 Pawan Faris1
Pawan Faris1 Claudio Casali2
Claudio Casali2 Sharon Negri1Lara Iengo3Marco Biggiogera2
Sharon Negri1Lara Iengo3Marco Biggiogera2 Angela Serena Maione3*†
Angela Serena Maione3*† Francesco Moccia1*†
Francesco Moccia1*†- 1Laboratory of General Physiology, Department of Biology and Biotechnology “Lazzaro Spallanzani”, University of Pavia, Pavia, Italy
- 2Laboratory of Cell Biology and Neurobiology, Department of Biology and Biotechnology “Lazzaro Spallanzani”, University of Pavia, Pavia, Italy
- 3Vascular Biology and Regenerative Medicine Unit, Centro Cardiologico Monzino, IRCCS, Milan, Italy
Nicotinic acid adenine dinucleotide phosphate (NAADP) is a newly discovered second messenger that gates two pore channels 1 (TPC1) and 2 (TPC2) to elicit endo-lysosomal (EL) Ca2+ release. NAADP-induced lysosomal Ca2+ release may be amplified by the endoplasmic reticulum (ER) through the Ca2+-induced Ca2+ release (CICR) mechanism. NAADP-induced intracellular Ca2+ signals were shown to modulate a growing number of functions in the cardiovascular system, but their occurrence and role in cardiac mesenchymal stromal cells (C-MSCs) is still unknown. Herein, we found that exogenous delivery of NAADP-AM induced a robust Ca2+ signal that was abolished by disrupting the lysosomal Ca2+ store with Gly-Phe β-naphthylamide, nigericin, and bafilomycin A1, and blocking TPC1 and TPC2, that are both expressed at protein level in C-MSCs. Furthermore, NAADP-induced EL Ca2+ release resulted in the Ca2+-dependent recruitment of ER-embedded InsP3Rs and SOCE activation. Transmission electron microscopy revealed clearly visible membrane contact sites between lysosome and ER membranes, which are predicted to provide the sub-cellular framework for lysosomal Ca2+ to recruit ER-embedded InsP3Rs through CICR. NAADP-induced EL Ca2+ mobilization via EL TPC was found to trigger the intracellular Ca2+ signals whereby Fetal Bovine Serum (FBS) induces C-MSC proliferation. Furthermore, NAADP-evoked Ca2+ release was required to mediate FBS-induced extracellular signal-regulated kinase (ERK), but not Akt, phosphorylation in C-MSCs. These finding support the notion that NAADP-induced TPC activation could be targeted to boost proliferation in C-MSCs and pave the way for future studies assessing whether aberrant NAADP signaling in C-MSCs could be involved in cardiac disorders.
1 Introduction
Nicotinic acid adenine dinucleotide phosphate (NAADP) has emerged as a the most powerful (already in the nanomolar concentration range) Ca2+-releasing second messenger in mammalian cells (Galione, 2015; Patel, 2015). NAADP elicits an increase in intracellular Ca2+ concentration ([Ca2+]i) by gating a novel family of intracellular Ca2+-releasing channels, known as two-pore channels (TPCs), which present two isoforms in mammals (i.e., TPC1 and TPC2) and mobilize endo-lysosomal (EL) Ca2+ into the cytosol (Patel, 2015; Galione, 2019; Jin et al., 2020). Jupiter microtubule-associated homolog 2 (JPT2) (Gunaratne et al., 2021) and the RNA-binding protein, Lsm2 (Zhang et al., 2021), serve as auxiliary protein to bind NAADP and thereby contribute to mediate TPC-mediated EL Ca2+ release. The Ca2+ response to NAADP may remain spatially confined in proximity of EL vesicles (Ruas et al., 2010; Vassileva et al., 2020) or it can be amplified into a regenerative Ca2+ wave through the Ca2+-dependent recruitment of juxtaposed ryanodine and inositol-1,4,5-trisphosphate (InsP3) receptors at membrane contact sites (MCSs) between lysosomes and endoplasmic reticulum (ER) (Kinnear et al., 2004; Davis et al., 2012; Kilpatrick et al., 2013; Penny et al., 2014). Lysosomal Ca2+ refilling is impaired by alkalinization of the EL lumen (Ronco et al., 2015), although the mechanisms whereby intraluminal pH recharges EL vesicles with Ca2+ remains a controversial issue (Morgan et al., 2011; Garrity et al., 2016; Faris et al., 2018).
NAADP has been recognized as the trigger of the cellular Ca2+ response to extracellular stimuli in multiple tissues (Galione, 2015; Patel, 2015), including the cardiovascular system (Fameli et al., 2017; Moccia et al., 2021a; Negri et al., 2021b). NAADP-induced Ca2+ release through TPC2 increases the Ca2+ content within the sarcoendoplasmic reticulum in ventricular (Macgregor et al., 2007) and atrial myocytes (Collins et al., 2011), both at rest (Macgregor et al., 2007; Collins et al., 2011) and during β-adrenergic receptor stimulation (Macgregor et al., 2007; Collins et al., 2011; Lewis et al., 2012; Capel et al., 2015). Likewise, a flurry of reports showed that NAADP-induced intracellular Ca2+ signals elicit contraction in multiple types of vascular smooth muscle cells (VSMCs) (Kinnear et al., 2004; Jiang et al., 2013; Fameli et al., 2014; Trufanov et al., 2019). For instance, NAADP gates TPC2 to promote the Ca2+-dependent recruitment of RyR3 and global cytosolic Ca2+ waves in pulmonary artery VSMCs stimulated with either endothelin-1 (Kinnear et al., 2004; Jiang et al., 2013) or angiotensin II (Lee et al., 2015). Finally, NAADP may serve as a trigger of the Ca2+ response to extracellular stimuli also in vascular endothelial cells (Favia et al., 2014; Zuccolo et al., 2019; Negri et al., 2021a) and circulating endothelial colony forming cells (ECFCs) (Balducci et al., 2021; Moccia et al., 2021b). Aberrant NAADP signalling in cardiac myocytes may result in arrhythmia (Nebel et al., 2013) and ischemia-reperfusion injury (Davidson et al., 2015), whereas it could lead to pulmonary artery hypertension in VSMCs (Jiang et al., 2018; Hu et al., 2021).
Once regarded as mere bystanders of the contractile function effected by neighbouring cardiac myocytes, cardiac mesenchymal stromal cells (C-MSCs) are required to maintain myocardial structure and function and, therefore, to ensure effective cardiac contraction (Brown et al., 2005; Camelliti et al., 2005). C-MSCs contribute to wound healing and fibrotic remodelling after ischemic injury (Jugdutt, 2003; Camelliti et al., 2005) and they have been put forward as a promising cellular substrate to induce cardiac repair (Bagno et al., 2018; Braunwald, 2018). Furthermore, C-MSCs could stimulate cardiac myocytes to undergo proliferation or hypertrophy depending on whether this interaction takes place during embryonic development or in the adult heart (Kakkar and Lee, 2010). Finally, C-MSCs exhibit significant immunomodulatory potential by attenuating the inflammatory response in the infarcted myocardium (Czapla et al., 2016; Diedrichs et al., 2019). In agreement with their contribution to the structural, biochemical and electro-chemical features of the myocardium, C-MSCs are involved in the pathogenic mechanisms of multiple cardiac diseases (Brown et al., 2005; Camelliti et al., 2005). For instance, C-MSCs provide a source of adipocytes (Sommariva et al., 2016; Stadiotti et al., 2017) and support fibrotic remodelling (Maione et al., 2021) in arrhythmogenic cardiomyopathy (ACM), a rare genetic disorder that is featured by fibro-fatty myocardium substitution, malignant arrhythmias, and heart failure and that can lead to sudden death in young individuals (Moccia et al., 2019). It has long been known that an increase in [Ca2+]i regulates multiple functions in human MSCs (Moccia et al., 2015; Forostyak et al., 2016; Jiang et al., 2017), including proliferation (Foreman et al., 2006), migration (Peng et al., 2016), gene expression (Kawano et al., 2006), and differentiation (Kawano et al., 2006; Tao et al., 2011). However, it is still unclear whether and how NAADP evokes intracellular Ca2+ signals and whether lysosomal-ER MCSs do exist in C-MSCs. This information could be extremely helpful to boost the design of alternative strategies to effectively target C-MSCs in a variety of life-threatening cardiac disorders. In the present investigation, we first provided the evidence that NAADP evokes robust lysosomal Ca2+ mobilization, which is amplified into a global increase in [Ca2+]i by InsP3 receptors (InsP3Rs). Transmitted electron microscopy (TEM) then revealed clearly discernible MCSs between lysosomes and ER membrane in C-MSCs. Finally, we found that NAADP-induced Ca2+-dependent crosstalk between lysosomes and ER triggers the intracellular Ca2+ signals whereby Fetal Bovine Serum (FBS) induces cell proliferation. The role of Ca2+ signalling in regulating proliferation and differentiation in MSCs confer these findings the potential to provide the molecular framework for further studies aiming at manipulating C-MSCs for therapeutic purposes.
2 Materials and Methods
2.1 Ethical Statement
This study complies with the WMA Declaration of Helsinki. The use of human cells from biopsy samples of healthy subjects (cardiomyopathies ruled out) was approved by IEO-CCM IRCCS Ethic Committee (project CCM1072). Written informed consent was obtained from all participants.
2.2 C-MSC Isolation and Culture
Cells were obtained from endomyocardial specimens and characterized as previously described (Pilato et al., 2018) and cultured with Iscove’s Modified Dulbecco’s Medium (Thermo Fisher Scientific, MA, United States) supplemented with 20% Fetal Bovine Serum (FBS), 10 ng/ml basic fibroblast growth factor, 10,000 U/ml Penicillin, 10,000 μg/ml Streptomycin, and 0.02 M L-Glutamine.
2.3 Solutions
Physiological salt solution (PSS) had the following composition (in mM): 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10 Glucose, 10 Hepes. In Ca2+-free solution (0Ca2+), Ca2+ was substituted with 2 mM NaCl, and 0.5 mM EGTA was added. Solutions were titrated to pH 7.4 with NaOH. The osmolality of the extracellular solution, as measured with an osmometer (Wescor 5500, Logan, UT, United States), was 300–310 mmol/kg.
2.4 [Ca2+]i Measurements and Statistics of Ca2+ Signals
C-MSCs were loaded with 2 µM fura-2 acetoxymethyl ester (fura-2/AM; 1 mM stock in dimethyl sulfoxide) in PSS for 30 min at room temperature (RT). The details of the Ca2+ recording set-up have been described in Moccia et al. (2021b) and are reported in the Supplementary Material. All the experiments were performed at RT. The amplitude of intracellular Ca2+ release in response to each agonist (NAADP or FBS) or drug [Gly-Phe β-naphthylamide (GPN), nigericin, bafilomycin A1, and cyclopiazonic acid (CPA)] was measured as the difference between the ratio at the peak of intracellular Ca2+ mobilization and the mean ratio of 1 min baseline before the peak. Pooled data are given as mean ± SE and statistical significance (p < 0.05) was evaluated by the Student’s t-test for unpaired observations or one-way Anova analysis followed by the post-hoc Dunnett’s test as appropriate (Negri et al., 2021a; Remigante et al., 2021). Data relative to Ca2+ signals are presented as mean ± SE, while the number of cells analysed is indicated in the corresponding bar histograms.
2.5 mRNA Extraction and qRT-PCR Assay
Cell cultures were lysed in RL lysis buffer (Norgen Biotek Corp., Thorold, ON, Canada). RNA was isolated from cells by using a Total RNA Purification kit (Norgen Biotek Corp., Thorold, ON, Canada). The quantification of the isolated RNA was determined by NanoDrop spectrophotometer (ND-1000, EuroClone, Milan, Italy). Reverse transcription was conducted with SuperScript III (Invitrogen, Carlsbad, CA, United States) following the manufacturer’s instructions. qRT-PCR was performed with the use of the iQTM SYBR Green Super Mix (Bio-Rad Laboratories, Hercules, CA, United States) and specific primers (reported in Table 1). All reactions were performed in a 96-well format with the 7900HT Fast Real-Time PCR System (Thermo Fisher Scientific, MA, United States). The relative quantities of specific mRNA were obtained with the use of the comparative Ct method and were normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Maione et al., 2021; Zuccolini et al., 2022). The expression of each target gene was assessed in triplicate (Ferrera et al., 2021; Maione et al., 2021).

TABLE 1. Primer sequences 5′-3′.
2.6 Protein Extraction and Western Blot Analysis
C-MSCs were lysed in cell lysis buffer (Cell Signalling Technology, Danvers, MA, United States) supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich, Saint Louis, MO, United States). Total protein extracts were subjected to SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad, CA, United States). The membranes were blocked for 1 h at room temperature in 5% non-fat dry milk in Wash Buffer (Tris Buffer Sulfate, 0.1% Tween-20) and then incubated O/N at 4°C with the appropriate primary antibodies (reported in Table 2). The membranes were incubated with peroxidase-conjugated secondary antibodies (GE Healthcare, Chicago, IL, United States) for 1 h. Signals were visualized using the LiteUP Western Blot Chemiluminescent Substrate (EuroClone, Milan, Italy). Images were acquired with the ChemiDocTM MP Imaging System (Bio-Rad, CA, United States), and densitometric analysis of membranes was performed using the ImageJ software (National Institutes of Health, Bethesda, MD, United States). C-MSC proteins were normalized according to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) signal.

TABLE 2. Primary antibodies.
2.7 Transmission Electron Microscopy
For transmission electron microscopy (TEM) analysis, following trypsinization cells were centrifuged at 800 rpm for 5 min and then fixed with 2.5% glutaraldehyde in culture medium, for 2 h at RT (Carriero et al., 2021). The cell pellet was then rinsed in PBS overnight, post-fixed in 1% aqueous OsO4 for 3 h at room temperature and rinsed in H2O. Cells were pre-embedded in 2% agarose in water, dehydrated in acetone and then embedded in epoxy resin (Electron Microscopy Sciences, EM-bed812). Ultrathin sections (60–80 nm) were cut on a Reichert OM-U3 ultramicrotome, collected on nickel grids and then stained with uranyl acetate and lead citrate. The specimens were observed with a JEM 1200 EX II (JEOL, Peabody, MA, United States) electron microscope operating at 100 kV and equipped with a MegaView G2 CCD camera (Olympus OSIS, Tokyo, Japan).
2.8 Cell Proliferation
C-MSCs were plated in 6-well plates (100,000 cells/well) and serum starved for 4 h. Cells were then stimulated with 20% FBS in the absence (Ctrl) or presence of 100 µM of NED-19, a selective TPC blocker (Galione, 2015; Jin et al., 2020). 24 and 48 h after stimulation with FBS, the medium was removed, cells detached from the plates, and counted.
2.9 Flow Cytometry
To evaluate whether blocking TPCs with NED-19 was able to induce apoptosis in C-MSCs, Annexin V Alexa Fluor™ 488 Dye (Thermo Fisher Scientific, MA, United States) has been used, according to the manufacturer’s instructions. Briefly, cells were detached using TrypLE™ Select Enzyme (Thermo Fisher Scientific, MA, United States) and incubated with Annexin V Alexa Fluor™ 488 Dye for 15 min at RT. The fluorescence emission at 530 nm corresponding to apoptotic cells has been measured using flow cytometry (Gallios, Beckman Coulter, Brea, CA, United States).
3 Results
3.1 Nicotinic Acid Adenine Dinucleotide Phosphate Induces Intracellular Ca2+ Signals by Mobilizing Lysosomal Ca2+ in Cardiac Mesenchymal Stromal Cells
In order to assess whether they are endowed with a NAADP-sensitive Ca2+ store, C-MSCs were loaded with Fura-2/AM (2 µM), a Ca2+ sensitive fluorophore, as shown elsewhere (Maione et al., 2020a). Human MSCs may exhibit spontaneous oscillations in [Ca2+]i (Kawano et al., 2002; Kawano et al., 2003; Kawano et al., 2006). Consistently, a fraction of C-MSCs (≈56.4%) exhibited a few (1-4) Ca2+ spikes in the absence of extracellular stimulation (Supplementary Figure S1). These cells were, therefore, discarded from subsequent analysis as shown elsewhere (Zuccolo et al., 2017; Zuccolo et al., 2019), since the spontaneous, unpredictable Ca2+ activity could mask or even prevent (in case of transient depletion of endogenous target organelle) the Ca2+ response to NAADP. We then assessed whether NAADP-AM, a membrane-permeable analogue of NAADP (Macgregor et al., 2007; Brailoiu et al., 2010), was able to increase the [Ca2+]i in C-MSCs. NAADP-AM (1 µM) evoked a short train of intracellular Ca2+ oscillations that declined ≈25 min after their onset in the presence of extracellular Ca2+ in 88 out of 164 cells (53.6%) (Figure 1A). In 58 out of 164 cells (35.4%), NAADP-AM (1 µM) induced a transient increase in [Ca2+]i that lasted ≈800 s returned to the baseline in the continuous presence of the agonist (Figure 1A). Eighteen cells (11%) were not responsive to NAADP-AM (1 µM). Under 0Ca2+ conditions, NAADP-AM (1 µM) induced only a transient increase in [Ca2+]i that was not followed by additional Ca2+ spikes (Figure 1B). Intriguingly, the duration of the elevation in [Ca2+]i was significantly shorter, i.e., ≈280 s, while the peak amplitude was higher (Figure 1C), as compared to the Ca2+ transient recorded in the presence of extracellular Ca2+. The subsequent restitution of extracellular Ca2+ after the full recovery of [Ca2+]i to the baseline resulted in a second Ca2+ signal that was due to extracellular Ca2+ entry (Figure 1B). NAADP-AM was removed from the perfusate 100 s before re-addition of extracellular Ca2+ (Figure 1B), which suggests that the Ca2+ entry pathway recruited downstream of NAADP-AM-induced Ca2+ release is provided by store-operated Ca2+ entry (SOCE), as more widely discussed below (Yamazaki et al., 2007; Sanchez-Hernandez et al., 2010; Negri et al., 2020). The statistical analysis of the two distinct components of the Ca2+ response to NAADP-AM (i.e., endogenous Ca2+ release and SOCE) is presented in Figure 1D. NAADP is recognized as a mobilizer of the lysosomal Ca2+ pool (Galione, 2015; Patel, 2015). Control experiments confirmed that adding back extracellular Ca2+ after 700 s exposure to 0Ca2+ conditions did not increase the in C-MSCs (Supplementary Figure S2). In accord, NAADP-AM-evoked intracellular Ca2+ release was significantly (p < 0.001) reduced by discharging the lysosomal Ca2+ store with the lysosomotropic compound, dipeptide glycyl-l-phenylalanine 2-naphthylamide (GPN; 200 μM, 30 min) (Kilpatrick et al., 2013; Yuan et al., 2021) (Figures 1E,F). Of note, GPN has recently been reaffirmed as a reliable pharmacological tool to mobilize lysosomal Ca2+ (Yuan et al., 2021). Furthermore, NAADP-AM-evoked endogenous Ca2+ mobilization was abolished by collapsing the lysosomal H+ gradient that maintains lysosomal Ca2+ refilling with the H+/K+ ionophore, nigericin (50 μM, 30 min) (Figures 1E,F), or with the v-ATPase inhibitor, bafilomycin A1 (1 μM, 30 min) (Figures 1E,F) (Morgan et al., 2011; Ronco et al., 2015; Faris et al., 2019; Yuan et al., 2021). Supplementary Figure S3 shows that GPN (200 µM), nigericin (50 µM), and bafilomycin A1 (1 µM) induced a remarkable reduction in Lysotracker Red fluorescence, thereby confirming that all of these drugs target lysosomal Ca2+ (Pandey et al., 2009; Faris et al., 2019; Yuan et al., 2021). In accord with these observations, ammonium chloride (NH4Cl), which disrupts the lysosomal Ca2+ pool by inducing intraluminal alkalinization (Christensen et al., 2002), also reduced Lysotracker Red Fluorescence and impaired NAADP-AM-evoked intracellular Ca2+ mobilization (Supplementary Figure S4).
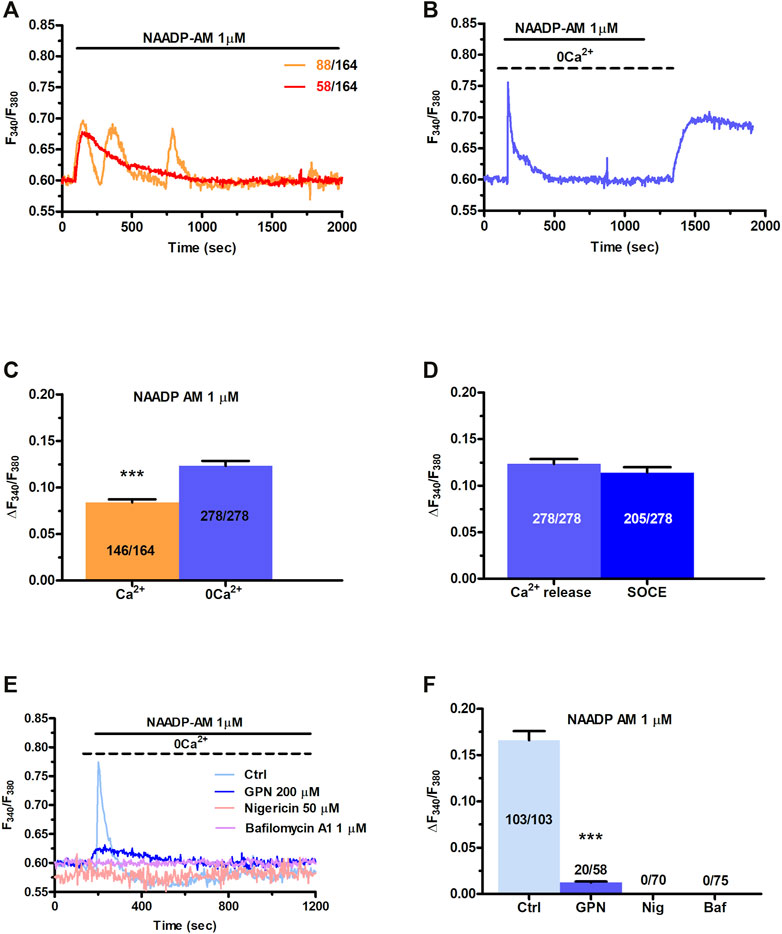
FIGURE 1. NAADP induces intracellular Ca2+ signals by mobilizing lysosomal Ca2+ in C-MSCs. (A) Exogenous administration of NAADP-AM (1 µM) indues either intracellular Ca2+ oscillations or a transient increase in [Ca2+]i. (B) In absence of external Ca2+ (0Ca2+), NAADP-AM (1 µM) induced only a transient increase in [Ca2+]i, whereas subsequent restitution of extracellular Ca2+ after the full recovery of [Ca2+]i to the baseline resulted in a second Ca2+ signal that was due to extracellular Ca2+ entry. (C) Mean ± SE of the amplitude of the peak Ca2+ response to NAADP in the presence and absence of extracellular Ca2+. Student’s t-test: ***p < 0.001. (D) Mean ± SE of the amplitude of NAADP-induced intracellular Ca2+ release and SOCE. (E) Disrupting the lysosomal Ca2+ store with GPN (200 μM, 30 min), nigericin (50 μM, 30 min) or bafilomycin A1 (1 μM, 30 min) severely affected the intracellular Ca2+ response to NAADP-AM. (F) Mean ± SE of the amplitude of the peak Ca2+ response to NAADP-AM in the absence and in the presence of GPN, nigericin (Nig), or bafilomycin A1 (Baf). One-Way Anova followed by the post-hoc Dunnett’s test: ***p < 0.001.
Overall, these findings provide the first evidence that NAADP may induce lysosomal Ca2+ release followed by extracellular Ca2+ entry in C-MSCs.
3.2 Nicotinic Acid Adenine Dinucleotide Phosphate-Induced Intracellular Ca2+ Release is Mediated by TPCs in Cardiac Mesenchymal Stromal Cells
TPCs mediate NAADP-induced intracellular Ca2+ release throughout the phylogenetic tree (Patel, 2015; Galione, 2019; Jin et al., 2020), including the cardiovascular system (Moccia et al., 2021a; Negri et al., 2021b). In accord, qRT-PCR analysis showed that both TPC1 and TPC2 transcripts are expressed in C-MSCs, although TPC1 mRNA is slightly more abundant (Figure 2A). Negative controls were performed by omitting reverse transcriptase from the reaction (not shown) (Faris et al., 2019). Immunoblotting confirmed that TPC1 and TPC2 are also expressed at protein level. Two single bands of, respectively, 94 and 85 kDa were found for TPC1 and TPC2 proteins (Figure 2B). C-MSCs are not amenable for lipofectamine-mediated transfection of selective small interfering RNAs (Maione, Sommariva, and Pompilio, unpublished results), which is the strategy we have recently employed to downregulate TPC1 expression in different cellular models (Faris et al., 2019; Moccia et al., 2021b). Therefore, we probed the effect of NED-19, a selective TPC inhibitor (Galione, 2015; Jin et al., 2020), which has been widely employed to inhibit NAADP-dependent TPC activation throughout the cardiovascular system (Macgregor et al., 2007; Jiang et al., 2013; Hu et al., 2021; Moccia et al., 2021a; Negri et al., 2021a). As predicted, NED-19 (100 μM, 30 min) fully suppressed NAADP-AM-evoked intracellular Ca2+ mobilization (Figures 2C,D). Likewise, NED-K (10 μM, 30 min), a chemically modified analogue of NED-19 that has recently been shown to selectively inhibit TPC1 (Davidson et al., 2015), and tetrandrine (10 μM, 30 min), a traditional Chinese herbal remedy that block both TPC1 and TPC2 (Sakurai et al., 2015; Moccia et al., 2021a), respectively, inhibited (p < 0.001) and abrogated NAADP-AM-evoked intracellular Ca2+ release (Figures 2C,D). In aggregate, these data demonstrate that NAADP stimulates TPCs to mobilize lysosomal Ca2+ in c-MSCs.
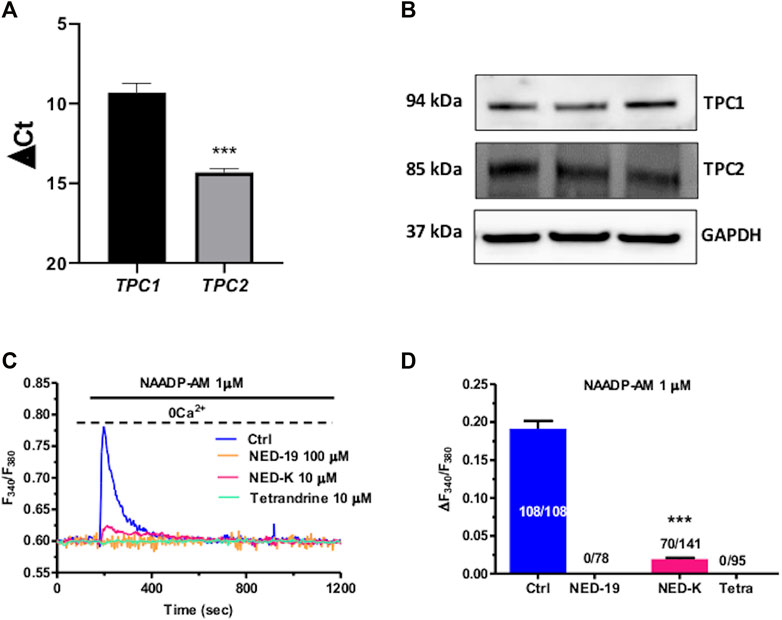
FIGURE 2. Two-pore channels (TPCs) mediate NAADP-induced lysosomal Ca2+ release in C-MSCs. (A) TPC1 and TPC2 gene expression in total RNA extracts of C-MSCs. qRT-PCR data are shown as transcript abundance (genes threshold cycles [Ct] with respect to the house-keeping gene GAPDH). n = 4/group. Student’s t-test: ***p < 0.001. (B) Western Blot analysis of TPC1 and TPC2 proteins in total cellular extracts. n = 3/group. (C) The Ca2+ response to NAADP-AM was suppressed by incubating the cells with the following TPC inhibitors: NED-19 (100 μM, 30 min), NED-K (10 μM, 30 min), and tetrandrine (10 μM, 30 min). (D) Mean ± SE of the amplitude of the peak Ca2+ response to NAADP in the absence (Ctrl) and in the presence of NED-19, NED-K and tetrandrine (Tetra). Student’s t-test: ***p < 0.001.
3.3 InsP3Rs at MCSs are Activated Downstream of NAADP-AM-Induced Intracellular Ca2+ Release in Cardiac Mesenchymal Stromal Cells
The local release of lysosomal Ca2+ evoked by NAADP has long been known to be amplified into a global increase in [Ca2+]i by the recruitment of juxtaposed InsP3Rs on the ER membrane (Churchill and Galione, 2001; Kinnear et al., 2004; Davis et al., 2012; Faris et al., 2019; Moccia et al., 2021b). To assess whether the ER Ca2+ store is required to maintain lysosomal Ca2+ release, we first exploited cyclopiazonic acid (CPA), an established inhibitor of Sarco-Endoplasmic reticulum Ca2+-ATPase activity, as recently shown elsewhere (Kilpatrick et al., 2013; Faris et al., 2019; Moccia et al., 2021b). In the absence of extracellular Ca2+ (0Ca2+), CPA (30 µM) induced a transient elevation in [Ca2+]i due to Ca2+ efflux into the cytosol through ER leakage channels followed by Ca2+ extrusion across the plasma membrane (Figure 3A). While NAADP-AM (1 µM) was able to induce robust Ca2+ release in not-treated cells (Figure 3B), it failed to evoke endogenous Ca2+ mobilization upon CPA-induced depletion of the ER Ca2+ store (Figure 3A). A preliminary characterization of the Ca2+ handling machinery revealed that C-MSCs express InsP3Rs, but not RyRs, and that InsP3-induced ER Ca2+ discharge activates SOCE (Maione et al., 2020a). To assess the contribution of InsP3Rs to NAADP-induced intracellular Ca2+ mobilization, we adopted a similar strategy to that described in (Kilpatrick et al., 2013; Kilpatrick et al., 2016; Faris et al., 2019; Moccia et al., 2021b). The transient increase in [Ca2+]i evoked by NAADP-AM (1 µM) was significantly (p < 0.001) reduced by blocking InsP3Rs with 2-Aminoethoxydiphenyl borate (2-APB) (50 μM, 30 min) (Figure 3C) (Kilpatrick et al., 2013; Kilpatrick et al., 2016) and was suppressed by inhibiting the basal production of InsP3 with U73122 (10 μM, 10 min) (Figure 3C), which selectively interferes with phospholipase C (PLC) activity (Moccia et al., 2006; Negri et al., 2021a). The statistical analysis of these data has been presented in Figure 3D. The lack of full inhibition of NAADP-AM-evoked intracellular Ca2+ mobilization could be due to the incomplete inhibition of InsP3Rs, as also reported in ECFCs (Moccia et al., 2021b), rat gastric smooth muscle cells (Pereira et al., 2014), and MDA-MB-231 breast cancer cells (Vismara et al., 2021). Therefore, InsP3Rs provide a robust source of Ca2+ during lysosomal Ca2+ mobilization and, based upon previous observations (Davis et al., 2012; Kilpatrick et al., 2013; Ronco et al., 2015; Kilpatrick et al., 2016; Faris et al., 2019; Moccia et al., 2021b), it can be concluded that they can be recruited by CICR upon NAADP-induced lysosomal Ca2+ release. TEM was then exploited to assess whether MCSs between lysosomal vesicles and ER cisternae can also be detected and thereby sustain the Ca2+-dependent cross-talk between the two organelles also in C-MSCs (Kilpatrick et al., 2013). For this purpose, after glutaraldehyde fixation cells have been post-fixed in aqueous OsO4 in order to darkly stain lipids and membranes, as described in Section 2. TEM micrographs indicated extensive ER-lysosome MCSs (<20 nm, 14.3 ± 1.13, n = 27 from five cells) with ultrastructural resolution (Figure 4). As also reported in human fibroblasts, in the regions of close appositions (e.g., Figure 4A), fibres that appear to tether lysosomes and ER membranes were clearly discernible. In addition, we detected regions where the apposing membranes appeared to be physically coupled with no visible space between them (e.g., Figures 4B,C). Quantification in random sections showed that 60.5% of lysosomes established contact sites with the ER. As also discussed in Kilpatrick et al. (2013), this is likely to be an underestimate as lysosomal diameter spans between 200 and 500 nm and is, therefore, predicted to extend over several sections above and below the selected plane, where additional contact sites might have been established. Of note, lysosomes could establish extensive contact sites both with the smooth (Figure 4D) and the rough (Figure 4E) ER. We also found that ER cisternae could come in direct contacts with more than one lysosome (Figure 4E). Overall, these findings provide the ultrastructural evidence that the architecture of lysosomes and ER MCSs is fully consistent with the recruitment of ER-embedded InsP3Rs by NAADP-induced lysosomal Ca2+ release through TPCs.
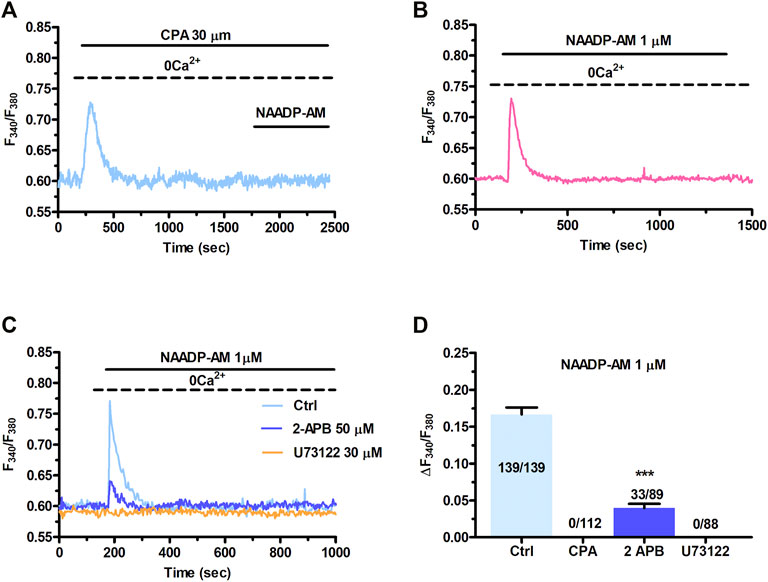
FIGURE 3. InsP3Rs support the Ca2+ response to NAADP. (A) Administration of NAADP-AM (1 µM) after the pharmacological depletion of the ER Ca2+ pool with CPA (30 μm, 30 min) failed to induce intracellular Ca2+ release. CPA induced a transient increase in [Ca2+]i that reflects passive ER Ca2+ leakage from the ER. (B) The intracellular Ca2+ transient evoked by NAADP-AM (1 µM) in the absence of extracellular Ca2+ (0Ca2+) under control conditions. (C) The intracellular Ca2+ release evoked by NAADP-AM (1 µM) under control (Ctrl) conditions was severely affected by blocking InsP3Rs with 2-APB (50 μm, 30 min) or inhibiting PLC activity with U73122 (10 μm, 10 min). (D) Mean ± SE of the amplitude of the peak intracellular Ca2+ response to NAADP-AM under the designated treatments. Student’s t-test: ***p < 0.001.
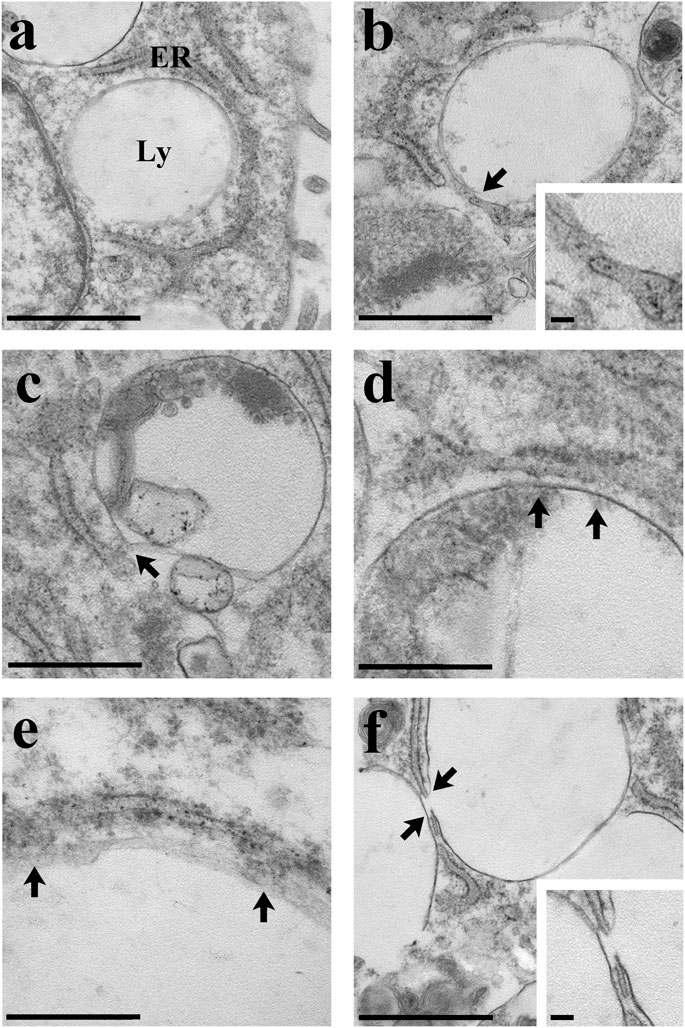
FIGURE 4. Ultrastructural analysis of C-MSCs. Several examples of membrane contacts between lysosomes (Ly) and ER are shown. (A) Note the closeness of the lysosomes with the cell nucleus and ER. Scale bar: 1 µm. (B) The lysosome-ER contact site is indicated (arrow and inset). Scale bar: 1 μm; inset scale bar: 100 nm. (C) The arrow indicates the membrane contact site. Scale bar: 500 nm. (D) Extensive contact (arrows) between the lysosomal and the ER membranes. Scale bar: 500 nm. (E) Note the contact (arrows) between the ribosomes-rich ER and the lysosome. Scale bar: 200 nm. (F) Two close lysosomes; the one on the right is in tight contact with the ER (arrows and inset). Scale bar: 1 μm; inset scale bar: 100 nm.
3.4 SOCE Maintains NAADP-AM-Evoked Intracellular Ca2+ Signals in Cardiac Mesenchymal Stromal Cells
Figure 1B clearly shows that NAADP-AM-induced mobilization of intercellularly stored Ca2+ resulted in extracellular Ca2+ entry even after the agonist washout from the perfusate. This feature clearly hints at SOCE as the Ca2+ entry pathway sustaining the long-lasting increase in [Ca2+]i evoked by NAADP in the presence of extracellular Ca2+. Indeed, InsP3-dependent ER Ca2+ mobilization results in SOCE activation virtually in all mammalian cells (Prakriya and Lewis, 2015; Emrich et al., 2021), including C-MSCs (Maione et al., 2020a). In order to assess whether NAADP-AM-induced lysosomal Ca2+ release can lead to SOCE via intermediate ER Ca2+ depletion, we repeated the “Ca2+ add-back” protocol described in Figure 1 in the absence and presence of BTP-2 or Pyr6, two selective blockers of SOCE (Schleifer et al., 2012; Moccia et al., 2016). This strategy has long been exploited to selectively evaluate the blocking effect of SOCE-targeting drugs on agonist-evoked extracellular Ca2+ entry rather than on the previous phase of endogenous Ca2+ mobilization (Sanchez-Hernandez et al., 2010; Jairaman et al., 2015; Rahman and Rahman, 2017; Scarpellino et al., 2019; Negri et al., 2020; Schach et al., 2020). The influx of Ca2+ secondary to Ca2+ restitution to the perfusate after removal of NAADP-AM (Figure 5A) from the perfusate was significantly (p < 0.001) attenuated by BTP-2 (20 μM, 20 min) and abrogated by Pyr6 (10 μM, 10 min) (Figure 5A). The statistical analysis of these data has been presented in Figure 5B. These observations demonstrate that NAADP-induced lysosomal Ca2+ mobilization in C-MSCs is functionally coupled to SOCE via InsP3-dependent ER Ca2+ release. Therefore, lysosomal Ca2+ release must induce depletion of ER Ca2+ via InsP3Rs, thereby leading to SOCE recruitment on the plasma membrane. To further support this conclusion, Supplementary Figure S5A shows that also the pharmacological depletion of the lysosomal Ca2+ store with nigericin (50 µM) induced both intracellular Ca2+ release and extracellular Ca2+ entry. Furthermore, the intracellular Ca2+ response to nigericin (50 µM) was significantly (p < 0.001) reduced by blocking InsP3Rs with 2-APB (50 μM, 30 min) (Supplementary Figures S5B,C) and by interfering with basal InsP3 production with U73122 (10 μM, 10 min) (Supplementary Figures S5B,C), as recently shown in primary cultures of colorectal cancer cells (Faris et al., 2019) and in circulating ECFCs (Moccia et al., 2021b). Finally, nigericin-evoked extracellular Ca2+ entry was significantly (p < 0.001) attenuated by blocking SOCE with either BTP-2 (20 μM, 20 min) or Pyr6 (10 μM, 10 min) (Figures 5C,D). This result is, therefore, consistent with the evidence reported above that NAADP-induced Ca2+ release through TPCs is able to induce ER Ca2+ depletion followed by SOCE activation.
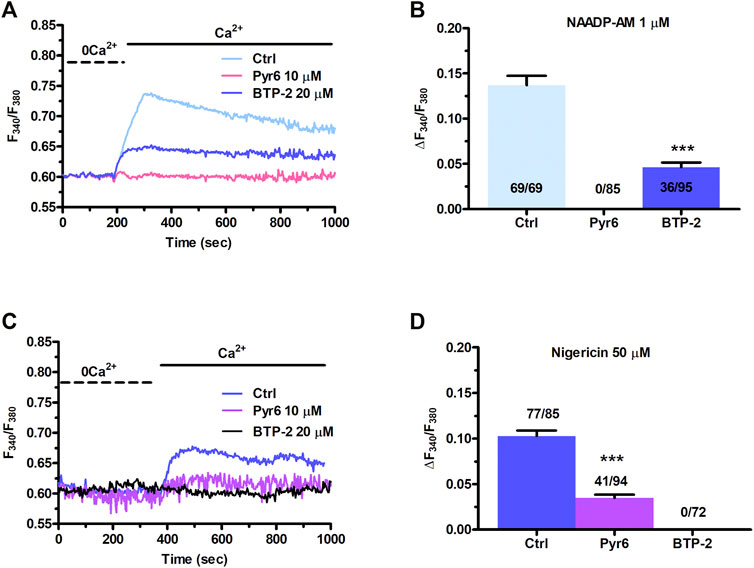
FIGURE 5. NAADP-AM-induced lysosomal Ca2+ mobilization is functionally coupled to SOCE in C-MSCs. (A) The influx of extracellular Ca2+ evoked by NAADP-AM (1 µM) upon depletion of intracellular Ca2+ stores (Ctrl) was severely affected by inhibiting SOCE with Pyr6 (10 μM, 10 min) or BTP-2 (20 μm, 20 min). The previous NAADP-AM-evoked endogenous Ca2+ release has not been shown. (B) Mean ± SE of the amplitude of NAADP-AM-evoked Ca2+ entry evoked by nigericin in the absence (Ctrl) and presence of Pyr-6 and BTP-2. Student’s t-test: ***p < 0.001. (C) Nigericin-evoked extracellular Ca2+ entry was attenuated or inhibited by, respectively, blocking SOCE with BTP-2 (20 μM, 20 min) or Pyr6 (10 μM, 10 min). (D) Mean ± SE of the amplitude of Ca2+ entry evoked by nigericin in the absence (Ctrl) and presence of Pyr-6 and BTP-2. Student’s t-test: ***p < 0.001.
3.5 Nicotinic Acid Adenine Dinucleotide Phosphate-Induced Lysosomal Ca2+ Release via TPCs Supports FBS-Induced Intracellular Ca2+ Oscillations in C-MSCs
FBS has been shown to induce intracellular Ca2+ signals to stimulate proliferation in primary MSCs harvested from rat bone marrow (Foreman et al., 2006). 20% FBS induced intracellular Ca2+ oscillations also in ≈26% of C-MSCs, whereas it promoted a transient increase elevation in [Ca2+]i in the remaining 74% cells (Figure 6A). Intracellular Ca2+ oscillations lasted for at least 30 min, while the transient Ca2+ signal took approximately 13 min to decline to pre-stimulation levels (Figure 6A). In the absence of extracellular Ca2+ (0Ca2+), 20% FBS induced a rapid (≈3 min) increase in [Ca2+]i that reflected endogenous Ca2+ mobilization. The subsequent re-addition of extracellular Ca2+, 100 s after FBS removal from the bath, resulted in a second bump in [Ca2+]i, which was due to extracellular Ca2+ entry and was likely to be mediated by SOCE (Figure 6B). FBS-induced intracellular Ca2+ signals are known to be triggered by InsP3-induced ER Ca2+ mobilization and maintained over time by SOCE (Foreman et al., 2006; Hu et al., 2009; Zuccolo et al., 2018b). Preliminary experiments confirmed that 20% FBS-induced intracellular Ca2+ release was abrogated by depleting the ER Ca2+ store with CPA (30 μM, 30 min) (Figures 6C,D), inhibiting InsP3Rs with 2-APB (50 μM, 30 min) (Figures 6C,D), and blocking PLC with U73122 (10 μM, 10 min) (Figures 6C,D). Furthermore, 20% FBS-induced extracellular Ca2+ entry was significantly (p < 0.001) reduced by inhibiting SOCE with BTP-2 (20 μM, 20 min) or Pyr6 (10 μM, 10 min) (Figures 6E,F).
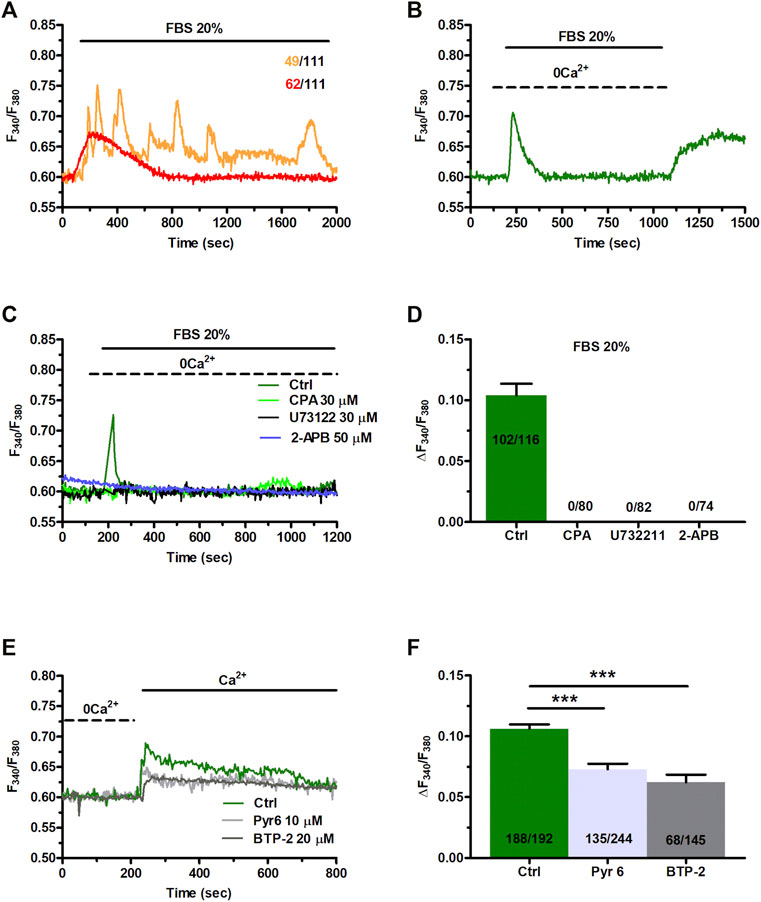
FIGURE 6. Fetal bovine serum (FBS)-induces intracellular Ca2+ release from endogenous stores and enhance SOCE. (A) 20% FBS induced oscillatory or transient increases in [Ca2+]i. (B) Under 0Ca2+ conditions, 20% FBS induced a transient elevation in [Ca2+]i. Subsequent re-addition of extracellular Ca2+, 100 s after agonist removal from the bath, resulted in a second bump in [Ca2+]i that was indicative of SOCE. (C) The intracellular Ca2+ release evoked by 20% FBS (Ctrl) was inhibited by depleting the ER Ca2+ pool with CPA (30 μM, 30 min), by blocking InsP3Rs with 2-APB (50 μM, 30 min), or inhibiting PLC with U73122 (10 μM, 10 min). (D) Mean ± SE of the amplitude of the peak intracellular Ca2+ response to 20% FBS under the designated treatments. (E) Subsequent to store depletion by 20% of FBS application (data are not shown her(E), FBS were washed out from bath, then extracellular Ca2+ added to the bath to bath in the presence and absence of SOCE inhibitors, Pyr6 (10 μm, 10 min) or BTP-2 (20 μm, 20 min). (F) Mean ± SE of the amplitude of Ca2+ entry evoked by 20% FBS in the absence (Ctrl) and presence of Pyr6 and BTP-2. The asterisk indicates ***p < 0.001.
The evidence reported above clearly showed that NAADP-induced lysosomal Ca2+ release via TPCs was able to promote InsP3-induced Ca2+ release from the ER, thereby resulting in SOCE activation on the plasma membrane. Therefore, we sought to assess the role of NAADP-induced lysosomal Ca2+ release in the Ca2+ response to 20% FBS (Figure 7A). The depletion of the lysosomal Ca2+ store with either GPN (200 μM, 30 min) (Figures 7A,B) or nigericin (50 μM, 30 min) (Figures 7A,B) abrogated FBS-induced intracellular Ca2+ mobilization. The same effect was achieved upon pharmacological blockade of TPCs with NED-19 (100 μM, 30 min) (Figures 7C,D), NED-K (10 μM, 30 min) (Figures 7C,D), and tetrandrine (Figures 7C,D). Therefore, NAADP plays a crucial role in igniting the Ca2+ response to 20% FBS in C-MSCs.
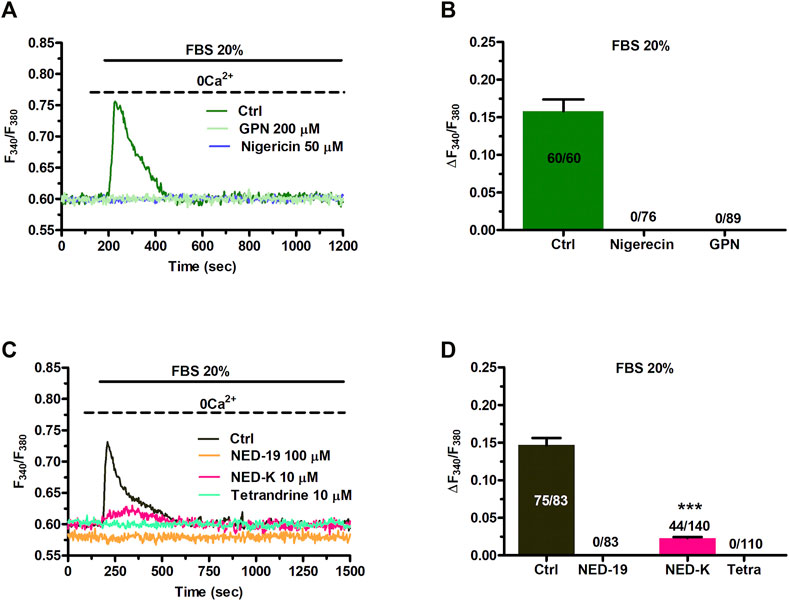
FIGURE 7. TPCs mediate 20% FBS-induced intracellular Ca2+ release. (A) Intracellular Ca2+ release induced by 20% FBS was abolished upon depletion of the lysosomal Ca2+ pool with either GPN (200 μM, 30 min) or nigericin (50 μM, 30 min). (B) Mean ± SE of the amplitude of the intracellular Ca2+ peak evoked by 20% FBS under the designated treatments. (C) 20% FBS induced an intracellular Ca2+ transient that was significantly reduced or inhibited by blocking TPCs with, respectively, NED-K (10 μM, 30 min), tetrandrine (10 μM, 30 min) or NED-19 (100 μM, 30 min). (D) Mean ± SE of the amplitude of the intracellular Ca2+ peak evoked by 20% FBS in the absence (Ctrl) or presence of NED-19, NED-K or tetrandrine (Tetra). Student’s t-test: ***p < 0.001.
3.6 TPCs Mediate 20% FBS-Induced Proliferation and ERK Phosphorylation in Cardiac Mesenchymal Stromal Cells
In order to assess the physiological role of NAADP-induced lysosomal Ca2+ release through TPCs, 20% FBS-induced C-MSC proliferation was evaluated in the absence (Ctrl) and presence of NED-19 (100 μM, 30 min). Figure 8A shows that the pharmacological blockade of TPCs significantly (p < 0.05) reduced the total cell number at 24 and 48 h, thereby showing the crucial role of TPCs in supporting C-MSC proliferation. Flow cytometric analysis of Annexin V fluorescence confirmed that pre-treating C-MSCs with NED-19 did not induce apoptosis (Supplementary Figure S6). In order to determine whether TPCs recruit mitogen-associated protein kinases (MAPKs), we evaluated the phosphorylated levels of the Ca2+-dependent extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) and of the survival kinase, Akt (Zuccolo et al., 2018a; Faris et al., 2019; Negri et al., 2021b). Figures 8B,C illustrate that 20% FBS-induced ERK1/2, but not Akt, phosphorylation was significantly (p < 0.05) inhibited by blocking TPCs with NED-19 (100 μM, 30 min). Overall, these findings demonstrate that NAADP-induced lysosomal Ca2+ release through TPCs stimulates C-MSC proliferation by engaging ERK1/2.
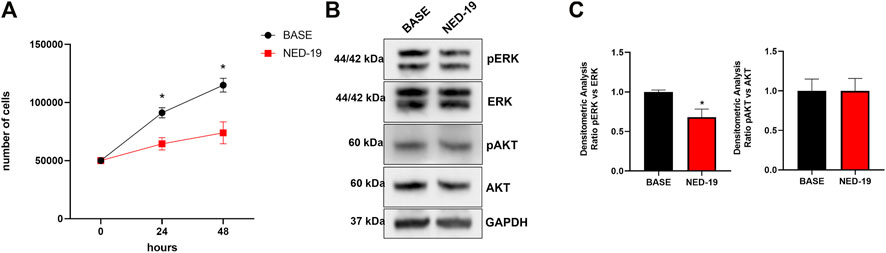
FIGURE 8. TPCs mediate 20% FBS-induced proliferation and ERK phosphorylation in C-MSCs. Following 4 h of growth without serum, cells were treated with NED-19 (100 μM, 30 min) and subsequently stimulated with 20% FBS. (A) Following 24 and 48 h of FBS stimulation, the medium was removed, cells detached from the plates, and counted by hemocytometer (n = 3/group). Student’s t-test: *p < 0.05. (B) The cells were lysate after 60 min of FBS stimulation. Total protein extract from treated cells was subjected to Western blot analysis to visualize active phosphorylated form and total of ERK and AKT using specific antibodies. Phospho-ERK1/2 and Phospho-AKT levels were corrected by total ERK1/2 and AKT densitometry respectively. (C) Western blot data are presented as the fold change of target protein expression. The results are expressed as mean ± SEM (n = 3/group). Student’s t-test: *p < 0.05.
4 Discussion
NAADP is emerging as a crucial regulator of intracellular Ca2+ signalling and Ca2+-dependent processes in the cardiovascular system (Macgregor et al., 2007; Collins et al., 2011; Fameli et al., 2017; Moccia et al., 2021a). C-MSCs represent the large majority of supportive cells in the heart, are critical to normal cardiac function and contribute to maladaptive cardiac remodelling under multiple pathological conditions. Herein, we showed for the first time that NAADP mobilizes EL Ca2+ via TPCs also in C-MSCs. NAADP-evoked intracellular Ca2+ signals are amplified by InsP3-sensitive ER Ca2+ release at lysosomes-ER C-MCSs followed by SOCE activation. The functional crosstalk between NAADP-evoked lysosomal Ca2+ release, InsP3-induced ER Ca2+ mobilization and SOCE sustains FBS-induced intracellular Ca2+ signals and proliferation by promoting ERK phosphorylation.
4.1 Nicotinic Acid Adenine Dinucleotide Phosphate Evokes Complex Ca2+ Signals in Cardiac Mesenchymal Stromal Cells
Intracellular Ca2+ signals tightly control a plethora of crucial functions in human MSCs from multiple sources, as reviewed in Moccia et al. (2015), Forostyak et al. (2016), and Jiang et al. (2017). According to the canonical model, the Ca2+ response evoked by chemical stimulation in human MSCs is triggered by InsP3-dependent ER Ca2+ and may be maintained over time by SOCE (Kawano et al., 2002; Peng et al., 2016; Kotova et al., 2018). Herein, we expanded our knowledge of the molecular mechanisms shaping intracellular Ca2+ signalling in these cells by focusing on the role played by NAADP in C-MSCs. The intracellular delivery of NAADP mobilizes acidic Ca2+ stores throughout the cardiovascular system (Moccia et al., 2021a), e.g., in guinea pig ventricular (Macgregor et al., 2007) and atrial (Collins et al., 2011) cardiomyocytes, rat pulmonary artery VSMCs (Kinnear et al., 2004), human aortic endothelial cells (Brailoiu et al., 2010), mouse brain endothelial cells (Zuccolo et al., 2019), and circulating ECFCs (Di Nezza et al., 2017; Moccia et al., 2021b). Likewise, NAADP-AM, a membrane-permeable analogue of NAADP, could induce either a transient elevation in [Ca2+]i or a burst of intracellular Ca2+ oscillations. This latter observation is in accord with the evidence that: 1) intracellular delivery of NAADP may induce oscillatory Ca2+ signals in human Jurkat T-lymphocytes (Berg et al., 2000), cytotoxic T lymphocytes (Davis et al., 2012), and human pancreatic β-cells (Johnson and Misler, 2002); 2) NAADP contributes to agonist-induced repetitive Ca2+ spikes in several types of endothelial cells (Zuccolo et al., 2019; Berra-Romani et al., 2020; Balducci et al., 2021), and that 3) NAADP induces intracellular Ca2+ oscillations in mouse cardiomyocytes during reperfusion injury (Davidson et al., 2015). Early work conducted on echinoderms first suggested that NAADP was able to elicit repetitive Ca2+ oscillations by promoting a Ca2+-dependent crosstalk between two different Ca2+ pools (Churchill and Galione, 2001), which were later shown to be located in acidic vesicles and ER (Churchill et al., 2002; Moccia et al., 2006).
4.2 Nicotinic Acid Adenine Dinucleotide Phosphate-Induced Intracellular Ca2+ Signals Are Triggered by Lysosomal Ca2+ Release via TPCs, Amplified by InsP3-Evoked ER Ca2+ Release and Maintained by SOCE
The Ca2+ response to NAADP in C-MSCs comprised an early phase of intracellular Ca2+ mobilization followed by a later phase of extracellular Ca2+ entry, which required the previous depletion of the endogenous Ca2+ pool but not the NAADP-AM presence in the perfusate. First, we found that GPN, nigericin, and bafilomycin A1, which provide three established pharmacological tools to mobilize acidic Ca2+ stores (Kilpatrick et al., 2013; Ronco et al., 2015; Morgan and Galione, 2021; Yuan et al., 2021), prevent NAADP-induced intracellular Ca2+ mobilization. In agreement with the hypothesis that the lysosomal compartment represents the primary source of this increase in [Ca2+]i, all of these drugs, as well as NH4Cl, induced a rapid reduction in Lysotracker Red fluorescence. Although a recent investigation questioned the documented GPN ability to release lysosomal Ca2+ (Atakpa et al., 2019), Patel’s group provided the clear-cut evidence that this compound mobilizes Ca2+ from acidic organelles and may, therefore, be safely exploited to probe the endogenous store primarily targeted by NAADP (Morgan et al., 2020; Yuan et al., 2021). We further showed that C-MSCs express both TPC1 and TPC2 transcripts and proteins, and that the Ca2+ response to NAADP was inhibited by blocking TPCs with two selective antagonists, such as NED-19 (Macgregor et al., 2007; Di Nezza et al., 2017; Jin et al., 2020; Moccia et al., 2021a) and NED-K (Davidson et al., 2015), and the traditional Chinese herbal remedy, tetrandrine, which can target both TPC1 and TPC2 (Sakurai et al., 2015; Moccia et al., 2021a). As recently reviewed in Moccia et al. (2021a) and Negri et al. (2021b), TPC1 and TPC2 are both present in mouse ventricular cardiomyocytes, but this is the first time that they were reported in any other cellular component of the human heart. As reviewed in Pitt et al. (2016), TPC1 presents a limited Ca2+ permeability, while TPC2 is predicted to release more Ca2+ upon activation. Nevertheless, it has been shown that even a small Ca2+ flux through TPC1 can generate a global increase in [Ca2+]i when lysosomal vesicles are juxtaposed to ER cisternae (Galione, 2019). For instance, TPC1 alone supports NAADP-induced intracellular Ca2+ oscillations in circulating ECFCs (Di Nezza et al., 2017; Moccia et al., 2021b) and in mouse cardiac myocytes undergoing the ischemia-reperfusion injury (Davidson et al., 2015). Furthermore, TPC1 was sufficient to maintain the intracellular Ca2+ response to nutrients or incretins in mouse pancreatic β cells deficient for TPC2 (Cane et al., 2016). Three pieces of evidence suggest that InsP3Rs in ER cisternae contribute to amplify NAADP-induced lysosomal Ca2+ release. First, depletion of the ER Ca2+ pool with CPA suppressed or attenuated the intracellular Ca2+ release evoked by both NAADP and the H+/K+ antiporter, nigericin. Second, NAADP-induced endogenous Ca2+ mobilization was impaired by inhibiting InsP3Rs with 2-APB and by blocking basal InsP3 production with U73122. Conversely, functional RyRs are absent in C-MSCs (Maione et al., 2020a). The requirement for InsP3Rs to sustain the increase in [Ca2+]i resulting from NAADP-AM-evoked Ca2+ release from lysosomal vesicles is in full agreement with previous work carried out on human fibroblasts (Kilpatrick et al., 2013), human ECFCs (Moccia et al., 2021b), COS-7 cells (Morgan and Galione, 2021), HeLa cells (Ronco et al., 2015), and human metastatic colorectal cancer cells (Faris et al., 2019). Third, TEM revealed clearly discernible ER-lysosomes MCSs, which closely resemble those previously described in human fibroblasts (Kilpatrick et al., 2013) and could provide the sub-cellular framework to enable InsP3R recruitment by local Ca2+ release through TPCs (Penny et al., 2014). Likewise, the MCSs between lysosomal vesicles and ER cisternae in C-MSCs are similar to the cytoplasmic nanojunctions between lysosomes and sarcoplasmic reticulum (SR) recently reported in rat aortic VSMCs (Fameli et al., 2014).
The different extent of coupling between lysosomal TPCs and ER-embedded InsP3Rs (due to changes in either their distribution or density at MCSs) could explain the onset of a long-lasting elevation in [Ca2+]i that replaces the intracellular Ca2+ oscillations in a fraction of C-MSCs. For instance, computational modelling indicated that TPC clustering within the microdomain could accelerate the frequency of InsP3Rs-driven Ca2+ oscillations (Penny et al., 2014), which could ultimately lead to the fusion of the Ca2+ spikes and the occurrence of a single, broader increase in [Ca2+]i (Bartlett et al., 2020).
Removal of extracellular Ca2+ shortened the duration of the Ca2+ response to NAADP-AM. Therefore, NAADP is predicted to gate a Ca2+-permeable pathway on the plasma membrane. This observation is supported by the evidence that restitution of extracellular Ca2+ following exposure to NAADP (or nigericin) under 0Ca2+ conditions, results in a second bump in [Ca2+]i that reflects extracellular Ca2+ entry. This influx of Ca2+ occurs after washout of the agonist from the bath and, therefore, it is exclusively coupled to the previous depletion of endogenous Ca2+ stores. As discussed elsewhere (Yamazaki et al., 2007; Sanchez-Hernandez et al., 2010; Negri et al., 2020), this feature hints at SOCE as being responsible for NAADP-induced extracellular Ca2+ entry. In agreement with this hypothesis, NAADP-evoked Ca2+ influx was remarkably reduced in the presence of either BTP-2 or Pyr6, two different inhibitors of Orai1 channels, which provide the pore-forming subunit of store-operated Ca2+ channels in non-excitable cells (Prakriya and Lewis, 2015; Emrich et al., 2021) and MSCs (Lee et al., 2016; Peng et al., 2016). SOCE activation ultimately results from the reduction of ER Ca2+ concentration (Emrich et al., 2021). As discussed elsewhere (Davis et al., 2012; Brailoiu and Brailoiu, 2016), the engagement of SOCE by NAADP (and nigericin) hints at the depletion of the ER Ca2+ content as the intermediate step between lysosomal Ca2+ release and extracellular Ca2+ entry. However, extracellular Ca2+ entry directly evoked by NAADP delivery was not always engaged during acidic Ca2+ signalling in the cell types where this functional interplay has been investigated (Faris et al., 2019; Moccia et al., 2021b). Therefore, it is conceivable that lysosomal Ca2+ release recruits ER sub-domains that are functionally coupled to the SOCE machinery in C-MSCs, but not in other cell types, as widely discussed in Parekh and Putney (2005). These observations hint at NAADP as a Ca2+-releasing second messenger that can trigger a functional crosstalk among multiple Ca2+ sources (lysosomes, ER, and plasma membrane) in C-MSCs. In these cells, NAADP may serve as a provider of the “trigger” Ca2+ response to extracellular stimulation that is subsequently amplified by InsP3Rs on the ER and maintained over time by SOCE activation on the plasma membrane, as previously reported in many mammalian cells, including human fibroblasts (Kilpatrick et al., 2013), human ECFCs (Moccia et al., 2021b) and brain microvascular endothelial cells (Zuccolo et al., 2019), human metastatic colorectal cancer cells (Faris et al., 2019), human primary CTL cells (Davis et al., 2012), and rat pulmonary artery VSMCs (Kinnear et al., 2004).
4.3 Lysosomal Ca2+ Release via TPCs is Crucial to FBS-Induced Intracellular Ca2+ Signalling and Proliferation in Cardiac Mesenchymal Stromal Cells
It has long been known that FBS stimulates proliferation through an increase in [Ca2+]i that can adopt either a biphasic (Faris et al., 2019) or an oscillatory pattern (Tao et al., 2011) in a variety of cell types, including rat bone marrow MSCs (Foreman et al., 2006). FBS-induced intracellular Ca2+ signals are known to impinge on the interplay between InsP3-induced Ca2+ release from the ER and SOCE (Foreman et al., 2006; Hu et al., 2009). Intriguingly, a recent investigation reported the first evidence that NAADP-evoked lysosomal Ca2+ release via TPC1 interacts with InsP3-dependent ER Ca2+ mobilization and SOCE to promote FBS-induced proliferation in human metastatic colorectal cancer cells (Faris et al., 2019). Unveiling the molecular mechanisms that drive C-MSC proliferation is crucial to improve the therapeutic outcome of regenerative strategies aiming at utilizing these cells to promote cardiac repair (Bagno et al., 2018; Braunwald, 2018). Preliminary analysis showed that FBS evoked a complex increase in [Ca2+]i also in C-MSCs, which displayed either an oscillatory or a biphasic Ca2+ signal. Pharmacological manipulation confirmed that the Ca2+ response to FBS comprised InsP3-induced ER Ca2+ mobilization followed by SOCE activation. Indeed, FBS-induced intracellular Ca2+ release was suppressed by inhibiting InsP3Rs with 2-APB, by blocking basal InsP3 production with U73122 and by depleting the ER Ca2+ store with CPA, whereas FBS-induced extracellular Ca2+ entry was remarkably attenuated by blocking SOCE with BTP-2 and Pyr6. Next, we provided the evidence that the NAADP-sensitive acidic Ca2+ store is crucial to FBS-induced intracellular Ca2+ signals and proliferation in C-MSCs. Indeed, FBS-induced intracellular Ca2+ release was abrogated by depleting the lysosomal Ca2+ store with either GPN or nigericin, as previously shown in human metastatic colorectal cancer cells (Faris et al., 2019). In agreement with these observations, the selective blockade of TPCs with NED-19, NED-K or tetrandrine also abolished the intracellular Ca2+ response to FBS. Therefore, NAADP-induced lysosomal Ca2+ release is indispensable to trigger the cytosolic Ca2+ response to FBS and this requires the functional recruitment of InsP3Rs on the ER via CICR at lysosomal-ER MCSs. That the ER is depleted via InsP3Rs-mediated ER Ca2+ release following NAADP-induced lysosomal Ca2+ mobilization in response to FBS is also suggested by FBS-induced SOCE activation, which requires a reduction in ER Ca2+ concentration (Brailoiu et al., 2009; Davis et al., 2012). The mechanism whereby FBS stimulation results to intracellular NAADP generation in C-MSCs, as well as in human metastatic cancer cells (Faris et al., 2019), remains to be elucidated. Nevertheless, FBS is likely to engage the multifunctional enzyme CD38, which catalyses the “base exchange” of the nicotinamide moiety of NADP with nicotinic acid, thereby resulting in NAADP production in most cell types (Galione, 2015), including cardiomyocytes (Negri et al., 2021b). A recent paper suggested that the dual NADPH oxidases, DUOX1 and DUOX2, contribute to NAADP biosynthesis in murine T lymphocytes (Gu et al., 2021), but their role in NAADP-dependent Ca2+ response to FBS is yet to be investigated.
The physiological role of NAADP-induced intracellular Ca2+ signals were further assessed by evaluating the effect of NED-19 on C-MSC proliferation. The pharmacological blockade of TPCs with NED-19 strongly reduced FBS-induced C-MSC proliferation at 24 and 48 h. Preliminary experiments indicated that the massive release of Ca2+ induced by nigericin per se resulted in C-MSC cell death already at 24 h from exposure to this lysosomotropic compound. While this observation is in accord with the reported effects of nigericin on various cell models (Murakami et al., 2012), it prevented us from probing its ability to interfere with FBS-induced proliferation. Previous work showed that NAADP-induced Ca2+ release may stimulate proliferation by recruiting the Ca2+-dependent ERK1/2 and Akt signalling pathways (Faris et al., 2019; Negri et al., 2021b). Consistently, FBS-induced ERK1/2 phosphorylation was impaired by the pharmacological blockade of TPCs with NED-19, whereas Akt engagement was unaffected. Interestingly, ERK1/2, but not Akt, was harnessed by intracellular Ca2+ oscillations to drive FBS-induced proliferation also in human bone marrow MSCs (Tao et al., 2011). Additionally, NAADP-induced intracellular Ca2+ oscillations could underpin another crucial function of C-MSCs, i.e., the regulation of extracellular matrix (ECM) composition (Maione et al., 2020b). For instance, bone marrow-derived human MSCs exhibited repetitive Ca2+ spikes during aligned collagen matrix formation (Gilchrist et al., 2019), whereas extracellular Ca2+ entry in human airway epithelial cells drives the expression and secretion of matrix-degrading enzymes, such as matrix metalloprotease 1 (Li et al., 2011). Interestingly, an increase in [Ca2+]i in cardiac fibroblasts may also regulate collagen remodelling in mouse hearts (Adapala et al., 2020). Therefore, future studies will have to assess the role of NAADP-induced Ca2+ signalling in the modulation of ECM composition by C-MSCs.
In conclusion, this study demonstrated that NAADP induces intracellular Ca2+ signals in C-MSCs by promoting lysosomal Ca2+ release via TPCs that is in turn amplified by ER-embedded InsP3Rs at lysosomal-ER MCSs. The following depletion of the ER Ca2+ pool activates SOCE, which prolongs the Ca2+ response to NAADP. FBS impinges on the NAADP-induced Ca2+-dependent crosstalk between lysosomes and ER to stimulate proliferation through the Ca2+-dependent ERK1/2 signalling pathway. These findings pave the way for future studies assessing whether NAADP signalling in C-MSCs could be targeted to favour cardiac repair upon an ischemic insult or to other pathologies associated to maladaptive cardiac remodelling, such as ACM, heart failure and cardiac fibrosis.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the IEO-CCM IRCCS Ethic Committee (project CCM1072). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
FM conceived and directed the project in collaboration with AM. FM, MB, and AM conceived the experiments. PF, CC, AM, SN, and LI performed the experiments and analysed the data. All authors contributed to the article and approved the submitted version.
Funding
The authors gratefully acknowledge financial support from: Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022)—Department of Biology and Biotechnology “L. Spallanzani,” University of Pavia (MB and FM); Fondo Ricerca Giovani from the University of Pavia (MB and FM); EU Horizon 2020 FETOPEN-2018-2020 Programme “LION-HEARTED,” grant agreement No. 828984 (FM); Italian Ministry of Health—RCXXXXXX—(AM); “Fondazione di Comunità di Milano” and “Fondo Giacomo Ponzone” (AM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors gratefully acknowledge the Laboratory of Electron Transmission Microscopy, Centro Grandi Strumenti of the University Pavia, for excellent technical and scientific support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.874043/full#supplementary-material
References
Adapala, R. K., Kanugula, A. K., Paruchuri, S., Chilian, W. M., and Thodeti, C. K. (2020). TRPV4 Deletion Protects Heart from Myocardial Infarction-Induced Adverse Remodeling via Modulation of Cardiac Fibroblast Differentiation. Basic Res. Cardiol. 115, 14. doi:10.1007/s00395-020-0775-5
Atakpa, P., Van Marrewijk, L. M., Apta-Smith, M., Chakraborty, S., and Taylor, C. W. (2019). GPN Does Not Release Lysosomal Ca2+ but Evokes Ca2+ Release from the ER by Increasing the Cytosolic pH Independently of Cathepsin C. J. Cel Sci 132. doi:10.1242/jcs.223883
Bagno, L., Hatzistergos, K. E., Balkan, W., and Hare, J. M. (2018). Mesenchymal Stem Cell-Based Therapy for Cardiovascular Disease: Progress and Challenges. Mol. Ther. 26, 1610–1623. doi:10.1016/j.ymthe.2018.05.009
Balducci, V., Faris, P., Balbi, C., Costa, A., Negri, S., Rosti, V., et al. (2021). The Human Amniotic Fluid Stem Cell Secretome Triggers Intracellular Ca2+ Oscillations, NF‐κB Nuclear Translocation and Tube Formation in Human Endothelial colony‐forming Cells. J. Cel Mol Med 25, 8074–8086. doi:10.1111/jcmm.16739
Bartlett, P. J., Cloete, I., Sneyd, J., and Thomas, A. P. (2020). IP3-Dependent Ca2+ Oscillations Switch into a Dual Oscillator Mechanism in the Presence of PLC-Linked Hormones. iScience 23, 101062. doi:10.1016/j.isci.2020.101062
Berg, I., Potter, B. V. L., Mayr, G. W., and Guse, A. H. (2000). Nicotinic Acid Adenine Dinucleotide Phosphate (Naadp+) Is an Essential Regulator of T-Lymphocyte Ca2+-Signaling. J. Cel Biol 150, 581–588. doi:10.1083/jcb.150.3.581
Berra-Romani, R., Faris, P., Pellavio, G., Orgiu, M., Negri, S., Forcaia, G., et al. (2020). Histamine Induces Intracellular Ca2+ Oscillations and Nitric Oxide Release in Endothelial Cells from Brain Microvascular Circulation. J. Cel Physiol 235, 1515–1530. doi:10.1002/jcp.29071
Brailoiu, G. C., Brailoiu, E., Parkesh, R., Galione, A., Churchill, G. C., Patel, S., et al. (2009). NAADP-mediated Channel 'chatter' in Neurons of the Rat Medulla Oblongata. Biochem. J. 419 (91-97), 91–97. doi:10.1042/BJ20081138
Brailoiu, G. C., and Brailoiu, E. (2016). Modulation of Calcium Entry by the Endo-Lysosomal System. Adv. Exp. Med. Biol. 898, 423–447. doi:10.1007/978-3-319-26974-0_18
Brailoiu, G. C., Gurzu, B., Gao, X., Parkesh, R., Aley, P. K., Trifa, D. I., et al. (2010). Acidic NAADP-Sensitive Calcium Stores in the Endothelium. J. Biol. Chem. 285, 37133–37137. doi:10.1074/jbc.c110.169763
Braunwald, E. (2018). Cell-Based Therapy in Cardiac Regeneration. Circ. Res. 123, 132–137. doi:10.1161/circresaha.118.313484
Brown, R. D., Ambler, S. K., Mitchell, M. D., and Long, C. S. (2005). The Cardiac Fibroblast: Therapeutic Target in Myocardial Remodeling and Failure. Annu. Rev. Pharmacol. Toxicol. 45, 657–687. doi:10.1146/annurev.pharmtox.45.120403.095802
Camelliti, P., Borg, T., and Kohl, P. (2005). Structural and Functional Characterisation of Cardiac Fibroblasts. Cardiovasc. Res. 65, 40–51. doi:10.1016/j.cardiores.2004.08.020
Cane, M. C., Parrington, J., Rorsman, P., Galione, A., and Rutter, G. A. (2016). The Two Pore Channel TPC2 Is Dispensable in Pancreatic β-cells for normal Ca2+ Dynamics and Insulin Secretion. Cell Calcium 59, 32–40. doi:10.1016/j.ceca.2015.12.004
Capel, R. A., Bolton, E. L., Lin, W. K., Aston, D., Wang, Y., Liu, W., et al. (2015). Two-pore Channels (TPC2s) and Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) at Lysosomal-Sarcoplasmic Reticular Junctions Contribute to Acute and Chronic β-Adrenoceptor Signaling in the Heart. J. Biol. Chem. 290, 30087–30098. doi:10.1074/jbc.m115.684076
Carriero, F., Martinelli, C., Gabriele, F., Barbieri, G., Zanoletti, L., Milanesi, G., et al. (2021). Berberine Photo-Activation Potentiates Cytotoxicity in Human Astrocytoma Cells through Apoptosis Induction. J. Pers Med. 11. doi:10.3390/jpm11100942
Christensen, K. A., Myers, J. T., and Swanson, J. A. (2002). pH-dependent Regulation of Lysosomal Calcium in Macrophages. J. Cel Sci 115, 599–607. doi:10.1242/jcs.115.3.599
Churchill, G. C., and Galione, A. (2001). NAADP Induces Ca2+ Oscillations via a Two-Pool Mechanism by Priming IP3- and cADPR-Sensitive Ca2+ Stores. EMBO J. 20, 2666–2671. doi:10.1093/emboj/20.11.2666
Churchill, G. C., Okada, Y., Thomas, J. M., Genazzani, A. A., Patel, S., and Galione, A. (2002). NAADP Mobilizes Ca2+ from Reserve Granules, Lysosome-Related Organelles, in Sea Urchin Eggs. Cell 111, 703–708. doi:10.1016/s0092-8674(02)01082-6
Collins, T. P., Bayliss, R., Churchill, G. C., Galione, A., and Terrar, D. A. (2011). NAADP Influences Excitation-Contraction Coupling by Releasing Calcium from Lysosomes in Atrial Myocytes. Cell Calcium 50, 449–458. doi:10.1016/j.ceca.2011.07.007
Czapla, J., Matuszczak, S., Wiśniewska, E., Jarosz-Biej, M., Smolarczyk, R., Cichoń, T., et al. (2016). Human Cardiac Mesenchymal Stromal Cells with CD105+CD34- Phenotype Enhance the Function of Post-Infarction Heart in Mice. PLoS One 11, e0158745. doi:10.1371/journal.pone.0158745
Davidson, S. M., Foote, K., Kunuthur, S., Gosain, R., Tan, N., Tyser, R., et al. (2015). Inhibition of NAADP Signalling on Reperfusion Protects the Heart by Preventing Lethal Calcium Oscillations via Two-Pore Channel 1 and Opening of the Mitochondrial Permeability Transition Pore. Cardiovasc. Res. 108, 357–366. doi:10.1093/cvr/cvv226
Davis, L. C., Morgan, A. J., Chen, J.-L., Snead, C. M., Bloor-Young, D., Shenderov, E., et al. (2012). NAADP Activates Two-Pore Channels on T Cell Cytolytic Granules to Stimulate Exocytosis and Killing. Curr. Biol. 22, 2331–2337. doi:10.1016/j.cub.2012.10.035
Di Nezza, F., Zuccolo, E., Poletto, V., Rosti, V., De Luca, A., Moccia, F., et al. (2017). Liposomes as a Putative Tool to Investigate NAADP Signaling in Vasculogenesis. J. Cel. Biochem. 118, 3722–3729. doi:10.1002/jcb.26019
Diedrichs, F., Stolk, M., Jürchott, K., Haag, M., Sittinger, M., and Seifert, M. (2019). Enhanced Immunomodulation in Inflammatory Environments Favors Human Cardiac Mesenchymal Stromal-like Cells for Allogeneic Cell Therapies. Front. Immunol. 10, 1716. doi:10.3389/fimmu.2019.01716
Emrich, S. M., Yoast, R. E., and Trebak, M. (2021). Physiological Functions of CRAC Channels. Annu. Rev. Physiol. 84:355. doi:10.1146/annurev-physiol-052521-013426
Fameli, N., Evans, A. M., and Van Breemen, C. (2017). Tissue Specificity: The Role of Organellar Membrane Nanojunctions in Smooth Muscle Ca2+ Signaling. Adv. Exp. Med. Biol. 993, 321–342. doi:10.1007/978-3-319-57732-6_17
Fameli, N., Ogunbayo, O. A., Van Breemen, C., and Evans, A. M. (2014). Cytoplasmic Nanojunctions between Lysosomes and Sarcoplasmic Reticulum Are Required for Specific Calcium Signaling. F1000Res 3, 93. doi:10.12688/f1000research.3720.1
Faris, P., Pellavio, G., Ferulli, F., Di Nezza, F., Shekha, M., Lim, D., et al. (2019). Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca2+ Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers (Basel) 11. E542. doi:10.3390/cancers11040542
Faris, P., Shekha, M., Montagna, D., Guerra, G., and Moccia, F. (2018). Endolysosomal Ca(2+) Signalling and Cancer Hallmarks: Two-Pore Channels on the Move. TRPML1 Lags behind!. Cancers (Base1) 11, 27. doi:10.3390/cancers11010027
Favia, A., Desideri, M., Gambara, G., D'alessio, A., Ruas, M., Esposito, B., et al. (2014). VEGF-induced Neoangiogenesis Is Mediated by NAADP and Two-Pore Channel-2-dependent Ca2+ Signaling. Proc. Natl. Acad. Sci. 111, E4706–E4715. doi:10.1073/pnas.1406029111
Ferrera, L., Barbieri, R., Picco, C., Zuccolini, P., Remigante, A., Bertelli, S., et al. (2021). TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase. Int. J. Mol. Sci. 22. doi:10.3390/ijms22168359
Foreman, M. A., Smith, J., and Publicover, S. J. (2006). Characterisation of Serum-Induced Intracellular Ca2+ Oscillations in Primary Bone Marrow Stromal Cells. J. Cel. Physiol. 206, 664–671. doi:10.1002/jcp.20521
Forostyak, O., Forostyak, S., Kortus, S., Sykova, E., Verkhratsky, A., and Dayanithi, G. (2016). Physiology of Ca2+ Signalling in Stem Cells of Different Origins and Differentiation Stages. Cell Calcium 59, 57–66. doi:10.1016/j.ceca.2016.02.001
Galione, A. (2019). NAADP Receptors. Cold Spring Harb Perspect. Biol. 11. doi:10.1101/cshperspect.a035071
Galione, A. (2015). A Primer of NAADP-Mediated Ca2+ Signalling: From Sea Urchin Eggs to Mammalian Cells. Cell Calcium 58, 27–47. doi:10.1016/j.ceca.2014.09.010
Garrity, A. G., Wang, W., Collier, C. M., Levey, S. A., Gao, Q., and Xu, H. (2016). The Endoplasmic Reticulum, Not the pH Gradient, Drives Calcium Refilling of Lysosomes. Elife 5. doi:10.7554/eLife.15887
Gilchrist, C. L., Leddy, H. A., Kaye, L., Case, N. D., Rothenberg, K. E., Little, D., et al. (2019). TRPV4-mediated Calcium Signaling in Mesenchymal Stem Cells Regulates Aligned Collagen Matrix Formation and Vinculin Tension. Proc. Natl. Acad. Sci. USA 116, 1992–1997. doi:10.1073/pnas.1811095116
Gu, F., Krüger, A., Roggenkamp, H. G., Alpers, R., Lodygin, D., Jaquet, V., et al. (2021). Dual NADPH Oxidases DUOX1 and DUOX2 Synthesize NAADP and Are Necessary for Ca2+ Signaling during T Cell Activation. Sci. Signal. 14, eabe3800. doi:10.1126/scisignal.abe3800
Gunaratne, G. S., Brailoiu, E., He, S., Unterwald, E. M., Patel, S., Slama, J. T., et al. (2021). Essential Requirement for JPT2 in NAADP-Evoked Ca2+ Signaling. Sci. Signal. 14. doi:10.1126/scisignal.abd5605
Hu, R., He, M.-l., Hu, H., Yuan, B.-X., Zang, W.-J., Lau, C.-P., et al. (2009). Characterization of Calcium Signaling Pathways in Human Preadipocytes. J. Cel. Physiol. 220, 765–770. doi:10.1002/jcp.21823
Hu, W., Zhao, F., Chen, L., Ni, J., and Jiang, Y. (2021). NAADP‐induced Intracellular Calcium Ion Is Mediated by the TPCs (Two‐pore Channels) in Hypoxia‐induced Pulmonary Arterial Hypertension. J. Cel Mol Med 25, 7485–7499. doi:10.1111/jcmm.16783
Jairaman, A., Yamashita, M., Schleimer, R. P., and Prakriya, M. (2015). Store-Operated Ca2+ Release-Activated Ca2+ Channels Regulate PAR2-Activated Ca2+ Signaling and Cytokine Production in Airway Epithelial Cells. J.I. 195, 2122–2133. doi:10.4049/jimmunol.1500396
Jiang, L.-H., Mousawi, F., Yang, X., and Roger, S. (2017). ATP-induced Ca2+-Signalling Mechanisms in the Regulation of Mesenchymal Stem Cell Migration. Cell. Mol. Life Sci. 74, 3697–3710. doi:10.1007/s00018-017-2545-6
Jiang, Y.-L., Lin, A. H. Y., Xia, Y., Lee, S., Paudel, O., Sun, H., et al. (2013). Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Activates Global and Heterogeneous Local Ca2+ Signals from NAADP- and Ryanodine Receptor-Gated Ca2+ Stores in Pulmonary Arterial Myocytes. J. Biol. Chem. 288, 10381–10394. doi:10.1074/jbc.m112.423053
Jiang, Y., Zhou, Y., Peng, G., Tian, H., Pan, D., Liu, L., et al. (2018). Two-pore Channels Mediated Receptor-Operated Ca2+ Entry in Pulmonary Artery Smooth Muscle Cells in Response to Hypoxia. Int. J. Biochem. Cel Biol. 97, 28–35. doi:10.1016/j.biocel.2018.01.012
Jin, X., Zhang, Y., Alharbi, A., Hanbashi, A., Alhoshani, A., and Parrington, J. (2020). Targeting Two-Pore Channels: Current Progress and Future Challenges. Trends Pharmacol. Sci. 41, 582–594. doi:10.1016/j.tips.2020.06.002
Johnson, J. D., and Misler, S. (2002). Nicotinic Acid-Adenine Dinucleotide Phosphate-Sensitive Calcium Stores Initiate Insulin Signaling in Human Beta Cells. Proc. Natl. Acad. Sci. 99, 14566–14571. doi:10.1073/pnas.222099799
Jugdutt, B. I. (2003). Ventricular Remodeling after Infarction and the Extracellular Collagen Matrix. Circulation 108, 1395–1403. doi:10.1161/01.cir.0000085658.98621.49
Kakkar, R., and Lee, R. T. (2010). Intramyocardial Fibroblast Myocyte Communication. Circ. Res. 106, 47–57. doi:10.1161/circresaha.109.207456
Kawano, S., Otsu, K., Kuruma, A., Shoji, S., Yanagida, E., Muto, Y., et al. (2006). ATP Autocrine/paracrine Signaling Induces Calcium Oscillations and NFAT Activation in Human Mesenchymal Stem Cells. Cell Calcium 39, 313–324. doi:10.1016/j.ceca.2005.11.008
Kawano, S., Otsu, K., Shoji, S., Yamagata, K., and Hiraoka, M. (2003). Ca2+ Oscillations Regulated by Na+ -Ca2+ Exchanger and Plasma Membrane Ca2+ Pump Induce Fluctuations of Membrane Currents and Potentials in Human Mesenchymal Stem Cells. Cell Calcium 34, 145–156. doi:10.1016/s0143-4160(03)00069-1
Kawano, S., Shoji, S., Ichinose, S., Yamagata, K., Tagami, M., and Hiraoka, M. (2002). Characterization of Ca2+ Signaling Pathways in Human Mesenchymal Stem Cells. Cell Calcium 32, 165–174. doi:10.1016/s0143416002001240
Kilpatrick, B. S., Yates, E., Grimm, C., Schapira, A. H., and Patel, S. (2016). Endo-lysosomal TRP Mucolipin-1 Channels Trigger Global ER Ca2+ Release and Ca2+ Influx. J. Cel Sci 129, 3859–3867. doi:10.1242/jcs.190322
Kilpatrick, B. S., Eden, E. R., Schapira, A. H., Futter, C. E., and Patel, S. (2013). Direct Mobilisation of Lysosomal Ca2+ Triggers Complex Ca2+ Signals. J. Cel Sci 126, 60–66. doi:10.1242/jcs.118836
Kinnear, N. P., Boittin, F.-X., Thomas, J. M., Galione, A., and Evans, A. M. (2004). Lysosome-Sarcoplasmic Reticulum Junctions. J. Biol. Chem. 279, 54319–54326. doi:10.1074/jbc.m406132200
Kotova, P. D., Bystrova, M. F., Rogachevskaja, O. A., Khokhlov, A. A., Sysoeva, V. Y., Tkachuk, V. A., et al. (2018). Coupling of P2Y Receptors to Ca2+ Mobilization in Mesenchymal Stromal Cells from the Human Adipose Tissue. Cell Calcium 71, 1–14. doi:10.1016/j.ceca.2017.11.001
Lee, S. H., Park, Y., Song, M., Srikanth, S., Kim, S., Kang, M. K., et al. (2016). Orai1 Mediates Osteogenic Differentiation via BMP Signaling Pathway in Bone Marrow Mesenchymal Stem Cells. Biochem. Biophysical Res. Commun. 473, 1309–1314. doi:10.1016/j.bbrc.2016.04.068
Lee, S., Paudel, O., Jiang, Y., Yang, X.-R., and Sham, J. S. K. (2015). CD38 Mediates Angiotensin II-Induced Intracellular Ca2+Release in Rat Pulmonary Arterial Smooth Muscle Cells. Am. J. Respir. Cel Mol Biol 52, 332–341. doi:10.1165/rcmb.2014-0141oc
Lewis, A. M., Aley, P. K., Roomi, A., Thomas, J. M., Masgrau, R., Garnham, C., et al. (2012). SS-Adrenergic Receptor Signaling Increases NAADP and cADPR Levels in the Heart. Biochem. Biophysical Res. Commun. 427, 326–329. doi:10.1016/j.bbrc.2012.09.054
Li, J., Kanju, P., Patterson, M., Chew, W.-L., Cho, S.-H., Gilmour, I., et al. (2011). TRPV4-mediated Calcium Influx into Human Bronchial Epithelia upon Exposure to Diesel Exhaust Particles. Environ. Health Perspect. 119, 784–793. doi:10.1289/ehp.1002807
Macgregor, A., Yamasaki, M., Rakovic, S., Sanders, L., Parkesh, R., Churchill, G. C., et al. (2007). NAADP Controls Cross-Talk between Distinct Ca2+ Stores in the Heart. J. Biol. Chem. 282, 15302–15311. doi:10.1074/jbc.m611167200
Maione, A. S., Faris, P. S., Bisonni, L., Lodola, F., Casella, M., Catto, V., et al. (2020a). Imbalance of Calcium-dependent Mechanisms in Cardiac Mesenchymal Stromal Cells from Arrhythmogenic Cardiomyopathy Patients. Vasc. Pharmacol. 132, 106732. doi:10.1016/j.vph.2020.106732
Maione, A. S., Pilato, C. A., Casella, M., Gasperetti, A., Stadiotti, I., Pompilio, G., et al. (2020b). Fibrosis in Arrhythmogenic Cardiomyopathy: The Phantom Thread in the Fibro-Adipose Tissue. Front. Physiol. 11, 279. doi:10.3389/fphys.2020.00279
Maione, A. S., Stadiotti, I., Pilato, C. A., Perrucci, G. L., Saverio, V., Catto, V., et al. (2021). Excess TGF-Beta1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-fibrotic Commitment in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 22. doi:10.3390/ijms22052673
Moccia, F., Lodola, F., Stadiotti, I., Pilato, C. A., Bellin, M., Carugo, S., et al. (2019). Calcium as a Key Player in Arrhythmogenic Cardiomyopathy: Adhesion Disorder or Intracellular Alteration? Int. J. Mol. Sci. 20. doi:10.3390/ijms20163986
Moccia, F., Negri, S., Faris, P., Perna, A., De Luca, A., Soda, T., et al. (2021a). Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol. 12, 629119. doi:10.3389/fphys.2021.629119
Moccia, F., Nusco, G. A., Lim, D., Kyozuka, K., and Santella, L. (2006). NAADP and InsP3 Play Distinct Roles at Fertilization in Starfish Oocytes. Develop. Biol. 294, 24–38. doi:10.1016/j.ydbio.2006.02.011
Moccia, F., Ruffinatti, F., and Zuccolo, E. (2015). Intracellular Ca2+ Signals to Reconstruct A Broken Heart: Still A Theoretical Approach? Cdt 16, 793–815. doi:10.2174/1389450116666141219121723
Moccia, F., Zuccolo, E., Di Nezza, F., Pellavio, G., Faris, P. S., Negri, S., et al. (2021b). Nicotinic Acid Adenine Dinucleotide Phosphate Activates Two‐pore Channel TPC1 to Mediate Lysosomal Ca2+ Release in Endothelial colony‐forming Cells. J. Cel Physiol 236, 688–705. doi:10.1002/jcp.29896
Moccia, F., Zuccolo, E., Poletto, V., Turin, I., Guerra, G., Pedrazzoli, P., et al. (2016). Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy. Cmc 23, 3450–3480. doi:10.2174/0929867323666160607111220
Morgan, A. J., and Galione, A. (2021). Lysosomal Agents Inhibit Store-Operated Ca2+ Entry. J. Cel Sci 134. doi:10.1242/jcs.248658
Morgan, A. J., Platt, F. M., Lloyd-Evans, E., and Galione, A. (2011). Molecular Mechanisms of Endolysosomal Ca2+ Signalling in Health and Disease. Biochem. J. 439, 349–378. doi:10.1042/bj20110949
Morgan, A. J., Yuan, Y., Patel, S., and Galione, A. (2020). Does Lysosomal Rupture Evoke Ca2+ Release? A Question of Pores and Stores. Cell Calcium 86, 102139. doi:10.1016/j.ceca.2019.102139
Murakami, T., Ockinger, J., Yu, J., Byles, V., Mccoll, A., Hofer, A. M., et al. (2012). Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc. Natl. Acad. Sci. 109, 11282–11287. doi:10.1073/pnas.1117765109
Nebel, M., Schwoerer, A. P., Warszta, D., Siebrands, C. C., Limbrock, A.-C., Swarbrick, J. M., et al. (2013). Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP)-mediated Calcium Signaling and Arrhythmias in the Heart Evoked by β-Adrenergic Stimulation. J. Biol. Chem. 288, 16017–16030. doi:10.1074/jbc.m112.441246
Negri, S., Faris, P., Maniezzi, C., Pellavio, G., Spaiardi, P., Botta, L., et al. (2021a). NMDA Receptors Elicit Flux-independent Intracellular Ca2+ Signals via Metabotropic Glutamate Receptors and Flux-dependent Nitric Oxide Release in Human Brain Microvascular Endothelial Cells. Cell Calcium 99, 102454. doi:10.1016/j.ceca.2021.102454
Negri, S., Faris, P., and Moccia, F. (2021b). Endolysosomal Ca2+ Signaling in Cardiovascular Health and Disease. Int. Rev. Cel Mol Biol 363, 203–269. doi:10.1016/bs.ircmb.2021.03.001
Negri, S., Faris, P., Pellavio, G., Botta, L., Orgiu, M., Forcaia, G., et al. (2020). Group 1 Metabotropic Glutamate Receptors Trigger Glutamate-Induced Intracellular Ca2+ Signals and Nitric Oxide Release in Human Brain Microvascular Endothelial Cells. Cel. Mol. Life Sci. 77, 2235–2253. doi:10.1007/s00018-019-03284-1
Pandey, V., Chuang, C.-C., Lewis, A. M., Aley, P. K., Brailoiu, E., Dun, N. J., et al. (2009). Recruitment of NAADP-Sensitive Acidic Ca2+ Stores by Glutamate. Biochem. J. 422, 503–512. doi:10.1042/bj20090194
Parekh, A. B., and Putney, J. W. (2005). Store-operated Calcium Channels. Physiol. Rev. 85, 757–810. doi:10.1152/physrev.00057.2003
Patel, S. (2015). Function and Dysfunction of Two-Pore Channels. Sci. Signal. 8, re7. doi:10.1126/scisignal.aab3314
Peng, H., Hao, Y., Mousawi, F., Roger, S., Li, J., Sim, J. A., et al. (2016). Purinergic and Store-Operated Ca2+ Signaling Mechanisms in Mesenchymal Stem Cells and Their Roles in ATP-Induced Stimulation of Cell Migration. Stem Cells 34, 2102–2114. doi:10.1002/stem.2370
Penny, C. J., Kilpatrick, B. S., Han, J. M., Sneyd, J., and Patel, S. (2014). A Computational Model of Lysosome-ER Ca2+ Microdomains. J. Cel Sci 127, 2934–2943. doi:10.1242/jcs.149047
Pereira, G. J. S., Hirata, H., Do Carmo, L. G., Stilhano, R. S., Ureshino, R. P., Medaglia, N. C., et al. (2014). NAADP-sensitive Two-Pore Channels Are Present and Functional in Gastric Smooth Muscle Cells. Cell Calcium 56, 51–58. doi:10.1016/j.ceca.2014.04.005
Pilato, C. A., Stadiotti, I., Maione, A. S., Saverio, V., Catto, V., Tundo, F., et al. (2018). Isolation and Characterization of Cardiac Mesenchymal Stromal Cells from Endomyocardial Bioptic Samples of Arrhythmogenic Cardiomyopathy Patients. J. Vis. Exp. 132, e57263. doi:10.3791/57263
Pitt, S. J., Reilly-O'donnell, B., and Sitsapesan, R. (2016). Exploring the Biophysical Evidence that Mammalian Two-Pore Channels Are NAADP-Activated Calcium-Permeable Channels. J. Physiol. 594, 4171–4179. doi:10.1113/jp270936
Prakriya, M., and Lewis, R. S. (2015). Store-Operated Calcium Channels. Physiol. Rev. 95, 1383–1436. doi:10.1152/physrev.00020.2014
Rahman, S., and Rahman, T. (2017). Unveiling Some FDA-Approved Drugs as Inhibitors of the Store-Operated Ca2+ Entry Pathway. Sci. Rep. 7, 12881. doi:10.1038/s41598-017-13343-x
Remigante, A., Zuccolini, P., Barbieri, R., Ferrera, L., Morabito, R., Gavazzo, P., et al. (2021). NS-11021 Modulates Cancer-Associated Processes Independently of BK Channels in Melanoma and Pancreatic Duct Adenocarcinoma Cell Lines. Cancers (Basel) 13. doi:10.3390/cancers13236144
Ronco, V., Potenza, D. M., Denti, F., Vullo, S., Gagliano, G., Tognolina, M., et al. (2015). A Novel Ca2+-Mediated Cross-Talk between Endoplasmic Reticulum and Acidic Organelles: Implications for NAADP-dependent Ca2+ Signalling. Cell Calcium 57, 89–100. doi:10.1016/j.ceca.2015.01.001
Ruas, M., Rietdorf, K., Arredouani, A., Davis, L. C., Lloyd-Evans, E., Koegel, H., et al. (2010). Purified TPC Isoforms Form NAADP Receptors with Distinct Roles for Ca2+ Signaling and Endolysosomal Trafficking. Curr. Biol. 20, 703–709. doi:10.1016/j.cub.2010.02.049
Sakurai, Y., Kolokoltsov, A. A., Chen, C.-C., Tidwell, M. W., Bauta, W. E., Klugbauer, N., et al. (2015). Two-pore Channels Control Ebola Virus Host Cell Entry and Are Drug Targets for Disease Treatment. Science 347, 995–998. doi:10.1126/science.1258758
Sánchez-Hernández, Y., Laforenza, U., Bonetti, E., Fontana, J., Dragoni, S., Russo, M., et al. (2010). Store-Operated Ca2+ Entry Is Expressed in Human Endothelial Progenitor Cells. Stem Cell Develop. 19, 1967–1981. doi:10.1089/scd.2010.0047
Scarpellino, G., Genova, T., Avanzato, D., Bernardini, M., Bianco, S., Petrillo, S., et al. (2019). Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers (Basel). 11, 766. doi:10.3390/cancers11060766
Schach, C., Wester, M., Leibl, F., Redel, A., Gruber, M., Maier, L. S., et al. (2020). Reduced Store‐operated Ca2+ Entry Impairs Mesenteric Artery Function in Response to High External Glucose in Type 2 Diabetic ZDF Rats. Clin. Exp. Pharmacol. Physiol. 47, 1145–1157. doi:10.1111/1440-1681.13300
Schleifer, H., Doleschal, B., Lichtenegger, M., Oppenrieder, R., Derler, I., Frischauf, I., et al. (2012). Novel Pyrazole Compounds for Pharmacological Discrimination between Receptor‐operated and Store‐operated Ca2+ Entry Pathways. Br. J. Pharmacol. 167, 1712–1722. doi:10.1111/j.1476-5381.2012.02126.x
Sommariva, E., Brambilla, S., Carbucicchio, C., Gambini, E., Meraviglia, V., Dello Russo, A., et al. (2016). Cardiac Mesenchymal Stromal Cells Are a Source of Adipocytes in Arrhythmogenic Cardiomyopathy. Eur. Heart J. 37, 1835–1846. doi:10.1093/eurheartj/ehv579
Stadiotti, I., Catto, V., Casella, M., Tondo, C., Pompilio, G., and Sommariva, E. (2017). Arrhythmogenic Cardiomyopathy: the Guilty Party in Adipogenesis. J. Cardiovasc. Trans. Res. 10, 446–454. doi:10.1007/s12265-017-9767-8
Tao, R., Sun, H.-Y., Lau, C.-P., Tse, H.-F., Lee, H.-C., and Li, G.-R. (2011). Cyclic ADP Ribose Is a Novel Regulator of Intracellular Ca2+ Oscillations in Human Bone Marrow Mesenchymal Stem Cells. J. Cel Mol Med 15, 2684–2696. doi:10.1111/j.1582-4934.2011.01263.x
Trufanov, S. K., Rybakova, E. Y., Avdonin, P. P., Tsitrina, A. A., Zharkikh, I. L., Goncharov, N. V., et al. (2019). The Role of Two-Pore Channels in Norepinephrine-Induced [Ca(2+)]i Rise in Rat Aortic Smooth Muscle Cells and Aorta Contraction. Cells 8. 1144. doi:10.3390/cells8101144
Vassileva, K., Marsh, M., and Patel, S. (2020). Two-pore Channels as Master Regulators of Membrane Trafficking and Endocytic Well-Being. Curr. Opin. Physiol. 17, 163–168. doi:10.1016/j.cophys.2020.08.002
Vismara, M., Zarà, M., Negri, S., Canino, J., Canobbio, I., Barbieri, S. S., et al. (2021). Platelet-derived Extracellular Vesicles Regulate Cell Cycle Progression and Cell Migration in Breast Cancer Cells. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1868, 118886. doi:10.1016/j.bbamcr.2020.118886
Yamazaki, D., Ohya, S., Asai, K., and Imaizumi, Y. (2007). Characteristics of the ATP-Induced Ca2+-Entry Pathway in the T-BBEC 117 Cell Line Derived from Bovine Brain Endothelial Cells. J. Pharmacol. Sci. 104, 103–107. doi:10.1254/jphs.sc0070080
Yuan, Y., Kilpatrick, B. S., Gerndt, S., Bracher, F., Grimm, C., Schapira, A. H., et al. (2021). The Lysosomotrope GPN Mobilises Ca2+ from Acidic Organelles. J. Cel Sci 134. doi:10.1242/jcs.256578
Zhang, J., Guan, X., Shah, K., and Yan, J. (2021). Lsm12 Is an NAADP Receptor and a Two-Pore Channel Regulatory Protein Required for Calcium Mobilization from Acidic Organelles. Nat. Commun. 12, 4739. doi:10.1038/s41467-021-24735-z
Zuccolini, P., Ferrera, L., Remigante, A., Picco, C., Barbieri, R., Bertelli, S., et al. (2022). The VRAC Blocker DCPIB Directly gates the BK Channels and Increases Intracellular Ca(2+) in Melanoma and Pancreatic Duct Adenocarcinoma (PDAC) Cell Lines. Br. J. Pharmacol., 1–18. doi:10.1111/bph.15810
Zuccolo, E., Di Buduo, C., Lodola, F., Orecchioni, S., Scarpellino, G., Kheder, D. A., et al. (2018a). Stromal Cell-Derived Factor-1α Promotes Endothelial Colony-Forming Cell Migration through the Ca2+-dependent Activation of the Extracellular Signal-Regulated Kinase 1/2 and Phosphoinositide 3-Kinase/AKT Pathways. Stem Cell Develop. 27, 23–34. doi:10.1089/scd.2017.0114
Zuccolo, E., Kheder, D. A., Lim, D., Perna, A., Nezza, F. D., Botta, L., et al. (2019). Glutamate Triggers Intracellular Ca2+ Oscillations and Nitric Oxide Release by Inducing NAADP‐ and InsP 3 ‐dependent Ca2+ Release in Mouse Brain Endothelial Cells. J. Cel Physiol 234, 3538–3554. doi:10.1002/jcp.26953
Zuccolo, E., Laforenza, U., Ferulli, F., Pellavio, G., Scarpellino, G., Tanzi, M., et al. (2018b). Stim and Orai Mediate Constitutive Ca2+ Entry and Control Endoplasmic Reticulum Ca2+ Refilling in Primary Cultures of Colorectal Carcinoma Cells. Oncotarget 9, 31098–31119. doi:10.18632/oncotarget.25785
Keywords: nicotinic acid adenine dinucleotide phosphate (NAADP), two-pore channels (TPCs), membrane contact sites, store operated Ca2+ entry, cardiac mesenchymal stem cells, proliferation
Citation: Faris P, Casali C, Negri S, Iengo L, Biggiogera M, Maione AS and Moccia F (2022) Nicotinic Acid Adenine Dinucleotide Phosphate Induces Intracellular Ca2+ Signalling and Stimulates Proliferation in Human Cardiac Mesenchymal Stromal Cells. Front. Cell Dev. Biol. 10:874043. doi: 10.3389/fcell.2022.874043
Received: 11 February 2022; Accepted: 24 February 2022;
Published: 15 March 2022.
Edited by:
Alessia Remigante, University of Messina, ItalyReviewed by:
Dmitry Lim, University of Eastern Piedmont, ItalyAlessandra Rossini, Eurac Research, Italy
Copyright © 2022 Faris, Casali, Negri, Iengo, Biggiogera, Maione and Moccia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Serena Maione, YW5nZWxhLm1haW9uZUBjYXJkaW9sb2dpY29tb256aW5vLml0; Francesco Moccia, ZnJhbmNlc2NvLm1vY2NpYUB1bmlwdi5pdA==
†These authors have contributed equally to this work and share last authorship