upstream of the Alu element. Notably, triplex-forming regions of bound genes were themselves enriched in Alu elements. These data suggest, from one side, that triplex formation may be the prevalent mechanism by which MIR205HG/LEADR selects and physically interacts with target DNA, from the other that direct or protein-mediated Alu (RNA)/Alu (DNA) interaction may represent a further functional requirement. We also found that triplex-forming regions were enriched in specific histone modifications, including H3K4me1 in the absence of H3K27ac, H3K4me3 and H3K27me3, indicating that in prostate basal cells MIR205HG/LEADR may preferentially bind to primed proximal regulatory elements. This may underscore the need for basal cells to keep MIR205HG/LEADR target genes repressed but, at the same time, responsive to differentiation cues.
Long noncoding RNAs (lncRNAs) are an interesting class of transcripts longer than 200 nt, which are devoid of protein-coding potential though exerting important regulatory functions in a variety of biological processes (Ulitsky and Bartel, 2013) and disease states (Rossi et al., 2020; Pandini et al., 2021; Tassinari and Gandellini, 2021; Cava et al., 2022). Differently from mRNAs, they can be expressed either in the nucleus or in the cytoplasm (or both), an aspect, that is, intimately linked with their mechanism of action. In fact, depending on their subcellular localization, their activities range from regulating the chromatin state and transcription in the nucleus to acting post-transcriptionally as sponges for microRNAs in the cytoplasm (Statello et al., 2021). This wide spectrum of regulation modalities is intrinsically linked to their RNA nature. Indeed, the interactome of RNA may include other nucleic acids, both DNA and RNA, to which RNA can bind through canonical Watson-Crick base pairing (as is the case of RNA/RNA duplexes or R-loops with the DNA) or Hoogsteen bonds, as in triple helices or triplexes. In addition, by folding into complex secondary and tertiary structures, RNA can provide scaffolds for interacting with proteins (Tassinari et al., 2021). Although examples for all the cited modes of action have been reported, most lncRNAs are still poorly characterized from the mechanistic point of view.
In this regard, MIR205HG is a lncRNA abundantly expressed in epithelia, the function of which has been acknowledged only recently. In physiological conditions, our group showed that, aside serving as host gene for miR-205 (Gandellini et al., 2012), MIR205HG acts as nuclear lncRNA able to restrain the differentiation of prostate basal cells, a finding that led to its reannotation as LEADR (Long Epithelial Alu-interacting Differentiation-related RNA) (Profumo et al., 2019). An oncogenic role has been proposed for MIR205HG in several cancers characterized by the expansion of basal cells, such as cervical (Li et al., 2019), lung squamous cell (Liu et al., 2020), head and neck squamous cell carcinomas (Di Agostino et al., 2018). A tumor-suppressive function has been instead reported in esophageal adenocarcinoma, where MIR205HG is downregulated (Song et al., 2021; Dong et al., 2022). From the mechanistic side, most of the studies suggest that MIR205HG would act post-transcriptionally as sponge for various miRNAs, among which miR-122-5p, miR-299-3p (Guo et al., 2021) and miR-590-3p, or by hindering the translation of HNRNPA0 mRNA (Dong et al., 2022). However, data from the lncAtlas (Mas-Ponte et al., 2017) (Supplementary Figure S1) and from our group (Profumo et al., 2019) indicate that the expression of MIR205HG is prevalently nuclear and chromatin-associated.
Here, to start elucidating MIR205HG/LEADR mechanism of functioning at the chromatin level, we performed Chromatin Isolation by RNA purification (ChIRP)-sequencing (Chu et al., 2011) of immortalized prostate epithelial cells, which express the lncRNA at high level. Our results show that MIR205HG/LEADR preferentially binds to primed proximal regulatory regions by forming DNA/RNA triplexes, through a mechanism that may imply the participation of Alu elements present in both the lncRNA and target genes.
ChIRP was performed on RWPE-1 cells in triplicate as described in Profumo et al. (2019). RWPE-1 cells were chosen as model of normal prostate basal epithelial cells, as they express the typical basal cytokeratins (KRT5 and KRT14) together with high levels of MIR205HG/LEADR (Profumo et al., 2019). A unique pool of ten 20-mer 3′-BiotinTEG-modified antisense probes covering the whole sequence of MIR205HG/LEADR transcript (1 probe/100 bp of RNA length) was used for the experiment, whereas a symmetrical set of probes against lacZ RNA was used as mock control (sequences are reported in Profumo et al., 2019). RNA was obtained from ChIRP-ed samples to check successful enrichment of MIR205HG/LEADR transcript upon precipitation with specific probes as compared to lacZ probes (Supplementary Figure S2A). DNA from the same samples was obtained for sequencing. High-throughput sequencing libraries from three independent ChIRP experiments were prepared and indexed using the ThruPLEX® DNA-seq Kit (Rubicon Genomics), purified on AMPure XP beads, then checked for quality and size range on Agilent Bioanalyzer using the High Sensitivity DNA Assay kit. Samples were then pooled and sequenced on Illumina HiSeq2000 with single read length of 100 bp (SR100). Raw reads were aligned to the human genome Hg19 using Bowtie2. Raw data and detailed procedures have been made publicly available on GEO, with accession GSE201567.
Peaks of each MIR205HG-ChIRP sample were called against LacZ signal using MACS2, with p-value cut-off of 1e-5. For each MACS2 peak, we filtered for regions sharing the same features in at least two of three independent experiments using the findConsensusPeakRegions function of the consensusSeekeR package v1.18.0 (Samb et al., 2015) in R environment (R version 4.0.3). We provided two inputs: peaks (files containing called peaks) and narrowPeaks (files containing called peak regions), both given by MACS2 as output. The package consensusSeekeR compares genomic positions and genomic ranges from multiple experiments to extract common regions. We took advantage of this tool and created consensus peaks extending the region of 500 bp on both sides of the position of the peak center. We shrank the region size, which is set by the extendingSize parameter, to fit the narrow peak regions of the peaks when all the regions were smaller than the consensus region through the shrinkToFitPeakRegion parameter. We finally obtained 5,064 consensus peaks, considered as the definitive peaks, characterized by an average length of 775 bp. Annotated consensus peaks are reported in Supplementary Table S1. The tracks of narrow peaks of single replicates and of consensus peaks are depicted in Supplementary Figure S2B. Enrichment of genomic features within consensus peaks was assessed using the annotatePeak function of ChipSeeker R package v1.26.2 giving −1,500; +500 from the transcription start site (TSS) as our custom definition of “proximal regions.” The TxDb was built from the Ensembl database, consequently the Ensembl annotation (EnsDb.Hsapiens.v75 v2.99.0) of transcripts was used. As a control, 1,000 lists of 5,000 random peaks of 775 bp of length (i.e., average length of MIR205HG peaks) were created with the bedtools toolset. We also run annotatePeaks.pl of HOMER software with default parameters (Human Genome as background) in order to compare results with the enrichments obtained by ChipSeeker.
We employed LOLA v1.24.0 (Sheffield and Bock, 2016), a Bioconductor package, to perform the enrichment analysis for genomic region sets and regulatory elements. We used the buildRestrictedUniverse function to build a universe based on query sets and to test for differential enrichment of regions against a background. As a control, a list of 500 random peaks of 775 bp of length falling in proximal regulatory regions (−1,500; +500 from the TSS) was created with the bedtools toolset. The tested Region Databases included LOLA standard features, collection of Alu sequences downloaded from Table Browser (hg19) and ChIP-seq data for histone modification patterns in RWPE-1 cells (GSE63094).
We used Regulatory Genomics Toolbox (RGT) (http://www.regulatory-genomics.org/tdf/basic-introduction/) (Kuo et al., 2019; Sentürk et al., 2019), an open-source python library, in order to find the triplex-forming potential between RNA and DNA regions of 15–20 base pair length (Kuo et al., 2019). We run the genomic region test function of the Triplex Domain Finder (TDF) tool, by providing the coordinates of consensus peaks in bed format and MIR205HG sequence (ENST00000429156.1). We also compared our results with different controls such as MEG3 peaks or a random list of 500 sequences spanning −1,500 to −500 bp from TSS. With the Sfold tool (https://sfold.wadsworth.org/cgi-bin/index.pl) (Ding and Lawrence, 2001; Ding and Lawrence, 2003; Zuker, 2003; Ding et al., 2004), we identified and then masked MIR205HG bases having less than 10% of probability of being single-stranded. The promoter test of TDF was used for the list of genes differentially expressed upon MIR205HG manipulation (GSE104003).
All bioinformatic analyses were performed in the R environment (R version 4.0.3). Statistical analysis was performed using Chi-square test to assess the difference between expected and observed frequencies and Jonckheere-Terpstra (JT) test to assess the monotone trend of different classes. When applying permutation analysis, empiric p-value was calculated as 1 divided for the number of times the observed percentage of a given feature in the set of random gene lists was statistically significantly different (based on multiple testing-corrected Chi-square p-value) from the percentage of the tested peak list. A threshold of 0.05 was considered statistically significant.
To map the chromatin occupancy of MIR205HG/LEADR at a genome-wide level in prostate basal cells, three independent MIR205HG-ChIRP experiments were performed on immortalized RWPE-1 cells, as described in Profumo et al. (2019) and subjected to high-throughput sequencing. Actual pull-down of MIR205HG (and not GAPDH) RNA with specific probes as compared to lacZ probes is reported for the three replicate experiments in Supplementary Figure S2A. Peaks in MIR205HG-ChIRP samples were called against the lacZ-ChIRP sample to correct for non-specific binding. Then consensus peaks shared by at least two out of three experiments were selected, resulting in 5,064 unique peaks (Supplementary Figure S2B, Supplementary Table S1). Such peaks were distributed all over the genome, however they overlapped more preferentially to regions proximal to gene TSS (−1,500 to +500 bp, hereafter defined as “proximal regions”) than to all other regions, when tested against 1,000 lists of 5,000 random regions of comparable length (775 bp) (∼1.3-fold enrichment: 10.23% in MIR205HG ChIRP peaks vs. 8.02% of averaged random regions, Chi-square test p = 0.000123; empirical p-value calculated upon permutation of random gene lists p = 0.0001) (Figure 1A). A similar analysis run with the HOMER tool confirmed significant (−logP = 7.3) enrichment of promoters (defined by the tool as −1,000 bp to +100 bp, so included in our “proximal regions”) and unearthed even higher enrichment (−logP = 1401) of SINE/Alu sequences, as compared to the background (Figure 1B). Overall, such observation is in trend with the enrichment of Alu sequences found in promoters of genes differentially expressed upon MIR205HG manipulation (Profumo et al., 2019). Notably, MIR205HG peaks were also significantly enriched in intronic regions, suggesting that the lncRNA may bind outside of proximal regulatory elements (Figure 1A). In this work, we decided to focus our attention on the specific features of MIR205HG binding to proximal regions.
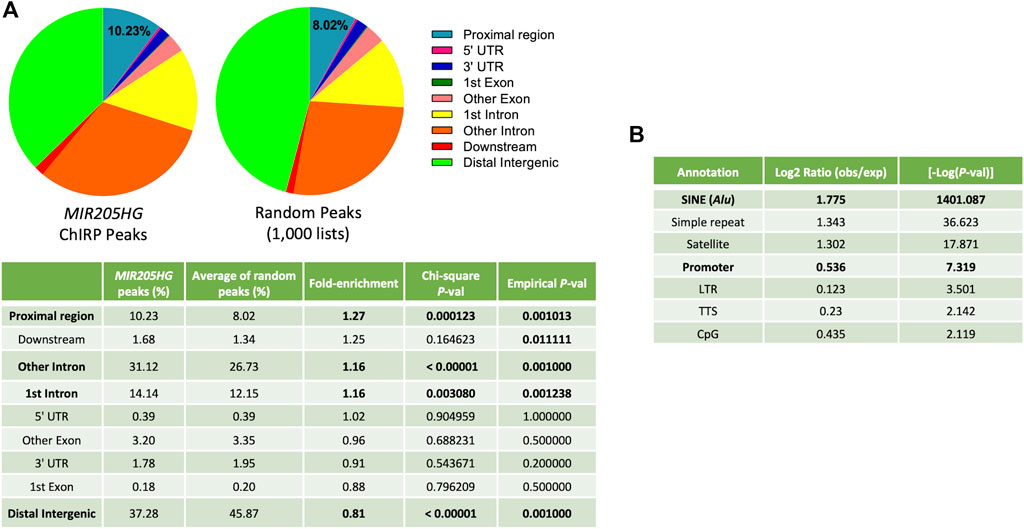
FIGURE 1. Genome distribution of MIR205HG ChIRP consensus peaks as compared to random regions. (A) Feature enrichment analysis performed using the annotatePeak function of ChipSeeker R package on 5,064 consensus peaks and on 1,000 lists of about 5,000 random peaks of equal nucleotide length. The pie plots were generated using GraphPad prism. Chi-square test p-values calculated against the average percentage of each feature in random gene lists and empirical p-values associated with permutation analyses are reported in the enclosed table. Proximal Region = −1,500 to +500 bp from gene TSS; Downstream = within 3,000 bp from gene termination. (B) Feature enrichment analysis performed using the annotatePeaks.pl function of HOMER software on 5,064 consensus peaks. The tables were generated using Microsoft Excel.
One of the emerging mechanisms by which lncRNAs seem to interact with chromatin is DNA/RNA triplexes (Figure 2A). In these structures, the single-stranded RNA of the lncRNA accommodates into the double helix of the DNA by forming Hoogsteen bonds [Reviewed in (Li et al., 2016)]. Therefore, we wondered whether MIR205HG could bind to the target DNA through the formation of triplexes. The analysis run with TDF tool (using 15 nt as the minimum triplex size) on the 518 peaks in proximal regions (corresponding to 448 genes), identified 349 regions able to form triplexes (the so-called DNA binding sites, DBS) with MIR205HG (Figure 2B), however their frequency was not significantly different from that of non-target regions.
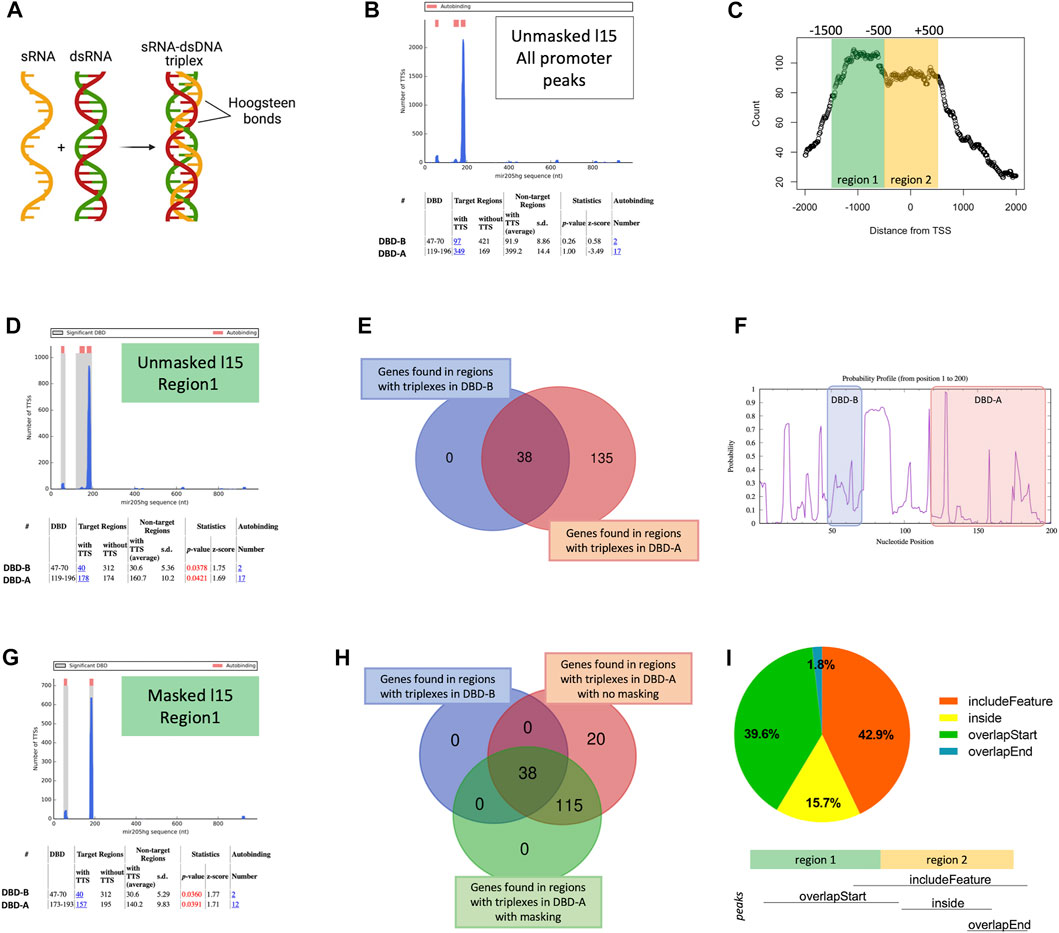
FIGURE 2. Enrichment of triplexes in MIR205HG proximal region peaks. (A) Representation of RNA/DNA triplex formation (created with BioRender.com). (B) TDF output for the unmasked analysis with minimum triplex length equal to 15 performed on 518 MIR205HG proximal region peaks. (C) Bimodal distribution of MIR205HG proximal region peaks around the TSS, generated using the binOverFeature function of the ChIPpeakAnno R package (v3.28.1). (D) TDF output of the unmasked analysis with minimum triplex length equal to 15 performed on “region 1”-cutpeaks. (E) Intersection between genes forming a triplex with 119–196 DBD-A (173) and genes forming a triplex with 47–70 DBD-B (38) of MIR205HG transcript. (F) Profile showing the probability of MIR205HG bases (form base position 1 to 200) of being single stranded. The DBD-B and DBD-A are highlighted. The image was generated by Sfold tool. (G) TDF output of the masked analysis with minimum triplex length equal to 15 performed on “region 1”-cutpeaks. (H) Intersection between genes forming a triplex with DBD-A (173) of MIR205HG with no masking, genes forming a triplex with DBD-B (38) and with 173–196 DBD-A with masking. (I) Degree of overlap between whole peaks and region 2 (−500/+500 bp from TSS) calculated using the findOverlappingPeaks function of the ChIPpeakAnno R package (v3.28.1). Results show that 42.9% of peaks included region 2 completely, though starting before (i.e., in region 1). This is indicated in the figure as “includeFeature”. 39.6% of peaks fell mainly in region 1, only overlapping with the start of region 2 (“overlapstart”); 15.7% fell completely within region 2 (“inside”); 1.8% of peaks overlapped with the end of region 2 (“overlapend”). Pie plot was generated using GraphPad prism. Venn diagrams were generated with the web accessible tool available at https://bioinformatics.psb.ugent.be/webtools/Venn/.
The analysis of ChIRP-seq read counts with respect to TSS however showed a bimodal distribution of proximal region peaks (Figure 2C), with higher density from −1,500 to −500 bp (region 1) and lower between −500 and +500 bp (region 2). Therefore, we generated “region 1” and “region 2” -cutpeaks as a result of the intersection between proximal region peaks and the abovementioned regions. Interestingly, TDF found significant enrichment of triplex-forming sequences in the first region (Figure 2D), and lack of enrichment in the latter (with even fewer triplexes than expected) (Supplementary Figure S3A). Specifically, DBS were found in 178 of 352 “region 1”-cutpeaks, corresponding to 173 genes. These DBS formed triplexes with 119–196 nt region of MIR205HG, hereafter referred to as putative DNA binding domain A (DBD-A). Notably, 40 out of 178 (=22.5%) “region 1”-cutpeaks forming a triplex with DBD-A were predicted to form an additional triplex with 47–70 nt region of MIR205HG (hereafter referred to as putative DBD-B), meaning that all DBS forming a triplex with DBD-B also form a triplex with the more downstream DBD-A, corresponding to 38 out of 173 genes (Figure 2E). Notably, DBD-A was confirmed to be significantly enriched in “region 1”-cutpeaks (49 DBS in 352 peaks) when the analysis was run using 20 nt as minimum triplex length, whereas DBD-B was lost (Supplementary Figure S3B).
Importantly, no significant enrichment of triplex-forming regions with MIR205HG was found in ChOP peaks of MEG3, an unrelated lncRNA known to form triplexes. Moreover, no triplex-forming regions with MEG3 were found in MIR205HG peaks spanning −1,500 to −500 bp from TSS. The other way around, triplex-forming regions with MIR205HG were not statistically enriched in a random list of 500 sequences spanning −1,500 to −500 bp from TSS.
It is known that lncRNAs may be highly structured and that some bases may not be available for triplex formation due to their involvement in RNA secondary structures (Matveishina et al., 2020). Therefore, we masked MIR205HG bases having a <10% likelihood of being single-stranded and thus being unavailable to form triplexes, and re-run TDF analysis. Notably, bases ranging from 47 to 70 of MIR205HG (DBD-B) all appeared to exceed the cut-off of accessibility, whereas DBD-A resulted to be highly accessible starting from base 174 (Figure 2F). TDF run using masked MIR205HG sequence again revealed enrichment of DBS in “region 1”-cutpeaks (Figure 2G), with 157 triplexes forming with DBD-A (all included in the 178 triplexes found in the “unmasked” analysis) and 40 (of 40 of the unmasked analysis) with DBD-B of MIR205HG (Figure 2H). It is to note that, according to this analysis, DBD-A appeared to be shorter, spanning bases 173–193 of MIR205HG sequence.
Overall these data suggest that MIR205HG peaks from −1,500 to −500 of TSS may account for a triplex-forming mechanism, whereas −500/+500 peaks may be alternatively region 1 peak tails or accounting for a different mechanism of binding. To explore this, we verified the degree of overlap between whole peaks and region 2. We found that 42.9% of peaks covered both regions (i.e., started in region 1 but included region 2 completely) whereas 39.6% and 17.5% (15.7% + 1.8%) of peaks fell specifically in region 1 and in region 2, respectively (Figure 2I). The existence of region-specific peaks may underscore the possibility that MIR205HG may bind to the DNA with different mechanisms, being however triplex formation the most prevalent modality (total peaks covering region 1 = 42.9% + 39.6% = 82.5%).
We then used LOLA to highlight features that could be specific to either bound region. We found exclusive enrichment of Alu sequences in “region 1”-cutpeaks and of transcription factor binding sites (TFBsite), DNAse, CpG islands, H3K4me3 in “region 2”-cutpeaks (Figure 3A). All other tested features resulted to be not significantly enriched overall in cutpeaks as compared to the background (Figure 3A). To exclude that feature enrichments could be region-specific and not associated with triplex formation, we run a comparative analysis between triplex-containing and triplex-less peaks in each region. In both cases, triplex-containing peaks were enriched in Alu and H3K4me1 and devoid of CpG, H3K27me3, and H3K4me3 (Figure 3B). In region 1, they were also enriched in H3K36me3 and devoid of TFBS. These results seem to confirm that some feature enrichments are specific of MIR205HG modality of binding (triplex yes vs. no), rather than being solely region-specific. Potentially, this may also underscore a dual mode of MIR205HG binding, the first and more prevalent involving triplex formation in proximity of Alu sequences and H3K4me1 modification, more frequently occurring in region 1; the latter, possibly mediated by the interaction with TFs and accompanied by H3K4me3 and H3K27me3, more frequently occurring in region 2.
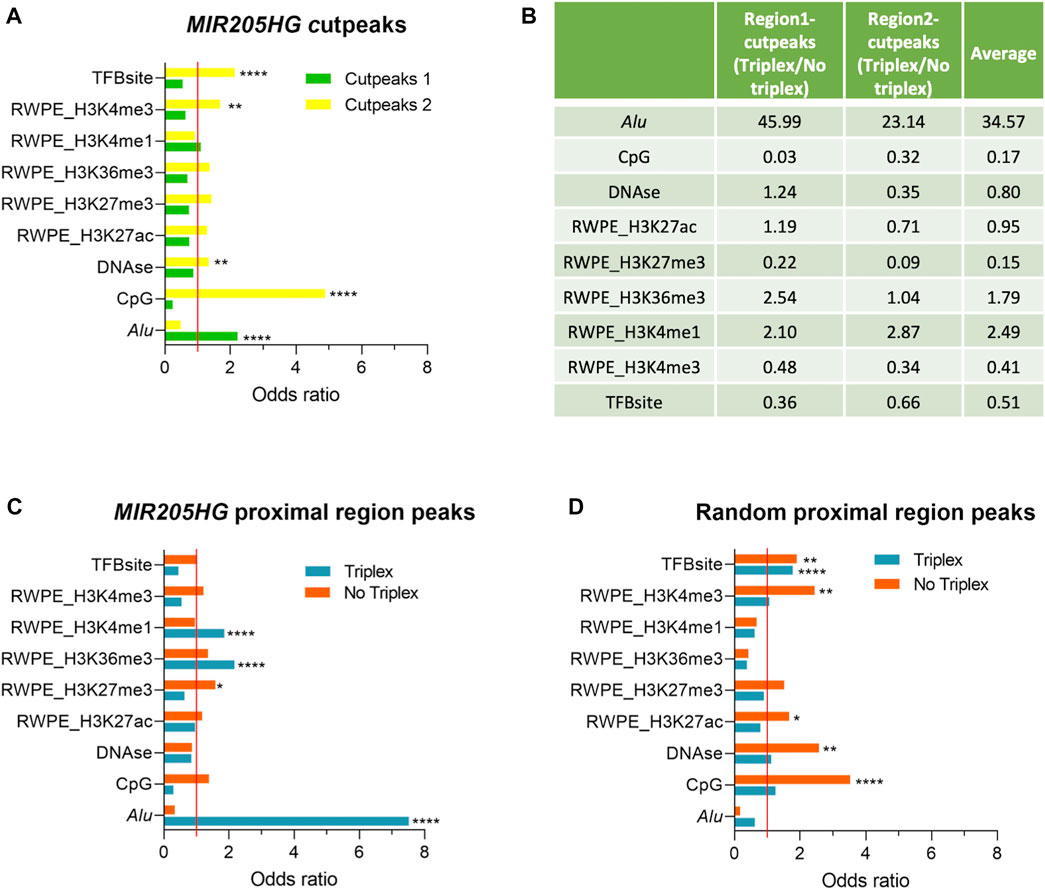
FIGURE 3. Enrichment of Alu and specific histone modifications in MIR205HG proximal region peaks. (A) The bar plot shows the odds ratios for the “region 1”- and “region 2”-cutpeaks for each LOLA library. Line set to 1 represents no enrichment as compared to the background. (B) For each LOLA library, the ratio of the odds ratios in triplex-containing and in triplex-less peaks among “region 1” and “region 2”-cutpeaks, together with their average, are reported to show the relative enrichment in presence and absence of the triplex. (C) The bar plot shows the odds ratios for MIR205HG proximal region peaks divided into triplex-forming and triplex-less peaks for each LOLA library. (D) The bar plot shows the odds ratios for random proximal region peaks divided into triplex-forming and triplex-less peaks for each LOLA library. The computed p-Value is reported (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Bar plots and the table were generated using GraphPad Prism and Microsoft Excel, respectively.
We focused on the triplex-mediated mechanism and to test it from a more general perspective, we went back to the analysis of all proximal region peaks. To increase specificity, we run TDF with 0.1 masking on whole peaks (not “cut” for regions). As previously observed in the unmasked analysis, we did not find any significant enrichment of triplexes overall, however we could detect 306 triplexes forming with site DBD-A (in 283 genes) and 97 with site DBD-B (in 93 genes, all included in those forming triplex with DBD-A) (Supplementary Figure S3C). Overall, 59% of peaks had at least a triplex, which accounts for 63% of genes.
The LOLA analysis run on all proximal region peaks based on the presence/absence of the triplex, regardless of peak position with respect to TSS, confirmed enrichment of Alu, H3K4me1 and H3K36me3 in triplex-containing peaks, and H3K27me3 in triplex-less peaks (Figure 3C).
As a further validation, we run TDF to find triplex-forming sequences in a list of 500 random regions in the range of −1,500/+500 from TSS and then analyzed with LOLA. Notably, no enrichment of Alu, H3K4me1 or H3K36me3 was found in this case (Figure 3D), suggesting that 1) triplexes predicted in MIR205HG peaks may be true and that 2) features associated with them are specifically linked to MIR205HG mode of binding. For example, striking enrichment of Alu was found exclusively in MIR205HG peaks with triplex and not in triplex-less MIR205HG peaks, nor in unrelated proximal regions regardless of potential predicted triplexes.
The results are reminiscent of the enrichment of Alu sequences that we found in proximal regions of genes deregulated upon MIR205HG manipulation, as from microarray analysis (Profumo et al., 2019; Percio et al., 2020). TDF analysis run on such gene lists revealed significant enrichment of triplex-forming sequences in bona fide MIR205HG target genes (i.e., “MIR205HG-core up”) as compared to non-target genes (Figure 4A), which formed triplexes with DBD element of the lncRNA. Moreover, the simultaneous presence of DBS and Alu element in gene proximal regions was prominent in most markedly modulated genes and tended to decrease with fold-change (Figures 4B,C).
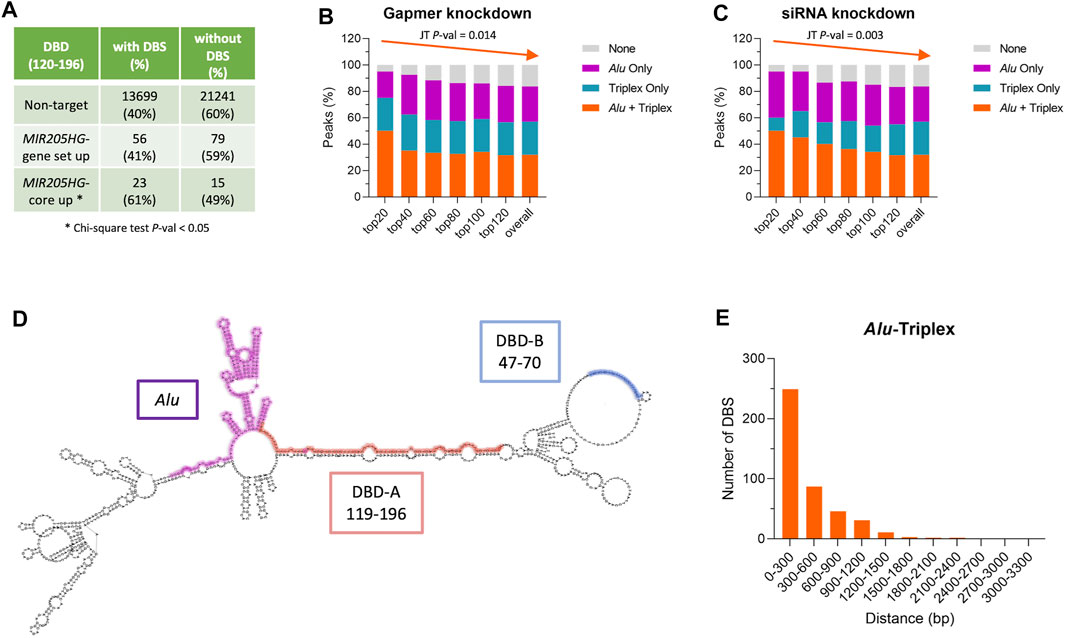
FIGURE 4. The presence of Alu and triplex-forming regions is a feature of both MIR205HG and its target genes. (A) Table showing the number and percentage of genes predicted to form or not a triplex with MIR205HG in lists of non-target genes (“Non-target”), genes commonly up-modulated after MIR205HG knockdown using either a siRNA or a gapmer (“MIR205HG-gene set up”), and genes commonly upmodulated after MIR205HG knockdown and coherently downmodulated after MIR205HG overexpression (“MIR205HG-core up”). Gene lists are from Profumo et al. (2019). (B) Bar plot showing the percentage of genes having a DBS (i.e., a triplex) and/or Alu element among genes differentially expressed after MIR205HG knockdown using a gapmer antisense oligonucleotide. Significance of the monotone trend of triplex + Alu percentage in regulated genes ranked for fold-change (top20 genes have the highest fold-change) was assessed by Jonckheere-Terpstra (JT) test. (C) Bar plot showing the percentage of genes having a DBS (i.e., a triplex) and/or Alu elements among genes differentially expressed after MIR205HG knockdown using a siRNA. Significance of the monotone trend of triplex + Alu percentage in regulated genes ranked for fold-change (top20 genes have the highest fold-change) was assessed by Jonckheere-Terpstra (JT) test. (D) MIR205HG predicted secondary structure (as from RNAfold); DBD-A, DBD-B and Alu elements are highlighted in the structure. (E) Distance between the triplex (DBS) and the Alu element in MIR205HG proximal region peaks forming a triplex. Bar plots and the table were generated using GraphPad Prism and Microsoft Excel, respectively.
Analysis of MIR205HG secondary structure (as from RNAfold web service, http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) (Gruber et al., 2008; Lorenz et al., 2011) revealed that the putative DBDs, which are located just upstream of the Alu element, are poorly structured (Figure 4D), thus making them prone to triplex formation. In contrast, the Alu element is highly structured, making the protein-mediated interaction with Alu sequence on target genes more plausible than the direct DNA/RNA pairing. Most MIR205HG target genes as from ChIRP-seq analysis revealed to have triplex/DBS at a distance of less than 300 nt from their Alu element (Figure 4E), which would allow interaction with DBD and Alu on MIR205HG without any need of bending of either the DNA or the RNA. This is exemplified in Supplementary Figure S4, where some examples of genes having MIR205HG peak in their proximal regions are reported, together with indication of their triplex-forming region (DBS) and Alu element.
Aside Alu, features enriched exclusively in MIR205HG triplex-forming regions (not in other regions, even not in triplex-forming regions of random proximal regions) were H3K4me1 and H3K36me3 (Figure 3C, Supplementary Figure S4). Other features previously found to be enriched in triplex-less MIR205HG peaks (CpG, H3K4me3, H3K27me3, Figure 3B) showed a tendency to be enriched in triplex-less regions also in random proximal regions (Figure 3D), thus not allowing us to draw specific conclusions on whether and how MIR205HG may work in a triplex-independent modality.
Understanding of lncRNA function passes through the identification of elements that allow RNA to interact with other molecules (Graf and Kretz, 2020; Ohyama et al., 2020). In some cases, such interaction is simply mediated by complementarity between nucleic acids, as in the miRNA sponging mechanism. In most circumstances, however, more complex secondary and tertiary structures need to form to allow interaction with double-stranded DNA or proteins (Tassinari et al., 2021). In this regard, the primary sequence of lncRNAs is only poorly informative of the function of a given lncRNA, and curiously also less conserved than higher-order structures (Johnsson et al., 2014). It is however conceivable that, just alike proteins, lncRNAs may act through discrete, modular functional elements (Przanowska et al., 2022). Recognition of such structural motifs is together the main obstacle and the key to define the exact mechanism of action of lncRNAs.
Our previous work demonstrated, for the first time, the pivotal role of the chromatin-associated lncRNA MIR205HG/LEADR in controlling the basal phenotype of prostate epithelial cells (Profumo et al., 2019). Understanding the mechanism of action of MIR205HG/LEADR will allow to dissect the processes governing differentiation of epithelial cells, which often appear deregulated in cancer (Ferrari and Gandellini, 2020), and eventually lay the foundations for the development of novel therapeutic approaches for tumors where MIR205HG/LEADR expression is deregulated (Di Agostino et al., 2018; Li et al., 2019; Liu et al., 2020; Song et al., 2021; Dong et al., 2022). For a discussion on the potential (yet to be disclosed) role of MIR205HG in prostate cancer refer to Profumo et al. (2019).
Here, by analyzing the genome-wide chromatin occupancy of MIR205HG/LEADR in prostate basal cells, we provide initial clues into MIR205HG/LEADR functional modules. Specifically, we recognized two regions potentially essential for interaction with chromatin: 1) a highly structured Alu (RNA) element that potentially binds to Alu (DNA) motifs on target gene proximal regions, likely through protein intermediates, and 2) a poorly structured DBD responsible to form triplexes with regions located upstream of target genes; such domain should allow the physical direct interaction with the DNA double helix and simultaneously provide specificity for target genes over the plethora of Alu elements in the human genome. While we had previously showed that deletion of Alu element from MIR205HG sequence abrogated its capability to regulate gene expression, at least in part by impairing binding to target gene proximal regions, the functionality and real contribution of the predicted DBDs to DNA binding through triplex formation remain to be validated through ad hoc experiments. These may include selective deletion of either DBDs followed by analysis of MIR205HG chromatin occupancy, as well as direct biochemical assays to assess formation of triplex with target genes (as in Mondal et al., 2015; Sentürk et al., 2019).
Accumulating evidence shows that the formation of triplexes, especially with target gene regulatory regions, is a mechanism shared by different nuclear lncRNAs, such as MEG3 (Mondal et al., 2015)—used as control in our experiments—HOTAIR (Kalwa et al., 2016) and KHPS1 (Blank-Giwojna et al., 2019). In this regard, Sentürk et al. (2019) have recently provided evidence of the existence and physiological relevance of DNA/RNA triplexes in vivo. In their work, the authors also showed that triplexes can form at active chromatin domains and in trans with distant genomic loci.
The possible role of Alu elements as functional domains of lncRNAs has been proposed in 2014 as the so-called RIDL hypothesis (where RIDL stands for Repeat Insertion Domain of LncRNAs), mainly based on the correlative evidence of enrichment of Alu in lncRNA exons (Johnson and Guigó, 2014; Kim et al., 2016). For some lncRNAs, the essential role of Alu domains in function has been proven by deletion experiments, as in the case of ANRIL (Holdt et al., 2013) and MIR205HG/LEADR (Profumo et al., 2019). From the mechanistic point of view, most of the literature focuses on the “cytoplasmic activities” of Alu RNA domains. Imperfect base-pairing between Alu elements in the 3′-UTR of mRNAs and Alu elements in cytoplasmic lncRNAs was shown to transactivate STAU1-mediated mRNA decay (Gong and Maquat, 2011). Alu-directed sense to antisense interaction was demonstrated to be the mechanism by which SINEUPs, a new functional class of natural antisense lncRNAs (Schein et al., 2016), select mRNAs with overlapping 5′-UTR to ultimately increase their translation, a process, that is, mediated by a distinct effector domain (Spinoza et al., 2021). Sequences enriched in Alu repeats have been also reported to drive nuclear localization of long RNAs in human cells (Lubelsky and Ulitsky, 2018), even if this is not the case of MIR205HG (Profumo et al., 2019). Regarding the “nuclear” activity of Alu elements, Alu/Alu direct pairing was proposed between APTR lncRNA and the promoter of CDKN1A/p21 (Negishi et al., 2014), as well as between the splicing-regulatory lncRNA 5S-OT and its targets (Hu et al., 2016). In contrast, Alu elements in the human lincRNA-p21 need to adopt a conserved secondary structure to regulate RNA function and localization (Chillón and Pyle, 2016). Also the Alu element of ANRIL was shown to form a stem-loop structure. Whether structured Alu domains are the scaffolds for the protein-mediated interaction with other Alu elements or more in general are the platforms for effector proteins is an aspect that still needs extensive investigation.
The possible link between triplex formation and the presence of the Alu in either the lncRNA or target genes is also a poorly developed issue. In this regard, Sentürk et al. (2019) showed that a large fraction of triplex-forming RNAs is enriched with repeat elements, thus suggesting that repeat-derived sequences may represent functional domains that target regulatory RNAs to distant genomic regions. In addition, triplex-forming DNA sequences have a propensity to harbor significantly more SINE and LTR elements than control DNA, with Alu and ERVL subclasses being predominant. This finding supports the notion that repetitive DNA sequences might serve an important function in tethering regulatory RNAs to specific genomic regions, as is for MIR205HG/LEADR. Partially in contrast with these and our results are the findings by Bai et al. (2021), who reported Alu enrichment in RNA:DNA hybrids (R-loops), but depletion in DNA:DNA and RNA:DNA triplexes.
Our analysis showed that triplex-containing MIR205HG/LEADR peaks were enriched in H3K4me1 in the absence of H3K27ac, H3K4me3 and H3K27me3, a feature that has been historically associated with primed enhancers, i.e., enhancers characterized by accessible chromatin conformation despite being transcriptionally repressed (Calo and Wysocka, 2013). This evidence is reminiscent of what has been reported for super-lncRNAs, a class of tissue-restricted lncRNAs that target and contribute to the local chromatin organization of the super-enhancers (i.e., regions in the genome containing multiple enhancers which drive transcription of genes involved in cell identity). Such lncRNAs harbor a single triplex-forming repeat domain, which forms an RNA:DNA triplex with multiple anchor DNA sites originating from transposable elements within the super-enhancers (Soibam, 2017). Therefore, preferential binding to enhancer-like regions through a triplex- and Alu-based mechanism seems to be a shared feature between super-lncRNAs and MIR205HG. Notably, Alu elements are found in most enhancers, which may suggest them as proto-enhancers in primates (Feschotte, 2008). In this regard, we should acknowledge that the mentioned histone modification pattern can be found in both distal and proximal enhancers, as well as in promoters (Bae and Lesch, 2020). As an alternative to the classical positional definition, promoters and enhancers may be considered as a single class of transcriptional elements distinguished by different levels of transcription, which are then associated with different ratios between H3K4me1 and other histone modifications. Whether MIR205HG-bound elements work as proximal enhancers or promoters of the neighboring genes should be assessed for each individual gene using the appropriate assays. From a global perspective, the “primed” histone modification pattern found to be associated with MIR205HG at proximal regions may underscore the need of basal cells to keep MIR205HG/LEADR target genes repressed but, at the same time, responsive to differentiation cues. Curiously, another triplex-forming lncRNA, MEG3, was found to preferentially interact with promoter-distal sites enriched in H3K4me1 and H3K27me3, which are instead characteristic of poised enhancers (Calo and Wysocka, 2013).
Further investigation will be required to understand whether MIR205HG/LEADR is itself responsible for the recruitment of histone modifying complexes at bound sites, thus directly regulating the chromatin state of target genes, and through which structural domain. In this regard, other triplex-forming lncRNAs, such as MEG3, HOTAIR and PARTICLE, were shown to recruit PRC2 complexes to promoters of target genes to drive epigenetic silencing (O'Leary et al., 2015). The Alu-containing lncRNA ANRIL was itself shown to regulate target gene expression through histone methylation complexes. In all of the mentioned cases, the lncRNA structural motifs responsible to recruit and transport necessary regulators to promoter/enhancers of target genes have been either yet not identified or shown to be distinct from the triplex-forming domain. It is not even unlikely that Alu elements may themselves participate in this process.
Overall, here we provided initial clues into MIR205HG/LEADR possible mechanism of action at the chromatin level, further corroborating the emerging role of Alu elements as functional RNA domains of lncRNAs and key regulatory DNA elements in proximal regions. We also confirmed triplex formation as a prominent mechanism of DNA binding for chromatin-interacting lncRNAs. Further experimental validation is warranted to assess the real contribution of the suggested functional elements and modality of DNA binding, as well as the mechanism by which MIR205HG is then able to directly modulate the expression of target genes. If successful, such experiments may inform on the role that MIR205HG/LEADR may have in the differentiation of basal cells from various tissue contexts (e.g., breast, skin) and stimulate interest regarding the contribution of aberrant differentiation programs to epithelial carcinogenesis.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE201567.
EB and GP performed computational analyses, helped drafting the manuscript, prepared the figures. BF performed wet experiment. SP performed part of statistical analyses. NZ contributed to supervise the work and draft the manuscript, and obtained funding. DD assisted in designing computational analyses. PG conceived and designed the study, obtained funding, supervised the work and wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by grants from: Italian Ministry of Health (GR-2013-02355625 to PG), CARIPLO Foundation (2015-0866 to PG) and I. Monzino Foundation (to NZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We authors would like to thank Veronica De Sanctis and Roberto Bertorelli from the Next Generation Sequencing Facility at CIBIO, University of Trento, for technical support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.909097/full#supplementary-material
Bae, S., and Lesch, B. J. (2020). H3K4me1 Distribution Predicts Transcription State and Poising at Promoters. Front. Cell Dev. Biol. 8, 289. doi:10.3389/fcell.2020.00289
Bai, X., Li, F., and Zhang, Z. (2021). A Hypothetical Model of Trans-acting R-Loops-Mediated Promoter-Enhancer Interactions by Alu Elements. J. Genet. Genomics 48 (11), 1007–1019. doi:10.1016/j.jgg.2021.07.005
Blank-Giwojna, A., Postepska-Igielska, A., and Grummt, I. (2019). lncRNA KHPS1 Activates a Poised Enhancer by Triplex-dependent Recruitment of Epigenomic Regulators. Cell Rep. 26 (11), 2904–2915. doi:10.1016/j.celrep.2019.02.059
Calo, E., and Wysocka, J. (2013). Modification of Enhancer Chromatin: what, How, and Why? Mol. Cell 49 (5), 825–837. doi:10.1016/j.molcel.2013.01.038
Cava, C., Armaos, A., Lang, B., Tartaglia, G. G., and Castiglioni, I. (2022). Identification of Long Non-coding RNAs and RNA Binding Proteins in Breast Cancer Subtypes. Sci. Rep. 12 (1), 693. doi:10.1038/s41598-021-04664-z
Chillón, I., and Pyle, A. M. (2016). Inverted repeatAluelements in the Human lincRNA-P21 Adopt a Conserved Secondary Structure that Regulates RNA Function. Nucleic Acids Res. 44 (19), gkw599–9471. doi:10.1093/nar/gkw599
Chu, C., Qu, K., Zhong, F. L., Artandi, S. E., and Chang, H. Y. (2011). Genomic Maps of Long Noncoding RNA Occupancy Reveal Principles of RNA-Chromatin Interactions. Mol. Cell 44 (4), 667–678. doi:10.1016/j.molcel.2011.08.027
Di Agostino, S., Valenti, F., Sacconi, A., Fontemaggi, G., Pallocca, M., Pulito, C., et al. (2018). Long Non-coding MIR205HG Depletes Hsa-miR-590-3p Leading to Unrestrained Proliferation in Head and Neck Squamous Cell Carcinoma. Theranostics 8 (7), 1850–1868. doi:10.7150/thno.22167
Ding, Y., Chan, C. Y., and Lawrence, C. E. (2004). Sfold Web Server for Statistical Folding and Rational Design of Nucleic Acids. Nucleic Acids Res. 32, W135–W141. doi:10.1093/nar/gkh449
Ding, Y., and Lawrence, C. E. (2003). A Statistical Sampling Algorithm for RNA Secondary Structure Prediction. Nucleic Acids Res. 31 (24), 7280–7301. doi:10.1093/nar/gkg938
Ding, Y., and Lawrence, C. E. (2001). Statistical Prediction of Single-Stranded Regions in RNA Secondary Structure and Application to Predicting Effective Antisense Target Sites and beyond. Nucleic Acids Res. 29 (5), 1034–1046. doi:10.1093/nar/29.5.1034
Dong, X., Chen, X., Lu, D., Diao, D., Liu, X., Mai, S., et al. (2022). LncRNA miR205HG Hinders HNRNPA0 Translation: Anti‐oncogenic Effects in Esophageal Carcinoma. Mol. Oncol. 16 (3), 795–812. doi:10.1002/1878-0261.13142
Espinoza, S., Bon, C., Valentini, P., Pierattini, B., Matey, A. T., Damiani, D., et al. (2021). SINEUPs: a Novel Toolbox for RNA Therapeutics. Essays Biochem. 65 (4), 775–789. doi:10.1042/EBC20200114
Ferrari, E., and Gandellini, P. (2020). Unveiling the Ups and Downs of miR-205 in Physiology and Cancer: Transcriptional and Post-transcriptional Mechanisms. Cell Death Dis. 11 (11), 980. doi:10.1038/s41419-020-03192-4
Feschotte, C. (2008). Transposable Elements and the Evolution of Regulatory Networks. Nat. Rev. Genet. 9 (5), 397–405. doi:10.1038/nrg2337
Gandellini, P., Profumo, V., Casamichele, A., Fenderico, N., Borrelli, S., Petrovich, G., et al. (2012). miR-205 Regulates Basement Membrane Deposition in Human Prostate: Implications for Cancer Development. Cell Death Differ. 19 (11), 1750–1760. doi:10.1038/cdd.2012.56
Gong, C., and Maquat, L. E. (2011). lncRNAs Transactivate STAU1-Mediated mRNA Decay by Duplexing with 3′ UTRs via Alu Elements. Nature 470 (7333), 284–288. doi:10.1038/nature09701
Graf, J., and Kretz, M. (2020). From Structure to Function: Route to Understanding lncRNA Mechanism. Bioessays 42 (12), 2000027. doi:10.1002/bies.202000027
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neubock, R., and Hofacker, I. L. (2008). The Vienna RNA Websuite. Nucleic Acids Res. 36, W70–W74. doi:10.1093/nar/gkn188
Guo, J., Gan, Q., Gan, C., Zhang, X., Ma, X., and Dong, M. (2021). LncRNA MIR205HG Regulates Melanomagenesis via the miR-299-3p/VEGFA axis. Aging 13 (4), 5297–5311. doi:10.18632/aging.202450
Holdt, L. M., Hoffmann, S., Sass, K., Langenberger, D., Scholz, M., Krohn, K., et al. (2013). Alu Elements in ANRIL Non-coding RNA at Chromosome 9p21 Modulate Atherogenic Cell Functions through Trans-regulation of Gene Networks. PLoS Genet. 9 (7), e1003588. doi:10.1371/journal.pgen.1003588
Hu, S., Wang, X., and Shan, G. (2016). Insertion of an Alu Element in a lncRNA Leads to Primate-specific Modulation of Alternative Splicing. Nat. Struct. Mol. Biol. 23 (11), 1011–1019. doi:10.1038/nsmb.3302
Johnson, R., and Guigó, R. (2014). The RIDL Hypothesis: Transposable Elements as Functional Domains of Long Noncoding RNAs. RNA 20 (7), 959–976. doi:10.1261/rna.044560.114
Johnsson, P., Lipovich, L., Grandér, D., and Morris, K. V. (2014). Evolutionary Conservation of Long Non-coding RNAs; Sequence, Structure, Function. Biochimica Biophysica Acta (BBA) - General Subj. 1840 (3), 1063–1071. doi:10.1016/j.bbagen.2013.10.035
Kalwa, M., Hänzelmann, S., Otto, S., Kuo, C.-C., Franzen, J., Joussen, S., et al. (2016). The lncRNA HOTAIR Impacts on Mesenchymal Stem Cellsviatriple Helix Formation. Nucleic Acids Res. 44 (22), 10631–10643. doi:10.1093/nar/gkw802
Kim, E. Z., Wespiser, A. R., and Caffrey, D. R. (2016). The Domain Structure and Distribution of Alu Elements in Long Noncoding RNAs and mRNAs. RNA 22 (2), 254–264. doi:10.1261/rna.048280.114
Kuo, C.-C., Hänzelmann, S., Sentürk Cetin, N., Frank, S., Zajzon, B., Derks, J.-P., et al. (2019). Detection of RNA-DNA Binding Sites in Long Noncoding RNAs. Nucleic Acids Res. 47 (6), e32. doi:10.1093/nar/gkz037
Li, Y., Syed, J., and Sugiyama, H. (2016). RNA-DNA Triplex Formation by Long Noncoding RNAs. Cell Chem. Biol. 23 (11), 1325–1333. doi:10.1016/j.chembiol.2016.09.011
Li, Y., Wang, H., and Huang, H. (2019). Long Non-coding RNA MIR205HG Function as a ceRNA to Accelerate Tumor Growth and Progression via Sponging miR-122-5p in Cervical Cancer. Biochem. Biophysical Res. Commun. 514 (1), 78–85. doi:10.1016/j.bbrc.2019.04.102
Liu, L., Li, Y., Zhang, R., Li, C., Xiong, J., and Wei, Y. (2020). MIR205HG Acts as a ceRNA to Expedite Cell Proliferation and Progression in Lung Squamous Cell Carcinoma via Targeting miR-299-3p/MAP3K2 axis. BMC Pulm. Med. 20 (1), 163. doi:10.1186/s12890-020-1174-2
Lorenz, R., Bernhart, S. H., Höner zu Siederdissen, C., Tafer, H., Flamm, C., Stadler, P. F., et al. (2011). ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26. doi:10.1186/1748-7188-6-26
Lubelsky, Y., and Ulitsky, I. (2018). Sequences Enriched in Alu Repeats Drive Nuclear Localization of Long RNAs in Human Cells. Nature 555 (7694), 107–111. doi:10.1038/nature25757
Mas-Ponte, D., Carlevaro-Fita, J., Palumbo, E., Hermoso Pulido, T., Guigo, R., and Johnson, R. (2017). LncATLAS Database for Subcellular Localization of Long Noncoding RNAs. Rna 23 (7), 1080–1087. doi:10.1261/rna.060814.117
Matveishina, E., Antonov, I., and Medvedeva, Y. A. (2020). Practical Guidance in Genome-wide RNA:DNA Triple Helix Prediction. Ijms 21 (3), 830. doi:10.3390/ijms21030830
Mondal, T., Subhash, S., Vaid, R., Enroth, S., Uday, S., Reinius, B., et al. (2015). MEG3 Long Noncoding RNA Regulates the TGF-β Pathway Genes through Formation of RNA-DNA Triplex Structures. Nat. Commun. 6, 7743. doi:10.1038/ncomms8743
Negishi, M., Wongpalee, S. P., Sarkar, S., Park, J., Lee, K. Y., Shibata, Y., et al. (2014). A New lncRNA, APTR, Associates with and Represses the CDKN1A/p21 Promoter by Recruiting Polycomb Proteins. PLoS One 9 (4), e95216. doi:10.1371/journal.pone.0095216
Ohyama, T., Takahashi, H., Sharma, H., Yamazaki, T., Gustincich, S., Ishii, Y., et al. (2020). An NMR-Based Approach Reveals the Core Structure of the Functional Domain of SINEUP lncRNAs. Nucleic Acids Res. 48 (16), 9346–9360. doi:10.1093/nar/gkaa598
O’Leary, V. B., Ovsepian, S. V., Carrascosa, L. G., Buske, F. A., Radulovic, V., Niyazi, M., et al. (2015). PARTICLE, a Triplex-Forming Long ncRNA, Regulates Locus-specific Methylation in Response to Low-Dose Irradiation. Cell Rep. 11 (3), 474–485. doi:10.1016/j.celrep.2015.03.043
Pandini, C., Garofalo, M., Rey, F., Garau, J., Zucca, S., Sproviero, D., et al. (2021). MINCR: A Long Non-coding RNA Shared between Cancer and Neurodegeneration. Genomics 113 (6), 4039–4051. doi:10.1016/j.ygeno.2021.10.008
Percio, S., Rotundo, F., and Gandellini, P. (2020). Gene Expression Dataset of Prostate Cells upon MIR205HG/LEADR Modulation. Data Brief 29, 105139. doi:10.1016/j.dib.2020.105139
Profumo, V., Forte, B., Percio, S., Rotundo, F., Doldi, V., Ferrari, E., et al. (2019). LEADeR Role of miR-205 Host Gene as Long Noncoding RNA in Prostate Basal Cell Differentiation. Nat. Commun. 10 (1), 307. doi:10.1038/s41467-018-08153-2
Przanowska, R. K., Weidmann, C. A., Saha, S., Cichewicz, M. A., Jensen, K. N., Przanowski, P., et al. (2022). Distinct MUNC lncRNA Structural Domains Regulate Transcription of Different Promyogenic Factors. Cell Rep. 38 (7), 110361. doi:10.1016/j.celrep.2022.110361
Rossi, T., Pistoni, M., Sancisi, V., Gobbi, G., Torricelli, F., Donati, B., et al. (2020). RAIN Is a Novel Enhancer-Associated lncRNA that Controls RUNX2 Expression and Promotes Breast and Thyroid Cancer. Mol. Cancer Res. 18 (1), 140–152. doi:10.1158/1541-7786.MCR-19-0564
Samb, R., Khadraoui, K., Belleau, P., Deschênes, A., Lakhal-Chaieb, L., and Droit, A. (2015). Using Informative Multinomial-Dirichlet Prior in a T-Mixture with Reversible Jump Estimation of Nucleosome Positions for Genome-wide Profiling. Stat. Appl. Genet. Mol. Biol. 14 (6), 517–532. doi:10.1515/sagmb-2014-0098
Schein, A., Zucchelli, S., Kauppinen, S., Gustincich, S., and Carninci, P. (2016). Identification of Antisense Long Noncoding RNAs that Function as SINEUPs in Human Cells. Sci. Rep. 6, 33605. doi:10.1038/srep33605
Sentürk Cetin, N., Kuo, C.-C., Ribarska, T., Li, R., Costa, I. G., and Grummt, I. (2019). Isolation and Genome-wide Characterization of Cellular DNA:RNA Triplex Structures. Nucleic Acids Res. 47 (5), 2306–2321. doi:10.1093/nar/gky1305
Sheffield, N. C., and Bock, C. (2016). LOLA: Enrichment Analysis for Genomic Region Sets and Regulatory Elements in R and Bioconductor. Bioinformatics 32 (4), 587–589. doi:10.1093/bioinformatics/btv612
Soibam, B. (2017). Super-lncRNAs: Identification of lncRNAs that Target Super-enhancers via RNA:DNA:DNA Triplex Formation. RNA 23 (11), 1729–1742. doi:10.1261/rna.061317.117
Song, J. H., Tieu, A. H., Cheng, Y., Ma, K., Akshintala, V. S., Simsek, C., et al. (2021). Novel Long Noncoding RNA miR205HG Functions as an Esophageal Tumor-Suppressive Hedgehog Inhibitor. Cancers 13 (7), 1707. doi:10.3390/cancers13071707
Statello, L., Guo, C.-J., Chen, L.-L., and Huarte, M. (2021). Gene Regulation by Long Non-coding RNAs and its Biological Functions. Nat. Rev. Mol. Cell Biol. 22 (2), 96–118. doi:10.1038/s41580-020-00315-9
Tassinari, M., and Gandellini, P. (2021). Noncoding RNAs in the Interplay between Tumor Cells and Cancer-Associated Fibroblasts: Signals to Catch and Targets to Hit. Cancers 13 (4), 709. doi:10.3390/cancers13040709
Tassinari, M., Richter, S. N., and Gandellini, P. (2021). Biological Relevance and Therapeutic Potential of G-Quadruplex Structures in the Human Noncoding Transcriptome. Nucleic Acids Res. 49 (7), 3617–3633. doi:10.1093/nar/gkab127
Ulitsky, I., and Bartel, D. P. (2013). lincRNAs: Genomics, Evolution, and Mechanisms. Cell 154 (1), 26–46. doi:10.1016/j.cell.2013.06.020
Keywords: long noncoding RNA, triplex, Alu, ChIRP, sequencing
Citation: Bezzecchi E, Pagani G, Forte B, Percio S, Zaffaroni N, Dolfini D and Gandellini P (2022) MIR205HG/LEADR Long Noncoding RNA Binds to Primed Proximal Regulatory Regions in Prostate Basal Cells Through a Triplex- and Alu-Mediated Mechanism. Front. Cell Dev. Biol. 10:909097. doi: 10.3389/fcell.2022.909097
Received: 31 March 2022; Accepted: 31 May 2022;
Published: 17 June 2022.
Edited by:
Giacomo Cossa, Julius Maximilian University of Würzburg, GermanyReviewed by:
Claudia Cava, National Research Council (IBFM-CNR), ItalyCopyright © 2022 Bezzecchi, Pagani, Forte, Percio, Zaffaroni, Dolfini and Gandellini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Gandellini, cGFvbG8uZ2FuZGVsbGluaUB1bmltaS5pdA==
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.