 Wanchen Wang1
Wanchen Wang1 Daisuke Kawauchi
Daisuke Kawauchi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 29 April 2022
Sec. Signaling
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.864035
This article is part of the Research Topic Hedgehog Signaling Pathway in Development and Cancer View all 8 articles
The sonic hedgehog (SHH) pathway regulates the development of the central nervous system in vertebrates. Aberrant regulation of SHH signaling pathways often causes neurodevelopmental diseases and brain tumors. In the cerebellum, SHH secreted by Purkinje cells is a potent mitogen for granule cell progenitors, which are the most abundant cell type in the mature brain. While a reduction in SHH signaling induces cerebellar structural abnormalities, such as hypoplasia in various genetic disorders, the constitutive activation of SHH signaling often induces medulloblastoma (MB), one of the most common pediatric malignant brain tumors. Based on the existing literature on canonical and non-canonical SHH signaling pathways, emerging basic and clinical studies are exploring novel therapeutic approaches for MB by targeting SHH signaling at distinct molecular levels. In this review, we discuss the present consensus on SHH signaling mechanisms, their roles in cerebellar development and tumorigenesis, and the recent advances in clinical trials for MB.
In 1980, Nüsslein-Volhard and Wieschaus first identified the hedgehog (hh) gene in a Drosophila mutant by screening (Nusslein-Volhard and Wieschaus, 1980). hh-mutant larvae exhibited a disrupted body plan and duplicated denticles (cuticle projections in the anterior half of each body segment). The appearance of lawns of denticles sticking out of the segments resembled that of a hedgehog spine, inspiring the gene name. In 1992, the Drosophila hh gene was cloned by three independent groups (Lee et al., 1992; Mohler and Vani, 1992; Tabata et al., 1992). Shortly thereafter, homologs of the hh gene have been reported in vertebrates (Krauss et al., 1993; Riddle et al., 1993). In mammals, the HH family of proteins consists of three members: Sonic hedgehog (SHH), Desert hedgehog, and Indian hedgehog. Among them, SHH signaling has been the best studied so far and is known to play a central role in the regulation of developmental processes, such as the formation and function of the limb bud, nervous system, skeleton, and skin.
The key components of canonical SHH signaling include the secreted ligand SHH, its twelve-pass transmembrane receptor patched1 (PTCH1), the class Frizzled seven-transmembrane helix G protein-coupled receptor, smoothened (SMO), and the downstream glioma-associated oncogene (GLI) family of transcription factors (Figure 1). After SHH binds to PTCH1, the primary cilium, a microtubule-based membrane protrusion functions as the central hub of signal initiation by releasing SMO from PTCH1-mediated signal repression. Immunolocalisation studies have revealed that the primary cilium harbors proteins required for signal transduction (Bangs and Anderson, 2017). Eventually, de-repressed SMO activates an intricate process mediated by the GLI family of zinc-finger transcription factors. In addition to these factors, several molecular mechanisms regulating these key components have been discovered at multiple levels of the non-canonical SHH pathway (Brennan et al., 2012), providing a deep understanding of how strictly and intricately SHH signaling is regulated in vertebrates.
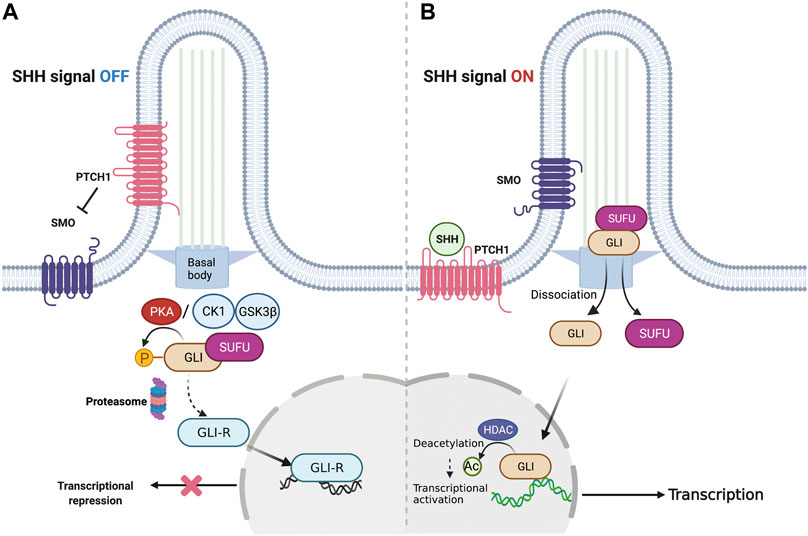
FIGURE 1. Overview of canonical SHH signaling. (A) Representative signaling cascade when SHH signaling is shut off. In the absence of the SHH ligand, PTCH1 gets localized to the plasma membrane in the primary cilium and inhibits SMO. This causes the GLI family proteins to stay restrained by SUFU to facilitate their phosphorylation by PKA. Consequently, the GLI proteins undergo proteolysis to generate GLI-R that suppresses the transcription of target genes. (B) Representative signaling cascade when the SHH signaling is switched on. In the presence of the SHH ligand, PTCH1 gets inhibited by the SHH ligand-dependent interaction. This activates SMO, allowing its translocation to the cilia. Subsequently, GLI proteins dissociate from SUFU to enter the cell nucleus and activate the target genes. SHH, Sonic hedgehog; PTCH1, Patched1; SMO, Smoothened; GLI, Glioma-associated oncogene; SUFU, Suppressor of fused; PKA, Protein kinase A.
The SHH signaling pathway in the vertebrate central nervous system has been studied well in mice (Chiang et al., 1996; Ruiz i Altaba et al., 2002). This pathway governs various neurodevelopmental processes within the brain. For example, SHH functions as a morphogen for cortical dorsal-ventral patterning and oligodendrogenesis in the early and later stages, respectively, and is required for the generation and maintenance of stem cell pools in postnatal and adult brains (Ahn and Joyner, 2005; Palma et al., 2005). The genetic ablation of ciliary genes induces abnormal radial astrocyte development and hypoplasia of the hippocampal dentate gyrus (Han et al., 2008). In the development of the spinal cord, SHH secreted from the notochord induces floor plate formation. Subsequently, SHH is secreted from the floor plate and functions as a long-range morphogen to trigger the differentiation of progenitor cells into spinal motor neurons and interneurons (Briscoe and Ericson, 2001; Danesin and Soula, 2017). SHH secreted from the floor plate also acts as a chemoattractant for axon pathfinding of commissural neurons (Charron et al., 2003). Thus, all these studies have confirmed the importance of SHH signaling in cell fate specification and axon pathfinding in the developing brain.
Another key role of SHH is to regulate cell proliferation in the dorsal brain as a potent mitogen (Dahmane et al., 2001). In particular, the cerebellum is one of the best-studied organs strongly affected by SHH signaling. Aberrant regulations of the signaling are related to neurodevelopmental diseases, including holoprosencephaly, Joubert syndrome (Aguilar et al., 2012), epilepsy (Hildebrand et al., 2016), autism spectrum disorder, and cancers, such as medulloblastoma (MB) (Northcott et al., 2012) and glioma (Clement et al., 2007). Thus, the importance of SHH signaling in the developing cerebellum has been increasingly recognized and research is focused on the pathway for the development of potential therapies for genetic diseases. In this article, we first review the roles of the principal components of SHH signaling pathways associated with normal cerebellar development and tumorigenesis at multiple levels of their regulatory mechanisms, and subsequently introduce potential therapeutic approaches with present clinical trials for SHH-driven cancer diseases based on recent accumulating knowledge.
The SHH gene encodes a 45-kDa precursor protein. After translation, it is catalyzed by an autocleavage reaction, yielding an N-terminal fragment (SHH-N) that triggers the activation of its downstream cascade and a C-terminal fragment whose function is not yet known. The maturation of SHH-N is completed by the covalent coupling of cholesterol to its C-terminal end and subsequent palmitoylation at its N-terminus by Hedgehog acyltransferase, a membrane-bound acyltransferase (Tukachinsky et al., 2016; Qi et al., 2018). The mature SHH-N is anchored to the plasma membrane by cholesterol and released under the regulation of the Dispatched protein (Burke et al., 1999). It has also been reported that a metalloprotease, A disintegrin and metalloproteinase 17 contribute to SHH shedding from the cell membrane in the presence of a specific extracellular heparan sulfate (Dierker et al., 2009). Thus, SHH undergoes heavy modifications to exert its function at multiple levels (Dierker et al., 2009).
PTCH1 is an SHH receptor that negatively regulates canonical SHH signaling. Its expression is strictly regulated by a negative feedback loop of transcriptional regulation (Goodrich et al., 1996). Subsequently, an increasing level of PTCH1, together with another negative regulator, Hedgehog interacting protein 1 (HHIP1), facilitates to capture the SHH ligand for endocytosis and lysosomal degradation (Incardona et al., 2002), limiting the range of SHH action (Jeong and McMahon, 2005). In contrast, SHH has been reported to enhance the endocytosis of PTCH1 via SMURF1 and two HECT-domain ubiquitin E3 ligases (Yue et al., 2014), indicating the ligand-mediated post-transcriptional regulation of PTCH1. The ciliary localization of PTCH1 is critical for its function. PTCH1 is distributed in the primary cilia to prevent its downstream signaling activity without SHH stimulation. Once the SHH protein binds to PTCH1, it leaves the cilia and loses its inhibitory ability for SMO, initiating a canonical SHH signaling cascade in the primary cilia (Rohatgi et al., 2007). Thus, multiple regulatory mechanisms for PTCH1 determine the intensity of SHH signaling under normal conditions.
In addition to PTCH1, other SHH co-receptors, such as CDON, BOC, and GAS1 are also essential for SHH pathway activation (Beachy et al., 2010; Allen et al., 2011). Several reports have identified their functions in central nervous system (CNS) development, which include cell fate specification, axon guidance, and cell proliferation (Okada et al., 2006; Izzi et al., 2011; Kim et al., 2020). In neural tube development, they show overlapping functions, and triple knockout (KO) causes complete loss of SHH-dependent ventral spinal cord specification. Interestingly, only Gas1 and Boc, not Cdon affect limb patterning, indicating a distinct requirement for co-factors in different tissues (Allen et al., 2011).
Signal transduction by SMO is a two-step process: ciliary localization and subsequent activation (Rohatgi et al., 2009). Similar to that of PTCH1, the ciliary localization of SMO is strictly regulated. SHH-mediated activation and constitutively active mutations of SMO promote its translocation to the cilia without changing its total expression level (Corbit et al., 2005). This translocation is mediated by the ArhGAP36-PKA-INVS axis (Zhang et al., 2019). In addition, the phosphorylation of the C-tail of SMO by casein kinase (CK) 1a and Grk2 under HH activation induces a conformational change, resulting in the ciliary accumulation of SMO (Zhao et al., 2007; Chen et al., 2010; Chen et al., 2011). As for the subsequent activation, given that the ciliary translocation of SMO is not sufficient for its activation (Rohatgi et al., 2009), it has long been a mystery as to how PTCH1 inhibits SMO without physical interactions. However, accumulating evidence has shown that a small fraction of membrane cholesterol, known as accessible cholesterol (Kinnebrew et al., 2019) could be a mediator between PTCH1 and SMO. Cholesterol acts as the endogenous ligand of SMO (Huang et al., 2016; Luchetti et al., 2016; Huang et al., 2018) via an ester bond on the Asp95 residue in its sequence (Xiao et al., 2017). Meanwhile, PTCH1 is capable of changing the distribution of cholesterol between the inner and outer leaflets of the plasma membrane (Zhang et al., 2018) and contributes to cholesterol cellular efflux as a cholesterol transporter (Bidet et al., 2011; Gong et al., 2018). Thus, multiple modification mechanisms influence the functions of SMO in SHH signaling.
The major target of activated SMO is the GLI family of zinc-finger transcription factors. In mammals, three members of the GLI family, GLI1, GLI2, and GLI3, mediate SHH-regulated transcription in an intricate manner (Ingham and McMahon, 2001). GLI1 acts as an amplifier and a downstream target of the SHH pathway (Marigo et al., 1996), whereas GLI2 and GLI3 exert dual effects on SHH activation depending on their post-transcriptional and post-translational modifications (Ruiz i Altaba et al., 2007). For example, in the absence of SHH signaling activation, a major negative regulator of the SHH pathway, suppressor of fused (SUFU) inhibits the GLI function by sequestering GLI in the cytoplasm (Humke et al., 2010). Subsequently, GLI2 and GLI3 undergo limited proteolysis by SCFbTrCP ubiquitin E3 ligase and transform into their repressor forms (GLI2-R and GLI3-R) after phosphorylation is initiated by PKA (Humke et al., 2010; Tuson et al., 2011), CK1, and glycogen synthase kinase 3 (GSK3) (Pan et al., 2006; Tukachinsky et al., 2010). HH stimulation recruits the SUFU-GLI complex at the tip of the cilia, where the GLI dissociates from SUFU and translocates to the nucleus to activate the SHH pathway (Figure 1; Humke et al., 2010; Tukachinsky et al., 2010). In this process, the PKA phosphorylation sites of GLI2/3 are dephosphorylated; however, other sites are phosphorylated instead. GLI phosphorylation remodeling is crucial for adjusting the ratio of the GLI activator form (GLI-A) to repressor form (GLI-R) in the cell (Niewiadomski et al., 2014).
In addition to the classic canonical SHH pathway, the importance of non-canonical activation of the SHH pathway in development and tumorigenesis has been highlighted in emerging studies. Non-canonical SHH signaling has two distinct subtypes: 1) GLI-independent HH signaling, which can be further subdivided into SMO-independent and SMO-dependent and 2) alternative mechanisms inducing GLI activity (Robbins et al., 2012; Pietrobono et al., 2019). GLI- and SMO-independent mechanisms are often referred to as type I non-canonical HH signaling (Robbins et al., 2012). PTCH1 can be characterized as a dependence receptor because it is capable of inducing apoptotic cell death in the absence of SHH via another signaling pathway. While the PTCH1 C-terminal domain (CTD) is sufficient to induce cell death even in the presence of the SHH ligand (Mille et al., 2009), the insertion of a mutation that prevents caspase-mediated cleavage inhibited PTCH1-induced cell death (Thibert et al., 2003; Mille et al., 2009), demonstrating the functional significance of the CTD (Thibert et al., 2003). As SMO overexpression does not rescue cell death, this process is independent of the canonical SHH pathway (Thibert et al., 2003; Chinchilla et al., 2010).
In addition to the involvement in apoptotic cell death, PTCH1 negatively regulates cell proliferation by interacting with phosphorylated CCNB1, a G2/M checkpoint regulator, followed by shuttling it into the cytoplasm in an SMO-independent manner (Barnes et al., 2001). Supporting this study, the PTCH1 harboring a nonsense mutation found in basal cell carcinoma (BCC) failed to interact with CCNB1 (Barnes et al., 2005) and was capable of the morphological transformation of NIH3T3 fibroblast cells. Moreover, the CTD of PTCH1 phys(BCC) ically interacts with the ULK complex through ATG101 and impairs autophagic flux as a tumor suppressor (Chen et al., 2018). Taken together, loss of type I non-canonical HH signaling potentially contributes to tumorigenesis.
The type II non-canonical HH pathway is SMO-dependent. SMO has been shown to function not only as a classic SHH signaling transducer but also as a G protein-coupled receptor that catalyzes the GDP–GTP exchange of the G inhibitory (Gi) family of G proteins (Riobo et al., 2006). Several studies have indicated that SMO mediates the activation of small GTPases, such as Rac1 and RhoA, followed by the rearrangement of the actin cytoskeleton for the proper regulation of cell migration, angiogenesis, tubulogenesis, and synaptogenesis (Bijlsma et al., 2007; Renault et al., 2010; Sasaki et al., 2010; Polizio et al., 2011a; Polizio et al., 2011b). SRC-family kinases are also involved in SHH-mediated axon guidance without the activation of canonical SHH signaling (Yam et al., 2009).
Apart from the type I and type II non-canonical HH pathways, the aberrant activation of GLI bypasses the SHH–PTCH1–SMO route, which is also considered to be a part of the non-canonical HH pathway. These mechanisms, such as the RAS-RAF-MEK-ERK, PI3K-AKT-mTOR, and TGF-β pathways and epigenetic modulation (Pietrobono et al., 2019) allow the direct transcriptional activation of GLI and post-transcriptional modification of GLI. Our recent study further found that ependymoma-specific ZFTA fusion genes directly bind to the GLI2 locus to activate it for tumor progression (Zheng et al., 2021). In addition, a study on SMO-resistant BCCs identified non-canonical GLI inducers, including AP-1, TGF-β, and their downstream Rho regulators (Yao et al., 2020). Thus, molecular analyses of cancerous cells provide novel insights into potential non-canonical SHH pathways.
The cerebellum is a prominent part of the hindbrain located in the dorsal brain stem and functions as the control center for motor coordination. It is known that the attenuation of SHH signaling by genetic mutations causes conditions of disrupted cerebellar development, such as hypoplasia, which is related to neurological diseases, including autism spectrum disorder (Filges et al., 2011), Joubert syndrome (De Mori et al., 2017), Dandy–Walker malformation (Zhang et al., 2021), and Down’s syndrome (Baxter et al., 2000; Roper et al., 2006). The aberrant activation of SHH signaling often induces extensive hyperplasia and results in MB, a common malignant pediatric brain tumor, arising in the cerebellum. Therefore, a profound understanding of how SHH signaling affects cerebellar development sheds light on novel therapeutic avenues for SHH-related genetic diseases.
Granule cells (GCs), interneurons essential for the coordination of afferent inputs and motor outputs of the cerebellum, represent the most prevalent neuronal type in the cerebellar system. During embryogenesis, GC progenitors (GCPs) arise from the upper rhombic lip, a germinal neuroepithelium in the most dorsal portion of the metencephalon, and migrate along the surface of the cerebellum to form the external granule layer (EGL) from E13 onward (Hatten and Heintz, 1995; Wang et al., 2005). After migration within the EGL at the postnatal stage, GCPs undergo a morphological transformation into round or polyhedral shapes and subsequent rapid proliferation, the strict regulation of which is critical for the development of the normal size and proper foliation of the cerebellum (Figure 2). SHH secreted from Purkinje cells (PCs) is the most efficacious mitogen for GCP expansion. Indeed, the local ablation of PCs inhibits GCP proliferation and induces thinning of the EGL (Smeyne et al., 1995). In contrast, the hyperactivation of SHH signaling due to a deficiency of Ptch1 in GCPs caused abnormal proliferation and MB formation (Figure 2) (Schüller et al., 2008; Yang et al., 2008), indicating that SHH mediated an interplay between PCs and GCPs in the developing cerebellum. Consistent with the observation in Ptch1-deficient mice, the forced activation of SMO resulted in the hyperproliferation of GCPs, eventually resulting in high penetrance of MB formation (Hatton et al., 2008; Dey et al., 2012). Also, the genetic ablation of Boc and Gas1 displayed their absolute contribution to the SHH-driven GCP proliferation by their direct interaction with PTCH1 (Izzi et al., 2011). In line with this report, elevated BOC levels enhanced SHH signaling activity in GCPs and mediated PTCH1-driven MBSHH formation via CCND1-dependent DNA damage (Mille et al., 2014). Of note, a recent study revealed that SHH- but not IGF1-induced GCP proliferation causes replication stress via an increased replication fork speed and active fork density, a proxy of replication origin firing, potentially increasing the likelihood of MB-inducing mutations (Tamayo-Orrego et al., 2020). Thus, GCPs are susceptible to transformation into tumor cells by the hyperactivation of SHH signaling, while proliferating properly by the stimulation of PC-derived SHH ligands.
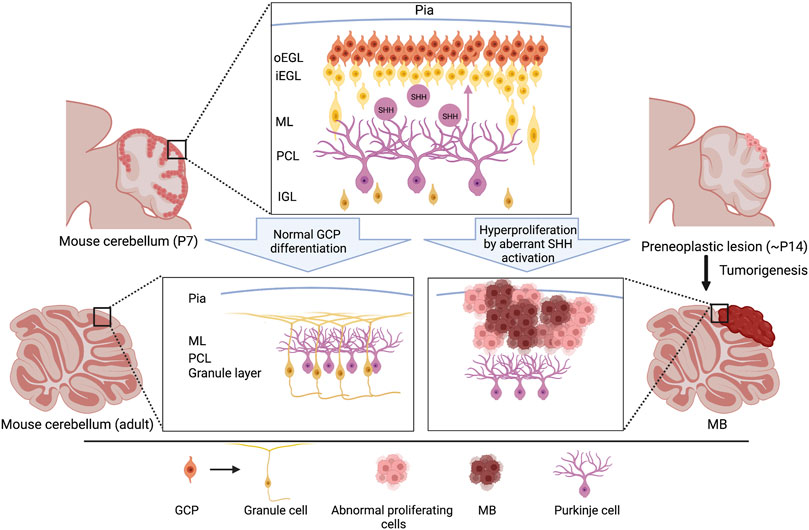
FIGURE 2. Schematic representation of cerebellar development and initiation of MBSHH. During the postnatal stage, GCPs normally proliferate in the outer edge of the EGL under the regulation of SHH secreted by the Purkinje cells. GCPs exit the cell cycle and subsequently migrate radially, toward the IGL, to mature into the iEGL. Somatic genetic mutations often cause aberrant SHH signaling that results in a failure to exit the cell cycle, leading to hyperproliferation. Abnormal differentiation of GCPs induces the formation of preneoplastic lesion and MBSHHs predominantly on the surface of the cerebellar hemispheres (bottom right). Please note that the locations of other subtypes of MBs, such as MBWNTs and MBGrp3/4s, are distinct from that of MBSHHs (Kawauchi et al., 2017), implying distinct cellular origins of these subgroups. GCP, granule cell precursor; oEGL, outer external granular layer; iEGL, inner external granular layer; ML, molecular layer; PCL, purkinje cell layer. IGL, internal granular layer.
The roles of SUFU in GCP differentiation have been evaluated in vivo recently. While SUFU functions as either a sustainer or repressor of SHH signaling through the context-dependent regulation of GLI proteins, the conditional KO of SUFU explains its repressor function in GCPs. GCP-specific KO induces enhanced proliferation and delayed cell cycle exit by upregulating GLI2 and suppressing GLI3R, coinciding with Fgf8 activation (Jiwani et al., 2020). However, unlike Ptch1, the loss of Sufu alone in GCPs is insufficient to cause MB development (Yin et al., 2019). This could be due to the destabilization of GLI2 in Sufu-KO GCPs. Indeed, the disruption of the Spop/Cul3-mediated proteasomal machinery for GLI2 remarkably enhanced MB formation at complete penetrance, indicating an essential role of GLI2 in tumorigenesis.
As a downstream molecule of GLI proteins, D-type cyclins have been identified as the proliferation machinery in GCPs (Kenney and Rowitch, 2000; Pogoriler et al., 2006; Tolosa et al., 2020). D-type cyclins interact with Cyclin-dependent kinase (CDK) 4 and phosphorylate retinoblastoma 1 (RB1) to inhibit its function, in turn allowing E2F-mediated gene expression for cell proliferation (Giacinti and Giordano, 2006). Indeed, Rb1 conditional KO mice showed a delay in GCP differentiation (Marino et al., 2003; Shakhova et al., 2006), indicating the involvement of a D-type cyclin-RB1 axis in GCP proliferation in vivo. Interestingly, a recent study proposed that the CDK4-CCND1 complex also phosphorylates ATOH1, a basic helix-loop-helix (bHLH) transcriptional factor required for the commitment of GCP cell identity, at the Ser309 residue, which could enhance ATOH1 stability to maintain the undifferentiated state of GCPs (Miyashita et al., 2021). However, whether this model is completely independent of RB1 remains to be confirmed using Ser309 mutation-carrying primary GCPs.
v-Myc myelocytomatosis viral related oncogene (MYCN), another direct downstream target of GLI proteins (Kenney et al., 2003), also boosts GCP proliferation by negatively regulating cyclin-dependent inhibitors CDKN1B and CDKN2C (Knoepfler et al., 2002; Zindy et al., 2006). The previous studies have highlighted the role of MYCN in MB formation (Swartling et al., 2010; Kawauchi et al., 2012; Swartling et al., 2012). Furthermore, MYCN activates miR-17–92 polycistronic miRNA clusters to maintain GCP proliferation (Northcott et al., 2009). Supporting this statement, mice lacking these miRNAs displayed decreased brain weight and abnormal foliation. In addition, Ptch1-driven MB formation was prevented by the conditional deletion of miR-17–92 in GCPs, while a retroviral infection of either D-type cyclins, Mycn, or miR-17–92 in Ptch1 heterozygous GCPs enhanced MB formation (Uziel et al., 2009; Kawauchi et al., 2012; Zindy et al., 2014), indicating that these miRNAs are required for the initiation of MB formation. In addition, the silencing of miR-17–92 and its orthologue miR-106a–25 clusters by 8-mer LNA-modified antimiR oligonucleotides attenuated tumor growth of PTCH1-driven MBs (Murphy et al., 2013), indicating that these miR clusters are also important for both tumor initiation and progression.
Consistent with a previous finding that the primary cilium is involved in mammalian SHH signaling (see previous sections), the loss of cilium-related genes Ift88 and Kif3a causes disrupted cerebellar development by the reduction of GCP expansion (Chizhikov et al., 2007; Spassky et al., 2008). A loss of ciliopathy genes, including CEP290, Zfp423, and Talpid3, has also been reported to attenuate SHH signaling activity, leading to cerebellar hypoplasia (Aguilar et al., 2012; Hong and Hamilton, 2016; Bashford and Subramanian, 2019). In contrast, Gαs, a G-coupled protein expressed at the primary cilium of the GCP, represses SHH signaling via the cAMP-dependent pathway and ciliary trafficking of GLI2 and SMO proteins (He et al., 2014). A recent study revealed that ATOH1 regulates ciliogenesis by upregulating Cep131 and restoring centriolar satellite integrity, indicating an indirect contribution of ATOH1 to SHH signaling (Chang et al., 2019). It is also known that cilium-related genes are involved in MB formation. For example, the removal of the primary cilia by the loss of either Ift88 or Kif3a inhibits either PTCH1- or SMO-driven MB formation and accelerates GLI2-induced tumorigenesis (Han et al., 2009; Wong et al., 2009; Han and Alvarez-Buylla, 2010; Barakat et al., 2013). Thus, ciliary formation affects tumorigenesis in an oncogenic driver-dependent manner. Furthermore, SHH-induced GCP proliferation has been reported to be enhanced by constitutive Gαi activation in the cilia (Barzi et al., 2011). Conversely, GCPs carrying deleted Gnas showed high proliferation and gave rise to MBSHH (He et al., 2014). Thus, along with previous implications (Niewiadomski et al., 2013; Kool et al., 2014), G proteins are also involved in GCP proliferation and MBSHH formation in the cilia.
Post-transcriptional regulation of SHH-related proteins has been reported to influence cerebellar development and tumorigenesis. Previous studies have shown that ERAP1 protects GLI from βTrCP-dependent degradation and activates the SHH pathway by binding to and sequestering USP47, a deubiquitylase enzyme of βTrCP. Consistent with this, the genetic or pharmacological inhibition of ERAP1 suppresses SHH-dependent tumor growth in vitro and in vivo (Bufalieri et al., 2019). In addition, the Itch/β-arrestin 2 complex has been reported to bind to SUFU and induce its K63-linked ubiquitylation. Without affecting the stability of SUFU, this process enhances the formation of the SUFU/GLI3 complex and consequently increases the amount of GLI3R, which keeps the SHH pathway switched off (Infante et al., 2018). Furthermore, histone deacetylase1 (HDAC1), a histone modifier, deacetylates GLI1 and GLI2 and enhances their transcriptional activation. Downstream of GLI proteins, MYCN is also regulated at the protein level by the E3 ubiquitin ligases HUWE1 and FBXW7 (Welcker et al., 2004; Zhao et al., 2008). While MYCN phosphorylated at Thr58 by GSK3b is recognized by FBXW7 for degradation (Yeh et al., 2004; Sjostrom et al., 2005), Aurora kinase A prevents FBXW7-mediated ubiquitination by directly interacting with MYCN. Intriguingly, the conditional inactivation of Huwe1 and Fbxw7 does not induce similar phenotypes in GCPs, which could imply differences in their other target proteins compared with MYCN (D'Arca et al., 2010; Jandke et al., 2011). In human MBSHHs, low FBXW7 expression levels were correlated with poor prognosis (Suryo Rahmanto et al., 2016). Based on these studies, inhibitors that alter the catalytic action of SHH-regulating enzymes are presently being investigated as potential drugs for SHH-driven cancers, including MB.
Epigenetic regulation is thought to be important for strict SHH signaling tuning. For example, the bromo and extra C-terminal (BET) bromodomain protein BRD4, a transcriptional facilitator at super-enhancer sites (Whyte et al., 2013), is involved in GLI1 and GLI2 expression (Tang et al., 2014). shRNA-based Brd4 knockdown and the pharmacological inhibition of BRD4 decreased GLI expression in SHH-driven MB cells (Tang et al., 2014). The histone modifier KDM6B has also been reported to regulate GCP proliferation via the epigenetic regulation of SHH-related genes. SHH signaling enhances the removal of a PRC2-mediated repressive H3K27me3 mark by KDM6B antagonizing PRC2 at the Gli1 locus, resulting in the upregulation of Gli1 and its downstream targets Ccnd1 and Mycn in GCPs (Shi et al., 2014). In contrast, the Kdm6b-deficient cerebellum displayed a decreased cerebellar size and lack of foliation, which is reminiscent of the Gli2-KO cerebellum. The loss of Kdm6b further prevented SMO-driven MB formation, coincident with SHH downstream genes (Shi et al., 2014). Thus, KDM6B could be a key player in the epigenetic regulation of SHH signaling, although how the signaling pathway regulates KDM6B and other unidentified epigenetic factors in GCPs requires further investigation.
In addition to histone modifiers, chromatin remodelers have also been implicated as important in GCP proliferation. A loss of Smarca4 (Zhan et al., 2011; Moreno et al., 2014; Shi et al., 2016), a critical component of the SWI/SNF chromatin remodeling complex, reduced EGL expansion and SMO-driven MB formation due to the lack of direct regulation of SHH-related genes. The conditional deletion of Smarcb1, another component of the SWI/SNF complex, did not upregulate SHH-, but WNT-related genes (Moreno et al., 2014) that impair GCP proliferation (Lorenz et al., 2011; Pei et al., 2012) and MB formation (Pöschl et al., 2014) by antagonizing SHH signaling. These results imply that these two SWI/SNF components may function differently in the GCPs.
Recent genome-wide data-mining studies on human MBSHHs have identified novel machinery that regulates SHH signaling. In addition to GNAS in the primary cilia (see previous sections), mutations in non-coding U1 small nuclear RNAs have been identified as potential regulators of SHH signaling in human MBSHHs (Suzuki et al., 2019). These mutations are strongly associated with the aberrant splicing of SHH-related genes, which in turn activates them abnormally. Thus, multi-disciplinary studies on human diseases would provide further insights into the unexpected regulatory mechanisms of SHH signaling pathways in the future.
Other types of cerebellar cells exhibit SHH signaling activity in Ptch1-, Gli1-, and Gli2-reporter mice (Bai et al., 2002; Corrales et al., 2004; Lewis et al., 2004). In addition to GCPs/GCs, TNC-positive GABAergic intermediate progenitors, astrocyte precursors in the white matter, and Bergmann glia exhibit SHH activity at postnatal stages. Indeed, SHH from PCs stimulates the proliferation of these white matter progenitors, and the conditional deletion of SMO in these progenitors reduces their proliferation and expansion (Fleming et al., 2013). Unlike that in white matter progenitors, the deletion of SMO in the Bergmann glia indirectly affects GCP proliferation with abnormal cytoarchitecture but not their own proliferative defects (Cheng et al., 2018). Whether these cell types are the cellular origin of MBSHH remains to be explored.
The dysregulation of potent mitogens often results in cancer. As mentioned previously, the aberrant activation of SHH signaling is strongly associated with the development of MBs, which account for approximately 25% of all brain tumors in children. Thus far, in addition to MBSHHs, MBs consist of the other three subgroups (WNT, group 3, and group 4) according to their molecular characteristics. MBSHHs account for 30% of all MBs (Maier et al., 2021). In patients with MBSHHs, mutations in genes involved in these pathways cause the constant activation of the SHH pathway (SHH gene amplification, 3%; PTCH1 loss-of-function mutation, 49.6%; SMO-activating mutation, 17.3%; SUFU loss-of-function mutation, 14.3%; GLI2 gene amplification, 7.5%; and MYCN gene amplification, 12.8%) (Kool et al., 2014). Interestingly, the frequency of these mutations depends on the age of the patient: PTCH1 mutations are found in all age groups, whereas SUFU mutations are mainly found in infants (0–3 years), and SMO mutations are predominantly detected in adults (18 years or older). Genetic amplification of GLI2 and MYCN is mainly observed in children (4–17 years) and is often accompanied by TP53 mutations, which are included in the high-risk group (Kool et al., 2014). Recent international multicenter MB predisposition studies have confirmed germline mutations in SUFU, PTCH1, and TP53 and discovered novel predisposition genes, such as ELP1 (14.4%), BRCA2 (2.5%), and PALB2 (1.0%), with the highest prevalence in MBSHHs patients (Waszak et al., 2018; Waszak et al., 2020). Whether these novel mutations affect SHH signaling and MB formation remains to be elucidated.
So far, several small molecules have been considered as a candidate drugs for SHH-driven tumors (Figure 3). Based on the high frequency of PTCH1 mutations in MBSHHs, SMO inhibitors are promising therapeutic drugs against SHH-mediated cancers. Vismodegib (GDC-0449) is an SMO inhibitor approved by the U.S. Food and Drug Administration (FDA) as a first-line drug for the treatment of BCCs, which are also driven by hyperactivation of the SHH pathway. Vismodegib binds to the transmembrane domain of SMO and inhibits its downstream signaling, and has been shown to regress tumors in an allogeneic transplantation model of tumor cells in the MBSHH mouse model (Robarge et al., 2009). Clinical trials of four drugs with potential clinical applications are presently underway (NCT01878617, NCT00939484, NCT01239316, and NCT00822458). In addition to vismodegib, sonidegib (LDE-225) has also been used in clinical trials as an SMO inhibitor that binds to the transmembrane domain (NCT01708174, NCT01208831, NCT00880308, NCT01125800, and NCT03434262). Sonidegib is more potent than vismodegib in inhibiting SMO and has been shown to cross the blood-brain barrier, which has attracted much attention (Pan et al., 2010). Given the present development of various SMO inhibitors in addition to vismodegib and sonidegib (Lospinoso Severini et al., 2020), it is likely that new SMO inhibitors will continue to enter clinical trials. The therapeutic effects of SMO inhibitors are often only transient, however. The SMO D473H mutation has been found in recurrent tumors after continuous treatment with vismodegib and has been shown to confer drug resistance to vismodegib (Yauch et al., 2009). In addition, mutations in genes downstream of SMO, such as SUFU, GLI, and MYCN, can confer resistance to SMO inhibitors in MBSHHs (Kool et al., 2014). Furthermore, the activation of the phosphatidylinositol-3 kinase pathway has been shown to attenuate the effect of sonidegib in mice models (Buonamici et al., 2010). Thus, although SMO inhibitors are expected to be therapeutic agents for MBSHHs, congenital or acquired drug resistance remains a major issue. To overcome this difficulty, it is necessary to further examine the mechanism of resistance acquisition to develop molecular-targeted compounds for combinatorial drug therapy.
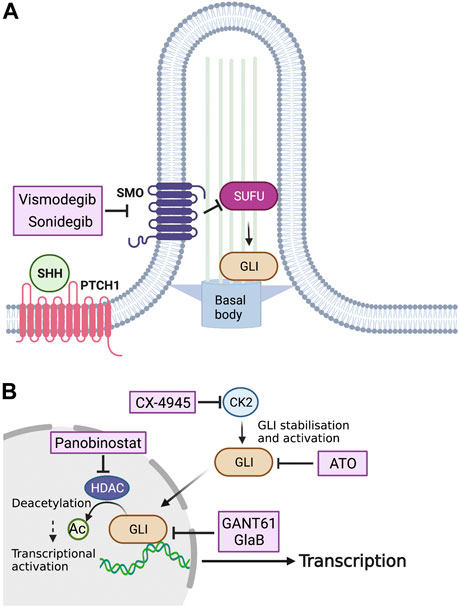
FIGURE 3. Potential inhibitors of SHH signaling for the treatment of MBs. (A) Vismodegib and sonidegib, drugs that inhibit SHH activation by SMO on the surface of the tumor cell, have been approved for treating MB patients. (B) Inside the tumor cell, silmitasertib (CX-4945) blocks the kinase activity of CK2, which results in dephosphorylation of GLI proteins, while GANT61, ATO, and GlaB are capable of preventing GLI-mediated transcription. Panobinostat, an HDAC inhibitor, reduces GLI activity via regulation of its acetylation. CK2, Casein kinase 2; GANT61, GLI antagonist 61; ATO, Arsenic trioxide; GlaB, Glabrescione B.
Because SMO is one of the most upstream targets of the SHH pathway, its inhibitors are not expected to have therapeutic effects on tumors with downstream genetic mutations. Therefore, in addition to SMO inhibitors, direct and indirect inhibitors of GLI proteins are presently under development. The first example of an indirect GLI inhibition is the inhibition of HDACs. As mentioned previously, GLI1 and GLI2 are known to increase their transcriptional activities via deacetylation by HDACs (Canettieri et al., 2010). A proof-of-concept validation using the MBSHH mouse model showed that the oral administration of mocetinostat, an HDAC inhibitor, drastically suppressed tumor growth (Coni et al., 2017). In addition, the HDAC inhibitor panobinostat (MTX110), which has been approved by the FDA for the treatment of cancer, has recently entered clinical trials for recurrent MBs (NCT04315064), and is expected to be approved for clinical applications.
The second method of indirect GLI inhibition is the blockade of CK2 that stabilizes the GLI2 protein and increases its transcriptional activity (Purzner et al., 2018). In vitro, CX-4945, a highly specific inhibitor of CK2, was shown to inhibit CK2 in mouse MBSHH cells and patient-derived tumor cells. In vivo, CX-4945 exhibits antitumor activity in orthotopic transplantation models of cancer cells derived from a vismodegib-resistant MBSHH mouse model (Purzner et al., 2018). CX-4945 is presently undergoing clinical trials (NCT03904862).
Although no direct inhibitors of GLI proteins have undergone clinical trials to date, some have already been utilized in cancer research. GLI antagonist 61 (GANT61) is a small molecule that binds to the groove between the zing-finger domain of GLI and directly inhibits its transcriptional activity (Agyeman et al., 2014). It has been reported that GANT61 inhibits migration, invasion, and proliferation of human MBSHH cell lines and promotes apoptosis (Lin et al., 2016). Glabrescione B (GlaB), an isoflavone naturally found in Derris glabrescens, also binds to the zinc-finger domain of GLI1 and interferes with its DNA-binding capability (Infante et al., 2015). It is also proven that GlaB inhibited MBSHH growth in vivo, when delivered by polymer micelles and intraperitoneal injections (Infante et al., 2021). Arsenic trioxide (ATO) is another drug that directly binds to and inhibits GLI1 (Beauchamp et al., 2011) and GLI2 (Kim et al., 2010) and is approved by the FDA for the treatment of patients with acute promyelocytic leukemia (Raffoux et al., 2003). ATO inhibits the SHH pathway in human MBSHH cell lines and prolongs the survival of a mouse model of MBSHH (Beauchamp et al., 2011). It is hoped that further preclinical studies using patient-derived tissue xenografts will allow these drugs to enter clinical trials as novel therapeutic agents against MBSHH.
Finally, we will discuss the therapeutic potential of CDK inhibitors in MBSHH. Palbociclib, an inhibitor of CDK4 and CDK6, is approved by the FDA for the treatment of patients with advanced breast cancer (Turner et al., 2018). The CDK4/6-Cyclin D-RB1 pathway is important for cell-cycle progression (Goel et al., 2018) and is expected to be a therapeutic agent for various types of cancers. As a downstream target of SHH signaling, the expression of CDK6 is elevated in human MBs and its pharmacological inhibition by palbociclib resulted in attenuated tumor growth and prolonged survival in an MBSHH mouse model (Raleigh et al., 2018). Palbociclib is presently in clinical trials for the treatment of several types of pediatric brain tumors, including MB (NCT 02255461). A recent phase I trial of palbociclib in pediatric brain tumor patients defined a dosage of 75 mg/m2 for 21 consecutive days as an optimal condition (Van Mater et al., 2021). In addition to palbociclib, a recent study has revealed that THZ-1, a covalent inhibitor of CDK7, antagonizes GLI1 and GLI2 transcription, preventing the growth of SHH-driven MBs and SMO inhibitor-resistant human tumor cell lines in vivo and in vitro (Liu et al., 2019). Thus, CDK inhibitor-based inhibition of the SHH pathway would be an alternative therapeutic strategy for SMO inhibitor-resistant tumors.
To date, tremendous efforts have been made to reveal the molecular mechanisms involved in the SHH pathway. Meanwhile, exciting new insights into the molecular mechanism underlying the SHH pathway are constantly being updated. For example, it has been reported that the SHH ligand is delivered by a tiny cellular projection called cytoneme, one of the most novel cellular compartments, using sophisticated live imaging technology (Hall et al., 2021). Despite such emerging new findings, we have encountered many obstacles in treating MBSHHs in the clinical field. The inter- and intra-tumor heterogeneity with different SHH-related mutations suggests the need for personalized treatment (Kool et al., 2014). MBSHH-specific mutations could interplay with SHH signaling to accelerate tumor progression. Actually, our recent study has reported that BCOR, a main component of PRC1.1 is frequently mutated in human MBSHHs; its deletion increases Igf2 transcriptional activation via histone H2A modification, causing enhanced tumor progression in SHH-driven murine tumors (Kutscher et al., 2020). Moreover, a recent single-cell genomics study identified Hes1 and Myod as markers for MBSHH cells responsive and resistant to an SHH inhibitor (Ocasio et al., 2019), respectively, implying the existence of unknown crosstalk between SHH signaling and these genes. Thus, recent advanced experimental techniques and multidisciplinary researches would offer the opportunities to understand the SHH signaling network more deeply, providing not only better biological insights into the normal development of organisms, but also opportunities to develop novel therapeutic avenues for many SHH-driven cancers.
WW, RS, and DK wrote the manuscript. All authors approved the submitted version of the manuscript.
This research was funded by grants from the Japan Agency for Medical Research and Development, AMED (JP20ck0106534), Suzuken Memorial Foundation (to DK), and Grant-in-Aid for the Japan Society for the Promotion of Science (JSPS) Fellows (20J13210) (to RS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to all the members of the Department of Biochemistry & Cellular Biology, NCNP for their feedback on the manuscript. We also thank Editage (www.editage.com) for English language editing. The figures in the manuscript were created with BioRender (biorender.com).
HH, Hedgehog; SHH, Sonic hedgehog, PTCH1, Patched1; SMO, Smoothened; GLI, Glioma-associated oncogene; SUFU, Suppressor of fused; HHIP1, Hedgehog Interacting Protein 1; PKA, Protein kinase A; MYCN, v-myc myelocytomatosis viral related oncogene; KO, Knockout; MB, Medulloblastoma, GCP, Granule cell progenitor, EGL, External granule layer; RB1, Retinoblastoma 1; FDA, Food and Drug Administration; CDK, Cyclin-dependent kinase; CK, Casein kinase; GANT61, GLI antagonist 61; ATO, Arsenic trioxide; GlaB, Glabrescione B; BCC, Basal cell carcinoma; PC, Purkinje cell.
Aguilar, A., Meunier, A., Strehl, L., Martinovic, J., Bonniere, M., Attie-Bitach, T., et al. (2012). Analysis of Human Samples Reveals Impaired SHH-dependent Cerebellar Development in Joubert syndrome/Meckel Syndrome. Proc. Natl. Acad. Sci. U.S.A. 109 (42), 16951–16956. doi:10.1073/pnas.1201408109
Agyeman, A., Jha, B. K., Mazumdar, T., and Houghton, J. A. (2014). Mode and Specificity of Binding of the Small Molecule GANT61 to GLI Determines Inhibition of GLI-DNA Binding. Oncotarget 5 (12), 4492–4503. doi:10.18632/oncotarget.2046
Ahn, S., and Joyner, A. L. (2005). In Vivo analysis of Quiescent Adult Neural Stem Cells Responding to Sonic Hedgehog. Nature 437 (7060), 894–897. doi:10.1038/nature03994
Allen, B. L., Song, J. Y., Izzi, L., Althaus, I. W., Kang, J.-S., Charron, F., et al. (2011). Overlapping Roles and Collective Requirement for the Coreceptors GAS1, CDO, and BOC in SHH Pathway Function. Develop. Cel 20 (6), 775–787. doi:10.1016/j.devcel.2011.04.018
Bai, C. B., Auerbach, W., Lee, J. S., Stephen, D., and Joyner, A. L. (2002). Gli2, but notGli1, Is Required for Initial Shh Signaling and Ectopic Activation of the Shh Pathway. Development 129 (20), 4753–4761. doi:10.1242/dev.129.20.4753
Bangs, F., and Anderson, K. V. (2017). Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb Perspect. Biol. 9 (5), a028175. doi:10.1101/cshperspect.a028175
Barakat, M. T., Humke, E. W., and Scott, M. P. (2013). Kif3a Is Necessary for Initiation and Maintenance of Medulloblastoma. Carcinogenesis 34 (6), 1382–1392. doi:10.1093/carcin/bgt041
Barnes, E. A., Heidtman, K. J., and Donoghue, D. J. (2005). Constitutive Activation of the Shh-Ptc1 Pathway by a Patched1 Mutation Identified in BCC. Oncogene 24 (5), 902–915. doi:10.1038/sj.onc.1208240
Barnes, E. A., Kong, M., Ollendorff, V., and Donoghue, D. J. (2001). Patched1 Interacts with Cyclin B1 to Regulate Cell Cycle Progression. EMBO J. 20 (9), 2214–2223. doi:10.1093/emboj/20.9.2214
Barzi, M., Kostrz, D., Menendez, A., and Pons, S. (2011). Sonic Hedgehog-Induced Proliferation Requires Specific Gα Inhibitory Proteins. J. Biol. Chem. 286 (10), 8067–8074. doi:10.1074/jbc.M110.178772
Bashford, A. L., and Subramanian, V. (2019). Mice with a Conditional Deletion of Talpid3 ( KIAA0586 ) - a Model for Joubert Syndrome. J. Pathol. 248 (4), 396–408. doi:10.1002/path.5271
Baxter, L. L., Moran, T. H., Richtsmeier, J. T., Troncoso, J., and Reeves, R. H. (2000). Discovery and Genetic Localization of Down Syndrome Cerebellar Phenotypes Using the Ts65Dn Mouse. Hum. Mol. Genet. 9 (2), 195–202. doi:10.1093/hmg/9.2.195
Beachy, P. A., Hymowitz, S. G., Lazarus, R. A., Leahy, D. J., and Siebold, C. (2010). Interactions between Hedgehog Proteins and Their Binding Partners Come into View. Genes Dev. 24 (18), 2001–2012. doi:10.1101/gad.1951710
Beauchamp, E. M., Ringer, L., Bulut, G., Sajwan, K. P., Hall, M. D., Lee, Y.-C., et al. (2011). Arsenic Trioxide Inhibits Human Cancer Cell Growth and Tumor Development in Mice by Blocking Hedgehog/GLI Pathway. J. Clin. Invest. 121 (1), 148–160. doi:10.1172/JCI42874
Bidet, M., Joubert, O., Lacombe, B., Ciantar, M., Nehmé, R., Mollat, P., et al. (2011). The Hedgehog Receptor Patched Is Involved in Cholesterol Transport. PLoS One 6 (9), e23834. doi:10.1371/journal.pone.0023834
Bijlsma, M. F., Borensztajn, K. S., Roelink, H., Peppelenbosch, M. P., and Spek, C. A. (2007). Sonic Hedgehog Induces Transcription-independent Cytoskeletal Rearrangement and Migration Regulated by Arachidonate Metabolites. Cell Signal. 19 (12), 2596–2604. doi:10.1016/j.cellsig.2007.08.011
Brennan, D., Chen, X., Cheng, L., Mahoney, M., and Riobo, N. A. (2012). Noncanonical Hedgehog Signaling. Vitam Horm. 88, 55–72. doi:10.1016/B978-0-12-394622-5.00003-1
Briscoe, J., and Ericson, J. (2001). Specification of Neuronal Fates in the Ventral Neural Tube. Curr. Opin. Neurobiol. 11 (1), 43–49. doi:10.1016/s0959-4388(00)00172-0
Bufalieri, F., Infante, P., Bernardi, F., Caimano, M., Romania, P., Moretti, M., et al. (2019). ERAP1 Promotes Hedgehog-dependent Tumorigenesis by Controlling USP47-Mediated Degradation of βTrCP. Nat. Commun. 10 (1), 3304. doi:10.1038/s41467-019-11093-0
Buonamici, S., Williams, J., Morrissey, M., Wang, A., Guo, R., Vattay, A., et al. (2010). Interfering with Resistance to Smoothened Antagonists by Inhibition of the PI3K Pathway in Medulloblastoma. Sci. Transl. Med. 2 (51), 51ra70. doi:10.1126/scitranslmed.3001599
Burke, R., Nellen, D., Bellotto, M., Hafen, E., Senti, K.-A., Dickson, B. J., et al. (1999). Dispatched, a Novel Sterol-Sensing Domain Protein Dedicated to the Release of Cholesterol-Modified Hedgehog from Signaling Cells. Cell 99 (7), 803–815. doi:10.1016/s0092-8674(00)81677-3
Canettieri, G., Di Marcotullio, L., Greco, A., Coni, S., Antonucci, L., Infante, P., et al. (2010). Histone Deacetylase and Cullin3-RENKCTD11 Ubiquitin Ligase Interplay Regulates Hedgehog Signalling through Gli Acetylation. Nat. Cel Biol 12 (2), 132–142. doi:10.1038/ncb2013
Chang, C.-H., Zanini, M., Shirvani, H., Cheng, J.-S., Yu, H., Feng, C.-H., et al. (2019). Atoh1 Controls Primary Cilia Formation to Allow for SHH-Triggered Granule Neuron Progenitor Proliferation. Develop. Cel 48 (2), 184–199. e185. doi:10.1016/j.devcel.2018.12.017
Charron, F., Stein, E., Jeong, J., McMahon, A. P., and Tessier-Lavigne, M. (2003). The Morphogen Sonic Hedgehog Is an Axonal Chemoattractant that Collaborates with Netrin-1 in Midline Axon Guidance. Cell 113 (1), 11–23. doi:10.1016/s0092-8674(03)00199-5
Chen, X., Morales-Alcala, C. C., and Riobo-Del Galdo, N. A. (2018). Autophagic Flux Is Regulated by Interaction between the C-Terminal Domain of PATCHED1 and ATG101. Mol. Cancer Res. 16 (5), 909–919. doi:10.1158/1541-7786.MCR-17-0597
Chen, Y., Li, S., Tong, C., Zhao, Y., Wang, B., Liu, Y., et al. (2010). G Protein-Coupled Receptor Kinase 2 Promotes High-Level Hedgehog Signaling by Regulating the Active State of Smo through Kinase-dependent and Kinase-independent Mechanisms in Drosophila. Genes Dev. 24 (18), 2054–2067. doi:10.1101/gad.1948710
Chen, Y., Sasai, N., Ma, G., Yue, T., Jia, J., Briscoe, J., et al. (2011). Sonic Hedgehog Dependent Phosphorylation by CK1α and GRK2 Is Required for Ciliary Accumulation and Activation of Smoothened. Plos Biol. 9 (6), e1001083. doi:10.1371/journal.pbio.1001083
Cheng, F. Y., Fleming, J. T., and Chiang, C. (2018). Bergmann Glial Sonic Hedgehog Signaling Activity Is Required for Proper Cerebellar Cortical Expansion and Architecture. Develop. Biol. 440 (2), 152–166. doi:10.1016/j.ydbio.2018.05.015
Chiang, C., Litingtung, Y., Lee, E., Young, K. E., Corden, J. L., Westphal, H., et al. (1996). Cyclopia and Defective Axial Patterning in Mice Lacking Sonic Hedgehog Gene Function. Nature 383 (6599), 407–413. doi:10.1038/383407a0
Chinchilla, P., Xiao, L., Kazanietz, M. G., and Riobo, N. A. (2010). Hedgehog Proteins Activate Pro-angiogenic Responses in Endothelial Cells through Non-canonical Signaling Pathways. Cell Cycle 9 (3), 570–579. doi:10.4161/cc.9.3.10591
Chizhikov, V. V., Davenport, J., Zhang, Q., Shih, E. K., Cabello, O. A., Fuchs, J. L., et al. (2007). Cilia Proteins Control Cerebellar Morphogenesis by Promoting Expansion of the Granule Progenitor Pool. J. Neurosci. 27 (36), 9780–9789. doi:10.1523/JNEUROSCI.5586-06.2007
Clement, V., Sanchez, P., de Tribolet, N., Radovanovic, I., and Ruiz i Altaba, A. (2007). HEDGEHOG-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr. Biol. 17 (2), 165–172. doi:10.1016/j.cub.2006.11.033
Coni, S., Mancuso, A. B., Di Magno, L., Sdruscia, G., Manni, S., Serrao, S. M., et al. (2017). Selective Targeting of HDAC1/2 Elicits Anticancer Effects through Gli1 Acetylation in Preclinical Models of SHH Medulloblastoma. Sci. Rep. 7, 44079. doi:10.1038/srep44079
Corbit, K. C., Aanstad, P., Singla, V., Norman, A. R., Stainier, D. Y. R., and Reiter, J. F. (2005). Vertebrate Smoothened Functions at the Primary Cilium. Nature 437 (7061), 1018–1021. doi:10.1038/nature04117
Corrales, J. D., Rocco, G. L., Blaess, S., Guo, Q., and Joyner, A. L. (2004). Spatial Pattern of Sonic Hedgehog Signaling through Gli Genes during Cerebellum Development. Development 131 (22), 5581–5590. doi:10.1242/dev.01438
Dahmane, N., Sànchez, P., Gitton, Y., Palma, V., Sun, T., Beyna, M., et al. (2001). The Sonic Hedgehog-Gli Pathway Regulates Dorsal Brain Growth and Tumorigenesis. Development 128 (24), 5201–5212. doi:10.1242/dev.128.24.5201
Danesin, C., and Soula, C. (2017). Moving the Shh Source over Time: What Impact on Neural Cell Diversification in the Developing Spinal Cord? Jdb 5 (2), 4. doi:10.3390/jdb5020004
D’Arca, D., Zhao, X., Xu, W., Ramirez-Martinez, N. C., Iavarone, A., and Lasorella, A. (2010). Huwe1 Ubiquitin Ligase Is Essential to Synchronize Neuronal and Glial Differentiation in the Developing Cerebellum. Proc. Natl. Acad. Sci. U.S.A. 107 (13), 5875–5880. doi:10.1073/pnas.0912874107
De Mori, R., Romani, M., D’Arrigo, S., Zaki, M. S., Lorefice, E., Tardivo, S., et al. (2017). Hypomorphic Recessive Variants in SUFU Impair the Sonic Hedgehog Pathway and Cause Joubert Syndrome with Cranio-Facial and Skeletal Defects. Am. J. Hum. Genet. 101 (4), 552–563. doi:10.1016/j.ajhg.2017.08.017
Dey, J., Ditzler, S., Knoblaugh, S. E., Hatton, B. A., Schelter, J. M., Cleary, M. A., et al. (2012). A Distinct Smoothened Mutation Causes Severe Cerebellar Developmental Defects and Medulloblastoma in a Novel Transgenic Mouse Model. Mol. Cel Biol 32 (20), 4104–4115. doi:10.1128/MCB.00862-12
Dierker, T., Dreier, R., Petersen, A., Bordych, C., and Grobe, K. (2009). Heparan Sulfate-Modulated, Metalloprotease-Mediated Sonic Hedgehog Release from Producing Cells. J. Biol. Chem. 284 (12), 8013–8022. doi:10.1074/jbc.M806838200
Filges, I., Röthlisberger, B., Blattner, A., Boesch, N., Demougin, P., Wenzel, F., et al. (2011). Deletion in Xp22.11: PTCHD1 Is a Candidate Gene for X-Linked Intellectual Disability with or without Autism. Clin. Genet. 79 (1), 79–85. doi:10.1111/j.1399-0004.2010.01590.x
Fleming, J. T., He, W., Hao, C., Ketova, T., Pan, F. C., Wright, C. C. V., et al. (2013). The Purkinje Neuron Acts as a central Regulator of Spatially and Functionally Distinct Cerebellar Precursors. Develop. Cel 27 (3), 278–292. doi:10.1016/j.devcel.2013.10.008
Giacinti, C., and Giordano, A. (2006). RB and Cell Cycle Progression. Oncogene 25 (38), 5220–5227. doi:10.1038/sj.onc.1209615
Goel, S., DeCristo, M. J., McAllister, S. S., and Zhao, J. J. (2018). CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cel Biol. 28 (11), 911–925. doi:10.1016/j.tcb.2018.07.002
Gong, X., Qian, H., Cao, P., Zhao, X., Zhou, Q., Lei, J., et al. (2018). Structural Basis for the Recognition of Sonic Hedgehog by Human Patched1. Science 361 (6402), eaas8935. doi:10.1126/science.aas8935
Goodrich, L. V., Johnson, R. L., Milenkovic, L., McMahon, J. A., and Scott, M. P. (1996). Conservation of the Hedgehog/patched Signaling Pathway from Flies to Mice: Induction of a Mouse Patched Gene by Hedgehog. Genes Dev. 10 (3), 301–312. doi:10.1101/gad.10.3.301
Hall, E. T., Dillard, M. E., Stewart, D. P., Zhang, Y., Wagner, B., Levine, R. M., et al. (2021). Cytoneme Delivery of Sonic Hedgehog from Ligand-Producing Cells Requires Myosin 10 and a Dispatched-BOC/CDON Co-receptor Complex. Elife 10, e61432. doi:10.7554/eLife.61432
Han, Y.-G., and Alvarez-Buylla, A. (2010). Role of Primary Cilia in Brain Development and Cancer. Curr. Opin. Neurobiol. 20 (1), 58–67. doi:10.1016/j.conb.2009.12.002
Han, Y.-G., Kim, H. J., Dlugosz, A. A., Ellison, D. W., Gilbertson, R. J., and Alvarez-Buylla, A. (2009). Dual and Opposing Roles of Primary Cilia in Medulloblastoma Development. Nat. Med. 15 (9), 1062–1065. doi:10.1038/nm.2020
Han, Y.-G., Spassky, N., Romaguera-Ros, M., Garcia-Verdugo, J.-M., Aguilar, A., Schneider-Maunoury, S., et al. (2008). Hedgehog Signaling and Primary Cilia Are Required for the Formation of Adult Neural Stem Cells. Nat. Neurosci. 11 (3), 277–284. doi:10.1038/nn2059
Hatten, M. E., and Heintz, N. (1995). Mechanisms of Neural Patterning and Specification in the Development Cerebellum. Annu. Rev. Neurosci. 18, 385–408. doi:10.1146/annurev.ne.18.030195.002125
Hatton, B. A., Villavicencio, E. H., Tsuchiya, K. D., Pritchard, J. I., Ditzler, S., Pullar, B., et al. (2008). The Smo/Smo Model: Hedgehog-Induced Medulloblastoma with 90% Incidence and Leptomeningeal Spread. Cancer Res. 68 (6), 1768–1776. doi:10.1158/0008-5472.CAN-07-5092
He, X., Zhang, L., Chen, Y., Remke, M., Shih, D., Lu, F., et al. (2014). The G Protein α Subunit Gαs Is a Tumor Suppressor in Sonic Hedgehog−driven Medulloblastoma. Nat. Med. 20 (9), 1035–1042. doi:10.1038/nm.3666
Hildebrand, M. S., Griffin, N. G., Damiano, J. A., Cops, E. J., Burgess, R., Ozturk, E., et al. (2016). Mutations of the Sonic Hedgehog Pathway Underlie Hypothalamic Hamartoma with Gelastic Epilepsy. Am. J. Hum. Genet. 99 (2), 423–429. doi:10.1016/j.ajhg.2016.05.031
Hong, C.-J., and Hamilton, B. A. (2016). Zfp423 Regulates Sonic Hedgehog Signaling via Primary Cilium Function. Plos Genet. 12 (10), e1006357. doi:10.1371/journal.pgen.1006357
Huang, P., Nedelcu, D., Watanabe, M., Jao, C., Kim, Y., Liu, J., et al. (2016). Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 166 (5), 1176–1187. e1114. doi:10.1016/j.cell.2016.08.003
Huang, P., Zheng, S., Wierbowski, B. M., Kim, Y., Nedelcu, D., Aravena, L., et al. (2018). Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell 175 (1), 295–297. doi:10.1016/j.cell.2018.09.003
Humke, E. W., Dorn, K. V., Milenkovic, L., Scott, M. P., and Rohatgi, R. (2010). The Output of Hedgehog Signaling Is Controlled by the Dynamic Association between Suppressor of Fused and the Gli Proteins. Genes Dev. 24 (7), 670–682. doi:10.1101/gad.1902910
Incardona, J. P., Gruenberg, J., and Roelink, H. (2002). Sonic Hedgehog Induces the Segregation of Patched and Smoothened in Endosomes. Curr. Biol. 12 (12), 983–995. doi:10.1016/s0960-9822(02)00895-3
Infante, P., Faedda, R., Bernardi, F., Bufalieri, F., Lospinoso Severini, L., Alfonsi, R., et al. (2018). Itch/β-arrestin2-dependent Non-proteolytic Ubiquitylation of SuFu Controls Hedgehog Signalling and Medulloblastoma Tumorigenesis. Nat. Commun. 9 (1), 976. doi:10.1038/s41467-018-03339-0
Infante, P., Malfanti, A., Quaglio, D., Balducci, S., De Martin, S., Bufalieri, F., et al. (2021). Glabrescione B Delivery by Self-Assembling Micelles Efficiently Inhibits Tumor Growth in Preclinical Models of Hedgehog-dependent Medulloblastoma. Cancer Lett. 499, 220–231. doi:10.1016/j.canlet.2020.11.028
Infante, P., Mori, M., Alfonsi, R., Ghirga, F., Aiello, F., Toscano, S., et al. (2015). Gli1/DNA Interaction Is a Druggable Target for Hedgehog‐dependent Tumors. EMBO J. 34 (2), 200–217. doi:10.15252/embj.201489213
Ingham, P. W., and McMahon, A. P. (2001). Hedgehog Signaling in Animal Development: Paradigms and Principles. Genes Dev. 15 (23), 3059–3087. doi:10.1101/gad.938601
Izzi, L., Lévesque, M., Morin, S., Laniel, D., Wilkes, B. C., Mille, F., et al. (2011). Boc and Gas1 Each Form Distinct Shh Receptor Complexes with Ptch1 and Are Required for Shh-Mediated Cell Proliferation. Develop. Cel 20 (6), 788–801. doi:10.1016/j.devcel.2011.04.017
Jandke, A., Da Costa, C., Sancho, R., Nye, E., Spencer-Dene, B., and Behrens, A. (2011). The F-Box Protein Fbw7 Is Required for Cerebellar Development. Develop. Biol. 358 (1), 201–212. doi:10.1016/j.ydbio.2011.07.030
Jeong, J., and McMahon, A. P. (2005). Growth and Pattern of the Mammalian Neural Tube Are Governed by Partially Overlapping Feedback Activities of the Hedgehog Antagonists Patched 1 and Hhip1. Development 132 (1), 143–154. doi:10.1242/dev.01566
Jiwani, T., Kim, J. J., and Rosenblum, N. D. (2020). Suppressor of Fused (Sufu) Controls Cerebellum Granule Cell Proliferation by Suppressing Fgf8 and Spatially Regulating Gli Proteins. Development 147 (3), dev170274. doi:10.1242/dev.170274
Kawauchi, D., Ogg, R. J., Liu, L., Shih, D. J. H., Finkelstein, D., Murphy, B. L., et al. (2017). Novel MYC-Driven Medulloblastoma Models from Multiple Embryonic Cerebellar Cells. Oncogene 36 (37), 5231–5242. doi:10.1038/onc.2017.110
Kawauchi, D., Robinson, G., Uziel, T., Gibson, P., Rehg, J., Gao, C., et al. (2012). A Mouse Model of the Most Aggressive Subgroup of Human Medulloblastoma. Cancer Cell 21 (2), 168–180. doi:10.1016/j.ccr.2011.12.023
Kenney, A. M., Cole, M. D., and Rowitch, D. H. (2003). Nmycupregulation by Sonic Hedgehog Signaling Promotes Proliferation in Developing Cerebellar Granule Neuron Precursors. Development 130 (1), 15–28. doi:10.1242/dev.00182
Kenney, A. M., and Rowitch, D. H. (2000). Sonic Hedgehog Promotes G 1 Cyclin Expression and Sustained Cell Cycle Progression in Mammalian Neuronal Precursors. Mol. Cel Biol 20 (23), 9055–9067. doi:10.1128/MCB.20.23.9055-9067.2000
Kim, H., Lee, S.-Y., Jeong, H.-J., Kang, J.-S., Cho, H., and Leem, Y.-E. (2020). Cdo Is Required for Efficient Motor Neuron Generation of Embryonic Stem Cells. Ijsc 13 (3), 342–352. doi:10.15283/ijsc20037
Kim, J., Lee, J. J., Kim, J., Gardner, D., and Beachy, P. A. (2010). Arsenic Antagonizes the Hedgehog Pathway by Preventing Ciliary Accumulation and Reducing Stability of the Gli2 Transcriptional Effector. Proc. Natl. Acad. Sci. U.S.A. 107 (30), 13432–13437. doi:10.1073/pnas.1006822107
Kinnebrew, M., Iverson, E. J., Patel, B. B., Pusapati, G. V., Kong, J. H., Johnson, K. A., et al. (2019). Cholesterol Accessibility at the Ciliary Membrane Controls Hedgehog Signaling. Elife 8, e50051. doi:10.7554/eLife.50051
Knoepfler, P. S., Cheng, P. F., and Eisenman, R. N. (2002). N-myc Is Essential during Neurogenesis for the Rapid Expansion of Progenitor Cell Populations and the Inhibition of Neuronal Differentiation. Genes Dev. 16 (20), 2699–2712. doi:10.1101/gad.1021202
Kool, M., Jones, D. T. W., Jäger, N., Northcott, P. A., Pugh, T. J., Hovestadt, V., et al. (2014). Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 25 (3), 393–405. doi:10.1016/j.ccr.2014.02.004
Krauss, S., Concordet, J.-P., and Ingham, P. W. (1993). A Functionally Conserved Homolog of the Drosophila Segment Polarity Gene Hh Is Expressed in Tissues with Polarizing Activity in Zebrafish Embryos. Cell 75 (7), 1431–1444. doi:10.1016/0092-8674(93)90628-4
Kutscher, L. M., Okonechnikov, K., Batora, N. V., Clark, J., Silva, P. B. G., Vouri, M., et al. (2020). Functional Loss of a Noncanonical BCOR-PRC1.1 Complex Accelerates SHH-Driven Medulloblastoma Formation. Genes Dev. 34 (17-18), 1161–1176. doi:10.1101/gad.337584.120
Lee, J. J., von Kessler, D. P., Parks, S., and Beachy, P. A. (1992). Secretion and Localized Transcription Suggest a Role in Positional Signaling for Products of the Segmentation Gene Hedgehog. Cell 71 (1), 33–50. doi:10.1016/0092-8674(92)90264-d
Lewis, P. M., Gritli-Linde, A., Smeyne, R., Kottmann, A., and McMahon, A. P. (2004). Sonic Hedgehog Signaling Is Required for Expansion of Granule Neuron Precursors and Patterning of the Mouse Cerebellum. Develop. Biol. 270 (2), 393–410. doi:10.1016/j.ydbio.2004.03.007
Lin, Z., Li, S., Sheng, H., Cai, M., Ma, L. Y. S., Hu, L., et al. (2016). Suppression of GLI Sensitizes Medulloblastoma Cells to Mitochondria-Mediated Apoptosis. J. Cancer Res. Clin. Oncol. 142 (12), 2469–2478. doi:10.1007/s00432-016-2241-1
Liu, F., Jiang, W., Sui, Y., Meng, W., Hou, L., Li, T., et al. (2019). CDK7 Inhibition Suppresses Aberrant Hedgehog Pathway and Overcomes Resistance to Smoothened Antagonists. Proc. Natl. Acad. Sci. U.S.A. 116 (26), 12986–12995. doi:10.1073/pnas.1815780116
Lorenz, A., Deutschmann, M., Ahlfeld, J., Prix, C., Koch, A., Smits, R., et al. (2011). Severe Alterations of Cerebellar Cortical Development after Constitutive Activation of Wnt Signaling in Granule Neuron Precursors. Mol. Cel Biol 31 (16), 3326–3338. doi:10.1128/MCB.05718-11
Lospinoso Severini, L., Ghirga, F., Bufalieri, F., Quaglio, D., Infante, P., and Di Marcotullio, L. (2020). The SHH/GLI Signaling Pathway: a Therapeutic Target for Medulloblastoma. Expert Opin. Ther. Targets 24 (11), 1159–1181. doi:10.1080/14728222.2020.1823967
Luchetti, G., Sircar, R., Kong, J. H., Nachtergaele, S., Sagner, A., Byrne, E. F., et al. (2016). Cholesterol Activates the G-Protein Coupled Receptor Smoothened to Promote Hedgehog Signaling. Elife 5, e20304. doi:10.7554/eLife.20304
Maier, H., Dalianis, T., and Kostopoulou, O. N. (2021). New Approaches in Targeted Therapy for Medulloblastoma in Children. Anticancer Res. 41 (4), 1715–1726. doi:10.21873/anticanres.14936
Marigo, V., Johnson, R. L., Vortkamp, A., and Tabin, C. J. (1996). Sonic Hedgehog Differentially Regulates Expression ofGLIandGLI3during Limb Development. Develop. Biol. 180 (1), 273–283. doi:10.1006/dbio.1996.0300
Marino, S., Hoogervoorst, D., Brandner, S., and Berns, A. (2003). Rb and P107 Are Required for normal Cerebellar Development and Granule Cell Survival but Not for Purkinje Cell Persistence. Development 130 (15), 3359–3368. doi:10.1242/dev.00553
Mille, F., Tamayo-Orrego, L., Lévesque, M., Remke, M., Korshunov, A., Cardin, J., et al. (2014). The Shh Receptor Boc Promotes Progression of Early Medulloblastoma to Advanced Tumors. Develop. Cel 31 (1), 34–47. doi:10.1016/j.devcel.2014.08.010
Mille, F., Thibert, C., Fombonne, J., Rama, N., Guix, C., Hayashi, H., et al. (2009). The Patched Dependence Receptor Triggers Apoptosis through a DRAL-Caspase-9 Complex. Nat. Cel Biol 11 (6), 739–746. doi:10.1038/ncb1880
Miyashita, S., Owa, T., Seto, Y., Yamashita, M., Aida, S., Sone, M., et al. (2021). Cyclin D1 Controls Development of Cerebellar Granule Cell Progenitors through Phosphorylation and Stabilization of ATOH1. EMBO J. 40 (14), e105712. doi:10.15252/embj.2020105712
Mohler, J., and Vani, K. (1992). Molecular Organization and Embryonic Expression of the Hedgehog Gene Involved in Cell-Cell Communication in Segmental Patterning of Drosophila. Development 115 (4), 957–971. doi:10.1242/dev.115.4.957
Moreno, N., Schmidt, C., Ahlfeld, J., Poschl, J., Dittmar, S., Pfister, S. M., et al. (2014). Loss of Smarc Proteins Impairs Cerebellar Development. J. Neurosci. 34 (40), 13486–13491. doi:10.1523/JNEUROSCI.2560-14.2014
Murphy, B. L., Obad, S., Bihannic, L., Ayrault, O., Zindy, F., Kauppinen, S., et al. (2013). Silencing of the miR-17∼92 Cluster Family Inhibits Medulloblastoma Progression. Cancer Res. 73 (23), 7068–7078. doi:10.1158/0008-5472.CAN-13-0927
Niewiadomski, P., Kong, J. H., Ahrends, R., Ma, Y., Humke, E. W., Khan, S., et al. (2014). Gli Protein Activity Is Controlled by Multisite Phosphorylation in Vertebrate Hedgehog Signaling. Cel Rep. 6 (1), 168–181. doi:10.1016/j.celrep.2013.12.003
Niewiadomski, P., Zhujiang, A., Youssef, M., and Waschek, J. A. (2013). Interaction of PACAP with Sonic Hedgehog Reveals Complex Regulation of the Hedgehog Pathway by PKA. Cell Signal. 25 (11), 2222–2230. doi:10.1016/j.cellsig.2013.07.012
Northcott, P. A., Fernandez-L, A., Hagan, J. P., Ellison, D. W., Grajkowska, W., Gillespie, Y., et al. (2009). The miR-17/92 Polycistron Is Up-Regulated in Sonic Hedgehog-Driven Medulloblastomas and Induced by N-Myc in Sonic Hedgehog-Treated Cerebellar Neural Precursors. Cancer Res. 69 (8), 3249–3255. doi:10.1158/0008-5472.CAN-08-4710
Northcott, P. A., Jones, D. T. W., Kool, M., Robinson, G. W., Gilbertson, R. J., Cho, Y.-J., et al. (2012). Medulloblastomics: the End of the Beginning. Nat. Rev. Cancer 12 (12), 818–834. doi:10.1038/nrc3410
Nüsslein-Volhard, C., and Wieschaus, E. (1980). Mutations Affecting Segment Number and Polarity in Drosophila. Nature 287 (5785), 795–801. doi:10.1038/287795a0
Ocasio, J., Babcock, B., Malawsky, D., Weir, S. J., Loo, L., Simon, J. M., et al. (2019). scRNA-Seq in Medulloblastoma Shows Cellular Heterogeneity and Lineage Expansion Support Resistance to SHH Inhibitor Therapy. Nat. Commun. 10 (1), 5829. doi:10.1038/s41467-019-13657-6
Okada, A., Charron, F., Morin, S., Shin, D. S., Wong, K., Fabre, P. J., et al. (2006). Boc Is a Receptor for Sonic Hedgehog in the Guidance of Commissural Axons. Nature 444 (7117), 369–373. doi:10.1038/nature05246
Palma, V., Lim, D. A., Dahmane, N., Sànchez, P., Brionne, T. C., Herzberg, C. D., et al. (2005). Sonic Hedgehog Controls Stem Cell Behavior in the Postnatal and Adult Brain. Development 132 (2), 335–344. doi:10.1242/dev.01567
Pan, S., Wu, X., Jiang, J., Gao, W., Wan, Y., Cheng, D., et al. (2010). Discovery of NVP-Lde225, a Potent and Selective Smoothened Antagonist. ACS Med. Chem. Lett. 1 (3), 130–134. doi:10.1021/ml1000307
Pan, Y., Bai, C. B., Joyner, A. L., and Wang, B. (2006). Sonic Hedgehog Signaling Regulates Gli2 Transcriptional Activity by Suppressing its Processing and Degradation. Mol. Cel Biol 26 (9), 3365–3377. doi:10.1128/MCB.26.9.3365-3377.2006
Pei, Y., Brun, S. N., Markant, S. L., Lento, W., Gibson, P., Taketo, M. M., et al. (2012). WNT Signaling Increases Proliferation and Impairs Differentiation of Stem Cells in the Developing Cerebellum. Development 139 (10), 1724–1733. doi:10.1242/dev.050104
Pietrobono, S., Gagliardi, S., and Stecca, B. (2019). Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors beyond Smoothened. Front. Genet. 10, 556. doi:10.3389/fgene.2019.00556
Pogoriler, J., Millen, K., Utset, M., and Du, W. (2006). Loss of Cyclin D1 Impairs Cerebellar Development and Suppresses Medulloblastoma Formation. Development 133 (19), 3929–3937. doi:10.1242/dev.02556
Polizio, A. H., Chinchilla, P., Chen, X., Kim, S., Manning, D. R., and Riobo, N. A. (2011a). Heterotrimeric Gi Proteins Link Hedgehog Signaling to Activation of Rho Small GTPases to Promote Fibroblast Migration. J. Biol. Chem. 286 (22), 19589–19596. doi:10.1074/jbc.M110.197111
Polizio, A. H., Chinchilla, P., Chen, X., Manning, D. R., and Riobo, N. A. (2011b). Sonic Hedgehog Activates the GTPases Rac1 and RhoA in a Gli-independent Manner through Coupling of Smoothened to G I ProteinsA Presentation from the 1st International HEALING Meeting: Hh-Gli Signaling in Development, Regeneration and Disease, Kolymbari, Crete, 23 to 25 June 2011. Sci. Signal. 4, pt7. doi:10.1126/scisignal.2002396
Pöschl, J., Bartels, M., Ohli, J., Bianchi, E., Kuteykin-Teplyakov, K., Grammel, D., et al. (2014). Wnt/β-catenin Signaling Inhibits the Shh Pathway and Impairs Tumor Growth in Shh-dependent Medulloblastoma. Acta Neuropathol. 127 (4), 605–607. doi:10.1007/s00401-014-1258-2
Purzner, T., Purzner, J., Buckstaff, T., Cozza, G., Gholamin, S., Rusert, J. M., et al. (2018). Developmental Phosphoproteomics Identifies the Kinase CK2 as a Driver of Hedgehog Signaling and a Therapeutic Target in Medulloblastoma. Sci. Signal. 11 (547), eaau5147. doi:10.1126/scisignal.aau5147
Qi, X., Schmiege, P., Coutavas, E., Wang, J., and Li, X. (2018). Structures of Human Patched and its Complex with Native Palmitoylated Sonic Hedgehog. Nature 560 (7716), 128–132. doi:10.1038/s41586-018-0308-7
Raffoux, E., Rousselot, P., Poupon, J., Daniel, M.-T., Cassinat, B., Delarue, R., et al. (2003). Combined Treatment with Arsenic Trioxide and All-Trans-Retinoic Acid in Patients with Relapsed Acute Promyelocytic Leukemia. Jco 21 (12), 2326–2334. doi:10.1200/JCO.2003.01.149
Raleigh, D. R., Choksi, P. K., Krup, A. L., Mayer, W., Santos, N., and Reiter, J. F. (2017). Hedgehog Signaling Drives Medulloblastoma Growth via CDK6. J. Clin. Invest. 128 (1), 120–124. doi:10.1172/JCI92710
Renault, M.-A., Roncalli, J., Tongers, J., Thorne, T., Klyachko, E., Misener, S., et al. (2010). Sonic Hedgehog Induces Angiogenesis via Rho Kinase-dependent Signaling in Endothelial Cells. J. Mol. Cell Cardiol. 49 (3), 490–498. doi:10.1016/j.yjmcc.2010.05.003
Riddle, R. D., Johnson, R. L., Laufer, E., and Tabin, C. (1993). Sonic Hedgehog Mediates the Polarizing Activity of the ZPA. Cell 75 (7), 1401–1416. doi:10.1016/0092-8674(93)90626-2
Riobo, N. A., Saucy, B., Dilizio, C., and Manning, D. R. (2006). Activation of Heterotrimeric G Proteins by Smoothened. Proc. Natl. Acad. Sci. U.S.A. 103 (33), 12607–12612. doi:10.1073/pnas.0600880103
Robarge, K. D., Brunton, S. A., Castanedo, G. M., Cui, Y., Dina, M. S., Goldsmith, R., et al. (2009). GDC-0449-a Potent Inhibitor of the Hedgehog Pathway. Bioorg. Med. Chem. Lett. 19 (19), 5576–5581. doi:10.1016/j.bmcl.2009.08.049
Robbins, D. J., Fei, D. L., and Riobo, N. A. (2012). The Hedgehog Signal Transduction Network. Sci. Signal. 5 (246), re6. doi:10.1126/scisignal.2002906
Rohatgi, R., Milenkovic, L., Corcoran, R. B., and Scott, M. P. (2009). Hedgehog Signal Transduction by Smoothened: Pharmacologic Evidence for a 2-step Activation Process. Proc. Natl. Acad. Sci. U.S.A. 106 (9), 3196–3201. doi:10.1073/pnas.0813373106
Rohatgi, R., Milenkovic, L., and Scott, M. P. (2007). Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 317 (5836), 372–376. doi:10.1126/science.1139740
Roper, R. J., Baxter, L. L., Saran, N. G., Klinedinst, D. K., Beachy, P. A., and Reeves, R. H. (2006). Defective Cerebellar Response to Mitogenic Hedgehog Signaling in Down's Syndrome Mice. Proc. Natl. Acad. Sci. U.S.A. 103 (5), 1452–1456. doi:10.1073/pnas.0510750103
Ruiz i Altaba, A., Mas, C., and Stecca, B. (2007). The Gli Code: an Information Nexus Regulating Cell Fate, Stemness and Cancer. Trends Cel Biol. 17 (9), 438–447. doi:10.1016/j.tcb.2007.06.007
Ruiz i Altaba, A., Palma, V., and Dahmane, N. (2002). Hedgehog-GLI Signaling and the Growth of the Brain. Nat. Rev. Neurosci. 3 (1), 24–33. doi:10.1038/nrn704
Sasaki, N., Kurisu, J., and Kengaku, M. (2010). Sonic Hedgehog Signaling Regulates Actin Cytoskeleton via Tiam1-Rac1 cascade during Spine Formation. Mol. Cell Neurosci. 45 (4), 335–344. doi:10.1016/j.mcn.2010.07.006
Schüller, U., Heine, V. M., Mao, J., Kho, A. T., Dillon, A. K., Han, Y.-G., et al. (2008). Acquisition of Granule Neuron Precursor Identity Is a Critical Determinant of Progenitor Cell Competence to Form Shh-Induced Medulloblastoma. Cancer Cell 14 (2), 123–134. doi:10.1016/j.ccr.2008.07.005
Shakhova, O., Leung, C., van Montfort, E., Berns, A., and Marino, S. (2006). Lack of Rb and P53 Delays Cerebellar Development and Predisposes to Large Cell Anaplastic Medulloblastoma through Amplification ofN-MycandPtch2. Cancer Res. 66 (10), 5190–5200. doi:10.1158/0008-5472.CAN-05-3545
Shi, X., Wang, Q., Gu, J., Xuan, Z., and Wu, J. I. (2016). SMARCA4/Brg1 Coordinates Genetic and Epigenetic Networks Underlying Shh-type Medulloblastoma Development. Oncogene 35 (44), 5746–5758. doi:10.1038/onc.2016.108
Shi, X., Zhang, Z., Zhan, X., Cao, M., Satoh, T., Akira, S., et al. (2014). An Epigenetic Switch Induced by Shh Signalling Regulates Gene Activation during Development and Medulloblastoma Growth. Nat. Commun. 5, 5425. doi:10.1038/ncomms6425
Sjostrom, S. K., Finn, G., Hahn, W. C., Rowitch, D. H., and Kenney, A. M. (2005). The Cdk1 Complex Plays a Prime Role in Regulating N-Myc Phosphorylation and Turnover in Neural Precursors. Develop. Cel 9 (3), 327–338. doi:10.1016/j.devcel.2005.07.014
Smeyne, R. J., Chu, T., Lewin, A., Bian, F., S.-Crisman, S., Kunsch, C., et al. (1995). Local Control of Granule Cell Generation by Cerebellar Purkinje Cells. Mol. Cell Neurosci. 6 (3), 230–251. doi:10.1006/mcne.1995.1019
Spassky, N., Han, Y.-G., Aguilar, A., Strehl, L., Besse, L., Laclef, C., et al. (2008). Primary Cilia Are Required for Cerebellar Development and Shh-dependent Expansion of Progenitor Pool. Develop. Biol. 317 (1), 246–259. doi:10.1016/j.ydbio.2008.02.026
Suryo Rahmanto, A., Savov, V., Brunner, A., Bolin, S., Weishaupt, H., Malyukova, A., et al. (2016). FBW7 Suppression Leads to SOX9 Stabilization and Increased Malignancy in Medulloblastoma. EMBO J. 35 (20), 2192–2212. doi:10.15252/embj.201693889
Suzuki, H., Kumar, S. A., Shuai, S., Diaz-Navarro, A., Gutierrez-Fernandez, A., De Antonellis, P., et al. (2019). Recurrent Noncoding U1 snRNA Mutations Drive Cryptic Splicing in SHH Medulloblastoma. Nature 574 (7780), 707–711. doi:10.1038/s41586-019-1650-0
Swartling, F. J., Grimmer, M. R., Hackett, C. S., Northcott, P. A., Fan, Q.-W., Goldenberg, D. D., et al. (2010). Pleiotropic Role for MYCN in Medulloblastoma. Genes Dev. 24 (10), 1059–1072. doi:10.1101/gad.1907510
Swartling, F. J., Savov, V., Persson, A. I., Chen, J., Hackett, C. S., Northcott, P. A., et al. (2012). Distinct Neural Stem Cell Populations Give Rise to Disparate Brain Tumors in Response to N-MYC. Cancer Cell 21 (5), 601–613. doi:10.1016/j.ccr.2012.04.012
Tabata, T., Eaton, S., and Kornberg, T. B. (1992). The Drosophila Hedgehog Gene Is Expressed Specifically in Posterior Compartment Cells and Is a Target of Engrailed Regulation. Genes Develop. 6 (12B), 2635–2645. doi:10.1101/gad.6.12b.2635
Tamayo-Orrego, L., Gallo, D., Racicot, F., Bemmo, A., Mohan, S., Ho, B., et al. (2020). Sonic Hedgehog Accelerates DNA Replication to Cause Replication Stress Promoting Cancer Initiation in Medulloblastoma. Nat. Cancer 1 (8), 840–854. doi:10.1038/s43018-020-0094-7
Tang, Y., Gholamin, S., Schubert, S., Willardson, M. I., Lee, A., Bandopadhayay, P., et al. (2014). Epigenetic Targeting of Hedgehog Pathway Transcriptional Output through BET Bromodomain Inhibition. Nat. Med. 20 (7), 732–740. doi:10.1038/nm.3613
Thibert, C., Teillet, M.-A., Lapointe, F., Mazelin, L., Le Douarin, N. M., and Mehlen, P. (2003). Inhibition of Neuroepithelial Patched-Induced Apoptosis by Sonic Hedgehog. Science 301 (5634), 843–846. doi:10.1126/science.1085405
Tolosa, E. J., Fernandez-Barrena, M. G., Iguchi, E., McCleary-Wheeler, A. L., Carr, R. M., Almada, L. L., et al. (2020). GLI1/GLI2 Functional Interplay Is Required to Control Hedgehog/GLI Targets Gene Expression. Biochem. J. 477 (17), 3131–3145. doi:10.1042/BCJ20200335
Tukachinsky, H., Lopez, L. V., and Salic, A. (2010). A Mechanism for Vertebrate Hedgehog Signaling: Recruitment to Cilia and Dissociation of SuFu-Gli Protein Complexes. J. Cel Biol 191 (2), 415–428. doi:10.1083/jcb.201004108
Tukachinsky, H., Petrov, K., Watanabe, M., and Salic, A. (2016). Mechanism of Inhibition of the Tumor Suppressor Patched by Sonic Hedgehog. Proc. Natl. Acad. Sci. U.S.A. 113 (40), E5866–E5875. doi:10.1073/pnas.1606719113
Turner, N. C., Slamon, D. J., Ro, J., Bondarenko, I., Im, S.-A., Masuda, N., et al. (2018). Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 379 (20), 1926–1936. doi:10.1056/NEJMoa1810527
Tuson, M., He, M., and Anderson, K. V. (2011). Protein Kinase A Acts at the Basal Body of the Primary Cilium to Prevent Gli2 Activation and Ventralization of the Mouse Neural Tube. Development 138 (22), 4921–4930. doi:10.1242/dev.070805
Uziel, T., Karginov, F. V., Xie, S., Parker, J. S., Wang, Y.-D., Gajjar, A., et al. (2009). The miR-17 ∼ 92 Cluster Collaborates with the Sonic Hedgehog Pathway in Medulloblastoma. Proc. Natl. Acad. Sci. U.S.A. 106 (8), 2812–2817. doi:10.1073/pnas.0809579106
Van Mater, D., Gururangan, S., Becher, O., Campagne, O., Leary, S., Phillips, J. J., et al. (2021). A Phase I Trial of the CDK 4/6 Inhibitor Palbociclib in Pediatric Patients with Progressive Brain Tumors: A Pediatric Brain Tumor Consortium Study (PBTC‐042). Pediatr. Blood Cancer 68 (4), e28879. doi:10.1002/pbc.28879
Wang, V. Y., Rose, M. F., and Zoghbi, H. Y. (2005). Math1 Expression Redefines the Rhombic Lip Derivatives and Reveals Novel Lineages within the Brainstem and Cerebellum. Neuron 48 (1), 31–43. doi:10.1016/j.neuron.2005.08.024
Waszak, S. M., Northcott, P. A., Buchhalter, I., Robinson, G. W., Sutter, C., Groebner, S., et al. (2018). Spectrum and Prevalence of Genetic Predisposition in Medulloblastoma: a Retrospective Genetic Study and Prospective Validation in a Clinical Trial Cohort. Lancet Oncol. 19 (6), 785–798. doi:10.1016/S1470-2045(18)30242-0
Waszak, S. M., Robinson, G. W., Gudenas, B. L., Smith, K. S., Forget, A., Kojic, M., et al. (2020). Germline Elongator Mutations in Sonic Hedgehog Medulloblastoma. Nature 580 (7803), 396–401. doi:10.1038/s41586-020-2164-5
Welcker, M., Orian, A., Jin, J., Grim, J. A., Harper, J. W., Eisenman, R. N., et al. (2004). The Fbw7 Tumor Suppressor Regulates Glycogen Synthase Kinase 3 Phosphorylation-dependent C-Myc Protein Degradation. Proc. Natl. Acad. Sci. U.S.A. 101 (24), 9085–9090. doi:10.1073/pnas.0402770101
Whyte, W. A., Orlando, D. A., Hnisz, D., Abraham, B. J., Lin, C. Y., Kagey, M. H., et al. (2013). Master Transcription Factors and Mediator Establish Super-enhancers at Key Cell Identity Genes. Cell 153 (2), 307–319. doi:10.1016/j.cell.2013.03.035
Wong, S. Y., Seol, A. D., So, P.-L., Ermilov, A. N., Bichakjian, C. K., Epstein, E. H., et al. (2009). Primary Cilia Can Both Mediate and Suppress Hedgehog Pathway-dependent Tumorigenesis. Nat. Med. 15 (9), 1055–1061. doi:10.1038/nm.2011
Xiao, X., Tang, J.-J., Peng, C., Wang, Y., Fu, L., Qiu, Z.-P., et al. (2017). Cholesterol Modification of Smoothened Is Required for Hedgehog Signaling. Mol. Cel 66 (1), 154–162. e110. doi:10.1016/j.molcel.2017.02.015
Yam, P. T., Langlois, S. D., Morin, S., and Charron, F. (2009). Sonic Hedgehog Guides Axons through a Noncanonical, Src-family-kinase-dependent Signaling Pathway. Neuron 62 (3), 349–362. doi:10.1016/j.neuron.2009.03.022
Yang, Z.-J., Ellis, T., Markant, S. L., Read, T.-A., Kessler, J. D., Bourboulas, M., et al. (2008). Medulloblastoma Can Be Initiated by Deletion of Patched in Lineage-Restricted Progenitors or Stem Cells. Cancer Cell 14 (2), 135–145. doi:10.1016/j.ccr.2008.07.003
Yao, C. D., Haensel, D., Gaddam, S., Patel, T., Atwood, S. X., Sarin, K. Y., et al. (2020). AP-1 and TGFss Cooperativity Drives Non-canonical Hedgehog Signaling in Resistant Basal Cell Carcinoma. Nat. Commun. 11 (1), 5079. doi:10.1038/s41467-020-18762-5
Yauch, R. L., Dijkgraaf, G. J. P., Alicke, B., Januario, T., Ahn, C. P., Holcomb, T., et al. (2009). Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 326 (5952), 572–574. doi:10.1126/science.1179386
Yeh, E., Cunningham, M., Arnold, H., Chasse, D., Monteith, T., Ivaldi, G., et al. (2004). A Signalling Pathway Controlling C-Myc Degradation that Impacts Oncogenic Transformation of Human Cells. Nat. Cel Biol 6 (4), 308–318. doi:10.1038/ncb1110
Yin, W.-C., Satkunendran, T., Mo, R., Morrissy, S., Zhang, X., Huang, E. S., et al. (2019). Dual Regulatory Functions of SUFU and Targetome of GLI2 in SHH Subgroup Medulloblastoma. Develop. Cel 48 (2), 167–183. e165. doi:10.1016/j.devcel.2018.11.015
Yue, S., Tang, L.-Y., Tang, Y., Tang, Y., Shen, Q.-H., Ding, J., et al. (2014). Requirement of Smurf-Mediated Endocytosis of Patched1 in Sonic Hedgehog Signal Reception. Elife 3. doi:10.7554/eLife.02555
Zhan, X., Shi, X., Zhang, Z., Chen, Y., and Wu, J. I. (2011). Dual Role of Brg Chromatin Remodeling Factor in Sonic Hedgehog Signaling during Neural Development. Proc. Natl. Acad. Sci. U.S.A. 108 (31), 12758–12763. doi:10.1073/pnas.1018510108
Zhang, B., Zhuang, T., Lin, Q., Yang, B., Xu, X., Xin, G., et al. (2019). Patched1-ArhGAP36-PKA-Inversin axis Determines the Ciliary Translocation of Smoothened for Sonic Hedgehog Pathway Activation. Proc. Natl. Acad. Sci. U.S.A. 116 (3), 874–879. doi:10.1073/pnas.1804042116
Zhang, Y.-w., Qu, H.-b., Long, N., Leng, X.-y., Liu, Y.-q., and Yang, Y. (2021). A Rare Mutant of OFD1 Gene Responsible for Joubert Syndrome with Significant Phenotype Variation. Mol. Genet. Genomics 296 (1), 33–40. doi:10.1007/s00438-020-01726-1
Zhang, Y., Bulkley, D. P., Xin, Y., Roberts, K. J., Asarnow, D. E., Sharma, A., et al. (2018). Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 175 (5), 1352–1364. e1314. doi:10.1016/j.cell.2018.10.026
Zhao, X., Heng, J. I.-T., Guardavaccaro, D., Jiang, R., Pagano, M., Guillemot, F., et al. (2008). The HECT-Domain Ubiquitin Ligase Huwe1 Controls Neural Differentiation and Proliferation by Destabilizing the N-Myc Oncoprotein. Nat. Cel Biol 10 (6), 643–653. doi:10.1038/ncb1727
Zhao, Y., Tong, C., and Jiang, J. (2007). Hedgehog Regulates Smoothened Activity by Inducing a Conformational Switch. Nature 450 (7167), 252–258. doi:10.1038/nature06225
Zheng, T., Ghasemi, D. R., Okonechnikov, K., Korshunov, A., Sill, M., Maass, K. K., et al. (2021). Cross-Species Genomics Reveals Oncogenic Dependencies in ZFTA/C11orf95 Fusion-Positive Supratentorial Ependymomas. Cancer Discov. 11 (9), 2230–2247. doi:10.1158/2159-8290.CD-20-0963
Zindy, F., Kawauchi, D., Lee, Y., Ayrault, O., Ben Merzoug, L., McKinnon, P. J., et al. (2014). Role of the miR-17∼92 Cluster Family in Cerebellar and Medulloblastoma Development. Biol. Open 3 (7), 597–605. doi:10.1242/bio.20146734
Keywords: sonic hedgehog, cerebellum, medulloblastoma, brain tumor, patched1, smoothened, GLI family
Citation: Wang W, Shiraishi R and Kawauchi D (2022) Sonic Hedgehog Signaling in Cerebellar Development and Cancer. Front. Cell Dev. Biol. 10:864035. doi: 10.3389/fcell.2022.864035
Received: 28 January 2022; Accepted: 28 March 2022;
Published: 29 April 2022.
Edited by:
Aimin Liu, The Pennsylvania State University (PSU), United StatesReviewed by:
Lucia Di Marcotullio, Sapienza University of Rome, ItalyCopyright © 2022 Wang, Shiraishi and Kawauchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daisuke Kawauchi, ZC5rYXdhdWNoaUBuY25wLmdvLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.