
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 20 April 2022
Sec. Cell Death and Survival
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.839041
This article is part of the Research Topic Inflammasomes: Cornerstone of Immunity View all 6 articles
 Sebastian Lillo1
Sebastian Lillo1 Maya Saleh1,2*
Maya Saleh1,2*The inflammasomes are critical regulators of innate immunity, inflammation and cell death and have emerged as important regulators of cancer development and control. Inflammasomes are assembled by pattern recognition receptors (PRR) following the sensing of microbial- or danger-associated molecular patterns (MAMPs/DAMPs) and elicit inflammation through the oligomerization and activation of inflammatory caspases. These cysteinyl-aspartate proteases cleave the proinflammatory cytokines IL-1β and IL-18 into their biologically active mature form. The roles of the inflammasomes and associated pro-inflammatory cytokines vary greatly depending on the cancer type. Here we discuss recent studies highlighting contrasting roles of the inflammasome pathway in curbing versus promoting tumorigenesis. On one hand, the inflammasomes participate in stimulating anti-tumor immunity, but they have also been shown to contribute to immunosuppression or to directly promote tumor cell survival, proliferation, and metastasis. A better understanding of inflammasome functions in different cancers is thus critical for the design of novel cancer immunotherapies.
Cancer development is a complex process that integrates tumor cell-intrinsic and -extrinsic signals that favor cellular transformation, unhinged growth, invasion and metastasis. Among the hallmarks of cancer (Hanahan and Weinberg, 2011), inflammation and tumor escape from immune destruction exert determining roles. Indeed, cancer control is intricately linked to the potency of the immune response. The fundamental understanding of immune surveillance, immune editing (Dunn et al., 2002) (a process in which the immune ‘pressure’ shapes tumor immunogenicity) and immune escape has resulted in breakthroughs in cancer immunotherapies, such as immune checkpoint inhibitors (ICI) or chimeric antigen receptor (CAR)-T cell therapy, among others. Such therapies have revolutionized cancer treatment by significantly improving patient survival and quality of life. Notably, the eligibility of patients to ICI immunotherapy has increased from 1.5% in 2011 to 43.6% in 2018, and >29 immunotherapies are currently approved. However, despite this impressive progress, the clinical reality reveals several unmet challenges: 1) relatively low proportions of patients exhibit objective responses to these therapies as a standalone treatment and a subset of patients develop a hyper-progressive disease, 2) immunotherapy efficacy is often associated with inflammatory toxicities that require clinical management or suspension of treatment, 3) a subset of responding patients receiving immunotherapy relapse and develop aggressive disease; 4) the efficacy of cancer therapies, including chemotherapies, ICI, adoptive T cell therapy and other immunotherapies requires signals from the microbiota; 5) systemic and cellular metabolisms impact cancer and anti-tumor immunity; and 6) immune responses promotes clonal selection, the transformation of the tumor microenvironment (TME) and the establishment of a pre-metastatic niche. The heterogeneous response of patients reflects gaps in knowledge, particularly in the understanding of organ-specific tumor macro- and micro-environments and the interactions between systems, namely that of the immune, metabolic and microbial systems in cancer control.
The innate immune system is our first line of defense, as it rapidly senses ‘danger’ signals (Matzinger, 2002) via pattern recognition receptors (PRR), e.g. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) including inflammasome receptors, and provides adjuvant effects to fully engage adaptive immunity. Concerning anti-tumor immunity, PRR ligands are considered as a promising avenue in cancer immunotherapy. For instance, three TLR agonists, namely Bacillus Calmette-Guérin (BCG), monophosphoryl lipid A (MPL) and imiquimod are approved for use in cancer patients, and additional PRR ligands, primarily nucleic acid mimetics, are currently in clinical trial.
The inflammasomes are central effectors of innate immunity and have been demonstrated to exert important, albeit contrasting, roles in tumorigenesis, anti-tumor immunity and response to cancer therapies. In this review, we discuss recent work examining inflammasome functions in cancer and the potential of developing inflammasome-based immunotherapies.
The inflammasome term was dubbed in 2002 by Dr. Jürg Tschopp, who first described the NLRP1 (Nod-like receptor with a pyrin domain) inflammasome as an intracellular multiprotein complex, consisting of the sensor NLRP1 (Nod-like receptor with a pyrin domain), the adaptor ASC (Apoptosis-associated speck-like protein containing a CARD) and both inflammatory caspases-1 and -5 (Martinon et al., 2002). Since, more than twelve different inflammasomes have been characterized, namely NLRP1, NLRP2, NLRP3, NLRP6, NLRP7, NLRP9, NLRP12, NLCR4/NAIP, AIM2, IFI16, CARD8 and PYRIN. Most inflammasome sensors belong to the Nod-like receptor (NLR) family, which consists of 22 members in humans and 34 in mice (Ariffin and Sweet, 2013), all harboring a central nucleotide-binding and oligomerization domain (NOD) and a C-terminal leucine-rich repeat (LRR). NLR family members are further classified into four subfamilies according to the nature of their N-terminal domain, an activation domain (NLRA), a baculovirus inhibitor of apoptosis protein (IAP) repeat (BIR) domain (NLRB), a caspase activation and recruitment domain (CARD) domain (NLRC) or a pyrin domain (NLRP). Except for NLRA, members of the other NLR subfamilies have been reported to assemble inflammasomes (Figure 1). While the putative microbial or host ligands have been identified for some inflammasomes, they remain elusive for the rest. Examples of known inflammasome ligands include cytoplasmic host or pathogen (bacterial or viral) double stranded DNA that activate Absent In Melanoma (AIM)2 (Bürckstümmer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009), nuclear viral DNA that triggers the AIM2-like receptors (ALRs) IFN-inducible factor IFI204 in mice and IFI16 in humans (Kerur et al., 2011), and intracellular bacterial proteins, namely flagellin and components of the bacterial type III secretion system such as needle and inner rod proteins, that engage NAIP/NLRC4 inflammasomes (Lightfield et al., 2008; Zhao et al., 2011; Rayamajhi et al., 2013; Yang et al., 2013; Reyes Ruiz et al., 2017; Tenthorey et al., 2017). On the other hand, the ligands of NLRP inflammasomes remain elusive. For instance, NLRP1 is activated following its proteosome-dependent “functional degradation” that liberates its C-terminal CARD to interact with caspase-1 and form an inflammasome. Such a functional degradation is induced by pathogen effectors such as Bacillus anthracis lethal factor (LF) (Klimpel et al., 1994; Fink et al., 2008), the protease component of lethal toxin, or Shigella flexneri E3 ubiquitin ligase IpaH7.8 (Sandstrom et al., 2019), and hence points to NLRP1 as a sensor of its own stability in response to pathogen activity. ATP depletion induced by infection with Toxoplasma gondii or Listeria monocytogenes also activates the NLRP1 inflammasome (Liao and Mogridge, 2013; Neiman-Zenevich et al., 2017). More recently, Bauernfried et al. demonstrated yet another activator of human but not mouse NLRP1, namely viral dsRNA, a bonafide ligand that binds to the leucine rich repeats (LRR) of NLRP1 (Bauernfried et al., 2021). As for NLRP1, the Pyrin inflammasome senses an “alteration” induced by infection, namely the inactivation of Rho family small GTPases by bacterial Rho-modifying toxins (Xu et al., 2014). Last, the ligands responsible for direct NLRP3 activation remain unknown. NLRP3 is activated by a wide range of MAMPs and DAMPs, including particulate matters, cholesterol crystals, cellular metabolites, lysosomal damage, defective mitophagy and ionic perturbations such as K+ efflux and Ca++ influx. NLRP3 activation also requires several post-translational modifications, i.e. ubiquitination, phosphorylation and sumoylation and the binding of NIMA-related kinase 7 (NEK7) to its LRRs (reviewed in (Swanson et al., 2019)). Activation of the non-canonical NLRP3 inflammasome is triggered by direct binding of cytosolic lipopolysaccharide (LPS) to caspase-11 in mice or caspase-4/-5 in humans (Shi et al., 2014). Oxidized phospholipid (oxPAPC) binds to caspase-11 and elicits inflammasome activation in dendritic cells (Zanoni et al., 2016) but competes with LPS binding and inhibits non-canonical inflammasome activation in macrophages conferring protection against Gram-negative bacterial sepsis (Chu et al., 2018). Last, although less well-studied, the mechanisms of activation of other NLRP inflammasomes, namely NLRP2 (Zhang et al., 2020), NLRP6 (Shen et al., 2021), NLRP7 (Radian et al., 2015), NLRP9 (reviewed in (Mullins and Chen, 2021)) and NLRP12 (reviewed in (Tuladhar and Kanneganti, 2020)) are emerging.
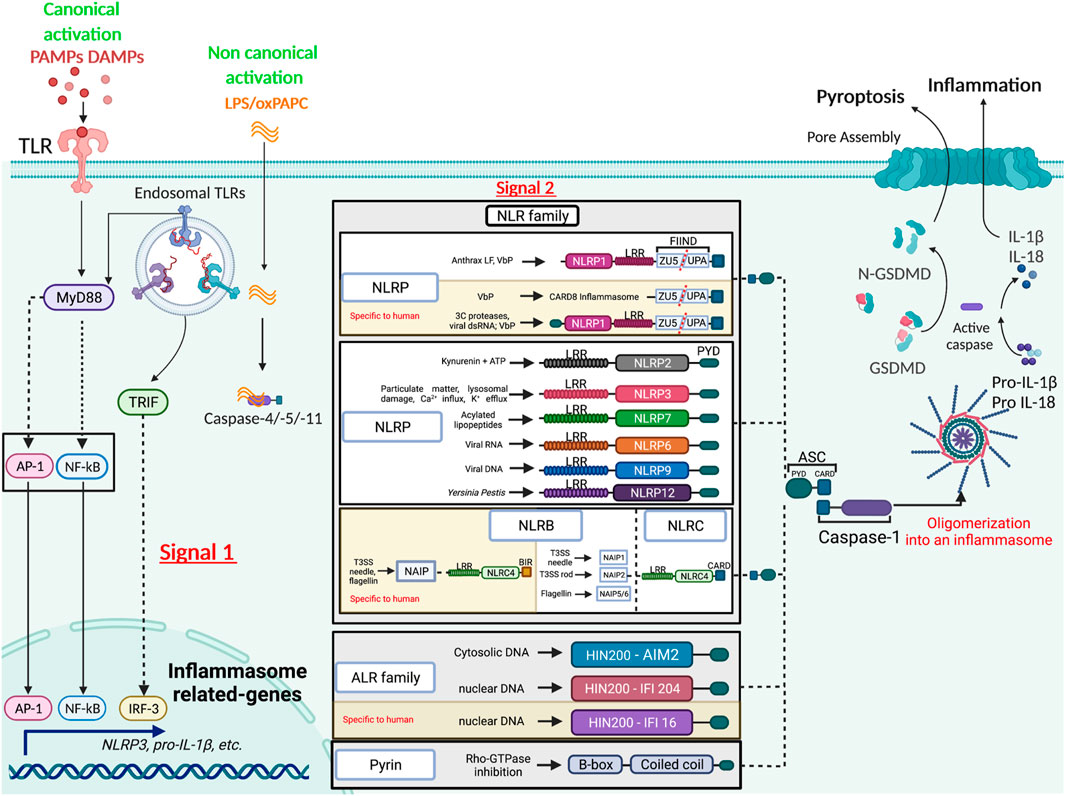
FIGURE 1. Diverse mechanisms of inflammasome activation. The activation of the inflammasome by the canonical pathway relies on two signals, the first or the priming signal allowing transcription of inflammasome components, and the second or activating signal triggering the assembly of the complex. The inflammasome scaffolding receptors belong to different families (NLR, ALR and PYRIN), each recognizing different MAMPs or DAMPs. Caspase-1 is central in catalyzing inflammasome functions, cleaving pro-inflammatory cytokines into their bioactive form and gasdermin D (GSDMD) into an N-terminal pore forming domain that elicits pyroptosis. The non-canonical route is mediated by direct binding of LPS or phospholipids to caspase-4/-5 in humans or -11 in mice.
Inflammasome assembly leads to the multi-oligomerization of the inflammasome sensor, the ASC adaptor and the proform of caspase-1 and/or −11/−4/−5 into a perinuclear aggregate known as the ASC “speck” with prionoid features (Franklin et al., 2014). Caspase activation promotes the release of bioactive IL-1β and IL-18, and in some instances, an inflammatory cell death termed pyroptosis via the cleavage of gasdermin-D into an N-terminal domain that generates pores in the plasma membrane leading to osmotic lysis [reviewed in (Broz et al., 2020)].
The inflammasomes are best known for their function in host defence against pathogens. However, aberrant inflammasome activation has also been linked to cancer development. The role of inflammasome signaling in cancer varies according to the type of cancer, cancer etiology and cells activating the inflammasome pathway within the tumor microenvironment. In this review, we provide a synthesis of the recent literature exploring the role of different inflammasomes in cancer progression or anti-tumor immunity. We discuss evidence generated from in vivo mouse models, either using pharmacological inhibitors or loss-of-function genetic models, as well as evidence from patients’ cohorts and clinical trials.
The role of the inflammasome in cancer promotion is often mediated by IL-1β, which is produced by various cells in the tumor, including tumor-associated macrophages (TAMs) and cancer-associated fibroblasts (CAFs). Indeed, myeloid- or CAF-specific deletion of genes involved in Nlrp3 inflammasome activation leads to decreased tumor growth. For example, the sphingolipid sphingosine-1-phosphate (S1P) receptor was shown to activate the Nlrp3 inflammasome in TAMs, and myeloid-specific deletion of S1pr reduced IL-1β-driven lymphangiogenesis and pulmonary metastasis in the polyoma middle T (PyMT) mouse model of breast cancer (Weichand et al., 2017). Using the same model, Ershaid et al. showed that CAF-specific genetic ablation of Nlrp3 or Il1b delayed tumor growth and attenuated lung metastasis (Ershaid et al., 2019). Mechanistically, IL-1β promoted tumor progression and metastasis by driving an immunosuppressive tumor microenvironment (TME) and inducing the expression of adhesion molecules on endothelial cells (Ershaid et al., 2019). Furthermore, Nlrp3 inflammasome-mediated IL-1β was shown to induce the production of the cancer-promoting cytokine IL-22 by memory CD4+ T cells. This was demonstrated in models of breast and lung cancers in which neutralization of IL-1R signaling with anakinra abrogated IL-22 production and reduced tumor growth (Voigt et al., 2017). IL-1β produced by TAMs or released from cancer cells can also boost the pro-tumorigenic activity of CAFs, highlighting its role as a master orchestrator of tumorigenesis (Brunetto et al., 2019). Besides its effects on the TME, IL-1β autocrine signaling was shown to act as a direct driver of cancer cell proliferation. This was demonstrated using the diethylnitrosamine (DEN) mouse model of hepatocellular carcinoma (HCC), in which the mitophagy effector FUNDC1 was specifically deleted in hepatocytes leading to aberrant activation of the Nlrp3 inflammasome and IL-1β-driven hepatocyte hyperproliferation (Wenhui Li et al., 2019). This was also reported in a mouse model of AML driven by the oncogenic KrasG12D pathway which activated the Nlrp3 inflammasome via Kras-RAC1-induced ROS production. Nlrp3 deletion reduced abnormal myeloproliferation and restored normal hematopoiesis (Hamarsheh et al., 2020). In addition to inducing proliferation and survival of cancer cells, IL-1β can also favor their migration and invasion. In a colorectal cancer (CRC) model, macrophage-derived IL-1β was shown to elicit the production of serum amyloid A1 (SAA1) by tumor cells, which in a feed-forward manner induced macrophages to upregulate metalloproteinase-9 secretion allowing cancer migration and establishment of metastasis (Sudo et al., 2021) (Figure 2A). Whereas the bulk of the evidence point to IL-1β as the main driver of inflammasome tumor-promoting effects, a recent study by Hofbauer et al., studying multiple myeloma in a murine model, described a role of Nlrp3-dependent IL-18 in bone destruction and multiple myeloma proliferation (Hofbauer et al., 2021).
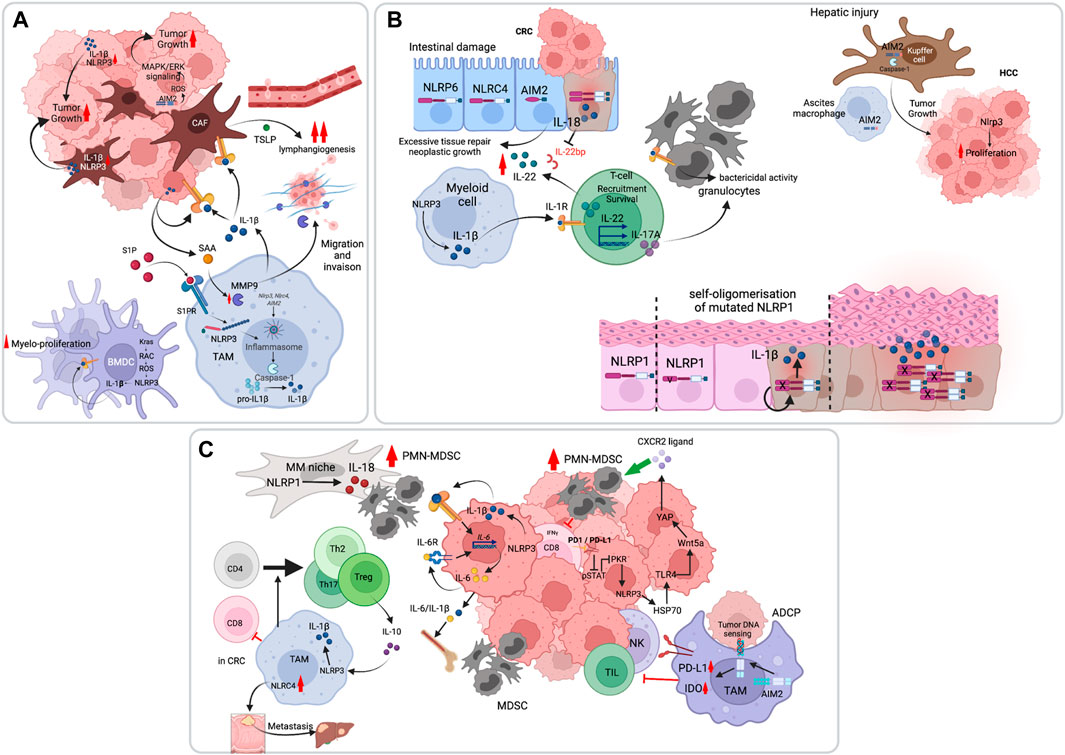
FIGURE 2. Pro-tumoral functions of the inflammasome pathway. (A) The inflammasome, primarily NLRP3, promotes tumor cell survival, proliferation and invasion through IL-1β. Inflammasome-independent functions of AIM2 have also been demonstrated, in which AIM2 controls mitochondrial dynamics and activate oncogenic ERK activation through reactive oxygen species (ROS). (B) The inflammasome can promote tumorigenesis through induction of a chronic inflammatory states. In the intestine, both IL-18 and IL-1β converge on IL-22 production which drives intestinal epithelial cell hyperproliferation. The chronic inflammatory state is also enacted by IL-17 production which recruits granulocytes to amplify the response. In the liver, hepatic injury is associated with AIM2 inflammasome activation both in Kupffer cells and in macrophages of the ascitic fluid. IL-1β in this case promotes hepatocytes hyper-proliferation. In the skin, germ-line mutations in NLRP1 result in chronic inflammation and tumorigenesis. (C) The inflammasomes can indirectly favor tumor development by suppressing anti-tumor immunity. IL-1β was shown to enhance myeloid-derived suppressor cells (MDSC) recruitment to the tumor site and to promote T cell tolerogenic differentiation.
Last, inflammasome-independent functions in tumor promotion have been reported. Qi et al. showed that while polydA:dT stimulation of three NSCLC cell-lines activated the AIM2 inflammasome, it did not have an impact on their growth or survival. In contrast, AIM2 promoted NSCLC tumor growth in vivo by regulating mitochondrial fusion dynamics and ERK activation (Qi et al., 2020).
In response to tissue damage, select inflammatory cytokines promote the repair process. When this response is aberrant, as in the context of chronic inflammation, excessive tissue repair can lead to neoplastic growth. This is best illustrated in colitis-associated CRC and HCC. We and others have shown early on that inflammasome-dependent IL-18 production is necessary for intestinal tissue repair in mouse models of colitis and that excessive inflammasome signaling promoted intestinal tumorigenesis [reviewed in (Saleh and Trinchieri, 2011)]. Huber et al. later showed that IL-18 promoted the biological activity of IL-22, an important reparative cytokine, by inhibiting the production of IL-22-binding protein (IL-22BP), a soluble form of the IL-22 receptor and a negative regulator of IL-22 (Figure 2B). Knock-out of Il22bp which resulted in increased IL-22/IL-22BP ratio accelerated tumorigenesis in the dextran sulfate sodium (DSS)-azoxymethane (AOM) colitis-associated CRC mouse model as well as in Apcmin/+ mice that develop intestinal adenomas (Huber et al., 2012). Besides IL-18, excessive IL-1β production in the gut drives IL-17 production by CD4+ Th17 cells and innate lymphocytes (ILC) 3, that recruit granulocytes and trigger intestinal inflammation (Coccia et al., 2012). More recently, Dmitrieva-Posocco O et al. demonstrated cell-type specific function of IL-1 signaling in intestinal tumorigenesis. Whereas T cell-specific deletion of Il1ra reduced IL-17A/IL-22-dependent tumorigenesis, Il1ra ablation in neutrophils impaired their function in bacterial control, leading to microbial tumor invasion, which elicited exacerbated inflammation and CRC (Dmitrieva-Posocco et al., 2019). As in CRC, chronic inflammation and cirrhosis induced by liver injury predispose to HCC. The AIM2 inflammasome was reported to be activated in patients with advanced cirrhosis, particularly in macrophages of the ascites (Lozano-Ruiz et al., 2015) (Figure 2B). This inflammasome was later shown to drive HCC in the DEN mouse model, where Aim2−/− or Casp1−/− mice had reduced carcinogenic liver injury and cancer growth (Martínez-Cardona et al., 2018). In the human skin, NLRP1 is the predominant inflammasome sensor. In 2016, Franklin et al. reported gain-of-function germline mutations in NLRP1 that lead to a familial skin inflammatory disease and associated carcinoma. The authors showed that inflammasome activation in keratinocytes leads to autocrine IL-1β that drives epidermal hyperplasia (Zhong et al., 2016).
Tumors use multiple means to evade anti-tumor immunity. For instance, they can usurp inflammatory pathways to establish an immunosuppressive environment. The inflammasome pathway is one such pathway demonstrated in some cancers to favor immunosuppression by promoting the genesis and recruitment of myeloid-derived suppressor cells (MDSC) or by inducing the differentiation of TAMs into a tolerogenic phenotype (Figure 2C). In 2010, using the poorly immunogenic B16-F10 melanoma model, van Deventer et al. showed that the Nlrp3 inflammasome suppressed the mouse response to dendritic cell tumor vaccines through MDSC recruitment (van Deventer et al., 2010). In a pancreatic ductal adenocarcinoma (PDAC) model, Daley et al. further implicated Nlrp3 signaling in TAM-mediated immunosuppression. They showed that deletion or inhibition of Nlrp3 inflammasome components was protective against PDAC. IL-1β mediated the tolerogenic function of Nlrp3 by regulating IL-10 production and T cell differentiation into Treg, Th2, and Th17 cells (Daley et al., 2017). Besides TAMs, PDAC tumor cells also activate the Nlrp3 inflammasome and act as a prominent source of IL-1β (Das et al., 2020). Tumor-derived IL-1β might also act via an IL-6-STAT3 inflammatory loop to expand MDSCs and induce immunosuppression (Tengesdal et al., 2021a). Tengesdal et al. have demonstrated this using the B16-F10 melanoma model and further showed that Nlrp3 inhibition with dapansutrile (OLT1177) abrogated the expression of immunosuppressives genes in PMN-MDSC and enhanced anti-tumor immunity in vivo (Tengesdal et al., 2021b). While NLRP3 appears to have a predominant role in dampening anti-tumor immunity, as illustrated above, other inflammasomes have also been implicated in this process. For instance, the NLRP1-IL-18 pathway was shown to promote the generation of mature MDSCs in multiple myeloma (Nakamura et al., 2018), and the Nlrc4 inflammasome in controlling CRC metastasis to the liver in non-alcoholic fatty liver disease (NAFLD) (Ohashi et al., 2019). Furthermore, the AIM2 inflammasome elicits immunosuppression following antibody-dependent cell phagocytosis (ADCP) and tumor cell DNA sensing, by upregulating PD-L1 and IDO expression, which inhibit anti-tumor immunity by NK and T cells (Su et al., 2018).
Notably, inflammasome-dependent cytokines such as IL-1β, can be produced in the TME independently of the inflammasomes. A recent work using mouse models of non-small cell lung cancer (NSCLC) and triple-negative breast cancer (TNBC) showed that IL-1β secretion by myeloid cells was independent of inflammasome activation and gasdermin D pore formation. IL-1β promoted immunosuppression by recruiting neutrophils to the tumor bed. The authors showed that tumor infiltration of neutrophils was abrogated in Il1b−/− mice, which resulted in loss of immunosuppression and restoration of anti-tumor immunity during treatment with antiangiogenic agents targeting VEGF. Although caspase-8 was activated in tumor-infiltrating myeloid cells, its deletion was not sufficient to completely block bioactive IL-1β release and neutrophil infiltration (Kiss et al., 2021). It is also worth noting that cytokine-independent functions of NLRP3 or AIM2 in immunosuppression have been documented. Petrilli and colleagues reported an immunosuppressive role of the Nlrp3-Asc-Caspase-1 pathway, that blunted NK cell tumor recruitment and activation, but that was independent of IL-1β, IL-18 or IL-1 receptor signaling. This was shown using orthotopic implantation of the breast cancer cell-line 4T1 in BALB/c mice or with MMTV-NeuV664E on the BALB/c background crossed to C57Bl/6 inflammasome components-deficient mice (Guey et al., 2020). In addition, Theivanthiran et al. have shown that anti-PD1 immunotherapy leads to the upregulation of PD-L1 on tumor cell surface, which converges on NLRP3 activation and downstream release of heat shock protein (HSP) 70. In an autocrine and paracrine manner, HSP70 sensing by TLR4 and the subsequent expression and secretion of Wnt5a promoted the production of CXCL5 that attracts PMN-MDSCs to the tumor (Theivanthiran et al., 2021).
Inflammation is a protective biological response induced physiologically to counter pathogenic infection or tissue damage. Early studies by us and others have shown that the inflammasome pathway has a protective role in DSS-induced injury by promoting intestinal tissue repair via IL-18-mediated IEC regeneration (Dupaul-Chicoine et al., 2010). Concordantly, loss of Nlrp3, Asc or caspase-1 led to severe colitis but also to colitis-associated CRC (CAC) (Allen et al., 2010). Similarly, a role of Nlrp6 (Elinav et al., 2011), Nlrc4 (Rauch et al., 2017), Aim2 (Man et al., 2015) and pyrin (Sharma et al., 2018) inflammasomes in maintaining intestinal homeostasis and countering CAC was reported (Figure 3A). Flavell, Elinav and colleagues showed that this was mediated by alterations in the intestinal microbial ecology caused by lack of IL-18-instructed production of antimicrobial peptides (AMP) (Elinav et al., 2011; Levy et al., 2015). Such microbiota dysbiosis promoted neutrophil infiltration, chronic inflammation and IL-6 driven intestinal epithelial cell (IEC) hyper-proliferation (Hu et al., 2013). Besides controlling tissue homeostasis by triggering compensatory proliferation, inflammasome signalling counters tumorigenesis by upregulating differentiation programs. In the MMTV-PyMT mouse model, Castaño Z et al. have demonstrated an anti-metastatic role of IL-1β. They showed that myeloid-derived IL-1β imposed a differentiation “block” on metastasis-initiating cells (MICs) by upregulating ZEB1-dependent differentiation that countered their proliferative capacity (Castaño et al., 2018) (Figure 3A).

FIGURE 3. Anti-tumoral functions of the inflammasome pathway. (A) The inflammasome can suppress tumor cell proliferation and promotes tissue homeostasis and cell differentiation. (B) Inflammasome activation favors anti-tumor immunity by enhancing NK and CD8 T cell tumorilytic activities. This is primarily mediated by IL-18. Inflammasome triggers enhance the efficacy of immune checkpoint inhibitors. (C) Pyroptotic cell death downstream of inflammasome activation establishes an immunogenic environment through the release of several DAMPs and immunostimulatory molecules. Pyroptosis is initiated by caspase-dependent processing of gasdermins that form pores leading to osmotic lysis of the cell. Certain chemotherapies induce toxicities because of pyroptotic death of healthy cells through gasdermins E cleavage by caspase-3.
While in most instances, the underlying inflammasome mechanisms involve IL-18 or IL-1β, inflammasome-independent roles have also been described. For instance, Aim2 has been shown to restrict CAC and sporadic colon cancer through suppression of Akt signalling (Wilson et al., 2015) (Figure 3A). Inflammasome-independent functions were also reported for Nlrc4 in a subcutaneous B16-F10 melanoma mouse model. Nlrc4 expression in TAMs promoted a protective anti-tumoral response, contrary to caspase-1 or Asc, suggesting that Nlrc4 acted through an alternative mechanism (Janowski et al., 2016).
Tumor development depends on the balance between the capacity of the tumor to escape immunosurveillance and antitumor immune responses. The inflammasome-dependent cytokines IL-18 and IL-1β play a central role in anti-tumor immunity. In 2009, Ghiringhelli et al. have first reported that ATP release from tumor cells is sensed by the P2X7 receptor on dendritic cells (DC) which activate the Nlrp3 inflammasome, triggering the release of IL-1β. Deficiency in caspase-1 or IL-1r impaired the priming of INFγ-producing CD8+ T cells (Ghiringhelli et al., 2009) (Figure 3B). Consistently, IL-1β conditioning allows a better response of adoptively transferred anti-tumor T-cells. Mechanistically, IL-1β promotes T-cell homing to the tumor site and improves T cell survival and activation (Lee et al., 2019). As for IL-1β, IL-18 is an essential inducer of anti-tumor immunity. On one hand, it enhances NK cell maturation and FasL-mediated lytic activity, as demonstrated in a model of CRC metastasis to liver (Dupaul-Chicoine et al., 2015) (Figure 3B). On the other, IL-18 increases the numbers of tumor-infiltrating lymphocytes (TIL), as shown in subcutaneous model of melanoma (Zhou et al., 2020). However, IL-18 has previously failed to demonstrate efficacy in cancer clinical trials. A potential cause could be the expression of IL-18BP, a negative regulator that quenches circulating IL-18. Indeed, an IL-18 variant resistant to IL-18BP was shown to elicit increased TIL frequency and function, and to expand intra-tumoral stem-like TCF1+ CD8+ T cells in mouse tumors (Zhou et al., 2020). Akin to its role in natural anti-tumor immunity, the inflammasome also mediates synthetic anti-tumor immunity driven by certain immune checkpoint inhibitors and small molecule inhibitors of ectonucleotidases. For instance, CD39 inhibition which results in extracellular ATP accumulation enhanced anti-PD-1 anti-tumoral immune responses by engaging the P2x7-Nlrp3-IL-18 pathway (Xian-Yang Li et al., 2019; Yan et al., 2020) (Figure 3B). Similarly, unleashing Nlrp3 activity through the inhibition of Transmembrane protein 176B (TMEM176B), an immunoregulatory cation channel, enhanced TILs antitumor activity in response to immune checkpoint inhibitors (Segovia et al., 2019). More recently, Kuchroo and colleagues demonstrated that inhibition of the immune checkpoint TIM-3 on migratory DCs, resulted in strong anti-tumor immunity, mediated by ROS-driven Nlrp3 inflammasome activation (Dixon et al., 2021) (Figure 3B).
Pyroptosis is a lytic and immunogenic regulated cell death that is induced by caspase activation. The main mechanism of pyroptosis involves GSDMD processing into a pore forming domain. The GSDM family is well conserved in vertebrates and is composed of six paralogs in humans, GSDMA, GSDMB, GSDMC, GSDMD, GSDME (DFNA5) and GSDMF (DFNB59). Mice lack a GSDMB homolog, and have three GSDMDA homologs, four GSDMC homologs, GSDMD, GSDME, and GSDMF [reviewed in (Zou et al., 2021)]. Pyroptosis of tumor cells release a spectrum of molecules allowing an efficient anti-tumoral immune response and tumor regression. This was elegantly demonstrated in a biorthogonal system in mice, which revealed that pyroptosis of less than 15% of tumor cells was sufficient to clear the entire tumor graft. Such an anti-tumoral response is mediated by a competent immune system as immunodeficient mice or T-cell deficient mice were unable to induce anti-tumor immunity (Wang et al., 2020). In addition to GSDMD, GSDME can trigger pyroptosis. Its cleavage by caspase-3 downstream of TNF or chemotherapy drugs was shown to trigger pyroptosis (Wang et al., 2017). Tumor cells, but not normal cells, tend to downregulate GSDME, which partly explains the off-target toxicity of some cancer therapies (Figure 3C). Consistently, Gsdme−/− mice were protected from chemotherapy-induced tissue damage (Wang et al., 2017). In contrast to GSDME, GSDMC is upregulated in tumor cells. Through its interaction with Stat3, PD-L1 controls GSDMC transcription. TNF released by TAMs activates caspase-8, which processes GSDMC leading to pyroptosis (Hou et al., 2020).
Multiple approaches can be used to target inflammasome activity, including the inhibition of upstream signaling pathways, inhibition of inflammasome components, or cytokine neutralization (Supplementary Table S1). Cleavage of inflammasome-dependent cytokines by caspase-1 renders it an attractive therapeutic target. Thalidomide, which targets caspase-1 is an efficient anti-inflammatory and anti-angiogenic drug. Its therapeutic use has been approved for the treatment of inflammatory skin diseases and certain types of cancer. In multiple myeloma, testing of Thalidomide in combination with other therapies have reached phase III or IV clinical trials. Other caspase-1 inhibitors used in inflammatory diseases, such as Pralnacasan or VX-765 (Belnacasan) are not yet in use in cancer clinical trials. Inhibition of specific Inflammasome receptors such as NLRP3 is also an interesting approach. Cytokine release inhibitory drug (CRID)3 also known as MCC950 and Dapansutrile (OLT1177) are NLRP3 inhibitors that have shown promising results in mice (Hamarsheh and Zeiser, 2020; Tengesdal et al., 2021b; Oizumi et al., 2021), and are expected to move into clinical trials. Nevertheless, in clinical studies, cytokine blockade is the most successful approach, and IL-1β appears to be one of the most promising targets. Based on the CANTOS trial evaluating the effect of canakinumab on preventing adverse cardiac events, Novartis is leading several clinical trials targeting IL-1β in cancer, including three in phase III in NSLC. CANOPY-A (NCT03447769) is a clinical trial combining canakinumab with cisplatin in NSLC patients following surgical resection to prevent relapse. CANOPY-1 (NCT03631199) assessed canakinumab as a first-line treatment for advanced or metastatic NSCLC in combination with pembrolizumab and platinum-based doublet chemotherapy. However, it did not reach the primary endpoints of overall survival (OS) and progression-free survival (PFS). CANOPY-2 (NCT03626545) investigated the combination of canakinumab with Docetaxel in second- or third-line therapy versus Docetaxel alone in NSCLC. However, it also did not reach its first endpoint. MABp1, a Janssen monoclonal antibody targeting IL-1α, has shown clinical benefit in a phase III clinical trial (NCT02138422), increasing the median survival of patients with CRC refractory to standard therapy. A phase II clinical trial from the Mayo Clinic (NCT00635154) investigated the effect of anakinra in patients with early-stage multiple myeloma. The results indicate that IL-1Ra suppresses markers of disease progression.
Another approach under investigation is that of stimulating anti-tumor immunity by activating the NLRP3 inflammasome. A phase III clinical trial (NCT03329846) is evaluating BMS-986299, a drug developed to activate NLRP3 inflammasome, in combination with Nivolumab in patients with advanced melanoma. The use of recombinant cytokines is also a way to promote anti-tumor activity, and SB-485232, a recombinant human IL-18 has been used in seven clinicals trials, albeit without showing efficacy.
The inflammasomes play double-edged roles in tumorigenesis and anti-tumor immunity. On one hand, they promote cancer growth and metastasis by instructing an immunosuppressive TME, mainly through IL-1β, and by stimulating the proliferation of tumor cells in autocrine and paracrine amplification loops. In addition, they elicit compensatory proliferation mechanisms in response to sustained tissue damage favoring tumorigenesis. On the other hand, inflammasome activation enhances the tumorilytic activity of CD8+ T cells and NK cells in an IL-18-dependent manner, and further promotes anti-tumor immunity through pyroptosis, an immunogenic cell death that activates antigen-presenting cells through the release of tumor antigens and adjuvants in the TME. In view of the different inflammasome components and their downstream inflammatory cytokines, several attractive inflammasome-based therapeutic targets are currently being explored. Nevertheless, much work is still needed prior to generalizing such potential immunotherapies, as the bulk of the pre-clinical work cited here requires validation in other cancer models, translational studies and early clinical trials. In addition, taken the pleiotropic roles of the inflammasomes and their cytokines, and as with other immunotherapies, particular attention is needed to potential inflammatory toxicities or deleterious immunosuppressive effects with these approaches.
SL and MS conceived the structure of this review. SL wrote the manuscript and MS edited the text and figures. All authors revised the manuscript and approved the final version.
MS is funded by grants from the ARC foundation Leader of Oncology Program, IDEX Bor-deaux, SIRIC BRIO and The New Aquitaine Region.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2022.839041/full#supplementary-material
Allen, I. C., TeKippe, E. M., Woodford, R. M., Uronis, J. M., Holl, E. K., Rogers, A. B., et al. (2010). The NLRP3 Inflammasome Functions as a Negative Regulator of Tumorigenesis during Colitis-Associated Cancer. J. Exp. Med. 207, 1045–1056. doi:10.1084/jem.20100050
Ariffin, J. K., and Sweet, M. J. (2013). Differences in the Repertoire, Regulation and Function of Toll-like Receptors and Inflammasome-Forming Nod-like Receptors between Human and Mouse. Curr. Opin. Microbiol. 16, 303–310. doi:10.1016/j.mib.2013.03.002
Bauernfried, S., Scherr, M. J., Pichlmair, A., Duderstadt, K. E., and Hornung, V. (2021). Human NLRP1 Is a Sensor for Double-Stranded RNA. Science 371, eabd0811. doi:10.1126/science.abd0811
Broz, P., Pelegrín, P., and Shao, F. (2020). The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat. Rev. Immunol. 20, 143–157. doi:10.1038/s41577-019-0228-2
Brunetto, E., De Monte, L., Balzano, G., Camisa, B., Laino, V., Riba, M., et al. (2019). The IL-1/IL-1 Receptor axis and Tumor Cell Released Inflammasome Adaptor ASC Are Key Regulators of TSLP Secretion by Cancer Associated Fibroblasts in Pancreatic Cancer. J. Immunotherapy Cancer 7, 45. doi:10.1186/s40425-019-0521-4
Bürckstümmer, T., Baumann, C., Blüml, S., Dixit, E., Dürnberger, G., Jahn, H., et al. (2009). An Orthogonal Proteomic-Genomic Screen Identifies AIM2 as a Cytoplasmic DNA Sensor for the Inflammasome. Nat. Immunol. 10, 266–272. doi:10.1038/ni.1702
Castaño, Z., San Juan, B. P., Spiegel, A., Pant, A., DeCristo, M. J., Laszewski, T., et al. (2018). IL-1β Inflammatory Response Driven by Primary Breast Cancer Prevents Metastasis-Initiating Cell Colonization. Nat. Cel Biol 20, 1084–1097. doi:10.1038/s41556-018-0173-5
Chu, L. H., Indramohan, M., Ratsimandresy, R. A., Gangopadhyay, A., Morris, E. P., Monack, D. M., et al. (2018). The Oxidized Phospholipid oxPAPC Protects from Septic Shock by Targeting the Non-canonical Inflammasome in Macrophages. Nat. Commun. 9, 996. doi:10.1038/s41467-018-03409-3
Coccia, M., Harrison, O. J., Schiering, C., Asquith, M. J., Becher, B., Powrie, F., et al. (2012). IL-1β Mediates Chronic Intestinal Inflammation by Promoting the Accumulation of IL-17A Secreting Innate Lymphoid Cells and CD4+ Th17 Cells. J. Exp. Med. 209, 1595–1609. doi:10.1084/jem.20111453
Daley, D., Mani, V. R., Mohan, N., Akkad, N., Pandian, G. S. D. B., Savadkar, S., et al. (2017). NLRP3 Signaling Drives Macrophage-Induced Adaptive Immune Suppression in Pancreatic Carcinoma. J. Exp. Med. 214, 1711–1724. doi:10.1084/jem.20161707
Das, S., Shapiro, B., Vucic, E. A., Vogt, S., and Bar-Sagi, D. (2020). Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 80, 1088–1101. doi:10.1158/0008-5472.CAN-19-2080
Dixon, K. O., Tabaka, M., Schramm, M. A., Xiao, S., Tang, R., Dionne, D., et al. (2021). TIM-3 Restrains Anti-tumour Immunity by Regulating Inflammasome Activation. Nature 595, 101–106. doi:10.1038/s41586-021-03626-9
Dmitrieva-Posocco, O., Dzutsev, A., Posocco, D. F., Hou, V., Yuan, W., Thovarai, V., et al. (2019). Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 50, 166–180. e167. doi:10.1016/j.immuni.2018.11.015
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., and Schreiber, R. D. (2002). Cancer Immunoediting: from Immunosurveillance to Tumor Escape. Nat. Immunol. 3, 991–998. doi:10.1038/ni1102-991
Dupaul-Chicoine, J., Arabzadeh, A., Dagenais, M., Douglas, T., Champagne, C., Morizot, A., et al. (2015). The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 43, 751–763. doi:10.1016/j.immuni.2015.08.013
Dupaul-Chicoine, J., Yeretssian, G., Doiron, K., Bergstrom, K. S., McIntire, C. R., LeBlanc, P. M., et al. (2010). Control of Intestinal Homeostasis, Colitis, and Colitis-Associated Colorectal Cancer by the Inflammatory Caspases. Immunity 32, 367–378. doi:10.1016/j.immuni.2010.02.012
Elinav, E., Strowig, T., Kau, A. L., Henao-Mejia, J., Thaiss, C. A., Booth, C. J., et al. (2011). NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell 145, 745–757. doi:10.1016/j.cell.2011.04.022
Ershaid, N., Sharon, Y., Doron, H., Raz, Y., Shani, O., Cohen, N., et al. (2019). NLRP3 Inflammasome in Fibroblasts Links Tissue Damage with Inflammation in Breast Cancer Progression and Metastasis. Nat. Commun. 10, 4375. doi:10.1038/s41467-019-12370-8
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J., and Alnemri, E. S. (2009). AIM2 Activates the Inflammasome and Cell Death in Response to Cytoplasmic DNA. Nature 458, 509–513. doi:10.1038/nature07710
Fink, S. L., Bergsbaken, T., and Cookson, B. T. (2008). Anthrax Lethal Toxin and Salmonella Elicit the Common Cell Death Pathway of Caspase-1-dependent Pyroptosis via Distinct Mechanisms. Proc. Natl. Acad. Sci. 105, 4312–4317. doi:10.1073/pnas.0707370105
Franklin, B. S., Bossaller, L., De Nardo, D., Ratter, J. M., Stutz, A., Engels, G., et al. (2014). The Adaptor ASC Has Extracellular and 'prionoid' Activities that Propagate Inflammation. Nat. Immunol. 15, 727–737. doi:10.1038/ni.2913
Ghiringhelli, F., Apetoh, L., Tesniere, A., Aymeric, L., Ma, Y., Ortiz, C., et al. (2009). Activation of the NLRP3 Inflammasome in Dendritic Cells Induces IL-1β-dependent Adaptive Immunity against Tumors. Nat. Med. 15, 1170–1178. doi:10.1038/nm.2028
Guey, B., Bodnar-Wachtel, M., Drouillard, A., Eberhardt, A., Pratviel, M., Goutagny, N., et al. (2020). Inflammasome Deletion Promotes Anti-tumor NK Cell Function in an IL-1/IL-18 Independent Way in Murine Invasive Breast Cancer. Front. Oncol. 10, 1683. doi:10.3389/fonc.2020.01683
Hamarsheh, S. a., Osswald, L., Saller, B. S., Unger, S., De Feo, D., Vinnakota, J. M., et al. (2020). Oncogenic KrasG12D Causes Myeloproliferation via NLRP3 Inflammasome Activation. Nat. Commun. 11, 1659. doi:10.1038/s41467-020-15497-1
Hamarsheh, S. a., and Zeiser, R. (2020). NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front. Immunol. 11, 1444. doi:10.3389/fimmu.2020.01444
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of Cancer: the Next Generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Hofbauer, D., Mougiakakos, D., Broggini, L., Zaiss, M., Büttner-Herold, M., Bach, C., et al. (2021). β2-microglobulin Triggers NLRP3 Inflammasome Activation in Tumor-Associated Macrophages to Promote Multiple Myeloma Progression. Immunity 54, 1772–1787. doi:10.1016/j.immuni.2021.07.002
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., et al. (2009). AIM2 Recognizes Cytosolic dsDNA and Forms a Caspase-1-Activating Inflammasome with ASC. Nature 458, 514–518. doi:10.1038/nature07725
Hou, J., Zhao, R., Xia, W., Chang, C. W., You, Y., Hsu, J. M., et al. (2020). PD-L1-mediated Gasdermin C Expression Switches Apoptosis to Pyroptosis in Cancer Cells and Facilitates Tumour Necrosis. Nat. Cel Biol 22, 1264–1275. doi:10.1038/s41556-020-0575-z
Hu, B., Elinav, E., Huber, S., Strowig, T., Hao, L., Hafemann, A., et al. (2013). Microbiota-induced Activation of Epithelial IL-6 Signaling Links Inflammasome-Driven Inflammation with Transmissible Cancer. Proc. Natl. Acad. Sci. 110, 9862–9867. doi:10.1073/pnas.1307575110
Huber, S., Gagliani, N., Zenewicz, L. A., Huber, F. J., Bosurgi, L., Hu, B., et al. (2012). IL-22BP Is Regulated by the Inflammasome and Modulates Tumorigenesis in the Intestine. Nature 491, 259–263. doi:10.1038/nature11535
Janowski, A. M., Colegio, O. R., Hornick, E. E., McNiff, J. M., Martin, M. D., Badovinac, V. P., et al. (2016). NLRC4 Suppresses Melanoma Tumor Progression Independently of Inflammasome Activation. J. Clin. Invest. 126, 3917–3928. doi:10.1172/JCI86953
Kiss, M., Vande Walle, L., Saavedra, P. H. V., Lebegge, E., Van Damme, H., Murgaski, A., et al. (2021). IL1β Promotes Immune Suppression in the Tumor Microenvironment Independent of the Inflammasome and Gasdermin D. Cancer Immunol. Res. 9, 309–323. doi:10.1158/2326-6066.CIR-20-0431
Klimpel, K. R., Arora, N., and Leppla, S. H. (1994). Anthrax Toxin Lethal Factor Contains a Zinc Metalloprotease Consensus Sequence Which Is Required for Lethal Toxin Activity. Mol. Microbiol. 13, 1093–1100. doi:10.1111/j.1365-2958.1994.tb00500.x
Lee, P.-H., Yamamoto, T. N., Gurusamy, D., Sukumar, M., Yu, Z., Hu-Li, J., et al. (2019). Host Conditioning with IL-1β Improves the Antitumor Function of Adoptively Transferred T Cells. J. Exp. Med. 216, 2619–2634. doi:10.1084/jem.20181218
Levy, M., Thaiss, C. A., Zeevi, D., Dohnalová, L., Zilberman-Schapira, G., Mahdi, J. A., et al. (2015). Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 163, 1428–1443. doi:10.1016/j.cell.2015.10.048
Li, W., Li, Y., Siraj, S., Jin, H., Fan, Y., Yang, X., et al. (2019). FUN14 Domain‐Containing 1-Mediated Mitophagy Suppresses Hepatocarcinogenesis by Inhibition of Inflammasome Activation in Mice. Hepatology 69, 604–621. doi:10.1002/hep.30191
Li, X.-Y., Moesta, A. K., Xiao, C., Nakamura, K., Casey, M., Zhang, H., et al. (2019). Targeting CD39 in Cancer Reveals an Extracellular ATP- and Inflammasome-Driven Tumor Immunity. Cancer Discov. 9, 1754–1773. doi:10.1158/2159-8290.CD-19-0541
Liao, K.-C., and Mogridge, J. (2013). Activation of the Nlrp1b Inflammasome by Reduction of Cytosolic ATP. Infect. Immun. 81, 570–579. doi:10.1128/IAI.01003-12
Lightfield, K. L., Persson, J., Brubaker, S. W., Witte, C. E., von Moltke, J., Dunipace, E. A., et al. (2008). Critical Function for Naip5 in Inflammasome Activation by a Conserved Carboxy-Terminal Domain of Flagellin. Nat. Immunol. 9, 1171–1178. doi:10.1038/ni.1646
Lozano-Ruiz, B., Bachiller, V., García-Martínez, I., Zapater, P., Gómez-Hurtado, I., Moratalla, A., et al. (2015). Absent in Melanoma 2 Triggers a Heightened Inflammasome Response in Ascitic Fluid Macrophages of Patients with Cirrhosis. J. Hepatol. 62, 64–71. doi:10.1016/j.jhep.2014.08.027
Man, S. M., Zhu, Q., Zhu, L., Liu, Z., Karki, R., Malik, A., et al. (2015). Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 162, 45–58. doi:10.1016/j.cell.2015.06.001
Martínez-Cardona, C., Lozano-Ruiz, B., Bachiller, V., Peiró, G., Algaba-Chueca, F., Gómez-Hurtado, I., et al. (2018). AIM2 Deficiency Reduces the Development of Hepatocellular Carcinoma in Mice. Int. J. Cancer 143, 2997–3007. doi:10.1002/ijc.31827
Martinon, F., Burns, K., and Tschopp, J. (2002). The Inflammasome. Mol. Cel. 10, 417–426. doi:10.1016/S1097-2765(02)00599-3
Matzinger, P. (2002). The Danger Model: A Renewed Sense of Self. Science 296, 301–305. doi:10.1126/science.1071059
Mullins, B., and Chen, J. (2021). NLRP9 in Innate Immunity and Inflammation. Immunology 162, 262–267. doi:10.1111/imm.13290
Nakamura, K., Kassem, S., Cleynen, A., Chrétien, M.-L., Guillerey, C., Putz, E. M., et al. (2018). Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 33, 634–648. doi:10.1016/j.ccell.2018.02.007
Neiman-Zenevich, J., Stuart, S., Abdel-Nour, M., Girardin, S. E., and Mogridge, J. (2017). Listeria Monocytogenes and Shigella Flexneri Activate the NLRP1B Inflammasome. Infect. Immun. 85, e00338–17. doi:10.1128/IAI.00338-17
Ohashi, K., Wang, Z., Yang, Y. M., Billet, S., Tu, W., Pimienta, M., et al. (2019). NOD‐like Receptor C4 Inflammasome Regulates the Growth of Colon Cancer Liver Metastasis in NAFLD. Hepatology 70, 1582–1599. doi:10.1002/hep.30693
Oizumi, T., Mayanagi, T., Toya, Y., Sugai, T., Matsumoto, T., and Sobue, K. (2021). NLRP3 Inflammasome Inhibitor OLT1177 Suppresses Onset of Inflammation in Mice with Dextran Sulfate Sodium-Induced Colitis. Dig. Dis. Sci. doi:10.1007/s10620-021-07184-y
Qi, M., Dai, D., Liu, J., Li, Z., Liang, P., Wang, Y., et al. (2020). AIM2 Promotes the Development of Non-small Cell Lung Cancer by Modulating Mitochondrial Dynamics. Oncogene 39, 2707–2723. doi:10.1038/s41388-020-1176-9
Radian, A. D., Khare, S., Chu, L. H., Dorfleutner, A., and Stehlik, C. (2015). ATP Binding by NLRP7 Is Required for Inflammasome Activation in Response to Bacterial Lipopeptides. Mol. Immunol. 67, 294–302. doi:10.1016/j.molimm.2015.06.013
Rauch, I., Deets, K. A., Ji, D. X., von Moltke, J., Tenthorey, J. L., Lee, A. Y., et al. (2017). NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity 46, 649–659. doi:10.1016/j.immuni.2017.03.016
Rayamajhi, M., Zak, D. E., Chavarria-Smith, J., Vance, R. E., and Miao, E. A. (2013). Cutting Edge: Mouse NAIP1 Detects the Type III Secretion System Needle Protein. J. Immunol. 191, 3986–3989. doi:10.4049/jimmunol.1301549
Reyes Ruiz, V. M., Ramirez, J., Naseer, N., Palacio, N. M., Siddarthan, I. J., Yan, B. M., et al. (2017). Broad Detection of Bacterial Type III Secretion System and Flagellin Proteins by the Human NAIP/NLRC4 Inflammasome. Proc. Natl. Acad. Sci. USA 114, 13242–13247. doi:10.1073/pnas.1710433114
Saleh, M., and Trinchieri, G. (2011). Innate Immune Mechanisms of Colitis and Colitis-Associated Colorectal Cancer. Nat. Rev. Immunol. 11, 9–20. doi:10.1038/nri2891
Sandstrom, A., Mitchell, P. S., Goers, L., Mu, E. W., Lesser, C. F., and Vance, R. E. (2019). Functional Degradation: A Mechanism of NLRP1 Inflammasome Activation by Diverse Pathogen Enzymes. Science 364. doi:10.1126/science.aau1330
Segovia, M., Russo, S., Jeldres, M., Mahmoud, Y. D., Perez, V., Duhalde, M., et al. (2019). Targeting TMEM176B Enhances Antitumor Immunity and Augments the Efficacy of Immune Checkpoint Blockers by Unleashing Inflammasome Activation. Cancer Cell 35, 767–781. e766. doi:10.1016/j.ccell.2019.04.003
Sharma, D., Malik, A., Guy, C. S., Karki, R., Vogel, P., and Kanneganti, T.-D. (2018). Pyrin Inflammasome Regulates Tight Junction Integrity to Restrict Colitis and Tumorigenesis. Gastroenterology 154, 948–964. doi:10.1053/j.gastro.2017.11.276
Shen, C., Li, R., Negro, R., Cheng, J., Vora, S. M., Fu, T.-M., et al. (2021). Phase Separation Drives RNA Virus-Induced Activation of the NLRP6 Inflammasome. Cell 184, 5759–5774. e5720. doi:10.1016/j.cell.2021.09.032
Shi, J., Zhao, Y., Wang, Y., Gao, W., Ding, J., Li, P., et al. (2014). Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 514, 187–192. doi:10.1038/nature13683
Su, S., Zhao, J., Xing, Y., Zhang, X., Liu, J., Ouyang, Q., et al. (2018). Immune Checkpoint Inhibition Overcomes ADCP-Induced Immunosuppression by Macrophages. Cell 175, 442–457. doi:10.1016/j.cell.2018.09.007
Sudo, G., Aoki, H., Yamamoto, E., Takasawa, A., Niinuma, T., Yoshido, A., et al. (2021). Activated Macrophages Promote Invasion by Early Colorectal Cancer via an Interleukin 1β‐serum Amyloid A1 axis. Cancer Sci. 112, 4151–4165. doi:10.1111/cas.15080
Swanson, K. V., Deng, M., and Ting, J. P.-Y. (2019). The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 19, 477–489. doi:10.1038/s41577-019-0165-0
Tengesdal, I. W., Dinarello, A., Powers, N. E., Burchill, M. A., Joosten, L. A. B., Marchetti, C., et al. (2021a). Tumor NLRP3-Derived IL-1β Drives the IL-6/STAT3 Axis Resulting in Sustained MDSC-Mediated Immunosuppression. Front. Immunol. 12, 661323. doi:10.3389/fimmu.2021.661323
Tengesdal, I. W., Menon, D. R., Osborne, D. G., Neff, C. P., Powers, N. E., Gamboni, F., et al. (2021b). Targeting Tumor-Derived NLRP3 Reduces Melanoma Progression by Limiting MDSCs Expansion. Proc. Natl. Acad. Sci. USA 118, e2000915118. doi:10.1073/pnas.2000915118
Tenthorey, J. L., Haloupek, N., López-Blanco, J. R., Grob, P., Adamson, E., Hartenian, E., et al. (2017). The Structural Basis of Flagellin Detection by NAIP5: A Strategy to Limit Pathogen Immune Evasion. Science 358, 888–893. doi:10.1126/science.aao1140
Theivanthiran, B., Haykal, T., Cao, L., Holtzhausen, A., Plebanek, M., DeVito, N. C., et al. (2021). Overcoming Immunotherapy Resistance by Targeting the Tumor-Intrinsic NLRP3-HSP70 Signaling Axis. Cancers 13, 4753. doi:10.3390/cancers13194753
Tuladhar, S., and Kanneganti, T.-D. (2020). NLRP12 in Innate Immunity and Inflammation. Mol. Aspects Med. 76, 100887. doi:10.1016/j.mam.2020.100887
van Deventer, H. W., Burgents, J. E., Wu, Q. P., Woodford, R.-M. T., Brickey, W. J., Allen, I. C., et al. (2010). The Inflammasome Component NLRP3 Impairs Antitumor Vaccine by Enhancing the Accumulation of Tumor-Associated Myeloid-Derived Suppressor Cells. Cancer Res. 70, 10161–10169. doi:10.1158/0008-5472.CAN-10-1921
Voigt, C., May, P., Gottschlich, A., Markota, A., Wenk, D., Gerlach, I., et al. (2017). Cancer Cells Induce Interleukin-22 Production from Memory CD4+ T Cells via Interleukin-1 to Promote Tumor Growth. Proc. Natl. Acad. Sci. USA 114, 12994–12999. doi:10.1073/pnas.1705165114
Wang, Q., Wang, Y., Ding, J., Wang, C., Zhou, X., Gao, W., et al. (2020). A Bioorthogonal System Reveals Antitumour Immune Function of Pyroptosis. Nature 579, 421–426. doi:10.1038/s41586-020-2079-1
Wang, Y., Gao, W., Shi, X., Ding, J., Liu, W., He, H., et al. (2017). Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 547, 99–103. doi:10.1038/nature22393
Weichand, B., Popp, R., Dziumbla, S., Mora, J., Strack, E., Elwakeel, E., et al. (2017). S1PR1 on Tumor-Associated Macrophages Promotes Lymphangiogenesis and Metastasis via NLRP3/IL-1β. J. Exp. Med. 214, 2695–2713. doi:10.1084/jem.20160392
Wilson, J. E., Petrucelli, A. S., Chen, L., Koblansky, A. A., Truax, A. D., Oyama, Y., et al. (2015). Inflammasome-independent Role of AIM2 in Suppressing colon Tumorigenesis via DNA-PK and Akt. Nat. Med. 21, 906–913. doi:10.1038/nm.3908
Xu, H., Yang, J., Gao, W., Li, L., Li, P., Zhang, L., et al. (2014). Innate Immune Sensing of Bacterial Modifications of Rho GTPases by the Pyrin Inflammasome. Nature 513, 237–241. doi:10.1038/nature13449
Yan, J., Li, X.-Y., Roman Aguilera, A., Xiao, C., Jacoberger-Foissac, C., Nowlan, B., et al. (2020). Control of Metastases via Myeloid CD39 and NK Cell Effector Function. Cancer Immunol. Res. 8, 356–367. doi:10.1158/2326-6066.CIR-19-0749
Yang, J., Zhao, Y., Shi, J., and Shao, F. (2013). Human NAIP and Mouse NAIP1 Recognize Bacterial Type III Secretion Needle Protein for Inflammasome Activation. Proc. Natl. Acad. Sci. 110, 14408–14413. doi:10.1073/pnas.1306376110
Zanoni, I., Tan, Y., Di Gioia, M., Broggi, A., Ruan, J., Shi, J., et al. (2016). An Endogenous Caspase-11 Ligand Elicits Interleukin-1 Release from Living Dendritic Cells. Science 352, 1232–1236. doi:10.1126/science.aaf3036
Zhang, Q., Sun, Y., He, Z., Xu, Y., Li, X., Ding, J., et al. (2020). Kynurenine Regulates NLRP2 Inflammasome in Astrocytes and its Implications in Depression. Brain Behav. Immun. 88, 471–481. doi:10.1016/j.bbi.2020.04.016
Zhao, Y., Yang, J., Shi, J., Gong, Y.-N., Lu, Q., Xu, H., et al. (2011). The NLRC4 Inflammasome Receptors for Bacterial Flagellin and Type III Secretion Apparatus. Nature 477, 596–600. doi:10.1038/nature10510
Zhou, T., Damsky, W., Weizman, O.-E., McGeary, M. K., Hartmann, K. P., Rosen, C. E., et al. (2020). IL-18BP Is a Secreted Immune Checkpoint and Barrier to IL-18 Immunotherapy. Nature 583, 609–614. doi:10.1038/s41586-020-2422-6
Keywords: Inflammasome, caspase, pyroptosis, tumor, immunotherapy, macrophages, immunosuppres-sion, metastasis
Citation: Lillo S and Saleh M (2022) Inflammasomes in Cancer Progression and Anti-Tumor Immunity. Front. Cell Dev. Biol. 10:839041. doi: 10.3389/fcell.2022.839041
Received: 19 December 2021; Accepted: 28 January 2022;
Published: 20 April 2022.
Edited by:
Deepika Sharma, University of Chicago, United StatesReviewed by:
Paras K. Anand, Imperial College London, United KingdomCopyright © 2022 Lillo and Saleh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maya Saleh, bWF5YS5zYWxlaEB1LWJvcmRlYXV4LmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.