 Laura L. Thompson1,2
Laura L. Thompson1,2 Kirk J. McManus
Kirk J. McManus
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 08 March 2022
Sec. Cell Growth and Division
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.859582
This article is part of the Research Topic Editors' Showcase 2021: Insights in Cell Growth and Division View all 11 articles
The S-phase Kinase-Associated Protein 1 (SKP1) is a core component of the SKP1, Cullin 1, F-box protein (SCF) complex, an E3 ubiquitin ligase that serves to poly-ubiquitinate a vast array of protein targets as a signal for their proteasomal degradation, thereby playing a critical role in the regulation of downstream biological processes. Many of the proteins regulated by SKP1 and the SCF complex normally function within pathways that are essential for maintaining genome stability, including DNA damage repair, apoptotic signaling, and centrosome dynamics. Accordingly, aberrant SKP1 and SCF complex expression and function is expected to disrupt these essential pathways, which may have pathological implications in diseases like cancer. In this review, we summarize the central role SKP1 plays in regulating essential cellular processes; we describe functional models in which SKP1 expression is altered and the corresponding impacts on genome stability; and we discuss the prevalence of SKP1 somatic copy number alterations, mutations, and altered protein expression across different cancer types, to identify a potential link between SKP1 and SCF complex dysfunction to chromosome/genome instability and cancer pathogenesis. Ultimately, understanding the role of SKP1 in driving chromosome instability will expand upon our rudimentary understanding of the key events required for genome/chromosome stability that may aid in our understanding of cancer pathogenesis, which will be critical for future studies to establish whether SKP1 may be useful as prognostic indicator or as a therapeutic target.
The SKP1 (S-phase Kinase-Associated Protein 1), CUL1 (Cullin 1), F-box protein complex (SCF complex) is an E3 ubiquitin ligase that regulates a vast array of cellular processes (e.g., cell cycle, DNA damage response, apoptosis and centrosome homeostasis) that are key to maintaining genome stability and ensuring proper segregation of genetic material into daughter cells. SKP1 is an invariable, core component of the SCF complex that functions as the adaptor protein responsible for binding CUL1 and recruiting various F-box proteins for SCF complex formation. This critical role of SKP1 enables the poly-ubiquitination of a diverse array of substrates targeted by the variable F-box proteins for subsequent proteolytic degradation by the 26S proteasome, making SKP1 activity essential to regulate the myriad of cellular processes governed by the SCF complex. Accordingly, genetic aberrations altering SKP1 expression and/or function will adversely impact the many biological processes normally required to maintain genome stability, and thus aberrant SKP1 expression is predicted to contribute to cancer pathogenesis. In support of this possibility, somatic alterations in SKP1, including mutations, deletions and mRNA misexpression occur frequently in a wide variety of cancer types.
Despite the many associations between altered SKP1 expression and cancer, the fundamental impact aberrant SKP1 expression and/or function has on oncogenesis remains unclear. This review describes how aberrant SKP1 expression and function impacts many biological pathways that are essential to maintain genome instability that when altered, are implicated in oncogenesis. Accordingly, these observations support the possibility that aberrant SKP1 expression may be a contributing pathogenic event, although definitive empirical data are still needed. First, we provide a historical background of mammalian SKP1, describing key characteristics at the gene/protein level as well as its relationship with orthologs from other species. We then discuss how SKP1 interacts with the other SCF complex members and their collective role within the ubiquitin proteasome system (UPS). Next, we describe the roles that SKP1 and the SCF complex have within three biological processes that are essential for maintaining genome stability, an enabling hallmark of cancer (Hanahan and Weinberg, 2011) including: 1) altered DNA damage response and apoptosis; 2) aberrant centrosome duplication and dynamics; and 3) chromosome stability. To further support a potential role in cancer pathogenesis, we detail the occurrence and frequency of SKP1 alterations within cancer patient samples. Finally, we conclude with a brief discussion on future therapeutic strategies that seek to exploit altered SKP1 expression and the downstream impacts of aberrant protein targeting and destruction.
Mammalian SKP1, also referred to as the Cyclin-A/Cyclin Dependent Kinase (CDK) 2-Associated Protein 19 (P19), was originally identified in 1980 within the guinea pig organ of corti by 2D polyacrylamide gel electrophoresis and was consequently named Organ of Corti Protein 2 (OCP2) (Thalmann et al., 1980; Thalmann et al., 2003). In the 1990s, a series of research groups independently investigated SKP1/P19 and its aliases OCP2 and TCEB1L as seemingly distinct genes. In 1995, Zhang and others (Zhang et al., 1995) determined that human SKP1/P19 interacted with the Cyclin A/CDK2 complex, suggesting a potential role in cell cycle regulation, and subsequently sequenced the SKP1/P19 DNA coding regions. Concurrently, Chen et al (Chen et al., 1995) sequenced human OCP2, while Sowden et al (Sowden et al., 1995) presented the cDNA sequence for a novel gene designated TCEB1L, suspected to encode a transcription elongation factor. Additionally, Bai and others (Bai et al., 1996) identified the yeast and human orthologs of SKP1 as a suppressor of cdc4 (cell division control 4) and as a Cyclin F-binding protein, respectively, in two independent lines of research. It was not until 1997, when Liang et al (Liang et al., 1997) noted that the coding sequences detailed above for human SKP1/P19, OCP2, and TCEB1L were identical and that the above genes encoding distinct roles in diverse cellular processes were in fact, one and the same.
The human SKP1 gene spans a region of 28,097 base pairs (bp) on chromosome 5q31.1 and encodes two protein coding mRNA transcripts of different lengths, 2,028 bp and 2,714 bp that are generated by alternative splicing. The transcripts are translated into two protein isoforms, 163 (Isoform B) and 160 (Isoform A) amino acids in size that differ at their carboxy-terminal regions (Figure 1A) (2009). Although Isoform B is considered the prototypic SKP1 protein (Schulman et al., 2000; Yamanaka et al., 2002; Kong et al., 2004), the potential functional differences between the two isoforms have yet to be fully explored. Nevertheless, a study in Saccharomyces cerevisiae revealed that the tryptophan residue at position 159 (Trp159), present only in human Isoform B (Figure 1A), is essential for its in vivo function. As Trp159 is evolutionarily conserved from yeast to humans, these experimental findings in S. cerevisiae suggest there may only be one functional human isoform (i.e., Isoform B) (Schulman et al., 2000). To test this possibility, isoform-specific studies must be designed to formally interrogate the functional differences and discern whether the non-prototypic SKP1 Isoform A has developed a de novo, Trp159-independent function during evolution.
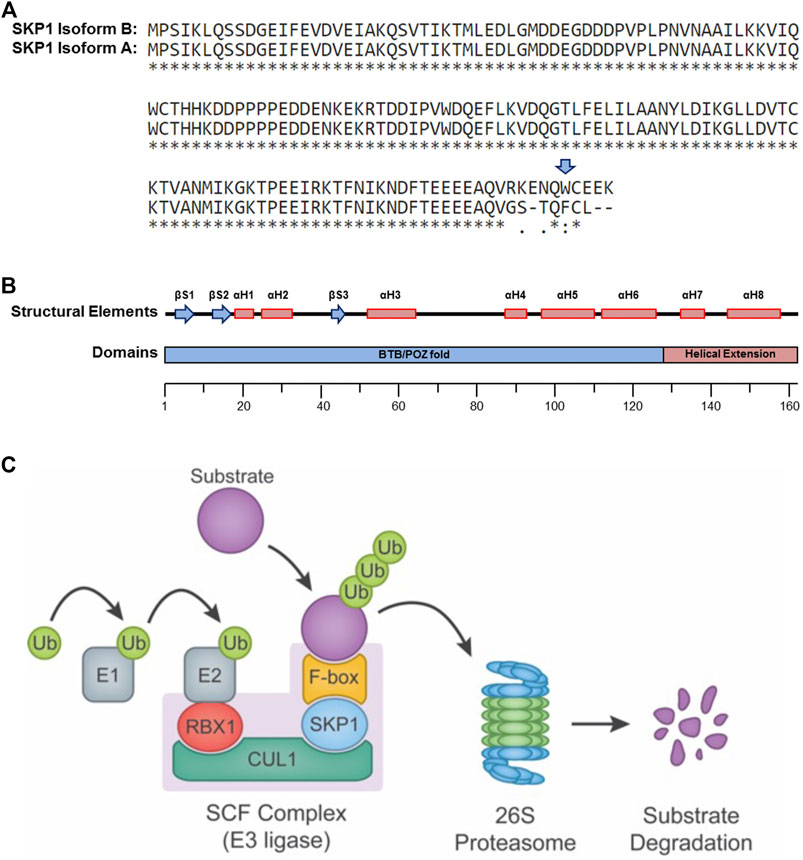
FIGURE 1. SKP1 Structure and Function. (A) Single amino acid sequence alignment of the two SKP1 (isoform A and isoform B) reveals sequence divergence within their carboxy-terminal tails. Sequence alignments performed using UniProt (Universal Protein Resource) (Altschul et al., 1990). Tryptophan 159 (W159), present only within Isoform B is highlighted by a blue arrow. A “*” identifies conserved amino acid positions, while “:” and “.” identify amino acid positions with similar or weakly similar properties, respectively. (B) Schematic depiction for the secondary structural elements (top) and protein domains (bottom) of SKP1 isoform B (βS, beta-sheet; αH, alpha-helix; BTB/POZ, broad complex, tramtrack and bric-à-brac(BTB)/poxviruses and zinc finger (POZ)). (C) Diagram depicting the SCF complex and its function in targeting protein substrates for poly-ubiquitination and proteolytic degradation by the 26S proteasome. The SCF complex consists of three invariable components (RBX1, CUL1, and SKP1) and one of 69 variable F-box proteins that confers substrate specificity. In general, ubiquitin (Ub) moieties are transferred to a protein substrate through the sequential actions of an E1 (activating) and an E2 (conjugating) enzymes in conjunction with an E3 (ligase) enzyme (e.g., SCF complex).
SKP1 Isoform B (Figure 1B) is ∼18 kDa and harbors a 128 residue domain at the amino-terminus resembling the α-helix/β-sheet structure of a BTB/POZ (broad complex, tramtrack and bric-à-brac/poxviruses and zinc finger) fold domain, but with an α-helical insertion (αH4) (Schulman et al., 2000). This domain is essential for heterodimerization and is required for the binding of SKP1 to the SCF complex scaffolding protein, CUL1. Additionally, SKP1 harbors a two-helix, carboxy-terminal extension (αH7 and αH48) that cooperates with elements of the BTB/POZ fold to create a variable interaction motif that binds F-box domains (Figure 1B). There are 69 distinct proteins containing F-box domains (i.e., F-box proteins) that have been identified in mammals (Jin et al., 2004), each with its own set of protein targets. Thus, SKP1 serves as an adaptor between CUL1 and one of 69 F-box proteins, playing a critical role in the formation of up to 69 distinct SCF complexes (Figure 1C) (Ng et al., 1998; Yoshida et al., 2011) and the regulation of a diverse set of protein targets and pathways.
The amino acid sequences and structural elements of human SKP1 share a significant degree of amino acid sequence similarity with its counterparts in model organisms including S. cerevisiae (98% similar; 43% identical), Mus musculus (100% similar; 99% identical), Drosophila melanogaster (100% similar; 77% identical), Caenorhabditis elegans (97% similar; 71% identical) and Arabidopsis thaliana (71% similar; 58% identical) (2009). Beyond these sequence and structural similarities, functional conservation is also readily apparent between humans and model organisms. For example, human SKP1 has been shown to functionally compensate for Skp1 deletion in S. cerevisiae (Bai et al., 1996). Although only one functional isoform is proposed to exist in humans, studies in C. elegans have identified at least 21 SKP1 paralogs or Skp1-related genes, each exhibiting varying degrees of sequence similarity with human SKP1 (Yamanaka et al., 2002). Similarly, D. melanogaster and A. thaliana harbor 7 and 19 Skp1-related genes, respectively (Yamanaka et al., 2002; Kong et al., 2004), which exhibit tissue-specific expression and unique binding specificities for both F-box and Cullin-family proteins. Furthermore, while the role of the shorter human SKP1 Isoform A has not yet been well-characterized, it remains possible that Isoform A may recognize alternate F-box proteins or be involved in SCF complex-independent functions. In general, the high degree of sequence and functional conservation throughout evolution underscores the key role SKP1 plays within the SCF complex and further emphasizes the importance of SKP1 in the regulation of fundamental cellular processes.
SKP1 and the SCF complex are arguably best understood for their roles in poly-ubiquitination, proteolytic degradation and the UPS. The UPS is a highly coordinated series of events involving the covalent attachment of ubiquitin molecules to protein targets and the subsequent degradation of these poly-ubiquitinated targets by the 26S proteasome. Substrate poly-ubiquitination is accomplished through the successive and repeated activities of three key enzymes (Figure 1C) that are generically referred to as the E1 ubiquitin (activating) enzyme, the E2 ubiquitin (conjugating) enzyme and the E3 ubiquitin (ligating) enzyme (reviewed in (Hershko and Ciechanover, 1998; Nakayama and Nakayama, 2006; Deshaies and Joazeiro, 2009)). Approximately 600–650 E3 ligases are predicted to exist within humans, which impart the extensive and requisite specificities to regulate the hundreds to thousands of protein targets believed to be modulated by the UPS, whereas only two E1 and approximately thirty E2 enzymes exist within the human genome (Deshaies and Joazeiro, 2009).
The E3 ubiquitin ligases are classically divided into three main families based on distinct structural motifs and include: 1) the Really Interesting New Gene (RING)-finger family containing ∼600 members in humans; 2) the Homologous to the E6-AP Carboxyl Terminus (HECT) family having ∼30 human members; and 3) the RING-between RING-RING (RBR) family with ∼12 members in humans (Morreale and Walden, 2016). The RING-finger family is further divided into sub-families, which includes the Cullin-RING ligase subfamily. The SCF complex is often considered the prototypic Cullin-based RING-finger E3 ubiquitin ligase and is comprised of three invariable core components (Figure 1C): 1) the RING-finger protein RBX1 (Ring-Box 1, also known as the regulator of cullins 1 [ROC1]) that recruits the E2 ubiquitin-conjugating enzyme; 2) CUL1, a scaffolding protein that complexes the E2 to the SCF complex; and 3) SKP1, the adaptor protein that physically connects the F-box protein and corresponding protein target with the core SCF complex.
F-box proteins are classified into three distinct families according to their substrate recognition domains, namely FBXW, FBXL, and FBXO family members, which harbor WD40 repeats (e.g., FBXW7), leucine-rich repeats (e.g., FBXL1/SKP2) or other domains (e.g., FBXO28), respectively, (Jin et al., 2004). As indicated above, it is the F-box protein that imparts the protein target specificity to the SCF complex, with F-box proteins often binding to phospho-activated targets. Once bound to the protein target, the F-box protein/protein target are subsequently recruited to the core SCF complex through an interaction with SKP1 to enable the transfer of ubiquitin from an E2 conjugating enzyme onto the protein target. It is the repeated covalent attachment of ubiquitin moieties (i.e., poly-ubiquitination) via specific linkages (lysine 48 [K48] linkages) that label the designated substrates for degradation by the 26S proteasome. Thus, it is the UPS that regulates the global and temporal abundance of an extensive array of protein targets within a given cell (Kulathu and Komander, 2012).
While there are potentially 69 distinct SCF complexes, the substrates and functions for many of these SCF complexes remain largely unknown. Nevertheless, there are a few well characterized F-box proteins/SCF complexes that target key proteins involved in a variety of cellular pathways such as DNA damage repair, apoptosis, centrosome biology and chromosome stability (discussed below), which highlights their innate roles in maintaining genome stability and preserving mitotic fidelity. As such, future studies aimed at functionally characterizing the complete cellular repertoire of SCF complexes will be essential to advance our rudimentary understanding of the specific impact each individual SCF complex has in normal cell physiology and genome stability. Perhaps even more important will be the fundamental and clinical studies aimed at determining the impact aberrant expression and function of SCF complex components have on disease development. Indeed, aberrant SKP1 expression and/or function is already associated with several human genetic disorders, including Sjögren’s syndrome (a chronic inflammatory autoimmune disease) (Sandhya and Danda, 2014), sporadic Parkinson’s disease (a neurological degenerative disorder) (Mandel et al., 2012) and cancer (Silverman et al., 2012). Thus, defining the underlying molecular etiology giving rise to SKP1 (and SCF complex) dysfunction will be critical to ultimately determine the individual and collective impacts on disease pathology, especially as it potentially relates to cancer development and progression.
The processes that regulate cell cycle progression and DNA damage response are intimately linked and are essential to maintain genome stability. In the presence of genotoxic stress or a stalled replication fork, a cell cycle arrest is invoked to facilitate repair prior to cell cycle re-entry with the ultimate goal of preventing genomic damage (mutations and alternations) from being propagated within daughter cells (Bassermann et al., 2014). These processes are highly dependent on appropriate protein turnover that is regulated by the UPS. Indeed, the SCF complex, and therefore SKP1, exhibit key roles within the DNA damage response, some of which are detailed below.
In general, following a DNA double strand break, a checkpoint kinase, either ATM (Ataxia Telangiectasia Mutated) or ATR (Ataxia Telangiectasia and Rad3 Related) is auto-phosphorylated, which initiates a series of cascading phosphorylation events on downstream targets. For example, ATM initiates a G1 arrest by phosphorylating Cyclin D1, which is subsequently ubiquitinated by SCFFBXO4 and targeted for proteolytic degradation. In turn, Cyclin D1 degradation promotes CDK2 inhibition by releasing P21 from CDK4 (Agami and Bernards, 2000), which ultimately prevents E2F transcription factor activation and cyclin expression (Silverman et al., 2012). Alternatively, an S-phase or G2 arrest can be invoked through ATR phospho-activation of CHEK1 (Checkpoint Kinase 1), which is mediated by the adaptor protein Claspin (Mamely et al., 2006) to hyperphosphorylate CDC25A, labeling it for SCFβTrCP(FBXW11) mediated targeting and proteolytic degradation to attenuate CDK activation (Busino et al., 2003). This CDK attenuation induces a cell cycle arrest, providing the requisite time for efficient DNA repair. Moreover, to ensure an adequate supply of deoxyribonucleotides for DNA repair, degradation of RRM2 (Ribonucleotide Reductase Regulatory Subunit 2) via SCFCyclinF(FBXO1) is inhibited by ATR-mediated Cyclin F degradation (D’Angiolella et al., 2012). Concurrently, the pre-replication complex component, CDT1 (Chromatin Licensing and DNA Replication Factor 1) is targeted for degradation by SCFSKP2(FBXL1) to prevent replication of damaged DNA (Kondo et al., 2004), while protein translation is reduced by the phospho-inactivation of the elongation factor, eEF2 (Eukaryotic Translation Elongation Factor 2) by eEF2K to prevent unnecessary energy expenditure during the DNA damage response. Once DNA repair is complete, SCFβTrCP directs eEF2K degradation to rapidly resume protein synthesis (Kruiswijk et al., 2012). SCFβTrCP also coordinates cell cycle re-entry by targeting phosphorylated Claspin for degradation, preventing CHEK1 activation by ATR, allowing for CDC25A reactivation of CDKs, while the increased abundance of CHEK1 is reduced by targeted degradation mediated by SCFFBXO6 (Silverman et al., 2012; Bassermann et al., 2014).
As the SCF complexes described above are crucial for DNA damage repair and maintaining genome stability, it is not difficult to envision how mutation, aberrant expression and/or function of SKP1 promotes genome instability and may contribute to cancer development and progression. For example, the siRNA-based silencing of βTrCP in S-phase cells exposed to ionizing radiation results in CDC25A accumulation (Jin et al., 2003), a defective S-phase check-point, failure to inhibit DNA replication and the propagation of DNA damage underlying genome instability and cancer (Bassermann et al., 2014).
In the event of excessive DNA damage, apoptosis is typically initiated to remove those cells from the population and prevent transmission of damaged DNA to daughter cells, which is a process normally regulated by the SCFFBXW7 complex. In response to DNA damage, GSK3 (Glycogen Synthase Kinase 3) phosphorylates the anti-apoptotic BCL2 (B-Cell Chronic Lymphocytic Leukemia/Lymphoma 2) family member MCL1 (Myeloid Cell Leukemia 1), allowing for SCFFBXW7-mediated MCL1 poly-ubiquitination and degradation. The cell death promoters BAX (BCL2 Associated X Protein) and BAK (BCL2 Antagonist/Killer) are released from MCL1 inhibition, which stimulates mitochondrial membrane permeabilization, caspase activation and apoptosis induction. Deletion of FBXW7 or its functional inactivation in acute lymphoblastic leukemia (ALL) cells, impairs MCL1 degradation in response to DNA-damaging agents, resulting in MCL1 overexpression and evasion of apoptosis (Inuzuka et al., 2011). In support of a role in oncogenesis, FBXW7 is somatically altered in >30% of human T-cell lymphomas, while T-cell-specific Fbxw7 knockout mice develop ALL (Crusio et al., 2010). Moreover, ∼20% of patients with colorectal adenocarcinoma have somatic FBXW7 mutations (Tate et al., 2019), with altered FBXW7 expression contributing to tumor development and progression, while loss-of-function mutations are predicted to be deleterious. Furthermore, ∼50% of somatic FBXW7 mutations occur at three hotspot codons (Arg465; Arg479; Arg505), which disrupt binding of FBXW7 to target substrates (Akhoondi et al., 2007; Cancer Genome Atlas Network, 2012; Grim, 2014), highlighting the critical role of the SCF complex and how dysregulation of key components may contribute to oncogenesis. Collectively, the above data demonstrate that SKP1 and the SCF complex are critical for coordinating a cellular response to DNA damage and facilitating either DNA repair or apoptosis depending on the extent of the damage.
As SKP1 is an invariable component of each SCF complex described above, SKP1 alterations such as mutations or copy number alterations (gains or losses) are predicted to impede DNA damage repair and foster cell survival by adversely impacting pro-apoptotic pathways leading to genome instability and perhaps promoting oncogenesis. This possibility is supported by the work of Piva and others (Piva et al., 2002), who generated and employed a transgenic mouse expressing a Cul1 deletion mutant (Cul1-N252) that sequesters and inactivates murine Skp1 (discussed further below). Interestingly, the in vivo inhibition of Skp1 function in a T-cell lineage corresponded with the development of T-cell lymphomas. Upon closer scrutiny, the authors also noted significant increases in micronucleus formation (DNA containing, extranuclear bodies indicative of DNA damage and genome instability (Bhatia and Kumar, 2013)), centrosome abnormalities, aberrant chromosome segregation and karyotypic heterogeneity. These data suggest SKP1 is critical to preserve the function of essential biological processes (e.g., DNA repair and apoptosis), while aberrant SKP1 expression and/or function disrupts these essential processes in a manner that may promote oncogenesis. Thus, it will be of tremendous interest to determine whether the accumulation of DNA damage within SKP1-deficient cancer cells or appropriate mouse models are associated with increased sensitivity towards genotoxic agents or whether these cells/models can be selectively targeted with immune checkpoint inhibitors or precision-based therapeutic strategies.
To ensure the accurate and faithful transmission of genetic material to daughter cells, chromosome dynamics are tightly regulated by the UPS, which coordinates centriole/centrosome duplication and separation. Centrosome aberrations lead to ongoing chromosome missegregation events and aneuploidy that are frequently observed in a myriad of cancer types. For example, one immunohistochemical study (Pihan et al., 1998) revealed that 93% (81/87 total) of human breast, prostate, lung, colon, brain, and metastatic cancer samples exhibit abnormal centrosome phenotypes including aberrant size, shape, and numbers relative to those in noncancerous adjacent tissues. Moreover, the aberrant phenotypes observed in tumor-derived cell lines are correlated with CIN (chromosome instability), a common form of genome instability characterized by ongoing changes in chromosome number and/or structure that is an established driver of cell-to-cell and genetic heterogeneity (reviewed in (Geigl et al., 2008; Lepage et al., 2019; Vishwakarma and McManus, 2020)). More recent studies have determined that SKP1 localizes to the centrosome throughout the cell cycle and that SCFCyclinF (D'Angiolella et al., 2010), SCFFBXW5 (Puklowski et al., 2011) and SCFβTRCP (Chan et al., 2008) exhibit key roles in centrosome dynamics (Gstaiger et al., 1999; D'Angiolella et al., 2010) that when disrupted with proteasome inhibitors (MG132), adversely impact centrosome formation and duplication. For example, during G2, the centriolar protein CCP110 (Centriolar Coiled-Coil Protein 110) that normally promotes centriole replication while inhibiting elongation, is targeted for proteolytic degradation by SCFCyclinF (Chen et al., 2002). Such timely CCP110 degradation prevents centriole over-duplication that would otherwise result in supernumerary centrosomes, chromosome missegregation events and aneuploidy. Indeed, D’Angiolella and others (D'Angiolella et al., 2010) determined that Cyclin F silencing induces centrosome over-duplication in G2 leading to multi-polar spindle formation, lagging chromosomes and an increase in micronucleus formation, all of which are hallmarks of CIN (Geigl et al., 2008; Lepage et al., 2019; Vishwakarma and McManus, 2020). As expected, co-silencing Cyclin F and CCP110 rescues these aberrant phenotypes effectively confirming the underlying mechanism leading to their formation.
Beyond CCP110, the centriolar scaffolding protein SASS6 (Spindle Assembly Protein 6) is also essential for centrosome formation and duplication, and is degraded in G2 by SCFFBXW5, which prevents over-duplication of centrosomes. FBXW5 is negatively regulated by APC/C (Anaphase-Promoting Complex/Cyclosome) and PLK4 (Polo-Like Kinase 4), which enables SASS6 to function appropriately during G1 and S-phase, respectively. As predicted, reduced FBXW5 expression corresponds with increasing SASS6 abundance and abnormally increased numbers of centrioles (Puklowski et al., 2011). Similarly, PLK4 promotes centriole duplication and separation, and is tightly regulated by SCFβTRCP (Guderian et al., 2010). Thus, aberrant PLK4 expression is associated with aberrant centriole numbers in human cancer cells (Habedanck et al., 2005), while βTrcp1 knockout in mouse embryonic fibroblasts corresponds with centrosome over-duplication and supernumerary centrosomes (Guardavaccaro et al., 2003). SCFβTRCP also contributes to centrosome homeostasis and chromosome stability by regulating the degradation of BORA (BORA Aurora Kinase A Activator), an activator Aurora Kinase A (AURKA). BORA regulates AURKA localization and kinase activity at the centrosome to ensure proper centrosome and mitotic spindle development, as overexpression of a SCFβTRCP-resistant form of BORA interferes with bipolar spindle formation as it adversely impacts AURKA localization and function (Chan et al., 2008). Based on these few examples, it is apparent that SKP1 and the SCF complex are critical for regulating centrosome dynamics and function, which is essential for chromosome transmission fidelity. Thus, further clinical studies into the types and prevalence of genomic aberrations affecting SKP1 expression are essential to better understand their impact on centrosome biology and gain a more holistic understanding of the potential downstream implications for disease development.
As an invariable component of the SCF complex, it is apparent that SKP1 is essential for the proper regulation of key substrates involved in many cancer-associated pathways. Despite this association, the potential pathophysiological impact aberrant SKP1 expression may have in cancer development is only beginning to emerge. This knowledge gap may in part, be attributed to the lack of transgenic or Skp1 knockout mouse models available for in vivo study (Zhou et al., 2013). Nevertheless, several transgenic mouse models do exist for the other SCF complex components (e.g., Cul1) that have provided key insight into SKP1 (and SCF complex) function, which includes the pathogenic implications for genomic instability and cancer associated with aberrant SCF complex expression and function. As indicated above, Piva et al. (Piva et al., 2002) developed a Cul1 deletion mutant (Cul1-N252) transgenic mouse model that inactivates Skp1 in vivo, leading to lymphoid organ hypoplasia, proliferation defects, supernumerary centrosomes, mitotic spindle aberrations and CIN. Following the initial proliferation reduction, >80% of Cul1-N252 mice develop T-cell lymphomas, suggesting Skp1 and SCF function are required to prevent lymphoid tumor development. Moreover, Cul1-N252 expression in a human cellular context (HEK293T cells) resulted in many aberrant phenotypes associated with CIN, including multinucleated cells, enlarged nuclei and increased micronucleus formation. Thus, their mouse and human work are consistent with aberrant Skp1/SKP1 function being an early etiological event underlying CIN and possibly contributing cancer pathogenesis. Moreover, these results highlight the utility of mouse models for studying the in vivo functions of SCF components and provide a means by which to investigate their potential roles in tumorigenesis. Their findings also underscore the paucity of clinically-relevant Skp1 mouse models, which are essential to clearly delineate and characterize any potential role for aberrant Skp1/SKP1 expression and/or function in oncogenesis.
Recently, several genetic studies have begun to identify potential pathogenic relationships between aberrant SCF complex expression/function and cancer (Thompson et al., 2020; Bungsy et al., 2021; Lepage et al., 2021). In particular, two studies focused on the impact reduced SKP1 expression has on CIN in colorectal (Thompson et al., 2020) and ovarian (Lepage et al., 2021) cancer contexts. First, Thompson et al (Thompson et al., 2020) performed a screen of 164 candidate genes whose diminished expression was suspected to underlie CIN. Using siRNA-based silencing and quantitative imaging microscopy, they determined that reduced SKP1 expression induced significant increases in CIN-associated phenotypes (Lepage et al., 2019), such as nuclear areas, micronucleus formation and chromosome numbers. They further showed that SKP1 silencing corresponded with increases in replication stress, DNA double strand breaks and chromothriptic events, or extensive chromosome shattering followed by reassembly in a single event (reviewed in (Ly and Cleveland, 2017)). Perhaps most importantly, they performed genetic rescue experiments and determined that the aberrant phenotypes were largely dependent on aberrant increases in Cyclin E1 levels, an established target of the SCF complex; however, as complete phenotypic rescues did not occur, they posited that additional protein targets must also be misregulated that contribute to the plethora of aberrant phenotypes observed. Given that ∼85% of sporadic colorectal cancers exhibit CIN (Lengauer et al., 1997; Cisyk et al., 2015; Cisyk et al., 2018), these findings are particularly important as they may shed new insight into the potential underlying molecular etiology driving colorectal cancer pathogenesis. A second study by Lepage and others (Lepage et al., 2021), assessed the impact that reduced SKP1 (and CUL1) expression has on CIN in non-transformed fallopian tube secretory epithelial cells, a cell of origin for high-grade serous ovarian cancer (Perets et al., 2013; Nakamura et al., 2018). Using a combination of siRNA and CRISPR/Cas9 approaches, they demonstrated that reduced expression corresponded with significant changes in nuclear areas, micronucleus formation and chromosome numbers. They further showed that CIN was prevalent and dynamic over an ∼3-month timeframe, which is key given recent evidence showing that CIN is both pervasive and dynamic in ascites (an accumulation of abdominal fluid containing tumor cells) and solid tumor samples isolated from patients with high-grade serous ovarian cancer (Penner-Goeke et al., 2017; Morden et al., 2021). Collectively, these data identify SKP1 as a novel CIN gene and further suggest that reduced expression may contribute to cancer pathogenesis. Accordingly, future fundamental and clinical studies are now essential to determine the extent and types of SKP1 genetic alterations that may drive disease development and progression, with potential downstream implications for treatment response and patient outcomes.
As SKP1 and the SCF complex normally function to regulate a multitude of essential cellular pathways required to maintain genome stability, genetic alterations impacting the invariable complex components (e.g., SKP1) are anticipated to promote cellular dysfunction, which may contribute to cancer development. As detailed above, several genetic studies performed in both malignant (Thompson et al., 2020) and non-malignant (Lepage et al., 2021) human cell contexts have established that reduced SKP1 expression induces CIN, an enabling hallmark of cancer (Hanahan and Weinberg, 2011) associated with cellular transformation, intra-tumoral heterogeneity, metastasis, drug resistance and poor patient outcomes (reviewed in (Geigl et al., 2008; Vishwakarma and McManus, 2020)). Unfortunately, Skp1 knockout mice do not exist, suggesting it may be an essential gene, a possibility supported by a CRISPR screen that identified SKP1 as an essential gene (Blomen et al., 2015); however, it should be noted that this work was conducted in a haploid malignant cancer cell line, and thus, the results may exhibit context-specific essentiality. Nevertheless, additional evidence comes from DepMap (Dependency Mapping), which is an online resource that identified SKP1 a common essential gene based on RNAi and CRISPR screens performed in a myriad of cell lines (Tsherniak et al., 2017; Dempster et al., 2019; Dharia et al., 2021; Pacini et al., 2021). Accordingly, while SKP1 appears to be an essential gene the functional impacts altered SKP1 expression has on various biological pathways are only beginning to emerge (Thompson et al., 2020; Lepage et al., 2021).
In support of reduced SKP1 expression and/or function harboring a potential pathogenic role in oncogenesis, in silico analyses of The Cancer Genome Atlas (TCGA) pan-cancer atlas patient data available through cBioPortal (Cerami et al., 2012; Gao et al., 2013) reveal that SKP1 is somatically altered in 12 common solid tumor cancer types (Figure 2) (Hoadley et al., 2018). Briefly, SKP1 mutations are rare with only 15 missense and 2 truncating mutations (one frameshift and one premature stop codon) identified within six of the 12 cancers assessed (Figure 2A) (Hoadley et al., 2018). Interestingly, and in agreement with SKP1 being a putative tumor suppressor gene, the mutational load is equally distributed (i.e., diffuse) across the entire coding sequence (Figure 2B), rather than a focal mutational load that is typical of an oncogene (Liu et al., 2011; Vogelstein et al., 2013; Sato et al., 2015). With respect to gene copy number alterations, both gains (oncogene-like) and losses (tumor suppressor-like) occur in all 12 cancer types; however, losses are more prevalent in 11 of 12 cancers evaluated (Figure 2C). Overall, SKP1 amplifications (two or more additional copies) are rare (0–1.0%), while gains (one additional copy) occur in all 12 cancers analyzed and range from 3.2 to 30.7% in uterine and liver cancers, respectively. Similarly, deep (i.e., homozygous) deletions are rare (0–1%), whereas shallow (i.e., heterozygous) deletions are present in all 12 cancer types and range from 6.3 to 43.8% in prostate and ovarian cancers, respectively. Collectively, these data show that large copy number alterations (amplifications or deep deletions) are rare, which suggests an expression threshold may exist whereby too much expression (i.e., gene amplification) may severely impact normal cellular physiology. Furthermore, complete loss (i.e., deep deletion) appears incompatible with viability further supporting the notion that SKP1 is an essential gene (Blomen et al., 2015; Tsherniak et al., 2017; Dempster et al., 2019; Dharia et al., 2021; Pacini et al., 2021).
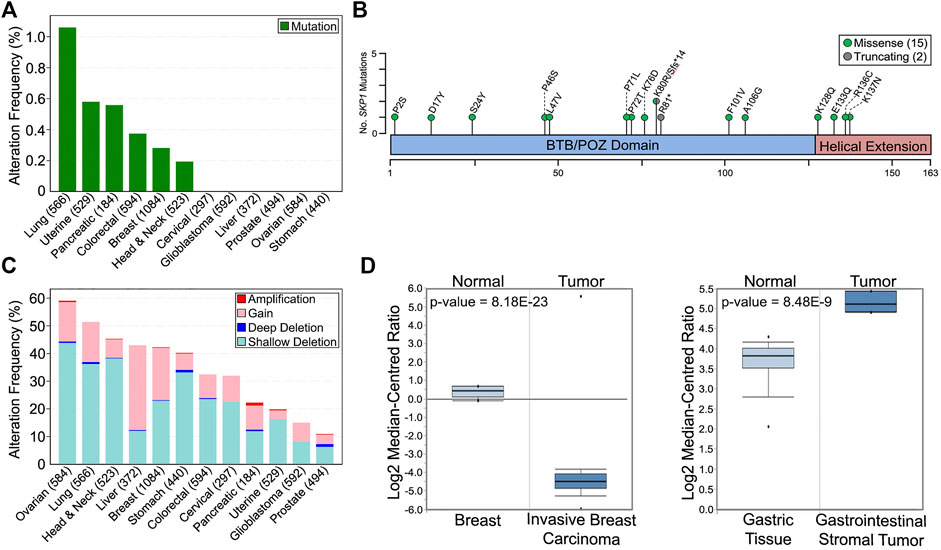
FIGURE 2. Prevalence and Impact of SKP1 Alterations in Cancer. (A) The frequency of total SKP1 mutations (missense; truncating; inframe; fusion) in 12 common, solid tumor cancer types (total cases) (Cerami et al., 2012; Gao et al., 2013). Note that only missense (15) and truncating (2) mutations were identified from the 12 pan-cancer TCGA datasets (Hoadley et al., 2018). (B) Schematic mapping the positions of the encoded SKP1 mutations across the SKP1 protein using the corresponding single amino acid codes (fs, frameshift; *, premature stop codon). (C) Prevalence of SKP1 copy number alterations (deep deletion; shallow deletion; gain; amplification) within the 12 common cancer types (total cases) (Cerami et al., 2012; Gao et al., 2013; Hoadley et al., 2018). (D) Box-and-whisker plots displaying SKP1 mRNA expression levels for normal and tumor tissues from invasive breast carcinoma (left) and gastrointestinal stromal tumor (right). Boxes display interquartile range, whiskers denote 10th and 90th percentiles, and the minimum/maximum values are displayed as black dots. Note that a significant >25-fold decrease in mean SKP1 expression occurs in invasive breast carcinoma relative to normal tissue, while a significant ∼3-fold increase in expression occurs in gastrointestinal stromal tumors. Data, graphs and statistical analyses were obtained from the Oncomine database (https://www.oncomine.org) (Rhodes et al., 2007).
A fundamental assumption of gene copy number alterations is that they induce corresponding changes in gene expression and that SKP1 copy number gains and losses are expected to underlie aberrant SCF complex activity leading to cellular dysfunction, genome instability and potentially tumorigenesis. Indeed, strong positive correlations exist between copy number changes and mRNA expression for all 12 cancer types investigated (Figure 3), and while the copy number alterations detailed above suggest SKP1 may encode both oncogene-like or tumor suppressor-like functions, these seemingly opposing activities are not specific to SKP1 and have been reported for other genes including TP53 (Lane, 1984; Jenkins et al., 1985; Finlay et al., 1989), USP22 (Jeusset and McManus, 2017), and RAD54B (McAndrew and McManus, 2017).
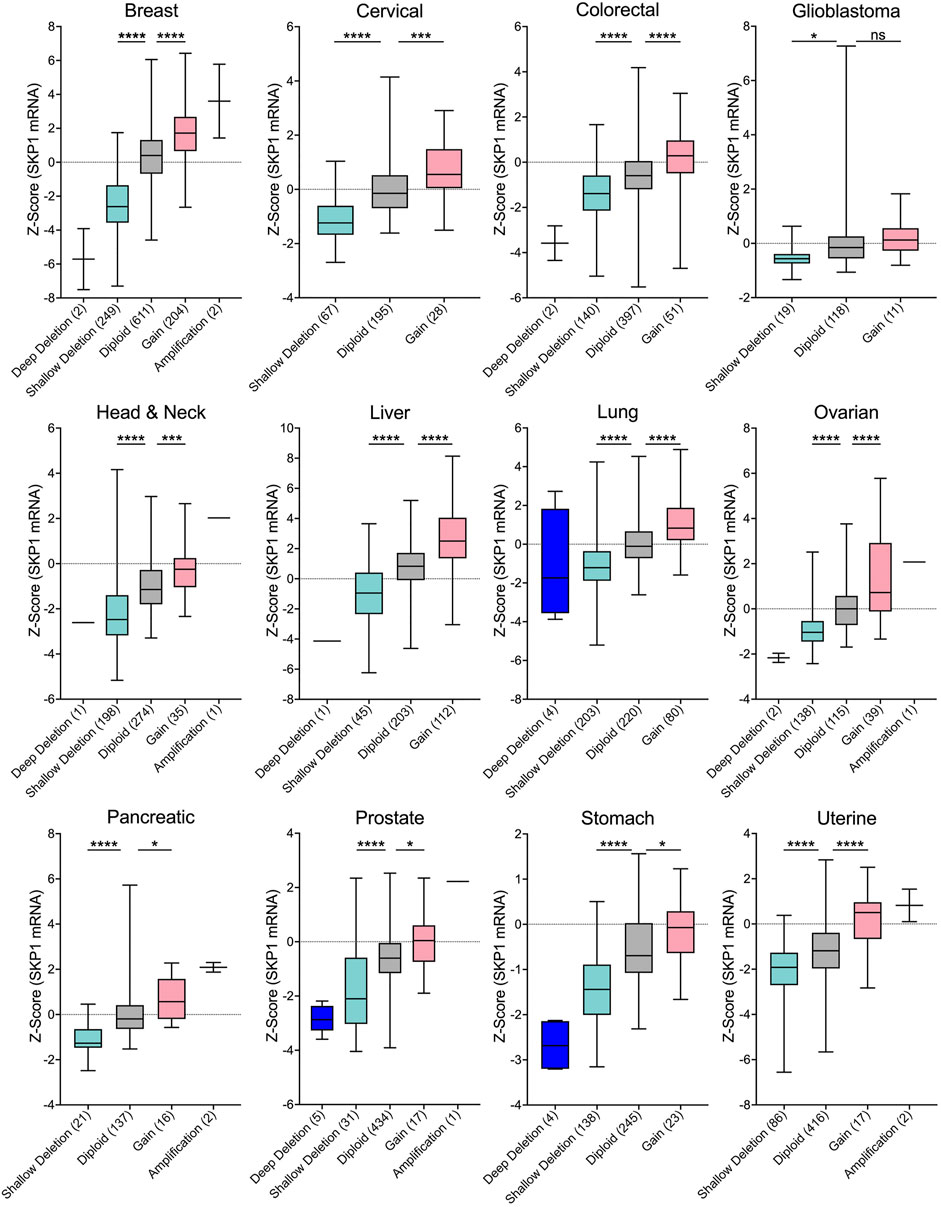
FIGURE 3. SKP1 Copy Number Alterations are Positively Correlated with mRNA Expression Levels in Cancer. Box-and-whisker plots of TCGA pan-cancer data from 12 common cancer types reveal linear correlations between SKP1 copy number alterations and mRNA expression levels (Cerami et al., 2012; Gao et al., 2013; Hoadley et al., 2018). Boxes identify interquartile ranges (25th, 50th, and 75th percentiles), while whiskers depict entire range. For orientation purposes, the dotted horizontal lines identify 0. Specific copy number alterations (deep deletion; shallow deletion; gain; amp) and diploid categories are presented along the x-axis with the total number of samples indicated within brackets. Unpaired t-tests were conducted comparing either Shallow Deletions or Gains with the corresponding Diploid control (ns [not significant] p-value >0.05; * p-value <0.05; *** p-value <0.001; **** p-value <0.0001). Note that in general, very few deep deletions or amplifications were identified in the 12 cancer types.
The potential for SKP1 to encode both tumor suppressor-like and oncogene-like activities is further bolstered by the many additional gene expression datasets available through various online resources. For example, while data contained within the In Silico Transcriptomics Online database (https://ist.medisapiens.com) (Kilpinen et al., 2008) show tremendous variation in SKP1 (ENSG00000113558) mRNA expression in both normal and tumor tissues (see (Thompson et al., 2021)), they also reveal that some cancers exhibit increases (head and neck; chronic lymphocytic leukemia; liver) or decreases (breast; ovarian; cervical; colorectal) in SKP1 expression relative to the corresponding normal tissues. Additionally, expression data from the Oncomine database (https://www.oncomine.org) (Rhodes et al., 2007) corroborate that SKP1 can be under or overexpressed within specific cancer types relative to normal tissues. For example, Figure 2D provides representative examples in which SKP1 is predicted to encode both tumor suppressor-like functions, as mRNA expression is significantly reduced (∼25-fold) within invasive breast carcinomas relative to normal tissues, or oncogene-like functions, as expression is significantly increased (∼3-fold) within gastrointestinal stromal tumors. Collectively, the data presented above support the possibility that SKP1 may encode either oncogene- or tumor suppressor gene-like capabilities depending on whether it is over or under-expressed, respectively.
Unfortunately, very little insight into SKP1 expression is available beyond transcriptomics, as only a single study has been performed in which SKP1 was assessed at the protein level. In 2015, Liu and others (Liu et al., 2015) employed western blots (64 matched cases) and immunohistochemistry (20 matched cases) to investigate SKP1 expression in non-small cell lung cancer and adjacent normal lung tissues. While both approaches revealed variable SKP1 expression in both cancer and matched tissues, 56% of cases showed significant increases in expression within tumors relative to control tissues. Furthermore, they determined that SKP1 expression was inversely correlated with survival as patients with high expression levels had significantly worse overall survival than those with low expression levels; however, the thresholds defining high versus low were not specified. Although the underlying genomic defects accounting for the increases in SKP1 expression observed in this study were not determined, this single example supports the possibility that aberrant SKP1 expression may be a pathogenic driver of cancer.
Collectively, the above data gleaned from a diverse array of patient-based genomic, transcriptomic and protein datasets show that SKP1 is frequently misexpressed in human cancers, which suggests aberrant SKP1 expression may harbor tumor suppressive or oncogenic functions depending on whether it is under- or over-expressed, respectively. These apparently opposing activities may simply reflect that as a core SCF complex member, SKP1 may function as a tumor suppressor or oncoprotein depending on the protein targeted for degradation suggesting SKP1 expression levels may need to be precisely regulated to maintain cellular homeostasis, preserve genome stability and prevent cancer development and progression. Thus, the patient-based findings presented above underscore the need for additional insight into SKP1, its protein targets and the underlying biological mechanisms and their potential impact for oncogenesis. In this regard, future studies should also assess the clinical utility of SKP1 as a potential prognostic indicator or a novel therapeutic target for cancers.
As the SCF complex regulates a diverse array of substrates involved in many biological pathways fundamental to genome stability, therapeutically targeting a core SCF component such as SKP1 may seem counter intuitive as there is the potential for increased toxicity and side effects. However, therapeutic success has been achieved with general proteasome inhibitors (e.g., Bortezomib (Robak et al., 2015)) and indirect SCF inhibitors (e.g., MLN4924 (Swords et al., 2015)) for the treatment of lymphoma, myeloma and leukemia lending support to use of broad-spectrum inhibitors targeting SKP1 and/or the SCF complex (Skaar et al., 2014). In fact, evidence shows cancer cells with a misregulated UPS are more sensitive to the broad-spectrum proteasome/SCF-targeting inhibitors than non-cancerous cells, which allows for the use of lower drug concentrations for effective outcomes and reduced side effects (Ludwig et al., 2005). Based on these findings, SKP1-targeted therapies designed to block SCF complex formation and function may represent promising treatment options. Rather than inhibiting global proteasomal degradation with agents like Bortezomib, or inactivating additional off-target Cullin family members with MLN4924, SKP1 inhibitors would specifically target the SCF complex, thereby reducing toxicity and ideally enhancing the therapeutic window (Silverman et al., 2012). Although a clinically administered dose would need to be strictly monitored, SKP1/SCF complex inhibitors could potentially be utilized in combination regimens with other chemotherapies to improve efficacy and/or help reduce the risk of drug resistance. For example, 5-fluorouracil, oxaliplatin, and irinotecan are first-line chemotherapies that induce DNA damage and cellular apoptosis (Longley et al., 2003). These drugs are often administered in combination for the treatment of colorectal cancer, with response rates from 40–50% and improved median survival (Douillard et al., 2000; Giacchetti et al., 2000; Longley et al., 2003). As the SCF complex is critical for eliciting an effective DNA damage response, perhaps co-treatment with a low-dose SKP1/SCF complex inhibitor would further sensitize cancer cells and synergize with standard chemotherapies to improve response rates and patient outcomes.
Considering the frequency of SKP1 copy number losses in cancer (Figure 2C), it remains plausible that a synthetic lethal (SL) paradigm may prove highly effective in a broad range of cancer types. Synthetic lethality is defined as a rare and lethal genetic interaction occurring between two unlinked genes. In practice, cells harboring a mutation in either gene alone remain viable, whereas the presence of both mutations within a single cell will induce lethality (Sajesh et al., 2013). Although a relatively new therapeutic concept, SL strategies have already begun to enter the clinic as breast and ovarian cancers harboring BRCA1/2 (Breast Cancer Type 1/2 Susceptibility Protein) defects are now being targeted with PARP1 (Poly [ADP-Ribose] Polymerase 1) inhibitors like Olaparib. Accordingly, genetic studies aimed at identifying SL interactors of SKP1 are highly warranted as the SL interactors are candidate drug targets that when inhibited are predicted to induce the selective killing of cancer cells harboring SKP1 defects. Beyond the genetic sensitization approaches detailed above, another promising strategy involves proteolysis-targeting chimeric molecules, or Protacs (reviewed in (Sakamoto et al., 2001; Burslem and Crews, 2020; Cecchini et al., 2021; Hughes et al., 2021)). The fundamental concept behind Protacs is that fusion proteins are created to link a specified target substrate to an F-box protein for SCF-mediated ubiquitination and degradation (Sakamoto et al., 2001). This approach would allow for conditional or tissue-specific degradation of overexpressed oncoproteins, suppression of tumor growth and cancer cell death.
Conceptualization LT, KR, CL, and KM; formal analyses, LT and KM; writing–original draft preparation LT, KR, CL, and KM; writing–review and editing, LT, KR, CL, and KM; supervision, KM; funding acquisition, KM. All authors have read and agreed to the published version of the manuscript.
Research in the McManus laboratory is/was generously supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Alexander Graham Bell Canadian Graduate Scholarship (LT), a Canadian Institutes of Health Research Canada Graduate (MSc) Scholarship (KR) a Research Manitoba/CancerCare Manitoba Studentship (KR), a Canadian Institutes of Health Research Canada Graduate Scholarship (CL), an NSERC Discovery Grant (KM; RGPIN: 2018-05007) and a CancerCare Manitoba Foundation Operating Grant (KM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge that the CancerCare Manitoba Research Institute and the University of Manitoba are located on original lands of Anishinaabeg, Cree, Oji-Cree, Dakota and Dene peoples, and on the homeland of the Métis Nation. We respect the Treaties that were made on these territories and acknowledge the harms and mistakes of the past. We dedicate ourselves to move forward in partnership with Indigenous communities in a spirit of reconciliation and collaboration. We thank members of the McManus laboratory for constructive criticisms during the writing of this review. We also acknowledge the ongoing support of the CancerCare Manitoba Research Institute and the CancerCare Manitoba Foundation.
Agami, R., and Bernards, R. (2000). Distinct Initiation and Maintenance Mechanisms Cooperate to Induce G1 Cell Cycle Arrest in Response to DNA Damage. Cell 102, 55–66. doi:10.1016/s0092-8674(00)00010-6
Akhoondi, S., Sun, D., Von Der Lehr, N., Apostolidou, S., Klotz, K., Maljukova, A., et al. (2007). FBXW7/hCDC4 Is a General Tumor Suppressor in Human Cancer. Cancer Res. 67, 9006–9012. doi:10.1158/0008-5472.can-07-1320
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410. doi:10.1016/s0022-2836(05)80360-2
Bai, C., Sen, P., Hofmann, K., Ma, L., Goebl, M., Harper, J. W., et al. (1996). SKP1 Connects Cell Cycle Regulators to the Ubiquitin Proteolysis Machinery through a Novel Motif, the F-Box. Cell 86, 263–274. doi:10.1016/s0092-8674(00)80098-7
Bassermann, F., Eichner, R., and Pagano, M. (2014). The Ubiquitin Proteasome System - Implications for Cell Cycle Control and the Targeted Treatment of Cancer. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1843, 150–162. doi:10.1016/j.bbamcr.2013.02.028
Bhatia, A., and Kumar, Y. (2013). Cancer Cell Micronucleus: an Update on Clinical and Diagnostic Applications. APMIS 121, 569–581. doi:10.1111/apm.12033
Blomen, V. A., Majek, P., Jae, L. T., Bigenzahn, J. W., Nieuwenhuis, J., and Staring, J. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. doi:10.1126/science.aac7557
Bungsy, M., Palmer, M. C. L., Jeusset, L. M., Neudorf, N. M., Lichtensztejn, Z., Nachtigal, M. W., et al. (2021). Reduced RBX1 Expression Induces Chromosome Instability and Promotes Cellular Transformation in High-Grade Serous Ovarian Cancer Precursor Cells. Cancer Lett. 500, 194–207. doi:10.1016/j.canlet.2020.11.051
Burslem, G. M., and Crews, C. M. (2020). Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 181, 102–114. doi:10.1016/j.cell.2019.11.031
Busino, L., Donzelli, M., Chiesa, M., Guardavaccaro, D., Ganoth, D., Valerio Dorrello, N., et al. (2003). Degradation of Cdc25A by β-TrCP during S Phase and in Response to DNA Damage. Nature 426, 87–91. doi:10.1038/nature02082
Cancer Genome Atlas Network (2012). Comprehensive Molecular Characterization of Human colon and Rectal Cancer. Nature 487, 330–337. doi:10.1038/nature11252
Cecchini, C., Pannilunghi, S., Tardy, S., and Scapozza, L. (2021). From Conception to Development: Investigating PROTACs Features for Improved Cell Permeability and Successful Protein Degradation. Front. Chem. 9, 672267. doi:10.3389/fchem.2021.672267
Cerami, E., Gao, J., Dogrusoz, U., Gross, B. E., Sumer, S. O., Aksoy, B. A., et al. (2012). The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2, 401–404. doi:10.1158/2159-8290.cd-12-0095
Chan, E. H. Y., Santamaria, A., Silljé, H. H. W., and Nigg, E. A. (2008). Plk1 Regulates Mitotic Aurora A Function through βTrCP-Dependent Degradation of hBora. Chromosoma 117, 457–469. doi:10.1007/s00412-008-0165-5
Chen, H., Thalmann, I., Adams, J. C., Avraham, K. B., Copeland, N. G., Jenkins, N. A., et al. (1995). cDNA Cloning, Tissue Distribution, and Chromosomal Localization of Ocp2, a Gene Encoding a Putative Transcription-Associated Factor Predominantly Expressed in the Auditory Organs. Genomics 27, 389–398. doi:10.1006/geno.1995.1068
Chen, Z., Indjeian, V. B., McManus, M., Wang, L., and Dynlacht, B. D. (2002). CP110, a Cell Cycle-dependent CDK Substrate, Regulates Centrosome Duplication in Human Cells. Dev. Cel. 3, 339–350. doi:10.1016/s1534-5807(02)00258-7
Cisyk, A. L., Penner-Goeke, S., Lichtensztejn, Z., Nugent, Z., Wightman, R. H., Singh, H., et al. (2015). Characterizing the Prevalence of Chromosome Instability in Interval Colorectal Cancer. Neoplasia 17, 306–316. doi:10.1016/j.neo.2015.02.001
Cisyk, A. L., Nugent, Z., Wightman, R. H., Singh, H., and McManus, K. J. (2018). Characterizing Microsatellite Instability and Chromosome Instability in Interval Colorectal Cancers. Neoplasia 20, 943–950. doi:10.1016/j.neo.2018.07.007
Crusio, K. M., King, B., Reavie, L. B., and Aifantis, I. (2010). The Ubiquitous Nature of Cancer: the Role of the SCF(Fbw7) Complex in Development and Transformation. Oncogene 29, 4865–4873. doi:10.1038/onc.2010.222
D'Angiolella, V., Donato, V., Vijayakumar, S., Saraf, A., Florens, L., Washburn, M. P., et al. (2010). SCF(Cyclin F) Controls Centrosome Homeostasis and Mitotic Fidelity through CP110 Degradation. Nature 466, 138–142. doi:10.1038/nature09140
D'Angiolella, V., Donato, V., Forrester, F. M., Jeong, Y.-T., Pellacani, C., Kudo, Y., et al. (2012). Cyclin F-Mediated Degradation of Ribonucleotide Reductase M2 Controls Genome Integrity and DNA Repair. Cell 149, 1023–1034. doi:10.1016/j.cell.2012.03.043
Dempster, R. J., Pacini, C., Pantel, S., Behan, F. M., Green, T., Krill-Burger, J., et al. (2019). Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun. 10, 5817. doi:10.1038/s41467-019-13805-y
Deshaies, R. J., and Joazeiro, C. A. P. (2009). RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 78, 399–434. doi:10.1146/annurev.biochem.78.101807.093809
Dharia, N. V., Kugener, G., Guenther, L. M., Malone, C. F., Durbin, A. D., Hong, A. L., et al. (2021). A first-generation pediatric cancer dependency map. Nat. Genet. 53, 529-538. doi:10.1038/s41588-021-00819-w
Douillard, J., Cunningham, D., Roth, A., Navarro, M., James, R., Karasek, P., et al. (2000). Irinotecan Combined with Fluorouracil Compared with Fluorouracil Alone as First-Line Treatment for Metastatic Colorectal Cancer: a Multicentre Randomised Trial. Lancet 355, 1041–1047. doi:10.1016/s0140-6736(00)02034-1
Finlay, C. A., Hinds, P. W., and Levine, A. J. (1989). The P53 Proto-Oncogene Can Act as a Suppressor of Transformation. Cell 57, 1083–1093. doi:10.1016/0092-8674(89)90045-7
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 6, pl1. doi:10.1126/scisignal.2004088
Geigl, J. B., Obenauf, A. C., Schwarzbraun, T., and Speicher, M. R. (2008). Defining 'chromosomal Instability'. Trends Genet. 24, 64–69. doi:10.1016/j.tig.2007.11.006
Giacchetti, S., Perpoint, B., Zidani, R., Le Bail, N., Faggiuolo, R., Focan, C., et al. (2000). Phase III Multicenter Randomized Trial of Oxaliplatin Added to Chronomodulated Fluorouracil-Leucovorin as First-Line Treatment of Metastatic Colorectal Cancer. Jco 18, 136. doi:10.1200/jco.2000.18.1.136
Grim, J. E. (2014). Fbxw7 Hotspot Mutations and Human colon Cancer: Mechanistic Insights from New Mouse Models. Gut 63, 707–709. doi:10.1136/gutjnl-2013-305144
Gstaiger, M., Marti, A., and Krek, W. (1999). Association of Human SCF(SKP2) Subunit p19(SKP1) with Interphase Centrosomes and Mitotic Spindle Poles. Exp. Cel Res. 247, 554–562. doi:10.1006/excr.1999.4386
Guardavaccaro, D., Kudo, Y., Boulaire, J., Barchi, M., Busino, L., Donzelli, M., et al. (2003). Control of Meiotic and Mitotic Progression by the F Box Protein β-Trcp1 In Vivo. Dev. Cel. 4, 799–812. doi:10.1016/s1534-5807(03)00154-0
Guderian, G., Westendorf, J., Uldschmid, A., and Nigg, E. A. (2010). Plk4 Trans-autophosphorylation Regulates Centriole Number by Controlling βTrCP-Mediated Degradation. J. Cel Sci. 123, 2163–2169. doi:10.1242/jcs.068502
Habedanck, R., Stierhof, Y.-D., Wilkinson, C. J., and Nigg, E. A. (2005). The Polo Kinase Plk4 Functions in Centriole Duplication. Nat. Cel Biol. 7, 1140–1146. doi:10.1038/ncb1320
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of Cancer: the Next Generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Hershko, A., and Ciechanover, A. (1998). The Ubiquitin System. Annu. Rev. Biochem. 67, 425–479. doi:10.1146/annurev.biochem.67.1.425
Hoadley, K. A., Yau, C., Hinoue, T., Wolf, D. M., Lazar, A. J., Drill, E., et al. (2018). Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–e6. doi:10.1016/j.cell.2018.03.022
Hughes, G. R., Dudey, A. P., Hemmings, A. M., and Chantry, A. (2021). Frontiers in PROTACs. Drug Discov. Today 26, 2377–2383. doi:10.1016/j.drudis.2021.04.010
Inuzuka, H., Shaik, S., Onoyama, I., Gao, D., Tseng, A., Maser, R. S., et al. (2011). SCF(FBW7) Regulates Cellular Apoptosis by Targeting MCL1 for Ubiquitylation and Destruction. Nature 471, 104–109. doi:10.1038/nature09732
Jenkins, J. R., Rudge, K., Chumakov, P., and Currie, G. A. (1985). The Cellular Oncogene P53 Can Be Activated by Mutagenesis. Nature 317, 816–818. doi:10.1038/317816a0
Jeusset, L. M., and McManus, K. J. (2017). Ubiquitin Specific Peptidase 22 Regulates Histone H2B Mono-Ubiquitination and Exhibits Both Oncogenic and Tumor Suppressor Roles in Cancer. Cancers (Basel) 9, 167. doi:10.3390/cancers9120167
Jin, J., Shirogane, T., Xu, L., Nalepa, G., Qin, J., Elledge, S. J., et al. (2003). SCFβ-TRCP Links Chk1 Signaling to Degradation of the Cdc25A Protein Phosphatase. Genes Dev. 17, 3062–3074. doi:10.1101/gad.1157503
Jin, J., Cardozo, T., Lovering, R. C., Elledge, S. J., Pagano, M., and Harper, J. W. (2004). Systematic Analysis and Nomenclature of Mammalian F-Box Proteins. Genes Dev. 18, 2573–2580. doi:10.1101/gad.1255304
Kilpinen, S., Autio, R., Ojala, K., Iljin, K., Bucher, E., Sara, H., et al. (2008). Systematic Bioinformatic Analysis of Expression Levels of 17,330 Human Genes across 9,783 Samples from 175 Types of Healthy and Pathological Tissues. Genome Biol. 9, R139. doi:10.1186/gb-2008-9-9-r139
Kondo, T., Kobayashi, M., Tanaka, J., Yokoyama, A., Suzuki, S., Kato, N., et al. (2004). Rapid Degradation of Cdt1 upon UV-Induced DNA Damage Is Mediated by SCF Complex. J. Biol. Chem. 279, 27315–27319. doi:10.1074/jbc.m314023200
Kong, H., Leebens-Mack, J., Ni, W., Depamphilis, C. W., and Ma, H. (2004). Highly Heterogeneous Rates of Evolution in the SKP1 Gene Family in Plants and Animals: Functional and Evolutionary Implications. Mol. Biol. Evol. 21, 117–128. doi:10.1093/molbev/msh001
Kruiswijk, F., Yuniati, L., Magliozzi, R., Low, T. Y., Lim, R., Bolder, R., et al. (2012). Coupled Activation and Degradation of eEF2K Regulates Protein Synthesis in Response to Genotoxic Stress. Sci. Signal. 5, ra40. doi:10.1126/scisignal.2002718
Kulathu, Y., and Komander, D. (2012). Atypical Ubiquitylation - the Unexplored World of Polyubiquitin beyond Lys48 and Lys63 Linkages. Nat. Rev. Mol. Cel. Biol. 13, 508–523. doi:10.1038/nrm3394
Lane, D. P. (1984). Oncogenic Intelligence: Cell Immortalization and Transformation by the P53 Gene. Nature 312, 596–597. doi:10.1038/312596a0
Lengauer, C., Kinzler, K. W., and Vogelstein, B. (1997). Genetic Instability in Colorectal Cancers. Nature 386, 623–627. doi:10.1038/386623a0
Lepage, C. C., Morden, C. R., Palmer, M. C. L., Nachtigal, M. W., and McManus, K. J. (2019). Detecting Chromosome Instability in Cancer: Approaches to Resolve Cell-To-Cell Heterogeneity. Cancers (Basel) 11, 226. doi:10.3390/cancers11020226
Lepage, C. C., Palmer, M. C. L., Farrell, A. C., Neudorf, N. M., Lichtensztejn, Z., Nachtigal, M. W., et al. (2021). Reduced SKP1 and CUL1 Expression Underlies Increases in Cyclin E1 and Chromosome Instability in Cellular Precursors of High-Grade Serous Ovarian Cancer. Br. J. Cancer 124, 1699–1710. doi:10.1038/s41416-021-01317-w
Liang, Y., Chen, H., Asher, J. H., Chang, C.-C., and Friedman, T. B. (1997). Human Inner Ear OCP2 cDNA Maps to 5q22-5q35.2 with Related Sequences on Chromosomes 4p16.2-4p14, 5p13-5q22, 7pter-Q22, 10 and 12p13-12qter. Gene 184, 163–167. doi:10.1016/s0378-1119(96)00590-2
Liu, H., Xing, Y., Yang, S., and Tian, D. (2011). Remarkable Difference of Somatic Mutation Patterns between Oncogenes and Tumor Suppressor Genes. Oncol. Rep. 26, 1539–1546. doi:10.3892/or.2011.1443
Liu, Y.-Q., Wang, X.-L., Cheng, X., Lu, Y.-Z., Wang, G.-Z., Li, X.-C., et al. (2015). Skp1 in Lung Cancer: Clinical Significance and Therapeutic Efficacy of its Small Molecule Inhibitors. Oncotarget 6, 34953–34967. doi:10.18632/oncotarget.5547
Longley, D. B., Harkin, D. P., and Johnston, P. G. (2003). 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 3, 330–338. doi:10.1038/nrc1074
Ludwig, H., Khayat, D., Giaccone, G., and Facon, T. (2005). Proteasome Inhibition and its Clinical Prospects in the Treatment of Hematologic and Solid Malignancies. Cancer 104, 1794–1807. doi:10.1002/cncr.21414
Ly, P., and Cleveland, D. W. (2017). Rebuilding Chromosomes after Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cel Biol. 27, 917–930. doi:10.1016/j.tcb.2017.08.005
Mamely, I., Van Vugt, M. A., Smits, V. A., Semple, J. I., Lemmens, B., Perrakis, A., et al. (2006). Polo-like Kinase-1 Controls Proteasome-dependent Degradation of Claspin during Checkpoint Recovery. Curr. Biol. 16, 1950–1955. doi:10.1016/j.cub.2006.08.026
Mandel, S. A., Fishman-Jacob, T., and Youdim, M. B. H. (2012). Targeting SKP1, an Ubiquitin E3 Ligase Component Found Decreased in Sporadic Parkinson's Disease. Neurodegener. Dis. 10, 220–223. doi:10.1159/000333223
McAndrew, E. N., and McManus, K. J. (2017). The Enigmatic Oncogene and Tumor Suppressor-like Properties of RAD54B: Insights into Genome Instability and Cancer. Genes Chromosomes Cancer 56, 513–523. doi:10.1002/gcc.22458
Morreale, F. E., and Walden, H. (2016). Types of Ubiquitin Ligases. Cell Res. 165, 248-248–e241. doi:10.1016/j.cell.2016.03.003
Morden, C. R., Farrell, A. C., Sliwowski, M., Lichtensztejn, Z., Altman, A. D., Nachtigal, M. W., et al. (2021). Chromosome Instability Is Prevalent and Dynamic in High-Grade Serous Ovarian Cancer Patient Samples. Gynecol. Oncol. 161, 769–778. doi:10.1016/j.ygyno.2021.02.038
Nakamura, K., Nakayama, K., Ishikawa, N., Ishikawa, M., Sultana, R., Kiyono, T., et al. (2018). Reconstitution of High-Grade Serous Ovarian Carcinoma from Primary Fallopian Tube Secretory Epithelial Cells. Oncotarget 9, 12609–12619. doi:10.18632/oncotarget.23035
Nakayama, K. I., and Nakayama, K. (2006). Ubiquitin Ligases: Cell-Cycle Control and Cancer. Nat. Rev. Cancer 6, 369–381. doi:10.1038/nrc1881
Ng, R. W. M., Arooz, T., Yam, C. H., Chan, I. W. Y., Lau, A. W. S., and Poon, R. Y. C. (1998). Characterization of the Cullin and F-Box Protein Partner Skp1. FEBS Lett. 438, 183–189. doi:10.1016/s0014-5793(98)01299-x
Penner-Goeke, S., Lichtensztejn, Z., Neufeld, M., Ali, J. L., Altman, A. D., Nachtigal, M. W., et al. (2017). The Temporal Dynamics of Chromosome Instability in Ovarian Cancer Cell Lines and Primary Patient Samples. Plos Genet. 13, e1006707. doi:10.1371/journal.pgen.1006707
Pacini, C., Dempster, J. M., Boyle, I., Goncalves, E., Najgebauer, H., Karakoc, E., et al. (2021). Integrated cross-study datasets of genetic dependencies in cancer. Nat. Commun. 12, 1661. doi:10.1038/s41467-021-21898-7
Perets, R., Wyant, G. A., Muto, K. W., Bijron, J. G., Poole, B. B., Chin, K. T., et al. (2013). Transformation of the Fallopian Tube Secretory Epithelium Leads to High-Grade Serous Ovarian Cancer in Brca;Tp53;Pten Models. Cancer Cell 24, 751–765. doi:10.1016/j.ccr.2013.10.013
Pihan, G. A., Purohit, A., Wallace, J., Knecht, H., Woda, B., Quesenberry, P., et al. (1998). Centrosome Defects and Genetic Instability in Malignant Tumors. Cancer Res. 58, 3974–3985.
Piva, R., Liu, J., Chiarle, R., Podda, A., Pagano, M., and Inghirami, G. (2002). In Vivo interference with Skp1 Function Leads to Genetic Instability and Neoplastic Transformation. Mol. Cel Biol. 22, 8375–8387. doi:10.1128/mcb.22.23.8375-8387.2002
Puklowski, A., Homsi, Y., Keller, D., May, M., Chauhan, S., Kossatz, U., et al. (2011). The SCF-FBXW5 E3-Ubiquitin Ligase Is Regulated by PLK4 and Targets HsSAS-6 to Control Centrosome Duplication. Nat. Cel Biol. 13, 1004–1009. doi:10.1038/ncb2282
Rhodes, D. R., Kalyana-Sundaram, S., Mahavisno, V., Varambally, R., Yu, J., Briggs, B. B., et al. (2007). Oncomine 3.0: Genes, Pathways, and Networks in a Collection of 18,000 Cancer Gene Expression Profiles. Neoplasia 9, 166–180. doi:10.1593/neo.07112
Robak, T., Huang, H., Jin, J., Zhu, J., Liu, T., Samoilova, O., et al. (2015). Bortezomib-Based Therapy for Newly Diagnosed Mantle-Cell Lymphoma. N. Engl. J. Med. 372, 944–953. doi:10.1056/nejmoa1412096
Sajesh, B., Guppy, B., and McManus, K. (2013). Synthetic Genetic Targeting of Genome Instability in Cancer. Cancers 5, 739–761. doi:10.3390/cancers5030739
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: Chimeric Molecules that Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. 98, 8554–8559. doi:10.1073/pnas.141230798
Sandhya, P., and Danda, D. (2014). Role of Vacuolar ATPase and Skp1 in Sjögren's Syndrome. Med. Hypotheses 82, 319–325. doi:10.1016/j.mehy.2013.12.019
Sato, M., Rodriguez-Barrueco, R., Yu, J., Do, C., Silva, J. M., and Gautier, J. (2015). MYC Is a Critical Target of FBXW7. Oncotarget 6, 3292–3305. doi:10.18632/oncotarget.3203
Schulman, B. A., Carrano, A. C., Jeffrey, P. D., Bowen, Z., Kinnucan, E. R. E., Finnin, M. S., et al. (2000). Insights into SCF Ubiquitin Ligases from the Structure of the Skp1-Skp2 Complex. Nature 408, 381–386. doi:10.1038/35042620
Silverman, J. S., Skaar, J. R., and Pagano, M. (2012). SCF Ubiquitin Ligases in the Maintenance of Genome Stability. Trends Biochem. Sci. 37, 66–73. doi:10.1016/j.tibs.2011.10.004
Skaar, J. R., Pagan, J. K., and Pagano, M. (2014). SCF Ubiquitin Ligase-Targeted Therapies. Nat. Rev. Drug Discov. 13, 889–903. doi:10.1038/nrd4432
Sowden, J., Morrison, K., Schofield, J., Putt, W., and Edwards, Y. (1995). A Novel cDNA with Homology to an RNA Polymerase II Elongation Factor Maps to Human Chromosome 5q31 (TCEB1L) and to Mouse Chromosome 11 (Tceb1l). Genomics 29, 145–151. doi:10.1006/geno.1995.1225
Swords, R. T., Erba, H. P., Deangelo, D. J., Bixby, D. L., Altman, J. K., Maris, M., et al. (2015). Pevonedistat (MLN4924), a First-In-Class NEDD8-Activating Enzyme Inhibitor, in Patients with Acute Myeloid Leukaemia and Myelodysplastic Syndromes: a Phase 1 Study. Br. J. Haematol. 169, 534–543. doi:10.1111/bjh.13323
Tate, J. G., Bamford, S., Jubb, H. C., Sondka, Z., Beare, D. M., Bindal, N., et al. (2019). COSMIC: the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 47, D941–D947. doi:10.1093/nar/gky1015
Thalmann, I., Rosenthal, H. L., Moore, B. W., and Thalmann, R. (1980). Organ of Corti-specific Polypeptides: OCP-I and OCP-II. Arch. Otorhinolaryngol. 226, 123–128. doi:10.1007/bf00455126
Thalmann, R., Henzl, M. T., Killick, R., Ignatova, E. G., and Thalmann, I. (2003). Toward an Understanding of Cochlear Homeostasis: the Impact of Location and the Role of OCP1 and OCP2. Acta Oto-Laryngol. 123, 203–208. doi:10.1080/0036554021000028100
Thompson, L. L., Baergen, A. K., Lichtensztejn, Z., and McManus, K. J. (2020). Reduced SKP1 Expression Induces Chromosome Instability through Aberrant Cyclin E1 Protein Turnover. Cancers (Basel) 12, 531. doi:10.3390/cancers12030531
Thompson, L. L., Rutherford, K. A., Lepage, C. C., and McManus, K. J. (2021). The SCF Complex Is Essential to Maintain Genome and Chromosome Stability. Ijms 22, 8544. doi:10.3390/ijms22168544
Tsherniak, A., Vazquez, F., Montgomery, P. G., Weir, B. A., Kryukov, G., Cowley, G. S., et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564-576–e516. doi:10.1016/j.cell.2017.06.010
UniProt Consortium (2015). UniProt: a Hub for Protein Information. Nucleic Acids Res. 43, D204–D212. doi:10.1093/nar/gku989
Vishwakarma, R., and McManus, K. J. (2020). Chromosome Instability; Implications in Cancer Development, Progression, and Clinical Outcomes. Cancers (Basel) 12, 824. doi:10.3390/cancers12040824
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A., and Kinzler, K. W. (2013). Cancer Genome Landscapes. Science 339, 1546–1558. doi:10.1126/science.1235122
Yamanaka, A., Yada, M., Imaki, H., Koga, M., Ohshima, Y., and Nakayama, K.-I. (2002). Multiple Skp1-Related Proteins in Caenorhabditis elegans: Diverse Patterns of Interaction with Cullins and F-Box Proteins. Curr. Biol. 12, 267–275. doi:10.1016/s0960-9822(02)00657-7
Yoshida, Y., Murakami, A., and Tanaka, K. (2011). Skp1 Stabilizes the Conformation of F-Box Proteins. Biochem. Biophys. Res. Commun. 410, 24–28. doi:10.1016/j.bbrc.2011.05.098
Zhang, H., Kobayashi, R., Galaktionov, K., and Beach, D. (1995). pl9skp1 and P45skp2 Are Essential Elements of the Cyclin A-CDK2 S Phase Kinase. Cell 82, 915–925. doi:10.1016/0092-8674(95)90271-6
Keywords: cancer, centrosome dynamics, chromosome instability, DNA damage response, Fbox protein, genome instability, SCF complex, SKP1
Citation: Thompson LL, Rutherford KA, Lepage CC and McManus KJ (2022) Aberrant SKP1 Expression: Diverse Mechanisms Impacting Genome and Chromosome Stability. Front. Cell Dev. Biol. 10:859582. doi: 10.3389/fcell.2022.859582
Received: 21 January 2022; Accepted: 22 February 2022;
Published: 08 March 2022.
Edited by:
Philipp Kaldis, Lund University, SwedenReviewed by:
Zhiwei Wang, Wenzhou Medical University, ChinaCopyright © 2022 Thompson, Rutherford, Lepage and McManus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kirk J. McManus, S2lyay5NY01hbnVzQHVtYW5pdG9iYS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.