 Jiaqi Yang
Jiaqi Yang Qianyun Guo
Qianyun Guo Xunxun Feng
Xunxun Feng Yang Liu
Yang Liu Yujie Zhou
Yujie Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 13 May 2022
Sec. Molecular and Cellular Pathology
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.841523
This article is part of the Research Topic Mitochondrial Dysfunction in Metabolic Diseases View all 6 articles
Cardiovascular diseases (CVDs) are serious public health issues and are responsible for nearly one-third of global deaths. Mitochondrial dysfunction is accountable for the development of most CVDs. Mitochondria produce adenosine triphosphate through oxidative phosphorylation and inevitably generate reactive oxygen species (ROS). Excessive ROS causes mitochondrial dysfunction and cell death. Mitochondria can protect against these damages via the regulation of mitochondrial homeostasis. In recent years, mitochondria-targeted therapy for CVDs has attracted increasing attention. Various studies have confirmed that clinical drugs (β-blockers, angiotensin-converting enzyme inhibitors/angiotensin receptor-II blockers) against CVDs have mitochondrial protective functions. An increasing number of cardiac mitochondrial targets have shown their cardioprotective effects in experimental and clinical studies. Here, we briefly introduce the mechanisms of mitochondrial dysfunction and summarize the progression of mitochondrial targets against CVDs, which may provide ideas for experimental studies and clinical trials.
Mitochondria are semiautonomous organelles located in the cytoplasm. Unlike other organelles, mitochondria have their own genomes (mtDNAs), double-stranded circular DNAs, and are coated by a double-layered membrane (Davis and Williams, 2012; van der Bliek et al., 2017). Mitochondria are responsible for the production of adenosine triphosphate (ATP) and the regulation of nutritional metabolism, calcium homeostasis, and cellular viability in several organs (Kim et al., 2008). Due to the huge demand for ATP in the heart, mitochondria are highly abundant in cardiac cells, especially in cardiomyocytes (CMs), occupying nearly 20–40% volume in adult CMs (Schaper et al., 1985). Approximately 6 kg of ATP is produced by cardiac mitochondria every day (Siasos et al., 2018). Thus, mitochondria play essential roles in the cardiovascular system.
The main source of energy obtained by oxidative phosphorylation (OXPHOS) comes from the oxidation of fatty acids in the adult heart (Stanley, 2005). ATP is synthesized in adult hearts following the steps below: fatty acyl-coenzyme A (CoAs) are synthesized with the help of acyl CoA syntheses. To enter the cardiac mitochondria, fatty acyl CoAs are converted to acylcarnitines by carnitine palmitoyl transferase 1 (CPT1) and are transferred to the inner membrane of mitochondria (IMM) where they are liberated to fatty acyl CoAs and initiate β-oxidation. Acetyl CoA, a product of β-oxidation, enters the Kerbs cycle and generates reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide (FADH2). NADH and FADH2 generated from the Kerbs cycle, and from β-oxidation, transfer electrons through the electron transport chain (ETC), which comprises four complexes on the IMM. The efflux of protons accompanied by the electronic flow on ETC activates ATP-synthase and produces ATP (shown in Figure 1) (Chen and Butow, 2005).
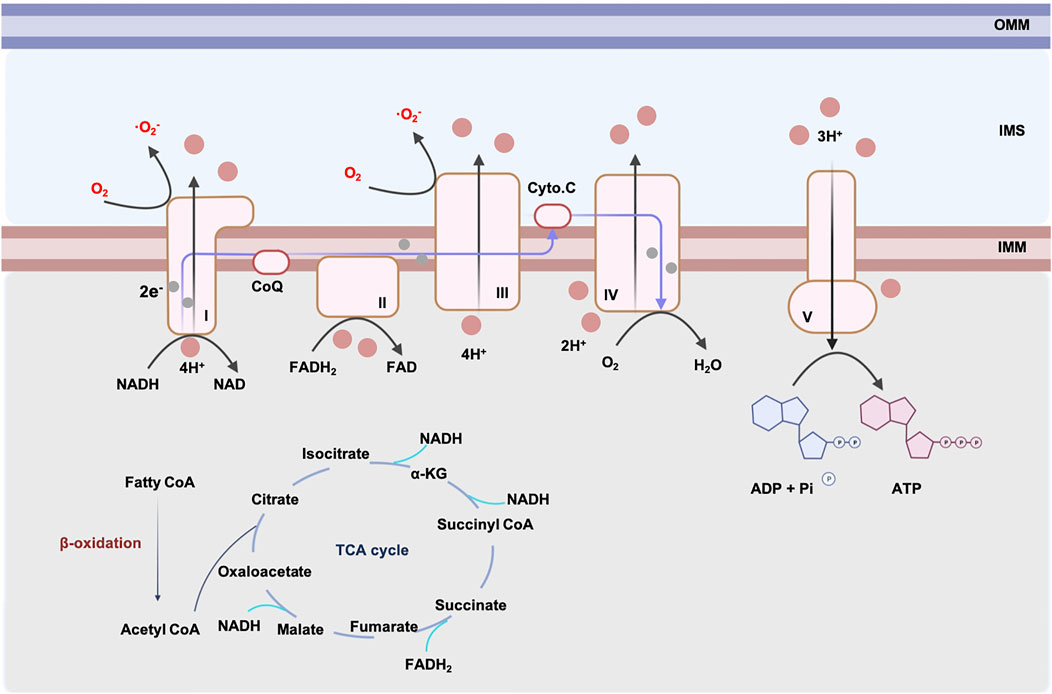
FIGURE 1. ATP generation and ROS production in cardiac mitochondrion. The major resource of cardiac mitochondria comes from β-oxidation. Generation of NADH and FADH2 from β-oxidation and TCA cycle are used by respiratory chain complexes. Respiratory chain complexes are located in IMM. Electrons generated from complex I, II pass through the respiratory chain and alter the concentration gradient of H+. Complex V produces ATP driven by H+ flow. During the generation of ATP, complex I and III produce O2−. Excessive ROS leads to mitochondrial dysfunction. Abbreviations: IMM, inner mitochondrial membrane, OMM, outer mitochondrial membrane, IMS, mitochondrial intermembrane space, ROS, reactive oxygen species, NADH, reduced nicotinamide adenine dinucleotide, FADH2, reduced flavin adenine dinucleotide, CoQ, coenzyme Q, Cyto. C, cytochrome C, ADP, adenosine diphosphate, ATP, adenosine-triphosphate triphosphate, TCA, tricarboxylic acid cycle, CoA, coenzyme A, α-KG, α-ketoglutarate.
However, the production of ATP through the respiratory chain is accompanied by an inevitable generation of ROS, especially superoxide anion (·O2−). It has been reported that nearly 1, 2% of electrons is related to superoxide production during ATP synthesis (Boveris et al., 1976). Complex I and III are the main sites of ROS production in mitochondria (Cadenas, 2018). Complex I is an L-shaped component made up of a hydrophilic arm and a hydrophobic arm, in which the semi-/reduced flavin mononucleotide (FMN) and N1a/N1b iron-sulfur clusters are the main sites for superoxide production (Efremov and Sazanov, 2011). Complex III generates superoxide mostly by ubisemiquinone formed at the Qo site, where ubisemiquinone transfers electrons to oxygen (Iwata et al., 1998). ROS generated from mitochondrial complex I and III is the main mechanism of ischemia/reperfusion (I/R) injury in the heart. Kang et al. reported that complex I is responsible for the generation of O2− in post-ischemic CMs, while damage to the N1a cluster causes FMN-produced superoxide (Kang et al., 2018). Meanwhile, Chen et al. found an increasing generation of antimycin A-enhanced superoxide in complex III ex vivo (Chen et al., 2008). Normal amounts of O2− can be converted to more stable hydrogen peroxide by superoxide dismutases (SODs) (Suzuki et al., 2013). However, excessive O2− can damage mtDNA, lipids, and proteins, leading to mitochondrial dysfunction, oxidative stress, and cell death (Haworth and Hunter, 1979; Peoples et al., 2019). In cardiovascular diseases (CVDs), it has been reported that excessive ROS and mitochondrial dysfunction are related to numerous cardiac diseases, such as atherosclerosis (AS), I/R injury, cardiac hypertrophy, heart failure (HF), and degenerative aortic valve disease (Peoples et al., 2019).
The classical perception is that ROS is the major driver of the mutations of mtDNA, while others are replicated errors of mtDNA, and present a less efficient mtDNA repair system than nuclear DNA. mtDNA, which is closed to ROS and ETC, causes the degradation of nucleic acid chemical components. The most common induction of mutations is via point mutation of purines and pyrimidines (Lee and Wei, 2007). An accumulation of multiple mutations leads to the exhaustion of mtDNA-encoded proteins and less efficient ATP production. It is also recognized that accumulated mtDNA damage can also increase ROS generation, resulting in a vicious circle (Płoszaj et al., 2010). Additionally, mitochondria trigger and sustain the chronic-sterile inflammation and immunity in the heart, which mostly occur after I/R damage (shown in Figure 2), and are related to the development of HF (Nakayama and Otsu, 2018). This phenomenon is induced by activated pattern recognition receptors (PRRs, e.g., toll-like receptor 9 [TLR9] and nod-like receptor pyrin domain containing 3 [NLRP3]) sensing damage-associated molecular patterns (DAMPs) (Rodrigues et al., 2015). As for the characteristics of mtDNA (close to the respiratory chain, sensitive to ROS and a relatively low level of methylation), the accumulation of mtDNA mutations represents the most well-known DAMP, which activates PRRs, recruits inflammatory cytokines, and activates the response of immune cells in the heart.
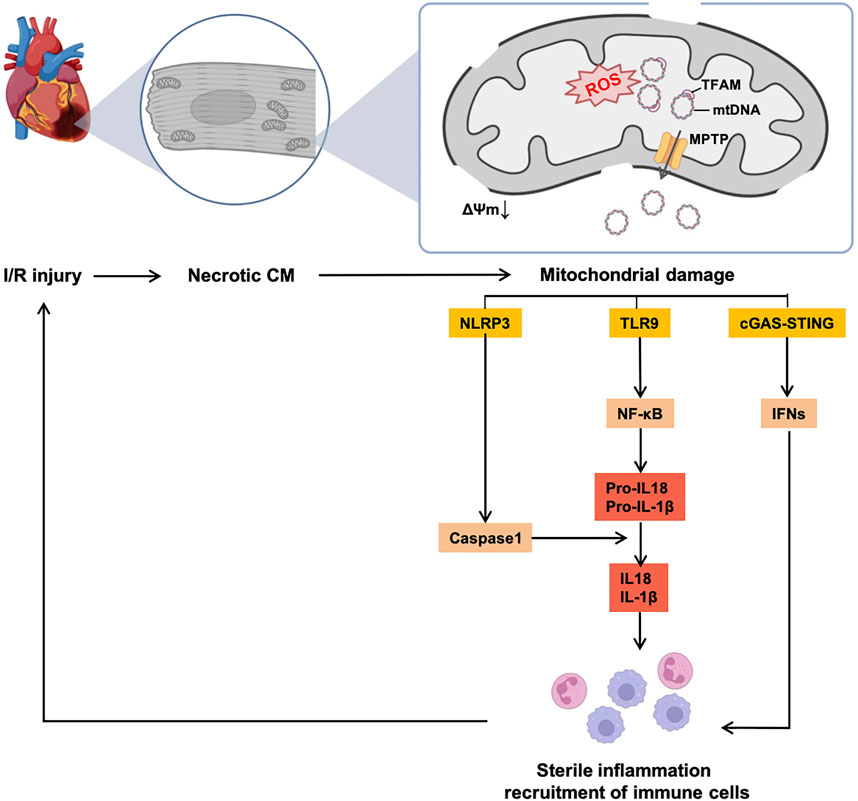
FIGURE 2. Vicious circle of mtDNA-induced sterile inflammation and mitochondrial dysfunction in the I/R heart. Accumulation of mutant mtDNAs caused by excessive ROS release from MPTP, thereby inducing consecutive sterile inflammatory responses and recruitment of immune cells, which continuously aggregate I/R injury and CM necrosis. Abbreviations: I/R, ischemia/reperfusion, CM, cardiomyocyte, NLRP3, nod-like receptor pyrin domain containing 3, TLR9, toll-like receptor 9, cGAS, cyclic GMP–AMP synthetase, STING, stimulator of interferon gene, NF-κB, nuclear factor kappa B, IFN, interferon, IL, interleukin.
Mechanisms of mtDNA release from mitochondria remain controversial. The classical theory believes that the mtDNA release is mediated by the opening of the mitochondrial permeability transition pore (MPTP) (Patrushev et al., 2004). The MPTP is an unspecific pore in the mitochondria initially found by Haworth et al. (Haworth and Hunter, 1979). MPTP is strongly related to acute I/R injury and is open under conditions of oxidative stress, overloaded [Ca2+]mito and altered phosphate level during ischemia (Crompton et al., 1987). Although the opening of the MPTP is not ROS-dependent, excessive ROS synergizes the opening of the MPTP with other factors (mentioned above), thereby leading to further mitochondrial injury (Haworth and Hunter, 1979). In addition, evidence has shown that inhibiting the MPTP is an essential mechanism of cardioprotective ischemic pre-/post-conditioning (iPre and iPost) in the heart (reviewed by Penna et al.) (Hausenloy et al., 2009). Opening of the MPTP causes the swelling of the mitochondrial matrix and damage to the outer mitochondrial membrane (OMM), therefore leading to the release of pro-apoptotic factors. Especially, the opening of the MPTP is the major way of releasing mtDNAs from mitochondria and induces sterile inflammation. Therefore, the MPTP is recognized as a promising target against mitochondrial dysfunction in the heart, and several studies from animal models and clinical trials have investigated this target (details are in Section 3.4).
It is also reported that mtDNA release is a voltage-dependent anion channel (VDAC) oligomers-regulated process. Mitochondrial outer membrane permeabilization (MOMP) is required for the release of the mtDNA. Kim et al. found that VDAC can oligomerize under the stimulation of oxidative stress, and induce the MOMP as well as the liberation of mtDNA (Kim et al., 2019). Riley et al. reported that the release of the mtDNA is also related to the mitochondrial inner membrane permeabilization (MIMP). A Bax/BAK (pro-apoptotic member B-cell lymphoma 2, Bcl-2 family)-induced expansion of OMM pores following behind MOMP causes extrusion of IMM from mitochondrion to cytoplasm and the occurrence of MIMP-mediated mtDNA release (Riley et al., 2018). Besides, mitochondrial-derived vesicles (MDVs) have been described as an additional regulator of mitochondrial homeostasis (the major mechanisms are mentioned in Section 2), which establish crosstalk between mitochondria and lysosomes. Dysfunctional mitochondria could lead to leakages of MDVs including their components, which trigger PRRs and activate inflammation. However, the hypothesis of MDVs-dependent mtDNA release remains under investigation (Picca et al., 2019; Picca et al., 2021).
In the next step, mutant mtDNA can be released into intracellular and extracellular spaces. For intracellular liberation, damaged mtDNAs released from disruptive OMM to the cytoplasm, which activates intracellular PRRs and initiates the non-infectious inflammation via interleukin 10 (IL-10) and IL-1β. mtDNA can also induce the activation of cyclic GMP–AMP synthetase (cGAS)/stimulator of interferon gene (STING)/type I interferon (IFN) pathways without inflammatory cytokines activation, especially in the I/R injured heart (White et al., 2014). Several pieces of evidence support the statement that mtDNA can also release out of cells, activating inflammation and stimulating an immune response. In necrotic cells, mtDNA leaks through the disruptive plasma membrane into extracellular space. There are some other mechanisms for the liberation of mtDNA from non-necrotic cells. Guescini et al. detected the existence of cell-free mtDNA in exosomes released by glioblastoma cells and astrocytes, which implies exosomes proceed with the extracellular release of mtDNA (Guescini et al., 2010). Lood et al. reported the release of neutrophils extracellular traps (NETs) is also accompanied by the liberation of mtDNA (Lood et al., 2016). Extracellular leakages of mtDNAs can be recognized by TLR9 through endocytosis or directly captured by TLR9 on the plasma membrane of some cells (e.g., resting B cells and peripheral blood monocytes) and activate inflammation (Eaton-Bassiri et al., 2004; Dasari et al., 2005).
It is not surprising to see that accumulated mtDNA damages and mtDNA-induced chronic inflammation are correlated to AS, I/R injury, hypertension and HF due to the high consumption of oxygen and ATP in the heart (Poznyak et al., 2020). Among them, AS is strongly related to mtDNA mutations. Primary AS (AS that cannot be explained by classical risk factors, such as diabetes, hyperlipidemia, and smoking) has been described as a result of mitochondrial disorder. A study analyzed twelve lipofibrous plaques of aorta intima compared to intact area, and reported four mtDNA mutations, including m.1555A > G, m.3256C > T, m.12315G > A and m.15059G > A, are relevant to atherosclerotic plaques (Sobenin et al., 2013). An analysis enrolled 65 blood samples and 23 atherosclerotic plaques from coronary artery disease (CAD) patients showed a higher deletion of 4977bp (26.2% vs 4.5%) as well as higher heteroplasmy rates (18–46%) than the control group. Interestingly, these deletions are independent of classical risk factors of AS and not inferred by baseline characteristics of patients (Sobenin et al., 2013). Furthermore, Sazonovo et al. showed the data from 225 myocardial infarction (MI) patients, that a mutation of m.5178C > A is related to the occurrence of MI. On the opposite, the other two variants of m.14846G > A and m.12315G > A are observed in healthy controls (Sazonova et al., 2018). A syndrome of Mitochondrial Encephalomyopathy, Lactic Acidosis, and at least one Stroke-like episode (MELAS) is one of the representative mitochondrial diseases carrying a mutant of m.3243A > G in mitochondrial tRNA leucine 1 (Manwaring et al., 2007). Numerous studies found patients with MELAS are also accompanied by dysfunctional endothelia, atherosclerotic carotid, and cerebral artery without potential risk factors of AF. These studies also provide indirect proof of the development of mutant mtDNA-related AS (Pek et al., 2019; Finsterer, 2020). Besides, mtDNA damages also play an essential role in the development of AS accelerated by traditional risk factors mentioned above. A review from Sobenin et al. well summarized the variants of mtDNA relevant to smoking, MELAS, and diabetes mellitus, which are the promoters of the development of AS (Sobenin et al., 2015).
On the other hand, chronic inflammatory and immune responses induced by mtDNA-related DAMPs are also involved in AS (Sobenin et al., 2015). mtDNA mutations affect many cell types in AS. In endothelial cells (ECs), mtDNA activates TLR9 and induces the production of interleukin-1β (IL-1β) through the NF-κB pathway. Moreover, the damage associated with ECs recruits inflammatory cells and alters vascular wall permeability, which initiates the development of AS (Jin et al., 2019). The release of mtDNA induces the activation of NF-κB in macrophages and is related to the synthesis of pro-inflammatory cytokines, uptake of oxidized low-density lipoprotein (oxLDL), and formation of foam cells (Tedgui and Mallat, 2006). Dysfunction of mitochondrial dynamics induced by damaged mtDNA in smooth muscle cells (SMCs) results in their energetic decline and apoptosis in plaque, which alters the stability of plaque in AS (Yu et al., 2017; Docherty et al., 2018). Besides, the release of mutant mtDNA from damaged mitochondria to the cytoplasm is a sign of the early phase of AS. Yu et al. detected mtDNA in both circulating and vascular cells in human atherosclerotic plaques even before other signs of AS occur (Yu et al., 2013). Together all, mtDNA mutations and mtDNA-induced sterile inflammations are essential targets/aspects for the detection and treatment of AS.
To avoid the accumulation of ROS and mitochondrial dysfunction, mitochondria sustain their homeostasis by mitochondrial biogenesis, mitochondrial dynamics, and mitophagy (summarized in Figure 3), all of which are potential targets for treatment.
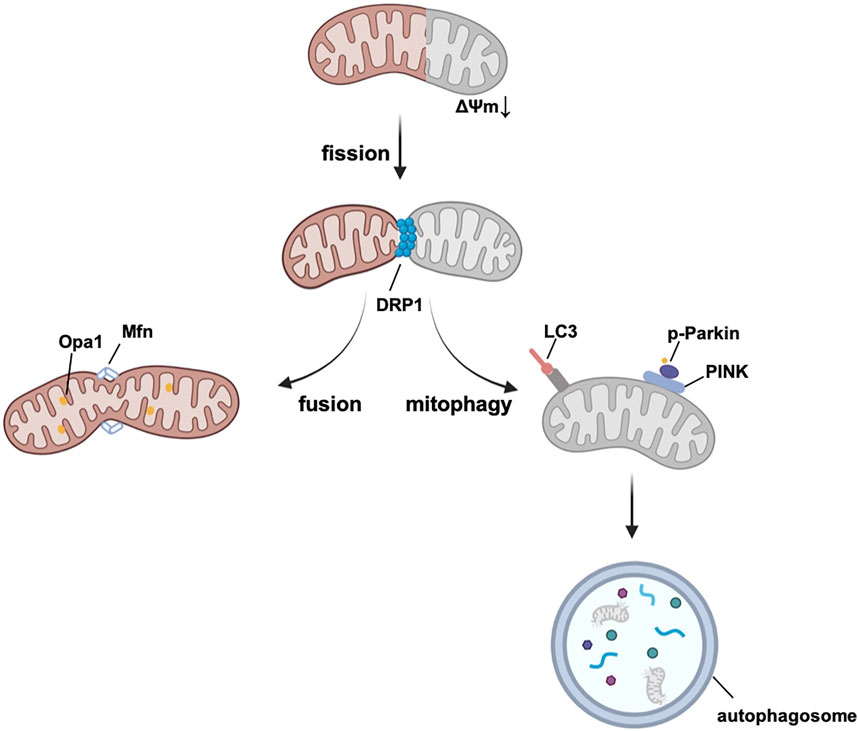
FIGURE 3. Mitochondrial quality control system. Damaged mitochondrial fragment splits from healthy mitochondrion by fission and then degrades by mitophagy. Mitochondrial fusion assembles healthy mitochondria and maintains the normal function of mitochondria. Abbreviations: Opa1, optic atrophy 1, Mfn, mitofusin, DRP1, Dynamin-related protein 1, LC3, microtubule-associated protein 1A/1B-light chain 3, PINK, PTEN-induced kinase 1, Δψm, mitochondrial membrane potential.
Mitochondrial biogenesis increases the number of healthy mitochondria in the heart, improves the replication and repair of mtDNA, and induces the synthesis of mitochondrial enzymes and proteins (Popov, 2020). It has been reported that mitochondrial transcription factor A (TFAM) and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) are central regulators of mitochondrial biogenesis (Jornayvaz and Shulman, 2010). PGC-1α activates TFAM, which is essential for the transcription, replication, and stability of mtDNA (Yokokawa et al., 2018). PGC-1α can also increase the expression of nuclear respiratory factor (NRF)-1 and NRF-2, thereby promoting the production of vital mitochondrial enzymes (Reznick et al., 2007). There are several pathways contributing to mitochondrial biogenesis. Jager et al. reported that adenosine 5‘-monophosphate-activated protein kinase (AMPK) is critical for mitochondrial biogenesis by regulating the phosphorylation of Thr177 and Ser538 (two sites of PGC1α) (Jäger et al., 2007). Sirtuins (SIRT) are a series of deacetylases and deacetylases that control energy metabolism. There is evidence that SIRT1 promotes the uptake of glucose and mitochondrial biogenesis by activating PGC-1α (Guarente, 2007). Wang et al. reported that SIRT1 can prevent heart aging by mediating deacetylation of PGC-1α and activation of mitochondrial biogenesis (Wang et al., 2020a). To summarize, morphological and quantitative changes in mitochondria have been observed in the heart, liver, and skeletal muscle in pathological conditions, and the regulation of mitochondrial biogenesis is a potential target to treat mitochondrial dysfunction.
Mitochondrial dynamics, represented by the balance of mitochondrial fission and fusion, are under the regulation of dynamin-related guanosine triphosphatase (GTPase) proteins (Hoppins et al., 2007). Dynamin-related protein 1 (DRP1) is the main protein of fission, which interacts with the receptors located in OMM, such as mitochondria fission factor (MFF), fission protein-1 (Fis1), and mitochondrial dynamics proteins 49 and 51 (MiD49/51) (Long et al., 2013; Richter et al., 2014; Kornfeld et al., 2018). Besides, MiD49/51 and MFF have also been reported to modulate GTPase-derived DRP1 constrictive activity (MiD49/51 inhibits GTPase, whereas, MFF promotes it) (Liu and Chan, 2015; Osellame et al., 2016). During the fission phase, dysfunctional fragments of mitochondria can be cut into small, spherical pieces and then degraded by mitophagy. Excessive mitochondrial fission is responsible for increasing the infarct area in I/R heart injury, and it induces both excessive mitophagy and cell death (Bi et al., 2019). Drugs such as metformin can block DRP1 and maintain the integrity of mitochondria according to the studies using diabetic animal models in vivo (Li et al., 2016; Wang et al., 2019). Recently, metformin is also demonstrated to protect against AS by reducing cardiac mitochondrial fission (it is most well recognized by its mechanism in the regulation of mitochondrial complex I, mainly through the AMPK pathway, discussed in 3.3) (Wang et al., 2017). Mitochondrial fusion is first mediated by mitofusin 1 (Mfn1) and Mfn2 in the OMM, and then by optic atrophy 1 (Opa1) in the IMM (Koshiba et al., 2004). Deletion of Mfn1 and Mfn2 in the embryo causes death in utero, indicating that Mfn1 and Mfn2 are strongly contributed to heart development (Chen et al., 2003; Chen et al., 2011). There is evidence from murine hearts that the ablation of Mfn1 and Mfn2 is related to the fragmentation and malfunction of mitochondria and induction of lethal cardiomyopathy (Papanicolaou et al., 2012). A decreasing level of Opa1 is found in post-MI hearts of both rats and humans and correlates with the downregulation of mtDNA and antioxidant genes (Chen et al., 2009). Mitochondrial fusion is a beneficial process in which mitochondria with tiny defects of proteins and DNA are complemented by functional parts of other mitochondria, and are merged into elongated, tubular, interconnected mitochondrial networks (Campello and Scorrano, 2010).
Mitophagy is a form of autophagy that selectively targets the degradation of dysfunctional mitochondria and serves as a control system of mitochondria, and autophagosomes are the main effectors during mitophagy (Lemasters, 2005). Together with mitochondrial biogenesis and dynamics, mitophagy sustains the homeostasis of mitochondria. At the cellular level, mitophagy shows effects on cell death of CMs (Shires and Gustafsson, 2015; Torrealba et al., 2017), and regulates the differentiation of cardiac fibroblasts (CFs), the main responding cell population in post-MI hearts, thereby controlling cardiac remodeling (Urban et al., 2015). Normal levels of mitophagy protect against I/R damage. Several studies have investigated that repeated and short iPre promotes the adaptation of cardiac mitochondria to I/R injury via mitophagy (Zhang et al., 2016; Zhang et al., 2017; Xiao et al., 2020). In addition, mitophagy contributes to the remission of other CVDs, such as hypertension, cardiac remodeling after MI and diabetic cardiomyopathy (more details are provided in the review by Morales et al.) (Morales et al., 2020). Several important pathways are involved in different phases of mitophagy. Mitophagy usually starts with mitochondrial imperfection and a reduction in mitochondrial membrane potential (Δψm). Regularly, PTEN-induced kinase 1 (PINK1) recognizes damaged mitochondria and accumulates in the OMM, thereby leading to the phosphorylation of Parkin E3 ligase. Parkin helps to ubiquitinate proteins of the OMM (Kondapalli et al., 2012). Mitochondrial cargo receptors (MCRs) guide these proteins to bind with microtubule-associated protein 1A/1B-light chain 3 (LC3), which can be recognized by autophagosomes and finally digested (Lazarou et al., 2015). Interestingly, DRP1 can also help selectively recognize damaged mitochondria from healthy population by mitochondrial fission. PINK sometimes cannot recognize the damaged part of mitochondrion easily due to the interconnected rapid balance of electrochemical gradients. However, transient reduction of Δψm can be a negative selective pressure for DRP1-induced fission. Healthy part of the mitochondrion will enter the fission-fusion cycle. Whereas, dysfunctional sectors which cannot recover back to normal Δψm can be negatively selected by mitophagy. Additionally, DRP1, together with Δψm, can also induce a positive feedback loop (reduction of Δψm recruits DRP1, recruited DRP1 ulteriorly reduces Δψm), which is helpful to selective mitophagy. (Cho et al., 2019). Other MCRs can directly stimulate mitophagy in ubiquitination-independent pathways. Cardiolipin is mostly synthesized on the IMM (Ren et al., 2014) and has effects on the respiratory chain and mitochondrial quality control system (mitochondrial dynamics and mitophagy) (Dudek, 2017; Paradies et al., 2019). Damage signals of mitochondria cause the translocation of cardiolipin to the OMM, which combines with LC3 and contributes to the recognition of dysfunctional (part of) mitochondria in the heart (Chu et al., 2013; Dudek et al., 2019). Furthermore, B-cell lymphoma-2 (BCL2)-interacting protein 3 (BNIP3) and FUN14 domain containing 1 (FUNDC1) have been reported to activate mitophagy via a PINK1-independent pathway and help to protect against oxidative injury and I/R in the heart (Liu et al., 2012; Zhang et al., 2016).
Based on the mechanisms of mitochondrial injury/repair mentioned above, we summarize mito-targeted modulators against mitochondrial dysfunction in CVDs.
Triphenylphosphonium (TPP+) is a lipophilic cation, which accumulates in mitochondria due to the Δψm. Drugs conjugated with TPP can concentrate up to 500-fold more in mitochondria than in the cytoplasm (Burns et al., 1995; Smith et al., 1999). Most of the antioxidants used for the treatment of mitochondrial dysfunction are TPP+-binding compounds, and MitoQ and SkQ are the representative TPP+-binding drugs.
MitoQ is co-enzyme Q10 (CoQ10) attached to TPP+ in C10 hydrophobic domain. CoQ10 has been well described as a global antioxidant and is beneficial to prevent hypertension and HF (Pepe et al., 2007). However, it is not clear how much CoQ10 can be uptake by mitochondria. MitoQ may solve the problem of non-targeted mitochondrial uptake, as it combines well with fatty acids and activates the transmembrane proton conduction in the IMM (Severin et al., 2010). Numerous studies have shown the antioxidant function of MitoQ and its mechanisms in HF. Junior et al. reported that MitoQ rescues the cardiac function of pressure-overloaded HF in a mouse model (Ribeiro Junior et al., 2018). Goh et al. described that MitoQ controls the expression levels of Chast and Mhrt and attenuates cardiac remodeling and prevents HF ex vivo (Goh et al., 2019). MitoQ also protects the function of Mfn2 by regulating redox-sensitive lncRNA-microRNA networks (e.g., Mfn2 targeted, Plscr4-miR-214) (Kim et al., 2020). Furthermore, MitoQ has been shown to alleviate I/R injury of mouse and rat hearts ex vivo (Adlam et al., 2005; Dare et al., 2015). Taken together, MitoQ displays impressive benefits in the treatment of aging diseases, I/R injury, HF, and obesity both in vitro and in vivo (Dietl and Maack, 2017; Braakhuis et al., 2018; Hu et al., 2018; Fink et al., 2021). As a potential target for treatment, numerous clinical studies (completed and ongoing) have attempted to investigate its effects on humans. Firstly, the safety and tolerability of MitoQ administration as a supplemental diet in young adults have been proven previously (Shill et al., 2016). Secondly, MitoQ shows mixed outcomes in different diseases but has the advantage of improving vascular function in aged adults. Rossman et al. reported their results of a clinical trial that administered MitoQ (up to 6 weeks), showing that MitoQ improved brachial artery flow-mediated dilation (FMD, a parameter of evaluating vascular function), and decreased aortic stiffness and plasma oxidized low-density lipoprotein (ox-LDL, a marker of oxidative stress); the underlying mechanism was the ability of MitoQ to ameliorate oxidative stress and promote endothelial function (Rossman et al., 2018). Furthermore, MitoQ treatment for up to 28 days in chronic hepatitis C showed decreased plasma alanine transaminase (ALT) aspartate aminotransferase (Gane et al., 2010). In contrast, another clinical study showed no improvement of MitoQ therapy on Parkinson’s disease (Snow et al., 2010). Currently, no completed clinical trial has focused on the effect of MitoQ on CVDs, especially in I/R injury in the heart. However, two clinical studies focused on cardiac function are ongoing (NCT03960073, NCT03586414). Regarding the cardioprotective function of MitoQ investigated in animal models and the positive clinical results of protecting vascular function against endothelial injury, MitoQ remains a promising avenue to be investigated in further studies.
Visomitin (SkQ1) is another TPP+-binding compound, which prevents the oxidation of mitochondrial cardiolipin, and it is a “rechargeable” antioxidant, which can be reduced by respiratory chain complexes (Skulachev, 2007). Among all the organelles, Skulachev et al. have only observed the exclusive fluorescence of SkQR1 (one of the derivatives of SkQ1) in mitochondria in vitro, indicating the high targeted ability of SkQ1 towards mitochondria (Skulachev et al., 2009). SkQ1, similar to MitoQ, exhibits high biological activity even in a nano-molar concentration. However, SkQ1 has even more remarkable anti-oxidation with lower pro-antioxidant activity and wider ranges of working antioxidant activity than MitoQ (Antonenko et al., 2008), which is the reason why SkQ1 is a promising drug target in the treatment of mitochondrial dysfunction. Manskikh et al. showed that SkQ1 alleviated fibrotic formation, rescued the cardiac function, and decreased the incidence of senescence-associated cardiomyopathy in the aged murine heart (Manskikh et al., 2015). Furthermore, a low amount of SkQ1 (0.02 nmol/kg) showed obvious regulation of cardiac rhythm after MI in mice, while relatively high concentrations of SkQ1 (125–250 nmol/kg) helped decrease the infarct area in an I/R model ex vivo (Bakeeva et al., 2008). Although there have been no SkQ1-related clinical trials in CVDs, scientists found its benefits in the context of eye diseases. Petrov et al. reported the success of phase II clinical study on SkQ1, showing that it improved the dry eye symptom, which is also consistent with the results in pre-clinical studies (Brzheskiy et al., 2015; Zernii et al., 2017).
Triphenylphosphonium chloride (mitoTEMPO) has been well-studied as a beneficial mitochondrial target in cardiac animal models. As an antioxidant, mitoTEMPO exhibits positive effects on the hearts of aged mice (Olgar et al., 2018). Dey et al. found that mitoTEMPO normalized ROS levels in all cell types and suppressed HF-induced cardiac remodeling in guinea pigs (Dey et al., 2018). In a study by Fang et al., mitoTEMPO rescued iron-dependent CMs death in doxorubicin (DOX)-treated and I/R-induced cardiomyopathy in a murine model (Fang et al., 2019). Besides, mitoTEMPO also has effects against overloaded pressure in a transverse aortic constriction (TAC) model by protecting the integrity of the respiratory chain (Hoshino et al., 2014).
Penetrating peptides, also called Szeto–Schiller peptides, have antioxidant properties in CVDs (Christodoulou et al., 2004). Penetrating peptides are small, transcellular peptides and can be uptake by mitochondria. The underlying mechanism of their antioxidant function may be attributed to tyrosine or dimethyltyrosine residue (Szeto, 2006). One of the representative penetrating peptides is SS-31 (also called elamipretide), which can be accumulated 1,000- to 5000-fold in the IMM and combined with cardiolipin to protect against the damage from ROS (von Hardenberg and Maack, 2017; Szeto, 2014). A previous study has shown the protective function of SS-31 against I/R in a mouse model (Cai et al., 2018). Recently, Whitson et al. observed the reversion of systolic cardiac function in aged mice after treatment with SS-31 for 8 weeks (Chiao et al., 2020). Further research has shown that the treatment with SS-31 together with nicotinamide mononucleotide (NMN, one of the nucleotide precursors of NAD+) increases the level of NAD(H) and improves cardiac function in aged mice (Whitson et al., 2020). Furthermore, SS-31 resists mitochondrial dysfunction by blocking the ROS-induced MPTP opening (Zhang et al., 2020). Pre-/clinical studies have also provided prospective outcomes, where SS-31 has been shown to exhibit positive signs of improving cardiac function after acute myocardial infarction (AMI) and angiotensin II infusion (Dai et al., 2011; Dai et al., 2013). Recent phase II clinical trials have focused on the use of SS-31 in the treatment of HF with reduced ejection fraction (HFrEF) or preserved ejection fraction (HFpEF) (Daubert et al., 2017; Butler et al., 2020). In these studies, SS-31 has been proven to have good safety and tolerability in humans. Unfortunately, in the PROGRESS phase II study, no improvement in cardiac function was observed in patients with HFrEF, although the number of enrolled patients was limited. Thus, the function of SS-31 needs to be proven through more clinical studies.
Whether acetyl-L-carnitine confers any cardiac protection remains controversial. In 1999, a double-blind randomized clinical study in 60 patients after AMI showed that L-carnitine had no significant effect in improving left ventricular function. Another study presented similar results after analyzing 4,000 patients (Tarantini et al., 2006); however, in that study, L-carnitine was reported to reduce acute mortality after AMI. In contrast, evidence of cardioprotection of acetyl-L-carnitine was investigated in 2002 by Heger et al., who demonstrated that oral administration of acetyl-L-carnitine promotes the process of β-oxidation and prevents aging-related oxidative stress in older murine hearts (Hagen et al., 2002). Further studies have reported that L-carnitine promotes the activation of SODs and AMPK proteins synthesis (Vacante et al., 2018), thereby protecting from mitochondrial dysfunction. Although acetyl-L-carnitine seems to have no positive influence on cardiac function according to the clinical studies, its role in enhancing cardiac mitochondrial function and protecting against heart aging cannot be estimated. This may represent an interesting target to treat mitochondrial dysfunction in the future.
A previous study has found that widely used angiotensin-converting-enzyme (ACE) inhibitors and angiotensin receptor-II blockers (ARB), which are used to treat hypertension and HF in the clinic, are also beneficial for mitochondrial functions (Muscari et al., 1998). A similar agent can also be found in β-blockers. Carvedilol is a non-selective β-blocker, which has similar properties to α-blockers. Carvedilol can increase cardiac mitochondrial biogenesis in vivo (Williams, 1999). Carvedilol improves the function of respiratory chain complexes and regulates ATP balance against cardiac damage in a rabbit model (Sanbe et al., 1995; Gvozdjáková et al., 1999). Notably, carvedilol is a prominent antioxidant compared to other β-blockers, such as atenolol and propranolol, which are not superior to carvedilol in terms of oxidation resistance. Quinn et al. investigated that propranolol inhibits NADH oxidase activity is associated with flavoprotein, whereas, atenolol is only observed a tiny inhibition of a relatively high concentration compared to propranolol in rat hearts. Other studies have demonstrated that atenolol failed to protect hearts under I/R injury by preserving cardiac mitochondria (Lu et al., 1990; Vandeplassche et al., 1991). Although propranolol presents better anti-oxidation than atenolol, propranolol does not affect Δψm and its protection against oxidase activity is negligible (Oliveira et al., 2004). Propranolol does not influence MPTP, and the potential mechanisms of protection come from its hemodynamic effect (Carreira et al., 2006). In contrast to propranolol, described above, carvedilol inhibits heart MPTP at a high-conductance level, and thus prevents swelling of mitochondria and efflux of Ca2+ (Oliveira et al., 2003). Oliveira et al. found that the anti-fibrotic function of carvedilol relies on the inhibition of MPTP and calcium overloading (Cheema et al., 2011). Together with its properties of unselected-β and α1-receptors, carvedilol has advantages in CVDs compared to other β-blockers. The underlying mechanisms of carvedilol-induced anti-oxidation are controversial. Yue et al. imply that oxidation resistance of carvedilol is mainly due to its carbazole moiety, and found a direct free radical scavenging pathway located in carbazole of carvedilol. On the opposite, Tadolini et al. demonstrated that the mechanism by which carvedilol scavenges ROS is catalyzed by Fe3+ (Tadolini and Franconi, 1998).
Sodium-dependent glucose transporter 2 (SGLT2) inhibitors decrease renal reabsorption of glucose (Heuvel et al., 2002), thereby increasing glucose excretion in the urine. Many clinical and real-world studies have investigated the advantages of a SGLT2 inhibitor in treating diabetic-induced cardiac diseases. In the EMPA-REG OUTCOME study, SGLT2 inhibitor administration exhibited benefits in reducing cardiovascular mortality, hospitalization due to HF, and nonfatal MI in patients with CVDs and diabetes (Zinman et al., 2015). According to experimental studies, SGLT2 inhibitors such as empagliflozin have a positive effect on mitochondrial biogenesis by activating SIRT1 and PGC-1α in a rat model (Maejima, 2019). Besides, inhibiting of SGLT2 rescues the expression level of Mfn2 and Opa1 in the murine heart (Takagi et al., 2018). Similarly, inhibition of Fis1 was observed following in treatment of SGLT2 inhibitor, which avoided ROS production through excessive mitochondrial fission, thereby reducing the infarct area in diabetic hearts in vivo (Mizuno et al., 2018). Besides, the SGLT2 inhibitor was also shown to maintain mitochondrial intracellular Ca+ homeostasis to improve the antioxidant ability of mitochondria and cardiac functions in a rat model (Olgar et al., 2020).
Another antioxidant found in grapes, resveratrol, has been investigated as an activator of SIRT1. According to Ma et al., resveratrol reversed the mitochondrial biogenesis in diabetic cardiomyopathy (DCM) mice model induced by deletion of SIRT1; the underlying mechanisms of the effect included resveratrol-induced activation of PGC-1α, and further activation of NRF-1, NRF-2, and TFAM (Ma et al., 2017; Kalliora et al., 2019). In addition, resveratrol has a similar function to SIRT3, which is the major regulator of acetylation and deacetylation in the heart (Bagul et al., 2018; Annesley and Fisher, 2019).
Mitochondrial division inhibitor 1 (Mdivi1) was first shown to inhibit DRP1 and effect on suppressing excessive mitochondrial fission by decreasing the activity of GTPase Drp1 enzymatic activity and improving mitochondrial fusion (Tanaka and Youle, 2008). However, this conclusion then was challenged by Bordt et al. who found no anti-fission impact of Mdivi on primary neuron cells, but instead, demonstrated its ability to inhibit complex I and reverse electron transfer-mediated ROS (Bordt et al., 2017). Considering the cardioprotective effect of inhibition of complex I in I/R injury (e.g., inhibition of complex I through mitoSNO protects the heart against I/R in vivo (Chouchani et al., 2013)), the mechanism of inhibiting either Drp1 or complex I could both contribute to the cardioprotective effect of Mdivi1 in I/R injury. Smith et al.‘s review summarized most of the in vitro results of studies on Mdivi1 by 2017. The majority of the results provided positive evidence of Mdivi1-regulated DRP1 fission. They also recommended using live imaging of fission and fusion rates to evaluate mitochondrial dynamics and suggested that further studies should be conducted under strict/suitable control of variables (such as the deletion of DRP1 in vitro and the control of the activity of complex I). In CVDs, Mdivi1 contributes to the alleviation of mitochondrial damage through suppressing excessive mitochondrial fission, apoptosis, and enhancing fusion, thereby reducing I/R damage and improving cardiac function (Rosdah et al., 2016). Ishikita et al. proved that the nanoparticle packaged-Mdivi1 decreases the infarct area of the murine heart after I/R damage; the underlying mechanism is that Mdivi1 inhibits DRP1 and DRP1-induced MOMP (Ishikita et al., 2016). The same positive effect of Mdivi1 on the improvement of cardiac function has also been shown in rat models (Ong et al., 2010). However, a clinically relevant pilot study indicated that Mdivi1 had no significant effect on either decreasing infarct area or improving cardiac function in close chest surgery in large rodents (e.g., pig model) (Ong et al., 2019). Mdivi1 also presents its effect on anti-proliferation and cytotoxicity, especially for hyper-proliferative cells. Although evidence in vivo showed its cardioprotective function in the heart, we should not ignore that most of these studies involved short-term administration of Mdivi1 (less than 2 h), which is much shorter than studies of cancer cells (ranges from 16 h to 4 days). The duration of treatment may be essential for Mdivi1 to exhibit its cytoprotective and cytotoxic effects (Rosdah et al., 2016). Taken together, the functions of Mdivi1 and its side effects remain to be further investigated.
Dynasore is a small molecule that inhibits the GTPase activity of dynamin and dynamin-related proteins (e.g., DRP1). Gao et al. reported that dynasore is beneficial against I/R injury in the murine heart by inhibiting excessive mitochondrial fission and mitophagy (Gao et al., 2013). It is worthy to notice that the potential pathways of dynasore are beyond GTPase activity. In the regulation of cellular cholesterol, Park et al. demonstrated that dynasore inhibits fluid-phase endocytosis in triple knock out (TKO) of dynamin1, dynamin2, and dynamin3 fibroblasts (Park et al., 2013). Treatment with dynasore presents unique regulation of micropinocytosis, which has not been seen in TKO fibroblasts (Nichols, 2003), suggesting that micropinocytosis is a dynasore-independent process. The function of dynasore on dynamin-like proteins or non-GTPase pathways should be further investigated in the heart.
Yue et al. provided the first evidence in mammals that non-proteolytic ubiquitylation of Mfn1/2 promotes mitochondrial fusion by inhibiting USP30 (a mitochondria-localized deubiquitinase); they also described a small natural molecule, diterpenoid derivative 15-oxospiramilactone (S3), which can promote mitochondrial fusion via inhibition of USP30 in Hela’s cells in vitro (Yue et al., 2014). However, the role of S3 in cardiac mitochondria remains to be explored.
NAD+ is a small molecule that plays an essential role in cellular metabolism and DNA repair (Cantó et al., 2015; Yaku et al., 2018). Recently, NAD+ has been shown to act as a coenzyme of sirtuins, which participate in multiple pathways of mitophagy (Palmeira et al., 2019). Nicotinamide riboside (NR) is one precursor of NAD+ and has been shown to improve cardiac function in a mouse model (Diguet et al., 2018; Liu et al., 2022). Yamaguchi et al. reported that oral NR is effective in increasing NAD+ and its intermediate nicotinic acid adenine dinucleotide (NAAD) safely and well-tolerated in the middle-aged and elderly (Yamaguchi and Yoshino, 2016). Several studies proved the safety of NR administration, which supports Yamaguchi’s finding (Mukherjee et al., 2017; Dollerup et al., 2018; Elhassan et al., 2019). Marten et al. investigated the cardioprotective function of NR in reducing systolic blood pressure (SBP) and aortic stiffness (Martens et al., 2018). Further studies are valuable to investigate the effects of NR on CVDs and additional precursors of NAD+.
Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) are a standard treatment for type II diabetes due to their hypoglycemic effect and favorable safety in the clinic (Association, 2020). Recent studies have shown beneficial effects of GLP-1 RAs on CVDs through both direct effects (improve vascular function) and indirect effects (control risk factors of CVDs) (Berndt et al., 2021). Liraglutide, a GLP-1 RAs, is an activator of protective mitophagy in rat hearts and injected liraglutide protects against ischemic damage and reduces the fibrotic formation and cardiac remodeling after MI in vivo through the SIRT1/Parkin/mitophagy pathway (Qiao et al., 2018).
In addition to the drugs with newly found cardioprotective functions and potential targets supported by clinical trials mentioned above, there are still some natural compounds found in plants or food that have been investigated as potential mitophagy regulators for protecting the heart. Urolithin A is a hydrolyzed metabolite of pomegranate, which is generated by enteric microbiota and represents a potential target of mitochondrial dysfunction in the cardiovascular system through promoting mitophagy (Oh et al., 2020). Urolithin A is the activator of PINK1/Parkin-dependent mitophagy and directly increases the expression of LC3, which is related to the formation of the autophagosome (Palikaras et al., 2018). In vivo models have shown the cardiac protection of Urolithin A in DCM through cardiac performance (Savi et al., 2017). Moreover, Tang et al. showed that Urolithin A alleviated I/R injury via the PI3K/Akt pathway. According to the research reported by Juan A et al., Urolithin A showed significant anti-atherosclerotic and antiangiogenic functions against CVDs by inhibiting the migration of ECs and suppressing the expression of chemokine (C–C motif) ligand 2 (CCL2) and interleukin-8 (IL-8) (Giménez-Bastida et al., 2012). As Urolithin A is derived from a natural compound and can be accumulated in the myocardium, it may represent a target agent for clinical treatment in the future (Savi et al., 2017).
Recent studies have demonstrated that another natural compound, spermidine, shows cardiac protection via enhancing mitochondrial mitophagy. Spermidine can be extracted from food such as broccoli, soybeans, and rice bran (Larqué et al., 2007). Eisenberg et al. showed that food intake of spermidine decreased SBP and delayed the occurrence of HF in Dahl salt-sensitive rats on a high-salt diet (Eisenberg et al., 2016). Oral administration of spermidine in aging mice led to a low level of interleukin-6 (IL-6) and alleviated AS by regulating mitophagy (Tyrrell et al., 2020). Eisenberg et al. found that spermidine was preferentially taken up and accumulated in murine CMs, indicating that spermidine may be a potential target for mitochondrial dysfunction (Nilsson and Persson, 2019). Wang et al. also reported that the function of spermidine in stimulating mitochondrial biogenesis was mediated by increasing SIRT1 and PGC-1α in aged hearts in vivo (Wang et al., 2020a). Spermidine prevents mitochondrial dysfunction in CVDs by multiple mechanisms, which makes it a potential substance against mitochondrial dysfunction in clinical studies.
Acacetin is a natural flavone that can be found in snow lotus and other plants. Recent studies have demonstrated its anti-arrhythmic function in different cardiac models in vitro and in vivo (Chang et al., 2017; Di Diego et al., 2020; Hong et al., 2021). Hong et al. showed that acacetin protected against cardiac senescence by stimulating the PINK1/Parkin pathway and increasing LC3II-critical pathways of mitophagy. Besides, acacetin can also activate SIRT1 and AMPK to revert mitochondrial dysfunction. Although there is not much research on acacetin in CVDs, it can also be a promising target for the treatment of MI (Hong et al., 2021).
Although compounds from food and traditional medicine show impressive cardioprotective results in pre-clinical studies, they are still far away from clinical trials. Firstly, intake from daily food is as high as the effective dosage for treatment and the safety and tolerability of high-dose preparations remain unknown. Secondly, the route of administration and drug delivery system plays essential roles in the pharmacokinetics response, and require further investigation in future studies.
AMPK is an exclusive kinase of eukaryotes and is activated by a low concentration of intracellular ATP in various organs (Hardie et al., 2012). AMPK regulates the balance of energy production/consumption by promoting ATP synthesis or restricting ATP depletion (Guigas and Viollet, 2016). AMPK is an essential kinase involved in the regulation of mitochondrial homeostasis, such as mitochondrial biogenesis (PGC-1α) (Guarente, 2007), mitochondrial dynamics (DRP1, MFF), and mitophagy (ULK1, PINK1–PARKIN pathway) (Kim et al., 2011; Scarpulla, 2011; Ducommun et al., 2015; Toyama et al., 2016; Wang et al., 2018). Due to the huge ATP demand, AMPK has a vital role in the prevention/treatment of CVDs. Hence, AMPK is a promising mitochondrial targeting molecule in the cardiovascular system.
Metformin, the most representative medication for type 2 diabetes, has been shown to have beneficial effects on CVDs (El Messaoudi et al., 2013). Various clinical studies have shown the cardioprotective effects of metformin related to diabetes-related HF, I/R, and mortality of AMI (discussed by Aguilar et al.) (Aguilar et al., 2011). Metformin is known as an inhibitor of complex I and restricts infarct area after I/R in vivo (Mohsin et al., 2019). In mechanistic studies, metformin has also been proved to be an activator of AMPK. According to Messaoudi et al., metformin can improve cardiac function in diabetic CVDs, which can be attributed not only to the antidiabetic treatment but also to the promotion of cardiac mitochondrial functions via AMPK-targeted mitochondrial protection (Whittington et al., 2013). Moheimani et al. showed the protective effects of metformin in I/R rat hearts ex vivo, with a significant decrease in the infarct area by modulating phosphorylation of AMPK (Moheimani et al., 2021). According to the review by Varjabedian et al., metformin is a safe clinical drug, with the most serious side effect being acidosis. However, based on the results of numerous clinical studies, the incidence of acidosis is rare and is not only related to the usage of metformin but also the complex conditions in patients with CVDs (Varjabedian et al., 2018). Taking all into consideration, metformin is a promising target against mitochondrial dysfunction in CVDs.
Melatonin is a regulator of circadian rhythms and has shown cardioprotective function in recent studies. Yu et al. showed that melatonin protected from I/R injury in DCM hearts in vivo by decreasing mitochondrial fission and enhancing mitochondrial biogenesis and mitophagy via SIRT6 and AMPK signaling (Yu et al., 2021). Melatonin alleviates the cardiotoxicity of DOX and protects against ROS and cell death in C57BL/6 mice by activating AMPK/PGC1α (Liu et al., 2018a).
Cyclosporine A (CsA), a classical immunosuppressant, is an effective inhibitor of MPTP opening and has shown promising results in pre-clinical studies (Griffiths and Halestrap, 1993). Cardioprotection of CsA is dose- and time-dependent. According to these studies, a dosage of 2.5 mg/kg CsA induces cardioprotective function in several (but not all) rodents (Huang et al., 2014). Treatment with CsA is only useful in the first 10–15 min of reperfusion in vivo because the MPTP opens transiently during reperfusion (Zalewski et al., 2015). In an early clinical study, Piot et al. reported that CsA is beneficial in ST-segment elevation myocardial infarction (STEMI) patients and decreases the infarct area in a small number of patients (Piot et al., 2008). However, in a phase II CYCLE clinical study, there were no benefits in long-term outcomes for patients with STEMI who underwent percutaneous coronary intervention (PCI) with CsA treatment. Moreover, results from the Phase III CIRCUS clinical study were also disappointing, showing no ability of CsA to decrease either the infarct area or the incidence of adverse events in the long term (Cung et al., 2015; Mewton et al., 2015). The relative homogeneity of the rodent and a repeatable diseased model cannot fully intimate the complicated homeostasis observed in humans, which may explain the failed transition of CsA from bench to bedside. Further clinical studies on CsA should be more precise and require a higher number of participants. CsA has unspecific functions not only targeting MPTP but also calcineurin. Cereghetti et al. believed that the cardioprotection of CsA is due to the regulation of calcineurin but not MPTP (Cereghetti et al., 2008). It also makes sense to explore more specific inhibitors of MPTP opening. Sanglifehrin-A (SFA) is an immunosuppressant, which shows more potent in inhibiting the MPTP opening than CsA. Additionally, SFA has no inhibitory effect on calcineurin (Clarke et al., 2002). Hausenloy et al., found that infusion of SFA during the initial period of reperfusion decreases the infarct area in rat heart and also increases the threshold of ROS for MPTP opening (Hausenloy et al., 2003). Further clinical studies of SFA on CVDs should be conducted.
The number of studies concentrated on noncoding RNAs (ncRNAs) as important regulators in CVDs has greatly increased in recent decades. Numerous studies have shown the positive effects of ncRNA-targeted treatments in pre-clinical studies (Matsui and Corey, 2017). Although these ncRNAs cannot be transcribed and translated into proteins, they have significant effects on chromosome modification, transcription, and post-transcriptional regulation, and thus alter biological functions (Lekka and Hall, 2018). Here, we discussed microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) as relevant targets for cardiac mitochondrial dysfunction.
Both nuclear microRNA (miRNA) and mitochondrial miRNA (mitoMir) have effects on mitochondrial homeostasis (Geiger and Dalgaard, 2017). Mito/miRNA-regulated mitochondrial functions are implicated in CVDs (Wojciechowska et al., 2017). Overexpression of miR-142 has been shown to inhibit protein kinase Cepsilon (PKCε)-mediated cardioprotection via targeting mitochondrial ATP-sensitive K+ channel, MPTP, ETC, leading to Δψm dissipation and CMs death (Budas and Mochly-Rosen, 2007; Liu et al., 2018b). Suppression of miR-874 alleviates CM necrosis and decreases the infarct area by regulating caspase-8 in vitro and in vivo (Wang et al., 2013). MitoMirs contribute to mitochondrial dynamics by regulating different targets. For example, miR-30 regulates p53-activated Drp1 (Li et al., 2010); miR-761 inhibits mitochondrial fission by modulation of MFF (Long et al., 2013); miR-499 regulates Drp1 with or without the p53 pathway, and PGC-1α (van Rooij et al., 2009; Wang et al., 2011; Liu et al., 2016), and miR-484 inhibits Fis1-induced excessive mitochondrial fission and apoptosis of CMs (Wang et al., 2012).
LncRNA cytoplasmic endogenous regulator of oxidative phosphorylation 1 (Cerox1) is the first reported lncRNA involved in regulating OXPHOS. Cerox1 is a cytoplasmic lncRNA and promotes the function of respiratory chain complex I in the regulation of proteins and enzymes. Ulteriorly, mitochondrial protection by Cerox1 has been proven both in human tissues and in mice. According to Sirey et al., Cerox1 is expressed in the heart (Sirey et al., 2019), although no study has investigated the cardiac protection of Cerox1. Cerox1 may be a potential target for cardiac mitochondrial dysfunction, but further investigations are needed. LncRNA urothelial carcinoma-associated 1 (UCA1) is specifically expressed in adult hearts (in normal tissue), shows low levels during early AMI, and increases 3 days after AMI. Hence, UCA-1 is believed to be a next-generation biomarker for the diagnosis and prognosis of AMI (Yan et al., 2016). Another study has shown that UCA-1 suppresses excessive ROS generation and cardiac mitochondrial dysfunction in vitro (Chen et al., 2019).
LncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) prevents CMs from mitochondria-dependent apoptosis induced by isoproterenol (ISO) after AMI. Specifically, MALAT1 enhances ULK1-mediated mitophagy in the mitochondria of CMs (Guo et al., 2019). In contrast, Zhang et al. reported another lncRNA, dachshund homolog 1 (DACH1), which aggravates ROS generation and mitochondrial dysfunction. The underlying mechanism involves the bond between DACH1 and SIRT3, which leads to the degradation of ubiquitination. Inhibition of DACH1 may be a promising target of anti-oxidation and may decelerate the progression of DCM (Zhang et al., 2021). Studies of lncRNA in cardiac mitochondria are still limited compared to those of miRNA. In general, ncRNAs show essential roles in cardiac mitochondrial regulation and CVDs. Research of ncRNAs will support new potential strategies from bench to bedside.
The heart is very dependent on the normal functioning of mitochondria due to its high energy consumption and ATP-sensitive characteristics. Scientists are becoming increasingly aware of the importance of mitochondrial targets for the treatment of CVDs. The topic is not only about the investigations of new targets and their mechanisms, but also about the delivery systems of these drugs. The targets against mitochondrial dysfunction mainly focus on antioxidants, repair of the respiratory chain, and mitochondrial homeostasis (including mitochondrial biogenesis, mitochondrial dynamics, and mitophagy). Furthermore, scientists have gradually accepted that ncRNAs play important regulatory roles in cardiac mitochondria, and future targeted therapies based on ncRNA may become a hot topic (all the targets mentioned in this review and their evidence are summarized in Table 1). However, there remain some issues related to mitochondria-targeted therapies. Firstly, some of the compounds have less effect on dysfunctional mitochondria than expected. For example, TPP+-linked modulators (such as MitoQ, MitoTEMPO, and SkQ1) can combine with the OMM due to their lipophilic characteristics, which rely on the accumulation of cationic molecules via Δψm (Jiang et al., 2009). However, Δψm is decreased in dysfunctional mitochondria (Zorova et al., 2018), which may reduce the therapeutic efficiency of these targets. Secondly, other targets are presenting accompanying effects during global treatment (tissue-unspecific treatment), the MPTP inhibitor, CsA, representing a good example. Namely, CsA, has shown cardioprotective function in different species/models (Onishi et al., 2012; Badavi et al., 2014; González Arbeláez et al., 2016; Lozano et al., 2020), however, CsA has a significant immunosuppressive effect at the same time (Frank et al., 1999). To solve the issues of low targeting efficiency and accompanying effects, pharmacologically specific targets such as NIM811, which is also an inhibitor of MPTP but has no immunosuppressive effect compared with CsA (Argaud et al., 2005), warrant further investigation. Another way is to establish a more precise way to transport compounds into the heart. Ikeda et al. synthesized nanoparticle-mediated CsA using poly-lactic/glycolic acid (PLGA) nanoparticles, which target injured organs and have shown promising effects on mouse heart models (Ikeda et al., 2016). Therapies based on nanoparticle-mounted targets (even intervention with small-interference RNA of ncRNA) may become a new direction for the treatment of mitochondrial dysfunction and CVDs. Furthermore, the impact of sterile inflammation and immunity induced by damaged mtDNA in CVDs cannot be ignored. The main solution now is to protect mtDNA against ROS damage. Besides, promising PRRs like NLRPs have been well-studied in animal models as a potential target involving inflammation response in CVDs (Molagoda et al., 2021). Clinical drugs, such as metformin and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor, also have the potential to suppress NLRP3-regulated inflammation in the heart (Wang et al., 2020b; Fei et al., 2020). However, targets towards mtDNA and inflammation response involve a vast amount of inflammatory/immune cytokines and pathways. The major problems at present are finding more efficient co-/interventional targets and specific drug delivery systems.
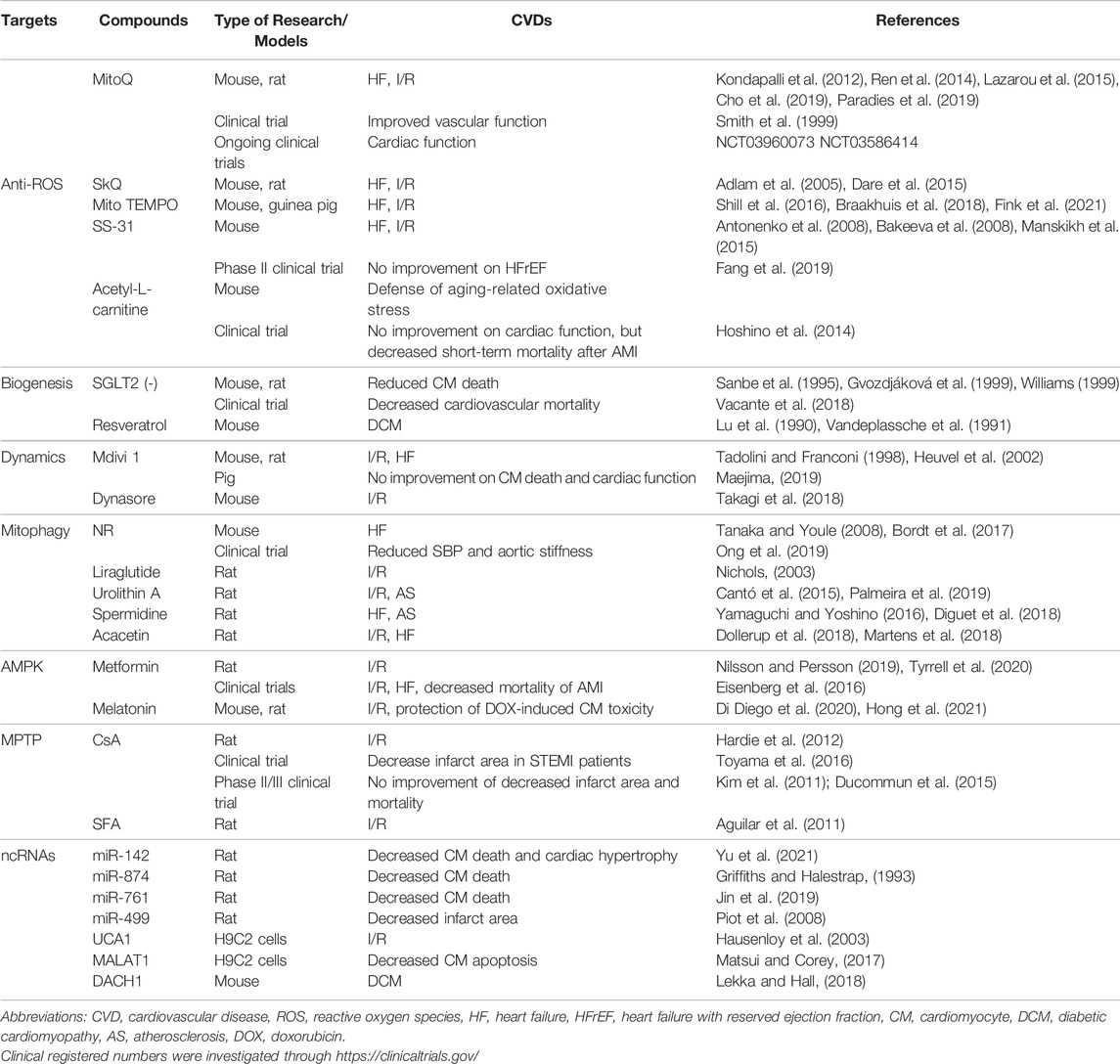
TABLE 1. Summary of pre-/clinical evidence of potential mito-targets in CVDs
YZ summarized the outline of this review. XF and YL selected the literature for this review. QG wrote the biological character of mitochondrion and its function in cardiovascular diseases. JY was responsible for the summary of targets of mitochondrial dysfunction.
This review was supported by Natural Science Foundation of Beijing, China (Grant Nos 7214223) and Beijing Hospitals Authority Youth Programme (Code No. QML20210601) to QG and Chinese Scholarship Council (CSC) scholarship (Grant Nos. 202008110200) to JY. YZ was supported by National Key Research and Development Program of China (2017YFC0908800), Natural Science Foundation of Beijing, China (Grant Nos. 7212027) and National Natural Science Foundation of China (Grant Nos. 82070293). All figures are created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adlam, V. J., Harrison, J. C., Porteous, C. M., James, A. M., Smith, R. A. J., Murphy, M. P., et al. (2005). Targeting an Antioxidant to Mitochondria Decreases Cardiac Ischemia‐reperfusion Injury. FASEB j. 19 (9), 1088–1095. doi:10.1096/fj.05-3718com
Aguilar, D., Chan, W., Bozkurt, B., Ramasubbu, K., and Deswal, A. (2011). Metformin Use and Mortality in Ambulatory Patients with Diabetes and Heart Failure. Circ. Heart Fail. 4 (1), 53–58. doi:10.1161/circheartfailure.110.952556
Annesley, S. J., and Fisher, P. R. (2019). Mitochondria in Health and Disease. Cells 8 (7). doi:10.3390/cells8070680
Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L. E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., et al. (2008). Mitochondria-targeted Plastoquinone Derivatives as Tools to Interrupt Execution of the Aging Program. 1. Cationic Plastoquinone Derivatives: Synthesis and In Vitro Studies. Biochem. Mosc. 73 (12), 1273–1287. doi:10.1134/s0006297908120018
Argaud, L., Gateauroesch, O., Muntean, D., Chalabreysse, L., Loufouat, J., Robert, D., et al. (2005). Specific Inhibition of the Mitochondrial Permeability Transition Prevents Lethal Reperfusion Injury. J. Mol. Cell Cardiol. 38 (2), 367–374. doi:10.1016/j.yjmcc.2004.12.001
Association, A. D. (2020). 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2020. Diabetes Care 43 (Suppl. 1), S98–s110. doi:10.2337/dc20-S009
Badavi, M., Sadeghi, N., Dianat, M., and Samarbafzadeh, A. (2014). Effects of Gallic Acid and Cyclosporine a on Antioxidant Capacity and Cardiac Markers of Rat Isolated Heart after Ischemia/reperfusion. Iran Red Crescent Med. J. 16 (6), e16424. doi:10.5812/ircmj.16424
Bagul, P. K., Katare, P. B., Bugga, P., Dinda, A. K., and Banerjee, S. K. (2018). SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 7 (12). doi:10.3390/cells7120235
Bakeeva, L. E., Barskov, I. V., Egorov, M. V., Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., et al. (2008). Mitochondria-targeted Plastoquinone Derivatives as Tools to Interrupt Execution of the Aging Program. 2. Treatment of Some ROS- and Age-Related Diseases (Heart Arrhythmia, Heart Infarctions, Kidney Ischemia, and Stroke). Biochem. Mosc. 73 (12), 1288–1299. doi:10.1134/s000629790812002x
Berndt, J., Ooi, S. L., and Pak, S. C. (2021). What Is the Mechanism Driving the Reduction of Cardiovascular Events from Glucagon-like Peptide-1 Receptor Agonists?-A Mini Review. Molecules 26 (16). doi:10.3390/molecules26164822
Bi, J., Zhang, J., Ren, Y., Du, Z., Li, Q., Wang, Y., et al. (2019). Irisin Alleviates Liver Ischemia-Reperfusion Injury by Inhibiting Excessive Mitochondrial Fission, Promoting Mitochondrial Biogenesis and Decreasing Oxidative Stress. Redox Biol. 20, 296–306. doi:10.1016/j.redox.2018.10.019
Bordt, E. A., Clerc, P., Roelofs, B. A., Saladino, A. J., Tretter, L., Adam-Vizi, V., et al. (2017). The Putative Drp1 Inhibitor Mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Developmental Cell 40 (6), 583–594. e6. doi:10.1016/j.devcel.2017.02.020
Boveris, A., Cadenas, E., and Stoppani, A. O. M. (1976). Role of Ubiquinone in the Mitochondrial Generation of Hydrogen Peroxide. Biochem. J. 156 (2), 435–444. doi:10.1042/bj1560435
Braakhuis, A. J., Nagulan, R., and Somerville, V. (2018). The Effect of MitoQ on Aging-Related Biomarkers: A Systematic Review and Meta-Analysis. Oxid Med. Cell Longev 2018, 8575263. doi:10.1155/2018/8575263
Brzheskiy, V. V., Efimova, E. L., Vorontsova, T. N., Alekseev, V. N., Gusarevich, O. G., Shaidurova, K. N., et al. (2015). Results of a Multicenter, Randomized, Double-Masked, Placebo-Controlled Clinical Study of the Efficacy and Safety of Visomitin Eye Drops in Patients with Dry Eye Syndrome. Adv. Ther. 32 (12), 1263–1279. doi:10.1007/s12325-015-0273-6
Budas, G. R., and Mochly-Rosen, D. (2007). Mitochondrial Protein Kinase Cepsilon (PKCepsilon): Emerging Role in Cardiac protection from Ischaemic Damage. Biochem. Soc. Trans. 35 (Pt 5), 1052–1054. doi:10.1042/BST0351052
Burns, R. J., Smith, R. A. J., and Murphy, M. P. (1995). Synthesis and Characterization of Thiobutyltriphenylphosphonium Bromide, a Novel Thiol Reagent Targeted to the Mitochondrial Matrix. Arch. Biochem. Biophys. 322 (1), 60–68. doi:10.1006/abbi.1995.1436
Butler, J., Khan, M. S., Anker, S. D., Fonarow, G. C., Kim, R. J., Nodari, S., et al. (2020). Effects of Elamipretide on Left Ventricular Function in Patients with Heart Failure with Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial. J. Card. Fail. 26 (5), 429–437. doi:10.1016/j.cardfail.2020.02.001
Cadenas, S. (2018). ROS and Redox Signaling in Myocardial Ischemia-Reperfusion Injury and Cardioprotection. Free Radic. Biol. Med. 117, 76–89. doi:10.1016/j.freeradbiomed.2018.01.024
Cai, J., Jiang, Y., Zhang, M., Zhao, H., Li, H., Li, K., et al. (2018). Protective Effects of Mitochondrion-Targeted Peptide SS-31 against Hind Limb Ischemia-Reperfusion Injury. J. Physiol. Biochem. 74 (2), 335–343. doi:10.1007/s13105-018-0617-1
Campello, S., and Scorrano, L. (2010). Mitochondrial Shape Changes: Orchestrating Cell Pathophysiology. EMBO Rep. 11 (9), 678–684. doi:10.1038/embor.2010.115
Cantó, C., Menzies, K. J., and Auwerx, J. (2015). NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab 22 (1), 31–53.
Carreira, R., Monteiro, P., Goncalves, L., and Providencia, L. (2006). Carvedilol: Just Another Beta-Blocker or a Powerful Cardioprotector? Chddt 6 (4), 257–266. doi:10.2174/187152906779010746
Cereghetti, G. M., Stangherlin, A., de Brito, O. M., Chang, C. R., Blackstone, C., Bernardi, P., et al. (2008). Dephosphorylation by Calcineurin Regulates Translocation of Drp1 to Mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105 (41), 15803–15808. doi:10.1073/pnas.0808249105
Chang, W., Wu, Q.-Q., Xiao, Y., Jiang, X.-H., Yuan, Y., Zeng, X.-F., et al. (2017). Acacetin Protects against Cardiac Remodeling after Myocardial Infarction by Mediating MAPK and PI3K/Akt Signal Pathway. J. Pharmacol. Sci. 135 (4), 156–163. doi:10.1016/j.jphs.2017.11.009
Cheema, Y., Sherrod, J. N., Zhao, W., Zhao, T., Ahokas, R. A., Sun, Y., et al. (2011). Mitochondriocentric Pathway to Cardiomyocyte Necrosis in Aldosteronism: Cardioprotective Responses to Carvedilol and Nebivolol. J. Cardiovasc. Pharmacol. 58 (1), 80–86. doi:10.1097/fjc.0b013e31821cd83c
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol 160 (2), 189–200. doi:10.1083/jcb.200211046
Chen, J., Hu, Q., Zhang, B.-f., Liu, X.-p., Yang, S., and Jiang, H. (2019). Long Noncoding RNA UCA1 Inhibits Ischaemia/reperfusion Injury Induced Cardiomyocytes Apoptosis via Suppression of Endoplasmic Reticulum Stress. Genes Genom 41 (7), 803–810. doi:10.1007/s13258-019-00806-w
Chen, L., Gong, Q., Stice, J. P., and Knowlton, A. A. (2009). Mitochondrial OPA1, Apoptosis, and Heart Failure. Cardiovasc. Res. 84 (1), 91–99. doi:10.1093/cvr/cvp181
Chen, Q., Moghaddas, S., Hoppel, C. L., and Lesnefsky, E. J. (2008). Ischemic Defects in the Electron Transport Chain Increase the Production of Reactive Oxygen Species from Isolated Rat Heart Mitochondria. Am. J. Physiology-Cell Physiol. 294 (2), C460–C466. doi:10.1152/ajpcell.00211.2007
Chen, X. J., and Butow, R. A. (2005). The Organization and Inheritance of the Mitochondrial Genome. Nat. Rev. Genet. 6 (11), 815–825. doi:10.1038/nrg1708
Chen, Y., Liu, Y., and Dorn, G. W. (2011). Mitochondrial Fusion Is Essential for Organelle Function and Cardiac Homeostasis. Mitochondrial Fusion Is Essential Organelle Function Cardiac Homeostasiscirc Res. 109 (12), 1327–1331. doi:10.1161/circresaha.111.258723
Chiao, Y. A., Zhang, H., Sweetwyne, M., Whitson, J., Ting, Y. S., Basisty, N., et al. (2020). Late-life Restoration of Mitochondrial Function Reverses Cardiac Dysfunction in Old Mice. Elife 9. doi:10.7554/eLife.55513
Cho, H. M., Ryu, J. R., Jo, Y., Seo, T. W., Choi, Y. N., Kim, J. H., et al. (2019). Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol. Cell 73 (2), 364–376. doi:10.1016/j.molcel.2018.11.009
Chouchani, E. T., Methner, C., Nadtochiy, S. M., Logan, A., Pell, V. R., Ding, S., et al. (2013). Cardioprotection by S-Nitrosation of a Cysteine Switch on Mitochondrial Complex I. Nat. Med. 19 (6), 753–759. doi:10.1038/nm.3212
Christodoulou, J., Malmendal, A., Harper, J. F., and Chazin, W. J. (2004). Evidence for Differing Roles for Each Lobe of the Calmodulin-like Domain in a Calcium-dependent Protein Kinase. J. Biol. Chem. 279 (28), 29092–29100. doi:10.1074/jbc.m401297200
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin Externalization to the Outer Mitochondrial Membrane Acts as an Elimination Signal for Mitophagy in Neuronal Cells. Nat. Cell Biol 15 (10), 1197–1205. doi:10.1038/ncb2837
Clarke, S. J., McStay, G. P., and Halestrap, A. P. (2002). Sanglifehrin A Acts as a Potent Inhibitor of the Mitochondrial Permeability Transition and Reperfusion Injury of the Heart by Binding to Cyclophilin-D at a Different Site from Cyclosporin A. J. Biol. Chem. 277 (38), 34793–34799. doi:10.1074/jbc.m202191200
Crompton, M., Costi, A., and Hayat, L. (1987). Evidence for the Presence of a Reversible Ca2+-dependent Pore Activated by Oxidative Stress in Heart Mitochondria. Biochem. J. 245 (3), 915–918. doi:10.1042/bj2450915
Cung, T. T., Morel, O., Cayla, G., Rioufol, G., Garcia-Dorado, D., Angoulvant, D., et al. (2015). Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 373 (11), 1021–1031. doi:10.1056/NEJMoa1505489
Dai, D.-F., Chen, T., Szeto, H., Nieves-Cintrón, M., Kutyavin, V., Santana, L. F., et al. (2011). Mitochondrial Targeted Antioxidant Peptide Ameliorates Hypertensive Cardiomyopathy. J. Am. Coll. Cardiol. 58 (1), 73–82. doi:10.1016/j.jacc.2010.12.044
Dai, D.-F., Hsieh, E. J., Chen, T., Menendez, L. G., Basisty, N. B., Tsai, L., et al. (2013). Global Proteomics and Pathway Analysis of Pressure-Overload-Induced Heart Failure and its Attenuation by Mitochondrial-Targeted Peptides. Circ. Heart Fail. 6 (5), 1067–1076. doi:10.1161/circheartfailure.113.000406
Dare, A. J., Logan, A., Prime, T. A., Rogatti, S., Goddard, M., Bolton, E. M., et al. (2015). The Mitochondria-Targeted Anti-oxidant MitoQ Decreases Ischemia-Reperfusion Injury in a Murine Syngeneic Heart Transplant Model. J. Heart Lung Transplant. 34 (11), 1471–1480. doi:10.1016/j.healun.2015.05.007
Dasari, P., Nicholson, I. C., Hodge, G., Dandie, G. W., and Zola, H. (2005). Expression of Toll-like Receptors on B Lymphocytes. Cell Immunol 236 (1-2), 140–145. doi:10.1016/j.cellimm.2005.08.020
Daubert, M. A., Yow, E., Dunn, G., Marchev, S., Barnhart, H., Douglas, P. S., et al. (2017). Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ. Heart Fail. 10 (12). doi:10.1161/CIRCHEARTFAILURE.117.004389
Davis, R. E., and Williams, M. (2012). Mitochondrial Function and Dysfunction: an Update. J. Pharmacol. Exp. Ther. 342 (3), 598–607. doi:10.1124/jpet.112.192104
Dey, S., DeMazumder, D., Sidor, A., Foster, D. B., and O’Rourke, B. (2018). Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 123 (3), 356–371. doi:10.1161/circresaha.118.312708
Di Diego, J. M., Patocskai, B., Barajas-Martinez, H., Borbáth, V., Ackerman, M. J., Burashnikov, A., et al. (2020). Acacetin Suppresses the Electrocardiographic and Arrhythmic Manifestations of the J Wave Syndromes. PLoS One 15 (11), e0242747. doi:10.1371/journal.pone.0242747
Dietl, A., and Maack, C. (2017). Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail. Rep. 14 (4), 338–349. doi:10.1007/s11897-017-0347-7
Diguet, N., Trammell, S. A. J., Tannous, C., Deloux, R., Piquereau, J., Mougenot, N., et al. (2018). Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 137 (21), 2256–2273. doi:10.1161/circulationaha.116.026099
Docherty, C. K., Carswell, A., Friel, E., and Mercer, J. R. (2018). Impaired Mitochondrial Respiration in Human Carotid Plaque Atherosclerosis: A Potential Role for Pink1 in Vascular Smooth Muscle Cell Energetics. Atherosclerosis 268, 1–11. doi:10.1016/j.atherosclerosis.2017.11.009
Dollerup, O. L., Christensen, B., Svart, M., Schmidt, M. S., Sulek, K., Ringgaard, S., et al. (2018). A Randomized Placebo-Controlled Clinical Trial of Nicotinamide Riboside in Obese Men: Safety, Insulin-Sensitivity, and Lipid-Mobilizing Effects. Am. J. Clin. Nutr. 108 (2), 343–353. doi:10.1093/ajcn/nqy132
Ducommun, S., Deak, M., Sumpton, D., Ford, R. J., Núñez Galindo, A., Kussmann, M., et al. (2015). Motif Affinity and Mass Spectrometry Proteomic Approach for the Discovery of Cellular AMPK Targets: Identification of Mitochondrial Fission Factor as a New AMPK Substrate. Cell Signal. 27 (5), 978–988. doi:10.1016/j.cellsig.2015.02.008
Dudek, J., Hartmann, M., and Rehling, P. (2019). The Role of Mitochondrial Cardiolipin in Heart Function and its Implication in Cardiac Disease. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1865 (4), 810–821. doi:10.1016/j.bbadis.2018.08.025
Dudek, J. (2017). Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 5, 90. doi:10.3389/fcell.2017.00090
Eaton-Bassiri, A., Dillon, S. B., Cunningham, M., Rycyzyn, M. A., Mills, J., Sarisky, R. T., et al. (2004). Toll-like Receptor 9 Can Be Expressed at the Cell Surface of Distinct Populations of Tonsils and Human Peripheral Blood Mononuclear Cells. Infect. Immun. 72 (12), 7202–7211. doi:10.1128/iai.72.12.7202-7211.2004
Efremov, R. G., and Sazanov, L. A. (2011). Respiratory Complex I: 'steam Engine' of the Cell? Curr. Opin. Struct. Biol. 21 (4), 532–540. doi:10.1016/j.sbi.2011.07.002
Eisenberg, T., Abdellatif, M., Schroeder, S., Primessnig, U., Stekovic, S., Pendl, T., et al. (2016). Cardioprotection and Lifespan Extension by the Natural Polyamine Spermidine. Nat. Med. 22 (12), 1428–1438. doi:10.1038/nm.4222
El Messaoudi, S., Rongen, G. A., and Riksen, N. P. (2013). Metformin Therapy in Diabetes: the Role of Cardioprotection. Curr. Atheroscler. Rep. 15 (4), 314. doi:10.1007/s11883-013-0314-z
Elhassan, Y. S., Kluckova, K., Fletcher, R. S., Schmidt, M. S., Garten, A., Doig, C. L., et al. (2019). Nicotinamide Riboside Augments the Aged Human Skeletal Muscle NAD+ Metabolome and Induces Transcriptomic and Anti-inflammatory Signatures. Cell Rep. 28 (7), 1717–1728. doi:10.1016/j.celrep.2019.07.043
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a Target for protection against Cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 116 (7), 2672–2680. doi:10.1073/pnas.1821022116
Fei, Q., Ma, H., Zou, J., Wang, W., Zhu, L., Deng, H., et al. (2020). Metformin Protects against Ischaemic Myocardial Injury by Alleviating Autophagy-ROS-NLRP3-Mediated Inflammatory Response in Macrophages. J. Mol. Cell Cardiol. 145, 1–13. doi:10.1016/j.yjmcc.2020.05.016
Fink, B. D., Yu, L., Coppey, L., Obrosov, A., Shevalye, H., Kerns, R. J., et al. (2021). Effect of Mitoquinone on Liver Metabolism and Steatosis in Obese and Diabetic Rats. Pharmacol. Res. Perspect. 9 (1), e00701. doi:10.1002/prp2.701
Finsterer, J. (2020). Atherosclerosis Can Be Mitochondrial: A Review. Cureus 12 (2), e6987. doi:10.7759/cureus.6987
Frank, D. A., Mahajan, S., and Ritz, J. (1999). Fludarabine-induced Immunosuppression Is Associated with Inhibition of STAT1 Signaling. Nat. Med. 5 (4), 444–447. doi:10.1038/7445
Gane, E. J., Weilert, F., Orr, D. W., Keogh, G. F., Gibson, M., Lockhart, M. M., et al. (2010). The Mitochondria-Targeted Anti-oxidant Mitoquinone Decreases Liver Damage in a Phase II Study of Hepatitis C Patients. Liver Int. 30 (7), 1019–1026. doi:10.1111/j.1478-3231.2010.02250.x
Gao, D., Zhang, L., Dhillon, R., Hong, T.-T., Shaw, R. M., and Zhu, J. (2013). Dynasore Protects Mitochondria and Improves Cardiac Lusitropy in Langendorff Perfused Mouse Heart. PLoS One 8 (4), e60967. doi:10.1371/journal.pone.0060967
Geiger, J., and Dalgaard, L. T. (2017). Interplay of Mitochondrial Metabolism and microRNAs. Cell. Mol. Life Sci. 74 (4), 631–646. doi:10.1007/s00018-016-2342-7
Giménez-Bastida, J. A., González-Sarrías, A., Larrosa, M., Tomás-Barberán, F., Espín, J. C., and García-Conesa, M.-T. (2012). Ellagitannin Metabolites, Urolithin A Glucuronide and its Aglycone Urolithin A, Ameliorate TNF-α-Induced Inflammation and Associated Molecular Markers in Human Aortic Endothelial Cells. Mol. Nutr. Food Res. 56 (5), 784–796. doi:10.1002/mnfr.201100677
Goh, K. Y., He, L., Song, J., Jinno, M., Rogers, A. J., Sethu, P., et al. (2019). Mitoquinone Ameliorates Pressure Overload-Induced Cardiac Fibrosis and Left Ventricular Dysfunction in Mice. Redox Biol. 21, 101100. doi:10.1016/j.redox.2019.101100
González Arbeláez, L. F., Ciocci Pardo, A., Fantinelli, J. C., and Mosca, S. M. (2016). Cyclosporine-A Mimicked the Ischemic Pre- and Postconditioning-Mediated Cardioprotection in Hypertensive Rats: Role of PKCε. Exp. Mol. Pathol. 100 (2), 266–275. doi:10.1016/j.yexmp.2016.01.009
Griffiths, E. J., and Halestrap, A. P. (1993). Protection by Cyclosporin A of Ischemia/reperfusion-Induced Damage in Isolated Rat Hearts. J. Mol. Cell Cardiol. 25 (12), 1461–1469. doi:10.1006/jmcc.1993.1162
Guarente, L. (2007). Sirtuins in Aging and Disease. Cold Spring Harbor Symposia Quantitative Biol. 72, 483–488. doi:10.1101/sqb.2007.72.024
Guescini, M., Genedani, S., Stocchi, V., and Agnati, L. F. (2010). Astrocytes and Glioblastoma Cells Release Exosomes Carrying mtDNA. J. Neural Transm. 117 (1), 1–4. doi:10.1007/s00702-009-0288-8
Guigas, B., and Viollet, B. (2016). Targeting AMPK: From Ancient Drugs to New Small-Molecule Activators. Exp. Suppl. 107, 327–350. doi:10.1007/978-3-319-43589-3_13
Guo, X., Wu, X., Han, Y., Tian, E., and Cheng, J. (2019). LncRNA MALAT1 Protects Cardiomyocytes from Isoproterenol‐induced Apoptosis through Sponging miR‐558 to Enhance ULK1‐mediated Protective Autophagy. J. Cell Physiol. 234 (7), 10842–10854. doi:10.1002/jcp.27925
Gvozdjáková, A., Šimko, F., Kucharská, J., Braunová, Z., Pšenek, P., and Kyselovič, J. (1999). Captopril Increased Mitochondrial Coenzyme Q10level, Improved Respiratory Chain Function and Energy Production in the Left Ventricle in Rabbits with Smoke Mitochondrial Cardiomyopathy. Biofactors 10 (1), 61–65. doi:10.1002/biof.5520100107
Hagen, T. M., Moreau, R., Suh, J. H., and Visioli, F. (2002). Mitochondrial Decay in the Aging Rat Heart. Ann. New York Acad. Sci. 959, 491–507. doi:10.1111/j.1749-6632.2002.tb02119.x
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: a Nutrient and Energy Sensor that Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol 13 (4), 251–262. doi:10.1038/nrm3311
Hausenloy, D., Duchen, M. R., and Yellon, D. M. (2003). Inhibiting Mitochondrial Permeability Transition Pore Opening at Reperfusion Protects against Ischaemia-Reperfusion Injury. Cardiovasc. Res. 60 (3), 617–625. doi:10.1016/j.cardiores.2003.09.025
Hausenloy, D. J., Ong, S.-B., and Yellon, D. M. (2009). The Mitochondrial Permeability Transition Pore as a Target for Preconditioning and Postconditioning. Basic Res. Cardiol. 104 (2), 189–202. doi:10.1007/s00395-009-0010-x
Haworth, R. A., and Hunter, D. R. (1979). The Ca2+-Induced Membrane Transition in Mitochondria. Arch. Biochem. Biophys. 195 (2), 460–467. doi:10.1016/0003-9861(79)90372-2
Heuvel, L. v. d., Assink, K., Williemsen, M., and Monnens, L. (2002). Autosomal Recessive Renal Glucosuria Attributable to a Mutation in the Sodium Glucose Cotransporter (SGLT2). Hum. Genet. 111 (6), 544–547. doi:10.1007/s00439-002-0820-5
Hong, Y.-X., Wu, W.-Y., Song, F., Wu, C., Li, G.-R., and Wang, Y. (2021). Cardiac Senescence Is Alleviated by the Natural Flavone Acacetin via Enhancing Mitophagy. Aging 13 (12), 16381–16403. doi:10.18632/aging.203163
Hoppins, S., Lackner, L., and Nunnari, J. (2007). The Machines that divide and Fuse Mitochondria. Annu. Rev. Biochem. 76, 751–780. doi:10.1146/annurev.biochem.76.071905.090048
Hoshino, A., Okawa, Y., Ariyoshi, M., Kaimoto, S., Uchihashi, M., Fukai, K., et al. (2014). Oxidative post-translational Modifications Develop LONP1 Dysfunction in Pressure Overload Heart Failure. Circ. Heart Fail. 7 (3), 500–509. doi:10.1161/circheartfailure.113.001062
Hu, Q., Ren, J., Li, G., Wu, J., Wu, X., Wang, G., et al. (2018). The Mitochondrially Targeted Antioxidant MitoQ Protects the Intestinal Barrier by Ameliorating Mitochondrial DNA Damage via the Nrf2/ARE Signaling Pathway. Cell Death Dis 9 (3), 403. doi:10.1038/s41419-018-0436-x
Huang, K., Lu, S.-J., Zhong, J.-H., Xiang, Q., Wang, L., and Wu, M. (2014). Comparative Analysis of Different Cyclosporine A Doses on protection after Myocardial Ischemia/reperfusion Injury in Rat. Asian Pac. J. Trop. Med. 7 (2), 144–148. doi:10.1016/s1995-7645(14)60011-0
Ikeda, G., Matoba, T., Nakano, Y., Nagaoka, K., Ishikita, A., Nakano, K., et al. (2016). Nanoparticle-Mediated Targeting of Cyclosporine A Enhances Cardioprotection against Ischemia-Reperfusion Injury through Inhibition of Mitochondrial Permeability Transition Pore Opening. Sci. Rep. 6, 20467. doi:10.1038/srep20467
Ishikita, A., Matoba, T., Ikeda, G., Koga, J., Mao, Y., Nakano, K., et al. (2016). Nanoparticle-Mediated Delivery of Mitochondrial Division Inhibitor 1 to the Myocardium Protects the Heart from Ischemia-Reperfusion Injury through Inhibition of Mitochondria Outer Membrane Permeabilization: A New Therapeutic Modality for Acute Myocardial Infarction. J. Am. Heart Assoc. 5 (7). doi:10.1161/JAHA.116.003872
Iwata, S., Lee, J. W., Okada, K., Lee, J. K., Iwata, M., Rasmussen, B., et al. (1998). Complete Structure of the 11-Subunit Bovine Mitochondrial Cytochrome Bc 1 Complex. Science 281 (5373), 64–71. doi:10.1126/science.281.5373.64
Jäger, S., Handschin, C., St.-Pierre, J., and Spiegelman, B. M. (2007). AMP-activated Protein Kinase (AMPK) Action in Skeletal Muscle via Direct Phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. U.S.A. 104 (29), 12017–12022. doi:10.1073/pnas.0705070104
Jiang, J., Stoyanovsky, D. A., Belikova, N. A., Tyurina, Y. Y., Zhao, Q., Tungekar, M. A., et al. (2009). A Mitochondria-Targeted Triphenylphosphonium-Conjugated Nitroxide Functions as a Radioprotector/mitigator. Radiat. Res. 172 (6), 706–717. doi:10.1667/rr1729.1
Jin, H., Ko, Y. S., Park, S. W., and Kim, H. J. (2019). P2Y2R Activation by ATP Induces oxLDL-Mediated Inflammasome Activation through Modulation of Mitochondrial Damage in Human Endothelial Cells. Free Radic. Biol. Med. 136, 109–117. doi:10.1016/j.freeradbiomed.2019.04.004
Jornayvaz, F. R., and Shulman, G. I. (2010). Regulation of Mitochondrial Biogenesis. Essays Biochem. 47, 69–84. doi:10.1042/bse0470069
Kalliora, C., Kyriazis, I. D., Oka, S. I., Lieu, M. J., Yue, Y., Area-Gomez, E., et al. (2019). Dual Peroxisome-Proliferator-Activated-Receptor-Alpha/gamma Activation Inhibits SIRT1-PGC1alpha axis and Causes Cardiac Dysfunction. JCI Insight 5 (17), e129556. doi:10.1172/jci.insight.129556
Kang, P. T., Chen, C.-L., Lin, P., Zhang, L., Zweier, J. L., and Chen, Y.-R. (2018). Mitochondrial Complex I in the post-ischemic Heart: Reperfusion-Mediated Oxidative Injury and Protein Cysteine Sulfonation. J. Mol. Cell Cardiol. 121, 190–204. doi:10.1016/j.yjmcc.2018.07.244
Kim, J.-a., Wei, Y., and Sowers, J. R. (2008). Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 102 (4), 401–414. doi:10.1161/circresaha.107.165472
Kim, J., Gupta, R., Blanco, L. P., Yang, S., Shteinfer-Kuzmine, A., Wang, K., et al. (2019). VDAC Oligomers Form Mitochondrial Pores to Release mtDNA Fragments and Promote Lupus-like Disease. Science 366 (6472), 1531–1536. doi:10.1126/science.aav4011
Kim, J., Kundu, M., Viollet, B., and Guan, K.-L. (2011). AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol 13 (2), 132–141. doi:10.1038/ncb2152
Kim, S., Song, J., Ernst, P., Latimer, M. N., Ha, C.-M., Goh, K. Y., et al. (2020). MitoQ Regulates Redox-Related Noncoding RNAs to Preserve Mitochondrial Network Integrity in Pressure-Overload Heart Failure. Am. J. Physiology-Heart Circulatory Physiol. 318 (3), H682–H695. doi:10.1152/ajpheart.00617.2019
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 Is Activated by Mitochondrial Membrane Potential Depolarization and Stimulates Parkin E3 Ligase Activity by Phosphorylating Serine 65. Open Biol. 2 (5), 120080. doi:10.1098/rsob.120080
Kornfeld, O. S., Qvit, N., Haileselassie, B., Shamloo, M., Bernardi, P., and Mochly-Rosen, D. (2018). Interaction of Mitochondrial Fission Factor with Dynamin Related Protein 1 Governs Physiological Mitochondrial Function In Vivo. Sci. Rep. 8 (1), 14034. doi:10.1038/s41598-018-32228-1
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M., and Chan, D. C. (2004). Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 305 (5685), 858–862. doi:10.1126/science.1099793
Larqué, E., Sabater-Molina, M., and Zamora, S. (2007). Biological Significance of Dietary Polyamines. Nutrition 23 (1), 87–95. doi:10.1016/j.nut.2006.09.006
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 524 (7565), 309–314. doi:10.1038/nature14893
Lee, H. C., and Wei, Y. H. (2007). Oxidative Stress, Mitochondrial DNA Mutation, and Apoptosis in Aging. Exp. Biol. Med. (Maywood) 232 (5), 592–606.
Lekka, E., and Hall, J. (2018). Noncoding RNA S in Disease. FEBS Lett. 592 (17), 2884–2900. doi:10.1002/1873-3468.13182
Lemasters, J. J. (2005). Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 8 (1), 3–5. doi:10.1089/rej.2005.8.3
Li, A., Zhang, S., Li, J., Liu, K., Huang, F., and Liu, B. (2016). Metformin and Resveratrol Inhibit Drp1-Mediated Mitochondrial Fission and Prevent ER Stress-Associated NLRP3 Inflammasome Activation in the Adipose Tissue of Diabetic Mice. Mol. Cell Endocrinol. 434, 36–47. doi:10.1016/j.mce.2016.06.008
Li, J., Donath, S., Li, Y., Qin, D., Prabhakar, B. S., and Li, P. (2010). miR-30 Regulates Mitochondrial Fission through Targeting P53 and the Dynamin-Related Protein-1 Pathway. Plos Genet. 6 (1), e1000795. doi:10.1371/journal.pgen.1000795
Liu, B.-l., Cheng, M., Hu, S., Wang, S., Wang, L., Tu, X., et al. (2018). Overexpression of miR-142-3p Improves Mitochondrial Function in Cardiac Hypertrophy. Biomed. Pharmacother. 108, 1347–1356. doi:10.1016/j.biopha.2018.09.146
Liu, D., Ma, Z., Di, S., Yang, Y., Yang, J., Xu, L., et al. (2018). AMPK/PGC1α Activation by Melatonin Attenuates Acute Doxorubicin Cardiotoxicity via Alleviating Mitochondrial Oxidative Damage and Apoptosis. Free Radic. Biol. Med. 129, 59–72. doi:10.1016/j.freeradbiomed.2018.08.032
Liu, J., Liang, X., Zhou, D., Lai, L., Xiao, L., Liu, L., et al. (2016). Coupling of Mitochondrial Function and Skeletal Muscle Fiber Type by a miR‐499/Fnip1/AMPK Circuit. EMBO Mol. Med. 8 (10), 1212–1228. doi:10.15252/emmm.201606372
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial Outer-Membrane Protein FUNDC1 Mediates Hypoxia-Induced Mitophagy in Mammalian Cells. Nat. Cell Biol 14 (2), 177–185. doi:10.1038/ncb2422
Liu, R., and Chan, D. C. (2015). The Mitochondrial Fission Receptor Mff Selectively Recruits Oligomerized Drp1. MBoC 26 (24), 4466–4477. doi:10.1091/mbc.e15-08-0591
Liu, X., Zhang, Y., Deng, Y., Yang, L., Ou, W., Xie, M., et al. (2022). Mitochondrial Protein Hyperacetylation Underpins Heart Failure with Preserved Ejection Fraction in Mice. J. Mol. Cell Cardiol. 165, 76–85. doi:10.1016/j.yjmcc.2021.12.015
Long, B., Wang, K., Li, N., Murtaza, I., Xiao, J.-Y., Fan, Y.-Y., et al. (2013). miR-761 Regulates the Mitochondrial Network by Targeting Mitochondrial Fission Factor. Free Radic. Biol. Med. 65, 371–379. doi:10.1016/j.freeradbiomed.2013.07.009
Lood, C., Blanco, L. P., Purmalek, M. M., Carmona-Rivera, C., De Ravin, S. S., Smith, C. K., et al. (2016). Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA Are Interferogenic and Contribute to Lupus-like Disease. Nat. Med. 22 (2), 146–153. doi:10.1038/nm.4027
Lozano, O., Silva-Platas, C., Chapoy-Villanueva, H., Pérez, B. E., Lees, J. G., Ramachandra, C. J. A., et al. (2020). Amorphous SiO2 Nanoparticles Promote Cardiac Dysfunction via the Opening of the Mitochondrial Permeability Transition Pore in Rat Heart and Human Cardiomyocytes. Part. Fibre Toxicol. 17 (1), 15. doi:10.1186/s12989-020-00346-2
Lu, H. R., Vandeplassche, G., Wouters, L., Flameng, W., and Borgers, M. (1990). Effects of β-adrenoceptor Antagonists on Cardiac Function in Ischemic-Reperfused Myocardium of the Isolated Working Rabbit Heart. Eur. J. Pharmacol. 184 (1), 65–74. doi:10.1016/0014-2999(90)90667-u
Ma, S., Feng, J., Zhang, R., Chen, J., Han, D., Li, X., et al. (2017). SIRT1 Activation by Resveratrol Alleviates Cardiac Dysfunction via Mitochondrial Regulation in Diabetic Cardiomyopathy Mice. Oxid Med. Cell Longev 2017, 4602715. doi:10.1155/2017/4602715
Maejima, Y. (2019). SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 6, 186. doi:10.3389/fcvm.2019.00186
Manskikh, V. N., Gancharova, O. S., Nikiforova, A. I., Krasilshchikova, M. S., Shabalina, I. G., Egorov, M. V., et al. (2015). Age-associated Murine Cardiac Lesions Are Attenuated by the Mitochondria-Targeted Antioxidant SkQ1. Histol. Histopathol 30 (3), 353–360. doi:10.14670/HH-30.353
Manwaring, N., Jones, M. M., Wang, J. J., Rochtchina, E., Howard, C., Mitchell, P., et al. (2007). Population Prevalence of the MELAS A3243G Mutation. Mitochondrion 7 (3), 230–233. doi:10.1016/j.mito.2006.12.004
Martens, C. R., Denman, B. A., Mazzo, M. R., Armstrong, M. L., Reisdorph, N., McQueen, M. B., et al. (2018). Chronic Nicotinamide Riboside Supplementation Is Well-Tolerated and Elevates NAD+ in Healthy Middle-Aged and Older Adults. Nat. Commun. 9 (1), 1286. doi:10.1038/s41467-018-03421-7
Matsui, M., and Corey, D. R. (2017). Non-coding RNAs as Drug Targets. Nat. Rev. Drug Discov. 16 (3), 167–179. doi:10.1038/nrd.2016.117
Mewton, N., Cung, T. T., Morel, O., Cayla, G., Bonnefoy-Cudraz, E., Rioufol, G., et al. (2015). Rationale and Design of the Cyclosporine to ImpRove Clinical oUtcome in ST-Elevation Myocardial Infarction Patients (The CIRCUS Trial). Am. Heart J. 169 (6), 758–766. doi:10.1016/j.ahj.2015.02.020
Mizuno, M., Kuno, A., Yano, T., Miki, T., Oshima, H., Sato, T., et al. (2018). Empagliflozin Normalizes the Size and Number of Mitochondria and Prevents Reduction in Mitochondrial Size after Myocardial Infarction in Diabetic Hearts. Physiol. Rep. 6 (12), e13741. doi:10.14814/phy2.13741
Moheimani, H. R., Amiriani, T., Alizadeh, A. M., Jand, Y., Shakiba, D., Ensan, P. S., et al. (2021). Preconditioning and Anti-apoptotic Effects of Metformin and Cyclosporine-A in an Isolated Bile Duct-Ligated Rat Heart. Eur. J. Pharmacol. 893, 173807. doi:10.1016/j.ejphar.2020.173807
Mohsin, A. A., Chen, Q., Quan, N., Rousselle, T., Maceyka, M. W., Samidurai, A., et al. (2019). Mitochondrial Complex I Inhibition by Metformin Limits Reperfusion Injury. J. Pharmacol. Exp. Ther. 369 (2), 282–290. doi:10.1124/jpet.118.254300
Molagoda, I. M. N., Athapaththu, A. M. G. K., Choi, Y. H., Park, C., Jin, C. Y., Kang, C. H., et al. (2021). Fisetin Inhibits NLRP3 Inflammasome by Suppressing TLR4/MD2-Mediated Mitochondrial ROS Production. Antioxidants (Basel) 10 (8), 1215. doi:10.3390/antiox10081215
Morales, P. E., Arias-Durán, C., Ávalos-Guajardo, Y., Aedo, G., Verdejo, H. E., Parra, V., et al. (2020). Emerging Role of Mitophagy in Cardiovascular Physiology and Pathology. Mol. Aspects Med. 71, 100822. doi:10.1016/j.mam.2019.09.006
Mukherjee, S., Chellappa, K., Moffitt, A., Ndungu, J., Dellinger, R. W., Davis, J. G., et al. (2017). Nicotinamide Adenine Dinucleotide Biosynthesis Promotes Liver Regeneration. Hepatology 65 (2), 616–630. doi:10.1002/hep.28912
Muscari, C., Pappagallo, M., Ferrari, D., Giordano, E., Capanni, C., Caldarera, C. M., et al. (1998). Simultaneous Detection of Reduced and Oxidized Glutathione in Tissues and Mitochondria by Capillary Electrophoresis. J. Chromatogr. B Biomed. Sci. Appl. 707 (1-2), 301–307. doi:10.1016/s0378-4347(97)00595-1
Nakayama, H., and Otsu, K. (2018). Mitochondrial DNA as an Inflammatory Mediator in Cardiovascular Diseases. Biochem. J. 475 (5), 839–852. doi:10.1042/bcj20170714
Nichols, B. (2003). Caveosomes and Endocytosis of Lipid Rafts. J. Cell Sci 116 (Pt 23), 4707–4714. doi:10.1242/jcs.00840
Nilsson, B.-O., and Persson, L. (2019). Beneficial Effects of Spermidine on Cardiovascular Health and Longevity Suggest a Cell Type-specific Import of Polyamines by Cardiomyocytes. Biochem. Soc. Trans. 47 (1), 265–272. doi:10.1042/bst20180622
Oh, C.-M., Ryu, D., Cho, S., and Jang, Y. (2020). Mitochondrial Quality Control in the Heart: New Drug Targets for Cardiovascular Disease. Korean Circ. J. 50 (5), 395–405. doi:10.4070/kcj.2019.0416
Olgar, Y., Degirmenci, S., Durak, A., Billur, D., Can, B., Kayki-Mutlu, G., et al. (2018). Aging Related Functional and Structural Changes in the Heart and Aorta: MitoTEMPO Improves Aged-Cardiovascular Performance. Exp. Gerontol. 110, 172–181. doi:10.1016/j.exger.2018.06.012
Olgar, Y., Tuncay, E., Degirmenci, S., Billur, D., Dhingra, R., Kirshenbaum, L., et al. (2020). Ageing‐associated Increase in SGLT2 Disrupts Mitochondrial/sarcoplasmic Reticulum Ca 2+ Homeostasis and Promotes Cardiac Dysfunction. J. Cell Mol Med 24 (15), 8567–8578. doi:10.1111/jcmm.15483
Oliveira, P. J., Esteves, T., Rolo, A. P., Monteiro, P., Gonçalves, L., Palmeira, C. M., et al. (2003). Carvedilol: Relation between Antioxidant Activity and Inhibition of the Mitochondrial Permeability Transition. Rev. Port Cardiol. 22 (1), 55–62.
Oliveira, P. J., Rolo, A. P., Sardão, V. A., Monteiro, P., Gonçalves, L., Providência, L. A., et al. (2004). Advantages in the Use of Carvedilol versus Propranolol for the protection of Cardiac Mitochondrial Function. Rev. Port Cardiol. 23 (10), 1291–1298.
Ong, S.-B., Subrayan, S., Lim, S. Y., Yellon, D. M., Davidson, S. M., and Hausenloy, D. J. (2010). Inhibiting Mitochondrial Fission Protects the Heart against Ischemia/reperfusion Injury. Circulation 121 (18), 2012–2022. doi:10.1161/circulationaha.109.906610
Ong, S. B., Kwek, X. Y., Katwadi, K., Hernandez-Resendiz, S., Crespo-Avilan, G. E., Ismail, N. I., et al. (2019). Targeting Mitochondrial Fission Using Mdivi-1 in A Clinically Relevant Large Animal Model of Acute Myocardial Infarction: A Pilot Study. Int. J. Mol. Sci. 20 (16). doi:10.3390/ijms20163972
Onishi, A., Miyamae, M., Kaneda, K., Kotani, J., and Figueredo, V. M. (2012). Direct Evidence for Inhibition of Mitochondrial Permeability Transition Pore Opening by Sevoflurane Preconditioning in Cardiomyocytes: Comparison with Cyclosporine A. Eur. J. Pharmacol. 675 (1-3), 40–46. doi:10.1016/j.ejphar.2011.11.040
Osellame, L. D., Singh, A. P., Stroud, D. A., Palmer, C. S., Stojanovski, D., Ramachandran, R., et al. (2016). Cooperative and Independent Roles of the Drp1 Adaptors Mff, MiD49 and MiD51 in Mitochondrial Fission. J. Cell Sci 129 (11), 2170–2181. doi:10.1242/jcs.185165
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of Mitophagy in Cellular Homeostasis, Physiology and Pathology. Nat. Cell Biol 20 (9), 1013–1022. doi:10.1038/s41556-018-0176-2
Palmeira, C. M., Teodoro, J. S., Amorim, J. A., Steegborn, C., Sinclair, D. A., and Rolo, A. P. (2019). Mitohormesis and Metabolic Health: The Interplay between ROS, cAMP and Sirtuins. Free Radic. Biol. Med. 141, 483–491. doi:10.1016/j.freeradbiomed.2019.07.017
Papanicolaou, K. N., Kikuchi, R., Ngoh, G. A., Coughlan, K. A., Dominguez, I., Stanley, W. C., et al. (2012). Mitofusins 1 and 2 Are Essential for Postnatal Metabolic Remodeling in Heart. Circ. Res. 111 (8), 1012–1026. doi:10.1161/circresaha.112.274142
Paradies, G., Paradies, V., Ruggiero, F. M., and Petrosillo, G. (2019). Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 8 (7). doi:10.3390/cells8070728
Park, R. J., Shen, H., Liu, L., Liu, X., Ferguson, S. M., and De Camilli, P. (2013). Dynamin Triple Knockout Cells Reveal off Target Effects of Commonly Used Dynamin Inhibitors. J. Cell Sci 126 (Pt 22), 5305–5312. doi:10.1242/jcs.138578
Patrushev, M., Kasymov, V., Patrusheva, V., Ushakova, T., Gogvadze, V., and Gaziev, A. (2004). Mitochondrial Permeability Transition Triggers the Release of mtDNA Fragments. Cmls, Cel. Mol. Life Sci. 61 (24), 3100–3103. doi:10.1007/s00018-004-4424-1
Pek, N. M. Q., Phua, Q. H., Ho, B. X., Pang, J. K. S., Hor, J.-H., An, O., et al. (2019). Mitochondrial 3243A > G Mutation Confers Pro-atherogenic and Pro-inflammatory Properties in MELAS iPS Derived Endothelial Cells. Cell Death Dis 10 (11), 802. doi:10.1038/s41419-019-2036-9
Peoples, J. N., Saraf, A., Ghazal, N., Pham, T. T., and Kwong, J. Q. (2019). Mitochondrial Dysfunction and Oxidative Stress in Heart Disease. Exp. Mol. Med. 51 (12), 1–13. doi:10.1038/s12276-019-0355-7
Pepe, S., Marasco, S. F., Haas, S. J., Sheeran, F. L., Krum, H., and Rosenfeldt, F. L. (2007). Coenzyme Q10 in Cardiovascular Disease. Mitochondrion 7 (Suppl. l), S154–S167. doi:10.1016/j.mito.2007.02.005
Picca, A., Calvani, R., Coelho-Junior, H. J., and Marzetti, E. (2021). Cell Death and Inflammation: The Role of Mitochondria in Health and Disease. Cells 10 (3). doi:10.3390/cells10030537
Picca, A., Guerra, F., Calvani, R., Bucci, C., Lo Monaco, M. R., Bentivoglio, A. R., et al. (2019). Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int. J. Mol. Sci. 20 (4). doi:10.3390/ijms20040805
Piot, C., Croisille, P., Staat, P., Thibault, H., Rioufol, G., Mewton, N., et al. (2008). Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. N. Engl. J. Med. 359 (5), 473–481. doi:10.1056/nejmoa071142
Płoszaj, T., Robaszkiewicz, A., and Witas, H. (2010). Oxidative Damage of Mitochondrial DNA: the Result or Consequence of Enhanced Generation of Reactive Oxygen Species. Postepy Biochem. 56 (2), 139–146.
Popov, L. D. (2020). Mitochondrial Biogenesis: An Update. J. Cell. Mol. Medi 24 (9), 4892–4899. doi:10.1111/jcmm.15194
Poznyak, A. V., Ivanova, E. A., Sobenin, I. A., Yet, S. F., and Orekhov, A. N. (2020). The Role of Mitochondria in Cardiovascular Diseases. Biology (Basel) 9 (6). doi:10.3390/biology9060137
Qiao, H., Ren, H., Du, H., Zhang, M., Xiong, X., and Lv, R. (2018). Liraglutide Repairs the Infarcted Heart: The Role of the SIRT1/Parkin/mitophagy Pathway. Mol. Med. Rep. 17 (3), 3722–3734. doi:10.3892/mmr.2018.8371
Ren, M., Phoon, C. K. L., and Schlame, M. (2014). Metabolism and Function of Mitochondrial Cardiolipin. Prog. Lipid Res. 55, 1–16. doi:10.1016/j.plipres.2014.04.001
Reznick, R. M., Zong, H., Li, J., Morino, K., Moore, I. K., Yu, H. J., et al. (2007). Aging-associated Reductions in AMP-Activated Protein Kinase Activity and Mitochondrial Biogenesis. Cell Metab. 5 (2), 151–156. doi:10.1016/j.cmet.2007.01.008
Ribeiro Junior, R. F., Dabkowski, E. R., Shekar, K. C., O´Connell, K. A., Hecker, P. A., and Murphy, M. P. (2018). MitoQ Improves Mitochondrial Dysfunction in Heart Failure Induced by Pressure Overload. Free Radic. Biol. Med. 117, 18–29. doi:10.1016/j.freeradbiomed.2018.01.012
Richter, V., Palmer, C. S., Osellame, L. D., Singh, A. P., Elgass, K., Stroud, D. A., et al. (2014). Structural and Functional Analysis of MiD51, a Dynamin Receptor Required for Mitochondrial Fission. J. Cell Biol 204 (4), 477–486. doi:10.1083/jcb.201311014
Riley, J. S., Quarato, G., Cloix, C., Lopez, J., O'Prey, J., Pearson, M., et al. (2018). Mitochondrial Inner Membrane Permeabilisation Enables mtDNA Release during Apoptosis. Embo j 37 (17). doi:10.15252/embj.201899238
Rodrigues, F., Fais, R., Tostes, R., and Carneiro, F. (2015). There Is a Link between Erectile Dysfunction and Heart Failure: it Could Be Inflammation. Cdt 16 (5), 442–450. doi:10.2174/1389450116666150420145757
Rosdah, A. A., K Holien, J., Delbridge, L. M., Dusting, G. J., and Lim, S. Y. (2016). Mitochondrial Fission - a Drug Target for Cytoprotection or Cytodestruction? Pharmacol. Res. Perspect. 4 (3), e00235. doi:10.1002/prp2.235
Rossman, M. J., Santos-Parker, J. R., Steward, C. A. C., Bispham, N. Z., Cuevas, L. M., Rosenberg, H. L., et al. (2018). Chronic Supplementation with a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 71 (6), 1056–1063. doi:10.1161/hypertensionaha.117.10787
Sanbe, A., Tanonaka, K., Kobayashi, R., and Takeo, S. (1995). Effects of Long-Term Therapy with ACE Inhibitors, Captopril, Enalapril and Trandolapril, on Myocardial Energy Metabolism in Rats with Heart Failure Following Myocardial Infarction. J. Mol. Cell Cardiol. 27 (10), 2209–2222. doi:10.1016/s0022-2828(95)91551-6
Savi, M., Bocchi, L., Mena, P., Dall’Asta, M., Crozier, A., Brighenti, F., et al. (2017). In Vivo administration of Urolithin A and B Prevents the Occurrence of Cardiac Dysfunction in Streptozotocin-Induced Diabetic Rats. Cardiovasc. Diabetol. 16 (1), 80. doi:10.1186/s12933-017-0561-3
Sazonova, M. A., Ryzhkova, A. I., Sinyov, V. V., Galitsyna, E. V., Melnichenko, A. A., Demakova, N. A., et al. (2018). Mitochondrial Genome Mutations Associated with Myocardial Infarction. Dis. Markers 2018, 9749457. doi:10.1155/2018/9749457
Scarpulla, R. C. (2011). Metabolic Control of Mitochondrial Biogenesis through the PGC-1 Family Regulatory Network. Biochim. Biophys. Acta (Bba) - Mol. Cell Res. 1813 (7), 1269–1278. doi:10.1016/j.bbamcr.2010.09.019
Schaper, J., Meiser, E., and Stämmler, G. (1985). Ultrastructural Morphometric Analysis of Myocardium from Dogs, Rats, Hamsters, Mice, and from Human Hearts. Circ. Res. 56 (3), 377–391. doi:10.1161/01.res.56.3.377
Severin, F. F., Severina, I. I., Antonenko, Y. N., Rokitskaya, T. I., Cherepanov, D. A., Mokhova, E. N., et al. (2010). Penetrating Cation/fatty Acid Anion Pair as a Mitochondria-Targeted Protonophore. Proc. Natl. Acad. Sci. U.S.A. 107 (2), 663–668. doi:10.1073/pnas.0910216107
Shill, D. D., Southern, W. M., Willingham, T. B., Lansford, K. A., McCully, K. K., and Jenkins, N. T. (2016). Mitochondria-specific Antioxidant Supplementation Does Not Influence Endurance Exercise Training-Induced Adaptations in Circulating Angiogenic Cells, Skeletal Muscle Oxidative Capacity or Maximal Oxygen Uptake. J. Physiol. 594 (23), 7005–7014. doi:10.1113/jp272491
Shires, S. E., and Gustafsson, Å. B. (2015). Mitophagy and Heart Failure. J. Mol. Med. 93 (3), 253–262. doi:10.1007/s00109-015-1254-6
Siasos, G., Tsigkou, V., Kosmopoulos, M., Theodosiadis, D., Simantiris, S., Tagkou, N. M., et al. (2018). Mitochondria and Cardiovascular Diseases-From Pathophysiology to Treatment. Ann. Transl. Med. 6 (12), 256. doi:10.21037/atm.2018.06.21
Sirey, T. M., Roberts, K., Haerty, W., Bedoya-Reina, O., Rogatti-Granados, S., Tan, J. Y., et al. (2019). The Long Non-coding RNA Cerox1 Is a post Transcriptional Regulator of Mitochondrial Complex I Catalytic Activity. Elife 8. doi:10.7554/eLife.45051
Skulachev, V. P. (2007). A Biochemical Approach to the Problem of Aging: "megaproject" on Membrane-Penetrating Ions. The First Results and Prospects. Biochem. Mosc. 72 (12), 1385–1396. doi:10.1134/s0006297907120139
Skulachev, V. P., Anisimov, V. N., Antonenko, Y. N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., et al. (2009). An Attempt to Prevent Senescence: a Mitochondrial Approach. Biochim. Biophys. Acta (Bba) - Bioenerg. 1787 (5), 437–461. doi:10.1016/j.bbabio.2008.12.008
Smith, R. A. J., Porteous, C. M., Coulter, C. V., and Murphy, M. P. (1999). Selective Targeting of an Antioxidant to Mitochondria. Eur. J. Biochem. 263 (3), 709–716. doi:10.1046/j.1432-1327.1999.00543.x
Snow, B. J., Rolfe, F. L., Lockhart, M. M., Frampton, C. M., O'Sullivan, J. D., Fung, V., et al. (2010). A Double-Blind, Placebo-Controlled Study to Assess the Mitochondria-Targeted Antioxidant MitoQ as a Disease-Modifying Therapy in Parkinson's Disease. Mov. Disord. 25 (11), 1670–1674. doi:10.1002/mds.23148
Sobenin, I. A., Sazonova, M. A., Postnov, A. Y., Bobryshev, Y. V., and Orekhov, A. N. (2013). Changes of Mitochondria in Atherosclerosis: Possible Determinant in the Pathogenesis of the Disease. Atherosclerosis 227 (2), 283–288. doi:10.1016/j.atherosclerosis.2013.01.006
Sobenin, I., Zhelankin, A., Mitrofanov, K., Sinyov, V., Sazonova, M., Postnov, A., et al. (2015). Mutations of Mitochondrial DNA in Atherosclerosis and Atherosclerosis-Related Diseases. Cpd 21 (9), 1158–1163. doi:10.2174/1381612820666141013133000
Stanley, W. C. (2005). Rationale for a Metabolic Approach in Diabetic Coronary Patients. Coron. Artery Dis. 16 Suppl 1 (Suppl. 1), S11–S15. doi:10.1097/00019501-200511001-00003
Suzuki, Y., Ali, M., Fischer, M., and Riemer, J. (2013). Human Copper Chaperone for Superoxide Dismutase 1 Mediates its Own Oxidation-dependent Import into Mitochondria. Nat. Commun. 4, 2430. doi:10.1038/ncomms3430
Szeto, H. H. (2006). Cell-permeable, Mitochondrial-Targeted, Peptide Antioxidants. AAPS J. 8 (2), E277–E283. doi:10.1007/bf02854898
Szeto, H. H. (2014). First-in-class Cardiolipin-Protective Compound as a Therapeutic Agent to Restore Mitochondrial Bioenergetics. Br. J. Pharmacol. 171 (8), 2029–2050. doi:10.1111/bph.12461
Tadolini, B., and Franconi, F. (1998). Carvedilol Inhibition of Lipid Peroxidation. A New Antioxidative Mechanism. Free Radic. Res. 29 (5), 377–387. doi:10.1080/10715769800300421
Takagi, S., Li, J., Takagaki, Y., Kitada, M., Nitta, K., Takasu, T., et al. (2018). Ipragliflozin Improves Mitochondrial Abnormalities in Renal Tubules Induced by a High-Fat Diet. J. Diabetes Investig. 9 (5), 1025–1032. doi:10.1111/jdi.12802
Tanaka, A., and Youle, R. J. (2008). A Chemical Inhibitor of DRP1 Uncouples Mitochondrial Fission and Apoptosis. Mol. Cell 29 (4), 409–410. doi:10.1016/j.molcel.2008.02.005
Tarantini, G., Scrutinio, D., Bruzzi, P., Boni, L., Rizzon, P., and Iliceto, S. (2006). Metabolic Treatment with L-Carnitine in Acute Anterior ST Segment Elevation Myocardial Infarction. Cardiology 106 (4), 215–223. doi:10.1159/000093131
Tedgui, A., and Mallat, Z. (2006). Cytokines in Atherosclerosis: Pathogenic and Regulatory Pathways. Physiol. Rev. 86 (2), 515–581. doi:10.1152/physrev.00024.2005
Torrealba, N., Aranguiz, P., Alonso, C., Rothermel, B. A., and Lavandero, S. (2017). Mitochondria in Structural and Functional Cardiac Remodeling. Adv. Exp. Med. Biol. 982, 277–306. doi:10.1007/978-3-319-55330-6_15
Toyama, E. Q., Herzig, S., Courchet, J., Lewis, T. L., Losón, O. C., Hellberg, K., et al. (2016). AMP-activated Protein Kinase Mediates Mitochondrial Fission in Response to Energy Stress. Science 351 (6270), 275–281. doi:10.1126/science.aab4138
Tyrrell, D. J., Blin, M. G., Song, J., Wood, S. C., Zhang, M., Beard, D. A., et al. (2020). Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 126 (3), 298–314. doi:10.1161/circresaha.119.315644
Urban, M. L., Manenti, L., and Vaglio, A. (2015). Fibrosis--A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 373 (1), 95–96. doi:10.1056/NEJMc1504848
Vacante, F., Senesi, P., Montesano, A., Frigerio, A., Luzi, L., and Terruzzi, I. (2018). L-carnitine: An Antioxidant Remedy for the Survival of Cardiomyocytes under Hyperglycemic Condition. J. Diabetes Res. 2018, 4028297. doi:10.1155/2018/4028297
van der Bliek, A. M., Sedensky, M. M., and Morgan, P. G. (2017). Cell Biology of the Mitochondrion. Genetics 207 (3), 843–871. doi:10.1534/genetics.117.300262
van Rooij, E., Quiat, D., Johnson, B. A., Sutherland, L. B., Qi, X., Richardson, J. A., et al. (2009). A Family of microRNAs Encoded by Myosin Genes Governs Myosin Expression and Muscle Performance. Developmental Cell 17 (5), 662–673. doi:10.1016/j.devcel.2009.10.013
Vandeplassche, G., Lu, H. R., Wouters, L., Flameng, W., and Borgers, M. (1991). Normothermic Ischemic Cardiac Arrest in the Isolated Working Rabbit Heart: Effects of Dl-Nebivolol and Atenolol. Basic Res. Cardiol. 86 (1), 21–31. doi:10.1007/bf02193868
Varjabedian, L., Bourji, M., Pourafkari, L., and Nader, N. D. (2018). Cardioprotection by Metformin: Beneficial Effects beyond Glucose Reduction. Am. J. Cardiovasc. Drugs 18 (3), 181–193. doi:10.1007/s40256-018-0266-3
von Hardenberg, A., and Maack, C. (2017). Mitochondrial Therapies in Heart Failure. Handb Exp. Pharmacol. 243, 491–514. doi:10.1007/164_2016_123
Wang, B., Nie, J., Wu, L., Hu, Y., Wen, Z., Dong, L., et al. (2018). AMPKα2 Protects against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 122 (5), 712–729. doi:10.1161/circresaha.117.312317
Wang, J.-X., Jiao, J.-Q., Li, Q., Long, B., Wang, K., Liu, J.-P., et al. (2011). miR-499 Regulates Mitochondrial Dynamics by Targeting Calcineurin and Dynamin-Related Protein-1. Nat. Med. 17 (1), 71–78. doi:10.1038/nm.2282
Wang, J., Li, S., Wang, J., Wu, F., Chen, Y., Zhang, H., et al. (2020). Spermidine Alleviates Cardiac Aging by Improving Mitochondrial Biogenesis and Function. Aging 12 (1), 650–671. doi:10.18632/aging.102647
Wang, K., Liu, F., Zhou, L.-Y., Ding, S.-L., Long, B., Liu, C.-Y., et al. (2013). miR-874 Regulates Myocardial Necrosis by Targeting Caspase-8. Cell Death Dis 4, e709. doi:10.1038/cddis.2013.233
Wang, K., Long, B., Jiao, J.-Q., Wang, J.-X., Liu, J.-P., Li, Q., et al. (2012). miR-484 Regulates Mitochondrial Network through Targeting Fis1. Nat. Commun. 3, 781. doi:10.1038/ncomms1770
Wang, Q., Zhang, M., Torres, G., Wu, S., Ouyang, C., Xie, Z., et al. (2017). Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 66 (1), 193–205. doi:10.2337/db16-0915
Wang, X., Li, X., Liu, S., Brickell, A. N., Zhang, J., Wu, Z., et al. (2020). PCSK9 Regulates Pyroptosis via mtDNA Damage in Chronic Myocardial Ischemia. Basic Res. Cardiol. 115 (6), 66. doi:10.1007/s00395-020-00832-w
Wang, Y., An, H., Liu, T., Qin, C., Sesaki, H., Guo, S., et al. (2019). Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 29 (6), 1511–1523. e5. doi:10.1016/j.celrep.2019.09.070
White, M. J., McArthur, K., Metcalf, D., Lane, R. M., Cambier, J. C., Herold, M. J., et al. (2014). Apoptotic Caspases Suppress mtDNA-Induced STING-Mediated Type I IFN Production. Cell 159 (7), 1549–1562. doi:10.1016/j.cell.2014.11.036
Whitson, J. A., Bitto, A., Zhang, H., Sweetwyne, M. T., Coig, R., Bhayana, S., et al. (2020). SS-31 and NMN: Two Paths to Improve Metabolism and Function in Aged Hearts. Aging Cell 19 (10), e13213. doi:10.1111/acel.13213
Whittington, H. J., Hall, A. R., McLaughlin, C. P., Hausenloy, D. J., Yellon, D. M., and Mocanu, M. M. (2013). Chronic Metformin Associated Cardioprotection against Infarction: Not Just a Glucose Lowering Phenomenon. Cardiovasc. Drugs Ther. 27 (1), 5–16. doi:10.1007/s10557-012-6425-x
Williams, R. S. (1999). Apoptosis and Heart Failure. N. Engl. J. Med. 341 (10), 759–760. doi:10.1056/nejm199909023411012
Wojciechowska, A., Braniewska, A., and Kozar-Kamińska, K. (2017). MicroRNA in Cardiovascular Biology and Disease. Adv. Clin. Exp. Med. 26 (5), 865–874. doi:10.17219/acem/62915
Xiao, Y., Chen, W., Zhong, Z., Ding, L., Bai, H., Chen, H., et al. (2020). Electroacupuncture Preconditioning Attenuates Myocardial Ischemia-Reperfusion Injury by Inhibiting Mitophagy Mediated by the mTORC1-ULK1-FUNDC1 Pathway. Biomed. Pharmacother. 127, 110148. doi:10.1016/j.biopha.2020.110148
Yaku, K., Okabe, K., and Nakagawa, T. (2018). NAD Metabolism: Implications in Aging and Longevity. Ageing Res. Rev. 47, 1–17. doi:10.1016/j.arr.2018.05.006
Yamaguchi, S., and Yoshino, J. (2016). The Pathophysiological Importance and Therapeutic Potential of NAD' Biosynthesis and Mitochondrial Sirtuin SIRT3 in Age-Associated Diseases. Nihon Rinsho 74 (9), 1447–1455.
Yan, Y., Zhang, B., Liu, N., Qi, C., Xiao, Y., Tian, X., et al. (2016). Circulating Long Noncoding RNA UCA1 as a Novel Biomarker of Acute Myocardial Infarction. Biomed. Res. Int. 2016, 8079372. doi:10.1155/2016/8079372
Yokokawa, T., Kido, K., Suga, T., Isaka, T., Hayashi, T., and Fujita, S. (2018). Exercise-induced Mitochondrial Biogenesis Coincides with the Expression of Mitochondrial Translation Factors in Murine Skeletal Muscle. Physiol. Rep. 6 (20), e13893. doi:10.14814/phy2.13893
Yu, E., Calvert, P. A., Mercer, J. R., Harrison, J., Baker, L., Figg, N. L., et al. (2013). Mitochondrial DNA Damage Can Promote Atherosclerosis Independently of Reactive Oxygen Species through Effects on Smooth Muscle Cells and Monocytes and Correlates with Higher-Risk Plaques in Humans. Circulation 128 (7), 702–712. doi:10.1161/circulationaha.113.002271
Yu, E. P. K., Reinhold, J., Yu, H., Starks, L., Uryga, A. K., Foote, K., et al. (2017). Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Atvb 37 (12), 2322–2332. doi:10.1161/atvbaha.117.310042
Yu, L. M., Dong, X., Xue, X. D., Xu, S., Zhang, X., Xu, Y. L., et al. (2021). Melatonin Attenuates Diabetic Cardiomyopathy and Reduces Myocardial Vulnerability to Ischemia-Reperfusion Injury by Improving Mitochondrial Quality Control: Role of SIRT6. J. Pineal Res. 70 (1), e12698. doi:10.1111/jpi.12698
Yue, W., Chen, Z., Liu, H., Yan, C., Chen, M., Feng, D., et al. (2014). A Small Natural Molecule Promotes Mitochondrial Fusion through Inhibition of the Deubiquitinase USP30. Cell Res 24 (4), 482–496. doi:10.1038/cr.2014.20
Zalewski, J., Claus, P., Bogaert, J., Driessche, N. V., Driesen, R. B., Galan, D. T., et al. (2015). Cyclosporine A Reduces Microvascular Obstruction and Preserves Left Ventricular Function Deterioration Following Myocardial Ischemia and Reperfusion. Basic Res. Cardiol. 110 (2), 18. doi:10.1007/s00395-015-0475-8
Zernii, E. Y., Gancharova, O. S., Baksheeva, V. E., Golovastova, M. O., Kabanova, E. I., Savchenko, M. S., et al. (2017). Mitochondria-Targeted Antioxidant SkQ1 Prevents Anesthesia-Induced Dry Eye Syndrome. Oxid Med. Cell Longev 2017, 9281519. doi:10.1155/2017/9281519
Zhang, H., Alder, N. N., Wang, W., Szeto, H., Marcinek, D. J., and Rabinovitch, P. S. (2020). Reduction of Elevated Proton Leak Rejuvenates Mitochondria in the Aged Cardiomyocyte. Elife 9. doi:10.7554/eLife.60827
Zhang, Q., Li, D., Dong, X., Zhang, X., Liu, J., Peng, L., et al. (2021). LncDACH1 Promotes Mitochondrial Oxidative Stress of Cardiomyocytes by Interacting with Sirtuin3 and Aggravates Diabetic Cardiomyopathy. Sci. China Life Sci. 2021. doi:10.1007/s11427-021-1982-8
Zhang, W., Ren, H., Xu, C., Zhu, C., Wu, H., Liu, D., et al. (2016). Hypoxic Mitophagy Regulates Mitochondrial Quality and Platelet Activation and Determines Severity of I/R Heart Injury. Elife 5, e21407. doi:10.7554/elife.21407
Zhang, W., Siraj, S., Zhang, R., and Chen, Q. (2017). Mitophagy Receptor FUNDC1 Regulates Mitochondrial Homeostasis and Protects the Heart from I/R Injury. Autophagy 13 (6), 1080–1081. doi:10.1080/15548627.2017.1300224
Zinman, B., Wanner, C., Lachin, J. M., Fitchett, D., Bluhmki, E., Hantel, S., et al. (2015). Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 373 (22), 2117–2128. doi:10.1056/nejmoa1504720
Keywords: cardiovascular diseases, mitochondrial dysfunction, reactive oxygen species, mitophagy, mitochondrial therapy
Citation: Yang J, Guo Q, Feng X, Liu Y and Zhou Y (2022) Mitochondrial Dysfunction in Cardiovascular Diseases: Potential Targets for Treatment. Front. Cell Dev. Biol. 10:841523. doi: 10.3389/fcell.2022.841523
Received: 22 December 2021; Accepted: 13 April 2022;
Published: 13 May 2022.
Edited by:
Ineke Luijten, University of Edinburgh, United KingdomReviewed by:
Alexander Nikolaevich Orekhov, Institute for Aterosclerosis Research, RussiaCopyright © 2022 Yang, Guo, Feng, Liu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yujie Zhou, YXp6eWoxMkAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.