
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 11 August 2022
Sec. Stem Cell Research
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.878311
This article is part of the Research Topic Muscular Dystrophies: Current Therapeutic Advances to Improve and Restore Muscle Homeostasis View all 7 articles
 Vittoria Marini1†
Vittoria Marini1† Fabiola Marino1,2†
Fabiola Marino1,2† Flaminia Aliberti1,3
Flaminia Aliberti1,3 Nefele Giarratana1
Nefele Giarratana1 Enrico Pozzo1
Enrico Pozzo1 Robin Duelen1
Robin Duelen1 Álvaro Cortés Calabuig4
Álvaro Cortés Calabuig4 Rita La Rovere5
Rita La Rovere5 Tim Vervliet5
Tim Vervliet5 Daniele Torella2Geert Bultynck5
Daniele Torella2Geert Bultynck5 Maurilio Sampaolesi1,6*
Maurilio Sampaolesi1,6* Yoke Chin Chai1*
Yoke Chin Chai1*Duchenne Muscular Dystrophy (DMD) is an X-linked neuromuscular disease which to date is incurable. The major cause of death is dilated cardiomyopathy however, its pathogenesis is unclear as existing cellular and animal models do not fully recapitulate the human disease phenotypes. In this study, we generated cardiac organoids from patient-derived induced pluripotent stem cells (DMD-COs) and isogenic-corrected controls (DMD-Iso-COs) and studied if DMD-related cardiomyopathy and disease progression occur in the organoids upon long-term culture (up to 93 days). Histological analysis showed that DMD-COs lack initial proliferative capacity, displayed a progressive loss of sarcoglycan localization and high stress in endoplasmic reticulum. Additionally, cardiomyocyte deterioration, fibrosis and aberrant adipogenesis were observed in DMD-COs over time. RNA sequencing analysis confirmed a distinct transcriptomic profile in DMD-COs which was associated with functional enrichment in hypertrophy/dilated cardiomyopathy, arrhythmia, adipogenesis and fibrosis pathways. Moreover, five miRNAs were identified to be crucial in this dysregulated gene network. In conclusion, we generated patient-derived cardiac organoid model that displayed DMD-related cardiomyopathy and disease progression phenotypes in long-term culture. We envision the feasibility to develop a more complex, realistic and reliable in vitro 3D human cardiac-mimics to study DMD-related cardiomyopathies.
Duchenne Muscular Dystrophy (DMD) is one of the most common muscular dystrophies which affects 1:5000 live male births (Yiu and Kornberg, 2015). It is a progressive X-linked genetic disorder caused by mutations within the DMD gene, which results in a complete absence of Dystrophin (DYS) protein expression (Muntoni et al., 2003; Flanigan, 2014; Loboda and Dulak, 2020). The absence of DYS leads to muscle weakness and wasting, owing to the loss of muscle membrane integrity and susceptibility to stress-induced damages (Lin et al., 2015). In recent years, the use of respiratory assist devices and non-invasive positive pressure ventilation have increased the life expectancy of DMD patients, nevertheless this has contributed to the rise of previously unknown late-stages DMD complications, such as dilated cardiomyopathy (DCM) (Kamdar and Garry, 2016; Breuls et al., 2021).
DMD-associated DCM is characterized by initial cardiomyocyte degeneration attributed to the inflammatory response, which leads to the replacement of heart muscle with fat and connective tissue (i.e. fibrosis of the left-ventricular (LV) myocardial wall) and thus the reduction of cardiac wall thickness (Finsterer and Stollberger, 2003; Law et al., 2020). Due to the latter, the myocardium becomes more sensitive to pressure overload causing LV dilatation, cardiac contractility reduction and eventually congestive heart failure (Luk et al., 2009; Fayssoil et al., 2010; McNally and Mestroni, 2017). Although DCM represents the major cause of mortality in DMD patients, no great research attention has been directed to DCM–partly due to limited accessibility to human cardiac tissues and the intrinsic limitation of two-dimensional (2D) cardiomyocyte culture in recapitulating human three-dimensional (3D) pathophysiology (Lin et al., 2015; Quattrocelli et al., 2015; Law et al., 2020). Similarly, DMD animal models (mdx mice and canine DMD models) do not fully resemble human DMD features and its disease progression, mainly due to inter-species variations. It is therefore imperative to develop 3D human cardiac-mimics of DMD-relevance to bridge this scientific gap (McGreevy et al., 2015; Filippo Buono et al., 2020; Jensen and Teng, 2020; Zhao et al., 2021).
Organoids are in vitro self-organized 3D cellular structures derived from either primary tissues or stem cells [e.g. embryonic (ESCs) or induced pluripotent stem cells (iPSCs), and primary stem cells] differentiated into designated functional cell types. They possess organotypic structures including the cytoarchitecture and the mechanisms involved in the cell behavior and fate within the specific tissue (Velasco et al., 2020; Heydari et al., 2021; Scalise et al., 2021). The advent of iPSC and CRISPR/Cas9 technologies represent a paramount breakthrough for patient-specific model generation, enabling the development of iPSC-derived cardiomyocyte (CM)-based 3D models and the isogenic controls, which are widely used to study patient-specific cardiac diseases in vitro (Filippo Buono et al., 2020; Richards et al., 2020). Although 3D cardiac models were used for investigating abnormal mechanical and electromechanical properties of DMD CMs (Caluori et al., 2019; Jelinkova et al., 2020), as to our knowledge, the organoid technology with proper isogenic control has not been used to model DMD cardiomyopathies. Given that, this study focused on the development of 3D cardiac organoids (COs) from DMD patient-derived iPSC (DMD-COs) and its mutation-corrected isogenic iPSC controls (DMD-Iso-COs), and studied if these human cardiac-mimics could reproduce DMD-related cardiomyopathy and disease progression in 3D via long-term culture.
Duchenne Muscular Dystrophy human iPSC (DMD-hiPSC) was obtained from DMD patient’s fibroblasts carrying a point mutation in exon 35 (c.4 996C>T; p.Arg1,666X) of the DMD gene that leads to a premature stop codon (Duelen et al., 2022). The DMD isogenic control (DMD-Iso-hiPSC) was generated through CRISPR/Cas9 gene editing from the S. pyogenes system (5’-NGG PAM) as previously described (Ran et al., 2013; Duelen et al., 2022). The healthy control iPSC (HC-hiPSC) line was a gift from Prof. P. Jennings (Medizinische Universität Innsbruck, Austria) to Stem Cell Institute Leuven, KU Leuven and generated by SeV-based reprogramming of male donor fibroblasts (SBAD2). Human iPSC lines were cultured feeder-free on Geltrex LDEV-Free hESC-Qualified Reduced Growth Factor Basement Membrane Matrix and maintained in Essential 8 Flex Basal Medium (Thermo Fisher Scientific) supplemented with Essential 8 Flex Supplement (50x, Thermo Fisher Scientific) and penicillin–streptomycin (0.1%, Thermo Fisher Scientific), at 37°C under normoxic conditions (21% O2 and 5% CO2). Colonies were routinely passaged non-enzymatically with 0.5 mM EDTA in Phosphate-Buffered Saline (PBS, Thermo Fisher Scientific). The use of human samples from DMD subjects for experimental purposes and protocols in the present study was approved by the Ethics Committee of the University Hospitals Leuven (S55438 and S65190, respectively).
DMD-, DMD-Iso- and HC-hiPSC lines were differentiated into functional cardiomyocytes (CMs) according to a monolayer-based cardiac differentiation protocol, as previously described (Burridge et al., 2014). Briefly, prior to differentiation, the DMD-, DMD-Iso- and HC-hiPSC lines were suspended into small colonies and subsequently cultured on Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix layer (Corning) in complete Essential 8 Flex Medium at 37°C under hypoxic conditions (5% O2 and 5% CO2) for 3 days, in order to obtain the pre-optimized targeted confluency of 85%. Mesoderm differentiation (day 0) was induced using 6 µM CHIR99021 (Axon Medchem) for 48 h in a chemically defined medium consisting of RPMI 1640 (Thermo Fisher Scientific), 500 μg/ml rice-derived recombinant human albumin and 213 μg/ml L-ascorbic acid 2-phosphate (Sigma-Aldrich). After 24 h of CHIR99021 stimulation, the cells were transferred from hypoxia to normoxia. On day 2 of differentiation, iPSC-derived mesodermal cells were fed with basal medium supplemented with 4 µM IWR-1 (Sigma-Aldrich) for 48 h, to induce cardiac progenitor cell differentiation. From day 4 onwards, medium was refreshed every 2 days with CM Maintenance Medium (RPMI 1640, rice-derived recombinant human albumin and L-ascorbic acid 2-phosphate). Contracting CMs appeared at day 8 or 9 of cardiac differentiation.
A 3% agarose (Invitrogen) gel solution was prepared in PBS. The powder was fully dissolved by heating in microwave oven and the agarose microwells were fabricated in sterile conditions. In brief, the heated agarose solution was added into a custom-made 3D printed micropillar molds (in 24-well plate format). Upon cooling at room temperature for 10 min, the agarose was removed from the molds, thus creating 24 culture inserts each consisting of 137 microwells (diameter × height = 500 × 700 μm). The culture inserts were transferred into a 24-well plate and equilibrated in PBS overnight at 37°C under normoxia conditions (5% O2 and 5% CO2).
After reaching confluency, the DMD-, DMD-Iso- and HC-hiPSC lines were detached using 0.5 mM EDTA at 37°C and resuspended in Essential 8TM medium supplemented with Revitacel™ Supplement (dilution 1:100, Thermo Fisher Scientific). After cell count, the hiPSCs were resuspended in 1 ml of Essential 8 Flex Basal Medium (Thermo Fisher Scientific) and were plated in agarose inserts at two different cell densities, 5 × 103 cells/microwell and 1 × 104 cells/microwell respectively. The plates were centrifuged for 10 min at 1,200 rpm to facilitate sedimentation of cells in the microwells. Subsequently, 1 ml of fresh Essential 8 Flex Basal Medium was added to completely cover the microwell area and incubated at 37°C under hypoxic conditions (5% O2 and 5% CO2) to promote embryoid bodies (EBs) formation. The medium was refreshed every day for 3 days and cardiac differentiation of the EBs into cardiac organoids (COs) was initiated as described above for the monolayer cardiomyocyte differentiation protocol. On day 5, the COs were transferred from the agarose molds to an ultra-low attachment 6-well plate (Costar, Corning) and dynamic culture was carried out using an orbital shaker at 75 rpm in CM maintenance medium until day 93. The media was changed every 2 days. Contracting COs start to appear from day 8 of the differentiation protocol. The samples were collected on day 10, 14, 28, 56 and 93 for subsequent analyses.
At different time points, the COs were fixed with 4% paraformaldehyde (PFA; Polysciences) for 30 min at room temperature and subsequently embedded in cryogel (Tissue- Tek ® O.C.T. ™ Compound). The samples were snap-frozen in liquid nitrogen and stored at −80°C until cryosectioning. The samples were sectioned at the thickness of 6 µm using the HM525 NX Cryostat (Thermo Scientific) and stored at −20°C prior to analysis. For H&E staining, the cryosections were stained in Harris hematoxylin solution (Sigma-Aldrich), counterstained in eosin solution (0.1% erithrosin extra bluish Sigma-Aldrich in 70% ethanol) and mounted with DPX mountant (Sigma) upon dehydration according to routine protocols. For PSR staining, the cryosections were stained for collagen content using the Vitro View™ Picro-Sirius Red Stain Kit (Cat. No. VB-3017) according to the manufacturer’s instructions (Giarratana et al., 2020). The nuclei were counterstained with Weigert’s Hematoxylin Solution and mounted with DPX mountant (Sigma-Aldrich). Lipid droplets deposition was detected by BODIPY staining. In brief, the BODIPY™ 493/503 4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene (Invitrogen) powder was dissolved in DMSO at the concentration of 1.3 mg/ml. The cryosections were incubated with the BODIPY solution, diluted 1:2500 in PBS, for 15 min at room temperature and subsequently mounted with Antifade Mounting Medium with DAPI (VECTASHIELD®). All images were acquired using Axiocam MRm microscope (Zeiss).
For quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR) assays, total RNA was isolated through Purelink® RNA mini kit (Thermo Fisher Scientific) and treated with TurboTM DNA-free kit (Thermo Fisher Scientific) to purify RNA samples. 1 μg RNA was reverse-transcribed using Superscript III Reverse Transcriptase First-Strand Synthesis SuperMix (Thermo Fisher Scientific). Thermal cycler setting: 25°C 10 min, 50°C 30 min, 85°C 5 min, 37°C 20 min incubation with E. Coli RNAse H. A 384-well plate was prepared using Platinum SYBR Green QPCR SuperMix-UDG (Thermo Fisher Scientific) as SYBR Green on 1:5 diluted cDNA. The RT-qPCR was performed by Viia7 384-plate reader (Thermo Fisher Scientific; final primer concentration, 100 nM; final volume, 10 μl; thermal profile, 95°C 15 s, 60°C 60 s, 40×). The oligonucleotide primer sequences are listed in Table 1. Delta Ct (ΔCt) values were calculated by subtracting the Ct values from the genes of interest with the Ct values of the housekeeping genes (GAPDH).

TABLE 1. List of primer used for gene expression analysis.
DMD- and DMD-Iso-COs at 14 days were dissociated using Collagenase A (1 U/ml) for 20 min at 37°C followed by 10 min incubation with Accutase® (Sigma-Aldrich). All flow cytometry procedures were performed according to the manufacturer’s instructions. PBS + 0.1% BSA (Sigma) was used as staining buffer. The cells were stained for the surface markers SIRPA, CD31 and PDGRFa specific for CMs, endothelial cells and fibroblast/adipose progenitors respectively. A total of 50.000 cells was recorded. After single cell gate selection, a cell number total between 15.000 and 20.000 cells was analysed for the used antibodies (CD31, PDGFRα, SIRPA). Fluorescence minus one (FMO) controls and compensations were included for appropriate gating. Samples were analysed using the FACS Canto II HTS (BD Biosciences) and the analysis was performed using FACS Diva Software. Table 2 provides a list of all flow cytometric antibodies used in this study.

TABLE 2. List of antibodies dilutions used for immunofluorescence analysis and flow cytometric analysis.
After three PBS washes, the cryosections were permeabilized for 1 h at room temperature using 0.1% Triton X-100 in PBS (Thermo Fisher Scientific). Non-specific antibody binding was blocked by incubation for 30 min with blocking solution containing 5% normal goat serum (NGS, Dako) at room temperature followed by overnight incubation at 4°C with different primary antibodies listed in Table 2. After washing in phosphate-buffered saline (PBS), the samples were incubated with respective secondary antibodies using Alexa Fluor 488 and 555-conjugated secondary antibody (4 μg/ml; Thermo Fisher Scientific). Nuclei were counterstained with Hoechst 33342 (1:1000, Thermo Scientific) for 7 min (Santoni de Sio et al., 2008). The sections were mounted with ProLong ™ Gold antifade reagent (Invitrogen) and stored in the dark at 4°C until imaging. All images were acquired using Axiocam MRm microscope (Zeiss). Confocal images were acquired using Nikon ECLIPSE Ti Microscope and NIS-Elements AR 4.11 software. Quantifications of immunofluorescent images were performed using ImageJ software tool by calculating the percentage of the signal area/positive nuclei with respect to the total organoid area/total nuclei respectively for each CO.
To assess the contractile properties of DMD-COs and DMD-Iso-COs, 3D cardiac organoids were live-imaged using the Dmi1 Microscope (Leica). The recorded videos were then analysed to determine the CO beating frequency by counting the number of spontaneous contractions per minute. The cardiac organoids growth area was measured at different time points using ImageJ software tool.
For Ca2+ imaging experiments, the DMD-, DMD-Iso- and HC-hiPSC monolayers were respectively plated on 35 mm dishes with four Chamber glass bottom. Following 14 days from cardiac induction, the DMD-, DMD-Iso- and HC- CM were incubated with 1 µM Fluo-4 AM solubilized in CM Maintenance Medium. Next, the cells were washed twice with CM Maintenance Medium after which de-esterification was allowed to occur for 45 min at 37°C and 5% CO2. The Ca2+ imaging experiments were performed in pre-warmed (37°C) modified Krebs-Ringer solution (135 mM NaCl, 6.2 mM KCl, 1.2 mM MgCl2, 12 mM HEPES, pH 7.3, 11.5 mM glucose and 2 mM CaCl2). Tetracaine was solubilized in the above modified Krebs-Ringer solution at 1 mM final concentration. For the KCl stimulus the modified Krebs-Ringer solution was prepared substituting the NaCl for 140 mM KCl. Imaging was performed using a Nikon eclipse Ti2 inverted fluorescence microscope (Nikon) equipped with excitation filter FF01-378/474/554/635 and dichroic mirror FF01-432/515/595/730 and emission filter 515/30 all from Semrock. Coolled pR-4000 (Coolled) was used for excitation at 470 nm. Acquisition of the fluorescent signal at 520 nM was performed at 10 Hz using a pco.edge 4.2bi sCMOS camera (pCO) (Nakamura et al., 2001). For analysis FIJI software was utilized. In each experiment a region of interest was drawn across spontaneously active cardiomyocytes. The fluorescence intensities were normalized to F0, where the F0 value was obtained after tetracaine administration.
RNA (>10 μg) extracted from DMD-COs and DMD-Iso-COs on day 56 was verified and processed by the Genomics Core (KU Leuven–UZ Leuven). As quality control, the RNA concentration was measured with Nanodrop and quality was checked with Bioanalyzer. The Lexogen QuantSeq 3′ mRNA-Seq library prep kit was used according to the manufacturer's protocol with 500 ng input. After the prep, the libraries were measured with Qubit and put on the Fragment analyser so the libraries could be pooled equimolar to 2 nM. The pool was then quantified with RT-qPCR and a final pool (2 nM) was made for single-read sequencing on the HiSeq4000 (Illumina Inc.). The settings were 51-8-8. The raw sequence files generated (.fastq files) underwent quality control analysis using FastQC v0.11.7 (Andrews, 2010). Adapters were filtered with ea-utils fastq-mcf v1.05 (Aronesty, 2011). Splice-aware alignment was performed with HiSat2 against the human reference genome hg38 using the default parameters. Reads mapping to multiple loci in the reference genome were discarded. Resulting BAM alignment files were handled with Samtools v1.5. (Li et al., 2009). Quantification of reads per gene was performed with HT-seq Count v2.7.14. Count-based differential expression analysis was done with R-based (The R Foundation for Statistical Computing, Vienna, Austria) Bioconductor package DESeq2 (Love et al., 2014). Reported p-values were adjusted for multiple testing with the Benjamini-Hochberg procedure, which controls false discovery rate (FDR). Gene Ontology (GO) and Biological Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were identified using g:Profiler (Raudvere et al., 2019). The GO Biological Process 2018 and KEGG 2016 of each tissue were determined. The significant terms and pathways were selected with the threshold of adjusted p-value < 0.05. Data has been deposited in the NCBI Gene Expression Omnibus (GEO) repository under accession code GSE194297.
The PPI network of differentially upregulated genes in DMD-COs was constructed by feeding a list of gene symbols and their log2fold changes into the NetworkAnalyst platform (http://www.networkanalyst.ca/) using the IMEx interactome database with Steiner Forest Network (SFN) reduction algorithm. Subsequently, the gene-miRNA interactions (Rotini et al., 2018) for the selected KEGG pathways were constructed based on the miRTarBase (v8.0) database, and the network was reduced using the SFN algorithm. The degree of each node was calculated based on its number of connections to other nodes. In the network, the area of an individual node indicates the degree, and the color represents the expression. The identified top five miRNAs were mapped out in the KEGG pathways to show their interactions with the genes of a particular pathway.
Data were statistically analysed using GraphPad Prism. All data were reported as mean ± standard deviation (SD). Differences between groups were examined for statistical significance using ANOVA and two-way ANOVA. Significance of the differences was indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Following the 2D monolayer differentiation protocol, we generated cardiomyocytes (CMs) from both DMD patient-derived hiPSC (DMD-CMs), the isogenic control hiPSC (DMD-Iso-CMs) and healthy control hiPSC (HC-CMs) monolayer cultures. Figure 1A shows representative images of 2D culture morphology of DMD-CMs and DMD-Iso-CMs on day 22. These cells started to develop contractile phenotype around day 8 and were morphologically similar in both conditions. On day 22, RT-qPCR analysis showed that the DMD-CMs expressed significantly lower dystrophin (DMD) than the isogenic controls and HC-CMs (Figure 1B), confirming the restoration of DMD expression in the isogenic controls, as described in Duelen et al. (2022). The DMD-CMs also expressed significantly lower sarcomeric α-actinin (ACTN2), the pacemaker gene HCN4, and the troponin-related genes (TNNI1, TNNC1 and TNNI3) but not the myosin light (MYL7, MYL2) or heavy chain (MYH6, MYH7) genes, than the isogenic controls (Figure 1C). Next, we established a cell physiological analysis of 14-day differentiated DMD-CMs and DMD-Iso-CMs. A hallmark of functional CMs is their ability to generate cytosolic Ca2+ signals that are driven by ryanodine receptors (RyRs), intracellular Ca2+ -release channels residing at the sarcoplasmic reticulum of CMs. Therefore, cytosolic Ca2+ imaging was performed in single-cell CMs loaded with Fluo-4. In the presence of extracellular Ca2+ (1.5 mM CaCl2), spontaneous Ca2+ oscillations were observed in both DMD-CMs and DMD-Iso-CMs that could be blocked by tetracaine, an inhibitor of RyR channels. However, spontaneous Ca2+ oscillations appeared to display a lower frequency and amplitudes in DMD-CMs compared to isogenic and healthy controls, indicating a defect in physiological Ca2+ signalling in dystrophic CMs that is corrected in the isogenic controls. Moreover, DMD-CMs displayed a lower Ca2+ response to KCl, which provokes membrane depolarization, compared to DMD-Iso-CMs (Figures 1D,E). These findings validated the dystrophic properties of DMD-CMs and their defects in physiological Ca2+ signalling, whereby both deficiencies could be reverted in DMD-Iso-CMs generated in this study.
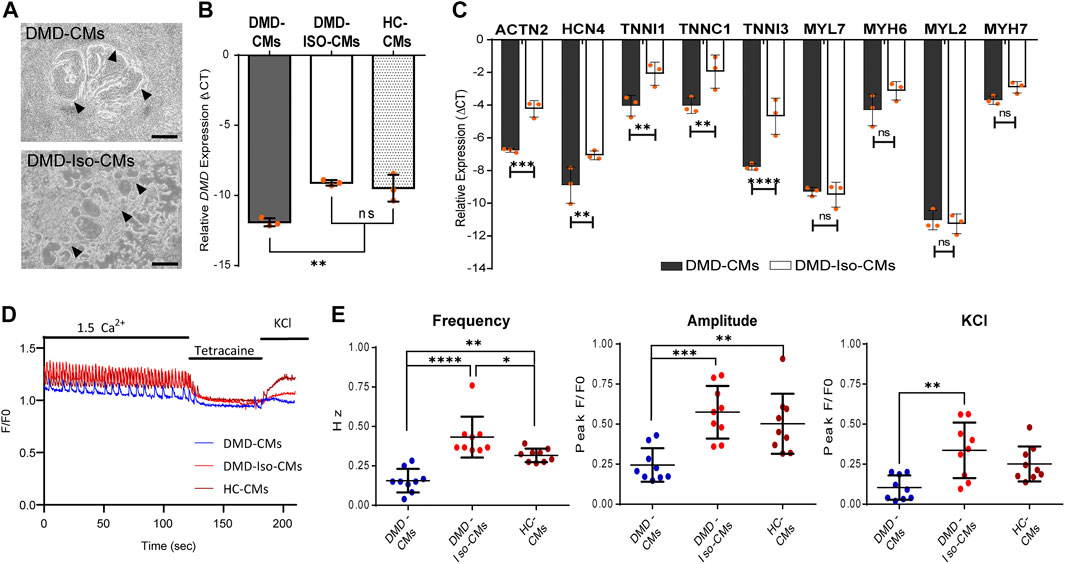
FIGURE 1. (A) Representative 2D culture morphology of differentiated cardiomyocytes from DMD patient-derived hiPSCs (DMD-CMs) and the isogenic controls (DMD-Iso-CMs) on day 22. Arrowheads indicated the contractile filaments. Scale bar = 200 μm. (B,C) Comparison of the expression of dystrophin (DMD) and key cardiac gene markers between DMD-CMs, DMD-Iso-CMs and HC-CMs 2D cultures on day 22. Data shown are mean ± s.d. (n = 3). Statistical analyses were performed by two-way ANOVA with Tukey’s multiple comparisons: **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s, not significant. (D,E) Intracellular Ca2+ imaging of DMD-CMs, DMD-Iso-CMs and HC-CMs 2D cultures on day 14 showing higher frequency and amplitude of spontaneous Ca2+ oscillation in DMD-CMs than the isogenic and healty controls, as well as a lower KCl response compared to DMD-Iso-CMs. Tetracaine was used to validate that Ca2+ oscillations were driven by RyR channels (n = 9). Statistical analyses were performed by two-way ANOVA with Tukey’s multiple comparisons: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We adapted the cardiomyocyte monolayer differentiation protocol to generate COs by direct differentiation of the embryoid bodies (EBs) (Figure 2A). By using the agarose microwell culture inserts, we could promote self-aggregation of the DMD-hiPSCs and DMD-Iso-hiPSCs into EBs at cell seeding number of 5,000 and 10,000 cells per microwell (Figure 2B). This allowed us to generate 137 EBs per insert per well of 24-well plate. On day 5 of cardiomyocyte differentiation, the resulting DMD-COs and DMD-Iso-COs were transferred to 6-well plate on orbital shaker for dynamic culture in the CM maintenance medium (Figure 2C). Contractile cardiomyocyte protrusions (Figure 2C, arrow) and self-organized cellular structures (Figure 2C, arrowheads) at the organoid periphery, both with specific spatial distribution of NKX2.5 and αACTN positivity, could be observed. The non-translucent organoid structure (#) was negative for both NKX2.5 and αACTN. Immunofluorescence staining showed abundant DYS localization in DMD-Iso-COs, which was undetectable in the DMD-COs (Figure 2D). The representative images of CO over 28 days of dynamic culture showed morphological changes and variations in organoid size (Figure 2E). Quantification of the organoid surface area over 28 days of dynamic culture showed no significant differences on the organoid size between the two cell seeding numbers within each cell line, however, the size of DMD-Iso-COs was significantly smaller than DMD-COs on day 14 and 28, respectively (Figure 2F). The DMD-COs displayed contraction on day 8 (± 19 per minute) which decreased over time and stopped contraction between day 14 and 18 (Figure 2G). The DMD-Iso-COs displayed contraction on day 12 (± 18 per minute) which persisted till day 28. FACS analysis was performed on DMD- and DMD-Iso-COs after 14 days to determine SIRPA (APC), CD31 (PE/Cy7) and PDGFRα (PE) positive cells (Supplementary Figures S1A,B). The analysis disclosed that both DMD- and DMD-Iso-COs accounted for 40/50% of SIRPA+ cells, while DMD-Iso-COs showed the double amount of the CD31+ cell (1.4% ± 0.2) population and the half of the PDGFRα+ cells (1.7% ± 0.2) compared to DMD-COs.
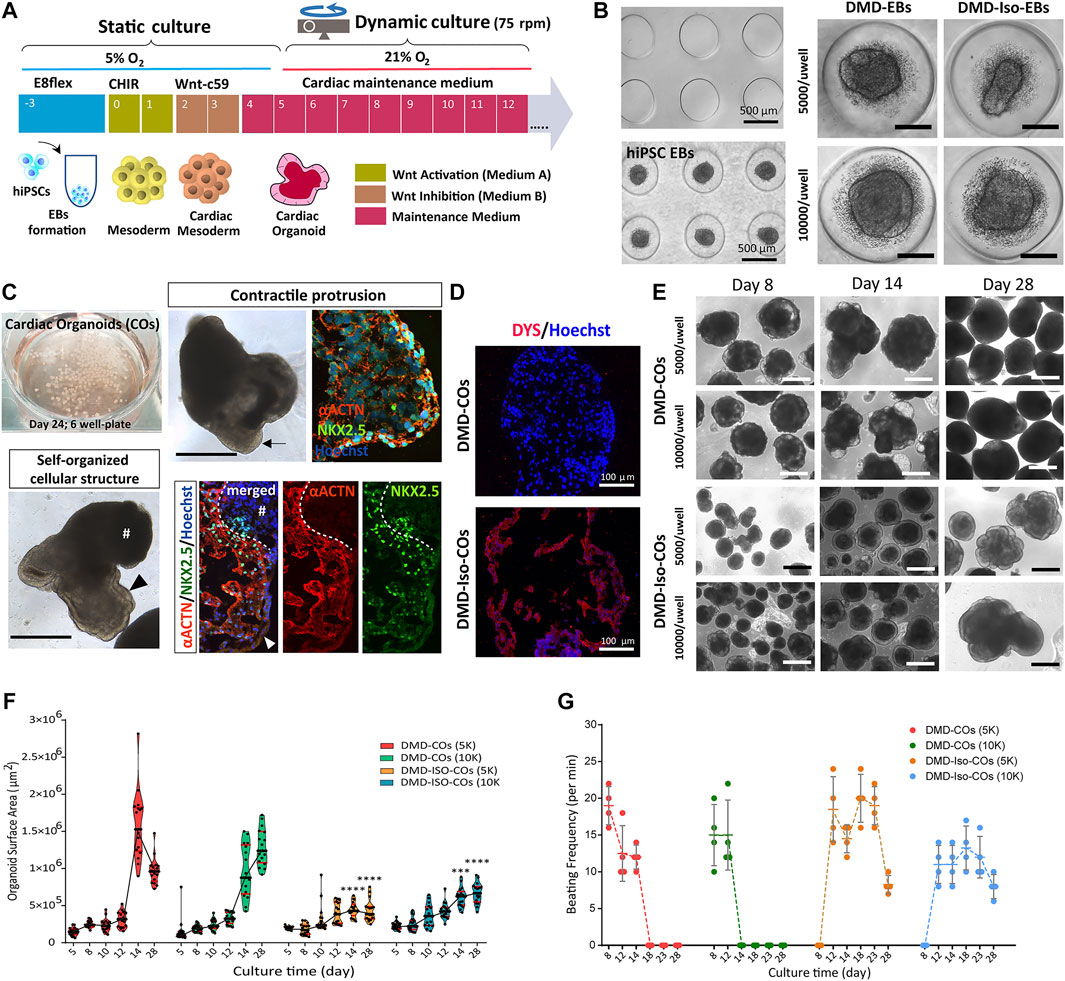
FIGURE 2. Generation of DMD-COs and DMD-Iso-COs from patient-derived hiPSC. (A) Schematic showing the culture protocol to generate DMD-COs and DMD-Iso-COs from patient-derived hiPSC embryoid bodies (EBs). (B) Brightfield images of the microwells of the agarose insert and the formed EBs. (C) EBs pooled from two agarose inserts inside a well of six-well plate for dynamic culture, and the morphology of COs at high magnification showing contractile cardiomyocyte protrusion (arrow) and distinct self-organized cellular structure at the organoid periphery (arrowhead) both with specific spatial distribution of NKX2.5 and αACTN positivity. Non-translucent organoid structure (#) was negative for NKX2.5 and αACTN. (D) Immunofluorescence staining showing the localization of DYS in DMD-Iso-COs, which was undetectable in DMD-COs. Nuclei were counterstained with Hoechst. (E) Representative images of CO morphology and the changes of organoid size (generated at two cells seeding numbers) over 28 days of dynamic culture. (F,G) Effect of cell seeding numbers on the growth (n = 17) and the beating frequency (n = 4) of DMD-COs or DMD-Iso-COs over 28 days. DMD-COs versus DMD-Iso-COs at 5K (5,000 cells/microwell) or 10K (10,000 cells/microwell). Statistical analyses were performed by two-way ANOVA with Tukey’s multiple comparisons: ***p < 0.001, ****p < 0.0001. CO, Cardiac organoid; DMD-COs and DMD-Iso-COs, cardiac organoids from DMD patient-derived hiPSC and isogenic corrected hiPSC, respectively. Scale bar = 1 mm or as stated in the figure.
We performed immunofluorescence staining for α-sarcoglycan (SCGA), sarcomeric α-actinin (αACTN), and NKX2.5 on day 10, 14 28, 56 and 93 in order to assess cardiac differentiation and contractile protein development within the organoids. The results showed abundant SCGA expression in DMD-COs on day 10, which became low on day 14 and undetectable from day 28 onwards (Figures 3A,C and Supplementary Figure S2A). Conversely, the SCGA expression in DMD-Iso-COs persisted till day 93. As reported in Supplementary Figure S3A also the β-, γ- and δ-sarcoglycans started to disappear from day 14 following the α-sarcoglycan trend. A transient expression of the early cardiac differentiation marker NKX2.5 was observed up to day 28 in both DMD-COs and DMD-Iso-COs, which became undetectable on day 56 and 93 (Figure 3C). Additionally, abundant αACTN, a cardiac contractile protein, was observed in both DMD-COs and DMD-Iso-COs on early time points, which remained detectable on day 93 (despite at lower expression level) in both conditions (Figures 3B,C and Supplementary Figure S2B). There was no distinguishable difference in the SCGA, αACTN and NKX2.5 expression between organoids generated from the two cell seeding numbers within a cell line. These results demonstrated a progressive loss of SCGA protein expression in DMD-COs (a member of the dystrophin associated complex, DAC) as compared to the isogenic controls. The sarcomeric pattern is shown at higher resolution and magnification in Supplementary Figure S3B which reports representative confocal images of αACTN and SCGA. In order to better characterize the cardiomyocyte population within the COs we performed the RT-qPCR for atrial/ventricular gene markers and TNNT2 at all the time points (Supplementary Figures S4A,B). Both atrial and ventricular markers showed a peak at the initial stage followed by a decline at later stages. Intriguingly IRX4, a ventricle-specific transcription factor, appeared to be always expressed in both samples until day 28 and declined at late stages as the majority as the other atrial and ventricle markers. However, the two major ventricular myosin heavy-chain genes MYL2 and MYH7 were sustained longer during the differentiation compared to the atrial ones (Supplementary Figure S4A). Likewise, TNNT2 expression reached the peak at day 14 and then decreased without showing striking differences between the two cell lines (Supplementary Figure S4B). Additionally, RT-qPCR analysis showed a significant upregulation of genes related to cardiac contractility in DMD-COs as compared to DMD-Iso-COs, in particularly from day 56 onwards (Figure 3D). These included ACTN1, IRX4, MYBPC3, MYL2, MYOM1, TNNC2 and TPM1.
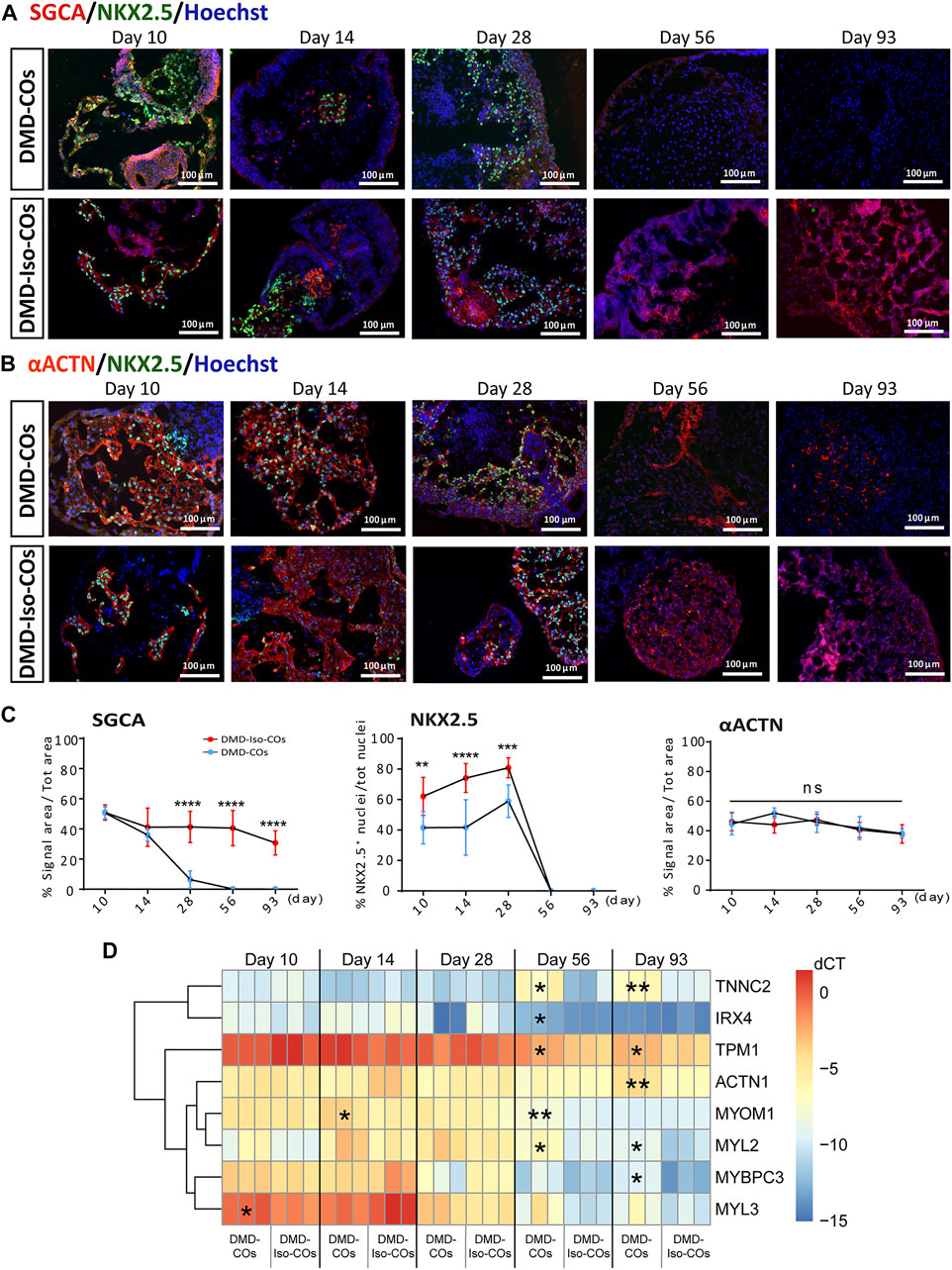
FIGURE 3. Assessment of cardiac differentiation and contractile proteins development in DMD-COs and DMD-Iso-COs over 93 days of dynamic culture. Representative immunofluorescence images for: (A) α-sarcoglycan (SCGA)/NKX2.5, and (B) sarcomeric α-actinin (αACTN)/NKX2.5 co-staining in DMD-COs and DMD-Iso-COs, respectively. Nuclei were counterstained with Hoechst. Data are representative of three independent experiments (n = 3). (Magnification: ×20). (C)Quantification of the immunofluorescence images for: SGCA, NKX2.5 and αACTN on day 10, 14 28, 56 and 93. Data shown are mean ± s.d. (n = 4, two-way ANOVA with Sidak’s multiple comparisons: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) RT-qPCR analysis of representative gene markers expression for cardiac contractility in DMD-COs and DMD-Iso-COs. Data shown are mean ± s.d. (n = 3, each pooled from ∼10 organoids). Statistical analyses were performed by two-way ANOVA with Tukey’s multiple comparisons: *p < 0.05, **p < 0.01.
We examined cell proliferation and apoptotic conditions within the DMD-COs and DMD-Iso-COs by immunostaining of the proliferation marker Ki67 and apoptotic marker cleaved caspase 3 (CCASP3). The results showed low Ki67 staining in DMD-COs but relatively higher signal in DMD-Iso-COs on day 10, while the signal became comparable on day 28 and 93 (Figures 4A,E and Supplementary Figure S2C). Though initially CCASP3 signal area was slightly more in DMD-Iso-COs, from day 28 it became significantly higher in DMD-COs (Figure 4E). However, no significant difference in Ki67 and CCASP3 staining was observed at both cell seeding densities for both COs conditions (data not shown). These data suggest that the DMD-COs was lacking an initial proliferative capacity at early time point while at later time points, they were more apoptotic. We then assessed the metabolic activity within the COs by immunostaining of the glycolytic marker phosphoglycerate kinase 1 (PGK1). The results showed high and comparable PGK1 staining in both COs conditions at all time points (Figures 4B,E and Supplementary Figure S2D), which was independent from the cell seeding densities (data not shown). These results suggest the glycolytic condition of immature COs in both CO conditions. The cellular stress was assessed by immunostaining of two known endoplasmic reticulum (ER) stress markers ARCN1 and GORASP2. Interestingly, we detected relatively higher level of ARCN1 in DMD-COs than DMD-Iso-COs at all the time points (Figures 4C,E and Supplementary Figure S2E), whereas GORASP2 increased progressively over the 28 days in DMD-COs, independently from the cell seeding densities (data not shown) and at higher level than that in DMD-Iso-COs at all the time points (Figures 4D,E and Supplementary Figure S2F). This finding indicated a high level of ER stress occurred in DMD-COs. To assess oxidative stress due to high NOX4 expression, we performed immunofluorescence staining on the organoid cryosections for NOX4. As shown in Supplementary Figure S5A, high NOX4 protein level was detected in the DMD-COs on day 14 which was barely present in DMD-Iso-COs.
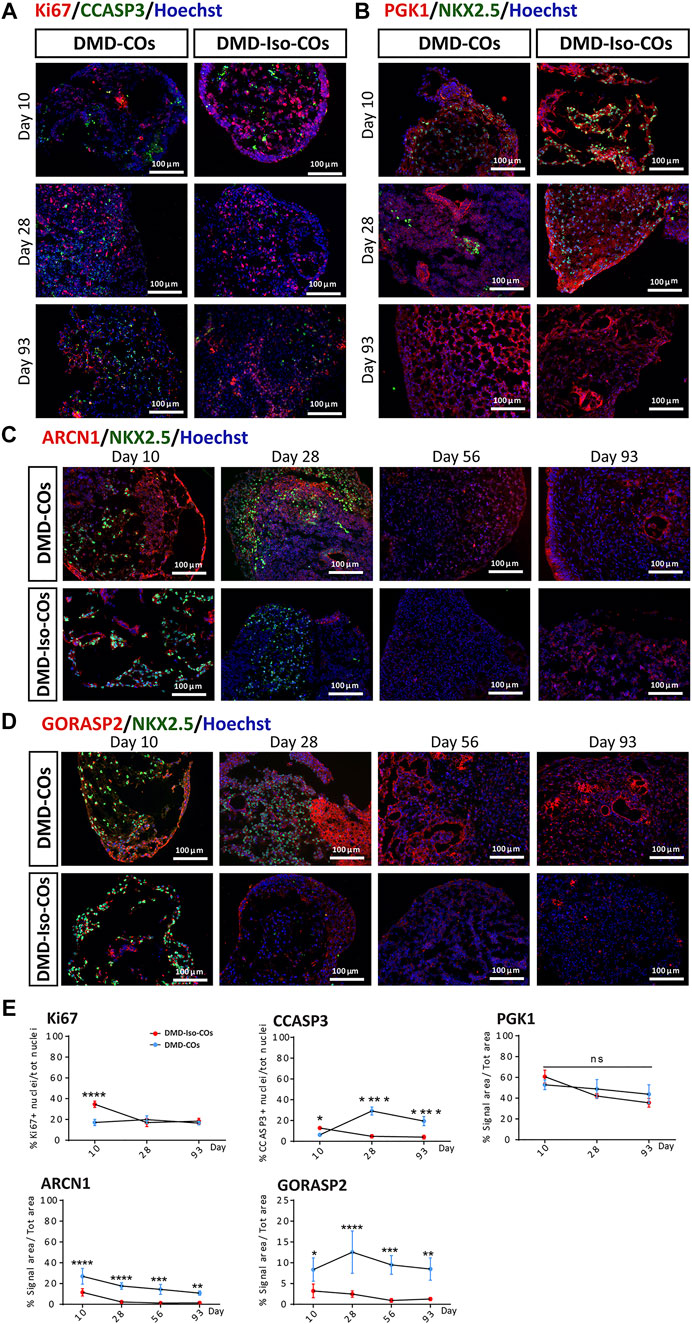
FIGURE 4. Assessment of cell proliferation, apoptosis and ER stress in DMD-COs and DMD-Iso-COs over 93 days of dynamic culture. Representative immunofluorescence images for: (A) Ki67/CCASP3, (B) PGK1/NKX2.5 on day 10, 28 and 93; (C) ARCN1/NKX2.5 and (D) GORASP2/NKX2.5 on day 10, 28, 56 and 93. Nuclei were counterstained with Hoechst. Data are representative of three independent experiments (n = 3). (Magnification: ×20). (E) Quantification of the immunofluorescence images for: Ki67, CCASP3 and PGK1 on day 10, 28 and 93; ARCN1 and GORASP2 on day 10, 28, 56 and 93. Data shown are mean ± s.d. (n = 4, two-way ANOVA with Sidak’s multiple comparisons: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We performed histological examination to assess any cytoarchitecture changes and DMD-related pathological progression within the COs over 93 days. The DMD-COs displayed normal cardiomyocyte-like structures similar to that of DMD-Iso-COs on day 10, which deteriorated on day 14 (indicated as “#”) and developed fibrotic-like structures (indicated as “f”) at later time points (Figure 5A; H&E staining on day 56, and Figure 5B; Picro-Sirius red staining for collagen deposition on day 93). These findings were corroborated by a significant upregulation of gene markers associated with fibrosis COL1A2, COL3A1 and FN1 in DMD-COs on day 56 and 93 as compared to DMD-Iso-COs (Figure 5C). Additionally, H&E staining also revealed adipose tissue formation in DMD-COs on day 28 (Figure 5D), confirmed by the detection of lipid droplets via BODIPY staining and immunolabelled PDGFRα+ cells (an adipocyte marker) in DMD-COs on day 28 and 56 (Figure 5E). Interestingly, GDF10 protein (an adipogenesis inhibitor) was also detected near the PDGFRα+ cells in DMD-COs (Figure 5F). These findings suggest that DMD-COs displayed an initial normal cardiac phenotype which deteriorated progressively and exhibited pro-fibrotic and adipogenic phenotypes upon long-term culture, resembling pathologic events associated with DMD cardiomyopathy.
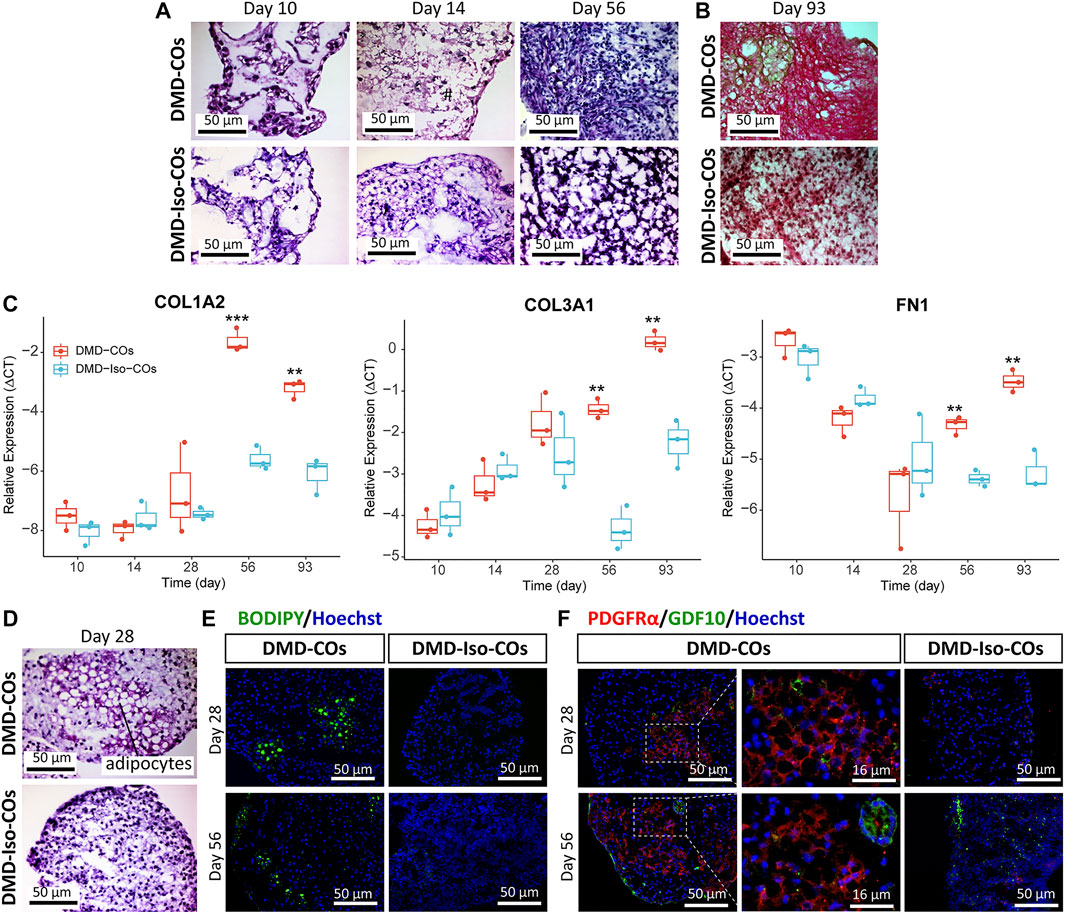
FIGURE 5. Assessment of fibrosis and adipogenesis in DMD-COs and DMD-Iso-COs over 93 days of dynamic culture. (A) H&E staining showing deterioration of cardiomyocyte tissue on day 14 and fibrosis on day 56 in DMD-COs. (Magnification: ×40). (B) Picro-Sirius red staining showing abundant collagen deposition in DMD-COs on day 93. (Magnification: ×40). (C) RT-qPCR analysis of representative fibrosis gene markers showing a significant upregulation of COL1A2, COL3A1 and FN1 expression in DMD-COs on day 56 and 93 as compared to DMD-Iso-COs. Data shown are mean ± s.d. (n = 3, each pooled from ∼10 organoids). Statistical analyses were performed by two-way ANOVA with Tukey’s multiple comparisons: **p < 0.01, ***p < 0.001. (D–F) Adipogenesis in DMD-COs as indicated by the formation of adipocytes with cytoplasmic vacuoles (H&E staining), lipid droplet deposition (BODIPY staining), and PDGFRα positivity on day 28 and 56. The adipogenesis inhibitor GDF10 was also detected near the PDGFRα+ cells in DMD-COs. Nuclei were counterstained with Hoechst. (Magnification: ×20 and ×40). Data are representative of three independent experiments (n = 3).
Principal component analysis of the RNA transcriptomic data showed a distinct separation between DMD-COs and DMD-Iso-COs clusters (PC1: 90%) with low intra-condition variance (PC2: 5%) (Figure 6A). Based on the enhanced volcano plot, out of 22,371 gene variables, 1,518 and 554 genes were differentially upregulated in DMD-COs and DMD-Iso-COs, respectively (Cut-off: log2 fold change = 1.5; −Log10p = 10–16) (Figure 6B). Among the top 30 most differentially upregulated genes in DMD-COs (Figure 6C), MGP, MYL1, COL1A2, HAPLN1 and OGN were the five most significant upregulated genes (Figure 6B and Table 3). In addition, we validated the upregulation of the majority of the genes in DMD-COs showed in Figure 6B by RT-qPCR in DMD-, DMD-Iso- and HC-COs at day 56 as additional control (Supplementary Figure S6A). Based on gProfiler analysis, gene ontologies that were significantly enriched for molecular function in extracellular matrix regulation (i.e. collagen and glycosaminoglycan; GO:MF), cardiac tissue structure formation (i.e. external encapsulating structure such as sarcolemma; GO:MM), and cardiovascular development (GO: BP) could be identified in DMD-COs (Figure 6D). Additionally, KEGG pathways associated with protein digestion and absorption, dilated and hypertrophic cardiomyopathy, ECM-receptor interaction, and cGMP-PKG signalling pathway (known to positively modulates cardiac contractility, hypertrophy and protects against apoptosis (Takimoto, 2012) were significantly enriched in DMD-COs (Table 4 and Figure 6E (i)). These findings were corroborated by the analysis on human phenotype ontology, whereby ontology related to abnormal cardiovascular system physiology, including abnormal left ventricular function, abnormal endocardium morphology, atrial arrhythmia and fibrillation, supraventricular arrhythmia, myopathy and cardiac arrest, as well as abnormal adipose tissue morphology and lipodystrophy were significantly enriched in DMD-COs as compared to DMD-Iso-COs (Figure 6E (ii)). Moreover, the gProfiler analysis also identified three top miRNA regulators for the differentially upregulated genes in DMD-COs, namely hsa-mir-335-5p, hsa-mir-29a-3p and hsa-mir-29b-3p. Altogether, the RNA sequencing data validated the histological observations described above on cardiomyocyte deterioration, adipogenesis and fibrosis at the transcriptomic level.
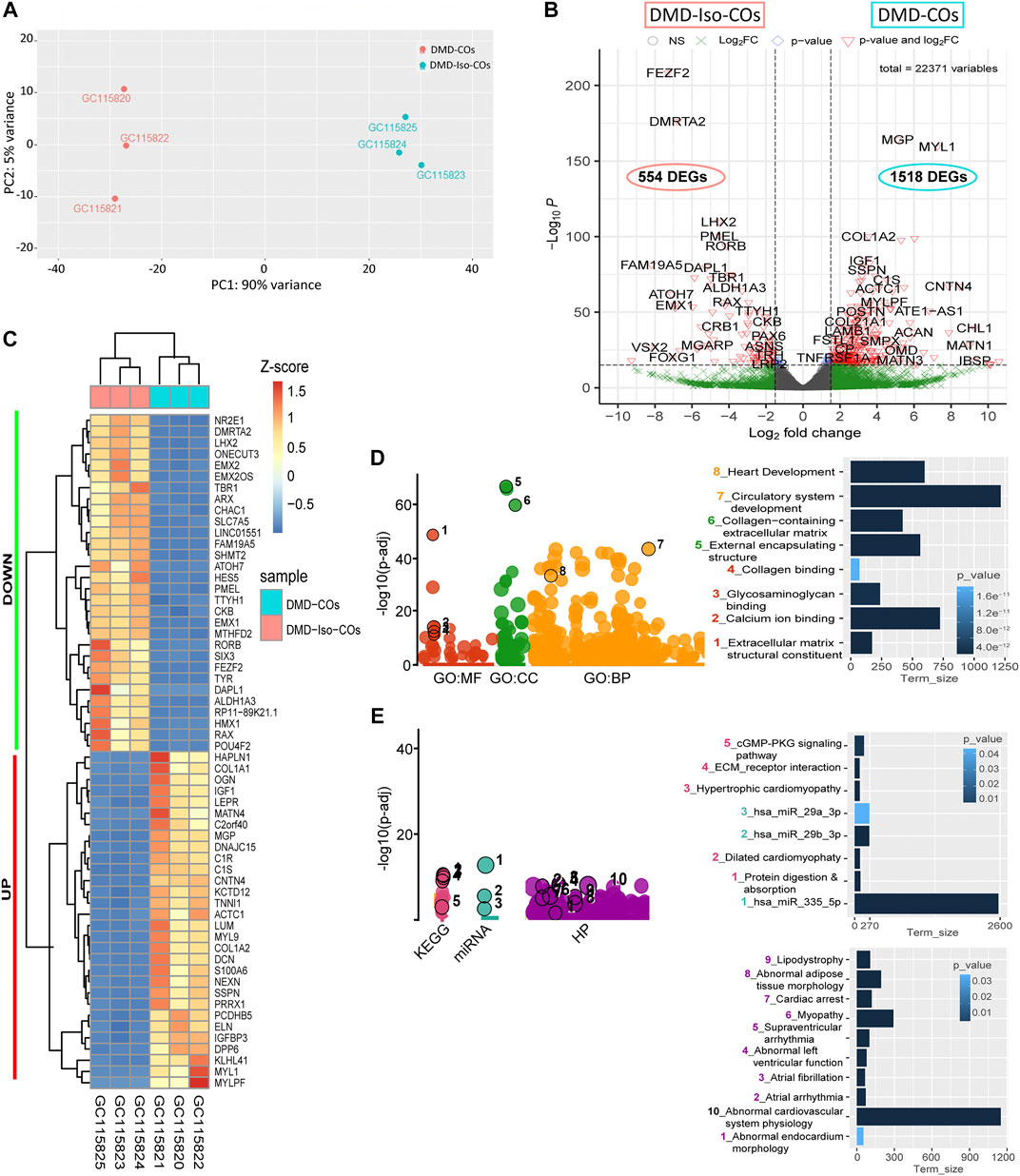
FIGURE 6. RNA sequencing analysis of DMD-COs and DMD-Iso-COs via DESeq2 method. (A) Principal component analysis (PCA) showing distinct separation of the DMD-COs and DMD-Iso-COs clusters (PC1: 90%) with low intra-condition variance (PC2: 5%) in both conditions, respectively. (B) Enhanced volcano plot showing the differentially expressed genes (DEGs) in DMD-COs versus DMD-Iso-COs. Cut-off log2 fold change = 1.5; Cut-off −Log10p = 10–16. (C) Heatmap showing the TOP30 DEGs in DMD-COs versus DMD-Iso-COs. (D,E) Functional enrichment analysis of the differentially upregulated genes in DMD-COs versus DMD-Iso-COs using gProfiler2 for Gene ontology, KEGG pathway, miRNA and human phenotype ontology. (All data shown: n = 3, each sample was a pooled of ∼10 organoids).
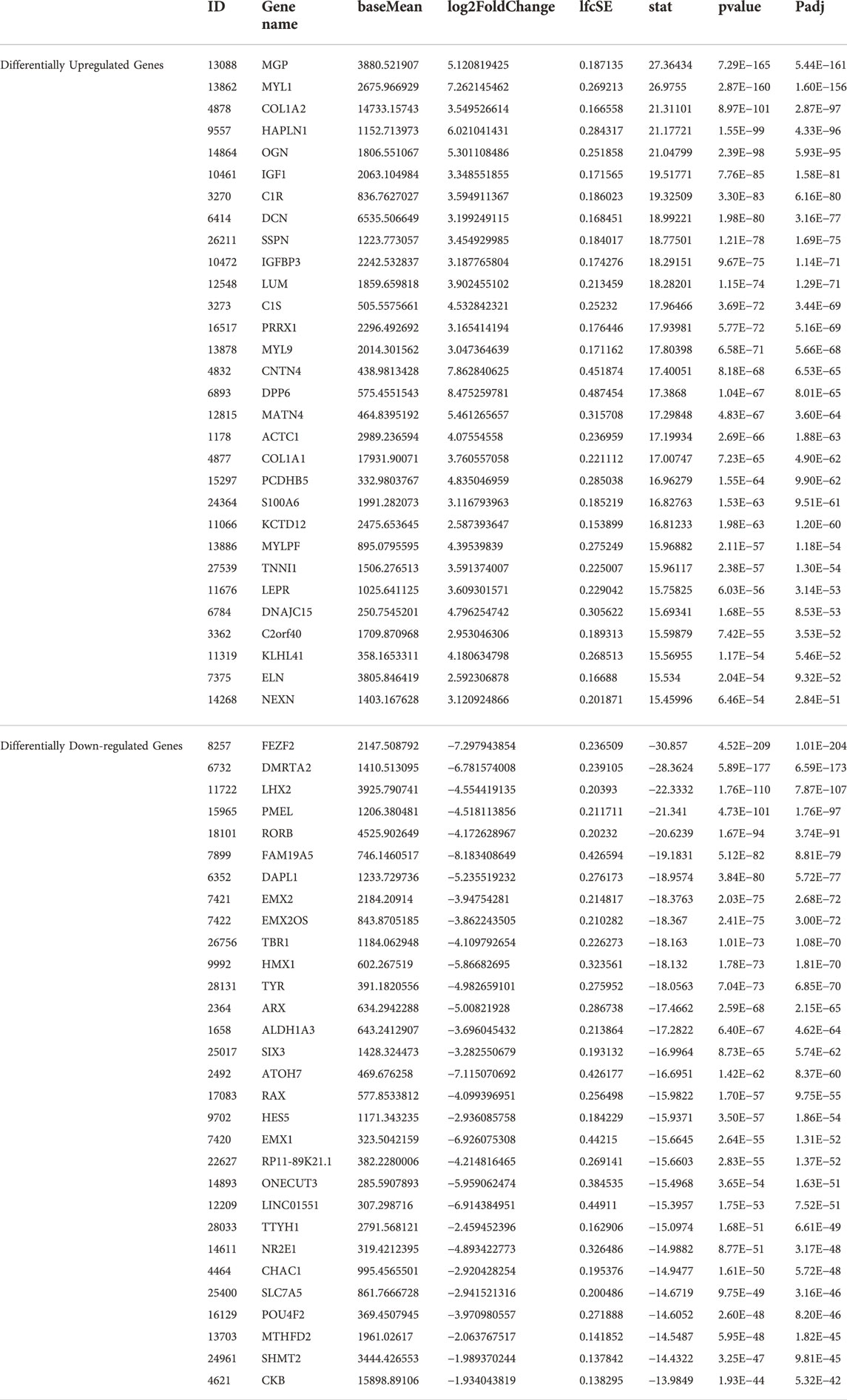
TABLE 3. Top30 differentially upregulated and down-regulated genes in DMD-COs versus DMD-Iso-COs.
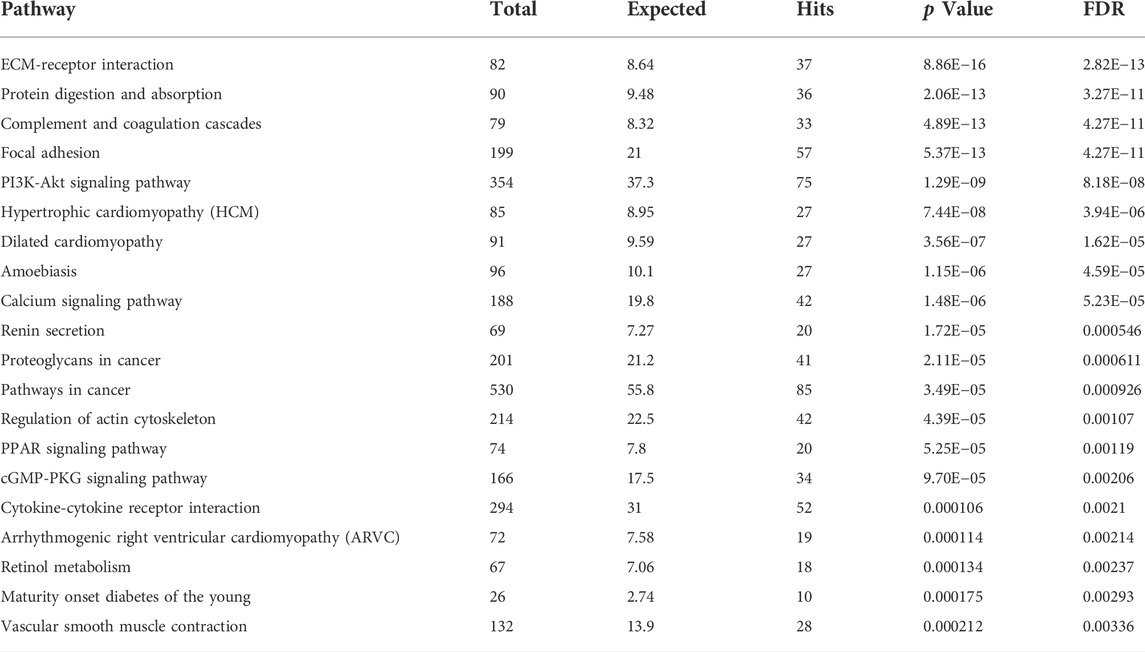
TABLE 4. List of identified TOP20 KEGG pathways based on the upregulated DEGs in DMD-COs (Cut-off log2FC > 1.5, Cut-off p-value <0.05).
PPI analysis of the differentially upregulated genes in DMD-COs revealed a gene network consisted of 2,289 nodes and 2,288 edges. According to the degree level (d), the top five hub nodes were HNF4A (d = 257), UBC (d = 108), UBD (d = 66), APP (d = 38) and EGR1 (d = 31) (Figure 7A). By exploring the miRNA database (i.e. mirTarBase v8.0), the top three miRNA regulators of this gene network were hsa-mir-335-5p, hsa-mir-124-3p, and hsa-mir-26b-5p. Together with the hsa-mir-29b-3p and hsa-mir-29a-3p identified by gProfiler2, we mapped out these miRNAs on the gene-miRNA regulatory networks for the selected KEGG pathways relevant to the DMD-COs phenotypes: 1) Hypertrophy cardiomyopathy, 2) Dilated cardiomyopathy, 3) Arrhythmogenic right ventricular cardiomyopathy (ARVC), 4) PPAR signalling pathway (for adipogenesis), and 5) PI3K-Akt signalling pathway (for cardiac fibrosis (Table 5) (Qin et al., 2021)). The results showed that hypertrophy and dilated cardiomyopathy networks shared the same gene set (50 nodes, 49 edges), miRNA interactions (Figure 7B) and 16 genes similarity with the ARVC network (Figure 7C). Except hsa-mir-124-3p, the other four top miRNAs were mapped in these three networks, respectively. The PPAR signalling gene-miRNA network consisted of 33 nodes and 43 edges (Figure 7D). In addition to hsa-mir-26b-5p and hsa-mir-355-5p, the hsa-mir-124-3p was mapped in the network and found interacts with the gene ACSL5 and ACADL. The has-mir-29b-3p, hsa-mir-26b-5p and hsa-mir-355-5p were the main miRNA regulators in the PI3K-Akt signalling (147 nodes, 146 edges), which interact with one of the two hub genes CCND2 (Figure 7E).
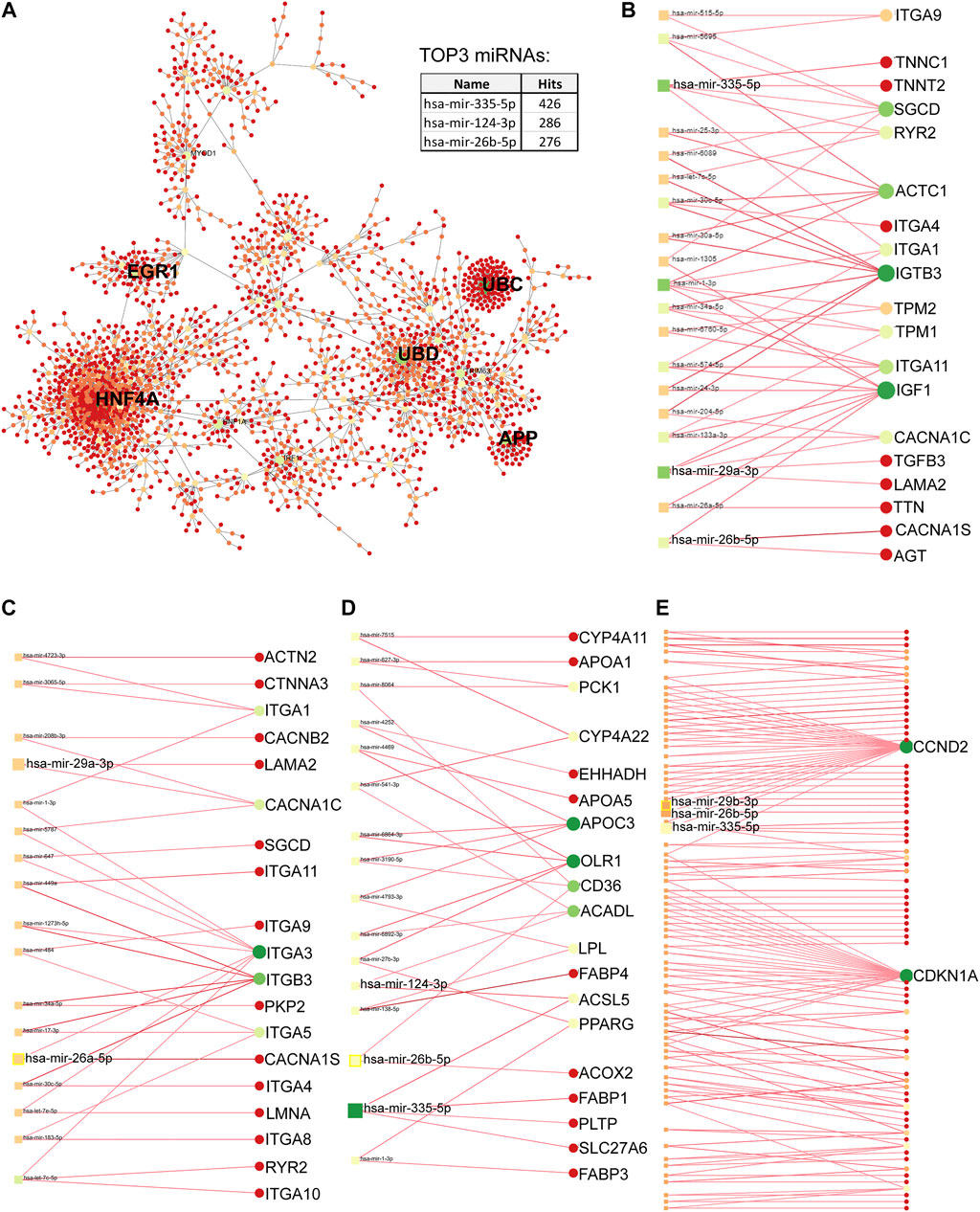
FIGURE 7. Protein-protein interaction network analysis based on differentially upregulated genes in DMD-COs using Network Analyst platform. (A) Top five main hub genes (HNF4A, UBC, UBD, APP and EGR1) were identified based on the degree levels. The gene-miRNA networks for hypertrophy/dilated cardiomyopathy (B), arrhythmogenic right ventricular cardiomyopathy (C), PPARgamma (D) and PI3K-Akt (E) signalling pathways, respectively. The colours indicate the degree of interaction (with lowest to highest degree from red to yellow to green), whereas the sizes of circles indicate the degree of betweenness of a node (larger size means higher degree of betweenness). The identified top three miRNAs by PPI and top two miRNAs by gProfiler2 analysis were mapped in each gene-miRNA network to indicate the genes they interact with.
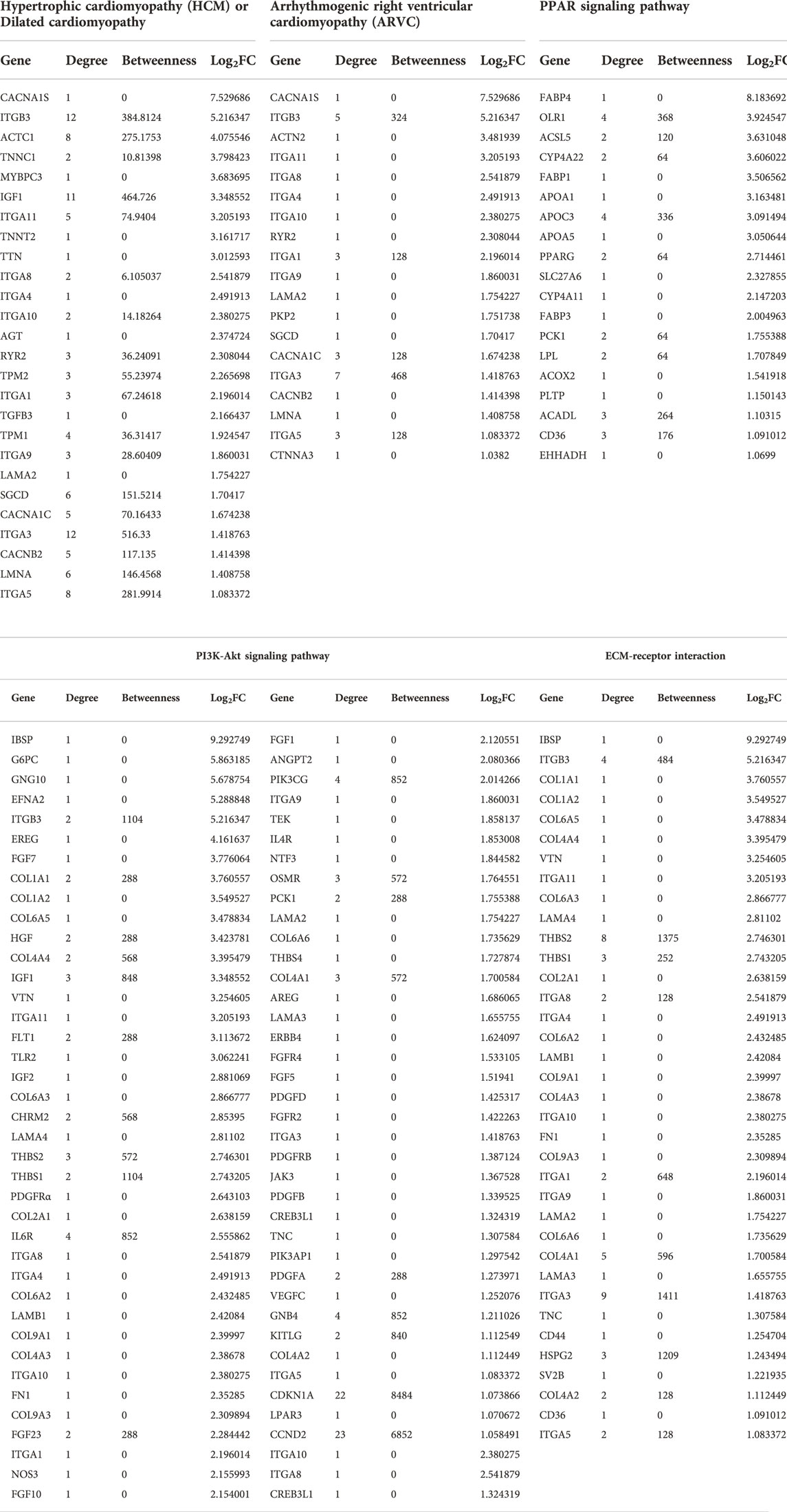
TABLE 5. List of genes identified for the selected KEGG pathways from Table 4, based on the upregulated DEGs in DMD-COs.
There is currently no cure for DMD patients. They are solely treated symptomatically via palliative therapies in combination with cardio-respiratory supporting devices in case of cardio-pulmonary complications–a major lethal cause in DMD patients. As DMD-related cardiomyopathy often manifested as hypertrophic or dilated heart due to cardiomyocyte deterioration followed by fibrosis and adipose tissue formation, novel therapeutic modality should be developed to prevent these pathological events from taking place in the heart. For this, gaining in-depth understanding on the human disease mechanisms is necessary. Unfortunately, limited accessibility to patient biopsy/autopsy and the inferiority of in vitro 2D cellular and animal models in fully recapitulating the human disease phenotype have precluded this scientific endeavour. Therefore, it is imperative to develop in vitro 3D human cardiac-mimics of DMD-relevance to bridge this scientific gap. The advent of hiPSC technology represents a paramount breakthrough for patient-specific model generation that can better mimic the individual phenotype. By using the isogenic-corrected controls (instead of healthy wild-type controls), we could compare the results at minimal genetic background variability.
In this study, we generated DMD-COs that functionally and transcriptionally modelled the cardiac evolution in DMD patients. Specifically, our model displayed a lack in proliferative capacity, cell death and a progressive deterioration of cardiomyocytes in early culture stages, followed by adipose tissue and fibrous-like tissue formation at later culture stage. We also observed a defect in physiological RyR-driven Ca2+ signals in DMD-CMs compared to isogenic-corrected and healthy controls. This further underpins the validity of our model since RyR dysfunction has also been implicated in dystrophic skeletal muscle cells (Andersson et al., 2012). In this work, dystrophic skeletal muscle was linked with leaky (skeletal muscle-type) RyR1 channels due to its oxidation. Hence, our work suggests that also the functional properties of (cardiac muscle-type) RyR2 channels may be affected in DMD patients, thereby contributing to cardiac pathophysiology. Probably as a consequence of the impaired Ca2+ handling and the dystrophic phenotype, DMD-COs showed a premature loss of the contractile properties compared to the controls independently from the CM:non-CM ratio, which is comparable in the two samples, as showed in the FACS analysis. These data also suggest the presence of a cardiomyocyte population within COs very close to the one physiologically contained in cardiac tissues (Lim, 2020).
Furthermore, DMD-COs displayed stable αACTN localization while α-, β-, γ- and δ-sarcoglycans became minimal present from day 14. In fact, the formed sarcoglycan complex possibly deteriorated within the DMD-COs over time due to its intrinsic DMD pathological phenotype. Although EHT models have been employed for investigating the DMD-CM contractile capability in the 3D context, the lack of several DAPC protein components, even in the healthy EHTs, hampers their usage as reliable genetic disease models (Gilbert et al., 2021). The fact that healthy organoids showed the presence of late cardiac differentiation markers as α-, β-, δ- and γ-sarcoglycans makes possible to model genetic cardiac diseases where sarcoglycans are missing. Furthermore, the cardiomyopathy phenotype development depends not only on the absence of dystrophin but also on the detrimental effects of non-cardiomyocyte subpopulations. Thus, 3D COs represent a superior in vitro model compared to EHTs in terms of cardiac maturation and environment.
These findings confirmed the formation of cardiac tissue within the organoids, which is also corroborated by the expression of atrial/ventricular markers and TNNT2 over time. Furthermore, the longer and sustained expression of the ventricular markers MYL2 and MYH7 suggests the ventricular-like identity of the CMs within these 3D models. The DMD-related phenotype is further confirmed by the detection of higher levels of NOX4 within the DMD-COs. Although NOX4 is constitutively active in healthy CMs at low levels inducing a cardioprotective effect, it has been demonstrated that high levels of NOX4 have severe detrimental effects. Indeed, the NOX4 accumulation contributes to the development of cardiovascular diseases by triggering an excessive ROS production. The endoplasmic reticulum stress marker GORASP2 increased over time in DMD-COs, while ARCN1 was more prominent in DMD-COs, but they were not co-localized with NKX2.5, suggesting other cell type than differentiating cardiomyocytes experienced high ER stress within the generated DMD-COs and DMD-Iso-COs. Moreover, we argued that the presence of GDF10 near the PDGFRα+ fibroblast/adipocyte progenitors could be a feedback regulation mechanism to inhibit pathological formation of adipose tissues in the DMD-COs, as GDF10 was not detected in DMD-Iso-COs where adipogenesis did not occur.
It is known by the literature that iPSC-derived CMs resemble foetal hearts from a metabolic standpoint and thus are qualitatively and quantitatively immature. After birth, the metabolism of CMs switches from glycolysis to a predominant use of oxidative phosphorylation to fulfil the energy demand of the contracting myocardium (Allen et al., 2016). In our model, the glycolytic marker PGK1 was strongly expressed in both DMD-COs and DMD-Iso-COs. We attempted to quantify the immunofluorescence signals for deleterious markers and correlated them with the aberrant fibrosis and adipogenesis observed only in DMD-COs. However, the results were affected by the heterogeneity in the cytoarchitectures, as the organoids were derived from hiPSC-EBs which might have undergone differential mesodermal induction. Still, comparing to pre-differentiated cardiomyocyte spheroids as well as engineered heart tissue (EHT) constructs, COs derived from hiPSC-EBs have the advantage of possibly containing other non-cardiogenic cells (as seen during heart development) that contribute to the adipogenesis and the fibrotic-like phenotype upon cardiomyocyte deterioration. Noteworthy that these pathological events were not observed in the DMD-Iso-COs controls. The reasons of the development of adipocytes and fibrotic-like tissues are still unclear and further experiments must be performed to elucidate the causes. We hypothesised that in the dystrophic environment the PDGFRα+ population, which includes fibroblast and adipose progenitors, could have a role in the onset of these detrimental events. In fact, the activation of the PDGFRα pathway in some adult cells must be sufficient to generate significant pro-fibrotic activity (Olson and Soriano, 2009). As reported in literature, dystrophic myocardium, due to the Ca2+ overload, is characterized by cell death and inflammatory response, which result not only in myocyte hypertrophy, atrophy/necrosis, fibrosis, but also in the replacement of heart muscle by connective tissue and fat (Flanigan, 2014). Intriguingly, contractile genes were upregulated in DMD-COs after 93 days of culture. The causes contributing to this upregulation in the diseased COs are still unknown. However, this phenomenon can be attributed to a compensatory mechanism, and we believe that other reasons are concurring with these results. Indeed, it is generally accepted that an abnormal elevation of the intracellular Ca2+ concentration in the dystrophin-deficient cardiomyocytes is a major secondary event, which contributes to disease progression and alters the contractile protein turnovers (Mareedu et al., 2021). Another possible explanation could be the foetal reprogramming occurring in diseased myocardium. This adaptive process is based on the suppression of adult and reactivation of foetal gene profile as a result of cardiac disease promoting the reversion of the contractile machinery to a more compliant foetal one (van der Pol et al., 2020). As reported in literature TNNC2, TPM1 and MYL2 are markers expressed at early developmental stages during cardiogenesis (Sheikh et al., 2015; England et al., 2017; Tsedeke et al., 2021). Furthermore, Cetinkaya et al. states that the ACTN1 expression in adult myocytes implies the activation of fetal pathways in patients affected by dilated cardiomyopathy (Cetinkaya et al., 2020). Moreover, MYBPC3 is not only expressed in adult but also in embryonic and neonatal hearts throughout the development (Jiang et al., 2015). These data could suggest the possible activation in DMD-COs of foetal genes in order to promote the turnover of sarcomeric proteins and counteract the cardiomyocyte derangement.
Through RNA sequencing analysis, we demonstrated that the DMD-COs generated on day 56 were valuable 3D cellular models to gain insight into the disease mechanism of DMD-associated hypertrophic/dilated cardiomyopathy, as well as adipogenesis and fibrosis. We focused on mapping out the functionally enriched pathways based on the differentially upregulated genes in DMD-COs as compared to DMD-Iso-COs, as well as their main miRNA regulators. Among the top five hub genes identified in the protein-protein interaction network, only HNF4A (log2FC = 1.89, p < 2.92e−5), UBD (log2FC = 2.69, p < 7.37e−5) and EGR1 (log2FC = 1.47, p < 5.31e−12) were significantly and differentially upregulated in DMD-COs. Despite HNF4A could be linked to cardiac differentiation and heart development (Duelen et al., 2017), we could not find in literature the association of these three hub genes with the development of cardiomyopathy, adipogenesis and fibrosis. We turned into looking at the identified miRNA regulators. The hsa-mir-335-5p was reported as a regulator of cardiac differentiation by upregulating cardiac mesoderm and cardiac progenitor commitments, potentially mediated through the activation of WNT and TGFβ pathways (Kay et al., 2019). In contrast, the upregulation of hsa-mir-335-5p was seen in fibrotic lung model (Honeyman et al., 2013). Additionally, a study showed that the hsa-mir-29a-3p and hsa-mir-29b-3p levels in cardiac tissue from patients with congenital heart disease was significantly increased, and the injection of miR-29b-3p into zebrafish embryos induced higher mortality and developmental disorders including cardiac malformation and dysfunction, as well as inhibition of cardiomyocyte proliferation by targeting NOTCH2 (Yang et al., 2020). Interestingly, delivery of miR-29a-3p has a beneficial effect in myocardial injury (Ren et al., 2021) and cardiac hypertrophy (Xie et al., 2020). Similarly, the hsa-mir-26a/b-5p was highly expressed in cardiac hypertrophy (Tang et al., 2020) and promoted myocardial infarction-induced cell death (Jung et al., 2021), yet overexpression of miR-26a/b attenuated cardiac fibrosis (Tang et al., 2017; Wang et al., 2019) and alleviated cardiac hypertrophy and dysfunction (Shi et al., 2021). Lastly, the hsa-mir-124-3p was reported to promote cardiac fibroblast activation and proliferation (Zhu et al., 2021), and its inhibition protects against acute myocardial infarction by suppressing cardiomyocyte apoptosis (Hu et al., 2019). Based on the duality effects of these miRNAs, the potential of these miRNAs as therapeutic targets for DMD-related cardiomyopathy need to be assessed carefully. Furthermore, the identified PI3K/Akt signalling pathway enriched in DMD-COs is interesting, as accumulating evidences showed that it plays a role in regulating the occurrence, progression and pathological cardiac fibrosis (Qin et al., 2021) and hypertrophy (Aoyagi and Matsui, 2011). These findings are encouraging and prompting us to investigate in future the potential of these miRNAs as therapeutic targets to inhibit the aberrant functional enrichments in DMD-COs. In turn, this will enable us to further validate DMD-COs as reliable in vitro 3D human cardiac models for DMD-related disease modelling, drug discovery and regenerative medicine.
In conclusion, we developed 3D human cardiac-mimics from DMD cell lines, which holds great relevance as these models reproduce in vitro, even if partially, the DMD-related cardiomyopathy (i.e. cardiomyocytes stress and deterioration) and disease progression (i.e. adipogenesis and fibrosis) in long-term cultures. By studying the transcriptomic dysregulations in DMD-COs versus the isogenic controls via RNA sequencing and in silico analysis, we have identified five miRNAs that were significantly and differentially expressed in late DMD-COs which could be associated with the functionally enriched hypertrophy and dilated cardiomyopathy, fibrosis and adipogenesis signalling pathways. These are encouraging findings showing the potential of these human cardiac-mimics as novel in vitro 3D cellular models for studying DMD cardiomyopathy. In future studies, adding endothelial cells to the EBs to generate 3D vascularized human cardiac models would also be highly valuable to investigate the cardiomyocyte-endothelial interplays in relation to DMD pathogenesis.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI [accession: GSE194297].
VM and FM performed all experiments. RL and TV performed calcium imaging experiment. VM, FM, NG, EP, AC, TV, FA, and YC analysed the data. VM, FM, RD, TV, GB, DT, MS, and YC designed the experiment, wrote and/or revised the manuscript. The authors gratefully acknowledge the VIB Bio Imaging Core (LIMONE) for the access to the Nikon C2 confocal microscopy and their support and assistance in this work.
This work was supported by FWO (#G066821N), INTERREG–Euregio Meuse-Rhine (GYM - Generate your muscle 2020-EMR116), and KU Leuven C1-3DMuSyC (C14/17/111) sustaining also YC. The authors gratefully acknowledge Sylvia Sauvage for technical assistance, Christina Vochten and Vicky Raets for the administrative assistance. RD is supported by KU Leuven Rondoufonds voor Duchenne Onderzoek (EQQ-FODUCH-O2010) and KU Leuven grant (C24/18/103).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.878311/full#supplementary-material
Allen, D. G., Whitehead, N. P., and Froehner, S. C. (2016). Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 96 (1), 253–305. doi:10.1152/physrev.00007.2015
Andersson, D. C., Meli, A. C., Reiken, S., Betzenhauser, M. J., Umanskaya, A., Shiomi, T., et al. (2012). Leaky ryanodine receptors in β-sarcoglycan deficient mice: a potential common defect in muscular dystrophy. Skelet. Muscle 2 (1), 9. doi:10.1186/2044-5040-2-9
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. [Online]. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. (Accessed April 26, 2010).
Aoyagi, T., and Matsui, T. (2011). Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 17 (18), 1818–1824. doi:10.2174/138161211796390976
Aronesty, E. (2011). ea-utils: Command-line tools for processing biological sequencing data. Available at: https://github.com/ExpressionAnalysis/ea-utils. (Accessed October 20, 2021).
Breuls, N., Giarratana, N., Yedigaryan, L., Garrido, G. M., Carai, P., Heymans, S., et al. (2021). Valproic acid stimulates myogenesis in pluripotent stem cell-derived mesodermal progenitors in a NOTCH-dependent manner. Cell Death Dis. 12 (7), 677. doi:10.1038/s41419-021-03936-w
Burridge, P. W., Matsa, E., Shukla, P., Lin, Z. C., Churko, J. M., Ebert, A. D., et al. (2014). Chemically defined generation of human cardiomyocytes. Nat. Methods 11 (8), 855–860. doi:10.1038/nmeth.2999
Caluori, G., Pribyl, J., Pesl, M., Jelinkova, S., Rotrekl, V., Skladal, P., et al. (2019). Non-invasive electromechanical cell-based biosensors for improved investigation of 3D cardiac models. Biosens. Bioelectron. 124-125, 129–135. doi:10.1016/j.bios.2018.10.021
Cetinkaya, A., Berge, B., Sen-Hild, B., Troidl, K., Gajawada, P., Kubin, N., et al. (2020). Radixin relocalization and nonmuscle α-actinin expression are features of remodeling cardiomyocytes in adult patients with dilated cardiomyopathy. Dis. Markers 2020, 9356738. doi:10.1155/2020/9356738
Duelen, R., Gilbert, G., Patel, A., de Schaetzen, N., De Waele, L., Roderick, L., et al. (2017). Activin A modulates CRIPTO-1/HNF4α+ cells to guide cardiac differentiation from human embryonic stem cells. Stem Cells Int. 2017, 4651238. doi:10.1155/2017/4651238
Duelen, R., Costamagna, D., Gilbert, G., De Waele, L., Goemans, N., Desloovere, K., et al. (2022). Human iPSC model reveals a central role for NOX4 and oxidative stress in Duchenne cardiomyopathy. Stem Cell Rep. 17 (2), 352–368. doi:10.1016/j.stemcr.2021.12.019
England, J., Granados-Riveron, J., Polo-Parada, L., Kuriakose, D., Moore, C., Brook, J. D., et al. (2017). Tropomyosin 1: Multiple roles in the developing heart and in the formation of congenital heart defects. J. Mol. Cell. Cardiol. 106, 1–13. doi:10.1016/j.yjmcc.2017.03.006
Fayssoil, A., Nardi, O., Orlikowski, D., and Annane, D. (2010). Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail. Rev. 15 (1), 103–107. doi:10.1007/s10741-009-9156-8
Filippo Buono, M., von Boehmer, L., Strang, J., Hoerstrup, S. P., Emmert, M. Y., Nugraha, B., et al. (2020). Human cardiac organoids for modeling genetic cardiomyopathy. Cells 9 (7), E1733. doi:10.3390/cells9071733
Finsterer, J., and Stollberger, C. (2003). The heart in human dystrophinopathies. Cardiology 99 (1), 1–19. doi:10.1159/000068446
Flanigan, K. M. (2014). Duchenne and Becker muscular dystrophies. Neurol. Clin. 32 (3), 671–688. doi:10.1016/j.ncl.2014.05.002
Giarratana, N., Conti, F., La Rovere, R., Gijsbers, R., Carai, P., Duelen, R., et al. (2020). MICAL2 is essential for myogenic lineage commitment. Cell Death Dis. 11 (8), 654. doi:10.1038/s41419-020-02886-z
Gilbert, G., Kadur Nagaraju, C., Duelen, R., Amoni, M., Bobin, P., Eschenhagen, T., et al. (2021). Incomplete assembly of the dystrophin-associated protein complex in 2D and 3D-cultured human induced pluripotent stem cell-derived cardiomyocytes. Front. Cell Dev. Biol. 9, 737840. doi:10.3389/fcell.2021.737840
Heydari, Z., Moeinvaziri, F., Agarwal, T., Pooyan, P., Shpichka, A., Maiti, T. K., et al. (2021). Organoids: a novel modality in disease modeling. Biodes. Manuf. 4, 689–716. doi:10.1007/s42242-021-00150-7
Honeyman, L., Bazett, M., Tomko, T. G., and Haston, C. K. (2013). MicroRNA profiling implicates the insulin-like growth factor pathway in bleomycin-induced pulmonary fibrosis in mice. Fibrogenes. Tissue Repair 6 (1), 16. doi:10.1186/1755-1536-6-16
Hu, G., Ma, L., Dong, F., Hu, X., Liu, S., SUn, H., et al. (2019). Inhibition of microRNA-124-3p protects against acute myocardial infarction by suppressing the apoptosis of cardiomyocytes. Mol. Med. Rep. 20 (4), 3379–3387. doi:10.3892/mmr.2019.10565
Jelinkova, S., Vilotic, A., Pribyl, J., Aimond, F., Salykin, A., Acimovic, I., et al. (2020). DMD pluripotent stem cell derived cardiac cells recapitulate in vitro human cardiac pathophysiology. Front. Bioeng. Biotechnol. 8, 535. doi:10.3389/fbioe.2020.00535
Jensen, C., and Teng, Y. (2020). Is it time to start transitioning from 2D to 3D cell culture? Front. Mol. Biosci. 33 (7). doi:10.3389/fmolb.2020.00033
Jiang, J., Burgon, P. G., Wakimoto, H., Onoue, K., Gorham, J. M., O'Meara, C. C., et al. (2015). Cardiac myosin binding protein C regulates postnatal myocyte cytokinesis. Proc. Natl. Acad. Sci. U. S. A. 112 (29), 9046–9051. doi:10.1073/pnas.1511004112
Jung, S. E., Kim, S. W., Jeong, S., Moon, H., Choi, W. S., Lim, S., et al. (2021). MicroRNA-26a/b-5p promotes myocardial infarction-induced cell death by downregulating cytochrome c oxidase 5a. Exp. Mol. Med. 53 (9), 1332–1343. doi:10.1038/s12276-021-00665-0
Kamdar, F., and Garry, D. J. (2016). Dystrophin-deficient cardiomyopathy. J. Am. Coll. Cardiol. 67 (21), 2533–2546. doi:10.1016/j.jacc.2016.02.081
Kay, M., Soltani, B. M., Aghdaei, F. H., Ansari, H., and Baharvand, H. (2019). Hsa-miR-335 regulates cardiac mesoderm and progenitor cell differentiation. Stem Cell Res. Ther. 10 (1), 191. doi:10.1186/s13287-019-1249-2
Law, M. L., Cohen, H., Martin, A. A., Angulski, A. B. B., and Metzger, J. M. (2020). Dysregulation of calcium handling in Duchenne muscular dystrophy-associated dilated cardiomyopathy: mechanisms and experimental therapeutic strategies. J. Clin. Med. 9 (2), E520. doi:10.3390/jcm9020520
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Lim, G. B. (2020). Complexity and plasticity of cardiac cellular composition. Nat. Rev. Cardiol. 17 (12), 759. doi:10.1038/s41569-020-00464-6
Lin, B., Li, Y., Han, L., Kaplan, A. D., Ao, Y., Kalra, S., et al. (2015). Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis. Model. Mech. 8 (5), 457–466. doi:10.1242/dmm.019505
Loboda, A., and Dulak, J. (2020). Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: past, present, and future. Pharmacol. Rep. 72 (5), 1227–1263. doi:10.1007/s43440-020-00134-x
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi:10.1186/s13059-014-0550-8
Luk, A., Ahn, E., Soor, G. S., and Butany, J. (2009). Dilated cardiomyopathy: a review. J. Clin. Pathol. 62 (3), 219–225. doi:10.1136/jcp.2008.060731
Mareedu, S., Million, E. D., Duan, D., and Babu, G. J. (2021). Abnormal calcium handling in Duchenne muscular dystrophy: mechanisms and potential therapies. Front. Physiol. 12, 647010. doi:10.3389/fphys.2021.647010
McGreevy, J. W., Hakim, C. H., McIntosh, M. A., and Duan, D. (2015). Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Model Mech. 8 (3), 195–213. doi:10.1242/dmm.018424
McNally, E. M., and Mestroni, L. (2017). Dilated cardiomyopathy: genetic determinants and mechanisms. Circ. Res. 121 (7), 731–748. doi:10.1161/CIRCRESAHA.116.309396
Muntoni, F., Torelli, S., and Ferlini, A. (2003). Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet. Neurol. 2 (12), 731–740. doi:10.1016/s1474-4422(03)00585-4
Nakamura, T. Y., Iwata, Y., Sampaolesi, M., Hanada, H., Saito, N., Artman, M., et al. (2001). Stretch-activated cation channels in skeletal muscle myotubes from sarcoglycan-deficient hamsters. Am. J. Physiol. Cell Physiol. 281 (2), C690–C699. doi:10.1152/ajpcell.2001.281.2.C690
Olson, L. E., and Soriano, P. (2009). Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev. Cell 16 (2), 303–313. doi:10.1016/j.devcel.2008.12.003
Qin, W., Cao, L., and Massey, I. Y. (2021). Role of PI3K/Akt signaling pathway in cardiac fibrosis. Mol. Cell. Biochem. 476 (11), 4045–4059. doi:10.1007/s11010-021-04219-w
Quattrocelli, M., Swinnen, M., Giacomazzi, G., Camps, J., Barthélemy, I., Ceccarelli, G., et al. (2015). Mesodermal iPSC-derived progenitor cells functionally regenerate cardiac and skeletal muscle. J. Clin. Invest. 125 (12), 4463–4482. doi:10.1172/JCI82735
Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F., et al. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8 (11), 2281–2308. doi:10.1038/nprot.2013.143
Raudvere, U., Kolberg, L., Kuzmin, I., Arak, T., Adler, P., Peterson, H., et al. (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47 (W1), W191–W198. doi:10.1093/nar/gkz369
Ren, S., Pan, L., Yang, L., Niu, Z., Wang, L., Feng, H., et al. (2021). miR-29a-3p transferred by mesenchymal stem cells-derived extracellular vesicles protects against myocardial injury after severe acute pancreatitis. Life Sci. 272, 119189. doi:10.1016/j.lfs.2021.119189
Richards, D. J., Li, Y., Kerr, C. M., Yao, J., Beeson, G. C., Coyle, R. C., et al. (2020). Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 4 (4), 446–462. doi:10.1038/s41551-020-0539-4
Rotini, A., Martínez-Sarrà, E., Pozzo, E., and Sampaolesi, M. (2018). Interactions between microRNAs and long non-coding RNAs in cardiac development and repair. Pharmacol. Res. 127, 58–66. doi:10.1016/j.phrs.2017.05.029
Santoni de Sio, F. R., Gritti, A., Cascio, P., Neri, M., Sampaolesi, M., Galli, C., et al. (2008). Lentiviral vector gene transfer is limited by the proteasome at postentry steps in various types of stem cells. Stem Cells 26 (8), 2142–2152. doi:10.1634/stemcells.2007-0705
Scalise, M., Marino, F., Salerno, L., Cianflone, E., Molinaro, C., Salerno, N., et al. (2021). From spheroids to organoids: the next generation of model systems of human cardiac regeneration in a dish. Int. J. Mol. Sci. 22 (24), 13180. doi:10.3390/ijms222413180
Sheikh, F., Lyon, R. C., and Chen, J. (2015). Functions of myosin light chain-2 (MYL2) in cardiac muscle and disease. Gene 569 (1), 14–20. doi:10.1016/j.gene.2015.06.027
Shi, H., Li, H., Zhang, F., Xue, H., Zhang, Y., Han, Q., et al. (2021). MiR-26a-5p alleviates cardiac hypertrophy and dysfunction via targeting ADAM17. Cell Biol. Int. 45 (11), 2357–2367. doi:10.1002/cbin.11685
Takimoto, E. (2012). Cyclic GMP-dependent signaling in cardiac myocytes. Circ. J. 76 (8), 1819–1825. doi:10.1253/circj.cj-12-0664
Tang, C. M., Zhang, M., Huang, L., Hu, Z. Q., Zhu, J. N., Xiao, Z., et al. (2017). CircRNA_000203 enhances the expression of fibrosis-associated genes by derepressing targets of miR-26b-5p, Col1a2 and CTGF, in cardiac fibroblasts. Sci. Rep. 7, 40342. doi:10.1038/srep40342
Tang, L., Xie, J., Yu, X., and Zheng, Y. (2020). MiR-26a-5p inhibits GSK3β expression and promotes cardiac hypertrophy in vitro. PeerJ 8, e10371. doi:10.7717/peerj.10371
Tsedeke, A. T., Allanki, S., Gentile, A., Jimenez-Amilburu, V., Rasouli, S. J., Guenther, S., et al. (2021). Cardiomyocyte heterogeneity during zebrafish development and regeneration. Dev. Biol. 476, 259–271. doi:10.1016/j.ydbio.2021.03.014
van der Pol, A., Hoes, M. F., de Boer, R. A., and van der Meer, P. (2020). Cardiac foetal reprogramming: a tool to exploit novel treatment targets for the failing heart. J. Intern. Med. 288 (5), 491-506. doi:10.1111/joim.13094
Velasco, V., Shariati, S. A., and Esfandyarpour, R. (2020). Microtechnology-based methods for organoid models. Microsyst. Nanoeng. 6, 76. doi:10.1038/s41378-020-00185-3
Wang, B., Zhang, A., Wang, H., Klein, J. D., Tan, L., Wang, Z. M., et al. (2019). miR-26a limits muscle wasting and cardiac fibrosis through exosome-mediated microRNA transfer in chronic kidney disease. Theranostics 9 (7), 1864–1877. doi:10.7150/thno.29579
Xie, Y., Hu, J., Zhang, X., Li, C., Zuo, Y., Xie, S., et al. (2020). Neuropeptide Y induces cardiomyocyte hypertrophy via attenuating miR-29a-3p in neonatal rat cardiomyocytes. Protein Pept. Lett. 27 (9), 878–887. doi:10.2174/0929866527666200416144459
Yang, Q., Wu, F., Mi, Y., Wang, F., Cai, K., Yang, X., et al. (2020). Aberrant expression of miR-29b-3p influences heart development and cardiomyocyte proliferation by targeting NOTCH2. Cell Prolif. 53 (3), e12764. doi:10.1111/cpr.12764
Yiu, E. M., and Kornberg, A. J. (2015). Duchenne muscular dystrophy. J. Paediatr. Child. Health 51 (8), 759–764. doi:10.1111/jpc.12868
Zhao, D., Lei, W., and Hu, S. (2021). Cardiac organoid - a promising perspective of preclinical model. Stem Cell Res. Ther. 12 (1), 272. doi:10.1186/s13287-021-02340-7
Zhu, P., Li, H., Zhang, A., Li, Z., Zhang, Y., Ren, M., et al. (2021). MicroRNAs sequencing of plasma exosomes derived from patients with atrial fibrillation: miR-124-3p promotes cardiac fibroblast activation and proliferation by regulating AXIN1. J. Physiol. Biochem. 78, 85–98. doi:10.1007/s13105-021-00842-9
Keywords: Duchenne muscular dystrophy, induced pluripotent stem cells, cardiomyopathy, cardiac organoids, disease modeling, aberrant adipogenesis, fibrosis
Citation: Marini V, Marino F, Aliberti F, Giarratana N, Pozzo E, Duelen R, Cortés Calabuig Á, La Rovere R, Vervliet T, Torella D, Bultynck G, Sampaolesi M and Chai YC (2022) Long-term culture of patient-derived cardiac organoids recapitulated Duchenne muscular dystrophy cardiomyopathy and disease progression. Front. Cell Dev. Biol. 10:878311. doi: 10.3389/fcell.2022.878311
Received: 17 February 2022; Accepted: 30 June 2022;
Published: 11 August 2022.
Edited by:
Francisco Hernandez-Torres, University of Granada, SpainReviewed by:
Valeria Ricotti, Solid Biosciences, United KingdomCopyright © 2022 Marini, Marino, Aliberti, Giarratana, Pozzo, Duelen, Cortés Calabuig, La Rovere, Vervliet, Torella, Bultynck, Sampaolesi and Chai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurilio Sampaolesi, bWF1cmlsaW8uc2FtcGFvbGVzaUBrdWxldXZlbi5iZQ==; Yoke Chin Chai, eW9rZWNoaW4uY2hhaUBrdWxldXZlbi5iZQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.