
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 24 February 2022
Sec. Cell Death and Survival
Volume 10 - 2022 | https://doi.org/10.3389/fcell.2022.838272
This article is part of the Research Topic Molecular and Cellular Biology of Podocytes View all 13 articles
 Fiammetta Ravaglia1*
Fiammetta Ravaglia1* Maria Elena Melica2
Maria Elena Melica2 Maria Lucia Angelotti2
Maria Lucia Angelotti2 Letizia De Chiara2
Letizia De Chiara2 Paola Romagnani2,3
Paola Romagnani2,3 Laura Lasagni2*
Laura Lasagni2*Podocytopathies are a group of proteinuric glomerular disorders driven by primary podocyte injury that are associated with a set of lesion patterns observed on kidney biopsy, i.e., minimal changes, focal segmental glomerulosclerosis, diffuse mesangial sclerosis and collapsing glomerulopathy. These unspecific lesion patterns have long been considered as independent disease entities. By contrast, recent evidence from genetics and experimental studies demonstrated that they represent signs of repeated injury and repair attempts. These ongoing processes depend on the type, length, and severity of podocyte injury, as well as on the ability of parietal epithelial cells to drive repair. In this review, we discuss the main pathology patterns of podocytopathies with a focus on the cellular and molecular response of podocytes and parietal epithelial cells.
Podocytes are differentiated epithelial cells whose number is determined shortly before birth as nephrogenesis ceases (Bertram et al., 2011). Podocytes have a limited capacity to complete successful cytokinesis as cell division requires a rearrangement of the podocyte actin cytoskeleton, disrupting the podocyte foot processes (Lasagni et al., 2013). Consequently, direct or indirect podocyte injury, which causes cytoskeleton rearrangement, poses a serious threat to kidney barrier function maintenance, which is reflected in glomerular proteinuria levels.
Podocyte injury can be caused by immunologic, infectious or toxic agents, as well as patient specific characteristics such as obesity or haemodynamic modifications (Kopp et al., 2020). In addition, genetic analysis techniques have broadened the known causes of podocytopathies adding genetic variants to the list. Currently, more than fifty podocyte-expressed genes have been identified as directly linked to podocytopathies as well as syndromal non-podocyte-specific genes, phenocopies with other underlying genetic abnormalities and susceptibility genes (i.e., apolipoprotein L1 (APOL1) variants) leading to a complete revision of carrier risk stratification (Freedman et al., 2014; Warejko et al., 2018; Landini et al., 2020). Podocyte injury can be observed in the setting of all forms of immune complex glomerulonephritis (i.e., lupus nephritis, membranous nephropathy) and in metabolic disorders (diabetes, amyloidosis) and is a key event in chronic kidney disease progression (Kopp et al., 2020). The latter will not be the topic of this review. We will rather focus on disorders that have podocyte as a primary target of injury and that are associated with a variety of lesion patterns that renal pathology struggles to classify (Ahn and Bomback, 2020). These histopathological lesion patterns range from 1) minimal changes (MC), traditionally referred to as “minimal change disease” (MCD) and defined as minimal alterations visible only by ultrastructural analysis; 2) focal segmental glomerulosclerosis (FSGS) where sclerotic lesions are restricted to a subset of the glomeruli; 3) diffuse mesangial sclerosis (DMS) characterized by mesangial matrix increase and podocyte hypertrophy; and lastly 4) collapsing glomerulopathy (CG) which presents as collapse of the glomerular capillaries and hyperplasia of parietal epithelial cells (PECs) migrating to the tuft forming “pseudocrescents” (Barisoni et al., 2007). These glomerular histopathological lesion patterns can be collectively viewed as podocytopathies and their progression to chronic kidney disease is related to the amount of podocyte loss (Kopp et al., 2020). In addition to podocytes and PECs, glomerular endothelial cells and mesangial cells likely contribute to the progression of podocytopathies (Chung et al., 2020). However, as a comprehensive analysis of all the cell types involved in the disease progression is beyond the scope of this review, we will focus our discussion on the PEC-podocyte axis.
Several lines of evidence support the podocyte depletion hypothesis (Kim et al., 2001; Wharram et al., 2005; Fukuda et al., 2012a). In particular, the Wiggins group (Wharram et al., 2005) elegantly showed the consequences of podocyte loss: less than 20% of podocyte loss is associated with a normal glomerulus at light microscopy or with mesangial expansion, whereas loss of more than 20% of podocytes leads to segmental denudation of glomerular basement membrane with consequent adhesions between the Bowman capsule and the glomerular capillary loop. Once the process is initiated sclerosis follows. Segmental sclerosis occurs if podocyte loss is less than 40%, while global sclerosis occurs if podocyte loss exceeds 60% (Wharram et al., 2005).
A subpopulation of PECs endowed with progenitor characteristics can replace, at least to some extent, lost podocytes (Peired et al., 2013; Zhang et al., 2013; Eng et al., 2015; Lasagni et al., 2015; Romoli et al., 2018; Kaverina et al., 2019). However, PECs may also have a detrimental role, as proliferation of activated PECs can also be a crucial determinant of glomerulosclerosis (Lasagni et al., 2015; Eymael et al., 2018).
Collectively, the aim of this review is to: 1) discuss the current concept of histopathological pattern recognition; 2) introduce the importance of PECs in the formation and identification of these peculiar lesions; 3) consider the relationship between PECs and podocytes as an important determinant of disease progression.
By definition, glomerular appearance observed by light microscopy in minimal changes is normal, while tubular compartment showed vacuolar, lipoid changes in the proximal tubules defined as lipoid nephrosis (Munk, 1918).
Ultrastructural examination is required to identify the only consistent glomerular pathologic feature of minimal changes, which is simplification of podocyte shape at ultrastructural level without glomerular abnormalities at light microscopy. By electron microscopy, this feature is visualized as effacement of the discrete foot processes and it may be associated to coarsening, i.e., retraction, shortening and widening of foot processes (De Vriese et al., 2018). Foot process simplification and effacement are the earliest morphological patterns of podocyte injury and are clinically associated with highly selective nephrotic-range proteinuria (Vivarelli et al., 2017). Extension of foot process effacement and coarsening is an issue. It has been hypothesized that the surface area extension of capillary loops involved in this process could differentiate the MC lesion pattern from FSGS (De Vriese et al., 2018). Specifically, MC lesion would show widespread foot process effacement and coarsening, involving at least more than half of the capillary loop surface area, while FSGS would show more segmental alterations (Deegens et al., 2008; da Silva et al., 2020) as in maladaptive FSGS increased fluid shear stress is typically a segmental phenomenon (Kriz and Lemley, 2017a). Nevertheless, such an assessment is prone to inaccuracy considering that the ultrastructural analysis is bidimensional and it is limited in the number of glomeruli analyzed (i.e., no more than one or two glomeruli). In addition, no correlation between foot process effacement measured by electron microscopy and proteinuria severity exists, suggesting that this evaluation alone is not sufficient to allow clinical-pathological correlations (Royal et al., 2020).
Foot process effacement is also associated with microfilament aggregation at the base of epithelial cells and filtration slit distortion, resulting in a reduction in the number of slit diaphragms (Patrakka et al., 2002). This, in turn, causes a displacement of the diaphragms in a ladder-like formation toward the pore apex (Patrakka et al., 2002). In addition, podocytes may show other non-specific structural alterations including hypertrophy, microvillous transformation, formation of vacuoles and the presence of resorption droplets (Royal et al., 2020). However, these alterations do not prevent podocytes from fully covering the glomerular basement membrane. Accordingly, in MC lesions, areas of glomerular basement membrane denudation (Lahdenkari et al., 2004) due to reduction in podocyte density or to net podocyte loss compared to healthy controls do not occur (Szeto et al., 2015) (Figure 1).
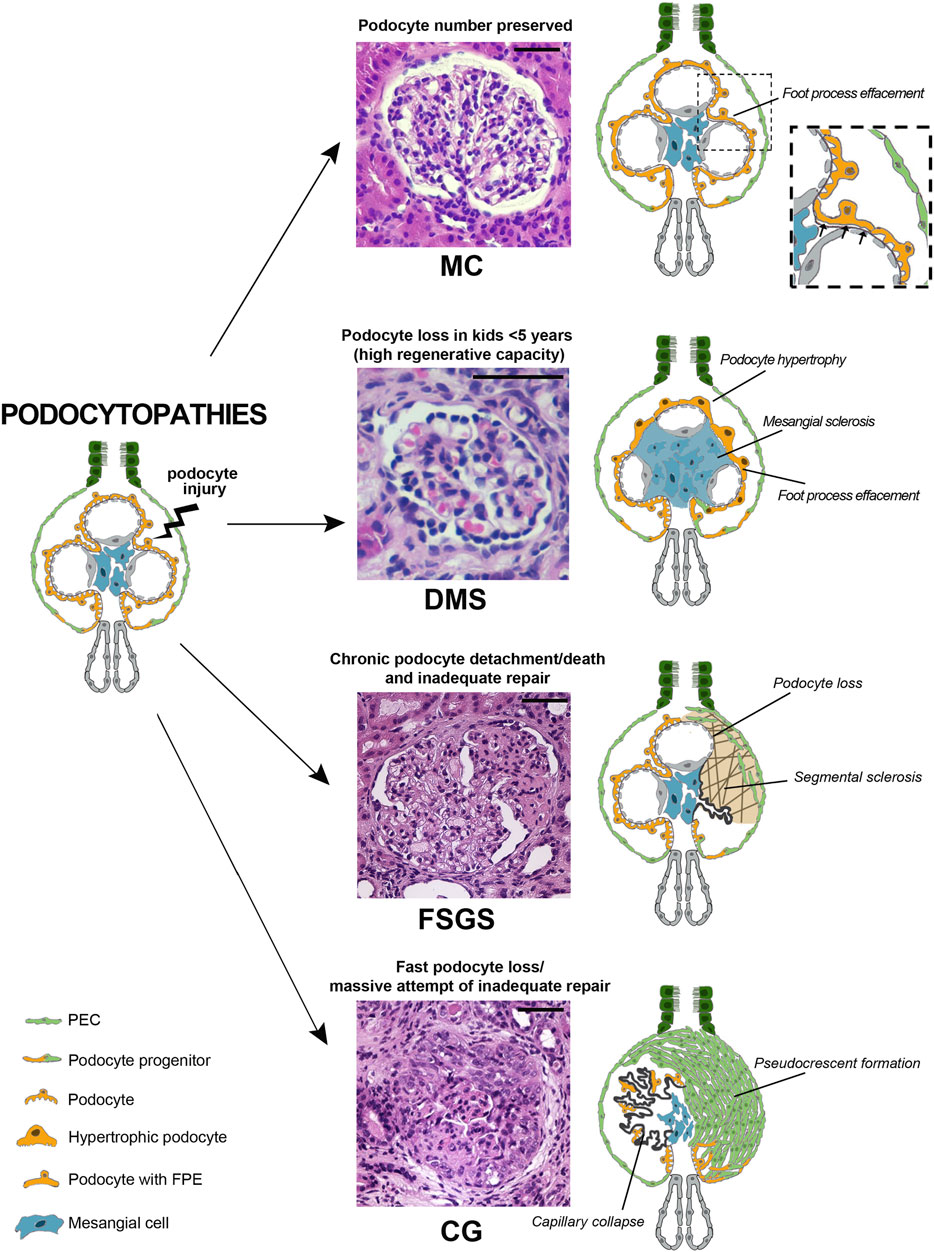
FIGURE 1. Podocytopathies result from the equilibrium between the nature of injury and the glomerular capacity of repair. When podocyte injury does not determine net cell loss, no changes are present on light microscopy and only foot process effacement is detectable, as in minimal changes (MC). In a setting of fast podocyte loss in kids younger than 5 years old, massive attempt of repair takes place. Immature podocytes are generated and are visible as a halo of hypertrophic podocytes overlying capillary loops as in diffuse mesangial sclerosis (DMS). When severity or chronicity of podocyte injury overcomes the capacity of PECs to replace detached or loss podocytes, glomerular basement membrane denudation triggers an injury cascade. This results in the segmental solidification of the tuft with accumulation of extracellular matrix characteristic of focal segmental glomerulosclerosis (FSGS). Finally, if fast podocyte loss occurs in individuals where the regenerative capacity is inadequate, generation of new podocytes is hampered and proliferating progenitors accumulate in Bowman space in the form of pseudocrescents resulting in collapsing glomerulopathy (CG) (Hematoxylin and eosin stains, magnifications 40x. Bars= 50 μm). (Abbreviations: MC= minimal changes, DMS= diffuse mesangial sclerosis, FSGS= focal segmental glomerulosclerosis, CG= collapsing glomerulopathy, PEC= parietal epithelial cell, FPE= foot process effacement).
Finally, immunofluorescence for immunoglobulins and complement fractions on kidney biopsy is generally negative (Vivarelli et al., 2017). However, recent findings of nephrin autoantibodies in a subset of patients with MC lesions at light microscopy and podocyte-associated punctate IgG at immunofluorescence provide support for an autoimmune etiology (Watts et al., 2021). Collectively, the lack of pathological features detectable by light microscopy and immunohistochemistry in biopsies of patients with MC lesions complicates the diagnostic process. Ongoing studies are attempting to identify new biomarkers to predict outcomes or individualize treatments. Nevertheless, podocyte injury at kidney biopsy may be difficult to identify and characterize, mostly due to the focal nature of the damage, inadequate sampling and specific shortcomings linked to traditional techniques, as previously mentioned. Recently, the combination of optical clearing techniques with state-of-the-art microscopy permitted morphometric analysis in thick tissues with a resolution up to nanoscale levels (Angelotti et al., 2021). This technique permits imaging of large tissue areas and to resolve fine structural details at once. Therefore, we can visualize the slit diaphragm 3D, giving direct evidence of structural changes or podocyte loss (Figure 2) (Artelt et al., 2018; Tesch et al., 2021).
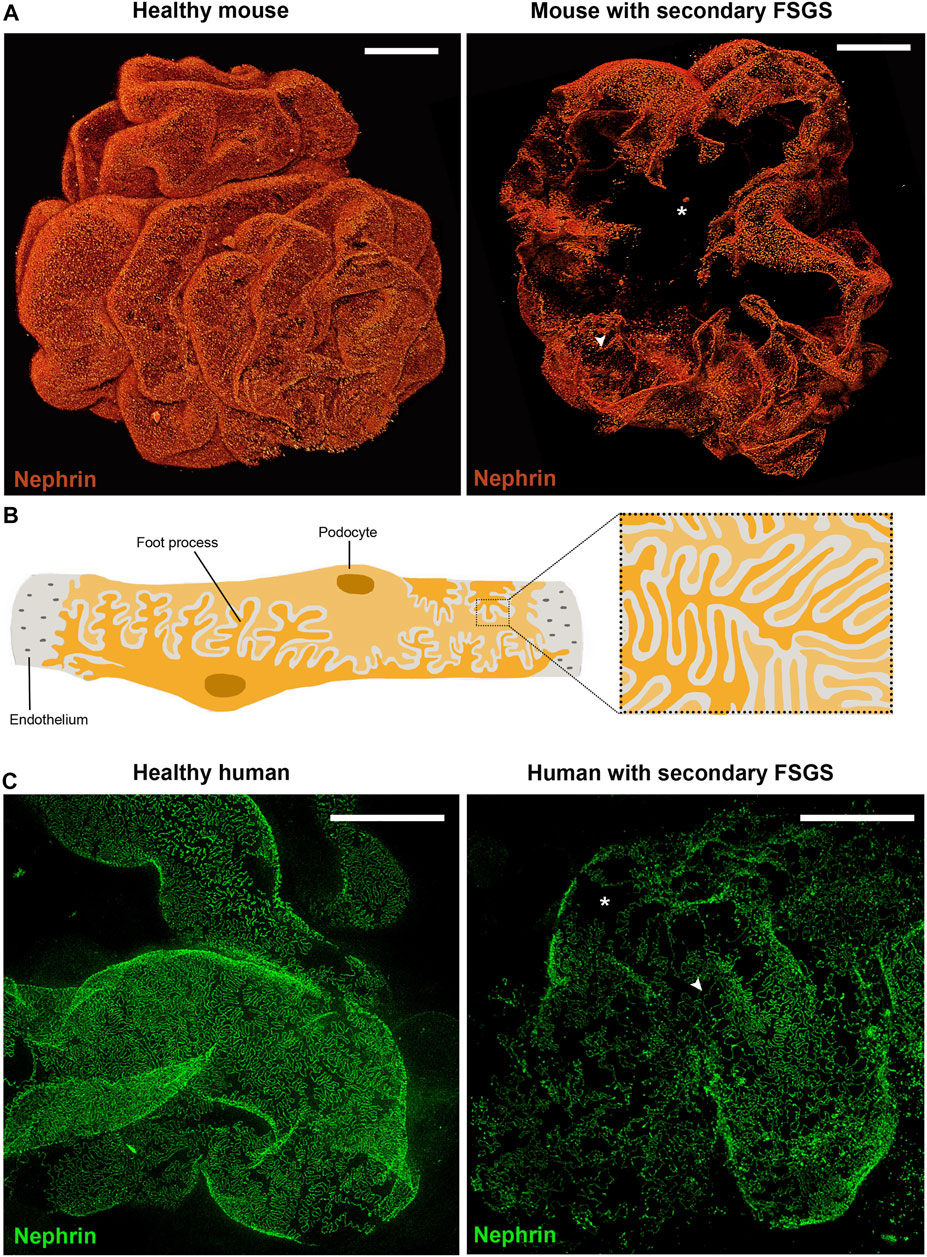
FIGURE 2. Podocytes are crucial for integrity of the filtration barrier. (A) Three-dimensional reconstruction of whole mouse glomeruli stained for nephrin upon optical tissue clearing by using confocal microscopy. The signal represents nephrin protein within the slit diaphragm. Z-series stacks were obtained from 80-μm kidney slices with images collected at 1 μ m intervals. On the left a representative glomerulus from a healthy mouse shows an intact filtration barrier. On the right a representative glomerulus from a mouse with secondary FSGS (obesity- related diabetic mouse, db/db mouse) shows large denudated areas with podocyte loss (asterisk) and foot process effacement (arrowhead). (B) Schematic drawing of representative podocytes, with their interdigitating foot processes, wrapped around a glomerular capillary loop. (C) Representative images of human podocyte foot processes by using STED-super resolution microscopy upon tissue clearing. Z-series stacks were obtained from 5-μm kidney slices. The green signal represents nephrin protein. On the left a representative image of a normal human kidney obtained from a patient who underwent nephrectomy for localized renal tumor. On the right a representative image of a kidney biopsy obtained from a patient with secondary FSGS, showing denudated areas with podocyte loss (asterisk) and foot process effacement (arrowhead) (Bars= 20 μm).
Podocyte foot process effacement is the ultrastructural hallmark of MC lesion, however, the process leading to podocyte effacement is not clear (Purohit et al., 2021). Immunologic dysregulations and modifications of the podocyte are thought to synergize in altering the integrity of the glomerular barrier and therefore determining proteinuria (Purohit et al., 2021). Animal models could potentially provide valuable insights into physiopathology of the MC lesion, but we lack an animal model fitting the specific MC characteristics, i.e., abrupt onset selective nephrotic-range proteinuria, diffuse foot process effacement but no podocytopenia, low rates of disease progression, and steroid sensitivity (Chugh et al., 2012). In fact, all mouse models of chronic and heavy proteinuria eventually develop FSGS following an initial phase with only diffuse podocyte foot process effacement, offering the opportunity to elucidate mechanisms of progression rather than that of acute renal damage. The models most broadly employed by researchers are the Puromycin Aminonucleoside (PAN) model, in which single low-dose injection of toxic agent directed against podocyte molecules induces transient proteinuria and foot process effacement, without inducing podocyte loss (Kim et al., 2001; Pippin et al., 2009) and the diphtheria toxin (DT) model, in which DT injection into rats expressing human diphtheria toxin receptor transgene results in dose-dependent podocyte depletion (Wharram et al., 2005). In particular, in the DT model, less than 20% of podocyte depletion results in mesangial expansion, transient proteinuria, and normal kidney function. The continuum between the MC lesion and FSGS in these models supports the hypothesis that in some patients these two lesion patterns represent different pathology manifestations of the same injury (Maas et al., 2016).
Long before animal models, the possible link between MC and FSGS lesions had already been suggested in the first clinical report of nephrotic syndrome in the 1970s (Habib and Kleinknecht, 1971). Afterwards, identification of FSGS in patients that previously exhibited MC biopsy pattern was reported in children who underwent repeated kidney biopsies during the course of steroid sensitive nephrotic syndrome (Tejani, 1985) and in serial post-transplantation biopsies of patients with FSGS recurrence (Charnaya and Moudgil, 2017), supporting evidence of an evolving process (Maas et al., 2016). During the progression from MC to FSGS lesion, the glomeruli significantly increase their volumes and podocyte hypertrophy appears as a distinguishing feature of FSGS vs. MC (Fogo et al., 1990). This would suggest that interventions aimed at regulating glomerular volume and podocyte hypertrophy could represent an effective strategy to sustain podocyte survival and to prevent podocytopenia (Puelles et al., 2019; Banu et al., 2021). However, this adaptive response of podocytes is only sustainable until a threshold is reached, after which the podocyte detaches and progression to FSGS occurs. Podocyte detachment is thought to occur through a substantial increase in the mechanical forces of fluid filtration (Saga et al., 2021). Evaluation of mechanical properties of podocytes and of their response to external mechanical stimuli has shown how circumferential wall tension as well as fluid shear stress play an important role in podocytopathies and progression from MC to FSGS lesion (Kriz and Lemley, 2017b; Embry et al., 2018; Calizo et al., 2019). It is also possible that in MC podocyte loss occurs but it is not detectable either because it does not differ from normal physiological turnover of healthy glomeruli or because the progenitors succeed in replacing lost podocytes. Indeed, many studies reported the PEC capacity to differentiate into mature podocytes after glomerular injury, replacing at least in part the lost podocytes (Peired et al., 2013; Zhang et al., 2013; Eng et al., 2015; Lasagni et al., 2015; Romoli et al., 2018; Kaverina et al., 2019).
If the cause of podocyte injury is reversible, we observe a return to normal of foot processes together with a complete proteinuria remission and a favorable prognosis. In case of irreversible podocyte injury, podocyte loss can still be partially compensated by progenitor replacement (Nagata, 2016; Kopp et al., 2020). When podocyte loss reaches its tipping point (more than 20% of podocytes are lost), PECs are unable to fully compensate for podocyte loss. In such cases, the MC lesion represents the first stage of a condition that will progress towards more severe patterns of injury characterized by more prominent podocyte loss (i.e., FSGS) (Figure 1 and Table 1).

TABLE 1. Animal models and clinical evidence supporting the proposed pathomechanisms for development of pathology lesion patterns associated with podocytopathies.
DMS is found in children younger than 5 years old with nephrotic syndrome progressing to end stage kidney disease during childhood (Wiggins, 2007). DMS is defined by the presence of diffuse mesangial sclerosis in kidney biopsy, with the deeper glomeruli being the least affected. The term DMS relies on the late appearance of the lesion, but does not give any clue on its pathogenesis (Barisoni et al., 2007).
Glomeruli in DMS appear as a halo-shaped distribution of podocytes surrounding a matrix-containing glomerular center (Figure 1). Initially, the matrix has a fibrillary appearance, but at later stages, the mesangial extracellular matrix accumulation becomes more prominent as capillary lumens obliterate with progressive tuft contraction. In parallel, podocytes show absence of foot processes and glomerular capillary loops tend to collapse with progressive podocyte hypertrophy and mild hyperplasia, remaining visible even in advanced disease stages (Barisoni et al., 2007). The tubules are dilated and atrophic, sometimes containing hyaline casts (Charles Jennette et al., 2015). Non-diagnostic deposits of IgM and C3 are seen in the mesangium of the less affected glomeruli and in the periphery of the sclerotic glomeruli (Charles Jennette et al., 2015). Electron microscopy shows mesangial collagen fibrils. The glomerular basement membrane is split and wavy because of zones of subepithelial lucent widening and segmental denudation due to foot process detachment (Charles Jennette et al., 2015).
Genetic mutations causing DMS have given an insight in podocyte biology. For instance, truncating mutations in phospholipase C epsilon (PLCε1) expressed by podocytes in the developing glomerulus; mutations in WT1, a major podocyte transcription factor present early in podocyte development, or mutations in b2laminin (LAMB2) gene involved in glomerular basement membrane expansion and assembly, are associated with the DMS pattern of injury in humans (Boyer et al., 2010; Lehnhardt et al., 2015; Matejas et al., 2010) and in mouse models (Atchison et al., 2020; Ratelade et al., 2010; Lin et al., 2018). These mutations result in arrested development of glomeruli (Hinkes et al., 2006; Yang et al., 1999) that are not able to prevent protein leakage while filtering, causing massive proteinuria responsible for fast podocyte loss and rapid progression to end stage kidney disease. In particular, glomeruli in kidneys of patients with DMS linked to WT1 mutations show a relatively less-differentiated podocyte phenotype and immunostaining reveals increased expression of the PEC progenitor marker Pax2 (Yang et al., 1999), suggesting migration and attempt of PECs to differentiate into podocytes. Interestingly, gene expression analysis of isolated glomeruli of DMS mouse model obtained introducing a heterozygous WT1 mutation, identified increased expression of Cyp26a1 in podocytes of mutated mice (Ratelade et al., 2010). This resulted in a decrease of the concentration of all-trans-retinoic acid—an inducer of PEC progenitor differentiation into podocytes (Peired et al., 2013; Zhang et al., 2012; Dai et al., 2017)—in the glomerular milieu, supporting the hypothesis that a hampered differentiation of PEC progenitors into podocytes could be involved in the development of DMS. In adult animals, mesangial expansion is the first indirect evidence of podocyte loss up to 20% of the podocyte compartment (Wharram et al., 2005). Indeed, in the attempt to maintain the capillary loop open despite the podocyte loss, the mesangium assumes a more prominent fibrillary appearance and expands (Wharram et al., 2005). Reduced number of podocytes, associated with excretion of urinary podocytes, was observed also in glomeruli with severe sclerosis in children with DMS (Ikezumi et al., 2014). Interestingly, some glomeruli showed hypercellular lesions with proliferating cells that stained positive for the PEC marker claudin-1 (Ikezumi et al., 2014), suggesting that podocyte loss and the consequent proliferation of PECs are common processes in the pathogenesis of DMS. Several studies have shown that around 10% of podocytes are formed after birth from PEC progenitors localized at the urinary pole of the Bowman capsule, that progressively differentiate into podocytes, migrating from the urinary pole towards the vascular pole of the glomerulus (Lasagni et al., 2015; Appel et al., 2009; Wanner et al., 2014) (Figure 3). PEC differentiation into podocytes happens substantially to support the increase of kidney dimension as the kidney grows, during childhood and adolescence in mouse models (Lasagni et al., 2015; Appel et al., 2009; Wanner et al., 2014) (Figure 3). Similarly in humans, the finding that podocytes increase in numbers during the first years after birth (Puelles et al., 2015), suggests the involvement of a podocyte progenitor subpopulation driving postnatal glomerular growth (Figure 3). These observations suggest that kidneys from children <5 years are endowed with a higher capacity to generate new podocytes from PECs deriving from the ongoing growth process. This explains why even severe podocyte loss driven by major genetic alterations of the podocyte in young children is associated with a pattern of injury characterized by mesangial expansion and the signs of a massive podocyte turnover. Indeed, in the context of repair attempt by PECs, the initial mesangial adaptive response paralleled by massive introduction of new podocytes along the glomerular basement membrane is enough to prevent scarring and maintain kidney function despite heavy proteinuria in young children (Figure 1 and Table 1). Unfortunately, due to the frequent underlying genetic cause connected to DMS, new podocytes originated by PECs carry the same functional problem of the lost ones, damping the regenerative potential of these patients and progressively leading first to FSGS and then to end stage kidney disease.

FIGURE 3. Progenitor cells generate novel podocytes during postnatal kidney growth. (A) The kidney is composed of functional units, nephrons, each of which is made of a glomerulus and a tubule. The glomerulus is composed of a tuft of capillaries covered by visceral epithelial cells, the podocytes, and surrounded by the Bowman capsule lined on the inner surface by flat epithelial cells, parietal epithelial cells (PEC). A subpopulation of PEC localized at the urinary pole is endowed with progenitor characteristics and progressively differentiate into podocytes toward the vascular pole of the glomerulus. This occurs as the kidney grows, during childhood and adolescence in mouse models and in humans. In (B,C) representative glomeruli from transgenic Pax2.rtTA;TetO.Cre;R26.Confetti mice, an established mouse model of renal progenitor cell lineage tracing. In this model, green, yellow, cyan or red fluorescent protein is randomly expressed by Pax2- expressing cells. Pax2 is expressed by PEC progenitor cells during kidney development but is lost upon their differentiation into mature podocytes in the post-natal kidney (magnification 63x). In (B) a representative glomerulus of Pax2.rtTA;TetO.Cre;R26.Confetti mouse, induced at postnatal day P5 (when the generation of new glomeruli from the metanephric mesenchyme has already ended) for 10 days and tracked until 5 weeks of age. Fluorescent Pax2+ cells are present in the parietal epithelium of the Bowman capsule as well as inside the glomerulus. These intraglomerular Pax2-derived cells expressed synaptopodin (cyan), demonstrating their podocyte nature. In (C) a representative glomerulus of a Pax2.rtTA;TetO.Cre;R26.Confetti adult mouse induced at 5 weeks of age for 10 days, showing Pax2+ cells only in Bowman capsule. Podocytes are not labeled. (D,E) In humans, the observation that the number of podocytes increases during glomerular growth and maturation in the early years after birth, suggest the involvement of a podocyte progenitor pool during postnatal kidney growth. In D a glomerulus of a 4 years old normal human kidney and in E a glomerulus of a 25 years old normal human kidney from two patients who underwent nephrectomy for localized renal tumor (Hematoxylin and eosin stain, magnification 40 x. Bars= 20 μm in B and C, bars= 50 μm in D and E).
The pathognomonic characteristic of FSGS is segmental solidification of the glomerular capillary tuft with hyalinosis and intracapillary foam cells, podocyte hypertrophy and extracellular matrix accumulation in the mesangium, often presented with an adhesion between the capillary tuft and the Bowman capsule (Rosenberg and Kopp, 2017) (Figure 1). Tubulointerstitial scarring is usually associated with glomerular disease and its presence in kidney biopsies with MC lesion suggests FSGS presence on unsampled glomeruli (Rosenberg and Kopp, 2017). Indeed, sampling constitutes an issue in FSGS, as distribution of segmental sclerosis starts in the juxtamedullary glomeruli and progresses towards the outer cortex at later disease stages (Rosenberg and Kopp, 2017). In addition, focality of sclerotic lesions is greater in children than in adults (Fogo, 2015).
Positive staining for IgM and C3 may be revealed by immunofluorescence, and it is believed to represent macromolecular trapping rather than specific deposition (De Vriese et al., 2021). On ultrastructural analysis, electron-dense material may be found in the mesangium and in the subendothelial compartment, consistent with hyalinosis (Barisoni et al., 2007).
FSGS is a pattern of injury shared by different diseases with variable clinical courses. To address this heterogeneity, the first attempt to classify FSGS relied on pathologic presentation describing five variants (D'Agati et al., 2004): the perihilar variant, with FSGS lesion located at the vascular pole; the tip lesion variant, with lesion located at the urinary pole; the cellular variant, characterized by endocapillary hypercellularity; the collapsing variant, characterized by collapse of the capillary tuft with epithelial cell hypertrophy and hyperplasia; and lastly FSGS not otherwise specified if lesions do not fit in the other variants mentioned (D'Agati et al., 2004). Unfortunately, with the exception of the tip lesion variant, which is usually associated with response to steroid treatment, the other variants have not provided sufficient help in patient stratification, mostly because the not otherwise specified variant represents by far the most frequent one. However, the common feature of FSGS is absolute or relative podocyte depletion as also demonstrated by the presence of podocyturia in FSGS patients (Szeto et al., 2015).
Podocyte injury is a major trigger of glomerulosclerosis but alone may not be sufficient to cause sclerosis as observed in the MC lesion. Additional cellular processes, including podocyte detachment are necessary to reach a critical threshold of podocyte depletion (Wharram et al., 2005), after which glomerulosclerosis occurs. The FSGS lesion is not due to a specific glomerular disease. Indeed, several conditions are well-described causative insults that lead to podocyte depletion such as hyperglycemia and insulin signaling, mechanical stress, angiotensin II, calcium signaling, viral infection, toxins, oxidants, and immunological injury (Zhong et al., 2019). Thus, a wide range of disease states can lead to the development of the FSGS injury pattern, the common denominator being that the initiating events take place in podocytes. FSGS animal models provided the proof-of-concept that podocyte depletion is a major factor mediating proteinuria and glomerulosclerosis. In particular, in the DT model, 21–40% podocyte depletion showed mesangial expansion, capsular adhesions, focal segmental glomerulosclerosis, mild persistent proteinuria and normal renal function, while more than 40% podocyte depletion showed segmental to global glomerulosclerosis with sustained high-grade proteinuria and reduced renal function (Wharram et al., 2005). Nevertheless, following podocyte loss, subsequent local responses are also critical for segmental sclerosis to occur. Indeed, following loss of podocyte coverage due to death or detachment, the uncovered glomerular basement membrane loses structural support by overlying podocytes at these sites (Jefferson and Shankland, 2014). Consequently, the capillary loop may bulge toward the Bowman capsule and an early connection, tuft adhesion, forms between PECs and the uncovered glomerular basement membrane or between podocytes and PECs (Löwen et al., 2021). As a response, PECs become focally activated, de novo express the specific markers CD9 and CD44 (Okamoto et al., 2013; Lazareth et al., 2019; Ito et al., 2020), migrate to a visceral location and deposit matrix. Interestingly, the CD44 marker is scarcely expressed by PECs in human MC lesion and may represent a useful marker to distinguish the MC and FSGS lesions in human biopsies (Smeets et al., 2014; Kuppe et al., 2015).
Several studies suggested that the presence of PECs on the glomerular tuft represents an attempt to replenish the podocyte pool (Romoli et al., 2018; Lasagni et al., 2015; Peired et al., 2013; Kaverina et al., 2019; Eng et al., 2015; Zhang et al., 2013; Kaverina et al., 2020). The capacity of a PEC subset to differentiate into podocytes and restore the podocyte number is the likely explanation for the clinical observations of remission and regression of functional parameters of chronic kidney disease in patients with diabetic and nondiabetic nephropathies (Cortinovis et al., 2016; Fioretto et al., 1998; Remuzzi et al., 2006; Takahashi et al., 2007), as well as for podocyte number restoration observed in response to pharmacological treatment in experimental models of diabetic and non-diabetic kidney disease (Zhang et al., 2012; Hudkins et al., 2020; Zhang et al., 2015). Recent results elucidated the podocyte-progenitor cross-talk revealing mediators of progenitor quiescence during homeostasis and mechanisms by which podocyte loss triggers the activation of the progenitor population in the setting of podocyte injury (Peired et al., 2021). In addition, they also provide possible explanation of why in certain conditions podocyte replacement by PECs may not be successful and lead to development of the FSGS lesion (Peired et al., 2021). In healthy conditions, the constitutive production of CXCL12 by podocytes maintains local podocyte progenitors in a quiescent state (Romoli et al., 2018), while the reduced expression of CXCL12 consequent to podocyte loss, promotes activation, migration and differentiation of PECs into podocytes (Romoli et al., 2018). Podocyte loss also permits the passage through the damaged glomerular filtration barrier of circulating retinol that is subsequently transformed into the Bowman space in retinoic acid that acts as an inducer of progenitor differentiation into podocytes (Peired et al., 2013; Zhang et al., 2012; Dai et al., 2017). The availability of retinoic acid for PECs is strongly reduced in the presence of a high-grade proteinuria, due to the retinoic acid sequestration operated by albumin in the Bowman space (Peired et al., 2013), with a consequent inefficient progenitor-to-podocyte differentiation. This observation likely explains the well-known clinical observation that high proteinuria associates with FSGS progression, while reduction of proteinuria by renin-angiotensin system blockers retards progression and restores podocyte number (Fogo, 2015; Boffa et al., 2003; Ma et al., 2005). Interestingly, increasing progenitor responsiveness to retinoic acid signaling through pharmacological approaches, such as administration of 6-bromo-indirubin-3′-oxime, mitigates glomerulosclerosis progression in non-diabetic (Lasagni et al., 2015) and diabetic mouse models (Motrapu et al., 2020), demonstrating that the progenitor-to-podocyte axis is a potential therapeutic target. PEC differentiation into podocytes appears strongly dependent on mechanical conditions present inside the glomerulus, such as its stiffness (Melica et al., 2019), that changes during the course of several diseases (Embry et al., 2018; Wyss et al., 2011) and precedes the appearance of glomerular sclerosis, as observed in the early phase of a HIV-associated nephropathy model (Embry et al., 2018). In conclusion, the FSGS pattern represents the pathology expression of a scar generated by a chronic podocyte loss (Motrapu et al., 2020) that exceeds the capacity for podocyte replacement provided by PECs, either because the podocyte loss is severe or because the capacity of PECs to differentiate into podocytes is hampered by an excessive proteinuria or an altered glomerular basement membrane stiffness (Table 1). Interestingly, juxtamedullary nephrons that show low numbers of PECs with progenitor capacity are particularly sensitive to development of sclerosis (Romoli et al., 2018) while superficial and mid-cortical nephrons harbor a higher number of podocyte progenitors, explaining the reported increased regenerative capacity of these glomeruli, and their resistance to development of FSGS lesions (Romoli et al., 2018). However, when podocyte loss overcomes PEC capacity of regeneration, glomerular basement membrane denudation occurs followed by synechia formation and ultimately sclerosis resulting in FSGS pattern (Figure 1).
CG is a pathology pattern characterized by the presence of segmental capillary tuft collapse (wrinkling and folding) in at least one glomerulus, in association with podocyte hypertrophy and/or hyperplasia (D'Agati et al., 2004). The Columbia classification had the merit to recognize the collapsing pattern as part of the same family of FSGS (D'Agati et al., 2004). Afterwards, terminology has evolved in collapsing glomerulopathy to underline the rapid and catastrophic collapsing nature of the pathological process. CG is histologically defined by the formation of a pseudocrescent, i.e., a massive proliferation of cuboidal undifferentiated epithelial cell in the Bowman space, leading to a collapse of the capillary loops, regardless of the extracellular matrix accumulation eventually leading to focal and global glomerulosclerosis (Muehlig et al., 2021) (Figure 1). This pattern of injury represents a common endpoint from multiple etiologies (APOL1 risk variants, infections, drugs, ischemia, hematologic neoplasia and autoimmune disease), suggesting a common pathological mechanism rather than a specific cause (Nicholas Cossey et al., 2017).
Immunohistochemistry and ultrastructural studies suggested that a primary damage to podocytes alone is sufficient to initiate the events underlying the formation of pseudocrescents (Henderson et al., 2008). Although the swollen and proliferating abnormal cells within the Bowman space involved in pseudocrescent formation lacked the expression of podocyte markers, they were previously regarded as “dysregulated or dedifferentiated” podocytes that had re-entered the cell cycle to proliferate (Barisoni et al., 1999). This assumption was mostly based on their occasional positivity for the cell cycle marker Ki67. However, positivity for Ki67 indicates cell cycle entry and not necessarily mitosis (Lazzeri et al., 2019). Indeed, after injury, podocytes can re-enter the S phase of the cell cycle (Frank et al., 2022) to undergo hypertrophy, and stain for Ki67 (Marshall and Shankland, 2006). However, if they are forced to bypass the G2/M cell cycle checkpoint, podocytes undergo aberrant mitosis and consequent detachment or death for mitotic catastrophe (Barisoni et al., 2000; Suzuki et al., 2009; Lasagni et al., 2013; Liapis et al., 2013; Al Hussain et al., 2017). Thus, consistent with their status of highly differentiated post-mitotic cells, staining for cell cycle markers in podocytes should not be interpreted as a sign of mitosis. On the contrary, it may be suggestive of an endoreplication process as shown in multiple organs (Lazzeri et al., 2019; Bischof et al., 2021).
A typical clinical condition associated with collapsing pattern of injury at kidney biopsy is viral-mediated nephropathy, such as that observed with HIV, Parvovirus and SARS-CoV-2 infection (Muehlig et al., 2021). Pseudocrescents, collapse of the capillary loops and podocyte multinucleation are predominant features of HIV-associated nephropathy (Barisoni et al., 2000; Suzuki et al., 2009; Lasagni et al., 2013; Liapis et al., 2013; Al Hussain et al., 2017). Indeed, virus-induced podocyte mitosis is catastrophic and induces podocyte death. In addition, immunohistochemical studies in idiopathic, HIV-associated, and pamidronate-associated CG have shown that cells comprising the pseudocrescents in human biopsies express proteins characteristic of PECs, such as cytokeratin, Pax2, CD24, and specific glycosylated isoform of CD133 (glycCD133), suggesting that cells within the pseudocrescents have a parietal epithelial rather than podocyte origin (Dijkman et al., 2005; Dijkman et al., 2006; Smeets et al., 2009). Lineage tracing by genetic tagging employing both podocyte and PEC-specific reporter mice in a model mimicking CG finally proved that hyperplastic cells were not podocyte-derived, but of PEC origin (Suzuki et al., 2009). Moreover, a recent multiomics study reported that podocyte-specific knockdown of Krüppel-like factor 4 contributes to podocyte loss triggering the activation of a distinct PEC subpopulation, suggesting that in this disorder PEC proliferation and pseudocrescent formation represent a response to podocyte injury and loss (Pace et al., 2021).
Experimental evidence demonstrated that, collapsing nephropathy and pseudocrescents originate from the proliferation of a specific PEC subpopulation expressing CD133 and Pax2 markers and representing renal progenitor cells that abnormally shift their reactions from reparative to detrimental (Yang et al., 2002; Smeets et al., 2004). It is unclear which factors are responsible for tilting the balance. It was proposed that pseudocrescents originate from PEC progenitors as a dysregulated response to the massive and fast podocyte detachment occurring in conditions of direct podocyte injury caused by drugs exposure, immune-mediated disorders or viral infections that cause a fast, massive podocyte loss leading to capillary collapse (Al Hussain et al., 2017; Kopp et al., 2020). In viral glomerulopathies, type I interferons (IFNs) are important mediators of viral infection (Anders et al., 2010). Indeed, in the biopsy of a patient with monogenic type I interferonopathy, MxA, a protein involved in antiviral immunity and induced by type I IFNs, was selectively expressed in CD133 positive PECs but not in podocytes (Fenaroli et al., 2021). In vivo, in a model of Adriamycin nephropathy, the injection of either IFN-α and IFN-β aggravated proteinuria and glomerulosclerosis and correlated not only with the triggering of local inflammation inside the glomerulus but also with a direct effect on podocytes and PECs (Migliorini et al., 2013). IFN- β specifically promoted podocyte loss by inducing mitotic catastrophe in podocytes (Migliorini et al., 2013). IFN- α affected PEC proliferation and migration (Migliorini et al., 2013). Both IFNs also impaired the differentiation of renal progenitors into mature podocytes, a mechanism that favors focal scarring over glomerular repair (Migliorini et al., 2013). Collapsing glomerulopathy has also been described in patients receiving exogenous IFN therapy administered for various medical conditions (Markowitz et al., 2010), further confirming that IFNs are critical mediators of the collapsing pattern of injury. Moreover, a growing body of evidence supports the role of IFNs as inducers of CG in individuals with the APOL1 high-risk genotype (Abid et al., 2020). APOL1 risk variants G1 and G2 are known to result in risk for kidney disease in patients of African ancestry and associate with a heterogeneous pattern of injury. Collapsing glomerulopathy is the most fulminant pattern of injury associated with APOL1-nephropathy (Abid et al., 2020). This form of glomerulopathy is observed mostly in diseases that have increased IFN levels, such as HIV infection and systemic lupus erythematosus (Larsen et al., 2013; Abid et al., 2020; Goyal and Singhal, 2021). APOL1 regulates PEC molecular phenotype through modulation of miR-193a expression through reciprocal feedback (Kumar et al., 2018). Indeed, PEC differentiation into podocytes is accompanied by a decrease in miR-193a expression (Kietzmann et al., 2015). Similarly, the suppression of miR-193a enhances APOL1 expression (Jessee and Kopp, 2018). Taken altogether, these observations suggest that the expression of APOL1 in PECs contributes to their differentiation into podocytes and the absence of APOL1 promotes PEC phenotype maintenance (Kumar et al., 2018). These data support the hypothesis that in the presence of massive and fast podocyte detachment observed in the collapsing pattern of injury, APOL1 risk variants aggravate the clinical outcome by hampering de novo formation of podocytes. Rapid and massive podocyte loss does not allow a mesangial adaptive response and abruptly stimulates the podocyte-progenitor feedback (Kietzmann et al., 2015; Jessee and Kopp, 2018; Kumar et al., 2018; Goyal and Singhal, 2021). The PEC compartment responds promptly with proliferation but it fails to complete the differentiation process towards mature podocytes. This results in obliteration of Bowman capsule with immature elements further compressing the glomerular tuft that lacks support from external and internal sides (Figure 1 and Table 1).
Altogether, these observations suggest that podocytopathies represent a complex group of disorders of the glomerular epithelial compartment, where the equilibrium between the nature, the length and the severity of podocyte injury as well as the efficiency of the repair response provided by PECs ultimately determines the pattern of injury observed at the biopsy as well as renal prognosis. A variety of genetic variants contributes to both podocyte injury and PEC repair response affecting kidney disease progression. Standard pathology techniques are not able to identify the ongoing evolution of these alterations but merely show the histological appearance that results from the process. In MC lesion only the slit diaphragm is damaged, and the structural alterations are reversible, either because the podocyte is not lost, or because PECs succeed in differentiating into new podocytes and maintaining full coverage of the filtration barrier. In FSGS, the balance between podocyte injury and replacement is lost, triggering a vicious circle where proteinuria prevents PEC progenitor cells from appropriately facing podocyte loss. In contrast, if PECs succeed in generating new podocytes, scar formation can be contained and limited to a certain extent. However, this high regenerative potential is restricted to a specific and relatively short age span, explaining why DMS is observed only in children. Finally, a fast and massive podocyte loss determining the collapse of the glomerular capillary loops is the key mechanism of CG. The ability of PECs to proliferate is retained, but the capacity to differentiate into mature podocytes is prevented, causing massive PEC activation, ultimately resulting in pseudocrescents that are typical of CG. Understanding the molecular and cellular alterations that lead to the generation of these patterns of injury can help the clinicians to convey the right diagnosis and the researchers to identify novel potential therapeutic targets for podocytopathies.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement N° 101019891, SIMPOSION). M.E.M. was supported by a FIRC-AIRC fellowship for Italy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abid, Q., Best Rocha, A., Larsen, C. P., Schulert, G., Marsh, R., Yasin, S., et al. (2020). APOL1-Associated Collapsing Focal Segmental Glomerulosclerosis in a Patient with Stimulator of Interferon Genes (STING)-Associated Vasculopathy with Onset in Infancy (SAVI). Am. J. Kidney Dis. 75, 287–290. doi:10.1053/j.ajkd.2019.07.010
Ahn, W., and Bomback, A. S. (2020). Approach to Diagnosis and Management of Primary Glomerular Diseases Due to Podocytopathies in Adults: Core Curriculum 2020. Am. J. Kidney Dis. 75, 955–964. doi:10.1053/j.ajkd.2019.12.019
Al Hussain, T., Al Mana, H., Hussein, M. H., and Akhtar, M. (2017). Podocyte and Parietal Epithelial Cell Interactions in Health and Disease. Adv. Anat. Pathol. 24, 24–34. doi:10.1097/pap.0000000000000125
Anders, H.-J., Lichtnekert, J., and Allam, R. (2010). Interferon-α and -β in Kidney Inflammation. Kidney Int. 77, 848–854. doi:10.1038/ki.2010.71
Angelotti, M. L., Antonelli, G., Conte, C., and Romagnani, P. (2021). Imaging the Kidney: from Light to Super-resolution Microscopy. Nephrol. Dial. Transpl. 36, 19–28. doi:10.1093/ndt/gfz136
Appel, D., Kershaw, D. B., Smeets, B., Yuan, G., Fuss, A., Frye, B., et al. (2009). Recruitment of Podocytes from Glomerular Parietal Epithelial Cells. J. Am. Soc. Nephrol. 20, 333–343. doi:10.1681/asn.2008070795
Artelt, N., Siegerist, F., Ritter, A. M., Grisk, O., Schlüter, R., Endlich, K., et al. (2018). Comparative Analysis of Podocyte Foot Process Morphology in Three Species by 3D Super-resolution Microscopy. Front. Med. 5, 292. doi:10.3389/fmed.2018.00292
Atchison, D. K., O’Connor, C. L., Menon, R., Otto, E. A., Ganesh, S. K., Wiggins, R. C., et al. (2020). Hypertension Induces Glomerulosclerosis in Phospholipase C-Ε1 Deficiency. Am. J. Physiology-Renal Physiol. 318, F1177–F1187. doi:10.1152/ajprenal.00541.2019
Banu, K., Lin, Q., Basgen, J. M., Planoutene, M., Wei, C., Reghuvaran, A. C., et al. (2021). AMPK Mediates Regulation of Glomerular Volume and Podocyte Survival. JCI Insight 6, e150004. doi:10.1172/jci.insight.150004
Barisoni, L., Kriz, W., Mundel, P., and D'Agati, V. (1999). The Dysregulated Podocyte Phenotype. J. Am. Soc. Nephrol. 10, 51–61. doi:10.1681/asn.v10151
Barisoni, L., Mokrzycki, M., Sablay, L., Nagata, M., Yamase, H., and Mundel, P. (2000). Podocyte Cell Cycle Regulation and Proliferation in Collapsing Glomerulopathies. Kidney Int. 58, 137–143. doi:10.1046/j.1523-1755.2000.00149.x
Barisoni, L., Schnaper, H. W., and Kopp, J. B. (2007). A Proposed Taxonomy for the Podocytopathies: a Reassessment of the Primary Nephrotic Diseases. Clin. J. Am. Soc. Nephrol. 2, 529–542. doi:10.2215/cjn.04121206
Bertram, J. F., Douglas-Denton, R. N., Diouf, B., Hughson, M. D., and Hoy, W. E. (2011). Human Nephron Number: Implications for Health and Disease. Pediatr. Nephrol. 26, 1529–1533. doi:10.1007/s00467-011-1843-8
Bischof, C., Mirtschink, P., Yuan, T., Wu, M., Zhu, C., Kaur, J., et al. (2021). Mitochondrial-cell Cycle Cross-Talk Drives Endoreplication in Heart Disease. Sci. Transl Med. 13, eabi7964. doi:10.1126/scitranslmed.abi7964
Boffa, J.-J., Lu, Y., Placier, S., Stefanski, A., Dussaule, J.-C., and Chatziantoniou, C. (2003). Regression of Renal Vascular and Glomerular Fibrosis: Role of Angiotensin II Receptor Antagonism and Matrix Metalloproteinases. J. Am. Soc. Nephrol. 14, 1132–1144. doi:10.1097/01.asn.0000060574.38107.3b
Boyer, O., Benoit, G., Gribouval, O., Nevo, F., Pawtowski, A., Bilge, I., et al. (2010). Mutational Analysis of the PLCE1 Gene in Steroid Resistant Nephrotic Syndrome. J. Med. Genet. 47, 445–452. doi:10.1136/jmg.2009.076166
Calizo, R. C., Bhattacharya, S., van Hasselt, J. G. C., Wei, C., Wong, J. S., Wiener, R. J., et al. (2019). Disruption of Podocyte Cytoskeletal Biomechanics by Dasatinib Leads to Nephrotoxicity. Nat. Commun. 10, 2061. doi:10.1038/s41467-019-09936-x
Charnaya, O., and Moudgil, A. (2017). Hypertension in the Pediatric Kidney Transplant Recipient. Front. Pediatr. 5, 86. doi:10.3389/fped.2017.00086
Chugh, S. S., Clement, L. C., and Macé, C. (2012). New Insights into Human Minimal Change Disease: Lessons from Animal Models. Am. J. Kidney Dis. 59, 284–292. doi:10.1053/j.ajkd.2011.07.024
Chung, J.-J., Goldstein, L., Chen, Y.-J. J., Lee, J., Webster, J. D., Roose-Girma, M., et al. (2020). Single-Cell Transcriptome Profiling of the Kidney Glomerulus Identifies Key Cell Types and Reactions to Injury. J. Am. Soc. Nephrol. 31, 2341–2354. doi:10.1681/asn.2020020220
Cortinovis, M., Ruggenenti, P., and Remuzzi, G. (2016). Progression, Remission and Regression of Chronic Renal Diseases. Nephron 134, 20–24. doi:10.1159/000445844
D'Agati, V. D., Fogo, A. B., Bruijn, J. A., and Jennette, J. C. (2004). Pathologic Classification of Focal Segmental Glomerulosclerosis: a Working Proposal. Am. J. Kidney Dis. 43, 368–382. doi:10.1053/j.ajkd.2003.10.024
da Silva, C. A., Monteiro, M. L. G. d. R., Araújo, L. S., Urzedo, M. G., Rocha, L. B., Dos Reis, M. A., et al. (2020). In Situ evaluation of Podocytes in Patients with Focal Segmental Glomerulosclerosis and Minimal Change Disease. PLoS One 15, e0241745. doi:10.1371/journal.pone.0241745
Dai, Y., Chen, A., Liu, R., Gu, L., Sharma, S., Cai, W., et al. (2017). Retinoic Acid Improves Nephrotoxic Serum-Induced Glomerulonephritis through Activation of Podocyte Retinoic Acid Receptor α. Kidney Int. 92, 1444–1457. doi:10.1016/j.kint.2017.04.026
De Vriese, A. S., Sethi, S., Nath, K. A., Glassock, R. J., and Fervenza, F. C. (2018). Differentiating Primary, Genetic, and Secondary FSGS in Adults: A Clinicopathologic Approach. J. Am. Soc. Nephrol. 29, 759–774. doi:10.1681/asn.2017090958
De Vriese, A. S., Wetzels, J. F., Glassock, R. J., Sethi, S., and Fervenza, F. C. (2021). Therapeutic Trials in Adult FSGS: Lessons Learned and the Road Forward. Nat. Rev. Nephrol. 17, 619–630. doi:10.1038/s41581-021-00427-1
Deegens, J. K. J., Dijkman, H. B. P. M., Borm, G. F., Steenbergen, E. J., van den Berg, J. G., Weening, J. J., et al. (2008). Podocyte Foot Process Effacement as a Diagnostic Tool in Focal Segmental Glomerulosclerosis. Kidney Int. 74, 1568–1576. doi:10.1038/ki.2008.413
Dijkman, H. B. P. M., Weening, J. J., Smeets, B., Verrijp, K. C. N., van Kuppevelt, T. H., Assmann, K. K. J. M., et al. (2006). Proliferating Cells in HIV and Pamidronate-Associated Collapsing Focal Segmental Glomerulosclerosis Are Parietal Epithelial Cells. Kidney Int. 70, 338–344. doi:10.1038/sj.ki.5001574
Dijkman, H., Smeets, B., van der Laak, J., Steenbergen, E., and Wetzels, J. (2005). The Parietal Epithelial Cell Is Crucially Involved in Human Idiopathic Focal Segmental glomerulosclerosis11See Editorial by Schwartz, P. 1894. Kidney Int. 68, 1562–1572. doi:10.1111/j.1523-1755.2005.00568.x
Embry, A. E., Liu, Z., Henderson, J. M., Byfield, F. J., Liu, L., Yoon, J., et al. (2018). Similar Biophysical Abnormalities in Glomeruli and Podocytes from Two Distinct Models. J. Am. Soc. Nephrol. 29, 1501–1512. doi:10.1681/asn.2017050475
Eng, D. G., Sunseri, M. W., Kaverina, N. V., Roeder, S. S., Pippin, J. W., and Shankland, S. J. (2015). Glomerular Parietal Epithelial Cells Contribute to Adult Podocyte Regeneration in Experimental Focal Segmental Glomerulosclerosis. Kidney Int. 88, 999–1012. doi:10.1038/ki.2015.152
Eymael, J., Sharma, S., Loeven, M. A., Wetzels, J. F., Mooren, F., Florquin, S., et al. (2018). CD44 Is Required for the Pathogenesis of Experimental Crescentic Glomerulonephritis and Collapsing Focal Segmental Glomerulosclerosis. Kidney Int. 93, 626–642. doi:10.1016/j.kint.2017.09.020
Fatima, H., Moeller, M. J., Smeets, B., Yang, H.-C., D’Agati, V. D., Alpers, C. E., et al. (2012). Parietal Epithelial Cell Activation Marker in Early Recurrence of FSGS in the Transplant. Clin. J. Am. Soc. Nephrol. 7, 1852–1858. doi:10.2215/cjn.10571011
Fenaroli, P., Rossi, G. M., Angelotti, M. L., Antonelli, G., Volpi, S., Grossi, A., et al. (2021). Collapsing Glomerulopathy as a Complication of Type I Interferon-Mediated Glomerulopathy in a Patient with RNASEH2B-Related Aicardi-Goutières Syndrome. Am. J. Kidney Dis. 78, 750–754. doi:10.1053/j.ajkd.2021.02.330
Fioretto, P., Steffes, M. W., Sutherland, D. E. R., Goetz, F. C., and Mauer, M. (1998). Reversal of Lesions of Diabetic Nephropathy after Pancreas Transplantation. N. Engl. J. Med. 339, 69–75. doi:10.1056/nejm199807093390202
Fogo, A. B. (2015). Causes and Pathogenesis of Focal Segmental Glomerulosclerosis. Nat. Rev. Nephrol. 11, 76–87. doi:10.1038/nrneph.2014.216
Fogo, A. B. (2001). Progression and Potential Regression of Glomerulosclerosis. Kidney Int. 59, 804–819. doi:10.1046/j.1523-1755.2001.059002804.x
Fogo, A., Hawkins, E. P., Berry, P. L., Glick, A. D., Chiang, M. L., MacDonell, R. C., et al. (1990). Glomerular Hypertrophy in Minimal Change Disease Predicts Subsequent Progression to Focal Glomerular Sclerosis. Kidney Int. 38, 115–123. doi:10.1038/ki.1990.175
Frank, C. N., Hou, X., Petrosyan, A., Villani, V., Zhao, R., Hansen, J. R., et al. (2022). Effect of Disease Progression on the Podocyte Cell Cycle in Alport Syndrome. Kidney Int. 101, 106–118. doi:10.1016/j.kint.2021.08.026
Freedman, B. I., Langefeld, C. D., Andringa, K. K., Croker, J. A., Williams, A. H., Garner, N. E., et al. (2014). End-Stage Renal Disease in African Americans with Lupus Nephritis Is Associated WithAPOL1. Arthritis Rheumatol. 66, 390–396. doi:10.1002/art.38220
Fukuda, A., Chowdhury, M. A., Venkatareddy, M. P., Wang, S. Q., Nishizono, R., Suzuki, T., et al. (2012b). Growth-dependent Podocyte Failure Causes Glomerulosclerosis. J. Am. Soc. Nephrol. 23, 1351–1363. doi:10.1681/asn.2012030271
Fukuda, A., Wickman, L. T., Venkatareddy, M. P., Sato, Y., Chowdhury, M. A., Wang, S. Q., et al. (2012a). Angiotensin II-dependent Persistent Podocyte Loss from Destabilized Glomeruli Causes Progression of End Stage Kidney Disease. Kidney Int. 81, 40–55. doi:10.1038/ki.2011.306
Goyal, R., and Singhal, P. C. (2021). APOL1 Risk Variants and the Development of HIV‐associated Nephropathy. FEBS J. 288, 5586–5597. doi:10.1111/febs.15677
Habib, R., and Kleinknecht, C. (1971). The Primary Nephrotic Syndrome of Childhood. Classification and Clinicopathologic Study of 406 Cases. Pathol. Annu. 6, 417–474.
Henderson, J. M., Al-Waheeb, S., Weins, A., Dandapani, S. V., and Pollak, M. R. (2008). Mice with Altered α-actinin-4 Expression Have Distinct Morphologic Patterns of Glomerular Disease. Kidney Int. 73, 741–750. doi:10.1038/sj.ki.5002751
Hinkes, B., Wiggins, R. C., Gbadegesin, R., Vlangos, C. N., Seelow, D., Nürnberg, G., et al. (2006). Positional Cloning Uncovers Mutations in PLCE1 Responsible for a Nephrotic Syndrome Variant that May Be Reversible. Nat. Genet. 38, 1397–1405. doi:10.1038/ng1918
Hudkins, K. L., Wietecha, T. A., Steegh, F., and Alpers, C. E. (2020). Beneficial Effect on Podocyte Number in Experimental Diabetic Nephropathy Resulting from Combined Atrasentan and RAAS Inhibition Therapy. Am. J. Physiology-Renal Physiol. 318, F1295–F1305. doi:10.1152/ajprenal.00498.2019
Ikezumi, Y., Suzuki, T., Karasawa, T., Kaneko, U., Yamada, T., Hasegawa, H., et al. (2014). Glomerular Epithelial Cell Phenotype in Diffuse Mesangial Sclerosis: a Report of 2 Cases with Markedly Increased Urinary Podocyte Excretion. Hum. Pathol. 45, 1778–1783. doi:10.1016/j.humpath.2014.03.017
Ito, N., Sakamoto, K., Hikichi, C., Matsusaka, T., and Nagata, M. (2020). Biphasic MIF and SDF1 Expression during Podocyte Injury Promote CD44-Mediated Glomerular Parietal Cell Migration in Focal Segmental Glomerulosclerosis. Am. J. Physiology-Renal Physiol. 318, F741–F753. doi:10.1152/ajprenal.00414.2019
Jefferson, J. A., and Shankland, S. J. (2014). The Pathogenesis of Focal Segmental Glomerulosclerosis. Adv. Chronic Kidney Dis. 21, 408–416. doi:10.1053/j.ackd.2014.05.009
Jessee, J., and Kopp, J. B. (2018). APOL1-miR-193 Axis as a Bifunctional Regulator of the Glomerular Parietal Epithelium. Am. J. Pathol. 188, 2461–2463. doi:10.1016/j.ajpath.2018.08.002
Kaverina, N. V., Eng, D. G., Freedman, B. S., Kutz, J. N., Chozinski, T. J., Vaughan, J. C., et al. (2019). Dual Lineage Tracing Shows that Glomerular Parietal Epithelial Cells Can Transdifferentiate toward the Adult Podocyte Fate. Kidney Int. 96, 597–611. doi:10.1016/j.kint.2019.03.014
Kaverina, N. V., Eng, D. G., Miner, J. H., Pippin, J. W., and Shankland, S. J. (2020). Parietal Epithelial Cell Differentiation to a Podocyte Fate in the Aged Mouse Kidney. Aging 12, 17601–17624. doi:10.18632/aging.103788
Kietzmann, L., Guhr, S. S. O., Meyer, T. N., Ni, L., Sachs, M., Panzer, U., et al. (2015). MicroRNA-193a Regulates the Transdifferentiation of Human Parietal Epithelial Cells toward a Podocyte Phenotype. J. Am. Soc. Nephrol. 26, 1389–1401. doi:10.1681/asn.2014020190
Kim, Y. H., Goyal, M., Kurnit, D., Wharram, B., Wiggins, J., Holzman, L., et al. (2001). Podocyte Depletion and Glomerulosclerosis Have a Direct Relationship in the PAN-Treated Rat. Kidney Int. 60, 957–968. doi:10.1046/j.1523-1755.2001.060003957.x
Kopp, J. B., Anders, H.-J., Susztak, K., Podestà, M. A., Remuzzi, G., Hildebrandt, F., et al. (2020). Podocytopathies. Nat. Rev. Dis. Primers 6, 68. doi:10.1038/s41572-020-0196-7
Kriz, W., and Lemley, K. V. (2017b). Mechanical Challenges to the Glomerular Filtration Barrier: Adaptations and Pathway to Sclerosis. Pediatr. Nephrol. 32, 405–417. doi:10.1007/s00467-016-3358-9
Kriz, W., and Lemley, K. V. (2017a). Potential Relevance of Shear Stress for Slit Diaphragm and Podocyte Function. Kidney Int. 91, 1283–1286. doi:10.1016/j.kint.2017.02.032
Kumar, V., Vashistha, H., Lan, X., Chandel, N., Ayasolla, K., Shoshtari, S. S. M., et al. (2018). Role of Apolipoprotein L1 in Human Parietal Epithelial Cell Transition. Am. J. Pathol. 188, 2508–2528. doi:10.1016/j.ajpath.2018.07.025
Kuppe, C., Gröne, H.-J., Ostendorf, T., van Kuppevelt, T. H., Boor, P., Floege, J., et al. (2015). Common Histological Patterns in Glomerular Epithelial Cells in Secondary Focal Segmental Glomerulosclerosis. Kidney Int. 88, 990–998. doi:10.1038/ki.2015.116
Lahdenkari, A.-T., Lounatmaa, K., Patrakka, J., Holmberg, C., Wartiovaara, J., Kestila, M., et al. (2004). Podocytes Are Firmly Attached to Glomerular Basement Membrane in Kidneys with Heavy Proteinuria. J. Am. Soc. Nephrol. 15, 2611–2618. doi:10.1097/01.asn.0000139478.03463.d9
Landini, S., Mazzinghi, B., Becherucci, F., Allinovi, M., Provenzano, A., Palazzo, V., et al. (2020). Reverse Phenotyping after Whole-Exome Sequencing in Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 15, 89–100. doi:10.2215/cjn.06060519
Larsen, C. P., Beggs, M. L., Saeed, M., and Walker, P. D. (2013). Apolipoprotein L1 Risk Variants Associate with Systemic Lupus Erythematosus-Associated Collapsing Glomerulopathy. J. Am. Soc. Nephrol. 24, 722–725. doi:10.1681/asn.2012121180
Lasagni, L., Angelotti, M. L., Ronconi, E., Lombardi, D., Nardi, S., Peired, A., et al. (2015). Podocyte Regeneration Driven by Renal Progenitors Determines Glomerular Disease Remission and Can Be Pharmacologically Enhanced. Stem Cel Rep. 5, 248–263. doi:10.1016/j.stemcr.2015.07.003
Lasagni, L., Lazzeri, E., J. Shankland, S., Anders, H.-J., and Romagnani, P. (2013). Podocyte Mitosis - a Catastrophe. Curr. Mol. Med. 13, 13–23. doi:10.2174/156652413804486250
Lazareth, H., Henique, C., Lenoir, O., Puelles, V. G., Flamant, M., Bollée, G., et al. (2019). The Tetraspanin CD9 Controls Migration and Proliferation of Parietal Epithelial Cells and Glomerular Disease Progression. Nat. Commun. 10, 3303. doi:10.1038/s41467-019-11013-2
Lazzeri, E., Angelotti, M. L., Conte, C., Anders, H.-J., and Romagnani, P. (2019). Surviving Acute Organ Failure: Cell Polyploidization and Progenitor Proliferation. Trends Mol. Med. 25, 366–381. doi:10.1016/j.molmed.2019.02.006
Lehnhardt, A., Karnatz, C., Ahlenstiel-Grunow, T., Benz, K., Benz, M. R., Budde, K., et al. (2015). Clinical and Molecular Characterization of Patients with Heterozygous Mutations in Wilms Tumor Suppressor Gene 1. Clin. J. Am. Soc. Nephrol. 10, 825–831. doi:10.2215/cjn.10141014
Liapis, H., Romagnani, P., and Anders, H.-J. (2013). New Insights into the Pathology of Podocyte Loss. Am. J. Pathol. 183, 1364–1374. doi:10.1016/j.ajpath.2013.06.033
Lin, M.-H., Miller, J. B., Kikkawa, Y., Suleiman, H. Y., Tryggvason, K., Hodges, B. L., et al. (2018). Laminin-521 Protein Therapy for Glomerular Basement Membrane and Podocyte Abnormalities in a Model of Pierson Syndrome. J. Am. Soc. Nephrol. 29, 1426–1436. doi:10.1681/asn.2017060690
Löwen, J., Gröne, E. F., Gross-Weissmann, M.-L., Bestvater, F., Gröne, H.-J., and Kriz, W. (2021). Pathomorphological Sequence of Nephron Loss in Diabetic Nephropathy. Am. J. Physiology-Renal Physiol. 321, F600–F616. doi:10.1152/ajprenal.00669.2020
Ma, L.-J., Nakamura, S., Aldigier, J. C., Rossini, M., Yang, H., Liang, X., et al. (2005). Regression of Glomerulosclerosis with High-Dose Angiotensin Inhibition Is Linked to Decreased Plasminogen Activator Inhibitor-1. J. Am. Soc. Nephrol. 16, 966–976. doi:10.1681/asn.2004060492
Maas, R. J., Deegens, J. K., Smeets, B., Moeller, M. J., and Wetzels, J. F. (2016). Minimal Change Disease and Idiopathic FSGS: Manifestations of the Same Disease. Nat. Rev. Nephrol. 12, 768–776. doi:10.1038/nrneph.2016.147
Markowitz, G. S., Nasr, S. H., Stokes, M. B., and D'Agati, V. D. (2010). Treatment with IFN-α, -β, or -γ Is Associated with Collapsing Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 5, 607–615. doi:10.2215/cjn.07311009
Marshall, C. B., and Shankland, S. J. (2006). Cell Cycle and Glomerular Disease: a Minireview. Nephron Exp. Nephrol. 102, e39–e48. doi:10.1159/000088400
Matejas, V., Hinkes, B., Alkandari, F., Al-Gazali, L., Annexstad, E., Aytac, M. B., et al. (2010). Mutations in the Human Laminin β2 (LAMB2) Gene and the Associated Phenotypic Spectruma. Hum. Mutat. 31, 992–1002. doi:10.1002/humu.21304
Melica, M. E., La Regina, G., Parri, M., Peired, A. J., Romagnani, P., and Lasagni, L. (2019). Substrate Stiffness Modulates Renal Progenitor Cell Properties via a ROCK-Mediated Mechanotransduction Mechanism. Cells 8, 1561. doi:10.3390/cells8121561
Migliorini, A., Angelotti, M. L., Mulay, S. R., Kulkarni, O. O., Demleitner, J., Dietrich, A., et al. (2013). The Antiviral Cytokines IFN-α and IFN-β Modulate Parietal Epithelial Cells and Promote Podocyte Loss. Am. J. Pathol. 183, 431–440. doi:10.1016/j.ajpath.2013.04.017
Motrapu, M., Świderska, M. K., Mesas, I., Marschner, J. A., Lei, Y., Martinez Valenzuela, L., et al. (2020). Drug Testing for Residual Progression of Diabetic Kidney Disease in Mice beyond Therapy with Metformin, Ramipril, and Empagliflozin. J. Am. Soc. Nephrol. 31, 1729–1745. doi:10.1681/asn.2019070703
Muehlig, A. K., Gies, S., Huber, T. B., and Braun, F. (2021). Collapsing Focal Segmental Glomerulosclerosis in Viral Infections. Front. Immunol. 12, 800074. doi:10.3389/fimmu.2021.800074
Munk, F. (1918). Pathologie und klinik der Nephrosen, Nephritiden und Schrumpfnieren. Berlin, Wien: Urban & Schwarzenberg.
Nagata, M. (2016). Podocyte Injury and its Consequences. Kidney Int. 89, 1221–1230. doi:10.1016/j.kint.2016.01.012
Nicholas Cossey, L., Larsen, C. P., and Liapis, H. (2017). Collapsing Glomerulopathy: a 30-year Perspective and Single, Large center Experience. Clin. Kidney J. 10, 443–449. doi:10.1093/ckj/sfx029
Okamoto, T., Sasaki, S., Yamazaki, T., Sato, Y., Ito, H., and Ariga, T. (2013). Prevalence of CD44-Positive Glomerular Parietal Epithelial Cells Reflects Podocyte Injury in Adriamycin Nephropathy. Nephron Exp. Nephrol. 124, 11–18. doi:10.1159/000357356
Pace, J. A., Bronstein, R., Guo, Y., Yang, Y., Estrada, C. C., Gujarati, N., et al. (2021). Podocyte-specific KLF4 Is Required to Maintain Parietal Epithelial Cell Quiescence in the Kidney. Sci. Adv. 7, eabg6600. doi:10.1126/sciadv.abg6600
Patrakka, J., Lahdenkari, A.-T., Koskimies, O., Holmberg, C., Wartiovaara, J., and Jalanko, H. (2002). The Number of Podocyte Slit Diaphragms Is Decreased in Minimal Change Nephrotic Syndrome. Pediatr. Res. 52, 349–355. doi:10.1203/00006450-200209000-00007
Peired, A., Angelotti, M. L., Ronconi, E., la Marca, G., Mazzinghi, B., Sisti, A., et al. (2013). Proteinuria Impairs Podocyte Regeneration by Sequestering Retinoic Acid. J. Am. Soc. Nephrol. 24, 1756–1768. doi:10.1681/asn.2012090950
Peired, A. J., Melica, M. E., Molli, A., Nardi, C., Romagnani, P., and Lasagni, L. (2021). Molecular Mechanisms of Renal Progenitor Regulation: How Many Pieces in the Puzzle. Cells 10, 59. doi:10.3390/cells10010059
Pippin, J. W., Brinkkoetter, P. T., Cormack-Aboud, F. C., Durvasula, R. V., Hauser, P. V., Kowalewska, J., et al. (2009). Inducible Rodent Models of Acquired Podocyte Diseases. Am. J. Physiology-Renal Physiol. 296, F213–F229. doi:10.1152/ajprenal.90421.2008
Puelles, V. G., van der Wolde, J. W., Wanner, N., Scheppach, M. W., Cullen-McEwen, L. A., Bork, T., et al. (2019). mTOR-mediated Podocyte Hypertrophy Regulates Glomerular Integrity in Mice and Humans. JCI Insight 4, e99271. doi:10.1172/jci.insight.99271
Puelles, V. G., Douglas-Denton, R. N., Cullen-McEwen, L. A., Li, J., Hughson, M. D., Hoy, W. E., et al. (2015). Podocyte Number in Children and Adults: Associations with Glomerular Size and Numbers of Other Glomerular Resident Cells. J. Am. Soc. Nephrol. 26, 2277–2288. doi:10.1681/asn.2014070641
Purohit, S., Piani, F., Ordoñez, F. A., de Lucas-Collantes, C., Bauer, C., and Cara-Fuentes, G. (2021). Molecular Mechanisms of Proteinuria in Minimal Change Disease. Front. Med. 8, 761600. doi:10.3389/fmed.2021.761600
Ratelade, J., Arrondel, C., Hamard, G., Garbay, S., Harvey, S., Biebuyck, N., et al. (2010). A Murine Model of Denys-Drash Syndrome Reveals Novel Transcriptional Targets of WT1 in Podocytes. Hum. Mol. Genet. 19, 1–15. doi:10.1093/hmg/ddp462
Remuzzi, G., Benigni, A., and Remuzzi, A. (2006). Mechanisms of Progression and Regression of Renal Lesions of Chronic Nephropathies and Diabetes. J. Clin. Invest. 116, 288–296. doi:10.1172/jci27699
Charles Jennette, J., Olson, J. L., Silva, F. G., and D'Agati, V. D. (2014). eds. 7th edition. 2014. Lippincott Williams8Wilki.
Romoli, S., Angelotti, M. L., Antonelli, G., Kumar Vr, S., Mulay, S. R., Desai, J., et al. (2018). CXCL12 Blockade Preferentially Regenerates Lost Podocytes in Cortical Nephrons by Targeting an Intrinsic Podocyte-Progenitor Feedback Mechanism. Kidney Int. 94, 1111–1126. doi:10.1016/j.kint.2018.08.013
Rosenberg, A. Z., and Kopp, J. B. (2017). Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 12, 502–517. doi:10.2215/cjn.05960616
Royal, V., Zee, J., Liu, Q., Avila-Casado, C., Smith, A. R., Liu, G., et al. (2020). Ultrastructural Characterization of Proteinuric Patients Predicts Clinical Outcomes. J. Am. Soc. Nephrol. 31, 841–854. doi:10.1681/asn.2019080825
Saga, N., Sakamoto, K., Matsusaka, T., and Nagata, M. (2021). Glomerular Filtrate Affects the Dynamics of Podocyte Detachment in a Model of Diffuse Toxic Podocytopathy. Kidney Int. 99, 1149–1161. doi:10.1016/j.kint.2020.12.034
Smeets, B., Angelotti, M. L., Rizzo, P., Dijkman, H., Lazzeri, E., Mooren, F., et al. (2009). Renal Progenitor Cells Contribute to Hyperplastic Lesions of Podocytopathies and Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. 20, 2593–2603. doi:10.1681/asn.2009020132
Smeets, B., Stucker, F., Wetzels, J., Brocheriou, I., Ronco, P., Gröne, H.-J., et al. (2014). Detection of Activated Parietal Epithelial Cells on the Glomerular Tuft Distinguishes Early Focal Segmental Glomerulosclerosis from Minimal Change Disease. Am. J. Pathol. 184, 3239–3248. doi:10.1016/j.ajpath.2014.08.007
Smeets, B., Te Loeke, N. A., Dijkman, H. B., Steenbergen, M. L., Lensen, J. F., Begieneman, M. P., et al. (2004). The Parietal Epithelial Cell: a Key Player in the Pathogenesis of Focal Segmental Glomerulosclerosis in Thy-1.1 Transgenic Mice. J. Am. Soc. Nephrol. 15, 928–939. doi:10.1097/01.asn.0000120559.09189.82
Suzuki, T., Matsusaka, T., Nakayama, M., Asano, T., Watanabe, T., Ichikawa, I., et al. (2009). Genetic Podocyte Lineage Reveals Progressive Podocytopenia with Parietal Cell Hyperplasia in a Murine Model of Cellular/collapsing Focal Segmental Glomerulosclerosis. Am. J. Pathol. 174, 1675–1682. doi:10.2353/ajpath.2009.080789
Szeto, C.-C., Wang, G., Chow, K.-M., Lai, F. M.-M., Ma, T. K.-W., Kwan, B. C.-H., et al. (2015). Podocyte mRNA in the Urinary Sediment of Minimal Change Nephropathy and Focal Segmental Glomerulosclerosis. Clin. Nephrol. 84, 198–205. doi:10.5414/cn108607
Takahashi, H., Ichihara, A., Kaneshiro, Y., Inomata, K., Sakoda, M., Takemitsu, T., et al. (2007). Regression of Nephropathy Developed in Diabetes by (Pro)renin Receptor Blockade. J. Am. Soc. Nephrol. 18, 2054–2061. doi:10.1681/asn.2006080820
Tejani, A. (1985). Morphological Transition in Minimal Change Nephrotic Syndrome. Nephron 39, 157–159. doi:10.1159/000183363
Tesch, F., Siegerist, F., Hay, E., Artelt, N., Daniel, C., Amann, K., et al. (2021). Super‐resolved Local Recruitment of CLDN5 to Filtration Slits Implicates a Direct Relationship with Podocyte Foot Process Effacement. J. Cel Mol Med 25, 7631–7641. doi:10.1111/jcmm.16519
Vivarelli, M., Massella, L., Ruggiero, B., and Emma, F. (2017). Minimal Change Disease. Clin. J. Am. Soc. Nephrol. 12, 332–345. doi:10.2215/cjn.05000516
Wanner, N., Hartleben, B., Herbach, N., Goedel, M., Stickel, N., Zeiser, R., et al. (2014). Unraveling the Role of Podocyte Turnover in Glomerular Aging and Injury. J. Am. Soc. Nephrol. 25, 707–716. doi:10.1681/asn.2013050452
Warejko, J. K., Tan, W., Daga, A., Schapiro, D., Lawson, J. A., Shril, S., et al. (2018). Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 13, 53–62. doi:10.2215/cjn.04120417
Watts, A., Keller, K., Lerner, G., Rosales, I., Collins, A., Sekulic, M., et al. (2021). Discovery of Autoantibodies Targeting Nephrin in Minimal Change Disease Supports a Novel Autoimmune Etiology. J. Am. Soc. Nephrol. 33, 238–252. doi:10.1681/asn.2021060794
Wharram, B. L., Goyal, M., Wiggins, J. E., Sanden, S. K., Hussain, S., Filipiak, W. E., et al. (2005). Podocyte Depletion Causes Glomerulosclerosis: Diphtheria Toxin-Induced Podocyte Depletion in Rats Expressing Human Diphtheria Toxin Receptor Transgene. J. Am. Soc. Nephrol. 16, 2941–2952. doi:10.1681/asn.2005010055
Wiggins, R.-C. (2007). The Spectrum of Podocytopathies: a Unifying View of Glomerular Diseases. Kidney Int. 71, 1205–1214. doi:10.1038/sj.ki.5002222
Wyss, H. M., Henderson, J. M., Byfield, F. J., Bruggeman, L. A., Ding, Y., Huang, C., et al. (2011). Biophysical Properties of normal and Diseased Renal Glomeruli. Am. J. Physiology-Cell Physiol. 300, C397–C405. doi:10.1152/ajpcell.00438.2010
Yang, H.-C., and Fogo, A. B. (2014). Mechanisms of Disease Reversal in Focal and Segmental Glomerulosclerosis. Adv. Chronic Kidney Dis. 21, 442–447. doi:10.1053/j.ackd.2014.04.001
Yang, Y., Gubler, M.-C., and Beaufils, H. (2002). Dysregulation of Podocyte Phenotype in Idiopathic Collapsing Glomerulopathy and HIV-Associated Nephropathy. Nephron 91, 416–423. doi:10.1159/000064281
Yang, Y., Jeanpierre, C., Dressler, G. R., Lacoste, M., Niaudet, P., and Gubler, M.-C. (1999). WT1 and PAX-2 Podocyte Expression in Denys-Drash Syndrome and Isolated Diffuse Mesangial Sclerosis. Am. J. Pathol. 154, 181–192. doi:10.1016/s0002-9440(10)65264-9
Zhang, J., Pippin, J. W., Krofft, R. D., Naito, S., Liu, Z.-H., and Shankland, S. J. (2013). Podocyte Repopulation by Renal Progenitor Cells Following Glucocorticoids Treatment in Experimental FSGS. Am. J. Physiology-Renal Physiol. 304, F1375–F1389. doi:10.1152/ajprenal.00020.2013
Zhang, J., Pippin, J. W., Vaughan, M. R., Krofft, R. D., Taniguchi, Y., Romagnani, P., et al. (2012). Retinoids Augment the Expression of Podocyte Proteins by Glomerular Parietal Epithelial Cells in Experimental Glomerular Disease. Nephron Exp. Nephrol. 121, e23–e37. doi:10.1159/000342808
Zhang, J., Yanez, D., Floege, A., Lichtnekert, J., Krofft, R. D., Liu, Z.-H., et al. (2015). ACE-inhibition Increases Podocyte Number in Experimental Glomerular Disease Independent of Proliferation. J. Renin Angiotensin Aldosterone Syst. 16, 234–248. doi:10.1177/1470320314543910
Keywords: podocytopathies, minimal change disease, focal segmental glomerulosclerosis, diffuse mesangial sclerosis, collapsing glomerulopathy, minimal change, parietal epithelial cells, renal progenitor
Citation: Ravaglia F, Melica ME, Angelotti ML, De Chiara L, Romagnani P and Lasagni L (2022) The Pathology Lesion Patterns of Podocytopathies: How and why?. Front. Cell Dev. Biol. 10:838272. doi: 10.3389/fcell.2022.838272
Received: 17 December 2021; Accepted: 07 February 2022;
Published: 24 February 2022.
Edited by:
Mario Ollero, INSERM U955 Institut Mondor de Recherche Biomédicale (IMRB), FranceReviewed by:
Aihua Zhang, Nanjing Children’s Hospital, ChinaCopyright © 2022 Ravaglia, Melica, Angelotti, De Chiara, Romagnani and Lasagni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fiammetta Ravaglia, cmF2YWdsaWEuZmlhbW1ldHRhQGdtYWlsLmNvbQ==; Laura Lasagni, bGF1cmEubGFzYWduaUB1bmlmaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.