
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 17 August 2022
Sec. Evolutionary Developmental Biology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.900321
 Cheryl W.Y. Shum1†
Cheryl W.Y. Shum1† Wenyan Nong1†
Wenyan Nong1† Wai Lok So1†
Wai Lok So1† Yiqian Li1†
Yiqian Li1† Zhe Qu1Ho Yin Yip1Thomas Swale2
Zhe Qu1Ho Yin Yip1Thomas Swale2 Put O. Ang3
Put O. Ang3 King Ming Chan4
King Ming Chan4 Ting Fung Chan5
Ting Fung Chan5 Ka Hou Chu4,6
Ka Hou Chu4,6 Apple P.Y. Chui4
Apple P.Y. Chui4 Kwok Fai Lau4
Kwok Fai Lau4 Sai Ming Ngai5
Sai Ming Ngai5 Fei Xu7
Fei Xu7 Jerome H.L. Hui1*
Jerome H.L. Hui1*Cnidarians including sea anemones, corals, hydra, and jellyfishes are a group of animals well known for their regeneration capacity. However, how non-coding RNAs such as microRNAs (also known as miRNAs) contribute to cnidarian tissue regeneration is poorly understood. Here, we sequenced and assembled the genome of the sea anemone Exaiptasia pallida collected in Hong Kong waters. The assembled genome size of E. pallida is 229.21 Mb with a scaffold N50 of 10.58 Mb and BUSCO completeness of 91.1%, representing a significantly improved genome assembly of this species. The organization of ANTP-class homeobox genes in this anthozoan further supported the previous findings in jellyfishes, where most of these genes are mainly located on three scaffolds. Tentacles of E. pallida were excised, and both mRNA and miRNA were sequenced at 9 time points (0 h, 6 h, 12 h, 18 h, 1 day, 2, 3, 6, and 8 days) from regenerating tentacles. In addition to the Wnt signaling pathway and homeobox genes that are shown to be likely involved in tissue regeneration as in other cnidarians, we have shown that GLWamide neuropeptides, and for the first time sesquiterpenoid pathway genes could potentially be involved in the late phase of cnidarian tissue regeneration. The established sea anemone model will be useful for further investigation of biology and evolution in, and the effect of climate change on this important group of animals.
Regeneration, in general, refers to the replacement of lost or damaged structures and is widespread in diverse groups of animals (Bideau et al., 2021). Revealing the capacities and underlying mechanisms in different animals is important to understand both the evolution of regeneration, as well as their potential contributions to animal adaptation to the environment.
The phylum Cnidaria comprises of anthozoans (sea anemones, corals), hydrozoans (hydra), and medusozoans (jellyfishes), and plays important roles in both freshwater and marine environments (Slobodkin & Bossert, 2001; Enríquez et al., 2005; Pitt and Purcell, 2009; Litsios et al., 2012). In general, tissue regeneration in cnidarians is comparatively understudied than other animals such as planarians and vertebrates, and most of the detailed studies on the cellular and molecular mechanisms are on hydrozoans (Amiel et al., 2015). Diverse mechanisms were revealed between species and tissues. For instance, cell proliferation is not required for head and foot regeneration to occur in Hydra (Park et al., 1970), but a surge of stem cell proliferation occurs subsequent to decapitation and is crucial for head regeneration in Hydractinia echinata (Bradshaw et al., 2015). On the other hand, decapitation in H. echinata led to blastema proliferation in the area where the head used to be, but no blastema formed in stolon regeneration (Bradshaw et al., 2015). In other cnidarians, such as the sea anemone Nematostella vectensis (Layden et al., 2016; Zimmermann et al., 2020), coral Favia favus (Oren et al., 2001), jellyfish Aurelia aurita and Clytia hemisphaerica (Abrams et al., 2015; Sinigaglia et al., 2020), capabilities of regeneration of lost body parts have also been documented. Nevertheless, how non-coding RNAs contribute to cnidarian regeneration is poorly understood.
MicroRNAs (commonly called miRNAs) are small non-coding RNAs that are around 21–23 nucleotides long and are sequence-specific post-transcription regulators of gene expression in animals (Moran et al., 2017). In cnidarians, miRNAs predominantly have highly complementary messenger RNA targets and result in transcripts cleavage (Moran et al., 2014; Praher et al., 2021). Despite the fact that miRNAs have demonstrated their indispensable roles in regeneration in vertebrates such as mice and zebrafish, how they contribute to cnidarian tissue regeneration remains poorly explored. In limited studies such as that carried out in H. magnipapillata, small RNA transcriptomic analyses revealed 10 differentially expressed miRNAs during the process of head regeneration (Krishna et al., 2013).
In this study, a high-quality genome assembly of the sea anemone Exaiptasia pallida collected from Hong Kong waters (Figure 1A) is provided to form the basis of miRNA annotation in this cnidarian species. Both messenger RNA and miRNA transcriptomes were sequenced from 9 time points with triplicates during its tentacle regeneration to shed light on how these crucial genetic components contribute to regeneration in this important group of animals.
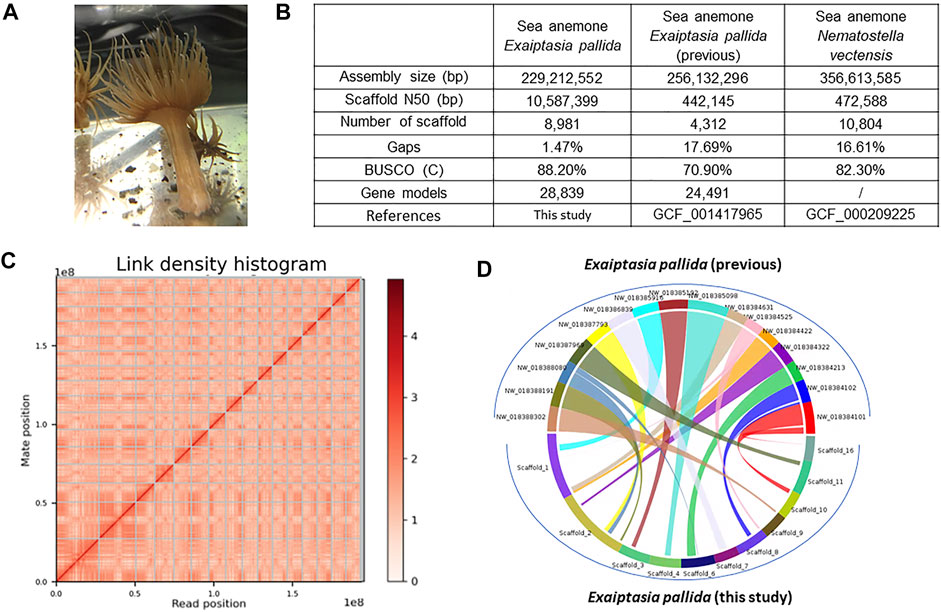
FIGURE 1. (A) Exaiptasia pallida individuals used in this study. (B) Genome sequencing information and statistics. (C) Hi-C information. The x- and y-axes give the mapping positions of the first and second read in the read pair respectively, grouped into bins. The color of each square gives the number of read pairs within that bin. Scaffolds less than 1 Mb are excluded; (D) Synteny blocks between the previous Exapitasia diaphana (GCF_001417965) genome and our genome.
E. pallida were collected from Simon F.S. Li Marine Science Laboratory, The Chinese University of Hong Kong (CUHK) and kept in 30 ppt artificial sea water at 22°C. All of the individuals were collected from different colonies 1 day prior to experiments. To observe tentacle regeneration in E. pallida, 30 individuals had their tentacles cut at the base of the tentacles. The course of regeneration was observed and documented up to 23 days using a stereomicroscope.
mRNA and small RNA extraction were done during 9 time points (0 h, 6 h, 12 h, 18 h, 1 day, 2, 3, 6, and 8 days) of E. pallida tentacle regeneration. Biological triplicates were done for each time point and each triplicate consisted of tissues excised from six individuals. Tissues from the base of tentacles were obtained for 0 h replicates and tissues from the tip of regenerating tentacles were obtained for the rest of the time points. mRNAs and small RNAs were extracted from the tissues obtained using TRIzol (Ambion) and miRVana Isolation kit respectively, and their concentrations were measured using NanoDrop™ One/One C Microvolume UV–Vis Spectrophotometer (Thermo Fisher Scientific, Fitchburg, WI). Agarose gel electrophoresis was used to assess the quality. Samples were stored at −80°C and sent to Novogene for transcriptome (Novaseq, PE151 platform for strand-specific library construction) and small RNA library (Novaseq, 50SE platform for strand-specific library construction) sequencing (Supplementary Tables S1,S4).
Transcriptome libraries were prepared using the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina®. In brief, after the purification of mRNA with poly-T oligo-attached magnetic beads, the mRNA was fragmented randomly. Using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H), the first cDNA strand was synthesized. The second cDNA strand was synthesized using DNA polymerase I and RNase H with the incorporation of dUTP. AMPure XP beads (Beckman Coulter, Beverly, United States) were used to purify the double-stranded cDNA to select cDNA fragments of 150–200 bp. Exonuclease/polymerase repaired the remaining overhangs of the cDNA into blunt ends. Subsequent to adenylation of 3′ ends of DNA fragments, NEBNext Adaptor was ligated to prepare for hybridization. The second strand of cDNA was digested by USER enzyme. The final library was generated by PCR amplification and purification of PCR products with AMPure XP beads.
Small RNA libraries were prepared using the NEBNext® Multiplex Small RNA Library Prep Set for Illumina®. In short, 3 and 5 adaptors were first ligated to the 3 and 5’ ends of small RNAs respectively. Subsequent to hybridization, the first strand of cDNA was synthesized with reverse transcription primers. Through PCR enrichment, the double-stranded library was generated. Purification was done to select fragments of 18–40 bp.
Genomic DNA was extracted from E. pallida using PureLink™ Genomic DNA Mini Kit (Invitrogen, Carlsbad, CA, United States). The oral disk was homogenized and digested for 2 h at 55°C. 10 times the amount suggested for PureLink™ Genomic Digestion Buffer, Proteinase K, RNase A, PureLink™ Genomic Lysis/Binding Buffer, and ethanol were used. The DNA was eluted with 35 μL ddH2O (concentration: 228.0 ng/μL; amount: 3.192 μg; OD260/280: 1.81; OD260/230: 1.91). The sample was then sent to Novogene for 10X Genomics linked-read sequencing and Dovetail Genomics for Chicago and Omni-C sequencing as described below. The details, including sequencing platform and prepared library size, are attached in Supplementary Table S2.
A Chicago library was made as described previously in Putnam et al. (2016). In short, high molecular genomic DNA was reconstructed into chromatin in vitro and fixed with formaldehyde solution. Resultant chromatin was then digested with the restriction enzyme DpnII. The 5’ overhangs were sealed with biotinylated nucleotides and the blunt ends were then ligated. The DNA was then purified from the crosslinked proteins and subject to the removal of biotins that were not located in the internal ligated fragments. Eventually, the DNA molecules were sheared to approximately 350 bp and a library was constructed using NEBNext Ultra enzymes and Illumina-compatible adaptors. Fragments that contain biotin were isolated by streptavidin beads prior to PCR enrichment of the library. The libraries were then sequenced on an Illumina HiSeq X platform to yield 161 million 2 × 150 bp paired end reads.
The Dovetail Omni-C library was prepared according to Manuals & Guides (https://dovetailgenomics.com/products/omni-c-product-page/). Briefly, the genomic DNA was fixed on site in the nucleus with formaldehyde solution prior to sample DNA extraction. Fixed chromatin was digested with DNAse I. The resultant chromatin ends were subject to end-repairing steps and ligated to biotinylated bridge adaptors and followed by proximity ligation. Afterwards, DNA was purified from the crosslinked proteins. The sequencing library was prepared the same way the Chicago library was. The sequencing platform produced 420 million 2 × 151 bp paired end reads in total, which provided 727x physical coverage of the genome (1–100 kbp).
The assembly was processed as described previously (Nong et al., 2020). Briefly, Kraken was used to check the 10X raw data for bacteria contamination (Wood & Salzberg, 2014). Using DSK (v 2.1.0), k-mers of the Illumina PE reads were counted with k = 21. In addition, based on a k-mer-based statistical approach in the GenomeScope webtool, estimation of genome size, heterozygosity, and repeat content were analyzed (Supplementary Table S2; GenomeScope.kmer_k21). Supernova (v 2.1.1; default parameters) generates phased and whole-genome de novo assemblies from a Chromium-prepared library (Weisenfeld et al., 2017). The de novo assembly and raw reads from the Chicago library were used as primary input data for HiRise (Putnam et al., 2016). Chicago library sequences were then aligned to the draft assembly using SNAP (http://snap.cs.berkeley.edu). The separation of Chicago read pairs mapped within draft scaffolds were analyzed by HiRise to generate a likelihood model for the estimation of genomic distance between read pairs, and the model was used to identify and break putative misjoints, score prospective joins, and make joins above a threshold. A similar procedure was adopted for OmniC data and eventually yielded a resultant genome.
The assembly and annotation were processed as described previously (Nong et al., 2020). Raw sequencing reads of the transcriptomes from different time points were quality-filtered by Trimmomatic (v0.33, with parameters “ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 SLIDINGWINDOW:4:5 LEADING:5 TRAILING:5 MINLEN:25”; Bolger, Lohse, & Usadel, 2014). Using Funannotate, sequences of nuclear genomes were cleaned and masked. To align RNA-seq data by HISAT2 (v2.0.5; Kim et al., 2019), the soft-masked assembly was used to run “funannotate train” with the parameters “--stranded RF--max_intronlen 350000”. A genome-guided assembly was then generated with Trinity (v2.9.1). Transcript abundance was estimated using “--est_method RSEM—aln_method bowtie” (v1.1.2) with the script of “align_and_estimate_abundance.pl” of Trinity. TransDecoder (v5.0.2) was used to annotate coding regions within transcripts (Supplementary Tables S6,S7). Trinotate (v3.1.1) was used to perform functional annotation and analysis.
Contigs of the genome-guided assembly were also used to run PASA for prediction of gene models. In the “funannotate predict” step, GeneMark-ES v4.32 was used for ab initio gene prediction and Augustus was trained using BUSCO. The gene models from the prediction sources “Augustus”, high-quality Augustus predictions (HiQ), “pasa”, “snap”, “GlimmerHMM”, and “GeneMark” were passed to Evidence Modeler [EVM Weights: ('GeneMark’: 1, ‘HiQ’: 2, ‘pasa’: 6, ‘proteins’: 1, ‘Augustus’: 1, ‘GlimmerHMM’: 1, ‘snap’: 1, ‘transcripts’: 1)] to generate the final annotation files. Finally, EVidenceModeler was used to combine data from protein alignments, transcript alignments, and ab initio predictions to construct high quality evidence-based gene models. PASA was then used to update EVM consensus predictions and add UTR annotations and models for alternatively spliced isoforms. The protein-coding gene models were then searched in the NCBI nr database using BLASTp and in the swissprot database using diamond (v0.9.24) with parameters “--more-sensitive--evalue 1e-3”, and mapped by HISAT2 (version 2.1.0) with transcriptome reads (Supplementary Table S3). Gene matrix count tables were then generated by StringTie (version 2.1.1) and used for further differential gene expression analyses. The gene models with no homology to any known protein in the GenBank nr database and no mRNA support were removed from the final version. The differential gene expression analyses were performed by Trinity’s run_DE_analysis.pl and analyze_diff_expr.pl scripts using the edgeR method, where at least three replicates had CPM values ≥ 50, and cut-off values were set to 0.05 p-values (FDR) and 2-fold changes. Heatmaps were generated by PtR of Trinity, using the parameter “--min_rowSums 10 --log2 –CPM” and default complete gene and sample clustering methods.
Putative gene sequences were retrieved from the established genome using the tBLASTn algorithm. The gene identity of each identified target was tested by comparing the sequences deposited in the NCBI nr database using the BLASTx algorithm. For neuropeptides, potential N-terminal signal peptide sequences were predicted by SignalP 3.0 (Bendtsen et al., 2004). The potential convertase cleavage sites were then manually checked and annotated based on the features of a canonical neuropeptide: various prohormone convertase cleavage (lysine/arginine) sites (KR, RR, R) in conjunction with a glycine residue (GKR, GRR, GR) for peptide C-terminal amidation. For phylogenetic analyses of different gene family sequences, amino acid sequences were aligned to other reference sequences extracted from NCBI; gapped sites were removed from alignments using the software MEGA 11 and phylogenetic trees (Neighbor-joining and Maximum likelihood) were constructed using MEGA 11 and IQ-TREE (http://www.iqtree.org/), with 1000 bootstrap replicates. The phylogenetic trees were further edited using iTOL (https://itol.embl.de/) and TBtools (Chen et al., 2020).
Potential homeobox genes were identified by searching homeodomain sequences from N. vectensis (Ryan et al., 2006), Branchiostoma floridae, Drosophila melanogaster, and Tribolium castaneum (retrieved from HomeoDB; Zhong & Holland, 2011) in our E. pallida genome and transcriptome and previously published cnidarian genomes (Fuller et al., 2020; Hu et al., 2020; Nong et al., 2020; Zimmermann et al., 2020; Xia et al., 2021; Simakov et al., 2022) using tBLASTn (Supplementary Table S5). Then, NCBI CD-Search (Lu et al., 2020) was used to validate the presence of homeodomains in the retrieved sequences. Identification of each putative gene was tested by comparison to sequences confirmed in the NCBI nr database using BLASTx and BLASTp, phylogenetic analysis, and syntenic analysis (Figure 2B). Syntenic relationships between E. pallida, N. vectensis, Acropora millepora, Rhopilema esculentum, and Hydra vulgaris were computed using MCScanX with default parameters (Chen et al., 2020).
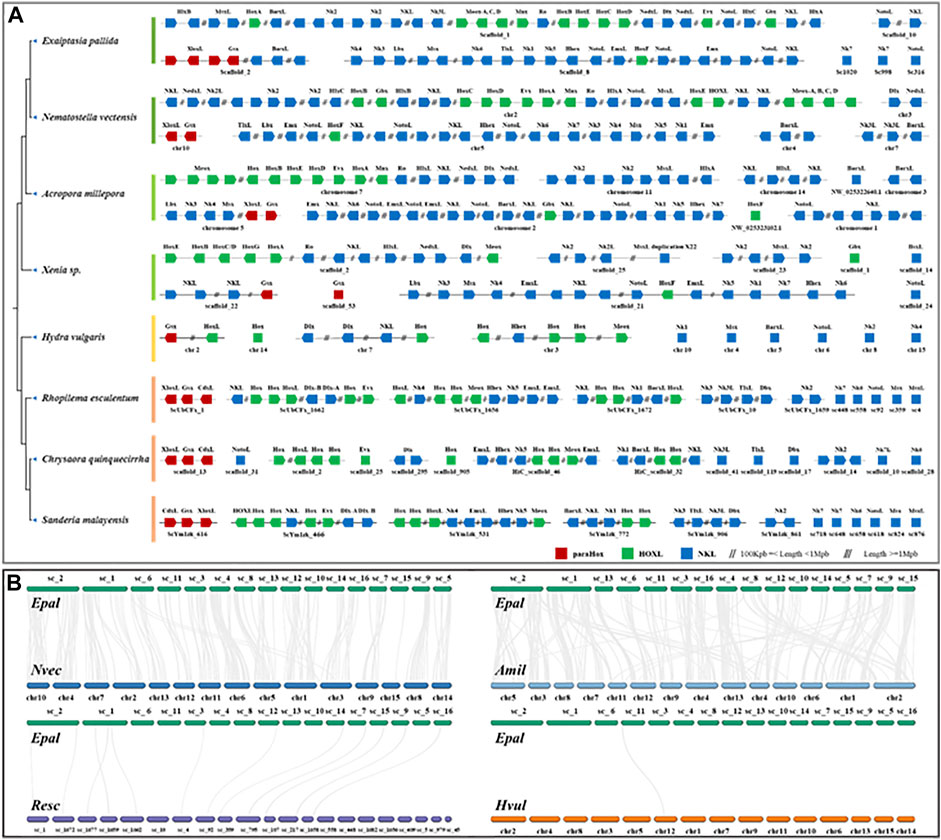
FIGURE 2. (A) Chromosomal organization of ANTP-class homeobox gene arrangement in cnidarian genomes. (B) Syntenic relationship between Exaiptasia pallida, Nematostella vectensis (sea anemone to sea anemone), Acropora millepora (sea anemone to coral), Rhopilema esculentum (sea anemone to jellyfish) and Hydra vulgaris (sea anemone to hydra).
Small RNA-sequencing reads had their adaptor sequences trimmed and those with Phred quality score lower than 20 were removed. Using the mapper.pl script of miRDeep2, reads of length 18 to 27 bp were then mapped to respective genomes. miRDeep2 was used to identify novel miRNAs, which were checked manually whether they fulfill the criteria of MirGeneDB (http://mirgenedb.org/information). miRNAs which fulfill the criteria of MirGeneDB were then annotated to known miRNAs in the miRBase database according to sequence similarity. Expression profiling and quantification were performed by the quantifier.pl script of miRDeep2. MapMi was utilized to find potential miRNA loci in the genomes of E. pallida (MapMi scorer cutoff = 15). Matrix count tables were generated by miRDeep2’s script quantifier.pl with 0 mismatches in mapping reads to precursors and other default parameters. Different gene expression analyses were performed by Trinity’s run_DE_analysis.pl and analyze_diff_expr.pl scripts using the edgeR method, where at least three replicates had CPM values ≥ 50, and cut-offs were set to 0.05 p-values (FDR) and 2-fold changes.
CIRCOS and synteny blocks were computed using SyMAP v5.0.6 (Synteny Mapping and Analysis Program) (Soderlund et al., 2011) with “mask_all_but_genes = 1’ to mask non-genic sequence” and other default parameters.
High molecular weight genomic DNA was extracted from one individual of E. pallida (Figure 1A) and sequenced on the Illumina short-read and 10X Genomics linked-read sequencing platforms. Chicago and Omni-C libraries were also established and sequenced on the Illumina platform (Supplementary Table S2). The genome sequences were primarily assembled with short raw reads. Subsequently, scaffolding was done using the Chicago and Omni-C reads. The genome assembly is 229.2 Mbp with a scaffold N50 of 10.6 Mbp (Figure 1B). This high physical contiguity is matched by high completeness, with 91.1% complete BUSCO score for metazoa_genes (version odb10) (Figure 1B). A total of 28,839 gene models including 454 tRNA genes and 28,385 protein coding genes, 95.78% and 98.48% of the encoded proteins, could hit the nr/swissprot databases and had mRNA support, respectively. The majority of the sequences assembled (∼84.18%) were contained in 16 pseudomolecules (Figure 1C). It is noticed that there is a chromosomal-level genome in the sea anemone N. vectensis available (Zimmermann et al., 2020), and our new resource represents the first near chromosomal-level genome generated for the species E. pallida.
Homeobox genes are essential players in body patterning and markers of large-scale genomic changes in evolution, and there is an increasing number of comparisons of homeobox genes between genomes in attempt to understand the evolution of these genes in animals (Chourrout et al., 2006; Holland et al., 2007; Holland, 2013; DuBuc et al., 2018; He et al., 2018). Analyses of homeobox gene organization across a range of species help to better understand the evolution of genome organization. The ANTP-class of homeobox genes is the largest group of homeobox genes in animals, including the ParaHox (Gsx, Xlox, and Cdx), Hox/Hox-linked (Hox genes, Gbx, Mnx, Meox, Evx, En, Ro, Mnx, Hhex, Moex, and Dlx), and NK/NK-linked clusters (NK1, NK3, NK4, NK5, NK6, NK7, Msx, Lbx, Tlx) (Holland, 2013; Ferrier, 2016). To investigate the evolution of gene clusters, we have annotated the ANTP-class of homeobox genes in a wide range of high-quality cnidarian genomes (Figure 2A, Supplementary Tables S2,S5).
In the genomes of bilaterians, the ANTP-class of homeobox genes are usually found in four chromosomal clusters (ParaHox, Hox, NK, and NK2) (Luke et al., 2003; Hui et al., 2012; Li et al., 2020). In a recent study, it was found that the Hox, NK, and Hox-like genes are clustered in two jellyfish genomes (Figure 2A; Nong et al., 2020), illustrating a divergent evolutionary pathway of the ANTP-gene cluster genes in cnidarians. Genomic analysis of the ANTP-class homeobox genes in E. pallida revealed that most of these genes are mainly located on three scaffolds similar to the jellyfish studies, including a ParaHox gene cluster (Xlox and Gsx) on Scaffold 2, Hox genes with HlxL, MsxL, Meox, Ro, NedxL, Dlx, NK2 genes on Scaffold 1, and Hox gene with NK cluster (NK1/3/4/5/6, Lbx, Msx, TlxL, and Hhex) on Scaffold 8 (Figure 2A). The ANTP-class homeobox genes organization in this anthozoan further supported the previous findings in jellyfishes, and refuted that it is due to lineage-specific rearrangement. Nevertheless, the hypothetical mega-homeobox cluster organization as originally proposed by Pollard and Holland (2000) remains to be revealed in an extant animal.
In E. pallida individuals cultured at 22°C, the wound closed at 12 hpa (hours post-amputation) (Figure 3). At 18 hpa, tentacle buds became visible and grew longer at 2 and 3 dpa (days post-amputation) (Figure 3). A total of 54 small RNA and messenger RNA datasets representing 9 time points during E. pallida tentacle regeneration were generated (Supplementary Table S1). Across the 9 time points, 127 and 58 mRNAs were upregulated and downregulated respectively, and 141 and 4 miRNAs were upregulated and downregulated respectively (Figures 4A,B, Supplementary Figures S2–S4, Supplementary Tables S8–S10). Most of the differentially expressed mRNAs were differentially regulated between 0 hpa and 6 hpa, while the majority of the differentially expressed miRNAs were differentially regulated between 18 hpa and 24 hpa (Figures 4A,B).
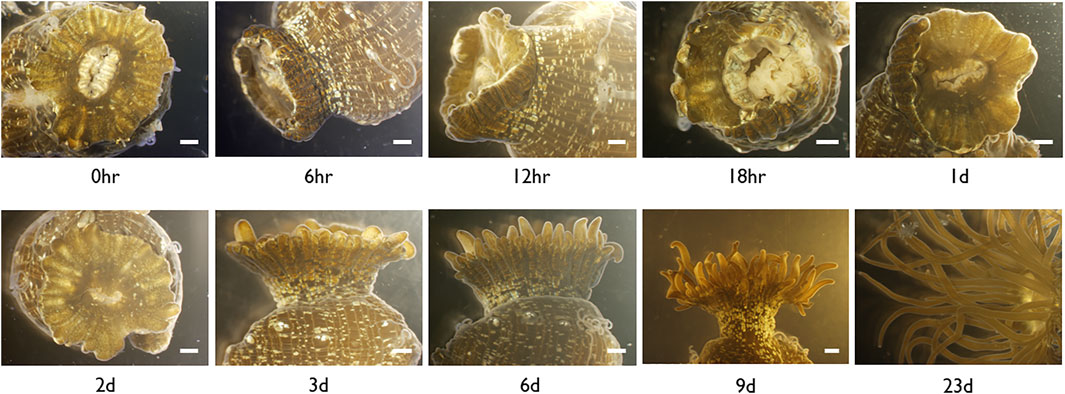
FIGURE 3. Microscopic images of Exaiptasia pallida cultured at 22°C with tentacles cut at the base from the moment of injury (0 h) to complete regeneration (Day 9). Scale bar, 1 mm.
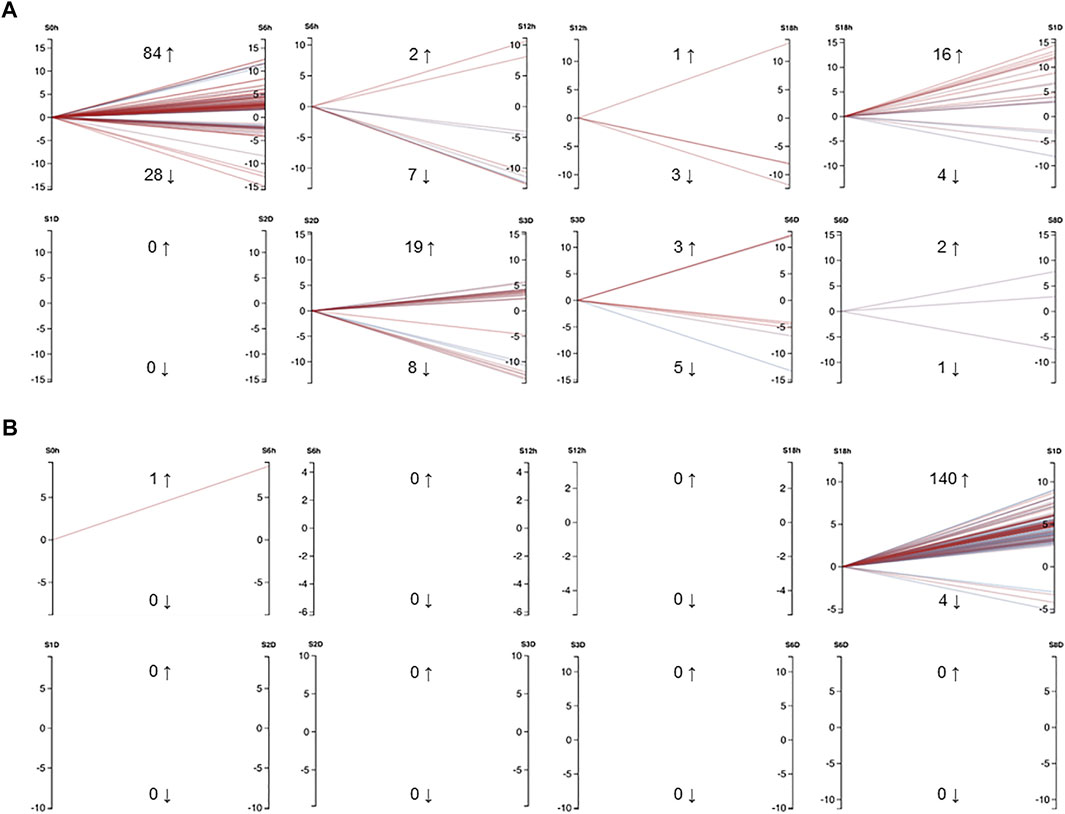
FIGURE 4. (A) Differentially expressed mRNAs in Exaiptasia pallida tentacle regeneration that had a minimum CPM of 50 in at least 3 samples. Cut-off values were set to FDR = 0.05, abs logFC = 1. (B) Differentially expressed miRNAs in E. pallida tentacle regeneration that had a minimum CPM of 5 in at least 3 samples. Cut-off values were set to FDR = 0.05, abs logFC = 1. Numbers adjacent to upwards and downwards arrows indicate the number of genes or miRNAs that were upregulated and downregulated respectively, e.g. from 0 hpa to 6 hpa, 84 mRNAs were upregulated and 28 mRNAs were downregulated. Each line is colored by FDR with red being more significant and blue less significant. S0h: 0 h post amputation (hpa), S6h: 6 hpa, S12h: 12 hpa, S18h: 18 hpa, S1D: 1 day post amputation (dpa), S2D: 2 dpa, S3D: 3 dpa, S6D: 6 dpa, S8D: 8 dpa. The graphs were generated using Degust (DOI: 10.5281/zenodo.3258932) (Powell, 2015).
We first investigated whether the key developmental and hormonal pathway genes were involved in E. pallida tentacle regeneration. Wnt signaling pathway genes are well known to be involved in the tissue regenerative processes in animals (e.g. Whyte et al., 2012). We found that the expression of Frizzled, LRP, Wnt, and other members in the Wnt signaling pathway were upregulated in the early- and mid-phase of tentacle regeneration (Supplementary Figures S5–S8, Supplementary Table S11).
Previous studies have identified the association of Hox gene expression in animal tissue regeneration (e.g. Novikova et al., 2016) so we investigated the expression of Hox/ParaHox genes during tentacle regeneration in E. pallida (Supplementary Figure S9). In E. pallida, a Hox cluster (HoxA-HoxE) together with Mox, Gbx, Rough, and Msx were identified on the same scaffold, while the ParaHox cluster, which consists of Gsx and 2 Cdx, was found on another scaffold (Figure 5A). During the regeneration of tentacles, a progressive upregulation of Hox genes was observed with HoxB and HoxE upregulated at 6 hpa, followed by the upregulation of HoxC and HoxD at 18 hpa (Figure 5B, Supplementary Table S12). HoxF, which is located on a different scaffold, was upregulated at 3 dpa (Figure 5B). Other homeobox genes including Mox, Cdx, and Gbx were upregulated between 18 hpa and 2 dpa, while HoxA, Gsx, Mnx, Rough, and Evx were downregulated shortly after tentacle amputation at 6 hpa (Figure 5B).
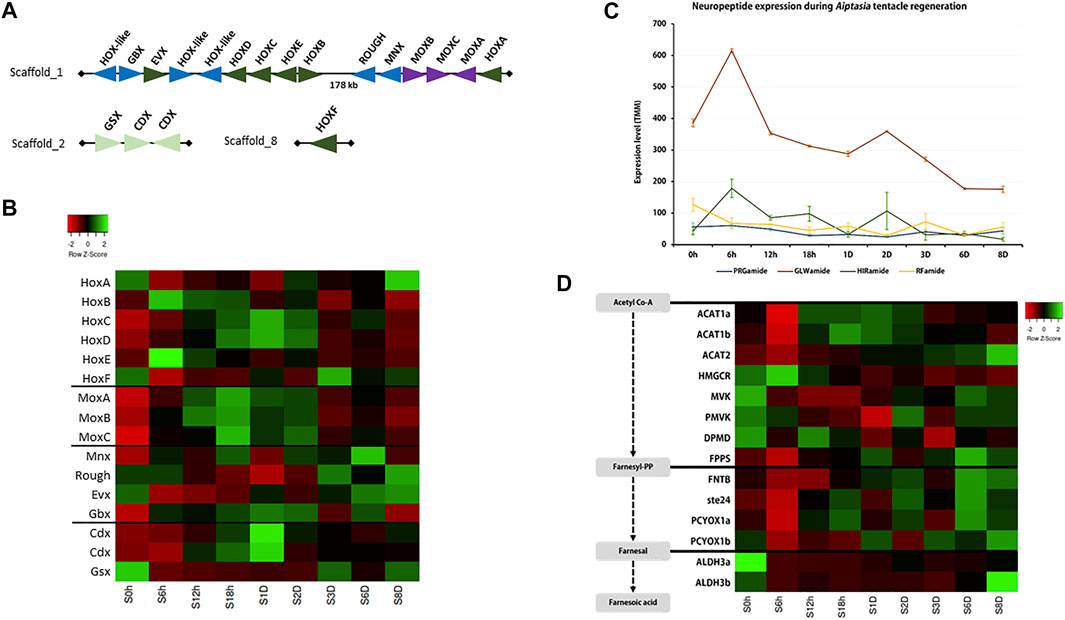
FIGURE 5. (A) Hox and other homeobox gene arrangement in the Exaiptasia pallida genome; (B–D) Expression of (B) Hox genes, (C) neuropeptides, and (D) sesquiterpenoid hormone pathway genes during tentacle regeneration.
Neuropeptides play crucial roles as hormones and neurotransmitters and are involved in the regulation of a variety of developmental processes (e.g. Takahashi, 2020). Here, we annotated 4 cnidarian conserved neuropeptides: PRGamide, GLWamide, HIRamide, and RFamide (Figure 5C, Supplementary Figure S10, Supplementary Table S13). Among the four neuropeptides, GLWamide was found to be associated with the tentacle regenerative process in E. pallida and was upregulated at 6 hpa.
In a recent study, the sesquiterpenoid hormonal system was identified in cnidarians (Nong et al., 2020). To test whether sesquiterpenoids could potentially be involved in tissue regeneration in cnidarians, we investigated the expression of sesquiterpenoid biosynthetic pathway genes during tentacle regeneration in E. pallida (Supplementary Figures S11–S22, Supplementary Table S14). Acetyl-CoA acetyltransferase (ACAT) and the isoprenylation pathway genes (FNTB, ste24, PCYOX1) were downregulated, while upregulation of gene expression was observed for HMGCR, MVK, PMVK, DPMD, FPPS, ALDH3a, and ALDH3b at 6 hpa. At 6 dpa, both terpenoid backbone pathway and isoprenylation pathway genes were upregulated (Figure 5D).
Cnidarians such as the hydroid Hydra and the sea anemone N. vectensis are conventional animal models to understand tissue and body regeneration (Holstein et al., 2003; Havrilak et al., 2021; Röttinger, 2021). Hox genes were found to be regulated by miRNAs in the sea anemone N. vectensis (Moran et al., 2014). Prior to this study, there have been other attempts at utilizing the sea anemone Exaiptasia in understanding the regeneration processes (e.g. Singer, 1971; van der Burg et al., 2020; Presnell et al., 2022). Nevertheless, how miRNAs contribute to cnidarian regeneration is relatively understudied in cnidarians (Krishna et al., 2013). This study represents the first understanding of the underlying genetic regulation of miRNAs during tentacle regeneration in the sea anemone E. pallida.
To better annotate the miRNAs, especially the novel miRNAs, we have re-sequenced the E. pallida genome with a population collected naturally in Hong Kong and sequenced new small RNA transcriptomes. To date, there are a total of 103 assemblies of cnidarian genomes at NCBI (5 July 2022), and we have tested them with BUSCO v5.3 and the metazoa_odb10 database with the same default parameters (Supplementary Table S15). Forty of them had BUSCO scores between 90.2% and 96.1%. Synteny analysis between the previous Exaiptasia diaphana (interchangeable name for E. pallida; GCF_001417965; Baumgarten et al., 2015) genome and the newly assembled genome showed that 78.35% of the sequences assembled in GCA_001417965 are syntenous with the 16 pseudomolecules of our genome assembly (Figure 1D, Supplementary Table S15), representing a significantly improved genome assembly of this species. We also performed OrthoFinder on the longest genes in both genomes and found a total of 47,712 orthogroups, of which 3.14% was only found in GCA_001417965.1, 6.70% was only found in our new genome, and the remaining 90.16% was common (Supplementary Table S15). Our new genome will be useful for future understanding of cnidarian biology, in particular, genome biology and evolution.
miRNAs found in Aiptasia (Baumgarten et al., 2018), N. vectensis (Moran et al., 2014), Sanderia malayensis (Nong et al., 2020), R. esculentum (Nong et al., 2020), Acropora digitifera (Gajigan & Conaco, 2017), and H. magnipapillata (Krishna et al., 2013) were searched in the new E. pallida genome assembly. Ten miRNAs, namely miR-100, miR-2022, miR-2023, miR-2025, miR-2026, miR-2030, miR-2036, miR-2037, miR-2050, and miR-9425, are found amongst most anthozoans (Praher et al., 2021). Amongst the ten miRNAs, only one miRNA, miR-9425, was not identified in our E. pallida genome (Supplementary Table S16). All of the miRNAs found in Aiptasia (Baumgarten et al., 2018) can be found in the new genome assembly, and we observed a dynamic miRNA conservation between different cnidarians.
We have identified Wnt signaling pathway genes and homeobox genes that participate in tentacle regeneration, which are congruent with other cnidarian regeneration studies (e.g. Schummer et al., 1992; Guder et al., 20066; Stierwald et al., 2004; Trevino et al., 2011; Ramirez et al., 2022). GLWamide, one of the most studied neuropeptides, is responsive in tentacle regeneration of E. pallida. GLWamide was previously revealed to be involved in the process of muscle contraction in the sea anemone genus Anthopleura and the coral Euphyllia ancora (Takahashi & Hatta, 2011; Shikina et al., 2020), metamorphosis in H. magnipapillata (Takahashi & Hatta, 2011), settlement in the coral Acropora palmata (Erwin & Szmant 2010; Takahashi & Hatta, 2011), and transition timing modulation of N. vectensis from planula larvae to polyps (Nakanishi & Martindale, 2018). Given its participation in cnidarian tissue regeneration, this study has now revealed that the level of pleiotropy of GLWamide is higher than previously thought. Another interesting finding of this study is that we have explored the potential roles of the newly identified sesquiterpenoid hormones in cnidarians (Nong et al., 2020). Notably, genes in both the terpenoid backbone pathway and isoprenylation pathway were upregulated in the latter phase of regeneration. This is the first study showing the potential roles of sesquiterpenoids in cnidarian regeneration.
Common DEGs among tentacle regeneration of E. pallida from our study and body regeneration of E. pallida from the study conducted by van der Burg et al. (2020) were identified and their expression levels at different time points were analysed. The study conducted by van der Burg et al. (2020) and this study investigated different regeneration time points, thus the most similar time points were compared. The expression levels of 9 common DEGs (genes with FDR smaller than or equal to 0.05 and abs logFC greater than or equal to 1) at 0 hpa (from both studies), 6 hpa (this study) VS. 8 hpa (van der Burg et al., 2020), 18 hpa (this study) VS. 20 hpa (van der Burg et al., 2020), 48 hpa (from both studies), and 72 hpa (from both studies) were compared. All 9 of these common genes were differentially expressed from 0 hpa to 6 hpa, or from 0 hpa to 8 hpa. No common DEGs were found in other time points. C3 and PZP-like alpha-2-macroglobulin domain-containing protein 8 (CPAMD8), protein naked cuticle homolog 2-like (nkd2l), and patched domain-containing protein 3 (PTCHD3) displayed significant upregulation from 0 hpa to 6/8 hpa (Supplementary Figures S23–S25). E. pallida displayed a similar trend in the expression of CPAMD8 and nkd2l across different time points in our study and the study conducted by van der Burg et al. (2020). For most of the transcripts, both genes displayed an upregulation from 0 hpa to 6 hpa, followed by gradual downregulation in subsequent time points. However, the read counts reported by van der Burg et al. (2020) are significantly lower than that by our study. The species displayed a somewhat similar trend in the expression of PTCHD3 across different time points in our study and the study conducted by van der Burg et al. (2020). Gene1884 and gene8054 were both annotated as PTCHD3 van der Burg et al. (2020). Gene1884 was upregulated from 0 hpa to 8 hpa and was gradually downregulated in subsequent time points van der Burg et al. (2020), which is the same trend as that observed in our study. On the other hand, gene8054 was downregulated from 0 hpa to 8 hpa and was upregulated until a downregulation at 72 hpa. Advillin, heme-binding protein 2 (HEBP2), and protein NLRC5 were significantly downregulated from 0 hpa to 6/8 hpa (Supplementary Figures S26–S28). E. pallida displayed a similar trend in the expression of advillin across time points in our study and the study conducted by van der Burg et al. (2020), with an exception observed from 18/20 hpa to 48 hpa. The general trend in the expression of HEBP2 observed in this study is also the same as that in the study conducted by van der Burg et al. (2020). A similar trend in the expression of protein NLRC5 across time points was observed, with an exception observed from 6/8 hpa to 18/20 hpa. TNF receptor-associated factor 3 (TRAF3) expression displayed opposite trends from 0 hpa to 6/8 hpa, with significant upregulation and downregulation (Supplementary Figure S29). Fibroblast growth factor receptor (FGFR) expression displayed opposite trends from 0 hpa to 6/8 hpa, with significant downregulation and upregulation (Supplementary Figure S30). La-related protein 6 (LARP6) displayed significant changes in expression in different time points in our study and the study conducted by van der Burg et al. (2020) (Supplementary Figure S31). The overall trend of expression is similar with an exception observed from 48 hpa to 72 hpa. None of the DEGs identified in oral and physal regeneration in N. vectensis (dissected halfway between the oral and aboral ends; Schaffer et al., 2016) were found in tentacle regeneration of E. pallida (this study) and whole body regeneration of E. pallida (dissected perpendicular to the oral-aboral axis; van der Burg et al. 2020).
CPAMD8 has a Kazal-type serine proteinase inhibitor/follistatin-like domain (Li et al., 2004). This gene has been found to be upregulated in regeneration-incompetent iris in newts and axolotls (Sousounis et al., 2013; Sousounis et al., 2014). Interestingly, CPAMD8 was significantly upregulated from 0 hpa to 6/8 hpa in both our study and the study done by van der Burg et al. (2020). Naked cuticle (Nkd) is a gene that targets Dishevelled to antagonize Wingless, a signal for the Wnt signaling pathway (Zeng et al., 2000; Rousset et al., 2001). It may also activate a second Wnt signaling pathway that controls planar cell polarity (Creyghton et al., 2005). Nkd2l was significantly upregulated at 6 hpa in our study and 8 hpa in the study conducted by van der Burg et al. (2020), suggesting that during these time points, nkd2l targeted Dishevelled to antagonize Wingless and the Wnt signaling pathway that controls planar cell polarity was activated. Patched is a receptor that mediates the Hedgehog (Hh) signaling pathway, which is involved in blastema cell proliferation, dorsoventral patterning, cartilage induction, and regulation of progenitor and stromal cell fate during regeneration (Schnapp et al., 2005; Ochoa et al., 2010). Patched has been shown to be crucial for posterior specification during planarian body regeneration (Yazawa et al., 2009). PTCHD3 was significantly upregulated at 6 hpa in our study and 8 hpa in the study conducted by van der Burg et al. (2020), suggesting that Hh signaling occurred during these time points. Advillin is a member of the gelsolin/villin family of actin regulatory proteins (Marks et al., 1998), and is involved in actin bundling (Rao et al., 2017). Actin cytoskeleton has been shown to be required for regeneration in previous studies (e.g. Lahne et al., 2015). Advillin is expressed at the tips of filopodia during axon regeneration in mice (Chuang et al., 2018). In our study and the study conducted by van der Burg et al., (2020), the expression level of the gene was significantly reduced post-amputation. HEBP2 facilitates necrotic cell death (Szigeti et al., 2006). This gene was significantly downregulated at 6 hpa in our study and 8 hpa in the study conducted by van der Burg et al. (2020), suggesting that necrosis was suppressed during these time points. Protein NLRC5 negatively regulates the NF-kappa-B and type I interferon signaling pathways in macrophages and decreases innate cytokine production and antiviral immunity (Cui et al., 2010; Tong et al., 2012). Immune genes have been found to be differentially expressed during regeneration in previous studies (e.g. Stewart et al., 2017). Protein NLRC5 was significantly downregulated at 6 hpa in our study and 8 hpa in the study conducted by van der Burg et al. (2020), suggesting that the immune system also has a vital role in regeneration in E. pallida. Degradative ubiquitination of TRAF3 during MyD88-depedent TLR signaling is crucial for the activation of mitogen-activated protein kinases (MAPKs) (Tseng et al., 2010), which have been found to play a role in regeneration in many regeneration studies (e.g. Jopling et al., 2012). TRAF3 also activates NF-kappa-B (Gardam et al., 2008) and regulates B cell survival (Gardam et al., 2008) and the production of type I interferons and proinflammatory cytokines (Tseng et al., 2010), demonstrating its importance in immunoresponses. Intriguingly, TRAF3 showed different trends of expression from 0 hpa to 6/8 hpa during tentacle regeneration (our study) and whole body regeneration (van der Burg et al. 2020) and its expression continued to increase until 18 hpa in our study, suggesting that MAPKs and immunoresponses may have a more important role in body regeneration and tentacle regeneration in E. pallida respectively. Fgfr2 and fgfr4 expression were detected in epicardial tissue during zebrafish heart regeneration (Lepilina et al., 2006). While wound fibrin was displaced with myocardium by 30 dpa in wild-type ventricles, dominant-negative Fgfr individuals retained considerable amounts of fibrin and developed scars that were large and rich in collagen, demonstrating that zebrafish heart regeneration is Fgf-dependent (Lepilina et al., 2006). FGFR was significantly downregulated and upregulated at 6 hpa in our study and at 8 hpa in the study conducted by van der Burg et al. (2020) respectively, suggesting that Fgfr signaling may be involved in whole body regeneration but not tentacle regeneration in E. pallida. LARP6 was found to be statistically upregulated during whole body regeneration in the sea anemone Calliactis polypus (Stewart et al., 2017). The gene is important for the interaction between collagen mRNAs and vimentin filaments and the stabilization of collagen mRNAs (Challa & Stefanovic, 2011). LARP6 was significantly upregulated at 6 hpa in our study and 8 hpa in the study conducted by van der Burg et al. (2020), suggesting that collagen mRNAs and vimentin filaments may also have important roles in regeneration in E. pallida.
An intriguing phenomenon we have observed in the 54 mRNA and miRNA transcriptomes is that the majority of the mRNAs were differentially expressed in the earliest phase of tentacle regeneration, while there was a delay of timing before most of the miRNAs were differentially regulated. These data imply that early tissue regeneration in E. pallida is controlled by mRNAs, and miRNAs mainly serve in the second phase of tissue regeneration by interacting with the mRNAs. Whether the roles of miRNAs in this second phase is “fine-tuning” the expression of mRNAs in the first phase, or act as a transitional phase to induce genes in another phase, or performing both functions, remains to be carefully examined. It is also unclear whether the interesting phenomenon in which mRNAs are differentially expressed in the earliest phase of regeneration and the differential regulation of miRNAs is delayed to a later phase is E. pallida specific or applicable to other cnidarians, which warrants further investigation.
This study has established the sea anemone E. pallida as a cnidarian model with a high-quality genome to study the contribution of miRNAs in tentacle regeneration. Sea anemones are well known to be sensitive to temperature changes and are more prone to bleach under increased temperatures. In Hydra and N. vectensis, temperature could affect their regeneration processes (Miljkovic-Licina et al., 2007; Kü et al., 2008; Amiel et al., 2015). Given the relatively fast tentacle regeneration time in E. pallida, alongside other conventional cnidarian models, we propose that this species can also be further used to test how physical parameters such as temperature, oxygen, nitrogen, and carbon dioxide content affect tissue regeneration and understand the effects of climate change on cnidarians.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Bioproject accession number: PRJNA631085.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by the TUYF Charitable Trust (No. 6904697), Hong Kong Research Grant Council Collaborative Research Grant (C4015-20EF), General Research Fund (14100919; 14100420), PROCORE France/Hong Kong Joint Research Scheme (F-CUHK402/18), the Open Collaborative Research Fund and Operation Fund from the Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (HKB L20200008), and the Direct Grant of The Chinese University of Hong Kong (4053489). CWYS was supported by the PhD studentship of The Chinese University of Hong Kong.
The authors would like to thank Michel Vevoort and Eve Gazave for discussion.
Author TS was employed by company Dovetail Genomics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.900321/full#supplementary-material
Abrams, M. J., Basinger, T., Yuan, W., Guo, C. L., and Goentoro, L. (2015). Self-repairing symmetry in jellyfish through mechanically driven reorganization. Proc. Natl. Acad. Sci. U. S. A. 112, 3365–3373. doi:10.1073/pnas.1502497112
Amiel, A. R., Johnston, H. T., Nedoncelle, K., Warner, J. F., Ferreira, S., and Röttinger, E. (2015). Characterization of morphological and cellular events underlying oral regeneration in the sea anemone, Nematostella vectensis. Int. J. Mol. Sci. 16, 28449–28471. doi:10.3390/ijms161226100
Baumgarten, S., Cziesielski, M. J., Thomas, L., Michell, C. T., Esherick, L. Y., Pringle, J. R., et al. (2018). Evidence for miRNA‐mediated modulation of the host transcriptome in cnidarian–dinoflagellate symbiosis. Mol. Ecol. 27 (2), 403–418. doi:10.1111/mec.14452
Baumgarten, S., Simakov, O., Esherick, L. Y., Liew, Y. J., Lehnert, E. M., Michell, C. T., et al. (2015). The genome of Aiptasia, a sea anemone model for coral symbiosis. Proc. Natl. Acad. Sci. U. S. A. 112, 11893–11898. doi:10.1073/pnas.1513318112
Bendtsen, J. D., Nielsen, H., Von Heijne, G., and Brunak, S. (2004). Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795. doi:10.1016/j.jmb.2004.05.028
Bideau, L., Kerner, P., Hui, J., Vervoort, M., and Gazave, E. (2021). Animal regeneration in the era of transcriptomics. Cell. Mol. Life Sci. 78, 3941–3956. doi:10.1007/s00018-021-03760-7
Bradshaw, B., Thompson, K., and Frank, U. (2015). Distinct mechanisms underlie oral vs aboral regeneration in the cnidarian Hydractinia echinata. Elife 4, e05506. doi:10.7554/eLife.05506
Challa, A. A., and Stefanovic, B. (2011). A novel role of vimentin filaments: Binding and stabilization of collagen mRNAs. Mol. Cell. Biol. 31 (18), 3773–3789. doi:10.1128/MCB.05263-11
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi:10.1016/j.molp.2020.06.009
Chourrout, D., Delsuc, F., Chourrout, P., Edvardsen, R. B., Rentzsch, F., Renfer, E., Technau, U., et al. (2006). Minimal ProtoHox cluster inferred from bilaterian and cnidarian Hox complements. Nature 442 (7103), 684–687. doi:10.1038/nature04863
Chuang, Y. C., Lee, C. H., Sun, W. H., and Chen, C. C. (2018). Involvement of advillin in somatosensory neuron subtype-specific axon regeneration and neuropathic pain. Proc. Natl. Acad. Sci. U. S. A. 115 (36), E855–E8566. doi:10.1073/pnas.1716470115
Creyghton, M. P., Roël, G., Eichhorn, P. J., Hijmans, E. M., Maurer, I., Destrée, O., et al. (2005). PR72, a novel regulator of Wnt signaling required for Naked cuticle function. Genes Dev. 19 (3), 376–386. doi:10.1101/gad.328905
Cui, J., Zhu, L., Xia, X., Wang, H. Y., Legras, X., Hong, J., Wang, R. F., et al. (2010). NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 141 (3), 483–496. doi:10.1016/j.cell.2010.03.040
DuBuc, T. Q., Stephenson, T. B., Rock, A. Q., and Martindale, M. Q. (2018). Hox and Wnt pattern the primary body axis of an anthozoan cnidarian before gastrulation. Nat. Commun. 9 (1), 2007–2012. doi:10.1038/s41467-018-04184-x
Enríquez, S., Méndez, E. R., and Iglesias-Prieto, R. (2005). Multiple scattering on coral skeletons enhances light absorption by symbiotic algae. Limnol. Oceanogr. 50 (4), 1025–1032. doi:10.4319/lo.2005.50.4.1025
Erwin, P. M., and Szmant, A. M. (2010). Settlement induction of Acropora palmata planulae by a GLW-amide neuropeptide. Coral Reefs 29 (4), 929–939. doi:10.1007/s00338-010-0634-1
Ferrier, D. E. (2016). Evolution of homeobox gene clusters in animals: The giga-cluster and primary vs. secondary clustering. Front. Ecol. Evol. 4, 36. doi:10.3389/fevo.2016.00036
Fuller, Z. L., Mocellin, V. J., Morris, L. A., Cantin, N., Shepherd, J., Sarre, L., Przeworski, M., et al. (2020). Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science 369 (6501), eaba4674. doi:10.1126/science.aba4674
Gajigan, A. P., and Conaco, C. (2017). A microRNA regulates the response of corals to thermal stress. Mol. Ecol. 26 (13), 3472–3483. doi:10.1111/mec.14130
Gardam, S., Sierro, F., Basten, A., Mackay, F., and Brink, R. (2008). TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity 28 (3), 391–401. doi:10.1016/j.immuni.2008.01.009
Guder, C., Philipp, I., Lengfeld, T., Watanabe, H., Hobmayer, B., and Holstein, T. W. (2006). The Wnt code: Cnidarians signal the way. Oncogene 25 (57), 7450–7460.
Havrilak, J. A., Al-Shaer, L., Baban, N., Akinci, N., and Layden, M. J. (2021). Characterization of the dynamics and variability of neuronal subtype responses during growth, degrowth, and regeneration of Nematostella vectensis. BMC Biol. 19, 104–119. doi:10.1186/s12915-021-01038-9
He, S., Del Viso, F., Chen, C. Y., Ikmi, A., Kroesen, A. E., and Gibson, M. C. (2018). An axial Hox code controls tissue segmentation and body patterning in Nematostella vectensis. Science 361 (6409), 1377–1380. doi:10.1126/science.aar8384
Holland, P. W., Booth, H. A. F., and Bruford, E. A. (2007). Classification and nomenclature of all human homeobox genes. BMC Biol. 5 (1), 47–28. doi:10.1186/1741-7007-5-47
Holland, P. W. (2013). Evolution of homeobox genes. Wiley Interdiscip. Rev. Dev. Biol. 2 (1), 31–45. doi:10.1002/wdev.78
Holstein, T. W., Hobmayer, E., and Technau, U. (2003). Cnidarians: An evolutionarily conserved model system for regeneration? Dev. Dyn. 226 (2), 257–267. doi:10.1002/dvdy.10227
Hu, M., Zheng, X., Fan, C. M., and Zheng, Y. (2020). Lineage dynamics of the endosymbiotic cell type in the soft coral Xenia. Nature 582 (7813), 534–538. doi:10.1038/s41586-020-2385-7
Hui, J. H., McDougall, C., Monteiro, A. S., Holland, P. W., Arendt, D., Balavoine, G., et al. (2012). Extensive chordate and annelid macrosynteny reveals ancestral homeobox gene organization. Mol. Biol. Evol. 29 (1), 157–165. doi:10.1093/molbev/msr175
Jopling, C., Suñè, G., Morera, C., and Izpisua Belmote, J. C. (2012). p38α MAPK regulates myocardial regeneration in zebrafish. Cell. cycle 11 (6), 1195–1201. doi:10.4161/cc.11.6.19637
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi:10.1038/s41587-019-0201-4
Krishna, S., Nair, A., Cheedipudi, S., Poduval, D., Dhawan, J., Palakodeti, D., et al. (2013). Deep sequencing reveals unique small RNA repertoire that is regulated during head regeneration in Hydra magnipapillata. Nucleic Acids Res. 41, 599–616. doi:10.1093/nar/gks1020
Kücken, M., Soriano, J., Pullarkat, P. A., Ott, A., and Nicola, E. M. (2008). An osmoregulatory basis for shape oscillations in regenerating hydra. Biophysical J. 95 (2), 978–985.
Lahne, M., Li, J., Marton, R. M., and Hyde, D. R. (2015). Actin-Cytoskeleton-and Rock-Mediated INM are required for photoreceptor regeneration in the adult zebrafish retina. J. Neurosci. 35 (47), 15612–15634. doi:10.1523/JNEUROSCI.5005-14.2015
Layden, M. J., Rentzsch, F., and Röttinger, E. (2016). The rise of the starlet sea anemone Nematostella vectensis as a model system to investigate development and regeneration. Wiley Interdiscip. Rev. Dev. Biol. 5, 408–428. doi:10.1002/wdev.222
Lepilina, A., Coon, A. N., Kikuchi, K., Holdway, J. E., Roberts, R. W., Burns, C. G., et al. (2006). A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. 127 (3), 607–619. doi:10.1016/j.cell.2006.08.052
Li, Y., Nong, W., Baril, T., Yip, H. Y., Swale, T., Hayward, A., Hui, J. H., et al. (2020). Reconstruction of ancient homeobox gene linkages inferred from a new high-quality assembly of the Hong Kong oyster (Magallana hongkongensis) genome. Bmc Genomics 21 (1), 1–17. doi:10.1186/s12864-020-07027-6
Li, Z. F., Wu, X. H., and Engvall, E. (2004). Identification and characterization of CPAMD8, a novel member of the complement 3/alpha2-macroglobulin family with a C-terminal Kazal domain. Genomics 83 (6), 1083–1093. doi:10.1016/j.ygeno.2003.12.005
Litsios, G., Sims, C. A., Wüest, R. O., Pearman, P. B., Zimmermann, N. E., and Salamin, N. (2012). Mutualism with sea anemones triggered the adaptive radiation of clownfishes. BMC Evol. Biol. 12 (1), 212–215. doi:10.1186/1471-2148-12-212
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: The conserved domain database in 2020. CDD/SPARCLE conserved domain database 20Nucleic acids Res. 48 (D1), D265–D268. doi:10.1093/nar/gkz991
Luke, G. N., Castro, L. F. C., McLay, K., Bird, C., Coulson, A., and Holland, P. W. (2003). Dispersal of NK homeobox gene clusters in amphioxus and humans. Proc. Natl. Acad. Sci. U. S. A. 100 (9), 5292–5295. doi:10.1073/pnas.0836141100
Marks, P. W., Arai, M., Bandura, J. L., and Kwiatkowski, D. J. (1998). Advillin (p92): A new member of the gelsolin/villin family of actin regulatory proteins. J. Cell. Sci. 111 (15), 2129–2136. doi:10.1242/jcs.111.15.2129
Miljkovic-Licina, M., Chera, S., Ghila, L., and Galliot, B. (2007). Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Dev. 134 (6), 1191–1201. doi:10.1242/dev.02804
Moran, Y., Agron, M., Praher, D., and Technau, U. (2017). The evolutionary origin of plant and animal microRNAs. Nat. Ecol. Evol. 1, 27–28. doi:10.1038/s41559-016-0027
Moran, Y., Fredman, D., Praher, D., Li, X. Z., Wee, L. M., Rentzsch, F., et al. (2014). Cnidarian microRNAs frequently regulate targets by cleavage. Genome Res. 24, 651–663. doi:10.1101/gr.162503.113
Nakanishi, N., and Martindale, M. Q. (2018). CRISPR knockouts reveal an endogenous role for ancient neuropeptides in regulating developmental timing in a sea anemone. Elife 7, e39742. doi:10.7554/eLife.39742
Nong, W., Cao, J., Li, Y., Qu, Z., Sun, J., Swale, T., et al. (2020). Jellyfish genomes reveal distinct homeobox gene clusters and conservation of small RNA processing. Nat. Commun. 11, 1–11. doi:10.1038/s41467-020-16801-9
Novikova, E. L., Bakalenko, N. I., Nesterenko, A. Y., and Kulakova, M. A. (2016). Hox genes and animal regeneration. Russ. J. Dev. Biol. 47, 173–180. doi:10.1134/s106236041604007x
Ochoa, B., Syn, W. K., Delgado, I., Karaca, G. F., Jung, Y., Wang, J., Diehl, A. M., et al. (2010). Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 51 (5), 1712–1723. doi:10.1002/hep.23525
Oren, U., Benayahu, Y., Lubinevsky, H., and Loya, Y. (2001). Colony integration during regeneration in the stony coral Favia favus. Ecology 82, 802–813. doi:10.2307/2680199
Park, H. D., Ortmeyer, A. B., and Blankenbaker, D. P. (1970). Cell division during regeneration in Hydra. Nature 227, 617–619. doi:10.1038/227617a0
K. A. Pitt, and J. E. Purcell (Editors) (2009), Jellyfish blooms: Causes, consequences and recent advances. (Springer Science & Business Media), 206.
Pollard, S. L., and Holland, P. W. (2000). Evidence for 14 homeobox gene clusters in human genome ancestry. Curr. Biol. 10 (17), 1059–1062. doi:10.1016/s0960-9822(00)00676-x
Praher, D., Zimmermann, B., Dnyansagar, R., Miller, D. J., Moya, A., Modepalli, V., et al. (2021). Conservation and turnover of miRNAs and their highly complementary targets in early branching animals. Proc. Biol. Sci. 288, 20203169. doi:10.1098/rspb.2020.3169
Presnell, J. S., Wirsching, E., and Weis, V. M. (2022). Tentacle patterning during Exaiptasia diaphana pedal lacerate development differs between symbiotic and aposymbiotic animals. PeerJ 10, e12770. doi:10.7717/peerj.12770
Putnam, N. H., O'Connell, B. L., Stites, J. C., Rice, B. J., Blanchette, M., Calef, R., et al. (2016). Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350. doi:10.1101/gr.193474.115
Rao, J., Ashraf, S., Tan, W., Van Der Ven, A. T., Gee, H. Y., Braun, D. A., et al. (2017). Advillin acts upstream of phospholipase C ϵ1 in steroid-resistant nephrotic syndrome. J. Clin. Invest. 127 (12), 4257–4269. doi:10.1172/JCI94138
Ramirez, A. N., Loubet-Senear, K., and Srivastava, M. (2022). A regulatory program for initiation of Wnt signaling during posterior regeneration. Cell Reports 32 (9), 108098.
Röttinger, E. (2021). Nematostella vectensis, an emerging model for deciphering the molecular and cellular mechanisms underlying whole-body regeneration. Cells 10, 2692. doi:10.3390/cells10102692
Rousset, R., Mack, J. A., Wharton, K. A., Axelrod, J. D., Cadigan, K. M., Fish, M. P., Scott, M. P., et al. (2001). Naked cuticle targets dishevelled to antagonize Wnt signal transduction. Genes Dev. 15 (6), 658–671. doi:10.1101/gad.869201
Ryan, J. F., Burton, P. M., Mazza, M. E., Kwong, G. K., Mullikin, J. C., and Finnerty, J. R. (2006). The cnidarian-bilaterian ancestor possessed at least 56 homeoboxes: Evidence from the starlet Sea anemone, Nematostella vectensis. Genome Biol. 7 (7), R64–R20. doi:10.1186/gb-2006-7-7-R64
Schaffer, A. A., Bazarsky, M., Levy, K., Chalifa-Caspi, V., and Gat, U. (2016). A transcriptional time-course analysis of oral vs. aboral whole-body regeneration in the Sea anemone Nematostella vectensis. Bmc Genomics 17 (1), 718–722. doi:10.1186/s12864-016-3027-1
Schnapp, E., Kragl, M., Rubin, L., and Tanaka, E. M. (2005). Hedgehog signaling controls dorsoventral patterning, blastema cell proliferation and cartilage induction during axolotl tail regeneration.
Schummer, M., Scheurlen, I., Schaller, C., and Galliot, B. (1992). HOM/HOX homeobox genes are present in hydra (Chlorohydra viridissima) and are differentially expressed during regeneration. The EMBO J. 11 (5), 1815–1823.
Shikina, S., Chiu, Y. L., Zhang, Y., Liu, T. Y., Tsai, P. H., Zatylny-Gaudin, C., et al. (2020). Involvement of GLWamide neuropeptides in polyp contraction of the adult stony coral Euphyllia ancora. Sci. Rep. 10 (1), 1–11. doi:10.1038/s41598-020-66438-3
Simakov, O., Bredeson, J., Berkoff, K., Marletaz, F., Mitros, T., Schultz, D. T., Rokhsar, D. S., et al. (2022). Deeply conserved synteny and the evolution of metazoan chromosomes. Sci. Adv. 8 (5), eabi5884. doi:10.1126/sciadv.abi5884
Singer, I. I. (1971). Tentacular and oral-disc regeneration in the sea anemone, Aiptasia diaphana: III. Autoradiographic analysis of patterns of tritiated thymidine uptake. Development 26, 253–270. doi:10.1242/dev.26.2.253
Sinigaglia, C., Peron, S., Eichelbrenner, J., Chevalier, S., Steger, J., Barreau, C., Leclère, L., et al. (2020). Pattern regulation in a regenerating jellyfish. Elife 9, e54868. doi:10.7554/eLife.54868
Slobodkin, L. B., and Bossert, P. E. (2001). “Cnidaria,” in Ecology and classification of North American freshwater invertebrates. Editor H. T. P. C. JamesAlan. Second edition (San Diego, California: Academic Press), 140.
Soderlund, C., Bomhoff, M., and Nelson, W. M. (2011). SyMAP v3. 4: A turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39 (10), e68. doi:10.1093/nar/gkr123
Sousounis, K., Athippozhy, A. T., Voss, S. R., and Tsonis, P. A. (2014). Plasticity for axolotl lens regeneration is associated with age‐related changes in gene expression. Regeneration 1 (3), 47–57. doi:10.1002/reg2.25
Sousounis, K., Looso, M., Maki, N., Ivester, C. J., Braun, T., and Tsonis, P. A. (2013). Transcriptome analysis of newt lens regeneration reveals distinct gradients in gene expression patterns. PLoS One 8 (4), e61445. doi:10.1371/journal.pone.0061445
Stewart, Z. K., Pavasovic, A., Hock, D. H., and Prentis, P. J. (2017). Transcriptomic investigation of wound healing and regeneration in the cnidarian Calliactis polypus. Sci. Rep. 7 (1), 1–11. doi:10.1038/srep41458
Stierwald, M., Yanze, N., Bamert, R. P., Kammermeier, L., and Schmid, V. (2004). The Sine oculis/Six class family of homeobox genes in jellyfish with and without eyes: development and eye regeneration. Develop. bio. 274 (1), 70–81.
Szigeti, A., Bellyei, S., Gasz, B., Boronkai, A., Hocsak, E., Minik, O., Gallyas Jr., F., et al. (2006). Induction of necrotic cell death and mitochondrial permeabilization by heme binding protein 2/SOUL. FEBS Lett. 580 (27), 6447–6454. doi:10.1016/j.febslet.2006.10.067
Takahashi, T. (2020). Comparative aspects of structure and function of cnidarian neuropeptides. Front. Endocrinol. 11, 339. doi:10.3389/fendo.2020.00339
Takahashi, T., and Hatta, M. (2011). The importance of GLWamide neuropeptides in cnidarian development and physiology. J. Amino Acids, 424501. doi:10.4061/2011/424501
Tong, Y., Cui, J., Li, Q., Zou, J., Wang, H. Y., and Wang, R. F. (2012). Enhanced TLR-induced NF-κB signaling and type I interferon responses in NLRC5 deficient mice. Cell. Res. 22 (5), 822–835. doi:10.1038/cr.2012.53
Trevino, M., Stefanik, D. J., Rodriguez, R., Harmon, S., and Burton, P. M. (2011). Induction of canonical Wnt signaling by alsterpaullone is sufficient for oral tissue fate during regeneration and embryogenesis in Nematostella vectensis. Develop. Dynamics 240 (12), 2673–2679.
Tseng, P. H., Matsuzawa, A., Zhang, W., Mino, T., Vignali, D. A., and Karin, M. (2010). Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 11 (1), 70–75. doi:10.1038/ni.1819
van der Burg, C. A., Pavasovic, A., Gilding, E. K., Pelzer, E. S., Surm, J. M., Smith, H. L., et al. (2020). The rapid regenerative response of a model sea anemone species Exaiptasia pallida is characterised by tissue plasticity and highly coordinated cell communication. Marine Biotech. 22 (2), 285–307.
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M., and Jaffe, D. B. (2017). Direct determination of diploid genome sequences. Genome Res. 27 (5), 757–767. doi:10.1101/gr.214874.116
Whyte, J. L., Smith, A. A., and Helms, J. A. (2012). Wnt signaling and injury repair. Cold Spring Harb. Perspect. Biol. 4, a008078. doi:10.1101/cshperspect.a008078
Xia, W. X., Li, H. R., Ge, J. H., Liu, Y. W., Li, H. H., Su, Y. H., et al. (2021). High-continuity genome assembly of the jellyfish Chrysaora quinquecirrha. Zool. Res. 42 (1), 130. doi:10.24272/j.issn.2095-8137.2020.258
Yazawa, S., Umesono, Y., Hayashi, T., Tarui, H., and Agata, K. (2009). Planarian Hedgehog/Patched establishes anterior–posterior polarity by regulating Wnt signaling. Proc. Natl. Acad. Sci. U. S. A. 106 (52), 22329–22334. doi:10.1073/pnas.0907464106
Zeng, W., Wharton, K. A., Mack, J. A., Wang, K., Gadbaw, M., Suyama, K., et al. (2000). Naked cuticle encodes an inducible antagonist of Wnt signalling. Nature 403 (6771), 789–795. doi:10.1038/35001615
Zhong, Y. F., and Holland, P. W. (2011). HomeoDB2: Functional expansion of a comparative homeobox gene database for evolutionary developmental biology. Evol. Dev. 13 (6), 567–568. doi:10.1111/j.1525-142X.2011.00513.x
Keywords: cnidarian, sea anemone, genome, regeneration, transcriptome, microRNA
Citation: Shum CWY, Nong W, So WL, Li Y, Qu Z, Yip HY, Swale T, Ang PO, Chan KM, Chan TF, Chu KH, Chui APY, Lau KF, Ngai SM, Xu F and Hui JHL (2022) Genome of the sea anemone Exaiptasia pallida and transcriptome profiles during tentacle regeneration. Front. Cell Dev. Biol. 10:900321. doi: 10.3389/fcell.2022.900321
Received: 20 March 2022; Accepted: 08 July 2022;
Published: 17 August 2022.
Edited by:
Maria Ina Arnone, Stazione Zoologica Anton Dohrn, ItalyReviewed by:
Carlos Herrera-Ubeda, University of Barcelona, SpainCopyright © 2022 Shum, Nong, So, Li, Qu, Yip, Swale, Ang, Chan, Chan, Chu, Chui, Lau, Ngai, Xu and Hui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerome H.L. Hui, aHVpLmplcm9tZUBnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.